DOI:
10.1039/D3NA00253E
(Paper)
Nanoscale Adv., 2023,
5, 3044-3052
Chitosan-coated ultrapure silicon nanoparticles produced by laser ablation: biomedical potential in nano-oncology as a tumor-targeting nanosystem†
Received
20th April 2023
, Accepted 28th April 2023
First published on 1st May 2023
Abstract
Ultrapure silicon nanoparticles (SiNPs) produced by femtosecond laser ablation in water have attracted great interest in the area of cancer therapy as they are efficient as photosensitizers in photodynamic therapy modality and can induce cell hyperthermia under radiofrequency radiation. Recently, we showed that these biocompatible nanoparticles were not able to reach tumors after intravenous injection in mice due to their rapid clearance from the bloodstream. In order to increase their half-life time and therefore their chances to reach and accumulate in tumors by an enhanced permeation retention (EPR) effect, a capping agent on SiNP surface acting as a colloidal stabilizer suspension is required. In this regard, this work focuses for the first time on the functionalization of SiNPs through the modification of their surface by chitosan (SiNPs-CH) in order to enhance their therapeutic properties in cancer therapy. Here, in vivo experiments were carried out during 15 days on nude mice developing a subcutaneously grafted malignant human brain tumor (glioblastoma). The characterization of SiNPs-CH showed an average hydrodynamic size of around 142 ± 65 nm as well as a relatively neutral charge (−5.2 mV) leading to a high colloidal suspension stability. The point of our work concerns the improvement of the biodistribution of SiNPs-CH with regard to tumors, the bloodstream, and organs. After the intravenous administration of 20 mg kg−1, all the studied parameters (animal behavior, organs' morphology, and histopathology) were in accord with the absence of toxicity due to SiNPs-CH, confirming their biocompatibility and even size and surface charge were modified compared to bare nanoparticles. Moreover an increased time in the bloodstream circulation of up to 7 days was observed, indicating the stealth of the nanoparticles, which could escape opsonization and premature elimination by macrophages and the reticuloendothelial system. As evidenced by silicon assessment, the interaction of the SiNPs-CH with the liver and spleen was significantly reduced compared to the bare nanoparticles. At the same time, SiNPs-CH were concentrated progressively in tumors from 12.03% after 1 day up to 39.55% after 7 days, confirming their uptake by the tumor microenvironment through the enhanced permeability retention effect. Subsequently, the silicon level declined progressively down to 33.6% after 15 days, evidencing the degradation of pH-sensitive SiNPs-CH under the acidic tumor microenvironment. Taken together, the stealthy SiNPs-CH exhibited an ideal biodistribution profile within the tumor microenvironment with a sustainable biodegradation and elimination profile, indicating their promising application in the nano-oncology field as a tumor-targeting system.
1 Introduction
According to the World Health Organization, cancer is the second leading cause of death in the world after cardiovascular disease. Cancer around the world caused more than 8.2 million deaths in 2010, or about 13% of global mortality, a figure that is constantly increasing and which could reach 13.1 million deaths p.a. by 2030.1 The fight against cancer has thus become a global issue. The most common types of cancer treatment are surgery, radiotherapy, and chemotherapy. Unfortunately, these conventional medical treatments can cause complications or even side effects that are often very severe for the patient.2 Advances in nanotechnology have raised hopes for new cancer treatments that may be more effective, better targeted, and better tolerated. Nanoparticles, due to their size, physical properties, and interactions with living tissues, can meet certain criteria and may be concentrated at a specific location in the body, notably in a tumor, thus promising significant advantages in medicine in terms of diagnosis and cancer therapy.3 Current research has shown that nanoparticles can efficiently cross biological membranes, particularly the vascular endothelium, and can reach tumors through the enhanced permeation retention (EPR) mechanism.4–7 Today nanomedicine can take place as an alternative therapy instead of conventional cancer treatment, through the use of nanoparticle-based drug-delivery systems, which can improve drug therapeutic efficacy while reducing the doses administered and the side effects of certain active ingredients on patients.8 Nevertheless, most of the organic nanoparticles currently used are sensitive to problems of instability under the acidic tumor microenvironment, which can lead to an uncontrolled release of encapsulated drugs.9–14 To overcome these drawbacks, several research studies have been focused on the use of inorganic nanoparticles as drug-delivery systems. Among these nano-objects, biocompatible metal nanoparticles, such as carbon nanotubes,15–17 titanate nanotubes,18,19 zinc oxide nanoparticles,20 gold nanoparticles,21 and MOFs,22 may potentially be applied to overcome drug resistance in cancer therapy. Out of all these metal nanoparticles, nanoengineering particles based on silicon (SiNPs) are the most important thanks to their uncontroversial biocompatibility and their large specific surface area with the presence of silanol groups, which makes them easy to functionalize.23–26 In addition, SiNPs are highly biocompatible since they are derived from the silicon element, which is not only essential for the body's health but is also found as a composite in collagen, elastin, and arterial walls.27 Moreover, when injected in the biological system, SiNPs are degraded into orthosilicic acid, which is devoid of toxicity and is eliminated from the body through the kidneys and urine.25,26,29 Several applications of SiNPs have been developed in nanomedicine, including drug-delivery systems28,29 and as a contrast agent in imaging due to their highly photoluminescence.30 Various methods have been described to produce silicon nanoparticles, such as the micro-emulsion technique,31,32 mechanochemical synthesis,33 post-fabrication treatments in acidic solutions,34,35 solution-phase reduction,36 and femtosecond laser ablation in water.25 The laser-ablation method (a green synthesis) is unique in producing ultrapure nanoparticles without any residual contamination on the surface and with the absence of any toxic by-products.25,31 Ultrapure SiNPs produced by femtosecond laser ablation in water have attracted great interest in the area of cancer therapy, since they are efficient photosensitizers in two-photon excited photodynamic therapy modality and can induce cell hyperthermia under radiofrequency radiation.37–39 In our previous work, we showed that these biocompatible nanoparticles were unable to specifically target a tumor. Indeed, after the intravenous injection of a 20 mg kg−1 in mice, SiNPs were rapidly coated by serum opsonins and cleared from the circulation by the reticuloendothelial system, mainly the liver and the spleen.25 Hence the presence of a capping agent on SiNPs surface acting as a colloidal stabilizer suspension is required to increase their half-life time in the bloodstream and therefore their chances to reach and accumulate in a tumor by the EPR effect. In this regard, several polymers were grafted on SiNPs surface, including polyethylene glycol (PEG), dextran, and chitosan (CH), thus making them able to efficiently avoid premature elimination by macrophages before reaching their target.40–42 In this context, we focus for the first time on the functionalization of laser-synthesized bare SiNPs through the modification of their surface by chitosan (SiNPs-CH) to enhance their biomedical potential as a tumor-targeting nanosystem. Here, in vivo experiments were carried out on nude mice developing a model of subcutaneously grafted malignant brain tumor (glioblastoma). The point of our work concerns the improvement of the biodistribution of SiNPs-CH with regard to tumors, the bloodstream, and organs, promising their application in nano-oncology as a tumor-targeting nanosystem.
2 Methods
2.1 Synthesis of SiNPs
SiNPs were synthesized by the laser ablation of silicon in 20 mL of deionized water, as described previously.25 Radiation from a Yb:KGW femtosecond laser (Amplitude Systems [Pessac, France], 1025 nm, 480 fs, 500 mJ, 1–5 kHz) was focused with the help of a 75 mm lens onto a silicon wafer target surface to provide the ablation of the material. After synthesis, 10 mL of the samples were centrifuged at 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm during 25 min, using a Beckman ultracentrifuge, in order to recover the SiNPs for surface modification with chitosan.
000 rpm during 25 min, using a Beckman ultracentrifuge, in order to recover the SiNPs for surface modification with chitosan.
2.2 Coating of SiNPs with the chitosan
Chitosan (≥75% deacetylated) was purchased from Sigma-Aldrich Tunisia. SiNPs-chitosan coating was performed as described in detail previously using mesoporous silicon nanoparticles.44 Briefly, chitosan solution was prepared by dissolving 0.15 g of chitosan in 1 wt% of acetic acid aqueous solution. Under an ultrasonic bath, 50 mg of ultrapure SiNPs was dispersed in 5 mL of chitosan acidic solution, stirred for 6 h at 60 °C, and then cooled in an ice water bath for 1 h as reported previously.44 The chitosan-coated nanoparticles (SiNPs-CH) were then washed to neutral pH with deionized water several times and dried at 50 °C overnight. After ultracentrifugation at 45000, the percentage of chitosan reacted with the nanoparticles surfaces was calculated as following: (initial concentration − final concentration in the supernatant)/initial concentration × 100. For the chitosan assay, a colorimetric assay kit (MBS169583, MyBioSource USA) was used.
2.3 Characterization of the SiNPs-CH
Several techniques were used to characterize the SiNPs-CH, including transmission electron microscopy (TEM), dynamic light scattering (DLS), zeta potential measurements, X-ray diffraction (XRD), and Fourier-transform infrared spectroscopy (FTIR).
2.3.1 TEM.
A sample of 20 μL of SiNPs-CH dispersed in water was observed by TEM using a Tecnai G2 transmission electron microscope at 200 kV (FEI, Netherlands), then the image was acquired with a Veleta camera (Olympus, Japan) as reported previously.1
2.3.2 DLS and zeta potential measurements.
The zeta potential and DLS measurements were performed on a Zetasizer Nano ZS instrument (Malvern Instruments, Orsay, France). The SiNPs-CH suspension was sonicated for 5 min, and then DLS and zeta potential measurements were performed at room temperature after equilibration for 10 min. The data were obtained as the average of three measurements.
2.3.3 X-ray diffraction (XRD).
X-ray diffraction (XRD) was performed at room temperature with an X-ray diffractometer (X'PertPRO MPD, PANalytical Co., Holland). Monochromatic Cu Kα-radiation (λ = 1.5418 Å) was obtained with a Ni-filtration and a system of diverging and receiving slides of 0.5° and 0.1 mm, respectively. The diffraction patterns were measured with a voltage of 40 kV and a current of 30 mA over a 2θ range of 3°–40° using a step size of 0.02° at a scan speed of 1 s per step.
2.3.4 Fourier-transform infrared spectroscopy (FTIR).
Fourier-transform infrared spectroscopy studies (FTIR) were recorded on SiNPs-CH powder using a Nicolet 6700 FTIR Thermo Scientific spectrometer in the 500–4000 cm−1 region.
2.4
In vivo experiments
2.4.1 Cell lines and cell cultures.
Human glioma cell lines U87-MG were provided by the American Type Culture Collection. U87-MG cells line were cultured in DMEM medium containing 10% fetal bovine serum, 1% (v/v) antibiotics (penicillin–streptomycin), and 1% glutamine in 25 cm2 flasks. Cells were incubated under 37 °C with 5% CO2 in a humidified incubator.18
2.4.2 Animal model of subcutaneously grafted glioblastoma.
Animal care procedures were conducted in conformity with the legislation for the protection of animals used for scientific purposes provided by the relevant Tunisian law and European Union Directive (Tunisian Legislative Decree 2009-2200 and 2010/63/EU) and the International Guiding Principles for the Biomedical Research Involving Animals (Council for the International Organizations of Medical Sciences, CH). Animals were subjected to experimental protocols approved by the Animal Ethics Committee of the Faculty of Veterinary Medicine of Sidi Thabet (Permit number CEBM 2.2021). All adequate measures were taken to minimize animal pain or discomfort and all surgery was performed under mild anesthesia. Thirty sex male athymic nude mice (Athymic Nude-Foxn1nu) were obtained from the central animal care facilities, Harlan, France. The mice were randomly divided into 8 groups of 6 animals and housed in standard cages in a filtered airflow at 25 °C on a 12 h light/dark cycle with water and food available. After 7 days of acclimatization, all the mice were subcutaneously injected with U87-MG (1 × 107 cells per mouse) in the right flank to form a solid tumor of glioblastoma. The tumor size of each animal was measured every three days using digital calipers. The tumor size was calculated by the equation: V = L × W2 × 0.523 (where V is the tumor volume, L is the length, and W is the width).43
2.4.3 Animal treatment and biodistribution.
After 14 days of subcutaneous injection, the tumor size grew to reach an average of 100 mm3. Then, the animals were randomly divided in to three groups of 6 mice: SiNPs-CH (1 d), SiNPs-CH (7 d), and SiNPs-CH (15 d). In these groups, the mice were intravenously injected in the tail vein with a single dose of 20 mg kg−1 of SiNPs-CH and then sacrificed at 1, 7, or 15 days following the treatment. At the same time, three control groups of 6 mice were intravenously injected in the tail vein with chitosan prepared in 1% acetic acid solution then diluted in 0.9% NaCl. All animals were placed individually in metabolic cages for 24 h before sacrifice to collect urine and feces. Animals were then anesthetized under isoflurane and sacrificed. The blood samples were collected by intra-cardiac puncture then inoculated in heparin tubes and centrifuged at 3600 rpm to separate the serum. Serum samples were aliquoted then stored under −80 °C until further analysis. Moreover, the organs, including liver, spleen, heart, lungs, kidneys, and brain, were extracted, washed with NaCl 0.9% at 4 °C for storing under −20 °C as reported previously.23 Likewise, the tumor was removed from each mouse.
2.5 Histology
For histological evaluation, the organs and solid tumors were excised and then fixed in 5% buffered neutral formalin and embedded in paraffin wax. Sections of 5 μm were cut from each as reported previously.23
2.6 TEM analysis of the tumors
For TEM analysis, fragments of the tumors were cut into 1 mm2 cubes and fixed by immersion with a 2.5% glutaraldehyde, 2% paraformaldehyde, 2 mM CaCl2, and 0.1% tannic acid initially prepared in 0.1 M cacodylate buffer (pH 7.4) as described previously.23 Ultrathin sections of 90 nm were cut from dried blocks with a diamond knife on an LBK Ultramicrotome Leica UCT and stained with 0.5% aqueous uranyl acetate followed by Reynold's lead citrate. The observation was done with a Tecnai G2 instrument at 200 kV (FEI, Netherlands) and images acquired with a Veleta camera (Olympus, Japan).
2.7 SiNPs-CH biodistribution
In order to determine the SiNPs-CH biodistribution profile, the serum, urine, feces, tumors, and organs, including the liver, spleen, lungs, heart, brain, and kidneys, were digested with nitric acid 4 N for silicon (Si) content determination by ICP-OES, as reported previously.23 (Quantification threshold was fixed at 0.1 μg mg−1 for tissues and feces and 0.06 μg μL−1 for urine and serum).
2.8 Statistics
The results are presented here as the mean ± standard deviation of the mean of at least three determinations (n = 3). Comparisons with the control were performed using the Tukey HSD test. All the analyzes were performed using the Statistical Package for the Social Science (SPSS) software (version 12 for Windows, SPSS Inc., Chicago, IL, USA). A value of p < 0.05 was considered statistically significant.
3 Results
3.1 SiNPs-CH characterization
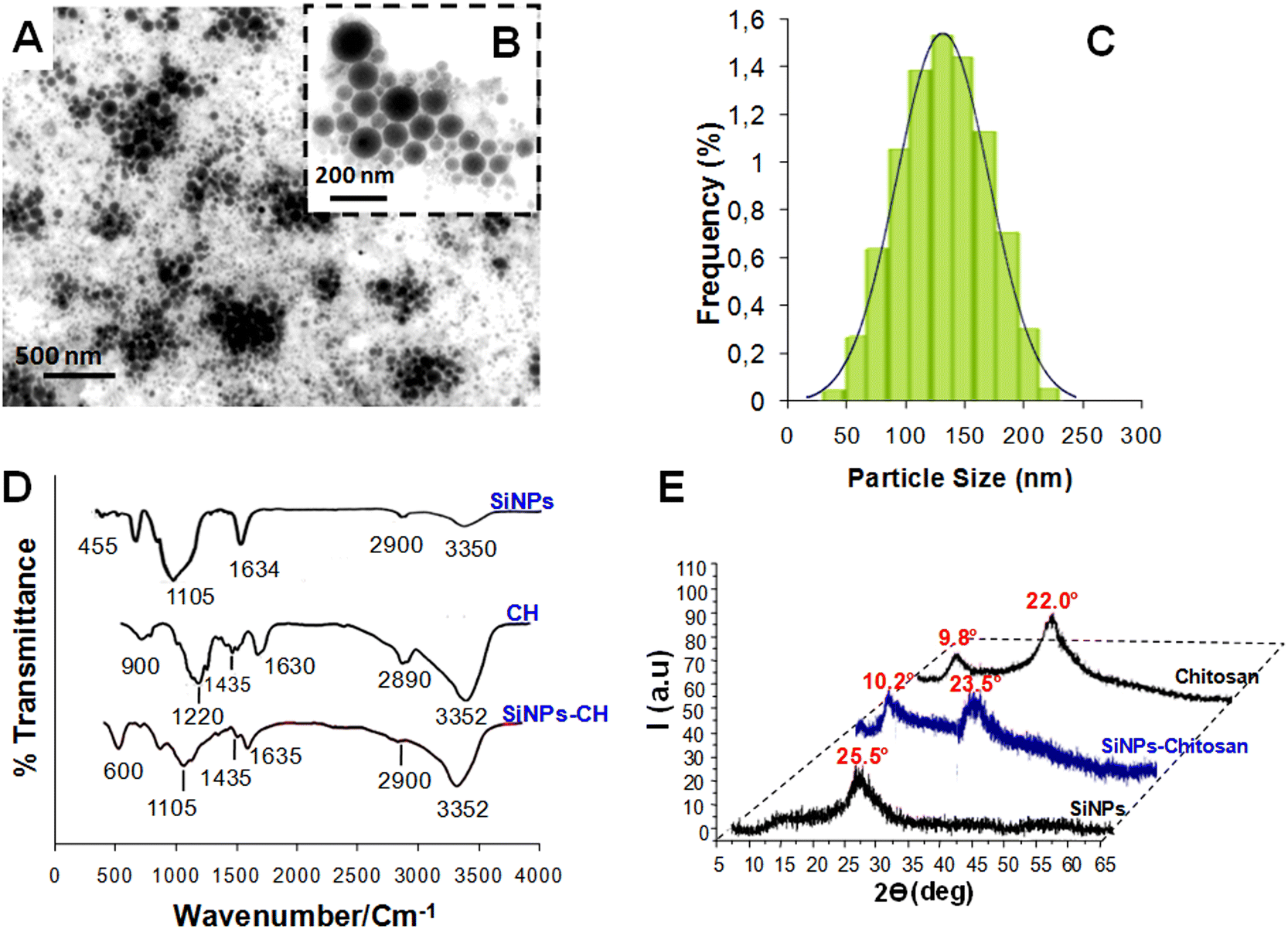
TEM showed that the SiNPs-CH shapes were spherical with sizes ranging from 50 nm up to 230 nm (Fig. 1A and B). The particle-size distribution measured by DLS (Fig. 1C) showed a mean hydrodynamic size of around 142 ± 65 nm, which was consistent with that measured by the TEM images. The SiNPs-CH aqueous suspension showed an homogeneous monodisperse nanoparticles system according to the polydispersity index value (PDI = 0.2). Moreover, according to the zeta potential measurement, the chitosan coating increased the nanoparticles surface charge from −35 mV for SiNPs to −5.2 mV for SiNPs-CH. Indeed, assessment of the chitosan reaction with the nanoparticles showed that around 60% of chitosan was capped on the silicon nanoparticles surface. The analysis of the FTIR spectrum of the pure SiNPs showed peaks at 3350 and 1634 cm−1, attributed to silanol OH stretching and water OH bending, respectively. Likewise, two other peaks were observed at 1105 and 455 cm−1, which were assigned to Si–O stretching and Si–O–Si bending stretching vibration, as reported previously.44 Regarding chitosan (Fig. 1D), its IR spectrum showed characteristic peaks at 3532 cm−1, attributed to OH− and N–H stretching overlapping, 2958 cm−1 attributed to C–H stretching, and at 1630, 1435, and 1220 cm−1 attributed to amide I, amide II (N–H), and amide III bands, respectively. Finally, a band at 900 cm−1 was observed due to the β-(1,4) glycosidic in chitosan, as has been reported previously.45
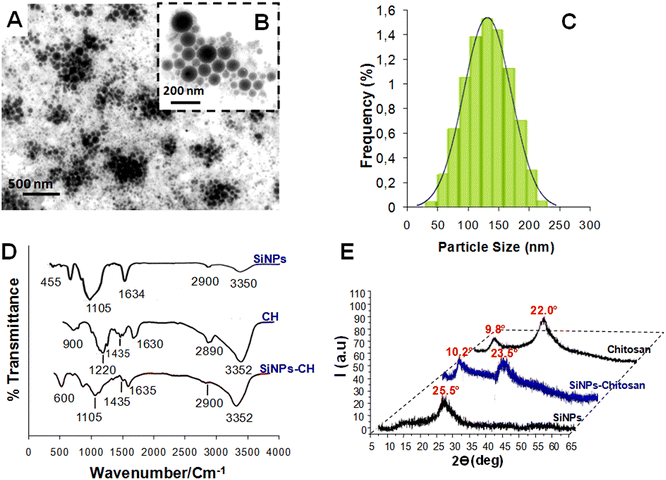 |
| | Fig. 1 (A and B) TEM images of SINPs-CH nanoparticles produced by laser ablation in water and then modified by chitosan (CH). (B) is a magnification of (A). (C) Particle-size distribution of the chitosan-coated nanoparticles (SiNPs-CH) determined by a Zetasizer Nano ZS instrument (DLS). (D) FTIR spectra of chitosan, SiNPs, and SiNPs-CH. (E) XRD patterns of SiNPs-CH compared to those of chitosan and uncoated SiNPs. | |
Concerning the SiNPs-CH, their FTIR spectrum compared to that of chitosan showed peaks at 3352 and 2900 cm−1 corresponding to OH− stretching and C–H stretching, respectively. In addition, two peaks at 1635 and 1435 cm−1 were observed, attributed to water OH and CH2 bending, respectively. Finally, peaks at 1105 and 500 cm−1 assigned to C–O–C bond and Si–O–Si bending, respectively, were observed. One can note that the peak intensity at 1635 cm−1 was decreased compared to that observed in chitosan, probably due to the reduction of the hydrophilicity of SiNPs-CH, as reported previously.35 The XRD pattern of the laser-synthesized SiNPs (Fig. 1E) matched well with the typical diffractogram of SiO2 nanoparticles, which presented a typical broad peak at 2θ = 25.5°.38 The XRD pattern of SiNPs-CH exhibited a similar broad peak observed for SiNPs, at 2θ = 23.5° and a minor diffraction peak at 2θ = 10.2°, which corresponded to the presence of chitosan, as confirmed by the XRD pattern of pure chitosan.46 Indeed, the XRD profile of chitosan showed a peak at 2θ = 9.8° with some shift compared to that of SiNPs-CH. Combined together, the XRD patterns, FTIR, DLS, and zeta potential analysis results showed that the decoration technique used for SiNPs coverage could efficiently produce highly chitosan-modified nanoparticle surfaces.
3.2 Mice behavior and safety of SiNPs-CH
Animal behavior, growth, and tumor size development were monitored from 1 to 15 days following SiNPs-CH intravenous injection. All the animals showed normal activity without any sign of lethargy or apathy. Animal growth as well as the organ weights of the SiNPs-CH treated groups increased similarly to those of the control groups (Fig. S1 and S2†), confirming the absence of toxic effects due to SiNPs-CH, as we reported for the uncoated SiNPs after the intravenous injection of 20 mg kg−1 in athymic nude mice.25
Moreover, the animal subcutaneously grafted malignant tumors grew slightly faster with no significant difference between the control and SiNPs-CH mice as evidenced by the macroscopic examination (Fig. 2A) at 15 days showing a volume of 340.2 ± 20.25 mm3 (Fig. S3†). Microscopic examination of the solid tumors tissues showed a high cell density with vascular thrombosis and necrosis areas (Fig. 2B, arrows) associated with a significant nuclear pleomorphism, as confirmed by TEM (Fig. 2C). These typical features of cancer cells characterized the malignant lesions of glioblastoma, as described previously.47 Additionally, histology analyses of the major organs of the SiNPs-CH-treated animals, including the liver, spleen, lungs, kidneys, heart, and brain, were similar to that of the organs of the control groups (Fig. 3, S4, S5, S6, S7, and S8,† respectively), confirming the absence of any systemic toxicity. Moreover, despite the presence of some SiNPs-CH clusters within hepatic Kupffer cells (Fig. 3, arrows) or within splenic macrophages (Fig. S3,† arrows) or in some lungs capillaries (Fig. S4,† arrows), these organs conserved their classical microscopic architecture as described previously.1
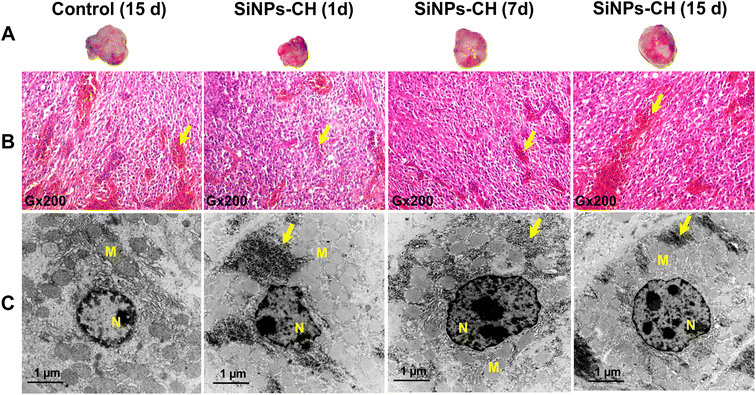 |
| | Fig. 2 Examination of the subcutaneous animal tumor tissue after 1, 7, and 15 days of the intravenous administration of SiNPs-CH (20 mg kg−1) in comparison with the control group. (A) Macroscopic morphology of a subcutaneous tumor. (B) Histological sections of tumor tissue stained with hematoxylin–eosin showing the nuclear pleomorphism, vascular thrombosis, and necrosis areas. (C) TEM images of the tumor tissue compared to the control group showing nuclear pleomorphism (N = nucleus, M = mitochondria). The arrows indicate SiNPs-CH taken up by the tumor tissue. | |
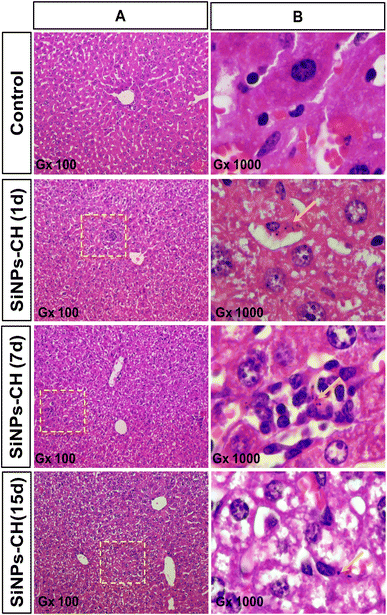 |
| | Fig. 3 Histology of mice livers at 1, 7, and 15 days after the intravenous administration of SiNPs-CH (20 mg kg−1) compared to the control group. Sections were stained with hematoxylin and eosin. (B) is a magnification of the square in (A). The arrows indicate SiNPs-CH taken up by Kupffer cells. | |
3.3 SiNPs-CH biodistribution and tumor targeting
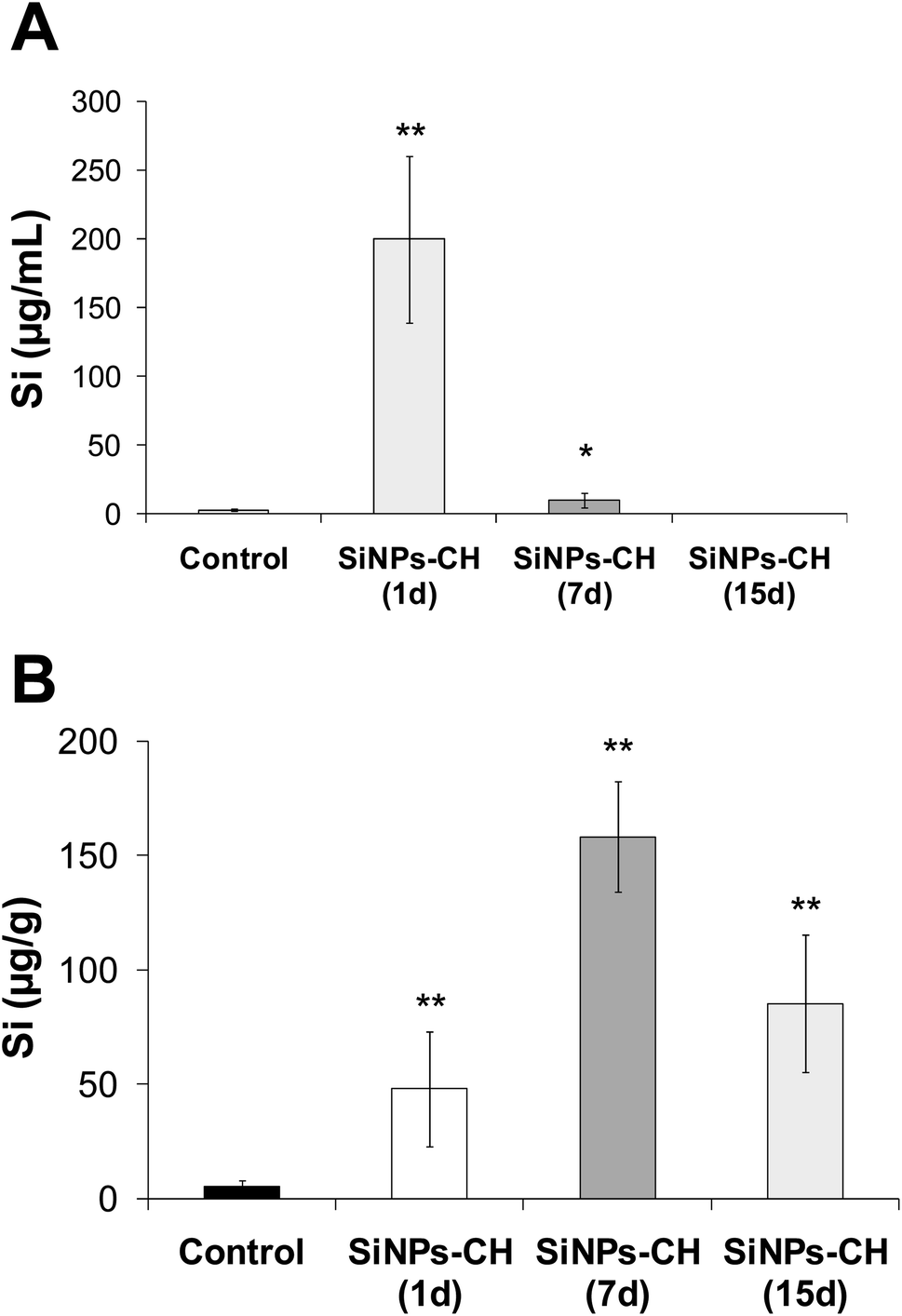
The biodistribution of SiNPs-CH was studied by quantifying the silicon concentration in several complex biological matrices, including serum, tumor, liver, spleen, lungs, heart, brain, kidneys, feces, and urine. The silicon level was first determined in the serum following 1, 7, and 15 days intravenous injection of SiNPs-CH, as summarized in Fig. 4A and Table 1. The percentage silicon level compared to the initially administered dose of SiNPs-CH increased significantly 1 day following the intravenous injection to reach 50.2%, and then declined progressively down to 2.55% after 7 days and was undetected after 15 days. At the same time, silicon was progressively concentrated in the tumors, reaching 12.05% after 1 day, and then 39.55 and 24.4% after 7 and 15 days, respectively (Fig. 4B and Table 1). These results were consistent with the increase in the bioavailability of the SiNPs-CH followed by their uptake in the tumor microenvironment, as confirmed by the TEM images, where the SiNPs-CH were observed as aggregate nanomaterials around the nucleus (Fig. 2C, arrows).
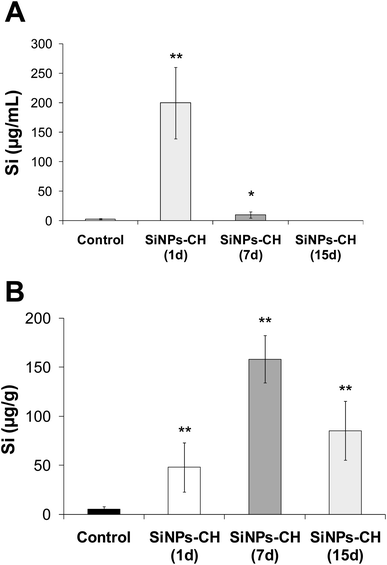 |
| | Fig. 4 Silicon concentration determined by ICP-OES in serum and tumor tissue at 1, 7, and 15 days following the intravenous administration of SiNPs-CH (20 mg kg−1). (A) Silicon concentration in serum. (B) Silicon concentration in the tumor. Results are representative of 6 animals (mean ± SD, n = 6). Statistical significance was determined by Tukey HSD test (*p < 0.05, **p < 0.01). | |
Table 1 Percentage of SiNPs-CH related to the initially intravenously administered dose (20 mg kg−1) measured in different organs, serum, urine, and feces at 1, 7, and 15 days following the intravenous administration. Results are representative of 6 animals (mean ± SD, n = 6)
| Time |
% of Si |
Total |
| Serum |
Tumor |
Liver |
Spleen |
Kidney |
Brain |
Urine |
| 1 days |
50.20 |
12.03 |
5.95 |
7.34 |
3.42 |
0.55 |
5.05 |
84.34 |
| 7 days |
2.55 |
39.55 |
15.81 |
11.55 |
7.08 |
2.74 |
6.33 |
85.61 |
| 15 days |
0.06 |
24.40 |
10.44 |
8.64 |
13.28 |
4.25 |
20.09 |
81.15 |
Regarding the lungs, heart, and feces, no significant difference in silicon levels was observed after 1, 7, and 15 days of the intravenous injection of SiNPs-CH as compared with the control group (Table S1†). However, the silicon level progressively increased in the liver, from 5.95% after 1 day to reach a maximum of 15.81% after 7 days, and then declined progressively down to 10.44% after 15 days. Likewise the percentage silicon level increased in the spleen, from 7.34% after 1 day to 8.64% at 15 days following the intravenous injection of SiNPs-CH. These results evidenced the uptake of SiNPs-CH by the reticuloendothelial system, notably in the liver and spleen. Meanwhile, the Si concentration progressively increased in the kidneys and urine after SiNPs-CH injection, reaching 13.28 and 20.09% after 15 days, indicating the elimination of nanoparticles into the urine. It should be noted that the Si percentage in the brain showed a four-fold increase after 15 days, showing that SiNPs-CH were able to cross the blood–brain barrier without any sign of toxicity, as confirmed by the histological examination of the brain tissues (Fig. S8†).
4 Discussion
This study aimed to develop a chitosan-coated silicon nanoparticles, and then to determine their toxicity and biodistribution profile following the intravenous administration of a relatively high dose (20 mg kg−1) to nude mice developing subcutaneously grafted glioblastoma as a model of malignant cancer. Prior to the in vivo experiments, the SiNPs-CH were characterized for assessing their suspension colloidal stability, size, chitosan-coating percentage, and surface charge. All the combined results obtained by FTIR, DLS, and zeta potential analyses were in accordance with the significant adsorption of chitosan on the ultrapure silicon nanoparticles surface. Indeed the colloidal stability of the SiNPs-CH suspension was highly increased compared to the bare SiNPs, revealing that the SiNPs-CH followed dynamics similar to monodisperse suspensions as confirmed by the polydispersity index (PDI). Several studies reported that chitosan coating dramatically prevented the aggregation of nanoparticles, showing much better colloidal stability under physiological conditions and enabling their long-time residence in the bloodstream circulation.48–50 Compared to the hydrodynamic sizes of bare SiNPs (20–80 nm),23 the sizes of the SiNPs-CH increased significantly due to the formation of a polymer coverage crown around the nanoparticles surface as confirmed by DLS analysis, which is known to represent the size of the nanoparticles as well as all the molecules bound to the surface.51 Surface nanoparticles coating depended on the chitosan's chemical properties, including its degree of acetylation and the pH of the solution, which play a crucial role in its solubility. Indeed when the degree of acetylation became greater than 60%, chitosan was completely insoluble in water at pH > 6, because of the formation of hydrogen bonds.51 Hence the passage of chitosan in acidic solution as we prepared in acetic acid solution was required to increase its solubility by transforming it into cationic form thanks to the protonation of its amine groups.52 In these conditions, the protonated amino substituents bind to the hydroxyl groups present in the SiNPs surface, thus forming a stable coverage crown, as described for mesoporous silicon nanoparticles.49 Moreover, the SiNPs-CH zeta potential compared to that of bare SiNPs23 shifted from negative to around neutral charge, involving the adsorption of the positive charge provided by chitosan. Yu et al. showed that the presence of amine groups on the surface of the silica particles reduces their toxicity compared with unmodified silica particles.53 In contrast with positively charged nanoparticles, relatively neutral charged nanoparticles promise greater biocompatibility because it reduces their interaction with biological compounds and serum opsonins, allowing them to cross the cell membranes easily without the requirement for specific bindings through internalization pathways.54–56 Nevertheless, characterization of the protein corona on the SiNPs-CH surface is a crucial issue to understand its impact on nanoparticle binding to the cell membrane and the subsequent cellular uptake57,58 as well as its effect on the colloidal stability. All the same, the mainly studied physical parameters of SiNPs-CH are suitable for their bioapplication, notably showing the chitosan is highly biodegradable by living organisms through its hydrolysis and metabolization by a variety of enzymes, such as chitosanase and lysosomes, providing fragments suitable for renal clearance.59 Concerning the toxicity of SiNPs-CH, all the studied parameters, including the healthy behavior of animals as well as the absence of acute and chronic toxicity in the kidneys, spleen, and liver, confirmed the biocompatibility of SiNPs-CH as reported in our previous study for bare SiNPs after the intravenous injection of a similar dose.23 Interestingly, no toxic effect was observed due to the increase in the SiNPs-CH hydrodynamic size, despite this physical parameter significantly influencing the interaction of nanoparticles with biological compounds. Several studies have shown that for diameters greater than 100 nm, the change in silicon particle size has little influence on cell viability, while for diameters less than 100 nm, the smaller particles become more toxic.60–62 In addition, some other authors have reported that nanoparticles with sizes ranging from 150 to 300 nm are considered generally safe when they are used as drug nanocarriers.63,64 However, the influence of the size must be modulated by the question of the dose, since changing the diameter of the particles will change the number of particles per unit of concentration. According to Napierska et al., the application of silicon nanoparticles in the biomedical field remains possible since under certain conditions, in vitro and in vivo, it is possible to obtain negligible or reversible toxicity after a period without exposure.62 On the other hand, our experiments confirmed that chitosan coverage improves the biodistribution of SiNPs-CH with regard to the tumor and the bloodstream circulation. Obviously, the percentage of SiNPs-CH increased significantly in the bloodstream 24 h and 7 days post injection, confirming the highly prolonged circulation time compared to that of bare SiNPs, which were eliminated only 3 h after intravenous injection of a similar dose.23 Indeed, chitosan coverage improved the stealth of the nanoparticles by forming a protective hydrophilic layer on their surface, thus enhancing their stability and minimizing their interaction with plasma opsonins to escape macrophages recognition and their rapid elimination by the reticuloendothelial system. Several reports have indicated that surface functionalization by chitosan reduces the interaction of nanoparticles with plasmatic proteins and their uptake by various reticular cells, macrophages, and reticuloendothelial system.26,28,65 In this regard, the low distribution of SiNPs-CH within the reticuloendothelial system, mainly the liver and spleen, was consistent with the stealth of the nanoparticles. Thereby the extended time period in circulation led to the accumulation of SiNPs-CH in the tumor, as we confirmed by the progressive increase in silicon in the subcutaneously grafted glioblastoma. In addition the level of SiNPs-CH remained concentrated in the tumor even after 15 days, evidencing their uptake in the tumor microenvironment mainly by the enhanced permeability retention (EPR) effect. Several factors related to the structural abnormalities tumor microenvironment and to the nanoparticles size are involved in increasing the nanoparticles tumor uptake. Indeed, defective angiogenesis develops a highly vessels fenestration in comparison with the normal vessels, enabling then an hyperpermeability to the passage of nanoparticles generally with a size lower than 200 nm.66,67 Beside the large fenestration of the tumor vascular wall, the poor lymphatic drainage of the tumor microenvironment associated with cancer proliferation allows not only the accumulation of nanoparticles but also the release of their contents to tumor cells, as described for other types of nanoparticles.68–70 Likewise, the nanoparticles size plays a crucial role in influencing the EPR effect, as larger particles (>200 nm) do not leak into tumor vessels and are more likely to be cleared by circulating macrophages.71,72 Importantly, the decrease in SiNPs-CH level in the tumors after 15 days of the intravenous injection indicated a degradation of the pH-sensitive nanoparticles under the acidic microenvironment rich in lactic acid, which was derived from the glycolytic metabolism of cancer cells.73,74 As we have shown, the bare SiNPs were degraded into orthosilicic acid and then eliminated through urine.23 The progressive increase in silicon levels in the urine and kidneys improved the slow degradation of SiNPs-CH and their elimination without inducing any signs of toxicity. The increase in silicon level in the brain indicated that the SiNPs-CH were able to cross the blood–brain barrier, which restricts the passage of large hydrophilic molecules into the cerebrospinal fluid while allowing the diffusion of hydrophobic molecules. Hence, we suggested that chitosan enhanced the retention of SiNPs-CH in the cerebrospinal fluid since it can be transformed into an hydrophobic form under the local brain pH ranging between 6.5 and 7.75 Additional experiments are required to confirm this hypothesis. Moreover, a longer time of experiments of more than 15 days is required to complete the nanoparticles clearance process. Altogether, the stealthy SiNPs-CH exhibited a perfect biodistribution profile within the tumor microenvironment with sustainable biodegradation and elimination. Our results clearly confirm the interest in the use of these biodegradable non-toxic nanoparticles for biomedical applications, notably in cancer therapy.
5 Conclusion
Ultrapure SiNPs produced by laser ablation were perfectly coated with chitosan in order to increase their time in the bloodstream circulation and improve their biodistribution within the tumor microenvironment after intravenous administration to nude mice developing subcutaneously grafted glioblastoma. The mainly physical parameters (size, shape, and electric charge) that can influence the toxicity and biodistribution of SiNPs-CH were studied and found to be suitable for the bioapplication of the nanoparticles. Thanks to the chitosan surface coating, the nanoparticles could efficiently escape macrophage recognition and premature elimination by the reticuloendothelial system. Altogether, stealthy SiNPs-CH were highly accumulated in the tumor microenvironment by the EPR effect and sustainably biodegraded and eliminated. This work clearly confirms the interest in the use of these biodegradable non-toxic nanoparticles in nano-oncology as a tumor-targeting nanosystem. Taken together, several works are to be considered to show the biomedical potential of SiNPs-CH in cancer therapy as delivery systems of therapeutic molecules or therapeutic tools for non-invasive treatment modalities.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
The work was supported by the Tunisian Ministry of Higher Education and Scientific Research (MHESR), grant number LR15INRAP02.
References
- R. L. Siegel, K. D. Miller and A. Jemal, CA Cancer J. Clin., 2020, 70, 7–30 CrossRef PubMed.
- J. S. Ross, D. P. Schenkein, R. Pietrusko, M. Rolfe, G. P. Linette, J. Stec, N. E. Stagliano, G. S. Ginsburg, W. F. Symmans, L. Pusztai and G. N. Hortobagyi, Am. J. Clin. Pathol., 2004, 122, 598–609 CrossRef CAS PubMed.
- R. Misra, S. Acharya and S. K. Sahoo, Drug Discovery Today, 2010, 15, 842–850 CrossRef CAS PubMed.
- F. Danhier, O. Feron and V. Préat, J. Controlled Release, 2010, 148, 135–146 CrossRef CAS PubMed.
- S. Acharya and S. K. Sahoo, Adv. Drug Deliv. Rev., 2011, 63, 170–183 CrossRef CAS PubMed.
- A. Dadwal, A. Baldi and R. N. Kumar, Artif. Cells Nanomed. Biotechnol., 2018, 46, 295–305 CrossRef CAS PubMed.
- D. Kalyane, N. Raval, R. Maheshwari, V. Tambe, K. Kalia and R. K. Tekade, Mater. Sci. Eng. C, 2019, 98, 1252–1276 CrossRef CAS PubMed.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS PubMed.
- J. O. Eloy, R. Petrilli, J. F. Topan, H. M. R. Antonio, J. P. A. Barcellos, D. L. Chesca, L. N. Serafini, D. G. Tiezzi, R. J. Lee and J. M. Marchetti, Colloids Surf. B, 2016, 141, 74–82 CrossRef CAS PubMed.
- H. Devalapally, Z. Duan, M. V. Siden and M. M. Amiji, Int. J. Cancer, 2007, 121, 1830–1838 CrossRef CAS PubMed.
- L. Fan, F. Li, H. Zhang, Y. Wang, C. Cheng, X. Li, C. H. Gu, Q. Yang, H. Wu and S. Zhang, Biomaterials, 2010, 31, 5634–5642 CrossRef CAS PubMed.
- P. Vaupel, F. Kallinowski and P. Okunieff, Cancer Res., 1989, 49, 6449–6465 CAS.
- C. Cuvier, L. Roblot-Treupel, J. M. Millot, G. Lizard, S. Chevillard, M. Manfait, P. Couvreur and M. F. Poupon, Biochem. Pharmacol., 1992, 44, 509–517 CrossRef CAS PubMed.
- J. W. Wojtkowiak, J. M. Rothberg, V. Kumar, K. J. Schramm, E. Haller, J. B. Proemsey, M. C. Lloyd, B. F. Sloane and R. J. Gillies, Cancer Res., 2012, 72, 3938–3947 CrossRef CAS PubMed.
- E. Luo, G. Song, Y. Li, P. Shi, J. Hu and Y. Lin, Curr. Drug Metab., 2013, 14, 879–890 CrossRef CAS PubMed.
- S. Y. Madani, N. Naderi, O. Dissanayake, A. Tan and A. M. Seifalian, Int. J. Nanomedicine, 2011, 6, 2963–2979 CAS.
- A. Bianco, K. Kostarelos and M. Prato, Curr. Opin. Chem. Biol., 2005, 9, 674–679 CrossRef CAS PubMed.
- T. Baati, B. B. Kefi, A. Aouane, L. Njim, F. Chaspoul, V. Heresanu, A. Kerkeni, F. Neffati and M. Hammami, RSC Adv., 2016, 6, 101688 RSC.
- T. Baati, M. B. Brahim, A. Salek, M. Selmi, L. Njim, P. Umek, A. Aouane, M. Hammami and K. Hosni, RSC Adv., 2022, 12, 5953–5963 RSC.
- M. Kundu, P. Sadhukhan, N. Ghosh, S. Chatterjee, P. Manna, J. Das and P. C. Sil, J. Adv. Res., 2019, 18, 161–172 CrossRef CAS PubMed.
- R. S. Riley and E. S. Day, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol., 2017, 9, e1449 CrossRef PubMed.
- D. Cunha, M. Ben Yahia, S. Hall, S. Miller, H. Chevreau, E. Elkaim, G. Maurin, P. Horcajada and C. Serre, Chem. Mater., 2013, 25, 2767–2776 CrossRef CAS.
- T. Baati, A. Al-Kattan, M. A. Esteve, L. Njim, Y. Ryabchikov, F. Chaspoul, M. Hammami, M. Sentis, A. V. Kabashin and D. Braguer, Sci. Rep., 2016, 6, 25400 CrossRef CAS PubMed.
- Y. Li, N. Li, W. Pan, Z. Yu and L. B. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 2123–2129 CrossRef CAS PubMed.
- Y. Wang, Q. Zhao, N. Han, L. Bai, J. Li, J. Liu, E. Che, L. Hu, Q. Zhang, T. Jiang and S. Wang, Nanomedicine, 2015, 11, 313–327 CrossRef CAS PubMed.
- J. Salonen and V. P. Lehto, Chem. Eng. J., 2008, 137, 162–172 CrossRef CAS.
- R. Jugdaohsingh, J. Nutr. Health Aging, 2007, 11, 99–110 CAS.
- J. L. Coffer, J. L. Montchamp, J. B. Aimone and R. P. Weis, Phys. Status Solidi A, 2003, 197, 336–339 CrossRef CAS.
- L. Vaccari, D. Canton, N. Zaffaroni, R. Villa, M. Tormen and E. Fabrizio, Microelectron. Eng., 2006, 83, 1598–1601 CrossRef CAS.
- L. Gu, D. J. Hall, Z. Qin, E. Anglin, J. Joo, D. J. Mooney, S. B. Howell and M. J. Sailor, Nat. Commun., 2013, 4, 2326 CrossRef PubMed.
- R. D. Tilley and K. Yamamoto, Adv. Mater., 2006, 18, 2053–2056 CrossRef CAS.
- Z. R. Yu, M. Aceves-Mijares and M. A. Cabrera, Nanotechnology, 2006, 17, 3962–3967 CrossRef CAS.
- A. S. Heintz, M. J. Fink and B. S. Mitchell, Adv. Mater., 2007, 19, 3984–3988 CrossRef CAS.
- H. Sugimoto, M. Fujii, K. Imakita, S. Hayashi and K. Akamatsu, J. Phys. Chem. C, 2013, 117, 11850–11857 CrossRef CAS.
- F. Sangghaleh, I. Sychugov, Z. Yang, J. G. C. Veinot and J. Linnros, ACS Nano, 2015, 9, 7097–7104 CrossRef CAS PubMed.
- D. Neiner, W. H. Chiu and S. M. Kauzlarich, J. Am. Chem. Soc., 2006, 128, 11016–11017 CrossRef CAS PubMed.
- A. Al-Kattan, L. M. A. Ali, M. Daurat, E. Mattana and M. Gary-Bobo, Nanomaterials, 2020, 10, 1462 CrossRef CAS PubMed.
- K. P. Tamarov, L. A. Osminkina, S. V. Zinovyev, K. A. Maximova, J. V. Kargina, M. B. Gongalsky, Y. V. Raybchikov, A. Al-Kattan, A. P. Sviridov, M. Sentis, A. V. Ivanov, V. N. Nikiforov, A. V. Kabashin and V. Y. Timoshenko, Sci. Rep., 2014, 4, 7034 CrossRef CAS PubMed.
- D. Rioux, M. Laferriere, A. Douplik, D. Shah, L. Lilge, A. V. Kabashin and M. Meunier, J. Biomed. Opt., 2009, 14, 021010 CrossRef PubMed.
- J. E. Fuller, G. T. Zugates, L. S. Ferreira, H. S. Ow, N. N. Nguyen, U. B. Wiesner and R. S. Langer, Biomaterials, 2008, 29, 1526–1532 CrossRef CAS PubMed.
- V. Mailaender and K. Landfester, Biomacromolecules, 2009, 10, 2379–2400 CrossRef CAS PubMed.
- M. R. Sayyid and A. Heydarinasab, ACS Sustainable Chem. Eng., 2017, 5, 10379–10386 CrossRef.
- Y. S. Ma, J. J. Lin, C. C. Lin, J. C. Lien, S. F. Peng, M. J. Fan, F. T. Hsu and J. G. Chung, Environ. Toxicol., 2018, 33, 1097–1104 CrossRef CAS PubMed.
- R. S. Dubey, Y. B. R. D. Rajesh and M. A. More, Mater. Today: Proc., 2015, 2, 3575–3579 CAS.
- M. R. Kasaai, Carbohydr. Polym., 2008, 71, 497–508 CrossRef CAS.
- M. T. Yen, J. H. Yang and J. L. Mau, Carbohydr. Polym., 2009, 75, 15–21 CrossRef CAS.
- Y. Yin, S. Qiu and Y. Peng, Gene, 2016, 576, 189–194 CrossRef CAS PubMed.
- S. K. Shukula, A. K. Mishra, O. A. Arotiba and B. B. Mamba, Int. J. Biol. Macromol., 2013, 59, 46–58 CrossRef PubMed.
- N. V. Majeti and K. Ravi, React. Funct. Polym., 2000, 46, 1–27 CrossRef.
- D. Zhao, S. Yu, B. Sun, S. Gao, S. Guo and K. Zhao, Polymers, 2018, 10, 462 CrossRef PubMed.
- R. Pecora, J. Nanopart. Res., 2000, 2, 123–131 CrossRef CAS.
-
A. Domard and M. Domard, «Chitosan: Structure–Properties Relationship and Biomedical Applications», Polymeric Biomaterials, ed. S. Dumitriu, Marcel Dekker, 2001, pp. 187–212 Search PubMed.
- T. Yu, K. Greish, L. D. McGill, A. Ray and H. Ghandehari, ACS Nano, 2012, 6, 2289–2301 CrossRef CAS PubMed.
- A. Villanueva, M. Cañete, A. G. Roca, M. Calero, S. Veintemillas-Verdaguer, C. J. Serna, M. Morales and R. Miranda, Nanotechnology, 2009, 20, 115103 CrossRef PubMed.
- C. R. Miller, B. Bondurant, S. D. McLean, K. A. McGovern and D. F. O'Brien, Biochemistry, 1998, 37, 12875–12883 CrossRef CAS PubMed.
- A. M. Bannunah, D. Vllasaliu, J. Lord and S. Stolnik, Mol. Pharm., 2014, 11, 4363–4373 CrossRef CAS PubMed.
- A. Verma and F. Stellacci, Small, 2010, 6, 12–21 CrossRef CAS PubMed.
- L. Shang, K. Nienhaus and G. U. Nienhaus, J. Nanobiotechnology, 2014, 12, 5 CrossRef PubMed.
- M. Izume, S. Nagae, H. Kawagishi, M. Mitsutomi and A. Ohtakara, Biosci. Biotechnol. Biochem., 1992, 56, 448–453 CrossRef CAS PubMed.
- T. Yu, K. Greish, L. D. McGill, A. Ray and H. Ghandehari, ACS Nano, 2012, 6, 2289–2301 CrossRef CAS PubMed.
- H. Jin, D. A. Heller, R. Sharma and M. S. Strano, ACS Nano, 2009, 3, 149–158 CrossRef CAS PubMed.
- D. Napierska, L. C. J. Thomassen, D. Lison, J. A. Martens and P. H. Hoet, Part. Fibre Toxicol., 2010, 7, 1–32 CrossRef PubMed.
- M. J. Ernsting, M. Murakami, A. Roy and S. D. Li, J. Control. Release, 2013, 172, 782–794 CrossRef CAS PubMed.
- S. W. Shin, I. H. Song and S. H. Um, Nanomaterials, 2015, 5, 1351–1365 CrossRef CAS PubMed.
- M. Longmire, P. L. Choyke and H. Kobayashi, Nanomedicine, 2008, 3, 703–717 CrossRef CAS PubMed.
- D. Fukumura and R. K. Jain, J. Cell. Biochem., 2007, 101, 937–949 CrossRef CAS PubMed.
- J. A. Nagy, S. H. Chang, S. C. Shih, A. M. Dvorak and H. F. Dvorak, Semin. Thromb. Hemostasis, 2010, 36, 321–331 CrossRef CAS PubMed.
- V. Torchilin, Adv. Drug Deliv. Rev., 2011, 63, 131–135 CrossRef CAS PubMed.
- V. P. Torchilin, AAPS J., 2007, 9, 128–147 CrossRef PubMed.
- P. Carmeliet and R. K. Jain, Nature, 2000, 407, 249–257 CrossRef CAS PubMed.
- A. C. Carita, J. O. Eloy, M. Chorilli, R. J. Lee and G. R. Leonardi, Curr. Med. Chem., 2018, 25, 606–635 CrossRef CAS PubMed.
- E. A. Sykes, J. Chen, G. Zheng and W. C. Chan, ACS Nano, 2014, 8, 5696–5706 CrossRef CAS PubMed.
- M. F. Burbridge, D. C. West, G. Atassi and G. C. Tucker, Angiogenesis, 1999, 3, 281–288 CrossRef CAS PubMed.
- E. K. Lim, B. H. Chung and S. J. Chung, Curr. Drug Targets, 2018, 19, 300–317 CrossRef CAS PubMed.
- H. Kadry, B. Noorani and L. Cucullo, Fluids Barriers CNS, 2020, 17, 69 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Imen
Chaabani
ab,
Abir
Salek
a,
Leila
Njim
c,
Mouna
Selmi
a,
Ahmed
Al-Kattan
*a,
Imen
Chaabani
ab,
Abir
Salek
a,
Leila
Njim
c,
Mouna
Selmi
a,
Ahmed
Al-Kattan