
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
NRAS DNA G-quadruplex-targeting molecules for sequence-selective enzyme inhibition†
Yoshiki Hashimoto,
Hiroki Kubo,
Keiko Kawauchi and
Daisuke Miyoshi *
*
Frontiers of Innovative Research in Science and Technology, Konan University, 7-1-20 Minatojima-minamimachi, Chuo-ku, Kobe, Hyogo 650-0047, Japan
First published on 27th September 2024
Abstract
Sequence-selective G-quadruplex ligands are valuable for controlling gene expression. Here, we established a new fluorescence displacement assay using a NRAS G-quadruplex selective fluorescent probe to identify sequence-selective DNA G-quadruplex ligands. These sequence-selective NRAS G-quadruplex ligands retained their binding affinity even in the presence of excessive human telomeric DNA G-quadruplex and regulated enzymatic activities in a sequence-selective manner.
Guanine-rich nucleic acid sequences can fold into G-quadruplex (G4) structures1,2 that play diverse roles in biological processes such as transcription, translation, and replication.3,4 Since G4 forming sequences are localized in biologically important genetic regions such as oncogenes and telomeres, G4s have garnered attention as targets for development of novel cancer treatment strategies to regulate oncogene expression. Various small molecules targeting G4 structures, G4 ligands, have been developed.5 These compounds are expected to stabilize G4s to control gene expression.
Since almost all genomic DNA folds into canonical duplexes in cells, the structure-selectivity of G4 ligands is crucial for their intracellular applications. There are several reports on the development of structure-selective G4 ligands such as pyridostatin and telomestatin.6,7 We also previously developed a screening system to identify structure-selective G4 ligands.8 However, to target a specific gene, not only structure-selectivity but also sequence-selectivity of G4 ligands is highly required, since approximately 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 potential G4-forming sequences exist in the human genome.9 Here, we define a sequence-selective G4 ligand as a small compound which binds to the target G4 but does not bind to other G4s.
000 potential G4-forming sequences exist in the human genome.9 Here, we define a sequence-selective G4 ligand as a small compound which binds to the target G4 but does not bind to other G4s.
Benzothiazole hydrazone analogues reported by Pradeepkumar's group showed approximately 3–20 times higher affinity for c-MYC G4 compared to other G4s.10 Madder's group developed a method for sequence-selective alkylation to a target G4 by conjugating an alkylation compound with the complementary sequence of the target DNA.11 Some sequence-selective G4 targeting by conjugated oligonucleotides has also been achieved.12,13 However, conjugation with nucleic acids could make G4 ligands difficult to deliver into living cells. Thus, G4 ligands with higher sequence-selectivity are still required. However, it is difficult to design such sequence-selective G4 ligands rationally. Here, we established a novel screening system for sequence-selective G4 ligands based on fluorescent intercalator displacement (FID).12 By utilizing this system, we identified new NRAS DNA G4 sequence-selective ligands. Importantly, it was demonstrated that the G4 ligands stabilized the target G4 and inhibited enzymatic activity in a sequence-selective manner.
A fluorescent probe for the FID is essential to screen NRAS DNA sequence-selective G4 ligands. We previously reported that an anionic phthalocyanine coordinating with Zn2+ (ZnAPC) can sequence-selectively bind to the NRAS mRNA G4,13 whereas it did not bind to telomeric DNA G4, which is the most abundant G4 in cells.14 We thus hypothesized that ZnAPC would bind to NRAS DNA G4 but not to telomeric DNA G4. Since the majority of G4 structures in cells are formed by telomere sequences, we evaluated the affinity and selectivity of ZnAPC with NRAS G4 against the telomeric DNA G4 as a proof of concept. We used two oligonucleotides, NRAS and TELO, which were derived from human NRAS and human telomere, respectively (Table S1, ESI†). Fig. S1 (ESI†) shows normalized UV melting curves at 295 nm and circular dichroism (CD) spectra of 10 μM NRAS and TELO. The hypochromic melting transition and the CD spectrum showed that NRAS and TELO folded into a parallel G4 and a mixed G4, respectively. Fig. S2A and B (ESI†) shows the absorption spectra of 20 μM ZnAPC with various concentrations of NRAS and TELO, respectively. An absorbance increase at 620 nm and a decrease at 680 nm were observed with increasing concentration of NRAS. Conversely, no significant change was observed with TELO, indicating the selective binding of ZnAPC with NRAS. In addition, we confirmed whether ZnAPC can maintain its affinity for NRAS even in the presence of 50 μM TELO (Fig. S2C, ESI†). Fig. S2D (ESI†) shows plots of Δabs680, (abs. at 680 nm with DNA) – (abs. at 680 nm without DNA), versus concentration of NRAS in the absence or presence of TELO. ZnAPC showed Kd values of 5.8 ± 0.7 μM and 6.0 ± 0.6 μM in the absence and presence of 50 μM TELO, respectively, at 25 °C (see ESI† for evaluation of the Kd values). These results demonstrate that the affinity of ZnAPC for NRAS was maintained even in the presence of excessive TELO. Higher concentrations of NRAS increased the fluorescence intensity of ZnAPC more than 20-fold with and without 50 μM TELO (Fig. S3A, ESI†), further confirming the sequence-selectivity of ZnAPC for NRAS (Fig. S3B, ESI†). It is not yet clear how ZnAPC can bind to the target NRAS G4 in a sequence-selective manner, although it is considered that not only the loop sequence but also the franking sequence are important for the sequence selective biding of ZnAPC.14 The increase in fluorescence intensity with sequence-selective binding shows that ZnAPC is a suitable probe for FID.
Fig. 1A shows a schematic illustration of a sequence-selective G4 ligand screening by ZnAPC displacement. In this assay, the affinity index (IAffinity), showing the degree of displacement by a tested compound, was evaluated from the F690 (fluorescence intensity of ZnAPC at 690 nm) decrease as the following: (F690 with the compound)/(F690 without the compound). Compounds showing lower IAffinity are expected to have stronger affinity for NRAS. We screened about 3000 compounds to identify sequence-selective ligands targeting NRAS. The obtained IAffinity values in the absence or presence of TELO were organized as shown in Fig. 1B. Compounds observed in the blue area lack sequence-selectivity, while compounds in the red area may possess sequence-selectivity. As shown in Fig. 1C, the screening results identified three compounds highlighted in red: pacritinib,15 osimertinib,16 and JNK-IN-817 (Fig. 2A), which exhibited the lowest IAffinity in the presence of TELO. It was demonstrated that the quenching of ZnAPC is not due to a direct interaction with the ligand (Fig. S4, ESI†). Of these three, pacritinib exhibited the lowest IAffinity in the presence of TELO, suggesting that it has the highest sequence-selectivity.
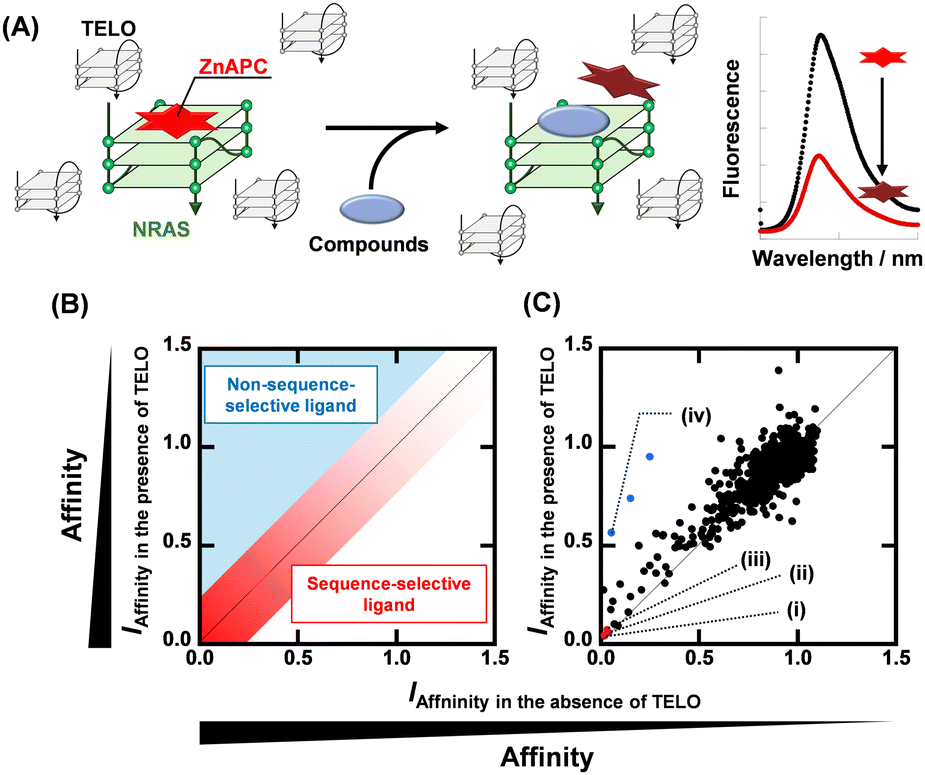 | ||
| Fig. 1 (A) Schematic illustration of sequence-selective G4 ligand screening by ZnAPC displacement. (B) Plots of IAffinity of ligands in the presence of 50 μM TELO versus IAffinity of ligands in the presence of 0 μM TELO. Plots located within the red area have sequence-selectivity. (C) Plots of IAffinity of compounds in the presence of 50 μM TELO versus IAffinity of compounds in the absence of 50 μM TELO. (i) Pacritinib, (ii) osimertinib, (iii) JNK-IN-8, and (iv) PDS. | ||
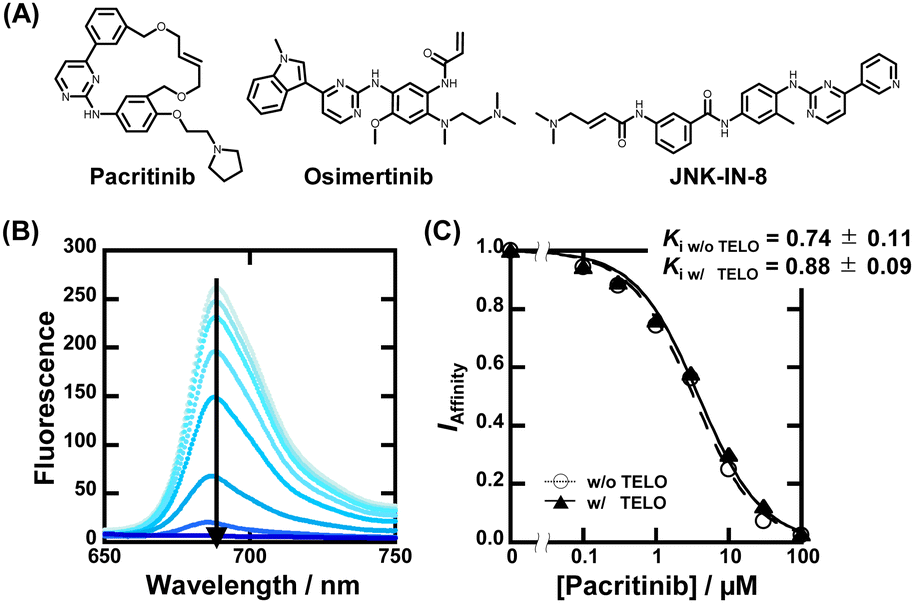 | ||
| Fig. 2 (A) Chemical structures of pacritinib, osimertinib, and JNK-IN-8. (B) Fluorescence spectra of 20 μM ZnAPC with 0, 0.1, 0.3, 1, 3, 10, 30, and 100 μM pacritinib in the buffer containing 50 mM MES-LiOH (pH 7.0), 100 mM KCl, and 1 μM NRAS at 25 °C. (C) Plots of IAffinity versus concentration of pacritinib. | ||
To confirm the binding affinity and selectivity of these compounds, we observed changes in IAffinity with varying concentrations of the compounds. Fig. 2B and Fig. S4A (ESI†) show the fluorescence spectra of 20 μM ZnAPC and 1 μM NRAS with various concentrations of pacritinib in the absence and presence of 50 μM TELO. The observed Kd (Ki) values of the ligands were evaluated from fluorescence changes of ZnAPC in the absence and presence of 50 μM TELO (Fig. 2C). The Ki values of pacritinib were 0.74 ± 0.11 μM and 0.88 ± 0.09 μM in the absence and presence of 50 μM TELO, respectively. The lack of significant change in these Ki values confirms the high sequence-selectivity of pacritinib. Similar results were obtained for osimertinib and JNK-IN-8 (Fig. S5B and C, ESI†). In contrast, the Ki values of pyridostatin (PDS), which was identified as a non-sequence-selective ligand (Fig. 1C(iv)), in the presence of 50 μM TELO (4.1 ± 0.1 μM) was about 10 times larger than that in the absence of 50 μM TELO (0.47 ± 0.08), confirming the lower sequence selectivity of PDS (Fig. S5D, ESI†). In addition, we carried out ThT displacement (TD) assays8 for TELO (see ESI† for evaluation of Ki, Fig. S6). The Ki values of pacritinib, osimertinib, and JNK-IN-8 were 77 μM, 89 μM, and 103 μM, respectively, for TELO, at 25 °C, which are about 100-fold larger than that for NRAS (Fig. S6C, ESI†). The Ki values support the high sequence-selectivity of pacritinib, osimertinib, and JNK-IN-8. In comparison, the Ki value of PDS for TELO was 0.64, which is almost the same as that for NRAS, confirming that PDS binds to G4s in a non-sequence-selective manner.
Next, we investigated the effect of these compounds on the structure and stability of NRAS. Fig. S6A (ESI†) shows CD spectra of 10 μM NRAS in the absence and presence of the three G4 ligands at 50 μM, suggesting that these compounds did not alter the structure of NRAS G4. The thermal stability of NRAS was studied using CD melting curves at 265 nm (Fig. S7B, ESI†). The Tm value of 10 μM NRAS was 41.0 ± 0.9 °C. Pacritinib, osimertinib, and JNK-IN-8 changed the Tm value to 72.7 ± 0.6 °C, 63.9 ± 0.6 °C, and 54.2 ± 0.3 °C, respectively (Fig. S7C, ESI†). These results indicate that the ligands not only bind sequence-selectively but also strongly stabilize NRAS.
With the identification of the sequence-selective G4 ligands, we performed T7 RNA polymerase (RNAP) stop assays to investigate the inhibitory effects of the G4 ligands on transcription. A template containing the NRAS G4-forming sequence (NRAS template, Table S1, ESI†) was utilized for the transcription reaction. The transcription products with G4 ligands in concentrations ranging from 0 to 100 μM were visualized using denaturing polyacrylamide gel electrophoresis (PAGE). When the transcription reaction proceeds completely, completed transcripts of 66 nucleotides can be observed (Fig. 3A). When the transcription reaction is inhibited by G4 ligands, arrested transcripts of 36 nucleotides are produced. Fig. 3B shows the results for pacritinib in the absence and presence of 50 μM TELO. A decrement of the completed transcript and an increment of the arrested transcript were observed with increasing concentration of pacritinib under both conditions. The IC50 values of pacritinib in the absence and presence of 50 μM TELO were 28 ± 2 μM and 32 ± 2 μM, respectively, at 37 °C (Fig. 3C) (see ESI† for evaluation of IC50). These results show that pacritinib can inhibit T7 RNAP activity in a sequence-selective manner. The significant difference between the values of the IC50 and the Ki was observed. Although the reason for the difference remains unclear, one of possible reasons is that the NRAS stabilized by the ligands competes the T7 RNAP binding to the substrate DNA, resulting in an unwinding of the DNA G4 structure. In addition, it was reported that not only their thermal stability but also the kinetics of the G4 formation and the ligand binding with G4 are also important for the inhibition of the polymerase activity.18 The kinetic controlling of the T7 RNAP activity may lead to the difference between the IC50 and the Ki values. The sequence-selective inhibition of T7 RNAP was also observed for osimertinib and JNK-IN-8 (Fig. S8, ESI†). These results demonstrate that the novel sequence-selective G4 ligands can sequence-selectively inhibit transcription activity mediated by stabilization of NRAS G4. Because of the direct inhibition of T7 RNAP activity by PDS at high concentrations in the absence of TELO, the experiments were conducted with PDS only up to 10 μM (Fig. S9, ESI†). Although PDS exhibited a high transcription inhibitory effect in the absence of TELO, the effect was hardly observed in the presence of TELO. To confirm that the transcription inhibitory effects of these compounds were not unique for T7 RNAP, we performed the T7 RNAP assay with a mutNRAS template (see Table S1 for sequence, ESI†), which does not form a G4 structure (Fig. S10, ESI†). Pacritinib, osimertinib, and JNK-IN-8 did not affect the transcription activity with the mutNRAS template, confirming that their inhibitory effect on transcription was mediated by G4 formation. In contrast, PDS decreased the total amount of transcripts in the absence of TELO, suggesting that a higher concentration of PDS directly inhibits the T7 RNAP activity.
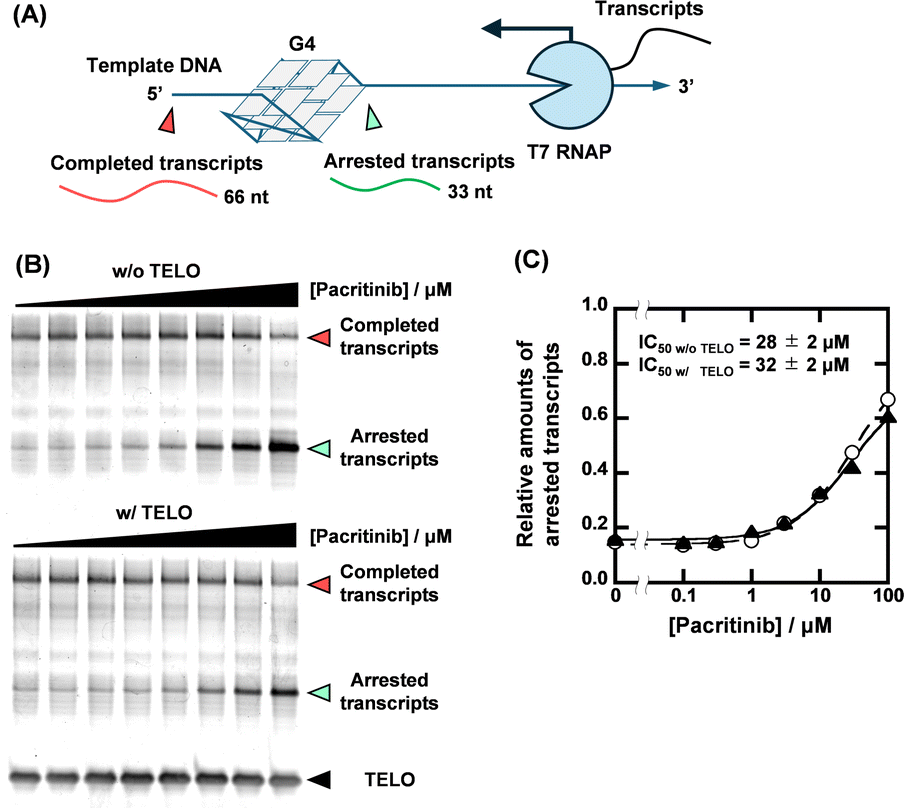 | ||
| Fig. 3 (A) Schematic illustration of T7 RNA polymerase stop assay with G4-forming template strand. (B) Transcripts from 1 μM NRAS template with 0, 0.1, 0.3, 1, 3, 10, 30, and 100 μM pacritinib in the absence (top) and presence (bottom) of 50 μM TELO. The transcription reactions were carried out in a buffer containing 1 mM KCl, 99 mM LiCl, 8 mM MgCl2, and 40 mM Tris-HCl (pH 7.2) at 37 °C for 2 hours. (C) Plots of relative amounts of arrested transcript with NRAS template versus concentration of pacritinib in the absence (circles with dotted line) or presence (triangles with continuous line) of 50 μM TELO. | ||
Given that the ligands showed high sequence-selectivity, it is expected that no inhibitory effects on enzymatic activity via formation of other G4s would be observed. We thus investigated the effects of pacritinib, osimertinib, JNK-IN-8, and PDS on telomere elongation by telomerase using the two-step telomeric repeat amplification protocol (tsTRAP) assay.19 This method allows the study of inhibitory effects of G4 ligands on the telomerase extension reaction without affecting the subsequent PCR stop.19 Fig. 4A shows a nondenaturing PAGE of tsTRAP products with 0, 1, 10, and 100 μM pacritinib, demonstrating the inhibitory effect of pacritinib on telomere elongation. The amount of the internal control by addition of pacritinib was constant, showing that the addition of pacritinib did not interfere with the PCR reaction. More importantly, no significant decrement of telomerase activity was observed upon addition of pacritinib up to 100 μM (Fig. 4B). These results confirm that the sequence-selective G4 ligands targeting NRAS did not inhibit enzymatic activity depending on G4 formation other than NRAS (Fig. 4B). Similar to pacritinib, osimertinib and JNK-IN-8 did not inhibit telomerase activity with increasing concentration (Fig. 4B and Fig. S11A, ESI†). In contrast, PDS strongly inhibited telomerase activity (Fig. 4B and Fig. S11B, ESI†) because of its non-sequence-selective binding to G4s. The sequence-selective NRAS G4 ligands, pacritinib, osimertinib, and JNK-IN-8, inhibited enzyme activity of the target G4 sequence, suggesting that developing target sequence-selective G4 ligands can control target enzymatic reactions. Such target selective enzyme inactivation is an important aspect for cellular applications of G4 ligands involving drug development.
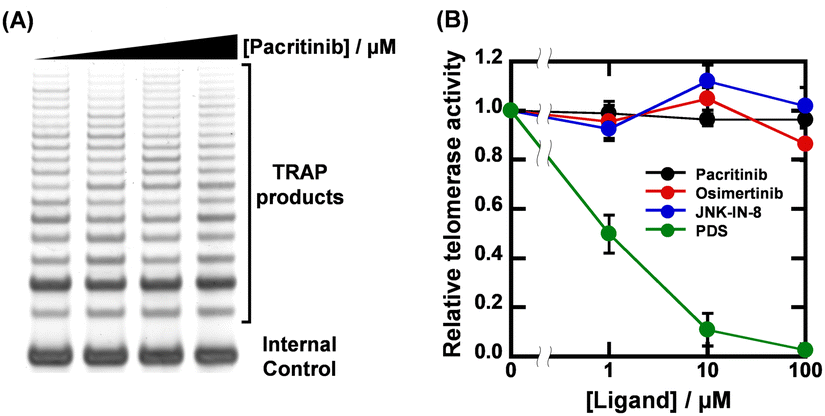 | ||
| Fig. 4 (A) Results of tsTRAP assay with 0, 1, 10, and 100 μM pacritinib. Telomerase elongation reactions were carried out in a buffer containing 200 mM Tris-HCl (pH 8.3), 15 mM MgCl2, 630 mM KCl, 0.5% Tween 20, and 10 mM EGTA for 30 min at 30 °C. (B) Plots of relative telomerase activity versus concentrations of pacritinib, osimertinib, JNK-IN-8, and PDS. | ||
In summary, we established a new screening method for obtaining NRAS sequence-selective G4 ligands. Pacritinib, osimertinib, and JNK-IN-8 were identified as novel sequence-selective G4 ligands that strongly stabilized NRAS. More importantly, the ligands showed sequence-selective regulation for enzymatic activity in the transcription reaction and telomere elongation. Although disease-related proteins have attracted attention as therapeutic targets, some of them, including NRAS, are often difficult to target with small molecule drugs.20–22 The use of sequence-selective G4 ligands enables the targeting of specific G4s, potentially allowing the control of specific biological reactions regardless of the protein structure. Although sequence-selective G4 ligands are useful for inhibit target enzyme inhibition, this screening method can be applied only for NRAS due to the properties of ZnAPC. For a screening of sequence-selective G4 ligands targeting other sequences, a sequence-selective fluorescent probe for the target G4 is required. As an alternative approach, performing of screenings for the target G4 and for the decoy G4 makes possible to obtain sequence-selective G4 ligands. The results obtained in this study may stimulate further studies for screening and designing sequence-selective G4 ligands.
Conceptualization, D. M.; methodology, D. M.; formal analysis, Y. H. and H. K.; investigation, Y. H. and H. K.; data curation, Y. H.; writing – original draft preparation, Y. H.; writing – review and editing, K. K. and D. M.; visualization, D. M.; supervision, K. K. and D. M.; project administration, D. M.; funding acquisition, Y. H., K. K. and D. M.
This work was supported by JSPS KAKENHI, grant numbers 24K21801, 21H02062, and 20K21259, a Research Grant of the Asahi Glass Foundation, Japan, the Hirao Taro Foundation and Junzo Tateno Foundation of Konan Gakuen for Academic Research, Japan, and JST SPRING, grant number JPMJSP2117.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Gellert, M. N. Lipsett and D. R. Davies, Proc. Natl. Acad. Sci. U. S. A., 1962, 48, 2013–2018 CrossRef CAS PubMed.
- D. Sen and W. Gilbert, Nature, 1988, 334, 364–366 CrossRef CAS PubMed.
- S. Balasubramanian, L. H. Hurley and S. Neidle, Nat. Rev. Drug Discovery, 2011, 10, 261–275 CrossRef CAS PubMed.
- D. Rhodes and H. J. Lipps, Nucleic Acids Res., 2015, 43, 8627–8637 CrossRef CAS PubMed.
- Y. H. Wang, Q. F. Yang, X. Lin, D. Chen, Z. Y. Wang, B. Chen, H. Y. Han, H. D. Chen, K. C. Cai, Q. Li, S. Yang, Y. L. Tang and F. Li, Nucleic Acids Res., 2022, 50, D150–D160 CrossRef CAS PubMed.
- R. Rodriguez, S. Müller, J. A. Yeoman, C. Trentesaux, J. F. Riou and S. Balasubramanian, J. Am. Chem. Soc., 2008, 130, 15758–15759 CrossRef CAS PubMed.
- M. Y. Kim, H. Vankayalapati, K. Shin-Ya, K. Wierzba and L. H. Hurley, J. Am. Chem. Soc., 2002, 124, 2098–2099 CrossRef CAS PubMed.
- Y. Hashimoto, Y. Imagawa, K. Nagano, R. Maeda, N. Nagahama, T. Torii, N. Kinoshita, N. Takamiya, K. Kawauchi, H. Tatesishi-Karimata, N. Sugimoto and D. Miyoshi, Chem. Commun., 2023, 59, 4891–4894 RSC.
- V. S. Chambers, G. Marsico, J. M. Boutell, M. Di Antonio, G. P. Smith and S. Balasubramanian, Nat. Biotechnol., 2015, 33, 877–881 CrossRef CAS PubMed.
- S. P. Pany, P. Bommisetti, K. V. Diveshkumar and P. I. Pradeepkumar, Org. Biomol. Chem., 2016, 14, 5779–5793 RSC.
- E. Cadoni, L. De Paepe, G. Colpaert, R. Tack, D. Waegeman, A. Manicardi and A. Madder, Nucleic Acids Res., 2023, 51, 4112–4125 CrossRef CAS PubMed.
- D. Monchaud, C. Allain and M. P. Teulade-Fichou, Bioorg. Med. Chem. Lett., 2006, 16, 4842–4845 CrossRef CAS PubMed.
- S. Kumari, A. Bugaut, J. L. Huppert and S. Balasubramanian, Nat. Chem. Biol., 2007, 3, 218–221 CrossRef CAS PubMed.
- K. Kawauchi, W. Sugimoto, T. Yasui, K. Murata, K. Itoh, K. Takagi, T. Tsuruoka, K. Akamatsu, H. Tateishi-Karimata, N. Sugimoto and D. Miyoshi, Nat. Commun., 2018, 9, 2271 CrossRef PubMed.
- S. Hart, K. C. Goh, V. Novotny-Diermayr, C. Y. Hu, H. Hentze, Y. C. Tan, B. Madan, C. Amalini, Y. K. Loh, L. C. Ong, A. D. William, A. Lee, A. Poulsen, R. Jayaraman, K. H. Ong, K. Ethirajulu, B. W. Dymock and J. W. Wood, Leukemia, 2011, 25, 1751–1759 CrossRef CAS PubMed.
- D. A. Cross, S. E. Ashton, S. Ghiorghiu, C. Eberlein, C. A. Nebhan, P. J. Spitzler, J. P. Orme, M. R. Finlay, R. A. Ward, M. J. Mellor, G. Hughes, A. Rahi, V. N. Jacobs, M. Red Brewer, E. Ichihara, J. Sun, H. Jin, P. Ballard, K. Al-Kadhimi, R. Rowlinson, T. Klinowska, G. H. Richmond, M. Cantarini, D. W. Kim, M. R. Ranson and W. Pao, Cancer Discovery, 2014, 4, 1046–1061 CrossRef CAS PubMed.
- T. Zhang, F. Inesta-Vaquera, M. Niepel, J. Zhang, S. B. Ficarro, T. Machleidt, T. Xie, J. A. Marto, N. Kim, T. Sim, J. D. Laughlin, H. Park, P. V. LoGrasso, M. Patricelli, T. K. Nomanbhoy, P. K. Sorger, D. R. Alessi and N. S. Gray, Chem. Biol., 2012, 19, 140–154 CrossRef CAS PubMed.
- S. Takahashi, A. Kotar, H. Tateishi-Karimata, S. Bhowmik, Z. F. Wang, T. C. Chang, S. Sato, S. Takenaka, J. Plavec and N. Sugimoto, J. Am. Chem. Soc., 2021, 143, 16458–16469 CrossRef CAS PubMed.
- H. Yaku, T. Murashima, D. Miyoshi and N. Sugimoto, J. Phys. Chem. B, 2014, 118, 2605–2614 CrossRef CAS PubMed.
- A. D. Cox, S. W. Fesik, A. C. Kimmelman, J. Luo and C. J. Der, Nat. Rev. Drug Discovery, 2014, 13, 828–851 CrossRef CAS PubMed.
- C. V. Dang, Cell, 2012, 149, 22–35 CrossRef CAS PubMed.
- L. Mabonga and A. P. Kappo, Biophys. Rev., 2019, 11, 559–581 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc03753g |
| This journal is © The Royal Society of Chemistry 2024 |