
On a seamlessly replicable circular photoreactor for lab-scale continuous flow applications†
Yi-Hsuan
Tsai
a,
Martin
Cattoen
 ab,
Guillaume
Masson
bc,
Gabrielle
Christen
b,
Lisa
Traber
b,
Morgan
Donnard
c,
Frédéric R.
Leroux
c,
Guillaume
Bentzinger
b,
Sylvain
Guizzetti
*b and
Jean-Christophe M.
Monbaliu
*ad
ab,
Guillaume
Masson
bc,
Gabrielle
Christen
b,
Lisa
Traber
b,
Morgan
Donnard
c,
Frédéric R.
Leroux
c,
Guillaume
Bentzinger
b,
Sylvain
Guizzetti
*b and
Jean-Christophe M.
Monbaliu
*ad
aCenter for Integrated Technology and Organic Synthesis, MolSys Research Unit, University of Liège, Sart Tilman, B-4000 Liège, Belgium. E-mail: jc.monbaliu@uliege.be; Web: http://www.citos.uliege.be
bNovAliX, Bio Parc, 850 Boulevard Sébastien Brant, 67405 Illkirch-Graffenstaden Cedex, France Web: https://novalix.com/
cUniversity of Strasbourg, University of Haute-Alsace, CNRS, UMR 7042-LIMA, ECPM, 67000 Strasbourg, France
dWEL Research Institute, Avenue Pasteur 6, B-1300 Wavre, Belgium
First published on 9th April 2024
Abstract
A cost effective and replicable continuous flow circular photoreactor system is introduced. All body parts of the reactor are 3D-printed, and the electronics are purchased from various mainstream suppliers. The reactive path of the reactor features a low-cost PFA coil assembly, which provides suitable chemical resistance and a wide wavelength window for scouting diverse photochemical reactions. The internal volume can be easily adapted from exploratory microscale experiments to multigram scale preparation of small libraries of compounds. The fabrication of the circular photoreactor was successfully reproduced and operated at 2 different locations. We showcase the versatility of the setup and its utility, as well as its suitability, to both academic and industrial environments. A total of 4 case studies relevant to pharmaceutical and medicinal chemists are demonstrated. The first case study is a photocatalyzed singlet oxygen oxidation of a thioether, using methionine as a model substrate and affording complete and selective conversion into the corresponding sulfoxide. Next, a photoredox application for the α-functionalization of a model tetrahydroisoquinoline is successfully optimized, with process conditions outclassing previous batch reports. Then, the preparation of a small library of (hetero)arylcycloalkylamines through XAT cross-electrophile coupling is carried out. Finally, photocatalyzed difluoroamidation of indoles is optimized and scaled up.
Introduction
The synergistic combination of flow technology and photochemistry is now well established among the chemistry and chemical engineering communities.1–3 These assets essentially stem from the small internal dimensioning and higher surface-to-volume ratio of flow reactors, which allow for enhanced and homogeneous irradiation within a controlled time frame.1–3 These properties positively impact both process acceleration and side-reaction reduction. Common general assets of flow technology also positively contribute to improving photochemical processes such as precise temperature control, which is critical for photoredox catalysis,4–7 high mass transfer efficiency for biphasic systems1 and scalability.3,8 Additional synergies with advanced automation, integration of design of experiment (DoE) and self-optimization tools have further contributed to rejuvenate photochemistry in flow.1,9–12In continuous flow settings, various methodologies have been employed to conduct photochemical reactions. These encompass a range of setups from plug flow13–15 to plate-based,16 falling film,17–19 vortex20,21 and continuous stirred tank reactors.22–25 These varied approaches have served as the foundation for developing both custom-built and commercial photoreactors, each differing in their unique approach to provide mixing and maximization of photon flux while minimizing light loss.26,27 These systems exhibit differences not only in the cooling mechanisms employed to prevent light source overheating, but also in the thermoregulation of the reactor and selection and placement of the light source with regard to the reactor.25–27
While continuous flow systems offer notable advantages, high costs associated with commercial continuous flow systems often divert researchers to familiar batch options, relying on readily available equipment. One way to expand the use of flow photochemistry is by using consumer-grade commercial technologies, which lower the barrier to entry for researchers: 3D printing enables the development of affordable photoreactor designs that can be iterated quickly to match specific constraints, and open-source electronic boards allow the programming of tailored control units.24,28–38 Nevertheless, it is important to note that replicating custom designs across labs may lead to inconsistencies due to slight differences in components like light sources, materials, or cooling systems.39 This emphasizes the importance of standardizing reactor designs to guarantee reliable operation and reproducibility, requiring user-friendly, versatile, and robust designs with interchangeable parts.
In this manuscript, we report the design and conception of a user-friendly and cost-effective 3D-printed flow photochemical reactor amenable to diverse photochemical reactions (Fig. 1). In its most elaborated version, the photochemical reactor features options to control temperature (range: 0–60 °C), to change internal volumes, and to adjust irradiation times at various wavelengths and intensities. We capitalize on the use of widely available parts to maximize its broad application as a low-cost premier step for lab-scale optimization and small-scale production. The reactor setup was then used to illustrate four photochemical applications, including homogeneous and gas–liquid reactions of industrial relevance. These examples include: (a) photooxidation with singlet oxygen, (b) photo α-alkylation of amines, (c) photocatalyzed cross-electrophile coupling via XAT and (d) photocatalyzed difluoroamidation. All four reactions were successfully optimized in the 3D-printed photoreactors, with applications (a) and (c) achieving results which were at least comparable to the literature precedent. Examples (b) and (d) showcase applications that are unprecedented under flow conditions.
 | ||
| Fig. 1 Design of the photoreactor (see Table 1). a. View of the complete system with the power supplies on the left-hand side. b. Close-up of the 3D-printed circular photoreactor with the outside enclosure. c. Close-up of the 3D-printed circular photoreactor without the outside enclosure. d. Top view of the 3D-printed circular photoreactor in operation. | ||
Results and discussion
Reactor development
At the outset of our project, different kinds of homemade photoreactors were described in the literature for batch,40 flow,13,14,23,41,42 or adaptable to both techniques.28,31,43 Since then, a number of alternative setups have been reported.15,24,31 Several considerations factored in our approach: ease of assembly (avoiding metal or glass printing), simplicity to reproduce across different sites, precise temperature control, and ideally avoiding expensive, dedicated thermal units such as cryostats.In this context, we were particularly interested in the work of Böse and co-workers31 describing a versatile 3D-printed photoreactor adapted to both batch and flow reactions. Their approach relies on commercial Peltier thermoelectric modules under the control of an Arduino microcontroller to accurately regulate the temperature of a 3D-printed enclosure. While sound and accessible, their design required some adaptation to match our needs, particularly to access a large range of flow reactor volume to perform both optimization and larger scale experiments. Further, the ability to control the system from a computer was a desirable step towards automated optimization of reaction conditions. Finally, as LED sources represent one of the main cost-drivers, we also aimed to build custom lamps from affordable commercial components (Table 1).
| Features | Böse31 | This work |
|---|---|---|
| a The Peltier module is associated with a circulating tap water cooling system. b 80 °C is the theoretical maximum temperature of operation based on the PETG transition temperature; process temperature above 60 °C is therefore not recommended. | ||
| Geometry | Square | Circular |
| Air flow | Horizontal | Vertical |
| LEDs | 2 commercial LEDs (18–45 W) | 3 custom built LEDs (50 W) |
| Power supply | One for each LED | Single power supply |
| Adjustable power | No | Yes |
| Thermoregulation | Peltier modulea + Arduino | |
| Temperature range | −17 to 80 °Cb | 0 °C to 80 °Cb |
| Batch reactors | 1–50 mL | None |
| Flow reactor | 7 mL | 2–20 mL |
To maximize the irradiation of our flow reactors, an elegant choice in terms of geometry was to design a circular reactor. This flow reactor consisted of fluorinated polymer (PFA or FEP) tubing with an internal diameter of 1/16′′ (ca. 1.59 mm). The diameter and height of the reactor were chosen to be able to host a coil with an internal volume of 20 mL (length = 1060 cm), but could be adapted to lower or higher capacity reactors depending on the tubing diameter and length. In our opinion, a maximum of 60 mL reactor volume (internal diameter = 3/16′′, length = 750 cm) could be considered, though this volume was beyond the scope of the present work. As no magnetic stirring was required for flow operation, the top and bottom of the reactor were free to accommodate the Peltier modules. One side of the Peltier module, facing away from the reactor, housed a water-cooling circuit, while on the side facing inside, a heatsink equipped with a fan was installed. Our design uses air as a heat transfer medium, offering the advantage of minimal light absorbance and avoiding issues with changes in the refraction index. While the heat capacity of air is low, the rapid movement of air inside of the reactor chamber provides sufficiently uniform temperature distribution. This design leaves the full circumference available for irradiation from three light sources placed at 120°.
Using 3D-printing technology significantly lowers the cost of the flow setup compared to any commercially available equipment. Nevertheless, the requirement for several high-power light sources remains a major cost-driver in the implementation of the photoreactor. This issue was addressed by designing a 3D-printed LED-support that can host LED COB (chip-on-board) of different wavelengths along with a heat sink to avoid overheating of the LEDs. A single adjustable power supply was designed to be able to adapt the light power to the different reactor volumes or reaction conditions.
The flow photoreactor described above met our expectations, especially regarding temperature control, with the setpoint being respected at ±0.1 °C in most cases. The additional ability to operate the photoreactor from a computer allowed remote monitoring of the temperature control. Due to the larger internal volume of the 3D printed reactor casing and the high light power (3 × 50 W), the lowest temperature achievable in our setup is around 0 °C.
After the maturation of the design and the validation of the assembly protocol including printing, assembly, and programming in one of the labs (Liège, Belgium) (see ESI,† Section S1), the setup was seamlessly replicated by another team in a partner facility (Illkirch, France). Once accomplished, our focus shifted towards showcasing the usefulness of our newly designed reactor by applying it to different reactions of interest for both groups.
Photocatalytic oxidation with singlet oxygen
The chemoselective oxidation of sulfides has attracted significant attention as it represents one of the most direct pathways to produce sulfoxides. These functional groups hold immense utility in organic synthesis,44 medicinal chemistry,45,46 and natural product research.47 To avoid overoxidation to sulfones and ensure a safe process,48,49 Monbaliu and colleagues showcased the photooxidation of sulfides with singlet oxygen, under scalable flow conditions in a Corning® Advanced-Flow™ (AFR) Lab Reactor.50 Their approach was performed in a continuous flow singlet oxygen generator to convert methionine (1) into its corresponding sulfoxide 2a (Fig. 2a). The optimal conditions for full conversion were determined to be 0.3 M of 1 in water as a solvent, adding as low as 0.1% of rose bengal (RB) as a photosensitizer. A slight excess of oxygen (1.1 equiv.) and a residence time of 1.4 min under either white or blue (405 nm) LED irradiation at 100% intensity were enough to reach quantitative conversion.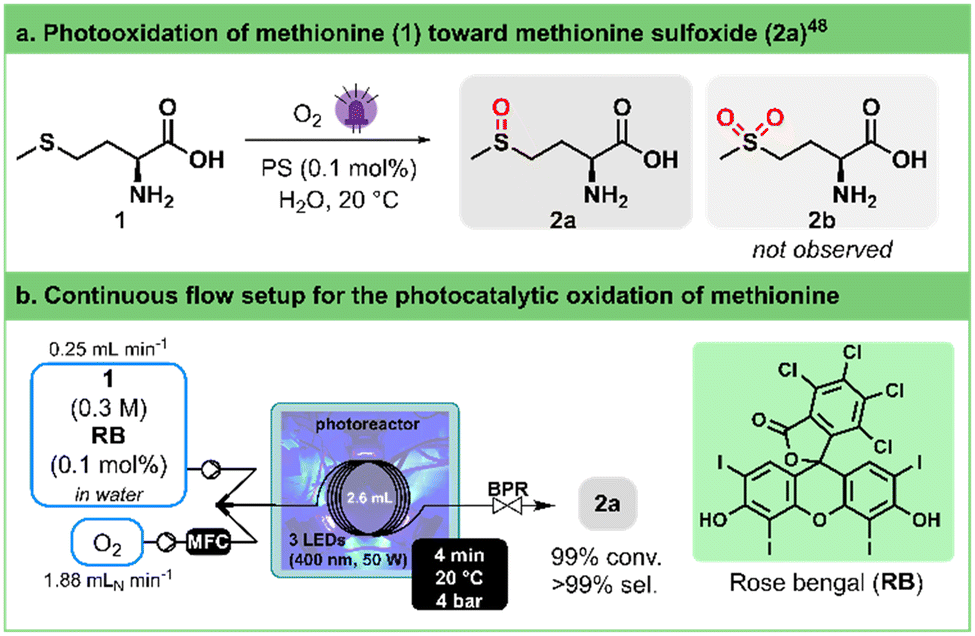 | ||
| Fig. 2 a. General conditions for the photogeneration of singlet oxygen and selective oxidation of methionine (1) toward methionine sulfoxide (2a). PS stands for photosensitizer. b. Continuous flow photooxidation of methionine (1) toward methionine sulfoxide (2a) using our 3D-printed circular photoreactor. MFC stands for mass flow controller. BPR stands for the back pressure regulator. See Table 2 for the optimization details. | ||
To evaluate our in-house photoreactor and facilitate comparison with the remarkable outcomes mentioned earlier, we standardized a reactor volume of ca. 2.6 mL (Fig. 2b), mirroring the internal volume of the Corning® AFR Lab Reactor glass fluidic module. To our delight, our experiments showed that complete conversion can be achieved within 2–4 min (entries 4 and 5, Table 2). Notably, this occurs even with the same low excess of oxygen (1.1 equiv.) and without as thorough and continuous mixing as observed in the AFR setup, demonstrating a parallel high selectivity toward the sulfoxide product. Here, a single high pressure static mixing element (IDEX high pressure static mixing tee, Fig. 2b) was used upstream the photoreactor. It is important to note that the applied back pressure significantly influences the conversion rate by directly modulating the solubility of oxygen in the solution (entries 1–4, Table 2).
| Entrya | Liquid flow rate (mL min−1) | O2 flow rate (mLN min−1) | BPR (bar) | t R (min) | Convb (%) |
|---|---|---|---|---|---|
| a Stock solution: aqueous (L)-methionine (1) (0.3 M) with 0.1 mol% rose bengal (RB) as a photosensitizer. The solution was irradiated with blue light (400 nm) and the photoreactor was set at 20 °C. b Average conversion of three samples, quantified by integration in 1H NMR (400 MHz). The solvent was evaporated under vacuum and the crude was dissolved in D2O. The following signals were used for the integration: 2.55 ppm (t, CH2) for (L)-methionine (1) and 2.65 ppm (s, CH3) for (L)-methionine sulfoxide (2a). | |||||
| 1 | 1 | 7.5 | 2.8 | 0.80 | 45 |
| 2 | 0.5 | 3.75 | 2.8 | 1.60 | 79 |
| 3 | 1 | 7.5 | 4 | 0.98 | 59 |
| 4 | 0.5 | 3.75 | 4 | 1.97 | 96 |
| 5 | 0.25 | 1.88 | 4 | 3.95 | >99 |
Photocatalyzed α-alkylation of amines
Cyclic amines are central moieties in numerous active pharmaceutical compounds.51 In the preparation of substituted saturated nitrogen heterocycles, a common approach involves functionalizing the less reactive but widely present C–H bonds at the α-position of the amine nitrogen atom.52–57Pandey and Reiser,58 as well as Yoon,59 independently reported the generation of α-amino radicals derived from tetrahydroisoquinolines in the presence of Ru(bpy)3Cl2 as a photocatalyst and their capture with Michael acceptors (Fig. 3a). Pandey and Reiser achieved a moderate yield of 75% after 24 h of irradiation with a blue LED at room temperature, while Yoon improved the yield to 90% within 5 h at 50 °C using a lower catalyst loading and a compact fluorescent light bulb. Later, Bergonzini and König demonstrated the impact of reaction temperature on the initial reaction rate, with higher temperatures leading to faster conversion.60
 | ||
| Fig. 3 a. General conditions for the photoredox α-functionalization of tetrahydroisoquinoline 3. b. Continuous flow photooxidation photoredox α-functionalization of tetrahydroisoquinoline 3 toward 5 using our 3D-printed circular photoreactor (illustration of entry 8, Table 3). BPR stands for the back pressure regulator. See Table 3 for the optimization details. | ||
Taking these findings into account and acknowledging the thermoregulation capability of our circular flow photoreactor, we aimed to optimize this reaction by exploring various residence times, temperatures, and catalyst loadings (Fig. 3b, Table 3). To ensure comparability with literature reports,60 we capped the maximum residence time at 15 min.
| Entrya | Cat. (mol%) | T (°C) | t R (min) | Flow rate (mL min−1) | Convb (%) |
|---|---|---|---|---|---|
| a Stock solution: tetrahydroisoquinoline 3 (0.25 M), methyl vinyl ketone (4, 2 equiv., 0.5 M), Ru(bpy)3Cl2 (catalyst loading as stated in Table 3) and TFA (1 equiv., 0.25 M) in acetonitrile. The solution was irradiated with blue light (400 nm) and the photoreactor was operated at different temperatures (as stated in Table 3). b Average conversion of two samples, quantified by integration in 1H NMR (400 MHz). Samples were prepared by neutralization of the crude with K2CO3 followed by filtration with a silica plug, solvent evaporation in vacuo and dissolution in CDCl3. NMR peaks used for the integration: 4.46 ppm (s, 2H) for compound 3 and 4.72 ppm (dd, 1H) for 5. | |||||
| 1 | 2 | 40 | 15.3 | 0.17 | 99 |
| 2 | 2 | 40 | 7.4 | 0.35 | 95 |
| 3 | 2 | 40 | 3.8 | 0.69 | 78 |
| 4 | 2 | 25 | 15.3 | 0.17 | 97 |
| 5 | 2 | 25 | 7.4 | 0.35 | 87 |
| 6 | 2 | 25 | 3.8 | 0.69 | 60 |
| 7 | 1 | 40 | 15.3 | 0.17 | 97 |
| 8 | 0.5 | 40 | 15.3 | 0.17 | 94 |
By contrasting entries 2 and 3 with 5 and 6 in Table 3, we validate the temperature's substantial influence, particularly on the initial reaction rate. Employing our in-house reactor, high conversion can be attained within approximately 15 min without requiring additional heating for the reaction (entry 4). Furthermore, operating at a higher temperature (40 °C, entry 2) allows shortened reaction times (7.4 min), possibly attributed to a more powerful light source and our reactor's design. Additionally, reducing the catalyst loading (0.5–1 mol%, entries 7 and 8) showcases no significant impact on the transformation's outcome. All in all, our photoreactor enabled us to significantly enhance conversion rates and to remarkably reduce reaction times when compared to previously reported batch protocols (minutes instead of hours).
Photocatalyzed cross-electrophile coupling via XAT
Among the different synthetic methodologies that have witnessed significant development thanks to the renewed interest in photocatalysis, elaboration of Csp2–Csp3 is undoubtedly one of the most attractive in the frame of medicinal chemistry programs.61–64 Cross-electrophile coupling between an aryl-halide and an alkyl-halide is a particularly interesting alternative to cross-coupling involving organometallic reagents as it avoids pre-formation of the reactive species. MacMillan has succeeded in developing elegant photocatalyzed protocols involving silyl radicals.65,66 The implementation of this strategy in flow required thorough experimentation because of the heterogeneity of the reaction mixture67 and involved the recourse to continuous stirred tank reactors68 or oscillatory plug flow photoreactors.69 More recently, Yatham described an alternative approach relying on halogen-atom transfer (XAT).70 The translation of the latter protocol in our circular photoreactor was attempted.A preliminary screening was conducted in batch to explore the reaction between aryl bromides 6a and 6b with iodide 7a (Fig. 4a). This evaluation encompasses various factors such as the nature of the photocatalyst, concentration, nickel source (NiBr2·glyme + dtbbpy or pre-formed NiBr(dtbbpy)), and solvent. 1,2,3,5-Tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) rapidly emerged as the most promising photosensitizer (see ESI,† Section S3.3.3.3). Consistent with Yatham's observations, a slight increase in yield was noted when utilizing a pre-formed catalyst. The poor reactivity of the bromide analogue (7b) of iodopiperidine was also confirmed, with a mere 3% yield toward the desired coupling product 8a (see ESI,† Section S3.3.3.4).
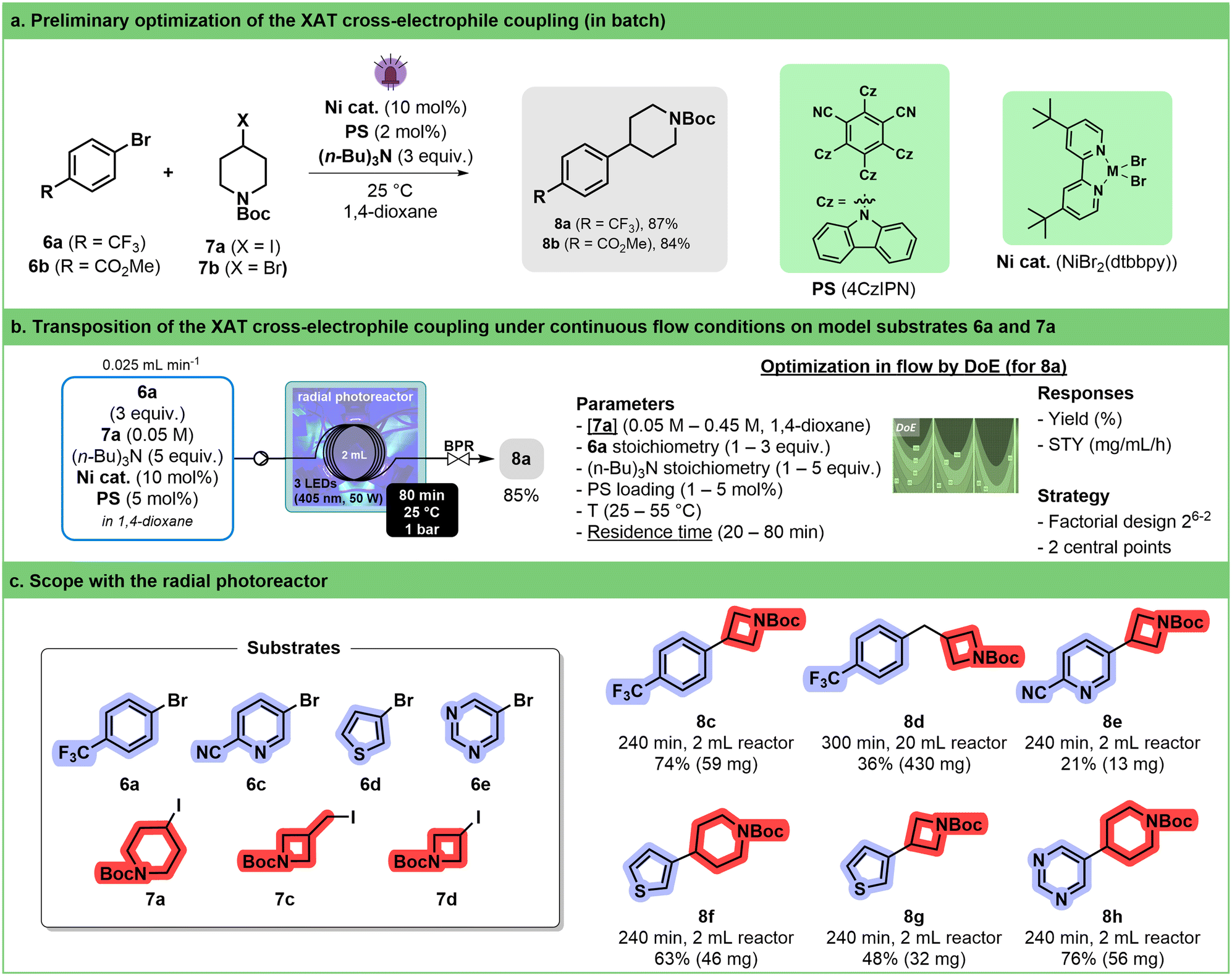 | ||
| Fig. 4 a. Optimization work on XAT cross-electrophile coupling in batch. Two aryl bromide substrates (6a and 6b) and two alkyl iodide substrates (7a and 7b) were selected as model compounds. b. Continuous flow XAT cross-electrophile coupling. The reaction conditions were optimized by DoE on aryl bromide 6a and alkyl iodide 7a toward 8a using our 3D-printed circular photoreactor (illustrated process conditions correspond to the optimum) for the continuous flow XAT cross-electrophile coupling. The two main factors are underlined. BPR stands for the back pressure regulator. c. Scope of the reaction under flow conditions. The optimized conditions for substrate 7a were used without reoptimization for other substrates (6c–6e and 7c and 7d). The total continuous operation (240 to 300 min), isolated yield and scale are indicated. | ||
Validation of these results in flow was performed by carrying out the reaction at 30 °C at different wavelengths with 60 min residence time (see ESI† Section S3.3.3.5). The desired product 8a was obtained in 25% yield. From there, a design of experiment (DoE) approach was followed to determine the most impactful parameters (stoichiometry, concentration, catalyst loading, residence time and temperature) on both yield and productivity (Fig. 4b, right). The two main factors impacting the reaction outcome were determined to be the concentration of alkyl iodide 7a and the residence time, which have opposite influences on both the yield and space time yield (STY). Increasing the stoichiometry of both tri-n-butylamine and aryl bromide has a positive impact on both responses. Under the best conditions (see ESI† Section S3.3.3.6), compound 8a was obtained in 85% yield, which corresponds to an STY of 21 mg mL−1 h−1 (Fig. 4b, left). Although fine-tuning of the conditions might have been necessary to increase productivity, these results compare favorably with Barham's optimized protocol,71 which is, to the best of our knowledge, the only protocol under flow conditions (STY = 3 mg mL−1 h−1 in a Vapourtec UV-150).
Therefore, we decided to use these conditions without reoptimization to establish a preliminary scope of the reaction (Fig. 4c). We were able to demonstrate that a handful of heteroaryl bromides including thiophene, pyridine and pyrimidine, as well as different alkyl halides, were potent coupling partners. Worthy of note is the fact that by increasing the volume of the tubing inside the reactor from 2 mL to 20 mL, several hundreds of mg of compound 8d could be obtained within 300 min of continuous operation.
Photocatalyzed difluoroamidation of indoles
Introduction of fluorinated moieties on organic scaffolds is a major topic within the pharmaceutical industry.72–74 This is particularly true for difluorinated compounds that are mainly accessed through deoxofluorination75,76 or metal-catalyzed cross-coupling reactions.77 Recently, different iridium-catalyzed protocols have appeared to introduce a difluoroacetamide group on (hetero)aromatic compounds.The reaction of 3-methylindole 9a with bromodifluoroacetamide 10a was chosen as a model for optimization study. A preliminary screening in batch was performed to identify flow compatible conditions and to find a suitable, more sustainable replacement for the iridium photocatalyst (Fig. 5a). The combination of 2,4,6-tris(diphenylamino)-3,5-difluorobenzonitrile (3DPA2FBN) as an organo-photocatalyst78 and N,N-dimethyl-p-toluidine in DMSO was found to be the most efficient. Initial studies in flow demonstrated that temperature has no effect on the outcome of the reaction and that 405 nm was the optimal wavelength (see ESI† Section S3.3.4.4).
 | ||
| Fig. 5 a. Preliminary optimization work on the photocatalyzed difluoroamidation of indole 9a toward product 11a. b. The reaction conditions were optimized by DoE on 3-methylindole (9a) and bromodifluoroacetamide (10a). The two main factors are underlined. c. Scalability trials for the photocatalyzed difluoroamidation of 9a in the circular 3D-printed photoreactor (20 mL internal volume). After 6 h of continuous operation and purification of the reactor effluent, 6.45 g of compound 11a were recovered. d. Telescoping and scope of the photocatalyzed difluoroamidation under flow conditions. Upstream formation of 2 different bromodifluoroacetamides (10b and 10c) was telescoped with the downstream photocatalyzed difluoroamidation with two different indoles (9a and 9b). Values in brackets are 19F NMR yield (internal standard: trifluorotoluene). | ||
A 25-2 fractional factorial design was used to evaluate five parameters (organo-photocatalyst concentration, 10a concentration, 9a stoichiometry, the amount of N,N-dimethyl-p-toluidine, and residence time, Fig. 5b, ESI† Section S3.3.4.6). This allowed us to highlight that substrate concentration and residence time have the highest impact on yield and productivity. While long residence time and low concentration led to high yield, they have a deleterious effect on space–time yield. Therefore, a response surface model on these two variables was established to find the best compromise between yield and productivity (see ESI† Section S3.3.4.7).
A compromise was made, and we decided to run the reaction at a moderate concentration of 0.125 M with a relatively short residence time (20 min); the latter condition gave the desired product with an 19F NMR yield (internal standard: trifluorotoluene) of 55% and a productivity of 65 mg mL−1 h−1 in a 2 mL reactor. These conditions were applied in our 3D-printed circular reactor equipped with a 20 mL coil at a flow rate of 1 mL min−1 over 6 h, providing more than 6 g of compound 11a after purification (Fig. 5c). Although the isolated yield after purification was lower than the 19F NMR yield (47% vs. 55%), it still corresponds to a satisfying STY (53.7 mg mL−1 h−1).
To further increase the interest in the transformation, we developed a two-step concatenated sequence including the upstream preparation of difluoroacetamides 10a and 10b from ethyl bromodifluoroacetate (12) and amines 13a and 13b, therefore allowing the application of the reaction to commercially available starting materials (Fig. 5d). The amidation takes place in DMSO in a relatively short residence time (26 min at 100 °C) and at high concentrations (1 M for each reactant). The obtained solution can be used in the photocatalyzed reaction with only a slight modification of the residence time being required to reach the same conversion as before. The best conditions were applied successfully to a set of two indoles (9a and 9b) and two amines (13a and 13b) paving the way for the synthesis of larger libraries of functionalized indoles.
Conclusion
This study reports a convenient and affordable circular photochemical reactor for applications under continuous flow conditions. The entire setup relies on 3D-printed parts and widely available components, thus contributing to its affordability (∼800 EUR per unit in average with 4 wavelengths; ∼400 EUR for the reactor body only, without LEDs). We demonstrate here its suitability not only for reproducing photochemical protocols from the primary literature, but also for exploring new photochemical avenues under flow conditions. This low footprint reactor can accommodate internal PFA coils between low (e.g., 2 mL) to larger internal volumes (e.g., 20 mL) to expedite the transfer from exploration to preparative scales. The operating wavelength is easily adaptable through a convenient design which enables to quickly change LEDs. The ease for replication was showcased through the reproduction of the same experimental protocols between two research teams. A thorough step-by-step user guide is available in the ESI† to make it available to other chemistry labs, facilitating its widespread adoption. We also foresee the adaption of such an approach for cohorts of students in the practice of modern organic photochemistry.Further information
Details for building the photoreactor are available in the ESI,† which includes links for purchasing all the required materials. Additionally, 3D print .3mf files, Arduino code, and .kicad_pcb files to order the circuit board online are all provided on our GitHub repository: https://github.com/CiTOS-Photoreactor/Circular-Photoreactor.Experimental
General information
All chemicals and solvents were purchased from commercial sources and used without further purification unless otherwise stated (ESI,† Section S3.1). Structural identity was confirmed by 1H, 13C and 19F NMR spectroscopy (400 MHz Bruker Avance spectrometer) in CDCl3 or DMSO-d6.Photocatalytic oxidation of methionine in flow
A solution of (L)-methionine (1 equiv., 0.3 M) and rose bengal (0.1 mol%, 0.3 mM) in deionized water was pumped at 0.25 mL min−1 and conveyed with a stream of oxygen flow set at 1.88 mLN min−1 (1.1 equiv.). Mixing and irradiation (3 blue LEDs, 400 nm, 50 W each) occurred along the entire reaction channel (2.6 mL internal volume) under 4 bars of back pressure. After stabilizing the system for 20 min, a sample was collected and concentrated in vacuo. Afterwards, the reaction mixture was redissolved in deuterated water and analyzed by 1H NMR. More than 99% conversion towards methionine sulfoxide (2a) was obtained.Photocatalyzed α-alkylation of amines
Under an inert atmosphere, a solution of 2-phenyl-1,2,3,4-tetrahydroisoquinoline (1 equiv., 0.25 M), methyl vinyl ketone (2.0 equiv., 0.50 M), Ru(bpy)3Cl2·6H2O (0.02 equiv., 0.005 M) and TFA (1 equiv., 0.25 M) was prepared in acetonitrile. The solvent was degassed by bubbling with N2 and sonicating for 30 min prior to use. The solution was pumped into the photoreactor at 0.17 mL min−1 and irradiated at 400 nm all along the coil reactor (2.6 mL internal volume), with the temperature set at 40 °C. After stabilizing the system for 23 min, a sample was collected for 10 min and neutralized with K2CO3 (118 mg, 2 equiv.). The mixture was filtered through a silica plug employing Et2O as the eluent and concentrated in vacuo. Redissolution of the crude in deuterated chloroform allowed us to determine a 99% conversion by 1H NMR.Photocatalyzed cross-electrophile coupling via XAT
A solution of aryl bromide 6 (3.0 equiv., 0.15 M), iodoalkane 7 (1.0 equiv., 0.05 M), n-Bu3N (5.0 equiv., 0.25 M), 4CzIPN (0.05 equiv., 5.1 mM) and NiBr2(dtbbpy) (0.1 equiv., 2.5 mM) in 1,4-dioxane was pumped through the photoreactor at 0.025 mL min−1, irradiated at 405 nm, for 160 min for equilibration and collected for 210 min into an Erlenmeyer flask. The collected fraction was diluted with water (10 mL) and extracted with EtOAc (3 × 15 mL). The combined organic layers were washed with brine (3 × 30 mL), dried over Na2SO4, filtered and concentrated in vacuo. The crude reaction mixtures were purified by flash chromatography to afford the coupling product 8.Photocatalyzed difluoroamidation of indoles, with the upstream concatenation toward difluoroacetamides 11a and 11b
A feed solution of amines 13a and 13b (1.1 equiv., 1.1 M) and ethyl 2-bromo-2,2-difluoroacetate (12) (1.0 equiv., 1 M) in DMSO was pumped at 0.040 mL min−1 into a 2.1 mL PFA coil reactor heated at 100 °C. The exit feed was cooled down to room temperature through a PFA loop thermostated in water. A solution of indoles 9a and 9b (1.25 equiv. 0.42 M), 3DPA2FBN (2 mol%, 0.007 M) and N,N-dimethyl-p-toluidine (1.5 equiv., 0.50 M) in DMSO was pumped at 0.119 mL min−1 and mixed with the reactor effluent from the upstream amidation. The resulting solution was pumped through the photoreactor irradiated at 405 nm (30 °C). The setup was equilibrated for 110 min and collected for 10 min into a vial at room temperature. The collected fraction was diluted with water (5 mL) and the layers separated. The aqueous layer was extracted with iPrOAc (3 × 3 mL). The combined organic layers were washed with brine (3 × 5 mL), dried over Na2SO4, filtered and concentrated in vacuo. The reaction crude was purified by column chromatography to afford compound 11.Author contributions
YHT designed and performed the experiments, analyzed the results, and wrote the first draft of the manuscript and ESI.† MC designed the 3D-printed reactor and assembled the various parts. GC executed the experiments on Csp2–Csp3 cross-coupling. LT carried out the experiments on difluoroamidation; GM designed and performed the experiments and analyzed the results from difluoroamidation. GB designed the experiments, supervised GC's and LT's work, and wrote the corresponding sections of the ESI.† MD and FL supervised the work of GM. SG and JCMM supervised the overall research program and wrote the manuscript and the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
YHT acknowledges the University of Liège (Belgium) and the Special Funds for Research for the IPD-STEMA post-doctoral fellowship. MC acknowledges the European Union for an MSCA post-doctoral fellowship (Flow&Iron for Pharma, Grant No. 892287). We thank Michaël Schmitz (CiTOS, University of Liège, Belgium) for his participation in the photoreactor design. The authors affiliated with NovAliX and LIMA sincerely thank the “Fondation de la Maison de la Chimie”, particularly the SME innovation support program, for its financial support including a postdoctoral fellowship for GM.Notes and references
- C. Sambiagio and T. Noël, Trends Chem., 2020, 2, 92–106 CrossRef CAS.
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef.
- L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Chem. Rev., 2022, 122, 2752–2906 CrossRef CAS PubMed.
- G. Cecere, C. M. König, J. L. Alleva and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 11521–11524 CrossRef CAS.
- F. Meng, Y. Liu, J. Wang, X. Tan, H. Sun, S. Liu and S. Wang, J. Colloid Interface Sci., 2018, 532, 321–330 CrossRef CAS PubMed.
- S. Costacurta, G. D. Maso, R. Gallo, M. Guglielmi, G. Brusatin and P. Falcaro, ACS Appl. Mater. Interfaces, 2010, 2, 1294–1298 CrossRef CAS PubMed.
- S. K. Pagire, A. Hossain and O. Reiser, Org. Lett., 2018, 20, 648–651 CrossRef CAS.
- Y. Su, N. J. W. Straathof, V. Hessel and T. Noël, Chem. – Eur. J., 2014, 20, 10562–10589 CrossRef CAS.
- E. Kayahan, M. Jacobs, L. Braeken, L. C. J. Thomassen, S. Kuhn, T. Van Gerven and M. E. Leblebici, Beilstein J. Org. Chem., 2020, 16, 2484–2504 CrossRef CAS.
- N. Holmes, G. R. Akien, R. J. D. Savage, C. Stanetty, I. R. Baxendale, A. J. Blacker, B. A. Taylor, R. L. Woodward, R. E. Meadows and R. A. Bourne, React. Chem. Eng., 2016, 1, 96–100 RSC.
- S. Mostarda, P. Filipponi, R. Sardella, F. Venturoni, B. Natalini, R. Pellicciari and A. Gioiello, Org. Biomol. Chem., 2014, 12, 9592 RSC.
- J. P. McMullen and B. M. Wyvratt, React. Chem. Eng., 2022, 8, 137–151 RSC.
- F. Lévesque, M. J. Di Maso, K. Narsimhan, M. K. Wismer and J. R. Naber, Org. Process Res. Dev., 2020, 24, 2935–2940 CrossRef.
- L. D. Elliott, M. Berry, B. Harji, D. Klauber, J. Leonard and K. I. Booker-Milburn, Org. Process Res. Dev., 2016, 20, 1806–1811 CrossRef CAS.
- J. G. H. Hermens, M. L. Lepage, A. Kloekhorst, E. Keller, R. Bloem, M. Meijer and B. L. Feringa, React. Chem. Eng., 2022, 7, 2280–2284 RSC.
- A. Steiner, P. M. C. Roth, F. J. Strauss, G. Gauron, G. Tekautz, M. Winter, J. D. Williams and C. O. Kappe, Org. Process Res. Dev., 2020, 24, 2208–2216 CrossRef CAS.
- T. H. Rehm, S. Gros, P. Löb and A. Renken, React. Chem. Eng., 2016, 1, 636–648 RSC.
- S. Naskar, D. Kowalczyk, S. Mal, S. Das, D. Mandal, P. Kumar and D. Ziegenbalg, React. Chem. Eng., 2023, 8, 2211–2222 RSC.
- O. Shvydkiv, K. Jähnisch, N. Steinfeldt, A. Yavorskyy and M. Oelgemöller, Catal. Today, 2018, 308, 102–118 CrossRef CAS.
- D. S. Lee, Z. Amara, C. A. Clark, Z. Xu, B. Kakimpa, H. P. Morvan, S. J. Pickering, M. Poliakoff and M. W. George, Org. Process Res. Dev., 2017, 21, 1042–1050 CrossRef CAS.
- J. Valdés, J. L. Domínguez-Juárez, R. Nava, Á. Cuán and C. M. Cortés-Romero, Processes, 2021, 9, 2237 CrossRef.
- J. D. Williams and C. O. Kappe, Curr. Opin. Green Sustainable Chem., 2020, 25, 100351 CrossRef.
- A. Pomberger, Y. Mo, K. Y. Nandiwale, V. L. Schultz, R. Duvadie, R. I. Robinson, E. I. Altinoglu and K. F. Jensen, Org. Process Res. Dev., 2019, 23, 2699–2706 CrossRef CAS.
- R. S. A. E. Ali, J. Meng and X. Jiang, Org. Process Res. Dev., 2023 DOI:10.1021/acs.oprd.3c00328.
- K. C. Harper, E. G. Moschetta, S. V. Bordawekar and S. J. Wittenberger, ACS Cent. Sci., 2019, 5, 109–115 CrossRef CAS.
- M. Zhang and P. Roth, Curr. Opin. Chem. Eng., 2023, 39, 100897 CrossRef.
- T. H. Rehm, ChemPhotoChem, 2020, 4, 235–254 CrossRef CAS.
- M. Rößler and M. A. Liauw, Chem.: Methods, 2021, 1, 261–270 Search PubMed.
- J. M. Aguirre-Cortés, A. I. Moral-Rodríguez, E. Bailón-García, A. Davó-Quiñonero, A. F. Pérez-Cadenas and F. Carrasco-Marín, Appl. Mater. Today, 2023, 32, 101831 CrossRef.
- M. R. Penny and S. T. Hilton, J. Flow Chem., 2023, 1–8 Search PubMed.
- F. Schiel, C. Peinsipp, S. Kornigg and D. Böse, ChemPhotoChem, 2021, 5, 431–437 CrossRef CAS.
- R. Zhou, R. Han, M. Bingham, C. O'Rourke and A. Mills, Photochem. Photobiol. Sci., 2022, 21, 1585–1600 CrossRef CAS PubMed.
- F. A. Kucherov, L. V. Romashov and V. P. Ananikov, Chem. Eng. J., 2022, 430, 132670 CrossRef CAS.
- M. B. Montaner, M. R. Penny and S. T. Hilton, Digital Discovery, 2023, 2, 1797–1805 RSC.
- M. J. Harding, S. Brady, H. O'Connor, R. Lopez-Rodriguez, M. D. Edwards, S. Tracy, D. Dowling, G. Gibson, K. P. Girard and S. Ferguson, React. Chem. Eng., 2020, 5, 728–735 RSC.
- M. C. Maier, A. Valotta, K. Hiebler, S. Soritz, K. Gavric, B. Grabner and H. Gruber-Woelfler, Org. Process Res. Dev., 2020, 24, 2197–2207 CrossRef CAS.
- V. Sans, Curr. Opin. Green Sustainable Chem., 2020, 25, 100367 CrossRef.
- D. Kowalczyk, P. Li, A. Abbas, J. Eichhorn, P. Buday, M. Heiland, A. Pannwitz, F. H. Schacher, W. Weigand, C. Streb and D. Ziegenbalg, ChemPhotoChem, 2022, 6, e202200044 CrossRef CAS.
- C. A. Hone and C. O. Kappe, Chem.: Methods, 2021, 1, 454–467 CAS.
- P. P. Lampkin, B. J. Thompson and S. H. Gellman, Org. Lett., 2021, 23, 5277–5281 CrossRef CAS.
- J. A. Manson, A. D. Clayton, C. G. Niño, R. Labes, T. W. Chamberlain, A. J. Blacker, N. Kapur and R. A. Bourne, Chimia, 2019, 73, 817–822 CrossRef.
- T. Wan, Z. Wen, G. Laudadio, L. Capaldo, R. Lammers, J. A. Rincón, P. García-Losada, C. Mateos, M. O. Frederick, R. Broersma and T. Noël, ACS Cent. Sci., 2022, 8, 51–56 CrossRef CAS PubMed.
- H. E. Bonfield, K. Mercer, A. Diaz-Rodriguez, G. C. Cook, B. S. J. McKay, P. Slade, G. M. Taylor, W. X. Ooi, J. D. Williams, J. P. M. Roberts, J. A. Murphy, L. Schmermund, W. Kroutil, T. Mielke, J. Cartwright, G. Grogan and L. J. Edwards, ChemPhotoChem, 2020, 4, 45–51 CrossRef CAS.
- G. Sipos, E. E. Drinkel and R. Dorta, Chem. Soc. Rev., 2015, 44, 3834–3860 RSC.
- A. S. Surur, L. Schulig and A. Link, Arch. Pharm., 2019, 352, e1800248 Search PubMed.
- E. Wojaczyńska and J. Wojaczyński, Curr. Opin. Chem. Biol., 2023, 76, 102340 CrossRef PubMed.
- Y. Hai, M. Y. Wei, C. Y. Wang, Y. C. Gu and C. L. Shao, Mar. Life Sci. Technol., 2021, 3(4), 488–518 CrossRef CAS.
- S. Gan, J. Yin, Y. Yao, Y. Liu, D. Chang, D. Zhu and L. Shi, Org. Biomol. Chem., 2017, 15, 2647–2654 RSC.
- R. Mello, A. Olmos, A. Alcalde-Aragonés, A. Díaz-Rodríguez, M. E. González Núñez and G. Asensio, Eur. J. Org. Chem., 2010, 2010, 6200–6206 CrossRef.
- N. Emmanuel, C. Mendoza, M. Winter, C. R. Horn, A. Vizza, L. Dreesen, B. Heinrichs and J. C. M. Monbaliu, Org. Process Res. Dev., 2017, 21, 1435–1438 CrossRef CAS.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- Y. He, Z. Zheng, J. Yang, X. Zhang and X. Fan, Org. Chem. Front., 2021, 8, 4582–4606 RSC.
- K. R. Campos, Chem. Soc. Rev., 2007, 36, 1069–1084 RSC.
- Z. Li and C. J. Li, J. Am. Chem. Soc., 2004, 126, 11810–11811 CrossRef CAS.
- T. Shono, Y. Matsumura and K. Tsubata, J. Am. Chem. Soc., 1981, 103, 1172–1176 CrossRef CAS.
- S. Seel, T. Thaler, K. Takatsu, C. Zhang, H. Zipse, B. F. Straub, P. Mayer and P. Knochel, J. Am. Chem. Soc., 2011, 133, 4774–4777 CrossRef CAS PubMed.
- J. W. Beatty and C. R. J. Stephenson, Acc. Chem. Res., 2015, 48, 1474–1484 CrossRef CAS.
- P. Kohls, D. Jadhav, G. Pandey and O. Reiser, Org. Lett., 2012, 14, 672–675 CrossRef CAS.
- L. Ruiz Espelt, E. M. Wiensch and T. P. Yoon, J. Org. Chem., 2013, 78, 4107–4114 CrossRef CAS PubMed.
- T. D. Svejstrup, A. Chatterjee, D. Schekin, T. Wagner, J. Zach, M. J. Johansson, G. Bergonzini and B. König, ChemPhotoChem, 2021, 5, 808–814 CrossRef CAS.
- R. Zhang, G. Li, M. Wismer, P. Vachal, S. L. Colletti and Z.-C. Shi, ACS Med. Chem. Lett., 2018, 9, 773–777 CrossRef CAS.
- A. W. Dombrowski, N. J. Gesmundo, A. L. Aguirre, K. A. Sarris, J. M. Young, A. R. Bogdan, M. C. Martin, S. Gedeon and Y. Wang, ACS Med. Chem. Lett., 2020, 11, 597–604 CrossRef CAS.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS.
- P. Li, J. A. Terrett and J. R. Zbieg, ACS Med. Chem. Lett., 2020, 11, 2120–2130 CrossRef CAS PubMed.
- P. Zhang, C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084–8087 CrossRef CAS PubMed.
- H. A. Sakai, W. Liu, C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2020, 142, 11691–11697 CrossRef CAS PubMed.
- H.-W. Hsieh, C. W. Coley, L. M. Baumgartner, K. F. Jensen and R. I. Robinson, Org. Process Res. Dev., 2018, 22, 542–550 CrossRef CAS.
- A. Pomberger, Y. Mo, K. Y. Nandiwale, V. L. Schultz, R. Duvadie, R. I. Robinson, E. I. Altinoglu and K. F. Jensen, Org. Process Res. Dev., 2019, 23, 2699–2706 CrossRef CAS.
- W. Debrouwer, W. Kimpe, R. Dangreau, K. Huvaere, H. P. L. Gemoets, M. Mottaghi, S. Kuhn and K. Van Aken, Org. Process Res. Dev., 2020, 24, 2319–2325 CrossRef CAS.
- G. S. Yedase, A. K. Jha and V. R. Yatham, J. Org. Chem., 2022, 87, 5442–5450 CrossRef CAS.
- X. Tian, J. Kaur, S. Yakubov and J. P. Barham, ChemSusChem, 2022, 15, e202200906 CrossRef CAS.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed.
- M. Klaus, F. Christoph and D. François, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- R. P. Singh and J. M. Shreeve, Synthesis, 2002, 17, 2561–2578 Search PubMed.
- T. Aggarwal, Sushmita and A. K. Verma, Org. Chem. Front., 2021, 8, 6452–6468 RSC.
- D.-Q. Dong, H. Yang, J.-L. Shi, W.-J. Si, Z.-L. Wang and X.-M. Xu, Org. Chem. Front., 2020, 7, 2538–2575 RSC.
- E. Speckmeier, T. G. Fischer and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. Details for the construction of the photoreactor, setups, additional experimental details and characterization of compounds. See DOI: https://doi.org/10.1039/d4re00109e |
| This journal is © The Royal Society of Chemistry 2024 |