
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic systems: reactions, mechanism, and applications
Farzaneh Mohamadpour * and
Ali Mohammad Amani*
* and
Ali Mohammad Amani*
Department of Medical Nanotechnology, School of Advanced Medical Sciences and Technologies, Shiraz University of Medical Sciences, Shiraz, Iran. E-mail: f_mohamadpour@sums.ac.ir; mohamadpour.f.7@gmail.com; amani_a@sums.ac.ir; aliamani@sums.ac.ir
First published on 1st July 2024
Abstract
The photocatalytic field revolves around the utilization of photon energy to initiate various chemical reactions using non-adsorbing substrates, through processes such as single electron transfer, energy transfer, or atom transfer. The efficiency of this field depends on the capacity of a light-absorbing metal complex, organic molecule, or substance (commonly referred to as photocatalysts or PCs) to execute these processes. Photoredox techniques utilize photocatalysts, which possess the essential characteristic of functioning as both an oxidizing and a reducing agent upon activation. In addition, it is commonly observed that photocatalysts exhibit optimal performance when irradiated with low-energy light sources, while still retaining their catalytic activity under ambient temperatures. The implementation of photoredox catalysis has resuscitated an array of synthesis realms, including but not limited to radical chemistry and photochemistry, ultimately affording prospects for the development of the reactions. Also, photoredox catalysis is utilized to resolve numerous challenges encountered in medicinal chemistry, as well as natural product synthesis. Moreover, its applications extend across diverse domains encompassing organic chemistry and catalysis. The significance of photoredox catalysts is rooted in their utilization across various fields, including biomedicine, environmental pollution management, and water purification. Of course, recently, research has evaluated photocatalysts in terms of cost, recyclability, and pollution of some photocatalysts and dyes from an environmental point of view. According to these new studies, there is a need for critical studies and reviews on photocatalysts and photocatalytic processes to provide a solution to reduce these limitations. As a future perspective for research on photocatalysts, it is necessary to put the goals of researchers on studies to overcome the limitations of the application and efficiency of photocatalysts to promote their use on a large scale for the development of industrial activities. Given the significant implications of the subject matter, this review seeks to delve into the fundamental tenets of the photocatalyst domain and its associated practical use cases. This review endeavors to demonstrate the prospective of a powerful tool known as photochemical catalysis and elucidate its underlying tenets. Additionally, another goal of this review is to expound upon the various applications of photocatalysts.
1 Introduction
The usage of fossil fuels is causing environmental damage and perhaps irreversible anthropogenic climate change as a result of the world's growing need for energy due to population increase and the fast industrialization of developing countries. Photocatalytic technologies have shown a lot of promise in recent years for reducing environmental pollution and energy crises.1,2 One notable benefit of visible-light-driven photocatalysis is its ability to effectively use solar radiation's enormous energy content as a clean, affordable, and renewable driving force. Because visible-light-active photocatalysts are so simple to make and recycle using basic chemical processes, they have garnered a lot of interest. These photocatalysts' band gap energy determines how much light they can absorb.1 Presently, visible-light photoredox catalysis and photocatalytic technology are regarded as two of the most significant methods for addressing the world's energy and environmental problems. Synthetic organic chemists are always working to create novel organic reactions that may be carried out in environmentally benign, sustainable, and green settings, drawing inspiration from photosynthesis in plants.3,4The field of green and sustainable chemistry witnesses the active participation of both scholars, and entrepreneurs in the endeavors to formulate chemical processes that are deemed environmentally sound, economically viable, and highly effective. The utilization of radical chemistry has emerged as a powerful methodology in fabricating supple open shell reactive entities, consequently engendering a highly efficient approach for the prompt synthesis of intricate organic compounds. Various processes such as thermolysis, radiation, photolysis, electrolysis, and redox systems can serve as means for initiating radical reactions. Photocatalysis is a field that has been widely utilized in radical chemistry and is considered to be the most pure and auspicious approach among them.5 Visible light photocatalysis has seen a sharp rise in research activity in recent years, indicating that using visible light as a reagent in conjunction with catalysts has become a compelling topic of study for the development of effective and selective chemical transformations.6 In the past, photochemical reactions used only UV light to excite substrates or reagents by involving high energy of these light sources, which due to insufficient absorption of visible light by some molecules did not cover all reactions. This has changed with the introduction of low-energy photon photocatalysts (PC) that can be triggered, opening the door for environmentally acceptable and non-hazardous visible light to drive sustainable chemical synthesis.7 Light is used as an energy input in photocatalysis to perform different transformations, and light-absorbing photocatalysts, such as metal-photocatalysts and organophotocatalysts, are usually used in catalytic proportions. A substance known as a photocatalyst absorbs light raises its energy level, and then transfers that energy to a substance that is reacting to cause a chemical reaction. New chemical transformations can be developed thanks to the photophysical characteristics of photoredox catalysts, which provide visible light-mediated access to excited species that act as both reductants and oxidants.6
The present study's conclusions can be juxtaposed with the resurgence of photo-redox catalysis using visible light in organic synthesis, which provides novel opportunities for the establishment of synthetic routes using efficacious light-triggered reactions. The aforementioned attributes contribute to the widespread utilization of photo-redox catalysis in the comprehensive synthesis of intricate organic compounds, as well as the generation of pharmaceuticals and fundamental components.8 The utilization of photoredox catalysts in organic synthesis has gained significant relevance as an expedient method for the formation of C–C and C–heteroatom bonds through the single-electron transfer (SET)/photo-induced electron transfer (PET) mechanism.9 When photoredox catalysts absorb photons in the visible portion of the electromagnetic spectrum, they form stable photo-excited states.9 Because it can produce open-shell species under extremely mild reaction conditions and use them in numerous transformations that aren't always possible via ground-state reactivities, visible-light photocatalysis is one of the most fascinating and thriving areas of organic synthesis.10 Crucially, the latter also entails the late-stage editing of intricate molecular architectures, such as medications. As a result, visible-light photocatalysis has transcended academic boundaries and grown into a potent tool for numerous pharmaceutical companies, enabling them to either produce compounds more effectively or obtain access to entirely new and patentable chemical entities.11 Furthermore, photoredox catalysis can be viewed as a green chemistry tool because it uses light, which is a free and renewable energy source, requires little in the way of high temperatures, pressures, or harsh conditions, and requires little energy consumption.7 Simultaneously, by reducing the production of undesirable byproducts, the high chemoselectivities and functional group orthogonality aid in the purification processes and trash disposal. Therefore, combining the inherent green qualities of photoredox catalysis with visible light will accelerate the development of organic processes that are sustainable and useful for the synthesis of both drugs and materials. Furthermore, photocatalytic devices' unique capacity to reveal novel chemical behaviors and propel atypical reaction pathways opens up new avenues for future developments in the area.
Since light is the best source of energy, photochemistry and sustainability are inextricably linked.12–14 However, the synthetic promise of photochemistry—which opens up novel reaction pathways not accessible through traditional ground-state reactivity—also drew the attention of chemists. This is because excited molecules' chemical reactivity is essentially different from that of the ground state.15 But up until recently, photochemistry remained a niche field with few real-world uses. The dispersed absorption characteristics of organic molecules and the requirement for specialized, high-energy light apparatuses prevented the broad development of universal photochemical processes. The introduction of photoredox catalysis has resulted in a significant shift in the situation during the last years.12,16 This rapidly expanding field of synthetic chemistry uses organic or metal catalysts that absorb light to capture the energy of visible light and use it to speed up chemical reactions. When photoexcited catalysts are used to activate substrates by various processes such as energy transfer, photo-induced electron transfer, and atom transfer, they can, for instance, make it easier for access to highly reactive radicals under moderate circumstances. Innovation in photocatalysis—in which light is directly captured by a catalytic intermediate (direct excitation) to generate unique reactivity molecules—was spurred by the interest in photoredox catalysis. The possibilities presented by photochemical catalytic reactions were promptly acknowledged by the organic chemistry community. To produce molecules effectively and sustainably, a large number of academic and industrial researchers are now working on light-driven processes, many of which lack an equivalent in the ground-state domain.12 The utilization of photochemical reactions catalyzed by high-energy UV light has been a prominent technique in organic synthesis for more than a century, as documented by various sources.17–19 The remarkable advancements observed in the realm of photocatalytic reactions propelled by visible light are causing a resurgence in the utilization of photochemistry for the synthesis of organic molecules, encompassing intricately structured natural products,17 environmental sustainability, as well as the biomedical and pharmacological sectors. Additionally, these developments hold significant potential for critical applications such as water treatment.
1.1 A study of some review articles in the field of photocatalysts
Numerous photocatalysts are employed in organic synthesis nowadays. Because of their potent ability to absorb both visible and ultraviolet light, metal nanoparticle photocatalysts have garnered attention recently.20 Many examples of effective reactions triggered by light at room temperature or mild temperatures using supported nanoparticles of pure metals and metal alloys are available today. Here, Peiris et al.20 give a summary of current studies on the direct photocatalysis of supported metal nanoparticles for organic synthesis in the presence of light and talk about important reaction processes that result from light irradiation. In addition, organic dyes have been used as a compelling substitute for transition metal-catalyzed photocatalysis in recent years.21 Because it is inexpensive and readily available, rose bengal, an organic dye, has been used extensively as a photocatalyst in organic synthesis. The main photophysical characteristics of the dye and its synthetic uses in the synthesis of C–C and C–X (N, O, S, P, Si, and Se) bonds are outlined in this review by Sharma et al.21 The other review provides a succinct overview of heterogeneous photocatalysts with an emphasis on the relationship between the structural architecture and the photocatalytic activity and stability. Qu et al. investigated the progress, challenge, and perspective of heterogeneous photocatalysts in their other study.22Numerous articles have also reviewed the different uses of photocatalysts. The application of visible light-responsive photocatalytic technology in water treatment has the potential to increase water supply by safely using non-traditional water sources and to improve purifying efficiency.23 The review article by Dong et al.23 summarizes the recent advancements in the design and fabrication of visible light-responsive photocatalysts through a variety of synthetic strategies. These include doping, dye sensitization, heterostructure formation, and π-conjugated architecture modifications of conventional photocatalysts, as well as the significant efforts made in the search for novel visible light-responsive photocatalysts. Additionally discussed in connection to water treatment are the photocatalytic capabilities of the generated visible-light-responsive photocatalysts, including their ability to degrade organic compounds and inorganic contaminants as well as perform photocatalytic disinfection. Using titanium dioxide as a “model” semiconductor, Ibhadon et al. (2013)24 conducted a review of the latest advancements in the study and use of heterogeneous semiconductor photocatalysis for the treatment of low-level concentrations of pollutants in water and air. Because it directly uses solar energy to remove a variety of contaminants, heterogeneous photocatalysis is a sophisticated oxidation method that has attracted a lot of interest in the field of environmental remediation.2 Wang et al.2 address the principles of heterogeneous photocatalysis in this review concerning the environmental remediation of various pollutants, such as gaseous pollutants, solid wastes, pathogenic microorganisms, and H2O pollutants. Furthermore, possible semiconductors and methods for modifying them are methodically covered.
An environmentally benign method that has shown promise in the breakdown of numerous organic contaminants is photocatalysis.25 Koe et al.25 provided an overview of photocatalytic degradation, including photocatalysts, processes, and the creation of photocatalytic membranes. The mechanism, synthesis techniques for biomass-supported photocatalysts, photocatalytic degradation of organic materials in actual wastewater, photocatalytic reactor designs and their operating parameters, and the most recent advancement in photocatalyst integrated membrane were all covered in this review.25 Examples of how different medications degrade in laboratories have been evaluated.26 Velempini et al. have examined several strategies for altering metal oxides and synthesis techniques to increase degrading efficiency.26 A compilation and comparison of the effects of operational and experimental parameters on the deterioration process has been conducted. Lastly, a brief synopsis of the pilot-scale deterioration of medications is also included. The photocatalytic degradation of six psychiatric medications—carbamazepine, sertraline, amisulpride, amitriptyline, diazepam, and alprazolam—was examined and evaluated in 2024 by Mohamadpour et al.27 They studied these photocatalytic degradations' effectiveness, route, and mechanism. The underlying mechanism of photocatalytic degradation of dyes has been elaborated in the other review article.28 The effects of various experimental parameters like the initial concentration of dyes, pH of the solution, and catalyst dosage on the performance of photocatalytic degradation have been discussed. In 2022, Saeed et al.28 reviewed the photocatalysts used for photodegradation of dyes in the aqueous medium.
In essence, water splitting is the electrolysis of water, which is the reduction of water to hydrogen at the cathode and its oxidation to oxygen at the anode.29 In the other study, Acar et al.30 address the state of the art and potential directions for future research in the development of photocatalysts for visible-light-driven water-splitting hydrogen generation. Results of a technical, financial, environmental, and health impact assessment and ranking of photocatalyst groups based on SrTiO3, TiO2, Zn/In/S, Ta/O, Cd/S/Zn, K/Ti/O, and Ga/Zn/O are reported.30
As presented above, most of the review articles published in the field of photocatalysts have studied one or a few fields of this vast field. There is a need to examine both the fundamental principles in the field of photocatalysts and photocatalytic reactions, as well as the study of reactions and the mechanism of activity, and also, mention some examples of the activities of photocatalysts in various fields such as biomedicine, environmental pollution management, and water splitting, etc. Our aim in this review is to show the potential of a powerful tool called photochemical catalysis and to clarify its basic principles, fundamentals of photocatalysts, mechanism of photochemical reactions, and recent advances in the development of their applications.
1.2. Summary
In recent years, photocatalytic processes and the use of solar energy and visible light as renewable energy sources are environmentally friendly methods that are consistent with universities and industry's environmental awareness when energy-related crises, the use of fossil fuels, and environmental pollution become pandemic problems. Photocatalysts in photocatalytic processes raise their energy level and start fresh chemical reactions by absorbing light. Because it needs moderate reaction conditions, few side products, renewable energy sources, and very little photocatalyst, this approach is exceptionally useful in the disciplines of organic synthesis, biomedicine, and environmental pollutant transformation. Furthermore, it doesn't need for high temperatures.2. History of photocatalysis
The term “photocatalysis” originates from the Greek language and is formed by combining two words: “photo” (which means “light” in Greek) and “catalysis” (derived from the Greek verb “katalyo”, meaning “to break apart” or “to decompose”).31 In the realm of scientific discourse, photocatalysis denotes a phenomenon wherein a photocatalyst is stimulated by light, leading to an expedited rate of chemical reaction without the need for direct participation of the photocatalyst. The term “photocatalysis” has been a subject of substantial confusion and debate regarding its precise definition. However, it has been officially determined by the International Union of Pure and Applied Chemistry (IUPAC) that the term “photocatalysis” is restricted to reactions that occur in the presence of a semiconductor and illumination. Photocatalysis was first documented in the book Allgemeine Photochemie by Plotnikow in 1936.32 After an elapsed period of nearly forty years, certain scholars initiated inquiries into the surface examination of photocatalysts, including but not limited to TiO2 and ZnO. During this period, certain individuals within the workforce postulated the feasibility of utilizing solar radiation as a potential source of power. In 1972, Fujishima and Honda33 performed an experiment involving the photolysis of water within a photoelectrochemical cell utilizing a semiconductor electrode composed of TiO2. This seminal work introduced significant impetus to the study of photocatalysis. This marked the genuine outset of the corresponding field. During the 1980s and 1990s, an extensive body of work was carried out to elucidate the underlying kinetics and to enhance the photocatalytic efficacy of titania. Recently, there has been a trend towards employing semiconductor materials as a photocatalyst to address issues concerning the purification of air and water sources, as well as in the elimination of toxic organic pollutants, and both industrial and medical applications.31,342.1. Homogeneous photocatalysts
The phenomenon wherein the reactant and photocatalyst are present in the identical phase is referred to as homogeneous photocatalysis in scientific discourse. Homogeneous photocatalysts, such as coordination compounds, dyes, and natural pigments, have been widely recognized as prevalent agents in catalytic processes.31 For organic synthesis, homogeneous photocatalysis is a tried-and-true method.35 Strada et al.36 provided a workable substitute for the employment of strong acids and unforgiving conditions by making use of the acidity of photo-excited molecules to promote C–C and C–S bond formation activities, as described in the reference article.35 Iron-based organometallic complexes have been used under visible light irradiation to reduce CO2 into CO.37 While ruthenium complexes with pyridine–quinoline or terpyridine ligands were used in the atom transfer radical addition of haloalkanes to olefins,38 the effects of certain reaction parameters were studied for the photodimerization of 2-anthracenecarboxylate catalyzed by platinum(II) complexes.392.2. Heterogeneous photocatalysts
In the present category, the reactant and photocatalyst are present in distinct phases. Heterogeneous photocatalysts, specifically transition metal chalcogenides, are widely prevalent and possess distinctive features.31 One example of an advanced oxidation process (AOP) that has received a lot of attention in recent years is heterogeneous photocatalysis, which is used to remove medicinal chemicals from aqueous media.40 In this environmental application, a semiconductor solid catalyst that is activated by light with a specific energy is present, and this accelerates the breakdown of contaminants (in the aqueous phase). Generally speaking, a wide range of medications can be oxidized by highly reactive species produced by heterogeneous photocatalysis, primarily hydroxyl radicals (˙OH), holes (h+), superoxide ions (O2˙−), and peroxide radicals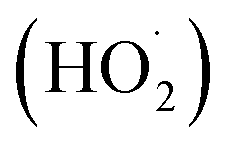 . The organic contaminant is successfully mineralized in its whole in several instances.41–43 Much study has been done on the subject in both water and municipal wastewater. Pharmaceuticals found in actual and artificial hospital wastewater have been removed so far using solid semiconductors such as tungsten oxide (WO3), zinc oxide (ZnO), zirconium oxide (ZrO2), titanium dioxide (TiO2), and cerium oxide (CeO2). In addition, g-C3N4 and g-C3N4/SrTiO3, immobilized TiO2, immobilized ZnO, doped TiO2, composite catalysts (cadmium sulfide (CdS)/TiO2), and graphene-based photocatalysts have also been employed.40
. The organic contaminant is successfully mineralized in its whole in several instances.41–43 Much study has been done on the subject in both water and municipal wastewater. Pharmaceuticals found in actual and artificial hospital wastewater have been removed so far using solid semiconductors such as tungsten oxide (WO3), zinc oxide (ZnO), zirconium oxide (ZrO2), titanium dioxide (TiO2), and cerium oxide (CeO2). In addition, g-C3N4 and g-C3N4/SrTiO3, immobilized TiO2, immobilized ZnO, doped TiO2, composite catalysts (cadmium sulfide (CdS)/TiO2), and graphene-based photocatalysts have also been employed.40
2.3. Coordination compounds
Metal complexes have long been recognized to be photosensitive.93,94 Scheele's (1772) publication on the impact of light on AgCl was the first to have some scientific qualities, and by the 1830 s, photography was starting to gain traction in some nations.95 Very early on, it was also recognized that other metal complexes, in particular [Fe(CN)6]4−, were light-sensitive.94 Research on Fe3+ and UO22+ complexes was conducted in the next years to find practical chemical actinometers.96 Some quantitative investigations on the photochemical behavior of [Fe(CN)6]4− (ref. 93) have also been published; however, any mechanistic explanation of the observed photoreactions was hindered by the absence of a theory regarding the absorption spectra and the nature of the excited states. The ligand field theory research97 and the initial attempts to explain the charge-transfer bands98,99 allowed for the beginning of the interpretation of the absorption spectra in the late 1950s. When a number of metal complexes of the bipyridine type were described spectroscopically in the late 1960s, [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) was found to have red luminescence, which was attributed to its lowest triplet metal-to-ligand charge-transfer excited state, 3MLCT.100,101 Not long later, it was discovered that [Ru(bpy)3]2+ may act as a photocatalyst for the breakdown of water into hydrogen and oxygen due to its photoredox characteristics.102 Coordination compound utilization as photocatalysts for organic pollutant degradation,103,104 CO2 reduction,105 and Cr(VI) reduction106,107 has been the subject of several publications published recently. When compared to conventional semiconductor photocatalysts such as TiO2, ZnO, Fe2O3, CdS, GaP, and ZnS, photocatalytic coordination compounds have several advantages. These include the ability to clarify the structure–property relationships of solid photocatalysts due to their well-defined crystalline structures; the ability to rationally design and fine-tune these catalysts at the molecular level due to their modular nature; and the ability to quickly transport guest molecules through open channels due to their intrinsic porosity and high surface area.103,104According to the findings examined in the literature research,105 coordination compound photochemistry has progressed through four phases thus far: (1) studies of intramolecular photochemical reactions (ligand photosubstitution, photoredox breakdown, photoisomerization); (2) finding of intrinsically photo-stable, luminescent compounds and their application in bimolecular energy and electron transfer processes; (3) integration of photostable and luminescent complexes in supramolecular (multi-component) systems to comprehend the importance of nuclear and electronic factors in dictating the rates of energy and electron transfer processes; (4) utilization of coordination compounds as fundamental building blocks for the bottom-up development of straightforward, light-powered nanoscale machines and devices.
Mixed ligands have been introduced to achieve a new level of rational design due to the synergistic interaction of diverse ligands with metals and subsequent networking, resulting in more complex and innovative structures. The solubilities of the ligands, their competition for coordination with the metal ions, thermodynamic and dynamic equilibria, and other factors are important concerns in such systems.108 In addition to their diverse coordination modes, multicarboxylate ligands like (H4qptb), (H2dczpb), and (H2odpa) have been employed as multifunctional organic ligands and counterions because of their capacity to function as hydrogen-bond donors and acceptors during the assembly of supramolecular structures.103,109–112
2.4. Dyes and pigments
Over 4000 years have passed since the beginning of the textile dying business.113 Natural sources provided the dyes for all except the last 150 years.114 The major shift in dyes occurred after William Henry Perkin discovered Mauveine in 1856 while attempting to figure out how to synthesize Quinine, a medication that treats malaria, a disease that plagues many tropical nations.115 Thus, Henry Perkin created a new type of dye in an attempt to manufacture quinine.116–119 The 21st century has seen a surge in the use of color prediction by several large pigment and dye manufacturers. Pigments are insoluble and stay in the form of particles, while dyes are synthetic organic compounds that dissolve in water or oil.120 Various organic dyes are used in textiles to color various items.121 Their chemical structures are numerous and include colors with highly distinct chemical and physical characteristics, such as azo and nitro dyes, phthalocyanine dyes, and diarylmethane dyes.122 Complex unsaturated chemical compounds called dyes are what absorb light and give the visible world color.123 Furthermore, a dye is a material with a chromophore—the ability to absorb a portion of the visible spectrum. The portion of light that is refracted rather than absorbed by the dye determines the hue. Conjugated double bonds provide a chemical structure that is advantageous for light absorption. Thus, aromatic amines are frequently found in colorants.124 The ability of dyes to absorb light in the visible range (380–750 nm) is what defines them. Chromatophoric groups, which are specific clusters of atoms, selectively absorb energy and change white light into colored light when it reflects off of a body or diffuses through the air. The chemical makeup of the various chemicals is related to how intensely colored they are.125 A dye is a substance that can absorb specific light radiations and then reflect the colors that are complimentary to them.126 Natural substances like beetroot are used to make colors before the middle of the 19th century.127–129 Plants, insects, and animals as well as minerals can be used to make natural dyes.130 They produce effluent that may be handled by biodegradation and are often less toxic and allergic than synthetic colors.131 The chromophore, autochrome, and matrix are the three key groups that make up the molecules of dyes, which are organic compounds.132 The chromophore, which is the dye's active site, can condense the spatial localization of atoms that absorb light energy. Chromophore-containing compounds are widely used in surgery, and synthetic dyes are becoming more and more common than naturally occurring optical adjuncts.113,133 Organic dyes are one of the numerous substances that have been and are currently used to assess photocatalytic activity. Typically, dyes are grouped based on their chromophores. The primary dye categories are included in Table 1,134 along with an estimate of the number of publications on the photocatalytic degradation of each dye under UV and visible light. Using “photocatalysis,” “visible light” (or “UV light”), and “a specific name of dye (in both its formal and its commercial name)” as keywords, the calculation is based on the SciFinderTM data source. The dyes that have been examined the most are thiazine dyes (methylene blue predominates in 37% of thiazine papers), followed by xanthenes (rhodamine B predominates in 30% of xanthenes manuscripts). Azo dyes rank only fourth in the world, although controlling between 50% and 70% of the market worldwide and, therefore, significantly contributing to the environmental problem. As can be seen from the chart, there are more papers on visible light photocatalysis of dyes than there are on UV light photocatalysis for every category of dye. For xanthenes (2.18) and thiazines (1.80), the ratio of manuscripts on visible light photocatalysis to UV light photocatalysis is greater. For azo dyes, the ratio is very low (1.56), as reported in ref. 134.| Class | UV | Visible |
|---|---|---|
| Anthraquinones | 238 | 390 |
| Azo dyes | 1285 | 2006 |
| Natural dyes | 187 | 303 |
| Thiazines | 7496 | 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 471 471 |
| Triarylmethanes | 1439 | 2758 |
| Xanthenes | 5625 | 12244 |
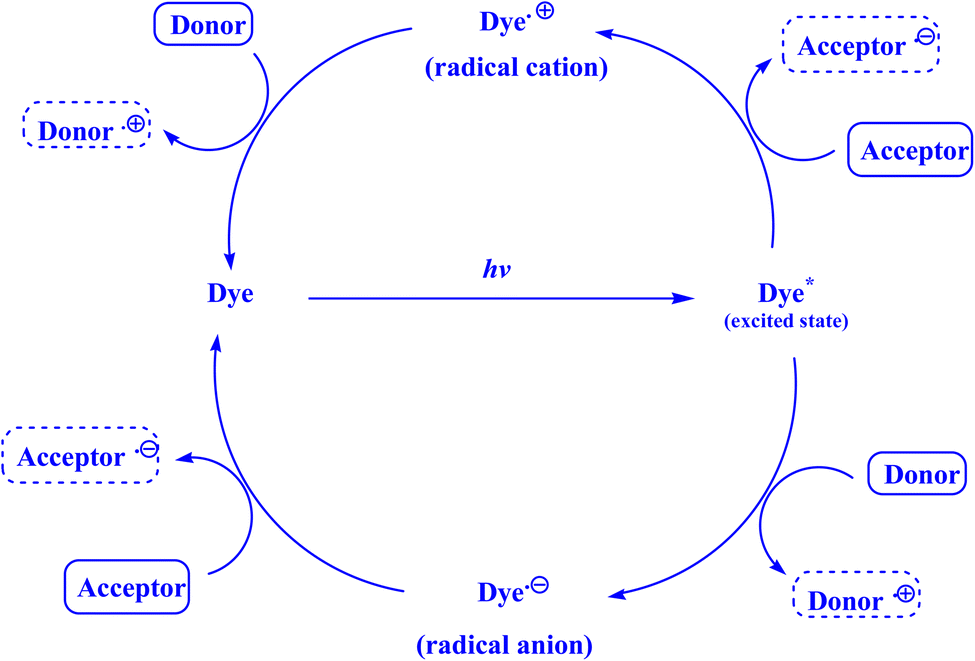 | ||
| Fig. 1 Photoredox cycle catalyzed by dye.135 | ||
2.5. Summary
The photocatalyst is stimulated by light radiation, which speeds up chemical reactions. Photocatalysts, both homogeneous and heterogeneous, are widely used to regulate bond formation, CO2 reduction, complex oxidation, breakdown of environmental pollutants, and extraction of medicinal components from wastewater. The unique properties of graphitic carbon nitride (g-CN), including its lack of metals, and metal–organic frameworks, or MOFs, are readily deformed by external stimuli due to its flexible structure and porous crystal nature. Coordination compounds, which are unsaturated organic substances that add color to the visible region by absorbing light, and dyes, which are unsaturated organic substances that facilitate the rapid transport of guest molecules through the open channel, are among the compounds whose photochemistry has been studied and have wide applications in photocatalytic processes.3. Visible light and photoredox catalysts
Approximately one hundred years prior, Ciamician realized that “light” possesses the potential to serve as a plentiful and sustainable means of energy for the execution of eco-friendly chemical reactions.13 Photochemistry and photocatalysis, facilitated by photoinduced electron transfer (PET) have demonstrated significant applicability in the field of organic synthesis.137–139 The constraints posed by the insufficient absorption of visible light by several organic molecules have impeded the widespread utilization of photochemical synthesis. The utilization of visible light-absorbing photocatalysts and their associated electron/energy transfer mechanisms to elicit photochemical reactions in organic molecules represents a promising approach to surmounting the aforementioned constraint.137 Recently, synthetic chemists have discovered that photoredox catalysts are effective instruments for harnessing the energy derived from the absorption of low-energy light in the visible spectrum to start a range of organic reactions. Synthetic chemists' perspectives on photochemistry and redox manipulations of organic molecules have undergone a paradigm shift in the last few years due to the development of techniques based on the single electron transfer characteristics of photoredox catalysts.140 Photoredox arylations mediated by visible light can occur in extremely mild settings, making them a desirable substitute.141 Using photoredox catalysts (PCs) made of precious metals, photoredox catalysis was a flexible method for creating difficult covalent bonds under mild reaction conditions. Therefore, the development of organic equivalents was necessary as long-term substitutes.142 With the absence of costly and hazardous transition metals, remarkable advancements have been achieved in the creation of organic photoredox catalytic cascade reactions under visible light irradiation. The in situ production of different radical species in the photoredox catalytic cycles is crucial to these conversions. Using this effective and long-lasting procedure, many building blocks with potential applications in chemistry and biology have been created.143 Strongly decreasing, visible-light organic PCs are still lacking, despite the introduction of numerous organic PCs recently.1423.1. Photochemical reactions caused by visible light
In recent years, the utilization of visible light-mediated reactions has become increasingly recognized as a highly effective approach for instigating diverse organic transformations in a gentle manner. Consequently, this methodology has attained considerable significance in the realm of sustainable chemistry.144 Photoredox catalysis has been widely recognized as a notable breakthrough in chemical synthesis, offering an effective approach for inducing radical cyclization6 as well as enabling the development of innovative pathways for C–C and C–X bond formation in heterocyclic chemistry.145,146 Furthermore, this element has been discovered to possess a multitude of significant utilities in various fields, including green synthesis,137 radical chemistry, and photochemistry. Photoredox catalysts leverage a sustainable energy source to facilitate chemical reactions by absorbing visible light radiation and transferring it to low-energy organic substrates via a mechanism known as single-electron transfer (SET).73.2. Summary
Ecologically friendly organic synthesis is made possible by photochemical and photocatalysis processes, which harness the power of light. Often, an electron transfer mechanism or energy transfer is used with a radical cyclization method to accomplish these syntheses. Due to their immense capacity to absorb visible light, convert solar energy into chemical energy, and then transfer that energy to low-energy organic substrates through a process known as SET, photoredox catalysts are now widely used in water splitting and solar energy storage. Additionally, photoredox catalysts create novel pathways for the synthesis of complex compounds with possible medicinal applications by generating C–C and C-X bonds in heterocyclic chemistry.4. Photochemical approaches
One potent tactic that enables the creation of a high degree of molecular complexity from relatively simple building blocks in a single step is the application of photochemical reactions. A fundamental characteristic of all changes induced by light is the presence of electrically excited states, which are produced when photons are absorbed. This results in the production of transient reactive intermediates and modifies a chemical compound's reactivity considerably. Thus, the energy input from light provides a way to create strained and distinct target compounds that are not amenable to assembly through thermal protocols. With the use of photochemical transformations, one may easily obtain scaffolds with a variety of structural and stereochemical properties. Complex polycyclic carbon skeletons with remarkable efficiency are potentially achievable by synthetic designs based on photochemical reactions, and are highly valuable in total synthesis.18 Promising options for environmentally benign organic synthesis are photoinitiated reactions. Making somewhat stable organic compounds reactive is the main goal of organic chemistry. This can be achieved by cleaving a covalent link, polarizing it (for example, by complexation or by generating a hydrogen bond), or weakening a covalent bond (for example, via complexation or adsorption on a catalyst surface). The deprotonation of a carbonyl or carboxyl derivative to form an enolate is an example of the final option, which requires more energy and control over the medium and is likely the most frequently employed method for the formation of the carbon–carbon bond.147 Since a chemical compound's electron configuration determines its reactivity, it is clear that the photochemical reaction conditions significantly increase the number of transformations of a compound family. Photochemical reactivity is frequently complementary to ground-state chemistry.148 A molecule's electron configuration changes as a result of light absorption. The utilization of light-emitting diodes (LEDs) exhibiting a capacity to generate high-intensity visible light in a limited spectral range for each color has facilitated the availability of cost-effective and energy-efficient lighting options for photocatalytic applications. This development has been acknowledged by the scientific community and has been documented extensively.149 Due to the lower energy content of visible light compared to ultraviolet irradiation, classical photochemical reactions often exhibit a higher degree of selectivity, predictability, and controllability.1494.1. Summary
The desire to employ visible light as a sustainable energy source and to optimize the energy efficiency of chemical reactions has led to an increase in the use of photocatalytic processes. The effectiveness of photocatalysis has been attributed to the use of light-emitting diodes (LEDs) that can produce high-intensity visible light in a defined spectral range for each color. These components will open the door for the process's industrial use by allowing photochemical reactions to happen more often and on a greater scale.5. Fundamentals of radical reactions
Radicals are known to be exceedingly active intermediate species that exhibit reactivity towards a wide range of organic molecules, including solvents. Notably, the characteristic that distinguishes radicals is their swift reactivity, commonly at rates regulated by diffusion, when they encounter one another. Thus, the greater the extent to which radicals are generated, the swifter their dissipation will occur. “Radicals are characterized by their impatience, as a succinct descriptor.” The positive aspect of impatience can be attributed to the proactive nature of individuals with radical views, who display a distinct inclination towards swift action and execution.150,151 Free radical species have long been recognized as intermediaries in a wide range of chemical processes. Due primarily to their lack of selectivity, their employment in the synthesis of specialty chemicals (homogeneous, low molecular weight compounds) has been restricted. Nonetheless, several carefully “designed” radical reactions that produce high yields of targeted chemicals have been found in recent years. It is now evident that free radical reactions have become an essential component of synthetic methods in many laboratories due to the significant growth in interest in this area of organic synthesis.152 Compared to more traditional ionic transformations, radical reactions are known to have a variety of benefits.153,154 As part of this, mild and neutral reaction conditions are used instead of basic or acidic ones, which might encourage the breakdown or epimerization of delicate organic molecules in ionic processes. Numerous effective radical reactions, such as tandem and cascade sequences, have been discovered.155 These reactions can produce many carbon–carbon bonds in quick “one-pot” transformations. Because of the misconception that all radical reactions are unpredictable and nonselective, the use of radicals in synthesis has frequently been overlooked despite these benefits. This is especially true for the stereoselective synthesis of asymmetric compounds, where the synthetic chemist's preferred tools are ionic and pericyclic processes. But things are slowly starting to change, and it turns out that there are a number of highly stereoselective radical reactions that potentially provide an alternative to traditional techniques.156 These advancements have been made feasible by our growing understanding of the key variables influencing radical processes. This involves a deeper comprehension of the significance of radical polarity, steric effects, stereoelectronic effects, and bond dissociation energies in radical reactions.153,1575.1. Summary
These days, radical chemistry and radical reactions have a special place in chemical processes and heterocyclic chemistry due to their high reactivity. From the point of view of chemical synthesis, radical reactions have two advantages: high yields and rapid addition to molecules. Because of this feature, free radical species have been known as intermediates of chemical reactions for years.6. Mechanism of activity of photoredox catalysts
When given the proper electronic structure, organic and organometallic complexes can function as visible-light photosensitizers (PS) or photocatalysts, capturing visible photons and facilitating the transfer of electrons or energy to an organic substrate or another catalyst for subsequent usage.158 In the past century, a multitude of unconventional bond formations in organic chemistry have been made possible by the identification, advancement, and use of light-mediated catalysis. The study of photocatalysis has had a notable resurgence in recent times, and this has led to the development of numerous novel bond-forming protocols and synthetic techniques.16 In the field of photocatalysis, photonic energy is directed toward a specially designed photon-absorbing catalyst (a photocatalyst), which, when excited, can cause a companion substrate, reagent, or secondary catalyst to participate in novel reaction pathways that were not previously possible under thermal control. This growing list of generic activation modes is known as photocatalysis. The most popular methods used by photocatalysts to selectively activate molecules while converting light into chemical energy are (i) energy transfer, (ii) organometallic excitation, (iii) light-induced atom transfer, and (iv) photoredox catalysis.16Transition metal complexes and organic dyes are two forms of photoredox catalysts (PC) that can valorize sunlight as a cheap, abundant, and clean renewable energy source. Visible light irradiation of the PC results in long-lifetime photoexcited states (PC*). The photoexcited state (PC*) can be readily involved in a bimolecular electron-transfer event to release or capture one electron to or from the molecule, in contrast to the ground state (PC).159,160 PC* took an electron from a reductant [Red] or a substrate [Sub] during the reductive quenching cycles, producing the radical anion [PC]˙− and then oxidizing it (Fig. 2a). Most of the time, photoredox processes involving oxidative quenching cycles are used to functionalize C–H bonds. In this case, the photocatalyst [PC]˙+ produces a radical cation that is subsequently reduced when PC* donates an electron to either the substrate [Sub] or an oxidant [Ox] in the reaction mixture (Fig. 2b). Such single electron transfer (SET) occurrences have led to improved radical generation and provide a fresh life to radical transformation in synthesis.159
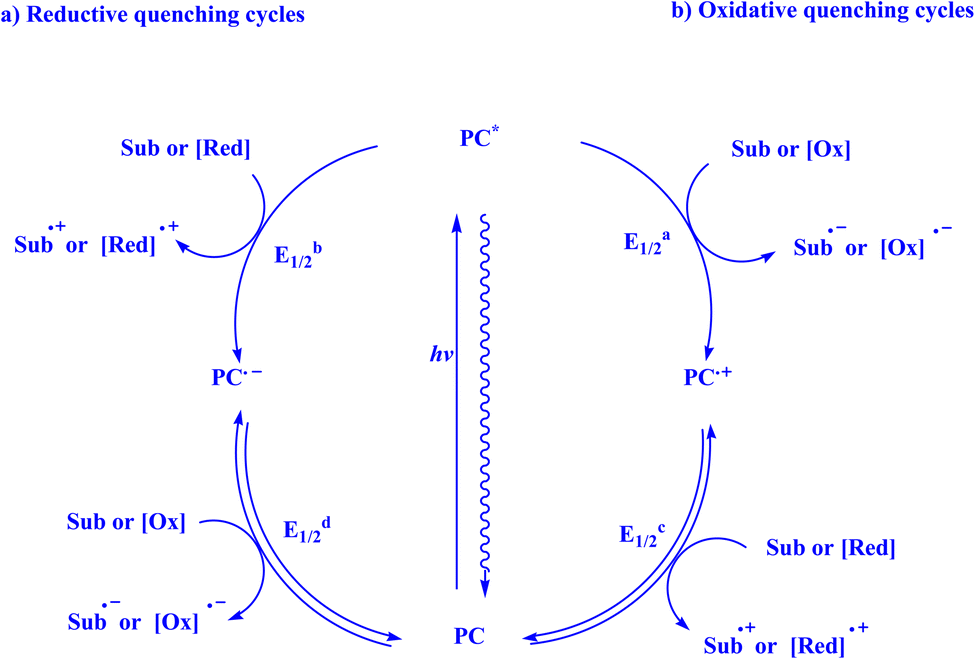 | ||
| Fig. 2 Quenching route of photoredox catalyst.159 | ||
6.1. Summary
The development of photocatalysis has made it possible to perform complex reactions and create new bonds. This occurs when the photocatalyst absorbs a photon of visible light and transitions from its ground state to its excited state, potentially involving the participation of a substrate, reagent, or secondary catalyst in the reaction pathways. A new partnership that was previously unattainable. The cheap and renewable energy of visible light is converted into chemical energy during this process.7. Fundamentals of photocatalytic reactions
The literature9,161 indicates that the use of photo-redox catalysts for the formation of C–C and C–heteroatom bonds via the single-electron transfer (SET)/photo-induced electron transfer (PET) pathway has become more significant in contemporary organic synthesis. Visible light absorption allows photoredox catalysts to reach their stable photoexcited states.9 Visible light and dual photosensitized electrochemical technology are used in a variety of flow reactors162 that have been found, leading to designs that are affordable, environmentally friendly, and extremely efficient.9,1637.1. Photoinduced-electron transfer (PET)
In contrast to ground states, electronically stimulated states simultaneously exhibit much higher reduction and oxidation characteristics.164 Marcus's study,165 which won him a Nobel Prize, contributed to the understanding of photo-induced electron transfer (PET) as a crucial stage in many photochemical processes.164 A charge-separated state made up of the acceptor radical anion and the donor radical cation is formed via photoinduced electron transfer (PET) from a donor (D) to an acceptor (A).166–168 D and A coexist in the same molecule during an intramolecular PET process, but they correspond to distinct molecules during an intermolecular PET.166 To effectively quench PET with a separation between donor and acceptor on the sub-nanometer length scale, contact formation (van der Waals contact) is required.169 Also, PET plays a crucial role in several chemical and biological contexts, such as photovoltaics,170 photoluminescence sensors and switches,171,172 as well as artificial and natural photosynthesis.166,173The present study174 delineates the general framework of photoinitiated PET (photoinduced electron transfer) processes used for synthetic purposes. This framework is illustrated by a catalytic triangle, as depicted in Fig. 3,174 and comprises three successive stages, specifically photoexcitation (induced either through direct means or by sensitization), electron transfer, and the re-establishment of the ground state by the catalytically active species. Electronically excited states possess unique properties, namely their ability to function as both superior oxidants and reductants. As such, the quenching of these states may transpire through either oxidative or reductive quenching pathways, as previously noted.174 The aforementioned adjectives pertain to the transformation of the catalyst/sensitizer into the respective ion radicals, namely the anion radical through reductive quenching and the cation radical via oxidative quenching. The acoustic emission spectroscopy data, as presented in Fig. 4,174 illustrates the phenomenon of oxidative quenching, which is observed alongside the reductive activation of the acceptor A.174 The process of terminal restoration can be initiated through the utilization of a sacrificial donor or through the application of negatively charged product molecules, which can effectively mitigate the catalyst radical cation, as reported in the literature174 (Fig. 5).
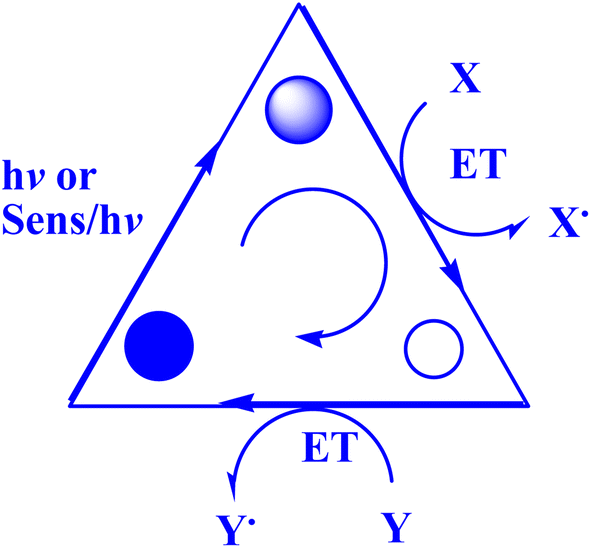 | ||
| Fig. 3 Photocatalytic triangle.174 | ||
 | ||
| Fig. 4 Oxidative quenching-reductive activation.174 | ||
 | ||
| Fig. 5 Reductive quenching-oxidative activation.174 | ||
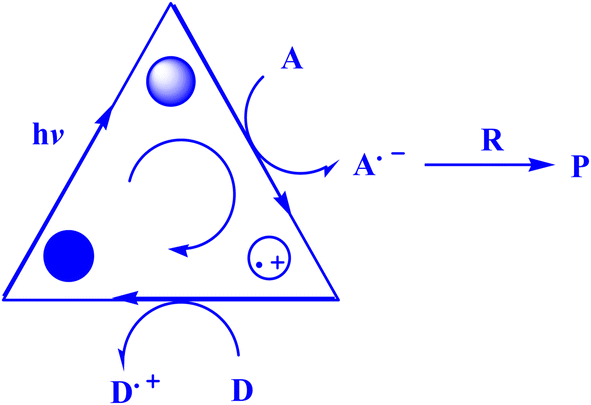
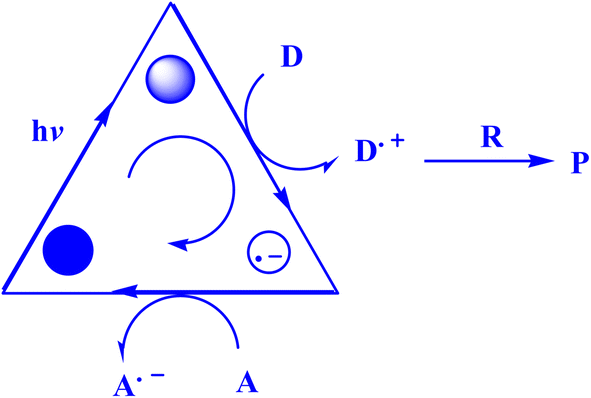
Recently, AYG has been used for the radical metal-free synthesis of tetrahydrobenzo[b]pyran scaffolds (Fig. 6),161 and synthesis of dihydropyrano[2,3-c]pyrazoles (Fig. 7).188
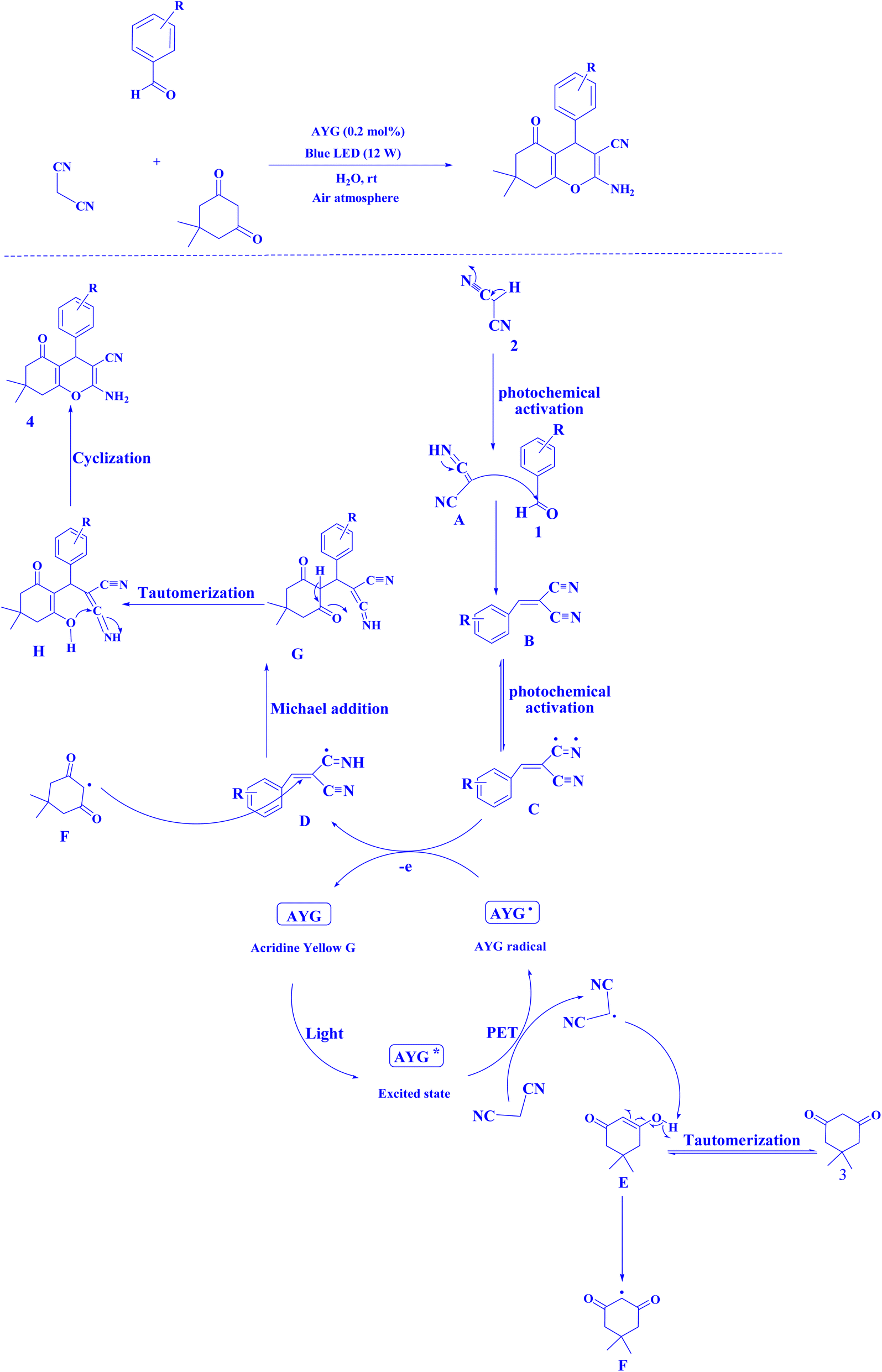 | ||
| Fig. 6 An approach to the synthesis of tetrahydrobenzo[b]pyran scaffolds.161 | ||
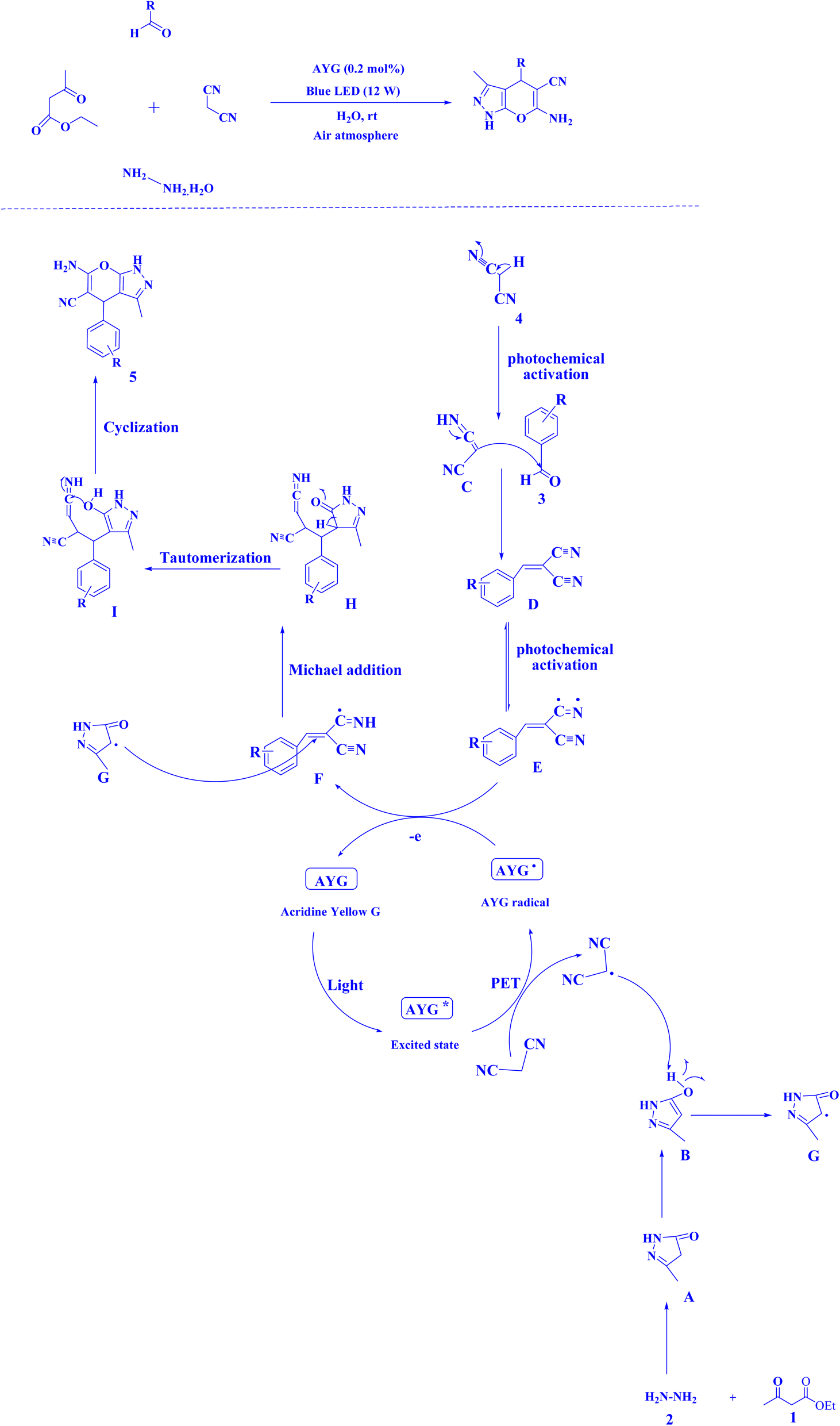 | ||
| Fig. 7 Synthesis of dihydropyrano[2,3-c]pyrazole scaffolds.188 | ||
7.2. Single-electron transfer (SET) and energy transfer (EnT)
To support a range of synthetic reactions, organic photocatalysis has flourishingly grown to rely on bimolecular energy transfer (EnT) or oxidative/reductive electron transfer (ET).189 Utilizing electron transfer (ET) vacancies as a coexisting strategy, EnT photo-organocatalysis has been able to get beyond the ET path's present restrictions on substrate redox potentials that are incompatible with those of excited photocatalysts.190 Over the last ten years, two distinct catalytic cycles have been united by the use of the combined EnT and ET methods in a unique photo-organocatalysis, enabling different activation modes using a single photocatalyst.189–191 By using visible light to drive organocatalysis, different aromatic C–H functionalizations imposed by photosensitized EnT and ET may be easily accessed along the energetically least expensive pathways.192,193 The issue is how to make up for the energy deficit between the input energies of visible light irradiation (about 400 nm) and the energy consumption of aromatic C–H bond dissociation.189 Consequently, mechanistic investigation efforts are desperately needed to give the structural and energetic foundation for comprehending the dynamic pathways of excitation-energy relaxation connected to structure evolution. However, in particular for dynamically coupled EnT and ET photocatalysis, the kinetic and thermodynamic assessments should be taken into account to reveal important parameters for managing photo-sensitized area C–H bond functionalization.189 The single electron transfer (SET) technique, which may be available in this instance, has been utilized to accomplish a number of beneficial chemical alterations in the hunt for new agents and medications.194,195 However, energy transfer photocatalysis has become a very effective method for forming new chemical bonds and activating substrates in organic synthesis.196 This method may help improve the current system's efficiency in using photons. There is much leeway for theoretical calculations because neither the SET nor the EnT process can yet be directly identified by experimentation. Ultimately, every piece of experimental data suggests that, for the optimal donor–acceptor pairings in SET- or EnT-mediated processes, a variety of parameters influence the yield of photocatalytic reactions. Merely focusing on thermodynamic elements, such as the energy correspondence between the donor and acceptor, is inadequate to comprehend the mechanism controlling photocatalytic efficiency. It is necessary to take into account kinetic considerations, which depend on the high-level treatment of the donor and acceptor's excited-state electronic structures.194000, along with a singlet lifespan of τf ∼1.0 ns.136 The triplet 3MB+* possesses a prolonged triplet lifetime, measured to be τf ∼32 μs,77 thereby indicating its superior stability as an excited state.9 In Fig. 8,9,197 the photocatalytic cycles involving methylene blue are illustrated.
 | ||
| Fig. 8 The feasibility of MB+ photocatalytic cycles is demonstrable.9 (a) Methylene blue photocatalytic quenching cycle during electron transfer (ET) processes;9 (b) methylene blue's photocatalytic cycle when exposed to energy transfer processes.9 | ||
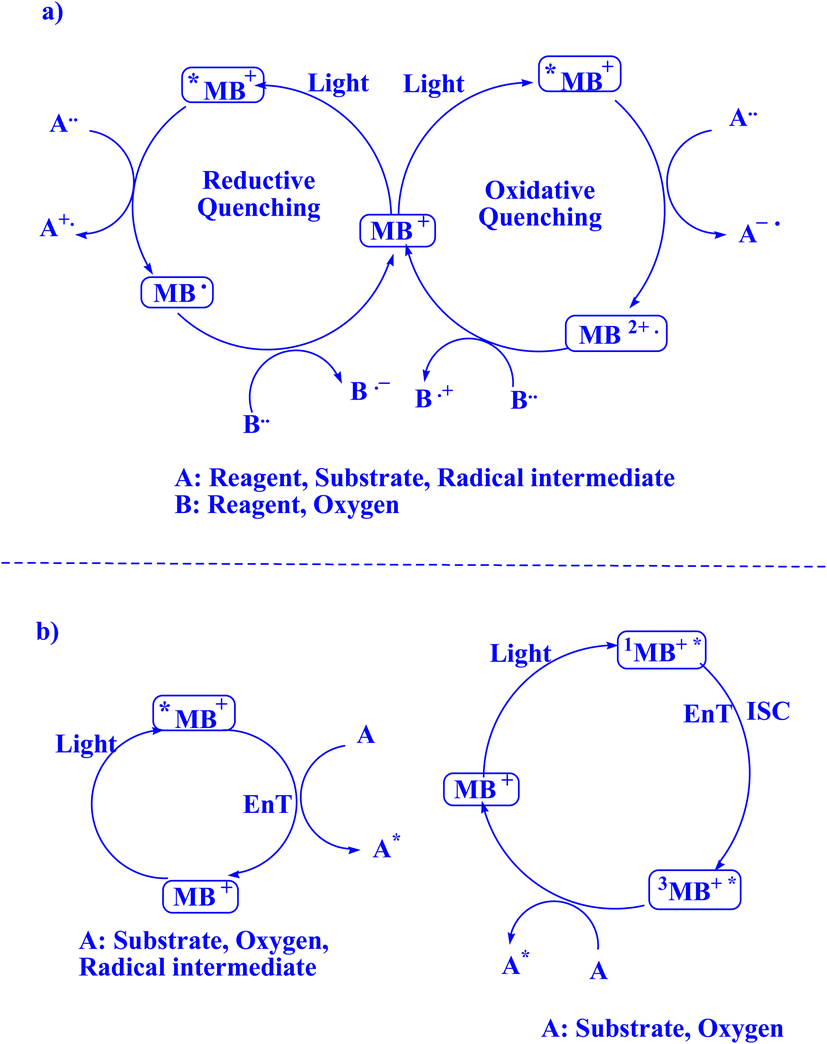
In contemporary literature, methylene blue (MB+) has emerged as a frequent agent of interest for research for the development of Knoevenagel-Michael cyclocondensation as a photo-redox catalyst (Fig. 9),197 synthesis of synthesis of polyfunctionalized dihydro-2-oxypyrroles (Fig. 10),201 in addition, MB+-catalysed decarboxylation of acids (Fig. 11),202 dehydrosulfurization of thioamides (Fig. 12),203 and the development of Friedländer hetero-annulation (Fig. 13).204
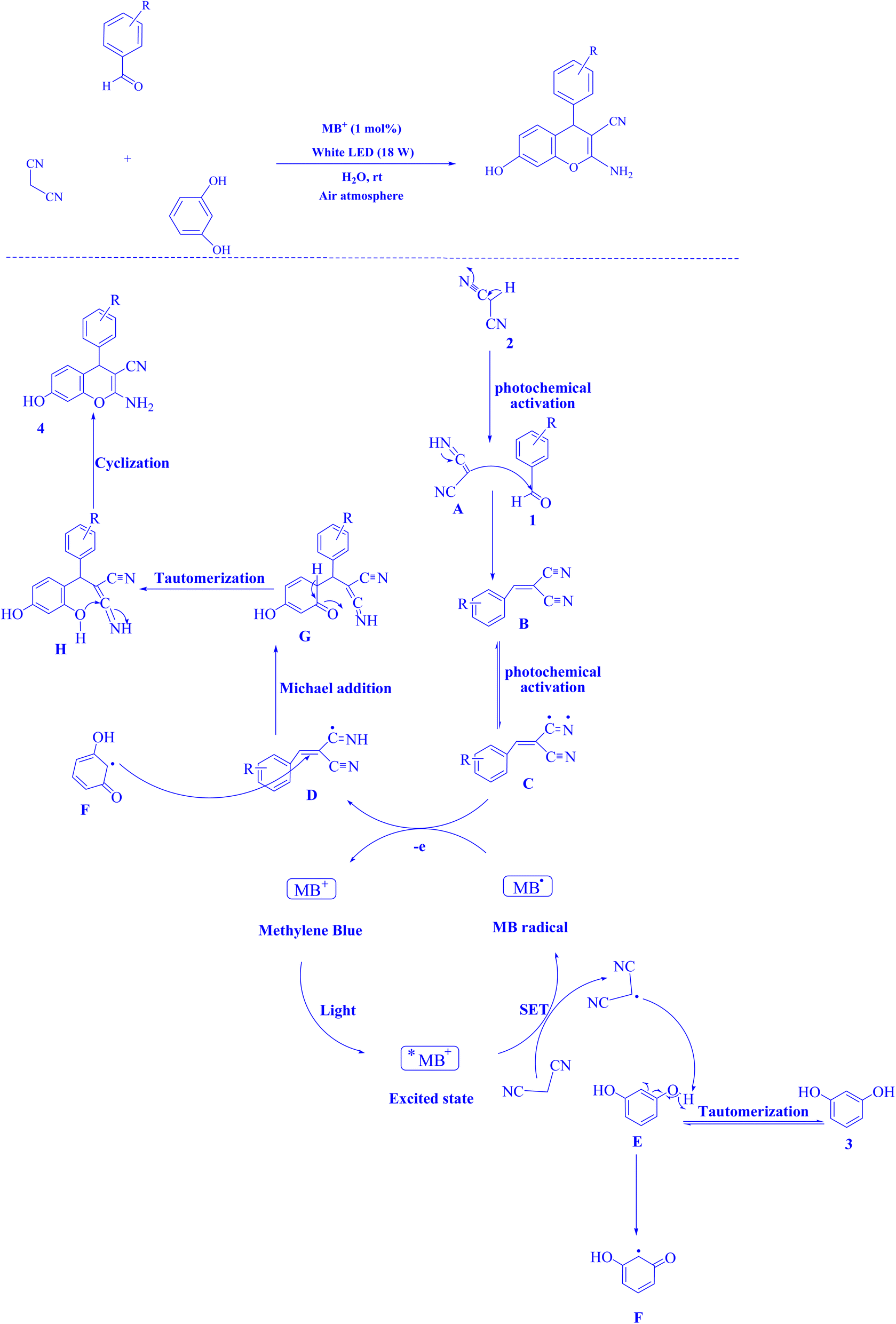 | ||
| Fig. 9 2-Amino-4H-chromene scaffolds synthesized.197 | ||
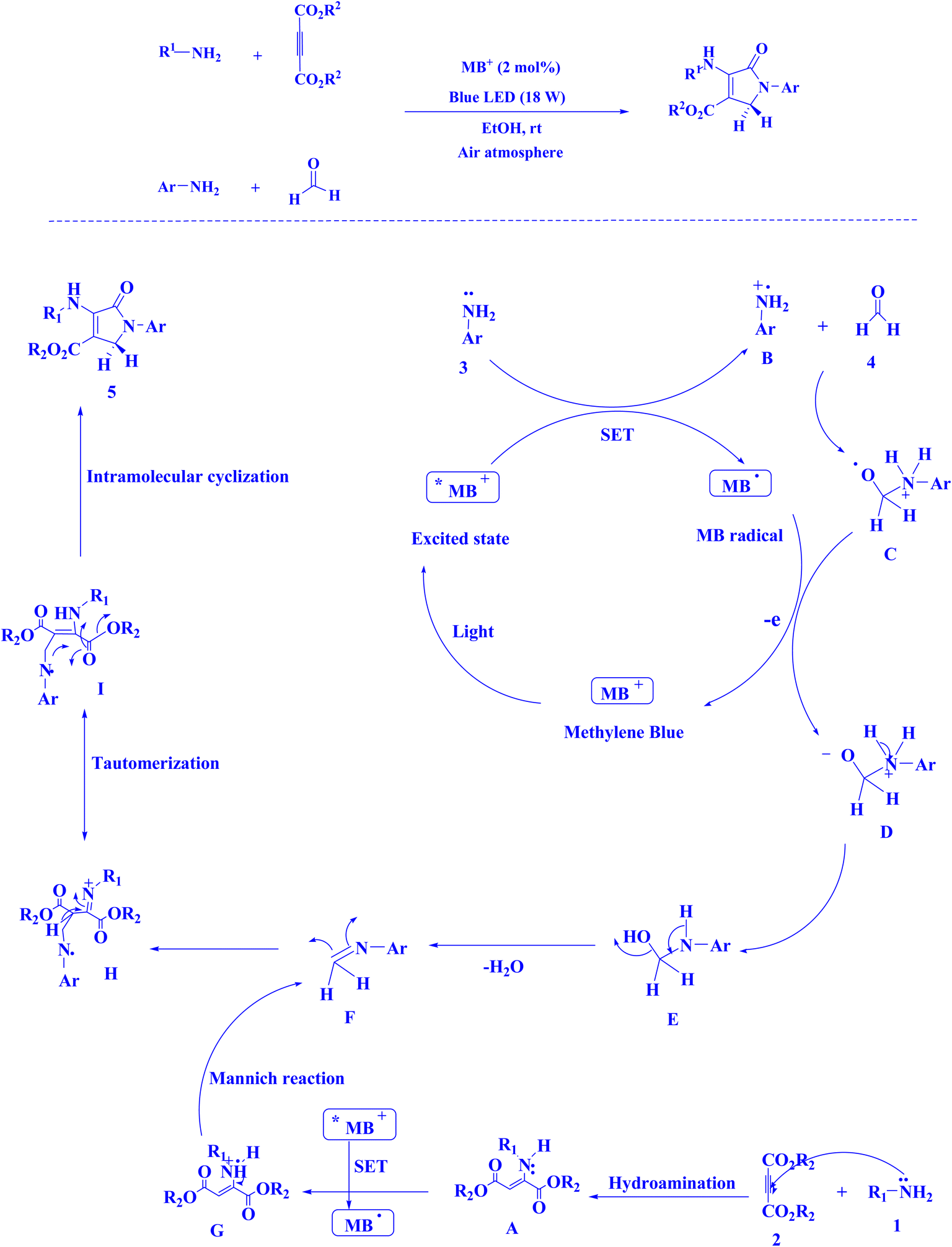 | ||
| Fig. 10 Polyfunctionalized dihydro-2-oxypyrroles synthesized.201 | ||
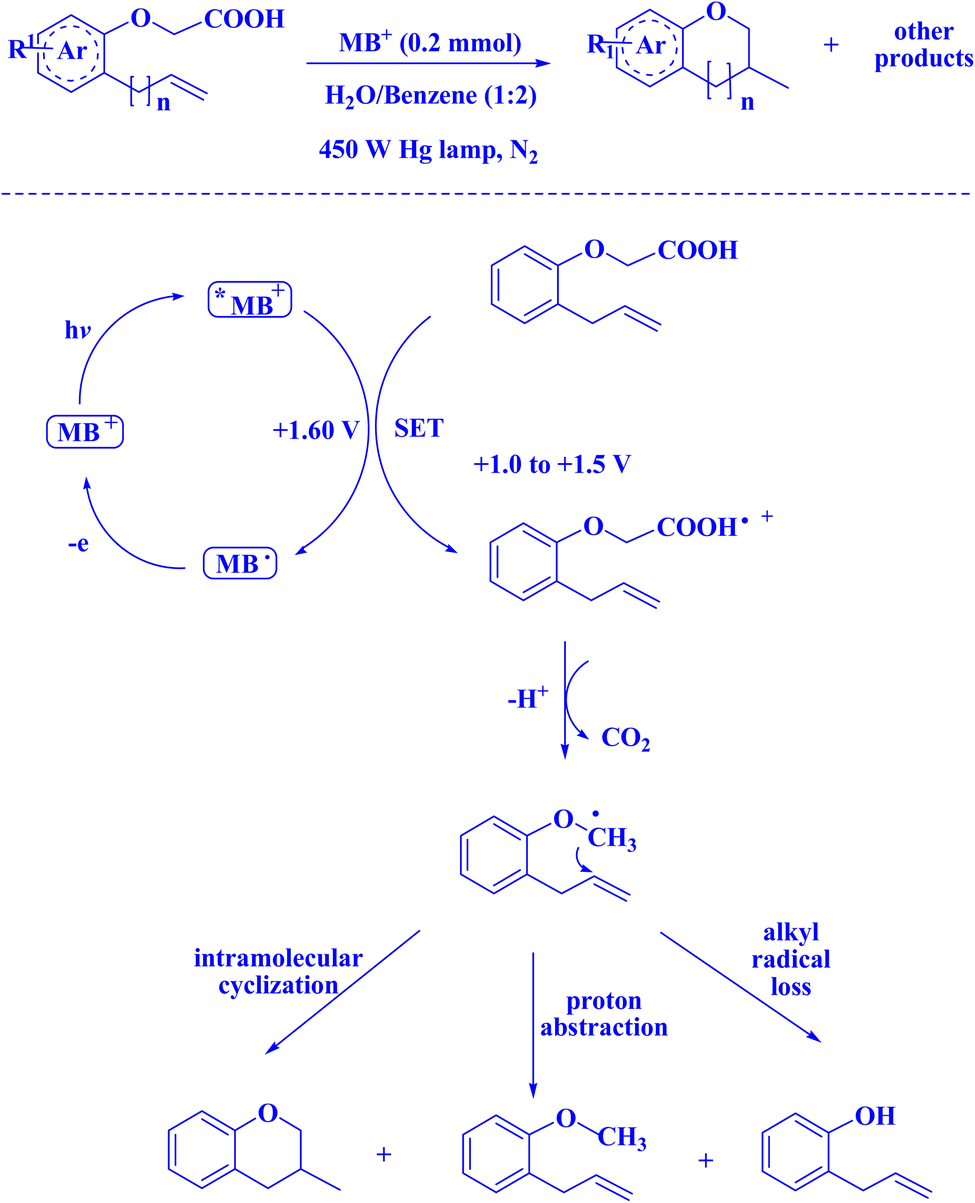 | ||
| Fig. 11 MB+-catalyzed decarboxylation of acids.202 | ||
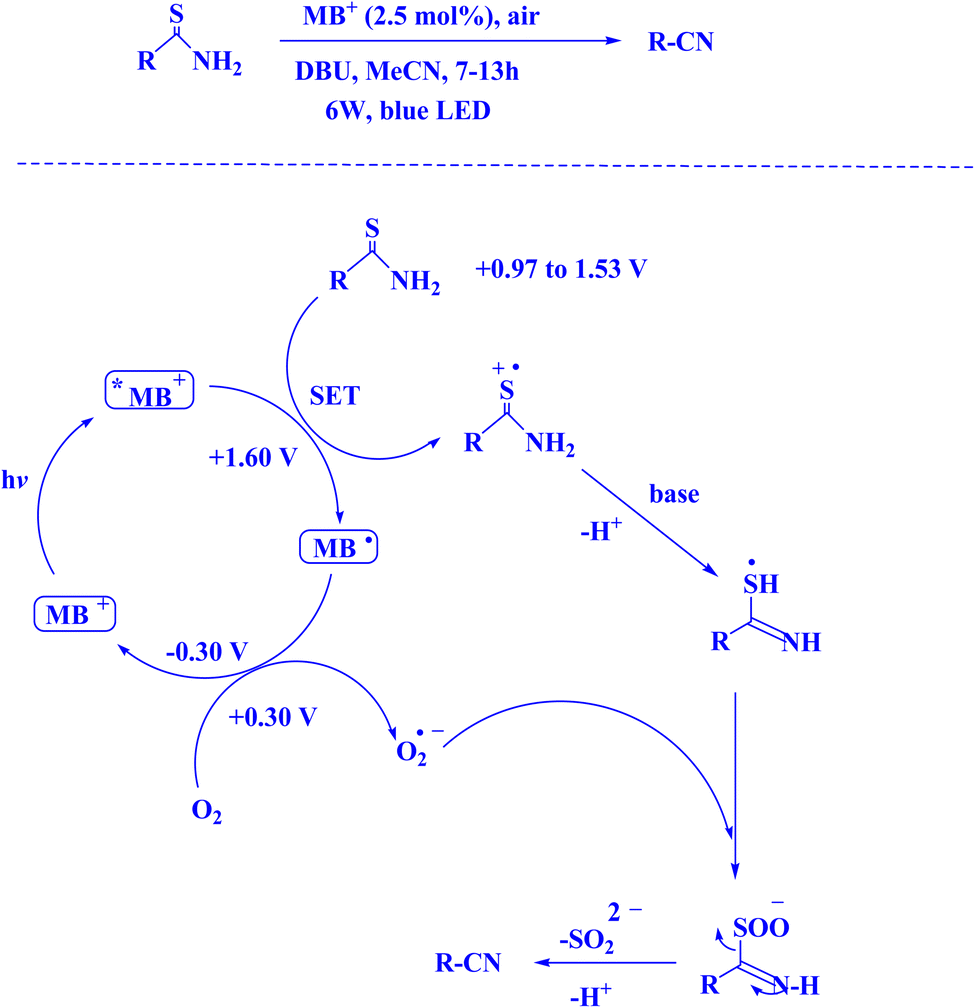 | ||
| Fig. 12 MB+ catalyzed dehydrosulfurization of thioamides for the synthesis of cyanides.203 | ||
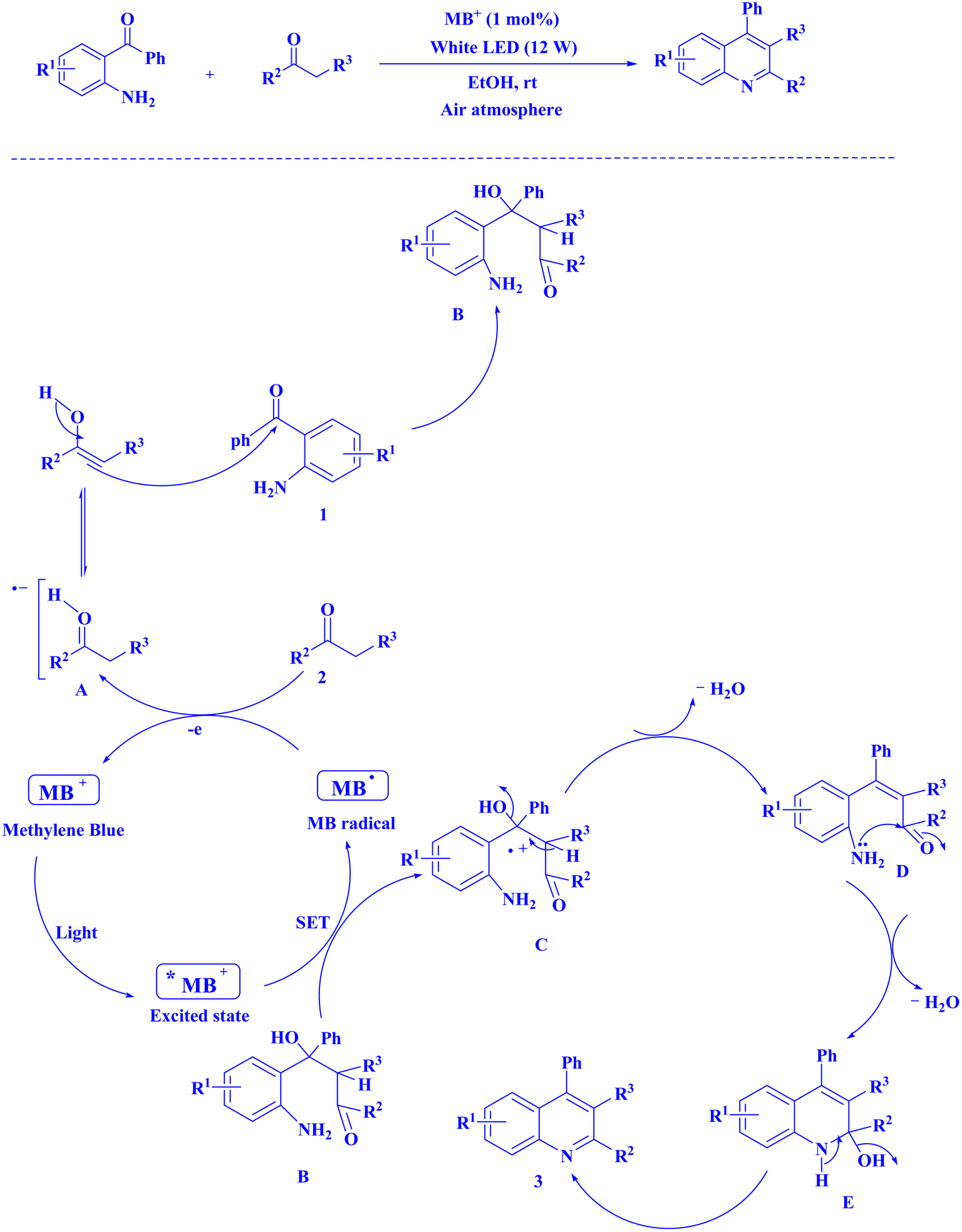 | ||
| Fig. 13 Polysubstitutedquinoline synthesis.204 | ||
7.3. Hydrogen atom transfer (HAT)
The process of hydrogen atom transfer (HAT) involves the simultaneous displacement of a proton and an electron between two substrates, occurring as a unified kinetic event.205,206 The overall reaction is explicitly stated in eqn (1).205
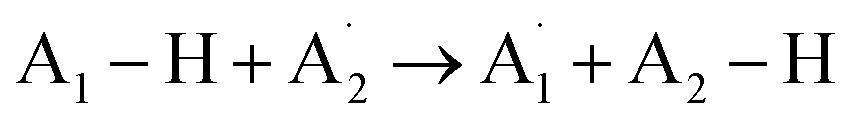 | (1) |
Theoretical inquiry reveals that hydrogen atom transfer (HAT) may be categorized as a subdivision of the broad spectrum of proton-coupled electron transfer (PCET) mechanisms. In PCET events, the proton and electron transitions are stimulated jointly, which results in the sharing of both the initial and terminal orbitals between the two entities.205,207,208 A multitude of research groups are currently engaged in the development of viable models aimed at comprehending the mechanism of HAT and forecasting the corresponding reaction rates. A conceptual framework for HAT using the Marcus theory approach has been proposed in previous literature.205,209 It has been demonstrated that the Valence Bond (VB) diagram model is capable of evaluating hydrogen-abstraction barriers and deriving general trends from accessible raw data, such as bond energies.205,210 The HAT process holds great significance in numerous chemical reactions, such as hydrocarbon combustion and aerobic oxidations, and also plays a crucial role in various atmospheric phenomena.205 In the field of biology, the utilization of several metalloenzymes has been reported to occur through a HAT step. A great deal of research has been conducted regarding the function of these processes in the detrimental effects of reactive oxygen species (ROS) within living organisms, as well as their involvement in the mechanism of action of antioxidants. Notably, numerous studies have been dedicated to this topic as evidenced by the existence of multiple publications.205–207,211–213 Additionally, it is noteworthy that HAT holds tremendous potential as a means of organic synthesis, since it enables the facile activation of (aliphatic) R–H bonds, frequently exhibiting remarkable selectivity. Consequently, it is plausible to circumvent the inclusion of a directing moiety within a given substrate and adjust the chemical reactivity through the judicious selection of reaction parameters such as a hydrogen abstractor, solvent, and so forth.205,214 In recent times, photocatalysis has made substantial contributions towards the advancements in the related field.215 The activation of the reaction partners, which results in the formation of reactive intermediates, is facilitated by an excited catalyst in said reactions. Subsequently, the catalyst is restored to its original state, thereby priming it to initiate another cycle.205
In recent years, eosin Y has been utilized in the process of chemical synthesis of 1,3,4-thiadiazole (Fig. 14),219 photochemical synthesis of pyrano[2,3-d]pyrimidine scaffolds (Fig. 15),220 synthesis of spiroacenaphthylenes (Fig. 16),221 and 1H-pyrazolo[1,2-b]phthalazine-5,10-diones (Fig. 17).221
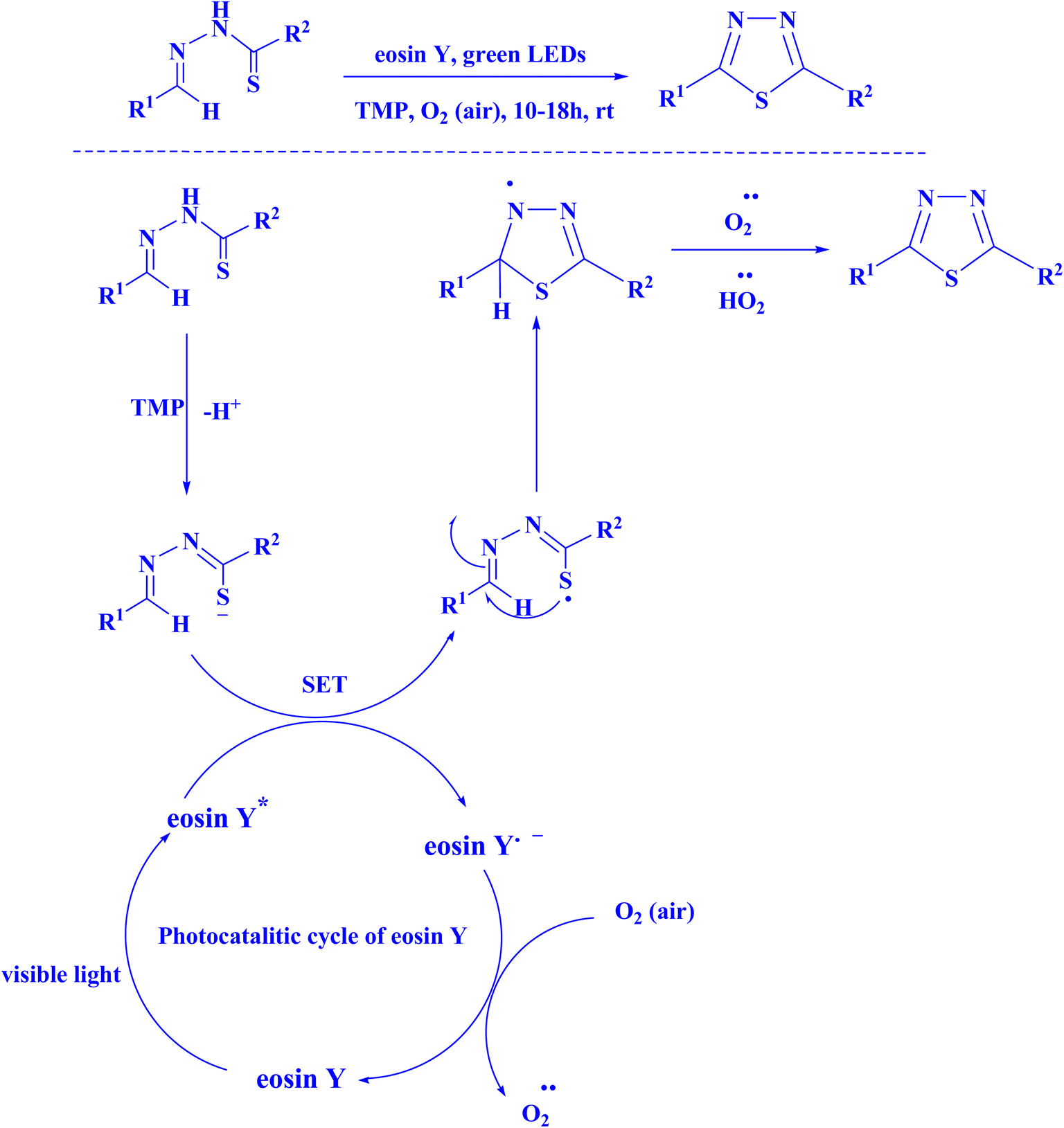 | ||
| Fig. 14 Eosin Y catalyzed visible light promoted synthesis of 1,3,4-thiadiazole.219 | ||
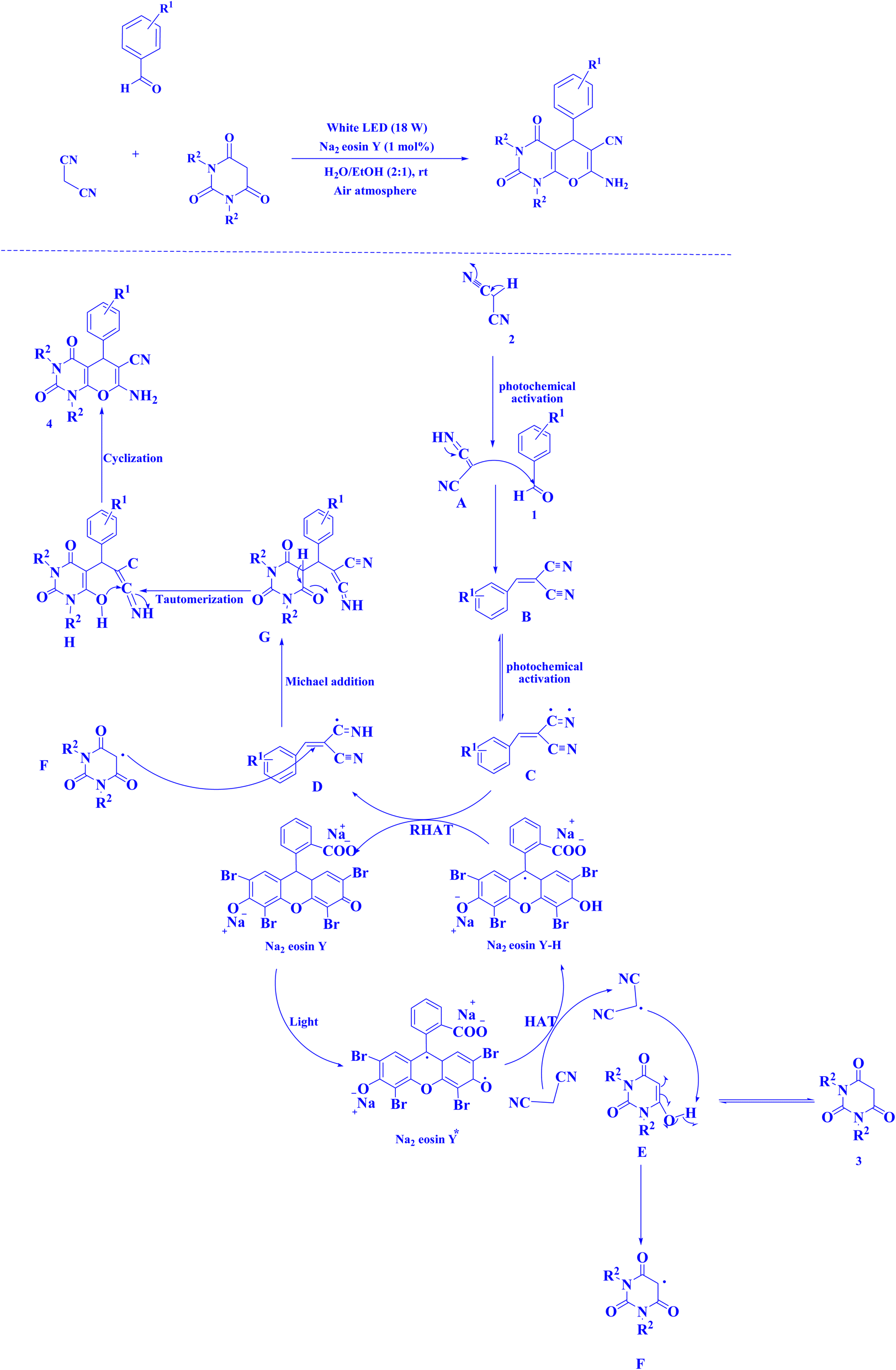 | ||
| Fig. 15 Synthesis of pyranopyrimidines.220 | ||
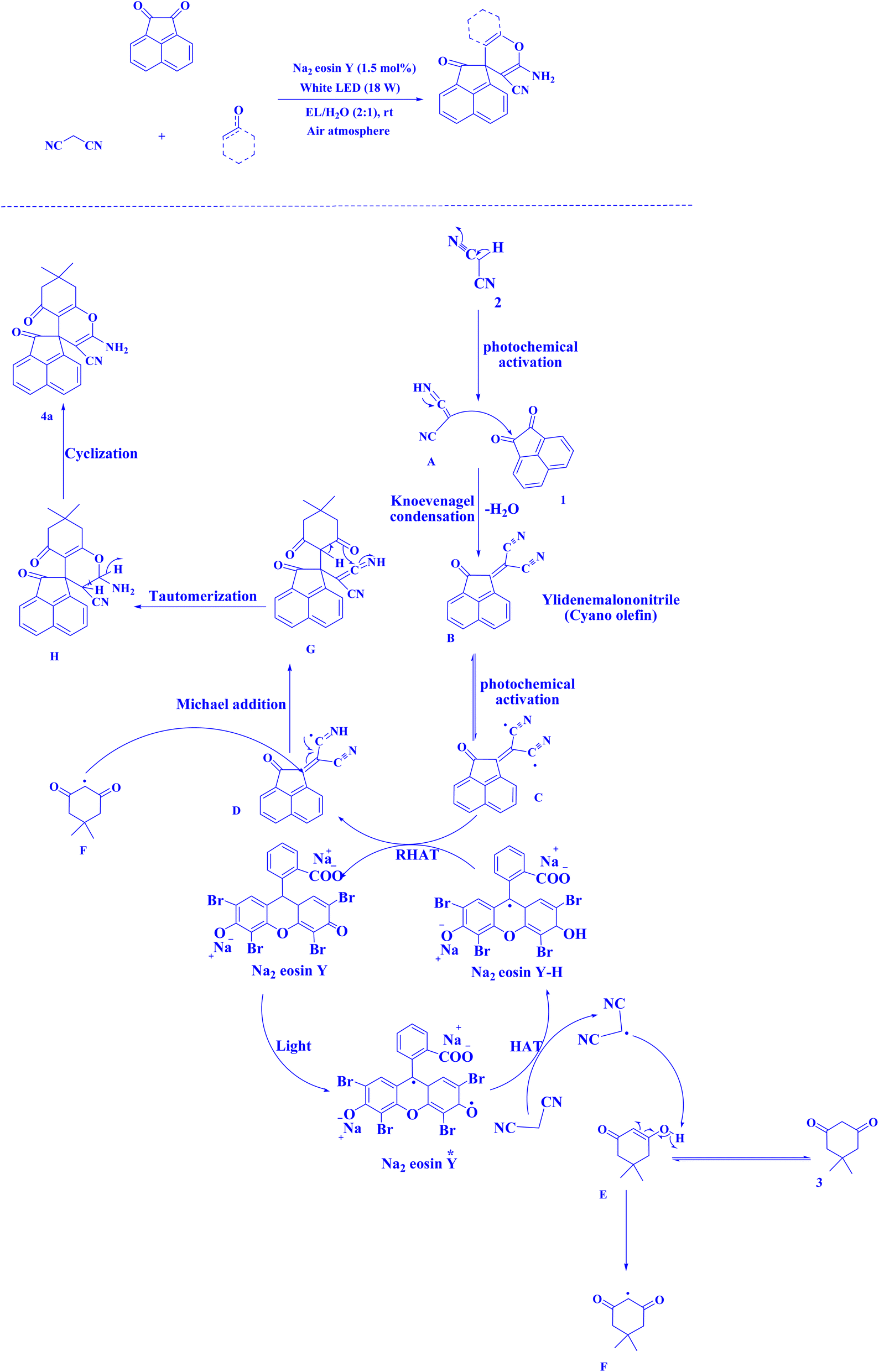 | ||
| Fig. 16 Synthesis of spiroacenaphthylenes.221 | ||
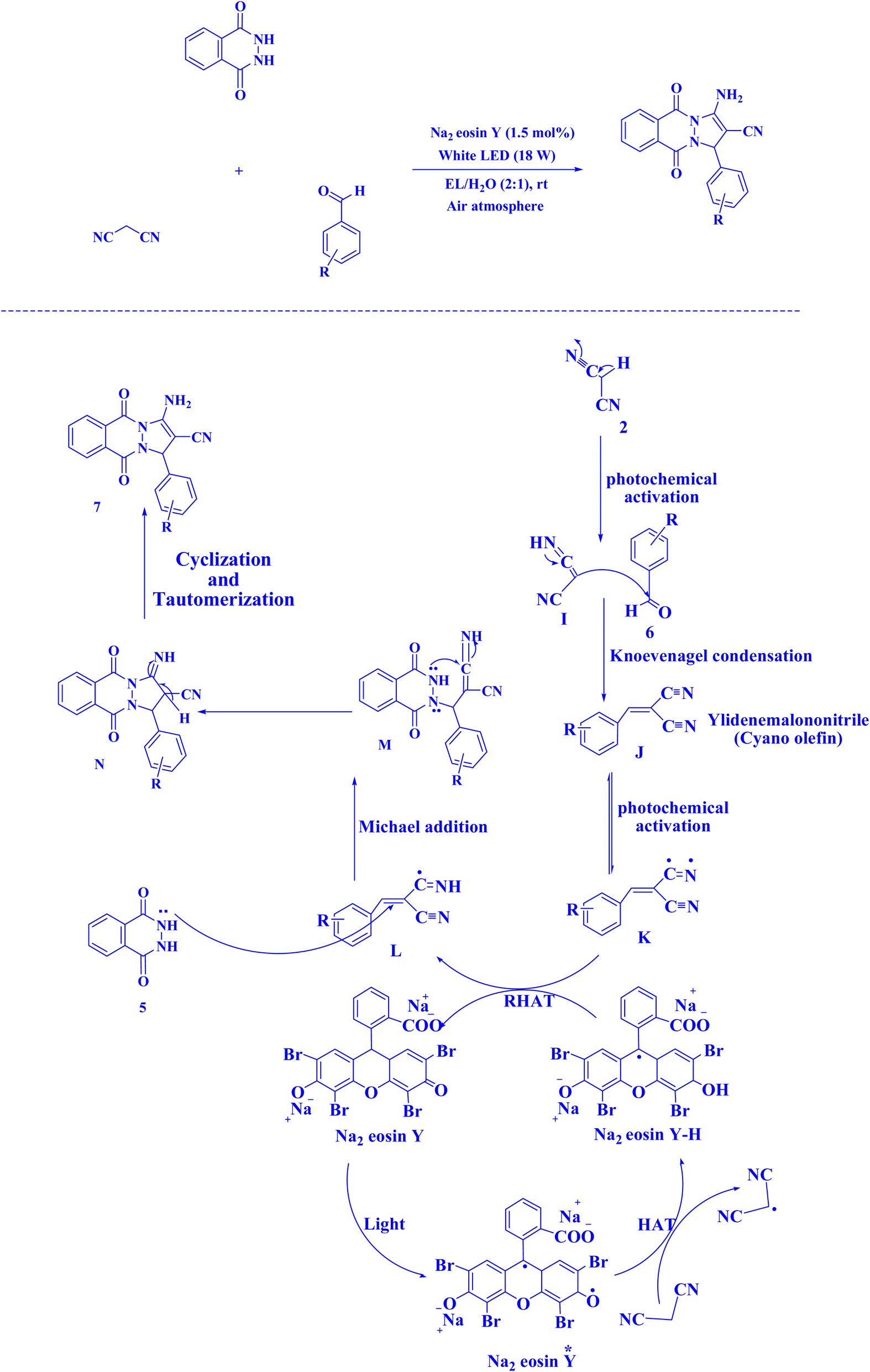 | ||
| Fig. 17 Synthesis of 1H-pyrazolo[1,2-b]phthalazine-5,10-diones.221 | ||
7.4. Summary
Photoredox catalysts achieve their stable photo-excited states by absorbing visible light. By use of photoinduced electron transfer (PET), a charge-separated state is created that consists of an acceptor radical anion and a donor radical cation from the donor (D) to the acceptor (A). Furthermore, the fundamentals of the fast growth of organic photocatalysis in recent years have been oxidative/reductive electron transfer (ET) or bimolecular energy transfer (EnT). Two different catalytic cycles have been merged using a novel photo-organocatalysis technique that combines EnT and ET technologies to enable energy-efficient solutions. The hydrogen atom transfer (HAT) process transfers a proton and an electron between two substrates in a single kinetic event. Theoretically, HAT can be considered a subset of the broad category of proton-coupled electron transfer (PCET) processes. Proton and electron transfer are activated simultaneously during PCET events, resulting in the sharing of beginning and final orbitals between the two entities. Photocatalysis has played a major role in recent advances in the linked discipline. In these reactions, an excited catalyst helps to activate reaction partners, which produces reactive intermediates. The catalyst is then returned to its initial state, ready for the start of a new cycle.8. Applications of photocatalysts and photocatalytic reactions
The discipline of photocatalysis encompasses a growing repertoire of activation modes that selectively harness photonic energy to activate a specifically designed photon-absorbing catalyst, known as a photocatalyst. Upon excitation, the photocatalyst facilitates the participation of accompanying substrates, reagents, or secondary catalysts in unique reaction pathways that were previously inaccessible under thermal control.16 “The present study16 explored the primary pathways utilized by photocatalysts to effectively facilitate the transfer of light energy into chemical reactivity and selectively activate targeted molecules.” Photocatalysis has garnered considerable attention as a viable alternative process for achieving a multitude of chemical reactions in the disinfection of potable water and safeguarding against microorganisms, owing to the potential utilization of solar energy.222 Photocatalytic technologies possess the capability for broad-spectrum applications, including utilization within indoor air and ecological health initiatives, as well as within sectors such as biological, medical, laboratory, hospital, pharmaceutical, and food industries. Furthermore, these technologies exhibit significant potential in the realm of plant protection.222–225 Photoredox catalysis has demonstrated significant utility in a multitude of applications.16 Photoredox catalysis has been extensively utilized in various domains, such as organic synthesis, biomedicine, water electrolysis,226 carbon dioxide reduction,227 and the exploration of innovative solar cell components.16,228 In the subsequent sections, we will scrutinize several of their implemented functions.8.1. Application of photochemical approaches in organic reactions
The production of materials, medicines, and agrochemicals is a common application of free radical chemistry.229,230 This is so because using radicals to create molecules is frequently an advantageous approach compared to using ionic pathways.155 Stochastic initiators, or stoichiometric oxidants or reductants, are necessary for the generation of radicals in conventional procedures.231 The applicability and generality of radical processes are greatly impacted by these relatively harsh conditions (high temperatures, UV light, toxic reagents, or strong redox-active reagents). More recently, milder and more efficient platforms for the production of open-shell intermediates have been made available by photoredox catalysis.232 In general, the mechanisms of electron transfer or atom abstraction are the main sources of support for these radical generation strategies.229 Chemists' interest in visible-light photocatalysis has grown recently due to its extensive use in organic synthesis and its importance for sustainable chemistry. Under mild reaction conditions, this catalytic method allows the formation of a variety of reactive species, often without the need for stoichiometric activation chemicals. Many synthetically valuable bond formations can be produced in a controllable way by manipulating these reactive intermediates.233 Because of its distinct mechanism, photocatalytic reaction, a synthetic technique that has emerged quickly in the last ten or so years, can be used to synthesize a variety of C–C, C–N, C–O, and C–P bonds with great efficiency and selectivity. The redox potential of the substrate and the photocatalyst's excitation potential, however, place restrictions on the application of photoredox catalysis. Additionally, a second terminal oxidant or reductant is needed as a sacrificial reagent to allow the catalytic cycle to be restarted to produce a net oxidation or reduction reaction. In addition to complicating the reaction system and making the addition of sacrificial reagents unfeasible, these sacrificial reagents have the potential to react with catalysts, substrates, reagents, or active intermediates to produce undesirable byproducts that reduce the efficiency of the primary reaction or prevent it from proceeding.234 Two categories of mechanisms are usually explored in relation to organic photochemical reactions.235 It is possible to move a hydrogen atom from a hydrogen donor to a species that is photochemically activated in a single step. Nearly simultaneously, the electron and the proton are exchanged (Fig. 18, eqn (2)).235 Alternatively, if the electron is transmitted first, a radical ion pair is formed. In an acid-base process (eqn (3)), the proton comes next.235 Both particles are transmitted from the same location of the hydrogen donor to the same location of the acceptor, in contrast to many other forms of proton-coupled electron transfer. This is referred to as “hydrogen atom transfer” in a restricted sense.236–238 The entire reaction is exergonic in the first example (eqn (2)), but the electron transfer is endergonic. Both stages are exergonic in the second scenario (eqn (3)). Beyond these processes, proton exchange may take place between the reaction medium and photochemically produced intermediates, such as a radical ion, which can also produce unexpected end products in comparison to analogous ground-state reactions.235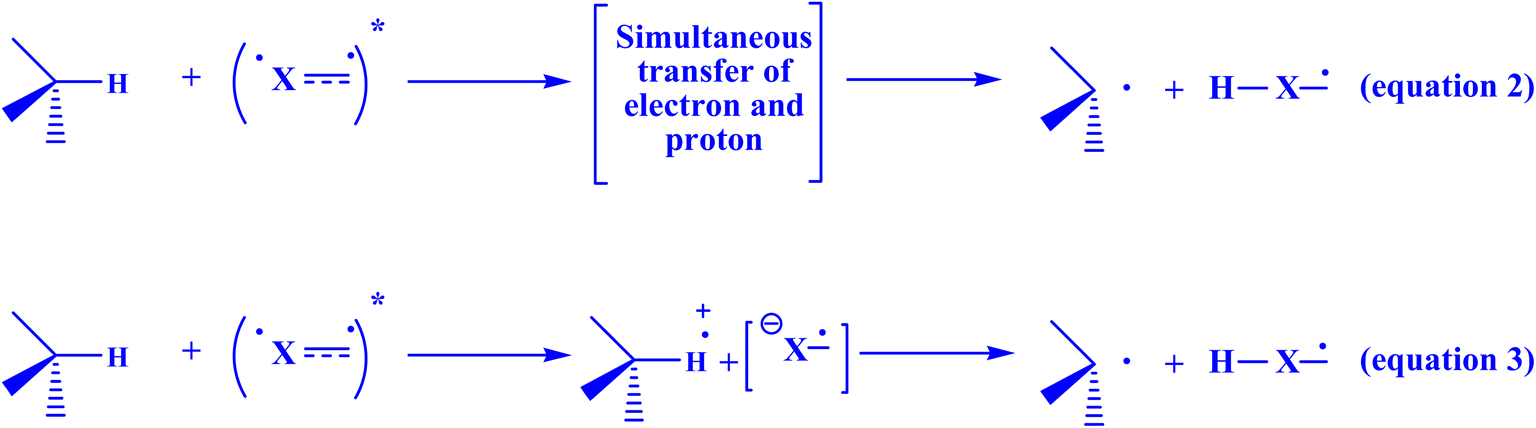 | ||
| Fig. 18 There are two ways to transfer hydrogen from a donor to an acceptor that is electrically stimulated.235 | ||
8.2. Development of drug design methods using photocatalytic reactions
The attractive properties of photoredox catalysts have attracted the attention of the chemical industry, which has recognized the possibility of photoredox methods to achieve efficient and environmentally friendly catalysis.7 The application of photoredox catalysis in various synthetic transformations plays a vital role in drug discovery and development, particularly in the pharmaceutical industry.7,238 An instance of interest pertains to the guidelines governing the direct and focused modification of pharmacologically relevant frameworks via the utilization of various chemical reactions, including alkylation, amination, halogenation, and perfluoroalkylation, which are widely employed in the field of organic chemistry. The utilization of photoredox methodologies has been observed in the installation of minuscule functionalities that have a direct impact on the ADME-tox aspects (i.e., absorption, distribution, metabolism, excretion, and toxicology) of a lead candidate.239 This technique employs non-toxic and conveniently accessible materials for the purpose.7,240 Many uses in drug discovery have been made possible by synthetic advances in visible-light photocatalysis and its naturally moderate conditions. This catalytic platform's biocompatibility, ability to use low-energy visible light, and moderate temperatures limit side reactivity and allow for broad functional group tolerance. In particular, these mild settings are essential to allow this platform to function in a biological environment without denaturing proteins or breaking DNA. The pharmaceutical industry has taken notice of these attributes and acknowledged the potential benefits of photoredox catalysis in medication research and discovery.241 Small-molecule medications are commonly utilized in daily life. The industrial production of small-molecule pharmaceuticals still faces several problems, including the utilization of costly metal catalysts, complicated reaction pathways, and non-recyclable catalysts. The advantages of photocatalytic organic synthesis over traditional methods include excellent selectivity, mild conditions, and environmental friendliness.11 To enhance photocatalytic performance, porous framework materials can accurately alter the electronic state and ligand structure of catalytic sites. Because of their distinctive shape, structural flexibility, high photocatalytic performance, distinctive recyclability, exceptional chemical stability, ease of synthesis, and low cost, MOFs, COFs, and PCCs-based photocatalysts have drawn a lot of attention from researchers. Thus, the creation of porous framework materials as photocatalysts for the synthesis of small-molecule medicines or drug precursors is an important field for future research.11For instance, because of their numerous pharmacological uses, the synthesis of benzothiophene derivatives has garnered a lot of interest recently.242 The benzothiophene core is included in a number of currently available medications. A radical annulation technique was used by Hari et al. (2012)242 to produce substituted benzothiophenes regioselectively through the photocatalytic reaction of o-methylthio-arenediazonium salts with alkynes. Photoredox catalysis was initiated by Eosin Y green light irradiation (Fig. 19).242
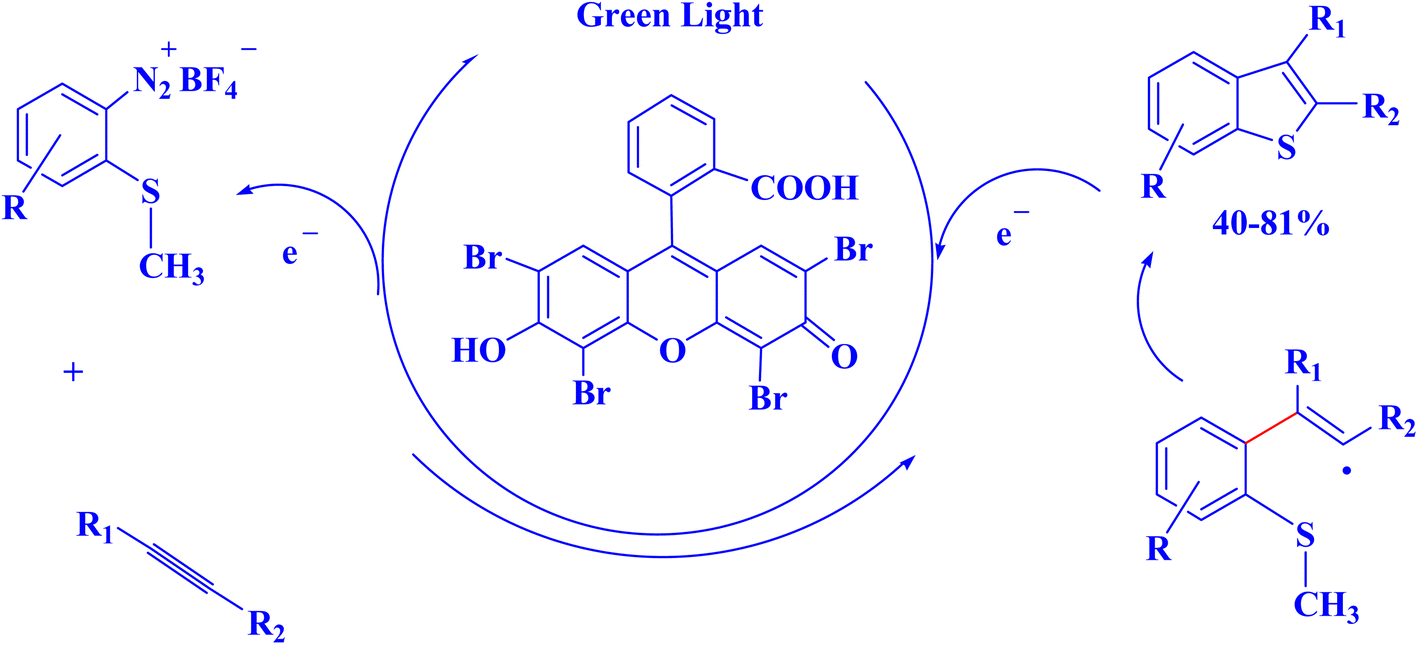 | ||
| Fig. 19 Photocatalytic synthesis of benzothiophenes.242 | ||
8.3. Investigating biomedical applications of nanophotocatalysts
The utilization of nanosized photocatalytic materials has become increasingly prevalent in various domains, including the mitigation of microbial infections. The utilization of nanoparticles as a means to regulate the development of biofilms in the oral cavity has been investigated due to their biocidal, antiadhesive, and delivery properties. Additionally, nanoparticles have been incorporated in prosthetic devices, topically administered agents, and dental materials for this purpose, as noted in the literature.222 The current research landscape is replete with explorations into the potential of a vast array of nanosized particles that have been subject to modification to exhibit meaningful antibacterial, antifungal, antitumor, and antiviral properties. This is largely due to their infinitesimal scale, exceptional surface-to-volume ratios, as well as the wide range of biological activities they are capable of eliciting.222 Compared to other traditional ways of eliminating germs or viruses from indoor and outdoor environments, the photocatalysis method of disinfection employing metal oxide semiconductors has considerable potential. Compared to employing chemicals, nano-photocatalysts may efficiently inactivate bacteria and viruses in the presence of light radiation in ambient circumstances without generating any additional byproducts.243–245 Numerous studies have been conducted on the inactivation of various bacteria and viruses using metal oxide semiconductor-based nano-photocatalysts, such as ZnO and TiO2.243 Currently, there is a lot of promise for nanophotocatalysts in biomedicine. Targeted drug delivery can be achieved with nanophotocatalysts; these catalysts have the potential to be applied in nuclear-targeted drug delivery as well as targeted administration of chemotherapy to cancer cells or tumor locations. However, nanophotocatalysts can be used in tissue engineering to create scaffolds that encourage tissue regeneration and cell development.246 The capacity of nanophotocatalysts to produce reactive oxygen species (ROS) when exposed to light is one of its main benefits in the field of biomedicine. Singlet oxygen and hydroxyl radicals are examples of ROS, which are very reactive substances that can cause a variety of biological effects, including the death of cells. This characteristic has been used to create photodynamic therapy (PDT), a non-invasive therapeutic method that targets and kills cancer cells specifically by using light-activated nanophotocatalysts. PDT is a viable substitute for or addition to conventional therapy techniques, as it has demonstrated encouraging outcomes in a number of cancer types.246–248 Their photoinduced inactivation is primarily caused by the photocatalytic generation of biocidal ROS that are short-lived yet effective, such as hydrogen peroxide (H2O2), superoxide (˙O2−), and hydroxyl radicals (•OH), by photochemical redox reactions in the presence of light.243,249,250 Further research is still needed, on a number of significant topics including photocatalysis performance, large-scale nanophotocatalyst manufacture, reaction/synthesis condition optimization, long-term biosafety concerns, stability, clinical translation, etc.246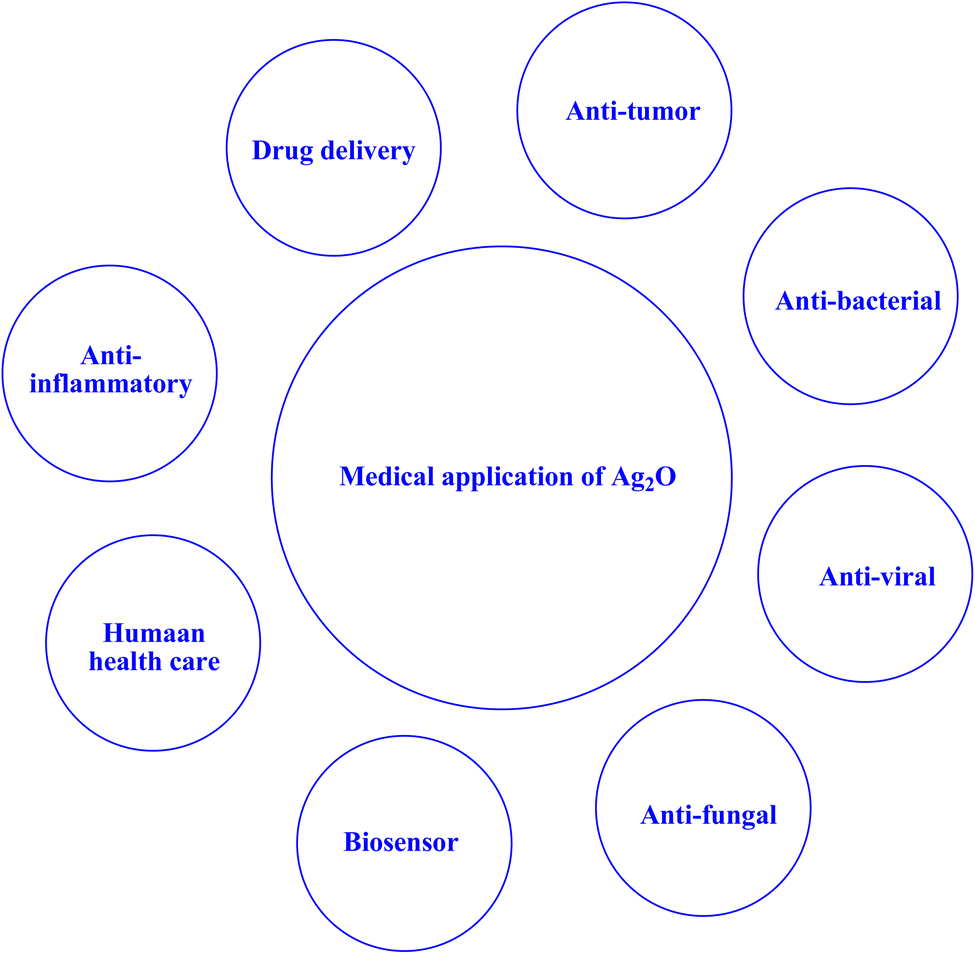 | ||
| Fig. 20 A schematic illustration of the medical applications associated with Ag2O.251 | ||
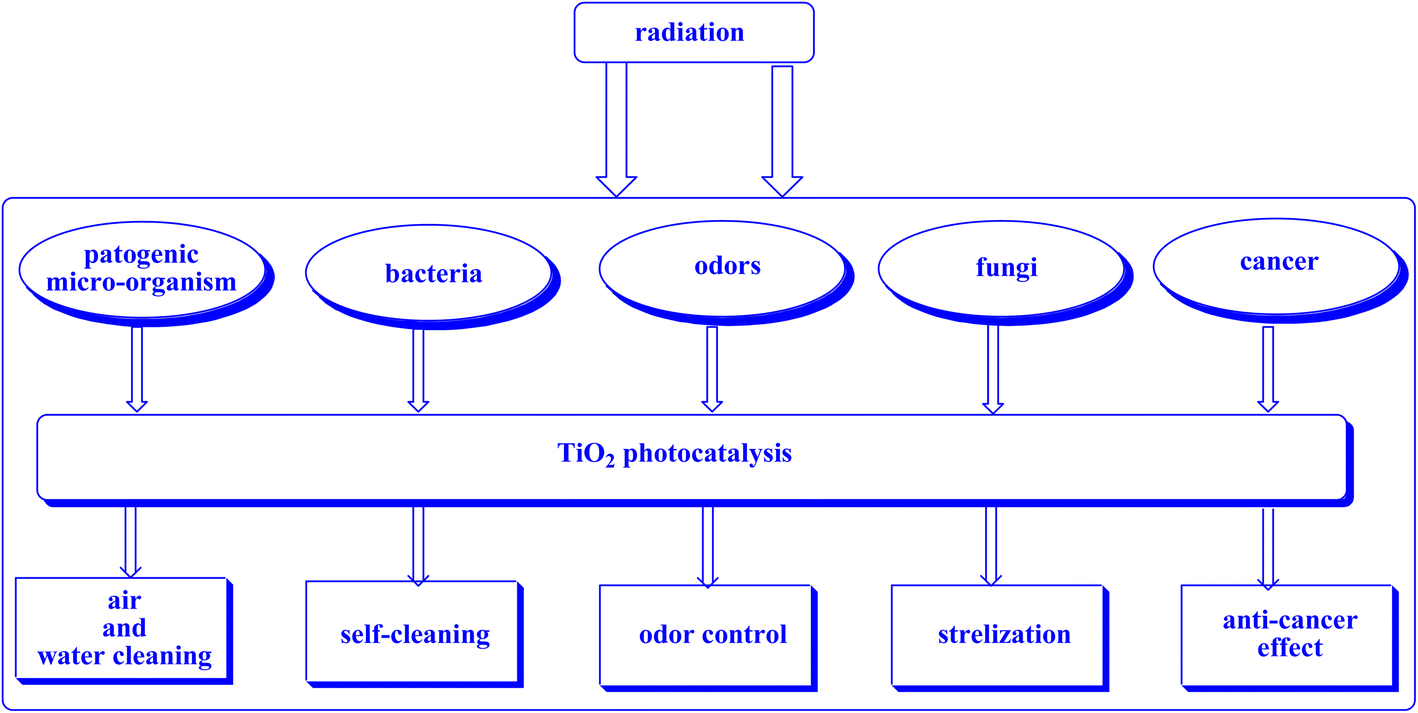 | ||
| Fig. 21 This study aims to elucidate the primary photocatalytic utilizations of TiO2.252 | ||
8.4. The use of a photocatalysis system for environmental pollution management
At present, the predominant clean energy sources that are widely available are visible light irradiation, solar energy, and heat emanating from the sun.253 The implementation of thorough research inquiries and the advancement of material technology intended for the proficient utilization of solar irradiation in addressing environmental pollution while preserving its eco-friendliness proves to be indispensable. The utilization of renewable solar energy to activate oxidation and reduction-based chemical reactions through photocatalysis presents a viable and sustainable technological approach to addressing environmental challenges, as corroborated by sources.253–255 The utilization of photocatalysis systems has garnered substantial attention within the scientific community as a highly promising approach for addressing environmental issues, particularly the removal of residual dye pollutants from wastewater streams.253 The present study concerns photocatalysis, which constitutes a photochemical process occurring at the surface of a metal oxide semiconductor. Specifically, this process denotes the occurrence of two concurrent reactions: the initial oxidation reaction, which ensues from photo-induced positive holes, and a subsequent reduction reaction, whose genesis stems from photo-induced negative electrons.253,256 Numerous photocatalysts exhibiting high potential have been extensively utilized in waste management systems with a focus on environmental sustainability. These include but are not limited to titanium dioxide (TiO2), zinc oxide (ZnO), iron(III) oxide (Fe2O3), zirconia (ZrO2), vanadium(V) oxide (V2O5), niobium pentoxide (Nb2O5) and tungsten trioxide (WO3).253,255,257,2588.4.1.1. Photodegradation of antibiotics. Antibiotics have been employed in many different industries and have become a vital component of people's lives during the past few decades.270 But antibiotics are easily overused and handled improperly, which allows them to build up in the environment and endanger the health of people and other living things.271 Antibiotic removal from the environment is therefore urgently needed, yet it remains a formidable task. Researchers have attempted to eradicate antibiotics using chemical treatment, biological filtering, traditional physical adsorption, and other techniques in the past several years.272–275 Unfortunately, the complexity of the processes involved, the expense of the facilities, and the possibility of secondary contamination make it challenging to put these procedures into reality.276–278 Antibiotics in the environment can cause a multitude of problems by encouraging the widespread development of antimicrobial resistance. Today, the most widely used methods for eliminating antibiotic pollutants from water are chemical oxidation, flocculation, and physical adsorption; however, these methods typically leave a large number of chemical reagents and polymer electrolytes in the water, making it difficult to post-treat large deposits. In addition, a variety of photocatalysts can be used to degrade pollutant residuals, providing several potential solutions to these problems.279
Chemotherapeutic drugs such as antibiotics are used to treat bacterial infections.280 Antibiotics in the environment are currently receiving more attention, which has led to a general search for potential containment strategies.281,282 Antibiotic-resistant genes and bacteria are produced as a result of this problem, hastening the development of antibiotic resistance and endangering both ecological systems and human health.281,282 As a result, the threat that antibiotic pollution poses to the integrity of our water resources in the twenty-first century is considered to be among the most significant environmental issues facing the globe, both in terms of environmental damage and possible health risks to humans.283 Since wastewater gathers output from hospitals, industries, and agriculture, wastewater treatment is typically thought of as the primary approach for handling these antibiotics.284 Nonetheless, several further investigations have verified that traditional methods are not very effective in eliminating these primarily water-soluble pollutants, which are neither volatile nor biodegradable.285 Much research has been focused on biotic elimination and non-biotic processes, such as sorption, hydrolysis, bacterial biodegradation, oxidation, and reduction.286 Yang et al. investigated the effectiveness of sulfonamide antibiotics for adsorption, desorption, and biodegradation in the presence of activated sludge both with and without NaN3 biocide.287 The outcomes of the experiment demonstrated that the antibiotics were removed by the activated sludge's sorption and biodegradation processes. Liu et al. studied four antibiotics as target antibiotics: azithromycin, roxithromycin, ofloxacin, and norfloxacin. They used UV254 photolysis, ozonation, and UV/O3 approaches to conduct nanofiltration disposal treatments, and they achieved the highest efficiency (>87%) in antibiotic elimination.279,288 However, these technologies' applicability was severely limited by their high cost, low stability, and inadequate recycling capacity. Because of this, researchers studying chemistry and environmental science have been looking for new ways to break down antibiotics in wastewater, with a particular focus on high-efficiency degradation strategies.279,289 As previously indicated, a variety of chemical techniques, including Fenton's oxidation, ozonation, and chlorination, have been developed for the treatment of antibiotic residues.290 Sadly, total mineralization is not always possible or would take unreasonably long. Due to their limited selectivity, these techniques occasionally have the potential to kill non-target species, resulting in unintentional harm.291,292 This approach also has significant operational and capital expenditures. When removing antibiotic residues from water, a combination of chemical and physical degradation methods can greatly lower the toxicity of treated effluents. However, these techniques are expensive and complicated.293 As an alternative, photocatalysis offers a wide range of potential applications in environmental remediation because of its special benefits, which include: (1) easily attainable reaction conditions (i.e., near ambient temperature and mostly ambient pressure); (2) the potential complete breakdown of organic pollutants into harmless inorganic molecules like carbon dioxide and water; and (3) strong redox ability, low cost, no adsorption saturation, and long durability.290 As a result, photocatalysis is gaining popularity all over the world and is being widely used in innovative methods for environmental management and energy extraction. One sophisticated oxidation method that has been used to remediate antibiotic residues is photocatalysis. Recent years have seen significant advancements.290
000 and 63000 tons annually in Europe304). This highlights the necessity of completely eliminating MPs from the environment. However, biodegradation, sophisticated oxidation techniques (such as photocatalysis), and catalysis may be able to remove the material permanently.295–297,300 Of these, photocatalysis is the most interesting since it is usually carried out under gentler circumstances than chemical degradation (catalysis) and has a substantially better degradation efficiency than biodegradation.294,300,302 Furthermore, as other self-propelled microrobot instances have shown, photocatalysis may be used in conjunction with waste plastic collecting to capture and degrade MPs.307–310 Furthermore, photocatalytic degradation can break down a variety of polymers into smaller plastic pieces, smaller organic molecules, and eventually CO2 since it lacks selectivity.295 Polystyrene (PS), polyvinyl chloride (PVC), polyethylene terephthalate (PET), polyamide fibers, poly(methylmethacrylate), and polyolefins including polyethylene (PE) and polypropylene (PP) are among the deteriorated plastics.295,302,303,306 Materials to be used as photocatalysts come in a wide variety. Metal oxides have been extensively studied as photocatalysts for the breakdown of microplastics.294,298,301,303 Examples of these oxides include TiO2 (ref. 311–314) and ZnO.315,316000 cases linked to particulate matter and over 16000 cases linked to ground-level ozone exposure.322 Controlling and lowering the content of NOx in the air is a focus of study because of its detrimental impact on human health.323 Heterogeneous photocatalysis, which combines semiconductor materials with light to produce reactive oxygen species (ROS) that break down developing pollutants, is one of the promising techniques for removing nitrogen oxides.324,325 The primary benefit of photocatalytic procedures over other NOx breakdown techniques is the ability to start the reaction at almost room temperature.323,326 The elimination of nitrogen oxides by photocatalysis has been thoroughly studied in recent years. The most well-known photocatalysts are made of composites or single materials made of bismuth, tungsten, carbon, or titanium compounds.327 Graphitic carbon nitride (g-C3N4), the most stable allotrope of carbon nitride under ambient conditions and an analog of graphite is an example of a feasible photocatalyst.328 Graphitic carbon nitride possesses unique physicochemical features such as an appropriate bandgap energy (∼2.7 eV), correlating to visible-light absorption.329 Therefore, g-C3N4 may be utilized as a visible-light-driven photocatalyst toward NOx elimination in the gas phase, which was proven in the previous investigations.323,330,3318.5. Photocatalytic water splitting and production of hydrogen
The clean fuel of the future is hydrogen. Currently, steam-reforming hydrocarbons, mostly derived from fossil fuels, are the method used in commercial hydrogen production. This process not only uses a lot of energy but also produces greenhouse gases. Alternative methods of producing hydrogen using nuclear, biomass, solar, and wind energy are also being investigated.29,332 The hydrogen synthesis by the above-outlined procedures suffers from efficiency losses due to the consumption of electricity for the electrolysis of water.333 The development of effective technologies to utilize naturally accessible solar energy directly to make hydrogen via water splitting utilizing solar energy has, consequently, emerged as a strong contender.33 In its most basic form, water splitting is the electrolysis of water, which is the reduction of water to hydrogen at the cathode and its oxidation to oxygen at the anode.29Various methodologies pertaining to solar hydrogen production have been documented.255
(i) Electrolytic water decomposition can be facilitated by an array of energy sources including but not limited to solar cells and hydroelectric power generators.
(ii) The process of biomass reformation.
(iii) Photocatalytic or photoelectrochemical water splitting is a process that resembles artificial photosynthesis.
The salient feature of water splitting through the employment of a powdered photocatalyst is its straightforwardness, as evidenced by the elucidation provided in Fig. 22.255 When photocatalyst powders are dispersed in a water pool and exposed to sunlight, hydrogen gas is easily generated. The requirement for the detachment of hydrogen produced from oxygen in the course of photocatalytic water splitting functions as a drawback in the said process. Nevertheless, the issue can be surmounted by adopting a Z-scheme photocatalyst configuration, as indicated by ref. 255.
 | ||
| Fig. 22 The generation of hydrogen from water through the utilization of a powdered photocatalyst powered by solar energy.255 | ||
In order to create holes in the valence band, excited electrons from the valence band jump to the conduction band when photons from the visible light spectrum (380–700 nm) are absorbed by semiconductor materials functioning as photocatalysts. Adsorbed water molecules are reduced and oxidized by the photo-induced electrons and holes that travel to the catalyst surface.29,334 Every stage in the photocatalytic water splitting process is essential to figuring out how efficient the process is overall for producing hydrogen. The stages that affect photocatalytic quantum efficiency include light harvesting, charge separation, charge transit, charge utilization, and surface adsorption capacity.29 The photocatalyst is an essential component of a photocatalytic water splitting reaction. In order to progress solar-hydrogen production via photocatalytic water-splitting in the future, it will be essential to create extremely stable visible-light-active photocatalytic materials and to construct cost-effective photoreactor systems.335 Because of their increased stability, increased charge carrier separation efficiency, and prolonged light harvest, several cocatalysts have altered semiconductor photocatalysts up to this point. Consequently, cocatalysts are required to enhance photocatalytic H2 evolution. The cocatalysts can be classified into the following groups: carbon-based cocatalysts, dual cocatalysts, Z-scheme cocatalysts, metal/alloy cocatalysts, metal phosphides cocatalysts, metal oxide/hydroxide cocatalysts, and MOFs cocatalysts.336
8.6. N2 fixation and CO2 reduction with the photocatalytic technique
Widespread research efforts to investigate renewable energy alternatives by sustainable technologies have been inspired by the ever-growing worries about energy shortages and climate change.337 An intricate artificial photosynthetic system has been developed, drawing inspiration from nature, to transform H2O, CO2, and N2 into products rich in energy, including H2, CO, CH4, CH3OH, NH3, and more.338–342 However, many electron transfers and large energy barriers occur during water splitting and the reduction of CO2 and N2, which results in sluggish reaction kinetics.343–346 Therefore, it would be ideal to create affordable catalysts to speed up these processes. Fujishima et al. presented the groundbreaking work of photoelectrocatalytic water splitting over TiO2 under UV irradiation in 1972,33 demonstrating the practical conversion of solar energy into chemical energy. Fe-doped TiO2 was initially shown by Schrauzer et al.347 as a proof-of-concept to convert N2 into NH3 when exposed to UV light. After a span of two years, Fujishima and colleagues348 succeeded in producing HCOOH, HCHO, and CH3OH by the direct reduction of CO2 on GaP, ZnO, CdS, GaP, and SiC. After these groundbreaking studies on photocatalytic water splitting, CO2 reduction,349 and N2 fixation, the subject has received more attention, and promising advancements have been achieved recently. The solar-to-chemical energy conversion version's state-of-the-art efficiency, however, still falls short of what is needed for real-world applications. Solar energy conversion systems are often strongly associated with the following three crucial phases in photocatalytic processes.350–353 Enhancing photogenerated charge usage, facilitating the separation and transfer of photogenerated charges, and enhancing catalysts' ability to absorb solar light are the three main goals.337 Among them, the interface chemical reaction—which mostly entails charge transfer and redox reaction at the interface and directly affects the effectiveness of photocatalytic reactions—is typically regarded as an important stage in the process of photocatalytic reactions. Here, on the surface of these catalytic processes, holes oxidize H2O to make O2, while electrons decrease water to produce H2. CO2 is converted to value-added fuels and chemicals. Furthermore, electrons have the ability to activate N2 and transform it into NH3.337 By timely utilizing electron–hole pairs to participate in the reaction, cocatalysts can inhibit photocorrosion and enhance photostability of the photocatalysts, as some visible light-responsive semiconductors may be oxidized by photogenerated holes and undergo self-decomposition. This information is supported by ref. 337. It is commonly known that when catalysts are combined with semiconductors or photosensitizers for photocatalytic performance, a number of factors, such as loading amount, particle size, shape, phase, dispersity, structure, and chemical composition, ultimately influence the activities of catalysts.337,3548.7. The effect of photocatalysts for the synthesis of natural compounds
The expedient synthesis of complex organic compounds entails employing a potent amalgamation of viable tactics and robust mechanisms for bond formation. The efficacy of strategies aimed at diminishing the step-count of synthesis comprises a reduction in processes that do not result in the formation of bonds inherent to the structure that is being targeted. Furthermore, such strategies aim to optimize the construction of multiple bonds of the target molecule with the employment of a singular step, whenever practicable.17 The complete synthesis of naturally-occurring compounds presents an optimal platform for assessing the pragmatic efficacy of novel chemical modifications, as well as their influence in facilitating succinct synthetic methodologies. The study presents findings indicative of the proliferating influence of photocatalytic reactions in the production of intricate, natural compounds.17 One notable benefit of the formation of C–C and C–N σ-bonds through free radical reactions is the ability to accommodate polar functional groups, including alcohols, thiols, carboxylic acids, as well as primary and secondary amines. These functional groups are frequently unsuited for bond constructions employing basic reagents but are commonly tolerated in free–radical reactions, as previously noted in scholarly literature.17,355–357 Recent progressions in visible-light photocatalysis have resulted in novel approaches to produce carbon- and nitrogen-based radicals, using both conventional and emerging substrates.17,358,359 Through the utilization of visible-light photocatalysis, the direct generation of carbon radicals from conventional functional groups has become a viable process, circumventing the need for supplementary measures to introduce “radical-generating” derivatives.17 The present discourse revolves around the exemplification of total syntheses wherein primary emphasis has been placed on the direct production of tertiary carbon radicals from tertiary alcohols,360,361 α-amino radicals derived from α-amino acids,362,363 ketones,364 and tertiary carbamates.365 Additionally, this discourse also takes into account the generation of nitrogen-centered radicals originating from secondary aryl sulfonamides366 and benzamides.17,367 As previously indicated, the complete synthesis of naturally occurring compounds and their derivatives serves as a motivating factor for organic chemists to discern and experiment with innovative synthetic approaches and novel techniques. In recent times, the utilization of visible light photoredox catalysis has significantly increased in its ability to facilitate selective bond formation in chemical reactions under moderately mild conditions, thereby proving to be a potent technology.368 This paper undertakes an examination of the utilization of photoredox catalysis, specifically the application of tris(bipyridyl)ruthenium(II) chloride ([Ru-(bpy)3Cl2]), in the production of (+)-gliocladin C, a natural product.Indole-derived alkaloids have long been regarded as intriguing and complex objectives for organic synthesis, spanning numerous periods in the history of this field.368 The intricate nature of their structures, as well as their compelling biological characteristics, have rendered them sought-after models for evaluating innovative synthetic approaches.368 Furst et al. (2011) presented a novel approach for the dehalogenation of C3-bromopyrroloindolines utilizing reductive photoredox catalysis facilitated by tris(bipyridyl)ruthenium(II) chloride [Ru-(bpy)3Cl2], without the requirement of tin-based reagents.368–370 The authors utilized a photoredox manifold closely linked to their initial findings as the focal point of their asymmetric synthesis of (+)-gliocladin C (1), a multifaceted natural compound exhibiting compelling cytotoxic properties.368,370 This synthesis is illustrated in Fig. 23.370 Significantly, the photoredox coupling procedure was executed on a natural product (1) at a gram-scale level by implementing seven highly efficient steps.368
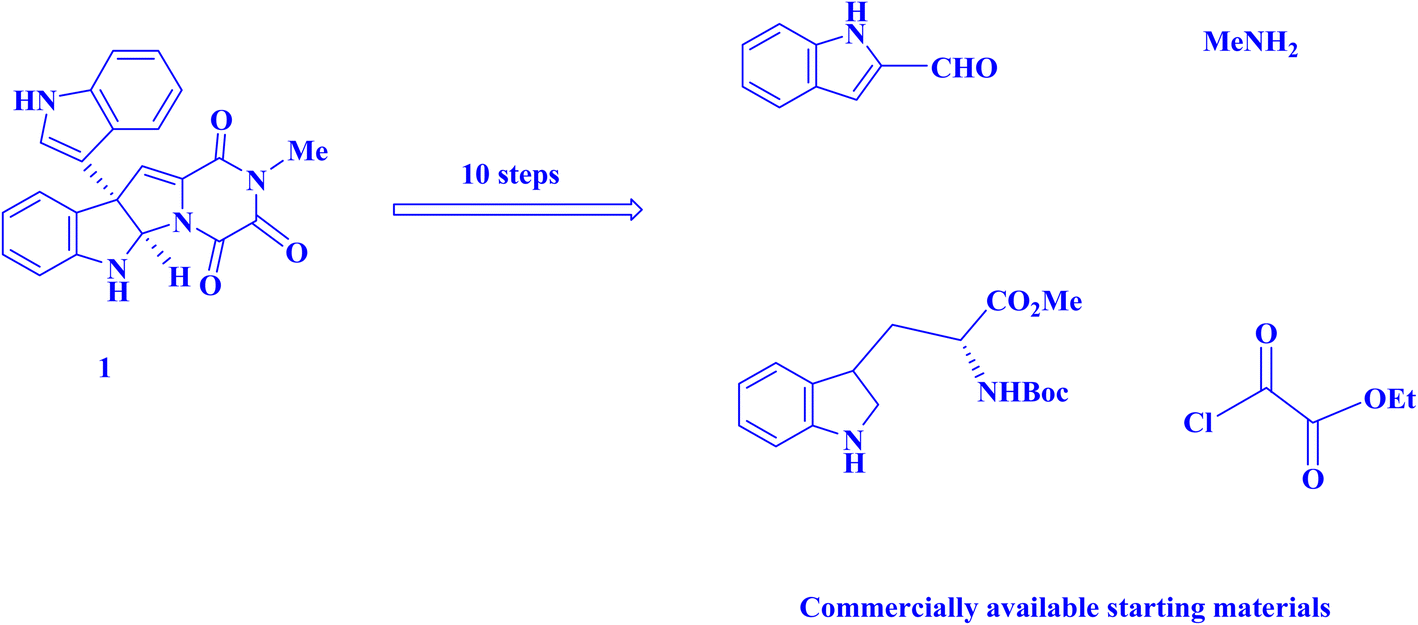 | ||
| Fig. 23 Total synthesis of gliocladin C (1) through the use of visible-light photoredox catalysis.370 | ||
8.8. The use of photocatalysts in the during the oxidation
An alternative to bioprocesses for the breakdown of hazardous or nonbiodegradable molecules is chemical oxidation.371–373 For this goal, many techniques have been employed, including ozonation,374,375 wet oxidation,376 hydrogen peroxide-based approaches,377 electrochemical,378,379 or photochemical oxidation.268 The remarkable capacity of metal oxide-based photocatalysts and visible light-driven photocatalysts to degrade organic pollutants through sophisticated oxidation processes, together with their cleaner processes, have garnered significant interest.380 The ability to create a mechanistically based treatment for each unique pollutant is one of the primary benefits of using organic photocatalysts. The variety of the involved chemical pathways is the reason behind this. Basically, two kinds of studies can yield pertinent information: (a) using time-resolved techniques (transient absorption spectroscopy, emission, etc.) to detect short-lived excited states or reactive intermediates, or (b) using steady-state photolysis (measured by a range of endpoints) and identifying the photoproducts obtained at various oxidation stages.268 We have highlighted two studies in this area in the following.8.9. The use of photocatalysts in the oxidative couplings
Green chemistry criteria are satisfied by oxidative coupling reactions, an interesting kind of photocatalytic transformation that may be used with predesigned organic materials and enable the use of O2 as an oxidant.383 We have highlighted two studies in this area in the following. acting as the photocatalyst, the current approach enables a variety of aryl bromides to undergo the reductive coupling smoothly, providing a pollutive reductant-free method for the synthesis of biaryl skeletons.384
acting as the photocatalyst, the current approach enables a variety of aryl bromides to undergo the reductive coupling smoothly, providing a pollutive reductant-free method for the synthesis of biaryl skeletons.3848.10. Summary
Studies show that there are several uses for photocatalysis. It has attracted a lot of interest due to its suitability as a substitute method for achieving various chemical reactions in the medical field, industrial setting, and drinking water purification. In addition, it offers a defense against microbes. These applications are always improving.Photocatalysis and photocatalytic reactions conducted under mild reaction conditions, often using light-emitting diodes that emit radiation in a well-defined wavelength range corresponding to the absorption spectrum of the desired photocatalyst and at room temperature, could greatly aid in the development of sophisticated chemical synthesis as well as the synthesis of natural compounds and their derivatives. These alterations are brought about by radical chain reactions. The pharmaceutical industry uses radical reactions in complex syntheses and photoreduction catalyst capacity in the search and development of novel medications. This necessitates using cost-effective techniques.
The use of nanoscale photocatalytic materials has become more widespread these days in a variety of fields, including the defense against microbial diseases. Their remarkable surface-to-volume ratio and minuscule size play a major role in this.
Numerous very promising photocatalysts have been widely used in waste management systems that prioritize environmental sustainability. In order to address environmental challenges, particularly the removal of residual pollutants from wastewater streams, photocatalysis, and renewable solar energy systems have been used to activate chemical reactions based on oxidation and reduction through photocatalysis. This is a viable and sustainable technological approach.
Over the past few decades, plastics and pharmaceuticals have become widely used and have finally become essential in people's lives. Compared to other plastics, microplastics (MPs) are more dangerous to the environment because of their tiny size, which makes them easily transportable. As a result, MPs have been discovered in sediments, water, air, soil, and live creatures. Furthermore, unmodified or metabolized organic molecules with a range of biological and toxicological properties that are present in pharmaceuticals may be released into wastewater. Even though they have been found at concentrations much below therapeutic levels, they may still have detrimental effects on people and/or animals since they have the ability to constantly impact organisms at different developmental stages. When photocatalysis occurs in the presence of visible light, it is the most effective method for removing microplastic contamination from wastewater and promoting photocatalytic degradation. Most air pollutants are mixes of harmful chemicals or other substances (gases, solid particles, and liquid droplets) whose concentration and atmospheric persistence time are high enough to cause harm to the environment and be ecotoxic. Among the many different kinds of air pollution are nitrogen oxides. NOx The removal of nitrogen oxides by photocatalysis has been the subject of much research in the last several years. The main advantage of photocatalytic technologies is their capacity to start the reaction at almost room temperature. In addition, nowadays, visible light photocatalysts have attracted much attention for the degradation of organic pollutants, including the degradation of hazardous or non-degradable molecules, through oxidation processes.
Studies on sustainable technology for renewable energy sources are becoming more and more numerous. Hydrogen is employed in the system for a number of reasons, including its versatility as a fuel for cells and its status as a clean energy source. Inspired by nature, the artificial photosynthesis system is made to transform H2O, CO2, and N2 into energy-dense compounds such as H2, CO, CH4, CH3OH, NH3, and so on.
9. Conclusion and future research
Over the course of the past few years, reactions induced by visible light have emerged as a potent technique for initiating a varied range of organic transformations under temperate conditions. Ultimately, they occupy a distinct position in the domain of sustainable chemistry. Photoredox catalysis represents a novel advancement, offering an optimal approach for inducing radical cyclization while simultaneously fragmenting the C–C/C–X bond formation route within heterocyclic chemistry. Moreover, its frequent application has been observed in the domain of green synthesis as well as in other fields encompassing radical chemistry and photochemistry. Photoredox catalysts leverage a renewable energy source to facilitate chemical reactions through the absorption of visible light radiation, which is subsequently transferred to low-energy organic substrates. Over the past few years, photoredox catalysis via visible light irradiation has garnered significant attention amongst the scientific community. The utilization of light as a tool for the creation of chemical bonds under mild reaction conditions has emerged as a particularly pertinent area of investigation. Photocatalysis is a rapidly developing technology that exhibits a diverse range of potential applications. The operational conditions of this process encompass ambient temperature and atmospheric pressure, thereby rendering it compatible with renewable energy resources. The photoredox catalyst exhibits several advantages in terms of stability that align with several principles of green chemistry, corroborating its position as a versatile tool for catalytic reactions.The utilization of photoredox catalysis presents a promising avenue toward increased efficacy and environmentally sustainable opportunities within the domains of industrial and academic research. In order to achieve enhanced energy efficiency and optimal atom economy, photoredox catalysis should ideally incorporate both sunlight and direct photochemistry. This dual approach not only accelerates the relevant processes but also circumvents the necessity for artificial light sources. In recent years, the utilization of photoredox processes and photoredox catalysis has garnered significant interest and is increasingly recognized as a potent new area in advanced synthesis and catalysis. The employment of photocatalysts in the capacity of an antimicrobial agent confers numerous benefits, including their ability to exterminate not only bacteria and fungi but also inhibit viral proliferation. Furthermore, they possess the capability to decompose hazardous and unsteady organic substances present in the atmosphere. In fact, a photocatalytic degradation reaction takes place whereby the formation of water and carbon dioxide act as by-products that are subsequently used in the photosynthesis process, thus leading to a stable and coordinated sequence of events. Photocatalytic techniques exhibit considerable potential for numerous wide-ranging applications, encompassing respiratory air and environmental health, biological, medical, laboratory, hospital, pharmaceutical, food industry, and plant protection. One of their applications lies in the treatment of sewage and effluent, as well as the disinfection of drinking water to mitigate the risk posed by microorganisms. In recent years, significant research endeavors have been undertaken to enhance the efficacy of photocatalytic systems, leading to the emergence of multiple commercial applications. Considering the emerging nature of this field requires more research in this field. According to the above, it is obvious that the research in the field of photocatalyst and applications related to photocatalyst is a promising research area and a perspective for future research. At the outset of this endeavor, a comprehensive exploration of this domain is imperative.
Data availability
All data generated or analyzed during this study are included in this published article.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
This work is financially supported by Iran National Science Foundation (INSF) (no. 4015618), financially supported by Iran's National Elites Foundation (no. 4015618), and also, Shiraz University of Medical Sciences.References
- A. Rahman, J. R. Jennings, A. L. Tan and M. M. Khan, ACS Omega, 2022, 7, 22089–22110 CrossRef CAS PubMed
.
- H. Wang, X. Li, X. Zhao, C. Li, X. Song, P. Zhang and P. Huo, Chin. J. Catal., 2022, 43, 178–214 CrossRef CAS
- P. P. Singh and V. Srivastava, RSC Adv., 2022, 12, 18245–18265 RSC
- Q. Xiao, Q. X. Tong and J. J. Zhong, Molecules, 2022, 27, 619 CrossRef CAS
- T. Y. Shang, L. H. Lu, Z. Cao, Y. Liu, W. M. He and B. Yu, Chem. Commun., 2019, 55, 5408–5419 RSC
- V. Srivastava, P. K. Singh and P. P. Singh, J. Photochem. Photobiol., C, 2022, 50, 100488 CrossRef CAS
- G. E. Crisenza and P. Melchiorre, Nat. Commun., 2020, 11, 803 CrossRef PubMed
- J. A. Leitch, H. R. Smallman and D. L. Browne, J. Org. Chem., 2021, 86, 14095–140101 CrossRef CAS PubMed
- R. I. Patel, A. Sharma, S. Sharma and A. Sharma, Org. Chem. Front., 2021, 8, 1694–1718 RSC
- C. Russo, F. Brunelli, G. Cesare Tron and G. Mariateresa, J. Org. Chem., 2023, 88, 6284–6293 CrossRef CAS PubMed
- Y. Zhao, J. Yuan, L. Zhu and Y. Fang, Chin. Chem. Lett., 2024, 35, 109065 CrossRef CAS
- P. Melchiorre, Chem. Rev., 2022, 122, 1483–1484 CrossRef CAS PubMed
- G. Ciamician, Science, 1912, 36, 385–394 CrossRef CAS PubMed
- A. Albini and M. Fagnoni, Green Chem., 2004, 6, 1–6 RSC
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, Sausalito, CA, 2010 Search PubMed
- M. H. Shaw, J. Twilton and D. W. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed
- S. P. Pitre and L. E. Overman, Chem. Rev., 2021, 122, 1717–1751 CrossRef PubMed
- M. D. Karkas and J. A. Porco Jr, Chem. Rev., 2016, 116, 9683–9747 CrossRef CAS PubMed
- P. Klán and J. Wirz, Photochemistry of Organic Compounds: from Concepts to Practice, John Wiley & Sons, 2009 Search PubMed
- S. Peiris, J. McMurtrie and H. Y. Zhu, Catal. Sci. Technol., 2016, 6, 320–338 RSC
- S. Sharma and A. Sharma, Org. Biomol. Chem., 2019, 17, 4384–4405 RSC
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC
- S. Dong, J. Feng, M. Fan, Y. Pi, L. Hu, X. Han, M. Liu, J. Sun and J. Sun, RSC Adv., 2015, 5, 14610–14630 RSC
- A. O. Ibhadon and P. Fitzpatrick, Catalysts, 2013, 3, 189–218 CrossRef CAS
- W. S. Koe, J. W. Lee, W. C. Chong, Y. L. Pang and L. C. Sim, Environ. Sci. Pollut. Res., 2020, 27, 2522–2565 CrossRef CAS
- T. Velempini, E. Prabakaran and K. Pillay, Mater. Today Chem., 2021, 19, 100380 CrossRef CAS
- F. Mohamadpour and F. Mohamadpour, Sustainable Environ. Res., 2024, 34, 8 CrossRef CAS
- M. Saeed, M. Muneer, A. U. Haq and N. Akram, Environ. Sci. Pollut. Res., 2022, 29, 293–311 CrossRef CAS PubMed
- T. K. Lee, Transactions of the Korean hydrogen and new energy society, 2001, 12, 21–27 Search PubMed
- C. Acar, I. Dincer and C. Zamfirescu, Int. J. Energy Res., 2014, 38, 1903–1920 CrossRef CAS
- R. Ameta and S. C. Ameta, Photocatalysis: Principles and Applications, Crc Press, 2016, pp. 1–2 Search PubMed
- J. Plotnikow, Allgemeine-photochemie, Berlin and Leipzig, Walter De Gruyter & Co, 1936 Search PubMed
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS
- V. C. Rengel, S. V. Khedkar and N. K. J. Thanvi, Int. J. Adv. Eng. Technol., 2012, 3, 31–35 Search PubMed
- F. Parrino and G. Palmisano, Molecules, 2020, 26, 23 CrossRef PubMed
- A. Strada, M. Fredditori, G. Zanoni and S. Protti, Molecules, 2019, 24, 1318 CrossRef PubMed
- Z. C. Fu, C. Mi, Y. Sun, Z. Yang, Q. Q. Xu and W. F. Fu, Molecules, 2019, 24, 1878 CrossRef CAS PubMed
- E. Voutyritsa, I. Triandafillidi, N. V. Tzouras, N. F. Nikitas, E. K. Pefkianakis, G. C. Vougioukalakis and C. G. Kokotos, Molecules, 2019, 24, 1644 CrossRef CAS PubMed
- M. Rao, W. Wu and C. Yang, Molecules, 2019, 24, 1502 CrossRef CAS PubMed
- M. Antonopoulou, C. Kosma, T. Albanis and I. Konstantinou, Sci. Total Environ., 2021, 765, 144163 CrossRef CAS PubMed
- M. Antonopoulou and I. Konstantinou, Appl. Catal., A, 2016, 515, 136–143 CrossRef CAS
- P. S. Konstas, C. Kosma, I. Konstantinou and T. Albanis, Water, 2019, 11, 2165 CrossRef CAS
- T. G. Vasconcelos, K. Kümmerer, D. M. Henriques and A. F. Martins, J. Hazard. Mater., 2009, 169, 1154–1158 CrossRef CAS PubMed
- N. Liu, T. Li, Z. Zhao, J. Liu, X. Luo, X. Yuan, K. Luo, J. He, D. Yu and Y. Zhao, ACS Omega, 2020, 5, 12557–12567 CrossRef CAS PubMed
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J. O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed
- F. K. Kessler, Y. Zheng, D. Schwarz, C. Merschjann, W. Schnick, X. Wang and M. J. Bojdys, Nat. Rev. Mater., 2017, 2, 1–7 Search PubMed
- B. V. Lotsch, M. Döblinger, J. Sehnert, L. Seyfarth, J. Senker, O. Oeckler and W. Schnick, Chem.–Eur. J., 2007, 13, 4969–4980 CrossRef CAS PubMed
- F. Fina, S. K. Callear, G. M. Carins and J. T. Irvine, Chem. Mater., 2015, 27, 2612–2618 CrossRef CAS
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed. Engl., 2012, 51, 68–89 CrossRef CAS PubMed
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S. T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed
- G. Liao, Y. Gong, L. Zhang, H. Gao, G. J. Yang and B. Fang, Energy Environ. Sci., 2019, 12, 2080–2147 RSC
- G. Gao, Y. Jiao, E. R. Waclawik and A. Du, J. Am. Chem. Soc., 2016, 138, 6292–6297 CrossRef CAS PubMed
- J. Barrio, D. Mateo, J. Albero, H. Garcia and M. Shalom, Adv. Energy Mater., 2019, 9, 1902738 CrossRef CAS
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed
- Y. Zeng, X. Liu, C. Liu, L. Wang, Y. Xia, S. Zhang, S. Luo and Y. Pei, Appl. Catal., B, 2018, 224, 1–9 CrossRef CAS
- F. Su, S. C. Mathew, G. Lipner, X. Fu, M. Antonietti, S. Blechert and X. Wang, J. Am. Chem. Soc., 2010, 132, 16299–16301 CrossRef CAS PubMed
- Y. Dai, C. Li, Y. Shen, T. Lim, J. Xu, Y. Li, H. Niemantsverdriet, F. Besenbacher, N. Lock and R. Su, Nat. Commun., 2018, 9, 60 CrossRef PubMed
- Q. Liu and J. Zhang, Langmuir, 2013, 29, 3821–3828 CrossRef CAS PubMed
- M. Wu, Q. Wang, Q. Sun and P. Jena, J. Phys. Chem. C, 2013, 117, 6055–6059 CrossRef CAS
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed
- H. Gao, S. Yan, J. Wang, Y. A. Huang, P. Wang, Z. Li and Z. Zou, Phys. Chem. Chem. Phys., 2013, 15, 18077–18084 RSC
- S. A. Younis, E. E. Kwon, M. Qasim, K. H. Kim, T. Kim, D. Kukkar, X. Dou and I. Ali, Prog. Energy Combust. Sci., 2020, 81, 100870 CrossRef
- D. Kukkar, K. Vellingiri, K. H. Kim and A. Deep, Sens. Actuators, B, 2018, 273, 1346–1370 CrossRef CAS
- M. Cheng, C. Lai, Y. Liu, G. Zeng, D. Huang, C. Zhang, L. Qin, L. Hu, C. Zhou and W. Xiong, Coord. Chem. Rev., 2018, 368, 80–92 CrossRef CAS
- L. Shen, R. Liang and L. Wu, Chin. J. Catal., 2015, 36, 2071–2088 CrossRef CAS
- S. I. Vasylevs’ kyy, G. A. Senchyk, A. B. Lysenko, E. B. Rusanov, A. N. Chernega, J. Jezierska, H. Krautscheid, K. V. Domasevitch and A. Ozarowski, Inorg. Chem., 2014, 53, 3642–3654 CrossRef PubMed
- L. Sun, M. G. Campbell and M. Dincă, Angew. Chem., Int. Ed. Engl., 2016, 55, 3566–3579 CrossRef CAS PubMed
- P. Kumar, A. Deep and K. H. Kim, TrAC, Trends Anal. Chem., 2015, 73, 39–53 CrossRef CAS
- A. Amini, S. Kazemi and V. Safarifard, Polyhedron, 2020, 177, 114260 CrossRef CAS
- V. Stavila, A. A. Talin and M. D. Allendorf, Chem. Soc. Rev., 2014, 43, 5994–6010 RSC
- L. Zeng, X. Guo, C. He and C. Duan, ACS Catal., 2016, 6, 7935–7947 CrossRef CAS
- T. Zhang, Y. Jin, Y. Shi, M. Li, J. Li and C. Duan, Coord. Chem. Rev., 2019, 380, 201–229 CrossRef CAS
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC
- N. Zaman, N. Iqbal and T. Noor, Arabian J. Chem., 2022, 15, 103906 CrossRef CAS
- Y. Yang, X. Chu, H. Y. Zhang, R. Zhang, Y. H. Liu, F. M. Zhang, M. Lu, Z. D. Yang and Y. Q. Lan, Nat. Commun., 2023, 14, 593 CrossRef CAS PubMed
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 1–9 Search PubMed
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W. H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed
- R. Chen, Y. Wang, Y. Ma, A. Mal, X. Y. Gao, L. Gao, L. Qiao, X. B. Li, L. Z. Wu and C. Wang, Nat. Commun., 2021, 12, 1354 CrossRef CAS PubMed
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef PubMed
- S. Li, L. Li, Y. Li, L. Dai, C. Liu, Y. Liu, J. Li, J. Lv, P. Li and B. Wang, ACS Catal., 2020, 10, 8717–8726 CrossRef CAS
- Y. Yang, D. He, X. Feng and X. Xiao, SmartMat, 2023, e1223 Search PubMed
- P. J. Waller, F. Gándara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef CAS PubMed
- X. Liu, D. Huang, C. Lai, G. Zeng, L. Qin, H. Wang, H. Yi, B. Li, S. Liu, M. Zhang and R. Deng, Chem. Soc. Rev., 2019, 48, 5266–5302 RSC
- Y. Shi, J. Song, F. Cui, X. Duli, Y. Tian, S. Jin, Q. Shu and G. Zhu, Nano Res., 2023, 16, 1507–1512 CrossRef CAS
- Z. Xiang, D. Cao and L. Dai, Polym. Chem., 2015, 6, 1896–1911 RSC
- Y. Pramudya and J. L. Mendoza-Cortes, J. Am. Chem. Soc., 2016, 138, 15204–15213 CrossRef CAS PubMed
- Z. Zhang, C. Kang, S. B. Peh, D. Shi, F. Yang, Q. Liu and D. Zhao, J. Am. Chem. Soc., 2022, 144, 14992–14996 CrossRef CAS PubMed
- Z. Guo, H. Wu, Y. Chen, S. Zhu, H. Jiang, S. Song, Y. Ren, Y. Wang, X. Liang, G. He and Y. Li, Angew. Chem., 2022, 134, e202210466 CrossRef
- M. X. Wu and Y. W. Yang, Chin. Chem. Lett., 2017, 28, 1135–1143 CrossRef CAS
- Q. Zhao, Y. Wang, M. Li, S. Zhu, T. Li, J. Yang, T. Lin, E. P. Delmo, Y. Wang, J. Jang and M. Gu, SmartMat, 2022, 3, 183–193 CrossRef CAS
- V. Balzani, G. Bergamini and P. Ceroni, Coord. Chem. Rev., 2008, 252, 2456–2469 CrossRef CAS
- V. Balzani and V. Carassiti, Photochemistry of Coordination Compounds, Aca-demic Press, London, 1970 Search PubMed
- D. H. John and G. T. Field, A Textbook of Photographic Chemistry, Chapman and Hall, London, 1963 Search PubMed
- W. G. Leighton and G. S. Forbes, J. Am. Chem. Soc., 1930, 52, 3139–3152 CrossRef
- F. Basolo and R. G. Pearson, Mechanisms of Inorganic Reactions, Wiley, NewYork, 1958 Search PubMed
- L. E. Orgel, Q. Rev., Chem. Soc., 1954, 8, 422–450 RSC
- C. K. Jorgensen, Absorption Spectra and Chemical Bonding in Complexes, Pergamon Press, Oxford, 1962 Search PubMed
- G. A. Crosby, W. G. Perkins and D. M. Klassen, J. Chem. Phys., 1965, 43, 1498–1503 CrossRef CAS
- G. A. Crosby, R. J. Watts and D. H. Carstens, Science, 1970, 170, 1195–1196 CrossRef CAS PubMed
- C. Creutz and N. Sutin, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 2858–2862 CrossRef CAS PubMed
- C. C. Wang, H. P. Jing, Y. Q. Zhang, P. Wang and S. J. Gao, Transition Met. Chem., 2015, 40, 573–584 CrossRef CAS
- C. C. Wang, J. R. Li, X. L. Lv, Y. Q. Zhang and G. Guo, Energy Environ. Sci., 2014, 7, 2831–2867 RSC
- C. C. Wang, Y. Q. Zhang, J. Li and P. Wang, J. Mol. Struct., 2015, 1083, 127–136 CrossRef CAS
- L. Shen, S. Liang, W. Wu, R. Liang and L. Wu, Dalton Trans., 2013, 42, 13649–13657 RSC
- R. Liang, L. Shen, F. Jing, W. Wu, N. Qin, R. Lin and L. Wu, Appl. Catal., B, 2015, 162, 245–251 CrossRef CAS
- Z. Yin, Y. L. Zhou, M. H. Zeng and M. Kurmoo, Dalton Trans., 2015, 44, 5258–5275 RSC
- X. T. Zhang, L. M. Fan, X. Zhao, D. Sun, D. C. Li and J. M. Dou, CrystEngComm, 2012, 14, 2053–2061 RSC
- Q. Chu, Z. Su, J. Fan, T. A. Okamura, G. C. Lv, G. X. Liu, W. Y. Sun and N. Ueyama, Cryst. Growth Des., 2011, 11, 3885–3894 CrossRef CAS
- D. R. Xiao, E. B. Wang, H. Y. An, Y. G. Li and L. Xu, Cryst. Growth Des., 2007, 7, 506–512 CrossRef CAS
- Y. Gong, J. Qin, T. Wu, J. Li, L. Yang and R. Cao, Dalton Trans., 2012, 41, 1961–1970 RSC
- S. Benkhaya, S. M'rabet and A. El Harfi, Inorg. Chem. Commun., 2020, 115, 107891 CrossRef CAS
- E. S. Ferreira, A. N. Hulme, H. McNab and A. Quye, Chem. Soc. Rev., 2004, 33, 329–336 RSC
- A. Abel, The History of Dyes and Pigments: from Natural Dyes to High Performance Pigments, InColour Design, Woodhead Publishing, 2012, pp. 557–587 Search PubMed
- R. G. Saratale, G. D. Saratale, J. S. Chang and S. P. Govindwar, J. Taiwan Inst. Chem. Eng., 2011, 42, 138–157 CrossRef CAS
- F. M. Rows, J. Soc. Dyers Colour., 1938, 54, 551–562 CrossRef
- D. Wesenberg, I. Kyriakides and S. N. Agathos, Biotechnol. Adv., 2003, 22, 161–187 CrossRef CAS PubMed
- V. Walsh, Resour. Policy, 1984, 13, 211–234 CrossRef
- F. Vazquez-Ortega, I. Lagunes and A. Trigos, Dyes Pigm., 2020, 176, 108248 CrossRef CAS
- S. Noreen, H. N. Bhatti, M. Iqbal, F. Hussain and F. M. Sarim, Int. J. Biol. Macromol., 2020, 147, 439–452 CrossRef CAS PubMed
- C. Belpaire, T. Reyns, C. Geeraerts and J. Van Loco, Chemosphere, 2015, 138, 784–791 CrossRef CAS PubMed
- A. Rehman, M. Usman, T. H. Bokhari, A. ul Haq, M. Saeed, H. M. Rahman, M. Siddiq, A. Rasheed and M. U. Nisa, J. Mol. Liq., 2020, 301, 112408 CrossRef CAS
- A. Bouchoul and M. Chaguer, Analyse, synthèse et étude solvatochromique de composés tinctoriaux, 2017 Search PubMed
- H. Nozet and J. Majault, Textiles Chimiques: Fibres Modernes, Eyrolles, 1976 Search PubMed
- A. Benaissa, Etude de la faisabilité d’élimination de certains colorants textiles par certains matériaux déchets d’origine naturelle, Université Abou Bakr Balkaid, Tlemcen-Algérie, 2012 Search PubMed
- V. Sivakumar, J. L. Anna, J. Vijayeeswarri and G. Swaminathan, Ultrason. Sonochem., 2009, 16, 782–789 CrossRef CAS PubMed
- A. K. Samanta and A. Konar, Dyeing of textiles with natural dyes, Natural Dyes, 2011, 30–56 Search PubMed
- A. K. Samanta and P. Agarwal, Indian J. Fibre Text. Res., 2009, 34, 384–399 CAS
- I. Zerin, Potentials of Natural Dyes for Textile Applications, in Encycl. Renew. Sustain. Mater, ed. S. Hashmi and I. A. Choudhury, Elsevier, Oxford, 2020, pp. 873–883 Search PubMed
- P. M. Pdos Santos Silva, T. R. Fiaschitello, R. S. de Queiroz, H. S. Freeman, S. A. da Costa, P. Leo, A. F. Montemor and S. M. da Costa, Dyes Pigm., 2020, 173, 107953 CrossRef
- A. D. Laurent, V. Wathelet, M. Bouhy, D. Jacquemin and E. Perpète, Simulation de la perception des couleurs de colorants organiques, 2010 Search PubMed
- E. A. Azzopardi, S. E. Owens, M. Murison, D. Rees, M. A. Sawhney, L. W. Francis, R. S. Teixeira, M. Clement, R. S. Conlan and I. S. Whitaker, J. Controlled Release, 2017, 249, 123–130 CrossRef CAS PubMed
- M. Rochkind, S. Pasternak and Y. Paz, Molecules, 2014, 20, 88–110 CrossRef PubMed
- H. Miyabe, Organic reactions promoted by metal-free organic dyes under visible light irradiation. InVisible-light Photocatalysis of Carbon-Based Materials, IntechOpen, 2017 Search PubMed
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed
- J. M. Narayanam and C. R. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC
- N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef CAS PubMed
- M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725–2756 CrossRef CAS PubMed
- J. W. Tucker, Y. Zhang, T. F. Jamison and C. R. Stephenson, Angew. Chem., Int. Ed. Engl., 2012, 51, 4144 CrossRef CAS PubMed
- I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49, 1566–1577 CrossRef CAS PubMed
- Y. Du, R. M. Pearson, C. H. Lim, S. M. Sartor, M. D. Ryan, H. Yang, N. H. Damrauer and G. M. Miyake, Chem–Eur. J., 2017, 23, 10962–10968 CrossRef CAS PubMed
- G. Q. Xu and P. F. Xu, Chem. Commun., 2021, 57, 12914–12935 RSC
- X. Deng, Z. Li and H. García, Chem–Eur. J., 2017, 23, 11189–11209 CrossRef CAS PubMed
- J. T. Yu and C. Pan, Chem. Commun., 2016, 52, 2220–2236 RSC
- R. Pawlowski, F. Stanek and M. Stodulski, Molecules, 2019, 24, 1533 CrossRef PubMed
- A. Albini, M. Fagnoni and M. Mella, Pure Appl. Chem., 2000, 72, 1321–1326 CrossRef CAS
- M. Oelgemöller and N. Hoffmann, Org. Biomol. Chem., 2016, 14, 7392–7442 RSC
- B. König, Eur. J. Org Chem., 2017, 2017, 1979–1981 CrossRef
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS PubMed
- C. Ferreri and C. Chatgilialoglu, Lipid isomerization, Encyclopedia of Radicals in Chemistry, Biol. Mater., 2012 DOI:10.1002/9781119953678.rad054
- M. Ramaiah, Tetrahedron, 1987, 43, 3541–3676 CrossRef CAS
- G. Bar and A. F. Parsons, Chem. Soc. Rev., 2003, 32, 251–263 RSC
- A. F. Parsons, An Introduction to Free Radical Chemistry, Blackwell Science, Oxford, 2000 Search PubMed
- H. Yorimitsu and K. Oshima, Radicals in Organic Synthesis, ed. P. Renaud and M. P. Sibi, WILEY-VCH, Weinheim, 2001 Search PubMed
- D. P. Curran, N. A. Porter and B. Giese, Stereochemistry of Radical Reactions, Concepts, Guidelines and Synthetic Applications, John Wiley & Sons, 2008 Search PubMed
- B. C. Gilbert and A. F. Parsons, J. Chem. Soc., Perkin Trans. 2, 2002, 367–387 RSC
- N. Kandoth, J. P. Hernández, E. Palomares and J. Lloret-Fillol, Sustain. Energy Fuels., 2021, 5, 638–665 RSC
- C. S. Wang, P. H. Dixneuf and J. F. Soulé, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed
- V. Balzani, G. Bergamini, S. Campagna and F. Puntoriero, Photochemistry and Photophysics of Coordination Compounds: Overview and General Concepts, In Photochemistry and Photophysics of Coordination Compounds I, Springer, Berlin, Heidelberg, 2007, pp. 1–36 Search PubMed
- F. Mohamadpour, Curr. Res. Green Sustainable Chem., 2023, 6, 100356 CrossRef CAS
- F. Politano and G. Oksdath-Mansilla, Org. Process Res. Dev., 2018, 22, 1045–1062 CrossRef CAS
- R. H. Verschueren and W. M. De Borggraeve, Molecules, 2019, 24, 2122 CrossRef PubMed
- A. G. Griesbeck, N. Hoffmann and K. D. Warzecha, Acc. Chem. Res., 2007, 40, 128–140 CrossRef CAS PubMed
- R. A. Marcus, Angew. Chem., Int. Ed. Engl., 1993, 32, 1111–1121 CrossRef
- D. Escudero, Acc. Chem. Res., 2016, 49, 1816–1824 CrossRef CAS PubMed
- P. Piotrowiak, M. A. Rodgers, J. Mattay and D. Astruc, Electron Transfer in Chemistry, ed. V. Balzani, Weinheim, Wiley-VCh, 2001 Search PubMed
- M. Natali, S. Campagna and F. Scandola, Chem. Soc. Rev., 2014, 43, 4005–4018 RSC
- A. A. Mohamed, M. A. Omar, S. M. Derayea, M. A. Elsayed and I. M. Mostafa, Spectrochim. Acta A Mol. Biomol. Spectrosc., 2024, 305, 123451 CrossRef CAS PubMed
- M. Grätzel, J. Photochem. Photobiol., C, 2003, 4, 145–153 CrossRef
- B. Daly, J. Ling and A. P. de Silva, Chem. Soc. Rev., 2015, 44, 4203–4211 RSC
- E. M. S. Stennett, M. A. Ciuba and M. Levitus, Chem. Soc. Rev., 2014, 43, 1057–1075 RSC
- L. Hammarstrom, Acc. Chem. Res., 2015, 48, 840–850 CrossRef PubMed
- M. Reckenthaeler and A. G. Griesbeck, Adv. Synth. Catal., 2013, 355, 2727–2744 CrossRef CAS
- G. J. Tangelder, C. J. Janssens, D. W. Slaaf, M. G. Oude Egbrink and R. S. Reneman, Am. J. Physiol.: Heart Circ. Physiol., 1995, 268, H909–H915 CrossRef CAS PubMed
- M. Teuber, M. Rögner and S. Berry, Biochim. Biophys. Acta, Bioenerg., 2001, 1506, 31–46 CrossRef CAS PubMed
- W. S. Vincent III and E. S. Goldstein, Anal. Biochem., 1981, 110, 123–127 CrossRef PubMed
- I. Y. Goryacheva, G. V. Mel’nikov and S. N. Shtykov, J. Anal. Chem., 2000, 55, 874–878 CrossRef CAS
- T. Pérez-Ruiz, C. Martínez-Lozano, V. Tomás and J. Fenoll, Anal. Bioanal. Chem., 2003, 375, 661–665 CrossRef PubMed
- T. Pérez-Ruiz, C. Martınez-Lozano, A. Sanz and M. T. San Miguel, J. Pharm. Biomed. Anal., 1997, 16, 249–254 CrossRef PubMed
- A. Arques, A. M. Amat, L. Santos-Juanes, R. F. Vercher, M. L. Marin and M. A. Miranda, Catal. Today, 2009, 144, 106–111 CrossRef CAS
- A. M. Amat, A. Arques, F. Galindo, M. A. Miranda, L. Santos-Juanes, R. F. Vercher and R. Vicente, Appl. Catal. B: Environ., 2007, 73, 220–226 CrossRef CAS
- F. Takemura, Bull. Chem. Soc. Jpn., 1962, 35, 1078–1086 CrossRef CAS
- P. Saint-Cricq, T. Pigot, S. Blanc and S. Lacombe, J. Hazard. Mater., 2012, 211, 266–274 CrossRef PubMed
- R. B. Webb, B. S. Hass and H. E. Kubitschek, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1979, 59, 1–3 CrossRef CAS PubMed
- L. O. Kostjukova, S. V. Leontieva and V. V. Kostjukov, Spectrochim. Acta, Part A, 2021, 249, 119302 CrossRef CAS PubMed
- C. Sengupta, P. Mitra, S. Chatterjee, G. Bhattacharjee, B. Satpati and S. Basu, J. Mol. Liq., 2018, 272, 198–208 CrossRef CAS
- F. Mohamadpour, J. Heterocycl. Chem., 2023, 60, 504–512 CrossRef CAS
- X. Zhang, L. Liu, W. Li, C. Wang, J. Wang, W. H. Fang and X. Chen, JACS Au, 2023, 3, 1452–1463 CrossRef CAS PubMed
- Q. Q. Zhou, Y. Q. Zou, L. Q. Lu and W. J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS PubMed
- J. R. Chen, D. M. Yan, Q. Wei and W. J. Xiao, ChemPhotoChem, 2017, 1, 148–158 CrossRef CAS
- F. Mohamadpour, Sci. Rep., 2023, 13, 13142 CrossRef CAS PubMed
- F. Mohamadpour, Sci. Rep., 2023, 13, 10262 CrossRef CAS PubMed
- Y. Yang, L. Liu, W. H. Fang, L. Shen and X. Chen, JACS Au, 2022, 2, 2596–2606 CrossRef CAS PubMed
- M. D. Kärkäs, B. S. Matsuura and C. R. Stephenson, Science, 2015, 349, 1285–1286 CrossRef PubMed
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC
- F. Mohamadpour, J. Photochem. Photobiol., A, 2022, 432, 114120 CrossRef CAS
- M. Wainwright and K. B. Crossley, J. Chemother., 2002, 14, 431–443 CrossRef CAS PubMed
- J. P. Tardivo, A. Del Giglio, C. S. De Oliveira, D. S. Gabrielli, H. C. Junqueira, D. B. Tada, D. Severino, R. de Fátima Turchiello and M. S. Baptista, Photodiagn. Photodyn. Ther., 2005, 2, 175–191 CrossRef CAS PubMed
- J. I. I. Clifton and J. B. Leikin, Am. J. Ther., 2003, 10, 289–291 CrossRef PubMed
- F. Mohamadpour, RSC Adv., 2022, 12, 10701–10710 RSC
- S. Das, T. L. Thanulingam, C. S. Rajesh and M. V. George, Tetrahedron Lett., 1995, 36, 1337–1340 CrossRef CAS
- T. Xu, T. Cao, Q. Feng, S. Huang and S. Liao, Chem. Commun., 2020, 56, 5151–5153 RSC
- F. Mohamadpour, Sci. Rep., 2022, 12, 7253 CrossRef CAS PubMed
- L. Capaldo and D. Ravelli, Eur. J. Org Chem., 2017, 2017, 2056–2071 CrossRef CAS PubMed
- A. Adhikary, A. Kumar, D. Becker and M. D. Sevilla, Encyclopedia of Radicals in Chemistry, Biology and Materials, Chichester, United Kingdom, John Wiley & Sons Ltd, 2012 Search PubMed
- J. T. Hynes, J. P. Klinman, H. H. Limbach and R. L. Schowen, Hydrogen-Transfer Reactions, Wiley-VCH Verlag GmbH & Co. KGaA, 2007 Search PubMed
- S. Hammes-Schiffer and A. A. Stuchebrukhov, Chem. Rev., 2010, 110, 6939–6960 CrossRef CAS PubMed
- J. M. Mayer, Acc. Chem. Res., 2011, 44, 36–46 CrossRef CAS PubMed
- Y. Zhang and M. Szostak, Chem.–Eur. J., 2022, 28, e202104278 CrossRef CAS PubMed
- P. R. Ortiz de Montellano, Chem. Rev., 2010, 110, 932–948 CrossRef CAS PubMed
- P. R. De Montellano, Cytochrome P450: Structure, Mechanism, and Biochemistry, Springer Science & Business Media, 2007 Search PubMed
- K. U. Ingold and D. A. Pratt, Chem. Rev., 2014, 114, 9022–9046 CrossRef CAS PubMed
- M. Salamone and M. Bietti, Acc. Chem. Res., 2015, 48, 2895–2903 CrossRef CAS PubMed
- D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed
- M. Majek, F. Filace and A. J. von Wangelin, Beilstein J. Org. Chem., 2014, 10, 981–989 CrossRef PubMed
- D. P. Hari and B. König, Org. Lett., 2011, 13, 3852–3855 CrossRef CAS PubMed
- A. G. Condie, J. C. González-Gómez and C. R. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464–1465 CrossRef CAS PubMed
- V. Srivastava, P. K. Singh and P. P. Singh, Croat. Chem. Acta, 2015, 88, 59–65 CrossRef CAS
- F. Mohamadpour, BMC Chem., 2023, 17, 2 CrossRef CAS PubMed
- F. Mohamadpour, Dyes Pigm, 2021, 194, 109628 CrossRef CAS
- B. Balli, A. Aygun and F. Sen, Medicinal applications of photocatalysts. Nanophotocatalysis and Environmental Applications: Detoxification and Disinfection, 2020, pp. 245–265 Search PubMed
- J. Gamage and Z. Zhang, Int. J. Photoenergy, 2010, 2010, 764870 CrossRef
- R. Ameta and S. C. Ameta, Photocatalysıs: Principles and Applications, Taylor & Francis, Boca Raton, 2017 Search PubMed
- J. G. McEvoy and Z. Zhang, J. Photochem. Photobiol., C, 2014, 19, 62–75 CrossRef
- M. Graetzel, Acc. Chem. Res., 1981, 14, 376–384 CrossRef CAS
- H. Takeda and O. Ishitani, Coord. Chem. Rev., 2010, 254, 346–354 CrossRef CAS
- K. Kalyanasundaram and M. Grätzel, Coord. Chem. Rev., 1998, 177, 347–414 CrossRef CAS
- E. de Pedro Beato, D. Mazzarella, M. Balletti and P. Melchiorre, Chem. Sci., 2020, 11, 6312–6324 RSC
- M. Yan, J. C. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed
- J. Lalevée and J. P. Fouassier, Overview of radical initiation, Encyclopedia of Radicals in Chemistry, Biology and Materials, 2012 Search PubMed
- F. Mohamadpour and A. M. Amani, Front. Chem., 2024, 12, 1304850 CrossRef CAS PubMed
- J. R. Chen, X. Q. Hu, L. Q. Lu and W. J. Xiao, Acc. Chem. Res., 2016, 49, 1911–1923 CrossRef CAS PubMed
- L. Qian and M. Shi, Chem. Commun., 2023, 59, 3487–3506 RSC
- N. Hoffmann, J. Phys. Org. Chem., 2015, 28, 121–136 CrossRef CAS
- M. H. V. Huynh and T. J. Meyer, Chem. Rev., 2007, 107, 5004–5064 CrossRef CAS PubMed
- C. T. Saouma and J. M. Mayer, Chem. Sci., 2014, 5, 21–31 RSC
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed
- R. Mannhold, H. Kubinyi and H. Timmerman, Lipophilicity in Drug Action and Toxicology, John Wiley & Sons, 2008 Search PubMed
- M. K. Bogdos, E. Pinard and J. A. Murphy, Beilstein J. Org. Chem., 2018, 14, 2035–2064 CrossRef CAS PubMed
- P. Li, J. A. Terrett and J. R. Zbieg, ACS Med. Chem. Lett., 2020, 11, 2120–2130 CrossRef CAS PubMed
- D. P. Hari, T. Hering and B. König, Org. Lett., 2012, 14, 5334–5337 CrossRef CAS PubMed
- J. Prakash, S. B. Krishna, P. Kumar, V. Kumar, K. S. Ghosh, H. C. Swart, S. Bellucci and J. Cho, Catalysts, 2022, 12, 1047 CrossRef CAS
- J. Prakash, J. Cho and Y. K. Mishra, Micro Nano Eng., 2022, 14, 100100 CrossRef CAS
- S. M. Zacarías, M. L. Satuf, M. C. Vaccari and O. M. Alfano, Chem. Eng. J., 2015, 266, 133–140 CrossRef
- S. Fooladi, M. H. Nematollahi and S. Iravani, Environ. Res., 2023, 231, 116287 CrossRef CAS PubMed
- A. Bigham, A. Zarepour, M. Safarkhani, Y. Huh, A. Khosravi, N. Rabiee, S. Iravani and A. Zarrabi, Nano Mater. Sci., 2024 DOI:10.1016/j.nanoms.2024.02.006
- Q. You, K. Zhang, J. Liu, C. Liu, H. Wang, M. Wang, S. Ye, H. Gao, L. Lv, C. Wang, L. Zhu and Y. Yang, Adv. Sci., 2020, 7, 1903341 CrossRef CAS PubMed
- G. W. Park, M. Cho, E. L. Cates, D. Lee, B. T. Oh, J. Vinjé and J. H. Kim, J. Photochem. Photobiol., B, 2014, 140, 315–320 CrossRef CAS PubMed
- Y. Si, Z. Zhang, W. Wu, Q. Fu, K. Huang, N. Nitin, B. Ding and G. Sun, Sci. Adv., 2018, 4, eaar5931 CrossRef PubMed
- S. Torabi, M. J. Mansoorkhani, A. Majedi and S. Motevalli, J. Coord. Chem., 2020, 73, 1861–1880 CrossRef CAS
- N. Lagopati, K. Evangelou, P. Falaras, E. P. Tsilibary, P. V. Vasileiou, S. Havaki, A. Angelopoulou, E. A. Pavlatou and V. G. Gorgoulis, Pharmacol. Ther., 2021, 222, 107795 CrossRef CAS PubMed
- K. M. Lee, C. W. Lai, K. S. Ngai and J. C. Juan, Water Res., 2016, 88, 428–448 CrossRef CAS PubMed
- N. S. Lewis, Science, 2007, 315, 798–801 CrossRef CAS PubMed
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC
- A. Fujishima, X. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS
- R. Vinu and G. Madras, J. Indian Inst. Sci., 2010, 90, 189–230 CAS
- K. Wenderich and G. Mul, Chem. Rev., 2016, 116, 14587–14619 CrossRef CAS PubMed
- A. Pavanello, D. Fabbri, P. Calza, D. Battiston, M. A. Miranda and M. L. Marin, J. Photochem. Photobiol. B: Biol., 2021, 221, 112250 CrossRef CAS PubMed
- E. B. Mordechay, J. Tarchitzky, Y. Chen, M. Shenker and B. Chefetz, Environ. Pollut., 2018, 232, 164–172 CrossRef PubMed
- T. Rasheed, M. Bilal, A. A. Hassan, F. Nabeel, R. N. Bharagava, L. F. Ferreira, H. N. Tran and H. M. Iqbal, Environ. Res., 2020, 185, 109436 CrossRef CAS PubMed
- C. R. Ohoro, A. O. Adeniji, A. I. Okoh and O. O. Okoh, Int. J. Environ. Res. Public Health, 2019, 16, 3026 CrossRef CAS PubMed
- D. R. Baker and B. Kasprzyk-Hordern, J. Chromatogr. A, 2011, 1218, 8036–8059 CrossRef CAS PubMed
- F. Pomati, S. Castiglioni, E. Zuccato, R. Fanelli, D. Vigetti, C. Rossetti and D. Calamari, Environ. Sci. Technol., 2006, 40, 2442–2447 CrossRef CAS PubMed
- P. Fernández-Castro, M. Vallejo, M. F. San Román and I. Ortiz, J. Chem. Technol. Biotechnol., 2015, 90, 796–820 CrossRef
- A. R. Ribeiro, O. C. Nunes, M. F. Pereira and A. M. Silva, Environ. Int., 2015, 75, 33–51 CrossRef CAS PubMed
- S. Giannakis, S. Rtimi and C. Pulgarin, Molecules, 2017, 22, 1070 CrossRef PubMed
- M. L. Marin, L. Santos-Juanes, A. Arques, A. M. Amat and M. A. Miranda, Chem. Rev., 2012, 112, 1710–1750 CrossRef CAS PubMed
- M. A. Fox and M. T. Dulay, Chem. Rev., 1993, 93, 341–357 CrossRef CAS
- X. Kong, L. Cao, Y. Shi, Z. Chen, W. Shi and X. Du, Molecules, 2023, 28, 5098 CrossRef CAS PubMed
- H. Wang, J. Li, M. Zhou, Q. Guan, Z. Lu, P. Huo and Y. Yan, J. Ind. Eng. Chem., 2015, 30, 64–70 CrossRef CAS
- N. Delgado, L. Bermeo, D. A. Hoyos, G. A. Peñuela, A. Capparelli, D. Marino, A. Navarro and J. C. Casas-Zapata, Water Res., 2020, 187, 116448 CrossRef CAS PubMed
- F. Chen, T. Ma, T. Zhang, Y. Zhang and H. Huang, Adv. Mater., 2021, 33, 2005256 CrossRef CAS PubMed
- M. Nemiwal, T. C. Zhang and D. Kumar, Sci. Total Environ., 2021, 767, 144896 CrossRef CAS PubMed
- H. Shen, J. Wang, J. Jiang, B. Luo, B. Mao and W. Shi, Chem. Eng. J., 2017, 313, 508–517 CrossRef CAS
- J. A. Nasir, Z. ur Rehman, S. N. Shah, A. Khan, I. S. Butler and C. R. Catlow, J. Mater. Chem. A, 2020, 8, 20752–20780 RSC
- P. Gao, Y. Yang, Z. Yin, F. Kang, W. Fan, J. Sheng, L. Feng, Y. Liu, Z. Du and L. Zhang, J. Hazard. Mater., 2021, 412, 125186 CrossRef CAS PubMed
- Q. Wu, Z. Li and H. Hong, Appl. Clay Sci., 2013, 74, 66–73 CrossRef CAS
- X. Bai, W. Chen, B. Wang, T. Sun, B. Wu and Y. Wang, Int. J. Mol. Sci., 2022, 23, 8130 CrossRef CAS PubMed
- M. H. Abdurahman, A. Z. Abdullah and N. F. Shoparwe, Chem. Eng. J., 2021, 413, 127412 CrossRef CAS
- M. J. Calvete, G. Piccirillo, C. S. Vinagreiro and M. M. Pereira, Coord. Chem. Rev., 2019, 395, 63–85 CrossRef CAS
- S. Shurbaji, P. T. Huong and T. M. Altahtamouni, Catalysts, 2021, 11, 437 CrossRef CAS
- Z. Wei, J. Liu and W. Shangguan, Chin. J. Catal., 2020, 41, 1440–1450 CrossRef CAS
- S. Shehu Imam, R. Adnan and N. H. Mohd Kaus, Toxicol. Environ. Chem., 2018, 100, 518–539 CrossRef CAS
- M. F. Alexandre-Franco and C. Fernández-González, Water, 2020, 12, 102 Search PubMed
- Q. Yang, Y. Gao, J. Ke, P. L. Show, Y. Ge, Y. Liu, R. Guo and J. Chen, Bioengineered, 2021, 12, 7376–7416 CrossRef PubMed
- S. F. Yang, C. F. Lin, A. Y. Lin and P. K. Hong, Water Res., 2011, 45, 3389–3397 CrossRef CAS PubMed
- P. Liu, H. Zhang, Y. Feng, F. Yang and J. Zhang, Chem. Eng. J., 2014, 240, 211–220 CrossRef CAS
- D. Li and W. Shi, Chin. J. Catal., 2016, 37, 792–799 CrossRef CAS
- X. Yang, Z. Chen, W. Zhao, C. Liu, X. Qian, M. Zhang, G. Wei, E. Khan, Y. H. Ng and Y. S. Ok, Chem. Eng. J., 2021, 405, 126806 CrossRef CAS PubMed
- L. V. de Souza Santos, A. M. Meireles and L. C. Lange, J. Environ. Manage., 2015, 154, 8–12 CrossRef PubMed
- M. S. Yahya, N. Oturan, K. El Kacemi, M. El Karbane, C. T. Aravindakumar and M. A. Oturan, Chemosphere, 2014, 117, 447–454 CrossRef CAS PubMed
- V. Homem and L. Santos, J. Environ. Manage., 2011, 92, 2304–2347 CrossRef CAS PubMed
- Y. He, A. U. Rehman, M. Xu, C. A. Not, A. M. Ng and A. B. Djurišić, Heliyon, 2023, 9, e22562 CrossRef CAS PubMed
- S. Chu, B. Zhang, X. Zhao, H. S. Soo, F. Wang, R. Xiao and H. Zhang, Adv. Energy Mater., 2022, 12, 2200435 CrossRef CAS
- J. Chen, J. Wu, P. C. Sherrell, J. Chen, H. Wang, W. X. Zhang and J. Yang, Adv. Sci., 2022, 9, 2103764 CrossRef PubMed
- W. Hamd, E. A. Daher, T. S. Tofa and J. Dutta, Front. Mar. Sci., 2022, 9, 885614 CrossRef
- A. Xie, M. Jin, J. Zhu, Q. Zhou, L. Fu and W. Wu, Catalysts, 2023, 13, 846 CrossRef CAS
- C. I. Pizarro-Ortega, D. C. Dioses-Salinas, M. D. Severini, A. D. López, G. N. Rimondino, N. U. Benson, S. Dobaradaran and G. E. De-la-Torre, Mar. Pollut. Bull., 2022, 176, 113474 CrossRef CAS PubMed
- Y. Pan, S. H. Gao, C. Ge, Q. Gao, S. Huang, Y. Kang, G. Luo, Z. Zhang, L. Fan, Y. Zhu and A. J. Wang, Environ. Sci. Ecotechnology, 2023, 13, 100222 CrossRef CAS PubMed
- R. Kumar, Adv. Sustainable Syst., 2023, 7, 2300033 CrossRef CAS
- W. Li, W. Zhao, H. Zhu, Z. J. Li and W. Wang, J. Mater. Chem. A, 2023, 11, 2503–2527 RSC
- A. U. Bacha, I. Nabi and L. Zhang, ACS ES&T Eng., 2021, 1, 1481–1501 Search PubMed
- B. Sarkar, P. D. Dissanayake, N. S. Bolan, J. Y. Dar, M. Kumar, M. N. Haque, R. Mukhopadhyay, S. Ramanayaka, J. K. Biswas, D. C. Tsang and J. Rinklebe, Environ. Res., 2022, 207, 112179 CrossRef CAS PubMed
- J. He, L. Han, F. Wang, C. Ma, Y. Cai, W. Ma, E. G. Xu, B. Xing and Z. Yang, Crit. Rev. Environ. Sci. Technol., 2023, 53, 504–526 CrossRef
- N. D. Dos Santos, R. Busquets and L. C. Campos, Sci. Total Environ., 2023, 861, 160665 CrossRef CAS PubMed
- S. M. Beladi-Mousavi, S. Hermanova, Y. Ying, J. Plutnar and M. Pumera, ACS Appl. Mater. Interfaces, 2021, 13, 25102–25110 CrossRef CAS PubMed
- K. Feng, J. Gong, J. Qu and R. Niu, ACS Appl. Mater. Interfaces, 2022, 14, 44271–44281 CrossRef CAS PubMed
- M. Urso and M. Pumera, Adv. Funct. Mater., 2022, 32, 2112120 CrossRef CAS
- L. Wang, A. Kaeppler, D. Fischer and J. Simmchen, ACS Appl. Mater. Interfaces, 2019, 11, 32937–32944 CrossRef CAS PubMed
- I. Nabi, K. Li, H. Cheng, T. Wang, Y. Liu, S. Ajmal, Y. Yang, Y. Feng and L. Zhang, Iscience, 2020, 23, 101326 CrossRef CAS PubMed
- J. M. Lee, R. Busquets, I. C. Choi, S. H. Lee, J. K. Kim and L. C. Campos, Water, 2020, 12, 3551 CrossRef CAS
- A. D. Vital-Grappin, M. C. Ariza-Tarazona, V. M. Luna-Hernández, J. F. Villarreal-Chiu, J. M. Hernández-López, C. Siligardi and E. I. Cedillo-González, Polymers, 2021, 13, 999 CrossRef CAS PubMed
- M. C. Ariza-Tarazona, J. F. Villarreal-Chiu, J. M. Hernández-López, J. R. De la Rosa, V. Barbieri, C. Siligardi and E. I. Cedillo-González, J. Hazard. Mater., 2020, 395, 122632 CrossRef CAS PubMed
- T. S. Tofa, K. L. Kunjali, S. Paul and J. Dutta, Environ. Chem. Lett., 2019, 17, 1341–1346 CrossRef CAS
- A. Uheida, H. G. Mejía, M. Abdel-Rehim, W. Hamd and J. Dutta, J. Hazard. Mater., 2021, 406, 124299 CrossRef CAS PubMed
- B. Rhimi, M. Padervand, H. Jouini, S. Ghasemi, D. W. Bahnemann and C. Wang, J. Environ. Chem. Eng., 2022, 10, 108566 CrossRef CAS
- M. D. Miah, M. F. Masum and M. Koike, Energy Policy, 2010, 38, 4643–4651 CrossRef CAS
- U. Gehring, O. Gruzieva, R. M. Agius, R. Beelen, A. Custovic, J. Cyrys, M. Eeftens, C. Flexeder, E. Fuertes, J. Heinrich and B. Hoffmann, Environ. Health Perspect., 2013, 121, 1357–1364 CrossRef PubMed
- D. Laohalertdecha, K. Theinnoi and S. Sittichompoo, The Prototype of Non-thermal Plasma After treatment System for Simultaneous Reduction of Nitrogen Oxide Emission in Flue Gas, InE3S Web of Conferences, EDP Sciences, 2021, vol. 302, p. 01010 Search PubMed
- Y. Yang, X. Zheng, W. Ren, J. Liu, X. Fu, S. Meng, S. Chen and C. Cai, Front. Environ. Sci. Eng., 2022, 16, 137 CrossRef CAS
- EEA (European Environment Agency), Air Quality in Europe: Report, Luxembourg, 2015 Search PubMed
- M. Kowalkińska, A. Fiszka Borzyszkowska, A. Grzegórska, J. Karczewski, P. Głuchowski, M. Łapiński, M. Sawczak and A. Zielińska-Jurek, Materials, 2022, 15, 633 CrossRef PubMed
- O. Carp, C. L. Huisman and A. Reller, Prog. Solid State Chem., 2004, 32, 33–177 CrossRef CAS
- M. A. Rauf and S. S. Ashraf, Chem. Eng. J., 2009, 151, 10–18 CrossRef CAS
- M. Xu, Y. Wang, J. Geng and D. Jing, Chem. Eng. J., 2017, 307, 181–188 CrossRef CAS
- V. H. Nguyen, B. S. Nguyen, C. W. Huang, T. T. Le, C. C. Nguyen, T. T. Le, D. Heo, Q. V. Ly, Q. T. Trinh, M. Shokouhimehr and C. Xia, J. Cleaner Prod., 2020, 270, 121912 CrossRef CAS
- J. Zhu, P. Xiao, H. Li and S. A. Carabineiro, ACS Appl. Mater. Interfaces, 2014, 6, 16449–16465 CrossRef CAS PubMed
- X. Zhang, X. Xie, H. Wang, J. Zhang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 18–21 CrossRef CAS PubMed
- X. Wu, J. Cheng, X. Li, Y. Li and K. Lv, Appl. Surf. Sci., 2019, 465, 1037–1046 CrossRef CAS
- N. Todorova, I. Papailias, T. Giannakopoulou, N. Ioannidis, N. Boukos, P. Dallas, M. Edelmannová, M. Reli, K. Kočí and C. Trapalis, Catalysts, 2020, 10, 1147 CrossRef CAS
- C. Acar and I. Dincer, Int. J. Hydrogen Energy, 2014, 39, 15362–15372 CrossRef CAS
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem, 2017, 1, 0003 CrossRef CAS
- A. A. Ismail and D. W. Bahnemann, Sol. Energy Mater. Sol. Cells, 2014, 128, 85–101 CrossRef CAS
- C. H. Liao, C. W. Huang and J. C. Wu, Catalysts, 2012, 2, 490–516 CrossRef CAS
- N. Xiao, S. Li, X. Li, L. Ge, Y. Gao and N. Li, Chin. J. Catal., 2020, 41, 642–671 CrossRef CAS
- W. Sun, J. Zhu, M. Zhang, X. Meng, M. Chen, Y. Feng, X. Chen and Y. Ding, Chin. J. Catal., 2022, 43, 2273–2300 CrossRef CAS
- H. Zhang, W. Tian, X. Duan, H. Sun, S. Liu and S. Wang, Adv. Mater., 2020, 32, 1904037 CrossRef CAS PubMed
- Y. Wang, X. Liu, X. Han, R. Godin, J. Chen, W. Zhou, C. Jiang, J. F. Thompson, K. B. Mustafa, S. A. Shevlin and J. R. Durrant, Nat. Commun., 2020, 11, 2531 CrossRef CAS PubMed
- R. Marschall, Eur. J. Inorg. Chem., 2021, 2021, 2435–2441 CrossRef CAS
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS
- D. Zhu, L. Zhang, R. E. Ruther and R. J. Hamers, Nat. Mater., 2013, 12, 836–841 CrossRef CAS PubMed
- X. Li, Z. Wang and L. Wang, Small Sci., 2021, 1, 2000074 CrossRef CAS
- W. Sun, J. Lin, X. Liang, J. Yang, B. Ma and Y. Ding, Acta Phys.-Chim. Sin., 2020, 36, 1905025 Search PubMed
- J. Li, P. Liu, Y. Tang, H. Huang, H. Cui, D. Mei and C. Zhong, ACS Catal., 2020, 10, 2431–2442 CrossRef CAS
- Z. Wang, J. Hong, S. F. Ng, W. Liu, J. Huang, P. Chen and W. J. Ong, Acta Phys.-Chim. Sin., 2021, 37, 2011022 Search PubMed
- G. N. Schrauzer and T. D. Guth, J. Am. Chem. Soc., 2002, 99, 7189–7193 CrossRef
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS
- B. Pan, Y. Wu, B. Rhimi, J. Qin, Y. Huang, M. Yuan and C. Wang, J. Energy Chem., 2021, 57, 1–9 CrossRef CAS
- R. Marschall, Adv. Funct. Mater., 2014, 24, 2421–2440 CrossRef CAS
- R. Godin, Y. Wang, M. A. Zwijnenburg, J. Tang and J. R. Durrant, J. Am. Chem. Soc., 2017, 139, 5216–5224 CrossRef CAS PubMed
- Y. Li, X. S. Hu, J. W. Huang, L. Wang, H. She and Q. Wang, Acta Phys.-Chim. Sin., 2021, 37, 2009022 Search PubMed
- J. Huang, T. Liu, R. Wang, M. Zhang, L. Wang, H. She and Q. Wang, J. Colloid Interface Sci., 2020, 570, 89–98 CrossRef CAS PubMed
- Y. Xu and R. Xu, Appl. Surf. Sci., 2015, 351, 779–793 CrossRef CAS
- B. Giese, Radicals in Organic Synthesis: Formation of Carbon-Carbon Bonds, CRC Press Reprints, 1986 Search PubMed
- C. P. Jasperse, D. P. Curran and T. L. Fevig, Chem. Rev., 1991, 91, 1237–1286 CrossRef CAS
- J. M. Smith, S. J. Harwood and P. S. Baran, Acc. Chem. Res., 2018, 51, 1807–1817 CrossRef CAS PubMed
- S. Crespi and M. Fagnoni, Chem. Rev., 2020, 120, 9790–9833 CrossRef CAS PubMed
- M. D. Karkas, ACS Catal., 2017, 7, 4999–5022 CrossRef
- Y. Slutskyy, C. R. Jamison, P. Zhao, J. Lee, Y. H. Rhee and L. E. Overman, J. Am. Chem. Soc., 2017, 139, 7192–7195 CrossRef CAS PubMed
- M. R. Garnsey, Y. Slutskyy, C. R. Jamison, P. Zhao, J. Lee, Y. H. Rhee and L. E. Overman, J. Org. Chem., 2017, 83, 6958–6976 CrossRef PubMed
- A. Noble and D. W. MacMillan, J. Am. Chem. Soc., 2014, 136, 11602–11605 CrossRef CAS PubMed
- H. Takeuchi, S. Inuki, K. Nakagawa, T. Kawabe, A. Ichimura, S. Oishi and H. Ohno, Angew. Chem., 2020, 132, 21396–21401 CrossRef
- D. Reich, A. Trowbridge and M. J. Gaunt, Angew. Chem., 2020, 132, 2276–2281 CrossRef
- J. B. McManus, N. P. Onuska and D. A. Nicewicz, J. Am. Chem. Soc., 2018, 140, 9056–9060 CrossRef CAS PubMed
- X. Y. Liu and Y. Qin, Acc. Chem. Res., 2019, 52, 1877–1891 CrossRef CAS PubMed
- C. Zhang, Y. Wang, Y. Song, H. Gao, Y. Sun, X. Sun, Y. Yang, M. He, Z. Yang, L. Zhan and Z. X. Yu, CCS Chem., 2019, 1, 352–364 CrossRef CAS
- T. P. Nicholls, D. Leonori and A. C. Bissember, Nat. Prod. Rep., 2016, 33, 1248–1254 RSC
- J. M. Narayanam, J. W. Tucker and C. R. Stephenson, Adv. Environ. Res., 2009, 131, 8756–8757 CAS
- L. Furst, J. M. Narayanam and C. R. Stephenson, Angew. Chem., Int. Ed. Engl., 2011, 50, 9655 CrossRef CAS PubMed
- C. Comninellis, A. Kapalka, S. Malato, S. A. Parsons, I. Poulios and D. Mantzavinos, J. Chem. Technol. Biotechnol., 2008, 83, 769–776 CrossRef CAS
- P. R. Gogate and A. B. Pandit, Adv. Environ. Res., 2004, 8, 501–551 CrossRef CAS
- P. R. Gogate and A. B. Pandit, Adv. Environ. Res., 2004, 8, 553–597 CrossRef CAS
- C. Gottschalk, J. A. Libra and A. Saupe, Ozonation of Water and Waste Water: A Practical Guide to Understanding Ozone and its Applications, John Wiley & Sons, 2010 Search PubMed
- S. J. Masten and S. H. Davies, Environ. Sci. Technol., 1994, 28, 180A–185A CrossRef CAS PubMed
- S. Imamura, Ind. Eng. Chem. Res., 1999, 38, 1743–1753 CrossRef CAS
- F. J. Beltrán, M. González and J. F. González, Water Res., 1997, 31, 2405–2414 CrossRef
- G. Chen, Sep. Purif. Technol., 2004, 38, 11–41 CrossRef CAS
- C. A. Martínez-Huitle and E. Brillas, Appl. Catal., B, 2009, 87, 105–145 CrossRef
- A. Pattnaik, J. N. Sahu, A. K. Poonia and P. Ghosh, Chem. Eng. Res. Des., 2023, 190, 667–686 CrossRef CAS
- Z. Zhang, X. Chen, H. Zhang, W. Liu, W. Zhu and Y. Zhu, Adv. Mater., 2020, 32, 1907746 CrossRef CAS PubMed
- Y. Zhang, N. Zhang, Z. R. Tang and Y. J. Xu, Chem. Sci., 2013, 4, 1820–1824 RSC
- A. Jiménez-Almarza, A. López-Magano, R. Mas-Ballesté and J. Alemán, ACS Appl. Mater. Interfaces, 2022, 14, 16258–16268 CrossRef PubMed
- X. Tian, Y. Guo, W. An, Y. L. Ren, Y. Qin, C. Niu and X. Zheng, Nat. Commun., 2022, 13, 6186 CrossRef CAS PubMed
- L. Huang, J. Zhao, S. Guo, C. Zhang and J. Ma, J. Org. Chem., 2013, 78, 5627–5637 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2024 |