
Additive-free molecular acceptor organic solar cells processed from a biorenewable solvent approaching 15% efficiency†
Zhifang
Du
 a,
Hoang Mai
Luong
a,
Sina
Sabury
b,
Pattarawadee
Therdkatanyuphong
c,
Sangmin
Chae
a,
Claire
Welton
d,
Austin L.
Jones
b,
Junxiang
Zhang
c,
Zhengxing
Peng
e,
Ziyue
Zhu
a,
Sadisha
Nanayakkara
f,
Veaceslav
Coropceanu
f,
Dylan G.
Choi
a,
Steven
Xiao
g,
Ahra
Yi
ah,
Hyo Jung
Kim
h,
Jean-Luc
Bredas
f,
Harald
Ade
e,
G. N. Manjunatha
Reddy
*d,
Seth R.
Marder
*c,
John R.
Reynolds
*b and
Thuc-Quyen
Nguyen
*a
a,
Hoang Mai
Luong
a,
Sina
Sabury
b,
Pattarawadee
Therdkatanyuphong
c,
Sangmin
Chae
a,
Claire
Welton
d,
Austin L.
Jones
b,
Junxiang
Zhang
c,
Zhengxing
Peng
e,
Ziyue
Zhu
a,
Sadisha
Nanayakkara
f,
Veaceslav
Coropceanu
f,
Dylan G.
Choi
a,
Steven
Xiao
g,
Ahra
Yi
ah,
Hyo Jung
Kim
h,
Jean-Luc
Bredas
f,
Harald
Ade
e,
G. N. Manjunatha
Reddy
*d,
Seth R.
Marder
*c,
John R.
Reynolds
*b and
Thuc-Quyen
Nguyen
*a
aCenter for Polymers and Organic Solids, Department of Chemistry and Biochemistry, University of California at Santa Barbara, Santa Barbara, CA 93106, USA. E-mail: quyen@chem.ucsb.edu
bSchool of Chemistry and Biochemistry, School of Materials Science and Engineering, Center for Organic Photonics and Electronics, Georgia Tech Polymer Network, Georgia Institute of Technology, Atlanta, Georgia 30332, USA. E-mail: reynolds@chemistry.gatech.edu
cUniversity of Colorado Boulder, Renewable and Sustainable Energy Institute, Boulder, CO 80303, USA. E-mail: sema1789@colorado.edu
dUniversity of Lille, CNRS, Centrale Lille Institut, Univ. Artois, UMR 8181, Unité de Catalyse et Chimie du Solide, F-59000, Lille, France. E-mail: gnm.reddy@univ-lille.fr
eDepartment of Physics and Organic and Carbon Electronics Laboratories (ORaCEL), North Carolina State University, Raleigh, NC 27695, USA
fDepartment of Chemistry and Biochemistry, The University of Arizona, Tucson, AZ 85721-0088, USA
g1-Material Inc, 2290 Chemin St-Francois, Dorval, Quebec H9P 1K2, Canada
hDepartment of Organic Material Science and Engineering, School of Chemical Engineering, Pusan National University, Busan 46241, Republic of Korea
First published on 5th October 2023
Abstract
We report on the use of molecular acceptors (MAs) and donor polymers processed with a biomass-derived solvent (2-methyltetrahydrofuran, 2-MeTHF) to facilitate bulk heterojunction (BHJ) organic photovoltaics (OPVs) with power conversion efficiency (PCE) approaching 15%. Our approach makes use of two newly designed donor polymers with an opened ring unit in their structures along with three molecular acceptors (MAs) where the backbone and sidechain were engineered to enhance the processability of BHJ OPVs using 2-MeTHF, as evaluated by an analysis of donor–acceptor (D–A) miscibility and interaction parameters. To understand the differences in the PCE values that ranged from 9–15% as a function of composition, the surface, bulk, and interfacial BHJ morphologies were characterized at different length scales using atomic force microscopy, grazing-incidence wide-angle X-ray scattering, resonant soft X-ray scattering, X-ray photoelectron spectroscopy, and 2D solid-state nuclear magnetic resonance spectroscopy. Our results indicate that the favorable D–A intermixing that occurs in the best performing BHJ film with an average domain size of ∼25 nm, high domain purity, uniform distribution and enhanced local packing interactions – facilitates charge generation and extraction while limiting the trap-assisted recombination process in the device, leading to high effective mobility and good performance.
 Thuc-Quyen Nguyen (left) and Seth R. Marder (right) | Congratulations on the 10th anniversary! We are honored to be part of the Materials Horizons family. We thank the RSC and in the particular the MH team for allowing some of us to play a role in shaping the journal. Seth was the Founding Chair of the MH Editor Board and Thuc-Quyen served as a Scientific Editor for 9 years and it has been a joy to see the journal becoming one of the most important journals in materials science. We wish MH continued success and we look forward to continuing our support as a member of the Advisory Board, as author, and a reader! |
New conceptsThe recent upsurge in the solar-to-electrical power conversion efficiency (PCE) associated with organic photovoltaics (OPVs) opens up new opportunities, particularly by means of their commercial viability. However, several challenges must be overcome. Among others, processability of OPVs using environmentally sustainable solvents without compromising much on the performance is paramount. This study makes use of newly designed small molecular acceptors (MAs) and donor polymers processed with a biomass-derived solvent (2-methyltetrahydrofuran, 2-MeTHF) to facilitate high performance of ∼15% PCE in bulk heterojunction (BHJ) OPVs without using halogenated additives. In doing so, this work provides unprecedented new insights into the molecular origins of moderate-to-high PCEs in the 9–15% range for the BHJ OPVs processed from 2-MeTHF. The synergism between synthesis, processing, characterization and device physics is used to explain the rationale for the large variation in the PCE values. This is further corroborated by morphology analysis at different length scales using microscopy, X-ray scattering and magnetic resonance spectroscopy techniques, which indicate that the domain size and purity, D–A intermix and distribution, and local packing interactions facilitate high performance in ‘green’ OPVs. |
1. Introduction
The continued push to optimize the properties associated with organic photovoltaic (OPV) cells has enabled high power conversion efficiencies (PCEs) approaching 20%, which can help encourage and facilitate the commercialization of devices based upon these materials.1–4 Most high-performance single junction OPV devices are processed from halogenated solvents such as chloroform (CF, which is highly volatile), chlorobenzene (CB), and additives such as 1,8-diiodooctane (DIO) and 1-chloronaphthalene (CN),5,6 which are hazardous to either human health or the environment, or both. Therefore, the use of such solvents (and additives) is an impediment to the large-scale commercialization of OPV modules and it is desirable to replace them with eco-friendly ‘green’ solvents generated from renewable agricultural resources.7,8 However, there usually is a trade-off between the use of these environmentally sustainable solvents and PCE, and the development of high-efficiency OPVs processed from green solvents necessitates new molecular design and BHJ processing approaches, as well as a comprehensive understanding of structure–property relationships. In this work, we focus on developing processable high-performance OPV BHJs, using an environmentally friendly and sustainable solvent 2-methyltetrahydrofuran (2-MeTHF).Although a large pool of non-halogenated solvents is available for processing of donor–acceptor molecules and their blends, a priori, screening of potential solvents can be obtained based on solubility, polarity, and dispersion, for example, by examining Hansen solubility parameters (HSP).9–12 Additives as morphology boosters also have a significant impact on the processability, efficiency, and stability of OPVs.13–22 Regarding the use of non-halogenated solvents for processing BHJ solar cells, ortho-xylene (o-xylene),23–27 toluene,26,28 tetrahydrofuran (THF),29 terpene bio-solvents,8 and 2-MeTHF have been reported. Among these solvents, 2-MeTHF is produced from agriculture byproducts and widely utilized for large-scale organic synthesis in industry.30,31 Furthermore, it has a low boiling point (78 °C) allowing for fast drying and processing BHJ mixtures into thin films32 for OPVs.28,33–38 For example in early work, 2-MeTHF treated BHJ-based OPVs involving a low bandgap polymer PPDPP and the PC71BM acceptor enabled a PCE of 4.18%.34 In addition, an all-polymer solar cell consisting of the PTzBI-Si donor and N2200 acceptor processed in non-halogenated solvents such as 2-MeTHF and cyclopentyl methyl ether (CPME) resulted in a PCE of ∼11%.33 Very recently, the utility of 2-MeTHF as a processing solvent is exemplified by work published during the preparation of this manuscript, where PTQ10 and FO6-T polymer donors blended with Y12 acceptor were used to attain organic solar cells (OSCs) with up to 14.5% and 11.4% PCE.35 Often, poor solubility of high-performance donor and acceptor molecules in non-halogenated solvents such as o-xylene and 2-MeTHF is a bottleneck for processing these molecules into BHJ thin films. Therefore, a considerable “molecular redesign” of the donor/acceptor components is necessary to achieve high solubility in 2-MeTHF. This enhanced miscibility and processability, while keeping optimal film morphology for charge generation and transport properties at the desired levels, which is crucial to formulate high-performance BHJ thin films. Understanding the molecular origins of the factors that influence the molecule packing in the BHJ morphology is important, as well as their role in governing device performance, which is yet to be addressed and compared for OPVs processed from 2-MeTHF.
In this work, we designed a series of donor:acceptor (D:A) blends based on the PM7 series of polymer donors with different backbone structures and Y-series molecule acceptors (MAs), in which the position of the sidechains on the polymer and the alkyl chain length on the MAs are used to control the solubility in 2-MeTHF without the use of additives. The replacement of the closed-ring diketone in PM6 and PM7 with an opened ring diester quaterthiophene moiety in PM7-D3 and PM7-D5 (structures are shown in Fig. 1a) provides backbone flexibility. A combination of characterization techniques is employed to resolve the BHJ morphology at different length scales ranging from sub-nanometer to micrometer distances using solid-state nuclear magnetic resonance spectroscopy (ssNMR), atomic force microscopy (AFM), grazing-incidence wide-angle X-ray scattering (GIWAXS), and resonant soft X-ray scattering (RSoXS) techniques. In addition, we characterized the optical properties, energetics, and electronic parameters that contribute to the different PCE values in these BHJ devices. The additive-free donor and MA blended PM7-D3:PTI04 device processed from 2-MeTHF demonstrates a PCE value of 14.9%, which is akin to PM6:Y6 reference devices39 processed from CF with additives. Our studies of the bulk and interfacial BHJ morphology indicate that a favorable intermixing of PM7-D3 and PTI04 results in appropriate MA domain sizes and phase purity. Furthermore, PM7-D3:PTI04 devices have high charge carrier mobilities and low charge recombination, enabling high PCEs. Our studies suggest that the underlying reasons for the lower performance of the other PM7-D5/PM7-D3:MA blends (PCEs in the 9–11% range) are an unfavorable local morphology, which we attribute to the positions of the ester sidechains on the donor polymers and elongated sidechains on the MAs.
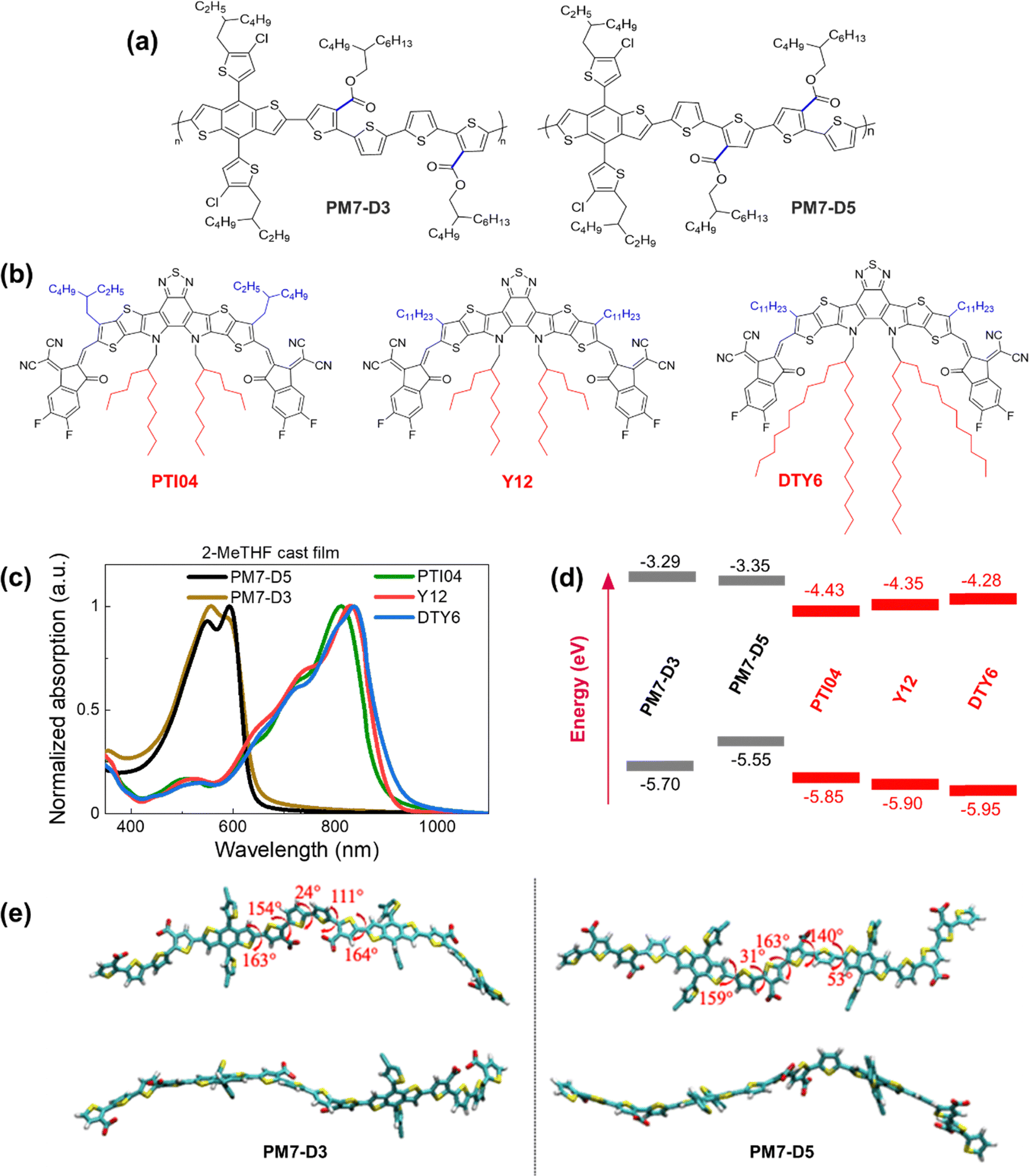 | ||
| Fig. 1 Chemical structures of (a) PM7-D3 and PM7-D5, and (b) PTI04, Y12, and DTY6. (c) Normalized absorption of thin films processed from 2-MeTHF. (d) Energy level diagram of polymers donors and acceptors. (e) Top and side views of the optimized geometries (at the ωB97X-D/6-31G(d,p) level of theory) for the symmetric dimeric models of PM7-D3 (left), and PM7-D5 (right) polymers. The dihedral angles between selected moieties are indicated in red. The long sidechains are omitted for clarity. | ||
2. Results and discussion
2.1. Molecular design, D–A interaction parameters, and optical properties
The repeat unit structures of polymer donors (PM7-D3 and PM7-D5), which differ only in the positions of the ester side chains on the thiophene rings of the backbone, and the structures of the Y-series MAs (Y12, DTY6, and PTI04) with different sidechain lengths and branching are shown in Fig. 1a and b. The synthesis of these polymers (ESI,† Schemes S1 and S2, and Fig. S1–S7) is simpler than the well-known high-performance polymers PM6 and PM7. From gel permeation chromatography (Fig. S8, ESI†), PM7-D3 has a higher molecular weight than PM7-D5 (Mn: 65 kg mol−1, Mw: 149 kg mol−1, Đ: 2.30 versus Mn: 26.1 kg mol−1, Mw: 55 kg mol−1, Đ: 2.09). More details of the material synthesis and molecular characterization are given in the ESI.†A comparison of the optical properties of neat donor polymer and MA thin films spin coated from 2-MeTHF solutions is given in Fig. 1c, while their ionization energies (IEs) and electron affinities (EAs) are shown in Fig. 1d. The IE and EA energies are estimated based on the extraction of the onset potentials using dynamic pulse voltammetry (DPV) versus ferrocene/ferrocenium (considering a conversion factor of −5.12 V vs. vacuum). To better represent solid-state characteristics and minimize any alterations in the film properties caused by electrolyte absorption, the initial voltammograms of fresh films are employed, and different films are utilized for the oxidation and reduction processes, so the onset for oxidation and reduction is not affected by the electrochemical history (Fig. 1d). A small variation in the IEs is observed, which may be attributed to different aggregation and packing behaviors, as will be thoroughly investigated below (vide infra).
The degree of assembly of the D-polymer or MA in the BHJ blend can be influenced by their solubility and aggregation tendency, which strongly depends on the backbone structures and sidechains. For instance, when a D-polymer is blended with a MA, the delayed solidification of the D-polymer results in a less favorable final morphology of the BHJ blend.40 While enhancing backbone planarity in conjugated polymers by utilizing fused aromatic rings is expected to improve transport properties and device performance, these fused-ring-core polymers may lead to reduced solubility and bring additional challenges to processability. Conversely, incorporating flexible connections (non-fused rings) into the backbone often leads to enhanced solubility, but the OPV performance may be compromised. By tailoring the backbone structure using fused ring (benzodithiophene part) and non-fused ring (diester quaterthiophene part) components in PM7-D3 and PM7-D5, together with the placement of the branched 2-butyloctyl sidechains on the ester functionalities, solubility in the green solvent 2-MeTHF can be adjusted and enhanced. The presence of two peaks or a peak-shoulder pattern in the absorption spectrum of both PM7-D3 and PM7-D5, respectively, suggests the presence of aggregates resulting from interchain π–π stacking. In particular, the larger 0-0/0-1 ratio (ratio between the absorbance of shoulder to main peak) in PM7-D5 points to a more planar backbone compared to PM7-D3.41 This observation is further supported by the 150 meV lower IE for PM7-D5 and the results of long-range corrected ωB97X-D/6-31G(d,p) density–functional theory (DFT) calculations based on symmetric dimers (Fig. 1e), where the PM7-D5 backbone conformation intrinsically exhibits a higher degree of planarity.
For the donor polymers, backbone flexibility in PM7-D3 and PM7-D5 provides the requisite solubility for processing films without compromising solid-state packing. PM7-D3 exhibits solubility of slightly greater than 10 mg mL−1 in 2-MeTHF while PM7-D5 shows a slightly lower solubility of approximately 7 mg mL−1 even at lower molecular weight, in line with the backbone planarity trend between the two polymers. While molecular weight differences between the two polymers can also play a role, the main factor contributing to the different solubilities and aggregation behaviors between these two structural isomeric polymers is believed to be the position of the diester sidechains in the quaterthiophene building block. One would expect PM7-D5 rather than PM7-D3 to have a higher solubility in 2-MeTHF if only considering the low molecular weight of PM7-D5. Thus, it is evident that the sidechain position in these polymers controls the conformation of the backbone as corroborated by the DFT calculations for symmetric dimers shown in Fig. 1e. The enhanced conformational degrees of freedom and less planar backbones in PM7-D3 coming from the position of the diester quaterthiophene are expected to contribute to the improved solubility in 2-MeTHF. It is worth noting that all three MAs exhibited ideal processing capabilities with a high solubility of ∼10 mg mL−1 in the eco-friendly solvent 2-MeTHF. For three MAs, similar high solubility of above 10 mg mL−1 in 2-MeTHF is observed, which is also supported by a solubility assessment that describes the origins of high solubility supported by Hansen solubility parameters (HSP). The HSP theory can evaluate the cohesive energy density (CED) of material/solvent considering the contribution of dispersion, polarity, and hydrogen bonding togetherly.8,11,12 From the CEDs of selected molecules and solvents, the molecule–solvent interaction parameters, χ, are calculated and summarized in the ESI† (Section 2, Tables S2 and S3). Smaller χ values of around 0.70 indicate small differences in the properties between three MAs and the host solvent, which further supports their high solubility in 2-MeTHF. Thus, PM7-Dx:MA blends are highly compatible with processability from 2-MeTHF.
2.2. Photovoltaic characteristics
To assemble OPV cells, the PM7-Dx:MA in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2 weight ratio BHJ active layer was cast from 2-MeTHF heated at 70 °C to accelerate the drying of the thin films during spin coating,42 and the resulting devices were tested. It is noteworthy that solvent additives were not used in order to eliminate the health and environmental hazards due to halogenated additives, which also simplifies the fabrication process for large-scale fabrication compatibility.20,43 The device structure of ITO/PEDOT:PSS/PM7-Dx:MA/PNDIT-F3N-Br/Ag was utilized and a more detailed description of the device fabrication procedure can be found in the ESI,† Section 3. The current density–voltage (J–V) characteristics of these solar cells are shown in Fig. 2a and Table 1. Due to this variation in IE values, it is expected that the PM7-D3 donor polymer with a higher IE (roughly speaking, a lower HOMO) can enhance the open-circuit voltage (Voc) of the PM7-D3:MA OPV device relative to PM7-D5:MA BHJ. Among the four BHJ blends with different combinations of the donor polymers and the MAs, we found the highest PCE of 14.91 ± 0.25% for the PM7-D3:PTI04 device, enabled by a relatively high fill factor, FF (0.69), Voc (0.90 V), and Jsc (23.88 mA cm−2), which is also one of the best performing systems processing from 2-MeTHF compared to recent eco-friendly solvent treated organic solar cells shown in Table S4 (ESI†). The integration of the external quantum efficiency (EQE) spectrum, Fig. 2b, results in a similar Jsc value for this system. By comparison, the other BHJ systems exhibited lower PCE values in the 9.61–11.28% range. We attribute this to the relatively large domain size and uneven distribution of donor and acceptor in the BHJ morphology of those devices, which will be further described by characterizing the photophysical parameters as well as performing a morphology analysis at different length scales.
:1.2 weight ratio BHJ active layer was cast from 2-MeTHF heated at 70 °C to accelerate the drying of the thin films during spin coating,42 and the resulting devices were tested. It is noteworthy that solvent additives were not used in order to eliminate the health and environmental hazards due to halogenated additives, which also simplifies the fabrication process for large-scale fabrication compatibility.20,43 The device structure of ITO/PEDOT:PSS/PM7-Dx:MA/PNDIT-F3N-Br/Ag was utilized and a more detailed description of the device fabrication procedure can be found in the ESI,† Section 3. The current density–voltage (J–V) characteristics of these solar cells are shown in Fig. 2a and Table 1. Due to this variation in IE values, it is expected that the PM7-D3 donor polymer with a higher IE (roughly speaking, a lower HOMO) can enhance the open-circuit voltage (Voc) of the PM7-D3:MA OPV device relative to PM7-D5:MA BHJ. Among the four BHJ blends with different combinations of the donor polymers and the MAs, we found the highest PCE of 14.91 ± 0.25% for the PM7-D3:PTI04 device, enabled by a relatively high fill factor, FF (0.69), Voc (0.90 V), and Jsc (23.88 mA cm−2), which is also one of the best performing systems processing from 2-MeTHF compared to recent eco-friendly solvent treated organic solar cells shown in Table S4 (ESI†). The integration of the external quantum efficiency (EQE) spectrum, Fig. 2b, results in a similar Jsc value for this system. By comparison, the other BHJ systems exhibited lower PCE values in the 9.61–11.28% range. We attribute this to the relatively large domain size and uneven distribution of donor and acceptor in the BHJ morphology of those devices, which will be further described by characterizing the photophysical parameters as well as performing a morphology analysis at different length scales.
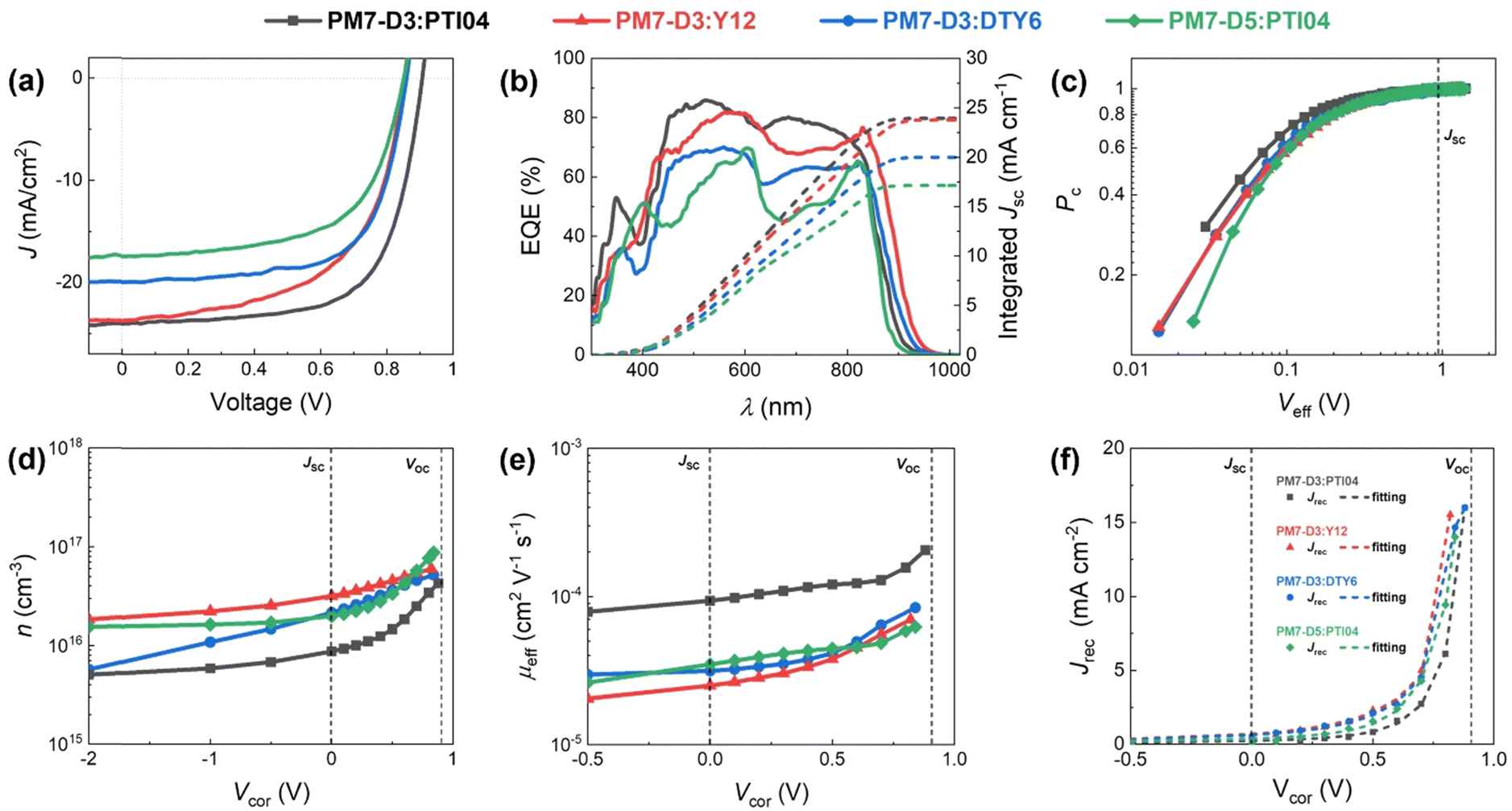 | ||
| Fig. 2 (a) J–V characteristics, (b) EQE spectra, (c) charge collection probability, (d) voltage-dependent charge carrier density, (e) voltage-dependent effective mobility, and (f) fitting of the recombination current density of PM7-D3:PTI04, PM7-D3:Y12, PM7-D3:DTY6, and PM7-D5:PTI04 under AM1.5G illumination at 100 mW cm−2. | ||
| Solvent | D:A | J sc (mA cm−2) | V oc (V) | FF | PCEavg (%) |
|---|---|---|---|---|---|
| 2-MeTHF | PM7-D3:PTI04 | 23.88 ± 0.12 | 0.90 ± 0.01 | 0.69 ± 0.01 | 14.91 ± 0.25 |
| PM7-D3:Y12 | 23.76 ± 0.61 | 0.86 ± 0.01 | 0.54 ± 0.02 | 11.06 ± 0.49 | |
| PM7-D3:DTY6 | 20.29 ± 0.37 | 0.86 ± 0.01 | 0.65 ± 0.02 | 11.28 ± 0.35 | |
| PM7-D5:PTI04 | 17.17 ± 0.51 | 0.87 ± 0.01 | 0.64 ± 0.02 | 9.61 ± 0.45 |
To gain a deeper understanding of the device performance, we studied the voltage-dependent charge collection probability (Pc) using the following equation,
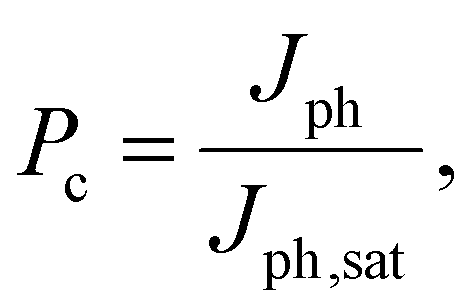 | (1) |
Next, impedance spectroscopy was carried out to determine the charge carrier density in the active layer upon varying operational biases and light intensities.45–52 To maintain the steady-state properties of the system during measurement under different illumination conditions, a DC bias in a range from −2 V to Voc, and a small AC signal (40 mV) were applied during the scanning. To account for the impact of the device series resistance and parasitic inductance of the connecting cables, a correction factor is included in the measured capacitance of the BHJ layer as represented in eqn (2):
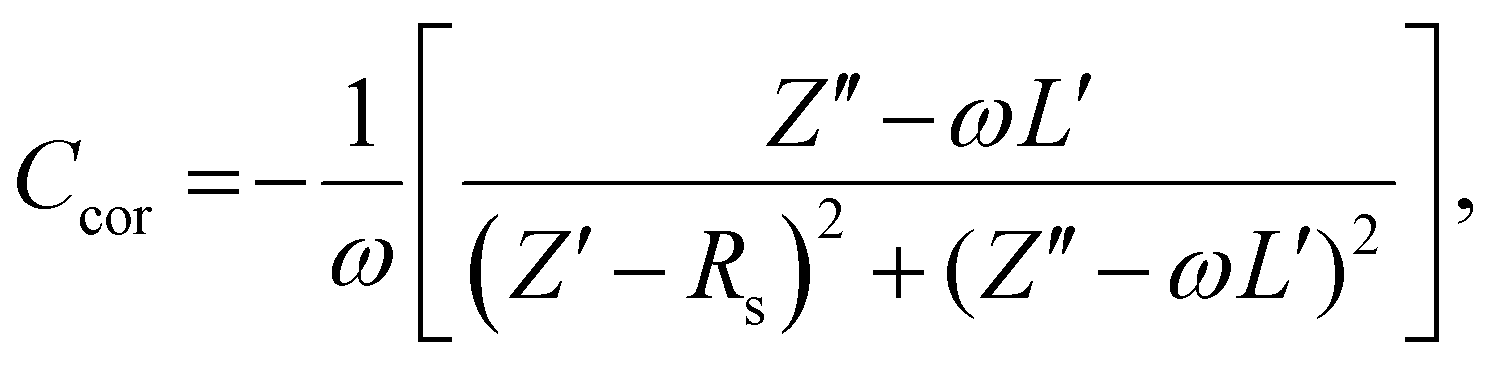 | (2) |
Here, L′ is the inductance of the connecting cables, 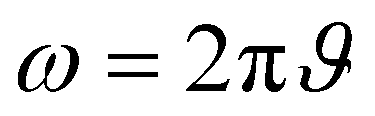 is the angular frequency of the AC signal, and Z′ and Z′′ are the real and imaginary components of impedance, respectively. In the dark, the corrected capacitance measured under a large reverse bias (−2 V) shows a horizontal line, which is equivalent to a frequency-independent capacitance. From this, one can determine the geometrical capacitance (Cg) of the active layer. Voltage-dependent impedance spectroscopy measurements were performed to obtain the charge carrier density n (Fig. 2d), via integration of the chemical capacitance Cchem using the following equations:24,46,53–55
is the angular frequency of the AC signal, and Z′ and Z′′ are the real and imaginary components of impedance, respectively. In the dark, the corrected capacitance measured under a large reverse bias (−2 V) shows a horizontal line, which is equivalent to a frequency-independent capacitance. From this, one can determine the geometrical capacitance (Cg) of the active layer. Voltage-dependent impedance spectroscopy measurements were performed to obtain the charge carrier density n (Fig. 2d), via integration of the chemical capacitance Cchem using the following equations:24,46,53–55
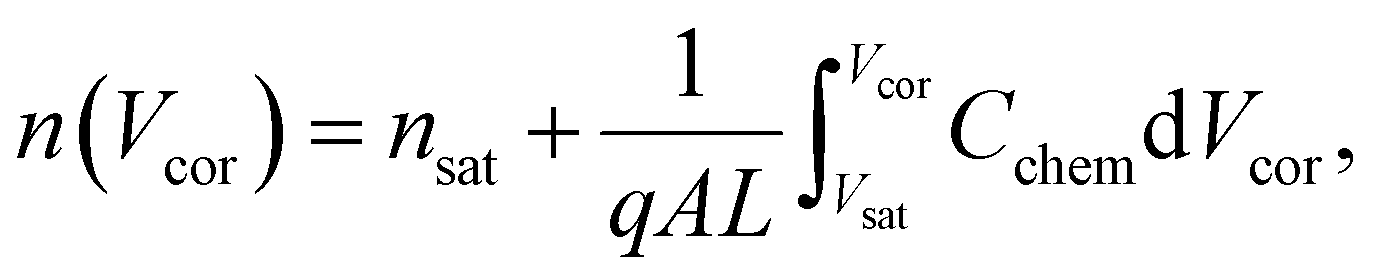 | (3) |
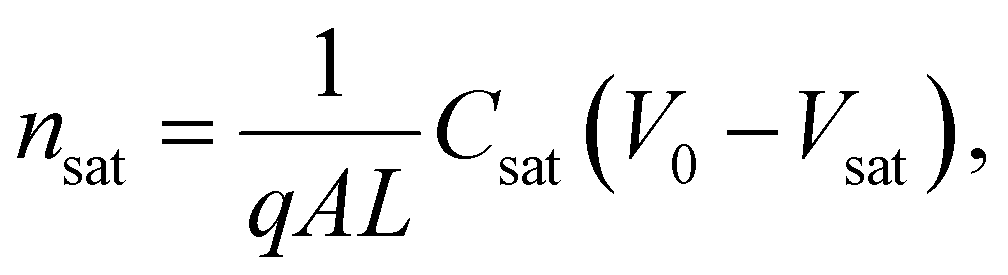 | (4) |
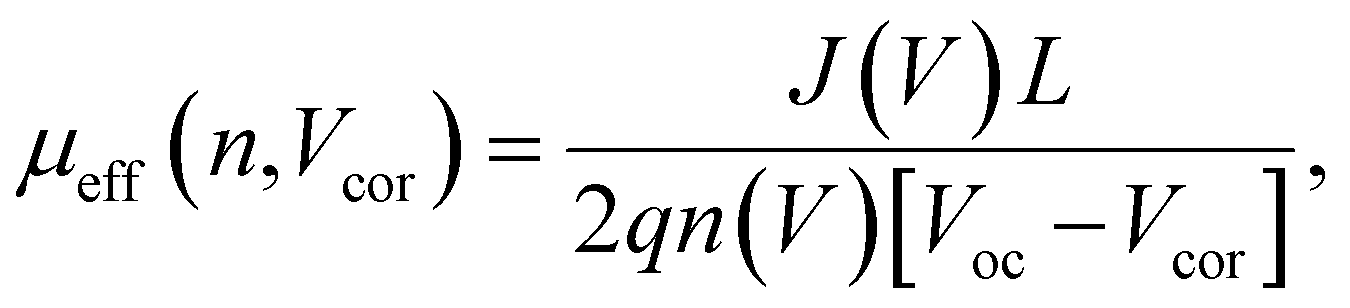 | (5) |
To obtain quantitative insights into the charge recombination dynamics, we employ a model that relates the recombination current density Jrec to the charge carrier density and compares it to experimental values of Jrec obtained from J–V curves, which can be achieved by a combination of J–V characteristics (in the dark and under illumination) and voltage-dependent impedance analysis.42,45 The density of bulk traps Nt,bulk, the density of surface traps Nt,surf, and the reduction factor (also known as Langevin prefactor) ξ are parameters used to fit the recombination current with the experimental data.56 A summary of the physical parameters used in the Jrec fitting model is presented in Table 2. Even though it shows a slightly higher Langevin prefactor correlating to bimolecular recombination due to its higher μeff, it is worth noting that both bulk and surface trap density dramatically decrease in PM7-D3:PTI04 system, suppressing charge carrier loss due to bulk and surface trap-assisted recombination in the device. Also, benefiting from the high mobility, the calculated τμ for the PM7-D3:PTI04 device exhibits the smallest value among the four systems, a feature that has been shown to correlate with a high FF in earlier reports.57,58 Next, the drift length (Ldr) and diffusion length (Ldiff) in the BHJ active layers are calculated based on n(V) and μeff, which are further normalized by the thickness of the film. Considering the relatively long Ldr (all above 600 nm) compared with the average thickness of the active layers (∼100 nm), longer Ldiff in the PM7-D3:PTI04 system makes a difference when the internal electric field is small. In other words, the drift length at JSC is much greater than the thickness of the active layer. However, the effective diffusion length is smaller than the active layer thickness, which can result in significant losses in carrier extraction when drift is not efficient.58 The charge transport and collection processes are mainly controlled by the diffusion process and benefit from the long Ldiff in the PM7-D3:PTI04 system when getting close to maximum power voltage (Vmp) and Voc conditions, which ultimately explains the higher PCE of the PM7-D3:PTI04 solar cells among the four blend systems. Key takeaway from the detailed analysis of photovoltaic parameters is that the PM7-D3:PTI04 system has lower surface- and bulk-trap assisted recombination and efficient charge extraction and collection resulting in a high FF and Jsc. Meanwhile, the lowest bulk traps in PM7-D3:PTI04 system also suppress the non-radiative voltage loss and lead to a high Voc. In contrast, the other PM7-D3:MA blends suffer from relatively high recombination rates and low FF, leading to poor performance in the OPV devices. The presence of significant bulk traps within the PM7-D3:DTY6 system results in serious non-radiative voltage losses, leading to a limitation in its Voc and overall device performance. The PM7-D5:PTI04 devices also suffer from these factors, indicating that both donor and acceptor molecules and their packing interactions are particularly important in order to steer the electronic properties of the BHJ blends toward high PCE values. This led us to investigate the BHJ morphology and molecular origins of the different photovoltaic properties, as discussed below.
| D:A | ξ | N t,bulk (cm−3) | N t,surf (cm−3) | τμ (cm2 V−1) | L dr (nm) | L dr/d | L diff (nm) | L diff/d |
|---|---|---|---|---|---|---|---|---|
| PM7-D3:PTI04 | 0.035 | 1.16 × 1012 | 6.97 × 1012 | 1.17 × 10−9 | 1044.03 | 10.44 | 54.77 | 0.55 |
| PM7-D3:Y12 | 0.021 | 1.56 × 1013 | 1.63 × 1014 | 8.83 × 10−10 | 742.37 | 7.35 | 47.62 | 0.47 |
| PM7-D3:DTY6 | 0.027 | 7.55 × 1014 | 6.68 × 1013 | 7.54 × 10−10 | 661.77 | 6.75 | 44.02 | 0.45 |
| PM7-D5:PTI04 | 0.028 | 4.02 × 1013 | 1.66 × 1013 | 6.64 × 10−10 | 600.11 | 6.00 | 41.30 | 0.41 |
2.3. Resolving BHJ morphology at different length scales
The charge generation, transport, recombination, and extraction processes in OPVs are closely linked to the BHJ morphology and packing arrangements of the donor–acceptor moieties.43,59–62 In particular, the processing solvent has a significant impact on the BHJ morphology.63–65 To gain further insights into the impact of the sidechain positions and lengths on the device performance of the four OPV systems processed from 2-MeTHF, the BHJ morphologies were further characterized with various techniques at different length scales (μm to sub-nm).66 At the micron to sub-micron length scales, AFM is employed to map the topography of PM7-D3:PTI04, PM7-D3:Y12, PM7-D3:DTY6, and PM7-D5:PTI04 blends under optimized conditions. As shown in Fig. 3a, AFM images of all BHJ blends at 5 μm × 5 μm reveal continuous networks and relatively smooth surfaces with RMS values from ∼1–3.06 nm.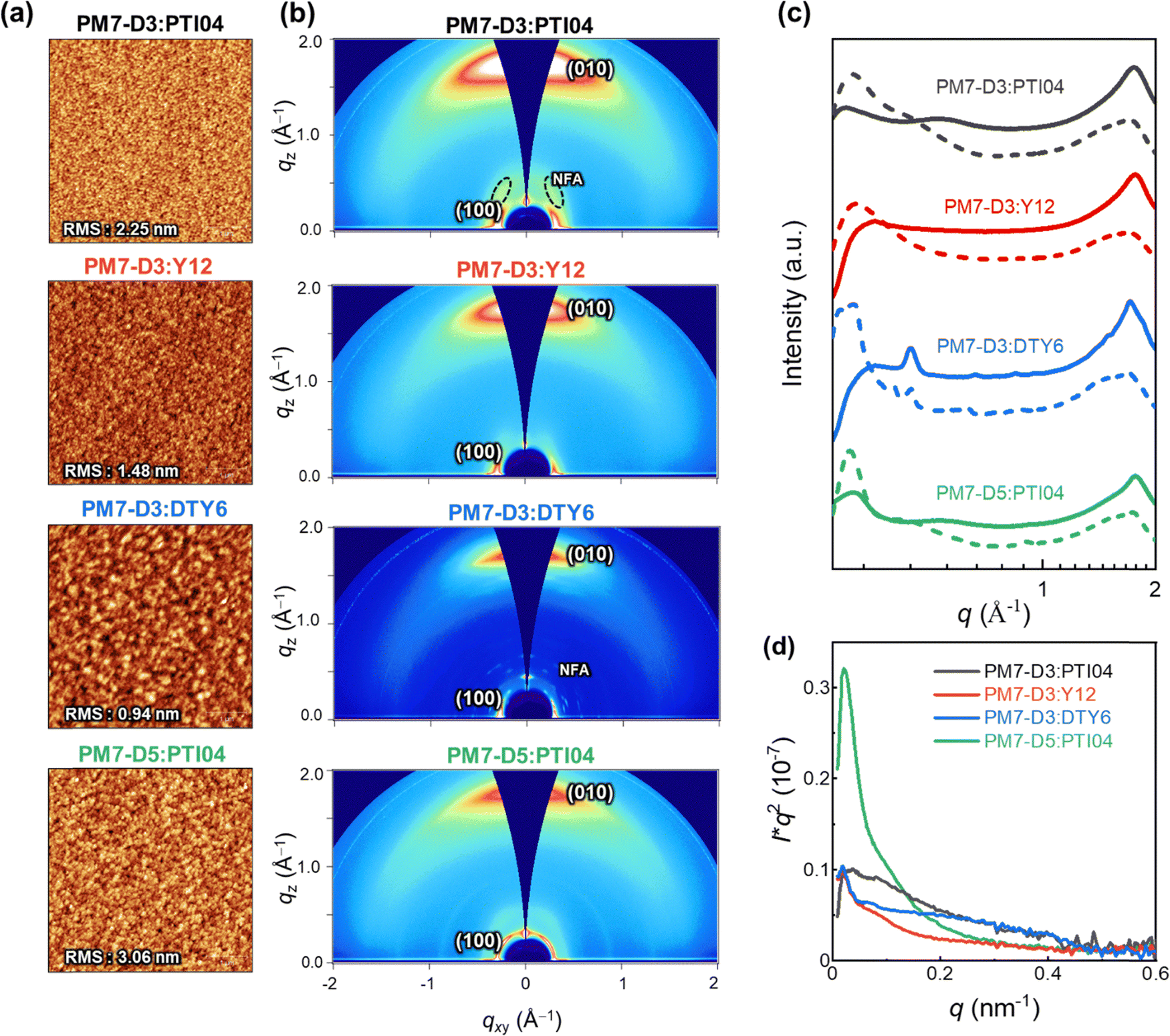 | ||
| Fig. 3 Surface and bulk morphology of the BHJ blends processed from 2-MeTHF: (a) AFM topography images 5 μm × 5 μm, (b) 2D GIWAXS patterns, (c) the corresponding log–log plot of in-plane (dash lines) and out-of-plane (solid lines) GIWAXS profiles, and (d) RSoXS profiles at 285.0 eV with film thickness normalization and Lorentz correction. | ||
GIWAXS measurements were conducted to gain insights into the molecular packing and orientation of the D and A molecules with respect to the substrate. The two-dimensional (2D) patterns of all blend films exhibit a combination of features from the donor polymers (PM7-D3 and PM7-D5) and MAs, as illustrated in Fig. 3b. The GIWAXS data for neat donor and acceptor are shown in Fig. S16 (ESI†). All the PM7-D3 systems displayed distinct (010) scattering patterns in the out-of-plane direction originating from the π–π stacking of the polymer donor and MAs, which indicates a preferred face-on orientation in an ordered structure (Fig. 3c). A comprehensive summary of all relevant parameters and details can be found in the ESI,† Table S5.
All blends exhibit a moderate coherence length (CL = 10–20 nm) for (100) scattering, which corresponds to length scale over which the sample scatter coherently and indicates the quality of the molecular packing. It is comparable to the exciton diffusion lengths in oligo-/polymeric organic semiconductors.67,68 Importantly, the molecular packing of PTI04 remained intact, as evidenced by the side scattering feature observed at qz = 0.36 Å−1, which corresponds to the characteristic scattering of neat PTI04. This observation suggests that the incorporation of PM7-D3 and PTI04 into the blend film does not disrupt the formation of their respective molecular packing during the blending process. In contrast, in the case of PM7-D3:Y12 system, although it exhibits a preferred face-on orientation indicative of ordered structures, the scattering feature corresponding to neat Y12 is undetected. This disruption of molecular packing upon mixing is consistent with the results obtained from ss-NMR measurements to be discussed. While the increased volume fraction of MAs in D:A domains can potentially enhance Jsc by facilitating efficient charge generation, it can also lead to a higher probability of recombination events, thereby adversely affecting the FF. These findings align well with the observed photovoltaic properties of the PM7-D3:Y12 system, which exhibits high Jsc values but low FF. Unlike the Y12 system, the PM7-D3:DTY6 system exhibits completely different behavior. In this case, the scattering feature originating from the DTY6 crystal remains in the blend. This intense scattering can be attributed to the poor miscibility between the donor and acceptor components supported by the value of the Flory–Huggins parameter, which will be further discussed below. While an improved molecular order of the acceptor component can enhance electron transport, an excessive presence of acceptor crystallites can have a detrimental effect on charge transport since the acceptor crystallite islands may work as traps. These findings provide an explanation for the observed high FF but low Jsc of the PM7-D3:DTY6 system. When PM7-D3 is replaced with PM7-D5, no significant changes are observed in the 2D GIWAXS analysis of PM7-D5:PTI04.
To gain detailed information on the amorphous mixed phases, RSoXS measurements were conducted on all four studied systems.69,70 Details of long period estimation by RSoXS measurements and analyses are presented in Fig. 3d, and ESI† (Table S6). These results suggest that all four blend films exhibit multi-length scale morphology with larger domains with a long period of ∼120–220 nm and smaller domains with a long period of ∼15–30 nm. The PM7-D3:PTI04 system contains the highest volume fraction (69%) of the smaller domains with an average size of 26 nm. For the other three systems, the volume fractions and long periods of the smaller domains are: PM7-D3:Y12 (63%, 51 nm), PM7-D3:PTI04 (56%, 17 nm), and PM7-D5:PTI04 (26%, 29 nm). Previously, strong correlations between FF and scattering intensity/domain purity have been observed in both single-mode and multi-mode morphologies, particularly if such morphologies were for the same material system and processing was varied.71–73 Similarly, domain size could be often correlated to Jsc.74 Using morphological parameters to explain relative device performance is more difficult and complex if devices are compared using different materials systems, as device performance depends on many parameters and materials’ intrinsic properties such as charge generation, charge carrier mobility, and recombination.75 Reasonable rough correlations between morphology and performance are observed here. For example, the high Jsc of PM7-D3:PTI04 blends agrees with the large volume fraction of small domains. However, no clear self-consistent, i.e. monotonic trend in FF or Jsc with a range of morphological parameters can be observed across all four material systems. This indicates that other parameters such as intrinsic mobilities of the materials, energetic offsets at interfaces, differences in interfacial structure, or vertical gradients make a significant contribution to the relative device performance.
The BHJ films were also characterized using depth-profiled X-ray photoelectron spectroscopy (XPS) to investigate the impact of sidechain length and position on the donor and acceptor distribution and composition in the vertical direction of the active layers.76 The distribution of the PM7-series donors and Y-series MAs can be tracked by the chlorine (Cl) signal and fluorine (F) signal, respectively, considering the fact that F is absent in the donors and Cl is absent in the acceptors. As a result of the different active layer thicknesses, the etching time to reach the bottom of the active layer is slightly different among the blends. The ratio of D/(D + A) for each blend was calculated from the representative elemental ratio of the polymers and MAs (Fig. 4). As shown in Fig. 4a, an overall decrease in the D/(A + D) molar ratio is observed in the vertical phase separation for all the blend films, and different trends in the depth profile are observed for the BHJ blends with different acceptors. In particular, the PM7-D3:PTI04 blend tends to be more uniform in the vertical phase arrangement with a gradual reduction of the D/(A + D) ratio from 0.43 to 0.31 upon going from top to bottom. In the other three blends, a dramatic drop of the donor component occurs after reaching 70% etching, and a region of ∼10 nm is dominated by MA molecules at the bottom of the active layers. These are illustrated by two different schematic diagrams (Fig. 4b and c) showing the vertical gradation trends in the microstructure of the PM7-D3:PTI04 blend and the other three systems. A significant number of acceptors accumulated at the bottom of the BHJ layer can have a negative impact on the device performance of the PM7-D3:Y12, PM7-D3:DTY6, and PM7-D5:PTI04 systems. Undesirable surface-trap-assisted recombination can occur as a result of the enrichment in acceptors at the interface between the PEDOT:PSS layer and the active layer. Compared with these blends, a uniform distribution in the vertical phase of the PM7-D3:PTI04 film implies higher miscibility of PM7-D3:PTI04 blend with PEDOT:PSS layer, which can contribute to reducing the density of traps and suppressing the trap-assisted recombination of charge carriers in the OPV device, especially surface-trap-assisted recombination, and thus lead to a more efficient charge collection process.76 The miscibility between PM7-D3 and PTI04 was further investigated by the Flory–Huggins interaction parameter χF–H derived from the contact angle measurements (CAM) (ESI,† Table S7). The polar and dispersive surface tension of the measured films was derived based on the CAM obtained by using water and glycerol as specific solvents, which describes the solubility parameters of the studied donor and MAs. And χF–H was calculated by comparing the difference of solubility parameters between different donor and acceptor materials. Typically, the better the miscibility between two studied materials, the smaller the value of χF–H.77,78 Among all four studied D:A blends, PM7-D3:PTI04 shows the smallest χF–H value of 0.036 as compared to the other three blends (0.233 for PM7-D3:Y12, 2.089 for PM7-D3:DTY6, and 0.177 for PM7-D5:PTI04). The lower χ value found for PM7-D3:PTI04 further confirms the better miscibility of D:A in the active layer. These results are not only consistent with the higher D:A interfacial areas from the RSoXS measurement, but also with the effective mobility and the recombination current fitting analysis discussed in the device physics part, which further confirms the origin of the surface trap in the studied systems. A favorable vertical gradient of the active layer increases the probability of excitons reaching and dissociating at the polymer and MA interfaces, promoting higher effective mobility (μeff ∼ 10−4 cm2 V−1 s−1), lower trap density (both in the bulk and at the surface), and lower non-radiative recombination loss in the PM7-D3:PTI04 system compared with the others, which is consistent with the higher observed PCE.
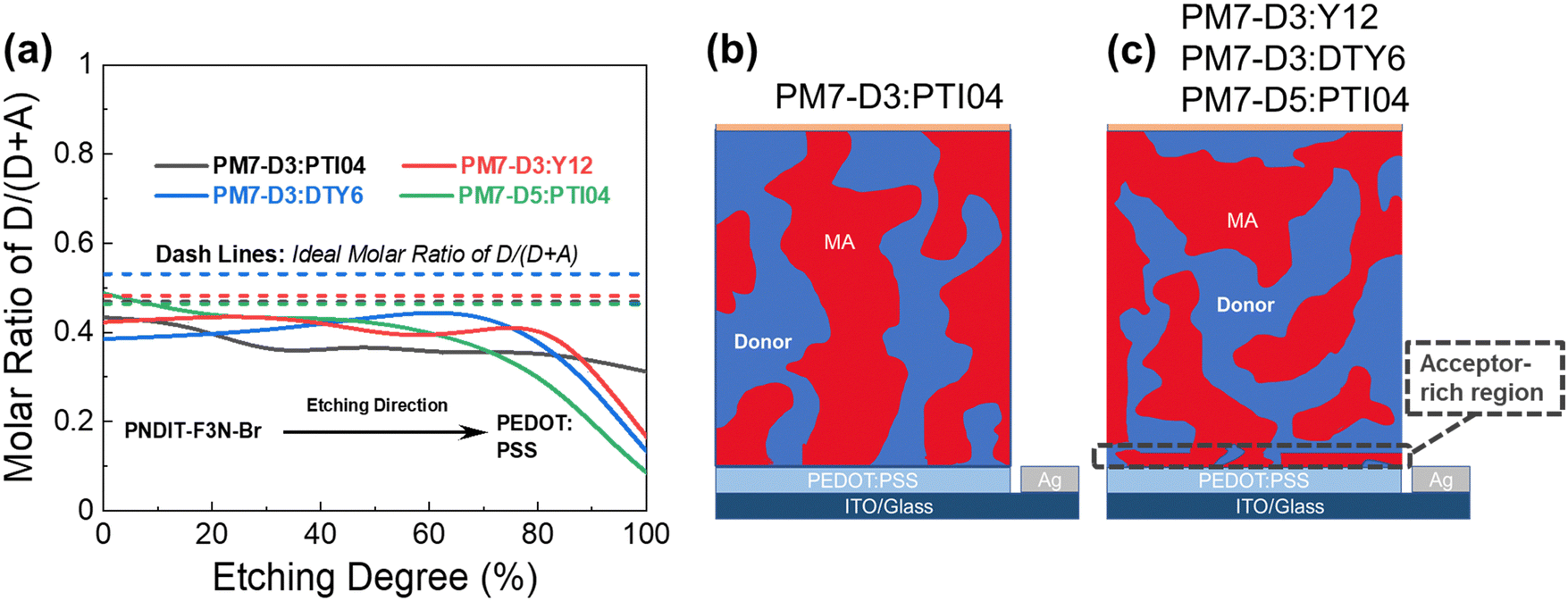 | ||
| Fig. 4 (a) XPS depth profile of PM7-D3:PTI04, PM7-D3:Y12, PM7-D3:DTY6, and PM7-D5:PTI04 BHJ films showing D/(A + D) molar ratio evolution as a function of etching degree, where etching begins at the top air/film interface. Dotted lines represent the ideal D/(D + A) molar ratio calculated from the original D:A ratio of 1:1.2 by weight. (b) Schematics of the PM7-D3:PTI04 blend with more uniform vertical phase separation and (c) of the other three blend films showing vertical phase gradation with an acceptor-rich region near the blend/PEDOT:PSS interface. | ||
Within the resolution capabilities of the above techniques, the analysis of long-range order at the surface, bulk, and interfacial BHJ morphology indicates that the acceptor domains exhibit different morphological features as observed by means of lamellar stacking peaks of blend films in GIWAXS plots and the XPS depth profile analysis. However, these data and observations do not resolve the molecular origins of the different acceptor morphology induced by the PTI04, DTY6, and Y12 molecules in the PM7-D3:MA BHJ blend films. In other words, changes in the bulk and interfacial morphology of Y12 and PTI04 molecules are expected to contribute to different performances in OPV devices. The local chemical environments of end groups and sidechains in acceptor morphology are known to be sensitive to solvent processing, which can be identified and distinguished by gaining access to atomic-level resolution enabled by ssNMR spectroscopy as discussed below.63
2.4. Local structures and intermolecular interactions
Molecular-level origins of the different local structures and packing interactions in neat materials and blends can be characterized by ssNMR spectroscopy.42,63,79–83 To resolve the different packing interactions in acceptor morphology that contribute to the different PCE values in OPV devices, we have selected the PM7-D3:PTI04 and PM7-D3:Y12 blends, owing to the differences in their PCE values (Table 1). For the neat PM7-D3, PTI04, and Y12 compounds and the BHJ blends, the short-range order and intermolecular interactions are examined by analyzing the 1H, 13C, and 19F chemical shifts (Fig. 5 and ESI,† Fig. S18, S19) and through-space dipole–dipole interactions between them. The local chemical environment of the 19F species in the 2-(5,6-difluoro-3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile (2FIC) terminal groups (henceforth referred to as end groups) in the PTI04 and Y12 molecules lead to different 19F chemical shifts;4,66 it is observed that PTI04 retains its local morphology in the BHJ blend but the Y12 molecules do not. The different 19F peaks are due to the different intermolecular interactions between the end groups and the sidechains and the fused ring core, as previously observed for Y-series molecules.63 Two-dimensional (2D) 19F–19F, 1H–13C and 1H–1H correlation NMR spectroscopy measurements further corroborate these results (Fig. 5 and Fig. S18; a detailed discussion of peak analysis is presented in ESI†). The local morphology of PTI04 is retained in the BHJ blend, likely due to the strong inter- and intramolecular interactions between the end-groups and sidechains of PTI04 (green color ovals in Fig. 5c, d, g and h). In contrast, subtle structural changes in the vicinity of end groups and sidechains are observed in Y12 molecules and the PM7-D3:Y12 BHJ blend leading to different chemical shifts and peak intensities, as highlighted by the red color ovals in Fig. 5e, f, i and j.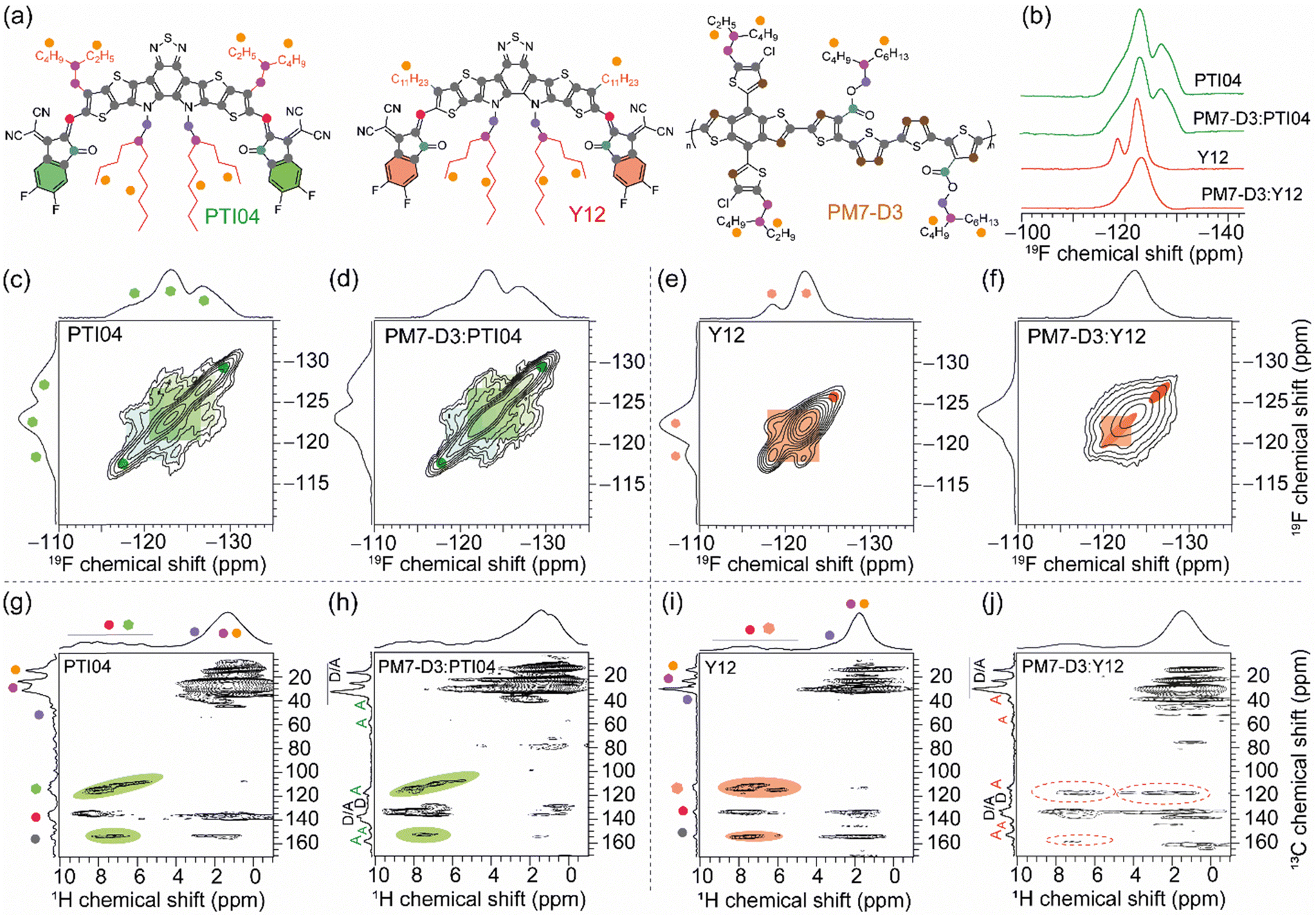 | ||
| Fig. 5 (a) Structures of PM7-D3, PTI04, and Y12 with color dots to guide the analysis of 2D NMR spectra. (b) Solid-state 1D 19F MAS NMR spectra of MAs and PM7-D3:MA blends. Solid-state 2D 19F–19F spin-diffusion NMR spectra of (c) neat PTI04 and (d) PM7-D3:PTI04 indicating the identical acceptor morphology, and (e) neat Y12 and (f) PM7-D3:Y12 blend films with different peak patterns highlighted in red. Solid-state 2D 1H–13C HETCOR NMR spectra of (g) neat PTI04 and (h) PM7-D3:PTI04 indicating the identical local acceptor morphology as depicted by green ovals, and (i) neat Y12 and (j) PM7-D3:Y12 BHJ blend films showing the difference in the local morphology as depicted in the red dashed ovals. | ||
Further insights into the local morphology and structural aspects of PM7-D3 moieties and MAs in neat compounds and in the BHJ blends are obtained by analyzing 2D 1H–1H double-quantum–single-quantum (DQ–SQ) spectra (ESI,† Fig. S20). The most notable aspect is that the changes in the inter- and intramolecular interactions among end-groups/sidechains in Y12 lead to the different 2D 1H–13C peaks. Therefore, local morphology changes in the Y12 molecules in the vicinity of the end groups as well as the branched sidechains (depicted in ovals), are expected to contribute to the performance deterioration in the PM7-D3:Y12-based OPVs. These observations are consistent with the AFM, GIWAXS, and XPS data that showed different surface and bulk morphologies with different lamellar packing distances and domain sizes of the D–A moieties in the 2-MeTHF processed BHJ blends, which impacts the charge generation, transport and extraction processes.
3. Conclusions
In summary, the green solvent processed PM7-D3:PTI04 device exhibits a PCE approaching 15%, comparable to the well-known PM6:Y6 system processed from traditional halogenated solvents and additives. In understanding the structure-processing-property relationships, the key learnings obtained from the device physics data and characterization at different length scales ranging from microns to sub-nanometer distances can be summarized as follows:1. The enhanced solubility of PM7-D3 polymer in 2-MeTHF due to its more flexible backbone structure leads to a better mixing and more optimal phase separation in the PM7-D3:PTI04 BHJ morphology. This results in enhanced J–V characteristics and FF and reduced bulk and surface trap-assisted recombinations. These factors combine to produce a high PCE value of 14.9% in the OPV devices. In contrast, the less favorable BHJ morphology in the other blends with MAs such as DTY6 and Y12, as well as PM7-D5:MA blends, leads to higher trap-assisted recombination dynamics and reduced performance in the OPV devices.
2. The surface and bulk BHJ morphologies consisting of favorable D/A separation and domain purity are paramount for the high efficiency achieved in the PM7-D3:PTI04 based OPV devices processed from 2-MeTHF solvent, as revealed by the AFM, 2D GIWAXS, and RSoXS data. The high-performance PTI04-based systems exhibit domain sizes of ∼26 nm vs. much larger domains for the other MAs in the BHJ blends.
3. At a few tens of nm scale, the long-range order associated with π–π stacked and lamellae stacked D and A moieties in the out-of-the-plane direction with respect to the substrates is expected to be beneficial for charge transport in all devices, although significant disruption in such interactions in PM7-D3:Y12 leads to its poorer efficiency.
4. The compositional and spatial distributions of the D and A domains in the vertical direction are important for charge generation and extraction. The PM7-D3:PTI04 systems exhibit favorable morphology while the PM7-D3:Y12 BHJ blend shows Y12 molecules phase-separated into larger domains at the photoactive/PEDOT:PSS interfaces, leading to higher charge recombination and therefore low FF.
5. Examining the local packing interactions at sub-nanometer distances, the backbone/sidechain interactions stabilize the donor and MAs morphology. The length of the branched sidechains attached to the core in PTI04 is particularly important in maintaining the self-assembly and phase separation in the BHJ morphology, as revealed by ssNMR.63
6. Overall, the molecular design associated with the donor polymers and MAs in this study including the core and sidechain engineering has a significant impact on the solubility, processability, morphology, and charge carrier properties of the 2-MeTHF processed OPV devices. A preferred average domain size of ∼25 nm with relatively high domain purity and more uniform distribution of D and A inter-mixing is observed in the PM7-D3:PTI04 BHJ film compared to the other D:A blends, which facilitates the charge generation and collection processes while limiting the trap-assisted recombination process in the device, leading to high effective mobility and remarkable performance.
Thus, this work provides insight into the structure-morphology-property relationships essential for developing environmentally friendly and commercially viable high-performance OPVs.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Z. D. and Z. Z. acknowledge funding from the Air Force Office of Scientific Research (AFOSR) Grant #FA9550-19-1-0348. D. C., S. C. and T.-Q. N. thank the Office of Naval Research (ONR) Grant #N00014-21-1-2181 through the Multidisciplinary University Research Initiative program. S. S., A. L. J., and J. R. R. acknowledge the Office of Naval Research (ONR) Grant #N00014-21-1-2144 through the Multidisciplinary University Research Initiative program. P. T., J. Z. and S. R. M. acknowledge the Department of the Navy, Office of Naval Research Multidisciplinary University Research Initiative Award #N00014-20-1-2167, and #N00014-21-1-2180. P. T. acknowledges the support from Vidyasirimedhi Institute of Science and Technology (VISTEC). Z. P. and H. A. acknowledge the support from the U.S. Office of Naval Research Grant #N00014-20-1-2155. RSoXS data were acquired at beamlines 11.0.1.2 at the Advanced Light Source, LBNL, which is supported by the Director of the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. C. W. and G. N. M. R. gratefully acknowledge the financial support from University of Lille, EU H2020 (grant no. 795091) and INFRANALYTICS FR 2040 for carrying out experiments at high field NMR facility at Lille, France. The work at the University of Arizona has been supported by the Office of Naval Research under Awards #N00014-21-1-2182 through the Multidisciplinary University Research Initiative program and #N00014-20-1-2110. The authors gratefully thank Dr Joachim Vollbrecht, Dr Tom Mates, and Dr Oleksandr Polonskyi for useful discussions.References
- A. J. Heeger, Adv. Mater., 2014, 26, 10–28 CrossRef CAS PubMed.
- J. Hou, O. Inganäs, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS PubMed.
- S. Albrecht, J. R. Tumbleston, S. Janietz, I. Dumsch, S. Allard, U. Scherf, H. Ade and D. Neher, J. Phys. Chem. Lett., 2014, 5, 1131–1138 CrossRef CAS PubMed.
- A. Karki, J. Vollbrecht, A. J. Gillett, S. S. Xiao, Y. L. Yang, Z. X. Peng, N. Schopp, A. L. Dixon, S. Yoon, M. Schrock, H. Ade, G. N. M. Reddy, R. H. Friend and T. Q. Nguyen, Energy Environ. Sci., 2020, 13, 3679–3692 RSC.
- Y. Cui, Y. Xu, H. Yao, P. Bi, L. Hong, J. Zhang, Y. Zu, T. Zhang, J. Qin, J. Ren, Z. Chen, C. He, X. Hao, Z. Wei and J. Hou, Adv. Mater., 2021, 33, e2102420 CrossRef PubMed.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C. C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- S. Zhang, L. Ye, H. Zhang and J. Hou, Mater. Today, 2016, 19, 533–543 CrossRef CAS.
- D. Corzo, D. Rosas-Villalva, G. Tostado-Blázquez, E. B. Alexandre, L. H. Hernandez, J. Han, H. Xu, M. Babics, S. De Wolf and D. Baran, Nat. Energy, 2023, 8, 62–73 CrossRef.
- M. Ghasemi, N. Balar, Z. Peng, H. Hu, Y. Qin, T. Kim, J. J. Rech, M. Bidwell, W. Mask, I. McCulloch, W. You, A. Amassian, C. Risko, B. T. O’Connor and H. Ade, Nat. Mater., 2021, 20, 525–532 CrossRef CAS PubMed.
- I. Burgués-Ceballos, F. Machui, J. Min, T. Ameri, M. M. Voigt, Y. N. Luponosov, S. A. Ponomarenko, P. D. Lacharmoise, M. Campoy-Quiles and C. J. Brabec, Adv. Funct. Mater., 2014, 24, 1449–1457 CrossRef.
- D. T. Duong, B. Walker, J. Lin, C. Kim, J. Love, B. Purushothaman, J. E. Anthony and T. Q. Nguyen, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1405–1413 CrossRef CAS.
- B. Walker, A. Tamayo, D. T. Duong, X. D. Dang, C. Kim, J. Granstrom and T. Q. Nguyen, Adv. Energy Mater., 2011, 1, 221–229 CrossRef CAS.
- Q. He, W. Sheng, M. Zhang, G. Xu, P. Zhu, H. Zhang, Z. Yao, F. Gao, F. Liu, X. Liao and Y. Chen, Adv. Energy Mater., 2021, 11, 2003390 CrossRef CAS.
- H. Zhang, Y. Li, X. Zhang, Y. Zhang and H. Zhou, Mater. Chem. Front., 2020, 4, 2863–2880 RSC.
- K. S. Wienhold, V. Körstgens, S. Grott, X. Jiang, M. Schwartzkopf, S. V. Roth and P. Müller-Buschbaum, ACS Appl. Mater. Interfaces, 2019, 11, 42313–42321 CrossRef CAS PubMed.
- Z. Zheng, E. He, J. Wang, Z. Qin, T. Niu, F. Guo, S. Gao, Z. Ma, L. Zhao, X. Lu, Q. Xue, Y. Cao, G. T. Mola and Y. Zhang, J. Mater. Chem. A, 2021, 9, 26105–26112 RSC.
- J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497–500 CrossRef CAS PubMed.
- C. V. Hoven, X. D. Dang, R. C. Coffin, J. Peet, T. Q. Nguyen and G. C. Bazan, Adv. Mater., 2010, 22, E63–E66 CrossRef CAS PubMed.
- J. T. Rogers, K. Schmidt, M. F. Toney, G. C. Bazan and E. J. Kramer, J. Am. Chem. Soc., 2012, 134, 2884–2887 CrossRef CAS PubMed.
- L. A. Perez, K. W. Chou, J. A. Love, T. S. Van Der Poll, D. M. Smilgies, T. Q. Nguyen, E. J. Kramer, A. Amassian and G. C. Bazan, Adv. Mater., 2013, 25, 6380–6384 CrossRef CAS PubMed.
- C. McDowell, M. Abdelsamie, M. F. Toney and G. C. Bazan, Adv. Mater., 2018, 30, 1707114 CrossRef PubMed.
- H. Gaspar, G. Bernardo and A. Mendes, Nanoenergy Adv., 2021, 2, 1–28 CrossRef.
- H. Yu, S. Luo, R. Sun, I. Angunawela, Z. Qi, Z. Peng, W. Zhou, H. Han, R. Wei, M. Pan, A. M. H. Cheung, D. Zhao, J. Zhang, H. Ade, J. Min and H. Yan, Adv. Funct. Mater., 2021, 31, 2100791 CrossRef CAS.
- Z. Du, M. Mainville, J. Vollbrecht, A. L. Dixon, N. Schopp, M. Schrock, Z. Peng, J. Huang, S. Chae, H. Ade, M. Leclerc, G. N. M. Reddy and T.-Q. Nguyen, Solar RRL, 2021, 5, 2100213 CrossRef CAS.
- S. Dong, T. Jia, K. Zhang, J. Jing and F. Huang, Joule, 2020, 4, 2004–2016 CrossRef CAS.
- D. Wang, G. Zhou, Y. Li, K. Yan, L. Zhan, H. Zhu, X. Lu, H. Chen and C.-Z. Li, Adv. Funct. Mater., 2022, 32, 2107827 CrossRef CAS.
- S. Zhang, H. Chen, P. Wang, S. Li, Z. Li, Y. Huang, J. Liu, Z. Yao, C. Li, X. Wan and Y. Chen, Solar RRL, 2023, 7, 2300029 CrossRef CAS.
- Z. Chen, L. Yan, J. J. Rech, J. Hu, Q. Zhang and W. You, ACS Appl. Polym. Mater., 2019, 1, 804–814 CrossRef CAS.
- H. Huang, X. J. Li, C. K. Sun, I. Angunawela, B. B. Qiu, J. Q. Du, S. C. Qin, L. Meng, Z. J. Zhang, H. Ade and Y. F. Li, J. Mater. Chem. C, 2020, 8, 7718–7724 RSC.
- V. Pace, P. Hoyos, L. Castoldi, P. Domínguez de María and A. R. Alcántara, ChemSusChem, 2012, 5, 1369–1379 CrossRef CAS PubMed.
- O. Al Musaimi, Y. E. Jad, A. Kumar, A. El-Faham, J. M. Collins, A. Basso, B. G. de la Torre and F. Albericio, Org. Process Res. Dev., 2018, 22, 1809–1816 CrossRef CAS.
- L. Hong, H. Yao, Z. Wu, Y. Cui, T. Zhang, Y. Xu, R. Yu, Q. Liao, B. Gao and K. Xian, Adv. Mater., 2019, 31, 1903441 CrossRef PubMed.
- Z. Li, L. Ying, P. Zhu, W. Zhong, N. Li, F. Liu, F. Huang and Y. Cao, Energy Environ. Sci., 2019, 12, 157–163 RSC.
- L. Bucher, L. Tanguy, N. Desbois, P. L. Karsenti, P. D. Harvey, C. P. Gros and G. D. Sharma, Solar RRL, 2018, 2, 1700168 CrossRef.
- J. Panidi, E. Mazzolini, F. Eisner, Y. Fu, F. Furlan, Z. R. Qiao, M. Rimmele, Z. Li, X. H. Lu, J. Nelson, J. R. Durrant, M. Heeney and N. Gasparini, ACS Energy Lett., 2023, 8, 3038–3047 CrossRef CAS PubMed.
- X. Chen, X. Liu, M. A. Burgers, Y. Huang and G. C. Bazan, Angew. Chem., Int. Ed., 2014, 53, 14378–14381 CrossRef CAS PubMed.
- S. V. Dayneko, A. D. Hendsbee and G. C. Welch, Chem. Commun., 2017, 53, 1164–1167 RSC.
- C. Liao, M. Zhang, X. Xu, F. Liu, Y. Li and Q. Peng, J. Mater. Chem. A, 2019, 7, 716–726 RSC.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- I. Pelse, A. L. Jones, L. J. Richter and J. R. Reynolds, Chem. Mater., 2021, 33, 657–667 CrossRef CAS.
- F. C. Spano and C. Silva, Annu. Rev. Phys. Chem., 2014, 65, 477–500 CrossRef CAS PubMed.
- N. Schopp, S. Sabury, T. Chaney, J. Zhang, H. Wakidi, B. M. Kim, R. Sankar, H. M. Luong, P. Therdkatanyuphong, V. V. Brus, S. Marder, M. F. Toney, J. R. Reynolds and T.-Q. Nguyen, ACS Energy Lett., 2023, 8(8), 3307–3313 CrossRef CAS.
- H.-C. Liao, C.-C. Ho, C.-Y. Chang, M.-H. Jao, S. B. Darling and W.-F. Su, Mater. Today, 2013, 16, 326–336 CrossRef CAS.
- N. Gasparini, A. Gregori, M. Salvador, M. Biele, A. Wadsworth, S. Tedde, D. Baran, I. McCulloch and C. J. Brabec, Adv. Mater. Technol., 2018, 3, 1800104 CrossRef.
- J. Vollbrecht, V. V. Brus, S. J. Ko, J. Lee, A. Karki, D. X. Cao, K. Cho, G. C. Bazan and T. Q. Nguyen, Adv. Energy Mater., 2019, 9, 1901438 CrossRef.
- V. V. Brus, C. M. Proctor, N. A. Ran and T. Q. Nguyen, Adv. Energy Mater., 2016, 6, 1502250 CrossRef.
- J. Vollbrecht and V. V. Brus, Adv. Electron. Mater., 2020, 6, 2000517 CrossRef CAS.
- H. M. Luong, S. Chae, A. Yi, K. Ding, J. Huang, B. M. Kim, C. Welton, J. Chen, H. Wakidi, Z. Du, H. J. Kim, H. Ade, G. N. M. Reddy and T.-Q. Nguyen, ACS Energy Lett., 2023, 8, 2130–2140 CrossRef CAS.
- L.-W. Feng, J. Chen, S. Mukherjee, V. K. Sangwan, W. Huang, Y. Chen, D. Zheng, J. W. Strzalka, G. Wang, M. C. Hersam, D. DeLongchamp, A. Facchetti and T. J. Marks, ACS Energy Lett., 2020, 5, 1780–1787 CrossRef CAS.
- I. Zonno, H. Zayani, M. Grzeslo, B. Krogmeier and T. Kirchartz, Phys. Rev. Appl., 2019, 11, 054024 CrossRef CAS.
- G. Garcia-Belmonte, P. P. Boix, J. Bisquert, M. Sessolo and H. J. Bolink, Sol. Energy Mater. Sol. Cells, 2010, 94, 366–375 CrossRef CAS.
- N. Schopp, H. M. Luong, B. R. Luginbuhl, P. Panoy, D. Choi, V. Promarak, V. V. Brus and T.-Q. Nguyen, ACS Energy Lett., 2022, 7, 1626–1634 CrossRef CAS.
- C. M. Proctor, S. Albrecht, M. Kuik, D. Neher and T. Q. Nguyen, Adv. Energy Mater., 2014, 4, 1400230 CrossRef.
- M. C. Heiber, T. Okubo, S. J. Ko, B. R. Luginbuhl, N. A. Ran, M. Wang, H. B. Wang, M. A. Uddin, H. Y. Woo, G. C. Bazan and T. Q. Nguyen, Energy Environ. Sci., 2018, 11, 3019–3032 RSC.
- N. A. Ran, J. A. Love, M. C. Heiber, X. C. Jiao, M. P. Hughes, A. Karki, M. Wang, V. V. Brus, H. Wang, D. Neher, H. Ade, G. C. Bazan and T. Q. Nguyen, Adv. Energy Mater., 2018, 8, 1701073 CrossRef.
- T. M. Burke, S. Sweetnam, K. Vandewal and M. D. McGehee, Adv. Energy Mater., 2015, 5, 1500123 CrossRef.
- D. Bartesaghi, I. D. C. Pérez, J. Kniepert, S. Roland, M. Turbiez, D. Neher and L. J. A. Koster, Nat. Commun., 2015, 6, 7083 CrossRef CAS PubMed.
- N. Tokmoldin, J. Vollbrecht, S. M. Hosseini, B. Sun, L. Perdigón-Toro, H. Y. Woo, Y. Zou, D. Neher and S. Shoaee, Adv. Energy Mater., 2021, 11, 2100804 CrossRef CAS.
- H. Cha, S. Wheeler, S. Holliday, S. D. Dimitrov, A. Wadsworth, H. H. Lee, D. Baran, I. McCulloch and J. R. Durrant, Adv. Funct. Mater., 2018, 28, 1704389 CrossRef.
- A. Karki, J. Vollbrecht, A. L. Dixon, N. Schopp, M. Schrock, G. N. M. Reddy and T.-Q. Nguyen, Adv. Mater., 2019, 31, 1903868 CrossRef CAS PubMed.
- G. Zhang, X.-K. Chen, J. Xiao, P. C. Y. Chow, M. Ren, G. Kupgan, X. Jiao, C. C. S. Chan, X. Du, R. Xia, Z. Chen, J. Yuan, Y. Zhang, S. Zhang, Y. Liu, Y. Zou, H. Yan, K. S. Wong, V. Coropceanu, N. Li, C. J. Brabec, J.-L. Bredas, H.-L. Yip and Y. Cao, Nat. Commun., 2020, 11, 3943 CrossRef CAS PubMed.
- A. L. Jones, C. H. Y. Ho, S. A. Schneider, J. Zhang, Y. Pei, J. Wang, X. Zhan, S. R. Marder, M. F. Toney, F. So, G. N. M. Reddy and J. R. Reynolds, Chem. Mater., 2022, 34(15), 6853–6867 CrossRef CAS.
- B. R. Luginbuhl, P. Raval, T. Pawlak, Z. Du, T. Wang, G. Kupgan, N. Schopp, S. Chae, S. Yoon, A. Yi, H. Jung Kim, V. Coropceanu, J. L. Bredas, T. Q. Nguyen and G. N. M. Reddy, Adv. Mater., 2022, 34, e2105943 CrossRef PubMed.
- J. Xin, H. Zhao, J. Xue, S. Seibt, B. A. Collins and W. Ma, Solar RRL, 2022, 6, 2200819 CrossRef CAS.
- L. Zhu, M. Zhang, G. Zhou, T. Hao, J. Xu, J. Wang, C. Qiu, N. Prine, J. Ali, W. Feng, X. Gu, Z. Ma, Z. Tang, H. Zhu, L. Ying, Y. Zhang and F. Liu, Adv. Energy Mater., 2020, 10, 1904234 CrossRef CAS.
- A. Karki, J. Vollbrecht, A. L. Dixon, N. Schopp, M. Schrock, G. M. Reddy and T. Q. Nguyen, Adv. Mater., 2019, 31, 1903868 CrossRef CAS PubMed.
- O. V. Mikhnenko, P. W. Blom and T.-Q. Nguyen, Energy Environ. Sci., 2015, 8, 1867–1888 RSC.
- M. T. Sajjad, A. Ruseckas and I. D. Samuel, Matter, 2020, 3, 341–354 CrossRef.
- L. Ye, Y. Xiong, S. Li, M. Ghasemi, N. Balar, J. Turner, A. Gadisa, J. Hou, B. T. O'Connor and H. Ade, Adv. Funct. Mater., 2017, 27, 1702016 CrossRef.
- E. Gann, A. T. Young, B. A. Collins, H. Yan, J. Nasiatka, H. A. Padmore, H. Ade, A. Hexemer and C. Wang, Rev. Sci. Instrum., 2012, 83, 045110 CrossRef CAS PubMed.
- L. Ye, H. W. Hu, M. Ghasemi, T. H. Wang, B. A. Collins, J. H. Kim, K. Jiang, J. H. Carpenter, H. Li, Z. K. Li, T. McAfee, J. B. Zhao, X. K. Chen, J. L. Y. Lai, T. X. Ma, J. L. Bredas, H. Yan and H. Ade, Nat. Mater., 2018, 17, 253–260 CrossRef CAS PubMed.
- S. Mukherjee, C. M. Proctor, J. R. Tumbleston, G. C. Bazan, T. Q. Nguyen and H. Ade, Adv. Mater., 2015, 27, 1105–1111 CrossRef CAS PubMed.
- L. Ye, S. Li, X. Liu, S. Zhang, M. Ghasemi, Y. Xiong, J. Hou and H. Ade, Joule, 2019, 3, 443–458 CrossRef CAS.
- B. A. Collins, Z. Li, J. R. Tumbleston, E. Gann, C. R. McNeill and H. Ade, Adv. Energy Mater., 2013, 3, 65–74 CrossRef CAS.
- O. M. Awartani, B. Gautam, W. Zhao, R. Younts, J. Hou, K. Gundogdu and H. Ade, J. Mater. Chem. A, 2018, 6, 12484–12492 RSC.
- X. G. Guo, N. J. Zhou, S. J. Lou, J. Smith, D. B. Tice, J. W. Hennek, R. P. Ortiz, J. T. L. Navarrete, S. Y. Li, J. Strzalka, L. X. Chen, R. P. H. Chang, A. Facchetti and T. J. Marks, Nat. Photonics, 2013, 7, 825–833 CrossRef CAS.
- M. Tambasco, J. Lipson and J. S. Higgins, Macromolecules, 2006, 39, 4860–4868 CrossRef CAS.
- L. Zhang, X. Zhu, D. Deng, Z. Wang, Z. Zhang, Y. Li, J. Zhang, K. Lv, L. Liu, X. Zhang, H. Zhou, H. Ade and Z. Wei, Adv. Mater., 2022, 34, e2106316 CrossRef PubMed.
- F. Etzold, I. A. Howard, N. Forler, D. M. Cho, M. Meister, H. Mangold, J. Shu, M. R. Hansen, K. Müllen and F. Laquai, J. Am. Chem. Soc., 2012, 134, 10569–10583 CrossRef CAS PubMed.
- N. C. Miller, E. Cho, M. J. Junk, R. Gysel, C. Risko, D. Kim, S. Sweetnam, C. E. Miller, L. J. Richter, R. J. Kline, M. Heeney, I. McCulloch, A. Amassian, D. Acevedo-Feliz, C. Knox, M. R. Hansen, D. Dudenko, B. F. Chmelka, M. F. Toney, J. L. Bredas and M. D. McGehee, Adv. Mater., 2012, 24, 6071–6079 CrossRef CAS PubMed.
- M. Seifrid, G. N. M. Reddy, B. F. Chmelka and G. C. Bazan, Nat. Rev. Mater., 2020, 5, 910–930 CrossRef CAS.
- A. L. Jones, C. H. Y. Ho, S. A. Schneider, J. Zhang, Y. Pei, J. Wang, X. Zhan, S. R. Marder, M. F. Toney, F. So, G. N. M. Reddy and J. R. Reynolds, Chem. Mater., 2022, 34, 6853–6867 CrossRef CAS.
- B. R. Luginbuhl, S.-J. Ko, N. A. Ran, H. Hu, S. M. Becwar, A. Karki, M. Seifrid, T. Okubo, M. Wang, H. W. Ade, B. F. Chmelka, G. C. Bazan, G. N. Manjunatha Reddy and T.-Q. Nguyen, Solar RRL, 2022, 6, 2200135 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh01133j |
| This journal is © The Royal Society of Chemistry 2023 |