
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-directed Pd-catalysed electrooxidative olefination of arenes†
Subir
Panja
a,
Salman
Ahsan
 b,
Tanay
Pal
a,
Simon
Kolb
c,
Wajid
Ali
a,
Sulekha
Sharma
b,
Chandan
Das
a,
Jagrit
Grover
a,
Arnab
Dutta
*a,
Daniel B.
Werz
*c,
Amit
Paul
*b and
Debabrata
Maiti
*a
b,
Tanay
Pal
a,
Simon
Kolb
c,
Wajid
Ali
a,
Sulekha
Sharma
b,
Chandan
Das
a,
Jagrit
Grover
a,
Arnab
Dutta
*a,
Daniel B.
Werz
*c,
Amit
Paul
*b and
Debabrata
Maiti
*a
aIIT Bombay, Department of Chemistry and IDP, Climate Studies, Powai, Mumbai 400076, India. E-mail: dmaiti@iitb.ac.in; arnabdutta@chem.iitb.ac.in
bIndian Institute of Science Education and Research (IISER) Bhopal, Department of Chemistry, Bhopal, Madhya Pradesh 462066, India. E-mail: apaul@iiserb.ac.in
cTechnische Universität Braunschweig, Institute of Organic Chemistry, Hagenring 30, 38106 Braunschweig, Germany. E-mail: d.werz@tu-braunschweig.de
First published on 5th August 2022
Abstract
The Fujiwara–Moritani reaction is a powerful tool for the olefination of arenes by Pd-catalysed C–H activation. However, the need for superstoichiometric amounts of toxic chemical oxidants makes the reaction unattractive from an environmental and atom-economical view. Herein, we report the first non-directed and regioselective olefination of simple arenes via an electrooxidative Fujiwara–Moritani reaction. The versatility of this operator-friendly approach was demonstrated by a broad substrate scope which includes arenes, heteroarenes and a variety of olefins. Electroanalytical studies suggest the involvement of a Pd(II)/Pd(IV) catalytic cycle via a Pd(III) intermediate.
Transition metal-catalysed C–H functionalisation reactions have increasingly gained importance over the last few decades since they allow direct and rapid installation of functionality. Regardless of the undeniable synthetic value of such transformations, the need for superstoichiometric quantities of expensive and hazardous oxidants (e.g., silver and copper salts) remains a major drawback from a sustainable chemistry perspective.1,2 Additionally, chemical oxidants often lead to the formation of by-products, hindering purification and decreasing atom economy. Nevertheless, very few reports were also reported in the literature wherein mild oxidant such as molecular oxygen can also serve as the oxidising agent.2j To make chemical processes and transformations intrinsically sustainable, organic chemists re-discovered synthetic electrochemistry as an environmentally friendly approach.3–6 In the domain of synthetic electrochemistry, the Lei group achieved a significant milestone and installed C–C bonds through a different cross-coupling strategy.1k,2f–h Electroorganic synthesis utilizes electric current to realize redox processes and thereby avoids the use of dangerous, expensive, and polluting chemical oxidising or reducing agents. Precise control of electrochemical reaction parameters often leads to commendable reactivity and chemoselectivity and hence to an improved atom economy. In addition, electrochemical processes fulfil the expectations of sustainability since electricity can be generated from renewable energy sources, such as wind, sunlight or biomass. Recent efforts in the field of electrochemical C–H activation resulted in significant progress towards efficient C–C and C–heteroatom bond formations.7–10 Hence, the utilization of electric current as an alternative oxidant in Pd-catalysed C–H functionalisations is emerging as an attractive alternative to stoichiometric reagents.11–13
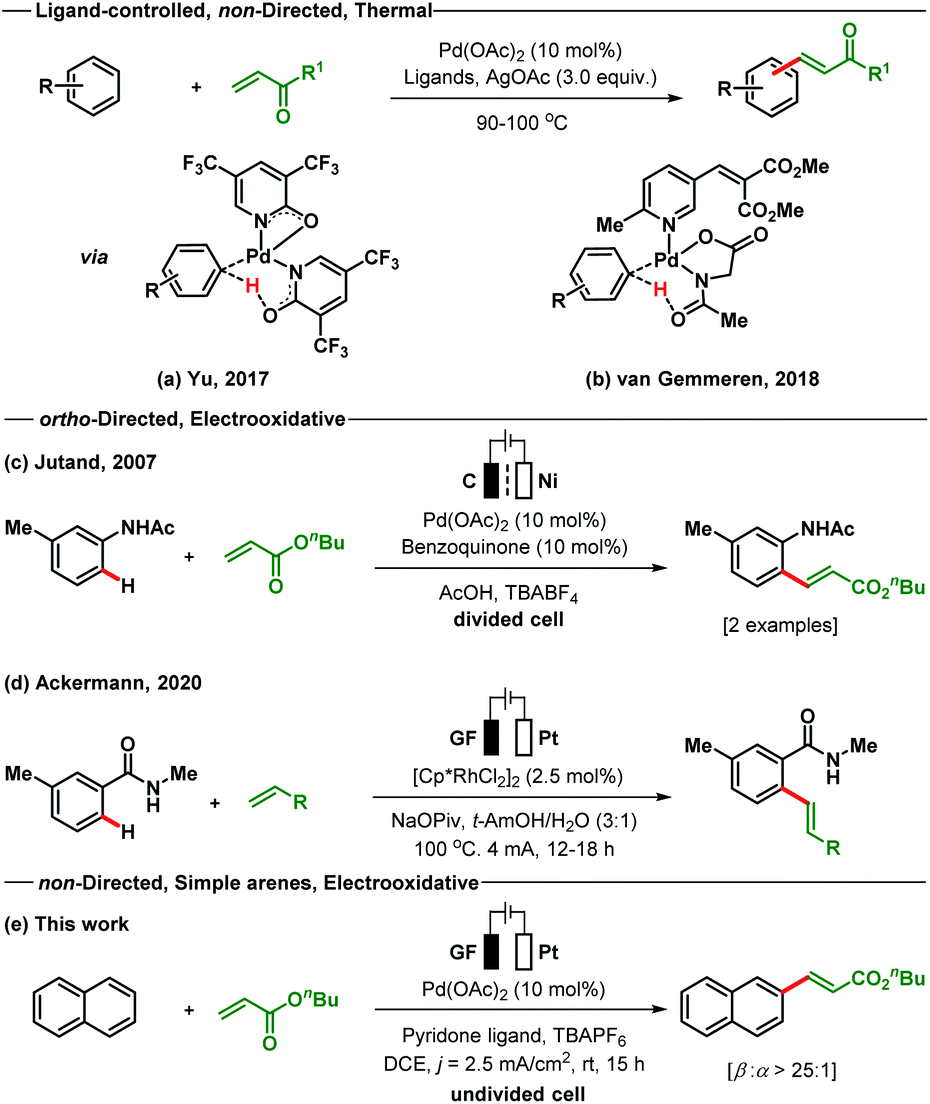
The Fujiwara–Moritani reaction is one of the earliest known examples of Pd-catalysed oxidative C–H functionalisations for C–C bond formation.14 This extraordinary C(sp2)-H alkenylation reaction avoids the use of prefunctionalised starting materials; however, it suffers from the drawbacks of regioselectivity, reactivity and use of excess arenes.15 Since its development, a number of modified strategies have been reported by different research groups to address the issue of reactivity and selectivity.16–21 In recent times, the ligand assisted oxidative C–H alkenylation of arenes without directing substituents has been established as one of the major strategies to overcome the reactivity issue and to elaborate the substrate scope.
However, regioselectivity for most of the sterically and electronically unbiased arenes is still not up to the mark. The most recent studies on the non-directed oxidative C–H olefination of arenes were reported independently by Yu and van Gemmeren (Scheme 1). The Yu group employed electron-deficient 2-pyridone as an X-type ligand for the olefination of both electron-rich and electron-poor arenes including heteroarenes as the limiting reagent (Scheme 1a).18 The pyridone ligand improves the selectivity in a non-directed approach as compared to the directed C–H olefination reaction by enhancing the influence of steric effects. On the other hand, the van Gemmeren group utilizes two complementary ligands N-Ac–Gly–OH and a 6-methylpyridine derivative in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio to accomplish the non-directed olefination reaction of arenes (Scheme 1b).20 Despite the indisputable advances made by these research groups in the area of non-directed oxidative C–H olefination of arenes, the use of superstoichiometric amounts of toxic and waste-generating oxidants (Ag salts) deciphers into a strong call for an environmentally responsive and atom-economic protocol. To address these shortcomings, we recently introduced Pd-photoredox catalysed olefination of non-directed arenes with excellent site selectivity under oxidant free conditions.21
:1 ratio to accomplish the non-directed olefination reaction of arenes (Scheme 1b).20 Despite the indisputable advances made by these research groups in the area of non-directed oxidative C–H olefination of arenes, the use of superstoichiometric amounts of toxic and waste-generating oxidants (Ag salts) deciphers into a strong call for an environmentally responsive and atom-economic protocol. To address these shortcomings, we recently introduced Pd-photoredox catalysed olefination of non-directed arenes with excellent site selectivity under oxidant free conditions.21
 | ||
| Scheme 1 Recent approaches to sustainable C–H alkenylation reactions. | ||
In 2007, Jutand reported the directing group assisted Pd-electracatalysed ortho-olefination of acetyl protected aniline in a divided cell by utilizing catalytic amounts of benzoquinone as a redox mediator (Scheme 1c).22a A Rh-catalysed ortho-C–H olefination of benzamide was developed through an electrooxidative pathway by the Ackermann group (Scheme 1d).22b Simple arenes that bear no directing groups are cheap, easily available and very desirable starting materials. However, the use of such arenes is significantly more challenging for selective functionalisation as transformations often result in the formation of complex product mixtures. With no report of an electrooxidative Pd-catalysed C(sp2)-H alkenylation of simple arenes present, we wish to present such a variant of the Fujiwara–Moritani reaction (Scheme 1e). The developed method proceeds through a non-directed pathway and is controlled by stereoelectronic factors. This protocol does not require additional chemical oxidizing agents and is executed using an operator-friendly undivided cell setup.
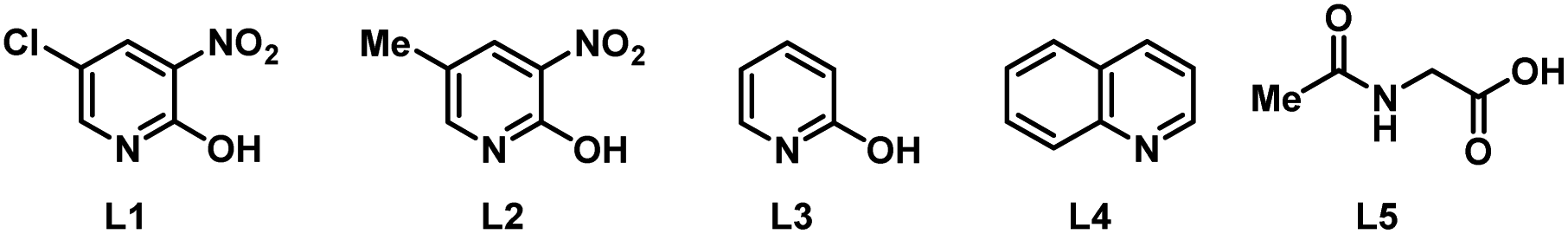
To start our study, naphthalene was chosen as a challenging substrate because of its ability to form α- and β-products. We examined various reaction conditions for the desired Pd-catalysed electrooxidative C–H alkenylation in a simple undivided cell setup (Table 1, and see ESI Tables S1–S8†) with n-butyl acrylate as the coupling partner. After rigorous optimisation, we found that naphthalene reacts with n-butyl acrylate in dichloroethane (DCE) in the presence of Pd(OAc)2 (10 mol%), ligand L1 (20 mol%), and the electrolyte tetra-n-butylammonium hexafluorophosphate (TBAPF6, 0.5 equiv.) while employing a graphite felt anode and a platinum cathode maintaining constant current electrolytic conditions (j = 2.5 mA cm−2, Table 1, entry 1). The desired β-olefinated product was formed in 70% yield and with >25:1 regioselectivity (β:α). Other transition metal catalysts such as Co(OAc)2·4H2O or [Ru(p-cymene)Cl2]2 as substitutes for Pd(OAc)2 were found to be completely ineffective (entries 2 and 3). Changes in the catalyst loading were not found to be beneficial (entries 4 and 5).
|
|
|||
|---|---|---|---|
| Entry | Alteration from standard conditions | Yield of 1b (%) | Selectivity (β:α) |
| a Standard reaction conditions: undivided cell, GF anode, Pt cathode, j = 2.5 mA cm−2, naphthalene (0.2 mmol), n-butyl acrylate (0.5 mmol), Pd(OAc)2 (10 mol%), L1 (20 mol%), TBAPF6 (0.5 equiv.), DCE (3 mL), 15 h, under air. b Yield determined by 1H-NMR of crude reaction mixture. NR = no reaction; TBAPF6 = tetra-n-butylammonium hexafluorophosphate. GF = graphite felt. Surface area of electrodes dipped in solution = 0.7 cm × 0.7 cm, current = 1.225 mA and current density = 2.5 mA cm−2 (electrochemical surface area = 1.23 cm2). | |||
| 1 | None | 70 | >25:1 |
| 2 | Co(OAc)2·4H2O instead of Pd(OAc)2 | 9 | 1:1 |
| 3 | [Ru(p-cymene)Cl2]2 instead of Pd(OAc)2 | NR | |
| 4 | Pd(OAc)2·(5 mol%) | 51 | >25:1 |
| 5 | Pd(OAc)2·(20 mol%) | 71 | >25:1 |
| 6 | L2 instead of L1 | 45 | 8:1 |
| 7 | L3 instead of L1 | 59 | 20:1 |
| 8 | L4 instead of L1 | 19 | 5:1 |
| 9 | L5 instead of L1 | 8 | 1:1 |
| 10 | Benzoquinone (10 mol%) | 68 | >25:1 |
| 11 | PivOH (1.0 equiv.) | 61 | 20:1 |
| 12 | Ni foam instead of Pt | 64 | >25:1 |
| 13 | GF instead of Pt | 49 | 15:1 |
| 14 | Steel instead of Pt | 31 | 13:1 |
| 15 | 6 mA cm−2 instead of 2.5 mA cm−2 | 27 | 11:1 |
| 16 | 24 h reaction time | 47 | 20:1 |
| 17 | 12 h reaction time | 56 | 21:1 |
| 18 | No electricity | NR | — |
| 19 | No Pd(OAc)2 | NR | — |
|
|||
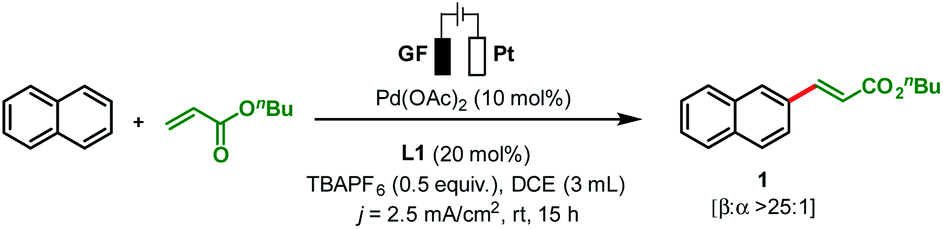
Notably, in the present transformation the ligand has a major influence on the reactivity and selectivity aspects (see the ESI, Table S4†). After studying a series of 2-pyridone, pyridine and amino acid-based ligands L2–L5 it was found that L1 is the optimal ligand since it provided superior yield and selectivity (entries 6–9). Addition of catalytic amounts of p-benzoquinone as a redox mediator (entry 10) or pivalic acid as an additive (entry 11, Table 1) had minimal influence on the reaction outcome. While nickel foam as a cathode material shows similar efficiency, the use of a carbon felt or a steel cathode led to lower reactivity and selectivity (entries 12–14). Increasing the electric current density (j = 6 mA cm−2) was detrimental since it provided the desired product in low yield (27%) and with decreased β:α-selectivity (11:1; entry 15). Additionally, longer and shorter reaction times resulted in lower yields of the olefinated product 1 (entries 16 and 17). Finally, control experiments confirmed that both the electric potential and Pd(OAc)2 are essential to furnish the reaction products (entries 18 and 19, Table 1).
With optimised reaction conditions in hand, reaction generality was explored by testing a range of substituted arenes and heteroarenes with n-butyl acrylate (Scheme 2). Following the olefination of naphthalene (68%, >25:1 β:α selectivity), 1,2,3,4-tetrahydronaphthalene was successfully reacted (52%, 11:1 β:α-selectivity). Next, we applied our standard reaction conditions to benzene and found them not to be equally effective as only 25% of the olefinated product 3 was obtained. As a result, further optimizations of electric current density and solvent were carried out to enhance the yield (see the ESI, Table S9†). To our satisfaction, the yield of product 3 increased to 63% when the electrolysis was carried out with an electric current density of j = 1.5 mA cm−2 and in a solvent mixture of DCE/HFIP (5:1). These modified reaction conditions were applied to the electrosynthesis of all other olefinated products 4–26 (Scheme 2). The olefination of 1,3,5-trimethoxybenzene and mesitylene with n-butyl acrylate proceeded smoothly under the revised reaction conditions to afford products 4–5 in up to 65% yield. The regioselectivity issue was more prominent for arenes bearing two or more electronically similar C–H bonds (e.g., electron-rich arenes: ortho vs. para). Dimethoxy benzene gives β-selective olefinated product 6 (β:α; 7:1). While toluene was converted with para-selectivity (7:1) to 7, phenol afforded olefinated product 8 with ortho-selectivity (o:others; 9:1, Scheme 2). On the other hand, subjecting TBDMS (tert-butyldimethylsilyl) protected phenol to the established protocol furnished 9 with 8:1 para-selectivity (Scheme 2). The TBDPS (tert-butyldiphenylsilyl) protected phenol afforded exclusively the para-olefinated product 10 which might be due to the steric repulsion caused by the bulky protecting group. Conversion of 2,6-diiso-propylphenol provided olefinated product 11 as a single para-olefinated isomer with 67% yield. Anisole and ethoxybenzene both reacted smoothly to produce 12 (72%, 15:1) and 13 (70%, 10:1) with ortho-selectivity (Scheme 2). The compatibility of the present transformation was further showcased by the olefination of N,N-dimethyl aniline in 70% yield (14) and 8:1 ortho-selectivity. Similarly, methyl ferrocene carboxylate and biologically active caffeine reacted smoothly with n-butyl acrylate to produce olefinated products 15 and 16 in good yields (Scheme 2). Moderately electron-withdrawing arenes such as a phenyl acetic acid derivative (17, 51%, o:others = 7:1), a homoveratric acid derivative (18, 48%, o:others = 15:1) or 4-methoxy acetophenone (19, 59%, m:others = 7:1) gave the corresponding products in satisfactory yields. The coupling of unsubstituted thiophene and furan with n-butyl acrylate afforded the olefinated products 20 and 21 (64% and 68%) with synthetically useful C2-selectivity, respectively (C2:others; 18:1 and C2:others; 19:1, Scheme 2). In contrast, thiophenes bearing a substituent at the C2 position such as 2-phenylthiophene and 1-(4-(thien-2-yl)phenyl)ethan-1-one reacted with high C5-selectivity (>20:1) to afford the arylated α,β-unsaturated esters 22 and 23 (76% and 73% yield). Conversion of 2-(2-nitrophenyl)thiophene delivered the desired product 24 in 64% yield with exclusive C5-selectivity. A C3-substituted thiophene also reacted with the acrylate to afford 25 in 72% yield (C5:others; 6:1 selectivity). Heteroarenes bearing electron-withdrawing substituents such as 2-acetyl thiophene (26) afforded the C5-olefinated product in moderate yield and selectivity (60%, C5:others = 8:1). However, aromatic rings bearing strong electron-withdrawing groups (–NO2, –CHO, –CF3, –F etc.) are not compatible under our present reaction conditions (see details in the ESI, Section 4.3†).
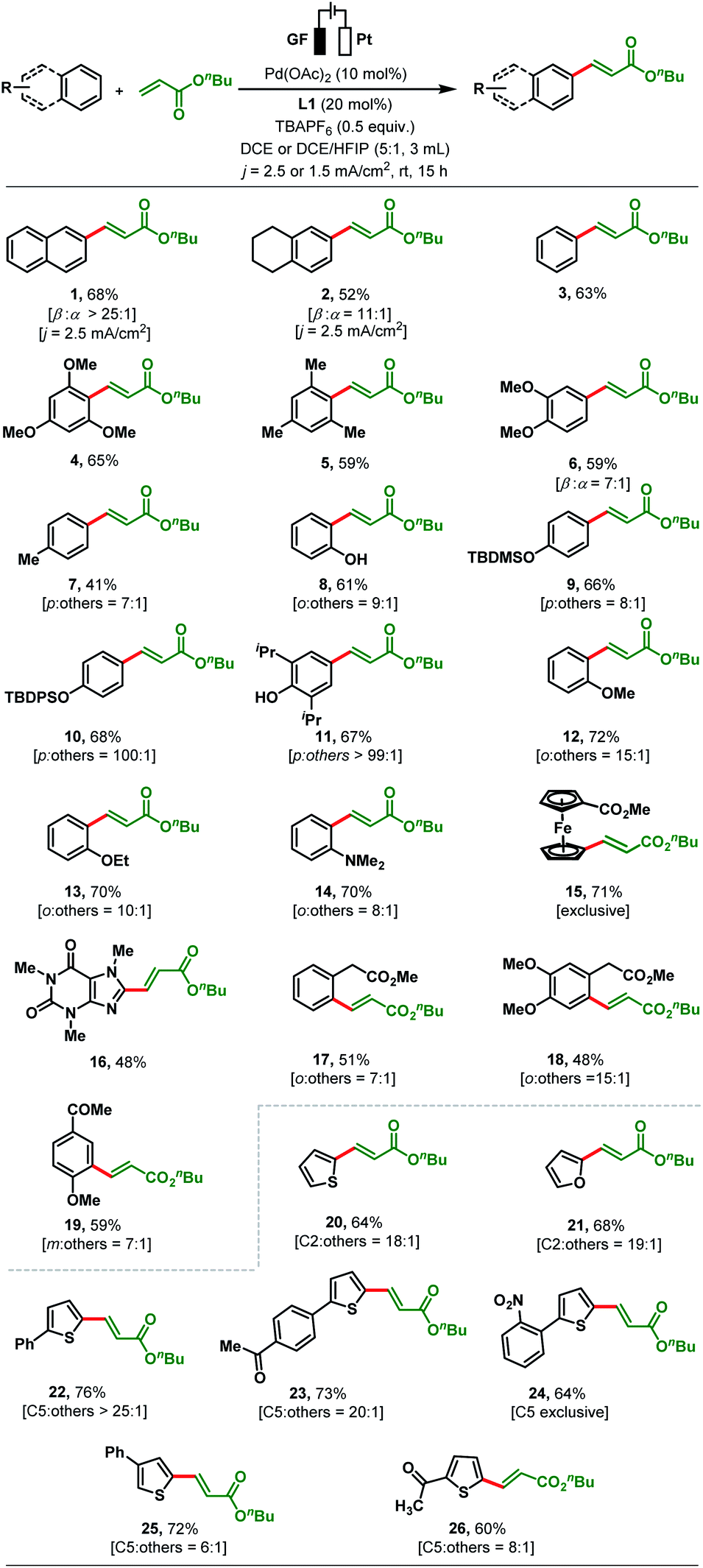 | ||
| Scheme 2 Evaluation of simple arenes and heteroarenes in the electrochemical olefination.a Reaction conditions: undivided cell, GF anode, Pt cathode, j = 2.5 mA cm−2 or j = 1.5 mA cm−2, corresponding arenes or heteroarenes (0.2 mmol), n-butyl acrylate (0.5 mmol), Pd(OAc)2 (10 mol%), L1 (20 mol%), TBAPF6 (0.5 equiv.), DCE (3 mL) or 5:1 ration of dichloroethane (DCE) and 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP), 15 h, under air. bYields of isolated products are reported. | ||
Next, we investigated the applicability of other olefins by reacting them with simple arenes (Scheme 3). In addition to other acrylates (methyl 27, ethyl 28 and tert-butyl 29), acrylic acid was successfully converted with naphthalene to its arylated product 30. Moderate yields (54–60%) and moderate to high β:α selectivities (up to >25:1) were obtained for all reactions. Coupling of methyl acrylate with benzene under adjusted electrochemical conditions (j = 1.5 mA cm2; DCE/HFIP mixtures) gave 62% of olefinated product 31. Other activated olefins such as methyl vinyl sulfone, and acrylonitrile were also amenable to the present olefination protocol. Subjecting these substrates in combination with different arenes to our protocol led to a variety of arylated products 32–35 in good yields and regioselectivities. α,β-Unsaturated ester derivatives of bioactive molecules such as δ-tocopherol and cholesterol were efficiently reacted with naphthalene to the olefinated products 36–37 in moderate yields. To further elaborate the scope of present protocol, un-activated olefins such as aliphatic olefins and styrene derivatives were tested. However, none of them afford olefinated products under our reaction conditions (see details in the ESI, Section 4.3†). To monitor the scalability of the present transformation, two reactions were performed with the model reaction at scales of 0.504 g (46%, β:α = 7:1) and 1.08 g (41%, β:α = 7:1; see ESISection 4.2†).
 | ||
| Scheme 3 Evaluation of other α,β-unsaturated systems in the electrochemical olefination of arenes. aReaction conditions: undivided cell, GF anode, Pt cathode, j = 2.5 mA cm−2 or j = 1.5 mA cm−2, corresponding arenes or heteroarenes (0.2 mmol), activated olefins (0.5 mmol), Pd(OAc)2 (10 mol%), L1 (20 mol%), TBAPF6 (0.5 equiv.), DCE (3 mL) or 5:1 ratio of dichloroethane (DCE) and 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP), 15 h, under air. bYields of isolated products are reported. | ||
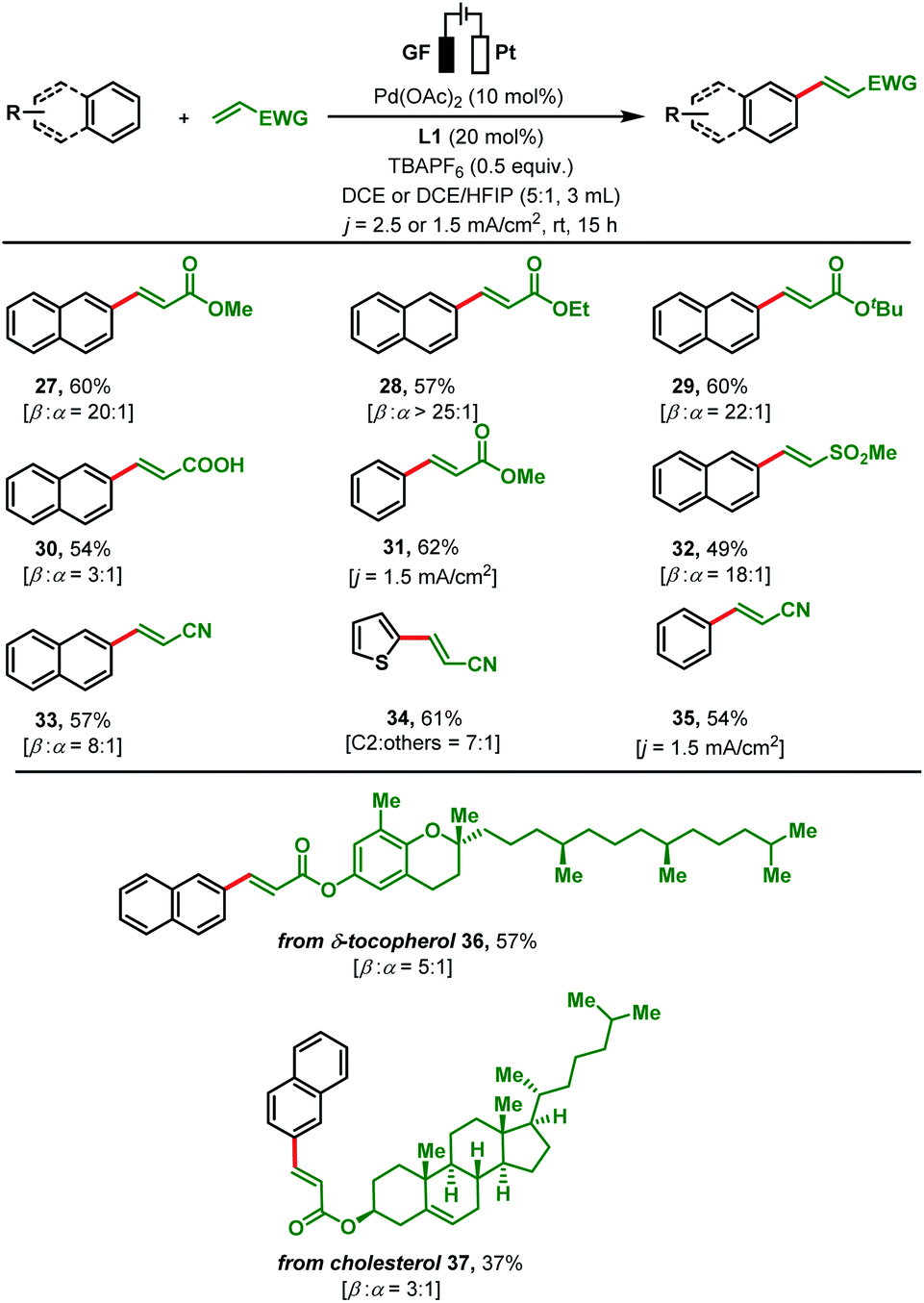
To gain insights into the catalytic mode of action, electrochemical and spectroelectrochemical experiments were performed. Cyclic voltammetry (CV) of Pd(OAc)2 in DCE revealed two oxidation waves at +1.42 V vs. NHE (Normal Hydrogen Electrode) and at +2.47 V vs. NHE (Fig. 1a) which might refer to the redox conversion of Pd(II/III) and Pd(III/IV).23Fig. 1b shows the CVs of naphthalene (substrate), ligand L1, n-butyl acrylate, and Pd(OAc)2. In comparison to the Pd(II/III) redox pair, a significantly higher oxidation potential (+2.16 V vs. NHE) was observed for naphthalene, which suggests that substrate activation is potentially induced by a Pd species with an oxidation state greater than +II (Fig. 1b). The CVs of other substrates followed the same pattern (see the ESI, Fig. S2†). According to an electrochemical study on approximate ranges of standard redox potentials for Pd intermediates in catalytic reactions, the oxidation of Pd(II) to Pd(IV) is usually observed in the range of +1.00–2.00 V (vs. Fc/Fc+ = ferrocene) or 1.63–2.63 V (vs. NHE).23 The CV profile of Pd(OAc)2 in the negative scan revealed two reduction waves at −0.23 V and at −1.06 V vs. NHE (Fig. S6†) which might refer to the redox conversion of Pd(II/I) and Pd(I/0). Taking these results into account, involvement of a Pd(II/IV) catalytic cycle during the present transformation appears to be likely as the negative scan rules out a Pd(II/0) cycle.23d,e
 | ||
| Fig. 1 (a) Cyclic voltammograms of Pd(OAc)2 and L1-Pd(OAc)2 (1 mM, 100 mV s−1 scan rate, glassy carbon, potential vs. NHE, 0.1 M TBAPF6 in DCE); (b) cyclic voltammogram of reactants (1 mM, 100 mV s−1 scan rate, glassy carbon, potential vs. NHE, 0.1 M TBAPF6 in DCE); (c) in situ UV-Vis spectroelectrochemical spectra of the reaction mixture during bulk electrolysis at +2.61 V vs. NHE; (d) in situ UV-Vis spectroelectrochemical spectra of the Pd-ligand complex during bulk electrolysis at +2.61 V vs. NHE. | ||
In order to obtain further evidence for this hypothesis, we examined the reaction mixture at a constant potential of +2.61 V (vs. NHE) spectroelectrochemically (SEC) to check any changes in optical features during the reaction. This in situ UV-visible analysis of the reaction mixture revealed the gradual decrease of an absorption band at 379 nm and a new peak (∼350 nm) appeared over time (Fig. 1c). Similar behaviour was observed for the Pd-ligand complex as a blue shift of optical bands was found from 368 nm to 352 nm at the same potential of +2.61 V (vs. NHE, Fig. 1d). The differences in the observed UV-Vis peak positions are presumably due to a change in the geometry of the Pd-complex upon oxidation in the analysed reaction mixtures.
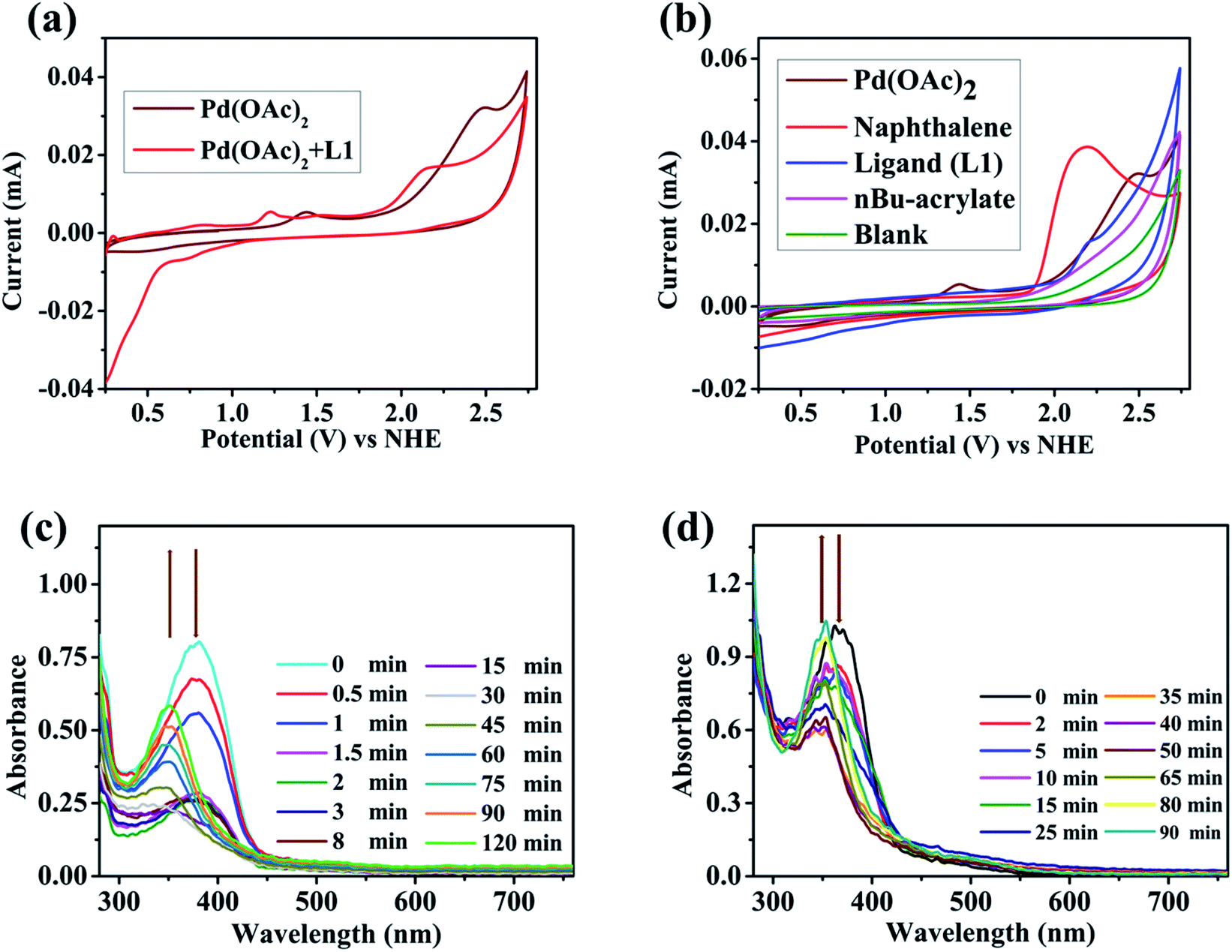
To further consolidate this hypothesis, the same SEC experiment was repeated with only Pd(OAc)2 which showed an absorption peak at 404 nm (Fig. S3†). Electrolysis of Pd(OAc)2 at +2.61 V (vs. NHE) also resulted in a blue shift with a new peak appearing at almost the same wavelength of 349 nm (Fig. S4†). All these results led us to postulate that the new peak was associated with a change in the oxidation state of the Pd(II) center. Moreover understand the nature of intermediates involved in the catalytic cycle, a series of electron paramagnetic resonance (EPR) experiments of the reaction mixture were conducted at different time intervals employing optimised reaction conditions. The EPR spectra (273 K) after 1 h showed a strong peak at g = 2.005 which was presumably due to the formation of an organic radical (Fig. 2a), however no naphthalene homo-coupled product was detected after different time intervals or under different conditions. At longer time intervals (4 h and 7 h), weak peaks at gx = 2.139, gy = 2.081 and gz = 2.055 arose due to the asymmetry of the electronic distribution. The appearance of rhombic signals suggested the formation of a Pd(III) intermediate having a d7 center (Fig. 2a).24 An enlarged version of the spectra for Pd(III) after 7 h is shown with simulated data in Fig. 2b. Time-dependent EPR spectra highlight that the build-up of Pd(III) was concomitant with the decreased formation of an organic radical (PdIII–R to PdIIR.) as the corresponding peak diminished. This implied that the catalytically active Pd(III) species got accumulated as the reaction approached towards completion. Furthermore, the EPR data in the absence of n-butyl acrylate (after 2 h) also revealed a very strong peak at g = 2.005; hence the formation of a radical species from the olefin was ruled out (Fig. S5†).
 | ||
| Fig. 2 (a) EPR spectrum of the reaction mixture under the standard reaction conditions at different time intervals (273 K); (b) enlarged EPR spectra of Pd(III) after 7 h of experiment at 273 K (experimental vs. simulated). | ||
Additionally, radical quenching experiments with TEMPO did not show any effects under the standard reaction conditions. Furthermore, electrochemical arene oxidation to generate organic radicals has been well reported in the literature.5f All these control experiments suggest that a phenoxy radical from L1 (C′) might be formed from intermediate C (Scheme 4).
 | ||
| Scheme 4 Proposed catalytic cycle for the electrooxidative olefination of arenes. | ||
All of the performed experiments give a strong indication that a Pd(II)/Pd(IV) cycle is involved in this electrochemical variant of the Fujiwara–Moritani reaction. Also, a palladium complex PdII(L1)4 was synthesised and characterised by X-ray crystallography (Fig. 3). This PdII(L1)4 complex was found to be a competent intermediate for the Pd-catalysed electrooxidative olefination of arenes.
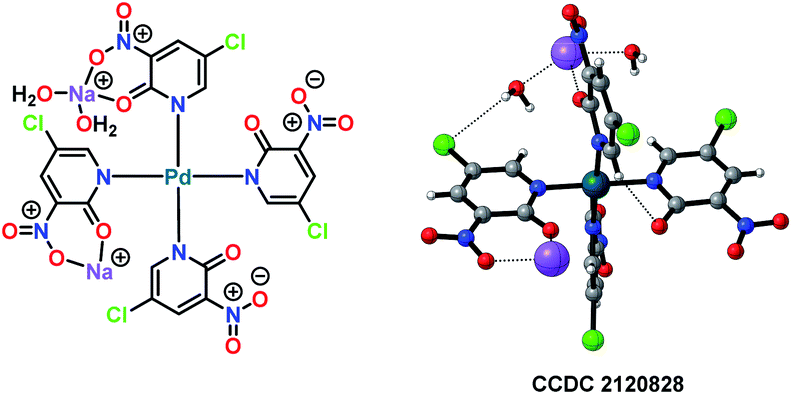 | ||
| Fig. 3 Single X-ray crystal structure of Pd-complex [Pd(L1)4].25 | ||
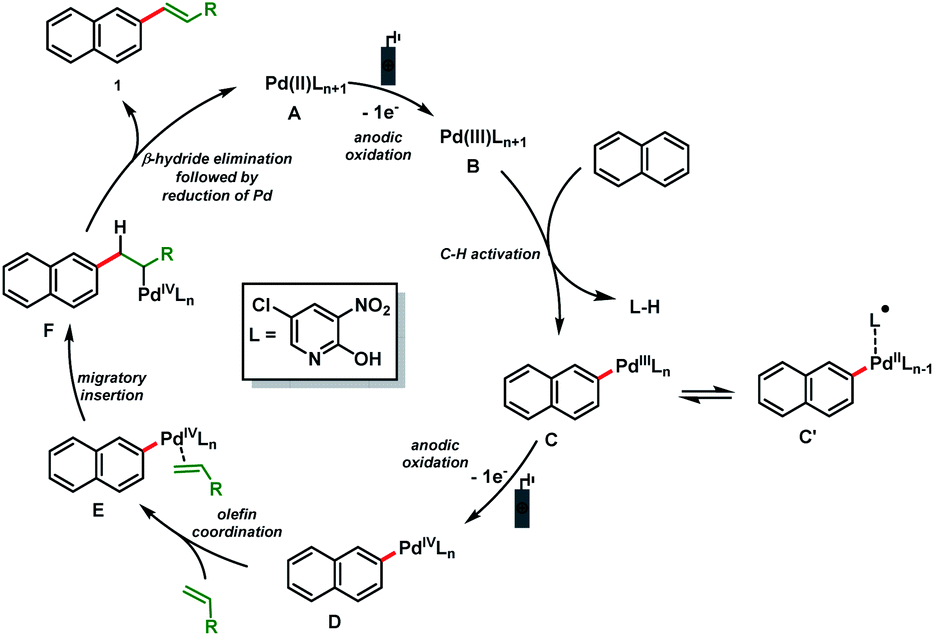
Based on these results and literature precedence,23 a plausible Pd(II/IV)-catalytic cycle is proposed for the electro-oxidative olefination of simple arenes (Scheme 4). The catalytic cycle starts with the anodic oxidation of the Pd(II) catalyst A to form a Pd(III) intermediate B. Arene C (sp2)–H bond activation delivers the organopalladium complex C which is converted to the Pd(IV) species D by anodic oxidation. Next, olefin coordination to form E followed by migratory insertion results in the formation of another organopalladium intermediate F. Finally, β-hydride elimination followed by reduction of Pd furnishes the olefinated product 1 and the Pd(II) catalyst A is regenerated.
Conclusions
In summary, we have demonstrated the first Pd-catalysed electrooxidative non-directed olefination of simple arenes. The developed transformation provides an alternative route to conventional Fujiwara–Moritani reactions by substituting toxic chemical oxidants with electric current. The applicability of this non-directed approach was proven by broad substrate scopes and high regioselectivities. Preliminary mechanistic investigations suggested the involvement of a Pd(II)/Pd(IV) catalytic cycle via a Pd(III) intermediate. Further investigations to expand the understanding of the reaction mechanism are currently underway in our laboratory.Data availability
All experimental data, and detailed experimental procedures are available in the ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The author declares no conflict of interest.Acknowledgements
Financial support received from SERB India is gratefully acknowledged (TTR/2021/000108, CRG/2020/002493). Financial support received from CSIR-India (fellowship to C. D.), PMRF-India (fellowship to T. P., J. G.), UGC-India (fellowship to S. S), IISER Bhopal (S. A.), and IIT Bombay (S. P., W. A.) is gratefully acknowledged. S. S. Anjana is greatly acknowledged for her help with PdII(L1)4 synthesis. D. M. and D. B. W. are grateful to the Alexander von Humboldt Foundation (Humboldt Research Fellowship for Experienced Researchers to D. M.). We thank G. A. Oliver for proofreading the manuscript and Mr Lalit Mohan Jha of CIF Facility, IISER Bhopal, for helping us in EPR measurements.Notes and references
-
(a) Ł. Woźniak, J.-F. Tan, A. Q.-H. Nguyen, M. du Vigné, V. Smal, Y.-X. Cao and N. Cramer, Chem. Rev., 2020, 120, 10516–10543 CrossRef PubMed
; (b) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788–1887 CrossRef CAS PubMed
-
(a) S. K. Sinha, S. Guin, S. Maiti, J. P. Biswas, S. Porey and D. Maiti, Chem. Rev., 2022, 122(6), 5682–5841 CrossRef CAS PubMed
-
(a) C. Ma, P. Fang, D. Liu, K.-J. Jiao, P.-S. Gao, H. Qiu and T.-S. Mei, Chem. Sci., 2021, 12, 12866–12873 RSC
-
(a) L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC
-
(a) G. S. Sauer and S. Lin, ACS Catal., 2018, 8, 5175–5187 CrossRef CAS
-
(a) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC
-
(a) T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chem, 2020, 6, 2484–2496 CrossRef CAS
-
(a) T. H. Meyer, L. H. Finger, P. Gandeepan and L. Ackermann, Trends Chem., 2019, 1, 63–76 CrossRef CAS
-
(a) Q.-L. Yang, P. Fang and T.-S. Mei, Chin. J. Chem., 2018, 36, 338–352 CrossRef CAS
-
(a) N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086–7103 CrossRef CAS
-
(a) U. Dhawa, C. Tian, T. Wdowik, J. C. A. Oliveira, J. Hao and L. Ackermann, Angew. Chem., 2020, 132, 13553–13559 (
Angew. Chem., Int. Ed.
, 2020
, 59
, 13451–13457
) CrossRef
-
(a) C. Ma, C.-Q. Zhao, Y.-Q. Li, L.-P. Zhang, X.-T. Xu, K. Zhang and T.-S. Mei, Chem. Commun., 2017, 53, 12189–12192 RSC
-
(a) F. Kakiuchi, T. Kochi, H. Mutsutani, N. Kobayashi, S. Ura-no, M. Sato, S. Nishiyama and T. Tanabe, J. Am. Chem. Soc., 2009, 131, 11310–11311 CrossRef CAS PubMed
- I. Moritani and Y. Fujiwara, Tetrahedron Lett., 1967, 8, 1119–1122 CrossRef
- J. Le Bras and Muzart, J. Chem. Rev., 2011, 111, 1170–1214 CrossRef CAS PubMed
-
(a) Y.-H. Zhang, B.-F. Shi and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 5072–5074 CrossRef CAS PubMed
-
(a) H. U. Vora, A. P. Silvestri, J. C. Engelin and J.-Q. Yu, Angew. Chem., 2014, 126, 2721–2724 (
Angew. Chem., Int. Ed.
, 2014
, 53
, 2683–2686
) CrossRef
- P. Wang, P. Verma, G. Xia, J. Shi, J. X. Qiao, S. Tao, P. T. W. Cheng, M. A. Poss, M. E. Farmer, K.-S. Yeung and J.-Q. Yu, Nature, 2017, 551, 489–493 CrossRef CAS PubMed
- B. Yin, M. Fu, L. Wang, J. Liu and Q. Zhu, Chem. Commun., 2020, 56, 3293–3296 RSC
- H. Chen, P. Wedi, T. Meyer, G. Tavakoli and M. van Gemme-ren, Angew. Chem., 2018, 130, 2523–2527 (
Angew. Chem., Int. Ed.
, 2018
, 57
, 2497–2501
) CrossRef
-
(a) A. Saha, S. Guin, W. Ali, T. Bhattacharya, S. Sasmal, N. Goswami, G. Prakash, S. K. Sinha, H. B. Chandrashekar, S. Panda, S. S. Anjana and D. Maiti, J. Am. Chem. Soc., 2022, 4, 1929–1940 CrossRef PubMed
-
(a) C. Amatore, C. Cammoun and A. Jutand, Adv. Synth. Catal., 2007, 349, 292–296 CrossRef CAS
-
(a) Y. H. Budnikova, Y. B. Dudkina and M. N. Khrizanforov, Inorganics, 2017, 5, 70–87 CrossRef
- N. P. Ruhs, J. R. Khusnutdinova, N. P. Rath and L. M Mirica, Organometallics, 2019, 38, 3834–3843 CrossRef CAS
- Deposition number 2120828 contains the supplementary crystallographic data for this paper. These data are provided free of charge by the Cambridge Crystallographic Data Centre http://https//www.ccdc.cam.ac.uk/structures.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2120828. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03288k |
| This journal is © The Royal Society of Chemistry 2022 |