
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom bench to industry, the application of all-inorganic solid base materials in traditional heterogeneous catalysis: a mini review
Zhixuan Zuo†
,
Yuchen Sha†
,
Peng Wang
 * and
Zhijian Da
* and
Zhijian Da
Sinopec Research Institute of Petroleum Processing CO.,Ltd, No. 18 Xueyuan Road, Haidian District, Beijing 100083, P.R. China. E-mail: wangpeng.ripp@sinopec.com; Tel: +86-10-82368650
First published on 4th March 2024
Abstract
Acids and bases generally occur in pairs as concepts, and a large number of catalytic reactions can be considered as interactions between acids and bases. Many chemical reactions are a combination of acid-catalyzed processes and base-catalyzed processes, and thus it is particularly important to study and explain the mechanisms of acid–base synergy or acid–base interactions. However, compared to the in-depth research on acid catalysts, there is a lack of research on solid bases. In addition to the application of basic materials to non-petroleum processes, recent studies have also applied basic materials to the catalytic cracking reaction process of heavy oils, providing new ideas for the processing of heavy oils. The formation of carbanions with the contribution of basicity is a critical stage in many fine chemical reactions, as well as in the hydrocarbon cracking reactions promoted by a base. Thus, herein, we summarize the research progress on the main types of all-inorganic solid base catalysts, including the types of catalysts used in non-petroleum processes and petroleum processes, their preparation, the properties of their basic sites, and their structure–performance correlation in the reactions. Also, we provide an outlook on the future research directions of all-inorganic solid base materials.
 Zhixuan Zuo | Zhixuan Zuo obtained his ME Degree in Chemical Engineering from the China University of Petroleum (Beijing) in 2021. He is currently working toward a PhD Degree under the direction of Prof. Zhijian Da in Chemical Technology with the Sinopec Research Institute of Petroleum Processing CO., LTD. His main research interests are the preparation and characterization of solid base catalysts, reaction mechanisms in hydrocarbon cracking, and applications in petrochemicals. |
 Yuchen Sha | Dr Yuchen Sha is an Associate Scientist at the Sinopec Research Institute of Petroleum Processing CO., LTD. He received his PhD from Wuhan University in 2021 working on the synthesis of nanocrystals and their application in organic synthesis. His current research interest mainly focuses on the development of the advanced catalysts for fluid catalytic cracking and deep catalytic cracking processes. |
 Peng Wang | Dr Peng Wang is a Scientist at the Sinopec Research Institute of Petroleum Processing CO., LTD. She received her PhD from the Sinopec Research Institute of Petroleum Processing CO., Ltd in 2006 working on the development of catalysts for fluid catalytic cracking. She is a senior expert on R&D of catalytic cracking catalysts and zeolite. She has rich experience in this field and developed more than 10 commercialized catalysts. Her number of authorized patents reached one hundred recently. |
 Zhijian Da | Dr Zhijian Da is a Senior Scientific and Technical Advisor and PhD supervisor at the Sinopec Research Institute of Petroleum Processing CO., LTD. He received his M.E and PhD degrees from the Sinopec Research Institute of Petroleum Processing CO., Ltd in 1988 and 1991, respectively. From 1996 to 1997, he undertook postdoctoral research at the CNRS and at the Organocatalytic Research Unit of the University of Poitiers. He has been engaged in the research and development of molecular sieve catalytic materials, FCC catalyst preparation, and refining-related processes. |
1 Introduction
Nowadays, environmental protection has become one of the most pivotal global issues. In terms of the chemical industry, the requirements of generating less pollutants and lower carbon emissions are becoming much more stringent, driving the further development of new processes and catalysts. Heterogeneous catalysis is the most critical process in the modern chemical industry.1,2 Generally, heterogeneous catalysts can be categorized into two parts, namely, solid acid catalyst and solid base catalyst. During the past few decades, both the fundamental research and industrial application of solid acid catalysts have been increasing rapidly and extensively with the booming petrochemical industry. In contrast, solid base materials have attracted less attention as catalysts probably due to their complicated preparation, propensity to be contaminated, etc.According to the Brønsted acid–base theory and Lewis acid–base theory, it is well-accepted that a solid base material can serve as a catalyst either by abstracting a proton from the reactant or donating electron pairs to the reactant.3,4 The history of solid base catalysts can be traced back to 1958 when Pines and co-workers showed that alumina-supported sodium metal could work as an effective catalyst for the double bond isomerization of alkenes.5 Previously, most of the applications of solid bases in catalytic processes were concentrated in the catalysis and synthesis of fine chemicals in non-refinery fields such as olefin and alkyne isomerization, aldol condensation, and Henry reaction, as shown in Fig. 1.6
 | ||
| Fig. 1 Some typical fine chemical reactions catalyzed by solid bases.6 Reprinted with permission from ref. 6. Copyright © 2015, The Royal Society of Chemistry. | ||
In recent years, several studies on the use of solid base catalysts for hydrocarbon catalytic cracking in oil refining have emerged; studies on catalytic cracking using solid base materials have also emerged recently due to the high residual carbon problems resulting from the increasing heaviness of oil around the world and the use of heterogeneous catalysts prepared with solid base materials that have better resistance to carbon deposition in petroleum processes than acid catalysts7–9
Solid base materials are used in a wide range of applications; however, the present preparation process of solidbase materials, especially solid superbases, is more complicated and expensive compared with that of solid acid catalysts. Generally, as-prepared solid base materials possess a small specific surface area, poor structural strength, and weak hydrothermal stability. Moreover, both CO2 and H2O in the air are easily adsorbed on the active sites of solid bases, leading to their undesirable passivation, thus reducing the catalytic activity of the catalyst.3,4,10,11
To date, there are several excellent reviews summarizing the application of solid bases in refining or non-refining fields.1,3,12–20 According to the analysis of the references in the literature, to date, the catalytic mechanism for base-catalyzed processes has not been explained in detail, and the structure–activity relationships between the physical and chemical properties of these catalysts and reactions have not been well established. Therefore, a systematic study on the mechanism of solid base catalysis is required to establish the structure–activity relationships between catalytic properties and physical and chemical properties of catalysts, which will be more helpful for the in-depth understanding and application improvement of solid bases. However, to date, a comprehensive description of the application of solid base catalysts in both fields has not been reported, and thus, herein, we summarize the general categories of all-inorganic solid base catalytic materials first, and then review the preparation, properties, and applications of metal oxide and zeolite-based solid base materials in non-refinery and refinery fields, which have been reported in numerous studies, to provide some guidance for the follow-up preparation and studies on improving these materials.
2 Categories of all-inorganic solid base catalysts
Different types of solid base catalysts and various basic sites generate different effects in multiple fine chemical processes and refining processes.13,21–23 These solid base catalysts range from single-component oxides to complex-component oxides and even zeolites. The most accepted definitions of solid surface acids and bases by researchers are those proposed by Brønsted and Lewis, where a Brønsted base is defined as the opposite of a Brønsted acid, namely, a substance that can take protons from reactants; a Lewis base is an electron donor that gives electron pairs.24 The type and strength of the base sites differ in catalytic reaction processes and results. Table 1 presents several types of commonly used all-inorganic solid base catalysts, which are grouped based on their composition and typical active components.3| Type | Main active components |
|---|---|
| Intrinsic metal oxide | MgO, CaO, Al2O3, ZrO2, La2O3, Rb2O |
| Mixed oxide | SiO2–MgO, CaO–SiO2, MgO–Al2O3 |
| Metal oxide loaded | Na2O/SiO2, MgO/SiO2, Cs2O/Zeolites |
| Alkali metal compound loaded | KF/Al2O3, K2CO3/Al2O3, KNO3/Al2O3 |
| Alkali metal loaded | Na/Al2O3, K/Al2O3, K/MgO, Na/Zeolites |
| Metal ion-exchanged zeolite | K, Rb, Cs-exchanged X, Y zeolites |
3 Metal oxide solid bases
At present, the research on metal oxide solid bases generally focuses on alkali and alkaline earth metal oxides, which are the earliest and most deeply researched solid base materials, but in the early stage of their industrial application.4,25,26 Recently, some commonly used methods for the preparation of metal oxide catalysts were summarized by Védrine.26 In the case of simple oxides, their preparation is usually based on three main series of conventional preparation routes, i.e., gas-phase polymerization, aqueous-phase precipitation, and hydrolytic or non-hydrolytic sol–gel processing. For mixed oxides, the most common technique is the co-precipitation method, where two metal salts are mixed in solution and precipitated at a given pH. Generally, supported oxide catalysts are prepared using ambient temperature, aqueous phase methods such as selective adsorption, impregnation and deposition–precipitation. Also, these methods are dominant on an industrial scale due to environmental and economic reasons. Further, their surface functionality can be adjusted by incorporating active sites in the walls of silica or depositing active substances on the inner surface of the materials to synthesize mesoporous silica-based materials with better physicochemical and textural properties and optimized catalytic properties.26,27 Therefore, some important routes for the preparation of metal oxide solid bases are provided in the following, which also include rare earth oxides and other oxides, while other oxides mainly include loaded oxides and mixed oxides.3.1 Alkaline earth metal oxides
Nevertheless, another important drawback of the aforementioned metal oxide solid base catalysts compared with zeolites or other catalysts for industrialization is their lower specific surface area. For example, the typical commercial MgO only has a low specific surface area of around 20 m2 g−1, resulting in a lower density of basic sites. Therefore, many studies have focused on enhancing its specific surface area. Recently, a route for the preparation of high specific surface area MgO from commercial MgO was reported, resulting in a specific surface area of up to 250 m2 g−1 and a basic site density of 2.5 mmol g−1, as determined by CO2-TPD.30 However, due to the complexity of this method, it is expensive and difficult to apply. Ordonez et al. reported a new thermal treatment method to prepare MgO, as shown in Fig. 2(a). This novel method involved the processes of hydration and dehydration of periclase, which addresses the conflict between surface area enhancement and environmental benignity.31 Various precursors and thermal treatments were investigated and the results indicated that the thermal treatment provided defects on the surface of magnesium oxide, which resulted in a large specific surface area (221 m2 g−1) and a significant concentration of surface basic sites (1.2 mmol g−1). Also, the equilibrium adsorption isotherms at 0–6.7 kPa and 50 °C, as shown in Fig. 2(b), indicated the presence of combined chemisorption and physisorption mechanisms. The large improvement in CO2 uptake confirmed that a suitable pathway for modifying MgO was reasonably achieved.
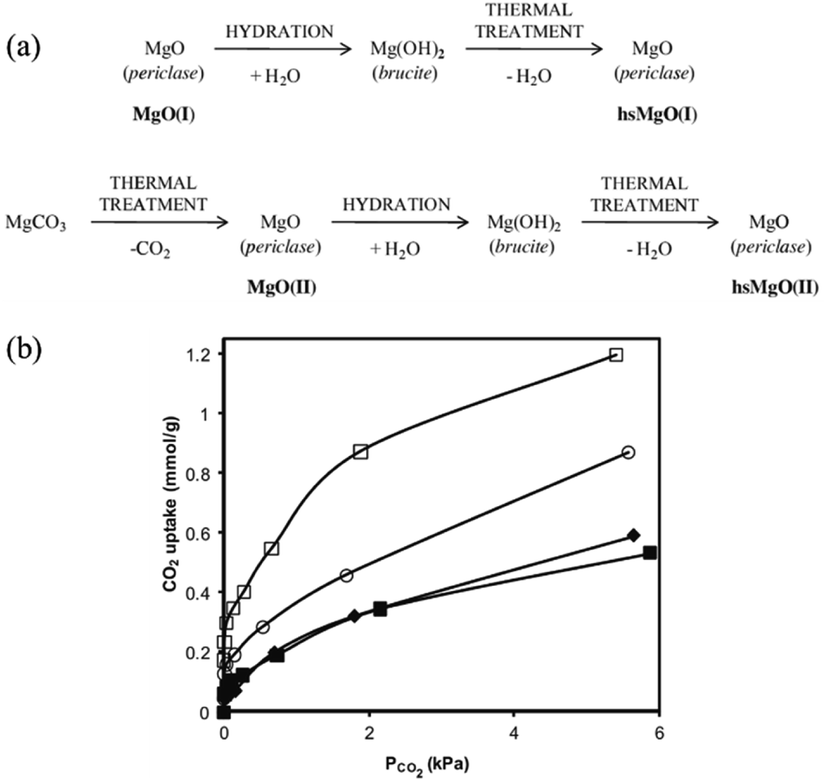 | ||
| Fig. 2 (a) Proposed activation steps and notation (bold) used for the different MgO. (b) CO2 adsorption isotherms at 50 °C and 6.7 kPa, measured by microcalorimetry for MgO(I) (◆), MgO(II) (■), hs MgO(I) (□), and hs MgO(II) (○).31 Reprinted with permission from ref. 31. Copyright © 2014, Elsevier. | ||
In general, the basic strength of alkali metal and alkaline earth metal oxide catalysts increases as their atomic number increases, and the calcination temperature and the types of precursors also significantly affect the basic strength of alkali metals and alkaline earth metal oxides. Specifically, different calcining temperatures generally lead to different basic strengths, and the active sites of metal oxides can vary from one preparation process to another. Moreover, the activity and stability of the basic sites can be affected by the chemical environment of the active sites.32 According to the above discussion, it can be seen that different treatment processes can regulate the base properties of metal oxide materials.
The active sites of metal oxides are mainly generated from the negatively charged lattice oxygen and hydroxyl groups formed by the adsorption of water on their surface. Anpo et al. discussed the different basic sites produced by various surface oxygen species on oxide materials and the electron paramagnetic resonance and photoluminescence techniques used for the characterization of paramagnetic (O−, O2−, and O3−) and diamagnetic surface oxygen species (O2,surf−), respectively.33
A classic example is shown in Fig. 3(a),34 where the crystal structure of the conventional magnesium oxide is halite (cubic) in shape, with its crystal lattice tightly bonded in an octahedral shape and having a lattice constant of a = 4.212 Å. A schematic representation of the irregularities on the surface of MgO with impurities removed and the terminology presented by Chizallet et al. are shown in Fig. 3(b) and Table 2, respectively. The O2− centers exist in different coordination numbers and at different positions and ion pairs with low coordination numbers are generally present at the corners of the MgO surface. Generally, these ion pairs possess the highest degree of unsaturation and the strongest basicity, while ion pairs with high coordination numbers are generally found on perfect crystalline surfaces or defect-free surface positions, which result in weaker basicity.35–39 Among the ion pairs with different coordination numbers, the triple-coordinated Mg–O ion pairs have the strongest ability to adsorb carbon dioxide and water and have high relative activity, and thus the calcining temperature must be increased to expose these ion pairs. The ion pairs with different coordination numbers are exposed to the surface in sequence by a step-up temperature, while the active sites with different coordination numbers have different adsorption properties for water and carbon dioxide, and the triple-coordinated Mg–O ion pair has the highest desorption temperature due to the highest reactivity. However, the most reactive ion pair is prone to rearrangement and annihilation at higher temperatures due to its instability.37 As the calcining temperature continues to increase, the highly unsaturated ion pairs will reach equilibrium between the process of generation and the process of disappearance of rearrangement, while the activity of MgO also reaches its maximum with an increase in the calcining temperature.35,37,39
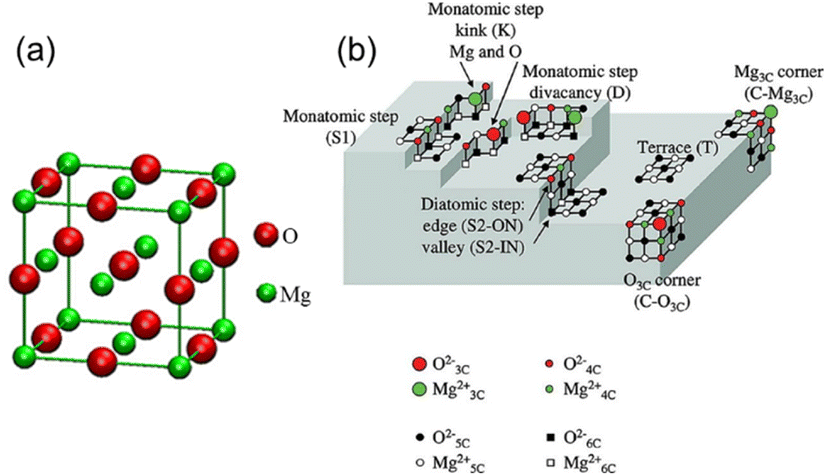 | ||
| Fig. 3 (a) Crystal structure of MgO.34 Reprinted with permission from ref. 34. Copyright © 2010, IOP. (b) Schematic representation of irregularities on the surface of MgO. The terminologies used are shown in Table 2. M3c2+: 3-coordinated Mg2+ on corners; O4c2−: 4-coordinated O2− on edges.35 Reprinted with permission from ref. 35. Copyright © 2011, Taylor & Francis. | ||
| Nature | Label | Formula |
|---|---|---|
| (100) plane | T | Mg13O14Mg17 |
| T′ | OMg5 | |
| Monatomic step | S1 | Mg20O14Mg10 |
| Diatomic step: edge | S2-ON | Mg10O10Mg10 |
| S2-ON′ | Mg10O16Mg22 | |
| Diatomic step: valley | S2-IN | Mg22O9 |
| Corner: O3c2−-terminated | C-O3c | Mg13O13Mg12 |
| Corner: Mg3c2+-terminated | C-Mg3c | Mg7O9Mg10 |
| Divacancy | D-Mg3c–O3c | Mg9O10Mg13 |
| Kink: O3c2−-terminated | K–O3c | Mg19O13Mg9 |
| Kink: Mg3c2+-terminated | K–Mg3c | Mg21O14Mg10 |
However, the strength of the basic sites required for solid base catalysis should be selected according to the change in the ease of extraction of protons from the reactants by the basic sites under the reaction conditions. For example, MgO requires a pretreatment temperature of 527 °C for catalytic 1-butene isomerization and 700 °C for CH4-D2 exchange, and different pretreatment temperatures correspond to different base strengths.19
In the 1-butene isomerization reaction, the O2− active site on the surface of the solid base adsorbs hydrogen protons from the reactant molecules to form anionic intermediates, and the adjacent metal ions stabilize the anionic intermediates for subsequent reactions. The lattice oxygen O2− on the surface of the solid base CaO serves as a base center to adsorb the H on the isopropyl tertiary carbon atom of isopropylbenzene (IPB), and the interaction between H and the lattice oxygen weakens the C–H bond of the tertiary carbon atom and lowers the activation energy for radical formation, promoting the formation of radicals.42 Another example is the alcohol-formaldehyde condensation of butyraldehyde, where the active center is not the surface hydroxyl group but the lattice oxygen O2−, which forms a stronger interaction with CO2 and H2O.43 In contrast, the relationship between the thermodynamic Brønsted basicity and kinetic basicity of magnesium oxide samples was investigated by comparing the results obtained from the deprotonation equilibrium of a protic molecule with the MBOH transformation as the model reaction. It was found that the hydroxylated surfaces are more reactive than the bare surfaces, despite their lower deprotonation capacity.44
Reaction path simulations of the nudged elastic band (NEB) method indicated that a bare Mg2+–O2− pair is available for MBOH adsorption and reaction occurs in the vicinity of the surface OH groups, which lowers the activation energy barrier.45
Although the OH groups do not interact directly with the MBOH reactant, they alter the basic reactivity of the bare Mg2+–O2− pairs in the vicinity where the adsorption and conversion of MBOH occur. Therefore, it can be seen that the same solid base catalyst has different active sites serving as base centers, and the active sites of the reaction are different for various reactions.
As consistently mentioned above, there are two other types of molecules that can be adsorbed, i.e., CO2 and H2O, which cannot be ignored. Generally, it is believed that they result in the passivation of the base center, but in some systems, this can be different, and the adsorption of both CO2 and H2O molecules on the solid base also has different effects. For example, the adsorption of CO2 and H2O has almost no effect on the reaction between nitromethane and propionaldehyde because the reactants are more easily adsorbed at the base center than CO2 and H2O, which inhibits them from poisoning the base sites, making it almost impossible to affect the catalyst activity.46 Another example is the addition of a small amount of water in the hydroxyl aldehyde condensation reaction to improve the catalytic activity of the solid base to a certain extent, where researchers believe that the adsorption of water on the surface of the solid base increases the surface hydroxyl or O2− ions in the solid base, increasing the density of the base center.47
As discussed in the previous two sections, it can be seen that to gain strong basicity via certain oxides in certain reactions, the previous strategy was the modification of oxides with alkaline metals to create surface vacancies or defects, which suffer from instability under the catalytic conditions. Alternatively, single-component basic oxides (e.g., magnesium oxide) are stable but have poor electron-withdrawing ability.
3.2 Rare earth oxides
In general, solid base catalysts promote the dehydrogenation of alcohols to aldehydes and ketones rather than the dehydration of alcohols in reactions, but rare earth oxides can show unique selectivity for alcohol dehydration reactions, e.g., when the reaction product is mainly 1-butene (2-butanol dehydration product) in the case of using rare earth metal oxides such as ThO2 as a solid base catalyst in the 2-butanol dehydration reaction.16,18,52,53
Rare earth oxides have not been widely used in base catalytic processes thus far because the mechanism of their base centers in the synthesis reactions of conventional fine chemicals as base catalysts is still unclear and poorly studied. However, researchers generally agree that the base centers on rare-earth oxides have significant catalytic properties for hydrogenation processes, especially given that many rare-earth oxides have strong ethylene hydrogenation ability at −78 °C.19,54 Simultaneously, given that rare earth oxides are mainly involved in the oxidative cracking process in oil treatment, the process inevitably generates COx (carbon oxygen compounds), which not only inhibits the production of low carbon olefins, but also tends to lead to the poisoning of base catalysts. Thus, the above-mentioned drawbacks also limit the research on the application of rare earth oxides in petroleum refining.
3.3 Other oxides
Presently, more studies on basic mixed oxides used in the field of fine chemicals are focused on mixed base metal oxides and magnesium-aluminum oxides prepared from calcined hydrotalcite. Generally, acidic oxides can be made significantly more acidic by mixing oxides, while unlike the results of acidic mixed oxides, the basicity of basic mixed oxides and their properties can usually be modified only to a limited extent by the addition of other component oxides. It has been found that mixing two oxides does not significantly improve their basicity. Noller et al. studied the basicity of MgO–Al2O3 and MgO–SiO2 by XPS and IR characterization of their hydroxyl groups, and also explored their catalytic activity in reactions.55 The results showed that the basicity of this mixed oxide is usually between the base strengths of the two oxides, and thus the basicity strength is considered to be consistent with Sanderson's average electronegativity theory, which can be used as a guidance for following research work.
The modification of the surface of solid bases and their basicity can be achieved not only by mixing two or more types of oxides but also by loading other components on the metal oxides or mixed oxides, which can change their basicity and surface properties to some extent. The presence of different metal ions in a solid base has a significant effect on its basicity, e.g., the introduction of metal ions in MgO. As shown in Fig. 4(a), the density of its basic sites increases if the radius of the metal ions is slightly larger than that of Mg2+, while the introduction of metal ions with a smaller radius than Mg2+ has no significant effect.56 A metal ion with an excessively large radius may not be able to dope the lattice of MgO, and therefore the change in basicity is small. Alternatively, a metal ion with a too small radius cannot elongate the bond length of the Mg–O bond and change the crystal structure, and therefore cannot effectively modulate the basicity of the oxide. Only when the metal ion radius is slightly larger than that of Mg2+ is causes lattice distortion, resulting in the elongation of the Mg–O bond and the electron delocalization effect of the lattice oxygen. The addition of an alkali metal to MgO can produce strong basic sites, as shown in Fig. 4(b). When an alkali metal atom is adsorbed on the surface of MgO it releases an electron to the alkali metal ion, the released electron is attracted to a single oxygen vacancy on the subsurface to form an electron F+ center or F center of a pair of electrons, and the three O2− ions present on the (111) crystal face adjacent to the F+ center or F center are the strongly basic sites (lattice O2−).57 After loading Li on MgO, the excess cations lead to the formation of oxygen vacancies on the surface of MgO, and oxygen in the gas phase under a high temperature atmosphere reacts with the vacancies to produce O2− and holes, which tend to be close to the O2− near Li+. Consequently, it generates a base center via the 2Li+O2− + X + 1/2O2 → 2[Li+O−] + O2− reaction to form the [Li+O−] center (X denotes an oxygen vacancy).35 Similar to MgO, there are intrinsic defects in the transition metal oxide TiO2, which can generate the corresponding oxygen vacancies to achieve the modulation of the other components introduced. The point defects such as oxygen vacancies and oxygen interstitials in TiO2 tend to attract and capture electrons to form electro–neutral complexes with the metal ions according to the theory of charge equilibrium and can be largely classified into the electron centers (F) and the electron vacancy centers (V0). The structure and formation process of various types of centers on TiO2 are shown in Fig. 5, in which there is no unpaired electron in the F center, there is an unpaired electron in the F+ center, and the two electrons in the F center tend to occupy the metal ions to form an F++ center with a positive charge.58
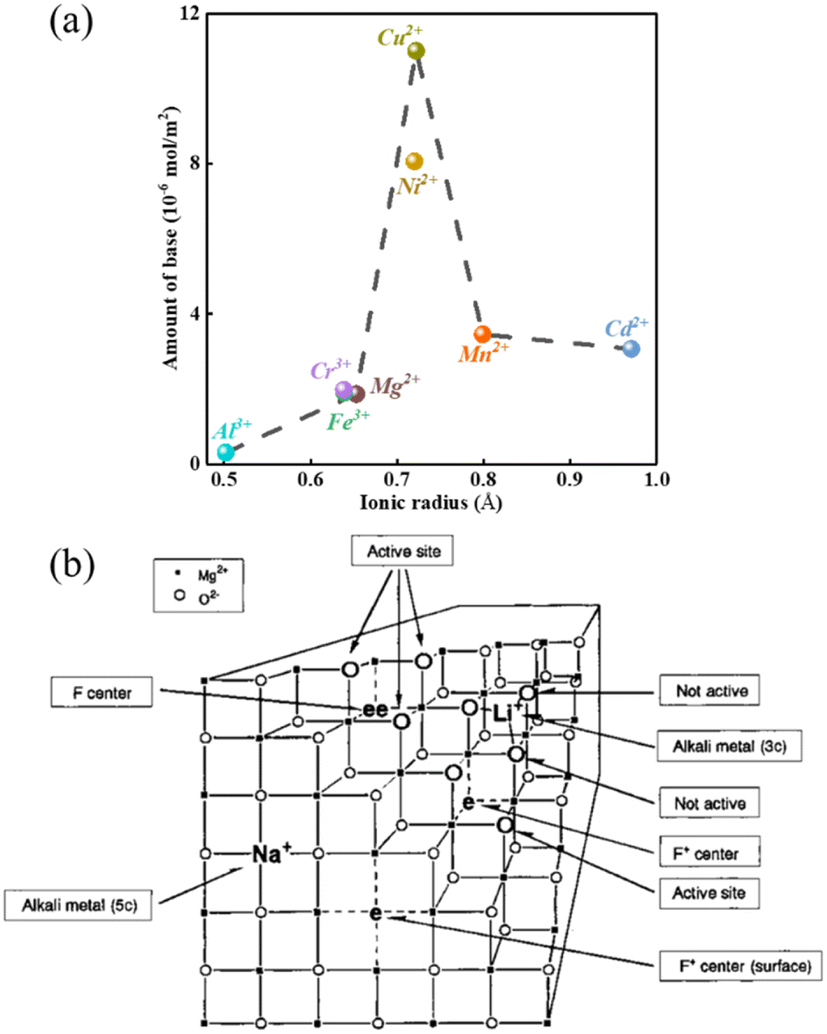 | ||
| Fig. 4 (a) Relationship between the amount of base (density) on the surface of MgO and the radius of the doped metal ions.56 Adapted with permission from ref. 56. Copyright © 2006, Oxford University Press. (b) Model for the super basic sites generated by the addition of alkali metal to MgO3c: 3-coordinated; F+ center: an electron attracted by a single oxygen vacancy; and F center: a pair of electrons attracted by a single oxygen vacancy.57 Reprinted with permission from ref. 57. Copyright © 2000, the American Chemical Society. | ||
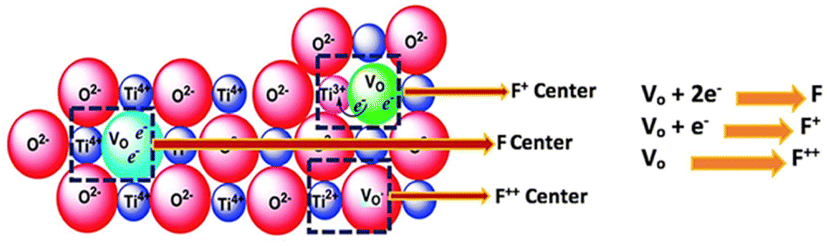 | ||
| Fig. 5 Schematic illustration and description of various F centers formed in TiO2. F center: four Ti4+ in the neighborhood with no unpaired electrons and F+ center: Tilattice3+ with an unpaired electron.58 Reprinted with permission from ref. 58. Copyright © 2018, MDPI. | ||
It is well known that catalytic reactions proceed at the defect sites in these oxide catalysts, e.g., the basic sites generated from metal oxide vacancies of lattice oxygen O2− or O−, a monotonic relationship does not always exist between the catalytic performance and the concentration of oxygen vacancies, lattice distortions, and defect-produced Lewis bases.59–62 In a pure oxide, the oxygen ions or vacancies usually have a high symmetric coordination with the cations. In recent years, researchers have paid more attention to the chemical environment of oxygen vacancies and have proposed “asymmetric oxygen vacancy sites,” which denote the asymmetric coordination of oxygen vacancies with cations, allowing oxygen to move more freely. The review article by Yu et al. well explained the “asymmetric oxygen vacancy site” and showed that the strength of cations of different sizes, electronic structures, oxidation states, and relatively stable coordination numbers can be adjusted to suit different chemical reactions, with the aim of equilibrating the adsorption and desorption of oxygen species.63 For example, in the fluorite-type bulk structure of CeO2, a simple way to break the oxygen/vacancy symmetry is to replace one of the cerium cations to form the asymmetric site M–O(–Ce)3 or M–□(–Ce)3, where □ stands for the oxygen vacancy, as shown in Fig. 6(a). These M1–O–M2 or M1–□–M2 sites coordinated to different cations can be denoted as “asymmetric oxygen vacancies” and the alternate formation of M1–O–M2 and M1–□–M2 can serve as active sites.64,65
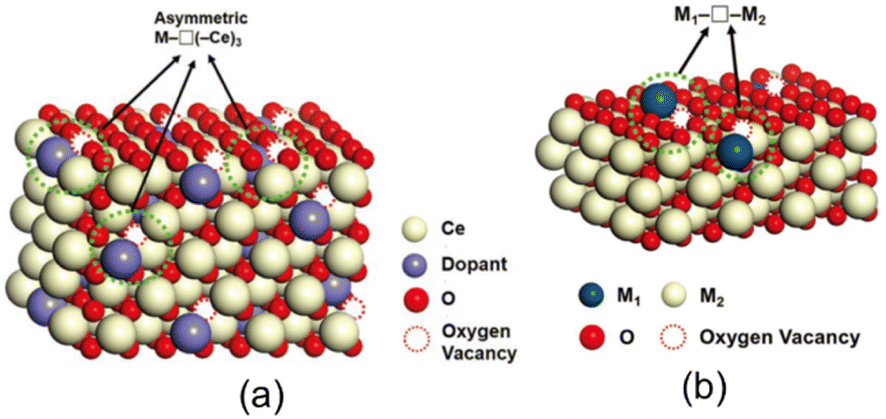 | ||
| Fig. 6 (a) Asymmetric oxygen vacancy sites of M–□(–Ce)3 in doped ceria. (b) Asymmetric oxygen vacancy sites on the surface of SACs.63 Reprinted with permission from ref. 63. Copyright © 2019, Wiley. | ||
For example, the dispersed metal atoms on the surface of SACs are usually isolated catalytically active sites, but the reaction may take place on surrounding atoms such as oxygen in the substrate and active asymmetric oxygen vacancies in these materials, as shown in Fig. 6(b). Nie et al. reported that the steam treatment of atomically dispersed Pt2+/CeO2 catalysts can effectively create active surface lattice oxygen, which was produced by filling the surface oxygen vacancies (Pt–□–Ce) derived from the substitution of Pt2+ for Ce4+.66 Unlike other materials, almost all the platinum atoms in the above-mentioned materials are located on the oxide surface and each platinum atom activates two or more surrounding oxygen atoms.
The selective oxidation of alcohol or dehydrogenation of alcohol to produce aldehyde also showed a demand for the use of base catalysts, where the base sites may be employed to remove H atoms from hydroxyl group and α-carbon.67–69 The first step in the selective oxidation of alcohols to produce the η1 (O)-configuration (M–O–C) requires either a weakly acidic site or a strongly basic site for the O–H bond to break rather than the C–O bond, which leads to dehydration.70 If the η2 (C, O)-configuration is formed, the weak basicity promotes π*-bond interactions, and the active site with fewer electrons will be favorable for coordination by the lone pair of electrons of the O atoms, thus reducing the stability of the C–O bond.2
More recently, Cho et al. investigated the effects of a Ca-promoter on the catalytic activity of MgO given that they observed very different catalytic activities of MgO in the oxidative coupling of methane (OCM) resulting from the company that supplied the precursors.71 The shift in the base properties of the surface lattice oxygen on the catalyst, as determined by CO2-TPD and shown in Fig. 7(a), indicated that MA99 (99% Mg with 1% Ca, mole fraction) contained the largest amounts of lattice oxygen with weak and medium basicity, while MA0 (100% Ca, mole fraction) contained the largest amount of lattice oxygen with strong basicity. Hence, the appropriate addition of Ca to the MgO catalyst would enrich the formation of oxygen vacancies and lattice oxygen with medium basicity, which facilitates the reaction of the adsorbed oxygen species and methane to produce the methyl radical, whereas lattice oxygen with strong basicity would be established by an excessive Ca content. The lattice oxygen with medium basicity facilitates the dehydrogenation of C2H6 to C2H4. On the contrary, lattice oxygen with strong basicity acts as a strong oxidant for the production of CO2 from various hydrocarbons. Due to the above-mentioned information between the catalytic activity and the properties, they proposed a process for the oxidative coupling of methane by surface oxygen species, as shown in Fig. 7(b).71
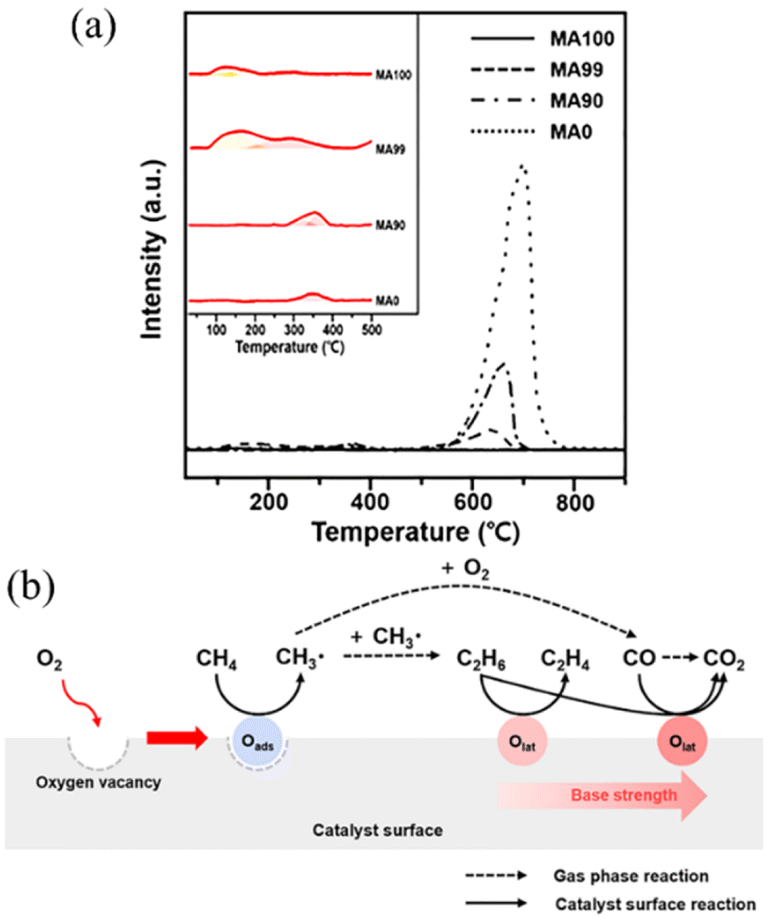 | ||
| Fig. 7 (a) CO2-TPD profiles of catalysts. MA0: 100% Ca; MA90: 10% Ca; MA99: 1% Ca; MA100: 100% Mg; mole fractions of the metal components were calculated as the mole ratio of the corresponding metal component (Mg or Ca) to the total metal components in the catalyst. (b) Proposed process for the oxidative coupling of methane by surface oxygen species. Oads: adsorbed oxygen and Olat: lattice oxygen.71 Reprinted with permission from ref. 71. Copyright © 2021, Elsevier. | ||
To enhance the basic strength of oxide-based solid base catalysts, Yang et al. developed Ga4B2O9 and evaluated its performance in reactions for the synthesis of α-aminonitriles (Strecker reaction) and the selective conversion of n-propanol. It exhibited strong Lewis basicity, and further its activity was compared with a type of mixed Ga–B oxide that exhibited Brønsted acidity.72 They discovered that Ga4B2O9 with the mullite-type structure exhibited intrinsic Lewis basicity, which was proved by CO2-TPD and DFT calculations despite its local superstructure, and then it was further proposed that the μ3-O atoms (linked to 3 metal atoms) linked with 5-coordinated Ga3+ exclusively are likely the active sites in basic catalytic reactions and the origin of the strong basicity due to the structure-induction. Nevertheless, Ga4B2O9 with fewer basic sites of 23.1 μmol g−1 exhibited superior catalytic efficiency to Ga-PKU-1 (mixed Ga–B oxide) with larger acidic sites of 68.9 μmol g−1, suggesting that the process via base catalysis is more efficient.72 Two years later, their group investigated the structure–performance correlation of Ga4B2O9 (prepared by high-temperature solid–state reaction) in the Knoevenagel condensation reaction (several aldehydes with malononitrile to form α,β-unsaturated compounds through nucleophilic addition) more in-depth, which showed a high yield of 90% and high stability.73 Their conclusions in earlier research were evidenced in this work by the structural comparison together with the catalytic activity among Ga4B2O9, GaBO3 and β-Ga2O3. Because the O2− in the distorted GaO5 (above-mentioned μ3-O) showed a relatively lower Bader charge, an electrophilic molecule, i.e., malononitrile, was adsorbed and activated, and then the corresponding carbanion emerged by seizing one proton from the reactants. Then the activated reactant underwent nucleophilic attack by the carbanion at the Ga site adjacent to μ3-O.73 Accordingly, it is evident that various strengths of basic sites can provide different catalytic performances when designing catalysts, and well-developed oxygen vacancies and an abundance of specific basic sites are critical factors in designing efficient and high-performance catalysts.
In contrast with the previous description, in many cases, the reaction mechanism of mixed oxides may be not only base-catalyzed but synergistic. However, this should not be confused with the coexistence of acidic and basic phases in a single material. For example, in MgO–SiO2, during the conversion of bioethanol to butadiene, the acidic sites on silica or magnesium silicate are involved in the dehydration step, while the basic sites on magnesium oxide catalyse the dehydrogenation and condensation steps. These catalysts are bifunctional and the acidity and basicity described need to be characterized independently, with a possible optimal ratio between the acid and basic phases.74 Moreover, the reaction mechanism for the ZrO2 hydrogenation-catalyzed aromatic carboxylic acid generation of aldehydes is shown in Fig. 8, where the basic site extracts a proton from the carboxylic acid, the hydrogen adsorbs on the adjacent ion pair, and then isomerizes, the carboxylic acid radical and the negatively electrostatically charged hydrogen ion generates an aldehyde molecule, and then desorbs, and the adsorbed O2− forms water with the two hydrogen protons adsorbed on the lattice oxygen, and then desorbs, showing a synergistic effect. Another example is the mixed oxide of MgO and TiO2, where the number of basic sites decreases and the number of acidic sites gradually increases with an increase in the specific gravity of TiO2. When this catalyst was used for the alkylation reaction, it was found that the best catalytic effect was achieved when the specific gravity of the two mixtures was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The mixture has both acidic and basic sites at this specific gravity, and thus it is considered that the use of MgO–TiO2 in this reaction causes it to proceed via an acid–base synergistic mechanism.75,77
:1. The mixture has both acidic and basic sites at this specific gravity, and thus it is considered that the use of MgO–TiO2 in this reaction causes it to proceed via an acid–base synergistic mechanism.75,77
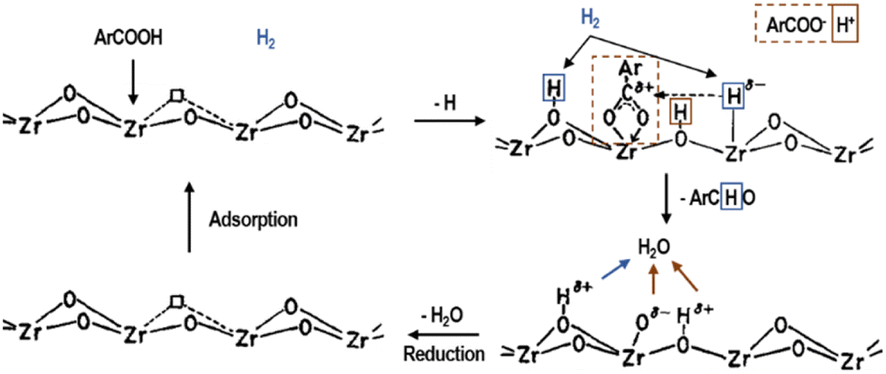 | ||
| Fig. 8 Reaction mechanism of aromatic carboxylic acids to aldehydes catalyzed on ZrO2 in the presence of hydrogen.75 Adapted with permission from ref. 75. Copyright © 1992, Elsevier. | ||
Wang et al. synthesized a series of Ni/CaxMgyO catalysts composed of Ni nanoparticles and CaO–MgO mixed oxides to upgrade n-butanol into branched 2-ethyl-1-hexanol. By varying the content of surface Ni0, the n-butanol conversion increased obviously from 31.1% to 62.0% with an increase in Ni0 concentration, evidencing the critical role of metal sites during the Guerbet reaction. By selectively poisoning the base sites using various amounts of benzoic acid, the conversion of n-butanol and the yield of 2-ethyl-1-hexanol decreased, while the yield of butyraldehyde increased slowly first, and then drastically. This indicates that the base sites serve as the active site in the Guerbet reaction, and also govern the catalytic activity in the aldolization. Research on the reaction duration and reaction temperature indicated that the highest conversion and yield of n-butanol dehydrogenation could reach 80.2% and 63.4%, respectively. Most notably, they confirmed that the dehydrogenation of n-butanol was the rate-determining step in the whole reaction process through the study of the structure–activity relationship and the reaction mechanism proposed, as shown in Fig. 9, in which the synergistic catalytic role of the Ni0 site (metal site) and the base site was obviously reflected.76
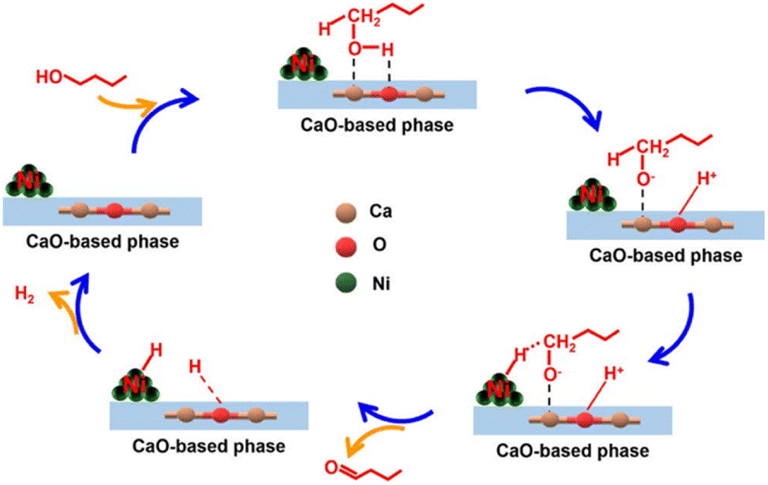
3.3.3.1 Low-carbon alkane catalytic cracking. Kolts et al. examined the catalytic performance of MgO solid bases loaded with Mn and Fe, respectively, using n-butane as the feedstock. It was found that the use of solid bases significantly increased the conversion of the reaction and improved the selectivity for ethylene and ethane, while decreasing the selectivity for propylene by about 20%. Based on the results of the EPR and XRD characterization, it was found that the number of ethyl radicals increased and the number of MnO crystals decreased in the system after using the solid base catalyst, and thus the mechanism of n-butane cracking reaction and catalyst reduction process was proposed, as shown in Fig. 10. It was hypothesized that the use of a solid base led to an increase in the content of major carbanions and charge transfer, thus facilitating the cracking reaction. The different product distributions that occur in cracking the two types of butanes also indicate differences in the stability of the carbanions produced by different feedstocks.78 Co/Al2O3 prepared by impregnation also provided high n-butane conversion in the oxidative cracking of n-butane, while N–Co/γ-Al2O3 could provide up to 82% n-butane conversion at 600 °C, and this conversion value was higher than that from thermal cracking at 800 °C. XRD characterization revealed that the introduction of N promoted the formation of oxynitride and enhanced the mobility of oxygen. Different types of oxygen were responsible for different hydrocarbon molecule conversion pathways, and the high activity of lattice oxygen in CoO is the main reason for promoting the base-catalyzed dehydrogenation process to produce olefins.83 In a study applying solid bases to the oxidative cracking of hexane, Li/MgO prepared using different methods exhibited different reaction properties, where the catalyst prepared by the sol–gel method provided about 28 mol% hexane conversion under the reaction condition at 575 °C and significantly improved the selectivity of low carbon olefins. Boyadjian et al. suggested that Li/MgO enhanced H capture by providing reactive oxygen sites, further increasing the production of olefins and free radicals, while Li/MgO prepared via the sol–gel method can further promote the oxidative cracking of hexane by providing a larger surface area and more active sites.85
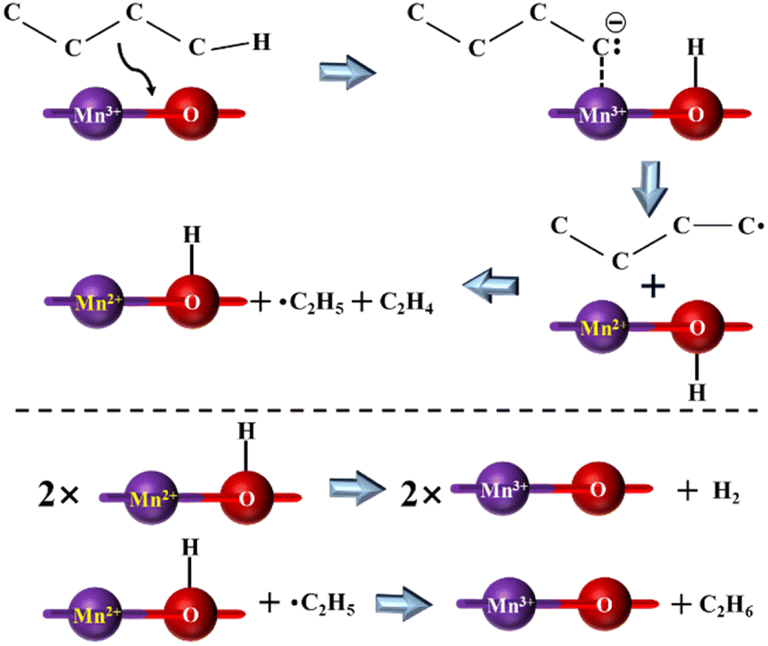 | ||
| Fig. 10 Proposed mechanism for the selective cracking of n-butane over the Mn–MgO catalyst.78 Adapted with permission from ref. 78. Copyright © 1986, AAAS. | ||
The recent study by Wang et al. suggested that the combination of metal sites and mobile oxygen species on Y2O3-modified Au–La2O3 catalysts provided superior activity for catalytic n-propane oxidative cracking and the synthesized catalysts showed stable activity over 24 h without significant deactivation.84 According to the H2-TPR results, as shown in Fig. 11(a), the combined sites arising from the metal-support interactions and 5Y-0.2Au–La2O3 (impregnation of calculated amount of 0.2Au–La2O3 in yttrium nitrate equivalent to 5 wt% yttrium oxide, Au (0.2 wt%) supported on La2O3 marked as 0.2Au–La2O3, where the same applies below) exhibited a high-intensity reduction peak. It is known that Y2O3 and La2O3 are basic in nature and can have intimate contact with gold metal nanoparticles to generate active sites.86 The O2-TPD results, as shown in Fig. 11(b), demonstrated three peaks, where the two main peaks are attributed to the desorption of different oxygen species (e.g., surface O2− and/or O22− species), surface and bulk lattice oxygen species of metal oxides from low-temperature to high-temperature, respectively. Among them, 5Y-0.2Au–La2O3 showed the highest desorption peaks, indicating the presence of more oxygen supplying centers. Meanwhile, the reaction activity improved after loading 1–5 wt% of Y2O3 on the carriers, suggesting that Y2O3 is a major factor in this improvement, as shown in Fig. 11(c and d). It was reported that the olefin molecules desorb faster over the basic oxides, which could halt olefin dissociation and improve the olefin selectivity.87 Consequently, 5Y-0.2Au–La2O3 delivered superior activity with the n-propane conversion of 78% and olefin selectivity of 72% due to its ease of reduction and more mobile oxygen species and metal sites.84
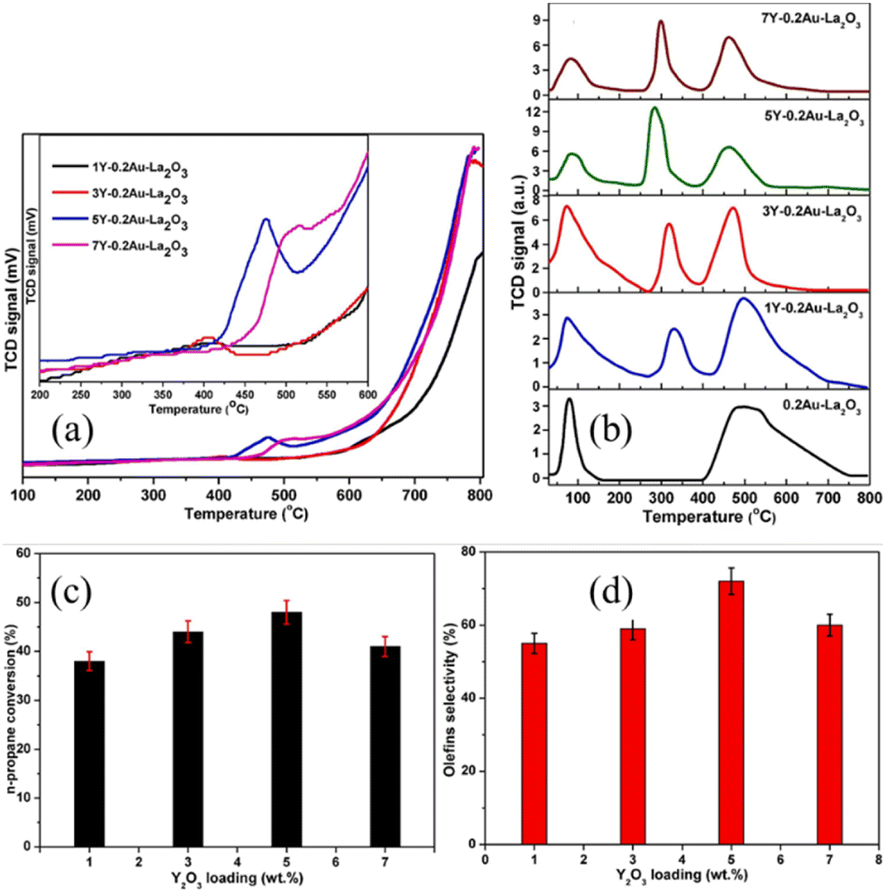 | ||
| Fig. 11 (a) H2-TPR and (b) O2-TPD patterns of synthesized samples. Reaction activity tests of the catalysts. The influence of Y2O3 loading on (c) conversion of n-propane and (d) selectivity to olefins. Reaction conditions: 500 °C; GHSV: 48000 h−1.84 Reprinted with permission from ref. 84. Copyright © 2020, Elsevier. | ||
However, the above-mentioned studies on the cracking of low-carbon alkanes using solid bases revealed that the production of COx can poison the catalysts and reduce the selectivity of low-carbon olefins, and the combustion of low-carbon olefins may occur during the oxidative cracking reaction, thus making it difficult to control the reaction process.84,88
3.3.3.2 Naphtha catalytic cracking. Jeong et al. used KVO3/Al2O3 prepared via the impregnation method, and then used it in the catalytic pyrolysis reaction of naphtha at 800 °C.89 They found that the use of this solid base significantly improved the yields of ethylene and propylene, while the yield of low carbon olefins was found to be almost the same when Al2O3 alone was used in the same reaction as that of the KVO3/Al2O3 catalyst. This indicates that the cracking performance is mainly dependent on the Al2O3 component, while KVO3 is responsible for reducing the carbon deposition, as shown in Table 3 for the COx products and carbon deposition of the series catalysts. Also, they suggested that the introduction of KVO3 increased the coking gasification rate to suppress carbon deposition.89 Since then, Jeong et al. introduced B2O3 in the above-mentioned solid base to investigate the anti-carbon deposition ability of these catalysts. The introduction of B2O3 was found to enhance the stability of the alkali metal K in the reaction according to the ICP-AES characterization, and the main reason for the stabilization of K by TPR was the strong interaction of B2O3 with KVO3.81
| Catalysts | Yield of COx (wt%) | Yield of coke (wt%) |
|---|---|---|
| α-Al2O3 | 1.96 | 0.51 |
| KVO3/α-Al2O3 | 2.87 | 0.17 |
| KVO3–B2O3/α-Al2O3 | 2.92 | 0.14 |
Previously, Pierre L. et al. studied the catalytic thermal cracking of naphtha with metal oxide mixtures as catalysts for the purpose of producing more low carbon olefins, with the main components of MgO, SiO2, ZrO2, and small amounts of Nd2O3, Al2O3, and CaO. The results showed that the catalysts were stable at the bed temperature of 650–900 °C and the corresponding pressure, with high ethylene yield and no carbon deposition, and the catalysts did not require regeneration.90 Thereafter, Lemonidou et al. showed that 12CaO–7Al2O3 had the highest yield of low carbon olefins and the lowest yield of COx in a study on n-hexane cracking using various mixed metal oxides, while the conversion of n-hexane started to decrease after reduction of the catalyst, which proved that the active oxygen in the catalyst acted as a base active center. This is also the reason why calcium aluminate catalysts have been more applied and studied in the research on solid base catalysts used in the field of catalytic cracking.91 When 12CaO–7Al2O3 was used for naphtha catalytic cracking, the reaction temperature could be reduced by 50 °C compared to the steam cracking process, and it also reduced the catalyst carbon deposition by promoting the gasification of coke, but the use of 12CaO–7Al2O3 did not affect the product distribution.92 Mukhopadhyay et al. introduced K2CO3 in 12CaO–7Al2O3 and found that the carbon deposition decreased with the loading of K2CO3, but the yield of low carbon olefins also decreased slightly, and they suggested that the combination of K and reactive oxygen species occupied the active site of the cracking.93 Kumar et al. studied the effect of calcium aluminate catalysts with K introduced by different preparation methods on the cracking performance of n-heptane and the amount of loaded metal, and found that calcium aluminate catalysts significantly improved the ethylene yield of n-heptane pyrolysis. Also, they showed that the introduction of K promoted the gasification of carbon deposits and the K-loaded calcium aluminate catalysts prepared by impregnation method had high cracking activity, but the amount of K loading was easily lost with an increase in the reaction time.94 T. Tomita et al. studied the naphtha cracking reaction of alkaline earth metal oxides or their mixtures co-sintered with alumina to prepare solid bases and concluded that alkaline earth metal oxides can inhibit the hydrocarbon dehydrogenation reaction and inhibit the thermal polymerization reaction. This is important for the inhibition of carbon deposition; meanwhile, CaO and Al2O3 co-crystals can form a spinel-like structure, which remains stable at 800 °C.95
The aforementioned studies on the application of mixed metal oxide catalysts in catalytic cracking almost came to a halt around the twentieth century, but due to their good resistance to carbon deposition, they are beginning to become important in the present environment of heavy and inferior crude oil.
3.3.3.3 Heavy oil catalytic cracking. Tian et al. used FCC catalysts with calcium aluminate catalysts for the catalytic cracking of vacuum residue oil and found that although the FCC catalysts resulted in higher conversion at the same temperature, their high dry gas yield and low liquid yield compared to calcium aluminate catalysts indicated that they may be more likely to lead to over-cracking of the oil, while having about twice the carbon deposition yield than calcium aluminate catalysts.96 Tang et al. compared the distribution of low-carbon olefin products after the catalytic cracking of vacuum residue oil with FCC catalysts and calcium aluminate catalysts, and then found that calcium aluminate had higher low-carbon olefin yields at 650 °C.97 They further investigated the catalytic ability of magnesium aluminate and calcium aluminate for cracking vacuum residue oil and found that both types of catalysts were favorable for improving the yield of low carbon olefins, while calcium aluminate was superior. They suggested that the catalysts promoted the generation of free radicals by hydrogen abstraction of the reactants, and thus improved the yield of low carbon olefins. Thus, to improve the base density of calcium aluminate, they created a porous structure in calcium aluminate by adding graphite and several types of organic substances, which improved the conversion of heavy oil to light oil, and they proposed that the porous structure formed on calcium aluminate facilitated the distribution of reactive oxygen species and increased the contact probability of reactants with the active center.98,99 Recently, Niwamanya et al. studied the catalytic cracking of heavy oil using the above-mentioned catalysts and found that the strong basicity of calcium aluminate promotes hydrocarbon dehydrogenation and its yield of low carbon olefins is higher compared to quartz sand, but its conversion, low-carbon olefin yield, and aromatic yield are lower compared to the acidic catalyst ZSM-5. Accordingly, they proposed the combination of calcium aluminate and ZSM-5 in an optimized ratio and concluded that only a suitable ratio can provide a suitable acid center concentration. Compared with the general FCC catalysts with ZSM-5 and Y-type zeolites as the main active components, these combinations can better balance the shape selectivity, cracking and hydrogen transfer activities of the catalysts, and thus obtain relatively high low-carbon olefin and aromatic yields.15
In summary, among the loaded oxides, researchers are currently focusing their efforts on calcium aluminate catalysts. Their early application was aimed at reducing carbon deposition in the steam cracking process, and their characteristics may make them suitable for the treatment of heavy and inferior heavy oils. Some studies on loaded solid base catalysts have confirmed that the use of these catalysts can indeed promote the cracking reaction of prolific olefins to some extent, and it is recognized that the introduction of alkali metal compounds in alkali metal oxides has a significant effect on the reduction of carbon deposition in catalysts. However, because of the aforementioned drawbacks, only a few loaded solid base catalysts that can catalyze the cracking reaction have been developed thus far, and their research has been stalled for a while. Besides, using mixed metal oxides in combination with zeolites may be another way forward for the efficient green treatment of heavy oil.
4 Zeolite-based solid bases
Zeolites have attracted significant attention because of their regular pore structure and high specific surface area, and they have also been studied by scientists from various countries because of their good shape-selective catalytic ability, and the selection of zeolites as carriers for basic shape-selective catalysis is an important step in the preparation of solid base catalysts and the realization of new green catalytic processes.174.1 Ion-exchange of alkali metals
In the majority of situations, the catalytic activity of the basic sites of zeolites after alkali metal ion exchange is positively correlated with the atomic number of alkali metal ions and negatively correlated with the silica-alumina ratio of the zeolite framework, and this can be explained by the change in the negative charge of the lattice oxygen. The negative charge number of the lattice oxygen can be calculated by the Sanderson electronegativity equilibrium theory, which is an effective method to determine the basicity strength of alkali metal ion-exchanged zeolites.103–105 The framework oxygen binding energy (BE) in the XPS characterization method corresponds to the electronic charge of the lattice oxygen, as shown in Fig. 12(a), demonstrating the relationship between the lattice oxygen binding energy of X- and Y-type zeolite and the electronegativity of the metal cation. The electronegativity of the exchange cation and the silica-alumina ratio of the framework are positively correlated with the binding energy BE of the lattice oxygen, respectively. The weaker the binding energy of the lattice oxygen, the stronger its electron-donating capacity and the basic strength. Meanwhile, Okamoto et al. argued that the basic sites of zeolites originate from the lattice oxygen after ion-exchange and proposed a bonding model of exchange ions, as shown in Fig. 12(b), where two possible binding states exist after ion exchange, covalent bonding between cation-oxygen and ionic bonding formed by the negative charge of the leaving group on cation-aluminium, and basicity is related to the ionic bonding mode, while the bonding mode in general is a mixture of these two types of bonding modes.4,100,106
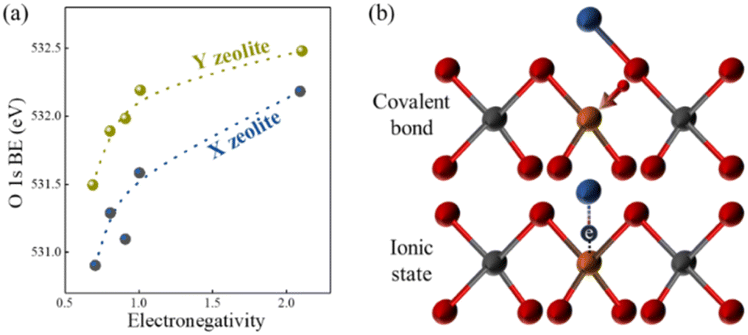 | ||
| Fig. 12 (a) Relationship between the O 1s binding energy of lattice oxygen and the electronegativity of cation-exchanged X and Y zeolites. (b) Proposed bonding models on zeolites after cation-exchange. Red sphere: O, grey sphere: Si, brown sphere: Al, and blue sphere: metal cation.100 Adapted with permission from ref. 100. Copyright © 1988, Elsevier. | ||
The zeolite exchanged by alkali metal ions has a negative oxygen charge number, and the order of its basicity is Cs, Rb, K, Na, and Li according to the negative charge number from the largest to the smallest, then the order of its basicity is Cs, Rb, K, Na, and Li from the largest to the smallest. The negative oxygen charge number of the zeolite exchanged by the same alkali metal ions increases with a decrease in the silica-alumina ratio of the framework, i.e., the smaller the silica-alumina ratio, the stronger the basicity of the modified zeolite. The basicity of zeolites is not only related to their intrinsic structure and the type of metal ions loaded but also their chemical environment, the distribution of aluminium on the framework, and the position of the metal cations.107
As can be seen from the above-mentioned investigation, alkali metal ion-exchanged zeolites have a lower base strength than that exchanged with alkaline earth metal oxide, and also lower than that of zeolites exchanged with an alkali metal compound, exceeding the ion exchange capacities of the zeolite. Although alkali metal ion-exchanged zeolites have a lower strength of basic sites, their base strength can be adjusted by different cation exchanges, and thus the zeolite after ion exchange can be better suited for multiple types of reactions.
The catalytic properties of alkali metal ion-exchanged zeolites depend on their weak base sites and regular pore channels. It was found that although the regular pore structure can produce shape-selectivity, it can also have an effect on the diffusion of reactants and products, especially in liquid-phase reactions involving solid bases, and thus researchers aimed to use exchange ions with different radii or electronegativities to modulate their basicity.109 In the application of X-type zeolites loaded with K+, Cs+, and Na+, respectively, in the reaction of dehydration and dehydrogenation of 2-propanol, the active sites of base catalysis were attributed to the loading of the three cations. Specifically, K+ and Cs+ were more likely to produce basic sites than Na+, and thus dehydrogenation mainly occurred on KX and CsX, while the dehydration reaction occurred on NaX.110,111 In the MPV (Meerwein–Ponndorf–Verley) reduction of aldehydes with 2-propanol to produce the corresponding 1-alcohol over NaX, the framework oxygen adjacent to Na+ abstracts a proton from 2-propanol, and the oxygen acts as the base center.112 The replacement of Si in NaX by Ge can produce relatively stronger basic sites, enhancing its catalytic activity in the Knoevenagel condensation reaction.113,114 However, Corma et al. suggested that the origin of the enhanced activities is the smaller T–O–T (intrinsic chemical bond formation with oxygen inside zeolite) bond angle, longer T–O bond, and larger cell size after the replacement of Si by Ge. The activity of the NaGe Faujasite is higher than that of pyridine and lower than that of piperidine, indicating that most of the basic sites on the NaGeX zeolite have a pKb of about 11.2 and basic sites with strength in the order of pKb of 13.3 should exist to abstract protons from ethyl malonate.115 In summary, although the ion-exchange can alter the negative charge of the zeolite framework, it is still difficult to prepare strong base materials via this method.
4.2 Loading alkali metals
Good catalytic activity was observed in side-chain alkenylation reactions at lower reaction temperatures on zeolites loaded with K, Na, and Cs with azide compounds as intermediates, while the reaction was difficult to occur on CsX zeolites loaded with CsOx.120 When the Michael addition reaction of ethyl acrylate with acetone was catalyzed using modified zeolites at 90 °C, Na/NaX showed the best catalytic activity, followed by CsOx/γ-Al2O3, Na/NaOH/γ-Al2O3, and CsOx/CsX, while NaX and CsX had no catalytic activity. The Hammett indicator of the modified zeolites was CsOx/γ-Al2O3 (>37), Na/NaOH/γ-Al2O3 (35–37), CsOx/CsX (17.2–18.4), and NaX (<9.3), and the base strength sequencing is consistent with the catalytic activity sequencing.121 The decomposition performance of a series of catalysts prepared with NaN3 as precursor loaded with Na on different zeolites and Al2O3 for the base-catalyzed reaction of MBOH decomposition to acetone followed the order of NaX > NaY > NaL > Naβ > Al2O3, and the difference in reactivity was attributed to the different types of zeolite carriers.3
According to the aforementioned studies, it can be concluded that the catalytic process of base catalysis is more sensitive to the properties of the basic site of the solid base rather than the strength and density of the base. It was demonstrated that some reactions could not proceed on zeolites with alkali metal ion exchange or loaded with alkali metal oxides, but good reactivity could be achieved via alkali metal loaded zeolites.
4.3 Loading alkali metal oxides
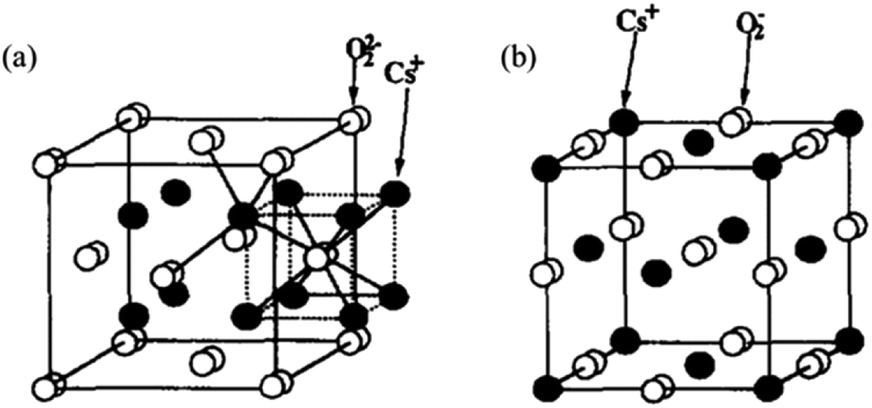
The basic sites of an X-type zeolite after ion exchange exceeding the exchange capacity of cesium acetate are Cs+ introduced by ion exchange or within the supercage of the zeolite and Cs2O loaded within its pore channel.124 Unlike Cs+ ion exchange, excess Na+ will be located in the hexagonal column away from the outer surface rather than inside the supercage. Yagi et al. conducted an in-depth study on the relationship between Cs content and basic sites on type X zeolites by 133Cs MAS NMR characterization and the results showed that if the number of Cs atoms in the supercage is greater than the exchangeable capacity of the X-type zeolite but not greater than 2.9, the Cs+ that is added greater than the exchangeable capacity exists in the pore cavity of the supercage on the zeolite. Alternatively, if it is more than 2.9, the Cs+ exceeding the exchangeable capacity part is distributed on the outer surface of the zeolite.125 The solid base obtained by ion exchange of Cs+ with the Y-type zeolite, and then impregnated with CsOH has Cs+ on the exchangeable ion sites in addition to the oxide species of Cs. Hunger et al. suggested that the Cs oxides within the zeolite did not form large clusters but highly dispersed oxide species on the surface of the zeolite.126 Further, the 18O2-TPD studies showed that the surface layer of Cs2O partially forms peroxide or superoxide such as Cs2O2, Cs2O3, and Cs2O4, which in turn may produce strongly basic sites. The oxides formed by these solid bases after the adsorption of oxygen all exhibit structural features of Cs+ bonding to O2−/O22− with negatively charged oxygen atoms in the adsorbed oxygen. The bonding model diagram of Cs2O2 and Cs2O4 generated after the adsorption of oxygen is shown in Fig. 13. The oxygen atom can be decoupled without breaking the chemical bond between the two oxygen atoms in the peroxide or superoxide when the peroxide and superoxide are reduced to Cs2O through the rearrangement of Cs and O.122,124 In summary, different pretreatment conditions can lead to different oxygen environments, and different chemistry states of cesium oxide may exist in solid bases such as superoxide CsO2, peroxide Cs2O2, oxide Cs2O, low oxygenated Cs7O, and metallic Cs.
4.4 Other loaded compounds
KF, KNO3, and K2CO3 are weakly basic and neutral metal salts, and it has been shown that these three compounds loaded with Al2O3 could produce strong solid base sites.136–138 For example, KF-loaded Al2O3 presented both stronger basicity and nucleophilicity than KOH/Al2O3, but its specific surface area remained at only about 31 m2 g−1 after treatment at 500 °C.139 Zeolites are good carriers for KF loading because of their extremely high specific surface area and good shape-selecting ability. NaY has a high specific surface area of 766 m2 g−1, and because silica-alumina zeolites have an elemental composition similar to Al2O3 and SiO2, NaY can react similarly to the loading of Al2O3 and SiO2 after loading KF, as shown in the following chemical eqn (1) and (2), which in turn produces a basic species similar to KOH.140
| 12KF + 3H2O + Al2O3 → 6KOH + 2K3AlF6 | (1) |
| 6KF + 2H2O + SiO2 → 4KOH + K2SiF6 | (2) |
Although KF does not destroy the structure of the zeolite during synthesis, as evidenced by XRD characterization, the interaction of KF with the zeolite framework intensifies during high-temperature pretreatment, and KF can react with the silicon component of the zeolite framework to form low-intensity silica-potassium compounds, which leads to the collapse of the framework at high temperatures.138,141 Also, because the zeolite contains a silicon component and the silicon component reacts preferentially with KF, resulting in a significantly lower base strength of KF/NaY (![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) < 9.3) than that of KF/Al2O3 ( > 17.2), some experiments have shown that the base strength of KF loaded on a mixture of Al2O3 and SiO2 is similar to that of KF/SiO2. This indicates that the presence of a silicon component in the zeolite hinders the generation of strong basic sites, and simultaneously reduces the stability of the zeolite in hydrothermal treatment.140
< 9.3) than that of KF/Al2O3 ( > 17.2), some experiments have shown that the base strength of KF loaded on a mixture of Al2O3 and SiO2 is similar to that of KF/SiO2. This indicates that the presence of a silicon component in the zeolite hinders the generation of strong basic sites, and simultaneously reduces the stability of the zeolite in hydrothermal treatment.140
Unlike KF modifications, KNO3 modification is not dependent on interaction with the zeolite carriers but can decompose itself to produce strongly basic sites, and thus KNO3 modification can greatly reduce the damage to the zeolite framework. The γ-Al2O3 framework has octahedral vacancies that can hold cations, which are important to facilitate the decomposition of KNO3 to promote the production of strongly basic sites.142,143 Meanwhile, KNO3/γ-Al2O3 activation at a high temperature of 600 °C produced a much more basic site than the MgO basicity with of about 27.0, but the specific surface area of KNO3/γ-Al2O3 after high-temperature activation was positively low, with only about 80 m2 g−1, and thus zeolites with a high specific surface area are employed to improve the specific surface area of these solid bases.137,144 However, the base amount of 21% KNO3/NaY was only 0.16 mmol g−1 after pretreatment at 600 °C. Some studies showed that the variation between NaY and γ-Al2O3 was due to the fact that NaY lacks important octahedral vacancies for embedding and adsorption of K+, and the zeolite framework and surface may change and reform during pretreatment, and thus although KNO3 can be highly dispersed on NaY, it is difficult to thermally decompose and form strong base sites.142
Not only loading different K predecessors can change the physicochemical properties and catalytic characteristics of the catalyst, but it is also necessary to choose a suitable carrier. For example, Y- and KL-type zeolites have different structures despite similar silica-alumina ratios, and KL zeolites were modified by KF to obtain a basic center with of only about 15 for KL zeolites.138 After pretreatment of KL zeolite loaded with KNO3 or K2CO3 at 600 °C at high temperature, it was found that the high-temperature decomposition of the compound on the surface of KL zeolite produced super basic sites with of about 27.0. Some studies showed that the loaded KNO3 started to decompose at 500 °C during high-temperature pretreatment. The characteristic peaks of NO2− and KOH were not found by FTIR characterization, confirming that K2O, not KNO2 and KOH, was produced when KNO3 was loaded on KL and pretreated at high temperature. The CO2-TPD characterization showed that the CO2 desorption temperature of 21% KNO3/KL was higher than that of KL zeolite, while a new high-temperature desorption peak appeared at 500 °C, indicating that the basicity of the carrier was significantly changed and enhanced.145,146
According to the research on alkali-modified zeolites used in the traditional fine-chemical industry, intrinsic solid base materials generally have a smaller specific surface area, poorer mechanical strength, and weaker hydrothermal stability, while they do not have the regular pore structure and very high specific surface area of zeolite-based catalysts. Thus, in the petroleum industry, based on the research experience of solid bases in the traditional fine-chemical industry, the combination of basic active components and materials with regular pore structure has received more attention from researchers.
The recent study by Singh et al. revealed the reason for the variation in the Knoevenagel condensation reaction activity, as shown in Fig. 14(b), due to the different surface amine sites after pretreatment of SBA-15-oxynitrides at different temperatures.147 According to this study, they proposed a completely different mechanism of ammonia attack on siloxanes, as shown in Fig. 14(a), than that reported by Asefa et al., enabling the production of secondary and even tertiary amines rather than just the production of primary amines.148 Notably, the advanced characterization method employed, as shown in Fig. 14(c), directly confirmed the predominant presence of secondary amines on SBA-15-N900 and tertiary amines on SBA-15-N1200. Solid-state NMR and XPS studies, as shown in Fig. 14(d), demonstrated that the differences in the formation of surface primary, secondary, and tertiary amines led to variations in reactivity, with the activity dependent on the complex factors among the total amine content, the fraction of primary amine content, and the available surface area, while the concentration of the surface amine depended on the nitriding temperature. The presence of a high primary amine content is associated with high catalytic efficiency and a higher fraction of secondary/tertiary amines contributes to a decreased TON.147
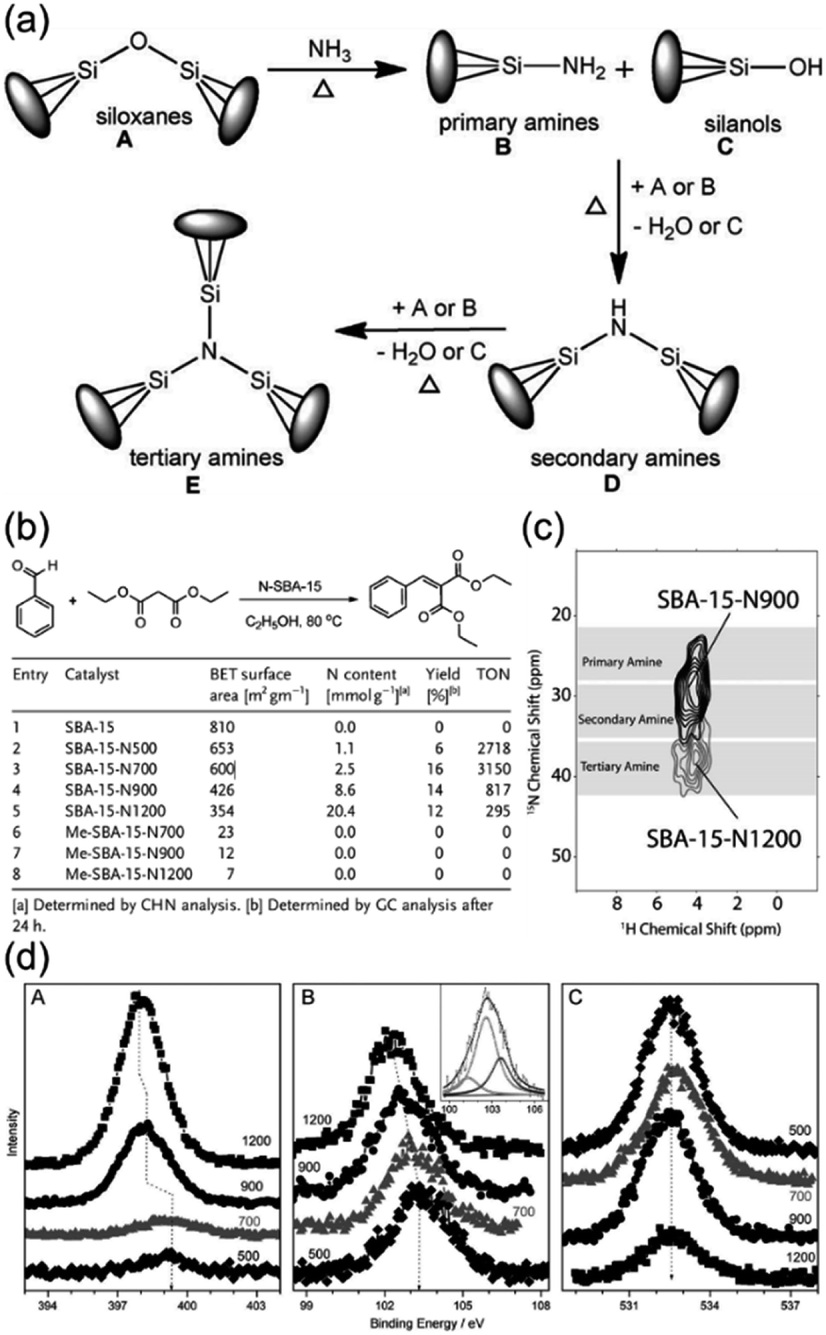 | ||
| Fig. 14 (a) Mechanism of nitridation during ammonolysis. (b) Catalytic activity SBA-15-NT (SBA-15 was nitridated at T °C) for the Knoevenagel condensation reaction between benzaldehyde and diethyl malonate. (c) 1H-detected 15N–1H correlation spectrum obtained for SBA-15-N1200 (grey) and SBA-15-N900 (black). (d) XPS spectra for (A) N 1s, (B) Si 2p, and (C) O 1s core levels of various SBA-15-oxynitrides. Binding energy changes observed are indicated by a dotted line.147 Reprinted with permission from ref. 147. Copyright © 2015, Wiley. | ||
Research on bio-oil have gained significant attention in recent years due to the growing focus on environmental protection globally. Several base-catalyzed condensation reactions, such as esterification, aldol condensation and ketonization, are important intermediate steps in the production of second-generation biofuels by exploiting the intrinsic reactivity of bio-oil components. Numerous mild base catalysts have been identified to exhibit a stable performance in aldol condensation such as supported alkali or alkaline earth metals and other supported metal oxides. Recently, Puértolas et al. investigated the deoxygenation via aldol condensation for bio-oil by the use of mild base catalysts and illustrated the activity, selectivity and stability properties of supported MgO catalysts in the vapor phase condensation of propionaldehyde, while this performance was maximized upon the mechanochemical activation of a siliceous USY zeolite (Si/Al = 405) containing 1 wt% Mg(OH)2.149 The IR studies on CO and CO2 adsorption, CO2-TPD, XRD and STEM-mapping characterization combined with the variation in the reaction performance due to different amounts of MgO, as shown in Fig. 15(a and b), revealed that the moderation of the basic strength is facilitated by the high dispersion of small amounts of MgO on the siliceous USY zeolites prepared by mechanochemical activation, as shown in Fig. 15(c and d), which is ascribed to the incorporation of Mg2+ in framework defects on the zeolite surface. The limited number of these defects leads to the formation of a undesirable MgO sub-phase with an increase in Mg content and fosters the non-selective conversion of propionaldehyde due to its strong basicity.149
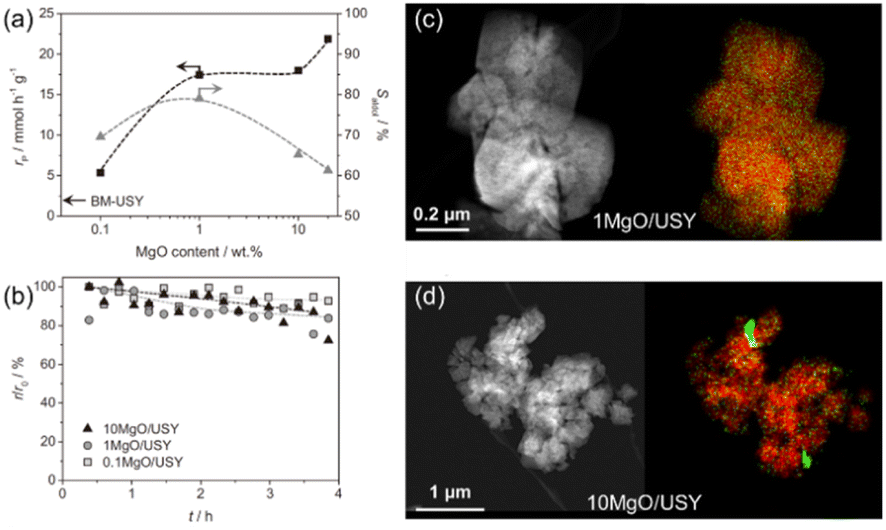 | ||
| Fig. 15 (a) Rate of propanal conversion (black) and selectivity to the aldol condensation products (grey) versus the MgO content over the MgO/USY zeolites. The arrow indicates the rate of propanal conversion over the BM-USY reference. Reaction conditions: 0.3 g, 400 °C, 6.25 kPa, He flow = 50 cm3 min−1. (b) Relative decrease in the reaction rate observed in the vapor phase condensation of propanal over MgO/USY zeolites with different MgO contents. HAADF-STEM images (left) and elemental maps (right) of Si (red) and Mg (green) of the (c) 1 MgO/USY and (d) 10 MgO/USY zeolite catalysts. The scale bars apply to both images in the same row.149 Reprinted with permission from ref. 149. Copyright © 2016, Elsevier. | ||
However, due to its carbonium ion mechanism which determines its susceptibility to hydrogen transfer, the possible carbon deposition on it can lead to a rapid decrease in catalyst activity or even rapid deactivation, and hence the high requirement for oil in the selection of the feedstock.152,153 If heavier or inferior feedstock is used, given that it has a higher asphaltene content and other impurities, this will lead to more carbon deposition on the catalyst, while the pore structure of the ZSM-5 zeolite will limit the access of macromolecules to its active center, and the feedstock tends to accumulate gradually on the catalyst surface, eventually inactivating the catalyst.155 Yoshimura et al. studied the effect of loading La2O3 and P (e.g., H3PO4) on ZSM-5 zeolite on the catalytic activity of naphtha cracking and the properties of these catalysts. It was shown that the La-loaded catalyst could inhibit the formation of BTX-like products, and thus promote the yields of ethylene and propylene to a certain extent. The loading of La and P did not affect the acidic properties of ZSM-5, while the inhibition of heavier products such as aromatics is due to the enhancement of the surface basicity by the introduction of La. Similar to the above-mentioned introduction of B2O3 to stabilize the K loading, as mentioned in Section 3.3.3, the addition of P could stabilize Al in the zeolite framework, thereby enhancing the hydrothermal stability of the catalyst and stabilizing its catalytic activity.12
Due to the correlation between the catalytic cracking performance of zeolites and their acidic property and pore structure, researchers have attempted to optimize the coking problem in the treatment of heavy oils by modifying the zeolites with alkaline metals in recent years. Zhang et al. introduced alkali metal oxides into a bifunctional catalyst prepared by zeolite to crack vacuum residue oil, as shown in Table 4, and the results showed that while over-cracking and the dry gas yield in the reaction were improved to certain extent by the introduction of bases, the coke yield was not significantly reduced, and it can be seen that the modification of zeolites by alkaline metals alone is not sufficient to solve the carbon deposition problem arising from the catalytic cracking of heavy oil.150
| Test no. | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| a FCC-T: FCC catalysts by hydrothermal treatment at T °C.b BFC-T: BFC catalysts (author's self-made bifunctional catalysts) by hydrothermal treatment at T °C. | |||||
| Catalyst | FCC-800a | BFC-800b | BFC-850 | BFC-800 | BFC-850 |
| Temp (°C) | 503 | 500 | 499 | 499 | 497 |
| Catalyst/oil ratio | 6.3 | 6.3 | 6.3 | 4.2 | 4.2 |
| Gas yield (wt%) | 10.3 | 19.4 | 11.2 | 13.4 | 7.3 |
| Liquid yield (wt%) | 82.4 | 71.0 | 79.1 | 78.2 | 84.0 |
| Coke yield (wt%) | 7.3 | 9.6 | 9.7 | 8.4 | 8.7 |
| Dry gas (vol%) | 36.0 | 32.0 | 38.0 | 31.2 | 42.9 |
| Conversion (%) | 98.7 | 100.0 | 100.0 | 99.0 | 94.9 |
|
|||||
| Distillation distribution of liquid | |||||
| Gasoline (wt%) | 38.5 | 58.4 | 43.8 | 55.4 | 29.8 |
| Diesel (wt%) | 42.3 | 38.0 | 43.9 | 33.3 | 34.7 |
| VGO (wt%) | 17.6 | 3.5 | 12.3 | 10.0 | 29.5 |
| Heavy oil (wt%) | 1.6 | 0.0 | 0.0 | 1.3 | 6.0 |
Recently, Wu et al. of CNOOC (China National Offshore Oil Corporation) developed DPC (direct petroleum cut to chemicals and materials) base-catalyzed technology for the catalytic cracking of heavy oil using base catalysts. All the base catalysts consisted of basic active components, additives, molding promoter, and carriers. Amorphous silicon oxide with a high specific surface area, silica-alumina composite oxide or SBA-15 carriers were selected to inhibit the adsorption of PAHs (polycyclic aromatic hydrocarbons) and non-hydrocarbon impurities. However, although these mesoporous structures could promote the macromolecular mass transfer in heavy oil, the heavy oil conversion and products yield such as LPG, gasoline and coke were all lower than acid catalysts. Tables 5 and 6 presents the natural properties of feedstock oil and the preparation information, reaction conditions and reaction results of this solid base, respectively.154,156 Compared with delayed coking technology, as reported by the inventor, DPC technology with a base catalyst could reduce the formation of coke and promote the yield of LPG, C=2–C=4, light naphtha and heavy naphtha.157
| Item | Vacuum residue oil | |
|---|---|---|
| Density, g cm−3 | 0.932 | |
| H–C ratio | 1.75 | |
| Residual carbon, wt% | 15.6 | |
| Basic nitride, ppm | 1925.1 | |
| Acid value, mg KOH% | 0.28 | |
| Element analysis, wt% | C | 87.23 |
| H | 11.44 | |
| S | 0.39 | |
| N | 0.94 | |
| Four components analysis, wt% | Saturated hydrocarbon, wt% | 28.8 |
| Aromatic hydrocarbon, wt% | 31.5 | |
| Colloid, wt% | 34.1 | |
| Asphaltene, wt% | 3.8 | |
| Catalysts | Cat. 1 | Cat. 2 | Cat. 3 |
|---|---|---|---|
| Carrier (wt%) | 38% high specific surface area SiO2 | 37.81% alumina-silica mixed oxide-0.06%TiO2–0.13%Mn | 37.81% SBA-15-0.06%TiO2–0.13%Mn |
| Alkaline earth metal (wt%) | 8%MgO | 8%CaO | 8%CaO |
| Rare earth oxide (wt%) | — | 1.5%CeO2 | 1.5%CeO2 |
| Preparation condition | Slurry water content 73 wt%. Calcined at 550 °C for 4 h | ||
| Aging condition | 100% hydrothermal treatment at 800 °C for 17 h | ||
| Reaction condition | Fluidized bed reactor, 0.2 MPa, 520 °C, steam/feed = 3:1(wt), ratio of catalyst to feed oil = 12, contact time = 2 s |
||
| Feed | VR | ||
| Dry gas (wt%) | 1.58 | 1.04 | 2.23 |
| LPG (wt%) | 6.68 | 4.95 | 6.86 |
| Gasoline (wt%) | 13 59 | 16.53 | 17.42 |
| Diesel (wt%) | 15.27 | 16.12 | 15.7 |
| Heavy oil (wt%) | 48.21 | 53.68 | 51.83 |
| Coke (wt%) | 14.67 | 7.68 | 5.96 |
5 Typical industrial applications
Acid catalysts have been well developed previously and many of them have been successfully utilized industrially in numerous industrial processes, such as naphtha cracking, isomerization, and alkylation. In contrast, the application of solid bases as catalysts came later than solid acids, and there were only a few examples of the successful application of solid bases as catalysts in the industry. One of the main reasons for this is that solid bases are sensitive to substances such as inherent moisture and carbon dioxide in the air. Meanwhile, the higher the strength of the base, the stronger the tendency to poisoning. Consequently, solid bases are difficult to prepare and store with stringent and demanding requirements, making it challenging to achieve convenient and inexpensive industrial applications.Additionally, unlike most lab research, the importance in industrial applications is to control the nature, morphology, textural properties (surface area and porosity) and thermal, chemical and mechanical stability of solid base catalysts, and it is challenging to tackle these aspects in solid bases. The conventional synthesis process for industrial catalysts is as follows: chemicals (control impurities), catalyst precursor (texture), form (texture, shape attrition resistance), and final catalyst (texture, attrition resistance).
However, due to the good catalytic properties of solid bases in some special base-catalyzed reactions (e.g., isomerization of alkenes and alkynes, transesterification, Michael addition, condensation reactions and anti-Markovnikov hydroamination and hydroalkoxylation), only a few solid bases has been industrially applied to date. An industrial process for the production of chemicals by catalytic cracking of crude oil using a novel solid base catalyst has been added to the list, as shown in Table 7, which summarizes the solid base catalysts for industrial applications reported by Hideshi Hattori in 2015.8,16 In recent years, the DPC catalyst was commercialized in the Huizhou refinery with an inventory of up to 26.5%, similar to an additive, given that the bottom cracking activity will be depressed with an increase in its proportion.8 The mechanism of the proposed carbon anion has not been described and proven to date, but the lower yield of coke should be related to the inhibition of hydrogen transfer and aromatic condensation reactions due to the reduction in the number of acid sites. Thus, there are still too many unknowns in the field of oil cracking catalyzed by solid base catalysts, and thus more in-depth basic research and systematic analytical validation are needed.
| Reaction | Catalyst | Year |
|---|---|---|
| Alkylation | ||
| Alkylation of phenol with methanol | MgO | 1970, 1985 |
| Alkylation of xylene with butadiene | Na/K2CO3 | 1995 |
| Alkylation of cumene with ethylene | Na/KOH/Al2O3 | 1988 |
|
||
| Isomerization | ||
| Isomerization of safrole to isosafrole | Na/NaOH/Al2O3 | 1988 |
| Isomerization of 2,3-dimethyl-1-butene | Na/NaOH/Al2O3 | 1988 |
| Isomerization of 3,5-vinylbicyclo [2,2,1] heptane | Na/NaOH/Al2O3 | 1988 |
| Isomerization of 1,2-propadiene to propyne | K2O/Al2O3 | 1996 |
|
||
| Dehydration/condensation | ||
| Dehydration of 1-cyclohexylethanol | ZrO2 | 1986 |
| Dehydration of propylamine-2-ol | ZrO2/KOH | 1992 |
| Isobutyraldehyde to diisopropyl ketone | ZrO2 | 1973 |
| Dehydrotrimerization of isobutyraldehyde | BaO–CaO | 1998 |
|
||
| Esterification | ||
| Esterification of ethylene oxide | Hydrotalcite | 1994 |
| Transesterification of triglycerides | ZnO–Al2O3 | 2006 |
|
||
| Catalytic cracking | ||
| Crude oil to chemicals | Alkali/alkaline earth metal oxide-highly mesoporous carrier | 2022 |
|
||
| Miscellaneous | ||
| Carboxylic acids to aldehydes | ZrO2–Cr2O3 | 1988 |
| Thiols from alcohols with hydrogen sulfide | Alkali/Al2O3 | 1988 |
| Cyclization of imine with sulfur dioxide | Cs-zeolite | 1995 |
It might be noteworthy that, as can be seen from Table 7, the solid base catalysts available for industrial applications currently exist mainly exclusively as metal oxides, whereas the catalytic activity of the zeolites named as basic-catalyst rarely reveals industrial or relevant applications in chemical processes thus far. Indeed, the structure–activity relationship for the microporous basic properties of zeolites remains an open discussion. It is obvious that the zeolite acid–base pair is different from that of the homogeneous acid–base compounds and some solid metal oxides, given that their strength is influenced by the type of exchangeable cations, aluminum content, structural dimension, and porosity.158,159
Nevertheless, several reviews and articles published in the last decade indicated that solid bases are recognized as a type of environmentally friendly catalyst, and therefore research on solid base materials and their catalytic reactions showed an accelerated development in recent years despite their many challenges.
6 Conclusion and outlook
Alkali metals, alkaline earth metals, rare earth metals, and their oxides and compounds are now widely used in the preparation and development of base catalysts. Solid base catalysts, which are expected to be environmentally friendly, are gaining increasing attention from researchers and may provide new ideas to improve the reaction process and cope with environmental requirements and treatment of inferior heavy oil. However, to date, the corresponding research on solid base catalysts is still less than that on solid acids catalysts, the knowledge is more superficial, and the catalytic mechanisms in different types of reactions need further study and elaboration. The lack of systematic research on the catalytic mechanism of bases is an important reason limiting the development of solid bases, and therefore the following are important:(1) Establishing a systematic study of solid bases. The relationship between the number of active sites in solid bases and their strength and the reaction mechanism is still lacking in regular studies, and further studies on the reaction processes occurring at the active sites and their reaction mechanism will provide a more effective way for the preparation and application of solid base catalysts.
(2) Modelling and analysis of active sites through computer analogs and calculations. Different models of active sites on the surface of solid base catalysts are constructed, and the structure–activity relationships between the active sites and reaction performance are established through theoretical calculations and analysis, which can assist experimental studies in designing different catalyst configurations for application to specific reaction processes.
(3) Emphasis is placed on the development of basic research work such as high-end characterization. Given that there are electron cloud shifts and charge transfer in solid base catalytic processes, the description of microstructure can better explain the reaction mechanism and structure–activity relationships. The combination of electron cloud theory and characterization at the atomic level can provide theoretical support for the development of new solid base catalysts.
(4) When the reaction does not require a strong base, oxides and zeolites modified with alkali metal can be considered. The solid base with Al2O3 as the carrier is stable in basicity and has good hydrothermal stability, and zeolites have good selectivity for the target products due to their intrinsic ability of selective shape and very high specific surface. Cs can be used to modify the oxides and zeolites when the reactions require superbases, but the cost of modification with Cs is high. Therefore, attention should be paid to the synthesis of solid base catalysts to improve the specific surface area and reduce the cost.
(5) Current studies have shown that solid base catalysts prepared with metal oxides can show a large change in basic strength, but their hydrothermal stability is poor and their specific surface area is low. Meanwhile, although solid base catalysts prepared with zeolites have a larger specific surface area and stronger hydrothermal stability, their basicity enhancement is limited. Therefore, the two components can be combined, which may achieve green conversion of inferior heavy oil, especially in the cracking process, where the fusion of the advantages of the two types of catalysts may be necessary.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Key Research & Development Program of China (grant number 2022YFB3504000) and the Contract Projects of China Petroleum & Chemical Corporation (SINOPEC Corp.) [grant number 123031].Notes and references
- S. Chen, F. Xiong and W. Huang, Surf. Sci. Rep., 2019, 74, 100471 CrossRef CAS.
- R. Lang, X. Du, Y. Huang, X. Jiang, Q. Zhang, Y. Guo, K. Liu, B. Qiao, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 11986–12043 CrossRef CAS.
- Y. Ono and H. Hattori, Solid Base Catalysis, Springer Science & Business Media, Germany, 2012 Search PubMed.
- H. Hattori, Chem. Rev., 1995, 95, 537–558 CrossRef CAS.
- H. Pines and W. Haag, J. Org. Chem., 1958, 23, 328–329 CrossRef CAS.
- L. B. Sun, X. Q. Liu and H. C. Zhou, Chem. Soc. Rev., 2015, 44, 5092–5147 RSC.
- Q. Wu, Pet. Refin. Eng., 2022, 52, 1–7 Search PubMed.
- A. X. He, L. G. Hou, F. Y. Jin, L. Xin and Q. Wu, Pet. Refin. Eng., 2023, 53, 34–38 Search PubMed.
- F. M. Alotaibi, S. González-Cortés, M. F. Alotibi, T. Xiao, H. Al-Megren, G. Yang and P. P. Edwards, Catal. Today, 2018, 317, 86–98 CrossRef CAS.
- T. Wei, M. H. Wang, W. Wei, Y. H. Sun and B. Zhong, Chemistry, 2002, 65, 594–600 CAS.
- X. Z. Li and Q. Jiang, Progress in catalysts for solid base, Low-carbon chemistry and chemical engineering, 2005, vol. 30, pp. 42–48 Search PubMed.
- Y. Yoshimura, N. Kijima, T. Hayakawa, K. Murata, K. Suzuki, F. Mizukami, K. Matano, T. Konishi, T. Oikawa and M. Saito, Catal. Surv. Asia, 2001, 4, 157–167 CrossRef.
- M. S. Gao, G. H. Zhang, L. Zhao, J. S. Gao and C. M. Xu, Ind. Eng. Chem. Res., 2023, 62, 1215–1226 CrossRef CAS.
- M. A. Alabdullah, A. R. Gomez, J. Vittenet, A. Bendjeriou-Sedjerari, W. Xu, I. A. Abba and J. Gascon, ACS Catal., 2020, 10, 8131–8140 CrossRef CAS.
- N. Niwamanya, J. Zhang, C. Gao, D. T. Sekyere, A. Barigye, J. Nangendo and Y. Tian, Fuel, 2022, 328, 125285 CrossRef CAS.
- H. Hattori, Appl. Catal., A, 2015, 504, 103–109 CrossRef CAS.
- H. Pines, Base-catalyzed Reactions of Hydrocarbons and Related Compounds, Academic Press, New York, 2012 Search PubMed.
- H. Hattori, in Studies in Surface Science and Catalysis, Elsevier, 1993, vol. 78, pp. 35–49 Search PubMed.
- K. Tanabe, M. Misono, Y. Ono and H. Hattori, New Solid Acids and Bases: Their Catalytic Properties, Kodansha, Elsevier, Japan, 1989, ch. 2, p. 5 Search PubMed.
- M. Aziz, A. A. Jalil, S. Wongsakulphasatch and D. N. Vo, Catal. Sci. Technol., 2020, 10, 35–45 RSC.
- M. Shirotori, S. Nishimura and K. Ebitani, J. Mater. Chem. A, 2017, 5, 6947–6957 RSC.
- T. C. Keller, K. Desai, S. Mitchell and J. Perez-Ramirez, ACS Catal., 2015, 5, 5388–5396 CrossRef CAS.
- K. Kajihara, N. Tez uka, M. Shoji, J. Wakasugi, H. Munakata and K. Kanamura, Bull. Chem. Soc. Jpn., 2017, 90, 1279–1286 CrossRef CAS.
- J. Dwyer and H. Schofield, NATO ASI Ser., Ser. C, 1994, 444, 13–32 CAS.
- K. Tanabe and W. F. Hölderich, Appl. Catal., A, 1999, 181, 399–434 CrossRef CAS.
- J. C. Védrine, Catalysts, 2017, 7, 341 CrossRef.
- I. Fechete and J. C. Vedrine, Nanotechnology in Catalysis: Applications in the Chemical Industry, Energy Development, and Environment Protection, 2017, pp. 57–90 Search PubMed.
- H. Hattori, K. Maruyama and K. Tanabe, J. Catal., 1976, 44, 50–56 CrossRef CAS.
- M. Mohri, K. Tanabe and H. Hattori, J. Catal., 1974, 32, 144–147 CrossRef CAS.
- M. Bhagiyalakshmi, J. Y. Lee and H. T. Jang, Int. J. Greenhouse Gas Control, 2010, 4, 51–56 CrossRef CAS.
- M. León, L. Faba, E. Díaz, S. Bennici, A. Vega, S. Ordonez and A. Auroux, Appl. Catal., B, 2014, 147, 796–804 CrossRef.
- K. Tanabe, M. Misono, Y. Ono and H. Hattori, in New Solid Acids and Bases, Elsevier, Oxford Amsterdam/New York/Tokyo, 1989, vol. 51 Search PubMed.
- M. Anpo, G. Costentin, E. Giamello, H. Lauron-Pernot and Z. Sojka, J. Catal., 2021, 393, 259–280 CrossRef CAS.
- X. Wang and J. D. Lee, Modell. Simul. Mater. Sci. Eng., 2010, 18, 85010 CrossRef.
- S. Arndt, G. Laugel, S. Levchenko, R. Horn, M. Baerns, M. Scheffler, R. Schlögl and R. Schomäcker, Catal. Rev.: Sci. Eng., 2011, 53, 424–514 CrossRef CAS.
- C. Chizallet, G. Costentin, H. Lauron-Pernot, J. M. Krafft, M. Che, F. O. Delbecq and P. Sautet, J. Phys. Chem. C, 2008, 112, 16629–16637 CrossRef CAS.
- S. Coluccia and A. J. Tench, Stud. Surf. Sci. Catal., 1981, 7, 1154–1169 CrossRef CAS.
- M. Che and A. J. Tench, in Advances in Catalysis, Elsevier, 1982, vol. 31, pp. 77–133 Search PubMed.
- H. Hattori, J. Jpn. Pet. Inst., 2004, 47, 67–81 CrossRef CAS.
- D. Cornu, H. Petitjean, G. Costentin, H. Guesmi, J. Krafft and H. Lauron-Pernot, Phys. Chem. Chem. Phys., 2013, 15, 19870–19878 RSC.
- G. A. Mutch, S. Shulda, A. J. McCue, M. J. Menart, C. V. Ciobanu, C. Ngo, J. A. Anderson, R. M. Richards and D. Vega-Maza, J. Am. Chem. Soc., 2018, 140, 4736–4742 CrossRef CAS.
- C. H. Xue, R. J. Wang and J. Y. Zhang, Acta Pet. Sin., 2002, 30–35 CAS.
- G. Zhang, H. Hattori and K. Tanabe, Bull. Chem. Soc. Jpn., 1989, 62, 2070–2072 CrossRef CAS.
- M. Bailly, C. Chizallet, G. Costentin, J. Krafft, H. Lauron-Pernot and M. Che, J. Catal., 2005, 235, 413–422 CrossRef CAS.
- H. Petitjean, H. Guesmi, H. Lauron-Pernot, G. Costentin, D. Loffreda, P. Sautet and F. Delbecq, ACS Catal., 2014, 4, 4004–4014 CrossRef CAS.
- K. Akutu, H. Kabashima, T. Seki and H. Hattori, Appl. Catal., A, 2003, 247, 65–74 CrossRef CAS.
- H. Hattori, Appl. Catal., A, 2001, 222, 247–259 CrossRef CAS.
- Y. Fukuda, H. Hattori and K. Tanabe, Bull. Chem. Soc. Jpn., 1978, 51, 3150–3153 CrossRef CAS.
- Y. Imizu, K. Sato and H. Hattori, J. Catal., 1982, 76, 65–74 CrossRef CAS.
- M. Utiyama, H. Hattori and K. Tanabe, J. Catal., 1978, 53, 237–242 CrossRef CAS.
- S. C. Shen, X. Chen and S. Kawi, Langmuir, 2004, 20, 9130–9137 CrossRef CAS.
- A. J. Lundeen and R. Vanhoozer, J. Org. Chem., 1967, 32, 3386–3389 CrossRef CAS.
- T. Tomatsu, N. Yoneda and H. Ohtsuka, J. Jpn. Oil Chem. Soc., 1968, 17, 236–245 CrossRef CAS.
- K. M. Minachev, Y. S. Khodakov and V. S. Nakhshunov, J. Catal., 1977, 49, 207–215 CrossRef CAS.
- H. Noller, J. A. Lercher and H. Vinek, Mater. Chem. Phys., 1988, 18, 577–593 CrossRef CAS.
- W. Ueda, T. Yokoyama, Y. Moro-Oka and T. Ikawa, Chem. Lett., 1985, 14, 1059–1062 CrossRef.
- H. Matsuhashi, M. Oikawa and K. Arata, Langmuir, 2000, 16, 8201–8205 CrossRef CAS.
- S. Jayashree and M. Ashokkumar, Catalysts, 2018, 8, 601 CrossRef.
- J. Jupille and G. Thornton, Defects at Oxide Surfaces, Springer, 2015 Search PubMed.
- D. He, D. Chen, H. Hao, J. Yu, J. Liu, J. Lu, F. Liu, G. Wan, S. He and Y. Luo, Appl. Surf. Sci., 2016, 390, 959–967 CrossRef CAS.
- T. Rajesh and R. N. Devi, J. Phys. Chem. C, 2014, 118, 20867–20874 CrossRef CAS.
- Z. Zhang, Y. Wang, J. Lu, J. Zhang, M. Li, X. Liu and F. Wang, ACS Catal., 2018, 8, 2635–2644 CrossRef CAS.
- K. Yu, L. L. Lou, S. Liu and W. Zhou, Adv. Sci., 2020, 7, 1901970 CrossRef CAS.
- K. Yu, D. Lei, Y. Feng, H. Yu, Y. Chang, Y. Wang, Y. Liu, G. Wang, L. Lou and S. Liu, J. Catal., 2018, 365, 292–302 CrossRef CAS.
- L. Qi, Q. Yu, Y. Dai, C. Tang, L. Liu, H. Zhang, F. Gao, L. Dong and Y. Chen, Appl. Catal., B, 2012, 119, 308–320 CrossRef.
- L. Nie, D. Mei, H. Xiong, B. Peng, Z. Ren, X. I. P. Hernandez, A. DeLaRiva, M. Wang, M. H. Engelhard and L. Kovarik, Science, 2017, 358, 1419–1423 CrossRef CAS PubMed.
- J. Shan, F. R. Lucci, J. Liu, M. El-Soda, M. D. Marcinkowski, L. F. Allard, E. C. H. Sykes and M. Flytzani-Stephanopoulos, Surf. Sci., 2016, 650, 121–129 CrossRef CAS.
- K. Otake, Y. Cui, C. T. Buru, Z. Li, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2018, 140, 8652–8656 CrossRef CAS.
- J. Xie, K. Yin, A. Serov, K. Artyushkova, H. N. Pham, X. Sang, R. R. Unocic, P. Atanassov, A. K. Datye and R. J. Davis, ChemSusChem, 2017, 10, 359–362 CrossRef CAS.
- C. Wang, G. Garbarino, L. F. Allard, F. Wilson, G. Busca and M. Flytzani-Stephanopoulos, ACS Catal., 2016, 6, 210–218 CrossRef CAS.
- J. Cho, D. Kwon, I. Yang, S. An and J. C. Jung, Mol. Catal., 2021, 510, 111677 CrossRef CAS.
- S. Hu, W. Wang, M. Yue, G. Wang, W. Gao, R. Cong and T. Yang, ACS Appl. Mater. Interfaces, 2018, 10(18), 15895–15904 CrossRef CAS.
- Y. Yang, D. Wang, P. Jiang, W. Gao, R. Cong and T. Yang, Mol. Catal., 2020, 490, 110914 CrossRef CAS.
- G. Pomalaza, P. A. Ponton, M. Capron and F. Dumeignil, Catal. Sci. Technol., 2020, 10, 4860–4911 RSC.
- T. Yokoyama, T. Setoyama, N. Fujita, M. Nakajima, T. Maki and K. Fujii, Appl. Catal., A, 1992, 88, 149–161 CrossRef CAS.
- J. Wang, W. Yang, C. Wu, Y. Gong, J. Zhang and C. Shen, ACS Sustain. Chem. Eng., 2020, 8, 16960–16967 CrossRef CAS.
- K. Tanabe, H. Hattori, T. Sumiyoshi, K. Tamaru and T. Kondo, J. Catal., 1978, 53, 1–8 CrossRef CAS.
- J. H. Kolts and G. A. Delzer, Science, 1986, 232, 744–746 CrossRef CAS.
- K. K. Pant and D. Kunzru, Chem. Eng. J., 2002, 87, 219–225 CrossRef CAS.
- K. K. Pant and D. Kunzru, Can. J. Chem. Eng., 1999, 77, 150–155 CrossRef CAS.
- S. M. Jeong, J. H. Chae, J. Kang, S. H. Lee and W. Lee, Catal. Today, 2002, 74, 257–264 CrossRef CAS.
- A. A. Lemonidou and I. A. Vasalos, Appl. Catal., 1989, 54, 119–138 CrossRef CAS.
- M. L. Kaliya and S. B. Kogan, Catal. Today, 2005, 106, 95–98 CrossRef CAS.
- K. Narasimharao and F. S. Al-Sultan, Fuel, 2020, 280, 118599 CrossRef CAS.
- C. Boyadjian, L. Lefferts and K. Seshan, Appl. Catal., A, 2010, 372, 167–174 CrossRef CAS.
- M. Nagao, H. Hamano, K. Hirata, R. Kumashiro and Y. Kuroda, Langmuir, 2003, 19, 9201–9209 CrossRef CAS.
- J. Sá, M. Ace, J. J. Delgado, A. Goguet, C. Hardacre and K. Morgan, ChemCatChem, 2011, 3, 394–398 CrossRef.
- F. Cavani, N. Ballarini and A. Cericola, Catal. Today, 2007, 127, 113–131 CrossRef CAS.
- S. M. Jeong, J. H. Chae and W. H. Lee, Ind. Eng. Chem. Res., 2001, 40, 6081–6086 CrossRef CAS.
- P. Lhonore, J. Quibel and M. Senes, US Pat., 3624176A, 1971 Search PubMed.
- A. A. Lemonidou, I. A. Vasalos, E. J. Hirschberg and R. J. Bertolacini, Ind. Eng. Chem. Res., 1989, 28, 524–530 CrossRef CAS.
- B. Basu and D. Kunzru, Ind. Eng. Chem. Res., 1992, 31, 146–155 CrossRef CAS.
- R. Mukhopadhyay and D. Kunzru, Ind. Eng. Chem. Res., 1993, 32, 1914–1920 CrossRef CAS.
- V. A. Kumar, K. K. Pant and D. Kunzru, Appl. Catal., A, 1997, 162, 193–200 CrossRef.
- T. Tomita, K. Kikuchi and T. Sakamoto, US Pat., 3767567A, 1973 Search PubMed.
- Y. Y. Tian, Y. J. Che, M. S. Chen, W. Feng, J. H. Zhang and Y. Y. Qiao, Energy Fuels, 2019, 33, 7297–7304 CrossRef CAS.
- R. Y. Tang, Y. Y. Tian, Y. Y. Qiao, G. M. Zhao and H. F. Zhou, Energy Fuels, 2016, 30, 8855–8862 CrossRef CAS.
- R. Y. Tang, Y. Y. Tian, J. L. Cai and Y. Y. Qiao, Chem. Eng. Oil Gas, 2015, 44, 23–27 CAS.
- R. Y. Tang, M. Yuan, K. Liu, H. F. Li, J. T. Zhang and Y. Y. Tian, J. Energy Inst., 2019, 92, 1936–1943 CrossRef CAS.
- Y. Okamoto, M. Ogawa, A. Maezawa and T. Imanaka, J. Catal., 1988, 112, 427–436 CrossRef CAS.
- J. Weitkamp, M. Hunger and U. Rymsa, Microporous Mesoporous Mater., 2001, 48, 255–270 CrossRef CAS.
- R. J. Davis, J. Catal., 2003, 216, 396–405 CrossRef CAS.
- D. Barthomeuf, J. Phys. Chem., 1984, 88, 42–45 CrossRef CAS.
- D. Barthomeuf, G. Coudurier and J. C. Vedrine, Mater. Chem. Phys., 1988, 18, 553–575 CrossRef CAS.
- R. T. Sanderson, J. Am. Chem. Soc., 1983, 105, 2259–2261 CrossRef CAS.
- W. Gruenert, M. Muhler, K. Schroeder, J. Sauer and R. Schloegl, J. Phys. Chem., 1994, 98, 10920–10929 CrossRef CAS.
- M. Sánchez-Sánchez and T. Blasco, J. Am. Chem. Soc., 2002, 124, 3443–3456 CrossRef PubMed.
- T. Yashima, K. Sato, T. Hayasaka and N. Hara, J. Catal., 1972, 26, 303–312 CrossRef CAS.
- A. Corma and R. M. Martin-Aranda, J. Catal., 1991, 130, 130–137 CrossRef CAS.
- T. Yashima, H. Suzuki and N. Hara, J. Catal., 1974, 33, 486–492 CrossRef CAS.
- A. Philippou, J. Rocha and M. W. Anderson, Catal. Lett., 1999, 57, 151–153 CrossRef CAS.
- J. Shabtai, R. Lazar and E. Biron, J. Mol. Catal., 1984, 27, 35–43 CrossRef CAS.
- A. Corma, V. Fornes, R. M. Martin-Aranda, H. Garcia and J. Primo, Appl. Catal., 1990, 59, 237–248 CrossRef CAS.
- D. Barthomeuf, in Studies in Surface Science and Catalysis, Elsevier, 1997, vol. 105, pp. 1677–1706 Search PubMed.
- A. Corma, R. M. Martin-Aranda and F. Sanchez, J. Catal., 1990, 126, 192–198 CrossRef CAS.
- L. R. Martens, P. J. Grobet and P. A. Jacobs, Nature, 1985, 315, 568–570 CrossRef CAS.
- P. J. Grobet, L. Martens, W. Vermeiren, D. Huybrechts and P. A. Jacobs, in Small Particles and Inorganic Clusters, Springer, 1989, pp. 37–40 Search PubMed.
- S. V. Bordawekar and R. J. Davis, J. Catal., 2000, 189, 79–90 CrossRef CAS.
- E. J. Doskocil and P. J. Mankidy, Appl. Catal., A, 2003, 252, 119–132 CrossRef CAS.
- E. J. Doskocil and R. J. Davis, J. Catal., 1999, 188, 353–364 CrossRef CAS.
- U. Meyer, H. Gorzawski and W. F. Hölderich, Catal. Lett., 1999, 59, 201–206 CrossRef CAS.
- F. Yagi and H. Hattori, Microporous Mater., 1997, 9, 247–251 CrossRef CAS.
- L. Martens, W. Vermeiren, P. J. Grobet and P. A. Jacobs, in Studies in Surface Science and Catalysis, Elsevier, 1987, vol. 31, pp. 531–542 Search PubMed.
- J. C. Kim, H. Li, C. Chen and M. E. Davis, Microporous Mater., 1994, 2, 413–423 CrossRef CAS.
- F. Yagi, N. Kanuka, H. Tsuji, S. Nakata, H. Kita and H. Hattori, Microporous Mater., 1997, 9, 229–235 CrossRef CAS.
- M. Hunger, U. Schenk and J. Weitkamp, J. Mol. Catal. A: Chem., 1998, 134, 97–109 CrossRef CAS.
- F. Yagi, H. Tsuji and H. Hattori, Microporous Mater., 1997, 9, 237–245 CrossRef CAS.
- J. Li and R. J. Davis, Appl. Catal., A, 2003, 239, 59–70 CrossRef CAS.
- J. H. Zhu, Y. Chun, Y. Wang and Q. H. Xu, Catal. Today, 1999, 51, 103–111 CrossRef CAS.
- C. B. Dartt and M. E. Davis, Catal. Today, 1994, 19, 151–186 CrossRef CAS.
- S. Ernst, T. Bongers, C. Casel and S. Munsch, in Studies in Surface Science and Catalysis, Elsevier, 1999, vol. 125, pp. 367–374 Search PubMed.
- K. R. Kloetstra and H. Van Bekkum, in Studies in Surface Science and Catalysis, Elsevier, 1997, vol. 105, pp. 431–438 Search PubMed.
- M. Ziolek, A. Michalska, J. Kujawa and A. Lewandowska, Nanoporous Materials III: Proceedings of the 3rd International Symposium on Nanoporous Materials, Elsevier Science Limited, Ottawa, Ontario, Canada, 2002, vol. 141, pp. 411 Search PubMed.
- R. M. Dessau, Zeolites, 1990, 10, 205–206 CrossRef CAS.
- K. Tanabe, Solid Acids and Bases : Their Catalytic Properties, Academic Press, New York and London, Kodansha, 1970 Search PubMed.
- J. H. Zhu, Y. Wang and Y. Tsutomu, Chin. J. Catal., 1996, 17, 286–290 CrossRef CAS.
- J. H. Zhu, Y. Chun, Y. Wang and M. Tu, Chin. Sci. Bull., 1997, 42, 1118–1119 CrossRef.
- J. H. Zhu, Y. Chun, Y. Wang and Q. H. Xu, Chin. Sci. Bull., 1999, 44, 897–902 Search PubMed.
- J. H. Zhu, H. Tsuji, H. Kabashima, H. Hattori and H. Kita, Chin. Chem. Lett., 1995, 6, 811–814 CAS.
- J. H. Zhu, Y. Chun, Y. Qin and Q. Xu, Microporous Mesoporous Mater., 1998, 24, 19–28 CrossRef CAS.
- Y. Chun, J. H. Zhu and Q. H. Xu, Chin. J. Catal., 1997, 18, 298–301 Search PubMed.
- J. H. Zhu, Y. Wang, Y. Chun and X. S. Wang, J. Chem. Soc., Faraday Trans., 1998, 94, 1163–1169 RSC.
- T. Yamaguchi, J. Zhu, Y. Wang, M. Komatsu and M. Ookawa, Chem. Lett., 1997, 26, 989–990 CrossRef.
- J. H. Zhu and Y. Wang, Chin. J. Catal., 1997, 18, 498–502 CAS.
- Y. Chun, J. H. Zhu, Y. Wang and Q. H. Xu, Chin. Sci. Bull., 1998, 43, 721–724 Search PubMed.
- J. H. Zhu, Y. Chun, Y. Wang and Q. H. Xu, Mater. Lett., 1997, 33, 207–210 CrossRef CAS.
- B. Singh, K. R. Mote, C. S. Gopinath, P. K. Madhu and V. Polshettiwar, Angew. Chem., Int. Ed., 2015, 54, 5985–5989 CrossRef CAS.
- T. Asefa, M. Kruk, N. Coombs, H. Grondey, M. J. MacLachlan, M. Jaroniec and G. A. Ozin, J. Am. Chem. Soc., 2003, 125, 11662–11673 CrossRef CAS.
- B. A. Puértolas, T. C. Keller, S. Mitchell and J. Pérez-Ramírez, Appl. Catal., B, 2016, 184, 77–86 CrossRef.
- Y. M. Zhang, D. P. Yu, W. L. Li, S. Q. Gao and G. W. Xu, Fuel, 2014, 117, 1196–1203 CrossRef CAS.
- A. Corma, E. Corresa, Y. Mathieu, L. Sauvanaud, S. Al-Bogami, M. S. Al-Ghrami and A. Bourane, Catal. Sci. Technol., 2017, 7, 12–46 RSC.
- R. Pujro, M. Falco and U. Sedran, J. Chem. Technol. Biotechnol., 2016, 91, 336–345 CrossRef CAS.
- R. B. Wang, Acta Pet. Sin., 2021, 37, 384–390 CAS.
- Q. Wu, B. Li, C. L. Guo, J. Z. Zang, Z. H. Sun, M. Y. He, J. X. Fan, J. Guo, F. Y. Jin and Y. P. Gong, CN Pat., 113509925A, 2021.
- N. Rahimi and R. Karimzadeh, Appl. Catal., A, 2011, 398, 1–17 CrossRef CAS.
- Q. Wu, J. Z. Zang, B. Li, J. X. Fan, Z. H. Sun, Y. Zhao, J. Guo, W. Zhou, Y. P. Gong and L. W. Hou, CN Pat., 113477247A, 2021.
- Q. Wu, Fuel, 2023, 332, 126132 CrossRef CAS.
- G. Busca, Microporous Mesoporous Mater., 2017, 254, 3–16 CrossRef CAS.
- V. Verdoliva, M. Saviano and S. De Luca, Catalysts, 2019, 9, 248 CrossRef.
Footnote |
| † These two authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |