DOI:
10.1039/D4DT02417F
(Perspective)
Dalton Trans., 2024,
53, 18400-18419
Revitalisation of group IV metal-oxo clusters: synthetic approaches, structural motifs and applications
Received
25th August 2024
, Accepted 15th October 2024
First published on 22nd October 2024
Abstract
Group (IV) metal oxo clusters represent a unique family of molecular species that are increasingly being utilized in applications ranging from catalysis and materials chemistry to electronics, and sensors. These clusters exhibit distinctive structural features, chemical reactivity, and electronic structure. Nevertheless, their full potential has yet to be fully realized due to the lack of deeper understanding regarding their structure and formation mechanisms, inherent traits, and intricacies in their design, which could ultimately enable significant customization of their properties and overall behaviour. Considering the recently observed reignited interest in the chemistry of group IV molecular species, the scope of this article is to bring to the readers the main chemical characteristics of the family of titanium, zirconium, and hafnium-based clusters, their structural features and their potential in future applications.
 Stamatis S. Passadis | Stamatis S. Passadis received his Bachelor's degree in Chemistry from the University of Ioannina, Greece, in 2015. He completed his Ph.D. in Chemistry at the University of Ioannina in 2022 under the supervision of Professor Themistoklis Kabanos. Since 2023, he has been a postdoctoral fellow at KU Leuven, working in the Laboratory of Bioinorganic Chemistry (LBC) group led by Professor Tatjana N. Parac-Vogt. His research focuses on the synthesis and characterization of metal-oxo clusters for photocatalytic applications. |
 Mark Gray | Mark completed his undergraduate studies in chemistry at the University of Glasgow in 2019, before going on to complete a research Masters in molecular magnetism under Prof. Euan Brechin at the University of Edinburgh. Upon completion of his masters, he then returned to the University of Glasgow in 2021 to undertake his PhD with Prof. Haralampos Miras in electro- and photochemical catalysis of inorganic clusters. |
 Tatjana N. Parac-Vogt | Tatjana N. Parac-Vogt is a full Professor of Chemistry and head of the Laboratory of Bioinorganic Chemistry at KU Leuven. She leads an interdisciplinary team performing research at the interface of inorganic chemistry, biochemistry, materials science, and catalysis. She is the recipient of the IUPAC 2023 Distinguished Women in Chemistry/Chemical Engineering award. Tatjana is also a Fellow of the Royal Society of Chemistry and has been elected as a Chemistry Europe Fellow (Class 2020/2021), the highest award given by an association of European chemical societies. |
 Anastasios D. Keramidas | Anastasios Keramidas studied Chemistry at the University of Ioannina and received his Ph.D. in 1992. He worked as a Post-Doc. fellow at the University of Colorado, USA, from 1994 up to 1997. Now he is Professor at the University of Cyprus, Cyprus. His research is mainly focused on the synthesis and the electrochemical and spectrochemical solid-state and solution characterization of early transition metal and lanthanide–actinide metal complexes. He is specialized in the investigation of the equilibria and dynamic phenomena of chemical reactions with applications in bioinorganic chemistry and catalysis. |
 Haralampos N. Miras | Haralampos N. Miras (FRSC, FHEA) is Professor in Inorganic and Supramolecular Chemistry at the University of Glasgow in UK. He obtained his PhD in 2005 (Ioannina, Greece) and subsequently held research fellowships in the United States and Glasgow from 2005 to 2010. In 2010, he received a prestigious five-year personal fellowship from the Royal Society of Edinburgh. He was a finalist in the European Young Chemist Award competition in 2012 and secured a lectureship at Glasgow in 2013 and promoted to Professor in 2023. Prof. Miras’ research is centred on the exploration of self-assembly and emergent behaviours within supramolecular chemical systems, as well as the elucidation of underlying mechanisms and the design of molecule-based catalytic materials for energy applications. |
 Themistoklis A. Kabanos | Themistoklis A. Kabanos received his bachelor's degree in chemistry, from the University of Thessaloniki, in 1976 and his Ph.D. from the Chemistry Department, and University of Ioannina, in 1983. He was a postdoctoral fellow in the Chemistry Department at the University of Queensland, from 1984 to 1987. After postdoctoral research at Queensland, he joined University of Ioannina as a Lecturer in 1988. In 2000 he promoted to a full Professor. He is currently a Professor Emeritus at the University of Ioannina. His recent research interests focus on the synthesis and physicochemical characterization of vanadium, uranium and group IV compounds with potential applications for environmental and energy-related applications. |
1. Introduction
Metal oxo clusters (MOCs) are a diverse class of molecules constructed by group III–VI metals in their highest oxidation state, connected by oxygen atoms and frequently stabilized by capping ligands. Compared to nanoparticles and 2D metal oxide materials, these molecular species exhibit two main advantages. Firstly, the detailed structural information of these crystalline materials can be revealed using X-ray techniques, which provide precise structural information and are crucial for the deeper understanding and elucidation of their formation mechanism and their mode of function in various applications. The rapid and vast improvements in single crystal data collection processes and instrumentation that have been observed in the last decades were a crucial factor for the development of new multinuclear structural motifs, allowing the research groups to not only focus on the design of new clusters, but to start shifting the focus towards the engineering of more complex molecular systems with multiple functionalities and tailored properties for specific applications. As a result, molecular metal oxides can be used as excellent models for investigating the surface chemistry of related 2D materials. They have been used to probe reaction pathways and structural intricacies related to doping processes or generation of appropriate vacancies that are crucial for the underlying processes of metal oxide-based catalysts. Secondly, solution processable metallic cores stabilized by various ligands which are soluble in organic solvents, and are appropriate for recrystallization and post-functionalization, can serve as building blocks for the bottom-up assembly of a variety of novel functional materials and engineering of complex multinuclear architectures giving rise to new properties and phenomena. Therefore, understanding the formation, structure characterization, and solution chemistry of MOCs is crucial for advancing molecule-based metal oxide materials and catalytic systems. Metal oxo-clusters garner a lot of attention due to their unique and interesting electronic properties, which can lead to different applications including catalysis, sensing, medicine and other industries.1,2 In marked contrast to polyoxometalates (POMs) which are a well-studied and diverse series of clusters based primarily on group V & VI metals in their highest oxidation state, group IV clusters are comparatively less explored. One key contrasting property between Ti, Zr and Hf clusters and other common molecular metal oxides is the absence of a terminal-oxo bond. The terminal oxo bond seen in species such as molybdate, tungstate and vanadate is conducive to the formation of discrete clusters as it leads to a distortion in geometry of the metal centre, in which the metal centre will adopt a “bent” geometry when condensing with other metal centres to form spherical or toroidal species. This absence in group IV metals encourages the formation of 2- and 3-dimensional polymers, and thus, careful choice of ligands and capping moieties is required. Organometallic species of Zr and Hf can serve to alleviate this to some extent, as species such as (Cp)2ZrCl2 and (Cp)HfCl3 (Cp− = cyclopentadienyl) have been shown to form molecular clusters due to the capping η5-cyclopentadienyl ligands preventing extensive growth in 3 dimensions (vide infra). Group IV metal–organic compounds (MOCs) have the tendency to form a number of neutral species in the presence of appropriate ligands. This characteristic influences their solubility and subsequent solution processability in marked contrast to other multinuclear clusters such as polyoxometalates (POMs).3 The photochemical properties of these metals are particularly intriguing, with some group IV group MOCs potentially being direct band gap semiconductors, unlike their bulk counterparts such as anatase, ZrO2 and HfO2, which typically have indirect band gaps.3,4 ZrO2 and HfO2 have similar bandgaps (5 eV for ZrO2, and 5.3–5.7 eV for HfO2) which are much larger than that of TiO2 (∼3.2 eV).5,6 By controlling speciation and nuclearities precisely, new Lewis acid catalysts and photocatalytic materials for various reactions could be created, including the degradation of organic pollutants, photochemical oxygen evolution and oxidation reactions.
2. Titanium(IV) chemistry
2.1. Synthetic considerations
Titanium(IV) ions tend to rapidly and spontaneously undergo hydrolysis in water, causing the uncontrolled precipitation of TiO2. Due to this issue, several synthetic methods have been developed to produce highly crystalline titanium-oxo clusters. The use of Ti(OR)4 in aqueous solutions often results in the formation of polymeric phases as a white precipitate, which complicates its use as a starting material for reactions involving water as a solvent for titanium oxo-cluster (TOC) formation. This is the main reason that there are only limited examples of Ti(OR)4 hydrolysis with small amounts of water in alcoholic solutions to prepare TOCs.7–9 One potential approach to utilize Ti(OR)4 in water is by employing acidic solutions to inhibit hydrolysis.10 Other titanium salts that have been used in the synthesis of TOCs in aqueous medium include TiCl4 and TiOSO4, which form highly acidic solutions upon dissolution.11,12 TiCl4 has also been used in the synthesis of TOCs in organic solvents under ambient conditions.13–15 As explained above, due to the facile hydrolysis of Ti(OR)4, many titanium oxo clusters have been synthesized under strict inert conditions. For their synthesis, dry solvents were used under nitrogen atmosphere, employing standard Schlenk line and glove box techniques.16–19
2.1.1. Solvothermal & ionothermal synthesis.
The solvothermal technique is the most commonly employed approach for synthesizing TOCs. Solvothermal synthesis is a method used to produce oxo-clusters, often nanoparticles or crystalline materials, in a solvent under high temperature and pressure. In this process, the solvent acts as both a medium for chemical reactions and a temperature-regulating environment. The reaction usually occurs in a sealed vessel, such as an autoclave, where temperatures above the solvent's boiling point can be achieved. This method allows for the use of a wider variety of starting materials, resulting in increased structural and functional diversity in the final products due to the high-temperature and high-pressure conditions.20–22 Moreover, the reaction parameters can be easily manipulated by adjusting the temperature at various stages of the reaction, and controlling the cooling time. One example of the solvothermal method is the reaction of salicylhydroxamic acid, acetic acid, isopropyl alcohol, and Ti(OiPr)4 in an autoclave at 80 °C for three days, which led to the formation of the cluster, Ti6(μ2-O)(μ3-O)2(OiPr)10(OAc)2(L)2 (H3L = salicylhydroxamic acid). Under the same conditions, using ethanol instead of isopropyl alcohol resulted in the formation of yellow crystals of the cluster Ti7(μ3-O)2(OEt)18(L)2.23 In many cases, the synthesis under inert atmosphere can be combined with the solvothermal method.16,24,25
Compared to traditional solvent-based synthesis, ionothermal synthesis involves the use of ionic liquids that can be customised based on specific needs through the adjustment of their physical properties like melting point, viscosity, solubility, and density.26 Ionic liquids can be used not only as solvents, but also as templates for the formation of desirable products.27 Moreover, ionic liquids can solubilize a large variety of starting materials at low temperatures, stabilize the Ti–O cores, and prevent disordered aggregation into precipitation by utilizing charge-balancing cations of appropriate size and functional groups. For example, two titanium(IV) oxo sulfate clusters, a toroidal shaped Ti26 and a capsule shaped Ti44 were synthesized by employing the ionothermal synthetic approach (Fig. 1).28
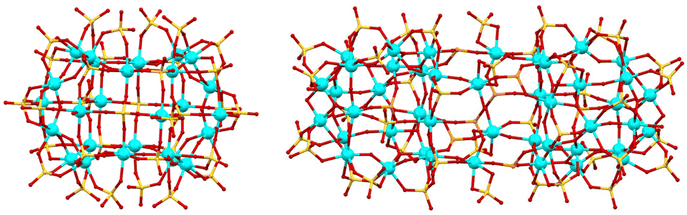 |
| | Fig. 1 Left: Toroidal shaped Ti26. Right: Capsule shaped Ti44. Hydrogen atoms are omitted for clarity. Colour code: Ti, cyan; S, yellow; O, red. | |
2.1.2. TOCs as precursors.
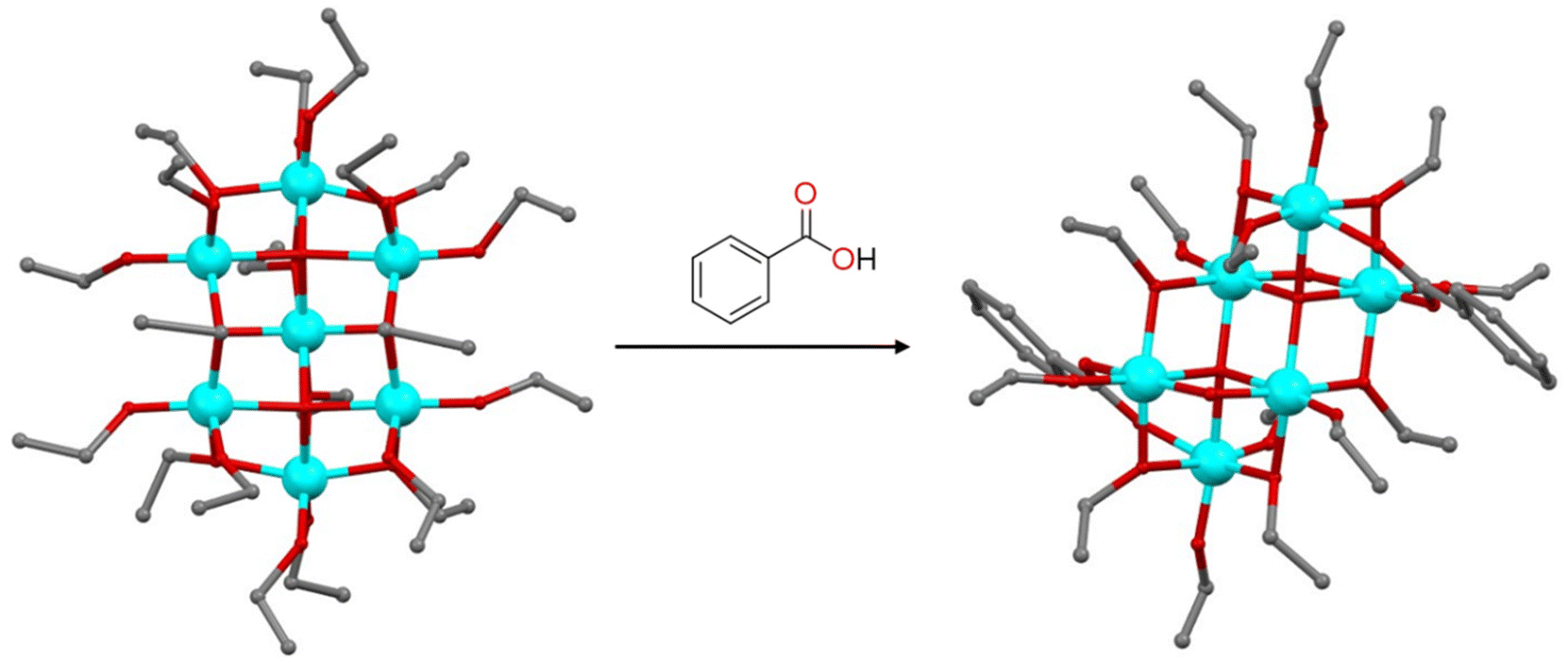
Besides the typical precursors like Ti(OR)4 and TiCl4, titanium oxo-clusters can also serve as precursors for the synthesis of TOCs. These clusters can undergo modification through a ligand exchange process in which the stability of the metal-oxo core depends on the balance between the reactivity and lability of the ligands. Metal oxo clusters tend to remain stable when the exchanging ligands possess the same charge, coordination ability, and geometry. Nevertheless, even if these conditions are met, the metal oxo core can still undergo rearrangement. The reaction of [Ti7O4(OEt)20] in the presence of benzoic acid leads to the formation of a new cluster, [Ti6O4(OEt)14(OOCPh)2], which adopts a substantially different configuration, due to the different coordination angles of the μ-OEt and μ-OOCPh ligands, which may significantly affect the overall structure (Fig. 2).29 On the contrary, the treatment of [Ti12O16(OiPr)16] and [Ti11O13(OiPr)18] with ethanol led to alkoxide exchange and the formation of [Ti12O16(OiPr)10(OEt)6] and [Ti11O13(OiPr)13(OEt)5] with retention of the parent structural features.8 Similarly, when [Ti16O16(OEt)32] reacts with linear aliphatic alcohols like n-propanol, only 8 terminal ethoxy groups attached to the more electrophilic titanium atoms are replaced.30 The efficiency of ligand exchange decreases with sterically hindered alcohols, while alcohols with higher acidity, such as phenol, lead to the substitution of all terminal ligands. This occurs because, in trans-alcoholysis, a nucleophilic substitution takes place, followed by a proton transfer from the entering ligand to the departing one. As the acidity of the entering ligand increases, the probability of ligand substitution also increases accordingly. TOCs can also be used as precursors in MOF synthesis. The solvothermal reaction of [Ti8Zr2O12(MeCOOH)16] in the presence of terephthalic acid as a linker led to the formation of a photoactive MOF.31 Using the preformed cluster as the starting material is crucial for obtaining a pure MOF, as it helps to avoid numerous competing side reactions. In contrast, synthesizing the MOF from a mixture of stoichiometric ZrIV and TiIV metal salts results in a low-crystallinity MOF with UiO-66 impurities.31 Additionally, an excess of carboxylic acid as a modulating agent is essential for achieving a highly crystalline product. The carboxylic acid competitively coordinates with the metals, slowing crystal growth and aiding in the production of highly crystalline materials.
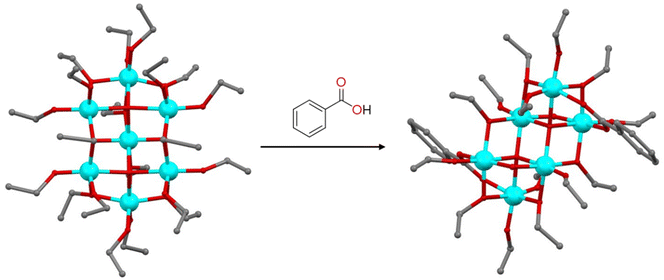 |
| | Fig. 2 Formation of [Ti6O4(OEt)14(OOCPh)2] upon the reaction of [Ti7O4(OEt)20] with benzoic acid. Hydrogen atoms are omitted for clarity. Colour code: Ti, cyan; O, red; C, grey. | |
2.2. Importance of ligand design: oxime and catechol stabilized titanium-oxo cores
Many TOCs have been synthesized by the coordination-delayed-hydrolysis (CDH) method using organic ligands that can prevent fast hydrolysis.32 The organic ligands coordinate to Ti(IV) centres, preventing water molecules from contacting the titanium ions and leading to a controlled hydrolytic aggregation and formation of crystalline products (Fig. 3). The proper choice of ligand can result in different hydrolysis processes of titanium ions, resulting not only in novel structural motifs but also in modification of the structure. The choice of appropriate ligands can prevent or diminish the impact of these challenges, paving the way for the stabilisation of new cores and the discovery of new phenomena. Recently our group made use of oxime and catechol-based ligands33,34 to give rise to new structural motifs not only in the Ti(IV) chemistry, but also in the chemistry of Zr(IV) and Hf(IV), which we will discuss below.
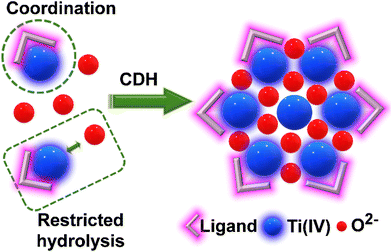 |
| | Fig. 3 Coordination-delayed-hydrolysis (CDH) method. | |
2.2.1. Oxime-substituted titanium oxo clusters.
A large number of the discovered titanium oxo-clusters are typically sensitive to moisture and subsequent hydrolysis due to the presence of alkoxy groups. One way to address this challenge is the utilization of organic chelating ligands to protect the metal-oxo core from uncontrolled hydrolysis. The oxime-substituted ligands also allow a greater structural variety due to the different coordination modes of the oxime moiety.23 Most synthesized TOCs are carboxylate-supported.35,36 The carboxylate groups apart from providing appropriate carboxylate-bridges, they also act as an in situ water source through esterification of the alkoxo leaving group.37 For example, the Ti42-oxo cluster, H6[Ti42(μ3-O)60(OiPr)42(OH)12], is formed from the reaction of Ti(OiPr)4 with isopropanol and formic acid under solvothermal conditions (Fig. 4a).21 Although formates do not appear as stabilizing ligands, they are essential for the cluster formation, possibly by controlling the rate of hydrolysis and regulating the water content of the reaction. Additional examples include the formation of the larger Ti44-oxo cluster, Ti44(μ2-OH)4(μ2-O)32(μ3-O)30(HPA)2(PA)46(HFA)2(FA)2(H2O)2 (PA− = propionate, FA− = formate),38 and the largest titanium-oxo cluster to date, Ti52(μ-OH)2(μ-O)14(μ3-O)50(μ4-O)8(PA)34(OiPr)28 (Fig. 4b).39
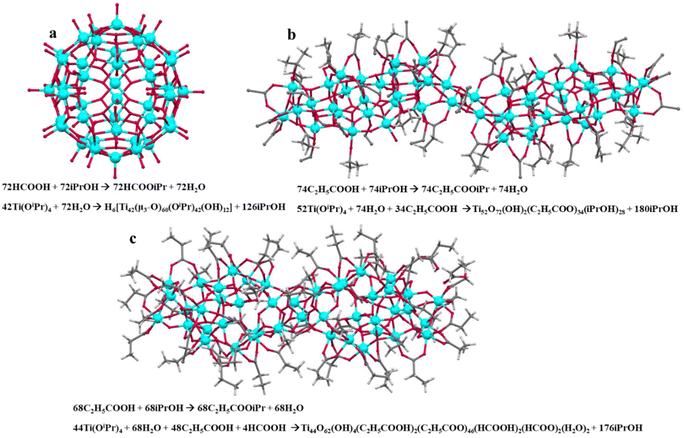 |
| | Fig. 4 Overview of the synthesis and structures of (a) Ti42, (b) Ti52, and (c) Ti44-oxo clusters utilizing carboxylates as hydrolysis delayed ligands (a proposed reaction process is depicted). This figure has been adapted/reproduced from ref. 32 with permission from American Chemical Society, copyright 2022. | |
On the contrary, even though N-doping can tune the band gap of TiO2, there are not many examples of TOCs with all-N donor ligands due to the low coordination affinity of nitrogen towards Ti(IV). Oximes are significant members of the mixed O/N-donor ligand family and can be used as hydrolysis-delaying ligands, with their nitrogen being slightly basic and the hydroxyl group slightly acidic. Both nitrogen and oxygen can exhibit different coordination modes. By modifying oxime ligands and the synthetic conditions, a series of crystalline TOCs from Ti4 to Ti18 were reported, with Ti18 being the highest nuclearity oxime-derived TOC to date (Fig. 5).23 The oxime ligands not only induce structural diversity to the system, but also influence to a great extent the cluster's light-harvesting and photocurrent responses, with the lower nuclearity TOCs typically performing better.
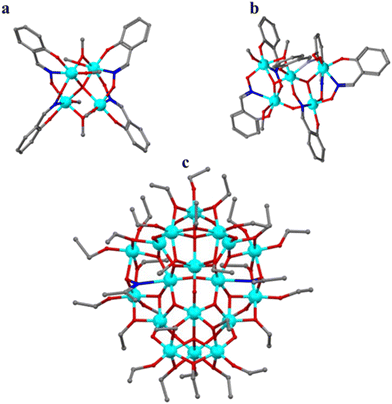 |
| | Fig. 5 Oxime-substituted titanium-oxo clusters: (a) Ti4, (b) Ti5, (c) Ti18. Hydrogen atoms are omitted for clarity. Colour code: Ti, cyan; O, red; N, blue; C, grey. | |
2.2.2. Catechol-based titanium oxo clusters.
In a similar manner, catechol adsorbed to TiO2 serves as a representative model for molecules that exhibit strong binding affinity with metal oxides, resulting in a significant electronic coupling between the localized catechol orbitals and the delocalised electron levels of the TiO2 conduction band.40,41 Catechol functions as a type II sensitizer, where charge injection occurs from the ground state of catechol to the TiO2 conduction band.42 Catecholate-functionalised titanium-oxo clusters have attracted significant interest as structural models for catechol–TiO2 systems, in order to provide atomic-scale representations of colloidal TiO2 nanoparticles modified with catechol.43–45 The two oxygens of catechol can have different coordination modes when they are bonded to titanium atoms, and up to now, five different coordination modes have been reported: (A) monodentate, (B) chelate bidentate, (C) doubly bridging chelate, (D) bridging chelate μ2-(O,O′), and (E) bridging (Fig. 6), with (B) and (E) being the most common binding modes.46 The coordination modes of catechol depend on several factors such as, the size of the cluster, synthetic conditions and defective surfaces.47 The catechol ligands have a significant influence on the photoelectrochemical and photocatalytic properties of titanium-oxo clusters, with charge transfer from catechol to the Ti–O metallic core playing an important role in enhanced photocurrent conversion under visible light irradiation.20,48
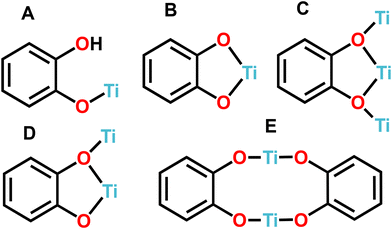 |
| | Fig. 6 Coordination modes of catechol (A–E). | |
2.2.3. Oxime-catechol-substituted titanium oxo clusters.
Our group envisaged that incorporating both catechol and oxime groups within the same chelating system, as in the organic ligand 2,3-dihydroxybenzaldehyde oxime (Fig. 7), could lead to the exploitation of the structural and electronic effects of both groups to modulate the material's band gap.15 Moreover, the electron-withdrawing –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–OH group on the aromatic ring is expected to greatly reduce the catechol protonation constant, making it more acidic and enhancing its effectiveness as a sequestering ligand at neutral or even acidic pH values. Indeed, the use of the catechol-oxime ligand led to the synthesis of the hexanuclear cluster, K2[TiIV6(μ3-O)2(μ-O)3(OCH3)4(HOCH3)2(μ-η1,η2,η1-Hdihybo-O,O′,N)6], (H3dihybo = 2,3-dihydroxy benzaldehyde oxime) under mild conditions (Fig. 7). This Ti6 species exhibits a rare structural motif with very interesting properties, such as a very low band gap value and metalloaromaticity.15
N–OH group on the aromatic ring is expected to greatly reduce the catechol protonation constant, making it more acidic and enhancing its effectiveness as a sequestering ligand at neutral or even acidic pH values. Indeed, the use of the catechol-oxime ligand led to the synthesis of the hexanuclear cluster, K2[TiIV6(μ3-O)2(μ-O)3(OCH3)4(HOCH3)2(μ-η1,η2,η1-Hdihybo-O,O′,N)6], (H3dihybo = 2,3-dihydroxy benzaldehyde oxime) under mild conditions (Fig. 7). This Ti6 species exhibits a rare structural motif with very interesting properties, such as a very low band gap value and metalloaromaticity.15
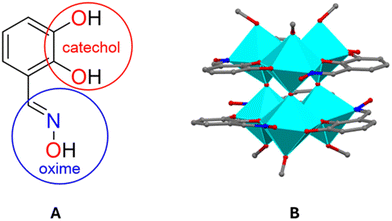 |
| | Fig. 7 2,3-Dihydroxybenzaldehyde oxime (H3dihybo) (A), and its Ti6 cluster (B). Hydrogens are omitted for clarity. Colour code: O, red; N, blue; C, grey. Polyhedral colour code: TiO6, cyan. | |
2.3. Structural diversity of Ti6-oxo clusters
The hexanuclear titanium-oxo clusters exhibit the greatest structural diversity among all TOCs,35 with many different skeletal arrangements reported to date (Fig. 8). The skeletal arrangements of Ti6-oxo clusters are: (i) two Ti6(μ2-O)9 configurations (Fig. 8a and b),49,50 (ii) two Ti6(μ2-O)2(μ3-O)2 configurations (Fig. 8c and d),51,52 (iii) Ti6(μ3-O)8 (Fig. 8e),53 (iv) Ti6(μ3-O)6 (Fig. 8f),54 (v) Ti6(μ2-O)4(μ3-O)2 (Fig. 8g),55 (vi) Ti6(μ2-O)(μ3-O)2 (Fig. 8h),56 (vii) Ti6(μ2-O)8 (Fig. 8i),57 (viii) Ti6(μ3-O)4 (Fig. 8j),58 (ix) Ti6(μ2-O)3(μ3-O)2 (Fig. 8k).14
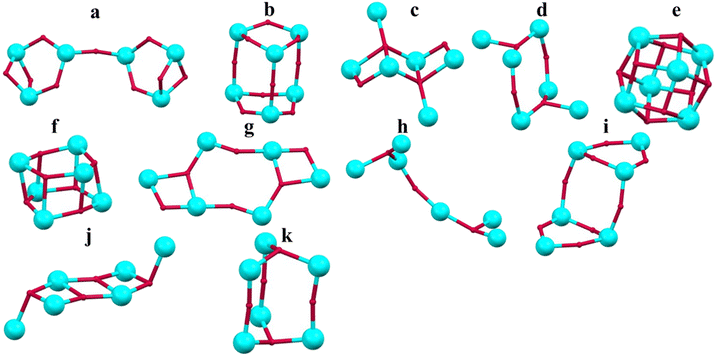 |
| | Fig. 8 Skeletal arrangements of the Ti6-oxo-metallic cores (a)–(k). This figure has been adapted/reproduced from ref. 14 with permission from Royal Society of Chemistry, copyright 2019. | |
3. Band gap in titanium-oxo clusters
The large band gap value of 3.2 eV for TiO2 limits its application in photocatalysis to the UV region.59,60 However, UV light constitutes only 5% of the irradiation reaching the Earth's surface. Therefore, reducing this band gap is crucial to expand its applications into the visible region. One effective method to reduce the band gap is to raise the valence band through a non-metal-ion doping.61 In contrast, metal-ion dopants introduce impurity levels within the band gap.62 Transition metal ions possess multiple valence states and unfilled d-electron orbitals, allowing them to accept additional electrons, create impurity levels within the band gap of TiO2, and act as shallow traps for photogenerated electrons or holes, which help to reduce the recombination of electron–hole pairs.63 Crystalline TOCs provide a chance to bridge the gap between dye-sensitized TiO2 and doped-TiO2 nanomaterials with theoretical models, considering both atomic structures and electronic states. Currently, there are only a limited number of studies focusing on band gap modulation of TOCs, especially without altering their metallic core. Coppens et al. reported a study of Ti11 nanoclusters with the formula Ti11(MX)O14(OiPr)17 (M = Mn(II), Fe(II), Co(II) and X = Cl−, Br−, I−), in which the iron-doped TOC exhibits the lowest band gap value due to the higher energy of the t2g β HOMO orbital, which is positioned midway between the valence and conduction bands. In contrast, the occupied β orbitals in the CoII complex are found at much lower energy levels, and as expected, these orbitals remain unoccupied in high-spin MnII.64 In the case of FeII and MnII clusters, the different halogens linked to the transition metals do not affect the band gap significantly. However, in another study, the absorption edge of the clusters, Ti11(CoX)O14(OiPr)17 and Ti11(NiX)O14(OiPr)17 (X = Cl−, Br−, I−) follows the order: Ti11MCl ≤ Ti11MBr < Ti11MI (M = CoII, NiII). The band gaps are all smaller than those of the pristine, [Ti12O16(OiPr)16], due to the appearance of new states that effectively hybridize with the O 2p orbitals. The halogen orbitals are mixed with metal orbitals, generating two new states just above the valence band maximum.65 The dimensions of materials can also influence their physical properties in various ways. Zhang et al. conducted a study on the precise assembly engineering of TOCs incorporating copper halide dopants66 and reported Ti6 clusters connected to various polynuclear copper halides, such as [Cu2X2], [Cu4X4], and [Cu4X4–DABCO–Cu4X4] (DABCO = 1,4-diazabicyclo[2.2.2]octane), leading to the formation of 1D chains, 2D layers, and 3D diamond frameworks (Fig. 9). The polymetallic doping and the different dimensions significantly affect the light absorption of TOCs.
 |
| | Fig. 9 (a) {Cu2Cl2} doped 1D Ti–O material, (b) {Cu2Cl4} doped 2D material, (c) {Cu4Br4} doped 3D material, (d) {Cu4I4-DABCO-Cu4I4} doped 3D material. Hydrogens are omitted for clarity. Colour code: Ti, cyan; Cu, green; Cl, light green; Br, brown; I, purple; P, pink; S, yellow; C, grey; O, red; N, blue. This figure has been adapted/reproduced from ref. 66 with permission from American Chemical Society, copyright 2017. | |
Moreover, the same group, exploited the labile coordination sites of the [Ti6O4(OiPr)10(O3P-Phen)2(OAc)2] cluster (O3P-Phen = phenylphosphonate), and conducted a study to investigate the band gap modulation by introducing various O-donor ligands, such as carboxylates, phosphonates, and sulfonates, along with additional transition metals (Fig. 10A).67 The incorporation of transition metals and the increasing electron-withdrawing effect of the ligands led to a reduction in the band gap values (Fig. 10B).
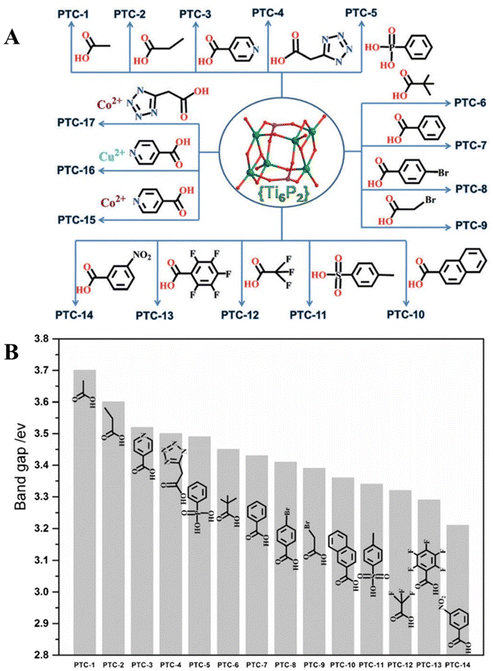 |
| | Fig. 10 O-donor ligands introduced in Ti6 clusters (A). Band gap values of PTC-1 to PTC-14 (B). This figure has been adapted/reproduced from ref. 67 with permission from WILEY-VCH Verlag GmbH & Co, copyright 2016. | |
4. Metalloaromaticity
Metalloaromaticity is a concept used to describe the aromaticity of all-metal ring-shaped cores.68–70 The phenomenon of metalloaromaticity was reported for the first time in titanium-oxo clusters by our group.14 Besides providing stability to the structure, this phenomenon can also contribute to reduced band gaps as discussed above. The concept of metalloaromaticity is important for a better understanding of the chemical bonds, electronic properties, and surface characteristics of metal oxides and mixed-metal clusters.71 A better understanding of metalloaromaticity has led to the discovery of new metalloaromatic species, including metallo-benzenes72 and clusters such as Al42− (ref. 73) and Au5Zn+,74 which have a wide range of applications in molecular electronics,75 aerospace engineering,76 drug development, and molecular magnets.77 In contrast to organic aromatic rings, where the electron delocalization occurs only through π molecular orbitals (MOs), in all-metal cyclic species, delocalization occurs through σ, π, δ, and φ MOs.77 In 2019, our group reported the first Ti-oxo cluster exhibiting metalloaromaticity.14 We used the nucleus-independent chemical shift (NICS) criterion, which is one of the most powerful and reliable methods available to study aromaticity. The NICSzz scan curves can be effective indicators of the aromaticity or antiaromaticity of cyclic metallic ring cores.78,79 The negative NICSzz(1) and NICSzz(−1) values, calculated to be −4.3 ppm and −23.2 ppm respectively at 1 Å above and below the centre of a molecular ring, suggest that the cyclo-Ti3(μ2-O)3(μ3-O) ring of the cluster, K6{TiIV6(μ3-O)2(μ-O)3(CH3O)6[μ2-η1,η1,η2-Hpidiox-O,N,O′]4[μ2-η1,η1,η2-pidiox-O,N,O′]2}, exhibits weak aromaticity (Fig. 11).14 We also calculated the NICSzz scan curves for the model system [TiIV3(μ3-O)(μ-Hdihybo)3(OCH3)3(OH)3], in which the NICSzz(0) and NICSzz(1) values are −14.5 ppm and −18.4 ppm, indicating that the metallic ring core is aromatic.15 Therefore, the magnitude of aromaticity in the cyclo-Ti3 trinuclear rings is affected and potentially modulated by the type of surrounding ligands.
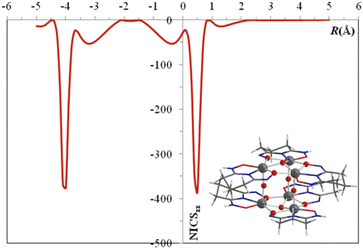 |
| | Fig. 11 NICSzz scan curves for the [cyclo-Ti3(μ2-O)3(μ3-O)]2. This figure has been adapted/reproduced from ref. 14 with permission from Royal Society of Chemistry, copyright 2019. | |
5. Applications of titanium-oxo clusters
TOCs can exhibit important chemical and physical properties such as solubility, stability, and photoactivity. Based on these properties, researchers investigate potential applications not only in the photo-related fields, but also in other areas. Consequently, numerous new potential applications of TOCs have been reported in recent years. In 2011, Chen et al. reported the synthesis of black titanium oxide (B-TiO2) using a hydrogen atmosphere.80 B-TiO2 exhibits a lower optical band gap and improved physicochemical properties compared to conventional TiO2. The altered properties of the black TiO2 nanomaterials have been attributed to the structural and/or chemical changes, such as surface lattice disorder, oxygen vacancies, and the presence of TiIII ions, TiIV–OH and TiIV–H groups.81 These differences naturally arise from the various synthetic methods used in producing black TiO2 nanomaterials. For example, B-TiO2 demonstrates better nonlinear optical (NLO) performance, with a nonlinear absorption coefficient β at least two orders of magnitude larger than that of conventional TiO2.82 This makes B-TiO2 a good candidate for techniques like Q-switching and mode-locking, which are used for generating short and intense laser pulses. One way to better understand the structure–property correlations of B-TiO2 materials is through the synthesis of black titanium oxo-clusters. These clusters are considered molecular analogues of B-TiO2 and are easier to study for mechanistic and theoretical investigations. B-TOCs are quite rare with only the first few examples reported in recent years.15,83,84 Our group reported the synthesis of a B-TOC in 2020 with the lowest band gap value of 1.48 eV for a homometallic TOC to date. This was accomplished by reacting TiCl4 with 2,3-dihydroxybenzaldehyde oxime (H3dihybo) under mild conditions.15 The cluster consists of [TiIV6(μ3-O)2(μ-O)3] core, in which the six titanium(IV) atoms are arranged in a trigonal-prismatic configuration (Fig. 7).
By increasing the concentration of catechol in the reaction system of Ti(OiPr)4, formic acid, and isopropanol, under solvothermal conditions, Zhang et al., synthesized three B-TOCs with a very low band gap and high stability in air, in different solvents, across a wide pH range, and even under laser irradiation (Fig. 12).83 The band gaps of the compounds decrease as the ratio of catechol/Ti increases, indicating the significant impact of the catechol. These compounds are also the first examples of homometallic TOCs with promising NLO properties. Recently, it was reported that the reaction of Ti(OiPr)4 with catechol and formic acid in toluene at 100 °C led to the formation of the first all-catechol protected Ti-oxo cluster (TOC), [Ti16(μ3-O)4(μ3-OH)8(μ2-O)4(Cat)20], in which the [Ti16O16] core is surrounded by 20 catechol ligands and consists of two [Ti8O6] cages linked together through four μ3-O atoms (Fig. 12).84 The Ti16 cluster exhibits high stability in different environments and very good photoelectric and photothermal properties, due to its very low band gap. The reported studies clearly demonstrate that B-TOCs are not only important for extending the chemistry of TOCs, but also because their low band gap value endows the clusters with many interesting properties.
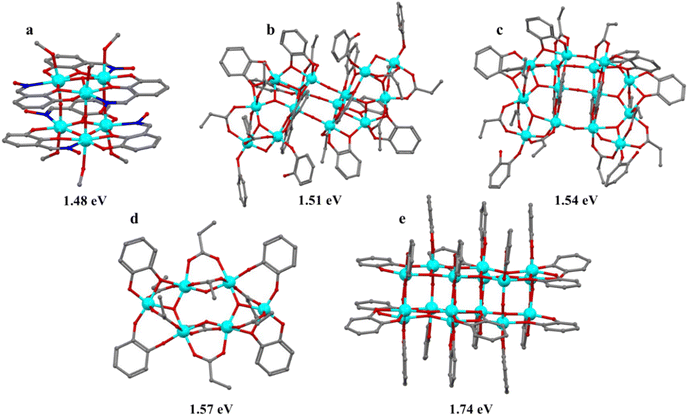 |
| | Fig. 12 Structures of black titanium oxo-clusters with their band gaps. (a) [TiIV6(μ3-O)2(μ-O)3(OCH3)4(HOCH3)2(Hdihybo)6], (b) Ti14(μ2-O)5(μ3-O)4(μ4-O)2(PA)10(Cat)10 (HCat)4, (c) Ti14(μ2-O)6(μ3-O)6(PA)10(Cat)10(HCat)2, (d) Ti6(μ2-O)2(μ3-O)2(PA)8(Cat)4, and (e) Ti16(μ3-O)4(μ3-OH)8(μ2-O)4(Cat)20. Hydrogens are omitted for clarity. Colour code: Ti, cyan; O, red; N, blue; C, grey. | |
5.1. Photocatalytic hydrogen production
Photocatalytic hydrogen production through water splitting offers a viable solution for addressing the global energy crisis. Despite the large number of reported titanium-oxo clusters, research on photocatalytic water splitting remains quite limited, with most experiments conducted under UV-visible light irradiation. Recently, the solvothermal reaction of Ti(OiPr)4 and H2SO4 in DMF and isopropanol led to the formation of the cluster [(CH3)2NH2]2[Ti21O29(OiPr)12(DMF)6(SO4)8], which consists of six pentagonal {Ti(Ti)5} motifs connected through corner-sharing or edge-sharing patterns.85 The Ti21 cluster is very stable and exhibits excellent UV/Vis light-driven water-splitting for H2 production, which might be enhanced due to the presence of {Ti(Ti5)} units. When the Ti21 cluster is irradiated by UV/Vis light, electrons are excited to the conduction band and the holes are generated in the valence band. H2PtCl6 acts as a co-catalyst to separate the photogenerated electrons, while triethanolamine (TEOA) acts as a hole scavenger (Fig. 13).
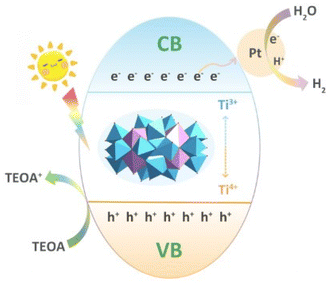 |
| | Fig. 13 Photocatalytic H2 production by the Ti21 cluster. This figure has been adapted/reproduced from ref. 85 with permission from Wiley-VCH GmbH, copyright 2023. | |
Some studies have shown that replacing oxygen with sulfur in specific TiO2-based nanomaterials enhances their photocatalytic activity under visible light,86,87 but there are limited examples of S-heterocyclic ligand modified TOCs. Two titanium-oxo clusters, [Ti4(nBuO)8(iPrO)4(1,2-TC4A)] and [Ti6(μ-O)(μ3-O)2(ox)(iPrO)12(c-TC4A)] (H4TC4A = p-tert-butylthiacalix[4]arene), where the ligand has 1,2-alternate and cone-shaped conformations (Fig. 14), respectively, were synthesized under solvothermal conditions.88 Both clusters exhibit photocatalytic H2 production under visible light, with the Ti4 cluster showing 5 times better performance. Additionally, the Ti4 cluster exhibits a better photocurrent response compared to the Ti6 cluster. DFT calculations indicate that the different conformations of TC4A4− are the reason for the varying performance, with the shorter Ti–S bond in Ti4 affecting not only the band gap values, but also the charge separation.
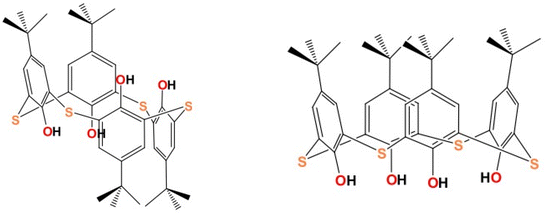 |
| | Fig. 14 Conformations of p-tert-butylthiacalix[4]arene. Left: 1,2-Alternate, right: cone. This figure has been adapted/reproduced from ref. 88 with permission from American Chemical Society, copyright 2020. | |
5.2. Adsorption
One of the applications for which TOCs have been used is the selective adsorption of dyes, though there are not many reports on this topic. In 2022, Zhu et al. synthesized the TOC compound, [Ti6O3(OiPr)14(TTFTC)]4 (TTFTC4− = tetrathiafulvalene–tetracarboxylate), in which four Ti6-oxo clusters are linked through four TTFTC linear connectors, forming a cyclic ring structure (Fig. 15a and b).16 This cluster self-condenses to form a polymeric organic–inorganic material that retains its cyclic structure. Given the material's porous structure and the high electron density of the TTF moiety, which can host and trap cationic guests, it was investigated for its ability to remove dye molecules through selective adsorption. The material demonstrates exceptional efficiency in adsorbing cationic dyes and can selectively capture methylene blue (MB) not only from solutions containing both cationic and anionic dyes, but also from those with mixed-cationic dyes. The suggested mechanism for the adsorption of MB is supported by several factors, including electrostatic interactions, cyclic effects, hydrogen bonding, and π⋯π stacking between the adsorbent and the MB dye (Fig. 15c). Control experiments indicated that the primary forces driving the adsorption are the strong electrostatic attraction and the cyclic effect of the adsorbent.
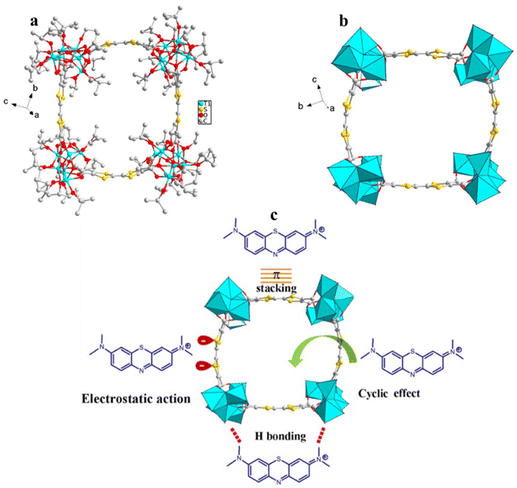 |
| | Fig. 15 (a) [Ti6O3(OiPr)14(TTFTC)]4, (b) cyclic ring structure of [Ti6O3(OiPr)14 (TTFTC)]4, (c) suggested mechanism for the adsorption of MB. Hydrogen atoms are omitted for clarity. This figure has been adapted/reproduced from ref. 16 with permission from American Chemical Society, copyright 2022. | |
Recently, the same group reported a rectangular ring TOC, [Ti6O6(OiPr)8(TTFTC)(phen)2]2 (phen = 1,10-phenanthroline), in which the width of the rectangle is 3.5 Å and the length is 11.32 Å (Fig. 16a).89 The cluster undergoes topotactic conversion via the condensation of isopropanol groups, preserving the ring structure and resulting in the formation of a stable TOC-MOF (Fig. 16b). The TOC-MOF can adsorb cationic dyes due to the rich electron density of the TTF moiety, and it exhibits selective adsorption of crystal violet among other cationic dyes, most likely due to the host–guest structure matching.
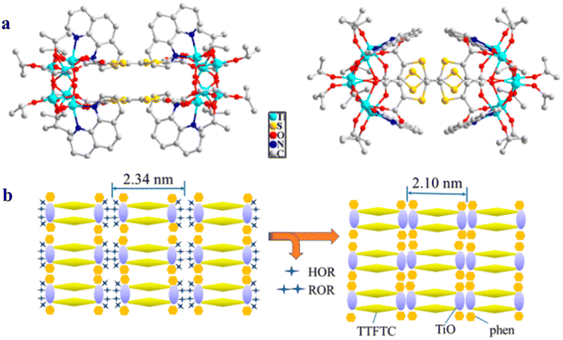 |
| | Fig. 16 (a) The structure of [Ti6O6(OiPr)8(TTFTC)(phen)2]2, (b) TOC-MOF formed by condensation of isopropanol groups. Hydrogen atoms are omitted for clarity. This figure has been adapted/reproduced from ref. 89 with permission from American Chemical Society, copyright 2024. | |
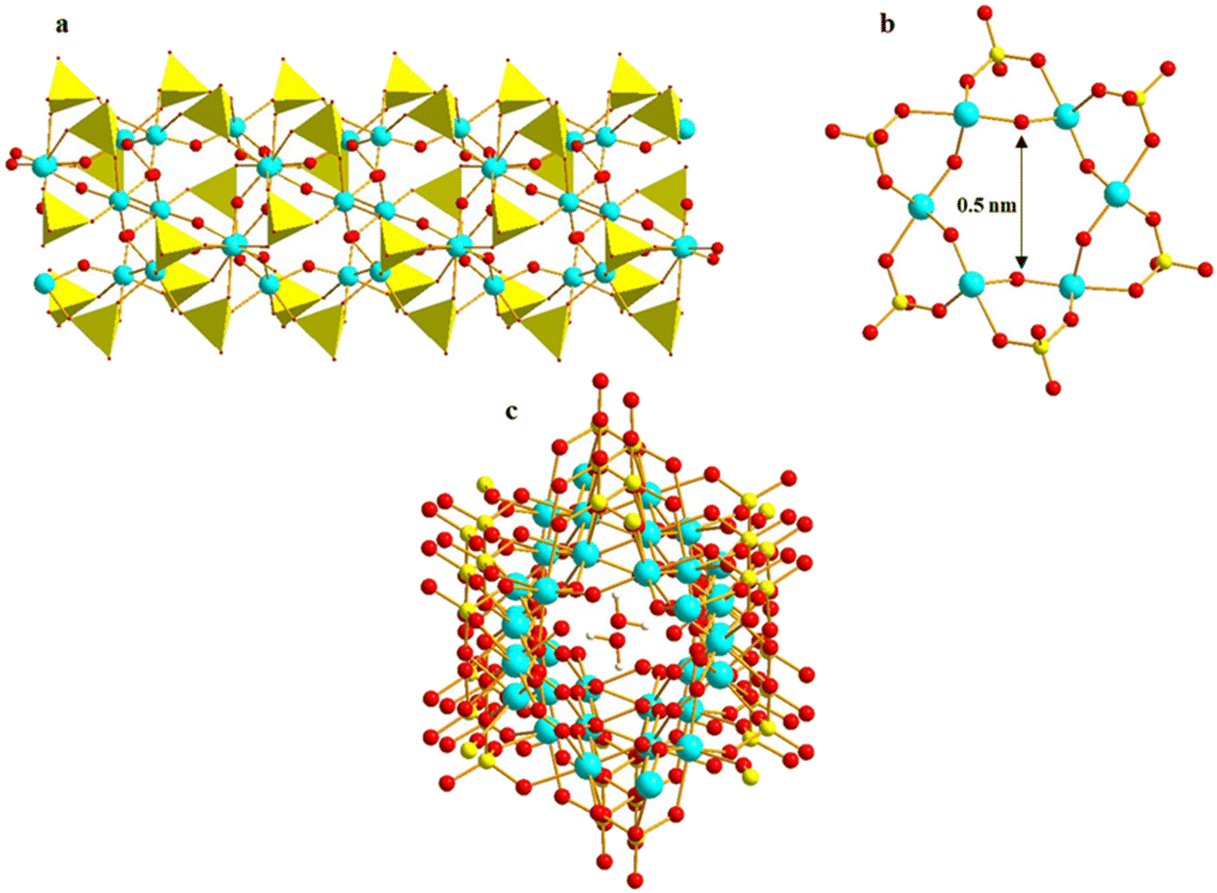
The ionothermal reaction of Ti(OiPr)4 and sulfuric acid in 1-ethyl-3-methylimidazolium ethyl ester sulfate ([EMIm]EtOSO3) ionic liquid led to the formation of the first molecular Ti–O nanotube, {(EMIm)3[(H2O)⊂Ti6O6(μ2-OH)3(SO4)6]}n (EMIm = 1-ethyl-3-methylimidazolium), which consists of hexagonal Ti6O6 rings supported by sulfate ligands and counterbalanced by EMIm cations (Fig. 17a and b).90 At room temperature, a rare phenomenon of water molecule nanoconfinement was observed within the pores of the Ti–O nanotubes (Fig. 17c). The nanotubes exhibit selective water adsorption over methanol and ethanol, indicating promising potential for separating miscible solvent mixtures, particularly in industrial applications such as bioethanol purification.
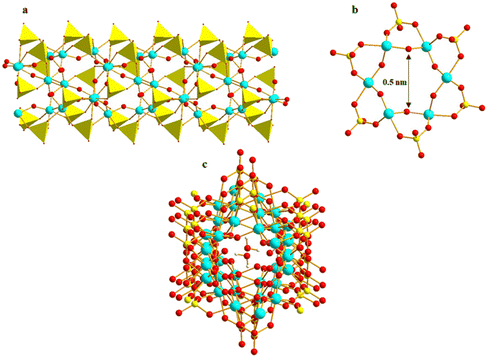 |
| | Fig. 17 (a) Ti–SO4–Ti nanotube array of [Ti6O6(μ2-OH)3(SO4)6]n, (b) [Ti6O6(SO4)6] hexagonal hole, (c) water molecule nanoconfinement within the pores of the Ti–O nanotubes. Colour codes: Ti, cyan; S, yellow; O, red; H, white; SO4, yellow polyhedron. | |
6. Zirconium(IV) and hafnium(IV) chemistry
6.1. Synthetic considerations
Zirconium(IV) and hafnium(IV) are classified as group IV elements and are known to be strong Lewis acids. They exhibit high affinity towards oxygen, which influences their chemical behaviour significantly. Similar to 3d metals, which tend to form polymeric and bulk oxides under basic conditions, these elements can undergo condensation reactions even at relatively low pH levels (>2).91 The formation of discrete oxo-clusters is further complicated by the limited structural diversity, as exemplified by [M6(O)4(OH)4(H2O)12]12+ and [M4(OH)8(H2O)16]8+ (M = Zr, Hf) which are among the most common structural motifs (see Fig. 18), especially in aqueous environments.92 This is to say that attempts to form novel architectures often instead results in a limited number of known structural motifs, due to their high thermodynamic stability. Thus, careful consideration must be given to the choice of ligands and reaction conditions to overcome this. These clusters are also resistant to further growth – dimers of M6 clusters are known, however M18 is not comprised of (M6)3 units for example, and adopts a different structure (vide infra).92,93 These metals exhibit very similar chemical behaviour due to their close proximity in the periodic table and nearly identical sizes resulting from lanthanide contraction (with ionic radii of 0.78 Å for Hf and 0.79 Å for Zr), often displaying comparable coordination numbers and geometries (typically 8, although this may vary).91 This trend significantly differs from most d-block elements that tend to become softer as they descend a group. Some of the main differences between Ti and Zr/Hf lies in their coordination number and size, and their reduction potential despite their similar reactivity, as Ti(III) species are somewhat stable and have been reported for various applications,94,95 whereas Zr(III)/Hf(III) are far scarcer (particularly outside of organometallic systems, however the binary halides have been reported for each).91 Zr/Hf metals are predominantly utilized in catalysis and in semiconductors as supports or functional components, along with niche applications in organic synthesis, like olefin polymerization.96–98 Some other factors to consider are the ceramic properties of the bulk materials and their tendency to react with atmospheric oxygen/water, leading to the spontaneous formation of stable clusters over time. This process is virtually irreversible in a laboratory setting, with the exception of the sulfate salts.91 As a result, there is a limited number of starting materials available to synthetic chemists for these transition metals.
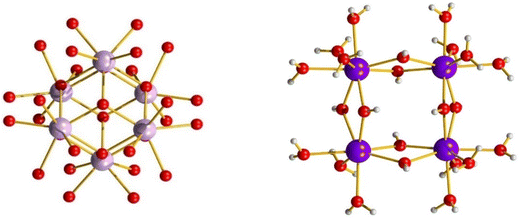 |
| | Fig. 18 Zr6 (left) and Hf4 (right). Colour code: Zr, light purple; Hf, dark purple; O, red; H, white. | |
In aqueous environments, the sulfate anion is typically preferred due to its stability in water, which prevents spontaneous cluster formation.92 On the other hand, chlorides and other halides, with or without Cp− ligands, are prone to hydrolysis, making them more suitable for use in organic solvents.91 Alkoxy salts are also commonly utilized in organic solvents because of their natural solubility and the absence of a need for additional base in reaction mixtures.92 When it comes to halides, their reaction with protic solvents in organic conditions will release HX (X− = Cl−, Br−, I−) and will necessitate additional base to form oxo/hydroxy clusters.91 However, an exception to this occurs, when amide solvents are used in a solvothermal setting, as the thermal degradation of the amide solvent releases the free amine in situ.99
Synthetic methodologies can be broadly categorized based on whether they are carried out in aerobic or anaerobic conditions, and whether they are conducted in aqueous or organic solvents.92 The common salts of Ti, Zr and Hf tend to polymerize, under mildly basic conditions, and thus, it is often necessary to employ inert techniques, like Schlenk or glovebox manipulation.91 The utilization of these techniques is crucial due to the reactivity and oxophilicity of the metals involved, allowing for the use of organometallic complexes and starting materials, and expanding the range of solvents/conditions and enhancing ease of handling.91 Alternatively, the predictability of hydrolysis products can be strategically utilized to form well-defined clusters as secondary building units (SBUs).92 This approach is frequently employed, for instance, in the UiO family of MOFs, which have been synthesized using various methods (such as solvothermal, mechanochemical) for a wide range of applications.100 These MOFs are comprised of the aforementioned Zr6 cluster, and variable dicarboxylate ligands of the general formula [Zr6O4(OH)4L6] (H2L = dicarboxylic acid). Due to their remarkable stability, the resulting MOFs have been used for a variety of applications, such as gas separation, host–guest catalytic systems etc.101–103 In Fig. 19, it is depicted a schematic diagram of the reaction path towards the formation Zr/Hf clusters discussed in this article.
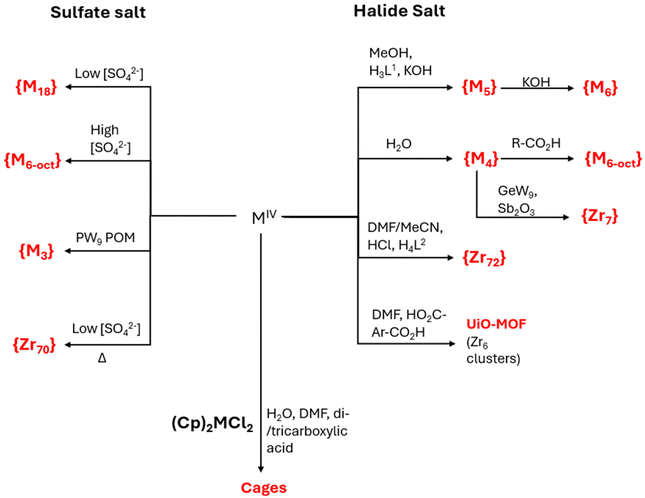 |
| | Fig. 19 Schematic diagram of the reaction path towards Zr/Hf clusters discussed in this article. The reaction path is applicable to both Zr(IV) and Hf(IV), (M = Zr, Hf), unless otherwise stated. L1 can be either H3dihybo or H3pidiox, L2 is 3,3′,3′′,3′′′-(anthracene-9,10-diylidenebis(methane-1,1-diyl-1-ylidene))tetrabenzoic acid. | |
6.2. Applications
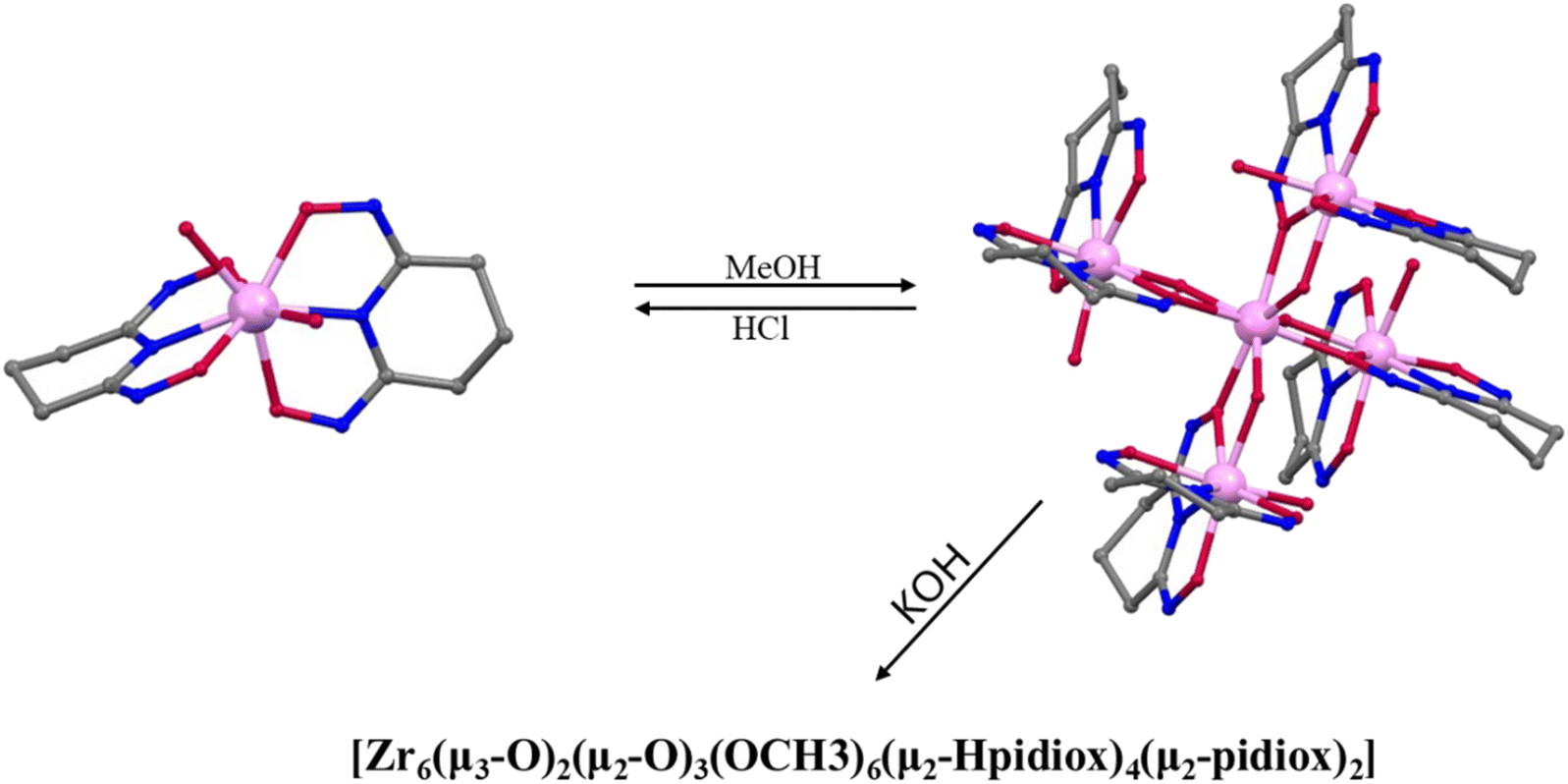
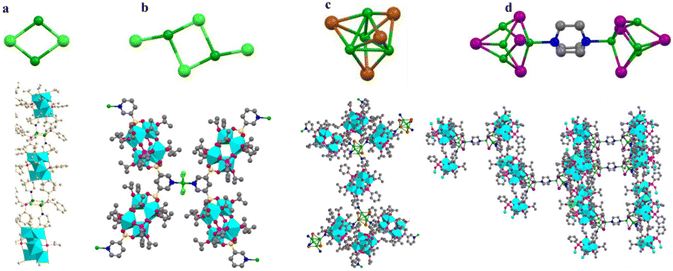
Prior work by our group led to publication of a series of complexes of group IV metals displaying reduced band gaps or metalloaromaticity as in the case of Ti discussed above.15,33,34 The complexes utilised ligands with multiple binding sites and modes (catecholate and hydroxamate) in order to make a series of layered trimers and distorted tetrahedra (Fig. 20). The two clusters featuring trigonal prismatic Hf6 motifs were isolated using triethylamine and pyridine as bases, exhibiting significantly low band gaps of 2.36 and 2.51 eV, respectively, positioning them as promising options for photocatalytic applications.34 Additionally, reacting the ligand (2Z,6Z)-piperidine-2,6-dione dioxime (H3pidiox) with ZrCl4 in methanol and adjusting the pH of the solution allowed the manipulation of the resulting products (Fig. 21).33 There are not many examples of pentanuclear Zr and Hf oxo-clusters reported in the literature, with the Hf5 cluster being the only one reported to date.34
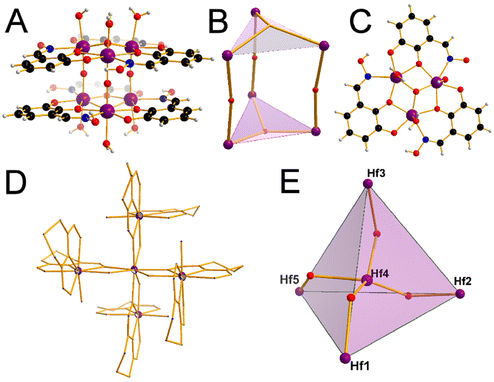 |
| | Fig. 20 Layered Hf trimers displaying metalloaromaticity. Isostructural Zr species were also reported. The structure of the catecholate–oxime Hf6 species (A). The Hf6O5 core (B). The top-down view of the coordination with catecholate–oxime ligands featuring the central μ3-oxo group (C). Hf5 species displaying a distorted tetrahedral geometry with a central octacoordinated Hf (D) and (E) respectively. Colour code: Hf, purple; C, black; N, blue; O, red; H, white. | |
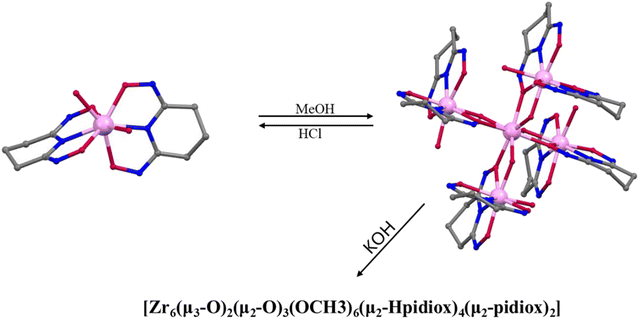 |
| | Fig. 21 pH-Dependent formation of Zr-oxo clusters. | |
The structures of discrete Zr4L6 and Zr4L4 clusters, shown in Fig. 22, are produced through the hydrolysis product of (Cp2)ZrCl2 reacting with di- and tricarboxylic acids to form tetrahedral clusters.104 The clusters can be considered as fragments or “pores” of the extended UiO MOF species, which, because of their molecular nature, can be dissolved in different organic solvents. This dissolution enables the examination of bulk structure properties and opens up possibilities for applications in homogeneous catalysis, solution dynamics studies, and easier characterizations using single crystal X-ray crystallography compared to their bulk forms. Furthermore, they maintain many of the appealing features of MOFs, such as modularity and reticularity. Hf(IV) congeners may be synthesised in an analogous manner due to their paralleled reactivity. Butyl substituted cyclopentadienyl clusters of the same type have also been shown to be a viable means for potential treatment of nerve agent poisoning/degradation,105 and control over linker molecules serves as a possible tool for catalysis in confined spaces as has been demonstrated in similar Ga4 tetrahedra.106 Substitution of the dicarboxylic acids for viologen derived carboxylates also resulted in a stable photo- and electrochromic material, with potential applications in smart windows, sensing, memory devices and optical switches (Fig. 23).107
 |
| | Fig. 22 Zr4 tetrahedra displaying control over pore size and aperture through ligand substitution, with potential applications in catalysis. | |
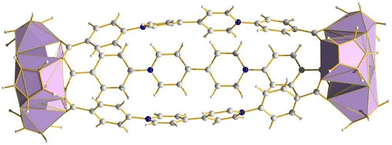 |
| | Fig. 23 (Cp3Zr3(μ3-O)(μ2-OH)3)2L3, L = 1,1′-bis(4-carboxyphenyl)-4,4′-bipyridinium dichloride, Cp = cyclopentadienyl. Colour code: Zr, light purple; C, grey; N, blue; H, white. Counter ions omitted for clarity. | |
Both Zr and Hf MOCs have been applied to promote selective protein hydrolysis, an important step in proteomics and biotechnology.108,109 Due to their high reactivity as Lewis acid catalysts, and their stability they have been shown to be site selective catalysts for the scission of various proteins such as equine skeletal muscle myoglobin, which Zr6 (Fig. 18, left) was able to selectively hydrolyse the protein within 24 hours yielding protein fragments of lower molecular weights,108 while Hf18 acted as heterogeneous site-selective catalysts for the hydrolysis of horse heart myoglobin (Fig. 24, left). Furthermore, due to the solubility of the Zr6 cluster, detailed mechanistic studies were able to be undertaken in an effort to elucidate the mechanism for Zr6 promoted hydrolysis of peptide bonds. In both cases, the species showed selectivity towards aspartame residues, which is believed to arise from combination of their potent Lewis acidity and their Brønsted acidic surfaces.109
 |
| | Fig. 24 Zr18 cluster (left), which is isostructural to Hf18. Lowering sulfate concentration and increasing temperature led to the rearrangement to the toroidal Zr70 cluster (right, top-down view). Colour code: Zr, light purple; S, yellow; O, red. Counter ions and solvate omitted for clarity. | |
6.3. Limitations in cluster nuclearity
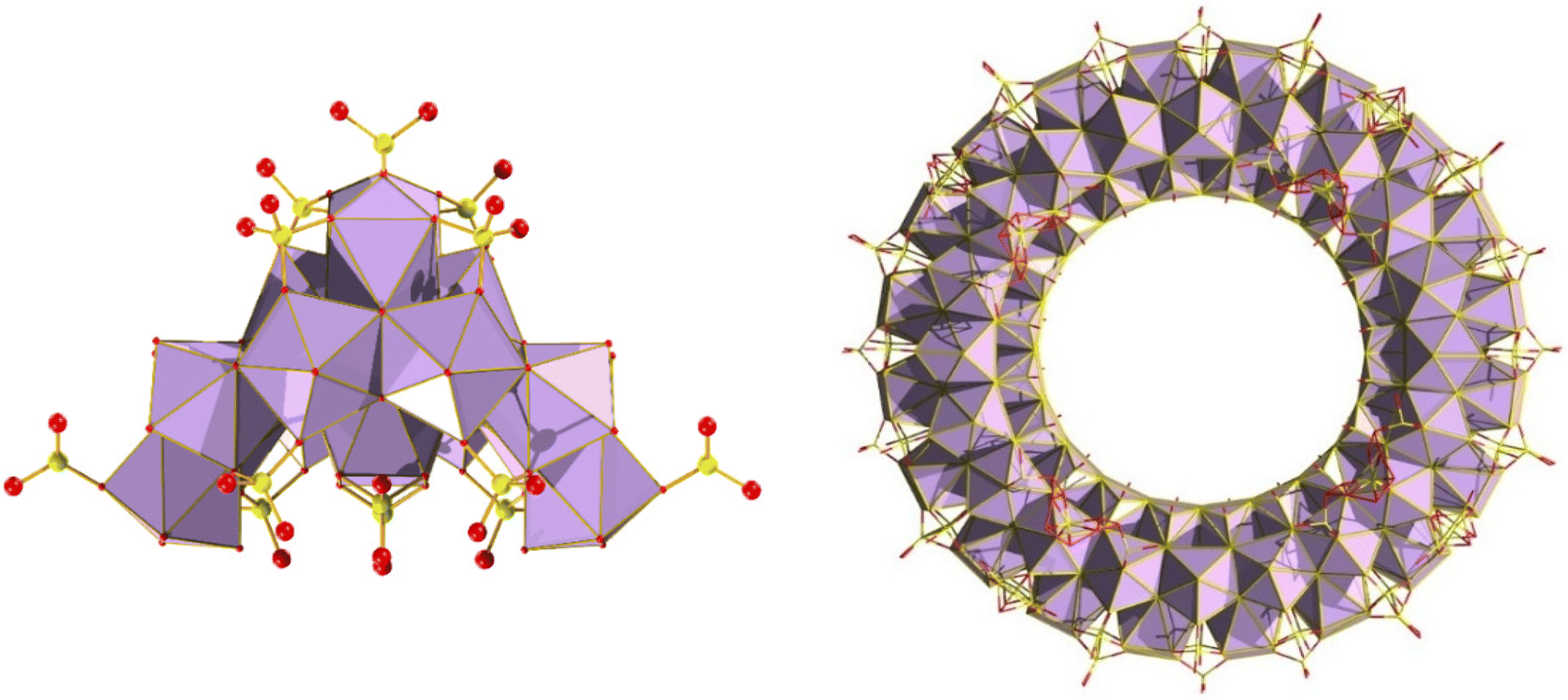
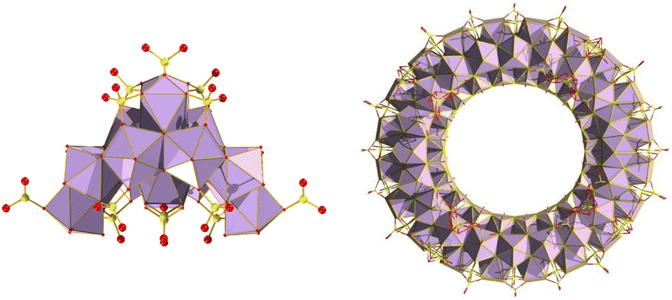
While POMs can display very high nuclearities of up to 368, such as Mo154,110 and Mo368 clusters,111 group IV clusters containing Zr and Hf typically exhibit lower nuclearities. Only a few instances of clusters with more than 20 metal centres have been observed. The largest known isolated Zr MOC, composed solely of zirconium oxo-clusters is Zr70(O/OH)146(SO4)58·xH2O (Zr70) (with capping sulfates).112 Interestingly, this species highlights the effect of temperature and sulfate concentration on nuclearity of the resulting clusters, as lower temperatures/higher sulfate concentration led only to the isolation of the previously reported Zr18 (Zr18O20(OH)26(H2O)23.2(SO4)12.7). It is believed that excess of sulfate anions in solution creates a sulfate “passivation” layer, preventing further growth. It is also worth noting the Zr70 species is the largest cluster bearing no organic motifs. A larger, Zr72 structure was reported, comprised of six (Zr12(μ3-O)8(μ3-OH)8(μ2-OH)6) cluster-based building blocks linked by twelve organic tetracarboxylates.113 The Zr12 clusters can be considered as a dimer of the Zr6 archetype bridged by six OH− groups. Interestingly, unlike Zr, superclusters of Hf have remained elusive. Given the relatively recent discovery of Zr superclusters such as Zr70/Zr72, it may yet be feasible to isolate such species. Thus, the largest Hf cluster reported to date is the Hf analogue of Zr18 (as shown in Fig. 24), which is formed under similar reaction conditions with the exception that it requires higher temperatures, which is a characteristic difference between Zr and Hf.92,114
At this point, it is worth noting that the largest titanium-oxo clusters to date are: (1) H6[Ti42(μ3-O)60(OiPr)42(OH)12], with a core diameter exceeding 1.5 nm.21 The core consists of 42 titanium atoms bridged by 60 μ3-O atoms and 12 pentagonal Ti(Ti5) units, forming a spherical structure (Fig. 4a). (2) Ti52(μ-OH)2(μ-O)14(μ3-O)50(μ4-O)8 (PA)34(OiPr)28, which is a 3.6 nm Ti52 cluster, with a boat-like interlayer structure, constructed from Ti6 units (Fig. 4b).39 (3) By adjusting the type and concentration of acid, it was synthesized a series of TOCs with varying nuclearities, including three Ti44 clusters with a large {Ti44O66} core of 2.8 nm, which exhibits the second-highest nuclearity among titanium-oxo cores (Fig. 4c).38 The previously mentioned studies represent a major step forward in developing large Ti-oxo nanoclusters that are comparable to TiO2 nanoparticles.
6.4. Entrapment of Zr(IV) and Hf(IV) metallic cores
The well-researched properties and abundant oxygen content of POMs make them an effective tool for not only capturing unique nuclearities of group IV metals, but also for enhancing the solubility of these species to improve their processability in solutions. Rather than simply substituting existing POMs with Zr/Hf centres, a range of nuclearities have been identified through the use of POMs and lacunary POMs as metalloligands.115–117 Moreover, the possibilities for post-synthetic modifications and synergistic catalysis present numerous potential future applications.118–121 In Fig. 25, the potential for trapping irregular nuclearities of group IV clusters is emphasized, where the solubility of the final product is also maintained. Interestingly, the authors point out that the central M3 species remain unsaturated, showcasing their potential as Lewis acid catalysts.122
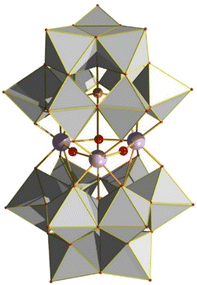 |
| | Fig. 25 Polyhedral representation of [M3(OH)3]9+ cluster sandwiched between two tri-lacunary Keggin POMs. M = Zr, Hf. Keggin = [A-α-PW9O34]9−. Colour code: W, grey; P, orange; Zr, light purple; O, red. The Hf congener is isostructural. | |
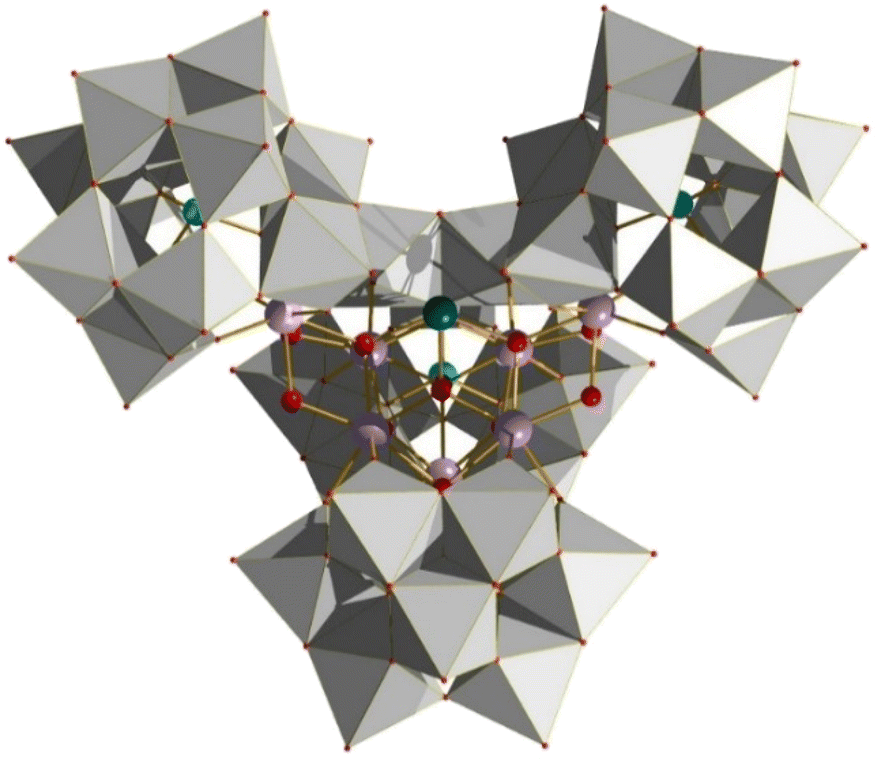
Another interesting example is the POM/MOC hybrid material (Fig. 26) which demonstrated superior photochemical oxidation towards methylene blue relative to its constituent materials, highlighting potential applications of such materials in remediation of organic pollution and the underlying synergistic effects that it may be possible to control.123 As depicted, the Zr6 cluster has been embellished with a sub-stoichiometric quantity of benzoate, resulting in the cationic nature of the cluster – a key driving force in the formation process in the presence of negatively charged SiW12 Keggin POM.123 Similarly, the isolation of a Zr7 cluster through the use of two tri-lacunary and two monolacunary Keggin ions was demonstrated to be a successful heterogeneous catalyst for the oxidation of sulfides to the corresponding sulfones with H2O2. The Zr7 core can be considered as a Sb-substituted Zr6, with the monolacunary Keggin-based building blocks serving to trap the additional two edge sharing Zr centres (Fig. 27).124
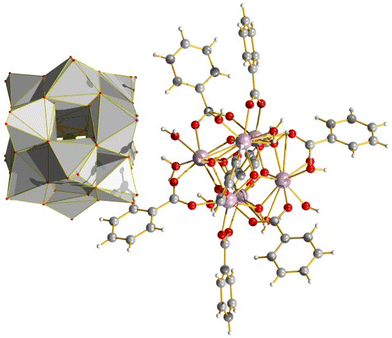 |
| | Fig. 26 The co-crystallisation of a Zr6 benzoate with a silicotungstate Keggin POM showed synergistic enhancements in photochemical properties. The crystallisation resulted in ionic channels in the bulk structure, which the authors note as being a key aspect of its properties in photocatalysis. | |
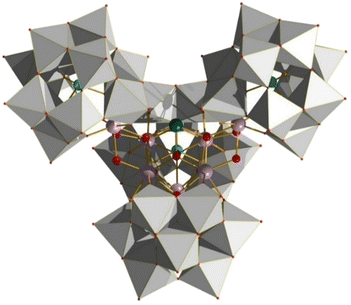 |
| | Fig. 27 Structure of the Zr7 species isolated through use of Keggin POM ions. Colour code: W, grey; Ge, light blue; Sb, dark blue; Zr, light purple; O, red. Counter ions and solvate omitted for clarity. | |
7. Conclusions and perspectives
Given the relatively underexplored nature of the heavier group IV elements, it becomes clear that there is a huge potential for development of their novel applications and undiscovered species. Overcoming synthetic challenges/limitations will be crucial to harnessing the untapped potential of group IV materials. Further control of ligands, rational design of constituents and careful consideration of system components are a necessary step towards the discovery of novel species with potentially interesting properties. Herein, we have highlighted some conceivable means of realising these new structures and given insight into possibly fruitful synergistic properties. As discussed, of particular interest are the photochemical and catalytic properties of these materials, considering an increasing trend to move towards sustainability and away from fossil fuels. Harnessing the potential of renewable energies, such as solar power, will be a vital and necessary step in this endeavour, and materials with appropriate band gaps and tunability for this purpose will become ever more sought after. Indeed, beyond solar power, applications of MOCs in catalysis, environmental remediation, proteomics and biotechnology are also very intriguing, while some unique properties they display, such as metalloaromaticity, could potentially pave the way towards the discovery of new functionalities. Further research into the speciation, solution dynamics, mechanistic aspects and reactivity of this unique class of compounds should enable the discovery and subsequent design of new materials with exciting properties.
Data availability
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
TPV thanks KU Leuven and FWO for funding.
References
- D. Van Den Eynden, R. Pokratath and J. De Roo, Chem. Rev., 2022, 122, 10538–10572 CrossRef CAS PubMed.
- H. N. Miras, J. Yan, D. L. Long and L. Cronin, Chem. Soc. Rev., 2012, 41, 7403–7430 RSC.
- J. W. Park, D. K. Lee, D. Lim, H. Lee and S. H. Choi, J. Appl. Phys., 2008, 104, 033521 CrossRef.
- U. S. Patel, K. H. Patel, K. V. Chauhan, A. K. Chawla and S. K. Rawal, Procedia Technol., 2016, 23, 336–343 CrossRef.
- E. Bersch, S. Rangan, R. A. Bartynski, E. Garfunkel and E. Vescovo, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 085114 CrossRef.
- C. Gionco, M. C. Paganini, E. Giamello, R. Burgess, C. Di Valentin and G. Pacchioni, J. Phys. Chem. Lett., 2014, 5, 447–451 CrossRef CAS PubMed.
- V. W. Day, T. A. Eberspacher, Y. Chen, J. Hao and W. G. Klemperer, Inorg. Chim. Acta, 1995, 229, 391–405 CrossRef CAS.
- V. W. Day, T. A. Eberspacher, W. G. Klemperer and C. W. Park, J. Am. Chem. Soc., 1993, 115, 8469–8470 CrossRef CAS.
- V. W. Day, T. A. Eberspacher, W. G. Klemperer, C. W. Park and F. S. Rosenberg, J. Am. Chem. Soc., 1991, 113, 8190–8192 CrossRef CAS.
- Y. Fu, Z. Xu and F. Zhang, J. Mol. Struct., 2008, 873, 168–172 CrossRef CAS.
- G. Zhang, C. Liu, D. L. Long, L. Cronin, C. H. Tung and Y. Wang, J. Am. Chem. Soc., 2016, 138, 11097–11100 CrossRef CAS PubMed.
- G. Zhang, J. Hou, C. H. Tung and Y. Wang, Inorg. Chem., 2016, 55, 3212–3214 CrossRef CAS PubMed.
- S. S. Passadis, S. Hadjithoma, P. Siafarika, A. G. Kalampounias, A. D. Keramidas, H. N. Miras and T. A. Kabanos, Molecules, 2021, 26, 5588 CrossRef CAS PubMed.
- S. S. Passadis, S. Hadjithoma, A. G. Kalampounias, A. C. Tsipis, S. Sproules, H. N. Miras, A. D. Keramidas and T. A. Kabanos, Dalton Trans., 2019, 48, 5551–5559 RSC.
- S. S. Passadis, M. G. Papanikolaou, A. Elliott, C. G. Tsiafoulis, A. C. Tsipis, A. D. Keramidas, H. N. Miras and T. A. Kabanos, Inorg. Chem., 2020, 59, 18345–18357 CrossRef CAS PubMed.
- C.-Y. Ge, J.-L. Hou, Z.-Y. Zhou, Q.-Y. Zhu and J. Dai, Inorg. Chem., 2021, 61, 486–495 CrossRef PubMed.
- S. E. Brown, I. Mantaloufa, R. T. Andrews, T. J. Barnes, M. R. Lees, F. De Proft, A. V. Cunha and S. D. Pike, Chem. Sci., 2022, 14, 675–683 RSC.
- N. Li, P. D. Matthews, J. J. Leung, T. C. King, P. T. Wood, H. K. Luo and D. S. Wright, Dalton Trans., 2015, 44, 19090–19096 RSC.
- T. Krämer, F. Tuna and S. D. Pike, Chem. Sci., 2019, 10, 6886–6898 RSC.
- H. T. Lv, H. M. Li, G. D. Zou, Y. Cui, Y. Huang and Y. Fan, Dalton Trans., 2018, 47, 8158–8163 RSC.
- M. Y. Gao, F. Wang, Z. G. Gu, D. X. Zhang, L. Zhang and J. Zhang, J. Am. Chem. Soc., 2016, 138, 2556–2559 CrossRef CAS.
- W. H. Fang, L. Zhang and J. Zhang, Dalton Trans., 2017, 46, 803–807 RSC.
- S. Chen, W. H. Fang, L. Zhang and J. Zhang, Inorg. Chem., 2018, 57, 8850–8856 CrossRef CAS PubMed.
- C.-Y. Luo, L.-J. Ma, W. Liu, Y.-C. Tan, R.-N. Wang, J.-L. Hou and Q.-Y. Zhu, Inorg. Chem., 2024, 63, 5961–5971 CrossRef CAS PubMed.
- Y. Y. Wu, X. W. Lu, M. Qi, H. C. Su, X. W. Zhao, Q. Y. Zhu and J. Dai, Inorg. Chem., 2014, 53, 7233–7240 CrossRef CAS PubMed.
- Q. Zhang, S. Zhang and Y. Deng, Green Chem., 2011, 13, 2619–2637 RSC.
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357–6426 CrossRef CAS PubMed.
- K. Chintakrinda, N. Narayanam, G. Chen, F. Wang, J. Zhang and L. Zhang, Dalton Trans., 2022, 6162–6165 RSC.
- I. Mijatovic, G. Kickelbick and U. Schubert, Eur. J. Inorg. Chem., 2001, 4, 1933–1935 CrossRef.
- G. Fornasieri, L. Rozes, B. Alonso, D. Massiot, M. Noe, M. Evain, K. Boubekeur, M. Curie, P. Cedex, C. De and M. Curie, J. Am. Chem. Soc., 2005, 127, 4869–4878 CrossRef CAS PubMed.
- S. Yuan, J. S. Qin, H. Q. Xu, J. Su, D. Rossi, Y. Chen, L. Zhang, C. Lollar, Q. Wang, H. L. Jiang, D. H. Son, H. Xu, Z. Huang, X. Zou and H. C. Zhou, ACS Cent. Sci., 2018, 4, 105–111 CrossRef CAS PubMed.
- L. Zhang, X. Fan, X. Yi, X. Lin and J. Zhang, Acc. Chem. Res., 2022, 55, 3150–3161 CrossRef CAS PubMed.
- S. S. Passadis, S. Hadjithoma, M. G. Papanikolaou, A. D. Keramidas, H. N. Miras and T. A. Kabanos, Dalton Trans., 2022, 51, 1806–1818 RSC.
- S. S. Passadis, S. Hadjithoma, N. J. Fairbairn, G. J. Hedley, N. A. G. Bandeira, A. C. Tsipis, H. N. Miras, A. D. Keramidas and T. A. Kabanos, Inorg. Chem., 2022, 61, 20253–20267 CrossRef CAS PubMed.
- U. Schubert, Chem. – Eur. J., 2021, 27, 11239–11256 CrossRef CAS PubMed.
- W. H. Fang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2018, 47, 404–421 RSC.
- M. Czakler, C. Artner and U. Schubert, Eur. J. Inorg. Chem., 2014, 2038–2045 CrossRef CAS PubMed.
- M. Y. Gao, L. Zhang and J. Zhang, Chem. – Eur. J., 2019, 25, 10450–10455 CrossRef CAS PubMed.
- W.-H. Fang, L. Zhang and J. Zhang, J. Am. Chem. Soc., 2016, 138, 7480–7483 CrossRef CAS PubMed.
- L. G. C. Rego and V. S. Batista, J. Am. Chem. Soc., 2003, 125, 7989–7997 CrossRef CAS PubMed.
- S. C. Li, J. G. Wang, P. Jacobson, X. Q. Gong, A. Selloni and U. Diebold, J. Am. Chem. Soc., 2009, 131, 980–984 CrossRef CAS PubMed.
- B. K. An, W. Hu, P. L. Burn and P. Meredith, J. Phys. Chem. C, 2010, 114, 17964–17974 CrossRef CAS.
- C. Chaumont, A. Chaumont, N. Kyritsakas, P. Mobian and M. Henry, Dalton Trans., 2016, 45, 8760–8769 RSC.
- C. Liu, J. Hu, F. Zhu, J. Zhan, L. Du, C. H. Tung and Y. Wang, Chem. – Eur. J., 2019, 25, 14843–14849 CrossRef CAS PubMed.
- J. B. Benedict and P. Coppens, J. Am. Chem. Soc., 2010, 132, 2938–2944 CrossRef CAS PubMed.
- J. Hou, Q. Zhang, Y. Wu, Y. Liu, L. Du, C. H. Tung and Y. Wang, Inorg. Chim. Acta, 2016, 443, 279–283 CrossRef CAS.
- D. Finkelstein-Shapiro, S. K. Davidowski, P. B. Lee, C. Guo, G. P. Holland, T. Rajh, K. A. Gray, J. L. Yarger and M. Calatayud, J. Phys. Chem. C, 2016, 120, 23625–23630 CrossRef CAS.
- J. Le Hou, P. Huo, Z. Z. Tang, L. N. Cui, Q. Y. Zhu and J. Dai, Inorg. Chem., 2018, 57, 7420–7427 CrossRef PubMed.
- A. Pérez-Redondo, A. Martín and C. Rizzoli, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2015, 71, m97 Search PubMed.
- L. Ni, D. Liang, Y. Cai, G. Diao and Z. Zhou, Dalton Trans., 2016, 45, 7581–7588 RSC.
- S. Yang, H. C. Su, J. Le Hou, W. Luo, D. H. Zou, Q. Y. Zhu and J. Dai, Dalton Trans., 2017, 46, 9639–9645 RSC.
- X. Fan, N. Narayanam, M. Gao, L. Zhang and J. Zhang, Dalton Trans., 2018, 47, 663–665 RSC.
- N. Singh and S. Bhattacharya, Dalton Trans., 2011, 40, 2707–2710 RSC.
- P. Piszczek, A. Radtke, T. Muzioł, M. Richert and J. Chojnacki, Dalton Trans., 2012, 41, 8261–8269 RSC.
- K. Hong, W. Bak and H. Chun, Inorg. Chem., 2014, 53, 7288–7293 CrossRef CAS PubMed.
- Y. Wu, W. Luo, Y. Wang, Y. Pu, X. Zhang, L. You, Q. Zhu and J. Dai, Inorg. Chem., 2012, 51, 8982–8988 CrossRef CAS PubMed.
- S. Aguado-Ullate, J. J. Carbó, O. González-Del Moral, M. Gómez-Pantoja, A. Hernán-Gómez, A. Martín, M. Mena, J. M. Poblet and C. Santamaría, J. Organomet. Chem., 2011, 696, 4011–4017 CrossRef CAS.
- T. J. Boyle, R. P. Tyner, T. M. Alam, B. L. Scott, J. W. Ziller and B. G. Potter, J. Am. Chem. Soc., 1999, 121, 12104–12112 CrossRef CAS.
- V. Etacheri, C. Di Valentin, J. Schneider, D. Bahnemann and S. C. Pillai, J. Photochem. Photobiol., C, 2015, 25, 1–29 CrossRef CAS.
- K. Liu, T. Wu, L. Xu, Z. Zhang, Z. Liu, L. Wang and Z. L. Wang, Nano Res., 2024, 17, 1173–1181 CrossRef CAS.
- S. In, A. Orlov, R. Berg, F. Garcı, S. Pedrosa-jimenez, M. S. Tikhov, D. S. Wright and R. M. Lambert, J. Am. Chem. Soc., 2007, 129, 13790–13791 CrossRef CAS PubMed.
- N. Li, P. D. Matthews, H. Luo and D. S. Wright, Chem. Commun., 2016, 52, 11180–11190 RSC.
- D. Zhang, J. Chen, Q. Xiang, Y. Li, M. Liu and Y. Liao, Inorg. Chem., 2019, 58, 12511–12515 CrossRef CAS PubMed.
- Y. Chen, K. N. Jarzembska, E. Trzop, L. Zhang and P. Coppens, Chem. – Eur. J., 2015, 21, 11538–11544 CrossRef CAS PubMed.
- J. Hu, L. Zhan, G. Zhang, Q. Zhang, L. Du, C. Tung and Y. Wang, Inorg. Chem., 2016, 55, 8493–8501 CrossRef CAS PubMed.
- W. H. Fang, J. F. Wang, L. Zhang and J. Zhang, Chem. Mater., 2017, 29, 2681–2684 CrossRef CAS.
- J. Liu, M. Gao, W. Fang, L. Zhang and J. Zhang, Angew. Chem., 2016, 128, 5246–5251 CrossRef.
- A. I. Boldyrev and L. S. Wang, Chem. Rev., 2005, 105, 3716–3757 CrossRef CAS PubMed.
- C. A. Tsipis, Coord. Chem. Rev., 2005, 249, 2740–2762 CrossRef CAS.
- L. J. Li, B. Ali, Z. Chen and Z. M. Sun, Chin. J. Chem., 2018, 36, 955–960 CrossRef CAS.
- X. Li, J. Sun, Y. Zeng, Z. Sun, S. Zheng and L. Meng, J. Phys. Chem. A, 2012, 116, 5491–5496 CrossRef CAS PubMed.
- Z. N. Chen, G. Fu, I. Y. Zhang and X. Xu, Inorg. Chem., 2018, 57, 9205–9214 CrossRef CAS PubMed.
- J. Higino Damasceno, J. N. Teixeira Rabelo and L. Cândido, Inorg. Chem., 2016, 55, 7442–7447 CrossRef CAS PubMed.
- H. Tanaka, S. Neukermans, E. Janssens, R. E. Silverans and P. Lievens, J. Am. Chem. Soc., 2003, 125, 2862–2863 CrossRef CAS PubMed.
- S. Shetty, R. Kar, D. G. Kanhere and S. Pal, J. Phys. Chem. A, 2006, 110, 252–256 CrossRef CAS PubMed.
- E. Nembach, Prog. Mater. Sci., 2000, 45, 275–338 CrossRef CAS.
- F. Feixas, E. Matito, J. Poater and M. Solà, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 105–122 CAS.
- P. Von Ragué Schleyer, M. Manoharan, Z. X. Wang, B. Kiran, H. Jiao, R. Puchta and N. J. R. Van Eikema Hommes, Org. Lett., 2001, 3, 2465–2468 CrossRef PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. von Ragué Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- X. Chen, L. Liu and F. Huang, Chem. Soc. Rev., 2015, 44, 1861–1885 RSC.
- R. F. Zhang, D. Z. Guo and G. M. Zhang, Appl. Surf. Sci., 2017, 426, 763–769 CrossRef CAS.
- M. Y. Gao, Z. Wang, Q. H. Li, D. Li, Y. Sun, Y. H. Andaloussi, C. Ma, C. Deng, J. Zhang and L. Zhang, J. Am. Chem. Soc., 2022, 144, 8153–8161 CrossRef CAS PubMed.
- J. Hou, N. Huang, D. Acharya, Y. Liu, J. Zhu, J. Teng, Z. Wang, K. Qu, X. Zhang and D. Sun, Chem. Sci., 2024, 15, 2655–2664 RSC.
- Y. Song, S. Y. Liu, C. H. Li, W. C. Chen, K. Z. Shao, C. Qin, X. L. Wang and Z. M. Su, Eur. J. Inorg. Chem., 2023, 26, e202300020 CrossRef CAS.
- T. Ohno, T. Mitsui and M. Matsumura, Chem. Lett., 2003, 32, 364–365 CrossRef CAS.
- X. Chen and C. Burda, J. Am. Chem. Soc., 2008, 130, 5018–5019 CrossRef CAS PubMed.
- X. Wang, Y. Yu, Z. Wang, J. Zheng, Y. Bi and Z. Zheng, Inorg. Chem., 2020, 59, 7150–7157 CrossRef CAS PubMed.
- C.-Y. Luo, L.-J. Ma, W. Liu, Y.-C. Tan, R.-N. Wang, J.-L. Hou and Q.-Y. Zhu, Inorg. Chem., 2024, 63, 5961–5971 CrossRef CAS PubMed.
- K. Chintakrinda, N. Narayanam, Y. Z. Li, F. Wang, C. Kashi, Q. H. Li, G. Xu, L. Zhang and J. Zhang, CCS Chem., 2020, 2, 209–215 CrossRef CAS.
-
N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Butterworth-Heinemann, Oxford, 1997, pp. 954–975 Search PubMed.
- Y. Zhang, F. De Azambuja and T. N. Parac-vogt, Coord. Chem. Rev., 2021, 438, 213886 CrossRef CAS.
- W. Mark and M. Hansson, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 1101–1108 CrossRef.
- L. Podvorica, E. Salvadori, F. Piemontesi, G. Vitale, G. Morini and M. Chiesa, Appl. Magn. Reson., 2020, 51, 1515–1528 CrossRef CAS.
- I. Sancho, M. Navarro, M. Montilla, P. Salvador, C. Santamaría, J. M. Luis and A. Hernán-Gómez, Inorg. Chem., 2023, 62, 14873–14887 CrossRef CAS PubMed.
- S. Melada, R. Rioda, F. Menegazzo, F. Pinna and G. Strukul, J. Catal., 2006, 239, 422–430 CrossRef CAS.
- X. Yang, X. Ma, X. Yu and M. Ge, Appl. Catal., B, 2020, 263, 118355 CrossRef CAS.
- H. G. Alt and A. Köppl, Chem. Rev., 2000, 100, 1205–1221 CrossRef CAS PubMed.
- R. S. Forgan, Chem. Sci., 2020, 11, 4546–4562 RSC.
- J. Winarta, B. Shan, S. M. Mcintyre, L. Ye, C. Wang, J. Liu and B. Mu, Cryst. Growth Des., 2020, 20, 1347–1362 CrossRef CAS.
- Y. Benseghir, A. Lemarchand, M. Duguet, P. Mialane, M. Gomez-Mingot, C. Roch-Marchal, T. Pino, M.-H. Ha-Thi, M. Haouas, M. Fontecave, A. Dolbecq, C. Sassoye and C. Mellot-Draznieks, J. Am. Chem. Soc., 2020, 142, 9428–9438 CrossRef CAS PubMed.
- A. H. Vahabi, F. Norouzi, E. Sheibani and M. Rahimi-Nasrabadi, Coord. Chem. Rev., 2021, 445, 214050 CrossRef CAS.
- J. Pan, B. Xiao, W. Zhu, Y. Yang and H. Huang, Nano Res., 2024, 17, 6713–6720 CrossRef CAS.
- G. Liu, Z. Ju, D. Yuan and M. Hong, Inorg. Chem., 2013, 52, 13815–13817 CrossRef CAS PubMed.
- P. Delgado, J. D. Martin-Romera, C. Perona, R. Vismara, S. Galli, C. R. Maldonado, F. J. Carmona, N. M. Padial and J. A. R. Navarro, ACS Appl. Mater. Interfaces, 2022, 14, 26501–26506 CrossRef CAS PubMed.
- C. M. Hong, R. G. Bergman, K. N. Raymond and F. D. Toste, Acc. Chem. Res., 2018, 51, 2447–2455 CrossRef CAS PubMed.
- Y. Luo, S. W. Ying, S. J. Li, L. K. Li, H. Y. Li, M. Asad, S. Q. Zang and T. C. W. Mak, Inorg. Chem., 2022, 61, 2813–2823 CrossRef CAS PubMed.
- K. Declerck, N. D. Savić, M. A. Moussawi, C. Seno, R. Pokratath, J. De Roo and T. N. Parac-Vogt, J. Am. Chem. Soc., 2024, 146, 11400–11410 CAS.
- J. Moons, F. de Azambuja, J. Mihailovic, K. Kozma, K. Smiljanic, M. Amiri, T. Cirkovic Velickovic, M. Nyman and T. N. Parac-Vogt, Angew. Chem., Int. Ed., 2020, 59, 9094–9101 CrossRef CAS PubMed.
- A. Müller and C. Serain, Acc. Chem. Res., 2000, 33, 2–10 CrossRef PubMed.
- A. Müller, E. Beckmann, H. Bögge, M. Schmidtmann and A. Dress, Angew. Chem., Int. Ed., 2002, 41, 1162–1167 CrossRef.
- S. Øien-Ødegaard, C. Bazioti, E. A. Redekop, Ø. Prytz, K. P. Lillerud and U. Olsbye, Angew. Chem., Int. Ed., 2020, 59, 21397–21402 CrossRef PubMed.
- C. Zhang, Y. Luo, Y. Li, B. Zhao, Z. Yang, X. Li, J. Duan, Y. Zhao, Z. Lin and W. Huang, Cryst. Growth Des., 2022, 22, 6384–6389 CrossRef CAS.
- A. Kalaji, S. Skanthakumar, M. G. Kanatzidis, J. F. Mitchell and L. Soderholm, Inorg. Chem., 2014, 53, 6321–6328 CrossRef CAS PubMed.
- Z. Zhang, H. Lou Li, Y. L. Wang and G. Y. Yang, Inorg. Chem., 2019, 58, 2372–2378 CrossRef CAS PubMed.
- H. Lou Li, Z. Zhang, Y. L. Wang and G. Y. Yang, Eur. J. Inorg. Chem., 2019, 2019, 486–491 CrossRef.
- C. N. Kato, A. Shinohara, K. Hayashi and K. Nomiya, Inorg. Chem., 2006, 45, 8108–8119 CrossRef CAS PubMed.
- Y. Saku, Y. Sakai, A. Shinohara, K. Hayashi, S. Yoshida, C. N. Kato, K. Yoza and K. Nomiya, Dalton Trans., 2009, 805–813 RSC.
- G. Al-Kadamany, S. S. Mal, B. Milev, B. G. Donoeva, R. I. Maksimovskaya, O. A. Kholdeeva and U. Kortz, Chem. – Eur. J., 2010, 16, 11797–11800 CrossRef CAS PubMed.
- C. N. Kato, W. Unno, S. Kato, T. Ogasawara, T. Kashiwagi, H. Uno, K. Suzuki and N. Mizuno, Catal. Lett., 2016, 146, 2119–2128 CrossRef CAS.
- D. Li, H. Han, Y. Wang, X. Wang, Y. Li and E. Wang, Eur. J. Inorg. Chem., 2013, 1926–1934 CrossRef CAS.
- Y. Saku, Y. Sakai, A. Shinohara, K. Hayashi, S. Yoshida, C. N. Kato, K. Yoza and K. Nomiya, Dalton Trans., 2009, 805–813 RSC.
- J. Napal, B. Artetxe, G. Beobide, O. Castillo, A. Luque, J. Pascual-Colino, S. Pérez-Yáñez and M. Perfecto-Irigaray, Inorg. Chem. Front., 2022, 9, 935–940 RSC.
- P. Y. Zhang, Y. Wang, L. Y. Yao and G. Y. Yang, Inorg. Chem., 2022, 61, 10410–10416 CrossRef CAS PubMed.
Footnote |
| † The authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Tatjana N.
Parac-Vogt
b,
Tatjana N.
Parac-Vogt