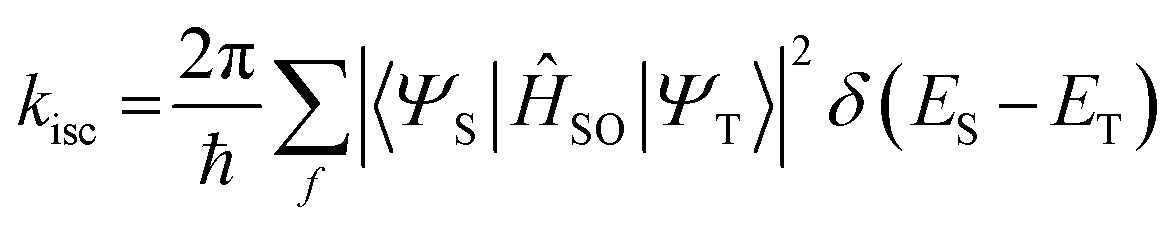DOI:
10.1039/D4TC00491D
(Paper)
J. Mater. Chem. C, 2024,
12, 9760-9772
Photophysical properties and excited-state dynamics of donor–acceptor–heavy-atom molecules and their application in triplet–triplet annihilation upconversion†
Received
5th February 2024
, Accepted 21st May 2024
First published on 11th June 2024
Abstract
In the field of triplet–triplet annihilation upconversion (TTA-UC), the design of an optimized UC system with a high quantum yield at a low excitation power density is a challenging issue. To achieve this goal, it is essential to improve the triplet quantum yields (ΦT) and the UC quantum yields (ΦUC) of the triplet photosensitizers. In this study, we synthesized a novel series of donor–acceptor–heavy-atom (D–A–H) molecules where we have focused on pyrene, BODIPY, and halogen atoms (Cl, Br, and I) as D, A, and H moieties, respectively. The TTA-UC properties of the D–A–H molecules were examined, and their electrochemical properties and excited-state dynamics were investigated. The highest singlet oxygen and UC quantum yields were estimated for PY-BDP-2I (PY = pyrene and BDP = BODIPY) with the values of 0.93 and 4.84%, respectively. In addition, the threshold intensities (Ith) of PY-BDP-2Cl, PY-BDP-2Br, and PY-BDP-2I (112, 27, and 42 mW cm−2, respectively) were lower than that of PY-BDP (406 mW cm−2). In UC quantum yields, the iodine-containing samples (PY-BDP-2I and BEN-BDP-2I) show higher quantum yields than other compounds, which demonstrates that the introduction of iodine into the D–A–H photosensitizer system is an effective strategy for improving the TTA-UC performance.
Introduction
A triplet photosensitizer is a compound that efficiently undergoes intersystem crossing (ISC), representing a generation of a molecule in a triplet excited state with a high quantum yield. Because the singlet and triplet states are strictly separated with a total spin angular momentum, the electronic transition between these two states requires a spin flip and typically shows low efficiencies. In this regard, with a merit on efficient spin-flip, the triplet photosensitizers exhibit high industrial utility and wide applicability in photovoltaics, photocatalytic organic reactions, photoinduced hydrogen production from water, the photoreduction of CO2, photodynamic therapy (PDT), and triplet–triplet annihilation upconversion (TTA-UC).1–8
Among the various applications of triplet photosensitizers, TTA-UC has received constant attention due to its capability for upconversion of photon energy.8 TTA-UC allows the emission of high-energy light from absorption of low-energy light. This conspicuous phenomenon is implemented with underlying energy transfer and subsequent fusion processes, which are activated by a combination of a triplet sensitizer and an annihilator (or called as an emitter). For several decades, metalloporphyrins have been intensively applied to TTA-UC systems.9 Recently, research studies on TTA-UC have paid attention to non-porphyrin-based triplet photosensitizers due to the intrinsic limitations of porphyrin series to practical applications, such as low light absorption ability in visible and NIR regions, a notable toxicity from their coordinated heavy metals, and high synthetic costs.10 Among these non-porphyrin series molecules, there is growing interest in boron-dipyrromethene (BODIPY) dyes with advantages of strong absorption in the visible region, excellent photostability, low toxicity, and versatile derivatization.11–13 Extensive studies have been conducted with BODIPY moieties on the development of triplet photosensitizers in donor–acceptor (D–A) geometry to reveal an effect of photoinduced electron transfer (PET) on ISC. Simultaneously, heavy atom effect (HAE) strategies have also been intensively investigated in this context.14–17 Nevertheless, the D–A sensitizers have typically showed not enough triplet quantum yields for TTA-UC and the heavy atoms in sensitizers have caused too short triplet lifetime to activate TTA-UC even if they facilitate ISC by a large spin–orbit coupling (SOC).
To overcome these issues, we recently suggested a series of photosensitizers in donor–acceptor–heavy atom (D–A–H) geometry that simultaneously implements both HAE and PET strategies and balances their ISC efficiency and triplet lifetime.18 Despite the enhanced ISC resulting from this synergistic effect, there remains potential for further refinement in triplet quantum yields and molecular characteristics related to TTA-UC. To the best of our knowledge, there have been no studies on the photophysical properties and excited-state dynamics of the proposed D–A–H sensitizers and an effect of change of HAE on them. It is, therefore, necessary to scrutinize and validate a complementary effect of PET and HAE depending on the type of heavy atoms in the D–A–H sensitizers.
In this regard, we herein synthesized a series of D–A–H systems where we have focused on pyrene, BODIPY, and halogen atoms (Cl, Br, and I) as D, A, and H moieties, respectively. We evaluated the effect of the heavy atom on the ISC efficiency and UC quantum yields. For a comparative analysis, we further prepared a D–A molecule (PY-BDP) without HAE. In addition, BEN-BDP-2I (BEN = benzene) was also synthesized for the attenuation of the PET effect by the replacement of a pyrene unit into the benzene unit. Subsequently, the electrochemical properties and excited-state dynamics of these series of triplet photosensitizers are investigated. The SOQY (ΦΔ) and triplet lifetime (τT) are measured to evaluate their ISC efficiency, UC quantum yield (ΦUC) and threshold intensity (Ith) in TTA-UC.
Experimental
General information
All commercial reagents and anhydrous solvents were purchased from Sigma-Aldrich, Alfa Aesar, and Tokyo Chemical Industry (TCI) and were used without further purification.
The 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance 500 spectrometer at 500 MHz or a Bruker Avance III HD spectrometer at 850 MHz using chloroform-d as the solvent. Tetramethylsilane (TMS) was used as an internal reference. The mass spectra were recorded using gas chromatography-high resolution mass spectrometry (GC-HRMS; JMS-700, JEOL; 6890 GC series, Agilent). Column chromatography was performed using silica gel 60 (0.040–0.063 mm). Analytical thin-layer chromatography (TLC) was performed on pre-coated silica plates (silica gel 60 F254 aluminum-backed, Merck). The absorption and fluorescence spectra were measured using a UV-vis spectrophotometer (UV-1900i, Shimadzu) and a fluorescence spectrometer (LS-55, PerkinElmer Co.), respectively. The fluorescence quantum yield was evaluated using an integrating sphere (QE-1100, Otsuka Electronics Co.). The time-resolved photoluminescence decay of each photosensitizer was measured using a fluorophotometer (Fluorolog-3, HORIBA Scientific). The delayed fluorescence and UC emission lifetime were obtained using a spectrofluorometer (FS5, Edinburgh).
Synthesis and characterization
The synthetic procedures and characterization details can be found in the ESI.†
Singlet oxygen quantum yield (SOQY)
Singlet oxygen generation occurs when a triplet species reacts with triplet oxygen and is one of the representative methods used to determine the efficiency of ISC. Therefore, to enhance the triplet quantum yield of the photosensitizer, improving the singlet oxygen quantum yield serves as an indispensable factor.19 The singlet oxygen quantum yield (ΦΔ) of each sample was measured using the indirect method in toluene. The SOQY values were calculated using the following equation:| |  | (1) |
where ΦΔ,sam is the SOQY of the sample and ΦΔ,std is the SOQY of the standard (ΦΔ,std = 0.83 in toluene).20 In addition, msam and mstd represent the slope of the absorbance change at 416 nm in the sample and in the standard, respectively, while nsam and nstd are the refractive indices of the sample and standard solutions, respectively. 1,3-Diphenylisobenzofuran (DPBF) was used as a singlet oxygen scavenger. All samples were irradiated with a laser (505 nm, 1.81 W cm−2). The DPBF concentration was fixed at 30 mM to prevent chain reactions. A detailed absorption spectrum of the mixture containing the photosensitizer and DPBF is shown in Fig. S18 (ESI†).
Femtosecond and nanosecond transient absorption spectroscopies
The femtosecond time-resolved transient absorption (fs-TA) spectrometer consisted of an optical detection system and an optical parametric amplifier (OPA; Palitra, Quantronix) pumped by a Ti:sapphire regenerative amplifier system (Integra-C, Quantronix) operating at a repetition rate of 1 kHz. The generated OPA pulses, which were used as pump pulses, had a pulse width of ∼150 fs and an average power of 100 mW in the range of 280–2700 nm. White light continuum (WLC) probe pulses were generated using a sapphire window (4 mm thick) by focusing a small portion of the fundamental 800 nm pulses, which were picked off using a quartz plate before entering the OPA. The time delay between the pump and probe beams was carefully controlled by allowing the pump beam to travel along a variable optical delay (ILS250, Newport). The intensities of the spectrally dispersed WLC probe pulses were monitored using a high-speed spectrometer (Ultrafast Systems) for both the visible and near-infrared measurements. To obtain the time-resolved transient absorption difference signal (ΔA) at a specific time, the pump pulses were chopped at 500 Hz and the intensities of the absorption spectra were saved alternately with or without pump pulses. Typically, 4000 pulses were used to excite the samples and obtain the fs-TA spectra at each delay time. The polarization angle between the pump and probe beams was set to the magic angle (54.7°) using a Glan-Laser polarizer with a half-wave retarder to prevent the generation of polarization-dependent signals. The cross-correlation full-width at half maximum in the pump–probe experiments was <200 fs. For measuring the nanosecond TA (ns-TA) measurements, we utilized a commercial ns-TA spectrometer (EOS, Ultrafast systems) with the same pump source. After the TA experiments, the absorption spectra of all compounds were carefully examined to determine the presence of artifacts caused by any degradation or photo-oxidation of the samples (Z-202308028841 at the Research Support Center for Bio-Bigdata Analysis and Utilization of Biological Resources).
Theoretical calculations
Geometry optimization of the ground and excited states was carried out using Gaussian 16. The structures of the compounds were optimized using the B3LYP hybrid functional and 6-31G+(d,p) basis set with toluene as the solvent (CPCM). The B3LYP/GEN basis set was used for the heavy atoms, including iodine. The natural transition orbital (NTO) and the overlap integral of the transition were analyzed using Multiwfn. The spin–orbit coupling matrix element (SOCME) was calculated using ORCA 5.0.1 software.
Triplet–triplet annihilation upconversion (TTA-UC)
The UC quantum yield (ΦUC) was measured in response to laser excitation at 532 nm. All samples were prepared in a deaerated environment after N2 purging for 15 min. The UCQY values were calculated using the following equation:| |  | (2) |
where A, I, and n represent the absorbance at the excitation wavelength, the integrated photoluminescence intensity, and the refractive index of the medium, respectively. The subscript terms “sam” and “std” denote the sample and the standard, respectively. 2,6-Diiodo-1,3,5,7-tetramethyl-8-phenyl-4,4-difluoroboradiazaindacene was used as the standard (Φstd = 0.043 in toluene).
The UC emission spectra were obtained using custom laser settings. The UC sample was excited using a 532 nm commercial diode laser with a 6 mm-diameter beam at an angle of ∼45°. The emitted light was modulated using an optical chopper (120 Hz) and irradiated using a monochromator (Oriel Cornerstone, Newport, UK) with a series of focusing lenses. The scattered laser light was removed using a 532 nm short-pass filter. The photon signals were detected using an Oriel photomultiplier tube, then converted into electrical signals using a lock-in amplifier (SR810 DSP, Stanford Research Systems) and subjected to analysis. The intensity of the incident laser was adjusted using a continuously variable neutral density filter and measured using a laser power meter (843-R, Newport).
Results and discussion
Molecular structures
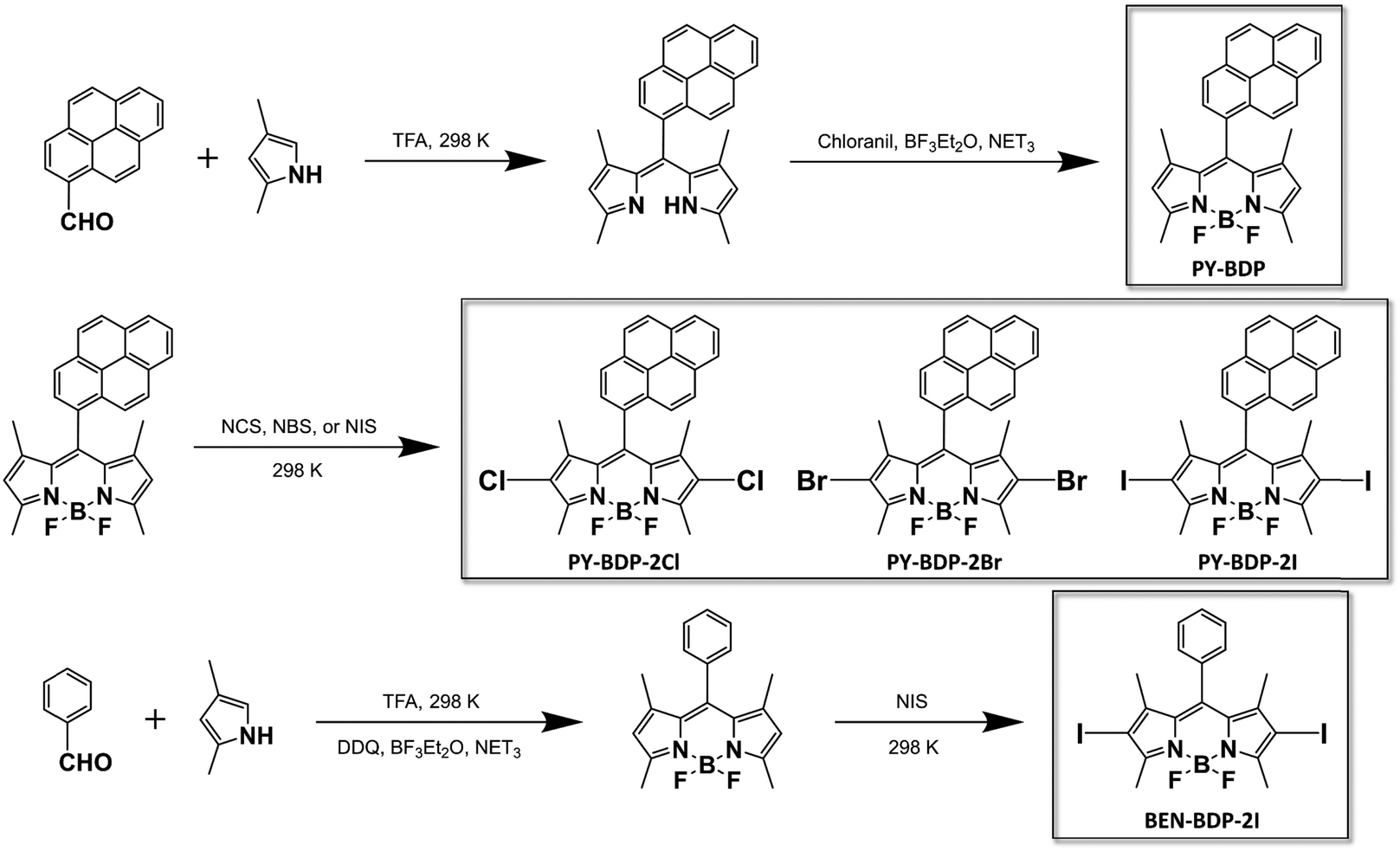
Three D–A–H molecules, namely PY-BDP-2Cl, PY-BDP-2Br, and PY-BDP-2I, were synthesized to evaluate the effect of the heavy atoms (chlorine, bromine, and iodine), where the BODIPY (BDP) and pyrene (PY) moieties were introduced as electron donor (D) and acceptor (A) units. A heavy atom-free D–A molecule (PY-BDP) was synthesized for comparison, along with BEN-BDP-2I, which is designed to minimize PET by the replacement of pyrene to the benzene moiety (Scheme 1). Here, to guarantee an orthogonal geometry between D and A in these molecules, the BODIPY moiety bearing four methyl substituents is directly bonded with the pyrene moiety. This conformation restricts the rotation of the moieties by steric hindrance, which suppresses the non-radiative decay channel of the lowest excited states (S1 and T1 states) and subsequently improves both triplet quantum yields and lifetimes.21,22
 |
| | Scheme 1 Preparation of the PY-BDP, PY-BDP-2Cl, PY-BDP-2Br, PY-BDP-2I, and BEN-BDP-2I molecules. | |
Photophysical properties
The UV-vis absorption and fluorescence emission spectra of the series of compounds were measured, as presented in Fig. 1. Apart from BEN-BDP-2I, all compounds exhibited two distinct absorption ranges corresponding to the pyrene absorption band (300–350 nm) and the BODIPY absorption band (450–550 nm). The photophysical properties of the compounds are listed in Table 1. The compounds possessing iodine showed higher molar absorption coefficients in the visible region, which is a prerequisite for high-performance triplet photosensitizers. Compared with the original PY-BDP structure, the introduction of a halogen atom resulted in a redshift (27–34 nm) in the maximum absorption wavelength due to the heavy atoms acting as electron acceptors that conjugate with the BODIPY core. In addition, it was found that the iodinated BODIPY formed a stronger conjugate than its brominated counterpart, absorbing at longer wavelengths as the HOMO–LUMO energy difference decreased. Upon examining the absorption and emission spectra in different solvents, it was apparent that the peak wavelengths remained relatively constant, and this was attributed to the suppression of the bathochromic shift by the methyl groups (Fig. S17, ESI†). Indeed, it has previously been reported that the incorporation of alkyl substituents, such as methyl groups, into BODIPY dyes leads to characteristics that are independent of the solvent polarity.23–26 These alkyl groups restrict the rotation of the pyrene unit, where the weak dipole moments present within the molecule constrain realignment in an organic solvent to produce minimal solvent effects. These observations indicate that fluorescence emission occurs in a relaxed Franck–Condon excited state rather than as a result of solvent relaxation.27 In addition, the fluorescence quantum yield was found to decrease as the solvent polarity gradually increased. This observation confirmed the occurrence of PET in the molecule, which led to charge separation (Table S1, ESI†). Under high-polarity conditions, non-radiative decay or quenching from the excited singlet state (S1) to the ground state (S0) occurs, and in the case of a structural configuration comprising a donor and an acceptor, charge transfer (CT) takes place to facilitate the transfer of electrons to the triplet state.28,29
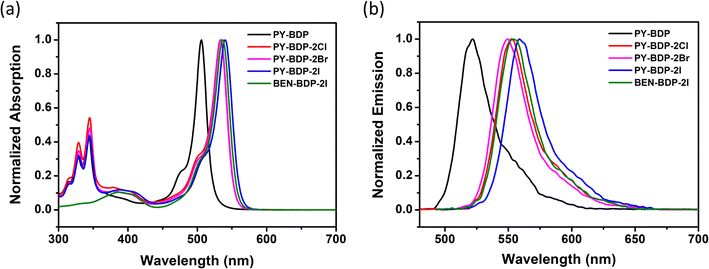 |
| | Fig. 1 (a) Normalized UV-vis absorption spectra, and (b) normalized fluorescence emission spectra of the prepared compounds in toluene (c = 1.0 × 10−5 M, T = 25 °C). | |
Table 1 Photophysical properties of compounds in toluene
| Compound |
λ
abs
(nm) |
ε
max
(M−1 cm−1) |
λ
emi
(nm) |
Φ
F
|
τ
F
(ns) |
Φ
Δ
|
τ
DF
(µs) |
|
Maximum absorption wavelength.
Molar absorption coefficient (1.0 × 10−5 M−1 cm−1).
Maximum fluorescence emission wavelength.
Fluorescence quantum yield.
Fluorescence lifetime.
Singlet oxygen quantum yield.
Delayed fluorescence.
|
|
PY-BDP
|
506 |
42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 610 610 |
522 |
0.766 |
5.476 |
0.05 |
110 |
|
PY-BDP-2Cl
|
533 |
32980 |
552 |
0.723 |
5.253 |
0.21 |
7031 |
|
PY-BDP-2Br
|
534 |
72970 |
549 |
0.109 |
0.939 |
0.76 |
3196 |
|
PY-BDP-2I
|
540 |
85610 |
558 |
0.020 |
0.167 |
0.93 |
940 |
|
BEN-BDP-2I
|
536 |
106900 |
553 |
0.043 |
0.322 |
0.83 |
415 |
Singlet oxygen quantum yield (SOQY)
Because singlet oxygens are produced through an energy transfer of excited species in a triplet state to neighboring abundant triplet ground oxygens, the singlet oxygen quantum yield (SOQY) serves as a crucial indicator for assessing ISC efficiency.30 Thus, to evaluate the ISC efficiency of the triplet photosensitizer, the SOQY of the D–A–H compounds was measured in toluene (Fig. 2), giving SOQY values of 0.05, 0.21, 0.76, 0.93, and 0.83 for the PY-BDP, PY-BDP-2Cl, PY-BDP-2Br, PY-BDP-2I, and BEN-BDP-2I systems, respectively (Table 1). In the case of PY-BDP, its notably low SOQY value can be comprehended with its D-A structure without heavy atoms. On the other hand, PY-BDP-2Cl, PY-BDP-2Br, and PY-BDP-2I showed the enhanced SOQYs compared to PY-BDP. Furthermore, their SOQYs increased gradually upon an increase of atomic number of heavy atoms (chlorine → bromine → iodine). This improvement in the SOQYs well describes that both PET and HAE in the D–A–H structure effectively promote the ISC efficiency. Indeed, the SOQY of PY-BDP-2I shows a remarkably high value of 0.93, which was significantly elevated efficiency among previously reported BODIPY-based triplet photosensitizers.31–34 In contrast, despite the same heavy atom of iodine, BEN-BDP-2I exhibited a reduced SOQY of 0.83 compared to PY-BDP-2I. Given that BEN-BDP-2I is a counterpart of PY-BDP-2I, where the replacement of the pyrene unit into benzene attenuates electron-donating properties of an adjacent BODIPY unit, the lower SOC efficiency of BEN-BDP-2I obviously verifies the synergetic effect of simultaneous PET and HAE on the ISC efficiency. It should be noted here that an increase in the atomic number leads to a corresponding augmentation in the magnitude of the effective nuclear charge. Moreover, the association with the SOC constant was confirmed through calculations of the SOCME, as discussed later.
 |
| | Fig. 2 Time dependence of ln([DPBF]t/[DPBF]0) on the irradiation time in the presence of a photosensitizer and DPBF (505 nm green laser excitation). | |
Delayed fluorescence analysis
To assess the triplet characteristics, we measured the photoluminescence decay of all compounds in deaerated toluene by nitrogen purging (Fig. 3). In this condition, all molecules show exceptionally long-lived photoluminescence in the range of micro- to milli-second, which indicates a delayed fluorescence (P-type delayed fluorescence) of all samples via a TTA mechanism. More specifically, this long-lived delayed fluorescence in an organic compound can be caused by T1 → S1 reverse intersystem crossing (thermally activated delayed fluorescence or E-type delayed fluorescence) or by the 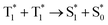 mechanism (TTA delayed fluorescence or P-type delayed fluorescence).35 Generally, achieving E-type delayed fluorescence requires a very small energy gap (ΔEST < 0.1 eV).36 Hence, the observed long-lived photoluminescence corresponds to P-type delayed fluorescence, i.e., TTA delayed fluorescence. It should be noted that TTA delayed fluorescence is not directly linked to the intrinsic triplet lifetime but is indirectly related to the real triplet lifetime, which enables a comparison to be carried out between the relative properties of triplet photosensitizers. As TTA-delayed fluorescence is directly proportional to the square of the triplet population, its magnitude is approximately half of the triplet lifetime.37
mechanism (TTA delayed fluorescence or P-type delayed fluorescence).35 Generally, achieving E-type delayed fluorescence requires a very small energy gap (ΔEST < 0.1 eV).36 Hence, the observed long-lived photoluminescence corresponds to P-type delayed fluorescence, i.e., TTA delayed fluorescence. It should be noted that TTA delayed fluorescence is not directly linked to the intrinsic triplet lifetime but is indirectly related to the real triplet lifetime, which enables a comparison to be carried out between the relative properties of triplet photosensitizers. As TTA-delayed fluorescence is directly proportional to the square of the triplet population, its magnitude is approximately half of the triplet lifetime.37
 |
| | Fig. 3 Triplet–triplet annihilation delayed fluorescence of PY-BDP, PY-BDP-2Cl, PY-BDP-2Br, PY-BDP-2I, and BEN-BDP-2I in deoxygenated toluene was measured at 580 nm (c = 1.0 × 10−5 M, T = 25 °C). | |
Taking a close look at Fig. 3, the time-resolved photoluminescence (TR–PL) curve exhibits the two distinguishable decay components, the initial rapid decay and subsequent slow long-lived decay lasting more than 1 ms. The slow decay component signifies delayed fluorescence. On the other hand, the rapid decay component that was commonly observed for all samples is unfortunately a result of the detection limit of our TR–PL spectrometer (FS5-MCS, Edinburgh instruments, reliable temporal detection range: 5 µs–10 s). The orange-colored decay curve for our instrument response function (IRF) is well matched with the rapid decay components of all samples. Given the ns-TA results of the samples (Fig. 6 and 7) discussed later, it is expected that these IRF signals in the TR–PL curve of all samples are dominated by their fluorescence decay signals. Notably, a significantly longer lifetime was measured for the D–A–H structures (PY-BDP-2Cl, PY-BDP-2Br, and PY-BDP-2I) than for the PY-BDP and BEN-BDP-2I systems. In addition, as mentioned above, PY-BDP-2I was found to exhibit a longer lifetime than BEN-BDP-2I, despite iodine being bound to the BODIPY dye in both structures. However, in the case of the D–A–H structure, a charge-separated state was formed because of the PET process, leading to an increase in the ISC efficiency, and facilitating electron transfer to the triplet excited state. Furthermore, PY-BDP exhibited the shortest measured lifetime compared to the other molecules capable of inducing HAE, and this was attributed to an enhancement of the ISC efficiency through HAE-facilitated SOC. Moreover, for the D–A–H structures examined herein, the lifetime diminished as the size of the heavy atom increased. These results are consistent with the previous observation, where molecules containing heavy atoms undergo rapid decay through a non-radiative pathway of electron transition from T1 to S0.38,39 Therefore, the lifetimes of PY-BDP-2Cl, PY-BDP-2Br, and PY-BDP-2I decreased to 7031, 3196, and 940 µs, respectively. However, despite this decrease, the described D–A–H structures exhibit longer lifetimes compared to the PY-BDP (110 µs) and BEN-BDP-2I (415 µs) systems.
Determination of the spin–orbit coupling matrix element (SOCME)
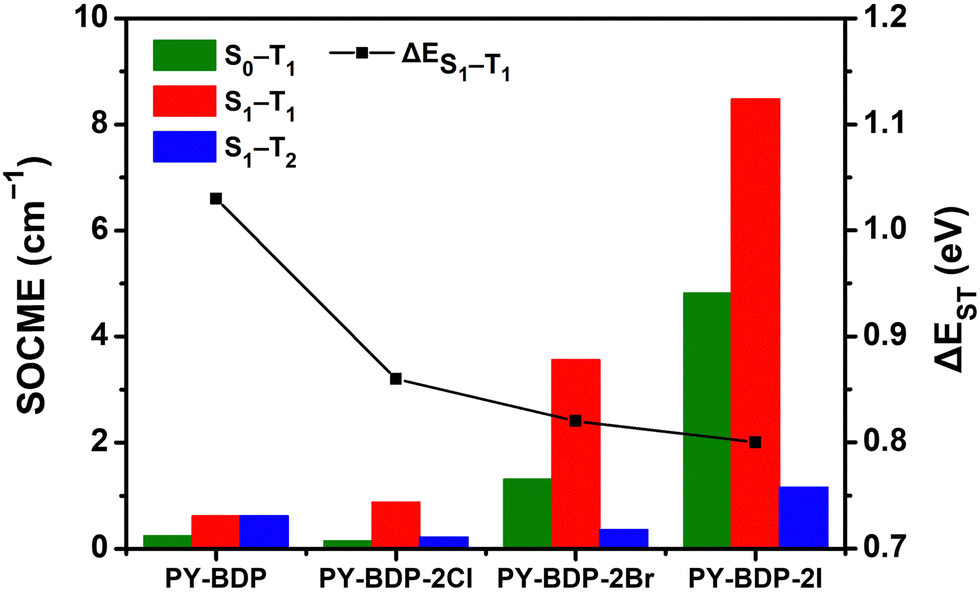
To elucidate the effect of the heavy atom on the SOC within the D–A–H structure, the SOCME value was calculated (Fig. 4). The SOC describes the modification in the electron spin along the up/down axis resulting from the magnetic interaction between the orbital momentum and the spin angular momentum of electrons orbiting around the atom. As the magnitude of the SOC is determined by the probability of an electron encountering a heavy atom, the magnitude of the SOC increases upon increasing the atomic number of the heavy atom.40,41 More specifically, with an increase in the atomic number, the orbitals contract toward the nucleus, reducing the distance from the core electrons and amplifying the significance of the spin–orbit interaction. The magnitude of the SOC can be computed using the SOCME, providing crucial information regarding the spin–orbit charge-transfer-induced intersystem crossing (SOCT-ISC) and influencing the efficiency of the SOQY. The SOCME values for the S1 ↔ T1 transitions of the PY-BDP, PY-BDP-2Cl, PY-BDP-2Br, and PY-BDP-2I structures were therefore determined to be 0.62, 0.88, 3.56, and 8.48 cm−1, respectively, which clearly indicate an increase upon increasing the atomic number. In contrast, ΔES1−T1 gradually decreased to 1.03, 0.86, 0.82, and 0.80 eV, respectively (Table 2).
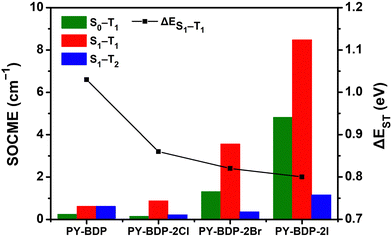 |
| | Fig. 4 Calculation of the magnitude of the spin–orbit coupling matrix element (SOCME) between Sn and Tm based on the type of heavy atom and the energy gap between the S1 and T1 states. | |
Table 2 Spin–orbit coupling matrix element (SOCME) values and singlet–triplet energy gaps calculated using ORCA 5.0.1 software
| Compound |
SOCME (cm−1) |
ΔESn–Tm (eV) |
| |〈ΨS0|ĤSO|ΨT1〉| |
|〈ΨS1|ĤSO|ΨT1〉| |
|〈ΨS1|ĤSO|ΨT2〉| |
ΔES0–T1 |
ΔES1–T1 |
|
PY-BDP
|
0.25 |
0.62 |
0.61 |
1.53 |
1.03 |
|
PY-BDP-2Cl
|
0.14 |
0.88 |
0.22 |
1.50 |
0.86 |
|
PY-BDP-2Br
|
1.31 |
3.56 |
0.36 |
1.51 |
0.82 |
|
PY-BDP-2I
|
4.82 |
8.48 |
1.16 |
1.52 |
0.80 |
The rate constant of ISC, according to Fermi's golden rule, can be expressed using the following simplified equation (eqn (3)):
| |  | (3) |
where |〈
ΨS|
ĤSO|
ΨT〉| denotes the SOC value between the singlet state (
ΨS) and the triplet state (
ΨT), and
ĤSO represents the spin–orbit Hamiltonian. Due to the increasing nuclear charge associated with larger atomic numbers, heavy atoms enhance the magnitude of the SOC. This necessitates an increase in the electron speed to prevent their collapse, and so implies an increase in the orbital angular momentum of the electrons, subsequently leading to an increase in
ĤSO. In other words, with the augmentation of the SOC, the ISC efficiency also increases, corresponding to the trend in the SOQY described above. Upon comparison of the SOCME values between the S
1 ↔ T
1 and S
1 ↔ T
2 transitions, the higher SOCME value obtained for S
1 ↔ T
1 indicates that SOCT-ISC will predominantly occur for this transition. Moreover, as molecules containing heavy atoms undergo non-radiative decay from T
1 to S
0, the triplet lifetime tends to decrease as the size of the heavy atom increases. This, in turn, can be attributed to the larger SOCME for the S
0 ↔ T
1 transition upon moving from chlorine to bromine and iodine (
i.e., 0.14, 1.31, and 4.82 cm
−1, respectively).
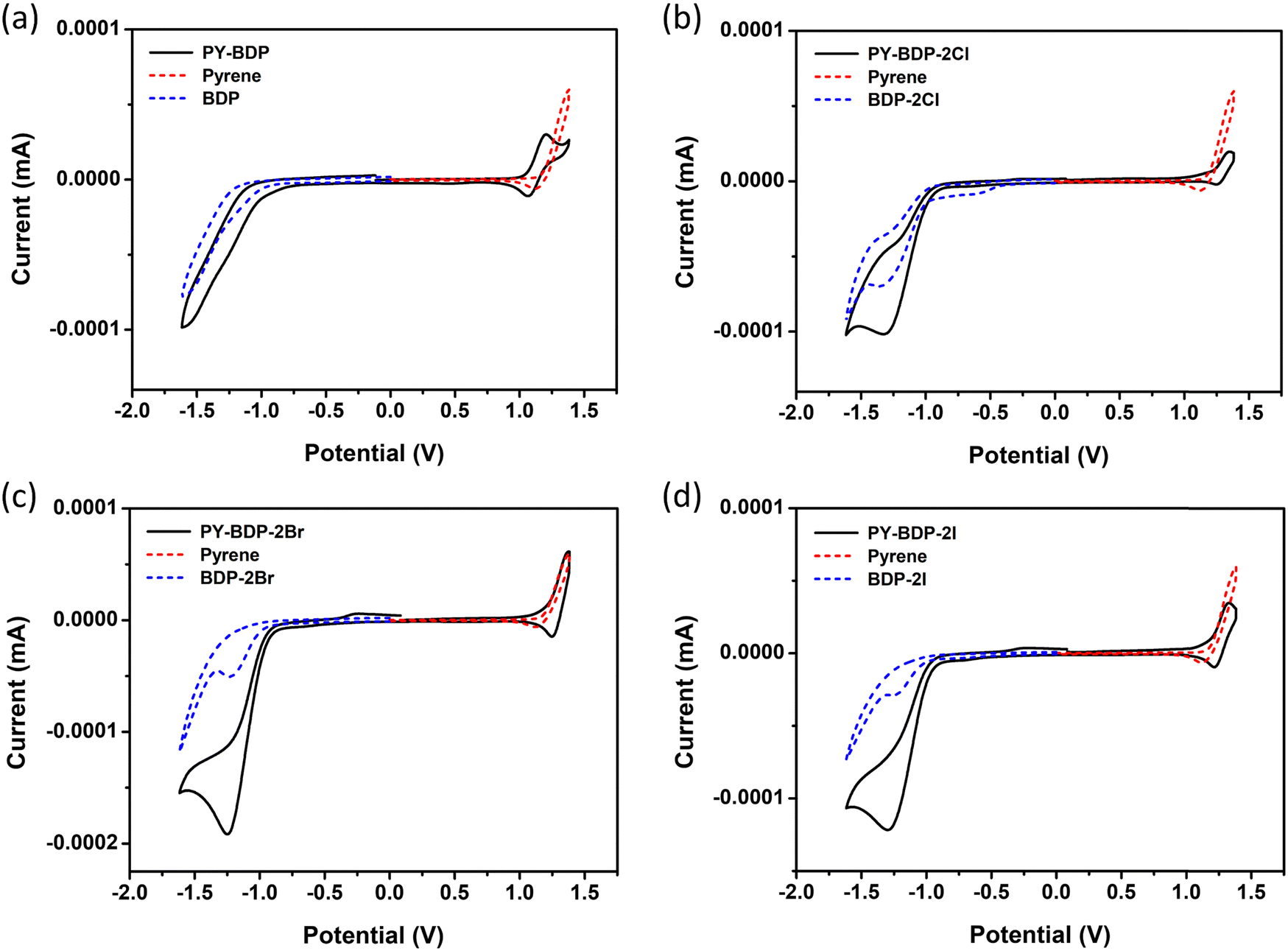
Electrochemical properties
Cyclic measurements were conducted to determine the electrochemical properties of the molecules (Table 3 and Fig. 5). More specifically, to compare the oxidation/reduction potentials of the photosensitizers, additional BODIPY moieties, including BDP, BDP-2Cl, BDP-2Br, and BDP-2I, were synthesized. It was deduced that the oxidation potential (EOX) of PY-BDP-2I is 1.18 V, while its reduction potential (ERED) is −0.98 V, closely matching the oxidation potential of pyrene at 1.19 V and reduction potential of BDP-2I at −1.01 V. These results obviously illustrate that the pyrene and BODIPY moieties dominantly work as an electron donor and acceptor, respectively, in the D–A–H structure. Similar findings were observed for the other compounds (Table S2, ESI†).
Table 3 Oxidation (EOX) and reduction (ERED) potentials, driving forces of charge separation (ΔGCS), and energy levels of the charge separated states (ECS) for the prepared compounds in n-hexane (HEX), toluene (TOL), dichloromethane (DCM), and acetonitrile (ACN)
| Compound |
E
OX (V) |
E
RED (V) |
ΔGCS (eV) |
E
CS (eV) |
| HEX |
TOL |
DCM |
ACN |
HEX |
TOL |
DCM |
ACN |
|
PY-BDP
|
1.09 |
−1.00 |
−0.40 |
−0.49 |
−0.73 |
−0.80 |
2.15 |
2.06 |
1.82 |
1.75 |
|
PY-BDP-2Cl
|
1.20 |
−1.00 |
−0.20 |
−0.25 |
−0.39 |
−0.43 |
2.11 |
2.06 |
1.92 |
1.88 |
|
PY-BDP-2Br
|
1.20 |
−0.97 |
−0.26 |
−0.30 |
−0.43 |
−0.47 |
2.06 |
2.02 |
1.89 |
1.85 |
|
PY-BDP-2I
|
1.18 |
−0.98 |
−0.31 |
−0.35 |
−0.46 |
−0.49 |
2.03 |
1.99 |
1.88 |
1.85 |
 |
| | Fig. 5 Cyclic voltammograms of (a) PY-BDP, (b) PY-BDP-2Cl, (c) PY-BDP-2Br, and (d) PY-BDP-2I in dichloromethane containing 0.10 M Bu4NPF6 as the supporting electrolyte and Ag/AgCl as the reference electrode. Ferrocene (Fc) was used as the external reference (E1/2 = +0.38 V, Fc+/Fc) (scan rate = 100 mV s−1, c = 1.0 × 10−3 M, T = 25 °C). | |
The magnitude of the energy transfer between the donor and acceptor can be calculated using the Rehm–Weller equation using the oxidation/reduction potential measured above.
| | | ΔGCS = e[EOX − ERED] − E00 + ΔGS | (4) |
| |  | (5) |
| | | ΔECS = e[EOX − ERED] + ΔGS | (6) |
In eqn (4)–(6), ΔGCS is the Gibbs free energy of charge separation, ΔGS is the static coulombic energy, and ECS is the energy level of the charge-separated states, where e is the elementary charge, EOX is the first oxidation of the electron donor unit, and ERED is the first reduction of the electron acceptor unit. In addition, E00 is the energy of the lowest excited state, RCC is the center-to-center separation distance between the donor (pyrene) and the acceptor (BODIPY moiety) determined using density functional theory (DFT) calculations, whereas RD and RA are the radii of the electron donor and acceptor, respectively, εS is the static dielectric constant of the solvent, ε0 is the vacuum permittivity, and εREF is the static dielectric constant of the dichloromethane solvent used in the electrochemical experiments. As indicated in Table 3, ΔGCS is negative in all solvents and increases as the solution polarity increases; in contrast, ECS gradually decreases. Although this solvent-dependent phenomenon does not alter the energy level in the locally excited (LE) state, the energy level changes in the CT state owing to its significant dependence on the dipole–dipole interactions among molecules. Consequently, the energy level gap between the LE state and the CT state gradually widens, which causes the PET magnitude to increase.42,43 In toluene, ΔGCS increased to −0.25, −0.30, and −0.35 for PY-BDP-2Cl, PY-BDP-2Br, and PY-BDP-2I, respectively. These results indicate that the halogen atoms serve as electron-withdrawing groups although the PET dominantly occurs between the pyrene and BODIPY moieties. In this regard, it can be suggested that the large halogen atoms, such as iodine, are beneficial to a realization of efficient D–A–H triplet photosensitizers due to their dual functionality to promote PET and HAE simultaneously.
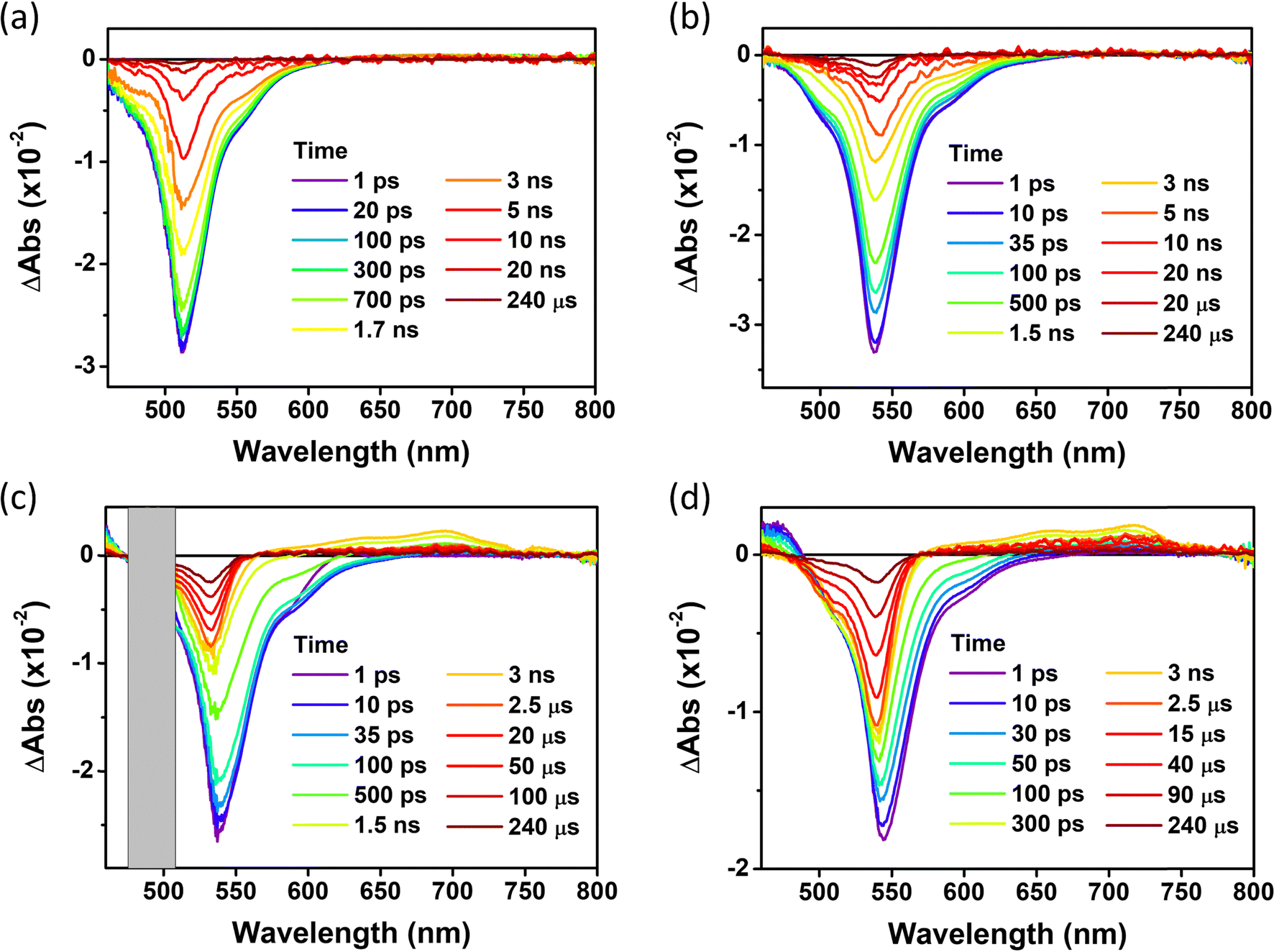
Excited-state dynamics
For the in-depth analysis of the effect of PET and HAE, we measured fs- and ns-transient absorption (TA) spectra of these series of compounds up to 240 µs under the Ar-bubbled condition (Fig. 6, 7, and Fig. S20, ESI†). In their TA results, all compounds show the long-lived negative signal, corresponding to ground state bleaching (GSB),44 over 240 µs, which clearly demonstrates their ISC to the T1 state. Their TA 2D maps displayed that the long-lived GSB signals became gradually grown from PY-BDP to PY-BDP-2Cl to PY-BDP-2Br to PY-BDP-2I, which apparently describes an enhancement of SOC upon an increase of atomic number of halogen atoms. This intensified SOC was quantitatively reflected in the TA decay profiles of PY-BDP-2Cl, PY-BDP-2Br, and PY-BDP-2I, where their ISC process was temporally estimated with time constants of 4.8 ns, 1 ns, and 70 ps, respectively. For BEN-BDP-2I, it also showed very efficient ISC due to its iodine substituents. It is noteworthy that the TA decay with a time constant of tens of µs was commonly observed in all compounds. Because molecular diffusion and Brownian motion can affect the excited-state dynamics longer than µs time scale, the commonly observed decay can be attributed to the collision of triplet molecules.45,46
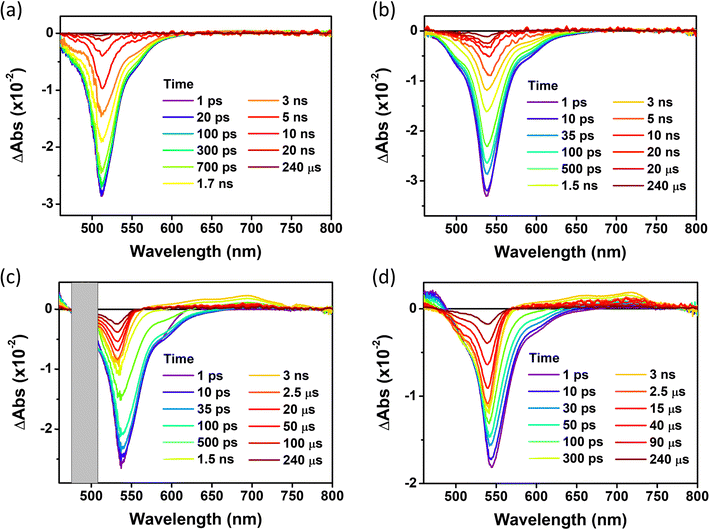 |
| | Fig. 6 fs-to-µs TA spectra for (a) PY-BDP, (b) PY-BDP-2Cl, (c) PY-BDP-2Br, and (d) PY-BDP-2I in toluene at different delay times (0–240 µs) (λex = 500 nm, T = 25 °C). | |
 |
| | Fig. 7 Fs-to-µs TA 2D map of (a) PY-BDP, (b) PY-BDP-2Cl, (c) PY-BDP-2Br, and (d) PY-BDP-2I in toluene under photoexcitation at 500 nm. TA decay profiles of (e) PY-BDP, (f) PY-BDP-2Cl, (g) PY-BDP-2Br, and (h) PY-BDP-2I at 505, 535, 535, and 540 nm, respectively. Because our measured time window, 240 µs, is not enough to measure the longest decay time constant, we estimated the other time constants with the last infinite time constant. | |
In a comparative analysis of TA data of PY-BDP-2Cl, PY-BDP-2Br, and PY-BDP-2I, they showed the fast TA decay with a time constant of 70–80 ps. The similar fast decay feature was observed in the TA results of PY-BDP. Thus, the common rapid decay of TA spectra clearly illustrates the PET behavior between the BODPY and pyrene moieties. Here, for PY-BDP-2I, its large HAE from the iodine substituents leads to highly efficient ISC which takes place as fast as its PET process. Notably, the rapid decay in the TA results of BEN-BDP-2I, corresponding to the PET process, was estimated with a time constant of 150 ps, which is almost doubly decelerated compared to that of PY-BDP-2I. Despite the presence of iodine, these significantly slow PET and ISC processes of BEN-BDP-2I obviously elucidate that the BODIPY and pyrene moieties in the D–A–H structure act as the main electron donor and acceptor, respectively. This further suggests that the PET nature accelerates the ISC process under the D–A–H structures by SOCT-ISC.17,42
Density functional theory (DFT) calculations
To understand the photophysical processes taking place in the triplet photosensitizer molecules, the energy levels corresponding to the singlet and triplet states were obtained using DFT calculations, while the energy level of 1CT was determined using electrochemical calculations. As reported previously, molecules in the D–A structure can efficiently generate PET, leading to the formation of a CT state and the induction of ISC from a singlet excited state to a triplet excited state.47 In other words, molecules with CT characteristics can reduce energy loss by narrowing the energy gap to the triplet excited state through electron transfer to the CT state by PET; ultimately, the ISC efficiency can be significantly enhanced. Due to the requirement for a clear investigation, the highest occupied and lowest unoccupied natural transition orbitals (HONTO and LUNTO, respectively) corresponding to each state were calculated using Multiwfn.48 As indicated in Scheme 2, in the PY-BDP-2I photosensitizer, photoexcitation induces the transition of electrons from a S0 state to a Sn state. In the S2 state, both the HONTO and the LUNTO are located on BODIPY and exhibit LE characteristics. PET occurs because of the D–A structure, leading to the generation of 1CT in the S1 state. In this state, the electrons of the HONTO are localized on pyrene, whereas those of LUNTO are distributed on the BODIPY moiety. For quantitative analysis, the overlap integral (Sr) between the HONTO and the LUNTO was introduced, giving values of 0.64 and 0.23 for the S2 and S1 states, respectively. These, in turn, correspond to the LE and CT characteristics, respectively.
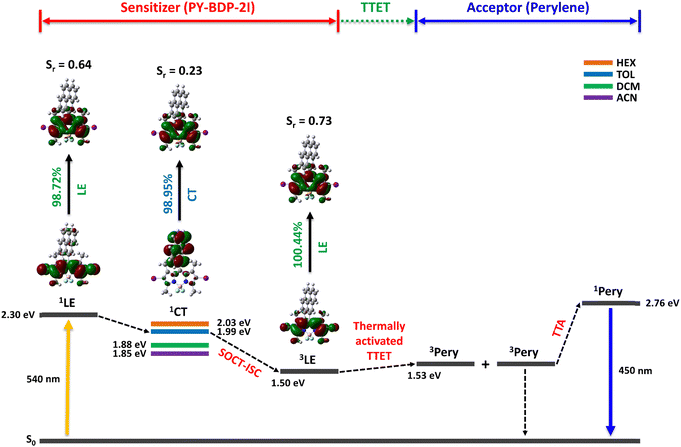 |
| | Scheme 2 Schematic of triplet–triplet annihilation upconversion (TTA-UC) using PY-BDP-2I as the triplet photosensitizer. The energy levels of the excited states were obtained using TD-DFT (LanL2DZ/6-31+G(d,p)) calculations. The orbital overlap between the HONTO and the LUNTO of the excited state was calculated using Multiwfn. The energy levels of the 1CT state were obtained using electrochemical calculations. | |
The electrochemical experiments confirmed that the CT state exhibits solvent-dependent characteristics as it relies significantly on the dipole–dipole interactions between molecules. Unlike general ISC, the ISC induced by PET is mainly caused by SOCT (i.e., SOCT-ISC),42 which involves the 1CT → 3LE transition. By analyzing the photophysical processes associated with the PY-BDP-2I system, it was verified that SOCT-ISC occurred efficiently. A reduction in ΔEST can therefore be achieved by adjusting the solvent polarity to lower the energy level of the 1CT state. Therefore, the optimization of the CT process is crucial for enhancing the SOCT-ISC efficiency.
Triplet–triplet annihilation upconversion (TTA-UC)
One of the major applications of triplet photosensitizers is TTA-UC, a unique photochemical process that generates higher-energy photons through a series of energy-transfer processes, which are outlined in Scheme 2. More specifically, in the first process, the excited singlet state of the sensitizer (1S*) is generated upon the absorption of the light source. A non-radiative transition then occurs from the excited singlet state to the triplet state (3S*) (ISC), and the excited triplet state of the acceptor (3A*) is produced by triplet–triplet energy transfer (TTET). Subsequently, TTA between two or more excited triplet states of the acceptor generates an excited singlet state (1A*) and emits UC emission. During these processes, appropriate energy levels are required for the sensitizer and the acceptor (1A* > 1S* > 3S* > 3A* and 2 × 3A* > 1A*). Thus, to apply the developed photosensitizers to the TTA-UC system, a perylene acceptor was selected, which is a representative acceptor for green-to-blue UC owing to its high fluorescence quantum yield.49,50 According to the calculated energy levels, the triplet energies of the developed photosensitizers are lower than those of perylene. The UC emission spectra of the various specimens were then recorded under 532 nm laser irradiation owing to the occurrence of thermally activated TTET.51 As mentioned earlier, ISC efficiency increases when heavy atoms are introduced, and this generates excited triplet states in perylene to enhance UC emission via TTA. The importance of ISC in the TTA-UC system can also be seen via the analysis of the acceptor triplet lifetimes (τT), and so the corresponding τT values were obtained by tail-fitting of the UC emission decays at 470 nm according to the relationship: exp(−t/τUC) = exp(−2t/τT), wherein τUC is the UC emission lifetime (Fig. S23, ESI†).52–54 With the exception of the PY-BDP-2Cl system, the calculated results tended to match the UC emission intensities. Although the long triplet lifetime of PY-BDP-2Cl provided a long triplet lifetime for the acceptor, sufficient TTA-UC was not achieved because the number of generated acceptor triplets was small because of its low ISC. Successful green-to-blue UC was also confirmed by the naked eye using a 500 nm short-pass filter (Fig. 8(b)). For the PY-BDP-2I and BEN-BDP-2I systems, the observed filter-free emission was white because of the intense UC emission (blue), the excitation source (green), and the fluorescence (yellow). In contrast, the remainder of the systems gave green or yellow emission attributed to their weak UC emissions. Thus, to evaluate the UC performances of the prepared systems in greater detail, the UC quantum yields were calculated using a relative method, and the observed trend (see Fig. 8(c)) is consistent with the UC emission spectra presented in Fig. 8(a). A maximum UC quantum yield of 4.84% was obtained for PY-BDP-2I (Table 4), which represents an improvement of >10% compared to our previously reported PY-BDP-2Br system (i.e., 4.4%).55 In addition, when compared to the state-of-the-art UC systems using other sensitizers containing heavy metals, our findings want for nothing. In similar conditions to our study (green-to-blue UC with perylene and toluene as the acceptor and solvent, respectively), a selenium-containing BODIPY sensitizer exhibited a UC quantum yield of 6%.16 In the case of PtOEP, it showed a UC quantum yield of 4.24%.56 These results demonstrate that our photosensitizer has excellent performance even without heavy metals. The advantage of the D–A–H photosensitizer was also confirmed by analyzing the power dependence of the UC emission intensity (Fig. 8(d)). In typical TTA-UC systems, an intensity of UC emission shows a gradual transition from quadratic to linear dependence in a double logarithm plot of the UC emission intensity versus the laser power density.57,58 This characteristic is caused by the change in the dominant mechanism for the depopulation of the excited triplet state of the acceptor (from pseudo first-order decay to TTA) with an increasing excitation power. Therefore, the quadratic-to-linear crossover point (i.e., the threshold intensity, Ith) indicates the power density at which sufficient TTA-UC occurs. Compared with the normal D–A photosensitizer (PY-BDP), the D–A–H photosensitizers (PY-BDP-2Cl, PY-BDP-2Br, and PY-BDP-2I) exhibited Ith values that were up to 15 times lower. It is worth noting that although the triplet lifetime of the photosensitizer decreased owing to the introduction of heavy atoms, the results of D–A–H photosensitizers exhibited superior UC performances due to their enhanced ISC.
 |
| | Fig. 8 (a) TTA-UC spectra of the triplet photosensitizers (10 µM) and perylene (56 µM) in deoxygenated toluene (532 nm green laser irradiation). (b) Photographic images of the UC processes for the various compounds with (right) and without (left) the use of a 500 nm short-pass filter. (c) Upconversion quantum yields and (d) upconversion intensities (log scale) as a function of incident laser power density (mW cm−2). The solid lines indicate the fitting results with the slopes of 2.0 and 1.0 in the low and high-power regions, respectively. | |
Table 4 TTA-UC properties of the prepared compounds in toluene
| Compound |
τ
UC (µs) |
Φ
UC (%) |
I
th (mW cm−2) |
|
PY-BDP
|
347 |
0.03 |
406 |
|
PY-BDP-2Cl
|
1489 |
0.93 |
112 |
|
PY-BDP-2Br
|
907 |
2.82 |
27 |
|
PY-BDP-2I
|
1500 |
4.84 |
42 |
|
BEN-BDP-2I
|
1115 |
3.21 |
55 |
Conclusions
To develop an optimized upconversion (UC) system, a molecule constructed from a donor–acceptor–heavy-atom (D–A–H) structure was designed to simultaneously incorporate both the photoinduced electron transfer (PET) and heavy atom effect (HAE) strategies. Following the preparation of the designed D–A–H molecules (PY-BDP-2Cl, PY-BDP-2Br, and PY-BDP-2I; PY, pyrene; BDP, BODIPY) containing different heavy atoms (i.e., chlorine, bromine, and iodine), the properties of the resulting systems were evaluated. It was found that the enlargement of the heavy atom in the D–A–H molecule resulted in a gradual improvement in both the triplet and triplet–triplet annihilation upconversion (TTA-UC) properties, signifying a highly meaningful outcome. When iodine was employed as the heavy atom to yield PY-BDP-2I, the singlet oxygen quantum yield (SOQY) was 0.93, while the upconversion quantum yield (UCQY) reached 4.84%. These data represent significantly higher values than the previously reported ones from BODIPY-based triplet photosensitizers without the D–A–H structure, which further suggests that the simultaneous PET and HAE leads to synergetic SOCT-ISC in the D–A–H structure of photosensitizers. Moreover, density functional theory (DFT) calculations and nanosecond transient absorption measurements were employed to elucidate the photophysical processes and behaviors of the excited states. These findings suggest that the introduction of iodine into the D–A–H structure evaluated herein is an effective strategy for improving the TTA-UC performance. Overall, this study provides a framework for the development of excellent novel triplet photosensitizers by enabling efficient ISC in molecules, thereby highlighting the significance of this newly proposed method.
Author contributions
J. H. Yoon and J. M. Park wrote the manuscript and performed the experiments. J. M. Lee, H. M. Kim, W. J. Choi, and H. K. Lee analyzed the data and proposed the research direction. S. Kim, W. S. Kim, M. S. Kim, Y. S. Kim, D. J. Lee, and Y. Noh conceived and planned the experiments. J. Oh, J. H. Kim, and J. P. Kim designed the research, supervised the project, and revised the manuscript. All authors read and approved the final manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean Government (MSIT) (2021R1F1A1063685, 2021R1A6A1A03039503, and 2021M3H4A4079509). This work was also supported by the ‘regional innovation mega project’ program through the Korea Innovation Foundation funded by the Ministry of Science and ICT (2023-DD-UP-0007). This work was supported by the 2023 BK21 FOUR Graduate School Innovation Support, funded by Pusan National University (PNU-Fellowship program).
References
- L. Huang, J. Zhao, S. Guo, C. Zhang and J. Ma, J. Org. Chem., 2013, 78, 5627–5637 CrossRef CAS PubMed
 .
.
- S. Dartar, M. Ucuncu, E. Karakus, Y. Hou, J. Zhao and M. Emrullahoglu, Chem. Commun., 2021, 57, 6039–6042 RSC .
- E. Bassan, A. Gualandi, P. G. Cozzi and P. Ceroni, Chem. Sci., 2021, 12, 6607–6628 RSC .
- H. Lee, M.-S. Lee, M. Uji, N. Harada, J.-M. Park, J. Lee, S. E. Seo, C. S. Park, J. Kim and S. J. Park, ACS Appl. Mater. Interfaces, 2022, 14, 4132–4143 CrossRef CAS PubMed .
- C.-F. Leung and T.-C. Lau, Energy Fuels, 2021, 35, 18888–18899 CrossRef CAS .
- S. Fukuzumi, T. Kobayashi and T. Suenobu, Angew. Chem., Int. Ed., 2011, 50, 728–731 CrossRef CAS PubMed .
- F. R. Dai, H. M. Zhan, Q. Liu, Y. Y. Fu, J. H. Li, Q. W. Wang, Z. Xie, L. Wang, F. Yan and W. Y. Wong, Chem. – Eur. J., 2012, 18, 1502–1511 CrossRef CAS PubMed .
- H.-L. Lee, J. H. Park, H.-S. Choe, M.-S. Lee, J.-M. Park, N. Harada, Y. Sasaki, N. Yanai, N. Kimizuka and J. Zhu, ACS Appl. Mater. Interfaces, 2019, 11, 26571–26580 CrossRef CAS PubMed .
- M. Imran, M. Ramzan, A. K. Qureshi, M. A. Khan and M. Tariq, Biosensors, 2018, 8, 95 CrossRef CAS PubMed .
- A. B. Ormond and H. S. Freeman, Materials, 2013, 6, 817–840 CrossRef CAS PubMed .
- K. Chen, W. Yang, Z. Wang, A. Iagatti, L. Bussotti, P. Foggi, W. Ji, J. Zhao and M. Di Donato, J. Phys. Chem. A, 2017, 121, 7550–7564 CrossRef CAS PubMed .
- Y. Wei, M. Zhou, Q. Zhou, X. Zhou, S. Liu, S. Zhang and B. Zhang, Phys. Chem. Chem. Phys., 2017, 19, 22049–22060 RSC .
- H. Liang, S. Sun, M. Zafar, Z. Yuan, Y. Dong, S. Ji, Y. Huo and J. Zhao, Dyes Pigm., 2020, 173, 108003 CrossRef CAS .
- J. Zou, Z. Yin, K. Ding, Q. Tang, J. Li, W. Si, J. Shao, Q. Zhang, W. Huang and X. Dong, ACS Appl. Mater. Interfaces, 2017, 9, 32475–32481 CrossRef CAS PubMed .
- Y. Hou, I. Kurganskii, A. Elmali, H. Zhang, Y. Gao, L. Lv, J. Zhao, A. Karatay, L. Luo and M. Fedin, J. Chem. Phys., 2020, 152, 114701 CrossRef CAS PubMed .
- M. Nakashima, K. Iizuka, M. Karasawa, K. Ishii and Y. Kubo, J. Mater. Chem. C, 2018, 6, 6208–6215 RSC .
- Z. Wang, M. Ivanov, Y. Gao, L. Bussotti, P. Foggi, H. Zhang, N. Russo, B. Dick, J. Zhao and M. Di Donato, Chem. – Eur. J., 2020, 26, 1091–1102 CrossRef CAS PubMed .
- J. M. Lee, J. M. Park, H. K. Lee, H. M. Kim, J. H. Kim and J. P. Kim, Dyes Pigm., 2021, 196, 109662 CrossRef CAS .
- Y. Hou, Q. Liu and J. Zhao, Chem. Commun., 2020, 56, 1721–1724 RSC .
- Y. Li, Y. Wei and X. Zhou, J. Photochem. Photobiol., A, 2020, 400, 112713 CrossRef CAS .
- N. Gupta, S. I. Reja, V. Bhalla, M. Gupta, G. Kaur and M. Kumar, J. Mater. Chem. B, 2016, 4, 1968–1977 RSC .
- Y. Dong, A. Elmali, J. Zhao, B. Dick and A. Karatay, ChemPhysChem, 2020, 21, 1388–1401 CrossRef CAS PubMed .
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, S. Callaghan, K. J. Flanagan, T. Wiesner, F. Laquai and M. O. Senge, ChemPhotoChem, 2018, 2, 606–615 CrossRef CAS .
- N. Kiseleva, M. A. Filatov, J. C. Fischer, M. Kaiser, M. Jakoby, D. Busko, I. A. Howard, B. S. Richards and A. Turshatov, Phys. Chem. Chem. Phys., 2022, 24, 3568–3578 RSC .
- S. Mukherjee and P. Thilagar, RSC Adv., 2015, 5, 2706–2714 RSC .
- Y. Yang, L. Zhang, C. Gao, L. Xu, S. Bai and X. Liu, RSC Adv., 2014, 4, 38119–38123 RSC .
- D. J. Stewart, M. J. Dalton, S. L. Long, R. Kannan, Z. Yu, T. M. Cooper, J. E. Haley and L.-S. Tan, Phys. Chem. Chem. Phys., 2016, 18, 5587–5596 RSC .
- Y. Lei, K. Chen, G. Tang, J. Zhao and G. G. Gurzadyan, J. Photochem. Photobiol., A, 2020, 398, 112573 CrossRef CAS .
- Z. Wang and J. Zhao, Org. Lett., 2017, 19, 4492–4495 CrossRef CAS PubMed .
- C. Schweitzer and R. Schmidt, Chem. Rev., 2003, 103, 1685–1758 CrossRef CAS PubMed .
- W. Hu, X.-F. Zhang, X. Lu, S. Lan, D. Tian, T. Li, L. Wang, S. Zhao, M. Feng and J. Zhang, Dyes Pigm., 2018, 149, 306–314 CrossRef CAS .
- A. A. Buglak, A. Charisiadis, A. Sheehan, C. J. Kingsbury, M. O. Senge and M. A. Filatov, Chem. – Eur. J., 2021, 27, 9934–9947 CrossRef CAS PubMed .
- R. C. Gonçalves, J. Pina, S. P. Costa and M. M. M. Raposo, Dyes Pigm., 2021, 196, 109784 CrossRef .
- Y. Dong, M. Taddei, S. Doria, L. Bussotti, J. Zhao, G. Mazzone and M. Di Donato, J. Phys. Chem. B, 2021, 125, 4779–4793 CrossRef CAS PubMed .
- P. Data and Y. Takeda, Chem. – Asian J., 2019, 14, 1613–1636 CrossRef CAS PubMed .
- H. Sun, Z. Hu, C. Zhong, X. Chen, Z. Sun and J.-L. Brédas, J. Phys. Chem. Lett., 2017, 8, 2393–2398 CrossRef CAS PubMed .
- Z. Wang, A. A. Sukhanov, A. Toffoletti, F. Sadiq, J. Zhao, A. Barbon, V. K. Voronkova and B. Dick, J. Phys. Chem. C, 2018, 123, 265–274 CrossRef .
- Z. Wang, A. Toffoletti, Y. Hou, J. Zhao, A. Barbon and B. Dick, Chem. Sci., 2021, 12, 2829–2840 RSC .
- X.-F. Zhang and X. Yang, J. Phys. Chem. B, 2013, 117, 5533–5539 CrossRef CAS PubMed .
-
N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern molecular photochemistry of organic molecules, University Science Books Sausalito, CA, 2010 Search PubMed .
- K. Shanavas, Z. S. Popović and S. Satpathy, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 165108 CrossRef .
- M. A. Filatov, Org. Biomol. Chem., 2020, 18, 10–27 RSC .
- H. Heitele, P. Finckh, S. Weeren, F. Pöllinger and M. Michel-Beyerle, J. Phys. Chem., 1989, 93, 5173–5179 CrossRef CAS .
- J. Kim, J. Oh, A. Osuka and D. Kim, Chem. Soc. Rev., 2022, 51, 268–292 RSC .
- E. Lee, X. Li, J. Oh, N. Kwon, G. Kim, D. Kim and J. Yoon, Chem. Sci., 2020, 11, 5735–5739 RSC .
- J. Baier, T. Maisch, M. Maier, M. Landthaler and W. Bäumler, J. Invest. Dermatol., 2007, 127, 1498–1506 CrossRef CAS PubMed .
- D. J. Gibbons, A. Farawar, P. Mazzella, S. Leroy-Lhez and R. M. Williams, Photochem. Photobiol. Sci., 2020, 19, 136–158 CrossRef CAS PubMed .
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed .
- J.-M. Park, H. Lee, H.-S. Choe, S.-K. Ahn, K.-Y. Seong, S. Y. Yang and J.-H. Kim, J. Mater. Chem. C, 2022, 10, 4584–4589 RSC .
- X. Cui, A. Charaf-Eddin, J. Wang, B. Le Guennic, J. Zhao and D. Jacquemin, J. Org. Chem., 2014, 79, 2038–2048 CrossRef CAS PubMed .
- L. Li, Y. Zeng, J. Chen, T. Yu, R. Hu, G. Yang and Y. Li, J. Phys. Chem. Lett., 2019, 10, 6239–6245 CrossRef CAS PubMed .
- F. Edhborg, A. Olesund and B. Albinsson, Photochem. Photobiol. Sci., 2022, 21, 1143–1158 CrossRef CAS PubMed .
- N. Yanai, M. Kozue, S. Amemori, R. Kabe, C. Adachi and N. Kimizuka, J. Mater. Chem. C, 2016, 4, 6447–6451 RSC .
- H. Kouno, T. Ogawa, S. Amemori, P. Mahato, N. Yanai and N. Kimizuka, Chem. Sci., 2016, 7, 5224–5229 RSC .
- J. M. Lee, J. M. Park, J. H. Yoon, J. H. Kim and J. P. Kim, ChemPhotoChem, 2023, 7, e202200326 CrossRef CAS .
- S. Liu, H. Liu, Y. Hu, C. Zhao, H. Huang, G. Yu, Z. Li, Z. Liu, Y. Chen and X. Li, Chem. Eng. J., 2023, 452, 139203 CrossRef CAS .
- Y. Y. Cheng, T. Khoury, R. G. Clady, M. J. Tayebjee, N. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, Phys. Chem. Chem. Phys., 2010, 12, 66–71 RSC .
- A. Monguzzi, J. Mezyk, F. Scotognella, R. Tubino and F. Meinardi, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 195112 CrossRef .
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Jeong-Min
Park‡
b,
Jae Moon
Lee
a,
Jeong-Min
Park‡
b,
Jae Moon
Lee
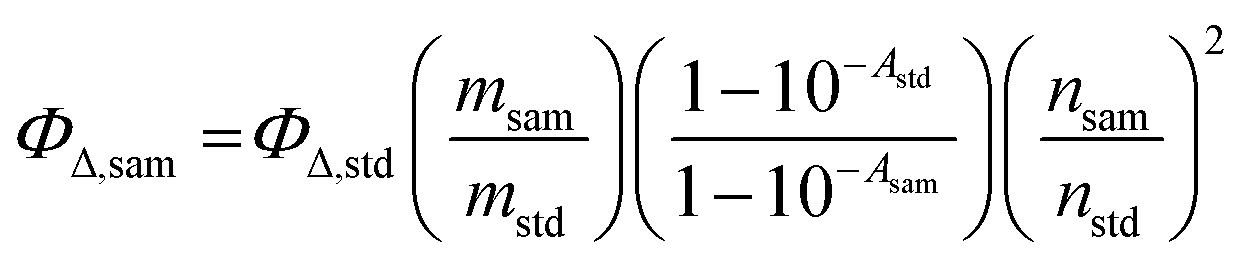



 mechanism (TTA delayed fluorescence or P-type delayed fluorescence).35 Generally, achieving E-type delayed fluorescence requires a very small energy gap (ΔEST < 0.1 eV).36 Hence, the observed long-lived photoluminescence corresponds to P-type delayed fluorescence, i.e., TTA delayed fluorescence. It should be noted that TTA delayed fluorescence is not directly linked to the intrinsic triplet lifetime but is indirectly related to the real triplet lifetime, which enables a comparison to be carried out between the relative properties of triplet photosensitizers. As TTA-delayed fluorescence is directly proportional to the square of the triplet population, its magnitude is approximately half of the triplet lifetime.37
mechanism (TTA delayed fluorescence or P-type delayed fluorescence).35 Generally, achieving E-type delayed fluorescence requires a very small energy gap (ΔEST < 0.1 eV).36 Hence, the observed long-lived photoluminescence corresponds to P-type delayed fluorescence, i.e., TTA delayed fluorescence. It should be noted that TTA delayed fluorescence is not directly linked to the intrinsic triplet lifetime but is indirectly related to the real triplet lifetime, which enables a comparison to be carried out between the relative properties of triplet photosensitizers. As TTA-delayed fluorescence is directly proportional to the square of the triplet population, its magnitude is approximately half of the triplet lifetime.37