
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpanding the poly(2-oxazoline) block copolymer possibilities through nitroxide mediated polymerisation†
James
Lefley
and
C. Remzi
Becer
 *
*
Department of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: Remzi.Becer@warwick.ac.uk
First published on 25th September 2024
Abstract
In recent years, poly(2-oxazoline)s (POx) have become a sought-after biomaterial to replace PEG. However, access to POx based block copolymers is rather limited and their combination with controlled radical polymerization (CRP) techniques is required. Herein, we report the combination of cationic ring opening polymerization (CROP) and nitroxide mediated radical polymerization (NMP) to enable block copolymerization of poly(2-oxazoline)s with styrenics, acrylics, 1,3-dienes, and acrylamides as the second block. A well-defined poly(2-ethyl-2-oxazoline) macroinitiator has been prepared via CROP and in situ termination via the carboxylic acid functional group of BlocBuilder alkoxyamine has been achieved with a functionalization efficiency of 78%. Four different monomers in each class have been copolymerized via NMP and gel permeation chromatography analysis allowed us to identify the suitable set of comonomers to be utilized in block copolymerization with POx in an efficient, facile, metal- and sulfur-free polymerization environment.
Introduction
Poly(2-oxazoline)s and their block copolymers are increasingly gaining interest as a major class of polymeric materials. They exhibit excellent water solubility,1 biocompatibility,2 and in vivo stealth properties,3–5 thus, making them highly suitable for biomedical applications. Additional features such as thermoresponsive behavior6,7 and the ability to form self-assembled nanostructures from amphiphilic copolymers8–10 further enhance their appeal. Their facile synthesis via CROP and extensive customizability has established POx as a versatile platform for block copolymer synthesis, allowing numerous combinations in block composition and order to fine tune properties for specific applications.11 Consequently, combining POx with other polymer classes can create novel block copolymers with potentially unique physical and chemical properties for further study.As a result, the combination of CROP with other CRP techniques has garnered significant attention in the past few years. We have recently published a review on the synthetic methods for conjugating POx with vinyl polymers by highlighting the reported combinations of CROP with CRP techniques, such as RAFT and ATRP.12 Of the main synthetic methods used, the macroinitiator route is one of the most common pathways for synthesizing POx-b-vinyl copolymers. In this method, a vinyl polymer is propagated from a POx-based macroinitiator synthesized via CROP and terminated with a compatible CRP initiator. Examples include terminating POx with functional CTAs or using a heterofunctional initiator for chain extension via RAFT.3,13–18 Similarly, terminating POx with a carboxylic acid-functionalized ATRP initiator or using a heterofunctional initiator allows chain extension via ATRP.19–21
Nitroxide mediated polymerization (NMP) is another type of CRP that traditionally offers excellent control over the polymerization of styrenic type monomers. Briefly, thermolysis of a unimolecular initiator yields the initiating radical species and a stable nitroxide radical. Reversible capping and uncapping of the nitroxide radical onto the propagating species affords very good control of the polymerization by limiting irreversible bimolecular termination of the propagating radicals. Seen as one of the first examples of a CRP technique,22 NMP is tolerant to a variety of functionalities, encompasses a broad range of monomers, and requires no metal catalysts or thio-containing CTAs,23 making it a powerful synthetic technique in polymer chemistry.
However, examples of combining CROP and NMP seldom exist in the literature. In 1997, Sogah and coworkers used heterofunctional initiators with NMP, CROP, and AROP initiating functionalities for consecutive polymerizations to synthesize comb polymers.24 A year later, they reported another heterofunctional NMP and CROP initiator for simultaneous controlled polymerizations, producing poly(styrene) (PS) and poly(2-phenyl-2-oxazoline) (PPhOx) block copolymers with high molecular weights (33.5 kDa) with moderate dispersities (Đ = 1.40).25 Nearly a decade later, Wang and Brittain adapted this procedure to create binary mixed homopolymer brushes via simultaneous polymerization of styrene (S) and 2-phenyl-2-oxazoline (PhOx).26 In 2009, the first POx-based NMP macroinitiator was reported by Ibrahim and Voit.27 They used an alkoxyamine chloride (TIPNO-Cl) initiator to synthesize poly(2-methyl-2-oxazoline) (PMeOx) via CROP, followed by chain extension with S via NMP, yielding PMeOx-b-PS diblock copolymers. However, TIPNO-Cl initiation resulted in poor control and broad dispersities for CROP (Đ = 2.1). To address this, Amiel and coworkers synthesized TIPNO-functionalized PMeOx macroinitiators using a more reactive alkoxyamine iodide initiator (TIPNO-I) or by terminating with TIPNO-amine.28 The termination strategy proved to be most successful, producing well-defined PMeOx-b-PS diblock copolymers with narrow molecular weight distributions (Đ = 1.2), though the maximum PS conversion was 23%.
BlocBuilder MA (also known as MAMA-SG1) was developed by Tordo and coworkers around the same time as the TIPNO alkoxyamine and offers similar monomer compatibility.29 The highly labile SG1-based alkoxyamine provides excellent control over styrenic and acrylate monomers with good control over diene30,31 and acrylamide32,33 monomers too. Additionally, BlocBuilder MA has been commercially available and produced on an industrial scale by Arkema since 2005.34 Therefore, its near-universal compatibility, commercial availability, and simple post-polymerization purification process makes NMP with BlocBuilder MA an ideal synthetic technique. Hence, the integration of CROP and NMP polymerization techniques through BlocBuilder MA presents a powerful avenue for the copolymerization of POx with a diverse array of vinylic monomers. This strategy holds promise for producing unique copolymer combinations, particularly by merging the stimuli-responsive properties of various vinyl polymers with the biocompatibility of POx for advanced drug delivery or gene therapy applications. Furthermore, in a biomedical context, utilizing a metal- and sulfur-free polymerization environment for the synthesis of such copolymers presents a significant advantage of using this system compared to other CRP techniques.
Herein, we report the synthesis of a well-defined, SG1-functionalized PEtOx macroinitiator via CROP. Termination of the living POx chain with the carboxylic acid moiety of BlocBuilder MA affords the SG1-terminated PEtOx macroinitiator and was characterized by nucelar magnetic resonance (NMR) and gel permeation chromatography (GPC) analysis. To demonstrate the versatility of the macroinitiator, various styrenic, acrylate, diene, and acrylamide monomers were chain extended using PEtOx-SG1 using fixed reaction conditions for every reaction unless stated otherwise (THF, 2.5M, 110 °C, 24 hours). All diblock copolymers were characterized by NMR and GPC analysis and their purification methods are outlined. The extensive copolymer library produced from a single macroinitiator represents one of the most comprehensive POx macroinitiators for conjugation with vinyl polymers ever reported.
Results and discussion
Synthesis of the PEtOx-SG1 macroinitiator is outlined in Scheme 1. It proceeds via CROP of EtOx followed by termination of the living chain with excess BlocBuilder MA. Optimizing the reaction conditions was crucial to ensure high functionalization efficiency with the alkoxyamine. Initial syntheses were conducted in acetonitrile and chlorobenzene, two solvents known to be effective for CROP of 2-oxazolines.35,36 However, due to the poor solubility of BlocBuilder MA in these solvents, termination efficiencies were low. Therefore, CROP of EtOx was performed in DCM at a lower temperature of 100 °C in a sealed microwave vial. Fig. 1A shows full conversion of EtOx ([M]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[I] = 19) in 90 minutes. Then, 4 equivalents of SG1-MAMA and DIPEA were added to terminate the polymerization and functionalize the ω-chain with the alkoxyamine. GPC analysis of PEtOx-SG1 shows a monomodal distribution with a narrow dispersity of 1.12 (Fig. 1B). The theoretical and obtained molecular weights for PEtOx-SG1 are in very good agreement with each other (Mn, theor = 2178.67 Da vs. Mn, GPC = 2200 Da) thus confirming DCM to be a suitable solvent for CROP of EtOx. 1H NMR spectroscopic analysis revealed a functionalization efficiency of 78% by comparing the integrals of the methyl –CH3 protons of the α-chain end and the ethoxy –CH3 protons of SG1 end group (Fig. 1A). The steric hinderance of the bulky terminating agent is ascribed to non-complete functionalization of the PEtOx chain. MALDI-ToF MS analysis was conducted to determine end group fidelity of PEtOx-SG1 but unfortunately none of the multiple distributions could be assigned due to the spontaneous dissociation of the nitroxide end group in mass spectrometry.37–39
:[I] = 19) in 90 minutes. Then, 4 equivalents of SG1-MAMA and DIPEA were added to terminate the polymerization and functionalize the ω-chain with the alkoxyamine. GPC analysis of PEtOx-SG1 shows a monomodal distribution with a narrow dispersity of 1.12 (Fig. 1B). The theoretical and obtained molecular weights for PEtOx-SG1 are in very good agreement with each other (Mn, theor = 2178.67 Da vs. Mn, GPC = 2200 Da) thus confirming DCM to be a suitable solvent for CROP of EtOx. 1H NMR spectroscopic analysis revealed a functionalization efficiency of 78% by comparing the integrals of the methyl –CH3 protons of the α-chain end and the ethoxy –CH3 protons of SG1 end group (Fig. 1A). The steric hinderance of the bulky terminating agent is ascribed to non-complete functionalization of the PEtOx chain. MALDI-ToF MS analysis was conducted to determine end group fidelity of PEtOx-SG1 but unfortunately none of the multiple distributions could be assigned due to the spontaneous dissociation of the nitroxide end group in mass spectrometry.37–39
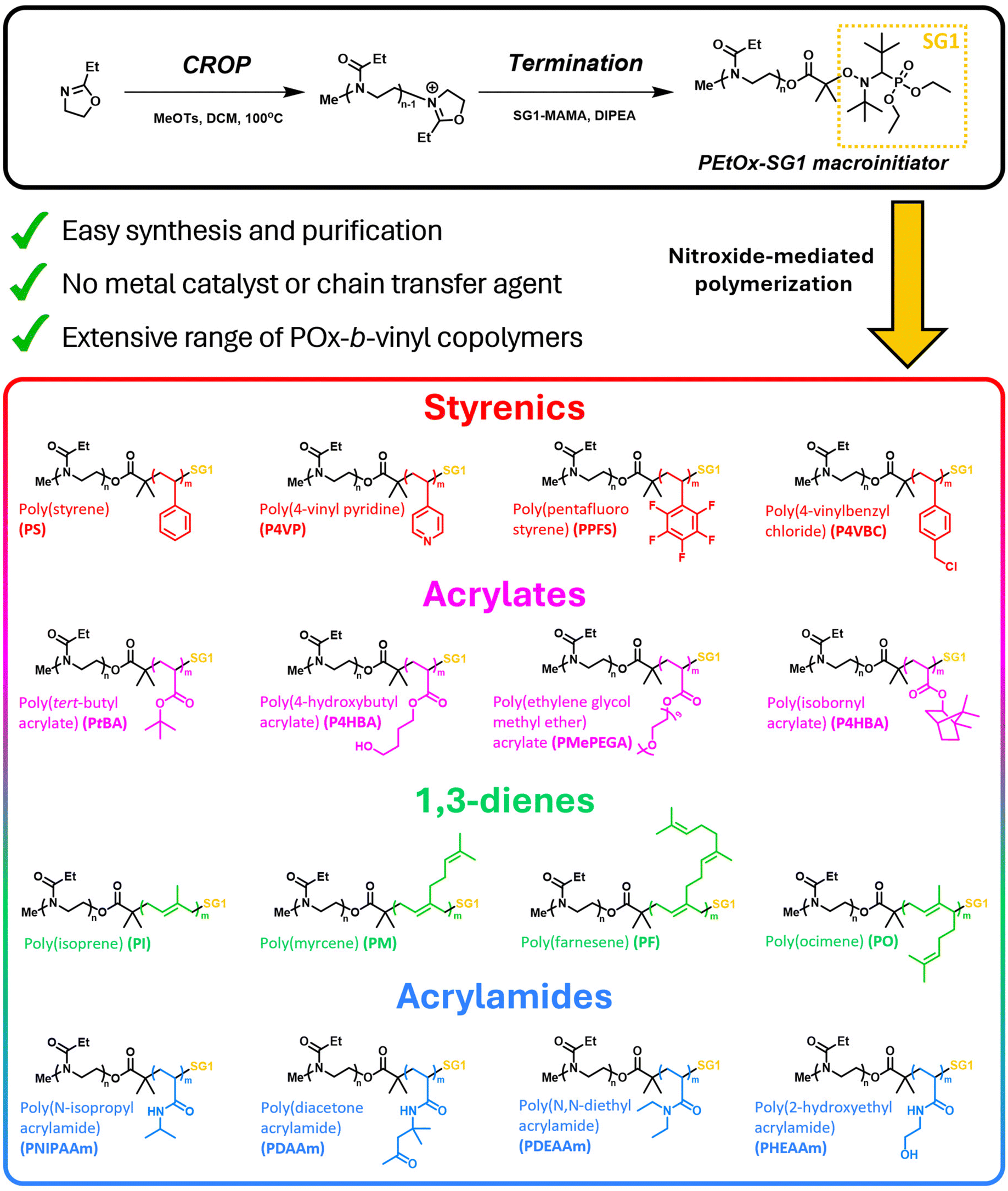 | ||
| Scheme 1 Synthesis of the PEtOx-SG1 macroinitiator and the diblock copolymers formed via nitroxide mediated polymerization with a variety of styrenic, acrylate, diene and acrylamide monomers. | ||
 | ||
| Fig. 1 (A) 1H NMR spectra of the reaction mixture before polymerization (t0 min), at 90 minutes showing full conversion of EtOx (t90 min) and the purified SG1-functionalized macroinitiator (purified). (B) GPC trace of the purified PEtOx-SG1. Measurement performed using THF (2% TEA and 0.01% BHT) as the eluent. PMMA standards were used for the calibration. | ||
Due to the versatility of BlocBuilder MA, a wide range of monomers can be polymerized in a controlled manner. Therefore, to demonstrate the compatibility of PEtOx-SG1, we selected four monomers from styrenic, acrylate, diene, and acrylamide monomer classes producing a 16-copolymer library. The conjugation of poly(2-oxazoline)s with styrenics, acrylates, and acrylamides have been previously reported. However, to the best of our knowledge, conjugation with dienes has not been documented and offers a unique new type of block copolymer combination, potentially opening the field to POxylated rubbers for medical implants or devices.
All polymerizations targeted a 100:1 monomer-to-macroinitiator ratio, producing block copolymers with large vinyl polymer mass fractions to simplify purification via precipitation into a non-solvent. For block copolymers where precipitation was unfeasible, dialysis or liquid–liquid extraction was utilized. To promptly screen suitable monomers, all polymerizations were conducted in THF at 110 °C for 24 hours with a 2.5M monomer concentration unless stated otherwise. Optimizing reaction conditions was beyond the scope of this study and will be addressed in future work in individual copolymer studies. Full characterization data of the copolymer library can be found in Table 1. GPC traces are shown in Fig. 2 and 1H NMR analysis of all copolymers can be found in the ESI (Fig. S1–S4†).
 | ||
| Fig. 2 GPC traces of PEtOx-styrenic diblock copolymers (A1–A4), PEtOx-acrylate diblock copolymers (B1–B4), PEtOx-diene diblock copolymers (C1–C4) and PEtOx-acrylamide diblock copolymers (D1–D4) synthesized via NMP. All samples ran using THF (2% TEA, 0.01% BHT) as the eluent apart from P4VP (A2), P4HBA (B4) and PHEAAm (D4) which were ran using DMF (5 mM NH4BF4) as the eluent. PMMA standards were used for the calibration. | ||
| Polymer | Conc. [M] | [M]:[PEtOx-SG1]a |
Conv.a (%) | DPtheob |
DPNMRa |
M
n, theoc (kDa) |
M n, GPC (kDa) | Đ |
|---|---|---|---|---|---|---|---|---|
|
a Determined by 1H NMR.
b Assuming 78% functionalization efficiency – calculated by ([M]:[PEtOx-SG1] × Conversion)/0.78.
c Calculated using DPtheor.
d Determined by THF GPC (2% TEA + 0.01% BHT).
e Determined by DMF GPC (5 mM NH4BF4).
f Unable to determine due to peak overlap in NMR.
|
||||||||
| PS | 2.5 | 102 | 86 | 112 | 142 | 13.9 | 15.6d | 1.18d |
| P4VP | 2.5 | 110 | 79 | 112 | 84 | 14.0 | 21.7e | 1.09e |
| PPFS | 2.5 | 106 | 91 | 123 | — | 26.0 | 18.2d | 1.14d |
| P4VBC | 2.5 | 110 | 73 | 103 | 140 | 17.9 | 17.0d | 1.72d |
| PtBA | 2.5 | 113 | 77 | 112 | 92 | 16.5 | 6.6d | 1.31d |
| PIBOA | 2.0 | 114 | 98 | 144 | 99 | 32.0 | 21.2d | 1.22d |
| PMePEGA | 1.0 | 50 | 70 | 45 | 33 | 23.8 | 7.4d | 1.35d |
| P4HBA | 2.0 | 185 | 95 | 225 | 244 | 34.6 | 21.5e | 1.94e |
| PI | 2.5 | 70 | 99 | 88 | 7 | 8.2 | 3.4d | 1.22d |
| PM | 2.5 | 97 | 75 | 94 | 65 | 15.0 | 11.1d | 1.21d |
| PO | 2.5 | 2.9d | 1.28d | |||||
| PF | 2.5 | 2.8d | 1.31d | |||||
| PNIPAAm | 2.5 | 111 | 28% | 40 | 38 | 6.7 | 5.5d | 1.30d |
| PDAAm | 2.5 | 97 | >99% | 124 | 23.1 | 14.9d | 2.41d | |
| PDEAAm | 2.5 | 116 | 92% | 137 | 19.6 | 9.6d | 1.32d | |
| PHEAAm | 1.5M | 107 | >99% | 137 | 18.0 | 12.4e | 1.69e | |
NMP is highly suitable for controlled polymerization of styrenic monomers, offering narrow dispersities and precise molecular weight control. Chain extension of PEtOx-SG1 with S, 4VP, and PFS resulted in narrow dispersities (Đ = 1.09–1.18) for PS, P4VP and PPFS (Fig. 2A1–3). Previous reports on POx-b-PS copolymers include Becer et al. who used a heterofunctional CROP/ATRP initiator for PEtOx and PS conjugation, yielding diblock copolymers with Đ = 1.27.21 Our approach using PEtOx-based NMP macroinitiators achieved better control, producing diblock copolymers with a narrower dispersity (Đ = 1.18). These findings align with other POx-b-PS systems synthesized via termination and chain extension using functional CRP initiators such as RAFT13 and NMP.28 Recently, de Menezes and Felisberti synthesized PEtOx-b-P4VP block copolymers via ATRP using a PEtOx macroinitiator. Despite achieving average termination efficiencies (38–54%), narrowly disperse block copolymers were synthesized (Đ = 1.20).19 In this study, the PEtOx-SG1 synthesis achieved a higher functionalization efficiency (78%), producing a PEtOx-b-P4VP diblock copolymer with superior dispersity (Đ = 1.09) and, consequently, less unfunctionalized PEtOx homopolymer remaining post polymerization. Although, a high molecular weight shoulder can be seen in the GPC trace of P4VP (Fig. 2A2), indicating some chain coupling reactions. A broader dispersity (Đ = 1.72) was observed for P4VBC (Fig. 2A4) indicating a loss of control of the polymerization. Under the set conditions, NMP of PFS with PEtOx-SG1 showed the highest monomer conversion (91%) among the four styrenic monomers. S and 4VP also achieved high conversions (79% and 86% respectively), while 4VBC had the lowest conversion (73%) after 24 hours. The large mass fraction of the styrenic block in these copolymers allows for easy removal of the unfunctionalized PEtOx homopolymer via precipitation into cold methanol or acetonitrile (Table S1†).
The development of the SG1 alkoxyamine in the early 2000s enabled controlled polymerization of acrylic monomers via NMP.29 Chain extension of PEtOx-SG1 with tBA, IBOA, and MePEGA yielded block copolymers PtBA, PIBOA, and PMePEGA with fairly narrow dispersities (Đ = 1.22–1.35) (Fig. 2B1–3). Chain extension of tBA via NMP using PEtOx-SG1 showed poorer control compared to RAFT using a trithiocarbonate functionalized PEtOx macroinitiator (Đ = 1.31 vs. Đ = 1.19) reported by Krieg et al.13 However, PMePEGA synthesized via NMP using PEtOx-SG1 had similar dispersities to PEtOx-b-PeTEGA copolymers synthesized via RAFT14 (Đ = 1.35 vs. Đ = 1.30), indicating comparable control for polymerizing oligo/polyethylene glycol acrylic monomers using RAFT and NMP functional POx macroinitiators. A high molecular weight shoulder observed for PIBOA (Fig. 2B2) indicates the presence of some chain coupling reactions. NMP of 4HBA produced P4HBA diblock copolymer with a broad dispersity (Đ = 1.94), indicating poor control and loss of polymerization livingness (Fig. 2B4). Like styrenic monomers, the propagation rates for acrylates using PEtOx-SG1 are generally fast, with high conversions (70–98%) achieved after 24 hours at 110 °C.
The versatility of the SG1 alkoxyamine is further demonstrated by its control over the polymerization of diene monomers. NMP of isoprene and β-myrcene with the PEtOx-SG1 macroinitiator produced narrowly disperse diblock copolymers PI and PM, with dispersity values Đ = 1.22 and Đ = 1.21 respectively. However, PI showed a significant discrepancy between theoretical DP (DP = 88) and NMR DP (DP = 7) (Table 1), likely due to the low boiling point of isoprene (34 °C) causing monomer loss during N2 purging and/or condensation of the monomer during the reaction. The GPC chromatogram of PI (Fig. 2C1) shows a small molecular weight shift, suggesting a low DP for the poly(isoprene) block. While PM (Fig. 2C2) shows a clear shift, indicating successful chain extension with high conversion (75%). The large poly(myrcene) block allowed easy purification via precipitation into acetonitrile (Table S1†). A key feature of poly(dienes) is the stereochemistry of the polymer backbone, yielding 1,4-, 3,4-, and 1,2-products. For radical polymerization, the 1,4-addition is predominantly favoured. Analysis of Fig. S5B† reveals PM consists of 92% of the 1,4-product, 6% of the 3,4-product, and 2% of the 1,2-product, which corroborates nicely with similar CRP studies of β-myrcene.40–42 The presence of a small percentage of pendant alkenyl groups in PM holds potential for further functionalization via thiol–ene click chemistry. Ocimene, an isomer of β-myrcene, was also polymerized with the PEtOx-SG1 macroinitiator but showed less successful propagation compared to β-myrcene (Fig. 2C3). The GPC chromatogram of the PO diblock copolymer closely matches the PEtOx-SG1 macroinitiator with a slight high molecular weight shoulder, suggesting that partial chain extension to oligomeric lengths was achieved within 24 hours. This slow propagation is likely attributed to the stability of the propagating radical. Ocimene produces a more stable secondary radical compared to the primary radical of β-myrcene, resulting in much slower propagation rates (Fig. S6†). The final diene monomer, farnesene, exhibited similar reactivity to ocimene. The GPC chromatogram of PF (Fig. 2C4) shows minimal peak shift, with only a small high molecular weight shoulder after 24 hours, indicating very poor conversion. This low conversion is likely due to the monomer's composition, which includes a mixture of farnesene isomers. Only β-farnesene, the primary propagating species, participates in the radical polymerization, while the other isomers do not or do so incredibly slowly. Further optimization of the reaction conditions would be necessary to achieve successful polymerization of isoprene, ocimene and farnesene.
Lastly, to showcase the extensive compatibility of the PEtOx-SG1 macroinitiator with a variety of monomers classes, acrylamides NIPAAm, DAAm, DEAAm, and HEAAm were polymerized via NMP. These block copolymer combinations could form the basis of a further study into their potentially interesting thermoresponsive solution behaviours. Chain extension of PEtOx-SG1 with these acrylamide monomers yielded varied results in overall control. Fig. 2D1 and D3 show that PNIPAAm and PDEAAm produced monomodal molecular weight distributions with moderate dispersities (Đ = 1.30 and Đ = 1.32, respectively). Although the polymerization was relatively controlled, NIPAAm conversion was surprisingly low (28%) after 24 hours, compared to the near-complete conversion for DEAAm (Table 1). Historically, it has been challenging to synthesize NIPAAm copolymers via NMP, often yielding broad molecular weight dispersities.43,44 Zetterlund et al. found that NMP of NIPAAm in DMF was hindered by chain transfer to solvent, limiting molar mass.45 The low NIPAAm conversion here may be due to chain transfer to THF. In contrast, using a RAFT functionalized PEtOx macroinitiator achieves greater control over the polymerization of NIPAAm.14 With very narrow dispersities (Đ < 1.10) obtained for PEtOx-b-PNIPAAm copolymers synthesized via RAFT, compared to NMP (Đ = 1.30). NMP of DAAm using the PEtOx-SG1 macroinitiator yielded PDAAm with a very broad dispersity (Đ = 2.41) (Fig. 2D2), indicating poor control over the polymerization. Despite this, full conversion was achieved within 24 hours, indicating a faster propagation rate compared to NIPAAm and DEAAm. Similarly, NMP of HEAAm showed a substantial lack of control, yielding a PHEAAm diblock copolymer with a broad dispersity (Đ = 1.69) but complete monomer conversion within 24 hours (Fig. 2D4). Due to the double hydrophilic nature of PNIPAAm, PDEAAm, and PHEAAm, dialysis was used to remove residual PEtOx homopolymer as their different solubilities could not be exploited. In contrast, PDAAm was sufficiently hydrophobic enough to be purified via DCM/H2O extraction (Table S1†).
Scheme 2 summarizes the current literature on POx-based macroinitiators functionalized with a CRP initiator. While the combination of CROP with RAFT remains the most extensively studied pairing of CROP with a CRP technique, we aim to demonstrate that combining CROP with NMP, using an SG1-functionalized POx macroinitiator, can offer a similarly versatile platform. This approach is compatible with a wide range of monomer types and has the added advantage of being carried out in a metal- and sulfur-free polymerization environment.
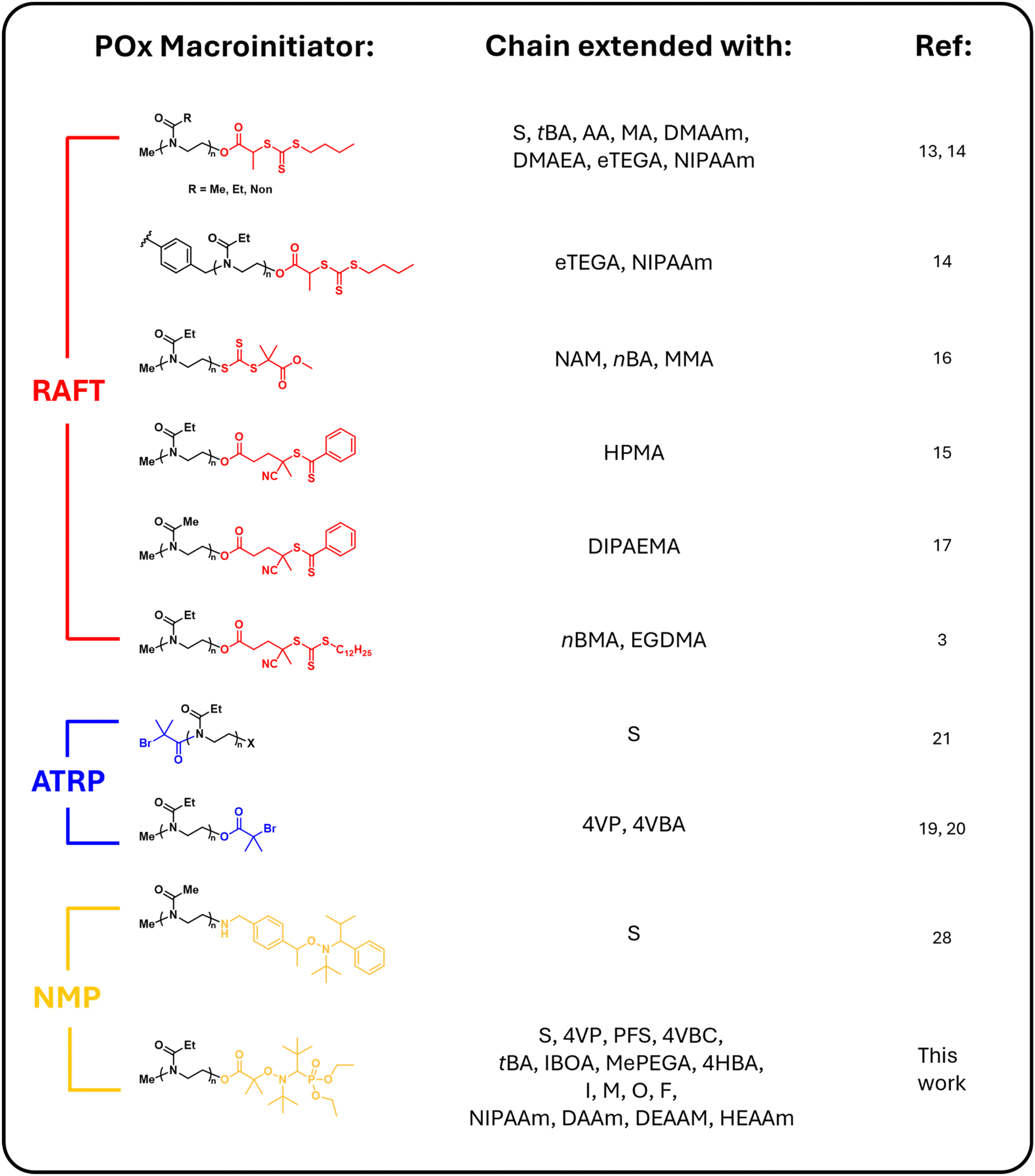 | ||
| Scheme 2 Overview of current POx-based macroinitiators used for combining CROP with CRP techniques such as RAFT, ATRP, and NMP. | ||
Conclusion
In conclusion, we have synthesized a well-defined PEtOx-based macroinitiator bearing an SG1 functional group for nitroxide-mediated polymerization. Chain extension with various styrenic, acrylate, diene, and acrylamide monomers demonstrated the versatility of this macroinitiator for metal catalyst-free and CTA-free CRP. Styrenic and acrylate monomers showed the highest compatibility with PEtOx-SG1, characterized by low dispersities and high conversions. Unique combinations of PPFS, PM, and PDEAAm with PEtOx yielded well-defined copolymers with narrow dispersities, offering the potential for novel future studies. This work may pave the way for future in-depth studies into new POx-b-vinyl diblock copolymers, with a focus on optimizing their synthesis and investigating their unique properties.Author contributions
JL performed all experimental work. CRB has supervised the work and supported the writing and editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- K. Mint, J. P. Morrow, N. M. Warne, X. He, D. Pizzi, S. Z. O. Shah, G. K. Pierens, N. Fletcher, C. Bell, K. J. Thurecht and K. Kempe, Polym. Chem., 2024, 15, 2662–2676 RSC.
- J. Kronek, Z. Kroneková, J. Lustoň, E. Paulovičová, L. Paulovičová and B. Mendrek, J. Mater. Sci.: Mater. Med., 2011, 22, 1725–1734 CrossRef CAS PubMed.
- M. N. Leiske, M. Lai, T. Amarasena, T. P. Davis, K. J. Thurecht, S. J. Kent and K. Kempe, Biomaterials, 2021, 274, 120843 CrossRef CAS PubMed.
- J. R. Finnegan, E. H. Pilkington, K. Alt, M. A. Rahim, S. J. Kent, T. P. Davis and K. Kempe, Chem. Sci., 2021, 12, 7350–7360 RSC.
- I. Muljajew, S. Huschke, A. Ramoji, Z. Cseresnyés, S. Hoeppener, I. Nischang, W. Foo, J. Popp, M. T. Figge, C. Weber, M. Bauer, U. S. Schubert and A. T. Press, ACS Nano, 2021, 15, 12298–12313 CrossRef CAS PubMed.
- R. Hoogenboom and H. Schlaad, Polym. Chem., 2017, 8, 24–40 RSC.
- R. Hoogenboom, H. M. L. Thijs, M. J. H. C. Jochems, B. M. van Lankvelt, M. W. M. Fijten and U. S. Schubert, Chem. Commun., 2008, 5758–5760, 10.1039/B813140F.
- T. Zhao, B. Drain, G. Yilmaz and C. R. Becer, Polym. Chem., 2021, 12, 6392–6403 RSC.
- G. Hayes, B. Drain, J. Lefley and C. R. Becer, Macromolecules, 2023, 56, 271–280 CrossRef CAS.
- J. Lefley, R. Terracciano, Z. Varanaraja, J. Beament and C. R. Becer, Macromolecules, 2024, 57(12), 5881–5891 CrossRef CAS.
- B. Verbraeken, B. D. Monnery, K. Lava and R. Hoogenboom, Eur. Polym. J., 2017, 88, 451–469 CrossRef CAS.
- B. A. Drain and C. R. Becer, Eur. Polym. J., 2019, 119, 344–351 CrossRef CAS.
- A. Krieg, C. Weber, R. Hoogenboom, C. R. Becer and U. S. Schubert, ACS Macro Lett., 2012, 1, 776–779 CrossRef CAS PubMed.
- M. Sahn, L. M. Stafast, M. Dirauf, D. Bandelli, C. Weber and U. S. Schubert, Eur. Polym. J., 2018, 100, 57–66 CrossRef CAS.
- D. Le, F. Wagner, M. Takamiya, I. L. Hsiao, G. Gil Alvaradejo, U. Strähle, C. Weiss and G. Delaittre, Chem. Commun., 2019, 55, 3741–3744 RSC.
- A.-K. Trützschler, M. N. Leiske, M. Strumpf, J. C. Brendel and U. S. Schubert, Macromol. Rapid Commun., 2019, 40, 1800398 CrossRef PubMed.
- P. Černoch, A. Jager, Z. Černochová, V. Sincari, L. J. C. Albuquerque, R. Konefal, E. Pavlova, F. C. Giacomelli and E. Jager, Polym. Chem., 2021, 12, 2868–2880 RSC.
- Y. C. Yu, H. S. Cho, W.-R. Yu and J. H. Youk, Polymer, 2014, 55, 5986–5990 CrossRef CAS.
- R. N. L. d. Menezes and M. I. Felisberti, Polym. Chem., 2021, 12, 4680–4695 RSC.
- R. N. L. de Menezes, W. Loh and M. I. Felisberti, Eur. Polym. J., 2023, 195, 112208 CrossRef CAS.
- C. R. Becer, R. M. Paulus, S. Höppener, R. Hoogenboom, C.-A. Fustin, J.-F. Gohy and U. S. Schubert, Macromolecules, 2008, 41, 5210–5215 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- R. D. Puts and D. Y. Sogah, Macromolecules, 1997, 30, 7050–7055 CrossRef CAS.
- M. W. Weimer, O. A. Scherman and D. Y. Sogah, Macromolecules, 1998, 31, 8425–8428 CrossRef CAS.
- Y. Wang and W. J. Brittain, Macromol. Rapid Commun., 2007, 28, 811–815 CrossRef CAS.
- S. Ibrahim and B. Voit, Macromol. Symp., 2009, 275–276, 59–66 CrossRef CAS.
- L. Marx, G. Volet and C. Amiel, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4785–4793 CrossRef CAS.
- D. Benoit, S. Grimaldi, S. Robin, J.-P. Finet, P. Tordo and Y. Gnanou, J. Am. Chem. Soc., 2000, 122, 5929–5939 CrossRef CAS.
- S. Harrisson, P. Couvreur and J. Nicolas, Macromolecules, 2011, 44, 9230–9238 CrossRef CAS.
- A. Métafiot, L. Gagnon, S. Pruvost, P. Hubert, J.-F. Gérard, B. Defoort and M. Marić, RSC Adv., 2019, 9, 3377–3395 RSC.
- T. Diaz, A. Fischer, A. Jonquières, A. Brembilla and P. Lochon, Macromolecules, 2003, 36, 2235–2241 CrossRef CAS.
- K. Schierholz, M. Givehchi, P. Fabre, F. Nallet, E. Papon, O. Guerret and Y. Gnanou, Macromolecules, 2003, 36, 5995–5999 CrossRef CAS.
- J.-L. Couturier, O. Guerret, D. Bertin, D. Gigmes, S. Marque, P. Tordo, F. Chauvin and P.-E. Dufils, France Pat., 2004/014926, 2004 Search PubMed.
- F. Wiesbrock, R. Hoogenboom, M. A. M. Leenen, M. A. R. Meier and U. S. Schubert, Macromolecules, 2005, 38, 5025–5034 CrossRef CAS.
- Q. Liu, M. Konas and J. S. Riffle, Macromolecules, 1993, 26, 5572–5576 CrossRef CAS.
- M. Girod, T. N. T. Phan and L. Charles, J. Am. Soc. Mass Spectrom., 2008, 19, 1163–1175 CrossRef CAS PubMed.
- M. Mazarin, M. Girod, S. Viel, T. N. T. Phan, S. R. A. Marque, S. Humbel and L. Charles, Macromolecules, 2009, 42, 1849–1859 CrossRef CAS.
- C. Barrère, C. Chendo, T. N. T. Phan, V. Monnier, T. Trimaille, S. Humbel, S. Viel, D. Gigmes and L. Charles, Chem. – Eur. J., 2012, 18, 7916–7924 CrossRef PubMed.
- N. Bauer, J. Brunke and G. Kali, ACS Sustainable Chem. Eng., 2017, 5, 10084–10092 CrossRef CAS.
- J. Zhang, C. Aydogan, G. Patias, T. Smith, L. Al-Shok, H. Liu, A. M. Eissa and D. M. Haddleton, ACS Sustainable Chem. Eng., 2022, 10, 9654–9664 CrossRef CAS PubMed.
- G. Moad, Polym. Int., 2017, 66, 26–41 CrossRef CAS.
- E. N. Savariar and S. Thayumanavan, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 6340–6345 CrossRef CAS.
- K. Kuroda and T. M. Swager, Macromolecules, 2004, 37, 716–724 CrossRef CAS.
- Y. Sugihara, P. O'connor, P. B. Zetterlund and F. Aldabbagh, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1856–1864 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR spectra, purification techniques for each block copolymer. See DOI: https://doi.org/10.1039/d4py00887a |
| This journal is © The Royal Society of Chemistry 2024 |