
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Film-electrochemical EPR spectroscopy to investigate electron transfer in membrane proteins in their native environment†
Davide
Facchetti
a,
Yunfei
Dang
a,
Maryam
Seif-Eddine
ab,
Blaise L.
Geoghegan
 a and
Maxie M.
Roessler
*a
a and
Maxie M.
Roessler
*a
aDepartment of Chemistry and Centre for Pulse EPR Spectroscopy (PEPR), Imperial College London, Molecular Sciences Research Hub, White City Campus, Wood Lane, London W12 0BZ, UK
bCNRS: Marseille, Bioénergétique et Ingénierie des Protéines (BIP), 31 Chemin Joseph Aiguier, 13009, Marseille, France. E-mail: m.roessler@imperial.ac.uk
First published on 23rd September 2024
Abstract
Film-electrochemical electron paramagnetic resonance spectroscopy (FE-EPR) enables simultaneous electrochemical and spectroscopic characterisation of paramagnetic electron-transfer centres, including in soluble proteins. We now report a modified set-up FE-EPR with tuneable macroporous working electrodes and demonstrate the feasibility to investigate electron transfer in membrane proteins in their native membrane environment.
Spectroelectrochemistry (SEC) is widely used to study electron transfer (ET) reactions, but its application to biological systems is still limited despite recent advances.1 Unlike other spectroscopic techniques, Electron Paramagnetic Resonance (EPR) spectroscopy is ‘blind’ to much of the complexity of the biological molecule, whilst sensing intrinsic unpaired electrons that offer detailed mechanistic insights into ET reactions.2 However, the detection and characterisation of such unpaired electrons can be challenging.3 We reported Film-Electrochemical EPR spectroscopy (FE-EPR) as a SEC method that enables characterisation of redox centres in proteins (ex situ PFE-EPR, where P stands for ‘protein’)4 as well as time-resolved studies of catalytic paramagnetic intermediates observable at room temperature (in situ FE-EPR).5 Proteins typically contain fast-relaxing paramagnetic metallic cofactors such as hemes6 and iron-sulphur clusters7,8 that are only detectable at cryogenic temperatures, especially with pulsed EPR techniques.9 Investigation of these systems thus requires an ex situ (or ‘quasi in situ’)10 set-up, in which the electrochemical reaction is performed inside the EPR tube, followed by snap-freezing and low-temperature EPR measurements. In PFE-EPR the protein of interest is anchored to the working electrode (WE) and potential control over buried redox centres is possible in relatively small soluble proteins (i.e. Cu, Zn superoxide dismutase).4 Direct electrochemical control avoids mass transport limitations seen in solution-based studies,11 allowing for the formation and detection of radical species that are difficult or impossible to identify with standard techniques.3 PFE-EPR requires only nanomoles of sample and directly detects these species on the working electrode.4,5
Purifying membrane proteins can disrupt their native structure12 or yield only the apo form, necessitating cofactor reconstitution that might alter their conformation.13 Additionally, some protein interactions and reaction intermediates are only detectable under in vivo-like conditions,14 such as with hydrophobic quinone species.15,16 Consequently, studies of purified membrane proteins often cannot reveal their in vivo conformations and reactions. Hence, extending PFE-EPR to proteins in membranes will provide a unique methodology to perform structural and functional studies on such complex biological systems. In our original PFE-EPR set-up,4 indium-tin oxide (ITO) nanoparticles were annealed around a coiled platinum wire, resulting in a porous cylindrical WE that can fit into a standard EPR tube. The main challenge with such a three-dimensional (3D) WE is the difficulty in controlling the pore size. Indeed, the structural integrity of the WE was insufficient17 when attempting assembly with macropores (750 nm) to create ‘inverse opal (IO)’ structures.18 Moreover, the high resistance within the porous ITO network and large Ohmic drop of the 3D set-up resulted in a non-optimal electrochemical response compared to a standard three-electrode electrochemical cell, manifested by a large separation between the cyclic voltammetry (CV) oxidation and the reduction peaks.4 To overcome these limitations and building on our development of in situ FE-EPR,5 we designed two-dimensional electrodes as shown in Fig. 1. High reproducibility of the electrode geometry was achieved by laser-cutting a titanium sheet into 4.5 × 0.1 cm strips, subsequently coated with a thin film (∼500 nm) of ITO (see Methods). To make the mesoporous structure with or without macropores (henceforth ‘IO-mesoITO’ and ‘mesoITO’, respectively), 10 μL of ITO nanoparticles with or without polystyrene beads were then drop cast onto both sides of each strip and annealed at 500 °C. Scanning electron microscopy (SEM) confirmed the flat homogeneous surface with randomly oriented nanochannels typical of mesoITO scaffolds19,20 and the macroporous structures in IO-mesoITO19,21 (Fig. 1b). Compared to the mesoITO, which mainly has mesopores (∼43 nm) on the surface, the hierarchical IO-mesoITO features both mesopores and interconnected macropores (∼640 nm) that extend into the ITO layer. This IO structure offers a significant increase in mesoporous surface area, approximately 40 times higher compared to the mesoITO structure (see ESI† Fig. S1 and S2), with macropores that can accommodate large membrane vesicles, as discussed below. To assess and benchmark the performance of our new FE-EPR set-up, electrodes were functionalised with a model redox couple amino-TEMPO.4,22 The compensated resistance (Rc) of the FE-EPR cell was minimised by moving the counter electrode (CE) as close as possible to the WE without touching it. The optimal distance between the reference electrode (RE) and WE (Fig. 1b) was identified by measuring the uncompensated resistance (Ru) with impedance spectroscopy (ZIR). The Ru (or Ohmic drop) of 4.47 ± 3 Ω was equivalent to a standard electrochemical cell (ESI† Table S1), leading to an acceptable peak separation in the FE-EPR cell compared to a standard electrochemical cell (ESI† Fig. S3c). Peak broadening in FE-EPR cell CV experiments was attributed to limited ET between the WE and CE caused by the reduced supporting electrolyte solution volume. To confirm this, the experiment was repeated at a higher ionic strength in the same buffer (300 mM) leading to significantly reduced broadening (ESI† Fig. S3d). However, ionic strength can have repercussions on protein stability.23 Since peak broadening has no influence on determining the midpoint potential (Em) and considering that our set-up was designed to be compatible with proteins and biological membranes, subsequent experiments used a lower more physiologically relevant ionic strength (150 mM). Next, we assessed the reproducibility of the amount of electroactive material on the WE by performing a potentiometric titration with amino-TEMPO. Mesoporous electrodes were used to enable direct comparison with 3D electrodes.4 Each amino-TEMPO functionalised WE was poised at a given potential with chronoamperometry inside the FE-EPR cell. The desired potential was reached in <30 s. The EPR tube with the FE-EPR cell was flash-frozen in liquid nitrogen after 60 s, without removing any of the electrodes, and continuous wave (CW) EPR spectra were acquired at 100 K. Double-integrals of the EPR spectra as a function of potential were fitted using the Nernst equation with Em = +0.853 V vs. SHE (as determined from CV) and resulted in an apparent ET constant (n) of only 0.13 (see ESI† Fig. S4). The TEMPO·EPR signals visible at potentials when only EPR-silent TEMPO+ should be present shows that some molecules did not respond to the applied potential (ca. 29%, see ESI† Fig. S5), likely because they did not react during functionalisation and yet remained trapped inside the pores. To account for the electrochemically inactive TEMPO·, double integrals of spectra from the samples poised at 1.07 and 1.12 V vs. SHE were averaged and subtracted from all remaining spectra (Fig. 2b inset). Fig. 2b shows the resulting Nernst plot, fitted with Em = +0.853 V vs. SHE and n = 1. Thus, accurate potential control is achieved and the amount of redox-active material inside the WEs is reproducible.
 | ||
| Fig. 1 Overview of FE-EPR set-up, working electrode assembly process and structure. (a) Production and assembly of mesoporous and inverse-opal mesoporous ITO working electrodes: 1 laser-cutting Ti strips out of Ti foil; 2 sputtering a layer of ITO onto Ti strips; 3 drop-casting the dispersion on to the sputtered strips; 4 annealing the strips at 500 °C; 5 resulting working electrode (WE) assembled in the FE-EPR set-up in a 4 mm O.D. EPR tube, with the reference electrode (RE) position at 2.5 cm from the bottom of the tube, at half the length of the WE; all the electrodes are submerged in 300 μL of electrolyte solution. (b) SEM images of the ITO structures of (i) mesoITO and (ii) IO-mesoITO working electrodes. | ||
 | ||
| Fig. 2 Benchmarking of the modified PFE-EPR set-up using amino-TEMPO. (a) Cyclic voltammograms in a standard electrochemical cell (black) and in the PFE-EPR cell (red) at 10 mV s−1 (b) Titration curve resulting from plotting the double integral of the EPR spectra (100 K) of different electrodes vs. the specific potentials that they were poised at. The dotted line represents the fit to the Nernst equation with n = 1 and Em = +0.853 V vs. SHE. The inset shows the averaged EPR spectra acquired at 100 K after subtraction of unreacted amino-TEMPO. | ||
To evaluate the applicability of the improved FE-EPR set-up with tuneable WE pore size to membrane proteins, we isolated inner membrane vesicles (IMVs) from E. coli with overexpressed methionine sulfoxide reductase Q (MsrQ, Fig. 3a and b). MsrQ was selected because its structure and function, particularly its interaction with endogenous quinones in the membrane, is not fully understood.24,25 Direct potential control is in principle possible in this system (ESI† Fig. S6) and would eliminate the need for mediators with overlapping g ∼ 2 EPR signals, such that this interaction could be elucidated by monitoring semiquinone intermediates. Similar to IMVs obtained from other Gram-negative bacteria such as R. sphaeroides,26 our IMVs had an average hydrodynamic diameter (AHD) of 150 nm with some aggregation occurring (AHD ∼ 400 nm), as shown in Fig. 3c. Such aggregation is common27,28 and ITO WEs were designed to have large macropores (∼640 nm) to facilitate IMV incorporation. Functionalisation of the ITO surface with amino-terminated phosphonic acid was essential (protonated at pH 7.0), providing a positive surface for the negatively charged IMVs to bind (see Methods and ESI† Fig. S7 for negative control). Successful functionalisation and IMV incorporation were confirmed by X-ray photoelectron spectroscopy (XPS) (Fig. 4a and ESI† Fig. S8): the functionalised WE (‘ITO-NH2’) exhibits N 1s signals that are absent in the blank (‘ITO’); following IMV drop casting (‘ITO-NH2 + IMVs’) C and N (main components of biological membranes) signals increase significantly. The disappearance of the Sn, In and Ti signals (and yet a similar intensity O 1s signal, also a component of biological membranes) from the drop-casted sample suggest that the IMV ‘film’ thickness exceeded 10 nm, making the ITO surface largely inaccessible to X-rays at these energies. EPR spectra of MsrQ-overexpressed IMVs (OE+) and not overexpressed (OE−), in buffer, were recorded prior to their incorporation into the WE (Fig. 4b). The same total protein concentration in the two samples is confirmed by the similar intensity g ∼ 6 signal arising from high-spin hemes present in other enzymes (not MsrQ) in the IMVs.29 Non-specific Fe3+ gives rise to a sharp signal at g = 4.3 with variable intensity, as is common in biological samples.30 In OE+ only, a heme b present in MsrQ gives rise to a low-spin Fe3+ signal with characteristic g values of 2.95 and 2.25.24 The much-increased g ∼ 2 signal intensity in OE+ suggests that an organic radical in the membranes depends on MsrQ overexpression, providing a first indication that these could be semiquinones of mechanistic interest. Fig. 4c (i) shows the EPR spectra of OE+ IMVs drop casted onto IO-mesoITO WEs. No potential was applied, to allow direct comparison with the solution EPR spectra. High-spin Fe3+ at g ∼ 6 is only visible when IMVs are present and the organic radical signal is significantly higher compared to the ‘blank’ electrode, further confirming successful incorporation of IMVs into the WE. Notably, the g ∼ 6 heme signals can be identified with EPR. The iron cofactors of such membrane-bound hemes are deeply buried inside the IMVs and thus invisible to XPS. MsrQ heme b signals could not be observed with IMV WEs due to insufficient sensitivity; the amount of IMVs adsorbed is an order of magnitude lower compared to the solution sample (based on the intensity difference of the g ∼ 6 signal). Similar spectra resulted when using mesoITO WEs (ESI† Fig. S9). Fig. 4c (ii) shows that IO-mesoITO WEs allowed incorporation of approximately twice as many IMVs compared to the mesoITO WEs, as judged by the intensity difference of the g ∼ 6 signal, when scaling the spectra by the intrinsic ITO signal at g ∼ 1.9.
 | ||
Fig. 3 MsrQ in bacteria, illustration of MsrQ IMVs and their characterisation. (a) Location of MsrQ in bacteria and electron transfer pathways involving MsrQ. (b) Schematic representation of inner membrane vesicles (IMVs) obtained from E. coli overexpressed with MsrQ. IMVs were isolated using a French press and sequential ultracentrifugations. (c) Dynamic Light Scattering measurement of IMVs. Inset: Transmission Electron Microscopy images of inner membrane vesicles at and 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× magnification. 000× magnification. | ||
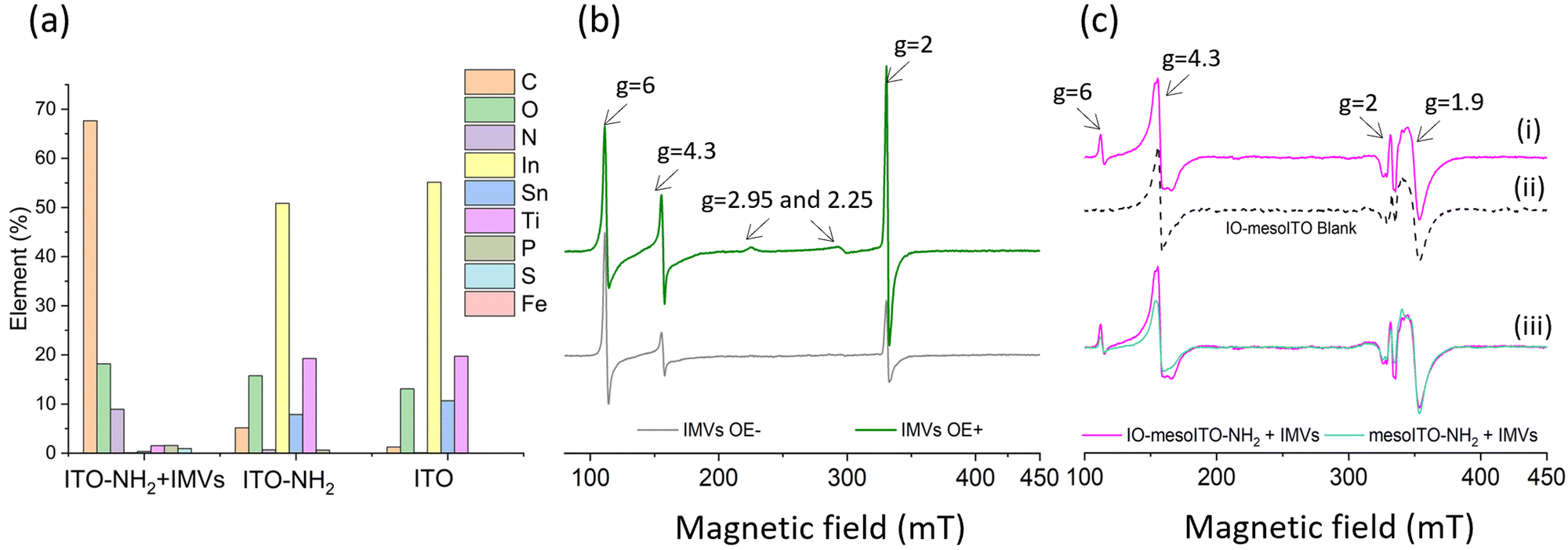 | ||
| Fig. 4 Characterisation of E. coli IMVs with MsrQ on IO-mesoITO working electrons. (a) XPS survey of IO-mesoITO electrodes at different steps of functionalisation; (b) CW EPR spectra of IMVs overexpressed (green) and not overexpressed (purple) at 30 mg ml−1 in MOPS 100 mM + EDTA 5 mM, pH 7.0; (c) CW EPR spectra of ITO electrodes after drop cast of 10 μL of the dispersion of IMVs at 30 mg ml−1 in MOPS 100 mM + EDTA 5 mM, pH 7.0: (i) IO-mesoITO-NH2 + IMVs, (ii) IO-mesoITO blank (iii) comparison between mesoITO-NH2 and IO-mesoITO-NH2 + IMVs. EPR spectra recorded at 12 K, 18 G modulation amplitude and 2 mW microwave power, 6 scans. | ||
In this work we have demonstrated extended capabilities of our FE-EPR method. Macroporous WEs and tuneable surface functionalisation allow incorporation of redox-active molecules of variable size – not only small soluble proteins4 but also large membrane vesicles. The improved set-up enables electrochemical experiments on par with a standard cell, but in an EPR tube. The feasibility of investigating proteins in their native membrane environment was exemplified through E. coli IMVs. We successfully incorporated IMVs into the macropores of the ITO electrode and confirmed EPR detectability of the paramagnetic centres in the membrane protein MsrQ. This paves the way for investigating other membrane proteins with PFE-EPR, although careful optimisation (e.g. WE pore size and surface charge, see ESI† Table S2) will be required in each case. Sensitivity challenges will need to be overcome to observe highly anisotropic EPR signals such as those of MsrQ heme b, which are already of low intensity in solution-based EPR spectra. Nonetheless, ex situ FE-EPR with IO-mesoITO WE constitutes a promising approach for investigating radical intermediates in ET reactions in membrane proteins. For MrsQ, this opens the door to investigating interactions of the enzyme with quinones in a native membrane environment. Future investigations will focus on the ET processes between the electrode and the IMVs, aiming to achieve full control of the potentials of membrane proteins in their native environments.
DF thanks Imperial College and Bruker for a PhD studentship. We are grateful to the EPSRC (EP/T031425/1 supporting PEPR and EP/W005794/1 to MMR) and the Leverhulme Trust (RPG-2018-183 to MMR) for funding.
Data availability
Data are available at the Imperial College London Research Data Repository DOI: https://doi.org/10.14469/hpc/14513.Conflicts of interest
There are no conflicts of interest to declare.References
- Y. Zhai, Z. Zhu, S. Zhou, C. Zhu and S. Dong, Nanoscale, 2018, 10, 3089–3111 RSC
.
- M. Roessler and E. Salvadori, Chem. Soc. Rev., 2018, 47, 2534–2553 RSC
- K. H. Richardson, M. Seif-Eddine, A. Sills and M. M. Roessler, Methods Enzymol., 2022, 666, 233–296 CAS
- K. Abdiaziz, E. Salvadori, K. P. Sokol, E. Reisner and M. M. Roessler, Chem. Commun., 2019, 55, 8840–8843 RSC
- M. Seif-Eddine, S. J. Cobb, Y. Dang, K. Abdiaziz, M. A. Bajada, E. Reisner and M. M. Roessler, Nat. Chem., 2024, 16(6), 1015–1023 CrossRef CAS PubMed
- F. A. Walker, Coord. Chem. Rev., 1999, 186, 471–534 CrossRef
- K. H. Richardson, J. J. Wright, M. Šimėnas, J. Thiemann, A. M. Esteves, G. McGuire, W. K. Myers, J. J. L. Morton, M. Hippler, M. M. Nowaczyk, G. T. Hanke and M. M. Roessler, Nat. Commun., 2021, 12(1), 5387 CrossRef CAS PubMed
- N. Le Breton, J. J. Wright, A. J. Y. Jones, E. Salvadori, H. R. Bridges, J. Hirst and M. M. Roessler, J. Am. Chem. Soc., 2017, 139, 16319–16326 CrossRef CAS PubMed
- J. Eisermann, M. Seif-Eddine and M. M. Roessler, Curr. Opin. Chem. Biol., 2021, 61, 114–122 CrossRef CAS PubMed
- S. A. Bonke, T. Risse, A. Schnegg and A. Brückner, Nat. Rev. Methods Primers, 2021, 1, 1–20 CrossRef
- J. N. Butt, L. J. C. Jeuken, H. Zhang, J. A. J. Burton and A. L. Sutton-Cook, Nat. Rev. Methods Primers, 2023, 3, 78 CrossRef
- J. J. Lacapère, E. Pebay-Peyroula, J. M. Neumann and C. Etchebest, Trends Biochem. Sci., 2007, 32, 259–270 CrossRef PubMed
- A. Pandey, K. Shin, R. E. Patterson, X. Q. Liu and J. K. Rainey, Biochem. Cell Biol., 2016, 94, 507–527 CrossRef CAS PubMed
- L. Reed, V. M. Arlt and D. H. Phillips, Carcinog, 2018, 39, 851–859 CrossRef CAS PubMed
- J. J. Wright, J. G. Fedor, J. Hirst and M. M. Roessler, BMC Biol., 2020, 18, 54 CrossRef CAS PubMed
- F. Melin and P. Hellwig, Chem. Rev., 2020, 120, 10244–10297 CrossRef CAS PubMed
- K. Abdiaziz, PhD Thesis, Queen Mary Univ. of London, 2019.
- K. P. Sokol, D. Mersch, V. Hartmann, J. Z. Zhang, M. M. Nowaczyk, M. Rögner, A. Ruff, W. Schuhmann, N. Plumeré and E. Reisner, Energy Environ. Sci., 2016, 9, 3698–3709 RSC
- D. Schaming, C. Renault, R. T. Tucker, S. Lau-Truong, J. Aubard, M. J. Brett, V. Balland and B. Limoges, Langmuir, 2012, 28, 14065–14072 CrossRef CAS PubMed
- A. Pohl and B. Dunn, Thin Solid Films, 2006, 515, 790–792 CrossRef CAS
- X. Fang, S. Kalathil, G. Divitini, Q. Wang and E. Reisner, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 5074–5080 CrossRef CAS PubMed
- P.
J. Hotchkiss, S. C. Jones, S. A. Paniagua, A. Sharma, B. Kippelen, N. R. Armstrong and S. R. Marder, Acc. Chem. Res., 2012, 45, 337–346 CrossRef CAS PubMed
- Y. Zhang, B. Zhang and H. S. White, J. Phys. Chem. B, 2006, 110, 1768–1774 CrossRef CAS PubMed
- C. Caux, B. Guigliarelli, C. Vivès, F. Biaso, M. Horeau, H. Hassoune, I. Petit-Hartlein, C. Juillan-Binard, S. Torelli, F. Fieschi and V. Nivière, ACS Chem. Biol., 2021, 16, 2547–2559 CrossRef CAS PubMed
- A. Gennaris, B. Ezraty, C. Henry, R. Agrebi, A. Vergnes, E. Oheix, J. Bos, P. Leverrier, L. Espinosa, J. Szewczyk, D. Vertommen, O. Iranzo, J. F. Collet and F. Barras, Nature, 2015, 528, 409–412 CrossRef CAS PubMed
- M. Lommen and J. Takemoto, J. Bacteriol., 1978, 136, 730–741 CrossRef CAS PubMed
- M. Futai, J. Membr. Biol., 1974, 15, 15–28 CrossRef CAS PubMed
- B. Rosen and T. Tsuchiya, Methods Enzymol., 1979, 56, 233–241 CAS
- C. Juillan-Binard, A. Picciocchi, J. P. Andrieu, J. Dupuy, I. Petit-Hartlein, C. Caux-Thang, C. Vivès, V. Nivière and F. Fieschi, J. Biol. Chem., 2017, 292, 2485–2494 CrossRef CAS PubMed
-
W. R. Hagen, Biomolecular EPR Spectroscopy, CRC Press, 1st Edn., 2008, p. 84 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc04013a |
| This journal is © The Royal Society of Chemistry 2024 |