
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDegradable thioester core-crosslinked star-shaped polymers†
Matthew
Laurel
a,
Daniel
MacKinnon
a,
Jonas
Becker
a,
Roberto
Terracciano
a,
Ben
Drain
 a,
Hannes A.
Houck
ab and
C. Remzi
Becer
*a
a,
Hannes A.
Houck
ab and
C. Remzi
Becer
*a
aDepartment of Chemistry, University of Warwick, CV4 7AL, Coventry, UK
bInstitute of Advanced Study, University of Warwick, CV4 7AL, Coventry, UK. E-mail: Remzi.Becer@Warwick.ac.uk
First published on 31st August 2022
Abstract
Degradable polymers are considered to present a promising solution to combat plastic pollution. However, many polymers are based on ester and amide bonds, which often require high temperatures and acidic/basic catalysis and hence do not easily degrade under typical environmental conditions. Thus, more readily degradable polymer structures that enable the use of milder conditions are highly sought after. Herein, degradable core-crosslinked star-shaped branched polymers have been synthesised via a two-step one-pot reversible addition–fragmentation chain transfer polymerisation. For the first time, a bifunctional thiomethacrylate crosslinker was used to prepare a range of star-shaped poly(methyl methacrylate) structures which were characterised by advanced viscometry gel permeation chromatography. The increased reactivity of thioesters over their oxoester analogues was exploited to degrade the resulting star-shaped polymers via amidation, as evidenced by the complete degradation of the crosslinked core upon heating in the presence of amines. The successful degradation of polymers containing thioesters shows the potential of thioester chemistry for producing more sustainable/degradable polymers with more complex and defined architectures.
Introduction
Branched polymers, which include dendrimers, stars and hyperbranched structures, have complex and more compact architectures than linear polymers, resulting in interesting properties, such as lower intrinsic viscosities.1–6 Hence, branched polymers have been used in many applications, including the biomedical field, catalysis, oils, and in paint adhesives.1,3–13 Dendrimers are mono-disperse, well-defined and highly symmetrical hyperbranched structures which contain a high density of functional surface groups resulting in advanced properties including good solubility and low viscosities.3 This makes the use of dendrimers advantageous in many applications including in drug delivery. However, the synthesis of dendrimers often includes multiple reaction steps and difficult purification processes.3–5 Therefore, (hyper)branched polymers such as stars present a more accessible alternative to dendrimers while still exhibiting sufficient complexity and compactness. Star polymers are a class of branched polymers which consist of 3 or more arms connected by a single core.4,8,12,14 Reversible deactivation radical polymerisation (RDRP) techniques, such as reversible addition–fragmentation chain transfer (RAFT) polymerisation, Cu(0)-mediated reversible-deactivation radical polymerisation (RDRP) and nitroxide-mediated polymerisation (NMP), have been widely used to synthesise star polymers.6,7,11,14–16The three main methodologies of synthesising stars are core-first approach, arm-first approach, and grafting onto approach.2,7 The core-first approach requires the use of a multi-functional initiator whereby the arms are grown from each initiating site during the polymerisation.2,7 The core-first approach can produce star polymers with a well-defined core, as the number of arms is known and typically equates to the number of initiating sites in the initiator.2,7,11 In contrast, the arm-first approach involves the synthesis of linear ‘living’ polymer arms that are interconnected upon the addition of a crosslinker (i.e., a multi-functional comonomer).2,7,15 A third method, the grafting onto approach, is less common but provides the highest level of control and involves the coupling of a multi-functional coupling reagent (core) and the arms.7 Similar to the core-first approach, the number of arms of stars formed by the grafting-onto method relates to the functionality of the core coupling reagent.7 However, coupling the multi-functional core to the long chain linear arms not only requires highly efficient reactions but also has to overcome the increased steric hindrance, resulting in longer reaction times and more difficult purification.7
Although synthetic pathways to branched polymers are well-established, implementing responsiveness is a highly desired feature. Indeed, in biomedical and drug delivery applications degradability is an important property for branched polymers.9,10 Whilst branched polymers offer ideal properties for use in the biomedical field, it is important that the polymers degrade to release the encapsulated drugs at the target site to be effective.10 Additionally, it is difficult for the body to eliminate such high molecular weight polymers.9 Degrading the star polymer once its function has been fulfilled, would hence allow for a more efficient excretion from the body. Several examples of degradable star polymers have been investigated based on a variety of chemistries, predominantly making use of hydrolysis.17 For example, McGuire et al. reported on the degradation of an ester-linked core-arm star polymer under acidic conditions at 100 °C.8 Our research group recently exploited amide hydrolysis of star-shaped glycopolymers that can be catalysed between pH 4–8 at 37 °C.9
In the search for ever milder conditions to degrade branched polymers, thioesters are an interesting platform to explore. Indeed, thioesters differ from conventional oxoesters as they contain a C–S bond that is weaker compared to the C–O bond of an oxoester.18–20 This is due to less delocalisation resulting from ineffective p-orbital overlap than in the case of the C–O bond.18–20 As a result, thioesters are more prone to hydrolysis, two orders of magnitude more reactive towards amidation, and have a lower activation barrier to undergo transthioesterification reactions compared to their oxoester analogues.18–23 Bowman and co-workers have extensively investigated thiol-thioester exchange as an alternative to transesterification for use in covalent adaptable networks.18,21,22 Transthioesterification was shown to be successful at room temperature, while transesterification of oxoester derivatives was slow even at temperatures above 200 °C.18 The reaction between thioesters and primary amines (S → N acyl shift) is also a crucial step in native chemical ligation as it renders the overall process irreversible.24
One way to incorporate thioesters into polymers is to polymerise thioester-containing monomers, such as thioacrylates and thiomethacrylates.20,25,26 Hadjichristidis and co-workers first reported the synthesis of thiomethacrylates by reacting thiols with methacryloyl chloride in aqueous sodium hydroxide followed by repeated distillations.27,28 Matsuda et al. reported the synthesis of a bi-functional thiomethacrylate, whilst Fila et al. used an aromatic bifunctional thiomethacrylate to form copolymers, with methyl methacrylate (MMA) and styrene.26,29 And yet, reports of vinyl thioester monomers remain rather limited. Aksakal et al. more recently described a four step synthetic route towards thioacrylate monomers, including a thioesterification and Wittig reaction, which could potentially revive their investigations in polymer science.20,23,27,28 For instance, polymers and block copolymers from thioacrylates became accessible via RAFT, NMP and Cu(0)-RDRP.20,23,30,31 Subsequently, post-polymerisation modifications of the thioester side chains, such as amidation to form polyacrylamides, have been successfully achieved.20,23,30,31 While amidation, hydrolysis, transthioesterification and oxidative hydrolysis have previously been reported for the complete and selective degradation of polymers containing thioesters in the backbone, they remain unexplored for the design of degradable core-crosslinked star-shaped polymers.18,21–23,30–32
Herein, we introduce thioester linkages into the crosslinked core of star-shaped polymers in order to exploit their more pronounced reactivity and degradability compared to ester derivatives. Core-crosslinked polymers were prepared from defined poly(methyl methacrylate) arms, obtained through RAFT polymerisation, upon the addition of a synthesised bi-functional thiomethacrylate crosslinker. The effects of the crosslinker equivalents, concentration of crosslinker, solvent and reaction time on the polymerisations were evaluated, while advanced viscometry gel permeation chromatography (GPC) was used to determine the degree of branching. Following their synthesis and characterisation, the degradation of the thioester-crosslinked polymers via amidation was finally evaluated relative to an oxoester-based branched reference polymer (Fig. 1).
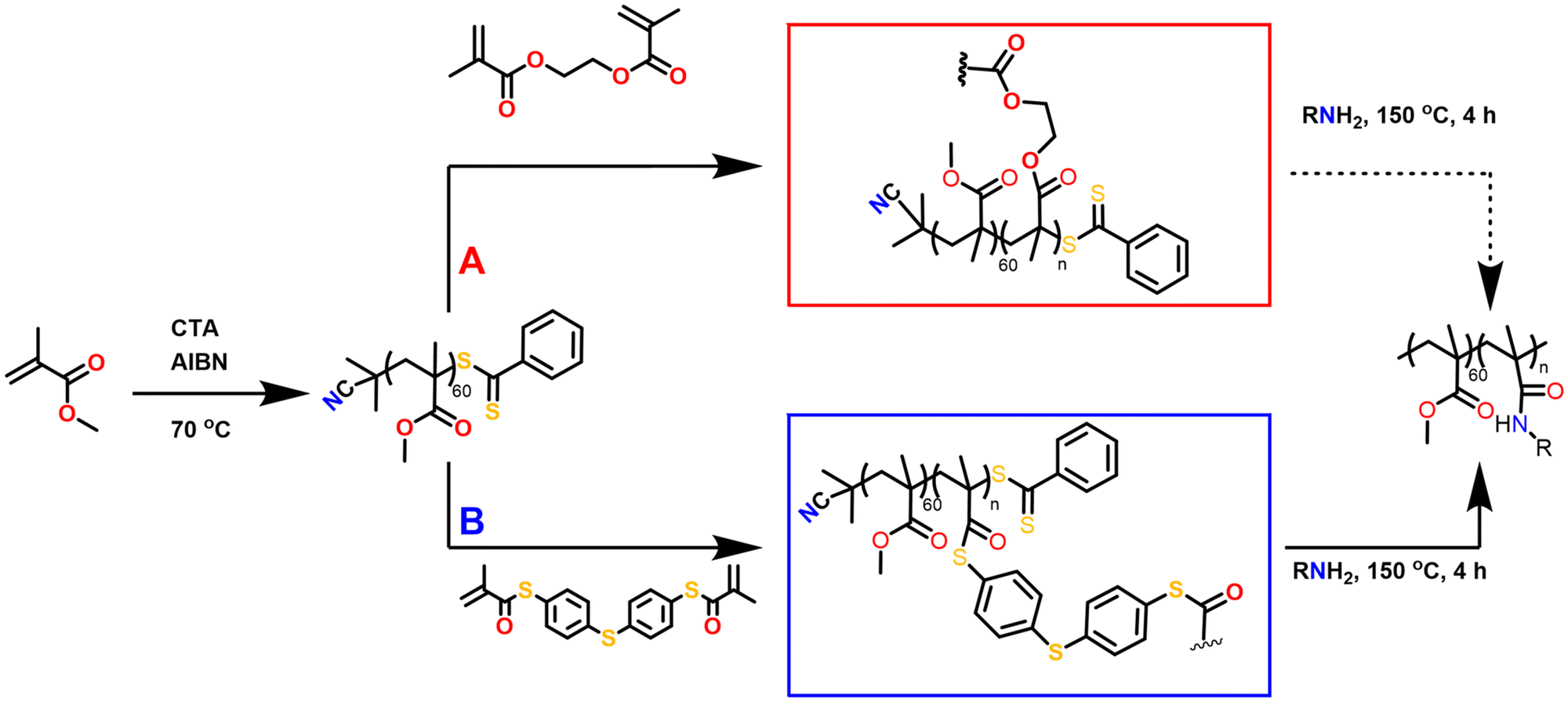 | ||
| Fig. 1 Reaction scheme for the formation and degradation of oxoester- and thioester-containing core-crosslinked star-shaped polymers, obtained via the core-crosslinking of RAFT-polymerisation prepared poly(methyl methacrylate) arms with (A) bis-methacrylate (bis-MA) and (B) bis-thiomethacrylate (bis-TMA), respectively. | ||
Results and discussion
Crosslinker synthesis
Firstly, the bifunctional aromatic thiomethacrylate (bis-TMA) crosslinker was synthesised,26 which could be readily incorporated as a core-crosslinker to access branched polymers. For this, a synthetic procedure towards phenyl thiomethacrylate was adopted using 4,4′-thiobisbenzenethiol as the bisthiol.27,28 Upon reaction with methacryloyl chloride, a crude yield of 70% bis-TMA was obtained. 1H and 13C NMR analysis (Fig. 2 and Fig. S1,† respectively) indicated the successful synthesis of the bifunctional thiomethacrylate. Notably, attempts to completely purify the bifunctional crosslinker, even in the presence of inhibitor, resulted in autopolymerisation and gelation. Nonetheless, the crude crosslinker was shown to be suitable during a test polymerisation with methyl methacrylate (MMA). Thus, the crude bis-TMA was directly used in the subsequent polymerisations.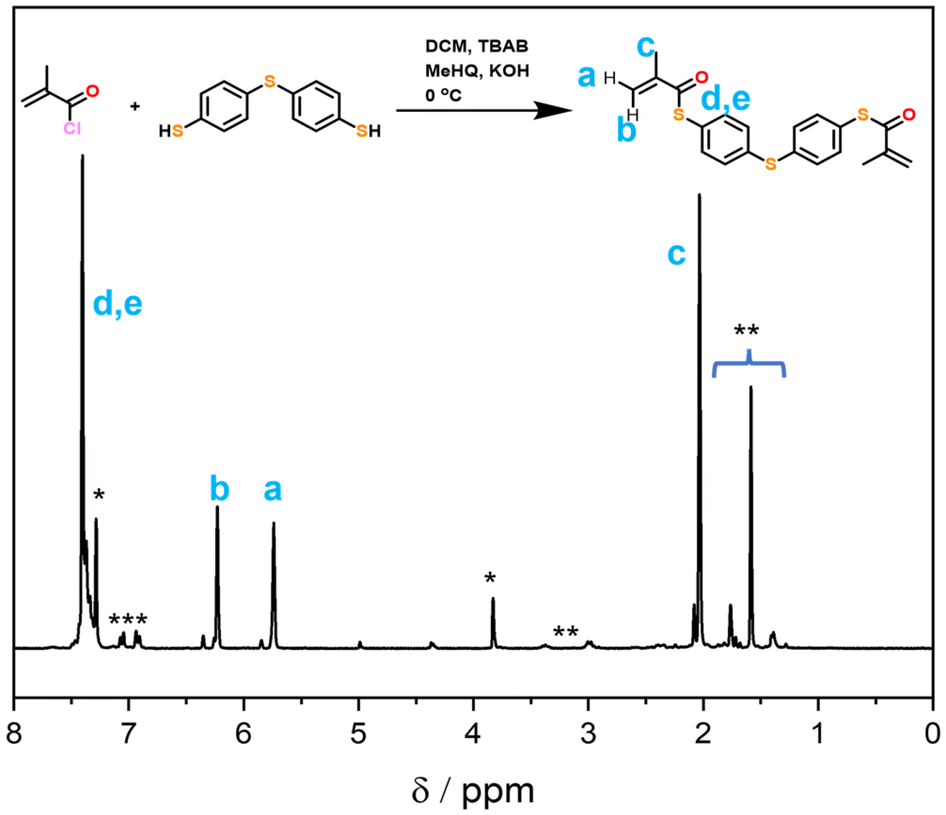 | ||
| Fig. 2 1H NMR (CDCl3) of the crude bifunctional thiomethacrylate crosslinker. Impurity peaks are identified as residual inhibitor p-methoxyphenol (MeHQ, *), tetrabutyl ammonium bromide (TBAB, **) and toluene (***). | ||
Star-shaped polymer formation, characterisation and analysis
Following the synthesis of bis-TMA, branched core-crosslinked polymers were successfully synthesised via a two-step, one-pot RAFT polymerisation arm-first approach. Two different crosslinkers were used to prepare a range of star-shaped polymers, bis-TMA and commercially available ethylene glycol dimethacrylate (bis-MA). Firstly, the poly(methyl methacrylate) arms (PMMA, target DP 60) were synthesised using 2,2′-azobis(2-methylpropionitrile) (AIBN) and 2-cyanoprop-2-yl dithiobenzoate as initiator and chain transfer agent, respectively. After a defined reaction time for the arm formation (t1), the crosslinker was added along with 0.1 eq. AIBN. The polymerisation was allowed to continue for a second reaction time (t2) before the polymerisations were air quenched and the polymers analysed. The effect of crosslinker equivalents, crosslinker concentration, solvent and reaction time were screened to determine optimal experimental conditions (Table 1). The conversion of the monomer and crosslinker were calculated via1H NMR integration (see Fig. S2–S5†) while the molecular weight increase was monitored by GPC analysis (Fig. 3). The GPC traces show the shift from the linear arms to the higher molecular weight core-crosslinked polymers. The small second peak at low molecular weight observed in the majority of polymerisations corresponds to the leftover linear arms. Notably, for the star polymers, Mp,GPC gave the most useful representation of the molecular weight, due to the effect of the leftover arm peak on the Mn,GPC value. The percentage of leftover arms was estimated via integration of the leftover arm peak and the combined area of the star and leftover arm peaks (Fig. S6†). This method provides an estimation which allows for a comparison between different stars, and later on also for tracking the degradation of these polymers.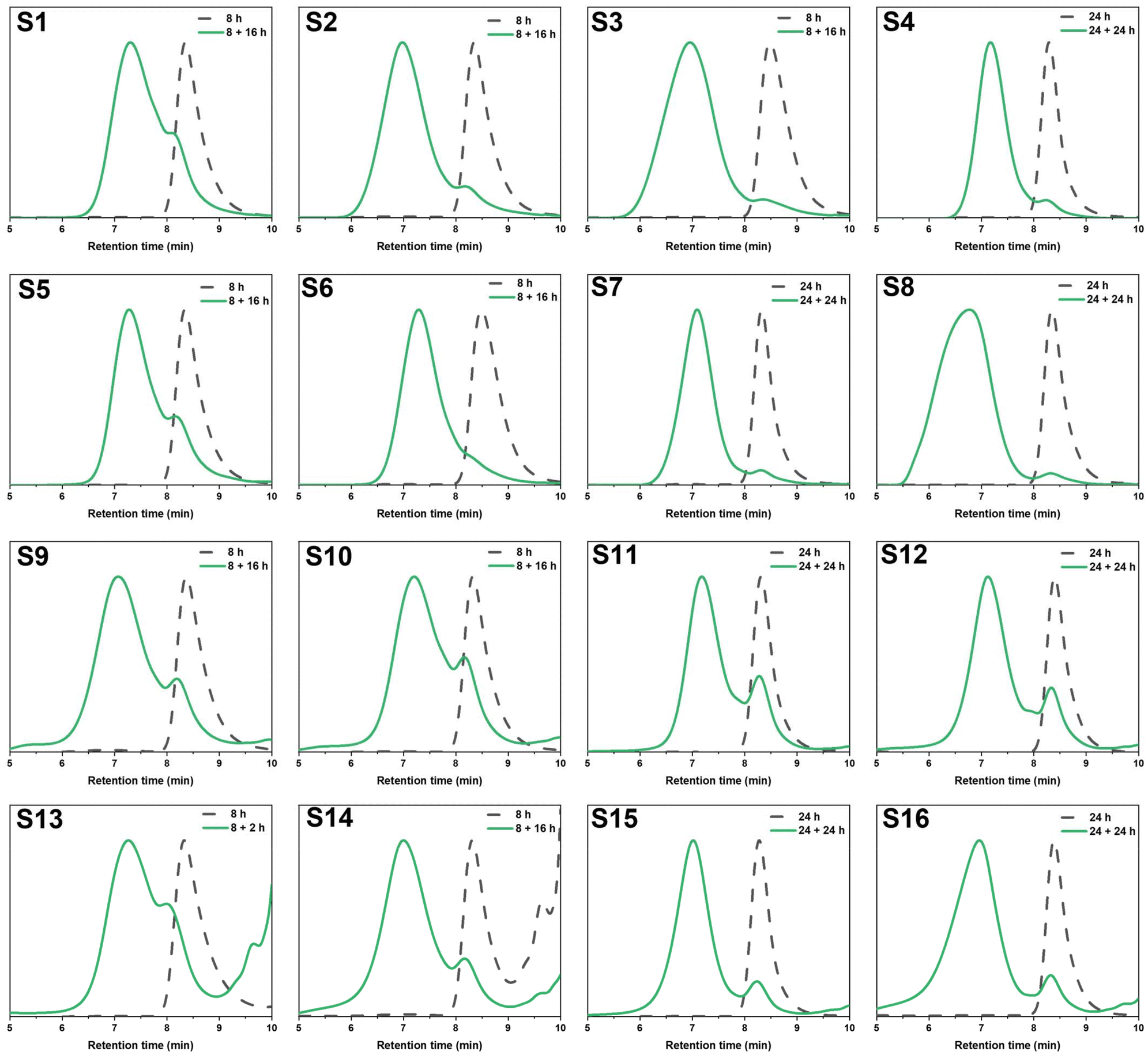 | ||
| Fig. 3 GPC traces of branched core-crosslinked polymers S1–S16 before (dashed) and after (solid) the addition of the crosslinker and their subsequent shifts to higher molecular weight. For S13, a sample is shown after 8 + 2 h due to gelation after the full reaction time. | ||
| Label | XL | XL eq. | XL conc (M) |
t
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (h) a (h) |
t
2b (h) |
Solvent | Convc MMAarm (%) | M n,th,arm (kDa) |
M
n,GPC,armd (kDa) |
Đ
armd |
Convc MMAstar (%) | Convc XLstar (%) |
M
p,GPC stard (kDa) |
Đ
stard,e |
Leftover armsf (%) |
g′g |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a t 1 is the time for the linear arm formation prior to crosslinker addition. b t 2 is the time of the core-crosslinking reaction. c Conversions calculated from 1H NMR. d Molecular weight data obtained from THF GPC as measured against PMMA standards. e The dispersity is for the combined star + leftover arm peaks. f Leftover arms calculated through integration of the leftover arm peak and the total star + arm peak in the GPC traces. g Calculated using viscometry data from the chloroform GPC and eqn (S1) and (S2).† h Star data for S13 was not measured due to gelation. | ||||||||||||||||
| S1 | Bis-MA | 5 | 0.199 | 8 | 16 | Toluene | 72 | 4.5 | 4.5 | 1.26 | 91 | 90 | 41.1 | 2.12 | 35 | 0.43 |
| S2 | Bis-MA | 5 | 0.144 | 8 | 16 | Toluene | 72 | 4.5 | 4.6 | 1.27 | 95 | 99 | 75.3 | 2.76 | 15 | 0.34 |
| S3 | Bis-MA | 5 | 0.144 | 8 | 16 | 1,4-Dioxane | 83 | 5.2 | 3.5 | 1.28 | 98 | 100 | 79.2 | 4.05 | 10 | 0.29 |
| S4 | Bis-MA | 5 | 0.144 | 24 | 24 | 1,4-Dioxane | 91 | 5.7 | 5.9 | 1.17 | 99 | 100 | 56.6 | 1.68 | 10 | 0.46 |
| S5 | Bis-MA | 5 | 0.099 | 8 | 16 | Toluene | 71 | 4.5 | 4.7 | 1.24 | 93 | 91 | 42.8 | 2.13 | 30 | 0.37 |
| S6 | Bis-MA | 5 | 0.099 | 8 | 16 | 1,4-Dioxane | 83 | 5.2 | 3.5 | 1.31 | 97 | 99 | 41.8 | 2.03 | 15 | 0.32 |
| S7 | Bis-MA | 5 | 0.168 | 24 | 24 | 1,4-Dioxane | 92 | 5.7 | 6.1 | 1.19 | 100 | 100 | 67.0 | 1.96 | 5 | 0.28 |
| S8 | Bis-MA | 10 | 0.144 | 24 | 24 | 1,4-Dioxane | 83 | 5.2 | 5.3 | 1.17 | 99 | 100 | 108.7 | 2.19 | 5 | 0.23 |
| S9 | Bis-TMA | 5 | 0.144 | 8 | 16 | Toluene | 72 | 4.5 | 6.5 | 1.25 | 95 | 99 | 63.0 | 3.71 | 25 | 0.30 |
| S10 | Bis-TMA | 5 | 0.144 | 8 | 16 | 1,4-Dioxane | 77 | 4.8 | 4.6 | 1.29 | 96 | 85 | 48.8 | 9.05 | 30 | 0.33 |
| S11 | Bis-TMA | 5 | 0.144 | 24 | 24 | 1,4-Dioxane | 95 | 5.9 | 6.3 | 1.19 | 99 | 97 | 52.3 | 2.56 | 25 | 0.31 |
| S12 | Bis-TMA | 5 | 0.168 | 24 | 24 | 1,4-Dioxane | 95 | 5.9 | 5.4 | 1.18 | 99 | 100 | 63.1 | 5.17 | 20 | 0.30 |
| S13 | Bis-TMA | 10 | 0.144 | 8 | 16 | Toluene | 41 | 2.7 | 4.5 | 1.29 | —h | —h | —h | —h | —h | —h |
| S14 | Bis-TMA | 10 | 0.144 | 8 | 16 | 1,4-Dioxane | 77 | 4.8 | 4.6 | 1.29 | 97 | 77 | 72.9 | 9.62 | 30 | 0.25 |
| S15 | Bis-TMA | 10 | 0.144 | 24 | 24 | 1,4-Dioxane | 94 | 5.9 | 6.3 | 1.19 | 99 | 93 | 69.8 | 2.59 | 15 | 0.26 |
| S16 | Bis-TMA | 10 | 0.168 | 24 | 24 | 1,4-Dioxane | 94 | 5.9 | 5.4 | 1.19 | 99 | 88 | 83.6 | 6.14 | 10 | 0.22 |
To further characterise the star-shaped polymers, information about the degree of branching was determined via advanced viscometry GPC. Whilst the molecular weight increases proportionally with the molecular size in solution for linear polymers, this is not the case for branched polymers. As the degree of branching increases, the molecular weight increases but the molecular size in solution does not proportionally increase.10,33 At a given molecular weight, the lower the intrinsic viscosity, the higher the degree of branching. Thus, advanced GPC measuring the viscosity and using that data in mathematical models allows for an evaluation of the degree of branching in the star-shaped polymers. By measuring samples of known concentration, information on the specific viscosity at different molecular weights can be obtained from the refractive index (RI) and viscosity (VS) detectors, respectively.10,33 Eqn (S1) and (S2) (cf. ESI†) were used to calculate the intrinsic viscosity (IV) and the branching factor g′ whilst eqn (S3) and (S4)† were used to estimate the average number of arms, f(n). The average g′ for all molecular weights was calculated and reported in Table 1. The molecular weight data obtained from the RI detector (Mp,GPC), the molecular weight data from the VS detector, using the universal calibration, (Mp,VS) and the average number of arms (f(n)) are displayed in Table S1.† It is noteworthy that the molecular weights obtained from the VS detector are significantly higher than the values derived from the RI detector. The VS detector gives a more accurate representation of the molecular weight as branched polymers have a smaller hydrodynamic volume than linear polymers of the same molecular weight. Conventional GPC provides a significant underestimation as it does not account for the effect of branching on the hydrodynamic volume.
The data can be illustrated on a Mark–Houwink plot of logIV vs. logM (Fig. 4A and C) and qualitative trends can be observed. All of the branched polymers contain a linear region at low molecular weights, corresponding to leftover arms. The viscosity then drops which is indicative of branching. The magnitude of the decrease in intrinsic viscosity from the linear reference corresponds to the degree of branching. The Mark–Houwink plots are also overlaid with the RI traces (dw/dlogM vs. logM) (Fig. S7 and S8†) to illustrate that the linear region of the Mark–Houwink plots overlay with the leftover arm peak on the GPC traces. The g′ values were also plotted against logM (Fig. 4B and D) to illustrate trends seen in the data. For a polymer with a higher degree of branching, the intrinsic viscosity will be lower and thus will have a lower g′ value. As can be seen in both Fig. 4B and D, the g′ vs. logM plots follow a similar trend for all of the branched polymers. At lower molecular weights, the degree of branching is lower, as indicated by the higher g′ values. As the molecular weight increases, g′ decreases and thus the degree of branching increases. This is expected as the higher molecular weights are obtained through the crosslinking of the linear polymer arms.
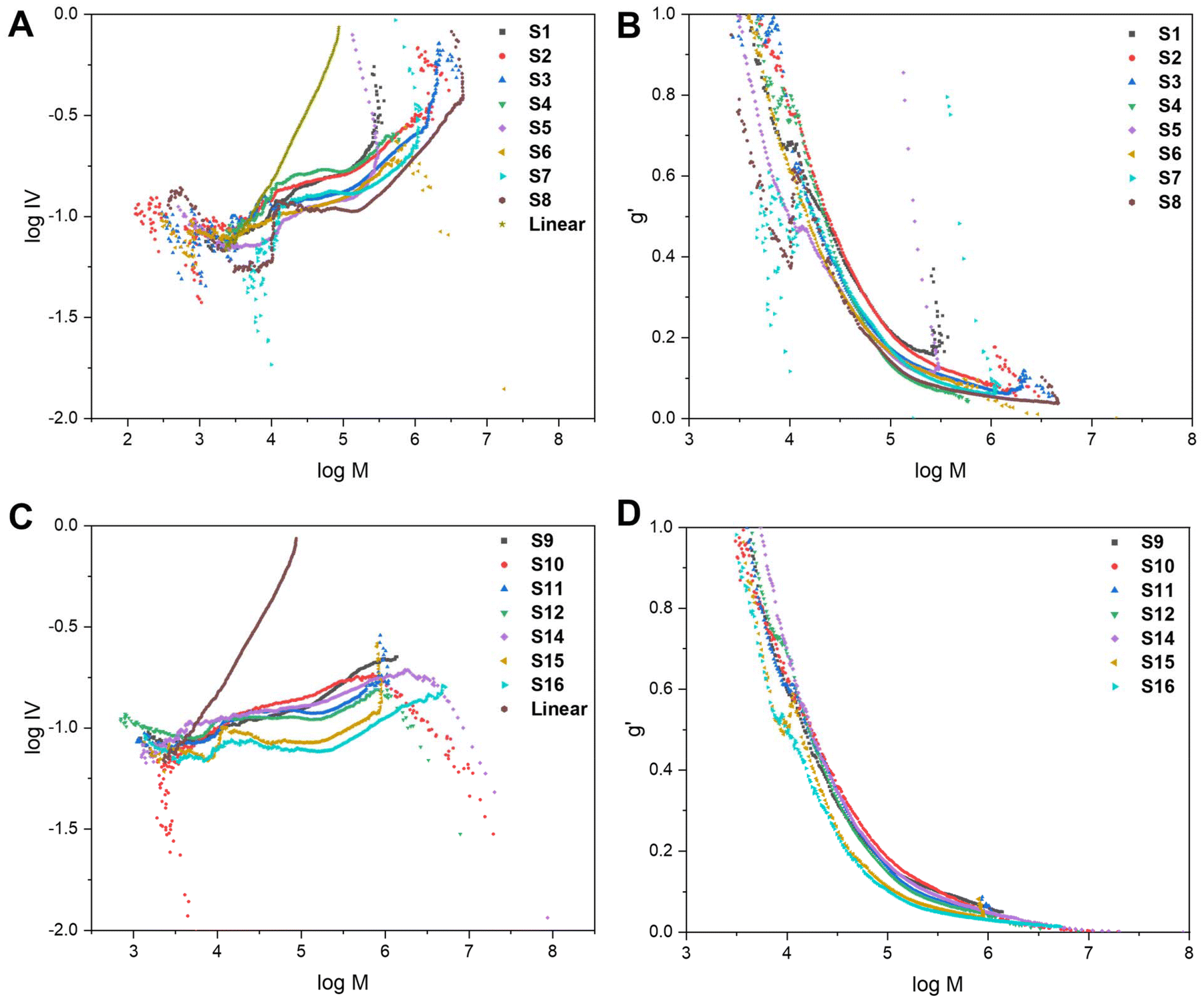 | ||
| Fig. 4 The Mark–Houwink plots and g′ vs. logM plots for the core-crosslinked branched polymers formed with a bis-methacrylate (A and B) and bis-thiomethacrylate (C and D) crosslinkers. | ||
Finally, due to the bis-TMA crosslinker being aromatic, the UV detector of the GPC was used to confirm the presence of the crosslinker. An example comparing the results obtained for branched polymers core-crosslinked with the bis-MA and bis-TMA crosslinkers is shown in Fig. S9.† Both show an absorbance at 309 nm due to the RAFT agent but only the bis-TMA polymer has an absorbance at 254 nm, originating from the aromaticity of the crosslinker.
Before core-crosslinking with bis-TMA, the core-crosslinking was first optimised with the bis-MA crosslinker (S1–S8). The initial conditions explored were 5 eq. of bis-MA, a crosslinker concentration of 0.199 M, t1 = 8 h, t2 = 16 h and toluene as the solvent (S1). In the arm formation step, a MMA conversion of 72% and a Mn,GPC of 4.5 kDa were recorded. After the core-crosslinking reaction, the molecular weight (Mp,star) significantly increased to 41.1 kDa whilst a 91% MMAstar conversion, 90% XLstar conversion and dispersity of 2.12 were obtained. The percentage of leftover arms was estimated as 35% and the g′ value was calculated as 0.43. Reducing the concentration of the crosslinker to 0.144 M (S2) resulted in increased MMAstar and XLstar conversions whilst the Mp,star of the star-shaped polymer was almost double that of S1. The percentage of leftover arms significantly decreased to 15% while g′ also decreased to 0.34. This is indicative of S2 having a higher degree of branching and increased arm incorporation. Substituting toluene for 1,4-dioxane as the solvent (S3), resulted in similar results to S2; however, the conversion of the arms after 8 h was significantly higher than when toluene was used. 1,4-Dioxane has previously been reported as a good solvent in similar polymerisations of star polymers using a diacrylate crosslinker.34 Thus, 1,4-dioxane was used as the solvent of choice for many of the subsequent reactions. There was increased arm incorporation (10% leftover arms) and a higher degree of branching (g′ value of 0.29) for S3 compared to S2 and S1. The trend of decreasing g′ from S1 to S3 can also be viewed from the Mark–Houwink plot (Fig. 4A) as the drop in viscosity is significantly greatest for S3.
However, the broad dispersity of S3 (4.05) indicated a lack of control of the core-crosslinking reaction which is not uncommon for core-crosslinked star polymers.35 The large amount of leftover MMA from the arm formation being incorporated into the core alongside the crosslinker could also play an effect on reducing the control in comparison to S1 and S2. To increase the arm conversion, the reaction time was increased (S4, t1 = 24 h; t2 = 24 h). The result was a significantly increased arm conversion of 91% and a much more well-defined star polymer with a dispersity of 1.68. Full conversions of MMA and crosslinker were observed for the star polymer, which did have a lower molecular weight of 56.6 kDa, fewer leftover arms (5%) and a higher g′ value (0.46). This indicates a lower degree of branching which results in a lower molecular weight and more well-defined star. The low degree of branching is also evident from the Mark–Houwink plot (Fig. 4A) as S4 has the smallest drop in viscosity of any of the stars S1–S8. Regardless of the low degree of branching and low molecular weight, the 48 h combined reaction time did have the advantage of significantly increasing the arm conversions.
The polymers S5 and S6 were prepared using the same conditions as S2 and S3 but with a more dilute crosslinker concentration of 0.099 M. The consequence of the reduction in concentration was a significant decrease in the molecular weight of the core-crosslinked polymers and an increase in the percentage of leftover arms. The lower molecular weights were attributed to a lower degree of branching as highlighted by increased g′ values. The decrease in concentration of crosslinker results in less crosslinker being incorporated into each core and thus branching was reduced. The effect of reducing the concentration was more pronounced for the reaction in toluene (S5) than 1,4-dioxane (S6) with a much poorer arm incorportation being observed (30% leftover arms compared to 15% for S6) which can be linked to a lower crosslinker conversion (91% vs. 97%). This was another reason that 1,4-dioxane was used for the majority of subsequent reactions. A final crosslinker concentration that was explored was 0.168 M (S7) using the 48 h combined reaction time. A similar trend, comparing S4 with S7, was seen as with S2/S5 and S3/S6. Full conversions for both MMA and XL were achieved for S7, along with a molecular weight of 67.0 kDa, 5% leftover arms and a g′ value of 0.28. For the majority of polymers in the series, increasing the crosslinker concentrations resulted in higher molecular weight polymers. The higher concentration results in the arms and crosslinker being in closer proximity thus allowing for more efficient crosslinker incorporation. This is also highlighted both by a reduced percentage of leftover arms and a lower g′ value (higher degree of branching).
The final polymer in this series, S8, was synthesised with 10 eq. of bis-MA at a crosslinker concentration of 0.144 M, with a reaction time of 24 h for both t1 and t2, and 1,4-dioxane as solvent. High conversions, a high molecular weight, good polymerisation control and a small percentage of leftover arms were observed. The molecular weight was significantly higher than for S1–S7 due to a vastly increased amount of crosslinker being present in the reaction, resulting in a higher degree of branching. This was supported the lowest g′ value calculated for S1–S8 (0.23) and the highest drop in viscosity illustrated on the Mark–Houwink plots (Fig. 4A).
Analogously, the core-crosslinking optimisation was then repeated with bis-TMA (S9–S16). S9 and S10 were synthesised with a 0.144 M crosslinker concentration, reaction times of 8 h and 16 h and with toluene and 1,4-dioxane as their respective solvents. Compared to the corresponding bis-MA polymers (S2 and S3) the polymerisations proceeded with lower conversions, reduced molecular weights, decreased control and a higher percentage of leftover arms. These comparisons are observed for several of the entries in Table 1. Several factors could contribute towards decreased crosslinker incorporation including the larger size of the crosslinker increasing the steric hindrance, the increased rigidity of the crosslinker or the behaviour of the crosslinker in solution. In contrast to the corresponding bis-MA polymerisations, due to solubility differences, toluene (S9) was shown to perform better than 1,4-dioxane (S10), with a very low crosslinker conversion, low molecular weight and high dispersity observed for S10. The g′ values of 0.3 (S9) and 0.33 (S10) are amongst the highest for the series S9–S16, while the Mark–Houwink plot (Fig. 4C) also indicates a low degree of branching. As for S4, when the reaction time was increased to 24 h for both t1 and t2 in 1,4-dioxane (S11), a significant improvement in the molecular weight control was observed, as indicated by the much narrower dispersity of 2.56. The crosslinker conversion was significantly improved (97% vs. 85% for S10) which resulted in a higher degree of branching (g′ = 0.31) and a reduced percentage of leftover arms (25%) which led to an increase in molecular weight. A final polymer with 5 eq. of the bis-TMA increased the crosslinker concentration to 0.168 M (S12). As with S7, the increase in concentration led to an increase in molecular weight due to increased crosslinker incorporation as indicated by a higher conversion (100%), reduced percentage of leftover arms (20%) and a decreased g′ value (0.30).
As for S8, when 10 eq. of the bis-TMA crosslinker were used, core-crosslinked polymers with significantly higher molecular weights were formed. When toluene was used as the solvent with 10 eq. of bis-TMA crosslinker with the shorter reaction time, (t1 = 8 h; t2 = 16 h) a gel was formed. This was attributed to the very low conversion (i.e., 41%) when forming the linear arms during the first step, thus providing enough leftover MMA monomer to induce crosslinking upon the addition of bis-TMA. The low arm conversion could suggest a larger number of dead polymer chains which would reduce the number of arms being available to be core-crosslinked. This corresponds to the observed trend of decreased control on the polymerisation as the arm conversion decreases. Indeed, a high MMA arm conversion typically led to narrower dispersities than the polymers formed with lower MMA arm conversions. Thus, care should be taken to keep the conversion during the arm formation sufficiently high to prevent an insoluble gel being formed. Similarly, the polymer formed with the shorter reaction times in 1,4-dioxane (S14) had a low arm conversion (77%) and a lower crosslinker conversion (77%). A molecular weight of 72.9 kDa was obtained as well as a percentage of leftover arms of 30% and a g′ value of 0.25. Even with a lower crosslinker conversion, a high molecular weight was observed due to the increased number of crosslinker equivalents (10 eq.). As for previous examples, increasing the reaction time (S15, t1 = 24 h; t2 = 24 h) resulted in an increased MMA arm conversion and a higher crosslinker conversion (93%). A slight decrease in molecular weight (69.8 kDa) was observed alongside a g′ value of 0.26 and a reduced percentage of leftover arms (15%). A final polymer that was synthesised, S16, was with an increased crosslinker concentration of 0.168 M. As for when 5 eq. were used (S12), the increase in concentration resulted in less control over the polymerisation and an increased molecular weight (83.6 kDa) due to increased branching (g′ = 0.22). The higher degree of branching for the polymers synthesised with 10 eq. is illustrated on the Mark–Houwink plots (Fig. 4B) with S15 and S16 having the greatest drops in viscosity.
In general, the star polymers formed with this bis-TMA crosslinker had lower g′ values and higher degrees of branching. Whilst this is not reflected fully in the Mp,GPC in Table 1, this is an underestimation of the molecular weight. It can be seen in Table S1,† that the molecular weight data using the viscometry detector, which takes branching into account, is higher for the series S9–S16 than for S1–S8.
Degradation studies
Following on from the formation of the branched polymer networks, the degradability of the core-crosslinked star-shaped polymers was explored. For this, degradation via amidation in tetrahydrofuran was investigated (Table 2). It was hypothesised that the polymers crosslinked with the thioester-containing bis-TMA would allow for the branched structures to degrade more readily, as compared to the conventionally used bis-MA ester-based crosslinker. Different variables were altered to study the degradation including the temperature, heating method, reaction time, and the structure and equivalents of amine. GPC was used as the main analytical technique to monitor the degradation progress as the disappearance of the core-crosslinked polymer peak and the simultaneous increase of the linear polymer arms is readily visible. The same method that was used to estimate the percentage of leftover arms in Table 1 was used to track the increase in the percentage of linear arms present as the degradation proceeded.| Run | Starting polymer | Heating method | Solvent | Time (h) | Temperature (°C) | Amine | Amine equivalents | Arms post-degradationa (%) |
|---|---|---|---|---|---|---|---|---|
| a Percentage arms calculated through integration of the GPC-RI leftover arm peak and the total star + arm peak. b Degradation of a bis-MA crosslinked star. c Degradations D16 and D17 conducted in bulk amine. | ||||||||
| D1 | S10 | Microwave | 4:1 THF:water |
1 | 70 | Hexylamine | 10 | 30 |
| D2 | S10 | Microwave | 4:1 THF:water |
1 | 90 | Hexylamine | 10 | 30 |
| D3 | S10 | Microwave | 4:1 THF:water |
1 | 110 | Hexylamine | 10 | 30 |
| D4 | S10 | Microwave | 4:1 THF:water |
1 | 130 | Hexylamine | 10 | 35 |
| D5 | S10 | Microwave | 4:1 THF:water |
1 | 150 | Hexylamine | 10 | 50 |
| D6 | S10 | Microwave | 4:1 THF:water |
12 | 150 | Hexylamine | 10 | 55 |
| D7 | S10 | Microwave | 4:1 THF:water |
4 | 150 | Benzylamine | 10 | 50 |
| D8 | S10 | Microwave | 4:1 THF:water |
4 | 150 | Hexylamine | 100 | 85 |
| D9 | S16 | Oil bath | THF | 4 | 150 | Hexylamine | 10 | 35 |
| D10 | S16 | Oil bath | THF | 4 | 150 | Hexylamine | 25 | 40 |
| D11 | S16 | Oil bath | THF | 2 | 150 | Hexylamine | 50 | 90 |
| D12 | S16 | Oil bath | THF | 2 | 150 | Hexylamine | 100 | 100 |
| D13 | S16 | Oil bath | THF | 2 | 150 | Benzylamine | 50 | 90 |
| D14 | S16 | Oil bath | THF | 3 | 150 | Ethanolamine | 50 | 95 |
| D15 | S16 | Oil bath | THF | 2 | 150 | 3-Amino-1-propanol | 50 | 100 |
| D16 | S16 | Oil bath | N/ac | 1 | 150 | Hexylamine | Bulk- 2492 | 100 |
| D17 | S2 | Oil bath | N/ac | 4 | 150 | Hexylamine | Bulk- 2588 | 20 |
Preliminary degradation reactions on the thioester-crosslinked star polymer S10 showed that little degradation occurred with 10 eq. of hexylamine at 25 °C over a 2-week period (Fig. S10†) but that significant degradation occurred at 70 °C over 9 days (Fig. S11†). In a bid to reduce the reaction times, degradation reactions of S10 in a microwave reactor were set up, whereby the temperature, choice of amine, amine equivalents, and reaction time were varied. The experimental conditions are summarised in Table 2 (D1–D8) and the GPC traces for the temperature screening (D1–D5) and the investigation into reaction time and amine equivalents (D5–D8) are illustrated in Fig. 5A and B, respectively. For reactions in the microwave reactor a 4:1 THF:water solvent was used; the addition of water was important in ensuring the reaction proceeded. The peak that occurs in the GPC traces after 9 min retention time is due to leftover amine from the degradation. Specifically, the molecular weight distribution at around a retention time of 7 minutes corresponds to the core-crosslinked polymer whilst leftover linear chains are observed at a retention time of 8 minutes. The success of the degradation can thus be monitored via the disappearance of the high molecular weight distribution and the simultaneous formation of lower molecular weight products. Further characterisation included 1H NMR and IR spectroscopy (Fig. 6 and Fig. S12†). Reactions D1–D5 aimed to explore the effect of temperature on the thioester degradation after 1 h with 10 equivalents of hexylamine. Whilst slight variations are observed for polymers D1–D4 subjected to 70–130 °C heating in the presence of hexylamine, only a minor reduction of the core-crosslinked polymers was observed. The percentage of leftover arms in that starting material S10 was 30% and the degraded polymers D1–D4 also contained 30–35% arms. This means there was less than a 5% increase in the number of arms being regenerated which suggests very little degradation occurred. Increasing the temperature to 150 °C (D5), however, did show a larger shift to higher retention times, thereby indicating a higher degree of degradation occurred. This is supported by an increase in the number of arms present in D5 to 50%. Following the temperature variations, other variables were changed for the degradations while keeping the temperature at 150 °C. The reaction time was increased from 1 h to 12 h (D6), which resulted in a percentage arm value of 55%. This was only a 5% increase from D5 even though the reaction time was significantly increased. Benzylamine (10 eq.) was used in a 4 h reaction at 150 °C (D7) which also resulted in little improvement on the results obtained from D5. Finally, the number of hexylamine equivalents was increased from 10 to 100, keeping the reaction mixture at 150 °C for 4 hours (D8). From the GPC traces shown in Fig. 5B, it can be deduced that D8 had a much higher degree of degradation than D6 or D7. The tailing observed in the GPC trace of D8 (which was also seen in other degradations) can be attributed to disulfide coupling following the aminolysis of the RAFT chain transfer agent end groups. This shows that increasing the amine equivalents was more effective in accelerating the degradation than increasing the reaction time or changing the nature of the amine. The conditions used in the degradation D8 led to the most successful degradation of the series D1–D8, with 85% arms being obtained. These results verified the importance of the amine in the degradation. When comparing D8 with D6, a higher degree of degradation was achieved with 100 equivalents of hexyl amine at 150 °C for 4 hours.
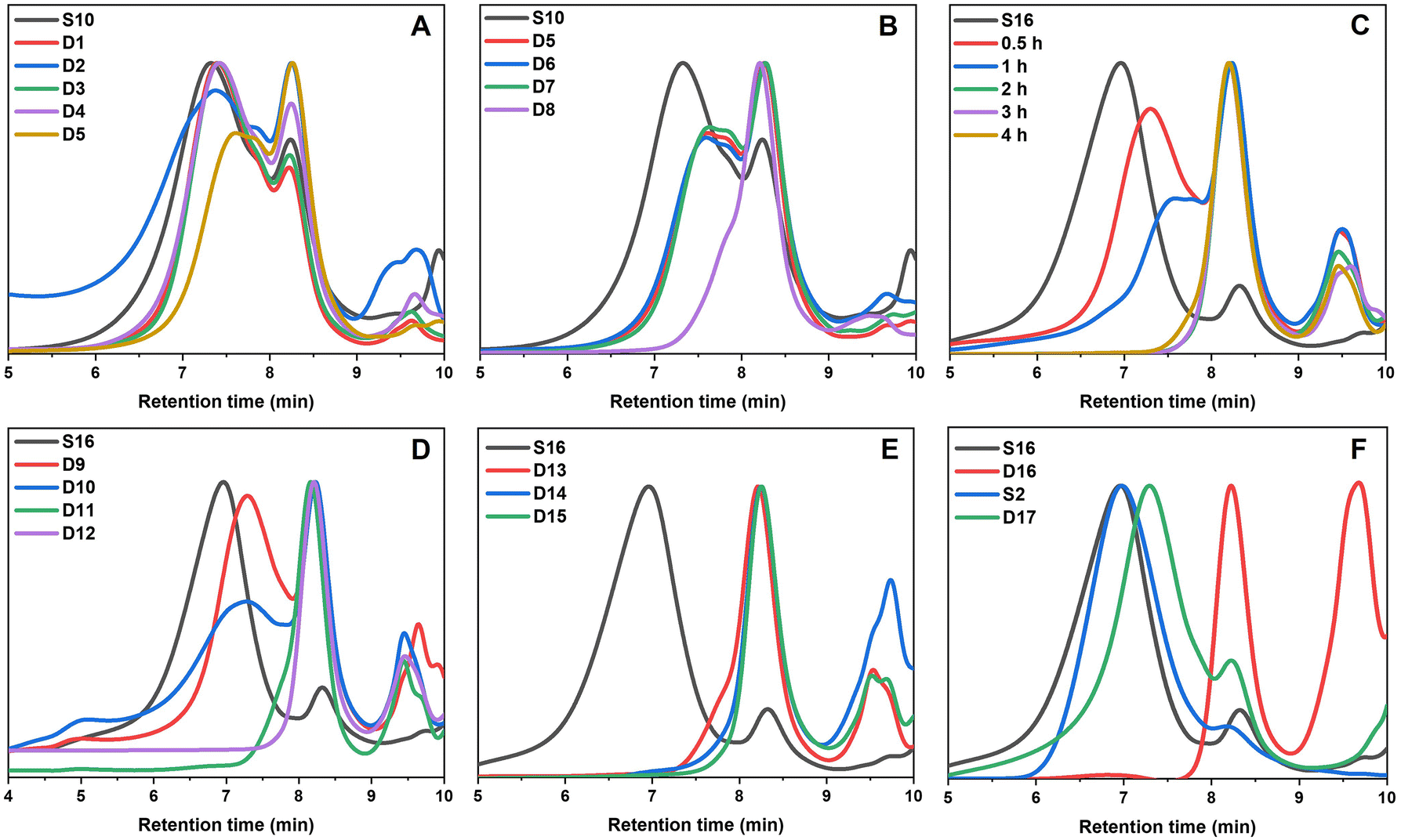 | ||
| Fig. 5 The GPC traces showing the degradation of the core-crosslinked branched polymers. (A) Temperature scanning in the microwave reactor between 70–150 °C. (B) Microwave degradations focused on reaction time (1 h, 4 h, 12 h) and amine equivalents (10 eq., 100 eq.). (C) The kinetics showing the progression of the degradation D12. (D) Differing equivalents of hexylamine (10 eq., 25 eq., 50 eq., 100 eq.). (E) Varying amines (benzyl amine, ethanolamine and 3-amino-1-propanol). (F) The bulk degradation of both the bis-thiomethacrylate and bis-methacrylate core-crosslinked polymers with hexylamine. | ||
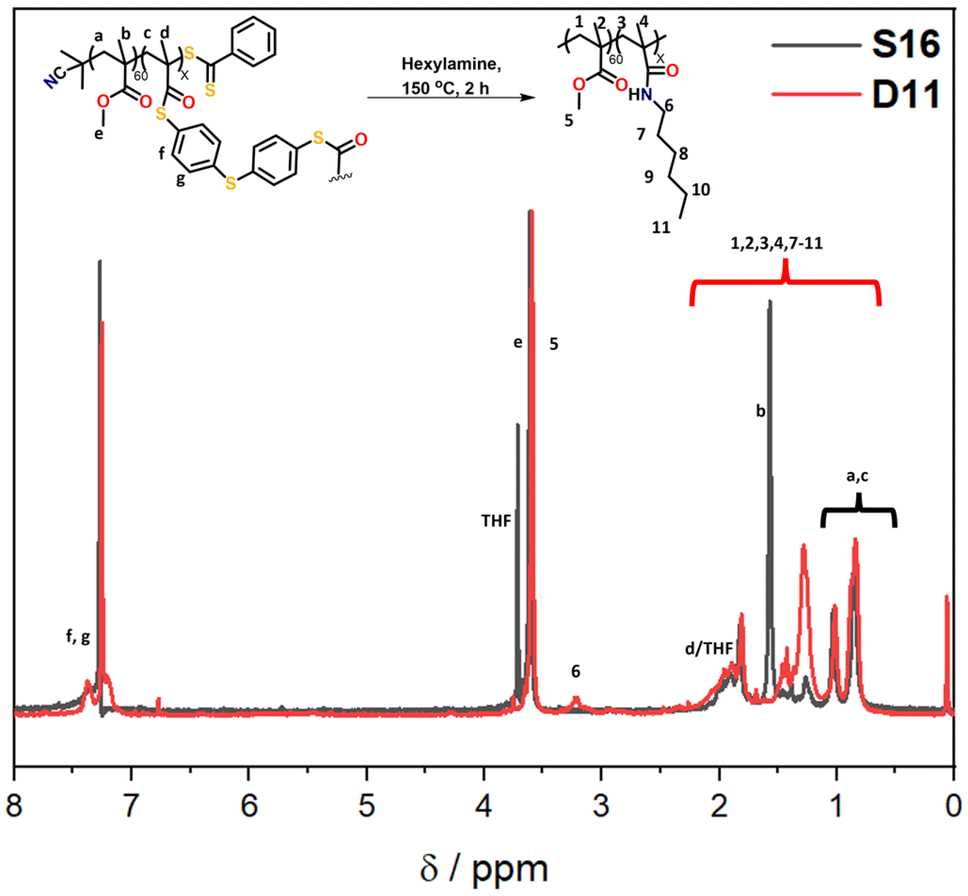 | ||
| Fig. 6 1H NMR spectra (CDCl3) of the starting polymer (S16) and crude hexylamine degraded product (D11), showing the formation of the amide product. | ||
From the temperature screening of the microwave degradation experiments, 150 °C was selected as most optimal reaction temperature. Although microwave conditions are typically useful to minimise reaction times, it seemed relevant to also investigate degradation of the thioester stars using a conventional heating method. Therefore, a series of experiments were conducted in an oil bath set at 150 °C using the starting material S16 (having 10% leftover arms). The first series of experiments investigated changing the number of equivalents of hexyl amine from 10 to 100 equivalents. The experimental conditions and GPC traces are displayed in Table 2 (D9–D12) and Fig. 5C and D. The first degradation that was conducted via conventional heating (D12) used the same experimental conditions that were successful in the microwave reactor (D8). The kinetics of this reaction was studied over 4 hours and the degradation was monitored by GPC (Fig. 5C). With 100 eq. of hexyl amine, incomplete degradation was observed within the first hour, but the degradation went to completion after 2 hours of heating (Fig. 5C). Similarly, with 50 equivalents of hexyl amine (D11), the degradation was successful within 2 hours (Fig. 5D). However, integration of the GPC traces estimated that the percentage of arms is only 90%. It was concluded that of those tested, 50 equivalents was the minimum amount required for a high degree of degradation within 2 hours. In addition to GPC analysis, 1H NMR of both the S16 starting material and the degraded product from conditions D11 was recorded (Fig. 6). The appearance of the CH2 proton resonance adjacent to the amide at around 3.20 ppm is indicative of successful amidation.
Lowering the amount of hexyl amine to 25 eq. (D10) and 10 eq. (D9) no longer resulted in complete degradation after 4 hours, as significant amounts of the core-crosslinked polymer were still observed in the GPC traces (Fig. 5D). Thus, significant amounts of amine are required in order to fully degrade the thioester cores. As mentioned previously for the microwave reactions, these results illustrate the importance of the amine. If the high temperature was the significant factor in the degradation, then even the samples with 10 and 25 equivalents would have been expected to result in significant star degradation.
Using the findings that 50 equivalents of amine led to a successful degradation, other amines were also investigated. The GPC traces before and after the reaction with benzylamine, ethanolamine and 3-amino-1-propanol (D13–D15) are shown in Fig. 5E whilst the degradation was also monitored at distinct time intervals (Fig. S13†). As was the case for the degradation with 50 equivalents of hexylamine, all were shown to be successful (>90% arms). The reactions with benzylamine and 3-amino-1-propanol were completed within 2 hours while the reaction with ethanolamine went to competition within 3 hours. In particular the latter two alkanolamines are of interest as these enable post-degradation functionalisation of the resulting linear polymer chains and their potential re-use to re-form core-crosslinked structures. Fig. S12† shows additional IR characterisation of the degraded product for D14. While in the S16 starting material, a small peak at around 1690 cm−1 corresponds to the thioester carbonyl stretch,36 this peak was absent for the bis-MA crosslinked star-shaped polymer (S2). For the degraded product, this carbonyl stretch shows a clear shift to around 1660 cm−1 further supporting the transformation of the thioester to an amide. A broad peak around 3100–3500 cm−1 was also present, indicating that the polymer contained functional hydroxyl groups.
As a final degradation experiment, the reaction of the core-crosslinked polymers with hexylamine was carried out under bulk conditions. The polymer core-crosslinked with the bis-TMA (S16) underwent complete degradation within one hour (D16, Fig. 5F). Contrastingly, a branched oxoester-containing polymer formed with the bis-MA crosslinker (S2) showed little degradation under the same conditions after 4 hours (D17, Fig. 5F). The starting polymer S2 contained 15% leftover arms, only increasing to 20% arms after being subjected to degradation conditions for 4 h, hence evidencing the higher reactivity and thus increased degradation potential of thioesters towards amidation compared to their oxoester derivatives.
Conclusion
A bifunctional thiomethacrylate crosslinker was synthesised which was then used to form PMMA star-shaped polymers via a core-crosslinked approach. The polymerisations were optimised in terms of the crosslinker equivalents, crosslinker concentration, reaction time and solvent. A 1,4-dioxane solvent and a combined reaction time of 48 hours resulted in higher conversion values whilst increasing the crosslinker equivalents and crosslinker concentration significantly increased the molecular weight of the branched polymers. The branched architectures were analysed via advanced viscometry GPC and their branching factor g′ was calculated. In general, the branched polymers synthesised with 10 equivalents of the bis-thiomethacrylate were shown to have lower g′ values and thus a higher degree of branching. The thioester crosslinked polymers were then degraded via amidation at high temperatures (150 °C) whilst the polymers crosslinked with an oxoester crosslinker derivative showed very little degradation. The choice of amine and equivalents thereof were shown to be of greater significance than the high reaction temperature, with 50 equivalents being the minimum amount of amine that resulted in successful degradation throughout our studies. In conclusion, we have successfully demonstrated that star-shaped polymers core-crosslinked with a bis-thiomethacrylate degraded significantly more readily through amidation than polymers core-crosslinked with a bis-methacrylate. This work is believed to foster opportunities to design other thio(meth)acrylate-based polymer structures that are not only susceptible to amine-induced degradation but also prone to hydrolysis and trans-thioesterification.Author contributions
M. L. conducted the experimental work, data analysis and writing. D. M. and R. T. carried out part of the polymer synthesis. J. B. helped with the monomer synthesis. B. D. assisted with the advanced GPC analysis. H. A. H. and R. B. supervised the work and contributed to writing and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. B. and R. T. has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No. 859416. H. A. H. acknowledges funding of his EUTOPIA-SIF fellowship received from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 945380.References
- T. K. Georgiou, Polym. Int., 2014, 63(7), 1130–1133 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Macromolecules, 2006, 39, 3154–3160 CrossRef CAS.
- R. M. England and S. Rimmer, Polym. Chem., 2010, 1(10), 1533–1544 RSC.
- A. Hirao, M. Hayashi, S. Loykulnant, K. Sugiyama, S. Ryu, N. Haraguchi, A. Matsuo and T. Higashihara, Prog. Polym. Sci., 2005, 30(2), 111–182 CrossRef CAS.
- L. I. F. Moura, A. Malfanti, C. Peres, A. I. Matos, E. Guegain, V. Sainz, M. Zloh, M. J. Vicent and H. F. Florindo, Mater. Horiz., 2019, 6(10), 1956–1973 RSC.
- W. Wu, W. Wang and J. Li, Prog. Polym. Sci., 2015, 46, 55–85 CrossRef CAS.
- J. M. Ren, T. G. McKenzie, Q. Fu, E. H. H. Wong, J. Xu, Z. An, S. Shanmugam, T. P. Davis, C. Boyer and G. G. Qiao, Chem. Rev., 2016, 116, 6743–6836 CrossRef CAS.
- T. M. McGuire, M. Miyajima, M. Uchiyama, A. Buchard and M. Kamigaito, Polym. Chem., 2020, 11(36), 5844–5850 RSC.
- A. Monaco, V. P. Beyer, R. Napier and C. R. Becer, Biomacromolecules, 2020, 21(9), 3736–3744 CrossRef CAS.
- A. Monaco, B. Drain and C. R. Becer, Polym. Chem., 2021, 12(36), 5229–5238 RSC.
- J. A. Syrett, D. M. Haddleton, M. R. Whittaker, T. P. Davis and C. Boyer, Chem. Commun., 2011, 47(5), 1449–1451 RSC.
- U. Wais, L. R. Chennamaneni, P. Thoniyot, H. Zhang and A. W. Jackson, Polym. Chem., 2018, 9(39), 4824–4839 RSC.
- J. T. Wiltshire and G. G. Qiao, J. Polym. Sci., Part A: Polym. Chem., 2009, 47(6), 1485–1498 CrossRef CAS.
- L. Xiang, M. Qiu, M. Shang and Y. Su, Polymer, 2021, 222, 123699 CrossRef.
- J. Ferreira, J. Syrett, M. Whittaker, D. M. Haddleton, T. P. Davis and C. Boyer, Polym. Chem., 2011, 2(8), 1671–1677 RSC.
- J. Skey, H. Willcock, M. Lammens, F. Du Prez and R. K. O'Reilly, Macromolecules, 2010, 43(14), 5949–5955 CrossRef CAS.
- J. Rosselgong, E. G. L. Williams, T. P. Le, F. Grusche, T. M. Hinton, M. Tizard, P. Gunatillake and S. H. Thang, Macromolecules, 2013, 46(23), 9181–9188 CrossRef CAS.
- C. Wang, S. Mavila, B. T. Worrell, W. Xi, T. M. Goldman and C. N. Bowman, ACS Macro Lett., 2018, 7(11), 1312–1316 CrossRef CAS PubMed.
- W. Yang and D. G. Drueckhammer, J. Am. Chem. Soc., 2001, 123, 11004–11009 CrossRef CAS PubMed.
- S. Aksakal, R. Aksakal and C. R. Becer, Polym. Chem., 2018, 9(36), 4507–4516 RSC.
- B. T. Worrell, S. Mavila, C. Wang, T. M. Kontour, C.-H. Lim, M. K. McBride, C. B. Musgrave, R. Shoemaker and C. N. Bowman, Polym. Chem., 2018, 9(36), 4523–4534 RSC.
- N. J. Bongiardina, K. F. Long, M. Podgórski and C. N. Bowman, Macromolecules, 2021, 54(18), 8341–8351 CrossRef CAS.
- S. Aksakal and C. R. Becer, Polym. Chem., 2016, 7(45), 7011–7018 RSC.
- N. Stuhr-Hansen, N. Bork and K. Stromgaard, Org. Biomol. Chem., 2014, 12(30), 5745–5751 RSC.
- B. F. M. Wanders and H. Haitjema, Int. Pat, WO 2007/094664A1, 2007 Search PubMed.
- K. Fila, M. Grochowicz and B. Podkościelna, J. Therm. Anal. Calorim., 2017, 133(1), 489–497 CrossRef.
- D. Kokkiaris, C. Touloupis and N. Hadjichristidis, Polymer, 1981, 22, 63–66 CrossRef CAS.
- N. Hadjichristidis, C. Touloupis and L. J. Fetters, Macromolecules, 1981, 14(1), 128–130 CrossRef CAS.
- T. Matsuda, Y. Funae, M. Yoshida and T. Yamamoto, Synth. Commun., 2000, 30(16), 3041–3045 CrossRef CAS.
- S. Aksakal, V. P. Beyer, R. Aksakal and C. R. Becer, Polym. Chem., 2019, 10(48), 6622–6629 RSC.
- S. Aksakal, R. Liu, R. Aksakal and C. R. Becer, Polym. Chem., 2020, 11(5), 982–989 RSC.
- S. Aksakal, R. Aksakal and C. R. Becer, Macromol. Rapid Commun., 2019, 40(16), 1900247 CrossRef.
- G. Hayes, B. Drain and C. R. Becer, Macromolecules, 2021, 55(1), 146–155 CrossRef.
- M. S. Rolph, A. Pitto-Barry and R. K. O'Reilly, Polym. Chem., 2017, 8(34), 5060–5070 RSC.
- J. Rosselgong, S. P. Armes, W. R. S. Barton and D. Price, Macromolecules, 2010, 43(5), 2145–2156 CrossRef CAS.
- I. Aped, Y. Mazuz and C. N. Sukenik, Beilstein J. Nanotechnol., 2012, 3, 213–220 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra, GPC traces and Mark–Houwink plots. See DOI: https://doi.org/10.1039/d2py00901c |
| This journal is © The Royal Society of Chemistry 2022 |