
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdsorption-based capture of iodine and organic iodides: status and challenges
Tingting
Pan
,
Kaijie
Yang
,
Xinglong
Dong
 and
Yu
Han
*
and
Yu
Han
*
Advanced Membranes and Porous Materials (AMPM) Center, Physical Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal, 23955-6900, Saudi Arabia. E-mail: yu.han@kaust.edu.sa
First published on 24th February 2023
Abstract
Nuclear energy is a sustainable low-carbon energy source that plays an increasingly important role in supporting the progress of human society. However, there are safety issues associated with the operation of nuclear reactors. In particular, volatile radioactive elements, primarily 129I and 131I, in the form of molecular iodine (I2) or organic iodides (e.g., CH3I and CH3CH2I), are harmful for the environment and human health and must be removed before discharging the off-gas. Adsorption processes employing porous solid adsorbents to capture radioactive iodine compounds have attracted considerable attention owing to their simple operation and low maintenance cost and because they avoid the use of highly corrosive solutions. Despite the efforts devoted to developing novel adsorbents for iodine capture, certain critical issues related to practical applications have been overlooked. This review summarizes the adsorption mechanisms employed to capture I2 and CH3I, focusing on the different adsorbent requirements. This review also compares the static and dynamic evaluation systems, analyzes the structure–function relationship under different testing conditions, and highlights the importance of using appropriate conditions to evaluate adsorbents. Moreover, the simultaneous capture of I2 and CH3I is discussed, which is quite challenging but has been largely ignored in previous studies. Finally, this review outlines the challenges and opportunities in this field from the perspective of materials design and system evaluation, indicating that properly designing adsorbents to provide sufficient chemisorption sites may be the only way to meet the practical application requirements.
1. Introduction
Nuclear power is one of the most important low-carbon energy sources, which is used to currently generate ∼10% of the global electricity.1 However, the reprocessing of used fuel rods or severe nuclear accidents can produce radioactive gaseous fission products such as 129/131I, 127Xe, and 85Kr.2–4 Therefore, technologies are required to efficiently capture these radioactive wastes, particularly iodine, because of its high volatility and well-established adverse effects on human health. Among the iodine radioisotopes, 129I has an extremely long half-life of 15.7 × 106 years and a lasting environmental impact. Although 131I has a relatively short half-life (approximately eight days), it requires to be immediately captured upon its release because it can damage the thyroid and interfere with the human metabolic process.5,6 The common forms of radioactive iodine in the off-gases of nuclear power plants include molecular iodine (I2), organic iodides such as methyl iodide (CH3I) and ethyl iodide (CH3CH2I). Inorganic radioactive iodine-containing compounds, such as HI, HOI, and ICN, may also be present, but usually in vary small amounts.7–10 Because of the different physicochemical properties of molecular iodine and organic iodides, specially designed absorption or adsorption systems are needed to achieve the desired capture efficiency and capacity.Liquid scrubbing processes have been used to capture radioactive I2 and other I-containing compounds from the off-gas, requiring highly corrosive solutions and high maintenance costs.11 Therefore, adsorption-based radioiodine capture has attracted considerable attention (Fig. 1). Activated carbon and zeolite materials are the most popular industrial adsorbents, but they have low adsorption capacities for iodine and organic iodides. Hence, they cannot meet the requirements of practical applications.12–15 Emerging porous materials, such as metal organic frameworks (MOFs),16–20 porous organic polymers (POPs),21–26 and covalent organic frameworks (COFs),27–29 provide new platforms for developing high-performance adsorbents because of their diverse structures, large surface areas, tunable pore sizes, and designable surface functionalities. Many studies have examined iodine capture based on these emerging adsorbent materials.30–32 However, the conditions most studies used to evaluate adsorbents were not related to practical applications. In particular, many studies measured the I2 adsorption capacity of the developed adsorbents using saturated I2 vapor, ignoring that the actual concentration of I2 in the off-gas stream is orders of magnitude lower. Furthermore, most studies have focused only on the I2 adsorption and ignored the coexisting organic iodides. Adsorbents are difficult to assess without standardizing the measurement conditions because the adsorption capacity is closely related to the type and concentration of the adsorbate. Moreover, although multiple review articles have summarized the adsorption capacities of various reported adsorbent materials, there has been no systematic analysis of the differences in the adsorption mechanism of iodine and organic iodides.
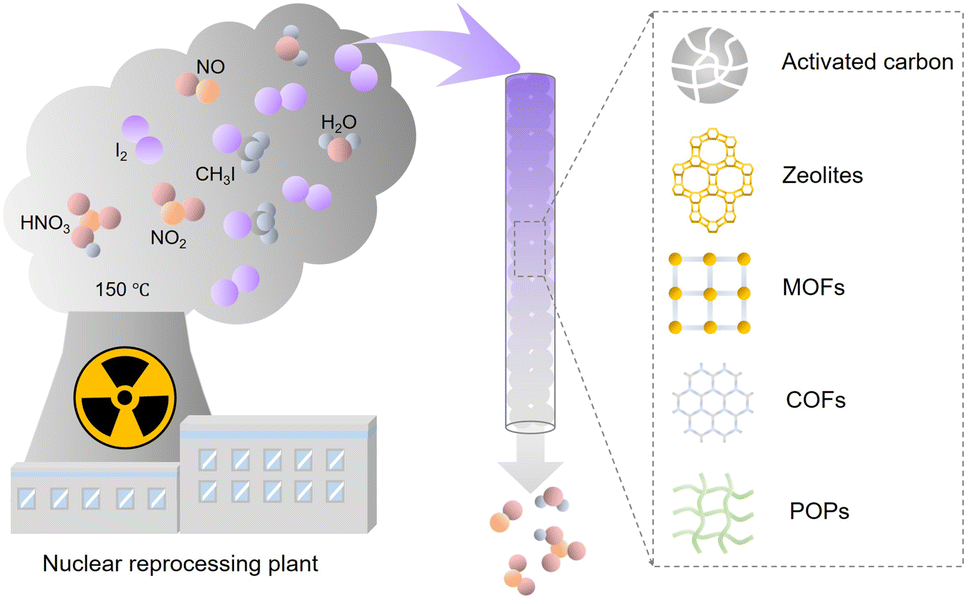 | ||
| Fig. 1 Schematic of the components in nuclear off-gas generated during the reprocessing process and emerging iodine adsorbents. | ||
This review article differs from the previous ones in the following ways. First, this article is structured based on the adsorption mechanism rather than the material type, in which iodine adsorption and organic iodide adsorption are separately discussed because of their different requirements for the adsorbent. Second, this article critically points out that the experimental conditions used in most studies were different from actual off-gas treatment applications and analyzes the origin of unreasonably high I2 adsorption capacities reported in the literature. Third, this article highlights the importance of evaluating adsorbent materials for simultaneous capture of iodine and organic iodides at low concentrations (<150 ppmv), high temperatures (∼150 °C), and dynamic conditions, which has largely been overlooked in previous original research and review articles. Lastly, this article discusses the challenges and future opportunities in this field from material design and performance evaluation perspectives.
2. Mechanisms for molecular iodine capture
Molecular iodine is the primary component of radioactive iodine species in off-gas, which can be captured chemically or physically using various mechanisms, including redox reactions, coordination interactions, electrophilic aromatic substitution, Lewis acid–base interactions, Coulomb interactions, hydrogen bonding, van der Waals interactions, and hydrophobic interactions.2.1 Redox reactions
Metal-containing adsorbents can capture I2via redox reactions. Ag is the most commonly used metal because it can specifically react with I2. Among the various support materials,13–15 zeolites are used widely because of their ion-exchange ability and inherent porosity, allowing for the easy loading and high dispersion of silver. The capture of I2 by Ag-zeolites involves the following reactions, depending on the state of Ag:33| I2 + 2Ag0 → 2AgI | (1) |
| I2 + 2Ag+ + H2O → AgI + AgIO + 2H+ | (2) |
| AgIO + O2 → AgIO3 | (3) |
The standard practice is to reduce the Ag species on the adsorbent to Ag0 prior to use for I2 capture, and studies have been performed to understand the respective roles of Ag0 and Ag+ in this process. For example, Nenoff et al. examined the distribution and structure of AgI formed in the Ag-MOR zeolite using a differential pair distribution function method.34 They reported that for reduced Ag-MOR (Ag0-MOR), two AgI phases are formed after I2 capture, i.e., α-AgI clusters in the zeolite pores and γ-AgI nanoparticles on the surface. For unreduced Ag-MOR (Ag+-MOR), all formed AgI was confined within the pores as α-AgI clusters (Fig. 2a). These results suggest that Ag+-MOR is superior to Ag0-MOR for the long-term storage of I2 because it can trap all captured I2 in the zeolite pores.
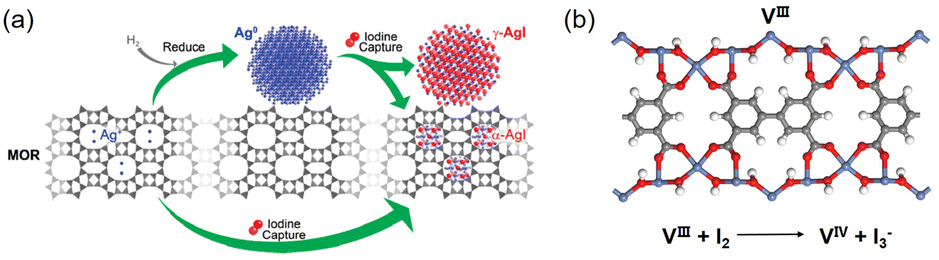 | ||
| Fig. 2 (a) Schematic of the I2 capture process using Ag-MOR zeolite as the adsorbent. For Ag0 sites, I2 adsorption leads to the formation of two AgI phases, i.e., α-AgI clusters in the zeolite pores and γ-AgI nanoparticles on the zeolite surface. For Ag+ sites, only α-AgI clusters are formed in the pores. Reproduced with permission from ref. 34. Copyright 2010, American Chemical Society. (b) Structure model of MOF MFM-300(VIII) and the redox reaction upon I2 adsorption. Color code: blue, VIII; red, O; gray, C; white, H.43 | ||
In addition to Ag+ and Ag0, Ag2O can capture I2.35,36 However, the reactivity of Ag2O towards I2 is controversial. Nan et al. suggested that the decreased adsorption capacity of Ag0-MOR above 150 °C in the presence of water is associated with the oxidation of Ag0 to Ag2O or AgOH.15 Holladay et al. attributed the adverse effects of NO2 on I2 adsorption to it slowly oxidizing Ag0 in Ag0-MOR.37 In general, the primary advantage of Ag-zeolites compared to other adsorbents for I2 capture is their high capacity at high temperatures because of the chemical reaction-based capture mechanism. Nevertheless, Ag-zeolites have limitations such as high cost and potential environmental toxicity.
Other metals have been assessed as alternatives to Ag for I2 capture. Huve et al. compared the Gibbs free energies of iodides and oxides of different metals (Ag, Cu, Hg, Fe, Tl, Sn, Cd, Pb, and Ti). They reported that oxides form preferentially over halides for all metals except Ag and Hg.38 In recent years, Bi-based materials are becoming promising alternatives to Ag-based adsorbents because they incur lower production costs while maintaining high I2 capture capacity.39–41 The reactions of Bi with I2 are as follows:
| Bi + 1.5I2 → BiI3 | (4) |
| 2BiI3 + O2 → 2BiOI + 2I2 | (5) |
| 5BiOI + O2 → Bi5O7I + 2I2 | (6) |
Yim et al. used thiol-functionalized mesoporous silica SBA-15 to immobilize Bi for capturing I2 vapor.39 The prepared Bi-SBA-15 outperformed Ag-zeolites in terms of I2 uptake capacity at 150 °C. However, as the temperature was increased to 250 °C, the I2 uptake of Bi-SBA-15 sharply decreased to ∼50% of Ag-X zeolite.42 This reversal in adsorption capacity may be attributed to two reasons. The chemical bond between bismuth and sulfur is less stable than that between Ag and zeolite, which breaks at 250 °C. Moreover, the Gibbs free energy of BiI3 is less than that of AgI at high temperatures.
Metal sites in redox-active MOFs can capture I2via redox reactions. Schröder et al. examined the forms of adsorbed iodine in MFM-300(VIII) and its oxidized analogue, MFM-300(VIV).43 A redox reaction occurred when MFM-300(VIII) was used as the adsorbent, as evidenced by the generation of I3− and VIV (Fig. 2b). However, I2 was only physically adsorbed when MFM-300 (VIV) was used as the adsorbent because the high-valence metal sites (VIV) could not be oxidized. In this study, the difference in the I2 adsorption capacity between the two adsorbents was insignificant because, under the measurement conditions used (343 K; high I2 concentrations), the I2 uptake was primarily determined by the pore volume of the adsorbent rather than the adsorbent/adsorbate interaction strength.
2.2 Coordination interactions
In addition to redox reactions, the metal sites in adsorbents can capture I2via coordination interactions if they are coordinately unsaturated. For example, Baladi et al. reported that polymers containing three-coordinated Cu+ are favorable for I2 adsorption, while those with similar structures but with four-coordinated Cu+ barely adsorb I2.44 Similarly, coordinately unsaturated (open) metal sites in MOF materials have been used for I2 capture. The MOF Co2(p-DOBDC) (p-DOBDC4− = 2,5-dioxo-1,4-benzenedicarboxylate) has open Co sites that can adsorb I2 in an end-on configuration with a Co–I–I angle of 123°, as determined via single-crystal X-ray diffraction (SXRD) (Fig. 3a).45 As per the studies of the I2 coordination behaviors in organometallics, I2 can act as a donor or an acceptor, depending on the nature of the metal center.46 When I2 acts as the acceptor, it bonds with the metal center collinearly using the σ*(I–I) orbital as the acceptor orbital. When I2 acts as the donor, it exhibits a bent coordination bond with the metal center using a p-type lone pair localized on one of the two iodine atoms as a dominant donation. For I2-loaded Co2(p-DOBDC), the determined bond angle suggested that I2 acted as the donor while the open Co sites acted as acceptors.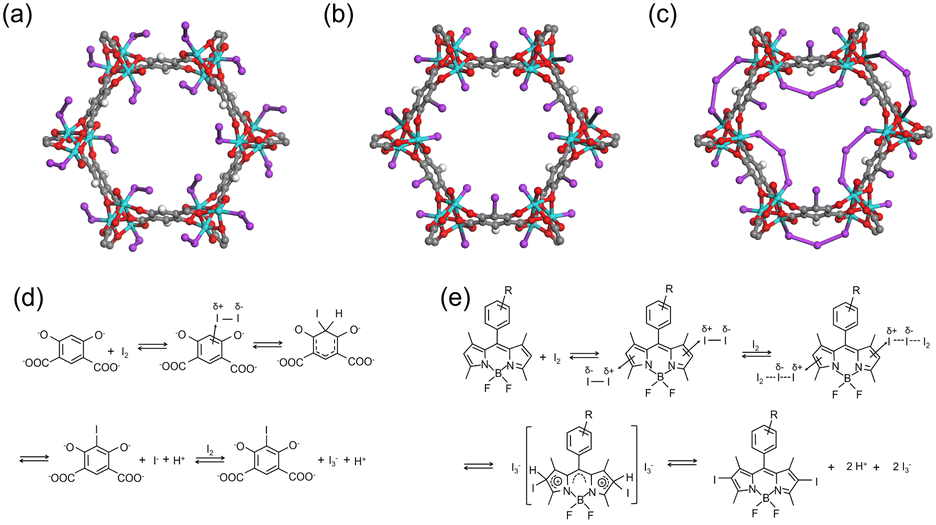 | ||
| Fig. 3 (a) Structure of Co2(p-DOBDC) (p-DOBDC4- = 2,5-dioxo-1,4-benzenedicarboxylate) with 10% I2 loading, where I2 coordinately interacts with open Co sites in an end-on configuration. Reproduced with permission from ref. 45. Copyright 2019, American Chemical Society. (b and c) Structures of Co2(m-DOBDC) (m-DOBDC4− = 4,6-dioxo-1,3-benzenedicarboxylate) with 10 wt% (b) and 100 wt% (c) I2 loading. Reproduced with permission from ref. 45. Copyright 2019, American Chemical Society. At 10 wt% I2 loading, m-DOBDC chemically adsorb I2via an electrophilic aromatic substitution reaction, with the remaining I− coordinated to the open Co site. At 100% I2 loading, additional I2 coordinates with residual open Co sites and bridges the neighboring I−, forming triiodides (I3−). (d) Scheme of the electrophilic aromatic substitution reaction between I2 and m-DOBDC4−. Reproduced with permission from ref. 45. Copyright 2019, American Chemical Society. (e) A reaction mechanism proposed for the iodination reaction between I2 and 2,6 position of BODIPY (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene). Reproduced with permission from ref. 49. Copyright 2013, Royal Society of Chemistry. Color code: cyan, Co; red, O; purple, I; gray, C; white, H. | ||
Because the real-world nuclear off-gas contains a considerable amount of water, Nenoff et al. examined the I2 adsorption behavior of HKUST-1 using a mixture of I2 and H2O.47 They reported that the coordinated water molecules impede the direct interaction of I2 with the open sites, and these water molecules “hold” I2 molecules via weak interactions with I⋯O distances of 3.46–3.84 Å. The adsorbed I2 molecules form a hydrophobic barrier that minimizes H2O sorption. To summarize, HKUST-1 exhibits an I2/H2O selectivity of 1.5 when considering the same concentrations of I2 and H2O in the mixture.
2.3 Electrophilic aromatic substitution
Electrophilic aromatic substitution reactions involve the replacement of a hydrogen atom on a benzene ring with an electrophile. As an electrophile, I2 can be captured by adsorbents containing suitable functional groups via such reactions, where the substituent groups on the aromatic ring directly affect the reactive site. For example, when the MOF Co2(m-DOBDC) (m-DOBDC4− = 4,6-dioxo-1,3-benzenedicarboxylate) was used to capture I2, in addition to the Co sites, the organic linker m-DOBDC could adsorb I2 chemically via an electrophilic aromatic substitution reaction (Fig. 3b).45 However, the organic linker in Co2(p-DOBDC) could not react with I2 (Fig. 3a) because the different distribution of substituent groups results in different reactivity, i.e., m-DOBDC is more electron-rich at the fifth position of the benzene ring than p-DOBDC.During I2 adsorption on Co2(m-DOBDC), the open Co sites and electron-rich ligands cooperatively polarize I2 to the [Iδ+–Iδ−] state at a low I2 loading. Subsequently, the aryl C5–H bond reacts with Iδ+ to form a C–I bond, and the remaining I− coordinated to the open Co site (Fig. 3b and d).45 At a higher I2 loading, additional I2 coordinates with the residual open Co sites and bridges the neighboring I−, thus forming triiodides (I3−) (Fig. 3c).
The 2,6-positions of 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) bearing a lower positive charge can undergo rapid iodination when exposed to I2 at room temperature (Fig. 3e).48 Therefore, Zhu et al. synthesized two BODIPY based conjugated porous polymers, BDP-CPP-1 and BDP-CPP-2.49 They hypothesized that in BDP-CPP-2, ethyl groups were substituted at the 2,6-positions of BODIPY, therefore these positions were not useful for electrophilic aromatic substitution. They observed that BDP-CPP-1 could adsorb additional I2 than BDP-CPP-2 (2.83 vs. 2.23 g g−1) and attributed this to the additional adsorption capacity of BDP-CPP-1 associated with electrophilic aromatic substitution. However, they ignored the difference in the number of functional groups per unit mass between the two polymers.
Although chemisorption based on covalent C–I bond formation prevents the release of adsorbed I2, it poses challenges to the regeneration of the organic adsorbent. In contrast, inorganic adsorbents that chemisorb I2 by forming AgI can be regenerated via calcination in a reducing atmosphere.50
2.4 Lewis acid–base interactions
Molecular iodine is widely used as a mild Lewis acid catalyst for various organic reactions. Therefore, I2 capture can be achieved using adsorbents with basic groups via Lewis acid–base interactions.51 Neutral charge-transfer complexes (D·I2) or charged polyiodides, such as I3− and I5−, can be generated depending on the donor ability of the Lewis bases (D).52X-ray photoelectron spectroscopy (XPS) and Raman spectroscopy are the most commonly used characterization tools for identifying the formed iodine species. However, the assignments of the XPS peaks for different iodine species (I2, I3− and I5−) are inconsistent in the literature. For example, peaks at 630.2 and 618.6 eV were assigned to I2 in certain studies49,53,54 but to I3− in others.55,56 By comparison, Raman spectroscopy is a more reliable approach for distinguishing the formed iodine species. When neutral D·I2 complexes are formed, the characteristic Raman band of solid I2 at 180 cm−1 is expected to move toward a lower frequency (Fig. 4a). When charged polyiodides are formed, I3− exhibits a symmetric stretching band at ∼110 cm−1 and an asymmetric stretching band at ∼140 cm−1, while the band around 160 cm−1 is usually assigned to I5− species (Fig. 4b).57
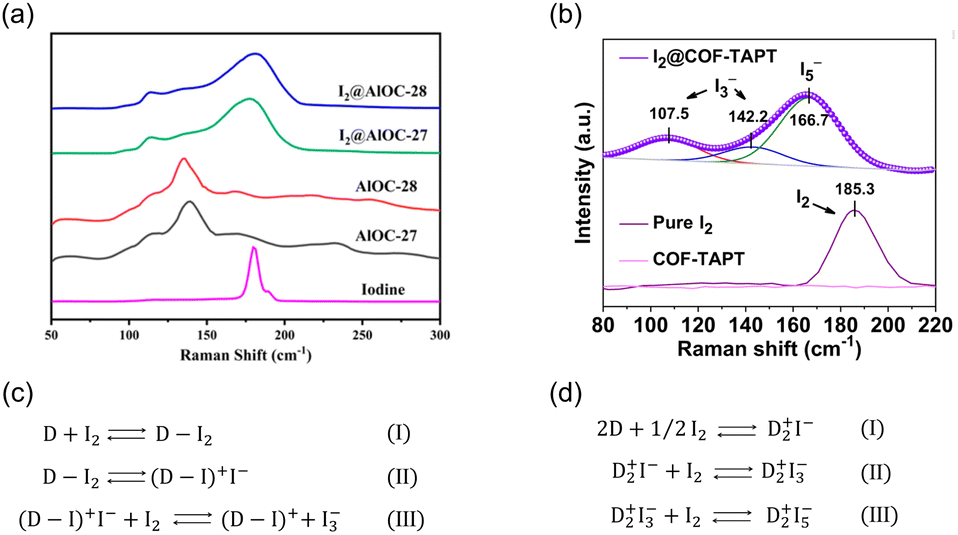 | ||
| Fig. 4 (a) Raman spectra of AlOC-27, AlOC-28, I2@AlOC-27-NC, I2@AlOC-28-NC, and solid I2. In the spectra of I2@AlOC-27-NC and I2@AlOC-28-NC, the peak at 180 cm−1 assigned to solid I2 was red-shifted, indicating the formation of neutral charge transfer complex D·I2. Reproduced with permission from ref. 73. Copyright 2021, American Chemical Society. (b) Raman spectra of pure I2, pristine COF-TAPT, and I2-saturated COF-TAPT. In the spectra of I2@COF-TAPT, peaks attributed to I3− and I5− appeared, indicating the formation of charged polyiodides in COF-TAPT. Reproduced with permission from ref. 99. Copyright 2022, Springer Nature. (c and d) The interaction mechanisms between electron donors and I2 proposed by different researchers.58–62 | ||
Although the generation of negatively charged polyiodides is generally accepted, there is no consensus on the species balancing of these negative charges. In previous studies, positively charged species, such as (D–I)+ (Fig. 4c)58–60 and D2+ (Fig. 4d),61,62 were proposed as counter cations.
The Lewis basic sites commonly incorporated into adsorbents for I2 capture can be divided into different categories (see Fig. 5), which are separately discussed below.
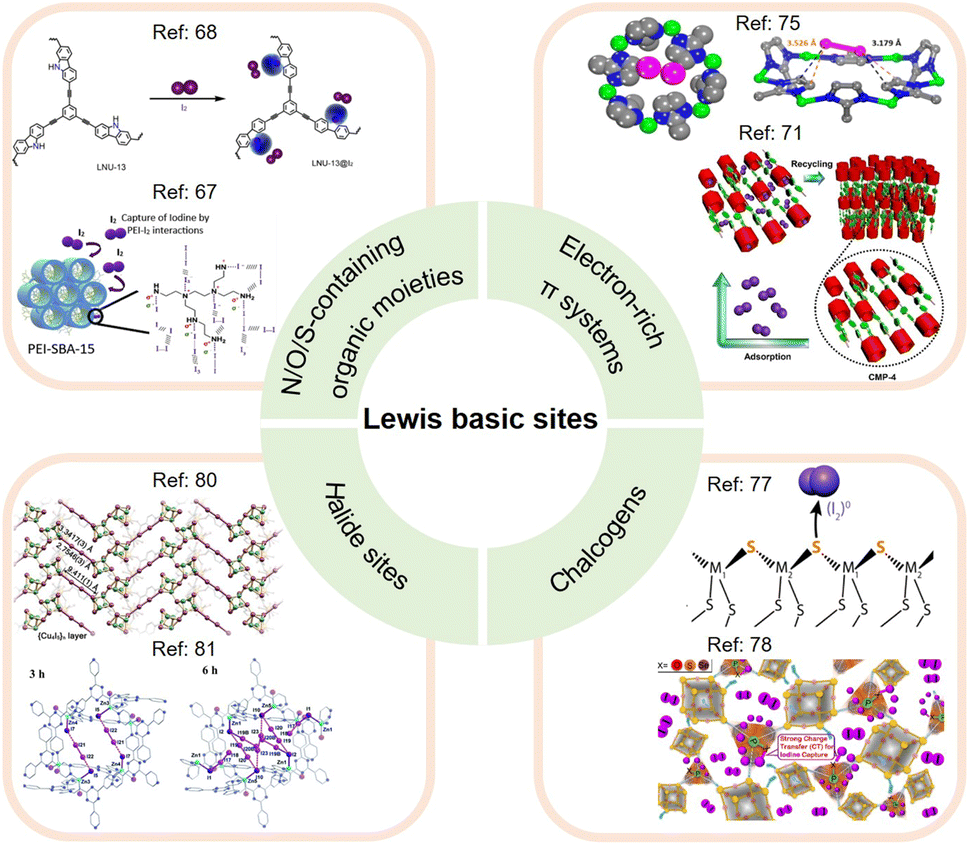 | ||
| Fig. 5 Classification of Lewis basic sites used for I2 capture, including N/O/S-containing organic moieties, electron-rich π systems, chalcogens, and halide sites. (Top left panel) The upper figure is reproduced with permission from ref. 68. Copyright 2021, MDPI. The lower figure is reproduced with permission from ref. 67. Copyright 2019, Elsevier. (Top right panel) The upper figure is reproduced with permission from ref. 75. Copyright 2013, American Chemical Society. The lower figure is reproduced with permission from ref. 71. Copyright 2021, Wiley. (Bottom right panel) The upper figure is reproduced with permission from ref. 77. Copyright 2013, American Chemical Society. The lower figure is reproduced with permission from ref. 78. Copyright 2020, Elsevier. (Bottom left panel) The upper figure is reproduced with permission from ref. 80. Copyright 2017, Wiley. The lower figure is reproduced from ref. 81 with permission from the Royal Society of Chemistry. | ||
Moreover, the pore geometry of the adsorbent material affects the distribution of adsorption sites and thus the binding strength to I2. For example, Nenoff et al. reported that each I2 molecule adsorbed in MOF ZIF-8 simultaneously interacts with two opposing 2-methylimidazolate linkers to form an iodine-aromatic charge-transfer complex because of the special pore size and shape of ZIF-8.75 The consequence of such multiple interactions is that ZIF-8 can firmly trap the adsorbed I2 until the framework decomposes at ∼575 K.76
2.5 Coulomb interactions
Another effective strategy to promote I2 adsorption is to generate ionic sites in the adsorbent, which can bind the dynamically formed polyiodide anions via Coulomb interactions.21,74,82 However, despite the enhanced affinity to I2, the generation of ionic sites in various amorphous POPs via post-synthesis modification usually leads to a greatly reduced surface area and limited improvement in the I2 adsorption capacity. Xie et al. introduced ionic sites into crystalline COFs using a “multivariate” strategy combined with a post-synthesis modification to overcome this issue.83 The as-prepared ionic COF (iCOF-AB-50) combined large surface areas, high pore volume, and abundant binding sites, resulting in excellent I2 capture performance under dynamic conditions (279 wt% at 25 °C). In particular, the significant promoting effect of the introduced ionic groups (quaternary ammonium) was attributed to their strong interactions with [I2Br]− and [2I2Br]− species via Coulomb forces.83A highly stable ionic guanidinium-based COF (TGDM) was developed recently (Fig. 6).84 TGDM has a high density of robust ionic sites owing to the reduced linker length and the unique stability of the guanidium moieties, which makes it particularly useful for I2 capture at high temperatures. At 150 °C and 150 ppmv of I2, TGDM exhibited an I2 adsorption capacity of ∼30 wt%, which was considerably higher than that of multiple benchmark adsorbents.
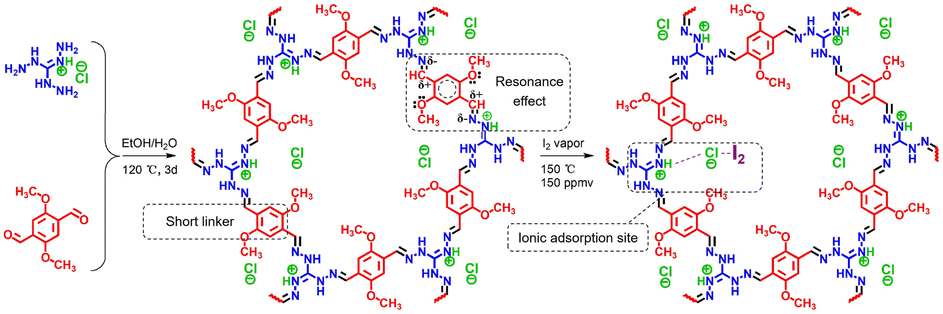 | ||
| Fig. 6 Schematic of the design and synthesis of ionic COF TGDM and explanation of its high stability and I2 uptake mechanism. Reproduced with permission from ref. 84. Copyright 2022, American Chemical Society. The ionic sites (quaternary ammonium) strongly interact with [I2Cl]− species through Coulomb forces. | ||
2.6 Hydrogen bonding
Hydrogen bonding has been used to facilitate I2 adsorption. The polyiodide I3− is readily formed in multiple adsorbents, which can dissociate to I2 and I−, resulting in the release of I2.85 Lu et al. fabricated a melamine-based polymer (MFP) to stabilize the formed I3− species.23 They claimed that the MFP could effectively capture I3−via multiple synergistic NH⋯I hydrogen bonds, and the binding energy was approximately four times higher than that of the conventional Ag–I ionic bond (Fig. 7a). Hydrogen bonding can occur between the proton and the electronegative end of molecular I2. Paul et al. analyzed the potential of three MOFs (MIL-53(Al), MIL-120(Al), and HKUST-1(Cu)) for I2 adsorption using periodic dispersion density functional theory. They reported that MIL-53 has stronger interactions with I2 than the other two MOFs.86 The infrared spectrum simulated via molecular dynamics calculations confirmed that this strong interaction originated from the hydrogen bonding between I2 and the hydroxyl groups of MIL-53 (Fig. 7b).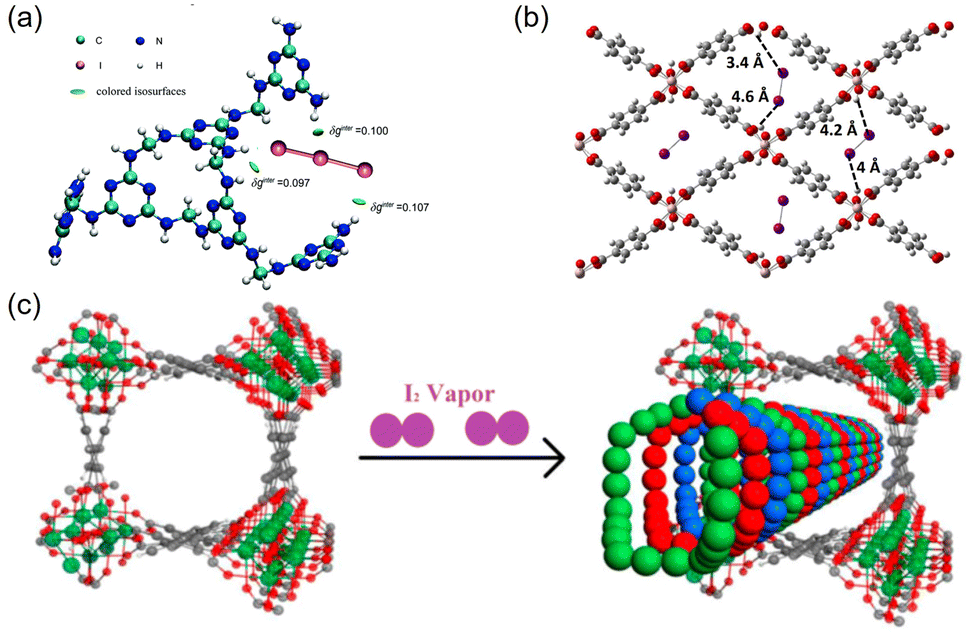 | ||
| Fig. 7 (a) DFT-calculated hexamer model of MFP bound with I3−, the green iso-surface represents strong hydrogen bonding between I and –NH–/–NH2–. Reproduced with permission from ref. 22. Copyright 2014, Royal Society of Chemistry. (b) Structure of MIL-53 (Al) with I2 adsorbed via hydrogen bonding with the hydroxyl groups. Reproduced with permission from ref. 86. Copyright 2017, American Chemical Society. (c) Schematic of triple-helical chains of iodine molecules formed within the channel of MOF MFM-300 (Sc) because of the strong inter-molecular interaction. Reproduced with permission from ref. 88. Copyright 2017, American Chemical Society. | ||
2.7 Van der Waals interactions
The van der Waals force is a relatively weak but major interaction force. As a nonpolar molecule, I2 does not have a permanent dipole. However, when close to a charged adsorbent framework, I2 can interact with the framework by inducing a transient dipole. Accordingly, Shetty et al. prepared porous polymers (covalent polycalix[4]arenes) and modified them by lithiation.87 Compared to the original polymers, lithiated polymers demonstrated faster adsorption kinetics and higher I2 uptake because of the charge-induced dipole interaction between Li+ and I2. In addition to ion-induced dipole interactions, the London dispersion force plays an important role in I2 capture. For I2 molecules, temporary dipoles can induce strong intermolecular interactions (dispersion forces), which are influenced by the size and shape of micropores in the adsorbent. For example, the suitable pore geometry MOF MFM-300(Sc) facilitates the self-aggregation of I2 molecules into an unusual triple helical chain through intermolecular interactions, thus resulting in the efficient packing of I2 with an exceptional storage density of 3.08 g cm−3 (Fig. 7c).882.8 Hydrophobic interactions
The real off-gas contains a significant amount of moisture, in addition to I2 and organic iodides. The presence of water molecules poses a significant challenge to I2 capture because the competitive adsorption of water can significantly reduce the I2 adsorption capacity. Many strategies used to promote I2 adsorption, such as Lewis acid–base interactions, Coulomb interactions, and hydrogen bonding, can promote water adsorption. Therefore, adsorption sites capable of preferentially adsorbing I2 over water are required. Hydrophobic porous materials can selectively capture I2 in the presence of water via hydrophobic interactions because I2 is a nonpolar hydrophobic molecule. However, such experiments are usually performed at low temperatures33,89 because relatively weak hydrophobic interactions cannot prevent I2 desorption at high temperatures.A chiral polymer zinc D-saccharate having two types of parallel channels, one hydrophilic and the other hydrophobic, has been investigated for I2 capture.89 Upon exposure to I2 vapor, water molecules in the hydrophobic channels are replaced with I2, while water molecules in the hydrophilic channels remain. This result shows that the preferential adsorption of I2 over water can be achieved by forming a hydrophobic environment in the adsorbent. Pham et al. synthesized a hydrophobicity-intensified silicalite-1 (HISL) zeolite and evaluated its I2 adsorption properties under various conditions relevant to practical off-gas treatment applications.33 HISL has similar I2 adsorption capacity in the presence and absence of water because of its super-hydrophobicity and demonstrates good tolerance to the presence of acids.33
3. Mechanisms for methyl iodide capture
In the off-gas from nuclear power plants, CH3I is produced from a reaction of I2 with volatile organic compounds (e.g., methane). Compared to I2, CH3I is more difficult to capture because of its considerably lower concentration and lack of intermolecular interactions.90,91 There are relatively few studies on CH3I capture, and the adsorption strategies employed can be classified broadly into catalytic decomposition reactions, coordination interactions, methylation reactions, halogen bonding, and hydrogen bonding.3.1 Catalytic decomposition reactions
As in the case of I2 adsorption, Ag-containing adsorbents, such as Ag-zeolites, can capture CH3I via chemical reactions. For example, Ag0-MOR zeolite was reported to catalyze the decomposition of CH3I, where the Brønsted acid sites of the zeolite act as catalytic centers. The surface methoxy species react with other components in the off-gas (e.g., H2O and NOx) to form volatile by-products. The liberated iodine reacts with Ag in the micropore to form sub-nm AgI clusters (Fig. 8a).92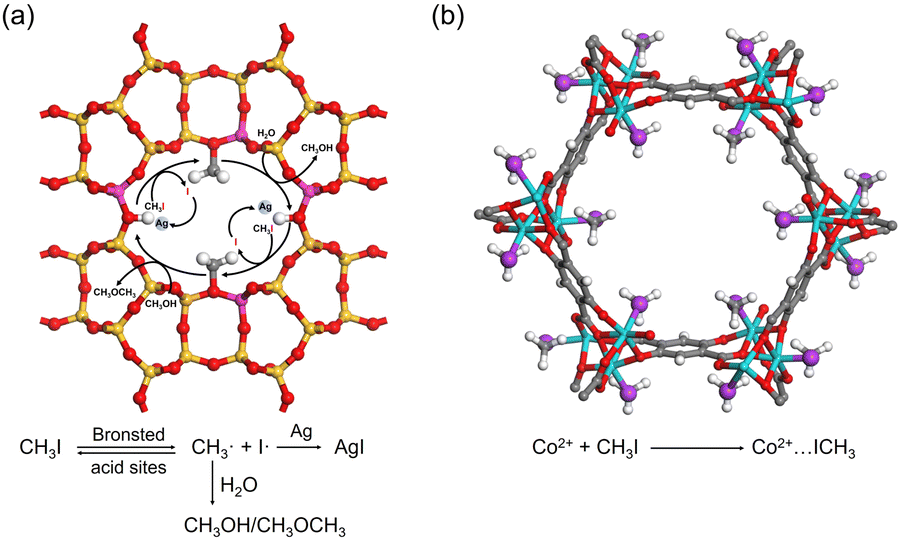 | ||
| Fig. 8 (a) Schematic of the mechanism of CH3I adsorption on the Ag0-MOR zeolite. Brønsted acid sites catalyze the decomposition of CH3I, and the formed I· reacts with Ag0 to form AgI, while the CH3· reacts with H2O to form by-products such as CH3OH or CH3OCH3. Color code: yellow, Si; red, O; pink, Al; gray, C; and white, H. (b) Crystal structures of CH3I@Co2(p-DOBDC), where CH3I reacts with open Co sites by coordination interaction. Reproduced with permission from ref. 95. Copyright 2020, Wiley. Color code: cyan, Co; red, O; purple, I; gray, C; and white, H. | ||
Multiple exchangeable metal cations, including Cu2+, Ag+, Pb2+, and Na+, have been introduced into FAU-type X and Y zeolites for CH3I adsorption.93 The adsorption capacity of these metal ions followed the order: Cu2+ > Ag+ ≫ Pb2+ > Na+. This suggests that Cu2+ has the highest CH3I capture efficiency. However, Ag–Y zeolite rather than Cu–Y zeolite was considered the best candidate for CH3I adsorption because the adsorption of CH3I on Cu2+ leads to undesirable I2 generation because of the following reaction:
| Cu2+ + 2I− → CuI2 → 2CuI + 1/2I2 | (7) |
3.2 Coordination interactions
The open metal sites in MOFs can adsorb CH3I via coordination interactions. For example, a mesoporous bimetallic organic framework ECUT-300-200-Ac was synthesized with purposely generated structural defects that render many open metal sites.94 XPS revealed a blue shift of the Cd3d and U4f binding energies in ECUT-300-200-Ac when exposed to CH3I, indicating the participation of these metal sites in adsorption. Similarly, SXRD indicated that MOF Co2(p-DOBDC) can adsorb CH3I at its open Co sites through coordination, with the I end of the CH3I molecule interacting with Co in a side-on configuration (Fig. 8b).953.3 Methylation reactions
Another effective strategy to capture CH3I is to functionalize the adsorbent with multiple amine groups that can bind CH3I via N-methylation reactions. For example, a number of nucleophilic amines, including triethylenediamine (TEDA), hexamethylenetetramine (HMTA), N,N′-dimethylethylenediamine, N,N′-dimethyl-1,3-propanediamine, and N,N′-dimethyl-1,4-butanediamine, were loaded onto activated carbon by impregnation to enhance their CH3I binding strength.12 Density functional theory (DFT) calculations and ab initio molecular dynamics simulations indicate that TEDA can reduce the dissociation activation barrier of CH3I and participate in the subsequent alkylation to form a quaternary ammonium salt (Fig. 9a).96 For example, tertiary amines, TEDA and HMTA, were grafted to the open metal sites of MIL-101 (Cr) for CH3I capture. The resulting MIL-101-Cr-TED exhibited an ultrahigh CH3I uptake of 71 wt% at 150 °C, which is more than three times that of Ag0-MOR.97 In addition to tertiary aliphatic amines, aromatic N species have been used to capture CH3I through N-methylation reactions, such as pyridine-N,98 aniline-N,25 pyrazole-N,94 imine-N, and triazine-N.99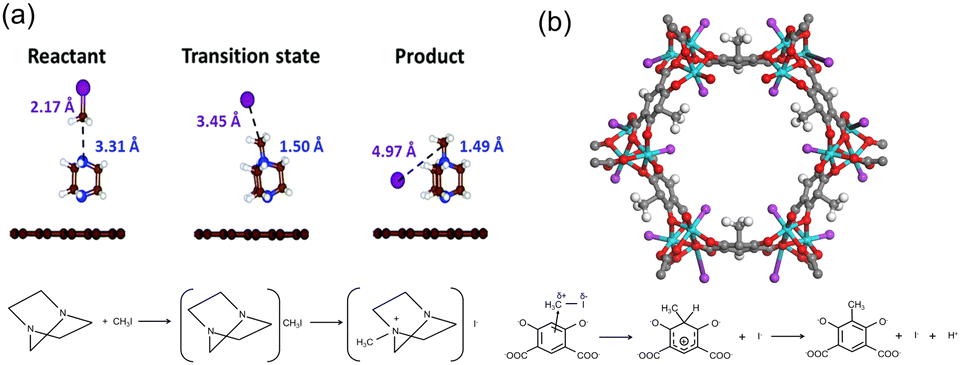 | ||
| Fig. 9 (a) Calculated geometries of the reactant, transition state, and product during the CH3I dissociation on the TEDA-modified activated carbon surface. TEDA lowers the dissociation energy of CH3I and forms quaternary ammonium salt with CH3I by N-methylation reaction. Reproduced with permission from ref. 96. Copyright 1999, Royal Society of Chemistry. (b) Crystal structures of CH3I@Co2(m-DOBDC) and the proposed mechanism for the reaction between m-DOBDC4− and CH3I. CH3I reacts with electron-rich m-DOBDC4− groups via electrophilic aromatic substitution reaction, while the dissociated I− ions coordinate to the open Co2+ sites. Reproduced with permission from ref. 95. Copyright 2020, Wiley. Color code: green, Co; red, O; purple, I; gray, C; and white, H. | ||
The adsorbent can also capture CH3I through electrophilic aromatic substitution reactions. Park et al. synthesized two MOFs, Co2(m-DOBDC) and Co2(p-DOBDC), and compared their structures after CH3I adsorption.95 SXRD indicated that CH3I reacted with electron-rich m-DOBDC4- groups, converting the aryl C–H bond to the C–C bond, with the dissociated I− coordinating to the open Co2+ sites (Fig. 9b). However, for the isostructural Co2(p-DOBDC) lacking electron-rich C, CH3I was coordinately adsorbed at the open Co2+ sites without undergoing electrophilic aromatic substitution (Fig. 8b).
3.4 Halogen bonding and hydrogen bonding
Quantum chemical calculations report that halogen bonds may play an important role in promoting CH3I adsorption when electron-rich moieties are present in the adsorbent. For example, CH3I can form weak halogen bonds with –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– in 1,2-dihydrophenazine in polymer MHP-P5Q, in which the bond length d[N⋯I] is 3.39 Å (Fig. 10a).25 A halogen bond between I and an electron-rich group appears counterintuitive because halogen atoms are electronegative. Brinck et al. theoretically explained this.100 They reported that the electrostatic potential of covalently bonded halogens is anisotropic and possesses positive regions at the tip of X (X = Cl, Br, and I). Therefore, halogen bonds can be formed between CH3I with nucleophiles in a head-on configuration.
N– in 1,2-dihydrophenazine in polymer MHP-P5Q, in which the bond length d[N⋯I] is 3.39 Å (Fig. 10a).25 A halogen bond between I and an electron-rich group appears counterintuitive because halogen atoms are electronegative. Brinck et al. theoretically explained this.100 They reported that the electrostatic potential of covalently bonded halogens is anisotropic and possesses positive regions at the tip of X (X = Cl, Br, and I). Therefore, halogen bonds can be formed between CH3I with nucleophiles in a head-on configuration.
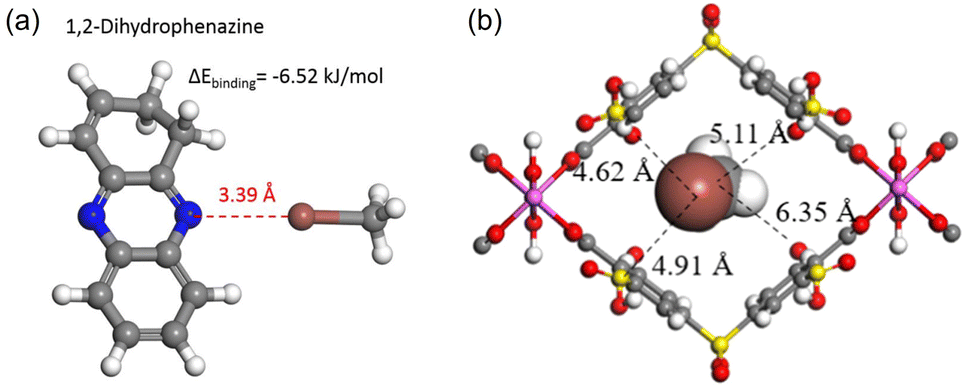 | ||
| Fig. 10 (a) Calculated structure of CH3I interacting with 1,2-dihydrophenazine, showing that CH3I forms weak halogen bonds with –CN– in a head-on configuration. Reprinted with permission from ref. 25. Copyright 2020, Springer Nature. Color code: gray, C; white, H; blue, N; brown, I. (b) DFT-optimized structure of CH3I in the CAU-11-SO3H pore channel, showing that CH3I interacts with the modified sulfonic acid by I⋯O and I⋯S electrostatic interactions. Color code: gray, C; white, H; red, O; pink, Al; brown, I and yellow, S. Reprinted with permission from ref. 101. Copyright 2021, American Chemical Society. | ||
In a purely theoretical study, Wu et al. screened a series of Al-based MOFs for CH3I capture from the simulated off-gas using Grand canonical Monte Carlo (GCMC) simulations and DFT calculations.101 Among the MOFs evaluated, CAU-11 with 1D narrow channels demonstrated the highest isosteric heat (Qst) of CH3I. Note that additional modification of CAU-11 with sulfonic acid groups enabled highly efficient capture of trace CH3I, which was attributed to the formation of I⋯O and I⋯S electrostatic interactions (Fig. 10b). Such interactions were not assigned as halogen bonds possibly because the calculated configuration of CH3I was “side-on” rather than “head-on”.
Endowing adsorbents with hydrogen bonding sites is another approach for promoting their CH3I capture capability. Unlike halogen bonding, when CH3I is immobilized by hydrogen bonding, the iodine atom acts as an electron donor.102 Chebbi et al. compared the CH3I capture capability of a number of MOFs, including ZIF-8 (Zn), MIL-53 (Al), MIL-100 (Al), UiO-66 (Zr), HKUST-1 (Cu), CAU-1 (Al), and MIL-120 (Al). They reported that MIL-120 (Al) had the strongest interaction with CH3I because of its abundant –OH groups, which promoted the formation of H-bonded complexes with CH3I.103 The captured CH3I could not be desorbed from MIL-120 (Al) by He evacuation.
4. Performance evaluation
The adsorbents developed for I2/CH3I capture can be evaluated under static or dynamic conditions or both. Static measurements are easy to operate but have multiple limitations. Whereas dynamic measurements require the development of gas circuits and the use of detectors, they provide more flexible test conditions relevant to practical applications. This section describes typically used static and dynamic systems and their applications for I2 and CH3I adsorption measurements.4.1 Static measurement
In most studies on I2 (or CH3I) capture, the adsorption capacity of the adsorbent is measured under static conditions using a closed system, in which I2 (or CH3I) and the adsorbent are heated simultaneously to the target temperature (Fig. 11a). After some time (assuming adsorption equilibrium has been achieved), the system is cooled to room temperature, and the adsorption capacity is determined based on the mass increase of the adsorbent. The static system is also used to determine the adsorption kinetics by recording the mass changes at different time intervals. The commonly used index for adsorption kinetics is K80%, which refers to the average adsorption rate before the adsorption capacity reaches 80%.104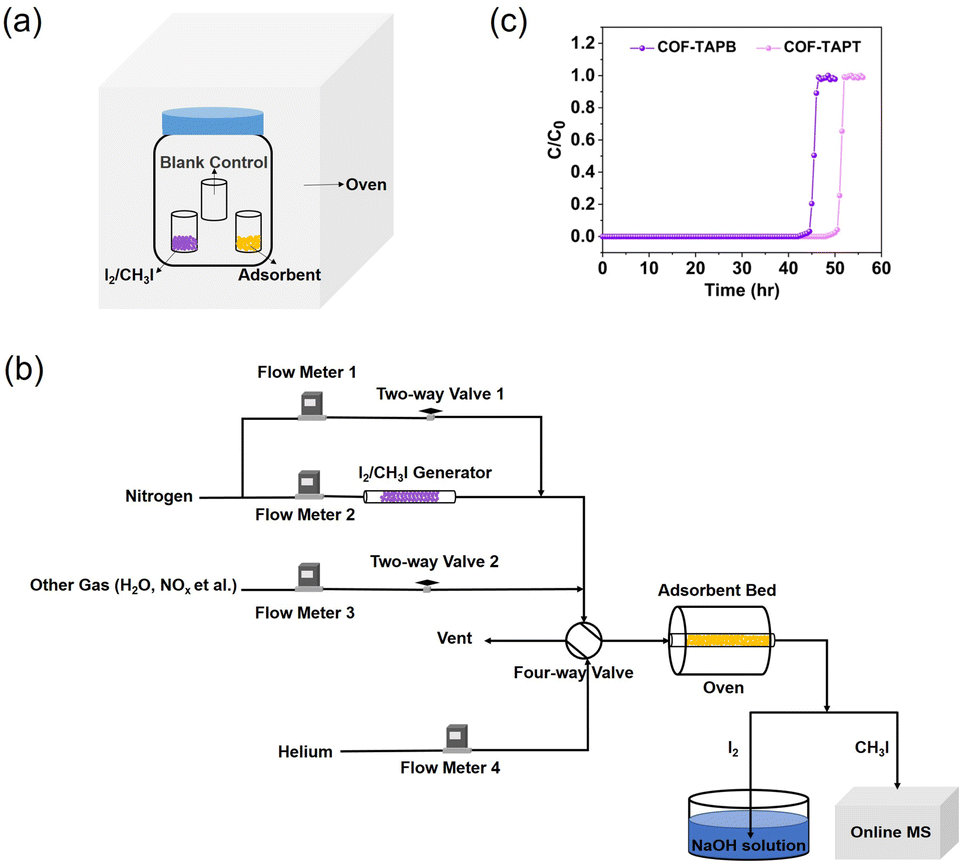 | ||
| Fig. 11 (a) Schematic of the static system used to measure the I2 or CH3I adsorption capacity of adsorbents. (b) Schematic of the dynamic system used to measure the I2 or CH3I adsorption capacity of adsorbents, which is based on a column breakthrough setup. The concentration of I2 in the effluent can be determined by collecting I2 using NaOH solution followed by elemental analysis using ICP-MS. (c) Typical breakthrough profiles obtained from the dynamic measurement to determine the adsorption capacity. Reproduced with permission from ref. 99. Copyright 2022, Springer Nature. | ||
Despite its simplicity and widespread use, there are several problems with such a static measurement system. For example, static I2 adsorption is typically conducted at 75 °C and ambient pressure; under such conditions, the partial pressure of I2 is 1.6 kPa. The corresponding I2 volumetric concentration is ∼1.6 × 104 ppmv, which is several orders of magnitude higher than the actual I2 concentration in the off-gas. Hence, the measured I2 adsorption capacity does not truly reflect the I2 capture performance in practical applications. Moreover, during the cooling process of the system, a large amount of I2 may condense on the surface of the adsorbent and the vial containing the adsorbent, resulting in a considerable overestimation of the adsorption capacity. Although using an empty vial as a blank control can eliminate this overestimation to a certain extent (Fig. 11a), the adsorption capacity determined in this manner remains unreliable. This can explain why certain reported adsorption capacities are considerably higher than the theoretical values calculated based on the adsorbent pore volume.105–109 Moreover, using a static measurement system, it is impossible to control the I2 concentration and adsorption temperature independently or co-feed other components, such as H2O and NOx, to simulate the actual off-gas conditions.
Considering these limitations and problems, using such a static measurement system to evaluate the performance of adsorbents for I2 capture is not recommended. Compared to I2, CH3I has weaker intermolecular forces and is less prone to condensation. Therefore, the CH3I adsorption capacity determined using a static system is relatively more reliable. However, when CH3I is adsorbed via chemical reactions (for example, when CH3I is adsorbed on Ag-zeolites), the adsorption capacity cannot be determined accurately by the mass change because of the production of volatile byproducts such as CH3OH and CH3OCH3.
Compared to the I2 capture, which has been extensively studied, there are only a few studies of CH3I capture. Xie et al. compared the different adsorption behavior of CH3I and I2 under static and dynamic conditions using specially designed COFs as adsorbents.99 They reported that I2 adsorption is dominated by intermolecular interactions under commonly used static evaluation conditions (saturated I2 vapor at 75 °C). The capacity is primarily determined by the textural properties (surface area and pore volume) of the adsorbent. However, the CH3I adsorption capacity depends on the number of strong binding sites of the adsorbent rather than its textural properties, thus exhibiting a positive correlation with the N content (strong binding sites) in the COFs. The observed one-to-one correspondence between CH3I and N suggests that CH3I molecules are only adsorbed on N sites possibly by forming salts.99 The adsorption kinetics of I2 and CH3I on these COFs demonstrate a similar trend, i.e., they are primarily determined by the textural properties and the number of strong binding sites, respectively.
4.2 Dynamic measurement
The problems associated with the static system can be circumvented by dynamic adsorption measurements using a fixed-bed setup (Fig. 11b). The dynamic measurement system allows free adjustment of the I2/CH3I concentration, precise control of the adsorbent temperature, and easy simulation of off-gas compositions.110 Importantly, it is close to the actual application scenario using adsorbent-packed columns to capture iodine.In a dynamic system, the carrier gas flows continuously through the I2/CH3I generator to produce I2/CH3I vapor, the concentration of which can be adjusted by the dilution gas. The adsorbent is separated from the vapor generator, and its temperature can be controlled independently using an oven. Other substances can be introduced into the system with independent gas lines to simulate the off-gas composition (Fig. 11b). Breakthrough curves can be plotted by analyzing the I2/CH3I content in the outlet gas via inductively coupled plasma mass spectrometry (ICP-MS) and online mass spectrometry, respectively (Fig. 11c). From this, a reliable adsorption capacity can be determined by curve integration (eqn (8)):
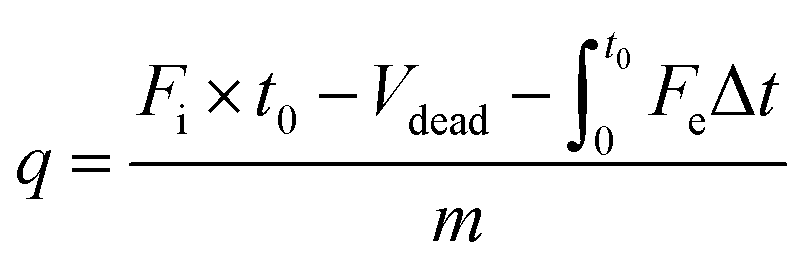 | (8) |
Han et al. used a dynamic system to measure the I2 uptake capacity of an ionic COF (iCOF-AB-50) at a low I2 concentration of 400 ppmv and 25 °C.111 They attributed the high I2 uptake capacity (2.79 g g−1) to the combination of high pore volume and abundant binding sites in iCOF-AB-50. Moreover, they examined the effects of competitive water adsorption on the I2 capture of iCOF-AB-50 by introducing water vapor (relative humidity: 50%) to the dynamic I2 adsorption system at 25 °C. The presence of water vapor caused only a slight decrease in the I2 uptake of iCOF-AB-50 to 2.70 g g−1 compared to that without water vapor addition (2.79 g g−1), during which a certain amount of water was adsorbed. This suggests that although iCOF-AB-50 is not completely water-repellant, most of its adsorptive sites bind preferentially to I2, thus leaving their I2 adsorption capacity almost intact under humid conditions.
In a more recent study, the same group developed a guanidinium-based ionic COF, termed TGDM, and evaluated its I2 capture ability using a dynamic measurement system.84 The tests were conducted at 150 ppmv of I2 and 150 °C to simulate the actual off-gas conditions. Under low-concentration and high-temperature conditions, TGDM exhibited significantly higher I2 uptake capacity than iCOF-AB-50 (0.3 vs. 0.08 g g−1) despite its lower pore volume. Compared with iCOF-AB-50, the superior high-temperature I2 capture performance of TGDM was attributed to its higher density of ionic groups and improved thermal stability.
The CH3I concentration in the off-gas of actual nuclear power plants is extremely low, typically <50 ppmv. However, most studies on CH3I capture evaluated the adsorbents at considerably higher CH3I concentrations (e.g., 2 × 105 ppmv), even with a dynamic system.94,97,98 Among the adsorbents investigated, mesoporous MOF ECUT-300-200-Ac demonstrated record CH3I uptake capacities of > 2.8 g g−1 at 25 °C and > 0.87 g g−1 at 150 °C. Its excellent CH3I capture ability was attributed to the combined effects of coordination interactions, methylation reactions, and hydrogen bonding.94 Han et al. compared the CH3I capture performance of multiple state-of-the-art adsorbents at 50 ppmv of CH3I and 25 °C.99 The CH3I adsorption capacities obtained were as follows: MIL-101-Cr-HMTA97 (0.51 g g−1) > COF-TAPT99 (0.39 g g−1) > TFPA-TAPT99 (0.18 g g−1) > COF-TAPB99 ≈ iCOF-AB-50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 111 (0.12 g g−1) > SCU-COF-298 (0.08 g g−1). These results confirm that the adsorption of CH3I is determined primarily by the type and number of binding sites and is not related significantly to the textural properties of the adsorbent. These results indicate that unlike I2 adsorption, ionic groups have little effect on CH3I adsorption, which may be because I2 readily forms charged polyiodide species, such as I3− and I5−, whereas CH3I cannot.
111 (0.12 g g−1) > SCU-COF-298 (0.08 g g−1). These results confirm that the adsorption of CH3I is determined primarily by the type and number of binding sites and is not related significantly to the textural properties of the adsorbent. These results indicate that unlike I2 adsorption, ionic groups have little effect on CH3I adsorption, which may be because I2 readily forms charged polyiodide species, such as I3− and I5−, whereas CH3I cannot.
Few studies have used dynamic systems to test the I2/CH3I capture performance of adsorbents. Table 1 lists the relevant results reported in the literature.
| Adsorbent name | Test temperature (°C) | Vapor concentration (ppmv) | Adsorption capacity (g g−1) | Adsorption mechanisms | Ref. | ||
|---|---|---|---|---|---|---|---|
| I2 capture | Inorganic materials | Ag0Z | 150 | 50 | 0.12 | Redox reaction | 112 |
| HISL | RT | 400 | 0.53 | Hydrophobic interaction | 33 | ||
| SL-1 | RT | 400 | 0.48 | Hydrophobic interaction | 33 | ||
| Si-BEA | RT | 400 | 0.47 | Hydrophobic interaction | 33 | ||
| AC | RT | 400 | 0.70 | Van der Waals interaction | 33 | ||
| 23Ag/Y | 100 | 1250 | 0.22 | Redox reaction | 113 | ||
| 35Ag/13X | 100 | 1250 | 0.46 | Redox reaction | 113 | ||
| C@ETS-10 | 20 | 32 | 0.04 | Van der Waals interaction | 114 | ||
| MOFs | HKUST-1 | RT | 400 | 0.38 | Hydrogen bonding | 33 | |
| ZIF-8 | RT | 400 | 0.03 | Lewis acid–base interaction | 33 | ||
| MIL-101-Cr-HMTA | 25 | 150 | 0.83 | Lewis acid–base interaction | 99 | ||
| COFs | iCOF-AB-50 | 25 | 400 | 2.79 | Coulomb interaction | 111 | |
| iCOF-AB-50 | 25 | 150 | 1.52 | Coulomb interaction | 99 | ||
| iCOF-AB-50 | 75 | 400 | 0.44 | Coulomb interaction | 111 | ||
| SCU-COF-2 | 25 | 400 | 0.98 | Lewis acid–base interaction | 98 | ||
| SCU-COF-2 | 25 | 150 | 0.49 | Lewis acid–base interaction | 99 | ||
| SCU-COF-2 | 75 | 400 | 0.35 | Lewis acid–base interaction | 98 | ||
| TGDM | 150 | 150 | 0.30 | Coulomb interaction | 84 | ||
| JUC-561 | 150 | 150 | 0.20 | Lewis acid–base interaction | 84 | ||
| COF-TAPT | 25 | 150 | 1.25 | Lewis acid–base interaction | 99 | ||
| COF-TAPB | 25 | 150 | 1.12 | Lewis acid–base interaction | 99 | ||
| TFPA-TAPT | 25 | 150 | 0.42 | Lewis acid–base interaction | 99 | ||
| POPs | POSS-TPPSe | 160 | 0.16 | 0.26 | Lewis acid–base interaction | 78 | |
| CH3I capture | Inorganic materials | 22.8Ag/Y | 35 | 1333 | 0.25 | Catalytic decomposition reaction | 115 |
| 22.8Ag/Y | 100 | 1333 | 0.223 | Catalytic decomposition reaction | 115 | ||
| 22.8Ag/Y | 250 | 1333 | 0.193 | Catalytic decomposition reaction | 115 | ||
| Ag+@13X | 150 | 200000 |
0.48 | Catalytic decomposition reaction | 97 | ||
| TED@AC | 150 | 200000 |
0.17 | Methylation reaction | 97 | ||
| HMTA@AC | 150 | 200000 |
0.14 | Methylation reaction | 97 | ||
| Ag+@ZSM-5 | 150 | 200000 |
0.24 | Catalytic decomposition reaction | 97 | ||
| HISL | 25 | 532967 |
0.42 | — | 33 | ||
| MOFs | MIL-101-Cr-TED | 30 | 200000 |
1.60 | Methylation reaction | 97 | |
| MIL-101-Cr-TED | 150 | 200000 |
0.71 | Methylation reaction | 97 | ||
| MIL-101-Cr-HMTA | 30 | 200000 |
1.74 | Methylation reaction | 97 | ||
| MIL-101-Cr-HMTA | 150 | 200000 |
0.62 | Methylation reaction | 97 | ||
| MIL-101-Cr-HMTA | 25 | 50 | 0.51 | Methylation reaction | 99 | ||
| ECUT-300-200-Ac | 25 | 200000 |
2.80 | Methylation reaction/hydrogen bonding/coordination interaction | 94 | ||
| ECUT-300-200-Ac | 150 | 200000 |
0.87 | Methylation reaction/hydrogen bonding/coordination interaction | 94 | ||
| MIL-53 | 35 | 1333 | 0.13 | Hydrogen bonding | 103 | ||
| MIL-120 | 35 | 1333 | 0.16 | Hydrogen bonding | 103 | ||
| HKUST-1 | 35 | 1333 | 0.43 | Coordination interaction | 103 | ||
| MIL-101-RSO3Ag | 30 | 20 | 0.16 | Coordination interaction | 116 | ||
| COFs | SCU-COF-2 | 25 | 200000 |
0.564 | Methylation reaction | 98 | |
| SCU-COF-2 | 75 | 200000 |
0.17 | Methylation reaction | 98 | ||
| SCU-COF-2 | 25 | 50 | 0.08 | Methylation reaction | 99 | ||
| iCOF-AB-50 | 25 | 200000 |
0.62 | — | 99 | ||
| iCOF-AB-50 | 25 | 50 | 0.11 | — | 99 | ||
| COF-TAPT | 25 | 200000 |
1.30 | Methylation reaction | 99 | ||
| COF-TAPT | 25 | 50 | 0.39 | Methylation reaction | 99 | ||
| COF-TAPB | 25 | 200000 |
0.71 | Methylation reaction | 99 | ||
| COF-TAPB | 25 | 50 | 0.12 | Methylation reaction | 99 | ||
| TFPA-TAPT | 25 | 50 | 0.18 | Methylation reaction | 99 |
5. Simultaneous capture of low-concentration I2 and CH3I
Molecular iodine and organic iodides (represented by CH3I) coexist in the off-gas of nuclear power plants at low concentrations, and both need to be captured. Because of their different properties, it is important to develop adsorbents that can capture both I2 and CH3I with high efficiency. However, few studies have evaluated the ability of adsorbents to simultaneously capture I2 and CH3I (Table 2), possibly because such measurements require dynamic adsorption systems that are difficult to operate and are less common.| Adsorbent name | Test temperature (°C) | I2 concentration (ppmv) | CH3I concentration (ppmv) | Total iodine uptake (g g−1) | Adsorption mechanisms | Ref. | |
|---|---|---|---|---|---|---|---|
| a The experiment was conducted under the conditions of simulated gas mixtures, including I2 (150 ppmv), CH3I (50 ppmv), humidity (RH = 95%), HNO3, and NOx. b The experiment was conducted with the mixed vapor of 150 ppm I2 and 50 ppm CH3I. | |||||||
| Zeolites | Ag0@MOR | 150 | 150 | 50 | 0.16a | Redox reaction/catalytic decomposition reaction | 97 |
| Ag0@MOR | 25 | 150 | 50 | 0.44a | Redox reaction/catalytic decomposition reaction | 97 | |
| HISL | 150 | 150 | 50 | 0.05a | Hydrophobic interaction | 97 | |
| HISL | 25 | 150 | 50 | 0.08a | Hydrophobic interaction | 97 | |
| MOFs | MIL-101-Cr-TED | 25 | 150 | 50 | 0.55a | Lewis acid–base interaction/methylation reaction | 97 |
| MIL-101-Cr-TED | 150 | 150 | 50 | 0.38a | Lewis acid–base interaction/methylation reaction | 97 | |
| MIL-101-Cr-HMTA | 25 | 150 | 50 | 0.44a | Lewis acid–base interaction/methylation reaction | 97 | |
| MIL-101-Cr-HMTA | 150 | 150 | 50 | 0.33a | Lewis acid–base interaction/methylation reaction | 97 | |
| MIL-101-Cr-HMTA | 25 | 150 | 50 | 1.08b | Lewis acid–base interaction/methylation reaction | 99 | |
| COFs | SCU-COF-2 | 25 | 150 | 50 | 0.56b | Lewis acid–base interaction/methylation reaction | 99 |
| iCOF-AB-50 | 25 | 150 | 50 | 1.59b | Coulomb interaction | 99 | |
| COF-TAPT | 25 | 150 | 50 | 1.51b | Lewis acid–base interaction/methylation reaction | 99 | |
| COF-TAPB | 25 | 150 | 50 | 1.17b | Lewis acid–base interaction/methylation reaction | 99 | |
| TFPA-TAPT | 25 | 150 | 50 | 0.47b | Lewis acid–base interaction/methylation reaction | 99 | |
Li et al. modified MOF MIL-101 (Cr) with various tertiary amines to simultaneously capture I2 and CH3I under simulated off-gas conditions.97 The tertiary amine groups can adsorb I2 through Lewis acid–base interactions and adsorb CH3I through methylation reactions. Consequently, amine-functionalized MIL-101 (Cr) MOFs exhibit high total iodine (I2 + CH3I) uptake when used for iodine capture from gas streams containing 150 ppmv I2 and 50 ppmv CH3I in the presence of HNO3, NOx, and water vapor. Specific iodine uptake depends on the density of the amine groups and test temperature (Table 2). At 150 °C, both MIL-101-Cr-TED and MIL-101-Cr-HMDA rendered a “decontamination factor” (DF) value as high as > 5000, which met the regulatory standards of nuclear processing facilities (DF > 3000).
Xie et al. reported the iodine capture performance of several COFs from gas streams containing 150 ppmv I2 and 50 ppmv CH3I at 25 °C.99 Two COF materials (iCOF-AB-50 and COF-TAPT) ranked the top two among all the adsorbents evaluated, with a total iodine uptake of 1.59 and 1.51 g g−1, respectively. These values are higher than the total iodine uptake of MIL-101-Cr-HMDA (1.08 g g−1) obtained under the same conditions. According to the single-component dynamic adsorption results, the ultrahigh total iodine uptake of iCOF-AB-50 is derived from the contribution of I2 adsorption because of the abundant ionic groups that effectively promote I2 adsorption via strong Coulomb interactions.
6. Summary and outlook
Over the past decade, many nanoporous materials, primarily COFs, POPs, and MOFs, have been reported as adsorbents for iodine capture. Compared to traditional industrial adsorbents, such as zeolites and activated carbon, these emerging adsorbents are characterized by easily regulated pore structures and abundant surface functionality, enabling the capture of molecular iodine and organic iodides via novel adsorption mechanisms.This review summarizes the common mechanisms for adsorption-based iodine capture. Molecular iodine can be adsorbed via redox reactions, coordination interactions, electrophilic aromatic substitution, Lewis acid–base interactions, Coulomb interactions, hydrogen bonding, van der Waals interactions, and hydrophobic interactions. As a representative of organic iodides, CH3I can be adsorbed via catalytic decomposition reactions, coordination interactions, methylation reactions, halogen bonding, and hydrogen bonding. In general, CH3I is extremely difficult to capture by physisorption than I2 because of the lack of strong intermolecular forces and its ultra-low concentration in the off-gas.
Despite the multiple studies on iodine capture in recent years, most did not consider the needs of practical applications. The adsorbents for capturing iodine from actual off-gas require to meet two requirements because the off-gas produced during the reprocessing of used fuel rods generally has high temperatures (∼150 °C), low concentrations of I2 (<150 ppmv) and organic iodides (∼50 ppmv), and various acidic (pH < 1) components and water vapor. First, they should be able to capture iodine at low concentrations and high temperatures. Second, they should be chemically and thermally stable under strongly acidic hydrothermal conditions.
However, previous studies did not consider these two important factors when examining whether the adsorbent has practical value. Indeed, a vast majority of “emerging” adsorbents, such as MOFs, COFs, and POPs, were evaluated at low temperatures (≤75 °C) with high I2 concentrations (using saturated I2 vapor). As high temperature and low I2 concentration are unfavorable factors for I2 adsorption, the I2 uptake of these adsorbents under the actual industrial conditions must be considerably lower than the reported values. Therefore, the reported adsorption capacity is meaningless for practical applications, and the static measurement method is unreliable (Section 4.1). Another neglected issue is the stability of the adsorbents under the hydrothermally acidic conditions of industrial off-gas. The intrinsic porous structures of organic or organic–inorganic hybrid adsorbents can be destroyed under harsh conditions with few exceptions.84,97,99 Structural damage to adsorbents indicates a significant reduction in adsorption capacity and loss of reusability.
Based on these considerations, although organic-based adsorbents are valuable for fundamental research, they are unlikely to be directly applied to capture iodine from industrial off-gas. They may be useful for capturing iodine from pretreated off-gas to remove moisture and reduce acidity at lower temperatures or under other mild-condition applications. It remains a considerable challenge to develop high-performance adsorbents for direct off-gas treatment that combine high iodine uptake with excellent structural stability and reusability. Considering the highly unfavorable adsorption conditions (i.e., high temperature of ∼150 °C and low iodine concentration), the desired iodine adsorption capacity is difficult to achieve by physisorption, whereas chemisorption may be the only option because of its high adsorption strength. Therefore, the ideal adsorbents for iodine capture from off-gas should have many easily regenerated chemisorption sites while maintaining structural integrity during adsorption and regeneration. Achieving this goal may require new adsorption strategies. For example, a single strong binding site can make regeneration difficult, but coupling several moderately strong binding sites may provide a balance between adsorption efficiency and recyclability. To avoid structural damage, it is necessary to understand the mechanisms responsible for the structural degradation of adsorbents caused by common species in nuclear off-gas. In this regard, quantum chemistry calculations and simulations would be a useful tool. The revealed adsorbate–adsorbent interaction modes can provide insight into the degradation mechanism, thereby guiding the design of stable iodine adsorbents.
Dynamic measurement systems are recommended for evaluating developed adsorbents because they can easily realize precise concentration and temperature control while allowing the incorporation of multiple components to simulate the actual application conditions and scenarios. For example, the experiments of simultaneously capturing I2 and CH3I can be easily performed using a dynamic system. Competitive adsorption between I2/CH3I and coexisting species in the off-gas, including H2O, NO, CO, CH3Cl, and Cl2, can be investigated using a dynamic system. There is still a lack of systematic research on the competitive adsorption of these components, except for a few studies focusing only on water. Finally, this study proposes to evaluate the adsorbents developed for iodine capture under conditions close to actual off-gas. Alternatively, researchers should discuss the correlation between the test conditions and specific application scenarios.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by the AMPM CCF fund (FCC/1/1972-43-01) to Y. H. from King Abdullah University of Science and Technology.Notes and references
- I. A. E. Agency, https://www.iaea.org/newscenter/news/the-use-of-nuclear-power-beyond-generating-electricity-non-electric-applications.
- E. Kintisch, Science, 2005, 310, 1406 CrossRef CAS PubMed.
- J. E. Ten Hoeve and M. Z. Jacobson, Energy Environ. Sci., 2012, 5, 8743–8757 RSC.
- P. A. Kharecha and J. E. Hansen, Environ. Sci. Technol., 2013, 47, 4889–4895 CrossRef CAS PubMed.
- F. C. Kupper, M. C. Feiters, B. Olofsson, T. Kaiho, S. Yanagida, M. B. Zimmermann, L. J. Carpenter, G. W. Luther, Z. L. Lu, M. Jonsson and L. Kloo, Angew. Chem., Int. Ed., 2011, 50, 11598–11620 CrossRef PubMed.
- A. Saiz-Lopez, J. M. C. Plane, A. R. Baker, L. J. Carpenter, R. von Glasow, J. C. G. Martin, G. McFiggans and R. W. Saunders, Chem. Rev., 2012, 112, 1773–1804 CrossRef CAS.
- K. Nakamura, M. Saeki and E. Tachikaw, J. Nucl. Sci. Technol., 1973, 10, 367–373 CrossRef CAS.
- E. C. Beahm, Y. M. Wang, S. J. Wisbey and W. E. Shockley, Nucl. Technol., 1987, 78, 34–42 CrossRef CAS.
- J. Paquette and B. L. Ford, Abstr. Pap. Am. Chem. Soc., 1988, 195, 88–Nucl Search PubMed.
- D. R. Haefner and T. J. Tranter, Report No. INL/EXT-07-12299; TRN: US0800243, 2007.
- S. U. Nandanwar, K. Coldsnow, V. Utgikar, P. Sabharwall and D. E. Aston, Chem. Eng. J., 2016, 306, 369–381 CrossRef CAS.
- C. M. Gonzalez-Garcia, J. F. Gonzalez and S. Roman, Fuel Process. Technol., 2011, 92, 247–252 CrossRef CAS.
- N. Mnasri, C. Charnay, L. C. de Menorval, Y. Moussaoui, E. Elaloui and J. Zajac, Microporous Mesoporous Mater., 2014, 196, 305–313 CrossRef CAS.
- Q. H. Cheng, Z. J. Li and T. W. Chu, Nucl. Sci. Technol., 2015, 26, 43–47 Search PubMed.
- Y. Nan, L. L. Tavlarides and D. W. DePaoli, AIChE J., 2017, 63, 1024–1035 CrossRef CAS.
- H. J. Choi and M. P. Suh, J. Am. Chem. Soc., 2004, 126, 15844–15851 CrossRef CAS PubMed.
- M. H. Zeng, Q. X. Wang, Y. X. Tan, S. Hu, H. X. Zhao, L. S. Long and M. Kurmoo, J. Am. Chem. Soc., 2010, 132, 2561–2563 CrossRef CAS PubMed.
- N. X. Zhu, C. W. Zhao, J. C. Wang, Y. A. Li and Y. B. Dong, Chem. Commun., 2016, 52, 12702–12705 RSC.
- B. Guo, F. Li, C. Wang, L. Zhang and D. Sun, J. Mater. Chem. A, 2019, 7, 13173–13179 RSC.
- T. Xu, J. T. Li, M. W. Jia, G. H. Li and Y. L. Liu, Dalton Trans., 2021, 50, 13096–13102 RSC.
- G. Das, T. Prakasam, S. Nuryyeva, D. S. Han, A. Abdel-Wahab, J. C. Olsen, K. Polychronopoulou, C. Plataslglesias, F. Ravaux, M. Jouiad and A. Trabolsi, J. Mater. Chem. A, 2016, 4, 15361–15369 RSC.
- T. Geng, Z. Zhu, W. Zhang and Y. Wang, J. Mater. Chem. A, 2017, 5, 7612–7617 RSC.
- J. Wang, Z. L. Li, Y. Wang, C. T. Wei, K. L. Ai and L. H. Lu, Mater. Horiz., 2019, 6, 1517–1525 RSC.
- W. Xie, D. Cui, S. R. Zhang, Y. H. Xu and D. L. Jiang, Mater. Horiz., 2019, 6, 1571–1595 RSC.
- K. C. Jie, Y. J. Zhou, Q. Sun, B. Li, R. Zhao, D. E. Jiang, W. Guo, H. Chen, Z. Z. Yang, F. H. Huang and S. Dai, Nat. Commun., 2020, 11, 1086 CrossRef CAS PubMed.
- M. Y. Xu, T. Wang, L. Zhou and D. B. Hua, J. Mater. Chem. A, 2020, 8, 1966–1974 RSC.
- Y. S. Lan, M. M. Tong, Q. Y. Yang and C. L. Zhong, Crystengcomm, 2017, 19, 4920–4926 RSC.
- J. H. Li, H. X. Zhang, L. Y. Zhang, K. Wang, Z. K. Wang, G. Y. Liu, Y. L. Zhao and Y. F. Zeng, J. Mater. Chem. A, 2020, 8, 9523–9527 RSC.
- L. Y. Zhang, J. H. Li, H. X. Zhang, Y. Liu, Y. M. Cui, F. C. Jin, K. Wang, G. Y. Liu, Y. L. Zhao and Y. F. Zeng, Chem. Commun., 2021, 57, 5558–5561 RSC.
- P. Wang, Q. Xu, Z. P. Li, W. M. Jiang, Q. H. Jiang and D. L. Jiang, Adv. Mater., 2018, 30, 1801991 CrossRef.
- Q. Zhao, L. Zhu, G. Lin, G. Chen, B. Liu, L. Zhang, T. Duan and J. Lei, ACS Appl. Mater. Interfaces, 2019, 11, 42635–42645 CrossRef CAS PubMed.
- T.-M. Geng, F.-Q. Wang, X.-C. Fang and H. Xu, Microporous Mesoporous Mater., 2021, 317, 111001 CrossRef CAS.
- T. C. T. Pham, S. Docao, I. C. Hwang, M. K. Song, D. Y. Choi, D. Moon, P. Oleynikov and K. B. Yoon, Energy Environ. Sci., 2016, 9, 1050–1062 RSC.
- K. W. Chapman, P. J. Chupas and T. M. Nenoff, J. Am. Chem. Soc., 2010, 132, 8897–8899 CrossRef CAS PubMed.
- Y. Lu, H. W. Liu, R. N. Gao, S. L. Xiao, M. Zhang, Y. F. Yin, S. Q. Wang, J. Li and D. J. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 29179–29185 CrossRef CAS PubMed.
- R. N. Gao, Y. Lu, S. L. Xiao and J. Li, Sci. Rep., 2017, 7, 4303 CrossRef PubMed.
- D. W. Holladay, Literature survey: methods for the removal of iodine species from off-gases and liquid waste streams of nuclear power and nuclear fuel reprocessing plants, with emphasis on solid sorbents, Oak Ridge National Lab.(ORNL), Report No. ORNL/TM-6350, 1979, DOI:10.2172/6394859.
- J. Huve, A. Ryzhikov, H. Nouali, V. Lalia, G. Auge and T. J. Daou, RSC Adv., 2018, 8, 29248–29273 RSC.
- J. H. Yang, Y. J. Cho, J. M. Shin and M. S. Yim, J. Nucl. Mater., 2015, 465, 556–564 CrossRef CAS.
- H. Zou, F. C. Yi, M. X. Song, X. Q. Wang, L. Bian, W. M. Li, N. Pan and X. Q. Jiang, J. Hazard. Mater., 2019, 365, 81–87 CrossRef CAS PubMed.
- A. T. Reda, M. Pan, D. X. Zhang and X. Y. Xu, J. Environ. Chem. Eng., 2021, 9, 105279 CrossRef.
- S. W. Kang, J. H. Yang and M. S. Yim, Nucl. Technol., 2020, 206, 1593–1606 CrossRef.
- X. R. Zhang, I. da Silva, R. Fazzi, A. M. Sheveleva, X. Han, B. F. Spencer, S. A. Sapchenko, F. Tuna, E. J. L. McInnes, M. Li, S. H. Yang and M. Schroder, Inorg. Chem., 2019, 58, 14145–14150 CrossRef CAS PubMed.
- E. Baladi, V. Nobakht, A. Tarassoli, D. M. Proserpio and L. Carlucci, Cryst. Growth Des., 2018, 18, 7207–7218 CrossRef CAS.
- B. Lee, Y. P. Chen, J. Park and J. Park, ACS Appl. Mater. Interfaces, 2019, 11, 25817–25823 CrossRef CAS PubMed.
- A. Y. Rogachev and R. Hoffmann, J. Am. Chem. Soc., 2013, 135, 3262–3275 CrossRef CAS PubMed.
- D. F. Sava, K. W. Chapman, M. A. Rodriguez, J. A. Greathouse, P. S. Crozier, H. Y. Zhao, P. J. Chupas and T. M. Nenoff, Chem. Mater., 2013, 25, 2591–2596 CrossRef CAS.
- J. H. Ye, Z. J. Hu, Y. X. Wang, W. C. Zhang and Y. Zhang, Tetrahedron Lett., 2012, 53, 6858–6860 CrossRef CAS.
- Y. Zhu, Y.-J. Ji, D.-G. Wang, Y. Zhang, H. Tang, X.-R. Jia, M. Song, G. Yu and G.-C. Kuang, J. Mater. Chem. A, 2017, 5, 6622–6629 RSC.
- T. R. Thomas, B. A. Staples, L. P. Murphy, Dry method for recycling iodine-loaded silver zeolite, US Pat. 4088737/A, 1978, http://inis.iaea.org/search/search.aspx?orig_q=RN:10456359 Search PubMed.
- J. A. Wang, K. L. Ai and L. H. Lu, J. Mater. Chem. A, 2019, 7, 16850–16858 RSC.
- A. J. Blake, F. A. Devillanova, R. O. Gould, W. S. Li, V. Lippolis, S. Parsons, C. Radek and M. Schroder, Chem. Soc. Rev., 1998, 27, 195–205 RSC.
- X. Qian, Z. Q. Zhu, H. X. Sun, F. Ren, P. Mu, W. Liang, L. Chen and A. Li, ACS Appl. Mater. Interfaces, 2016, 8, 21063–21069 CrossRef CAS PubMed.
- X. W. Pan, C. H. Ding, Z. M. Zhang, H. Z. Ke and G. E. Cheng, Microporous Mesoporous Mater., 2020, 300, 110161 CrossRef CAS.
- P. Chen, X. H. He, M. B. Pang, X. T. Dong, S. Zhao and W. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 20429–20439 CrossRef CAS.
- T. H. Niu, C. C. Feng, C. Yao, W. Y. Yang and Y. H. Xu, ACS Appl. Polym. Mater., 2021, 3, 354–361 CrossRef CAS.
- P. Deplano, F. A. Devillanova, J. R. Ferraro, F. Isaia, V. Lippolis and M. L. Mercuri, Appl. Spectrosc., 1992, 46, 1625–1629 CrossRef CAS.
- N. S. Rao, G. B. Rao and D. Ziessow, Spectrochim. Acta, Part A, 1990, 46, 1107–1124 CrossRef.
- M. Hasani and M. Shamsipur, J. Inclusion Phenom. Macrocyclic Chem., 2004, 48, 135–139 CrossRef CAS.
- N. Alizadeh and A. Roomiani, J. Chil. Chem. Soc., 2012, 57, 1130–1133 CrossRef CAS.
- E. M. Nour, Spectrochim. Acta, Part A, 2000, 56, 167–170 CrossRef PubMed.
- X. Q. Yan, H. Wang, W. Di Chen and W. J. Jin, Anal. Sci., 2014, 30, 365–370 CrossRef CAS PubMed.
- T. M. Geng, S. N. Ye, Z. M. Zhu and W. Y. Zhang, J. Mater. Chem. A, 2018, 6, 2808–2816 RSC.
- A. Hijazi, B. Azambre, G. Finqueneisel, F. Vibert and J. L. Blin, Microporous Mesoporous Mater., 2019, 288, 109586 CrossRef CAS.
- S. Xiong, X. Tang, C. Pan, L. Li, J. Tang and G. Yu, ACS Appl. Mater. Interfaces, 2019, 11, 27335–27342 CrossRef CAS PubMed.
- N. Liu, J. Chen, Z. Wu, P. Zhan, L. Zhang, Q. Wei, F. Wang and L. Shao, ACS Appl. Polym. Mater., 2021, 3, 2178–2188 CrossRef CAS.
- Z. J. Yan, B. Cui, T. Zhao, Y. F. Luo, H. C. Zhang, J. L. Xie, N. Li, N. S. Bu, Y. Yuan and L. X. Xia, Molecules, 2021, 26, 5263 CrossRef CAS PubMed.
- X. H. Xu, Y. X. Li, L. Zhou, N. Liu and Z. Q. Wu, Chem. Sci., 2022, 13, 1111–1118 RSC.
- B. Jiang, Y. Qi, X. F. Li, X. H. Guo, Z. M. Jia, J. Zhang, Y. Li and L. J. Ma, Chin. Chem. Lett., 2022, 33, 3556–3560 CrossRef CAS.
- K. C. Jie, Y. J. Zhou, E. R. Li, Z. T. Li, R. Zhao and F. H. Huang, J. Am. Chem. Soc., 2017, 139, 15320–15323 CrossRef CAS PubMed.
- C. C. Feng, G. J. Xu, W. Xie, S. R. Zhang, C. Yao and Y. H. Xu, Polym. Chem., 2020, 11, 2786–2790 RSC.
- D. H. Dai, J. Yang, Y. C. Zou, J. R. Wu, L. L. Tan, Y. Wang, B. Li, T. Lu, B. Wang and Y. W. Yang, Angew. Chem., Int. Ed., 2021, 60, 8967–8975 CrossRef CAS PubMed.
- S. Y. Yao, W. H. Fang, Y. Y. Sun, S. T. Wang and J. Zhang, J. Am. Chem. Soc., 2021, 143, 2325–2330 CrossRef CAS PubMed.
- Z. J. Yan, Y. Yuan, Y. Y. Tian, D. M. Zhang and G. S. Zhu, Angew. Chem., Int. Ed., 2015, 54, 12733–12737 CrossRef CAS PubMed.
- J. T. Hughes, D. F. Sava, T. M. Nenoff and A. Navrotsky, J. Am. Chem. Soc., 2013, 135, 16256–16259 CrossRef CAS PubMed.
- D. F. Sava, M. A. Rodriguez, K. W. Chapman, P. J. Chupas, J. A. Greathouse, P. S. Crozier and T. M. Nenoff, J. Am. Chem. Soc., 2011, 133, 12398–12401 CrossRef CAS PubMed.
- B. J. Riley, J. Chun, W. Um, W. C. Lepry, J. Matyas, M. J. Olszta, X. H. Li, K. Polychronopoulou and M. G. Kanatzidis, Environ. Sci. Technol., 2013, 47, 7540–7547 CrossRef CAS PubMed.
- W. Zhang, Y. Mu, X. He, P. Chen, S. Zhao, C. Huang, Y. Wang and J. Chen, Chem. Eng. J., 2020, 379, 122365 CrossRef CAS.
- Y. Q. Hu, M. Q. Li, Y. Y. Wang, T. Zhang, P. Q. Liao, Z. P. Zheng, X. M. Chen and Y. Z. Zheng, Chem.–Eur. J., 2017, 23, 8409–8413 CrossRef CAS PubMed.
- H. C. Hu, F. Y. Chen, Z. Y. Zhang, D. C. Liu, Y. N. Liang and Z. L. Chen, Front. Chem., 2022, 10, 864131 CrossRef CAS PubMed.
- G. Brunet, D. A. Safin, M. Z. Aghaji, K. Robeyns, I. Korobkov, T. K. Woo and M. Murugesu, Chem. Sci., 2017, 8, 3171–3177 RSC.
- Y. Z. Tang, H. L. Huang, J. Li, W. J. Xue and C. L. Zhong, J. Mater. Chem. A, 2019, 7, 18324–18329 RSC.
- C. Y. Yan and T. C. Mu, Phys. Chem. Chem. Phys., 2014, 16, 5071–5075 RSC.
- Z. Zhang, X. Dong, J. Yin, Z. G. Li, X. Li, D. Zhang, T. Pan, Q. Lei, X. Liu, Y. Xie, F. Shui, J. Li, M. Yi, J. Yuan, Z. You, L. Zhang, J. Chang, H. Zhang, W. Li, Q. Fang, B. Li, X. H. Bu and Y. Han, J. Am. Chem. Soc., 2022, 144, 6821–6829 CrossRef CAS PubMed.
- J. H. Liu, Y. J. Qi, D. Zhao, H. H. Li and S. T. Zheng, Inorg. Chem., 2019, 58, 516–523 CrossRef CAS PubMed.
- S. Chibani, F. Chiter, L. Cantrel and J. F. Paul, J. Phys. Chem. C, 2017, 121, 25283–25291 CrossRef CAS.
- D. Shetty, J. Raya, D. S. Han, Z. Asfari, J. C. Olsen and A. Trabolsi, Chem. Mater., 2017, 29, 8968–8972 CrossRef CAS.
- X. R. Zhang, I. da Silva, H. G. W. Godfrey, S. K. Callear, S. A. Sapchenko, Y. Q. Cheng, I. Vitorica-Yrezabal, M. D. Frogley, G. Cinque, C. C. Tang, C. Giacobbe, C. Dejoie, S. Rudic, A. J. Ramirez-Cuesta, M. A. Denecke, S. H. Yang and M. Schroder, J. Am. Chem. Soc., 2017, 139, 16289–16296 CrossRef CAS PubMed.
- B. F. Abrahams, M. Moylan, S. D. Orchard and R. Robson, Angew. Chem., Int. Ed., 2003, 42, 1848–1851 CrossRef CAS PubMed.
- A. Karhu, Methods to prevent the source term of methyl iodide during a core melt accident, Report No. NKS-13, ISBN 87-7893-063-4, VTT Energy, Finland, 1999 Search PubMed.
- S. H. Bruffey, R. T. Jubin and J. A. Jordan, Report No. FCRD-MRWFD-2016-000357; ORNL/TM-2016/568, 2016.
- T. M. Nenoff, M. A. Rodriguez, N. R. Soelberg and K. W. Chapman, Microporous Mesoporous Mater., 2014, 200, 297–303 CrossRef CAS.
- B. Azambre, M. Chebbi and A. Hijazi, Chem. Eng. J., 2020, 379, 122308 CrossRef CAS.
- H. P. Zhang, L. L. Gong, M. J. Yin, X. H. Xiong, Q. Y. Zhang, X. F. Feng, F. Luo, J. B. Carney and Y. F. Yue, Cell Rep. Phys. Sci., 2022, 3, 100830 CrossRef CAS.
- B. Lee, D. Moon and J. Park, Angew. Chem., Int. Ed. Engl., 2020, 59, 13793–13799 CrossRef CAS PubMed.
- H. Chun, J. Kang and B. Han, Phys. Chem. Chem. Phys., 2016, 18, 32050–32056 RSC.
- B. Y. Li, X. L. Dong, H. Wang, D. X. Ma, K. Tan, S. Jensen, B. J. Deibert, J. Butler, J. Cure, Z. Shi, T. Thonhauser, Y. J. Chabal, Y. Han and J. Li, Nat. Commun., 2017, 8, 485 CrossRef PubMed.
- L. W. He, L. Chen, X. L. Dong, S. T. Zhang, M. X. Zhang, X. Dai, X. J. Liu, P. Lin, K. F. Li, C. L. Chen, T. T. Pan, F. Y. Ma, J. C. Chen, M. J. Yuan, Y. G. Zhang, L. Chen, R. H. Zhou, Y. Han, Z. F. Chai and S. Wang, Chem, 2021, 7, 699–714 CAS.
- Y. Q. Xie, T. T. Pan, Q. Lei, C. L. Chen, X. L. Dong, Y. Y. Yuan, W. Al Maksoud, L. Zhao, L. Cavallo, I. Pinnau and Y. Han, Nat. Commun., 2022, 13, 2878 CrossRef CAS PubMed.
- T. Brinck, J. S. Murray and P. Politzer, Int. J. Quantum Chem., 1992, 44, 57–64 CrossRef.
- X. Y. Wu, L. J. Chen, E. J. Amigues, R. Y. Wang, Z. F. Pang and L. F. Ding, Acs Omega, 2021, 6, 18169–18177 CrossRef CAS PubMed.
- A. M. S. Riel, R. K. Rowe, E. N. Ho, A. C. C. Carlsson, A. K. Rappe, O. B. Berryman and P. S. Ho, Acc. Chem. Res., 2019, 52, 2870–2880 CrossRef CAS PubMed.
- M. Chebbi, B. Azambre, C. Volkringer and T. Loiseau, Microporous Mesoporous Mater., 2018, 259, 244–254 CrossRef CAS.
- X. Guo, Y. Li, M. Zhang, K. Cao, Y. Tian, Y. Qi, S. Li, K. Li, X. Yu and L. Ma, Angew. Chem., Int. Ed. Engl., 2020, 59, 22697–22705 CrossRef CAS PubMed.
- Y. Z. Liao, J. Weber, B. M. Mills, Z. H. Ren and C. F. J. Faul, Macromolecules, 2016, 49, 6322–6333 CrossRef CAS.
- Z. J. Yin, S. Q. Xu, T. G. Zhan, Q. Y. Qi, Z. Q. Wu and X. Zhao, Chem. Commun., 2017, 53, 7266–7269 RSC.
- X. M. Li, G. Chen, J. T. Ma and Q. Jia, Sep. Purif. Technol., 2019, 210, 995–1000 CrossRef CAS.
- G. J. Xu, Y. Zhu, W. Xie, S. R. Zhang, C. Yao and Y. H. Xu, Chem.–Asian J., 2019, 14, 3259–3263 CrossRef CAS PubMed.
- L. Shao, N. Liu, L. Wang, Y. Sang, H. Wan, P. Zhan, L. Zhang, J. Huang and J. Chen, Chemosphere, 2022, 288, 132499 CrossRef CAS PubMed.
- N. Soelberg and T. Watson, FY-2016 methyl iodide higher NOx adsorption test report, Report No. FCRD-MRWFD-2016-000352, INL/EXT-15-36817, Idaho National Laboratory, 2015 Search PubMed.
- Y. Q. Xie, T. T. Pan, Q. Lei, C. L. Chen, X. L. Dong, Y. Y. Yuan, J. Shen, Y. C. Cai, C. H. Zhou, I. Pinnau and Y. Han, Angew. Chem., Int. Ed., 2021, 60, 22432–22440 CrossRef CAS PubMed.
- S. Choi, Y. Nan and L. L. Tavlarides, AIChE J., 2021, 67(67), e17182 CAS.
- B. Azambre, M. Chebbi, O. Leroy and L. Cantrel, Ind. Eng. Chem. Res., 2018, 57, 1468–1479 CrossRef CAS.
- S. U. Nandanwar, K. Coldsnow, M. Green, V. Utgikar, P. Sabharwall and D. E. Aston, Chem. Eng. J., 2016, 287, 593–601 CrossRef CAS.
- M. Chebbi, B. Azambre, C. Monsanglant-Louvet, B. Marcillaud, A. Roynette and L. Cantrel, J. Hazard. Mater., 2021, 409, 124947 CrossRef CAS PubMed.
- G. Y. Cha, S. E. Sivan, M. Lee, K. R. Oh, A. H. Valekar, M. K. Kim, H. Jung, D. Y. Hong and Y. K. Hwang, J. Hazard. Mater., 2021, 417, 125904 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |