
Efficient and strategical installations of quaternary ammonium groups in metal–organic frameworks for hydroxide conductivity†
Ho Jeong
Choi‡
a,
Seungpyo
Hong‡
a,
Younghu
Son‡
 b,
Ki Tae
Kim
a,
Cheoljae
Kim
a,
Minyoung
Yoon
*b and
Min
Kim
*a
b,
Ki Tae
Kim
a,
Cheoljae
Kim
a,
Minyoung
Yoon
*b and
Min
Kim
*a
aDepartment of Chemistry, Chungbuk National University, Cheongju, 28644 Republic of Korea. E-mail: minkim@chungbuk.ac.kr
bDepartment of Chemistry and Green-Nano Materials Research Center, Kyungpook National University, Daegu, 41566 Republic of Korea. E-mail: myyoon@knu.ac.kr
First published on 6th January 2023
Abstract
Positively charged aromatic quaternary ammonium and aliphatic quaternary ammonium groups were successfully installed in metal–organic frameworks (MOFs) for hydroxide conductivity studies. Two representative solid-state MOF functionalization strategies, post-synthetic covalent modification (PSM) and post-synthetic ligand exchange (PSE), were used and their charge-loading efficiencies were compared. PSE processes were successfully performed for both aromatic and aliphatic quaternary ammonium groups, and the PSM technique was restricted to the aromatic ammonium functionalization of MOFs. Although all ammonium installations induced significantly enhanced hydroxide conductivities compared to the bare MOF, the aliphatic ammonium group via the triazole linker from the CuAAC click chemistry showed the best performance. This study discusses the practicality of PSE for MOF functionalization and the additional effects of functional group modification.
Design, System, ApplicationThree-dimensional and porous metal–organic frameworks (MOFs) possess not only great structural diversities from various combinations of metal–ligand coordination, but also wide chemical tunability in organic ligands and metal nodes. A variety of applications such as molecular storage, separation, catalysis, and energy chemistry has been targeted, especially with functionalized MOFs. The chemical tags in MOFs could be installed into the pore of MOFs through pre-functionalization and post-synthetic methods. The post-synthetic strategies have successfully expanded the range of functionalizations in MOF. Herein, strategical approaches to install positively-charged ammonium groups in MOFs have been investigated. Two representative post-synthetic methods, post-synthetic modification and post-synthetic exchange, were directly compared with aliphatic and aromatic systems. This study summarizes the systemized functionalization of MOFs with target functionalities for specific applications (hydroxide conductivity in this case). |
Introduction
Metal–organic frameworks (MOFs) are porous organic/inorganic hybrid materials based on the coordination bonds between metal clusters and organic ligands.1,2 The diversity of metal nodes and coordinative organic ligands allows a variety of strategic and logical combinations for MOFs, which are crucial for achieving target performance. Particularly, organic functional groups in MOFs pores depend on their chemical and physical properties. The representative applications of MOFs, such as molecular separation, molecular storage, molecular delivery, and catalysis, are highly dependent on the chemical tags in the organic ligands of MOFs.3–9 Additionally, physical and chemical stabilities, such as water stability, are correlated with the hydrophobicity of MOFs owing to ligand functionalization. In the last two decades, various functional groups have been investigated for MOF functionalizations.10–14Pre-functionalization and post-synthetic covalent modification (PSM) have been studied to integrate organic functionalities into MOFs. Target functional groups can be installed in the organic ligand before MOF formation (i.e., pre-functionalization) or the desired organic group can be incorporated into the MOF after MOF formation using solid-state PSM. A variety of functional groups and simple chemical transformations in MOFs have been investigated.15,16 Recently, post-synthetic ligand exchange (PSE) processes have been widely studied as functional group installation strategies.17–19 Non-functionalized MOFs (i.e., pre-synthesized) were incubated with functionalized ligand solutions and solid-solution exchanges allowed the functionalized MOFs to be washed. The PSE method enables the installation of thermally unstable or coordinative functional groups.20,21
Among the target functionalities in MOFs, charged functional groups are important for selective molecular separation or storage because they facilitate strong interactions between the frameworks and guest molecules.22–24 Owing to their coordinative nature, a few MOFs can exhibit permanent charges on their cluster of frameworks (e.g., from the combination of positive metal clusters and neutral organic ligands) and charged organic ligands, such as imidazolinium (for positive charge) and sulfonate (for negative charge), have been installed in the ligands of MOFs. However, the derivatization of charged MOFs are restricted for each case.22 Therefore, the generalized charge installations on neutral MOFs are necessary for target applications (Scheme 1).
 | ||
| Scheme 1 Strategical installations of aromatic and aliphatic quaternary ammonium groups into MOFs via PSM and PSE processes. | ||
Results and discussion
Aromatic, quaternary ammonium groups in MOFs (strategies I and II)
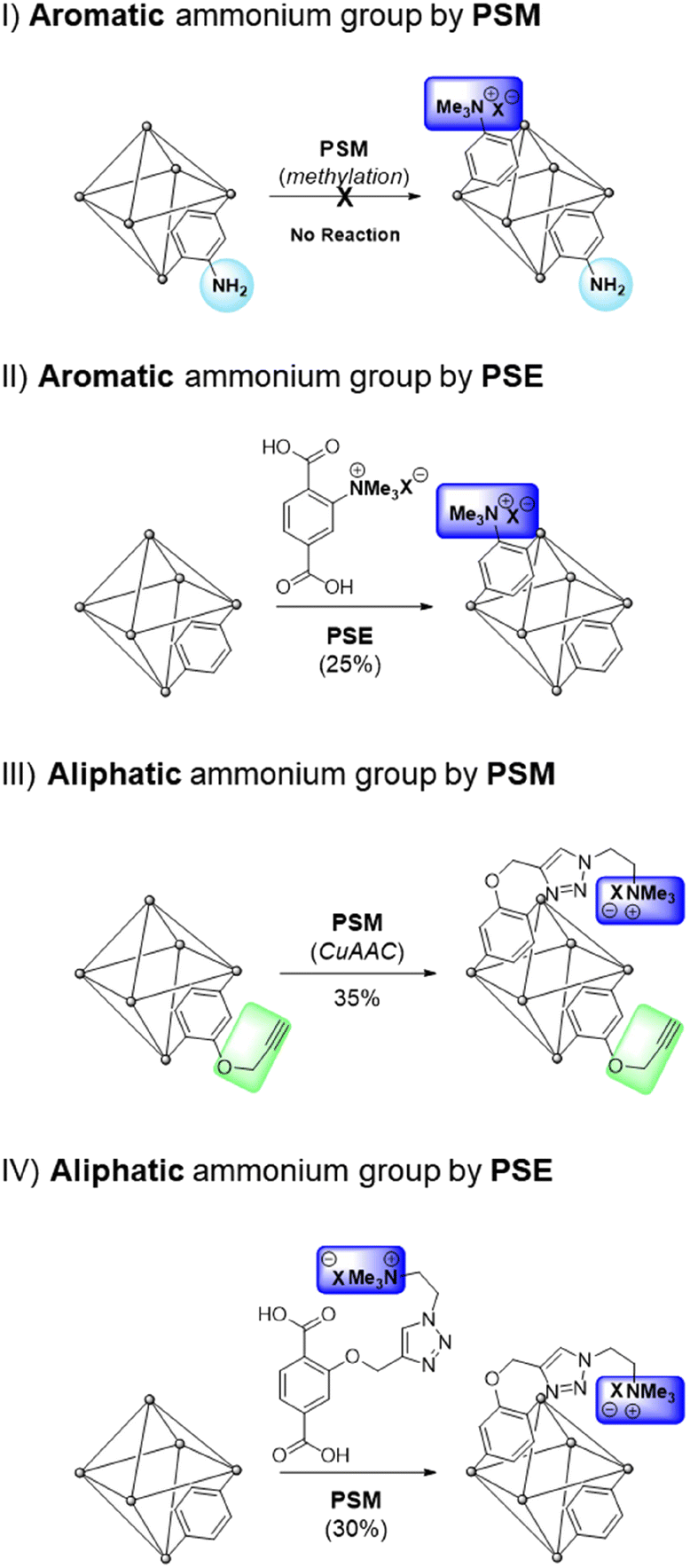
The practical application of MOFs for ion-selective systems requires chemically and physically stable MOFs. Therefore, zirconium-based UiO-66 (UiO = University of Oslo) is suitable for the proposed model system.25 The strong oxophilic character of zirconium contributes to the great stability of UiO-66s, including water and acidic solutions. Benzene-1,4-dicarboxylic acid (BDC or terephthalic acid) is the most common organic ligand used in MOF studies. Therefore, the derivatization and various applications of Zr and BDC-based MOFs have been extensively investigated.26–28The direct alkylation of aniline functionalities in UiO-66-NH2 was performed using PSM for the aromatic ammonium group (strategy I in Scheme 1). Although ammonium formations without alkyl groups like Ar-NH3+X− have been reported with Zr-based MOFs for separation and catalysis,29,30 alkylated ammoniums in MOF pores are relatively rare in the literature. Previous methylation of the aniline group in MOFs was reported by Wang's group with Zn-based IRMOF-3 and DMOF (Dabco MOF).31,32 The dialkylation converted the Ar-NH2 group in MOFs to Ar-NMe2, and additional methylation of the ammonium group (Ar-NMe3+X−) was performed using a one-pot system for IRMOF-3. For DMOFs, a tertiary amine MOF, DMOF-1-NMe2, was synthesized and converted to a quaternary ammonium group. However, both IRMOF-3 and DMOF are unstable under aqueous conditions and moisture; therefore, the practical applications of these MOFs are limited. With these references, direct methylation of the pre-synthesized UiO-66-NH2 was performed using PSM. Powder X-ray diffraction (PXRD) pattern comparison confirmed that the structure of the framework was maintained after methylation (Fig. 1). However, direct methylation of UiO-66-NH2 failed to form UiO-66-NMe3+X− with methyl triflate. Extensive reaction screenings, including elevating the PSM temperature and excess treatment of the methylation reagent, did not efficiently form UiO-66-NMe3+X− in a PSM manner (1H NMR after acid digestion, Fig. S1†). Other methylating reagents, such as iodomethane and dimethyl sulfate, are ineffective for ammonium group installation. The preparation of UiO-66-NMe2 and additional methylation to UiO-66-NMe3+X− (one-step methylation from the tertiary amine to quaternary ammonium) was not performed because the C–N bond cleavage on the zirconium-based UiO-66 was reported under solvothermal conditions.33
 | ||
| Fig. 1 Installation of the aromatic ammonium group in UiO-66 through (a) PSM and (b) PSE along with their PXRD patterns. | ||
Furthermore, the installation of a pre-synthesized aromatic ammonium group-containing ligand BDC-NMe3+X− (A) installation was performed using the ligand exchange process, i.e., PSE for the aromatic ammonium group (strategy II in Scheme 1). The target aromatic ammonium group-containing BDC was first synthesized via a three-step methylation (Scheme S1†). The first methylation step was performed using dimethyl sulfate, which is a strong methylating reagent. The second methylation occurred with iodomethane under basic NaH conditions for deprotonation of the secondary amine. The last methylation was carried out using methyl triflate. Three-step methylation was achieved with the methyl diester form of BDC (BDCE, benzene-1,4-dicarboxylate methyl ester), and the final hydrolysis produced the desired aromatic ammonium-functionalized A (Scheme S1†).
The desired positively charged ligand A was dissolved in an aqueous KOH solution that was neutralized to pH 7 with 1 M HCl. After adding the non-functionalized parent UiO-66 to the ligand solution, the mixture was incubated for 24 h at room temperature for PSE. The crystallinity of UiO-66 was completely retained after PSE, as indicated by the PXRD results (Fig. 1 and S2†). The size and morphology of UiO-66-NMe3+X− was checked by scanning electron microscopy (SEM) images (Fig. S3†). The incorporation ratio was determined using 1H NMR spectroscopy after acid digestion. Consequently, 25% of the positively charged aromatic ammonium group-functionalized A was incorporated into UiO-66-NMe3+X− (Fig. S4†). The porosity of the positively charged UiO-66 was confirmed using N2 adsorption at 77 K and compared with that before the PSE (Fig. S5 and Table S1†). The pore size distribution (PSD) also supported the incorporation of charged ligands into the pores (Fig. S5†). Finally, the thermal stability of the desired MOF was confirmed using thermogravimetric analysis (TGA, Fig. S6†).
Generally, PSM or the direct methylation of the aniline group is not an efficient method for installing aromatic ammonium groups into Zr-based MOFs. However, PSE or the exchange process provides the positively charged Zr-based UiO-66 MOFs with 25% of the ligand ratio. Furthermore, PSE can be performed at room temperature and applied to various BDC-based MOFs.
Aliphatic, quaternary ammonium groups in MOFs (strategies III and IV)
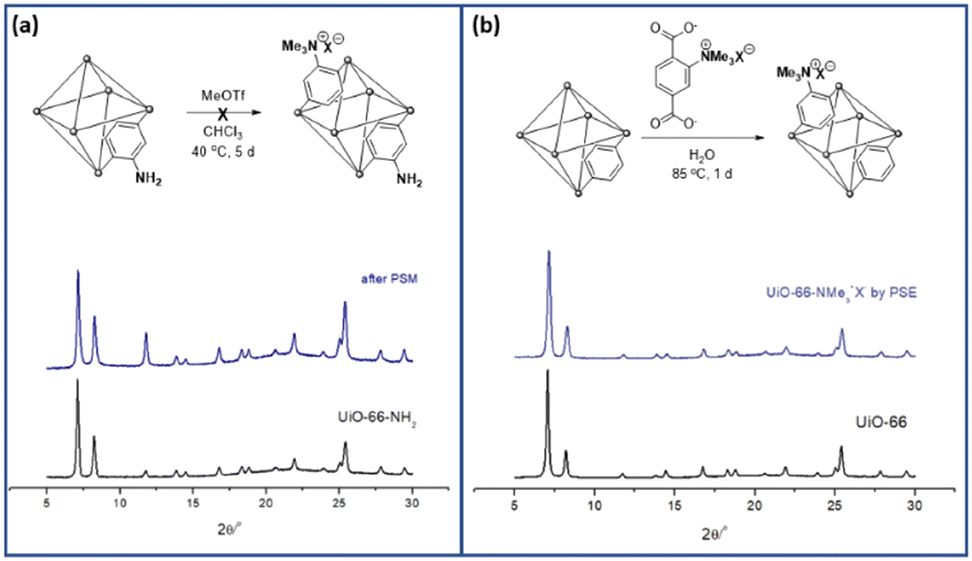
PSM is a versatile and powerful strategy for MOF functionalization. The reactive chemical handles, such as amino and hydroxy groups in MOF pores, were converted to the desired functionalities via solid-state functional group transformations.16,34 Recently, “click chemistry” has been widely studied for the PSM technique, and copper-catalyzed azide-alkyne cycloaddition (CuAAC) reactions have been investigated for PSM-type MOF functionalization with both azide- and alkyne-functionalized MOFs.35To install aliphatic ammonium groups, the CuAAC click reaction was performed with ammonium-containing azide- and alkyne-functionalized MOFs: PSM for aliphatic ammonium groups (strategy III in Scheme 1). First, alkyne-functionalized UiO-66s were prepared from O-propagyl-containing BDC ligands (BDC-OCH2CCH, B). The BDC-OCH2CCH ligand was synthesized via a simple Sandmeyer reaction and nucleophilic substitution from commercially available BDC-NH2 (Scheme S1†). However, the standard UiO-66 synthetic solvothermal conditions failed to form UiO-66-OCH2CCH with alkyne functionality. The C–O bond cleavage occurred with Zr under solvothermal conditions at 120 °C. After extensive screening for the low-temperature synthesis of UiO-66, the desired UiO-66-OCH2CCH was successfully obtained at 65 °C using formic acid modulators. The PXRD patterns are fully commensurate with those reported for UiO-66 (Fig. 2 and S2†), and the alkyne group was determined using 1H NMR after acid digestion (Fig. S7†). Thereafter, the corresponding aliphatic ammonium-containing azide was prepared via an SN2 reaction (see ESI† for detailed procedure), and the CuAAC reaction was performed in a PSM manner. A mixture of copper sulfate and sodium ascorbate successfully formed CuAAC triazoles at 50 °C. After modification, the retained crystallinity of UiO-66-triazole-NMe3+X− (by PSM) was confirmed using PXRD (Fig. 2 and S2†). SEM images confirmed the morphology of UiO-66-triazole-NMe3+X− (by PSM) (Fig. S8†). The modification ratio was determined to be 35% by acid digestion 1H NMR (Fig. S7†). The PSM ratio did not increase from 35% when increasing the PSM temperature, reaction time, and amount of azide reagent. The final MOF was decorated with 65% O-propargyl and 35% aliphatic quaternary ammonium groups. The porosity of the obtained MOF was analyzed using N2 adsorption (Fig. S9†), and thermogravimetric analysis (TGA) was used to determine the thermal stability of the parent MOF (Fig. S6†).
 | ||
| Fig. 2 Installation of aliphatic ammonium groups using (a) PSM and (b) PSE along with their PXRD patterns. | ||
Finally, the aliphatic ammonium group was installed via ligand exchange with UiO-66 MOF and PSE for the aliphatic ammonium group (Strategy IV in Scheme 1). The aliphatic ammonium group-containing ligand (C) was successfully synthesized using a synthetic strategy similar to strategy III (BDCE-O-propargyl preparation and CuAAC reaction with ammonium-containing azide, Scheme S1†). Thereafter, the obtained aliphatic ammonium group-containing BDC ligand and positively charged C was dissolved in aqueous KOH solution and neutralized to pH 7. After incubation of the ligand C solution with non-functionalized UiO-66 for 24 h, the desired positively charged UiO-66-triazole-NMe3+X− (by PSE) was obtained by simple centrifugation and washing steps. The slight broadening of the PXRD pattern may suggest a decrease in the crystalline size, but no change in the peak position and intensity proves the structural integrity and stability after the PSE process (Fig. 2 and S2†). Again, SEM images confirmed the morphology of UiO-66-triazole-NMe3+X− (by PSE) (Fig. S10†). 1H NMR after acid digestion indicated a PSE ratio of 30% (Fig. S11†). The decreased surface area of the pristine UiO-66 was confirmed by N2 adsorption experiments at 77 K (Fig. S12†), and TGA showed thermal stability similar to that of aliphatic ammonium-functionalized UiO-66 from PSM (Fig. S6†).
In addition, thermogravimetric-mass spectrometry (TG-MS) analysis was attempted to analyze weight loss (Fig. S13 and S14†). A bar mode of TG-MS data shows that the weight loss ∼100 °C of the MOFs corresponded to the solvent removal (water and methanol, Fig. S13†). In addition, the positively charged functional groups were stable at the activation condition of MOFs, which was also confirmed by TG-MS analysis. The framework decomposition over 300 °C was confirmed due to the CO2 signal appearing in TG-MS data (Fig. S14†).
For aliphatic quaternary ammonium, the desired UiO-66-triazole-NMe3+X− was successfully obtained using both PSM and PSE techniques. The incorporated ratios of the charged ammonium functional groups were as follows: 35% for PSM and 30% for PSE (Table 1). Because only the PSE process was successful for aromatic ammonium group installation, PSE showed generality as an installation technique for the charged functional groups in Zr-MOFs and BDC-type MOFs (Table 1).
| Aromatic ammonium | Aliphatic ammonium | |
|---|---|---|
| PSM | No reaction | 25% |
| PSE | 35% | 30% |
Structural analysis of the positively charged UiO-66s
The structure of MOFs can be analyzed by the PXRD and SEM data, along with destructive NMR data. The destructive NMR data clearly shows the amount and ratio of the ligands (Fig. S4, S7, and S11†). In addition, the SEM data can show the external structural integrity of the MOFs (Fig. S3, S8, and S10†). The inner structure of the MOFs should be explored using PXRD data (Fig. S2†). When analyzing PXRD, there are two critical points for understanding structural change: diffraction peak position and broadening. The similar diffraction profile of all MOFs suggests that the MOFs are isostructural frameworks. However, a careful comparison of the peak position of MOFs with UiO-66 shows a shift of diffraction peaks to higher 2θ values when the functional group is attached to the ligand of MOFs, suggesting the expansion of the unit cell. Because of the attachment of functional groups, the MOF structure was expanded to accommodate the functional groups inside the pores. Interestingly, the pore occupancy of MOFs by functional groups estimated by the surface area was proportional to the degree of MOF unit cell expansion.At the same time, peak broadening that can be estimated by the full width at half maximum (FWHM) was observed for the MOFs functionalized by PSE (Fig. S2†). The peak broadening of PXRD suggests that the MOF's crystallite size decreased during the ligand exchange process of PSE. The PSE may produce cracks inside the MOF crystals and smaller crystallites resulting in the diffraction peak broadening (Fig. S2, S3, and S10†).
Hydroxide conductivity of the positively charged UiO-66s
MOF-type materials are insulators for ion conduction. Therefore, additional charged guest molecules are required to change their properties from insulator to conductor or to maximize their ion conductivity.36,37 For hydroxide ion conductivity, alkali hydroxides (e.g., NaOH and KOH), EVIm-OH (EVIm = ethyl vinyl imidazolium salt), and TBAH ((n-Bu)4NOH, tetra-n-butylammonium hydroxide) were primarily utilized as guest molecules.38,39 In previous studies, MOFs containing both cations and anions in the pores were considered. In this study, PSM and PSE caused the MOF to act as stationary counter-cations for the accommodation of mobile hydroxide anions by anion exchange with hydroxide ions. Both strong NaOH and KOH showed structural impacts on the ammonium-functionalized UiO-66 MOFs, and the PXRD pattern changes confirmed the decomposition of UiO-66 frameworks after 4 h of exposure to NaOH or KOH aqueous solutions. For TBAH, the dependence of MOF stability on the concentration of the base media was studied. Both aromatic and aliphatic ammonium-containing MOFs maintained their crystallinity after soaking in 3.9 wt% TBAH solution (pH = 13.1) for 24 h (Fig. S15†). Therefore, the main hydroxide conductivity studies were conducted using TBAH with relative humidity (RH) controls (Fig. 3).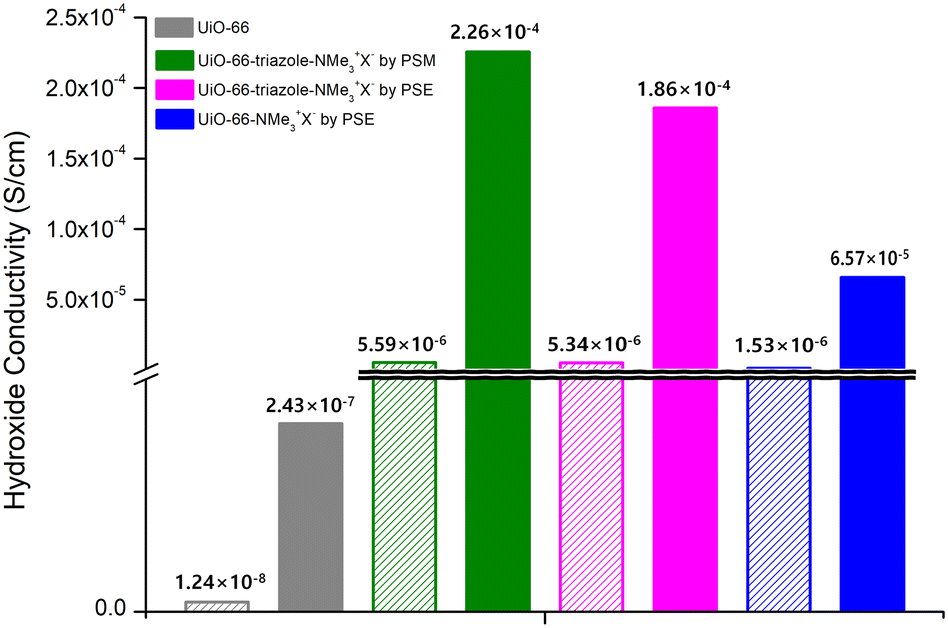 | ||
| Fig. 3 Hydroxide conductivities of quaternary ammonium-functionalized UiO-66s without TBAH (empty bar) and with TBAH (filled bar). | ||
As a control experiment, bare UiO-66 (without functional groups) was used to measure hydroxide conductivity. Although pristine UiO-66 is an insulating material, the employment of TBAH in the pores increased the hydroxide conductivity from 1.24 × 10−8 S cm−1 to 2.43 × 10−7 S cm−1 at 95 RH% (Fig. 3, S16 and Table S2†). For aromatic ammonium-functionalized UiO-66-NMe3+X− (by PSE), an increased hydroxide conductivity was observed without TBAH when compared to bare UiO-66. In addition, the addition of TBAH certainly increased the hydroxide conductivity under humid conditions. The maximum value of 6.57 × 10−5 S cm−1 was obtained at 95% RH at 25 °C and 3.72 × 10−4 S cm−1 at 95% RH at 55 °C (Fig. 3, S17 and Table S3†).
For both aliphatic ammonium group-containing MOFs (UiO-66-triazole-NMe3+X− by PSM and PSE), similar TBAH effects were observed (Fig. S18 and S19†). However, at the same time, the hydroxide conductivities without TBAH were also increased compared to bare UiO-66 (5.59 × 10−6 S cm−1 for UiO-66-triazole-NMe3+X− by PSM at 95 RH% and 5.34 × 10−6 S cm−1 for UiO-66-triazole-NMe3+X− by PSE in 95 RH% at 25 °C; 1.24 × 10−8 S cm−1 for UiO-66 in 95 RH% at 25 °C; 100 times increase by aliphatic ammonium groups, Fig. 3, Tables S4 and S5†), respectively. The maximum hydroxide conductivity was presented by both UiO-66-triazole-NMe3+X− from PSM and PSE (3.13 × 10−4 S cm−1 for UiO-66-triazole-NMe3+X− by PSM in 95 RH% at 45 °C and 4.09 × 10−4 S cm−1 for UiO-66-triazole-NMe3+X− by PSE in 95 RH% at 45 °C). This is a significant number for room-temperature hydroxide conductivity, except for the EVIm-OH study on FJU-66 (0.057 S cm−1 at 95 RH% at 30 °C).38 Further hydroxide ion derivatizations and more functionalization techniques on Zr-based MOFs are necessary for the development of advanced ion-conductive materials.
Herein, the cationic functional group-containing MOFs work as stationary supports for the accommodation of hydroxide anions, which also provide a channel structure for facile transport. The large population of ions transported inside stationary porous materials allows for high hydroxide conductivity. Among the two types of MOFs, the aromatic ammonium groups in the MOF pores are more rigid compared to the aliphatic ammonium groups in UiO-66 MOFs. Therefore, the flexible long alkyl chain (through methylene, triazole, and ethylene) could increase the probability of charged sites meeting and transferring hydroxide ions. Additionally, the effect of heterocycles (triazole) should be considered. Heterocyclic functionalities are important components of ion conductivity.40,41 The CuAAC chemistry permitted the installation of an ammonium group in the aliphatic chain and incorporation of the triazole moiety into MOFs. No significant differences were observed between UiO-66-triazole-NMe3+X− from PSM and PSE. Because UiO-66-triazole-NMe3+X− by PSM retained the remaining 65% O-propargyl group and UiO-66-triazole-NMe3+X− by PSE retained 70% of the non-functionalized BDC (from bare UiO-66), the existence of O-propargyl groups and the pore size differences were not affected by the hydroxide conductivity.
Conclusions
Both aromatic and aliphatic quaternary ammonium groups were employed to prepare positively charged MOFs for hydroxide conductivity studies of Zr- and BDC-based MOFs. Two representative MOF functionalization techniques, PSM and PSE, were investigated and compared for efficient charge loading on the ligand part of the MOF frameworks. For the aromatic quaternary ammonium groups, PSM failed to install the ammonium group in the MOFs; however, PSE was successfully performed on non-functionalized UiO-66 MOF with BDC-NMe3+X− ligands. PSE showed more generality compared to PSM for charged ligands in neutral MOFs.For the aliphatic quaternary ammonium group, CuAAC click chemistry was employed for the main reaction and both PSM and PSE were successfully performed using the Zr-based UiO-66 MOFs. The CuAAC reaction in a PSM manner at alkyne functionalization allowed 35% loading of positively charged aliphatic ammonium groups, and PSE at non-functionalized UiO-66 induced a 30% exchange ratio to aliphatic ammonium group installation.
In ion conduction applications, all aromatic and aliphatic ammonium group-containing UiO-66 MOFs displayed increased hydroxide conductivity compared with bare UiO-66, and the additional hydroxide ion effects from TBAH were enhanced for all positively charged MOFs. Particularly, the aliphatic ammonium group-containing UiO-66-triazole-NMe3+X− exhibited the best hydroxide conductivity owing to its flexible functionalities and effect of the triazole linkers. Generally, aliphatic ammonium group installation is more efficient than aromatic ammonium group installation for hydroxide conductivity and both PSM and PSE are suitable because they are based on CuAAC triazole chemistry. However, the PSE strategy can be utilized for both aromatic and aliphatic ammonium groups, and the PSM technique is only suitable for aliphatic CuAAC functionalization. This study summarizes the systemized functionalization of MOFs with charged functionalities and the effects of positively charged functional groups in the MOF pores on hydroxide conductivity.
Author contributions
H. J. Choi and S. Hong synthesized the compounds. Y. Song performed the conductivity experiment. K. T. Kim and C. Kim designed the preparation of ligands. M. Yoon and M. Kim prepared the overall manuscript and supervised the research. All authors contributed to the manuscript writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Chungbuk National University Korea National University Development Project (2021).Notes and references
- S. L. James, Chem. Soc. Rev., 2003, 32, 276–288 RSC.
- L. Jiao, J. Y. R. Seow, W. S. Skinner, Z. U. Wang and H. L. Jiang, Mater. Today, 2019, 27, 43–68 CrossRef CAS.
- R. B. Lin, Z. Zhang and B. Chen, Acc. Chem. Res., 2021, 54, 3362–3376 CrossRef CAS PubMed.
- S. Jeoung, S. Kim, M. Kim and H. R. Moon, Coord. Chem. Rev., 2020, 420, 213377 CrossRef CAS.
- L. Feng, G. S. Day, K. Y. Wang, S. Yuan and H. C. Zhou, Chem, 2020, 6, 2902–2923 CAS.
- Z. Ji, H. Wang, S. Canossa, S. Wuttke and O. M. Yaghi, Adv. Funct. Mater., 2020, 30, 2000238 CrossRef CAS.
- J. Lee, S. Hong, J. Lee, S. Kim, J. Kim and M. Kim, Bull. Korean Chem. Soc., 2021, 42, 359–368 CrossRef CAS.
- Y. Su, K.-I. Otake, J.-J. Zheng, S. Horike, S. Kitagawa and C. Gu, Nature, 2022, 611, 289–294 CrossRef CAS PubMed.
- C. Gu, N. Hosono, J. J. Zheng, Y. Sato, S. Kusaka, S. Sakaki and S. Kitagawa, Science, 2019, 363, 387–391 CrossRef CAS PubMed.
- F. A. A. Paz, J. Klinowski, S. M. F. Vilela, J. P. C. Tomé, J. A. S. Cavaleiro and J. Rocha, Chem. Soc. Rev., 2012, 41, 1088–1110 RSC.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed.
- S. A. A. Razavi and A. Morsali, Coord. Chem. Rev., 2019, 399, 213023 CrossRef.
- D. Kim, M. Kang, H. Ha, C. S. Hong and M. Kim, Coord. Chem. Rev., 2021, 438, 213892 CrossRef CAS.
- K. P. Samarakoon, M. S. Yazdanparast, V. W. Day and T. Gadzikwa, Mol. Syst. Des. Eng., 2020, 5, 804–808 RSC.
- S. M. Cohen, J. Am. Chem. Soc., 2017, 139, 2855–2863 CrossRef CAS PubMed.
- M. Kalaj and S. M. Cohen, ACS Cent. Sci., 2020, 6, 1046–1057 CrossRef CAS PubMed.
- Z. Yin, S. Wan, J. Yang, M. Kurmoo and M. H. Zeng, Coord. Chem. Rev., 2019, 378, 500–512 CrossRef CAS.
- M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS PubMed.
- M. Kim, J. F. Cahill, Y. Su, K. A. Prather and S. M. Cohen, Chem. Sci., 2011, 3, 126–130 RSC.
- H. Park, S. Kim, B. Jung, M. H. Park, Y. Kim and M. Kim, Inorg. Chem., 2018, 57, 1040–1047 CrossRef CAS PubMed.
- D. Lee, S. Lee, Y. Son, J. Y. Kim, S. Cha, D. Kwak, J. Lee, J. Kwak, M. Yoon and M. Kim, Bull. Korean Chem. Soc., 2022, 43, 912–917 CrossRef CAS.
- S. N. Zhao, Y. Zhang, S. Y. Song and H. J. Zhang, Coord. Chem. Rev., 2019, 398, 113007 CrossRef CAS.
- H. He, L. Hashemi, M. L. Hu and A. Morsali, Coord. Chem. Rev., 2018, 376, 319–347 CrossRef CAS.
- A. Karmakar, A. V. Desai and S. K. Ghosh, Coord. Chem. Rev., 2016, 307, 313–341 CrossRef CAS.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- A. Burrows, S. Cohen, M. Kim, S. M. Cohen, T. Ahnfeldt, D. Gunzelmann, J. Wack, J. Senker, N. Stock, A. D. Burrows and L. L. Keenan, CrystEngComm, 2012, 14, 4096–4104 RSC.
- D. Zou and D. Liu, Mater. Today Chem., 2019, 12, 139–165 CrossRef CAS.
- J. Winarta, B. Shan, S. M. McIntyre, L. Ye, C. Wang, J. Liu and B. Mu, Cryst. Growth Des., 2020, 20, 1347–1362 CrossRef CAS.
- H. C. Woo, D. K. Yoo and S. H. Jhung, ACS Appl. Mater. Interfaces, 2020, 12, 28885–28893 CrossRef CAS PubMed.
- T. M. Rayder, A. T. Bensalah, B. Li, J. A. Byers and C. K. Tsung, J. Am. Chem. Soc., 2021, 143, 1630–1640 CrossRef CAS PubMed.
- X. Zhou, Y. Zhang, X. Yang, L. Zhao and G. Wang, J. Mol. Catal. A: Chem., 2012, 361–362, 12–16 CrossRef CAS.
- H. Hahm, S. Kim, H. Ha, S. Jung, Y. Kim, M. Yoon and M. Kim, CrystEngComm, 2015, 17, 8418–8422 RSC.
- H. Hahm, H. Ha, S. Kim, B. Jung, M. H. Park, Y. Kim, J. Heo and M. Kim, CrystEngComm, 2015, 17, 5644–5650 RSC.
- D. Kim, H. Yoo, K. Kim, D. Kim, K. T. Kim, C. Kim, J. Y. Kim, H. R. Moon and M. Kim, Chem. Commun., 2022, 58, 5948–5951 RSC.
- B. Gui, X. Meng, H. Xu and C. Wang, Chin. J. Chem., 2016, 34, 186–190 CrossRef CAS.
- M. Sadakiyo, H. Kasai, K. Kato, M. Takata and M. Yamauchi, J. Am. Chem. Soc., 2014, 136, 1702–1705 CrossRef CAS PubMed.
- M. Sadakiyo and H. Kitagawa, Dalton Trans., 2021, 50, 5385–5397 RSC.
- Z. Li, Z. Zhang, Y. Ye, K. Cai, F. Du, H. Zeng, J. Tao, Q. Lin, Y. Zheng and S. Xiang, J. Mater. Chem. A, 2017, 5, 7816–7824 RSC.
- S. S. Nagarkar, B. Anothumakkool, A. V. Desai, M. M. Shirolkar, S. Kurungot and S. K. Ghosh, Chem. Commun., 2016, 52, 8459–8462 RSC.
- C. Jin, S. Zhang, Y. Cong and X. Zhu, Int. J. Hydrogen Energy, 2019, 44, 24954–24964 CrossRef CAS.
- J. Ponce-González, D. K. Whelligan, L. Wang, R. Bance-Soualhi, Y. Wang, Y. Peng, H. Peng, D. C. Apperley, H. N. Sarode, T. P. Pandey, A. G. Divekar, S. Seifert, A. M. Herring, L. Zhuang and J. R. Varcoe, Energy Environ. Sci., 2016, 9, 3724–3735 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2me00238h |
| ‡ H. J. Choi, S. Hong, and Y. Son equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2023 |