
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid microwave synthesis of sustainable magnetic framework composites of UTSA-16(Zn) with Fe3O4 nanoparticles for efficient CO2 capture†
John Luke
Woodliffe
 a,
Amy-Louise
Johnston
b,
Michael
Fay
c,
Rebecca
Ferrari
b,
Rachel L.
Gomes
b,
Ed
Lester
a,
Ifty
Ahmed
a and
Andrea
Laybourn
*a
a,
Amy-Louise
Johnston
b,
Michael
Fay
c,
Rebecca
Ferrari
b,
Rachel L.
Gomes
b,
Ed
Lester
a,
Ifty
Ahmed
a and
Andrea
Laybourn
*a
aAdvanced Materials Research Group, Faculty of Engineering, University of Nottingham, Nottingham, NG7 2RD, UK. E-mail: andrea.laybourn@nottingham.ac.uk
bFood Water Waste Research Group, Faculty of Engineering, University of Nottingham, Nottingham, NG7 2RD, UK
cNanoscale and Microscale Research Centre (nmRC), Cripps South Building, University of Nottingham, Nottingham, NG7 2RD, UK
First published on 10th October 2023
Abstract
Metal–organic frameworks (MOFs) have shown excellent potential for carbon dioxide capture applications due to their high sorption capacities and selectivities. However, MOFs are typically thermally insulating, and so thermal CO2 regeneration is challenging, especially on the large scales required in industry. This limitation can be overcome by inclusion of magnetic nanoparticles within the MOF structure, enabling rapid and energy efficient CO2 regeneration using induction heating. To this end we have developed novel magnetic framework composites (MFCs) comprised of MOF UTSA-16(Zn) (UTSA: University of Texas at San Antonio, a Zn-based MOF with citrate linkers) and Fe3O4 nanoparticles. Our work also addresses the sustainability and scalability challenges faced by MFCs required for industrial application, considering the use of inexpensive and widely-available materials. Herein we report a two-step procedure for preparing the MFCs. Firstly, a scalable single-step continuous hydrothermal synthesis method is used to produce highly pure, stable, and crystalline citrate-coated Fe3O4 nanoparticles (62% yield). The nanoparticles exhibit a uniform particle size (19 ± 11 nm) and a very high saturation magnetisation (78 emu g−1) compared with previously published citrate-coated Fe3O4 nanoparticles. Next, various concentrations (2.6–18.7 wt%) of these nanoparticles were incorporated into UTSA-16(Zn) via a rapid microwave-assisted direct-growth strategy (10 min) to form the MFCs (81–83% yield). The MFCs demonstrate high CO2 adsorption capacities (2.8–3.3 mmol g−1) and recyclability. In addition, the MFCs heat rapidly in an applied magnetic field for CO2 release, reaching regeneration temperatures in remarkably short times (e.g. 60 °C in 8 seconds). The MFCs developed in this work combine strong CO2 adsorption profiles and substantial regeneration heating capabilities, whilst being produced in a scalable and sustainable manner. The methods developed to prepare MFCs herein are also applicable to other MOFs, opening routes for a variety of sustainable MFCs to deliver impact for a range of applications across carbon capture and triggered release of other guest molecules.
1 Introduction
Since the industrial revolution, a sharp increase in atmospheric concentrations of the greenhouse gas carbon dioxide (CO2) has been observed from 180–280 ppm to over 400 ppm.1–3 This increase has significant negative implications for the environment and society,4 likely resulting in 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 additional deaths worldwide annually between 2030 and 2050.5 Governments around the world are setting net zero CO2 emissions targets by 2050–2070 in line with the recent report from the Intergovernmental Panel on Climate Change (IPCC),6 and have affirmed their commitments in Glasgow 2021 at COP26.7 Addressing these IPCC targets requires immediate action, including preventing CO2 from entering the atmosphere.
000 additional deaths worldwide annually between 2030 and 2050.5 Governments around the world are setting net zero CO2 emissions targets by 2050–2070 in line with the recent report from the Intergovernmental Panel on Climate Change (IPCC),6 and have affirmed their commitments in Glasgow 2021 at COP26.7 Addressing these IPCC targets requires immediate action, including preventing CO2 from entering the atmosphere.
The largest contributor to global anthropogenic CO2 emissions are coal-fired power plants (10.1 Gt in 2018, 30% of total emissions).8,9 Post-combustion CO2 capture facilities can be retrofitted into these existing power stations to capture the CO2 at source.10 However, the current aqueous amine absorption technologies for this process have high energy requirements for separation and purification, increasing a power plant's energy demand by 25–40%.11 Alternatives have been investigated such as physical sorbents, membranes, chemical looping and solid sorbents (e.g. porous carbons, zeolites, alumina and metal–organic frameworks (MOFs)11). Of these alternatives, solid sorbents possess many advantages for industrial-scale carbon capture. One major advantage is a reduced energy for CO2 regeneration, as the CO2 is physically adsorbed to the material surface, rather than bound by chemical bonds which would need to be broken to release the CO2.12 Other advantages include a greater uptake capacity and selectivity for CO2.12
Metal–organic frameworks are porous materials containing metal ions/clusters and organic linkers.13 MOFs are of particular interest to researchers due to their tuneable natures, high porosities and internal surface areas.14 These properties result in the potential for excellent CO2 adsorption profiles, optimisable for specific flue gas conditions.15 The next phase of development for carbon capture MOFs is consideration of sustainability, scalability, and industrial process design.15–17 This stage requires cost-effective materials with high thermal and chemical stabilities and the development of energy-efficient processes. During the carbon capture process, MOFs can undergo a temperature swing adsorption procedure.18 Once saturated with CO2, the MOFs are heated to release the CO2 after each capture cycle. This regenerates the material for re-use in the next cycle. However, the thermally insulating nature of MOFs make thermal regeneration challenging, especially at scale.19 A newly emerging approach is to incorporate magnetic nanoparticles (MNPs) in MOFs forming magnetic framework composites (MFCs), enabling rapid and highly energy-efficient regeneration of the composites by localised magnetic induction heating.20 This process, coined magnetic induction swing adsorption (MISA),21 requires remarkably low carbon capture energy costs (1.29 MJ per kgCO2,22cf. 3–4 MJ per kgCO2 for amine solvents23). MFCs have also found application across adsorption, sensing, biomedicine, catalysis, and environmental monitoring and water remediation among others.24–31
Aside from their use in MFCs, MNPs are an important class of nanomaterials which have been widely studied. The unique properties of their magnetism, size and surface area/volume ratio have led to applications across biomedicine,32,33 nanomedicine34 and sensing.35 Surface functionalisation/coating of MNPs is hugely beneficial for their thermodynamic and colloidal stability.36 Citrate coatings have been extensively used in the preparation of aqueous-stable MNPs due to the electrostatic, biocompatible, and anti-oxidising properties of citrates, alongside their low cost.37,38 Citrates also have chelating properties which can control the crystallite growth of the MNPs and allow them to conjugate to other molecules such as biomolecules and drugs, through the surface carboxyl groups.37,38 In this work, a further reason for choosing a citrate coating was the hypothesis that surface citrate groups on the MNPs would promote a binding interaction between the chosen MOF (which contains citrate linkers) and the MNPs.
To be viable for widespread industrial adoption, MFCs (including both the MOF and MNP components) must be produced via sustainable and economical routes, with minimal impact on the environment. Typically, the synthesis of MNPs require harsh batch conditions such as high temperatures or reflux for >8 h, often under air sensitive environments such as N2.22,39–41 Subsequent production of MFCs often requires lengthy MNP surface-functionalisation reactions (e.g. 24 h) required for MOF attachment. This is typically followed by prolonged high temperature autoclave MOF syntheses (e.g. 125 °C for 15 h under Ar) with toxic solvents such as DMF.22,39,42–44 The main alternative to autoclave routes is the labour intensive layer-by-layer MOF growth method,39,40,45 with a difficult outlook for scale-up due to the amount of time taken and number of steps required to grow the thin MOF layers. The use of expensive MOF linker components and moisture sensitive MOFs is also ubiquitous in MFCs.22,39,40,42,43,45 The development of manufacturing processes that reduce product cost and environmental burden of MNPs and MFCs is critical for successful transfer of these materials from the laboratory to industry.
In this paper, we first report a continuous-flow hydrothermal synthesis method for producing highly magnetic citrate-coated Fe3O4 nanoparticles (MNP-CA) (mean diameter: 19 nm, saturation magnetisation: 78.3 emu g−1) via a single step process, with high colloidal stability (zeta potential: −47.3 mV), purity and crystallinity. This method overcomes the previously mentioned issues of batch reactors, i.e. air sensitive environments and lengthy surface-functionalisation steps. We then report a rapid (10 min) microwave-assisted direct-growth strategy to encapsulate various quantities of MNP-CA (2.6–18.7 wt%) in UTSA-16(Zn) (C12H8KO14Zn3), a zinc and citrate based MOF chosen for its high CO2 uptake, good stability to moisture and acid gases and very low reagent costs (<1 USD g−1 MOF).46 The subsequent MFCs are sustainably produced with high yield and CO2 adsorption capacities (81–83%, 2.8–3.3 mmol g−1). The MFCs also show leading processing capabilities for carbon capture, including induction heating up to 150 °C in 30 s and no decrease in adsorption capacity over 12 CO2 adsorption–desorption cycles. The MNP-CA and the MFC syntheses methods developed herein are transferrable to other MOFs, allowing for the production of a variety of MFCs with wide-reaching impact in alternative applications.
2. Experimental section
2.1 Materials
All chemicals and reagents were used as received. Zinc acetate dihydrate (≥99.0%), citric acid monohydrate (≥99.0%), pristine magnetite (<5 μm, 95%) and potassium hydroxide (86.7%) were obtained from Sigma Aldrich. Ammonium iron(III) citrate (14.5–16% Fe basis) was bought from Honeywell. Methanol (≥99.9%) was purchased from Fisher Scientific. Ethanol (absolute, SpS grade) was purchased from Scientific Laboratory Supplies Ltd. All water used was deionised.2.2 Synthesis of citrate-coated Fe3O4 nanoparticles
Magnetic magnetite nanoparticles with a citrate coating (MNP-CA) were synthesised using a counter-current continuous hydrothermal reactor set-up (Fig. 1), first reported by Lester et al.47 The reactor set-up has been described in more detail for the synthesis of other materials, including barium strontium titanate48 and layered double hydroxides.49 Briefly, aqueous ammonium ferric citrate (0.05 M) was fed un-heated via the upflow pump into the reactor at a pre-determined flow rate (10 mL min−1). Separately, deionised water was heated to 435 °C in a 2 kW band heater and fed via the downflow pump into the top of the reactor at a pre-determined flow rate (20 mL min−1). The pressure was maintained in the system at a maximum of 24 MPa by a back-pressure regulator (Swagelok®). The reaction stream was then cooled through a counter-current water heat exchanger and the product in suspension collected at the outlet. Finally, the MNP-CA was allowed to settle, separated by gravity and magnetic attraction, washed with deionised water (3 × 25 mL) then stored in deionised water as a 5 w/v% suspension (yield: 62%).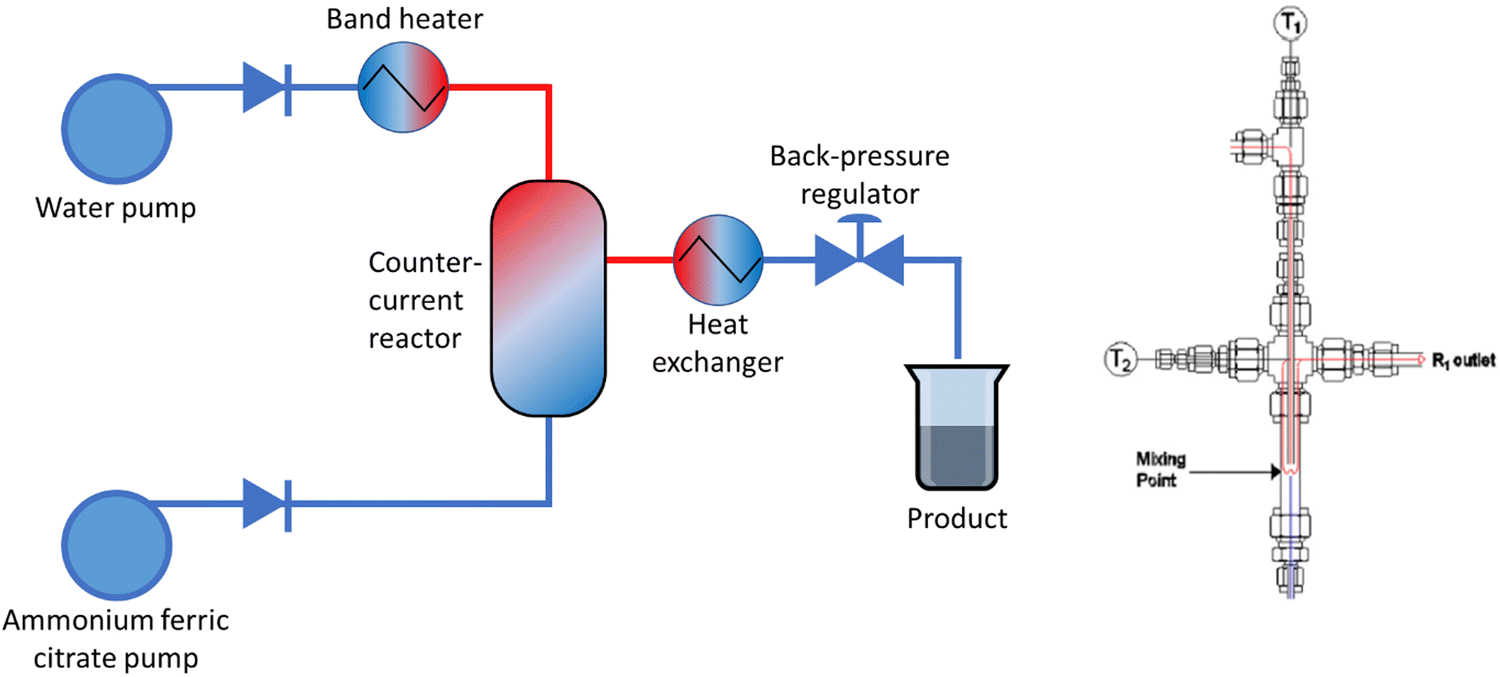 | ||
| Fig. 1 Left: Schematic of the counter-current flow reactor set-up for the synthesis of MNP-CA. Right: Schematic of the counter-current reactor. T1 and T2: thermocouples in the system. | ||
2.3 Synthesis of MFCs UTSA-16(Zn)@MNP-CA
The synthesis conditions for UTSA-16(Zn) were optimised to inform the conditions for the MFCs. Pristine UTSA-16(Zn) was also used as a comparison to the MFCs (Fig. S3, ESI†). The reagents, Zn(AcO)2·2H2O (0.878 g, 4.0 mmol), citric acid monohydrate (0.840 g, 4.0 mmol) and KOH (0.67 g, 10.4 mmol) were dissolved in H2O (6–10 mL) in a 35 mL microwave vial. A MNP-CA in H2O (5 w/v%) solution was added (0–4 mL), bringing the total H2O quantity to 10 mL. EtOH (10 mL) was added and stirred (10 min) before the vial was sealed and irradiated under autogenous pressure with stirring at a maximum forward power of 300 W (commercial CEM discover microwave reactor, dynamic mode, T = 60 °C, t = 10 min). After the reaction, the vial was cooled in the microwave cavity with air to <50 °C, then the product was collected by washing out the vial with H2O:EtOH 1:1 (20 mL) followed by centrifugation. The product was then washed and centrifuged with MeOH (3 × 30 mL, 20 mins) before drying in an oven (50 °C, >4 h).
2.4 Characterisation
3. Results and discussion
3.1 Hydrothermal synthesis of citrate-coated Fe3O4 magnetic nanoparticles (MNP-CA)
Citrate-coated magnetic nanoparticles (MNP-CA) were synthesised using a continuous-flow hydrothermal reactor, as described in the experimental section. This method is considered a significant improvement on other commonly reported techniques as it avoids the need for lengthy multi-step batch reactions, organic solvents, or air sensitive environments.22,39–41 The continuous-flow reactor design is also inherently scalable,47,48 providing a sustainable pathway for large-scale production. The formation of magnetite from ammonium ferric citrate is thought to proceed via an initial reduction of an amount of Fe(III) to Fe(II) by CO formed in the partial thermal decomposition of ammonium citrate.52 Both Fe(III) and Fe(II) then undergo hydrolysis upon reaction with water, before dehydration and precipitating out the metal oxide (Fe3O4).53 A simplified hypothetical reaction scheme is therefore proposed:54The nanoparticles produced herein were composed of highly crystalline magnetite (Fe3O4) as confirmed via PXRD (Fig. 2A), matching the powder diffraction file database (PDF-01-071-6336). The nanoparticles were produced in high purity and exhibit high stability, observed in the PXRD pattern (Fig. 2A) showing no oxidation to the less magnetic hematite (α-Fe2O3) or maghemite (γ-Fe2O3) phases over 8 days (storage at room temperature).55 This stability and purity is beneficial for applications relying on strong magnetic properties such as heating or separation.56,57 The size distribution of the nanoparticles was determined by TEM analyses of 302 particles. The resulting histogram is displayed in Fig. 2B, showing an average particle size of 19 ± 11 nm. The small size of the nanoparticles cause quantum size effects and large surface areas, resulting in superparamagnetic behaviour,58 with very high saturation magnetisation (78.3 emu g−1) and low remanence (4.2 emu g−1). These MNP-CA are comparable in size and shape (Fig. 3A and B) to reported citrate-coated Fe3O4 nanoparticles, which are synthesised via heated batch co-precipitation methods (19 nm,36 21 nm,59 25 nm60). Applying the Scherrer equation to the powder diffraction pattern of MNP-CA (Fig. 2A) gave a mean crystallite diameter of 29.1 nm, averaged over 6 peaks. This result indicates that each nanoparticle contains a single crystallite with one coherently scattering domain, which is also confirmed in the TEM images showing the diffracting planes in a single nanoparticle (Fig. 3C).
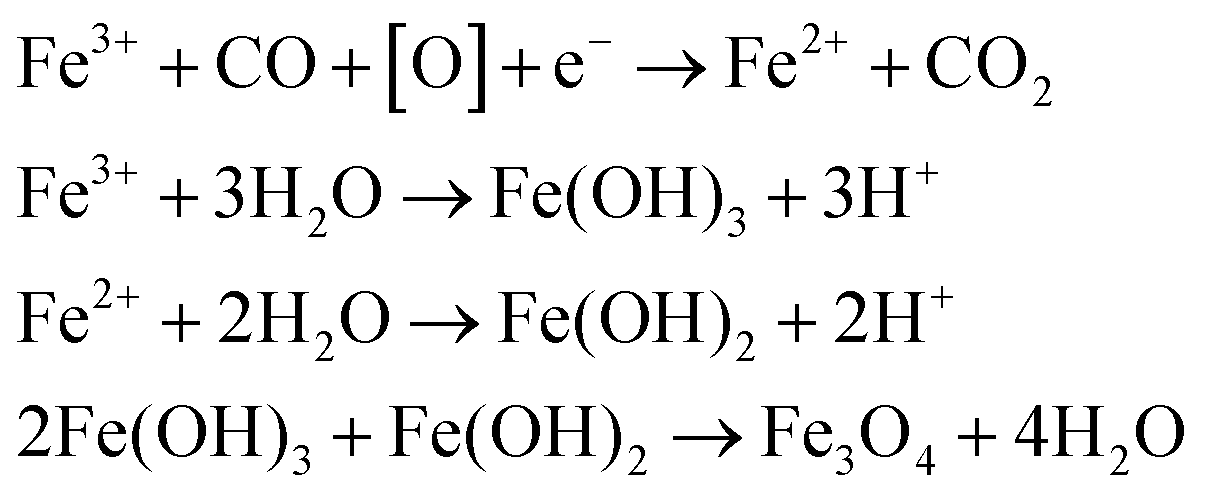
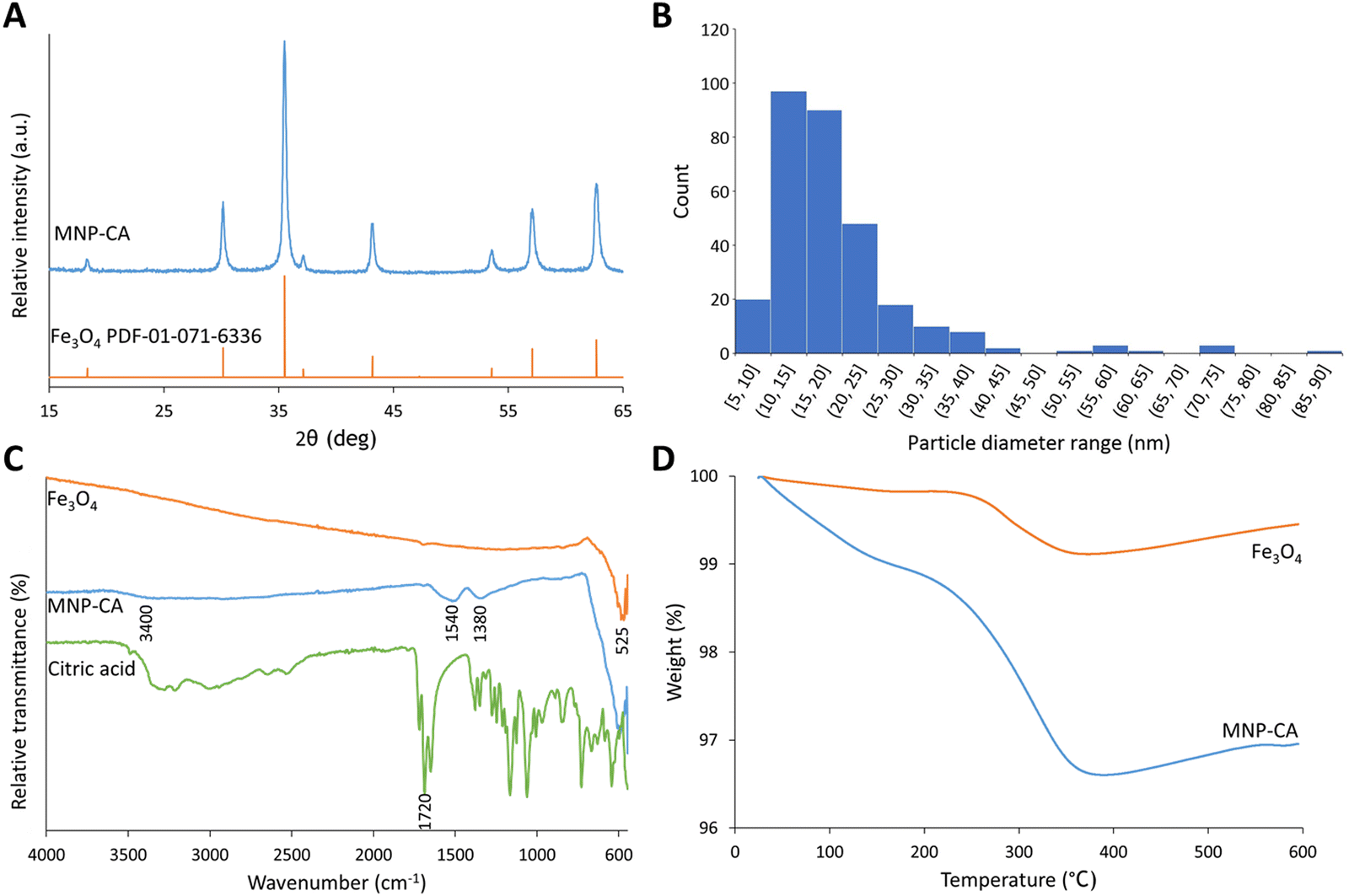 | ||
| Fig. 2 (A) PXRD patterns of synthesised MNP-CA and pristine Fe3O4 from the powder diffraction file database (PDF-01-071-6336).50 (B) Histogram showing the size distribution of MNP-CA from TEM image analyses. (C) FTIR spectra for pristine Fe3O4, synthesised MNP-CA, and citric acid. (D) Graph showing weight loss on heating during TGA for pristine Fe3O4 and synthesised MNP-CA. | ||
 | ||
| Fig. 3 (A) and (B) TEM images showing the approximate size and shape of MNP-CA. (B) TEM image showing the crystalline magnetite domains and the amorphous citrate coating on single nanoparticles. (D) TEM image showing the highly crystalline magnetite region, with an interplanar d-spacing of 0.30 nm corresponding to the (220) planes at 30° 2θ.64,65 (E) 20 s magnetic separation of MNP-CA in deionised water. | ||
As discussed in the introduction, MNP surface functionalisation is important not just for stability, but also allows the conjugation of other molecules to the MNPs. The presence of a citrate coating on the synthesised MNP-CA was confirmed by FTIR spectroscopy (Fig. 2C). The citric acid peak at 1720 cm−1 arises from C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O symmetric stretching from the COOH group.36 This peak is shifted down to 1540 cm−1 in the MNP-CA material owing to chemisorption of the carboxylate on the citrate ion with Fe at the MNP surface.36,38,61,62 Complexation likely weakens the CO bond, introducing partial single bond character and lowering the stretching frequency.38 The neighbouring peak at 1380 cm−1 in MNP-CA is ascribed to an asymmetric stretch of C–O from the carboxylate group,36,59,62 and the broad absorbance at 3400 cm−1 from OH groups.59 The low intensity of these peaks compared to pristine citric acid is common in other reports, and is possibly due to the citrate complexes forming a relatively close-packed layer on the NPs, restricting molecular motion.61 The low intensity of the peaks may also be attributed to the small surface quantities (2.5 wt% by TGA) relative to the bulk MNPs. Finally, the peak at 525 cm−1 in MNP-CA and pristine Fe3O4 materials arises from an Fe–O stretching vibrational mode of Fe3O4.36,38,59,62
O symmetric stretching from the COOH group.36 This peak is shifted down to 1540 cm−1 in the MNP-CA material owing to chemisorption of the carboxylate on the citrate ion with Fe at the MNP surface.36,38,61,62 Complexation likely weakens the CO bond, introducing partial single bond character and lowering the stretching frequency.38 The neighbouring peak at 1380 cm−1 in MNP-CA is ascribed to an asymmetric stretch of C–O from the carboxylate group,36,59,62 and the broad absorbance at 3400 cm−1 from OH groups.59 The low intensity of these peaks compared to pristine citric acid is common in other reports, and is possibly due to the citrate complexes forming a relatively close-packed layer on the NPs, restricting molecular motion.61 The low intensity of the peaks may also be attributed to the small surface quantities (2.5 wt% by TGA) relative to the bulk MNPs. Finally, the peak at 525 cm−1 in MNP-CA and pristine Fe3O4 materials arises from an Fe–O stretching vibrational mode of Fe3O4.36,38,59,62
The presence of citrate at the surface of the MNPs was further shown by zeta potential, highlighting the effect of citrate on the MNP properties. A zeta potential of −47.3 mV was obtained in deionised water (Fig. S2, ESI†). The large negative charge arises from electrostatic stabilisation from the strong adsorption of citrate ions on the MNP surface, providing excellent colloidal stability.36 The mass fraction of the citrate layer was determined by TGA to be 2.5 wt% (Fig. 2D). MNP-CA contains physically adsorbed (removed at <150 °C, approx. 1 wt%) and chemically bound (removed up to 375 °C, approx. 1.5 wt%) citrates. The amorphous citrate coating on the MNPs was also observed as an approximately 1 nm thick layer in the TEM images, see Fig. 3C. This result corroborates previous studies of citrate coatings on gold nanoparticles, with layer thicknesses of 0.8–1.0 nm.63 The TEM images also show the highly crystalline magnetite regions (Fig. 3C and D). TEM image analysis of one nanoparticle (Fig. 3D) gave an interplanar d-spacing of 0.30 nm (average of 25 measurements), corresponding to the (220) planes at 30° 2θ.64,65
Magnetisation of the synthesised MNP-CA was measured by VSM (Fig. 5A). The saturation magnetisation reached 78.3 emu g−1 at 10 kOe, which is considerably higher than other published citrate-coated magnetite nanoparticles, which typically range from 30–60 emu g−1.36–38,61,62 The higher saturation magnetisation for MNP-CA highlights its high purity as distortion/lattice deformation and micro-strain in nanoparticles can lead to reduced magnetisation.66 The magnetic and inductive heating properties of Fe3O4 nanoparticles are also size-dependant, with the specific absorption rate reported to increase with size up to around 28 nm due to susceptibility then hysteresis loss.67 The low remanent magnetisation (4.2 emu g−1) and intrinsic coercivity (42 Oe) of MNP-CA (Fig. 5A) are similar to other reports.36–38,60 These low values are advantageous as they result in a high permeability and low energy required to switch the spin directions, beneficial for applications involving magnetic separation, such as in environmental remediation.
High quality citrate-coated magnetite nanoparticles have thus been synthesised in a sustainable and scalable manor, ready for use in a variety of applications such as biomedicine,33,58 environmental remediation,68,69 catalysis70,71 and, as demonstrated in this work, in the synthesis of magnetic framework composites.72
3.2 Preparation of magnetic framework composites by encapsulation of nanoparticles
Magnetic framework composites (MFCs) were prepared using the synthesised MNP-CA and a direct growth encapsulation strategy, based on the development of a rapid (10 min) microwave synthesis for MOF UTSA-16(Zn) (Fig. S3, ESI†). As previously mentioned, MOF UTSA-16(Zn) was selected for its promise in carbon capture applications due to its high CO2 uptake and selectivity at relevant partial pressures for post combustion CO2 capture (0.15 bar), good stability to moisture and acid gases, and low production cost due to the very cheap and abundant starting reagents.46,73 A direct growth encapsulation strategy was selected here due to its ease and scalability compared with laborious methods such as layer-by-layer growth.25 The direct growth method also still ensures extensive mixing and strong attachment between the components compared to other strategies such as direct mixing.25 The method developed herein, involved adding various concentrations of MNP-CA to the dissolved MOF precursors in water, followed by the addition of an antisolvent (ethanol) and microwave irradiation at 300 W for 10 min at 60 °C (see Experimental section for further details). Fig. 4A shows the powder diffraction patterns of the composites, where the gradual decrease in the UTSA-16(Zn) peaks (e.g. at 7 and 14° 2θ) and increase in the MNP-CA peaks (e.g. at 36 and 63° 2θ) are observed with increasing MNP-CA content. PXRD data also confirm the crystallinity of both the MOF and MNP-CA components in the UTSA16(Zn)@MNP-CA MFCs. The exact quantities of MNP-CA in the MOF, determined using saturation magnetisation measurements22 (Fig. 5A), were between 2.6–18.7 wt%. This range is similar to other reported MFCs, including 0.1–7.8 wt% Fe3O4 in Mg-MOF-74,74 1–10 wt% MgFe2O4 in Al-fumarate,75 1.3–7.3 wt% Fe3O4 in PCN-250,76 0.1–15.0 wt% Fe3O4 in Co-MOF-7477 and 2.0–4.1 wt% MgFe2O4 in UiO-66.21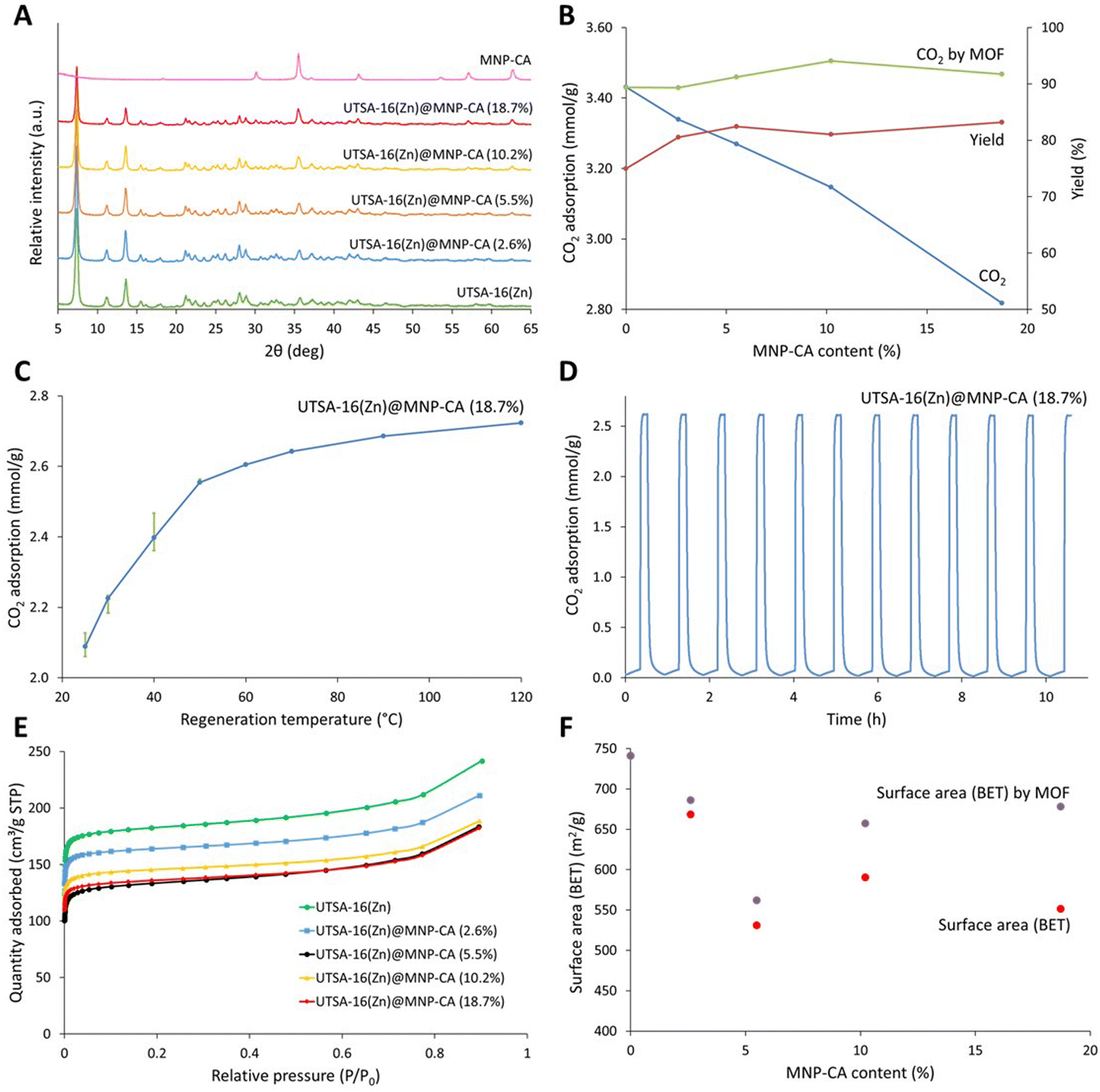 | ||
| Fig. 4 (A) Powder X-ray diffraction patterns between 5 and 45° 2θ of pristine UTSA-16(Zn), MNP-CA and UTSA-16(Zn)@MNP-CA MFCs containing various quantities of MNP-CA. (B) CO2 adsorption capacity and product yield of MFCs plotted against MNP-CA content. CO2 adsorption of the MOF is plotted by subtracting the mass contribution from MNP-CA. (C) Regeneration temperature plotted against CO2 adsorption capacity for UTSA-16(Zn)@MNP-CA (18.7%), results are averages of 4 repeat cycles. Error bars show the decrease in capacity between the first and fourth cycle. (D) CO2 adsorption capacity for UTSA-16(Zn)@MNP-CA (18.7%) over 12 adsorption–desorption cycles. Regeneration was performed at 60 °C. (E) N2 adsorption isotherms of UTSA-16(Zn) and UTSA-16(Zn)@MNP-CA MFCs, measured at 77 K. (F) Apparent BET surface area (Rouquerol)88 plotted against MNP-CA content for UTSA-16@MNP-CA MFCs. Surface area ‘by MOF’ was determined by subtracting the mass contribution from MNP-CA. | ||
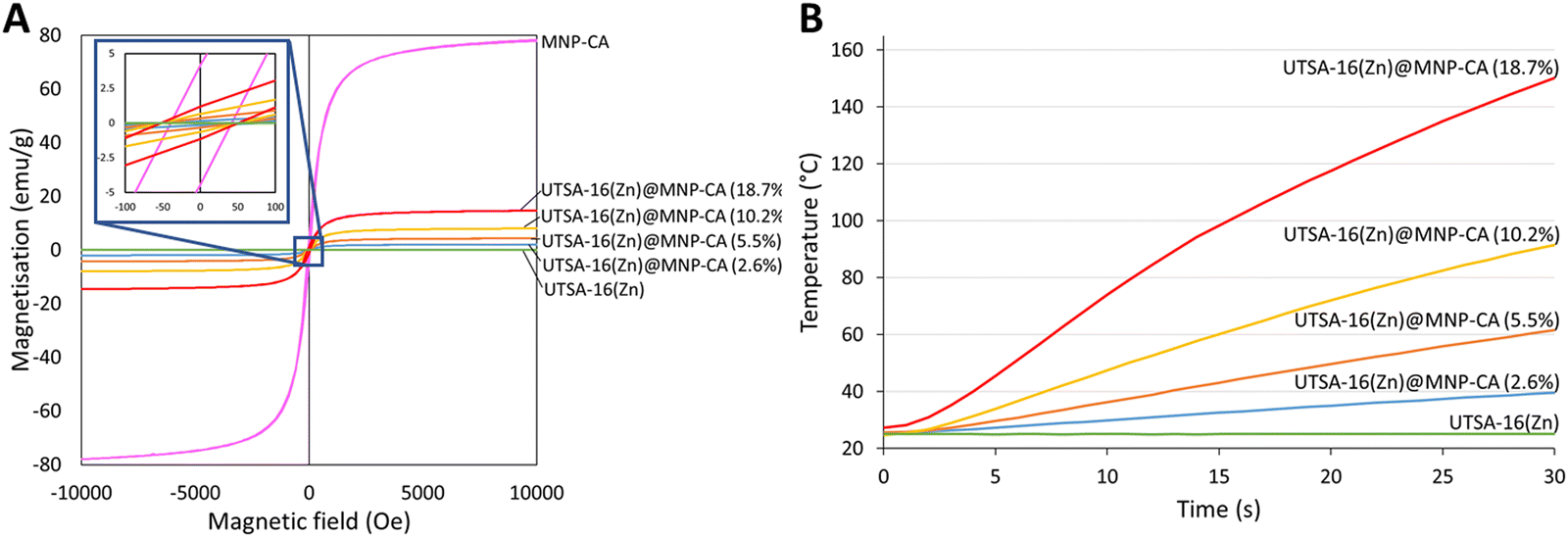 | ||
| Fig. 5 (A) VSM measurements of UTSA-16(Zn), UTSA-16(Zn)@MNP-CA MFCs, and MNP-CA taken at room temperature with a field strength up to 10 kOe. (B) Induction heating temperature plotted against time for UTSA-16(Zn) and UTSA-16(Zn)@MNP-CA MFCs at 500 W over 30 s. | ||
The impact of increasing MNP-CA content in the UTSA-16(Zn)@MNP-CA MFCs on yield and CO2 adsorption (measured gravimetrically by TGA78) is shown in Fig. 4B. It should be noted that although there is a decrease in CO2 capacity from 3.4 to 2.8 mmol g−1 with increasing MNP-CA content (as expected, owing to MNP-CA not adsorbing CO2), there was no decrease in the adsorption capacity of the MOF when accounting for the MNP-CA mass in the various MFCs. These results indicate that MNP-CA has no detrimental effect on the MOF synthesis (as crystal structure is maintained) or ability of the MOF to adsorb CO2, which could potentially arise from pore-blocking.28,79 The CO2 capacities observed for UTSA-16(Zn) and the MFCs were similar to those reported elsewhere for pristine UTSA-16(Zn) (3.5 mmol g−1)73 and other high performing MOFs/MOF composites,80,81 though were not as high as the absolute maximum reported value for pristine UTSA-16(Zn) (4.7 mmol g−1).46 However, a balance must be struck between how the material is prepared and its performance when considering industrial viability. Compared to the 4.7 mmol g−1 literature method, the synthesis time for preparing the pristine MOF (and associated MFCs) was reduced by a factor of 24, the washing/solvent exchange was reduced from 14 washes over 3 days to 3 washes over 1 h, diethyl ether was removed as a washing solvent, all anhydrous/dry washing solvents were replaced with standard commercial solvents, and 90 °C vacuum drying activation was reduced to 50 °C without a vacuum. These changes resulted in a significant improvement towards the sustainability and scalability of MOF synthesis, especially with regards to the washing/solvent exchange and activation procedure. The additional advantage of the MFCs is the magnetic induction heating capability for rapid and low-energy regeneration.
The overall product yield benefited from the presence of MNP-CA (Fig. 4B), as an increase from 77% (pristine MOF) to 81–83% (MFCs, 0.8–0.9 g) was observed. This increase in yield may result from higher localised temperatures around MNP-CA due to enhanced interactions with the microwave field.82,83 The impact of increasing MNP-CA content in the MFCs on N2 adsorption isotherms and apparent BET surface area was also evaluated (Fig. 4E and F). The adsorption isotherms in Fig. 4E of the UTSA-16(Zn) and MFCs match those found in the literature for the MOF, showing a type I isotherm due to the microporous structure.46 The surface areas are also similar to those previously reported for the MOF,46,73 though decreased more than expected purely from the mass contribution of the MNP-CA, from 741 m2 g−1 without any MNP-CA down to 551 m2 g−1 at 18.7% MNP-CA. The trendline between different MNP-CA concentrations in the MFCs was also less clear than observed with CO2 adsorption capacity (Fig. 4F and Fig. S4, ESI†). This result highlights the importance of testing the sorption capacity of new materials and composites for sorption applications, rather than depending on the apparent BET surface area as a sorption capacity analogue for comparisons between materials. Further considerations and guidance for using the BET method for surface area measurements of MOFs have also been recently reported.84,85
The impact of regeneration temperature on CO2 capacity was investigated, in order to maximise the efficiency of the sorbent regeneration process (releasing the adsorbed CO2 for sorbent reuse). Fig. 4C displays results between 25 and 120 °C for one of the MFCs, UTSA-16(Zn)@MNP-CA (18.7%). The graph shows the average CO2 adsorption capacity over 4 adsorption–desorption cycles at each regeneration temperature. The error bars show the decrease in capacity between the first (top error bar) and fourth (bottom error bar) cycle. Increasing regeneration temperatures result in larger CO2 adsorption capacities as expected, with the trend showing a steeper increase at lower temperatures (up to 50 °C), which slowly levels off at higher temperatures (up to 120 °C). The decrease in capacity over 4 cycles at each regeneration temperature is also largest at the lower regeneration temperatures (up to 40 °C). This drop in capacity is likely due to insufficient energy being supplied in each desorption step to remove all the adsorbed CO2 from the previous adsorption step. These are important considerations for industrial process designs, where energy costs from higher heating must be balanced against the requirement for more material to capture the same amount of CO2, or faster adsorption–desorption cycles.16 This balance will likely vary for different applications and scales, depending on availability and recoverability of heat, capture unit size restraints, and other factors. Overall, the regeneration temperatures required for the UTSA-16(Zn)@MNP-CA (18.7%) MFC were remarkably low (50–60 °C) compared with the temperatures required for regeneration of other adsorbents, including carbon-based materials (120–230 °C),86 zeolites (200 °C)87 and other MFCs (130–145 °C).22 For UTSA-16(Zn)@MNP-CA (18.7%), 60 °C was selected as a representative low regeneration temperature to test the CO2 capacity over 12 cycles of adsorption–desorption (Fig. 4D). No decrease in the CO2 adsorption capacity over the 12 cycles was observed (cycle 1: 2.58 mmol g−1, cycle 12: 2.59 mmol g−1). It is also worth noting here that UTSA16(Zn)@MNP-CA MFCs are all thermally stable up to 250 °C, which is the same as the pristine MOF, indicating that MNP-CA has no detrimental effect on the thermal stability of UTSA-16(Zn) (Fig. S5, ESI†).
As previously introduced, the primary benefit of forming magnetic composites with MOFs for carbon capture applications is the ability of the material to undergo rapid and energy-efficient induction heating for CO2 regeneration. Induction heating is the process by which energy is transferred from an H-field applicator to a material placed in the magnetic field. Heating occurs in magnetic conductive materials through both resistance to induced eddy currents and losses from the magnetisation-demagnetisation cycles in magnetic domains.89 Therefore, the magnetisation and induction heating properties of UTSA16(Zn)@MNP-CA MFCs were measured (Fig. 5 and Table 1). The results for magnetisation of the MFCs as measured by VSM were compared to pristine MNP-CA. Increasing concentrations of MNP-CA resulted in increasing saturation (2.0–14.7 emu g−1) and remanent (0.2–1.2 emu g−1) magnetisations, with intrinsic coercivity remaining roughly constant between samples (50 Oe). The saturation magnetisation values for the MFCs cover a similar range to the 1.5–4 emu g−1 reported for MgFe2O4 in UiO66,21 0.1–3.9 emu g−1 for Fe3O4 in Mg-MOF-7474 and 3.1–18.4 emu g−1 for Fe3O4 in PCN-250.76 The low relative values of the remanent magnetisations compared to saturation magnetisations, and the low intrinsic coercivities, are again advantageous for applications involving magnetic separation, as mentioned earlier for the pristine MNP-CA. There was an approximate direct proportionality between the saturation magnetisation of UTSA16(Zn)@MNP-CA MFCs and the temperature increase observed over a 30 s period of induction heating at 500 W, magnetic field: 164 A m−1 (factor of 7–8.4).
| Sample | Saturation magnetisation (emu g−1, Msat) | Remanent magnetisation (emu g−1, Mr) | Intrinsic coercivity (Oe, Hc) | Temperature increase over 30 s heating (°C, 500 W) |
|---|---|---|---|---|
| a MNP-CA heated very rapidly so was only heated for 8 s (not in triplicate). | ||||
| UTSA-16(Zn) | 0.0 | 0.0 | 0 | 0 |
| UTSA-16(Zn)@MNP-CA (2.6%) | 2.0 | 0.2 | 50 | 14 |
| UTSA-16(Zn)@MNP-CA (5.5%) | 4.3 | 0.3 | 50 | 36 |
| UTSA-16(Zn)@MNP-CA (10.2%) | 8.0 | 0.7 | 50 | 67 |
| UTSA-16(Zn)@MNP-CA (18.7%) | 14.7 | 1.2 | 50 | 123 |
| MNP-CA | 78.3 | 4.2 | 42 | 232a |
These magnetisation and heating results highlight another significant consideration for industrial process design, where heating/cycle durations can be balanced against concentration of MNP-CA in the MFCs (i.e. a higher concentration of MNP-CA in the MFCs results in a lower CO2 capacity, but a shorter heating/cycle time for regeneration. e.g. UTSA16(Zn)@MNP-CA (18.7%): CO2 capacity of 2.8 mmol g−1, reaches 60 °C in 8 s heating. UTSA16(Zn)@MNP-CA (10.2%): CO2 capacity of 3.1 mmol g−1, reaches 60 °C in 15 s heating). A higher power of induction heating would also result in a shorter heating/cycle time, but with a higher energy requirement. Fig. 5B shows the induction heating profiles of the UTSA16(Zn)@MNP-CA MFCs, demonstrating their ability to reach the temperatures required for regeneration (i.e. 60 °C) in only 8–30 seconds, even at low MNP-CA concentrations (5.5–18.7%) and low induction heating power (500 W). Typical regeneration times for alternative adsorbents such as activated carbon and zeolites are reported to be around 20–40 min to achieve >90% CO2 recovery, using conventional or microwave heating,90,91 and other MFCs have reported regeneration times as low as 4–11 min via magnetic induction heating.21,22,75
To examine the distribution of MNP-CA within UTSA-16(Zn) for the UTSA16(Zn)@MNP-CA MFCs, TEM and SEM imaging with EDX elemental analyses were conducted on UTSA-16(Zn)@MNP-CA (18.7%). The average particle size of the MFC was determined by SEM image analyses on 100 particles, showing a mean diameter of 55 ± 16 nm (standard deviation). Results of the TEM analyses are shown in Fig. 6. UTSA-16(Zn)@MNP-CA (18.7%) was sonicated to break down the powder into primary agglomerates, where it was observed that good mixing between the MOF and the MNP-CA had occurred, i.e. primary agglomerates contained MOF and MNP-CA nanoparticles bound together. This result was further corroborated by SEM and EDX analyses, where Zn from UTSA-16(Zn) (green) and Fe from MNP-CA (pink) could be clearly seen (Fig. S6, ESI†). The uniform distribution of MNP-CA and UTSA-16(Zn) nanoparticles in primary agglomerates meant that a high wt% MOF loading (81–97%) could be used in the MFCs, whilst still ensuring effective induction heating throughout the material. These composite structures differ from the thin MOF layers grown on individual nanoparticles (core–shell structures) frequently observed for other MFCs in the literature, such as HKUST-1 on Fe3O440,45 or ZIF-8 on Fe3O4.41 These core–shell structures are usually prepared by lengthy layer-by-layer syntheses methods, with much lower wt% MOF loadings in the composites.40,45 Although they demonstrate efficacy in catalysis applications,40 the low MOF loadings would result in low gravimetric capacities in adsorption applications. The MNP-CA and UTSA-16(Zn) nanoparticle primary agglomerates also differ in morphology from MFCs synthesised by lengthy direct-growth methods, where nanoparticles are embedded in large MOF crystals, such as MgFe2O4 in Mg-MOF-7422 or Fe3O4 in Co-MOF-7477 prepared via 15 h and 2.5 day syntheses, respectively.
 | ||
| Fig. 6 (A) and (D) TEM images of agglomerate particles of MFC UTSA-16(Zn)@MNP-CA (18.7%), containing both UTSA-16(Zn) and MNP-CA nanoparticles. (B), (C), (E) and (F) TEM EDX images of the MFC showing the distribution of Zn (green) from UTSA-16(Zn) and Fe (pink) from MNP-CA in the agglomerate particles. | ||
This work shows that MFCs containing UTSA-16(Zn) and MNP-CA can be synthesised in a facile, low-cost, rapid, and sustainable manner, with varying concentrations of MNP-CA for applications with different magnetic property requirements. The high CO2 adsorption capacity (3.4–3.5 mmol g−1) of the MOF in the MFCs was unaffected by the incorporation of MNP-CA, and the resulting MFCs showed highly effective thermal regeneration at temperatures as low as 60 °C, which could be attained in seconds via the energy efficient induction heating process.
4. Conclusions
In this work, the synthesis, characterisation, and testing of sustainable and novel UTSA-16(Zn)@MNP-CA MFCs for post-combustion carbon-capture applications is described. Firstly, a single-step and scalable continuous-flow hydrothermal method was developed for producing citrate-coated Fe3O4 nanoparticles (MNP-CA), mean diameter: 19 ± 11 nm. MNP-CA showed a very high saturation magnetisation (78 emu g−1) compared to previously published alternatives, due to its very high purity and crystallinity. This method not only avoided the use of harsh and lengthy batch synthesis conditions, but also citrate coated the nanoparticles in situ, removing the need for additional functionalisation steps.Next, various concentrations of MNP-CA (2.6–18.7 wt%) were incorporated into UTSA-16(Zn) MOF via a rapid 10 min 60 °C microwave direct-growth synthesis to produce innovative UTSA-16(Zn)@MNP-CA MFCs (magnetisation: 2–15 emu g−1). The yield of the MFCs was shown to slightly benefit from MNP-CA incorporation compared to the pristine MOF (81–83% for the MFCs, 77% for the pristine MOF). TEM and XRD analyses revealed that the MFCs contain agglomerates of bound crystalline MOF and MNP-CA nanoparticles, which maintain high CO2 adsorption capacities (2.8–3.3 mmol g−1) and recyclability, with no loss of MOF CO2 capacity with increasing NMP-CA loading in the MFCs. Regeneration temperature had an impact on both total CO2 capacity and capacity loss over multiple cycles. Remarkably low temperatures and short timescales were sufficient for regeneration of the MFCs compared with other solid adsorbents. The MFCs also exhibited the desired rapid magnetic induction heating (e.g. 60 °C in 8 seconds), overcoming the challenge of rapid and low energy thermal CO2 regeneration in CO2 capture applications.
These MFCs have demonstrated leading adsorption profiles and regeneration heating capabilities for CO2 release, whilst being produced in a scalable and sustainable manner from cheap and widely available materials. The combination of these properties represents a significant advancement in the field of MOFs/MFCs for CO2 capture, making the next step toward industrial application. The MFCs should next be produced and tested on a pilot-scale carbon capture unit prior to industrial use. In addition, since the MNP-CA and the MFC synthesis method can also be applied to other MOFs, a variety of sustainable MFCs could also by synthesised for impact in wide-reaching applications.
Author contributions
John Luke Woodliffe: conceptualization, formal analysis, validation, funding acquisition (via Henry Royce Institute), investigation, methodology, writing – original draft, writing – review & editing, visualization. Amy-Louise Johnston: investigation, writing – original draft, visualization. Michael Fay: investigation. Rebecca Ferrari: conceptualization, writing – review & editing. Rachel L. Gomes: writing – review & editing. Ed Lester: resources, writing – review & editing. Ifty Ahmed: conceptualization, supervision, writing – review & editing. Andrea Laybourn: conceptualization, project administration, supervision, writing – review & editing, funding acquisition (EPSRC CDT).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors give thanks to and acknowledge funding from the Engineering and Physical Sciences Research Council (EPSRC) and Science Foundation Ireland (SFI) Centre for Doctoral Training in Sustainable Chemistry: Atoms-2-Products an Integrated Approach to Sustainable Chemistry (Grant no. EP/S022236/1). The authors acknowledge the support of the Henry Royce Institute for advanced materials for John Luke Woodliffe through the Student Equipment Access Scheme enabling access to QD MPMS 3 facilities at Royce Discovery Centre, the University of Sheffield; EPSRC Grant Number EP/R00661X/1 & EP/P02470X/1. SEM and TEM analyses in this work were supported by the Engineering and Physical Sciences Research Council (EPSRC) [under grants EP/L022494/1 and EP/W006413/1] and the University of Nottingham. The authors thank the Nanoscale and Microscale Research Centre (nmRC) at the University of Nottingham for providing access to SEM and TEM instrumentation. The authors also thank Venus So and Promethean Particles for access to a Malvern Zetasizer and Micromeritics Tristar II 3020 instruments.References
- D. Lüthi, M. Le Floch, B. Bereiter, T. Blunier, J.-M. Barnola, U. Siegenthaler, D. Raynaud, J. Jouzel, H. Fischer, K. Kawamura and T. F. Stocker, Nature, 2008, 453, 379–382 CrossRef PubMed.
- J. R. Petit, J. Jouzel, D. Raynaud, N. I. Barkov, J.-M. Barnola, I. Basile, M. Bender, J. Chappellaz, M. Davis, G. Delaygue, M. Delmotte, V. M. Kotlyakov, M. Legrand, V. Y. Lipenkov, C. Lorius, L. Pépin, C. Ritz, E. Saltzman and M. Stievenard, Nature, 1999, 399, 429–436 CrossRef CAS.
- Global Monitoring Laboratory, https://www.esrl.noaa.gov/gmd/ccgg/trends/, (accessed 31 August 2023).
- IPCC, Climate Change 2014: Impacts, Adaptation, and Vulnerability. Part A: Global and Sectoral Aspects. Contribution of Working Group II to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2014.
- World Health Organization, Quantitative risk assessment of the effects of climate change on selected causes of death, 2030s and 2050s, ed. S. Hales, S. Kovats, S. Lloyd and D. Campbell-Lendrum, Geneva, 2014 Search PubMed.
- IPCC, Global Warming of 1.5 °C. An IPCC Special Report on the impacts of global warming of 1.5 °C above pre-industrial levels and related global greenhouse gas emission pathways, in the context of strengthening the global response to the threat of climate change, Cambridge University Press, Cambridge, UK and New York, NY, USA, 2018.
- United Nations, Report of the Conference of the Parties on its twenty-sixth session, held in Glasgow from 31 October to 13 November 2021, 2022.
- IEA, Global Energy & CO2 Status Report 2019, Paris, 2019.
- IEA, CO2 Emissions from Fuel Combustion, Paris, 2019 Search PubMed.
- S. Sjostrom and H. Krutka, Fuel, 2010, 89, 1298–1306 CrossRef CAS.
- D. M. D’Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- A. Samanta, A. Zhao, G. K. H. Shimizu, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2012, 51, 1438–1463 CrossRef CAS.
- O. M. Yaghi and H. Li, J. Am. Chem. Soc., 1995, 117, 10401–10402 CrossRef CAS.
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- Z. Hu, Y. Wang, B. B. Shah and D. Zhao, Adv. Sustain. Syst., 2019, 3, 1800080 CrossRef.
- J. L. Woodliffe, R. S. Ferrari, I. Ahmed and A. Laybourn, Coord. Chem. Rev., 2021, 428, 213578 CrossRef CAS.
- J. Yu, L.-H. Xie, J.-R. Li, Y. Ma, J. M. Seminario and P. B. Balbuena, Chem. Rev., 2017, 117, 9674–9754 CrossRef CAS PubMed.
- B. L. Huang, Z. Ni, A. Millward, A. J. H. McGaughey, C. Uher, M. Kaviany and O. Yaghi, Int. J. Heat Mass Transf., 2007, 50, 405–411 CrossRef CAS.
- V. Rudnev, D. Loveless and R. L. Cook, Handbook of Induction Heating, CRC Press, 2017 Search PubMed.
- M. M. Sadiq, H. Li, A. J. Hill, P. Falcaro, M. R. Hill and K. Suzuki, Chem. Mater., 2016, 28, 6219–6226 CrossRef CAS.
- M. M. Sadiq, K. Konstas, P. Falcaro, A. J. Hill, K. Suzuki and M. R. Hill, Cell Reports Phys. Sci., 2020, 1, 100070 CrossRef.
- S. Valluri and S. K. Kawatra, Fuel Process. Technol., 2021, 213, 106691 CrossRef CAS.
- B. E. Meteku, J. Huang, J. Zeng, F. Subhan, F. Feng, Y. Zhang, Z. Qiu, S. Aslam, G. Li and Z. Yan, Coord. Chem. Rev., 2020, 413, 213261 CrossRef CAS.
- Y. J. Ma, X. X. Jiang and Y. K. Lv, Chem. – Asian J., 2019, 14, 3515–3530 CrossRef CAS PubMed.
- G. Zhao, N. Qin, A. Pan, X. Wu, C. Peng, F. Ke, M. Iqbal, K. Ramachandraiah and J. Zhu, J. Nanomater., 2019, 2019, 1454358 Search PubMed.
- M. M. Sadiq, K. Suzuki and M. R. Hill, Chem. Commun., 2018, 54, 2825–2837 RSC.
- Q. Yang, Q. Xu and H.-L. Jiang, Chem. Soc. Rev., 2017, 46, 4774–4808 RSC.
- F. Maya, C. Palomino Cabello, R. M. Frizzarin, J. M. Estela, G. Turnes Palomino and V. Cerdà, TrAC, Trends Anal. Chem., 2017, 90, 142–152 CrossRef CAS.
- P. Falcaro, R. Ricco, A. Yazdi, I. Imaz, S. Furukawa, D. Maspoch, R. Ameloot, J. D. Evans and C. J. Doonan, Coord. Chem. Rev., 2016, 307, 237–254 CrossRef CAS.
- R. Taghavi, S. Rostamnia, M. Farajzadeh, H. Karimi-Maleh, J. Wang, D. Kim, H. W. Jang, R. Luque, R. S. Varma and M. Shokouhimehr, Inorg. Chem., 2022, 61, 15747–15783 CrossRef CAS PubMed.
- X. Han, K. Xu, O. Taratula and K. Farsad, Nanoscale, 2019, 11, 799–819 RSC.
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167–R181 CrossRef CAS.
- K. Wu, D. Su, J. Liu, R. Saha and J.-P. Wang, Nanotechnology, 2019, 30, 502003 CrossRef CAS PubMed.
- L. Gloag, M. Mehdipour, D. Chen, R. D. Tilley and J. J. Gooding, Adv. Mater., 2019, 31, 1904385 CrossRef CAS PubMed.
- M. A. Dheyab, A. A. Aziz, M. S. Jameel, O. A. Noqta, P. M. Khaniabadi and B. Mehrdel, Sci. Rep., 2020, 10, 10793 CrossRef CAS PubMed.
- L. Maurizi, F. Bouyer, J. Paris, F. Demoisson, L. Saviot and N. Millot, Chem. Commun., 2011, 47, 11706 RSC.
- S. Nigam, K. C. Barick and D. Bahadur, J. Magn. Magn. Mater., 2011, 323, 237–243 CrossRef CAS.
- W. Xie and F. Wan, Fuel, 2018, 220, 248–256 CrossRef CAS.
- J. Li, H. Gao, L. Tan, Y. Luan and M. Yang, Eur. J. Inorg. Chem., 2016, 4906–4912 CrossRef CAS.
- T. Zhang, X. Zhang, X. Yan, L. Kong, G. Zhang, H. Liu, J. Qiu and K. L. Yeung, Chem. Eng. J., 2013, 228, 398–404 CrossRef CAS.
- J. Li, J. Wang, Y. Ling, Z. Chen, M. Gao, X. Zhang and Y. Zhou, Chem. Commun., 2017, 53, 4018–4021 RSC.
- T. Xu, X. Hou, S. Liu and B. Liu, Microporous Mesoporous Mater., 2018, 259, 178–183 CrossRef CAS.
- M. Saikia, D. Bhuyan and L. Saikia, New J. Chem., 2015, 39, 64–67 RSC.
- F. Ke, L.-G. Qiu, Y.-P. Yuan, X. Jiang and J.-F. Zhu, J. Mater. Chem., 2012, 22, 9497–9500 RSC.
- S. Gaikwad, S.-J. Kim and S. Han, J. Ind. Eng. Chem., 2020, 87, 250–263 CrossRef CAS.
- E. Lester, P. Blood, J. Denyer, D. Giddings, B. Azzopardi and M. Poliakoff, J. Supercrit. Fluids, 2006, 37, 209–214 CrossRef CAS.
- P. W. Dunne, C. L. Starkey, A. S. Munn, S. V. Y. Tang, O. Luebben, I. Shvets, A. G. Ryder, Y. Casamayou-Boucau, L. Morrison and E. H. Lester, Chem. Eng. J., 2016, 289, 433–441 CrossRef CAS.
- I. Clark, P. W. Dunne, R. L. Gomes and E. Lester, J. Colloid Interface Sci., 2017, 504, 492–499 CrossRef CAS PubMed.
- S. Gates-Rector and T. Blanton, Powder Diffr., 2019, 34, 352–360 CrossRef CAS.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- T. Adschiri, Y. Hakuta and K. Arai, Ind. Eng. Chem. Res., 2000, 39, 4901–4907 CrossRef CAS.
- T. Adschiri, K. Kanazawa and K. Arai, J. Am. Ceram. Soc., 1992, 75, 1019–1022 CrossRef CAS.
- P. S. Shah, PhD thesis, Imperial College London, 2020.
- R. M. Cornell and U. Schwertmann, The Iron Oxides, Wiley, 2003, pp. 111–137 Search PubMed.
- E. A. Kwizera, S. Stewart, M. M. Mahmud and X. He, J. Heat Transfer, 2022, 144, 030801 CrossRef CAS PubMed.
- J. H. Li, P. Santos-Otte, B. Au, J. Rentsch, S. Block and H. Ewers, Nat. Commun., 2020, 11, 4259 CrossRef PubMed.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed.
- D. Singh, R. K. Gautam, R. Kumar, B. K. Shukla, V. Shankar and V. Krishna, J. Water Process Eng., 2014, 4, 233–241 CrossRef.
- L. Li, K. Y. Mak, C. W. Leung, K. Y. Chan, W. K. Chan, W. Zhong and P. W. T. Pong, Microelectron. Eng., 2013, 110, 329–334 CrossRef CAS.
- C. Hui, C. Shen, T. Yang, L. Bao, J. Tian, H. Ding, C. Li and H.-J. Gao, J. Phys. Chem. C, 2008, 112, 11336–11339 CrossRef CAS.
- M. Arefi, M. Kazemi Miraki, R. Mostafalu, M. Satari and A. Heydari, J. Iran. Chem. Soc., 2019, 16, 393–400 CrossRef CAS.
- J.-W. Park and J. S. Shumaker-Parry, J. Am. Chem. Soc., 2014, 136, 1907–1921 CrossRef CAS PubMed.
- Z. Li, H. Chen, H. Bao and M. Gao, Chem. Mater., 2004, 16, 1391–1393 CrossRef CAS.
- V. E. Noval and J. G. Carriazo, Mater. Res., 2019, 22, e20180660 CrossRef CAS.
- Y. Hadadian, H. Masoomi, A. Dinari, C. Ryu, S. Hwang, S. Kim, B. Ki Cho, J. Y. Lee and J. Yoon, ACS Omega, 2022, 7, 15996–16012 CrossRef CAS PubMed.
- J. Mohapatra, F. Zeng, K. Elkins, M. Xing, M. Ghimire, S. Yoon, S. R. Mishra and J. P. Liu, Phys. Chem. Chem. Phys., 2018, 20, 12879–12887 RSC.
- P. Xu, G. M. Zeng, D. L. Huang, C. L. Feng, S. Hu, M. H. Zhao, C. Lai, Z. Wei, C. Huang, G. X. Xie and Z. F. Liu, Sci. Total Environ., 2012, 424, 1–10 CrossRef CAS PubMed.
- M. Hua, S. Zhang, B. Pan, W. Zhang, L. Lv and Q. Zhang, J. Hazard. Mater., 2012, 211–212, 317–331 CrossRef CAS PubMed.
- A.-H. Lu, W. Schmidt, N. Matoussevitch, H. Bönnemann, B. Spliethoff, B. Tesche, E. Bill, W. Kiefer and F. Schüth, Angew. Chem., Int. Ed., 2004, 43, 4303–4306 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- Q.-L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468–5512 RSC.
- S. B. Peh, S. Xi, A. Karmakar, J. Y. Yeo, Y. Wang and D. Zhao, Inorg. Chem., 2020, 59, 9350–9355 CrossRef CAS PubMed.
- H. Li, M. M. Sadiq, K. Suzuki, R. Ricco, C. Doblin, A. J. Hill, S. Lim, P. Falcaro and M. R. Hill, Adv. Mater., 2016, 28, 1839–1844 CrossRef CAS PubMed.
- M. M. Sadiq, M. Rubio-Martinez, F. Zadehahmadi, K. Suzuki and M. R. Hill, Ind. Eng. Chem. Res., 2018, 57, 6040–6047 CrossRef CAS.
- H. Li, M. M. Sadiq, K. Suzuki, C. Doblin, S. Lim, P. Falcaro, A. J. Hill and M. R. Hill, J. Mater. Chem. A, 2016, 4, 18757–18762 RSC.
- L. Melag, M. M. Sadiq, S. J. D. Smith, K. Konstas, K. Suzuki and M. R. Hill, J. Mater. Chem. A, 2019, 7, 3790–3796 RSC.
- M. G. Plaza, C. Pevida, B. Arias, J. Fermoso, A. Arenillas, F. Rubiera and J. J. Pis, J. Therm. Anal. Calorim., 2008, 92, 601–606 CrossRef CAS.
- P. Railey, Y. Song, T. Liu and Y. Li, Mater. Res. Bull., 2017, 96, 385–394 CrossRef CAS.
- M. Ding, X. Cai and H.-L. Jiang, Chem. Sci., 2019, 10, 10209–10230 RSC.
- Z. Li, P. Liu, C. Ou and X. Dong, ACS Sustainable Chem. Eng., 2020, 8, 15378–15404 CrossRef CAS.
- J. Zhu, Z. Yang, X. Li, S. Qi and M. Jia, Energy Sci. Eng., 2018, 6, 548–562 CrossRef.
- D. K. Kim, M. S. Amin, S. Elborai, S.-H. Lee, Y. Koseoglu, M. Zahn and M. Muhammed, J. Appl. Phys., 2005, 97, 10J510 CrossRef.
- P. Sinha, A. Datar, C. Jeong, X. Deng, Y. G. Chung and L.-C. Lin, J. Phys. Chem. C, 2019, 123, 20195–20209 CrossRef CAS.
- J. W. M. Osterrieth, J. Rampersad, D. Madden, N. Rampal, L. Skoric, B. Connolly, M. D. Allendorf, V. Stavila, J. L. Snider, R. Ameloot, J. Marreiros, C. Ania, D. Azevedo, E. Vilarrasa-Garcia, B. F. Santos, X. Bu, Z. Chang, H. Bunzen, N. R. Champness, S. L. Griffin, B. Chen, R. Lin, B. Coasne, S. Cohen, J. C. Moreton, Y. J. Colón, L. Chen, R. Clowes, F. Coudert, Y. Cui, B. Hou, D. M. D’Alessandro, P. W. Doheny, M. Dincă, C. Sun, C. Doonan, M. T. Huxley, J. D. Evans, P. Falcaro, R. Ricco, O. Farha, K. B. Idrees, T. Islamoglu, P. Feng, H. Yang, R. S. Forgan, D. Bara, S. Furukawa, E. Sanchez, J. Gascon, S. Telalović, S. K. Ghosh, S. Mukherjee, M. R. Hill, M. M. Sadiq, P. Horcajada, P. Salcedo-Abraira, K. Kaneko, R. Kukobat, J. Kenvin, S. Keskin, S. Kitagawa, K. Otake, R. P. Lively, S. J. A. DeWitt, P. Llewellyn, B. V. Lotsch, S. T. Emmerling, A. M. Pütz, C. Martí-Gastaldo, N. M. Padial, J. García-Martínez, N. Linares, D. Maspoch, J. A. Suárez del Pino, P. Moghadam, R. Oktavian, R. E. Morris, P. S. Wheatley, J. Navarro, C. Petit, D. Danaci, M. J. Rosseinsky, A. P. Katsoulidis, M. Schröder, X. Han, S. Yang, C. Serre, G. Mouchaham, D. S. Sholl, R. Thyagarajan, D. Siderius, R. Q. Snurr, R. B. Goncalves, S. Telfer, S. J. Lee, V. P. Ting, J. L. Rowlandson, T. Uemura, T. Iiyuka, M. A. van der Veen, D. Rega, V. Van Speybroeck, S. M. J. Rogge, A. Lamaire, K. S. Walton, L. W. Bingel, S. Wuttke, J. Andreo, O. Yaghi, B. Zhang, C. T. Yavuz, T. S. Nguyen, F. Zamora, C. Montoro, H. Zhou, A. Kirchon and D. Fairen-Jimenez, Adv. Mater., 2022, 34, 2201502 CrossRef CAS PubMed.
- A. E. Creamer and B. Gao, Environ. Sci. Technol., 2016, 50, 7276–7289 CrossRef CAS PubMed.
- S. J. Chen, M. Zhu, Y. Fu, Y. X. Huang, Z. C. Tao and W. L. Li, Appl. Energy, 2017, 191, 87–98 CrossRef CAS.
- J. Rouquerol, P. Llewellyn and F. Rouquerol, in Characterization of Porous Solids VII, Studies in Surface Science and Catalysis, ed. P. Llewellyn, F. Rodriquez-Reinoso, J. Rouqerol and N. Seaton, Elsevier, 2007, vol. 160, pp. 49–56 Search PubMed.
- M. Mehdizadeh, Microwave/RF Applicators and Probes, Elsevier, 2015, pp. 219–268 Search PubMed.
- M. M. Yassin, J. A. Anderson, G. A. Dimitrakis and C. F. Martín, Sep. Purif. Technol., 2021, 276, 119326 CrossRef CAS.
- R. Morales-Ospino, R. G. Santiago, R. M. Siqueira, D. C. S. de Azevedo and M. Bastos-Neto, Adsorption, 2020, 26, 813–824 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00351e |
| This journal is © The Royal Society of Chemistry 2023 |