
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlatinum-based nanodendrites as glucose oxidase-mimicking surrogates†
Jose I.
Garcia-Peiro‡
abcd,
Javier
Bonet-Aleta‡
 abcd,
Maria L.
Tamayo-Fraile
ab,
Jose L.
Hueso
*abcd and
Jesus
Santamaria
*abcd
abcd,
Maria L.
Tamayo-Fraile
ab,
Jose L.
Hueso
*abcd and
Jesus
Santamaria
*abcd
aInstituto de Nanociencia y Materiales de Aragon (INMA); CSIC-Universidad de Zaragoza, Campus Rio Ebro, Edificio I+D, C/Poeta Mariano Esquillor, s/n, 50018, Zaragoza, Spain. E-mail: jlhueso@unizar.es; jesus.santamaria@unizar.es
bDepartment of Chemical and Environmental Engineering; University of Zaragoza, Spain, Campus Rio Ebro, C/María de Luna, 3, 50018 Zaragoza, Spain
cNetworking Research Center in Biomaterials, Bioengineering and Nanomedicine (CIBER-BBN), Instituto de Salud Carlos III, 28029 Madrid, Spain
dInstituto de Investigación Sanitaria (IIS) de Aragón, Avenida San Juan Bosco, 13, 50009 Zaragoza, Spain
First published on 1st August 2023
Abstract
Catalytic conversion of glucose represents an interesting field of research with multiple applications. From the biotechnology point of view, glucose conversion leads to the fabrication of different added-value by-products. In the field of nanocatalytic medicine, the reduction of glucose levels within the tumor microenvironment (TME) represents an appealing approach based on the starvation of cancer cells. Glucose typically achieves high conversion rates with the aid of glucose oxidase (GOx) enzymes or by fermentation. GOx is subjected to degradation, possesses poor recyclability and operates under very specific reaction conditions. Gold-based materials have been typically explored as inorganic catalytic alternatives to GOx in order to convert glucose into building block chemicals of interest. Still, the lack of sufficient selectivity towards certain products such as gluconolactone, the requirement of high fluxes of oxygen or the critical size dependency hinder their full potential, especially in liquid phase reactions. The present work describes the synthesis of platinum-based nanodendrites as novel enzyme-mimicking inorganic surrogates able to convert glucose into gluconolactone with outstanding selectivity values above 85%. We have also studied the enzymatic behavior of these Pt-based nanozymes using the Michaelis–Menten and Lineweaver–Burk models and used the main calculation approaches available in the literature to determine highly competitive glucose turnover rates for Pt or Pt–Au nanodendrites.
Introduction
The valorization of glucose, carbohydrates and biomass into chemical building blocks of interest represents one of the most currently explored green strategies in biotechnology. Gas phase reactions (oxidation, reduction, reforming) at mild or moderated reaction temperatures lead to the generation of complex oxidized by-products and green hydrogen energy vectors. Liquid phase reactions also lead to multiple added-value products in the form of gluconic acid, hydrogen peroxide, gluconates or complex esters.1 Natural enzymes such as glucose oxidase (GOx) may yield a highly selective conversion of glucose into hydrogen peroxide and gluconolactone. First, the glucose molecule binds to the active site of the enzyme, which contains a flavin adenine dinucleotide (FAD) cofactor. This binding causes a conformational change in the active site, which promotes the transfer of electrons from the glucose molecule to the FAD cofactor and forms FADH2. Glucose oxidation leads to an aldehyde group at the C1 position, forming glucono-1,5-lactone or gluconolactone. Then, the electrons are transferred to a bound molecular oxygen molecule, which causes it to be reduced to H2O2 and the FADH2 is oxidized back to FAD (Fig. 1a). Finally, the H2O2 produced in the second step is released from the enzyme and the gluconolactone product is usually further hydrolyzed to gluconic acid.2,3 The main drawbacks of natural enzymes such as GOx are related to their instability and cost, their difficult reusability and their highly specific operation window conditions that hinder their potential application in biomedical applications.4–8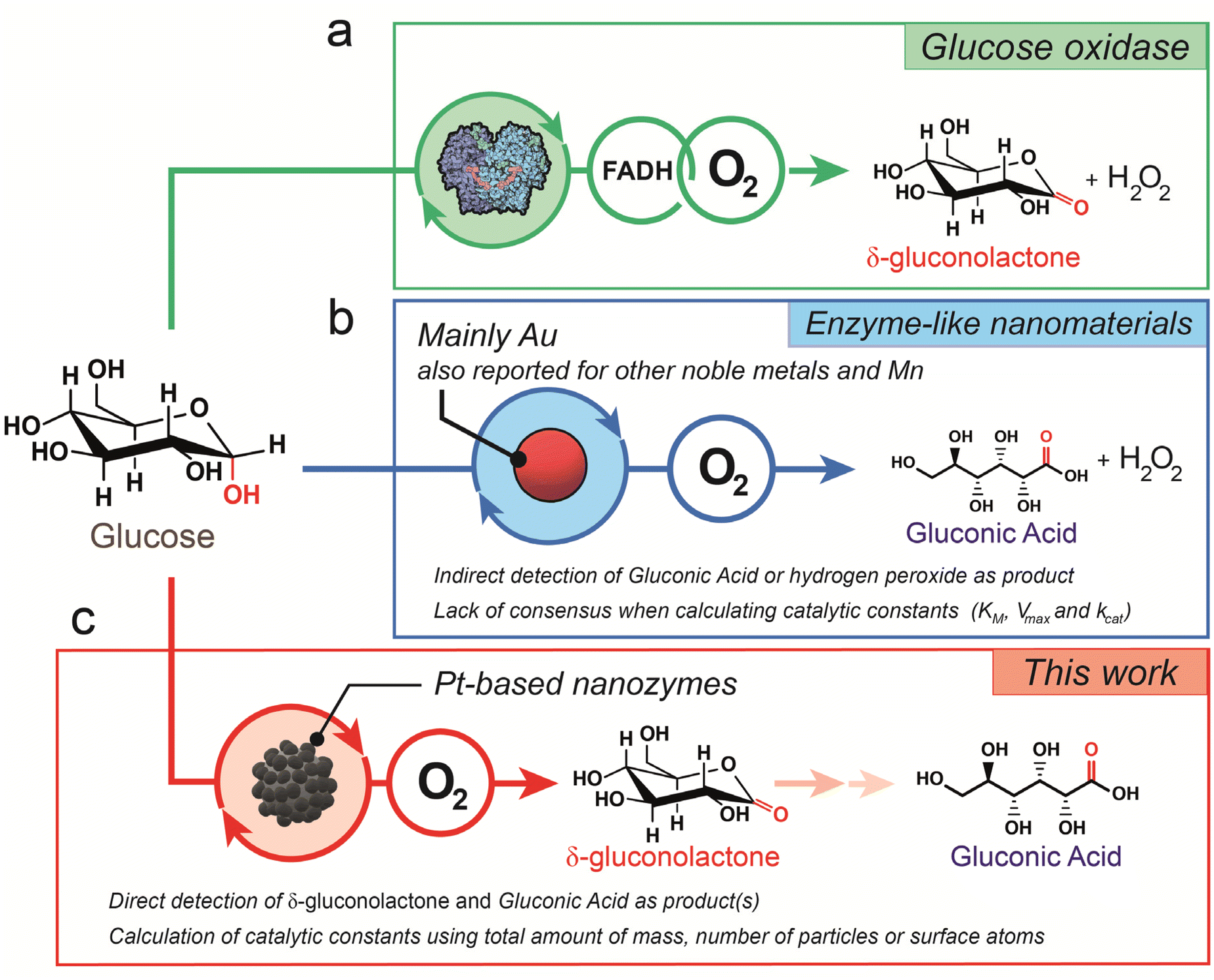 | ||
| Fig. 1 Comparison of glucose oxidase mechanisms: (a) schematic illustration of glucose oxidation catalyzed by the glucose oxidase (GOx) enzyme. The enzymatic reaction involves the FADH cofactor where glucose interacts with the active site to form δ-gluconolactone and transfers the electrons to FAD to form FADH. O2 is used to transfer electrons from FADH and form H2O2; (b) most reported mechanism of glucose oxidation in the presence of nanozymes such as noble metals or Mn. In this case, the reaction involves molecular oxygen and forms H2O2 and gluconic acid instead; (c) proposed mechanism for Pt-based nanodendrites described in this work where O2 leads to glucose oxidation towards δ-gluconolactone as the main glucose oxidation product. | ||
In recent years, nanozymes have emerged as a different kind of nanomaterial with a catalytic response mimicking the activity of enzymes.9–13 Moreover, nanozymes have been classified into two different types. Type 1 nanozymes were more dominant in the first decade of this century and refer to immobilized catalysts in nanomaterials. They were intensely studied by academic researchers such as Rotello and co-workers,14 showing potential applications in biomedicine and intracellular catalysis.15 Type 2 nanozymes are based on inorganic nanomaterials with surface catalytic properties and was the dominant type in the last decade.4
The term nanozyme refers to very different catalytic entities: from molecular catalysts entrapped in polymeric scaffolds to entire nanoparticles. This wide range of possibilities makes it challenging to establish a standardized methodology to quantify and reliably compare the catalytic performance among different nanozymes.5,7,16 Although it is generally accepted that a nanozyme implies the entire nanoparticle, a nanozyme may have more than one active site on the surface. In the literature, different normalization entities such as the number of surface sites, the number of particles, total mass, or total metal atom concentration have been used to define the nanozyme unit and to calculate or compare the turnover rate (kcat).17 The lack of a clear definition of a nanozyme unit can be misleading when attempting to properly evaluate the differences in activity that may vary over 106-fold for the same system.7,16–19 These four ways of defining a nanozyme unit can all be useful depending on the application scenarios and all of them should be provided in order to facilitate comparison for further studies in the literature.17
Most of the studies that deal with this issue use peroxidase-like nanozymes as examples for catalytic comparison. However, much less attention has been paid to GOx-mimicking reactions.20,21 GOx nanozymes offer great potential in industrial processes, including the food and beverage, pharmaceutical, and biosensing sectors. Since the early 2000s, Au NPs have been extensively evaluated as inorganic enzyme mimics for the specific purpose of catalyzing the oxidation of glucose to render gluconic acid and hydrogen peroxide (Fig. 1b).22 Initially, Pd23 and Pt24–26 nanoparticles were also evaluated as catalysts in the catalytic oxidation of glucose with O2. However, the burst of Au systems buried the potential of Pt and Pd until recently, where a series of recent advances have made them attractive again, especially for potential cancer therapy applications.8 Noble metals such as Pd and especially Pt have been in the spotlight in the last few years due to their appealing physicochemical properties, including those with a dendritic and porous structure.27–31 In particular, the catalase-like activity of Pt-based nanozymes may help in alleviating tumor hypoxia in the tumor microenvironment (TME) using overexpressed hydrogen peroxide as the oxygen source. This in situ oxygen supply facilitates glucose oxidation at low O2 concentrations.8 Moreover, Pt nanozymes also hold great potential for therapeutic synergy due to their response to electromagnetic radiation such as NIR light heating,32 X-ray absorption33 and electric fields.8,34
A good understanding of GOx enzyme-mimicking surrogates in comparison with natural enzymes is necessary. Understanding glucose adsorption or oxygen activation is key for further optimization of glucose oxidation catalysis.35 In addition, monitoring the selectivity in enzymatic reactions may also help in elucidating the involved mechanism in the nanozyme-mimicking alternative pathway. Nanozymes catalyze reactions by providing surface sites for substrate binding and promoting the transfer of electrons, thereby facilitating the formation of reaction intermediates. It has been commonly reported how reactive oxygen species (ROS) such as ˙O or ˙OH in the presence of O2 or H2O2 are key to perform oxidase-like reactions on organic substrates such as TMB or OPD.8 However, in the last few years, it has also been reported how the oxidation of glucose does not solely rely on the oxidation ability of O2.20 Dong's group studied the mechanism of glucose oxidation by noble metals. They studied the oxidation reaction of glucose that involves the dehydrogenation of the hydroxyl groups on glucose to form aldehyde groups. In addition, they reported that the reaction might proceed by abstracting hydrogen from glucose to form an M–H intermediate and further transferring H to different substrates to generate sub-H adducts. The rate-determining step involved the breaking of the C–H bond. Moreover, in Au-catalyzed oxygen reduction, the energy barrier for breaking the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond is higher, leading to a predominant 2e− pathway. However, other noble metal nanoparticles undergo a similar catalytic process, although O2 is typically reduced to water instead by using the 4e− pathway. In summary, both GOx and noble metal NPs can catalyze electron transfer from glucose to other electron acceptors such as O2.20
O double bond is higher, leading to a predominant 2e− pathway. However, other noble metal nanoparticles undergo a similar catalytic process, although O2 is typically reduced to water instead by using the 4e− pathway. In summary, both GOx and noble metal NPs can catalyze electron transfer from glucose to other electron acceptors such as O2.20
In this work, we present a novel type 2 Pt nanozyme with GOx-like activity. To evaluate the catalytic activity of the nanozyme, we defined the nanozyme unit using different approximations reported in the recent literature and compared them. We also evaluated the activity of core–shell bimetallic NPs (Au and Pt) to compare the activity of different, though analogous, systems. Regardless of the methodology to calculate the catalytic turnovers, we found that Pt nanodendrites exhibit a GOx-like behavior that can be fitted to the Michaelis–Menten and Lineweaver–Burk models, with outstanding selectivity towards gluconolactone (Fig. 1c) and highly competitive kcat values in comparison with the existing Au counterparts.
Results and discussion
Synthesis and characterization of Pt-based nanozymes
A templated polymer-assisted method was employed to synthesize core–shell Au–Pt nanodendrites and Pt nanodendrites (NDs), respectively, as depicted by transmission electron microscopy (TEMs) image (Fig. 2a and b). The process began by completely dissolving Pluronic F-127, and then mixing in metal precursors and ascorbic acid (A.A.). The reaction was performed for 4 days. X-ray diffraction (XRD) analysis displayed patterns that correspond to the cubic structure of Pt and/or Au (Fig. 2c).36,37 XPS analysis (Fig. 2d and e) of the Pt 4f region further confirmed the Pt-rich composition of the surface of both the AuPt hybrid and Pt-NDs. For the AuPt-NDs, the Au at% increased towards the inner part only after an etching treatment (data not shown) and microwave plasma atomic emission spectroscopy (MP-AES) revealed an overall composition of 43% and 57% of Au and Pt, respectively. Finally, the optical properties of the Pt-NDs were characterized using a UV-vis spectrophotometer. Broad absorption bands spreading towards the visible-near infrared window were identified, especially when Au NPs were present in the nanomaterials38,39 (Fig. 2f).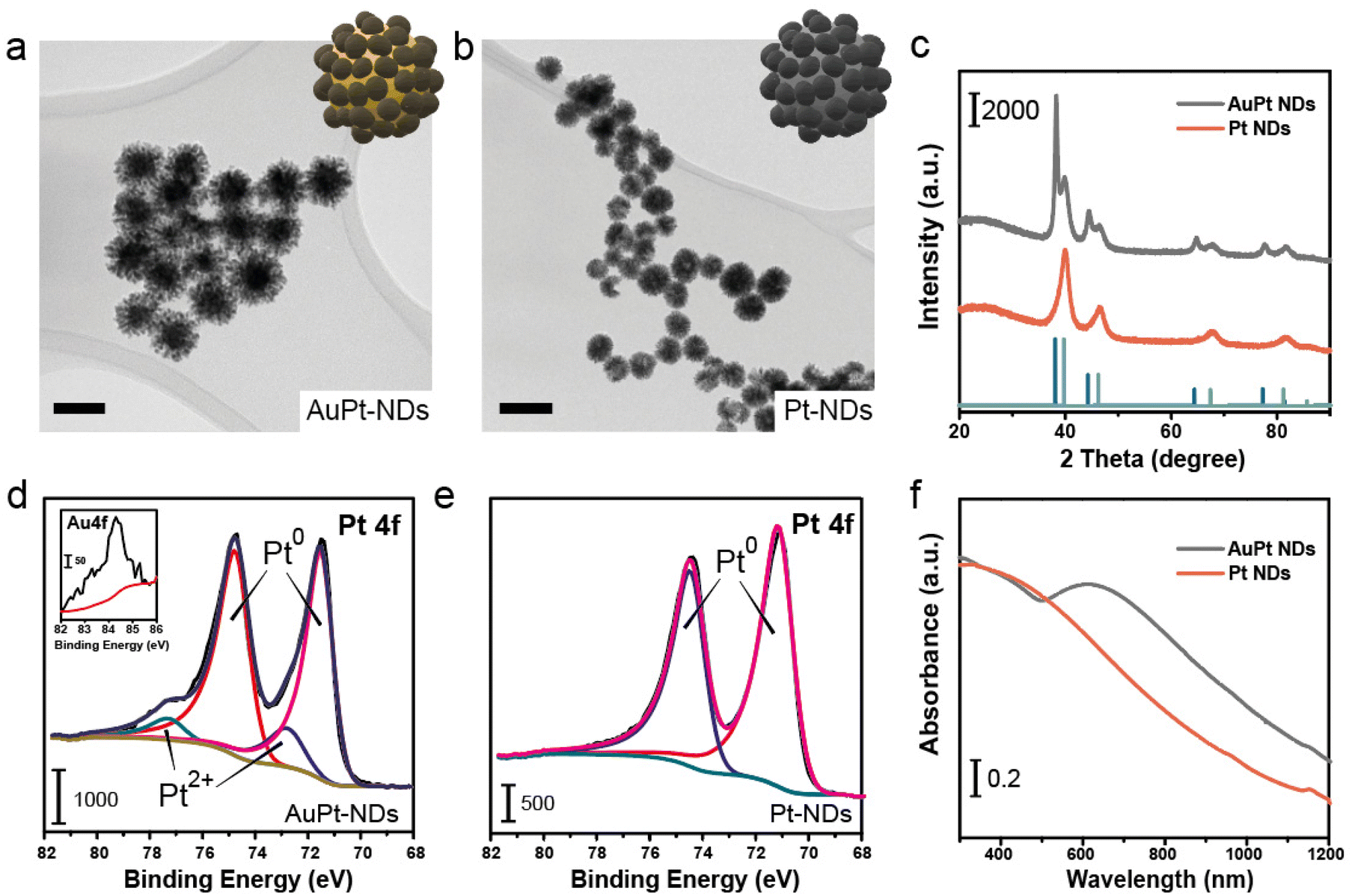 | ||
| Fig. 2 Characterization of AuPt and Pt nanodendrites: (a) TEM image of AuPt-NDs, revealing the solid Au core and dendritic Pt shell of the nanoparticles, inset: schematic illustration of AuPt-NDs; scale bar = 50 nm; (b) TEM image of Pt-NDs with a dendritic shape, inset: schematic illustration of Pt-NDs; scale bar = 100 nm; (c) X-ray diffraction patterns of AuPt-NDs and Pt-NDs and their comparison with those of Pt, Au and Pt; (d) X-ray photoemission spectra of the Pt 4f region, revealing the Pt0 and Pt2+ species in AuPt NDs; the inset shows the limited presence of Au in the Au 4f region; (e) X-ray photoemission spectra of Pt0 in Pt NDs, (f) UV-vis spectra of AuPt and Pt-NDs. We observe a continuous decreasing absorption band related to Pt NPs. AuPt-NDs also possess an intense absorption peak at 650 nm related to the plasmonic properties of the spherical Au core. | ||
Glucose oxidase-mimicking activity of Pt-based nanozymes: calculation of KM and Vmax
We evaluated the glucose oxidase-like properties of both AuPt-NDs and Pt-NDs. To have a meaningful comparison to the natural enzyme GOx, we carried out the glucose oxidation reaction under mild conditions, i.e. pH close to 7 and room temperature (Fig. 3a) and analyzed the supernatant by UPLC-mass spectroscopy (MS). Our main goal was to quantify how much glucose was oxidized at early reaction times and identify the by-products generated. Then, we evaluated the initial reaction rates (V0, mM s−1) at different glucose levels to build up Michaelis–Menten and Lineweaver–Burk plots to determine two key parameters of the nanozyme: the maximum initial rate (Vmax, mM s−1) related to the maximum substrate concentration a nanozyme can afford, and the Michaelis–Menten constant (KM, mM), which is a well-established descriptor of the affinity between the substrate and the nanozyme.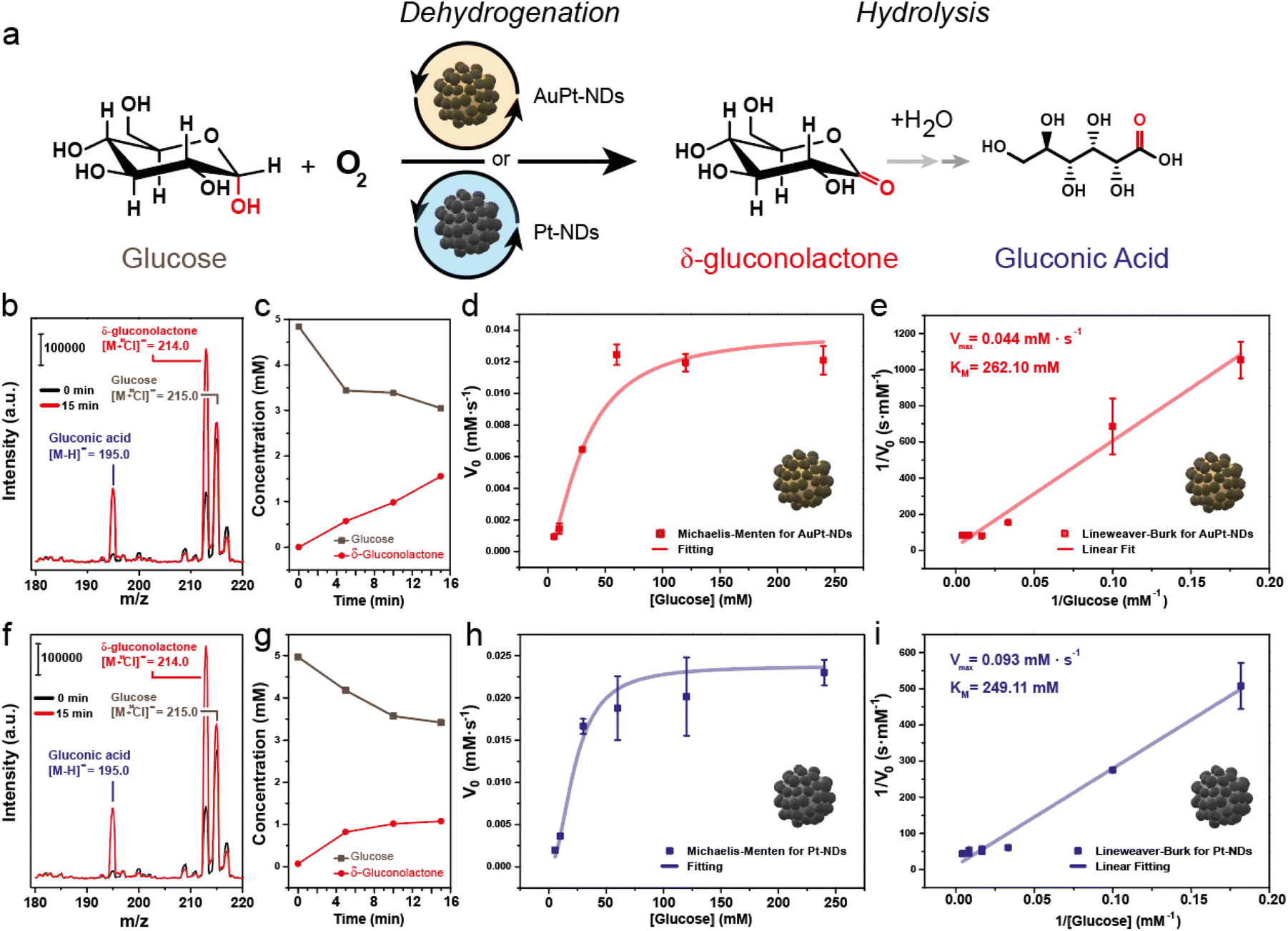 | ||
| Fig. 3 Glucose oxidase-mimicking response of AuPt and Pt nanodendrites: (a) schematic illustration of the glucose oxidation reaction with Pt-based NDs. Oxygen is used as an electron acceptor, leading to δ-gluconolactone as the main reaction product; (b) catalytic reaction of AuPt-NDs monitored by MS analysis after 0 and 15 minutes of reaction. [Glu-Cl] adduct with an m/z = 215.0, [δ-glu-Cl] adduct with an m/z = 214.0, [G.A.-H] with an m/z = 195.0; (c) evolution of glucose and δ-gluconolactone with the reaction time for AuPt-NDs; (d) Michaelis–Menten curve of the AuPt-NDs for GOx-like activity; (e) Lineweaver–Burk plot obtained from the glucose oxidation of AuPt-NDs used to determine enzymatic constants (Vmax, KM); (f) catalytic reaction of Pt-NDs monitored by MS analysis after 0 and 15 minutes of reaction. [Glu-Cl] adduct with an m/z = 215.0, [δ-glu-Cl] adduct with an m/z = 214.0, [G.A.-H] with an m/z = 195.0; (g) evolution of glucose and δ-gluconolactone with the reaction time for Pt-NDs; (h) Michaelis–Menten curve of the Pt-NDs for GOx-like activity; (i) Lineweaver–Burk plot obtained from the glucose oxidation of Pt-NDs used to determine enzymatic constants (Vmax, KM). Results are expressed as the average ± standard error measurement (S.E.M. – n = 2). | ||
After 15 minutes of reaction between glucose and AuPt NDs, we could detect the generation of two MS adducts by MS: [M − H]− = 195 and [M + Cl]− = 213.15, together with the unreacted glucose ([M + Cl]− = 215.15) (Fig. 3b). The detected ions suggested the generation of (i) gluconic acid, one of the most reported by-products of glucose oxidase-like nanozymes40,41 and (ii) δ-gluconolactone, the resulting product from the dehydrogenation reaction of glucose. With the aid of standards, we could confirm their generation under our reaction conditions (Fig. ESI-3†). The quantification of both glucose and δ-gluconolactone by UPLC-MS revealed a fast consumption at early reaction times (Fig. ESI-4a and b†), but the δ-gluconolactone levels were much higher than those of the generated gluconic acid (Fig. 3c). We calculated a selectivity towards δ-gluconolactone of about 86.0%, and only 8.2% towards gluconic acid. Increasing the initial glucose concentration led to a linear increase of V0 until the value leveled off around a glucose concentration of 60 mM. Data were correctly adjusted to the Michaelis–Menten plot, indicating an enzyme-like behavior of AuPt-NDs (Fig. 3d). The Lineweaver–Burk plot yielded a good linear fitting (R2 > 0.99), which allowed us to determine Vmax and KM to be 0.044 mM s−1 and 262.10 mM, respectively (Fig. 3e).
Analogous results were found for the Pt-NDs with both gluconic acid and δ-gluconolactone detected after 15 minutes of reaction (Fig. 3f) with similar patterns of glucose consumption and gluconolactone generation (Fig. 3g and Fig. ESI-4c, d†). In this case, selectivity towards δ-gluconolactone was 70.1% and that towards gluconic acid reached 13.1%. This indicated that the presence of Pt on two different catalysts, AuPt-NDs and Pt-NDs drive the reaction towards dehydrogenation preferentially rather than the direct generation of gluconic acid. This was initially unexpected, since most of the reported GOx-like nanomaterials typically report gluconic acid as the main reaction product.40,41
We searched the literature for previous studies on glucose oxidase-mimicking nanozymes, with special attention to the methods used to detect reaction products and the calculated kinetic parameters (Table 1). Most works used the as-generated H2O2 using HRP or ABTS to indirectly confirm the glucose oxidase-like activity. In the case of detection of gluconic acid, the most widely employed method was indirect detection through derivatization with NH2OH and subsequent complexation with Fe3+ to form a red-colored product, which was finally detected by UV-vis spectroscopy (Fig. ESI-5a†). We performed a control experiment using δ-gluconolactone in the same derivatization reaction, which yielded an identical signal corresponding to gluconic acid (Fig. ESI-5b†), indicating that this derivatization cannot be reliably used to distinguish among the possible reaction products. In this work instead, we provide a direct detection of δ-gluconolactone as a product of glucose oxidation when using Pt-based nanoplatforms. However, since the derivatization does not allow to discriminate gluconolactone from gluconic acid, it is possible that other nanozymes may have also yielded δ-gluconolactone but has gone undetected due to the lack of a protocol such as the one reported in this work.
| Nanozyme | V max (M s−1) | K M (mM) | Detection methodology | Ref. |
|---|---|---|---|---|
| AuNPs | 6.3 × 10−7 | 7.0 | G.A.: titration with NaOH | 41 |
| H2O2: colorimetric reaction (HRP and ABTS) | ||||
| AuNP-SBA15 | 5.3 × 10−7 | 26.2 | G.A.: complex with Fe3+ | 40 |
| H2O2: colorimetric reaction (TMB and low pH) | ||||
| Au-MIP | 2–6 × 10−5 | 0.18 | O2 decay measurement using an oximeter | 42 |
| Au-MIP-PFOB | 3.6 × 10−5 | 0.11 | H2O2: colorimetric reaction (HRP and ABTS) | |
| Au-APBA | 2.9 × 10−6 | 0.25 | G.A.: complex with Fe3+ | |
| AuNPs | 5.2 × 10−7 | 0.42 | ||
| AuNPs | 8.7 × 10−9 | 0.42 | H2O2: colorimetric reaction (HRP and ABTS) | 43 |
| AuPt | 1.8 × 10−8 | 0.33 | ||
| AuPt-MIP | 4.6 × 10−7 | 0.11 | ||
| Au-Fe-MPSN | 2.5 × 10−5 | 0.16 | G.A.: complex with Fe3+ and titration with NaOH | 44 |
| H2O2: colorimetric reaction (TMB, low pH and Fe3O4) | ||||
| Au-MCM41 | 1.8 × 10−5 | 55 | G.A.: complex with Fe3+ | 45 |
| H2O2: colorimetric reaction (HRP and ABTS) | ||||
| UCNPs_MOF_Au | 1.3 × 10−6 | 44.3 | H2O2: assay kit (Abcam) | 46 |
| G.A.: pH values | ||||
| AuNPs | 5.7 × 10−7 | 24.6 | H2O2: colorimetric reaction (HRP and DPD) | 47 |
| HMSN_Au | 4.0 × 10−7 | 50.7 | G.A.: complex with Fe3+ | 48 |
| H2O2: colorimetric reaction (HRP and TMB) | ||||
| BiogenAuNPs | 1.3 × 10−7 | 0.089 | H2O2: colorimetric reaction (TMB and low pH) | 49 |
| AuPt-Silk | 1.3 × 10−7 | 0.25 | Glucose: colorimetric reaction (DNS reagent) | 50 |
| Au-Hydrogel | 8.0 × 10−7 | 4.98 | H2O2: colorimetric reaction (HRP and TMB) | 51 |
| G.A.: colorimetric pH indicator (methyl red) | ||||
| AuPd-Aerogel | 13 × 10−8 | 0.19 | G.A.: complex with Fe3+ and a pH meter | 52 |
| H2O2: colorimetric reaction (HRP and TMB) | ||||
| AuPt-NDs | 4.5 × 10−5 | 262.1 | UPLC-MS | |
| Pt-NDs | 9.3 × 10−5 | 249.1 | O2: oximeter |
In terms of enzyme-like behavior, the absence of Au in the nanoparticle core did not seem to have a detrimental effect on the activity of Pt-NDs (Fig. 3h), suggesting that the catalytic behavior is mainly controlled by the dominant presence of Pt on the external surface of the particles. Overall, the catalytic activity of Pt-NDs outperformed their bimetallic AuPt counterparts when comparing both catalysts at their maximum conversion performance. The Lineweaver–Burk plot of Pt-NDs (R2 = 0.99) gave values for the enzymatic parameters KM and Vmax as 249.11 mM and 0.093 mM s−1, respectively (Fig. 3i). This means that the Vmax values obtained for Pd-NDs were roughly doubled in comparison with those obtained for their Pd–Au counterparts. In this regard, it is interesting to consider the reaction mechanism proposed by Chen et al.20 where a first H atom is transferred from glucose to the noble metal-based nanoparticle surface, then to an acceptor molecule. In our case, molecular O2 dissolved in solution may act as an electron sink as we could monitor its consumption during the reaction for both nanozymes (Fig. ESI-6†). Under this scenario, the Pt surface will abstract an H from glucose to form δ-gluconolactone, and then will transfer it to a dissolved O2 molecule. The presence of Au in the core of the particles could potentially limit the rate of this latter H transfer since the electronegativity of Au is higher than that of Pt, thus acting as an electron sink and making the H transfer more energetically unfavorable; further studies and detailed modeling (mainly DFT) will be needed to elucidate this trend.
Finally, when attempting to compare the obtained Vmax and KM values with the existing literature, we found large differences in the case of Pt-NDs (Table 1). However, it must be noted that Vmax values are dependent on the amount of the nanozyme employed in each experiment. Therefore, to provide a more accurate comparison, we proceeded to normalize Vmax by the number of active centers employed, using kcat.
Calculation of kcat assuming the ND concentration determined by different methods
k cat is a key parameter used to compare the catalytic efficiencies of different enzymes or nanozymes. It represents the maximum number of substrate molecules that can be converted to products by each active site of the enzyme/nanozyme per unit of time, under saturating substrate concentration conditions. kcat was calculated by dividing Vmax, obtained from the Lineweaver–Burk plot, by the concentration of active enzyme sites ([E]) for a given enzyme-catalyzed reaction (eqn (1)).Calculation of kcat for a given reaction catalyzed by an enzyme/nanozyme:
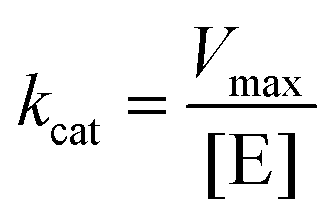 | (1) |
Although the determination of Vmax using the Lineweaver–Burk plot is well established in the nanozyme-related literature, assigning a value to the concentration of active enzyme sites is the subject of considerable controversy, but cannot be avoided since it is necessary to determine properly the value of kcat. In a recent review published by Zandieh et al.16 in 2021, different approaches to calculate the nanozyme concentration were presented, they are schematized in Fig. 4.
 | ||
| Fig. 4 Different approaches to define the nanozyme active units to calculate kcat: (a) total number of Pt atoms used as a reference. Each Pt atom is considered a specific active site independently of whether it is on the surface of Pt-NDs or in inner layers; (b) total number of Pt-NDs in solution used as a reference. Each Pt-ND is considered a specific active site independently of the number of active centers on its surface; (c) total number of surface Pt atoms used as a reference. Each surface Pt atom is considered a specific active site independently of its activity for the reaction with glucose. | ||
The central issue is of course establishing a clear definition of active sites. It is also necessary to find a consensus in the way [E] is calculated, since different assumptions can yield differences in activity values up to 8–10 orders of magnitude.7,16 Success in such standardization efforts would be invaluable, providing a productive way to link nanozymes work with natural enzymes and heterogeneous catalysis. In this case we have determined [E] employed in the catalytic glucose oxidation following the different assumptions proposed by Zandieh et al.16
The most straightforward methodology to calculate [E] consists of considering the total catalyst mass per unit volume (mg L−1) to obtain kcat. However, this approach can lead to huge differences among nanoparticles with no substantial variations in the size or shape. Moreover, this method can lead to a potential overestimation of [E] since it considers every atom of the nanoparticles as active, even those in the inner layers of the structure that probably remain unavailable for the catalytic reaction. In our nanosystems, this methodology would be equivalent to consider the total mass of Pt in both AuPt-NDs and Pt-NDs assuming Pt as the active element in the glucose oxidation reaction. Obviously, the turnover values obtained by this method are considerably lower compared to those of natural enzymes and also makes it hard to compare turnover rates among different nanozymes found in the literature, since factors such as the shape or aspect ratio have a strong influence on the fraction of exposed metal atoms. To follow this approach, the total Pt amount of AuPt-NDs and Pt-NDs was determined using MP-AES, and the calculated [Pt] value of AuPt-NDs and Pt-NDs was 0.292 and 0.512 mM, respectively (see also Table 2, vide infra).
| Calculation method | AuPt-NDs | Pt-NDs | ||
|---|---|---|---|---|
| [Pt] (mM) | k cat (s−1) | [Pt] (mM) | k cat (s−1) | |
| Total Pt mass | 0.292 | 0.15 | 0.512 | 0.18 |
| Total number of NPs | 8.80 × 10−8 | 5.1 × 105 | 2.17 × 10−8 | 4.3 × 106 |
| Total number of surface Pt atoms (TEM) | 1.08 × 10−2 | 4.2 | 6.70 × 10−3 | 13.8 |
| Total number of surface Pt atoms (NTA) | 2.88 × 10−2 | 1.6 | 3.50 × 10−2 | 2.7 |
| Total number of surface Pt atoms (BET) | 4.73 × 10−2 | 0.94 | 5.93 × 10−3 | 15.6 |
Another common approach is to consider the whole nanoparticles as a nanozyme unit (Fig. 4). This method is equivalent to assuming that each particle provides only one active site and therefore leads to an overestimation of the catalytic activity of nanozymes with results considerably higher than those of natural enzymes. The number of nanoparticles per unit volume was determined by NTA for AuPt-NDs and Pt-NDs. For a given concentration of 0.1 mg mL−1, the nanoparticle concentration for AuPt-NDs and Pt-NDs was 5.33 × 1010 and 1.31 × 1010 particles per mL or 8.80 × 10−8 and 2.17 × 10−8 if expressed in mM, respectively (see Table 2).
The third method normalizes activity by the number of surface active sites (Fig. 4). This seems the most reasonable approach as it would not consider internal atoms that are generally not available for the catalytic process but accounts for all the surface atoms potentially in contact with the substrate. This methodology would also allow to compare between nanozymes with different morphologies and shapes by taking into account the exposed atoms. Depending on the nanoparticle structure, the number of surface active sites can be determined by two methods: (i) calculation of the surface per particle using the nanoparticle radii, determined by direct measurement using TEM or NTA16 or (ii) measurement of the total surface per gram of material using BET (see the ESI† for details and Fig. 4). We believe the first method can only be applied to non-porous and spherical nanoparticles as it could otherwise underestimate the number of active sites exposed to the reaction. We calculated [E] with every method to provide an appropriate comparison of the obtained results (see the ESI† and Fig. 4). The average radius determined by TEM for AuPt-NDs and Pt-NDs was 27 and 40 nm (Fig. 1) and yielded a concentration of Pt of ca. 1.08 × 10−2 and 6.7 × 10−3 mM, respectively. However, NTA radii increase up to 44 and 96 nm, respectively, yielding [Pt] = 2.88 × 10−2 mM for AuPt-NDs and 3.50 × 10−3 mM for Pt-NDs, respectively (normalized by the number of particles, see Fig. ESI-1†). NTA employs a laser to irradiate photons on the particles and measures the scattered light, which can be related to the particle size. Using particles with high extinction coefficient in the range of the employed laser, as noble metal-based nanostructures like AuPt-NDs or Pt-NDs, can artificially decrease the light that finally reaches the detector and affect the final outcome.53 In addition, NTA measures the hydrodynamic size of aggregates but these aggregates are not dense structures, but an agglomeration of individual particles, whose surfaces are still at least partly available. For these reasons we believe that TEM is more appropriate, especially when low-porosity noble-metal nanoparticles are considered.
Finally, calculations of surface atoms from the BET data can be more appropriate when using porous or surface irregular materials as they provide a direct measurement of the nanoparticle surface. In contrast, TEM or NTA analysis is more prone to error when facing nanostructures with rough surfaces or a high polydispersity index. The margin of uncertainty will depend on how much the real particle surface deviates from an ideal spherical surface. On the other hand, BET analysis requires a higher amount of sample, in contrast to the low quantity necessary for TEM. Calculated [Pt] sites for AuPt and Pt using BET were 4.73 × 10−2 and 5.93 × 10−3 mM, respectively (see Table 2 for comparison and the ESI† for detailed calculations). We believe that this method is more appropriate for our Pt-NDs and AuPt-NDs since N2 adsorption gives a more reliable estimation of the number of surface atoms for textured surfaces, such as the dendrites prepared in this work.
Another approach proposed by Zandieh et al.16 refines the calculations by discriminating the non-active surface atoms. This is especially important in mixed-element nanoparticles such as Fe3O4, where oxygen surface atoms do not participate in catalysis as active sites. However, this method will not be considered in this work as we assume both particles only contain Pt on their outer surface layers. The final [Pt] and kcat (calculated by eqn (1)) values determined by the described method are summarized in Table 2 (vide infra).
For the sake of comparison, we have summarized representative values of kcat for Au-based nanozymes found in the literature in Table 3. The first example of Au as a GOx-like material reported by Comotti et al.54 found a kcat value of 13.9 s−1, calculated considering [E] as the total number of Au atoms. However, in their work the pH of the reaction was fixed to 9.5. This increases the reaction rate, since alkaline pH values favor the reaction, according to the mechanism reported recently by Chen et al.20 The kcat values calculated for our AuPt-NDs following the same methodology are comparable (0.94–16.2 s−1) even though they were obtained at pH = 6.5, which is likely affected by lower kinetics, given the importance of –OH ions in the reaction.20 For Pt-NDs, the kcat values normalized per Pt surface atom are larger, indicating an even better performance (Table 2). However, in other cases, a meaningful comparison to our results is challenging. As explained above, estimating [E] as the total number of NPs gives rise to extremely high kcat values, which are probably overestimated. This is for example the case of Luo et al.,41 who reported a kcat value of 18.52 s−1 assuming each AuNP as one enzyme unit.
In summary, the Pt-NDs prepared in this work present high kcat values that can be compared to the best values reported for nanozymes in the literature. However, a realistic comparison among works carried out under different conditions is highly challenging. It seems necessary to reach a consensus not only regarding the way [E] is calculated, but also under the conditions (e.g. pH) of glucose oxidation experiments and even on the techniques employed to quantify the reaction products.
Experimental
Chemicals and materials
Platinum(IV) chloride H2PtCl6 acid solution, gold(III) chloride hydrate (50% Au basis), L-ascorbic acid (99%), Pluronic F-127 (F-127), glucose (98%), and PBS pellets were purchased from Sigma-Aldrich (Darmstadt, Germany) and used without further purification.Synthesis of Pt nanozymes
Pt NPs were synthetized following a novel protocol. In a typical synthesis, 60 mg of Pluronic F-127 were completely dissolved in 2 mL of distilled water. Then, H2PtCl6 (1 mL, 100 mM) was added to the previous solution to a total volume of 4 mL. The mixture was ultrasonicated for 1 min. Then, L-ascorbic acid (2 mL, 0.25 M) was added to the reaction mixture. The mixture was allowed to settle at room temperature for 3 days without further stirring to favor the growth of Pt dendrites. The solution color turned from yellow to intensely black, indicating the reduction of the Pt4+ precursor to Pt0. The final product was then purified by centrifugation (7500 rpm for 7 min, two cycles, room temperature), and finally resuspended in 1 mL of mili-Q H2O. Au@Pt NPs were synthetized mixing H2PtCl6 (0.5 mL, 100 mM) and HAuCl3 (0.5 mL, 100 mM) and allowed to settle at room temperature for 1 day without further stirring. The synthesis of these materials has been performed at the Platform of Production of Biomaterials and Nanoparticles of the NANBIOSIS ICTS, more specifically by the Nanoparticle Synthesis Unit of the CIBER in BioEngineering, Biomaterials and Nanomedicine (CIBER-BBN, Madrid, Spain).Characterization techniques
Transmission electron microscopy (TEM) was performed using a FEI TECNAI T20 microscope (Tecnai, Eindhoven, The Netherlands) operated at 200 keV. Samples were prepared by drop-casting 3–5 μL of the NP suspension onto a holey carbon TEM grid. The specific surface area was calculated based on the Brunauer–Emmett–Teller (BET) method. NTA analysis (Nanosight NS200, Malvern Panalytical) was used to determine the diameter and concentration (expressed in particles per mL) of Pt NZ. The amount of Pt in the samples was quantified by using a quadrupole ICP mass spectrometer (4100 MP-AES, Agilent Technologies, USA). Particle size distribution was measured with ImageJ and using TEM images. X-ray diffraction patterns were obtained on an Empyrean instrument (Malvern-PANalytical, Malvern, UK) in Bragg–Brentano configuration using CuKα radiation and equipped with a PIXcel1D detector. The surface composition of the samples was analyzed by X-ray photoelectron spectroscopy (XPS) with an Axis Ultra DLD (Kratos Tech.). Spectra were excited with a monochromatic Al Kα source (1486.6 eV) operating at 12 kV and 10 mA and a step energy of 20 eV was used. The binding energies were referenced to the internal C 1s standard (284.3 eV). Peak analyses were performed with CasaXPS software using a weighted sum of Lorentzian and Gaussian component curves after the Shirley background subtraction.Glucose oxidase-mimicking reactions
Pt NPs were used in order to mimic GOx. The glucose oxidation reaction was exposed to air and monitored over time using UPLC-MS. Briefly, 2 mL of the reaction mixture with a variable amount of glucose was stirred in a vial at room temperature at pH = 6.5 (CH3COO−/CH3COOH 0.05 M). Subsequently, 20 μL of the nanozyme stock solution was added to the reaction mixture and the solution was incubated for 5, 10 and 15 minutes. Then, 20 μL were sampled from the reaction mixture for the acquisition of samples and mixed with 980 μL of a H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ACN mixture. The diluted solution was filtered and analyzed by UPLC-MS. Chromatographic separation was performed using a BEH AMIDE® UPLC column at 85 °C and 0.5 mL min−1 flow consisting of a starting composition of 90% acetonitrile and 10% milli Q water. Then, water concentration increased for 3 min until 65% ACN was reached. To modify the mobile phase, 0.1% v/v of 10 mM ammonium chloride in ammonium solution was added to both solvents. Cl− ions in the solvent solution interacted with glucose and δ-gluconolactone to form an [M − Cl] – adduct, which was detected using a coupled ACQUITY QDa mass detector (MS). In the case of gluconic acid, the detected ion was at m/z = 195 [M − H]−.
:ACN mixture. The diluted solution was filtered and analyzed by UPLC-MS. Chromatographic separation was performed using a BEH AMIDE® UPLC column at 85 °C and 0.5 mL min−1 flow consisting of a starting composition of 90% acetonitrile and 10% milli Q water. Then, water concentration increased for 3 min until 65% ACN was reached. To modify the mobile phase, 0.1% v/v of 10 mM ammonium chloride in ammonium solution was added to both solvents. Cl− ions in the solvent solution interacted with glucose and δ-gluconolactone to form an [M − Cl] – adduct, which was detected using a coupled ACQUITY QDa mass detector (MS). In the case of gluconic acid, the detected ion was at m/z = 195 [M − H]−.
Conclusions
AuPt and Pt nanodendrites synthesized with Pluronic F-127 have demonstrated a strong GOx-mimicking response. These catalysts displayed a high selectivity towards gluconolactone as the main by-product and were able to operate under close to neutral pH conditions. In this work, we have also addressed an important issue that is still under discussion: the lack of consensus regarding the way to calculate catalytic turnovers in enzyme-mimicking systems, a question that boils down to the very definition of active sites for these entities. Finally, we also advocate direct chromatography analysis as the most accurate detection method to report true reaction selectivities, avoiding potentially misleading confusions between gluconolactone and gluconic acid due to the lack of discrimination of colorimetric probes often used for indirect analyses. All these questions need to be fully addressed in order to establish a clear comparison between different nanozyme systems. Finally, we have been able to establish the excellent performance of Pt-based nanosystems as GOx surrogates able to provide glucose conversion with high selectivity to gluconolactone, while at the same time providing catalase-like activity, an extremely useful feature in oxygen-deprived, H2O2-rich environments.Author contributions
Conceptualization: J. I. G. P., J. B., M. T., J. S. and J. L. H.; data curation: J. I. G. P., M. T., J. B. and J. L. H.; formal analysis: J. I. G. P., J. B. and J. L. H.; methodology: J. I. G. P., J. B., and M. T.; validation: J. I. G. P., J. B. and J. L. H.; investigation: J. I. G. P., J. B., M. T., J. S. and J. L. H.; funding acquisition: J. S.; supervision: J. L. H. and J. S.; writing – original draft: J. B., J. I. G. P. and J. L. H.; writing – review and editing: J. B., J. I. G. P., J. S. and J. L. H.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the European Research Council (ERC-Advanced Grant CADENCE number 742684) and the Spanish Research Agency (LAERTES- PID2020-114926RB-I00) is acknowledged. The TEM measurements were conducted at the Laboratorio de Microscopias Avanzadas, ICTS ELECMI, Spain. The synthesis of materials has been performed by the Platform of Production of Biomaterials and Nanoparticles of the NANBIOSIS ICTS, more specifically by the Nanoparticle Synthesis Unit of the CIBER in BioEngineering, Biomaterials and Nanomedicine (CIBER-BBN). J. I. G.-P. and J. B-A. acknowledge the Aragon Regional Government and the Spanish Government, respectively, for their predoctoral contracts.References
- C. Della Pina, E. Falletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077–2095 RSC
.
- A. Guiseppi-Elie, S.-H. Choi and K. E. Geckeler, J. Mol. Catal. B: Enzym., 2009, 58, 118–123 CrossRef CAS
- S. B. Bankar, M. V. Bule, R. S. Singhal and L. Ananthanarayan, Biotechnol. Adv., 2009, 27, 489–501 CrossRef CAS PubMed
- H. Wang, K. Wan and X. Shi, Adv. Mater., 2019, 31, 1805368 CrossRef CAS PubMed
- A. Robert and B. Meunier, ACS Nano, 2022, 16, 6956–6959 CrossRef CAS PubMed
- J. Wu, X. Wang, Q. Wang, Z. Lou, S. Li, Y. Zhu, L. Qin and H. Wei, Chem. Soc. Rev., 2019, 48, 1004–1076 RSC
- M. Zandieh and J. Liu, Adv. Mater., 2023, e2211041 CrossRef PubMed
- J. I. Garcia-Peiro, J. Bonet-Aleta, J. Santamaria and J. L. Hueso, Chem. Soc. Rev., 2022, 51, 7662–7681 RSC
- S. Dong, Y. Dong, Z. Zhao, J. Liu, S. Liu, L. Feng, F. He, S. Gai, Y. Xie and P. Yang, J. Am. Chem. Soc., 2023, 145, 9488–9507 CrossRef CAS PubMed
- R. Zhao, Y. Zhu, J. Zhou, B. Liu, Y. Du, S. Gai, R. Shen, L. Feng and P. Yang, ACS Nano, 2022, 16, 10904–10917 CrossRef CAS PubMed
- Z. Chen, Z. Wang, J. Ren and X. Qu, Acc. Chem. Res., 2018, 51, 789–799 CrossRef CAS PubMed
- Z. Chen, C. Zhao, E. Ju, H. Ji, J. Ren, B. P. Binks and X. Qu, Adv. Mater., 2016, 28, 1682–1688 CrossRef CAS PubMed
- J. Zhou, D. Xu, G. Tian, Q. He, X. Zhang, J. Liao, L. Mei, L. Chen, L. Gao, L. Zhao, G. Yang, W. Yin, G. Nie and Y. Zhao, J. Am. Chem. Soc., 2023, 145, 4279–4293 CrossRef CAS PubMed
- S. Fedeli, J. Im, S. Gopalakrishnan, J. L. Elia, A. Gupta, D. Kim and V. M. Rotello, Chem. Soc. Rev., 2021, 50, 13467–13480 RSC
- Z. Chen, H. Ji, C. Liu, W. Bing, Z. Wang and X. Qu, Angew. Chem., Int. Ed., 2016, 55, 10732–10736 CrossRef CAS PubMed
- M. Zandieh and J. Liu, ACS Nano, 2021, 15, 15645–15655 CrossRef CAS PubMed
- M. Zandieh and J. Liu, Langmuir, 2022, 38, 3617–3622 CrossRef CAS PubMed
- H. Wei, L. Gao, K. Fan, J. Liu, J. He, X. Qu, S. Dong, E. Wang and X. Yan, Nano Today, 2021, 40, 1016 CrossRef
- B. Jiang, D. Duan, L. Gao, M. Zhou, K. Fan, Y. Tang, J. Xi, Y. Bi, Z. Tong, G. F. Gao, N. Xie, A. Tang, G. Nie, M. Liang and X. Yan, Nat. Protoc., 2018, 13, 1506–1520 CrossRef CAS PubMed
- J. Chen, Q. Ma, M. Li, D. Chao, L. Huang, W. Wu, Y. Fang and S. Dong, Nat. Commun., 2021, 12, 3375 CrossRef CAS PubMed
- H. Zhang, X. Liang, L. Han and F. Li, Small, 2018, 14, 1803256 CrossRef PubMed
- S. Biella, L. Prati and M. Rossi, J. Catal., 2002, 206, 242–247 CrossRef CAS
- H. Zhang, L. Lu, K. Kawashima, M. Okumura, M. Haruta and N. Toshima, Adv. Mater., 2015, 27, 1383–1388 CrossRef CAS PubMed
- M. Comotti, C. Della Pina and M. Rossi, J. Mol. Catal. A: Chem., 2006, 251, 89–92 CrossRef CAS
- H. Zhang and N. Toshima, Appl. Catal., A, 2011, 400, 9–13 CrossRef CAS
- X. Jin, M. Zhao, J. Shen, W. Yan, L. He, P. S. Thapa, S. Ren, B. Subramaniam and R. V. Chaudhari, J. Catal., 2015, 330, 323–329 CrossRef CAS
- Y. Zhu, R. Zhao, L. Feng, C. Wang, S. Dong, M. V. Zyuzin, A. Timin, N. Hu, B. Liu and P. Yang, ACS Nano, 2023, 17, 6833–6848 CrossRef CAS PubMed
- C. Ge, R. Wu, Y. Chong, G. Fang, X. Jiang, Y. Pan, C. Chen and J.-J. Yin, Adv. Funct. Mater., 2018, 28, 1801484 CrossRef
- A. Cheng, Y. Wang, L. Ma, L. Lin and H. Zhou, Nanotechnology, 2020, 31, 435403 CrossRef CAS PubMed
- R. Wu, Y. Xue, X. Qian, H. Liu, K. Zhou, S. H. Chan, J. N. Tey, J. Wei, B. Zhu and Y. Huang, Int. J. Hydrogen Energy, 2013, 38, 16677–16684 CrossRef CAS
- T. Yang, Y. Ma, Q. Huang, M. He, G. Cao, X. Sun, D. Zhang, M. Wang, H. Zhao and Z. Tong, ACS Appl. Mater. Interfaces, 2016, 8, 23646–23654 CrossRef CAS PubMed
- Q. Wang, H. Wang, Y. Yang, L. Jin, Y. Liu, Y. Wang, X. Yan, J. Xu, R. Gao, P. Lei, J. Zhu, Y. Wang, S. Song and H. Zhang, Adv. Mater., 2019, 31, e1904836 CrossRef PubMed
- Y. Li, K. H. Yun, H. Lee, S. H. Goh, Y. G. Suh and Y. Choi, Biomaterials, 2019, 197, 12–19 CrossRef CAS PubMed
- T. Gu, Y. Wang, Y. Lu, L. Cheng, L. Feng, H. Zhang, X. Li, G. Han and Z. Liu, Adv. Mater., 2019, 31, e1806803 CrossRef PubMed
- R. Long, K. Mao, X. Ye, W. Yan, Y. Huang, J. Wang, Y. Fu, X. Wang, X. Wu, Y. Xie and Y. Xiong, J. Am. Chem. Soc., 2013, 135, 3200–3207 CrossRef CAS PubMed
- W. He, X. Han, H. Jia, J. Cai, Y. Zhou and Z. Zheng, Sci. Rep., 2017, 7, 40103 CrossRef CAS PubMed
- A. Higareda, S. Kumar-Krishnan, A. F. García-Ruiz, J. Maya-Cornejo, J. L. Lopez-Miranda, D. Bahena, G. Rosas, R. Pérez and R. Esparza, Nanomaterials, 2019, 9, 1644 CrossRef CAS PubMed
- Q. Yang, J. Peng, Y. Xiao, W. Li, L. Tan, X. Xu and Z. Qian, ACS Appl. Mater. Interfaces, 2018, 10, 150–164 CrossRef CAS PubMed
- H. Ataee-Esfahani, L. Wang, Y. Nemoto and Y. Yamauchi, Chem. Mater., 2010, 22, 6310–6318 CrossRef CAS
- Y. Lin, Z. Li, Z. Chen, J. Ren and X. Qu, Biomaterials, 2013, 34, 2600–2610 CrossRef CAS PubMed
- W. Luo, C. Zhu, S. Su, D. Li, Y. He, Q. Huang and C. Fan, ACS Nano, 2010, 4, 7451–7458 CrossRef CAS PubMed
- L. Fan, D. Lou, H. Wu, X. Zhang, Y. Zhu, N. Gu and Y. Zhang, Adv. Mater. Interfaces, 2018, 5, 1801070 CrossRef
- F. Lin, T. Yushen, L. Doudou, W. Haoan, C. Yan, G. Ning and Z. Yu, Anal. Methods, 2019, 11, 4586–4592 RSC
- X. Wang, T. Xiong, M. Cui, X. Guan, J. Yuan, Z. Wang, R. Li, H. Zhang, S. Duan and F. Wei, Appl. Mater. Today, 2020, 21, 100827 CrossRef
- M. C. Ortega-Liebana, J. Bonet-Aleta, J. L. Hueso and J. Santamaria, Catalysts, 2020, 10, 333 CrossRef CAS
- L. He, Q. Ni, J. Mu, W. Fan, L. Liu, Z. Wang, L. Li, W. Tang, Y. Liu, Y. Cheng, L. Tang, Z. Yang, Y. Liu, J. Zou, W. Yang, O. Jacobson, F. Zhang, P. Huang and X. Chen, J. Am. Chem. Soc., 2020, 142, 6822–6832 CrossRef CAS PubMed
- H. Pezhhan, M. Akhond and M. Shamsipur, J. Lumin., 2020, 228, 117604 CrossRef CAS
- M. Chen, J. Song, J. Zhu, G. Hong, J. An, E. Feng, X. Peng and F. Song, Adv. Healthcare Mater., 2021, 10, 2101049 CrossRef CAS PubMed
- A. R. Deshmukh, H. Aloui and B. S. Kim, Chem. Eng. J., 2021, 421, 127859 CrossRef CAS
- R. Yang, S. Fu, R. Li, L. Zhang, Z. Xu, Y. Cao, H. Cui, Y. Kang and P. Xue, Theranostics, 2021, 11, 107–116 CrossRef CAS PubMed
- L. Jiao, W. Xu, H. Yan, Y. Wu, W. Gu, H. Li, D. Du, Y. Lin and C. Zhu, Chem. Commun., 2019, 55, 9865–9868 RSC
- R. Xu, X. Tan, T. Li, S. Liu, Y. Li and H. Li, Microchim. Acta, 2021, 188, 362 CrossRef CAS PubMed
- T. Zheng, S. Bott and Q. Huo, ACS Appl. Mater. Interfaces, 2016, 8, 21585–21594 CrossRef CAS PubMed
- M. Comotti, C. Della Pina, R. Matarrese and M. Rossi, Angew. Chem., Int. Ed., 2004, 43, 5812–5815 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr02026f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |