
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrophobicity as a tool for programming sequential mesophase transitions of enzyme-responsive polymeric amphiphiles†
Shahar
Tevet
 abc and
Roey J.
Amir
*abcd
abc and
Roey J.
Amir
*abcd
aDepartment of Organic Chemistry, School of Chemistry, Faculty of Exact Sciences, Tel-Aviv University, Tel-Aviv, Israel. E-mail: amirroey@tauex.tau.ac.il
bThe Center for Nanoscience and Nanotechnology, Tel-Aviv University, Tel-Aviv, Israel
cADAMA Center for Novel Delivery Systems in Crop Protection, Tel-Aviv University, Tel-Aviv, Israel
dThe Center for Physics and Chemistry of Living Systems, Tel-Aviv University, Tel-Aviv, Israel
First published on 25th September 2024
Abstract
The ability of polymeric assemblies to undergo programmable cascades of mesophase transitions is prevalent in many systems in nature, where structural and functional features are tightly bound to maximize activity. In this study, we have examined the ability to program the mesophase transition rates of co-assembled enzyme-responsive polymeric micelles, through fine adjustments of the hydrophobicity of their amphiphilic components. We have utilized the different reactivities of di- and tri-block amphiphiles toward enzymatic degradation as a tool for programming formulations to undergo sequential enzymatically induced transitions from micelles to hydrogels and finally to dissolved polymers. By varying the aliphatic end-groups of PEG-dendron di-block and tri-block amphiphiles, we could demonstrate the remarkable impact of minor modifications to the di-block amphiphiles’ structure and hydrophobicity on the transition rates between the different mesophases, ranging from a few hours to a week. Additionally, the study reveals how altering the relative hydrophobicity of its amphiphilic components influences the formulation ratio and enzymatic selectivity, as well as the stability and degradation rate of the resulting hydrogels. The findings underscore the importance of molecular architecture and hydrophobicity as key parameters in the design of programmable enzyme-responsive polymeric assemblies, offering insights into the ability to precisely control multi-step mesophase transitions for tailored functionality.
Introduction
Stimuli-responsive polymeric amphiphiles and their assemblies have gained considerable attention over the past decades due to their great potential in various fields, including biomedical applications such as drug delivery systems, imaging, and theranostics.1–12 These ‘Smart’ assemblies can be designed to react to desired specific stimuli, such as light,13,14 pH,15–17 temperature,18,19 or enzymes.20–23 Reacting to such stimuli generates a change in the physical and chemical properties of the amphiphiles, leading mostly either to the disassembly or aggregation of the altered polymers. The ability to design such systems to respond to various chemical and biological cues underscores their potential to serve as innovative bio-reactive systems. Notably, among the various types of stimuli, the high specificity and overexpression of disease-associated enzymes in diseased tissues make enzymes highly promising stimuli for triggering the selective activation of polymeric assemblies, such as micellar nanocarriers.20–22,24,25 However, while enzymes can play a key role in controlling these systems’ behaviour under biological conditions, it is important to note the unique challenges enzymes face when interacting with the hydrophobic domains of polymeric assemblies. Unlike dimensionless stimuli such as light13,14 and temperature,18,19 or low molecular weight species such as in pH-15–17 and oxidative-responsive systems,26–28 enzymes and other proteins have relatively large dimensions, which are of the same order of the dimensions of polymeric micelles. Hence, enzymes are limited in their ability to penetrate into the polymeric micelles and interact with the hydrophobic substrates, which are hidden inside the hydrophobic core.27 Consequently, enzymes can effectively engage with amphiphilic polymers only in their unimer form, thus making the delicate equilibrium between the micellar and unimer states a key factor in dictating the interaction of enzymes with their substrates.29 This balance, therefore, determines the responsiveness of the assembled system, adding another layer of complexity to the design and application of enzyme-responsive polymeric assemblies.Over the past decade, our group has investigated intensively the factors that govern the reactivity of enzyme-responsive polymeric micellar systems. Using dendritic amphiphiles, composed of a hydrophilic linear polymer and hydrophobic dendron, allowed us to gain the high molecular precision that is needed to explore the effects of small adjustments in the amphiphiles’ structure on the responsiveness of their micelles toward enzymatic degradation. Changes in the hydrophobicity,30–32 molecular weight,33,34 or architecture28,35 of the polymeric amphiphiles, were found to greatly impact the stability and responsiveness of the formed micellar assemblies towards enzymatic degradation. Interestingly, while a major part of the research on enzyme-responsive polymeric assemblies, including ours, has been focused on their degradation and disassembly, enzymatic stimuli can also be used to induce self-assembly or aggregation of polymers.36–49 While using enzymes to trigger either the disassembly or aggregation of polymeric amphiphiles can enable the utilization of such systems for various applications, many assembled structures in nature show a much more complex behaviour, as they are capable of shifting between several different phases. Hence, the ability to program materials to undergo multiple transitions between several mesophases can be extremely valuable for developing next generation materials for advanced applications such as drug delivery systems.50–53
Aiming toward the development of programable materials that can undergo several mesophase transitions, our group has recently developed an enzymatically responsive polymeric system, which can transform from micelles to hydrogels, and then to hydrolyzed hydrophilic polymers in the presence of a single stimulus.54 This polymeric system was based on a crucial understanding – while PEG-dendron di-block amphiphiles (DBA) tend to self-assemble into micelles in aqueous media, PEG-based tri-block amphiphiles (TBA) with identical hydrophilic to hydrophobic ratios have an inherent tendency to form hydrogels in solution.35 However, when mixed together, the two amphiphiles can co-assemble into micelles, which are stabilized by the di-block amphiphile.54 In these co-assembled micelles, despite their identical hydrophilic to hydrophobic ratios, the architectural differences between the di-block and tri-block amphiphiles, together with the different molecular weight, highly affect their micelle–unimer exchange rates. Consequently, the DBA was found to be more susceptible to enzymatic degradation, similarly to other micellar assemblies recently reported by our group.28 Therefore, upon exposure of the co-assembled micelles to the activating enzyme, the DBA was degraded into soluble polymers, which could no longer stabilize the TBA that remained nearly intact. This change in the composition of the micelles resulted in a mesophase transition from mixed micelles to a TBA-based hydrogel, which could then undergo a second mesophase transition into hydrophilic polymers upon its further enzymatic degradation.
In our previous publication,54 we established that different DBA to TBA ratios in the formulation can be applied as a tool for programming the kinetics of the first micelle to hydrogel mesophase transition. Herein, we aim to demonstrate that the DBA hydrophobicity can be utilized as a parallel programming tool to control the timeframe of this first mesophase transition. To achieve this goal, we wished to gradually modify the DBA's hydrophobicity by minor structural adjustments of the hydrophobic dendritic block. Based on previous studies on DBA-only micellar systems,30,31 we assumed that this seemingly minor increase in hydrophobicity would profoundly impact the amphiphiles’ exchange rate and the enzymatic degradation kinetics, subsequently slowing down the mesophase transition into a hydrogel. In addition, we wished to demonstrate the ability to use the modulation of the TBA hydrophobicity to program the enzymatic degradation rate of the hydrogel. We hypothesized that increasing its hydrophobicity would lead to the formation of a more stable hydrogel that will undergo slower enzymatic degradation.
Results and discussion
To carefully evaluate the impact of hydrophobicity on the transition kinetics of these mixed assembled systems, three DBAs were designed and precisely modified to establish different degrees of hydrophobicity (Fig. 1A). The three DBAs were composed of a 5 kDa polyethylene glycol monomethyl ether (mPEG5k) as a hydrophilic block linked to a four-armed hydrophobic dendron. Our synthetic methodology was aimed to be modular and step-efficient, while allowing high molecular control over the degree of hydrophobicity by a gradual tuning of the length of the aliphatic end-groups. The amphiphiles were synthesized in only two high-yielding steps, first by conjugating PEG-amine (mPEG5k–NH2) with an activated para-nitrophenol ester of a di-propargyl branching unit to yield a di-propargyl-functionalized PEG (mPEG5k–di-yne). The latter was subsequently reacted by thiol–yne reaction with three different thiol-containing esters: 2-mercaptoethyl hexanoate, 2-mercaptoethyl heptanoate, and 2-mercaptoethyl octanoate, yielding the final Hex-, Hep- and Oct-DBA amphiphiles, respectively. This minimal structural change of the hydrophobic end-groups allowed us to generate a set of well-defined DBAs with gradually decreasing hydrophilic to hydrophobic ratios. Importantly, the hydrophobic dendrons contain four aliphatic end-groups, which are linked through ester bonds, and can hence serve as hydrophobic substrates for an esterase enzyme.30 Upon enzymatic activation, these hybrids could be expected to degrade into fully soluble PEG-tetraol and the associated fatty acids. Similarly, a TBA amphiphile was synthesized by following the same methodology using a 10 kDa polyethylene glycol di-amine (NH2–PEG10k–NH2) as a central hydrophilic block and divergent growing of two hydrophobic dendrons on both sides (Fig. 1B and Fig. S9, ESI†). As we wished to examine a more hydrophobic TBA compared to the previously reported hexanoate-based system,54 we used octanoate-based end-groups. 1H NMR, size exclusion chromatography (SEC), and high-performance liquid chromatography (HPLC) measurements were used to verify the synthetic conversion, purity, and polydispersity of the amphiphiles, and the experimental results showed excellent correlations with the expected values, as can be seen in the ESI† (Fig. S1–S11) and Table 1. | ||
| Fig. 1 Molecular design and synthesis of enzyme-responsive DBA (A) and TBA (B) amphiphiles. | ||
| Amphiphile | End-group | M n (kDa) | Đ | M n (kDa) | Weight ratioc |
c![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) LogPd LogPd |
|---|---|---|---|---|---|---|
| a Measured by SEC using PEG commercial standards. b Calculated based on commercial PEG (5 kDa or 10 kDa) and the expected exact mass of the synthesized dendrons. c Weight ratio of the dendritic group to PEG. d Calculated for only the dendritic group of the amphiphile via ChemDraw Version 21.0. | ||||||
| DBA-Hex | Hexanoate | 6.0 | 1.05 | 6.05 | 0.21 | 11.6 |
| DBA-Hep | Heptanoate | 6.6 | 1.05 | 6.10 | 0.22 | 13.7 |
| DBA-Oct | Octanoate | 7.0 | 1.08 | 6.16 | 0.23 | 15.8 |
| TBA-Oct | Octanoate | 12.5 | 1.04 | 12.31 | 0.23 | 15.8 |
Once the three DBAs and the octanoate-based TBA were synthesized, we used them to formulate three DBA-TBA co-assembled micellar systems, differing in the hydrophobicity of their DBA stabilizers (Table 2). We first ensured the formation of hydrogel when dissolving the TBA alone directly in PBS (Fig. S16, ESI†), or in the presence of the fatty acids that are expected to be formed by the enzymatic cleavage (Fig. S17, ESI†). Notably, in our previously reported system, in which both DBA and TBA had identical hexanoate end-groups, a 1:1 weight ratio allowed the stabilization of the TBA in co-assembled mixed micelles. However, for the currently reported formulations, once we increased the relative hydrophobicity of the TBA, in comparison to the DBA, the 1:1 ratio didn’t allow the full solubilization of the TBA and the solution remained opaque. By slowly increasing the relative amount of the DBA, we found that a ratio of 3:2 (DBA:TBA w/w) yielded a clear solution of the desired mixed micelles for a DBA-Hex/TBA-Oct based system. We assumed that this ratio would be acceptable also for the two other DBAs containing heptanoate and octanoate end-groups, as they are more hydrophobic than the tested DBA-Hex and, therefore, should act as better stabilizers for the TBA. Indeed, all three synthesized DBAs were found to successfully stabilize the TBA as micellar assemblies in the mentioned ratio. Additionally, all three types of co-assembled micelles were found to have similar CMC values of around 4 μM (Table 2 and Fig. S12, ESI†) and similar diameters of around 13 nm were observed by DLS (Table 2 and Fig. S13, ESI†) and TEM (Fig. S15, ESI†). Compared to micellar systems based on DBA alone, the mixed micelles showed slightly lower CMC values, whereas the systems’ diameter remained similar (Table 2 and Fig. S12 and S13, ESI†). The lower CMC values were expected and can be attributed to the addition of the TBA, which has a higher tendency to aggregate than the DBA35,54 and, therefore, induces the assembly of micelles at lower concentrations.
| Formulationa | CMCb (μM) | D H (nm) |
|---|---|---|
|
a DBA:TBA formulation ratio 3:2 w/w.
b Determined using the Nile red method. [Nile Red] = 1.25 μM.
c Hydrodynamic diameter measured by DLS. [Amphiphiles mixture] = 17.5 mg mL−1.
|
||
| DBA-Hex/TBA-Oct | 5 ± 1 | 12 ± 2 |
| DBA-Hep/TBA-Oct | 4 ± 1 | 14 ± 2 |
| DBA-Oct/TBA-Oct | 4 ± 1 | 13 ± 3 |
| DBA-Hex only | 8 ± 1 | 14 ± 2 |
| DBA-Hep only | 8 ± 1 | 15 ± 1 |
| DBA-Oct only | 7 ± 1 | 15 ± 3 |
Encouraged by the successful formation of co-assembled micelles by mixing the TBA and each of the three DBAs, as indicated by the DLS and TEM, we proceeded to study their enzymatic degradation and mesophase transitions. Porcine liver esterase (PLE) was selected as a model enzyme, which can cleave the ester-containing aliphatic end-groups, to generate the equivalent fatty acids (hexanoic, heptanoic, and octanoic acids) as well as a soluble PEG-dendron with four hydrophilic hydroxyl end-groups. We decided to start with the DBA-Hex/TBA-Oct co-assembled micelles, to allow a direct comparison to our previous study of mixed micelles composed of DBA and TBA having both hexanoate-based end-groups. This comparison between DBA-Hex/TBA-Oct and DBA-Hex/TBA-Hex should reveal better how a change in the TBA's hydrophobicity will affect the enzymatically induced mesophase transitions of the assembled system and the properties of the TBA-based hydrogel that was expected to form after the first mesophase transition.
Upon incubation of the co-assembled DBA-Hex/TBA-Oct micellar solution with the activating enzyme under physiological pH at 37 °C, we observed a transition from clear to opaque solution within the first few hours, indicating the mesophase transition from nano-sized micellar assemblies to micro-particles. This transition was followed by their gradual aggregation into a bulk hydrogel at the bottom of the vial, which further shrank over a few days until reaching its final dimensions (Fig. 2A and B). To have a better understanding of the enzymatically induced transition, HPLC was used to directly monitor the molecular composition of the solution at different time points. Initially, HPLC analysis of the clear solution of the mixed micelles showed the appearance of two peaks corresponding to the two types of amphiphiles with the expected 3:2 ratio (Fig. 2C). Notably, the difference in the architecture, and the lower hydrophobicity and molecular weight of the DBA-Hex in comparison with the TBA-Oct, resulted in complete selectivity of the enzyme toward the degradation of the DBA-Hex over the TBA-Oct, and while DBA-Hex was fully hydrolyzed after 3 hours, TBA-Oct stayed intact (Fig. 2C and D). Only after the DBA-Hex was fully hydrolyzed, a sudden drop in the area of the peak related to the TBA-Oct was observed. As the disappearance of the TBA's peak was not accompanied by the appearance of a new peak for the hydrolyzed amphiphile, the sudden decrease in its concentration, suggested a mesophase transition from co-assembled micellar solution into TBA-based hydrogels, rather than enzymatic degradation of the amphiphile. This phenomenon was supported by the visual images of the HPLC vials, showing the formation of a bulk hydrogel at the bottom of the vial (Fig. 2B). In addition, DLS measurement of the upper solution, showed sizes of around 4 nm, indicating the full transition of the micellar system and degradation of the DBA into soluble hydrophilic polymers (Fig. S14, ESI†). These results demonstrate the enzymatic selectivity towards the DBA-Hex over the TBA-Oct, which are in good agreement with our previous reports of splittable TBA,28 gemini amphiphiles,34 and the recently reported hexanoate based DBA/TBA co-assembled systems.54 Our observations highlight yet again that the DBA-Hex could rapidly exchange between the micellar and unimer states, thus being highly accessible to the activating enzyme, while the higher molecular weight, different architecture, and increased hydrophobicity of the TBA-Oct, make its exchange rate significantly slower and, hence, unaffected by the activating enzyme during the initial micellar state. Interestingly, when looking at the HPLC data (Fig. 2C and D), it seems as if the gelation process occurs only after the full degradation of the DBA already happened, suggesting that the TBA might be stable in the micellar state without the presence of its DBA stabilizer. However, when looking at the visual images of the experiment vials (Fig. 2B), we can see a transition from clear to opaque solution within the first three hours, which only then gradually shows the formation of a hydrogel at the bottom of the vial. We hypothesize that the autosampler needle of the HPLC might sample both micelles and the larger hydrogel microparticles. Thus, while the disappearance of the TBA peak in the chromatogram could indicate the second step of the mesophase transition, which is the precipitation of a suspension of hydrogel microparticles into a bulk hydrogel, the HPLC analysis did not allow us to monitor the first step of the mesophase transition. This first step involves the transition from nano-sized micelles to a suspension of microparticles, which can then undergo aggregation into bulk hydrogel in the second step as mentioned above. To overcome this limitation and obtain kinetic data on the first step of the mesophase transition from micelles to hydrogel microparticles, we conducted a parallel experiment to evaluate the change in the turbidity of the solution as additional support for the visual findings. The absorbance data showed an initial increase in turbidity starting after one hour, which kept increasing until reaching a maximal value after three hours (Fig. 2D, yellow line). This data correlated well with the mentioned visual observations and the analyzed degradation profile of the DBA, suggesting that once its concentration decreases a certain threshold of about 25% of its original concentration in the formulation, the mesophase transition from micelles to microgels begins. As mentioned before, it then takes a few more hours for the formed hydrogel microparticles to further aggregate and precipitate into a hydrogel at the bottom of the vial, which then undergoes another shrinking process over a few more days until reaching its final dimensions (Fig. 2B). Importantly, throughout this mesophase transition and long gelation process, the hydrolyzed TBA was not observed in the solution, indicating its high stability toward enzymatic degradation under these conditions and timeframe.
 | ||
| Fig. 2 Enzymatic degradation and induced gelation of the DBA-Hex/TBA-Oct mixed-micellar system. (A) Schematic illustration of the enzymatically induced mesophase transition from DBA/TBA co-assembled micelles into TBA-based hydrogels. (B) Photos of the experimental vial over time, demonstrating these transitions. Overlay of (C) HPLC chromatograms and (D) analyzed kinetic data, for the enzymatic degradation of DBA (blue), accompanied by TBA peak disappearance (red) and a change in the absorbance (yellow), indicating the occurrence of such mesophase transition. | ||
To analyze the molecular composition of the formed hydrogel, the aqueous supernatant was discarded, and the precipitated hydrogel was fully dissolved in acetonitrile, which is a good solvent for both the PEG and dendron-based hydrophobic blocks, and the sample was analyzed by HPLC. The chromatogram showed the hydrogel was composed solely of the parent TBA, without any presence of hydrolyzed TBA or DBA derivatives (Fig. S21, ESI†). When a similar experiment was previously conducted using DBA and TBA, both with the same hexanoate end-groups,54 the hydrogel contained mostly the TBA (80%) alongside the original and partially hydrolyzed DBA. The fact that, in the current case, the generated hydrogel was composed solely from TBA, emphasizes our hypothesis regarding the ability to use hydrophobicity to obtain greater selectivity of the DBA component toward enzymatic degradation. Next, to characterize the mechanical properties of the formed gel and further evaluate its aging process, rheological measurements were conducted at several time points after the gel was settled. All rheological measurements at the different time points showed typical characteristics of a hydrogel mesophase (Fig. 3A and Fig. S22, ESI†). As could be expected based on the visual shrinking process of the formed hydrogels, an increase in the storage modulus was observed throughout the first few days of incubation. After the fourth day of incubation, the hydrogels’ properties stayed constant throughout an additional week of measurements, indicating that the gel completed its aging process within the first four days. Noticeably, these hydrogels remained stable for over a few months under these experimental conditions ([PLE] = 0.7 μM, pH 7.4, 37 °C, Fig. S24, ESI†).
 | ||
| Fig. 3 Analyzed hydrogel aging process by rheology measurements (A) amplitude sweep tests of the obtained hydrogels after an additional incubation period of 0, 1, 4, 7, and 10 days after the gel was settled (storage modulus (G′) is presented in graph (i), and the loss tangent (G′′/G′) is presented in graph (ii)). (B) visual images of the extracted samples and the experiment vials. | ||
To demonstrate that the formed hydrogels can undergo further enzymatic degradation, which can be a critical factor towards their potential application as a depot for slow and sustained drug release, we incubated them with bovine serum albumin (BSA) and a 20-fold higher concentration of the activating enzyme PLE. BSA is a transport protein that is known to have non-specific interactions with hydrophobic moieties and can hence induce the disassembly of polymeric aggregates. As mentioned above, the hydrogel seemed to be stable when incubated with the lower concentration of PLE, which was used to induce the transition from micelles to hydrogel. On the other hand, once the hydrogel samples were incubated with the higher concentration of PLE, BSA, and PLE with BSA, we could observe a slow transition to yield clear solutions after nearly two months of incubation. HPLC analysis of the solutions (Fig. S24, ESI†) revealed that for both PLE and PLE with BSA, only degraded TBA was observed, demonstrating the ability of the enzyme to hydrolyze the cleavable ester-containing end-groups of the TBA. It was striking to see that the sample containing solely BSA, showed only the presence of intact TBA and no degradation products were observed. The absence of a hydrogel mesophase in this sample can be attributed to the high (50 mg mL−1) concentration of BSA, which can interact with the hydrophobic block of the TBA leading to destabilization of the TBA based hydrogel.55 It is important to note that as hypothesized earlier, the degradation of the TBA-Oct-based hydrogel was much slower and required a higher concentration of both enzyme and BSA compared to our previously reported TBA-Hex-based hydrogel,54 which was composed of TBA with shorter and less hydrophobic alkyl end-groups.
Following the demonstration of the full cascade of mesophase transitions from micelles to hydrogel and finally to degraded polymers, we wished to examine whether the total concentration of the DBA and TBA would affect the programmed mesophase transition from micelles to hydrogel. To do this, we repeated the enzymatic degradation experiments while using half (8.75 mg mL−1) and double (35 mg mL−1) the total amount of the DBA-Hex/TBA-Oct in the original formulation (17.5 mg mL−1) while maintaining the same DBA:TBA ratio (3:2 w/w). We were pleased to find that the mesophase transition occurred at these concentrations, as shown in Fig. S19 (ESI†). Interestingly, we observed that as the concentration of amphiphiles increased while the concentration of the enzyme was not changed, the timeframe for the full degradation of DBA-Hex also increased as could be expected due to the higher DBA to enzyme ratio. In contrast, the increase in the TBA concentration led to faster formation of microparticle suspensions and the aggregation of TBA into settled hydrogel.
After confirming the ability of the DBA-Hex/TBA-Oct formulations to undergo the cascade mesophase transitions from micelles to hydrogel and eventually to fully degraded hydrophilic polymers, we wished to evaluate the DBA-Hep and DBA-Oct based formulations.
Based on our previous reports, which showed that increasing the length of the hydrophobic end-groups of the DBAs led to significantly slower degradation of their micellar assemblies,30 we expected to see an elongation of the transition timeframe from micelles to hydrogel as the hydrophobicity of the DBA increased. Towards this, we have repeated the enzymatic-degradation experiment for the formulations containing the TBA-Oct and either DBA-Hep or DBA-Oct as co-assembled stabilizers. The degradation profiles were monitored by HPLC, where parallel measurements in a spectrophotometer were conducted to assess a change in the turbidity of the solution as an indication of the mesophase transition, as was described earlier.
For the DBA-Hep-based co-assembled micelles, we observed the selective enzymatic degradation of the DBA-Hep (Fig. 4A), and after 6 hours, a sudden increase in the absorbance was observed (Fig. 4B), indicative of the formation of a suspension of hydrogel microparticles. This was followed by a sharp drop in the associated TBA peak in the HPLC chromatograms, indicating the mesophase transitions from microparticles suspension to settled hydrogels. These phenomena were also supported by visual observations (Fig. S20, ESI†). As we hypothesized, the elongation of only one carbon in each of the four end-groups of the DBA, resulted in slower enzymatic degradation of the DBA and consequently elongated the timeframe of the mesophase transition into a hydrogel from around 4 hours to nearly 18 hours. The final TBA-based hydrogel composition was further analyzed by HPLC by dissolving the hydrogel in acetonitrile, and the chromatogram showed that it was composed solely of the TBA (Fig. S21, ESI†). Rheology measurements of the hydrogel were conducted after allowing an additional aging period of one week, and indicated it had similar mechanical properties to the hydrogel that was generated from the DBA-Hex/TBA-Oct co-assembled system (Fig. S22, ESI†).
 | ||
| Fig. 4 Enzymatic degradation and induced gelation of the DBA-Hep/TBA-Oct and DBA-Oct/TBA-Oct mixed micellar system: (A) + (C) overlay of HPLC chromatograms and (B) + (D) analyzed kinetic data, demonstrating the enzymatic degradation of DBA (blue), accompanied by TBA peak disappearance (red) and a change in the absorbance (yellow), indicating the occurrence of the enzymatically induced sequential mesophase transitions. Graphs A and B refer to the DBA-Hep/TBA-Oct co-assembled micellar system; graphs C and D refer to the DBA-Oct/TBA-Oct system. | ||
As for the DBA-Oct-based formulation, the further elongation of an additional methylene unit in each of the four end-groups of the DBA, yielded additional significant change in the kinetics of the first mesophase transition, as the degradation of the DBA and gelation process occurred only after four days (Fig. 4 and Fig. S20, ESI†). These results highlight that while all three systems exhibited similar mesophase transitions, the alteration in the hydrophilic to hydrophobic ratio of the DBA stabilizer significantly influenced the timeframes of these transitions.
Interestingly, unlike the DBA-Hex and DBA-Hep co-assembled formulations, for the DBA-Oct-based formulation we observed a slight degradation (∼15%) of the TBA by HPLC (Fig. 4C and D). We assume that the slower degradation kinetics for the DBA-Oct in comparison with the DBA-Hex and DBA-Hep allowed the limited hydrolysis of the TBA in parallel to the DBA degradation before reaching the gelation point. These results correlate well with our previous findings for DBA-Hex/TBA-Hex formulation,54 suggesting that a similar hydrophilic to hydrophobic ratio of the two amphiphiles could allow a greater degree of interaction of the TBA with the enzyme, as the difference in the unimer–micelle exchange kinetics of the two is somewhat smaller. Nonetheless, the DBA-Oct based formulation still showed high selectivity toward the degradation of the DBA over the TBA and showed the desired transformation of TBA into a hydrogel mesophase upon enzymatic activation.
As mentioned for the other two co-assembled systems, HPLC and rheology measurements were conducted to assess the generated TBA-based hydrogel composition and properties, respectively. HPLC analysis indicated that the hydrogel was composed mainly (>90%) of the parent TBA and a small amount of DBA and hydrolyzed DBA (Fig. S21, ESI†). Notably, the presence of a small amount of DBA and hydrolyzed DBA-Oct species in the formed hydrogel, which was observed only for the DBA-Oct based formulation, could be attributed, as mentioned above, to the slightly decreased selectivity for the DBA in comparison with the TBA in this formulation. This finding also correlates well with our previous finding on a DBA-Hex/TBA-Hex based formulation, in which both types of amphiphiles had similar hydrophilic to hydrophobic ratios.54 These findings highlight the importance of the stabilizers’ hydrophobicity not only in programming the kinetics of those mesophase transitions, but also in governing the selectivity of the system toward enzymatic degradation, and consequently the composition of the formed hydrogel. Despite the small change in the hydrogel composition, rheological measurements showed that this hydrogel had similar mechanical properties to the hydrogels generated from DBA-Hex- and DBA-Hep-based systems (Fig. S22, ESI†).
The direct comparison of the timeframes of DBA degradation, transition of the TBA containing micelles into a suspension of hydrogel microparticles, and their final aggregation into bulk hydrogel for the three formulations is presented in Fig. 5. The results clearly highlight how minor structural changes of only a few carbons in the molecular structure of the stabilizing DBAs could highly impact the kinetic profile of the mesophase transitions of these formulations. By elongating the hydrophobic end-groups of the DBA from six to eight carbons, we were able to adjust the rate of these transitions from a few hours to several days. Furthermore, to gain quantitative insight into the influence of the DBAs’ hydrophobicity on their enzymatic degradation rates, the natural log of the normalized experimental data was plotted against time (Fig. 6A). This provided a linear equation consistent with the rate of a first-order reaction: ln([A]t/[A]o) = −kt. The calculated k values for the three different formulations were plotted against the dendrons’ cLogP values (Fig. 6B). Although it is clear that the overall hydrophobicity of the amphiphile should be lower due to the presence of the hydrophilic PEG block, the dendrons’ cLogP values provide a quantitative parameter that is a key component of the amphiphile's total hydrophobicity. The results indicate an exponential correlation between the cLogP values of the dendrons and the reaction rates, showing over a 100-fold increase in the reaction rate when transitioning from DBA-Hex to DBA-Oct, thereby emphasizing the importance of the amphiphile's hydrophobicity in enzymatic degradation.
 | ||
| Fig. 5 Enzymatic degradation and induced gelation of co-assembled polymeric systems with different degrees of hydrophobicity: a comparative representation of the timeframes of DBA complete degradation (blue), transition of the TBA containing micelles into hydrogel microparticles suspension (yellow), and their initial timepoint for aggregation into bulk hydrogel (red), as was monitored by HPLC analysis and by the change in absorbance, for all three formulations. DBA-Hex/TBA-Oct (full bar); DBA-Hep/TBA-Oct (dotted bar); DBA-Hex/TBA-Oct (dashed bar). | ||
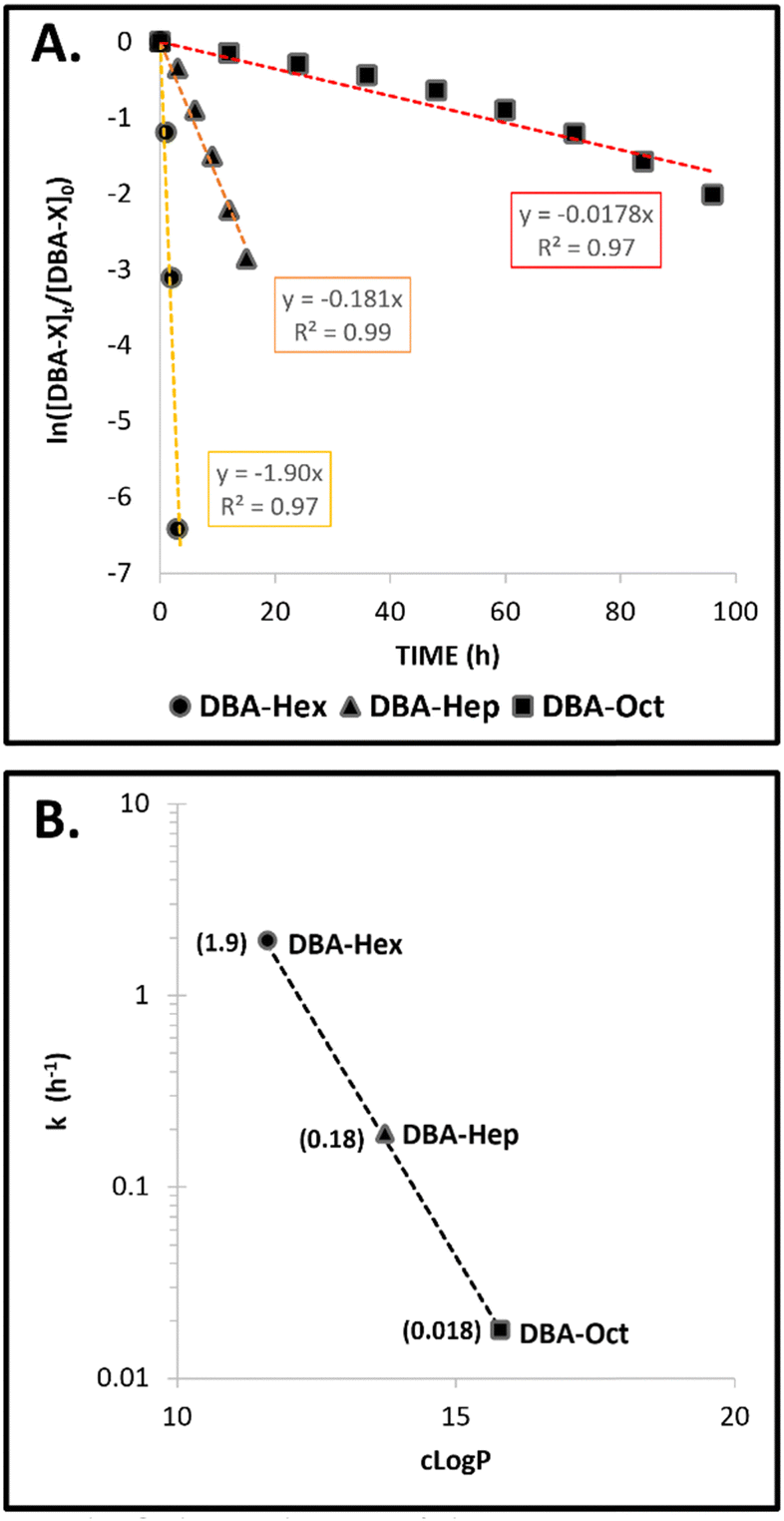 | ||
| Fig. 6 Kinetic evaluation of DBA-X enzymatic degradation rates: (A) natural log of the normalized experimental kinetic data of DBA degradation in different DBA-X/TBA-Oct formulations. (B) Logarithmic representation of the calculated rate constants plotted against the amphiphiles cLogP values. Calculated k values are shown in parentheses for each amphiphile. | ||
This newfound ability to utilize the hydrophobicity of the DBA for programming the timeframes of micelle to hydrogel mesophase transition with minor changes in the stabilizing amphiphiles’ hydrophobicity, coupled with the capability to control the enzymatic degradation rate of the hydrogel mesophase through adjustments in the hydrophobicity of the TBA component, shows promise for developing stimuli-responsive polymeric systems tailored to specific therapeutic and biomedical needs. These systems could potentially be used as multifunctional drug delivery systems, which can be administered as liquid micellar formulations that can accumulate at desired disease sites due to the enhanced permeability and retention (EPR) effect. Once there, the DBA amphiphiles can interact with the activating enzyme and generate a TBA-based macroscopic biodegradable hydrogel in situ. This hydrogel could then act as a reservoir for sustained drug release, gradually degrading via disease-associated enzymes. Once the TBA-based hydrogel completes its role as a drug depot, it can undergo the last mesophase transition into soluble polymers, thus facilitating their potential excretion from the body. Ongoing studies are exploring these applications and developing additional molecular tools to further program cascades of mesophase transitions.
Conclusions
To conclude, in this study, we demonstrated the ability to control the kinetics of mesophase transitions in enzyme-responsive polymeric amphiphiles by fine-tuning their hydrophobicity through structural adjustments. We designed three types of PEG-dendron di-block amphiphiles with varied hydrophobic end-groups and examined their co-assembled micellar formulations in response to enzymatic degradation. Our findings revealed that small modifications in the di-block amphiphiles’ hydrophobicity significantly influenced the timeframes of transitions from micelles to hydrogels and ultimately to fully soluble polymers, with transition durations varying from a few hours to several days. Notably, the hydrophilic to hydrophobic ratio affected the selectivity of enzymatic degradation, impacting the hydrogel's molecular composition. By enhancing the hydrophobicity of the tri-block amphiphiles, we achieved a more stable hydrogel that undergoes slow degradation for nearly two months. This research highlights the potential of using hydrophobicity as a strategic key tool for programming the kinetics of mesophase transitions in polymeric assemblies, paving the way for engineered formulations with specific structures and functionalities for various biomedical applications.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. J. A. thanks the ISRAEL SCIENCE FOUNDATION (grant No. 413/22) for the support of this research. S. T. thanks the ADAMA Center for Novel Delivery Systems in Crop Protection, Tel-Aviv University, for the financial support. TEM and HRSEM measurements were carried out with the help of Vered Holdengreber, Head of the electron microscopy unit at Tel Aviv University, Israel. S. T. and R. J. A. are thankful for the kind help from the Lihi Adler-Abramovich lab for the use of their rheometer.Notes and references
- A. P. Blum, J. K. Kammeyer, A. M. Rush, C. E. Callmann, M. E. Hahn and N. C. Gianneschi, J. Am. Chem. Soc., 2015, 137, 2140–2154 CrossRef CAS PubMed.
- Y. Tu, F. Peng, A. Adawy, Y. Men, L. K. E. A. Abdelmohsen and D. A. Wilson, Chem. Rev., 2016, 116, 2023–2078 CrossRef CAS PubMed.
- J. Kost and R. Langer, Adv. Drug Delivery Rev., 2012, 64, 327–341 CrossRef.
- E. Busseron, Y. Ruff, E. Moulin and N. Giuseppone, Nanoscale, 2013, 5, 7098–7140 RSC.
- J. F. Lutz, J. M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 1–14 Search PubMed.
- A. Harada and K. Kataoka, Prog. Polym. Sci., 2006, 31, 949–982 CrossRef CAS.
- S. S. Das, P. Bharadwaj, M. Bilal, M. Barani, A. Rahdar, P. Taboada, S. Bungau and G. Z. Kyzas, Polymers, 2020, 12, 1397 CrossRef CAS.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- R. Keogh, L. D. Blackman, J. C. Foster, S. Varlas and R. K. O’Reilly, Macromol. Rapid Commun., 2020, 41, 1900599 CrossRef CAS.
- Z. M. Png, C. G. Wang, J. C. C. Yeo, J. J. C. Lee, N. E. Surat’man, Y. L. Tan, H. Liu, P. Wang, B. H. Tan, J. W. Xu, X. J. Loh and Q. Zhu, Mol. Syst. Des. Eng., 2023, 8, 1097–1129 RSC.
- N. R. B. Boase, E. R. Gillies, R. Goh, R. E. Kieltyka, J. B. Matson, F. Meng, A. Sanyal and O. Sedláček, Biomacromolecules, 2024, 25, 5417–5436 CrossRef CAS.
- G. Whitton and E. R. Gillies, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 148–172 CrossRef CAS.
- J. F. Gohy and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7117–7129 RSC.
- Y. Wang, Y. Deng, H. Luo, A. Zhu, H. Ke, H. Yang and H. Chen, ACS Nano, 2017, 11, 12134–12144 CrossRef CAS PubMed.
- J. Ko, K. Park, Y. S. Kim, M. S. Kim, J. K. Han, K. Kim, R. W. Park, I. S. Kim, H. K. Song, D. S. Lee and I. C. Kwon, J. Controlled Release, 2007, 123, 109–115 CrossRef CAS.
- S. S. Müller, T. Fritz, M. Gimnich, M. Worm, M. Helm and H. Frey, Polym. Chem., 2016, 7, 6257–6268 RSC.
- E. R. Gillies and J. M. J. Fréchet, Bioconjugate Chem., 2005, 16, 361–368 CrossRef CAS.
- C. Wang, G. Zhang, G. Liu, J. Hu and S. Liu, J. Controlled Release, 2017, 259, 149–159 CrossRef CAS.
- J. Qiao, L. Qi, Y. Shen, L. Zhao, C. Qi, D. Shangguan, L. Mao and Y. Chen, J. Mater. Chem., 2012, 22, 11543–11549 RSC.
- R. de la Rica, D. Aili and M. M. Stevens, Adv. Drug Delivery Rev., 2012, 64, 967–978 CrossRef CAS.
- A. J. Harnoy, I. Rosenbaum, E. Tirosh, Y. Ebenstein, R. Shaharabani, R. Beck and R. J. Amir, J. Am. Chem. Soc., 2014, 136, 7531–7534 CrossRef CAS PubMed.
- M. Zelzer, S. J. Todd, A. R. Hirst, T. O. McDonald and R. V. Ulijn, Biomater. Sci., 2012, 1, 11–39 RSC.
- H. L. Sullivan, Y. Liang, K. Worthington, C. Luo, N. C. Gianneschi and K. L. Christman, Biomacromolecules, 2023, 24, 4695–4704 CrossRef CAS.
- J. Mu, J. Lin, P. Huang and X. Chen, Chem. Soc. Rev., 2018, 47, 5554–5573 RSC.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- B. Sun, C. Luo, H. Yu, X. Zhang, Q. Chen, W. Yang, M. Wang, Q. Kan, H. Zhang, Y. Wang, Z. He and J. Sun, Nano Lett., 2018, 18, 3643–3650 CrossRef CAS.
- P. Zhang, H. Zhang, W. He, D. Zhao, A. Song and Y. Luan, Biomacromolecules, 2016, 17, 1621–1632 CrossRef CAS.
- G. Slor, S. Tevet and R. J. Amir, ACS Polym. Au, 2022, 2, 380–386 CrossRef CAS PubMed.
- G. Slor and R. J. Amir, Macromolecules, 2021, 54, 1577–1588 CrossRef CAS PubMed.
- M. Segal, R. Avinery, M. Buzhor, R. Shaharabani, A. J. Harnoy, E. Tirosh, R. Beck and R. J. Amir, J. Am. Chem. Soc., 2017, 139, 803–810 CrossRef CAS.
- G. Slor, A. R. Olea, S. Pujals, A. Tigrine, V. R. De La Rosa, R. Hoogenboom, L. Albertazzi and R. J. Amir, Biomacromolecules, 2021, 22, 1197–1210 CrossRef CAS.
- S. Tevet, S. S. Wagle, G. Slor and R. J. Amir, Macromolecules, 2021, 54, 11419–11426 CrossRef CAS PubMed.
- G. Slor, N. Papo, U. Hananel and R. J. Amir, Chem. Commun., 2018, 54, 6875–6878 RSC.
- I. Rosenbaum, R. Avinery, A. J. Harnoy, G. Slor, E. Tirosh, U. Hananel, R. Beck and R. J. Amir, Biomacromolecules, 2017, 18, 3457–3468 CrossRef CAS PubMed.
- N. Edelstein-Pardo, M. Molco, P. Rathee, G. Koren, S. Tevet, S. Z. Sharabani, R. Beck, R. J. Amir and A. Sitt, Chem. Mater., 2022, 34, 6367–6377 CrossRef CAS.
- X. Li, Y. Wang, Y. Zhang, Z. Yang, J. Gao and Y. Shi, J. Mater. Chem. B, 2022, 10, 3242–3247 RSC.
- H. Wang, Z. Feng, Y. Wang, R. Zhou, Z. Yang and B. Xu, J. Am. Chem. Soc., 2016, 138, 16046–16055 CrossRef CAS PubMed.
- M. P. Chien, A. S. Carlini, D. Hu, C. V. Barback, A. M. Rush, D. J. Hall, G. Orr and N. C. Gianneschi, J. Am. Chem. Soc., 2013, 135, 18710–18713 CrossRef CAS PubMed.
- A. Fakhari and J. Anand Subramony, J. Controlled Release, 2015, 220, 465–475 CrossRef CAS.
- F. E. Alemdaroglu, J. Wang, M. Börsch, R. Berger and A. Herrmann, Angew. Chem., Int. Ed., 2008, 47, 974–976 CrossRef CAS PubMed.
- M. P. Chien, A. M. Rush, M. P. Thompson and N. C. Gianneschi, Angew. Chem., Int. Ed., 2010, 49, 5076–5080 CrossRef CAS PubMed.
- M. E. Hahn and N. C. Gianneschi, Chem. Commun., 2011, 47, 11814–11821 RSC.
- D. B. Wright, A. Ramírez-Hernández, M. A. Touve, A. S. Carlini, M. P. Thompson, J. P. Patterson, J. J. De Pablo and N. C. Gianneschi, ACS Macro Lett., 2019, 8, 676–681 CrossRef CAS.
- A. S. Carlini, M. F. Cassidy and N. C. Gianneschi, Methods Mol. Biol., 2022, 2371, 427–448 CrossRef CAS.
- T. H. Ku, M. P. Chien, M. P. Thompson, R. S. Sinkovits, N. H. Olson, T. S. Baker and N. C. Gianneschi, J. Am. Chem. Soc., 2011, 133, 8392–8395 CrossRef CAS PubMed.
- B. Y. Wang, H. Xu and X. Zhang, Adv. Mater., 2009, 21, 2849–2864 CrossRef.
- M. Dergham, S. Lin and J. Geng, Angew. Chem., Int. Ed., 2022, 61, e202114267 CrossRef CAS PubMed.
- Y. Li, B. Xue and Y. Cao, ACS Macro Lett., 2020, 9, 512–524 CrossRef CAS.
- Y. Liu, H. Wang, S. Li, C. Chen, L. Xu, P. Huang, F. Liu, Y. Su, M. Qi, C. Yu and Y. Zhou, Nat. Commun., 2020, 11, 1–12 CrossRef CAS PubMed.
- S. Doppalapudi, A. Jain, A. J. Domb and W. Khan, Expert Opin. Drug Delivery, 2016, 13, 891–909 CrossRef CAS.
- T. Manouras and M. Vamvakaki, Polym. Chem., 2016, 8, 74–96 RSC.
- R. Langer and D. A. Tirrell, Nature, 2004, 428, 487–492 CrossRef CAS PubMed.
- P. Rathee, N. Edelstein-Pardo, G. Koren, R. Beck and R. J. Amir, Biomacromolecules, 2024, 25(6), 3607–3619 CrossRef CAS PubMed.
- P. Rathee, N. Edelstein-Pardo, F. Netti, L. Adler-Abramovich, A. Sitt and R. J. Amir, ACS Macro Lett., 2023, 12, 814–820 CrossRef CAS PubMed.
- N. Feiner-Gracia, M. Buzhor, E. Fuentes, S. Pujals, R. J. Amir and L. Albertazzi, J. Am. Chem. Soc., 2017, 139, 16677–16687 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01587h |
| This journal is © The Royal Society of Chemistry 2024 |