
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA covalent inhibitor of the YAP–TEAD transcriptional complex identified by high-throughput screening†
Kayla
Nutsch‡
 a,
Lirui
Song‡
b,
Emily
Chen
b,
Mitchell
Hull
b,
Arnab K.
Chatterjee
b,
Jian Jeffery
Chen
*b and
Michael J.
Bollong
*a
a,
Lirui
Song‡
b,
Emily
Chen
b,
Mitchell
Hull
b,
Arnab K.
Chatterjee
b,
Jian Jeffery
Chen
*b and
Michael J.
Bollong
*a
aDepartment of Chemistry, The Scripps Research Institute, La Jolla, CA 92037, USA. E-mail: mbollong@scripps.edu
bCalibr, A Division of Scripps Research, La Jolla, CA 92037, USA. E-mail: jianjchen@scripps.edu
First published on 29th August 2023
Abstract
Yes-associated protein (YAP), the master transcriptional effector downstream of the Hippo pathway, regulates essential cell growth and regenerative processes in animals. However, the activation of YAP observed in cancers drives cellular proliferation, metastasis, chemoresistance, and immune suppression, making it of key interest in developing precision therapeutics for oncology. As such, pharmacological inhibition of YAP by targeting its essential co-regulators, TEA domain transcription factors (TEADs) would likely promote tumor clearance in sensitive tumor types. From a fluorescence polarization-based high throughput screen of over 800![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 diverse small molecules, here we report the identification of a pyrazolopyrimidine-based scaffold that inhibits association of YAP and TEADs. Medicinal chemistry-based optimization identified mCMY020, a potent, covalent inhibitor of TEAD transcriptional activity that occupies a conserved, central palmitoylation site on TEADs.
000 diverse small molecules, here we report the identification of a pyrazolopyrimidine-based scaffold that inhibits association of YAP and TEADs. Medicinal chemistry-based optimization identified mCMY020, a potent, covalent inhibitor of TEAD transcriptional activity that occupies a conserved, central palmitoylation site on TEADs.
Introduction
Genetic screens for candidate tumor suppressor genes in Drosophila melanogaster revealed a conserved regulatory network that governs cellular proliferation and organ size, termed the Hippo pathway after the first discovered gene Hpo.1–3 At its core, the Hippo pathway canonically consists of a linear kinase cascade in which the serine/threonine protein kinases macrophage stimulating 1 and 2 (MST1/2) and large tumor suppressor kinase 1 and 2 (LATS1/2) along with their scaffolding proteins Salvador (SAV) and monopolar spindle-one-binder proteins (MOB) result in the phosphorylation of YAP in response to extracellular signals, cell density, cell polarity, mechanotransduction, and cellular stress. In environments of high cellular density, Hippo pathway activation results in LATS1/2 mediated phosphorylation of YAP, sequestering it in the cytoplasm via interactions with 14-3-3 proteins. When the Hippo pathway is inactivated, LATS remains unphosphorylated and does not phosphorylate YAP, allowing nuclear translocation of YAP where it binds transcriptional coactivator TEA-domain (TEAD) proteins and executes a pro-proliferative transcriptional program consisting of several dozen target transcripts.4 In addition to its role as a central regulator of cellular proliferation, YAP can be constitutively activated in human cancers, enabling tumor cell intrinsic transcriptional programs such as proliferation, stemness, and metastasis, as well as tumor microenvironmental cues such as recruitment and activation of cancer-associated fibroblasts and immune evasion.5–16 Inactivating mutations in the Hippo pathway, which activate YAP, are extensive in malignant mesotheliomas and a rare schwannoma, NF217 but are not the driving factor in many cancers. Upregulation of YAP in most malignant tumors is induced following anti-cancer treatment, driving therapy resistance, likely due to the crosstalk between the Hippo pathway and other oncogenic drivers.12,18 Tumorigenic cells derived from dysregulated Hippo signaling can become uniquely dependent on YAP for cell growth, making it a key target for pharmacological therapeutic intervention.19YAP is an intrinsically disordered protein, making it difficult to target directly with pharmacological agents. Accordingly, targeting the interface of YAP and TEADs with drug like small molecules has become a promising strategy. Upon binding to TEAD, YAP inducibly forms secondary structures at three distinct interfaces.20 It has been shown that interface 1 is dispensable for the binding of YAP and TEAD, while disruption of binding at interfaces 2 and 3 lead to disassociation of the complex.20–22 In addition to these protein interaction sites, TEAD contains a deep hydrophobic pocket with a surface accessible cysteine that is palmitoylated in the active transcriptional species. Typically, palmitoylation of proteins is an enzyme catalyzed modification that results in the trafficking of proteins to cell membranes. However, TEAD is one of few proteins that undergoes auto-palmitoylation in the presence of cellular palmitoyl-CoA pools, which is essential for its transcriptional activity and stability.23–25 Whether palmitoylation affects the association of YAP and TEADs is unclear and there are conflicting reports to this end in the literature.24,26,27 The development of novel TEAD selective small molecule inhibitors would provide a tool to uncover the regulatory mechanisms of TEAD palmitoylation and have the potential to be further developed into therapeutic agents for cancer treatment.
Here, we performed a fluorescence polarization (FP) high-throughput screen to identify novel small molecules capable of disrupting the interaction of YAP and TEAD. A library of >826000 diverse small molecules was screened, resulting in the identification of a novel TEAD inhibitor scaffold. Selected hit molecules were further optimized to produce a covalent sub-micromolar inhibitor, mCMY020, that prevents palmitoylation, decreases YAP-driven transcription, and selectively slows proliferation of Hippo deficient cancer cells.
Results
Fluorescence polarization screen identifies inhibitors of the YAP–TEAD interaction
To identify small molecules capable of disrupting the interaction between YAP and TEAD, we first optimized a miniaturized fluorescence polarization-based assay to enable high throughput chemical screening. As interface three is crucial for the stabilization of the YAP–TEAD complex, we utilized a seventeen amino-acid peptide from the sequence of YAP mimicking the omega loop (residues 84–100) which contained several potency increasing mutations (VP{3-Cl-Phe}{Hcy}LRK{NIe}PASFCKPPE with a disulfide bond between Hcy and Cys) and an N-terminal tetramethylrhodamine (TMR)-based fluorophore (TMR-YAP)28 (Fig. 1A). In optimized conditions (50 nM of TMR-YAP) the approximate binding affinity of the non-acylated YAP binding domain (YBD) of TEAD4 (TEAD4-YBD) was determined to be 186.3 nM (Fig. 1B). An unlabeled YAP peptide containing the same amino acid sequence without TMR (unlabeled-YAP), was able to competitively inhibit TMR-YAP binding to TEAD4-YBD with an IC50 of 6.3 μM (Fig. 1C). Using unlabeled-YAP as a positive competition control, a Z-score of 0.86 with a standard deviation of 2.61 and coefficient of variant of 0.12 for 16 replicates, could be achieved in 384-well format and was deemed suitable for a high throughput screening campaign (Fig. 1D).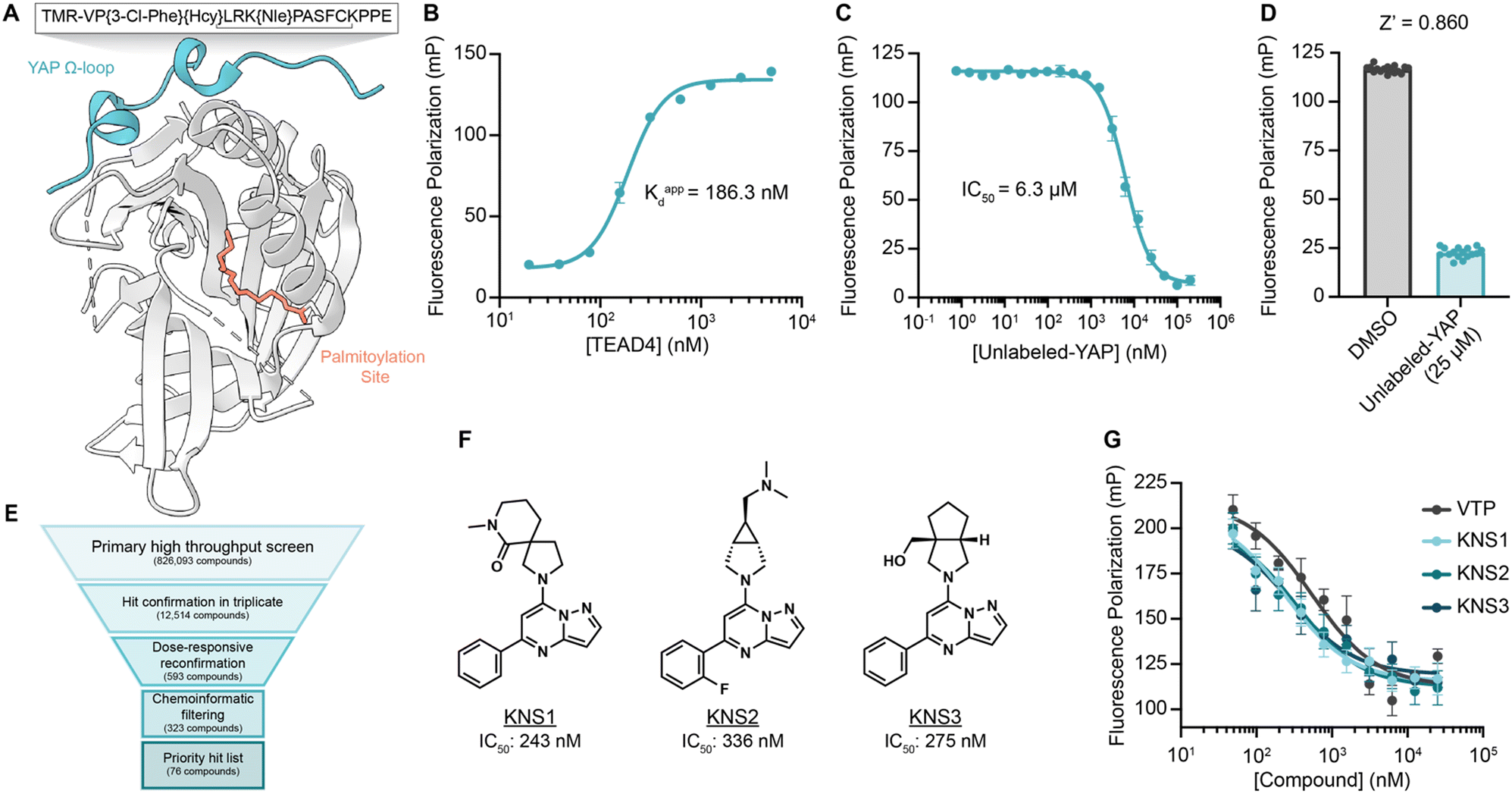 | ||
| Fig. 1 A fluorescence polarization-based screen for YAP–TEAD interaction inhibition identifies a series of efficacious pyrazolopyrimidines. (A) Schematic depicting TEAD4-YBD protein and TMR-YAP peptide used in the FP assay. Hcy, homocysteine; Nle, norleucine. (B) Binding of TMR-YAP (50 nM) to TEAD4-YBD in FP assay demonstrates an approximate Kd of 186.3 nM (n = 10, mean ± SEM). (C) Fluorescence polarization (mP) of TMR-YAP (50 nM) and TEAD4-YBD (1 μM) in response to increasing concentrations of unlabeled YAP peptide with an IC50 of 6.27 μM (n = 3, mean ± SEM). (D) Z-Score of a representative FP assay in 384-well format with 1 μM TEAD4-YBD and 50 nM TMR-YAP (n = 16, mean ± SEM). (E) Screening funnel depicting the high throughput screening campaign. (F) Structures of KNS1, KNS2, and KNS3 with their respective IC50 values. Structure of VTP can be found in Fig. S1 (ESI†). (G) Fluorescence polarization values in response to the indicated concentrations of KNS1, KNS2, KNS3, and VTP in 384-well format (n = 3, mean ± SEM). | ||
The FP assay was further miniaturized to 1536-well format and then screened at a single concentration (10 μM) against >826000 compounds: a library consisting of a 588000-compound proprietary collection, 215000 commercially obtained compounds, a 10000-compound bioactive collection, and the 12000-compound ReFRAME Library.29 This effort yielded 323 compounds with dose-responsive inhibitory activity in the primary screening assay. Among these, 76 culled hit compounds were manually selected for further study, as many screening hits contained fluorophores, known fluorescent quenching moieties (e.g., anilines, diazo motifs), or screening PAINS-like structures (e.g., catechols). Notably, among these, we identified several porphyrin containing molecules (kCAW794, kCBN853, and kCAS717, Fig. S1A, ESI†) that displayed low micromolar inhibitory potencies (9.6 μM, 6.9 μM, and 4.7 μM respectively, Fig. S1B, ESI†) in this assay. Interestingly, these molecules resemble the structure of reported YAP–TEAD interaction inhibitor Verteporfin30 (VTP), although VTP was considerably more potent in this assay (IC50 = 544 nM). Additionally, we identified a pyrazolopyrimidine-based scaffold that was the most prevalent among identified hit classes. From experiments performed in 1536-well format using library material, 11 of these compounds displayed inhibitory activity in the FP assay ranging from 5.9–26 μM (Fig. S1C and D, ESI†). Given this activity as well as the observation that no pyrazolopyrimidine has yet been described as possessing YAP inhibitory activity, we pursued this scaffold further. We next purchased three representative commercially available analogs (KNS1, KNS2, and KNS3) and found that each displayed sub-micromolar potency in the FP assay (243 nM, 336 nM, and 275 nM respectively, Fig. 1F and G), performing similarly to VTP in this context.
Identification of efficacious TEAD inhibitor mCMY020
To confirm the identified compounds bound to TEAD4, we measured the thermal stability of TEAD4-YBD upon treatment using a fluorescence-based thermal denaturation assay (Life Technologies). KNS1, KNS2, and KNS3 were all confirmed to engage TEAD4, shifting the thermal melting temperature by 3.71 °C, 1.88 °C, and 1.47 °C respectively (Fig. S2, ESI†). Next, to validate the capacity of this chemical scaffold to inhibit the YAP and TEAD interaction in cells, we tested the compounds in a Gal4-responsive reporter in which an exogenously supplied Gal4-TEAD4 fusion protein drives transcription of luciferase in the presence of a functional interaction with expressed YAP protein (Fig. S3A and B, ESI†). Consistent with an ability to inhibit this required protein–protein interaction, KNS1 and KNS2 (20 and 50 μM) were found to inhibit luciferase signal to a similar magnitude as that observed for VTP (10, 20, and 50 μM) over a 24 hour period (Fig. S3C, ESI†). In contrast, KNS3 displayed a more modest activity in cells that was not statistically significant, suggesting that this molecule likely does not efficiently engage TEAD4 in cells. While KNS1 and KNS2 display a more modest potency, these data confirm that the identified pyrazolopyrimidine scaffold functionally antagonizes the YAP–TEAD transcriptional complex in cells.To date, two reported mechanisms have been shown to disrupt the interaction between YAP and TEAD, either by inhibiting the protein–protein interaction at interface 2 or 3 (alpha-helix or omega loop respectively) or by occupying the hydrophobic core resulting in an allosteric change in affinity for YAP.27,28,31 As our fluorescence polarization screen was unbiased for detecting an inhibitor that acted by either mechanism, we next sought to determine if the identified pyrazolopyrimidine series could bind the hydrophobic core of TEAD. We reasoned that binders to the palmitoylation site could likely be identified from experiments in which the non-enzymatic modification of TEAD4 by an alkyne modified palmitoyl-CoA (Fig. S4A, ESI†), might be completed by pre-treatment with compound. Accordingly, we found that KNS1, KNS2, and KNS3 (10 and 100 μM) could compete with alkyne palmitoyl-CoA for labeling of TEAD 4 in vitro (Fig. S4B and C, ESI†). This data strongly suggested that the identified compounds bind to the palmitoylation pocket of TEAD, likely resulting in an allosteric disassociation of YAP.
To better understand the potential binding mode of the pyrazolopyrimidine scaffold within the hydrophobic pocket, KNS1, KNS2, and KNS3 were computationally docked into the palmitoylation site of an existing co-crystal structure of TEAD4 (PDB: 5OAQ) using Schrodinger XP Glide32 (Fig. S5A–C, ESI†). Evaluating the highest scoring docking pose for each molecule, it was observed that the hydrophobic phenyl moiety of each compound was buried within the more lipophilic palmitoyl binding pocket whereas the more polar northern substituents of the scaffold (as depicted in Fig. 1F) faced the solvent accessible pocket entrance near the conserved acylated cysteine (Fig. S5D, ESI†). A salient observation from this work was that the full depth of this pocket was not fully engaged in the binding mode adopted by the scaffold. Likewise, the scaffold docked most efficiently near the conserved cysteine (2.86 Å, 3.70 Å, or 6.40 Å for KNS1, KNS2, or KNS3 respectively). We reasoned that an elongated molecule that more effectively occupies the lipid binding site and engages the active site cysteine might more potently inhibit TEAD activity in cells. As such, from several iterative medicinal chemistry-based efforts, we designed and synthesized mCMY020 (Fig. 2A), which bears a western cyclohexyl substituent as well as a 2-fluoro acrylamide, a reactivity handle introduced to covalently engage the active site cysteine. Docking studies with mCMY020 and TEAD4 suggested it fully occupied the palmitoylation pocket and demonstrated a favorable orientation of the covalent warhead to Cys367 (Fig. 2A and Fig. S5E; covalent docking was not capable in this context, ESI†).
 | ||
| Fig. 2 mCMY020 is a covalent inhibitor of TEAD lipidation. (A) Structure of mCMY020. (B) Fluorescence signal from a thermal denaturation assay with recombinant TEAD4-YBD in the presence or absence of mCMY020. (C) Intact mass spectrometry of recombinant TEAD4-YBD in the presence (blue) or absence (black) of mCMY020 after 1 hour treatment. (D) Right, representative fluorescent gel scans of rhodamine-azide labeled palmitoylated TEAD1–4 after 1 hour treatment with the indicated concentrations of mCMY020. Left, densitometry-based quantification of competitive labeling from rhodamine gels (n = 3, mean ± SEM). (E) Representative fluorescent gel scan of rhodamine-azide labeled immunoprecipitated FLAG-TEAD4 from HEK293T cells treated with the indicated concentrations of mCMY020 and alkyne palmitate. | ||
We next confirmed that mCMY020 bound to TEAD4-YBD in vitro, inducing a thermal shift of 3.17 °C in thermal denaturation assays (Fig. 2B). We additionally demonstrated that mCMY020 formed a covalent adduct with TEAD4, as observed by intact mass spectrometry (Fig. 2C). Binding of mCMY020 to the palmitoylation site was additionally confirmed, as mCMY020 was found to dose dependently compete for labeling (IC50 of 91.9 nM) when recombinant TEAD4 protein was exposed to alkyne palmitoyl-CoA in vitro (Fig. 2D). The ability to bind the lipid site of TEAD4 was recapitulated in cells, as mCMY020 was capable of also dose dependently inhibiting (IC50 of 190 nM) the labeling of exogenously expressed FLAG-TEAD4 protein in HEK293T cells exposed to alkyne palmitate (Fig. 2E). In addition to TEAD4, mCMY020 was also found to occupy the palmitoylation sites (Fig. 2D) and form covalent adducts with recombinant preparations of TEAD1, TEAD2, and TEAD3 (Fig. S6, ESI†), which collectively suggests that mCMY020 is likely an efficacious inhibitor of the four TEAD proteins.
Due to the covalent nature of mCMY020, we also assessed its selectivity in binding TEADs relative to other proteins bearing reactive cysteine residues. The targets of the three most recently approved cysteine targeting covalently acting drugs, the epidermal growth factor receptor (EGFR), Bruton's tyrosine kinase (BTK), and SARS-CoV-2 main protease (MPro), were evaluated for enzymatic activity in vitro. Unlike representative inhibitors of each target (osimertinib, ibrutinib, and nirmatrelvir respectively), mCMY020 did not significantly inhibit the catalytic activity of these enzymes (Fig. S7A–C, ESI†). We also observed no observable effect at global cysteine labeling when cells were pre-treated with increasing concentrations of mCMY020 followed by labeling by iodoacetamide biotin (Fig. S7D, ESI†). These experiments further indicated that mCMY020 selectively binds TEADs and its activity is not driven by non-specific covalent modification of cysteines.
mCMY020 inhibits TEAD-dependent transcriptional program and cell proliferation
We next sought to understand if occupying the hydrophobic core of TEADs by mCMY020 would negatively influence YAP-driven transcription. An MCF-7 cell line with a stably integrated cassette encoding luciferase downstream of eight copies of the TEAD-binding element (8x-GTII-LUC; TEAD-LUC reporter) was used to assess YAP driven transcription in a cellular context. mCMY020 treatment (24 hours) was found to dose-dependently decrease the TEAD-LUC signal in this assay with an IC50 of 162.1 nM (Fig. 3A). Additionally, treatment with mCMY020 (20 μM) in the Gal4-TEAD4 assay resulted in a 96% decrease in YAP-driven luciferase signal, as compared to an 88% decrease at the same concentration of verteporfin (20 μM; Fig. 3B). To verify the compound could functionally inhibit the endogenous YAP driven transcriptional program, we next assessed NF2 deficient NCI-H226 cells by RT-qPCR. As anticipated, mCMY020 was found to markedly decrease the levels of the YAP-controlled transcripts CYR61, CTGF, and ANKRD1 by up to 75% over a 24 hour period (Fig. 3C). RNA-sequencing (RNA-seq) further revealed that treatment of NCI-H226 cells with mCMY020 selectively decreased conserved YAP targets (Fig. 3D), as transcripts shown to promote proliferation and cell growth in relation with YAP, NPPB, F3, SLC7A5, and IGFBP3, were the most significantly downregulated genes.33–36 Gene set enrichment analysis (GSEA) further demonstrated that treatment with mCMY020 decreased expression of annotated YAP transcriptional targets (Fig. 3E). Evaluating the top 200 most dynamically regulated transcripts by Gene Ontology term analysis revealed genes related to DNA replication and repair, cell cycle, growth factors, and p53 signaling were downregulated while transcripts associated with immune activation were upregulated upon treatment (Fig. S8, ESI†). | ||
| Fig. 3 mCMY020 inhibits YAP driven transcription in cells. (A) Relative luminance signal from TEAD-LUC MCF-7 cells treated with the indicated concentrations of mCMY020 for 24 hours (n = 3, mean ± SEM). (B) Relative UAS-LUC signal from a Gal4 assay with TEAD4 in HEK293T cells treated with the indicated concentrations of mCMY020 or verteporfin (VTP) for 24 hours (n = 10, mean ± SEM). Significance was determined by Welch and Brown–Forsythe ANOVA; ****p < 0.0001. (C) Relative transcript levels of CYR61, CTGF, and ANKRD1 from H226 cells treated with 10 μM mCMY020 for 24 hours. (n = 3, mean ± SEM). Significance was determined by two-tailed t-test; ***p < 0.001. (D) Volcano plot of RNA-sequencing data depicting the fold change of transcripts in response to 24 hour treatment with mCMY020 (10 μM). (E) GSEA plots of YAP-dependent gene sets (curated from Zhao et al.4 and Cordenonsi et al.9) in response to mCMY020. | ||
We next assessed if the ability of mCMY020 to decrease TEAD driven transcription might inhibit the proliferation of Hippo mutant cancer cells. NCI-H226 (NF2-deficient) and NCI-H2052 (homozygous for NF2 inactivating mutations) malignant mesotheliomas displayed strong sensitivity to mCMY020 treatment, as growth was potently inhibited (IC50 = 261.3 nM and 228.7 nM, respectively) over the course of five days in culture (Fig. 4A and B). This contrasts with NF2 wildtype NCI-H2452 or neuroblastoma IMR32 cells, which did not display any measurable decrease in proliferation at concentrations up to 10 μM (Fig. 4C and D).
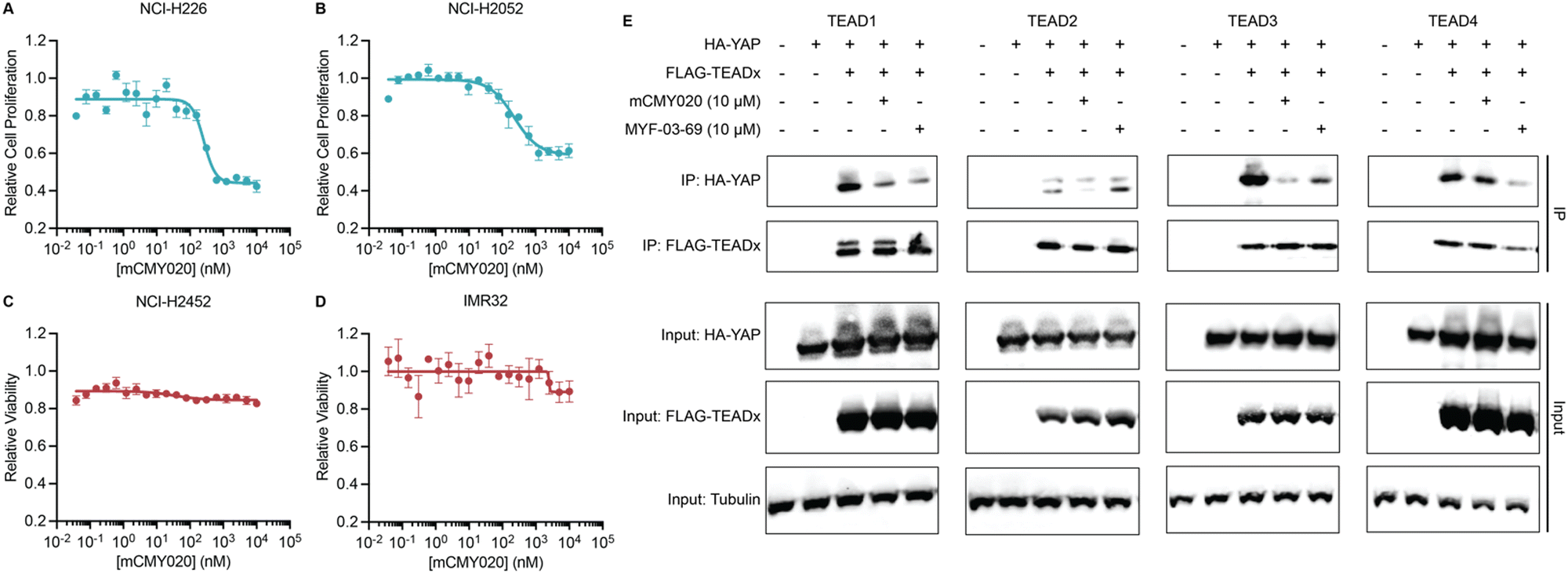 | ||
| Fig. 4 mCMY020 inhibits proliferation in Hippo pathway deficient cancers. Relative viability of NF2-deficient NCI-H226 (A), NF2-heterozygous mutant NCI-H2052 (B), NF2-wildtype NCI-H2452 (C) and NF2 independent neuroblastoma IMR32 cells (D) treated with the indicated concentrations of mCMY020 (n = 3, mean ± SEM). (E) Relative HA-YAP precipitated protein content from HEK293T cells expressing the indicate FLAG-TEAD transgene and then treated with mCMY020 or MYF-03-69. | ||
Lastly, we sought to understand the mechanism by which engagement of the TEAD palmitoylation site results in decreased proliferation and transcription. Inhibitors occupying the hydrophobic core of TEADs have been shown to cause allosteric disassociation of YAP and TEAD, like that reported by the small molecule inhibitor MYF-03-69, which has been hypothesized as essential for its inhibitory activities.37 To understand if mCMY020 treatment disassociates the YAP:TEAD complex, HEK293T cells were co-transfected with FLAG-TEAD (TEAD1–4) and HA-YAP overexpression plasmids and then treated with 10 μM mCMY020 or MYF-03-69 for 24 hours. Anti-FLAG co-immunoprecipitation studies evaluating the capacity to pull down the HA-YAP transgene revealed that both MYF-03-69 and mCMY020 robustly inhibited the association with TEAD1 transgene (Fig. 4E). Interestingly, whereas mCMY020 additionally was found to inhibit the association of YAP with TEAD2 and TEAD3 but showed no inhibitory activity towards the TEAD4 complex. This contrasts with MYF-03-69, which strongly inhibited YAP's association with TEAD4 while displaying a lesser inhibitory effect on TEAD2 and none towards TEAD3 (Fig. 4E).
Discussion
Here, we performed an unbiased fluorescence polarization-based screen to discover novel pharmacological inhibitors of the interaction between YAP and TEAD. This assay used the YAP binding domain (YBD) of TEAD4 in conjunction with the essential binding sequence of YAP, the omega loop at interface 3, to unbiasedly evaluate a large chemical library for mechanisms that might functionally inhibit this protein–protein interaction. Of identified hits, a small subset of molecules that mimicked the structure of the reported YAP inhibitor verteporfin were discovered, giving us confidence in the validity of our screening method (Fig. S1B, ESI†). Among the other hit molecules was a larger subset of compounds containing a previously undescribed pyrazolopyrimidine core, a scaffold which we pursued here as novel chemical matter to inhibit TEADs.We hypothesized that hit compounds likely acted either as direct PPI inhibitors or as inhibitors of TEAD auto-palmitoylation. Using an in vitro assay with recombinant TEAD4 protein and a set of representative pyrazolopyrimidines (KNS1–3), we found that the identified scaffold functionally inhibits TEAD auto-palmitoylation, albeit with modest cell-based potency. Evaluating the binding mode of KNS1–3 demonstrated the scaffold most likely did not efficiently occupy the palmitoylation site. Accordingly, we sought improve the pharmacological properties of the scaffold by developing an optimal chemical probe of TEAD palmitoylation using medical chemistry. From this effort we identified mCMY020, a compound with an extended lipophilic ‘tail’, bearing a terminal cyclohexyl group, that by docking, more efficiently occupies the depth of the palmitoyl cavity. Additionally, mCMY020 bears a 2-fluoro acrylamide (2FA), which results in efficient covalent targeting of the conserved active site cysteine in all 4 TEADs. While several acrylamide-based inhibitors of the TEADs have been developed, this is, to our knowledge, the first example of using a 2FA to effectively and covalently inhibit TEADs. As addition of the withdrawing group to the 2 position of the acrylamide endows a reversible covalent modification of target proteins, there is the potential that 2FA use can increase selectivity of the desired interaction. Using 2FA has shown to be of benefit in increasing the efficacy, pharmacokinetics, and reactivity of other covalent chemotypes in in oncology (e.g., KRAS G12C38,39 and FGFR440,41). Future work evaluating the proteome wide selectivity, pharmacokinetics, and safety of mCMY020 will necessarily be required to understand if altering the covalent targeting group results in a superior TEAD targeting phenotype.
Here we have shown that mCMY020 results in the efficient engagement of all four TEAD proteins covalently, likely acting as a pan-TEAD inhibitor in cells. Interestingly, however, from co-immunoprecipitation experiments, we have shown that mCMY020 inhibits the association of YAP with TEADs1–3 with varying degrees of efficacy and with no effect on TEAD4. This contrasts with the reported pan-TEAD inhibitor MYF-03-69, which shows preferentially activity at inhibiting the association of YAP with TEAD4, and to a lesser extent TEAD1 and TEAD3. While clearly there must be an association between YAP and TEADs for positively affecting transcription, it is unclear if inhibiting TEAD activity pharmacologically necessarily requires a decreased association with YAP and TEADs. Indeed, the lipid pocket on TEAD is not near the protein–protein interface of YAP and TEAD, and it is not functionally understood how preventing palmitoylation of TEAD leads to decreased YAP-target gene expression. Reports on if palmitoylation is essential for the YAP and TEAD interaction are contradictory and may be TEAD specific.23,24,26 For example, de-palmitoylated TEAD1 is unable to bind to YAP but remains stable and can bind to the co-repressor VGLL4.24,25,42 In contrast, if TEAD2 is de-palmitoylated, its protein levels are reduced dramatically.23 Future experiments aimed at understanding the structural, transcriptional, and interactome-level changes that occur in each TEAD in response to a diverse set of lipid site binding small molecules will likely be required to address this question. Given its unique chemotype as well as the divergent interactions it elicits with TEADs in cells, mCMY020 will likely be a useful chemical tool to investigate this hypothesis.
Recent reports have shown that YAP plays key roles in maintaining the tumor immune microenvironment. In tumor cells, YAP positively affects cytokines that recruit myeloid-derived suppressor cells and M2 macrophages while directly suppressing PD-L1 expression. YAP additionally suppresses activation and differentiation of T cells, as YAP-deficient CD4+ and CD8+ T cells dramatically suppress syngeneic tumor burden and display increased infiltrative capacity.43 Our RNA-sequencing data demonstrated that canonical inflammatory response related transcripts are upregulated when NF2 deficient tumor cells are treated with mCMY020, as assessed by Gene Ontology analysis (Fig. S8, ESI†). To the extent that inhibiting TEADs with compounds like mCMY020 can recapitulate the impressive phenotypic effects of deleting YAP will be of keen interest to the field.
YAP is also essential for the self-renewal and tumor-initiation capacities of cancer stem cells (CSCs).13–15,44 CSCs have been shown to be responsible for chemoresistance, metastasis, and recurrence in cancer patients leading to an increase of mortality rates.45 The Hippo pathway has been shown to induce CSCs in many cancers including breast,9,10 liver,46 ovary,47 lung,48 brain,49 colon,50 and pancreas.51 Of interest will be future work exploring if inhibition of TEAD by mCMY020 leads to a reduction in tumor stemness as a primary or combinatorial treatment. Others have recently uncovered that cancer cells can enter a senescence-like dormancy following treatment with epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKI). These cells display high YAP activity, engage in epithelial-to-mesenchymal transition, and avoid apoptosis, leading to recurrence.52 This shift in dependency on YAP following treatment with targeted therapy has also been observed in multiple contexts and provides an attractive use for TEAD inhibitors, such as mCMY020, as a combination therapy to abolish dormant cells, likely preventing recurrence or increasing the efficacy of frontline targeted therapies in solid tumors.
Methods and materials
Protein expression and purification
The coding region of human TEAD4-YBD (residues 216–434, Uniprot Q15561) was codon optimized and inserted into the pET-28a(+)-TEV vector between restriction sites NcoI and XhoI. The final construct contained a TEV cleavable His6-tag at the N-terminal and was verified by DNA sequencing. For protein expression, pET-28a-TEV-TEAD4-YBD was transformed into BL21 and cultured in LB supplemented with Kanamycin at 37 °C until O.D. 600 reached 0.6–0.7. Expression was induced by adding IPTG to a final concentration of 0.1 mM and cultured for 18 hours at 18 °C. Harvested cells were resuspended and lysed by sonication in lysis buffer (50 mM Tris–HCl, 300 mM NaCl, 10 mM imidazole, 10% glycerol, 1% TritonX-100, pH 7.4). The protein was first purified by metal ion affinity chromatography by batch binding with Ni-NTA resin equilibrated in lysis buffer. The solution was transferred to a column and washed with buffer A (50 mM Tris–HCl, 500 mM NaCl, 10% glycerol, 20 mM imidazole, pH 7.4) and eluted with buffer B (50 mM Tris–HCl, 500 mM NaCl, 10% glycerol, 200 mM imidazole, pH 7.4). The protein was further purified by size exclusion chromatography on a sephadex 200 increase gel filtration column with FPLC buffer (50 mM Tris–HCl, 100 mM NaCl, 2 mM MgCl2, 1 mM TCEP, 5% glycerol, pH 8). Protein purity was analyzed by SDS-PAGE and protein folding was analyzed by protein thermal shift. Purified protein was frozen in liquid nitrogen and stored at −80 °C. TEAD1-YBD (residues 207–426, Uniprot P28347), TEAD2-YBD (residues 217–447, Uniprot Q15562), and TEAD3-YBD (residues 216–435, Uniprot Q99594) were prepared using the same methods.Synthesis of YAP peptide and TMR-labeled YAP peptide probe
Unlabeled-YAP peptide was purchased from GenScript. TMR-YAP peptide probe was purchased from InnoPep. Both were analyzed by reverse-phase HPLC and MS.Fluorescence polarization (FP) measurement
All FP measurements for 384-well plates were performed on a CLARIOstar plate reader (BMG) in black opaque 384-well microplates (Corning), with 10 μL of the assay solution per well. All FP measurements for 1536-well screening were performed on a PHERAstar plate reader (BMG) in black opaque 1536-well microplates (Greiner), with 5 μL of the assay solution per well. The FP values were calculated according to the following equation:| FP (mP) = 1000 × (FS − FP)/(FS + FP) |
FP binding and competition assay
Recombinant TEAD4-YBD protein, serially diluted from 5 μM (final concentration) for the binding assay or at 1 μM (final concentration) for the competition assay, was prepared in 50 mM Tris–HCl buffer and 4 μL per well was pipetted into a black low volume 384-well plate (Corning 3281) using a matrix pipette. 2 μL of water, or unlabeled YAP peptide dissolved in water, was added to each well and incubated covered for 1 hour at room temperature. Following incubation, 4 μL of TMR-YAP at a final concentration of 50 nM in HBT buffer (20 mM HEPES, 150 mM NaCl, 0.1% BSA, 0.01% Tween-20) was added to each well and incubated covered overnight at room temperature.High-throughput screening
For high-throughput screening, 2.5 nL of 2 mM DMSO stock concentrations of compound were prespotted using an Echo Acoustic Liquid Handler instrument (Labcyte) before the addition of 5 μL of solution containing 1 μM TEAD4-YBD and 50 nM TMR-YAP in screening buffer (50% 50 mM Tris–HCl and 50% HBT buffer) per well and incubated overnight at room temperature before reading FP.Thermal denaturation assay
Master mixes containing 2 μM TEAD4-YBD and/or 100 μM compound (final concentrations) diluted in water to 75 μL were prepared. 5 μL of protein thermal shift buffer (4461146, Thermo Fisher Scientific) added per well in a 384-well MicroAmp Optical Reaction Plate (Life Technologies), followed by 12.5 μL of appropriate master mix or water, and 2.5 μL freshly diluted 8× Protein Thermal Shift Dye. Reaction mixed well via pipet and centrifuged at 1000 rpm for 1 minute before running a melt curve on a Viia7 system (Thermo Fisher Scientific).Gal4 assay
HEK293T cells were trypsinized, resuspended in growth medium, and diluted to a density of 125000 cells per mL. 40 μL of cell suspension (5000 cells) was dispensed per well in a white 384-well plate (Corning). The cells were then transfected with 25 ng UAS-LUC reporter, pCMX-Gal4-TEAD4, pCI-HA-YAP, and/or pCMV6-Entry (for a total of 100 ng DNA) per well using 200 ng P.E.I. transfection reagent complexed in 10 μL of Opti-MEM medium (Gibco). After 24 hours incubation at 37 °C, 5% CO2 10 μL of compounds diluted in growth medium were added per well and incubated an additional 24 hours followed by the addition of 30 μL of BrightGlo (Promega; diluted 1:3 in water) to each well and luminescence values recorded using an EnVision plate reader.
Cell-free TEAD palmitoylation assay
2 μg of purified recombinant TEAD-YBD was incubated with compounds at the indicated concentration for 1 hour at room temperature followed by the addition of 100 nM alkyne palmitoyl-CoA (Cayman Chemical, 36470) in a total volume of 100 μL. After 1 hour at room temperature the reaction was quenched with 1% SDS followed by click chemistry reaction with rhodamine-azide (Sigma-Aldrich, 760757). Palmitoylated TEAD and total TEAD protein were detected by rhodamine fluorescence followed by Coomassie or silver staining.Cell-based TEAD palmitoylation assay
FLAG-TEAD4 expression plasmid transfected HEK293T cells were treated with DMSO or 50 μM alkynyl palmitic acid and DMSO or compound for 24 hours. FLAG-TEAD4 protein was immunoprecipitated with anti-flag beads and subjected to click chemistry. Palmitoylated TEAD was detected by rhodamine fluorescence and total TEAD was detected by western blot.Docking
TEAD4 (PDB Code: 5OAQ) was prepared using the protein preparation wizard implemented in the Schrodinger software package. The bond orders were assigned, and hydrogens were added after removal of original hydrogens. Missing loops and side chains were filled in using Prime and the protonation states were generated using Epik with pH value 7.4 ± 2.0. Hydrogen bond networks were optimized, and their restrained minimization was carried out using OOPLS4 force fields. KNS1, KNS2, KNS3, and mCMY020 were prepared using LigPrep implemented in the Schrodinger software package. Ionization states were generated at target pH of 7.4 ± 2 using Epik. The docking grid was centered on the myristate ligand with a scaling factor value of 1. Docking was performed using Glide from the Schrodinger software suite with extra precision (XP) and all other default parameters, exporting 5 poses per molecule. The top scoring pose was chosen to illustrate the binding pose.Intact mass spectrometry
mCMY020 (100 μM) was incubated with TEAD4-YBD (10 μM) in PBS for 1 hour at 37 °C. The samples were diluted 10-fold with PBS prior to being analyzed on an Agilent 6230 ESI-TOF mass spectrometer coupled to an Agilent 1260 LC stack using an Agilent PLRP-S LC column. Signal was deconvoluted using Agilent Masshunter Bioconfirm software.YAP reporter assay
MCF-7 TEAD-LUC reporter cells were obtained from BPS Bioscience, Inc. and cultured in DMEM medium (Corning, 10-013-CV) supplemented with 10% fetal bovine serum (Gibco, 10438-026), 1% penicillin–streptomycin (Gibco, 15070063), 1% non-essential amino acids (Gibco, 15070063), and 10 μg mL−1 insulin (Sigma, I0516). Cells were trypsinized, resuspended in growth medium, and diluted to a concentrated of 100000 cells per mL. 5000 cells, 50 μL of cell suspension, per well were then plated in a white 384-well plate (Corning) and incubated at 37 °C, 5% CO2. After 24 hours incubation, 100 nL of test compounds in DMSO, 20-point 1:2 dilution starting from 10 μM (final concentration) in triplicate, were transferred to each well using an Agilent Bravo instrument outfitted with a pintool head. Cells were incubated at 37 °C, 5% CO2 for 24 hours followed by the addition of 30 μL of BrightGlo (Promega; diluted 1:3 in water) to each well and luminescence values recorded using an EnVision plate reader.
RNA extraction, qPCR, and RNA-sequencing
NCI-H226 cells were plated at 500000 cells per well in 6-well dishes in growth medium. After 24 hour incubation, cells were subjected to 10 μM mCMY020 or DMSO delivered as 0.1% DMSO final solutions for an additional 24 hours. After removing culture medium, cells were washed once with PBS, trypsinized with 500 μL TrypLE Express dissociation reagent (Gibo) and neutralized with 500 μL of growth medium. Pellets were collected by centrifugation for 5 minutes at 300 g and RNA was then extracted using RNeasy Plus Mini Kit (Qiagen).
For qPCR studies complementary DNA was reverse-transcribed using a high-capacity cDNA reverse transcription kit (Thermo Fisher Scientific, 4368813). qRT-PCR was performed using the SYBR green method using a PowerSYBR Green PCR master mix (Thermo Fisher Scientific, 4367659) along with transcript specific primers in the table below on the Viia7 system (Thermo Fisher Scientific). Samples were normalized to GAPDH levels and transcript abundance was calculated using the standard comparative Ct method.
RNA sequencing was performed at BGI using the DNAseq Technology platform. STAR53 was used to align to the reference transcriptome and estimate transcript abundance. Differential gene expression analysis (DESeq2) was carried out using R. Differentially expressed genes were defined as having an adjusted P value <0.05 and fold change >±0.5. Gene set enrichment analysis (GSEA, Broad Institute) was performed using the Java application, and results replotted using R. Gene Ontology analysis was conducted on selected genes using the DAVID program.54,55
| Transcript | Forward primer | Reverse primer |
|---|---|---|
| CTGF | TGGAGATTTTGGGAGTACGG | CAGGCTAGAGAAGCAGAGCC |
| ANKRD1 | GTGTAGCACCAGATCCATCG | CGGTGAGACTGAACCGCTAT |
| CYR61 | CCCGTTTTGGTAGATTCTGG | GCTGGAATGCAACTTCGG |
| GAPDH | AATGAAGGGGTCATTGATGG | AAGGTGAAGGTCGGAGTCAA |
Cell proliferation assays
NCI-H226, NCI-H2052, and NCI-H2452 cells were trypsinized, resuspended in growth medium, and diluted to a concentrated of 10000 cells per mL. 500 cells, 50 μL of cell suspension, per well were then plated in a white 384-well plate (Corning) and incubated at 37 °C, 5% CO2. After 24 hours incubation, 100 nL of test compounds in DMSO, 20-point 1:2 dilution starting from 20 μM (final concentration) in triplicate, were transferred to each well using an Agilent Bravo instrument outfitted with a pintool head. Cells were incubated at 37 °C, 5% CO2 for 5 days followed by the addition of 30 μL of Cell TiterGlo (Promega; diluted 1:6 in water) to each well and luminescence values recorded using an EnVision plate reader.
IMR32 cells were trypsinized, resuspended in growth medium, and diluted to a concentrated of 20000 cells per mL. 1000 cells, 50 μL of cell suspension, per well were then plated in a white 384-well plate (Corning) and incubated at 37 °C, 5% CO2. After 24 hours incubation, 100 nL of test compounds in DMSO, 20-point 1:2 dilution starting from 20 μM (final concentration) in triplicate, were transferred to each well using an Agilent Bravo instrument outfitted with a pintool head. Cells were incubated at 37 °C, 5% CO2 for 3 days followed by the addition of 30 μL of Cell TiterGlo (Promega; diluted 1:6 in water) to each well and luminescence values recorded using an EnVision plate reader.
Co-immunoprecipitation
HEK293T cells were plated at a density of 106 cells per well in 6-well dishes in growth medium. The cells were then transfected with 1 μg each of expression plasmids FLAG-TEADx and HA-YAP, or equivalent empty vector per well using 8 μL of FugeneHD (Promega) transfection reagent complexed in 100 μL of Opti-MEM medium (Gibco). After 24 hour incubation, the medium was changed. After a total 48 hour incubation cells were subjected to compound treatment for an additional 24 hours. After removing medium and washing once with PBS, lysates were collected with the addition of 250 μL of ice-cold 1× RIPA buffer (Millipore) and scraping. After brief tip sonication, lysates were clarified by centrifugation (5 min at 16000 g) and protein abundance was determined by absorbance measurements at 250 nm. Samples were normalized and split into two groups: Lysate and IP.
The lysate samples were immediately separated by SDS-PAGE using 4–12% Bis-Tris gels (12-well gels, Invitrogen) in 0.9× MOPS buffer (Invitrogen) and then transferred to PVDF membranes (Bio-Rad) using a semi-dry transfer apparatus (Bio-Rad). Membranes were blocked in 5% non-fat dry milk in TBST (Tris-buffered saline with 0.1% Tween-20). Antibody was incubated overnight at 4 °C (see antibody specific information below). After 30 minutes of washing in TBST, secondary antibody conjugated to HRP (HA) or IRDye 680RD (Tubulin and FLAG) was incubated for an hour in 5% non-fat dry milk in TBST. After an additional hour of washing in TBST, HRP treated membranes were exposed to SuperSignal West Dura Substrate (Thermo Fisher Scientific, 34076) and signals recorded via chemiluminescence on a ChemiDoc (Bio-Rad), and 680RD treated membranes were imaged on an Odyssey imaging system (Li-COR).
20 μL of Anti-FLAG M2 Magnetic beads (Sigma-Aldrich, M8823) were added per sample and incubated overnight at 4 °C with rotation. Using a magnetic rack, the beads were washed twice with 500 μL of 1× RIPA, once with 500 μL of PBS and resuspended in 55 μL of PBS containing 250 μg mL−1 FLAG peptide (DYKDDDDK, Sino Biological Inc., PP101274) to elute proteins bound to the FLAG-beads. These IP samples were then immediate separated by SDS-PAGE and subjected to the same western blot protocol as described above.
| Antigen | Supplier | Catalog number | Host species | Dilution |
|---|---|---|---|---|
| HA | Cell Signaling Technologies | 3724S | Rabbit | 1:1000 (BSA) |
| FLAG | Sigma | F1804 | Mouse | 1:2000 (milk) |
| Tubulin | Sigma | T6557 | Mouse | 1:2000 (milk) |
| Rabbit-HRP | Thermo Scientific | SA1-200 | Donkey | 1:3333 (milk) |
| Mouse IRDye 680RD | Li-COR | 926-68072 | Donkey | 1:1000 (milk) |
Cell culture
HEK293T and IMR32 cells were obtained from American Type Culture Collection (ATCC, CRL-3216 and CCL-127) and cultured in DMEM medium (Corning, 10-013-CV) supplemented with 10% fetal bovine serum (Gibco, 10438-026) and 1% penicillin–streptomycin (Gibco, 15070063). NCI-H226, NCI-H2052, and NCI-H2452 cells were obtained from American Type Culture Collection (ATCC, CRL-5826, CRL-5915, and CRL-5946) and cultured in RPMI 1640 with L-glutamine (Corning, 10-040-CV) supplemented with 10% fetal bovine serum (Gibco, 10438-026) and 1% penicillin–streptomycin (Gibco, 15070063). MCF7 TEAD-LUC reporter cells were obtained from BPS Bioscience, Inc. and cultured in DMEM medium (Corning, 10-013-CV) supplemented with 10% fetal bovine serum (Gibco, 10438-026), 1% penicillin–streptomycin (Gibco, 15070063), 1% non-essential amino acids (Gibco, 15070063), and 10 μg mL−1 insulin (Sigma, I0516). Cultures were routinely evaluated for the presence of mycoplasma contamination via an in-house ELISA-based core service.Reporter plasmids
| Vector name | Transgene | Epitope tag | Source |
|---|---|---|---|
| pCMV6-Entry | Empty Vector | None | Origene PS10001 |
| pCMV6-FLAG-TEAD1 | TEAD1 | N-Terminal FLAG | Origene RC215492 |
| pCMV6-FLAG-TEAD2 | TEAD2 | N-Terminal FLAG | Origene RC203526 |
| pCMV6-FLAG-TEAD3 | TEAD3 | N-Terminal FLAG | Origene RC210621 |
| pCMV6-FLAG-TEAD4 | TEAD4 | N-Terminal FLAG | Origene RC219686 |
| pCI-HA-YAP | YAP | N-Terminal HA | Addgene, plasmid #27007 |
| pCMX-Gal4-TEAD4 | TEAD4 | N-Terminal Gal4 | Addgene, plasmid #33105 |
| GAL4UAS-LUC Reporter | GAL4UAS | C-Terminal luciferase | Addgene, plasmid #64125 |
Chemicals
KNS1 (99089239), KNS2 (36910679), and KNS3 (56220347) were purchased from Chembridge. Verteporfin (17334), Ibrutinib (16274), and Nirmatrelvir (35257) were purchased from Cayman Chemical. Osimertinib was purchased from MedChemExpress (HY-15772). Commercial chemicals were dissolved as DMSO solutions and used in biological experiments without further purification.Iodoacetamide-biotin labeling
NCI-H226 cells were plated at a density of 150000 cells per well in a 6-well dish in growth medium and incubated for 24 hours. Cells were then subjected to the indicated concentrations of mCMY020 treatment for 1 hour at 37C followed by 250 iodoacetamide–PEG–biotin (250 μM; Sigma, 762024) treatment for 1 hour at 37C. After removing medium and washing once with PBS, lysates were collected with the addition of 250 μL of ice-cold 1× RIPA buffer (Millipore) and scraping. After brief tip sonication, lysates were clarified by centrifugation (5 min at 16000 g) and protein abundance was determined by absorbance measurements at 250 nm. Samples were normalized and immediately separated by SDS-PAGE using 4–12% Bis-Tris gels (15-well gels, Invitrogen) in 0.9× MOPS buffer (Invitrogen) on two gels. One gel was Coomassie stained while the other was then transferred to PVDF membranes (Bio-Rad) using a semi-dry transfer apparatus (Bio-Rad). The membrane was blocked in 5% non-fat dry milk in TBST (Tris-buffered saline with 0.1% Tween-20) and incubated in Streptavidin-680 antibody (Li-COR, 926-68079) 1:1000 in 5% milk in TBST overnight at 4 °C. After 30 minutes of washing in TBST, the membrane was imaged on an Odyssey imaging system (Li-COR).
EGFR kinase assay
In a white 384-well plate (Corning), 4 ng of EGFR enzyme, 5 μM ATP, 1 μg Poly E4Y1 substrate, and 5× indicated concentration of mCMY020, Osimertinib, or 5% DMSO were diluted in Tyrosine Kinase Buffer and added to each well as indicated by the Promega EGFR Kinase Enzyme System Kit (Promega, V3831). After 1 hour incubation at room temperature, 5 μL of ADP-Glo reagent (Promega, V9101) was added and incubated at room temperature for 40 minutes. Subsequently 10 μL of Kinase Detection Reagent was added per well and incubated at room temperature for 30 minutes before luminescence values were recorded using an EnVision plate reader.BTK kinase assay
In a white 384-well plate (Corning), 3 ng of EGFR enzyme, 50 μM ATP, 1 μg Poly E4Y1 substrate, and 5× indicated concentration of mCMY020, Ibrutinib, or 5% DMSO were diluted in Kinase Buffer and added to each well as indicated by the Promega BTK Kinase Enzyme System Kit (Promega, V2941). After 2 hours incubation at room temperature, 5 μL of ADP-Glo reagent (Promega, V9101) was added and incubated at room temperature for 40 minutes. Subsequently 10 μL of Kinase Detection Reagent was added per well and incubated at room temperature for 30 minutes before luminescence values were recorded using an EnVision plate reader.SARS-CoV-2 main protease assay
In the provided black 96-well plate, 50 μL of SARS-CoV-2 Main Protease Assay Buffer, 20 μL of SARS-CoV-2 Main Protease Enzyme, and 10 μL of 10× indicated concentration of mCMY020, Nirmatrelvir, or DMSO diluted in assay buffer were added to each well as described by the Cayman SARS-CoV-2 Main Protease Inhibitor Screening Assay Kit (Cayman Chemical, #701960) and incubated for 30 minutes at room temperature. 20 μL of substrate was then added to each well and incubated for 2 hours at room temperature before fluorescence values were recorded using 340 nm excitation and 490 nm emission on a SpectraMax iD3 plate reader.Synthesis of mCMY020
 :MeOH (10:2) to afford 5-(4-cyclohexylphenyl)pyrazolo[1,5-a]pyrimidin-3-amine (80 mg, 57.66% yield) as light yellow solid.
:MeOH (10:2) to afford 5-(4-cyclohexylphenyl)pyrazolo[1,5-a]pyrimidin-3-amine (80 mg, 57.66% yield) as light yellow solid.
Author contributions
M. J. B. conceived of and supervised the project. K. N. designed and conducted experiments. L. S. designed medicinal chemistry-based optimization and carried out small molecule syntheses. E. C. conducted high throughput-screening and hit selection. M. H. supervised the high throughput-screening. J. J. C. and A. K. C. supervised the project. M. J. B. and K. N. wrote the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the NIH (GM146865 to MJB). HEK293T cells were obtained from American Type Culture Collection (ATCC; CRL-3216).References
- R. W. Justice, O. Zilian, D. F. Woods, M. Noll and P. J. Bryant, Genes Dev., 1995, 9, 534–546 CrossRef CAS PubMed.
- T. Xu, W. Wang, S. Zhang, R. A. Stewart and W. Yu, Development, 1995, 121, 1053–1063 CrossRef CAS PubMed.
- M. Kango-Singh, R. Nolo, C. Tao, P. Verstreken, P. R. Hiesinger, H. J. Bellen and G. Halder, Development, 2002, 129, 5719–5730 CrossRef CAS PubMed.
- B. Zhao, X. Ye, J. Yu, L. Li, W. Li, S. Li, J. Yu, J. D. Lin, C.-Y. Wang, A. M. Chinnaiyan, Z.-C. Lai and K.-L. Guan, Genes Dev., 2008, 22, 1962–1971 CrossRef CAS PubMed.
- T. Moroishi, C. G. Hansen and K.-L. Guan, Nat. Rev. Cancer, 2015, 15, 73–79 CrossRef CAS PubMed.
- F. Zanconato, M. Cordenonsi and S. Piccolo, Nat. Rev. Cancer, 2019, 19, 454–464 CrossRef CAS PubMed.
- R. Cunningham and C. G. Carsten, Clin. Sci., 2022, 136, 197–222 CrossRef CAS PubMed.
- S. W. Chan, C. J. Lim, K. Guo, C. P. Ng, I. Lee, W. Hunziker, Q. Zeng and W. Hong, Cancer Res., 2008, 68, 2592–2598 CrossRef CAS PubMed.
- M. Cordenonsi, F. Zanconato, L. Azzolin, M. Forcato, A. Rosato, C. Frasson, M. Inui, M. Montagner, A. Maria, A. Poletti, S. Dupont, G. Basso, S. Bicciato and S. Piccolo, Cell, 2011, 147, 759–772 CrossRef CAS PubMed.
- M. Bartucci, R. Dattilo, C. Moriconi, A. Pagliuca, M. Mottolese, G. Federici, A. D. Benedetto, M. Todaro, G. Stassi, F. Sperati, M. I. Amabile, E. Pilozzi, M. Patrizii, M. Biffoni, M. Maugeri-Saccà, S. Piccolo and R. De Maria, Oncogene, 2015, 34, 681–690 CrossRef CAS PubMed.
- M. H. Kim, C. G. Kim, S.-K. Kim, S. J. Shin, E. A. Choe, S.-H. Park, E.-C. Shin and J. Kim, Cancer Immunol. Res., 2018, 6, 255–266 CrossRef CAS PubMed.
- M. H. Kim and J. Kim, Cell. Mol. Life Sci., 2017, 74, 1457–1474 CrossRef CAS PubMed.
- H. Hayashi, T. Higashi, N. Yokoyama, T. Kaida, K. Sakamoto, Y. Fukushima, T. Ishimoto, H. Kuroki, H. Nitta, D. Hashimoto, A. Chikamoto, E. Oki, T. Beppu and H. Baba, Cancer Res., 2015, 75, 4985–4997 CrossRef CAS PubMed.
- M. Escoll, R. Gargini, A. Cuadrado, I. M. Anton and F. Wandosell, Oncogene, 2017, 36, 3515–3527 CrossRef CAS PubMed.
- Y.-W. Li, J. Xu, G.-Y. Zhu, Z.-J. Huang, Y. Lu, X.-Q. Li, N. Wang and F.-X. Zhang, Cell Death Discovery, 2018, 4, 105 CrossRef PubMed.
- X. Ni, J. Tao, J. Barbi, Q. Chen, B. V. Park, Z. Li, N. Zhang, A. Lebid, A. Ramaswamy, P. Wei, Y. Zheng, X. Zhang, X. Wu, P. Vignali, C.-P. Yang, H. Li, D. Pardoll, L. Lu, D. Pan and F. Pan, Cancer Discovery, 2018, 8, 1026–1043 CrossRef CAS PubMed.
- T. A. Yap, J. G. Aerts, S. Popat and D. A. Fennell, Nat. Rev. Cancer, 2017, 17, 475–488 CrossRef CAS PubMed.
- C. D. K. Nguyen and C. Yi, Trends Cancer, 2019, 5, 283–296 CrossRef CAS PubMed.
- F. Zanconato, G. Battilana, M. Forcato, L. Filippi, L. Azzolin, A. Manfrin, E. Quaranta, D. Di Biagio, G. Sigismondo, V. Guzzardo, P. Lejeune, B. Haendler, J. Krijgsveld, M. Fassan, S. Bicciato, M. Cordenonsi and S. Piccolo, Nat. Med., 2018, 24, 1599–1610 CrossRef CAS PubMed.
- Z. Li, B. Zhao, P. Wang, F. Chen, Z. Dong, H. Yang, K.-L. Guan and Y. Xu, Genes Dev., 2010, 24, 235–240 CrossRef CAS PubMed.
- Y. Mesrouze, F. Bokhovchuk, M. Meyerhofer, P. Fontana, C. Zimmermann, T. Martin, C. Delaunay, D. Erdmann, T. Schmelzle and P. Chène, eLife, 2017, 6, e25068 CrossRef PubMed.
- L. Chen, S. W. Chan, X. Zhang, M. Walsh, C. J. Lim, W. Hong and H. Song, Genes Dev., 2010, 24, 290–300 CrossRef CAS PubMed.
- C. L. Noland, S. Gierke, P. D. Schnier, J. Murray, W. N. Sandoval, M. Sagolla, A. Dey, R. N. Hannoush, W. J. Fairbrother and C. N. Cunningham, Structure, 2016, 24, 179–186 CrossRef CAS PubMed.
- P. Chan, X. Han, B. Zheng, M. Deran, J. Yu, G. K. Jarugumilli, H. Deng, D. Pan, X. Luo and X. Wu, Nat. Chem. Biol., 2016, 12, 282–289 CrossRef CAS PubMed.
- N.-G. Kim and B. M. Gumbiner, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9877–9882 CrossRef CAS PubMed.
- Y. Mesrouze, M. Meyerhofer, F. Bokhovchuk, P. Fontana, C. Zimmermann, T. Martin, C. Delaunay, A. Izaac, J. Kallen, T. Schmelzle, D. Erdmann and P. Chène, Protein Sci., 2017, 26, 2399–2409 CrossRef CAS PubMed.
- Y. Li, S. Liu, E. Y. Ng, R. Li, A. Poulsen, J. Hill, A. V. Pobbati, A. W. Hung, W. Hong, T. H. Keller and C. Kang, Biochem. J., 2018, 475, 2043–2055 CrossRef CAS PubMed.
- W. Zhou, Y. Li, J. Song and C. Li, Anal. Biochem., 2019, 586, 113413 CrossRef CAS PubMed.
- J. Janes, M. E. Young, E. Chen, N. H. Rogers, S. Burgstaller-Muehlbacher, L. D. Hughes, M. S. Love, M. V. Hull, K. L. Kuhen, A. K. Woods, S. B. Joseph, H. M. Petrassi, C. W. McNamara, M. S. Tremblay, A. I. Su, P. G. Schultz and A. K. Chatterjee, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10750–10755 CrossRef CAS PubMed.
- Y. Liu-Chittenden, B. Huang, J. S. Shim, Q. Chen, S. J. Lee, R. A. Anders, J. O. Liu and D. Pan, Genes Dev., 2012, 26, 1300–1305 CrossRef CAS PubMed.
- T. Lu, Y. Li, W. Lu, T. Spitters, X. Fang, J. Wang, S. Cai, J. Gao, Y. Zhou, Z. Duan, H. Xiong, L. Liu, Q. Li, H. Jiang, K. Chen, H. Zhou, H. Lin, H. Feng, B. Zhou, C. L. Antos and C. Luo, Acta Pharm. Sin. B, 2021, 11, 3206–3219 CrossRef CAS PubMed.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
- J. Canon, K. Rex, A. Y. Saiki, C. Mohr, K. Cooke, D. Bagal, K. Gaida, T. Holt, C. G. Knutson, N. Koppada, B. A. Lanman, J. Werner, A. S. Rapaport, T. San Miguel, R. Ortiz, T. Osgood, J. R. Sun, X. Zhu, J. D. McCarter, L. P. Volak, B. E. Houk, M. G. Fakih, B. H. O'Neil, T. J. Price, G. S. Falchook, J. Desai, J. Kuo, R. Govindan, D. S. Hong, W. Ouyang, H. Henary, T. Arvedson, V. J. Cee and J. R. Lipford, Nature, 2019, 575, 217–223 CrossRef CAS PubMed.
- O. M. Yu, J. A. Benitez, S. W. Plouffe, D. Ryback, A. Klein, J. Smith, J. Greenbaum, B. Delatte, A. Rao, K.-L. Guan, F. B. Furnari, O. M. Chaim, S. Miyamoto and J. H. Brown, Oncogene, 2018, 37, 5492–5507 CrossRef CAS PubMed.
- A. V. Pobbati, S. W. Chan, I. Lee, H. Song and W. Hong, Structure, 2012, 20, 1135–1140 CrossRef CAS PubMed.
- C. G. Hansen, Y. L. D. Ng, W.-L. M. Lam, S. W. Plouffe and K.-L. Guan, Cell Res., 2015, 25, 1299–1313 CrossRef CAS PubMed.
- M. Fan, W. Lu, J. Che, N. P. Kwiatkowski, Y. Gao, H. S. Seo, S. B. Ficarro, P. C. Gokhale, Y. Liu, E. A. Geffken, J. Lakhani, K. Song, M. Kuljanin, W. Ji, J. Jiang, Z. He, J. Tse, A. S. Boghossian, M. G. Rees, M. M. Ronan, J. A. Roth, J. D. Mancias, J. A. Marto, S. Dhe-Paganon, T. Zhang and N. S. Gray, eLife, 2022, 11, 78810 CrossRef PubMed.
- J. Hallin, L. D. Engstrom, L. Hargis, A. Calinisan, R. Aranda, D. M. Briere, N. Sudhakar, V. Bowcut, B. R. Baer, J. A. Ballard, M. R. Burkard, J. B. Fell, J. P. Fischer, G. P. Vigers, Y. Xue, S. Gatto, J. Fernandez-Banet, A. Pavlicek, K. Velastagui, R. C. Chao, J. Barton, M. Pierobon, E. Baldelli, E. F. Patricoin, 3rd, D. P. Cassidy, M. A. Marx, I. I. Rybkin, M. L. Johnson, S. I. Ou, P. Lito, K. P. Papadopoulos, P. A. Janne, P. Olson and J. G. Christensen, Cancer Discovery, 2020, 10, 54–71 CrossRef CAS PubMed.
- J. B. Fell, J. P. Fischer, B. R. Baer, J. F. Blake, K. Bouhana, D. M. Briere, K. D. Brown, L. E. Burgess, A. C. Burns, M. R. Burkard, H. Chiang, M. J. Chicarelli, A. W. Cook, J. J. Gaudino, J. Hallin, L. Hanson, D. P. Hartley, E. J. Hicken, G. P. Hingorani, R. J. Hinklin, M. J. Mejia, P. Olson, J. N. Otten, S. P. Rhodes, M. E. Rodriguez, P. Savechenkov, D. J. Smith, N. Sudhakar, F. X. Sullivan, T. P. Tang, G. P. Vigers, L. Wollenberg, J. G. Christensen and M. A. Marx, J. Med. Chem., 2020, 63, 6679–6693 CrossRef CAS PubMed.
- W. Deng, X. Chen, K. Jiang, X. Song, M. Huang, Z. C. Tu, Z. Zhang, X. Lin, R. Ortega, A. V. Patterson, J. B. Smaill, K. Ding, S. Chen, Y. Chen and X. Lu, ACS Med. Chem. Lett., 2021, 12, 647–652 CrossRef CAS PubMed.
- J. M. Bradshaw, J. M. McFarland, V. O. Paavilainen, A. Bisconte, D. Tam, V. T. Phan, S. Romanov, D. Finkle, J. Shu, V. Patel, T. Ton, X. Li, D. G. Loughhead, P. A. Nunn, D. E. Karr, M. E. Gerritsen, J. O. Funk, T. D. Owens, E. Verner, K. A. Brameld, R. J. Hill, D. M. Goldstein and J. Taunton, Nat. Chem. Biol., 2015, 11, 525–531 CrossRef CAS PubMed.
- Z. Lin, H. Guo, Y. Cao, S. Zohrabian, P. Zhou, Q. Ma, N. VanDusen, Y. Guo, J. Zhang, S. M. Stevens, F. Liang, Q. Quan, P. R. van Gorp, A. Li, C. Dos Remedios, A. He, V. J. Bezzerides and W. T. Pu, Dev. Cell, 2016, 39, 466–479 CrossRef CAS PubMed.
- E. Stampouloglou, N. Cheng, A. Federico, E. Slaby, S. Monti, G. L. Szeto and X. Varelas, PLoS Biol., 2020, 18, e3000591 CrossRef PubMed.
- I. Lian, J. Kim, H. Okazawa, J. Zhao, B. Zhao, J. Yu, A. Chinnaiyan, M. A. Israel, L. S. B. Goldstein, R. Abujarour, S. Ding and K.-L. Guan, Genes Dev., 2010, 24, 1106–1118 CrossRef CAS PubMed.
- T. Shibue and R. A. Weinberg, Nat. Rev. Clin. Oncol., 2017, 14, 611–629 CrossRef PubMed.
- H. Wu, Y. Liu, Z. Liao, J. Mo, Q. Zhang, B. Zhang and L. Zhang, Biomark Res, 2022, 10, 42 CrossRef PubMed.
- H. Song, K. K. Mak, L. Topol, K. Yun, J. Hu, L. Garrett, Y. Chen, O. Park, J. Chang, R. M. Simpson, C.-Y. Wang, B. Gao, J. Jiang and Y. Yang, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1431–1436 CrossRef CAS PubMed.
- P. Thirusangu, U. Ray, S. Sarkar Bhattacharya, D. B. Oien, L. Jin, J. Staub, N. Kannan, J. R. Molina and V. Shridhar, Oncogene, 2022, 41, 4003–4017 CrossRef CAS PubMed.
- K. Masliantsev, L. Karayan-Tapon and P. O. Guichet, Cells, 2021, 10, 184 CrossRef CAS PubMed.
- Y. Ohta, M. Fujii, S. Takahashi, A. Takano, K. Nanki, M. Matano, H. Hanyu, M. Saito, M. Shimokawa, S. Nishikori, Y. Hatano, R. Ishii, K. Sawada, A. Machinaga, W. Ikeda, T. Imamura and T. Sato, Nature, 2022, 608, 784–794 CrossRef CAS PubMed.
- S. Barman, I. Fatima, A. B. Singh and P. Dhawan, Int. J. Mol. Sci., 2021, 22, 9 Search PubMed.
- K. J. Kurppa, Y. Liu, C. To, T. Zhang, M. Fan, A. Vajdi, E. H. Knelson, Y. Xie, K. Lim, P. Cejas, A. Portell, P. H. Lizotte, S. B. Ficarro, S. Li, T. Chen, H. M. Haikala, H. Wang, M. Bahcall, Y. Gao, S. Shalhout, S. Boettcher, B. H. Shin, T. Thai, M. K. Wilkens, M. L. Tillgren, M. Mushajiang, M. Xu, J. Choi, A. A. Bertram, B. L. Ebert, R. Beroukhim, P. Bandopadhayay, M. M. Awad, P. C. Gokhale, P. T. Kirschmeier, J. A. Marto, F. D. Camargo, R. Haq, C. P. Paweletz, K. K. Wong, D. A. Barbie, H. W. Long, N. S. Gray and P. A. Janne, Cancer Cell, 2020, 37, 104–122e112 CrossRef CAS PubMed.
- A. Dobin, C. A. Davis, F. Schlesinger, J. Drenkow, C. Zaleski, S. Jha, P. Batut, M. Chaisson and T. R. Gingeras, Bioinformatics, 2013, 29, 15–21 CrossRef CAS PubMed.
- B. T. Sherman, M. Hao, J. Qiu, X. Jiao, M. W. Baseler, H. C. Lane, T. Imamichi and W. Chang, Nucleic Acids Res., 2022, 50, W216–221 CrossRef CAS PubMed.
- W. Huang da, B. T. Sherman and R. A. Lempicki, Nat. Protoc., 2009, 4, 44–57 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cb00044c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |