
The difference between photo-iniferter and conventional RAFT polymerization: high livingness enables the straightforward synthesis of multiblock copolymers†
Anne-Catherine
Lehnen
 ab,
Jan A. M.
Kurki
a and
Matthias
Hartlieb
*ab
ab,
Jan A. M.
Kurki
a and
Matthias
Hartlieb
*ab
aInstitute of Chemistry, University of Potsdam, Karl-Liebknecht-Straße 24-25, 14476, Potsdam, Germany. E-mail: mhartlieb@uni-potsdam.de
bFraunhofer Institute for Applied Polymer Research (IAP), Geiselbergstraße 69, 14476, Potsdam, Germany
First published on 17th February 2022
Abstract
Photo-iniferter (PI)–RAFT polymerization, the direct activation of chain transfer agents via light, is a fascinating polymerization technique, as it overcomes some restriction of conventional RAFT polymerization. As such, we elucidated the role of reversible deactivation in this context using a monomer–CTA pair with low chain transfer capabilities. Tests with varying targeted degrees of polymerization (DP) or monomer concentrations revealed no significant improvement of polymerization control using the PI-process. Control can however be achieved via slow monomer addition, increasing the number of activation/deactivation events per monomer addition. More importantly, the livingness of the polymerization was found to be extraordinarily high, enabling the straightforward and rapid synthesis of multiblock copolymers with up to 20 blocks and a high number of repeating units per block (DP = 25–100) maintaining an overall excellent definition (Mn = 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 g mol−1, Đ = 1.29). This study highlights the enormous potential of PI–RAFT polymerization for the synthesis of polymeric materials.
300 g mol−1, Đ = 1.29). This study highlights the enormous potential of PI–RAFT polymerization for the synthesis of polymeric materials.
Introduction
The control of polymerization processes via light is a fascinating strategy, as it offers a high level of control (spatial and temporal)1–3 and decouples the reaction from the temperature as an external parameter.4 In addition, light is an abundant energy source, and it has been shown that polymerization reactions can be controlled precisely via photo-induced processes.5 As radical polymerization reactions are relatively tolerant toward various conditions and the presence of functional groups, controlling them by light is particularly worthwhile.6Besides light driven atom transfer radical polymerization (ATRP),7,8 single-electron transfer living radical polymerization (SET-LRP)9,10 or nitroxide mediated polymerization (NMP)11,12 reversible-addition fragmentation chain-transfer (RAFT) polymerization has been adapted to utilize light as an energy source.13–15
There are various strategies, which can be employed to control a RAFT process via light. For instance, photo-active initiators can be used in order to fuel a RAFT polymerization.6,16 However, as such initiators represent an exogenous radical source, intrinsic limitations of the RAFT process remain in place. For example, dead chains, unable to be reactivated will accumulate over the course of the polymerization and their share will be determined by the concentration of initiator used.17
The direct photo-activation of the chain transfer agent (CTA) offers a solution to this dilemma. In such a scenario, the thiocarbonylthio moiety is activated by a photo-mediated process and, subsequently, fragments to form a carbon-based transient radical, as well as a thio-based persistent radical (Scheme 1A). There are in principle two strategies to accomplish such an activation of the CTA: photo-electron/energy-transfer (PET)–RAFT polymerization16,18,19 and photo-iniferter (PI)–RAFT polymerization.20
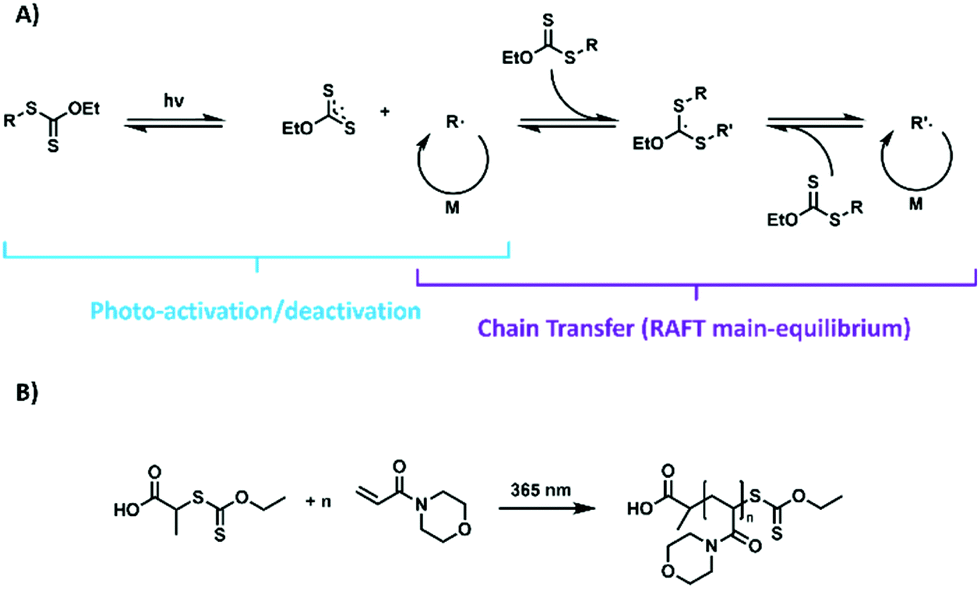 | ||
| Scheme 1 A) Interlink of photo-iniferter mechanism and RAFT main equilibrium; (B) schematic representation of the UV-light mediated polymerization of NAM initiated by a xanthate. | ||
PET–RAFT polymerization requires the addition of a photo-catalyst, which in turn transfers the harvested light energy to the CTA. As a large variety of different catalysts have been described, polymerizations can be performed at various wavelengths,21 and oxygen tolerant methodologies have been introduced.22
In contrast, PI–RAFT polymerization, which will be the focus of this contribution describes the direct interaction of the CTA with light. The thiocarbonyl functionality of typical CTAs can either be activated by a π–π* transition or by a spin forbidden n–π* transition.23,24 Despite the latter band being much weaker, it can be utilized efficiently in PI–RAFT, as its quantum yield for the desired β-fragmentation is higher in comparison.25 Activation is followed by a homolytic dissociation of the R–S bond of the CTA, thus liberating a carbon centred R-radical and a thiocarbonylthio radical. While the former is able to initiate a polymerization reaction, the latter is relatively stable, but reactive enough to deactivate growing chains in a reversible fashion (Scheme 1A). The resulting macro-CTA can in turn be activated again, rendering the process highly living.23
Xanthate CTAs are particularly interesting in this context as their n–π* band is located in the UV region of the spectrum (usually around 350 nm).23 It has been shown that this leads to a highly efficient activation,26 when compared to other CTAs like dithiocarbamates27 or trithiocarbonates.28,29
After the initial development of the iniferter process by Otsu in 1982,30 PI–RAFT has been developed into a useful tool in synthetic polymer chemistry. The process can now be driven by visible light,24,31 and under the presence of oxygen.32,33 It can also be used to produce ultra-high molecular weight polymers,23 to inverse the order of monomers in block copolymers,29 or control different monomer families with a single CTA.34 Also application like 3D-printing or self-healing benefit from the PI–RAFT mechanism.35,36
One defining trait of PI–RAFT is the reversible deactivation and the resulting increased chain end fidelity of the method when compared to conventional RAFT polymerization. While it was demonstrated that irreversible termination is still present,37 in a typical PI–RAFT process reversible deactivation is deemed to be the main fate of growing radical. Indeed, it is even hypothesized that this mechanism is able to control a polymerization.23 It is reasonable to assume that a sufficient deactivation by corresponding thiocarbonylthio radical can exert a certain control over the polymerization (like deactivation in NMP or ATRP). The persistent radical effect38 could play an important role here, as the termination between carbon based transient radicals (R-group or polymer chain) is suppressed by the accumulation of the persistent radicals (thiocarbonylthio-based radicals) upon termination (even though for certain CTAs disulfide formation is found as well).39
The question remains if this deactivation is sufficiently fast and if it is able to suppress irreversible termination processes in a sufficient manner. If so, this effect could be further used to push the limits of RAFT polymerization.
Results and discussion
Fascinated by PI–RAFT polymerization, we set out to elucidate the role of reversible deactivation within such a process and search for its limits. The combination of a xanthate (2-((ethoxycarbonothioyl)thio)propionic acid, (Xan)) and an acrylamide (N-acryloyl morpholine (NAM)) (Scheme 1B) seemed an ideal model system as the chain transfer coefficient (Ctr) of this monomer–CTA combination is expected to be relatively low, hence leading to comparably high dispersities (Đ > 1.5) of resulting polymers, while still enabling fast processing due to the high kp of the monomer.Consequently, an increased control as for instance provided by a reversible deactivation mechanism can be detected via a narrowing of the molecular weight distribution. Sumerlin et al. described ultra-high molecular weight polymers with low dispersities when using acrylamides in water.23 Thus, water was chosen as a reaction medium, alongside DMF as an organic solvent for comparison.
The Ctr for both systems were determined by comparing CTA consumption with monomer conversion in kinetic experiments (Fig. S2 and S3†).40–42 For determination of Ctr polymerizations were fuelled by a thermal azo initiator. In PI–RAFT polymerization, Ctr is not accessible as here, activation of the CTA happens directly via photolysis and not exclusively by chain transfer. As expected, the respective values were with 0.49 in DMF and 0.34 in water relatively low leading to broad molecular weight distributions. A lower Ctr in water seems plausible as it has been described that the kp of acrylamide monomers is increased in an aqueous medium.43
To screen the control of the polymerization process over a range of targeted polymer lengths various xanthate concentrations were used with a constant concentration of monomer (2 mol L−1), and conversion, as well as molecular weight distribution were determined via nuclear magnetic resonance (NMR) spectroscopy and size exclusion chromatography (SEC) respectively (Fig. 1). The polymerization was induced by UV-light (365 nm, 2 W, Fig. S1†) and control experiments, fuelled by an exogenous radical source (azo-initiator) were conducted as well. Here, the ratio of CTA to initiator was set to 5 for all samples. The difference in outcome between PI–RAFT and thermal polymerization might be able to hint on the impact of reversible deactivation, as in the case of conventional RAFT polymerization, chain transfer is the sole control mechanism. It should however be noted that variables like radical flux (and respective apparent kp), radical concentration, reaction temperature, reaction time, etc. that can also influence the outcome cannot be matched between the methods. Thus, any difference might also be associated to these parameters.
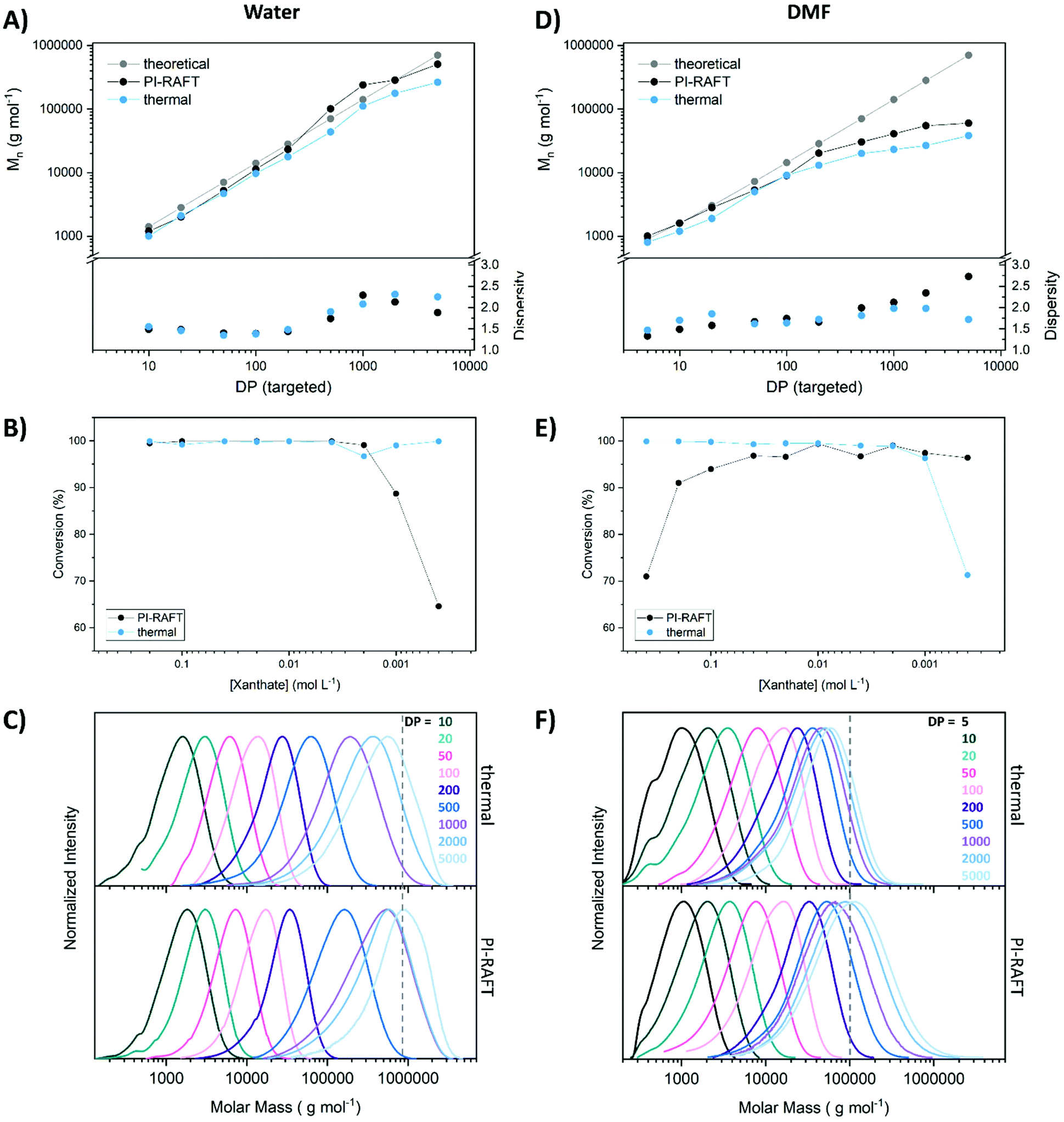 | ||
| Fig. 1 Polymerization of NAM using Xan as CTA under UV-light (365 nm, 1 h) or presence of azo-initiator (thermal) in water and DMF respectively. Various CTA/monomer ratios were tested and Mn and Đ (A and D) were determined by analysis of SEC curves (C and F) (eluent: THF, poly(styrene) calibration). Theoretical Mn is based on PI RAFT considering the respective conversion. Conversion (B and E) was determined from 1H-NMR measurements in D2O or CDCl3 respectively. | ||
When comparing the theoretical molecular weight based on the monomer/CTA ratio with the obtained values it seems that PI–RAFT is better able to achieve the desired polymer length in both solvents. The loss of control at high DPs is more obvious in DMF, where above a targeted degree of polymerization (DP) of 200, the measured Mn values fall short of expectations. This effect could be a result of an increased kp of the monomer in water, making it possible to achieve higher molecular weights within a certain time frame. It is likely that monomer propagation is not fast enough in DMF to produce high molecular weight polymers within 1 h of reaction time.
Still, this drop-of is more pronounced in the case of conventional RAFT polymerization, illustrating a potential advantage of reversible deactivation. In water, obtained Mn values are closer to the theoretical expectation based on the ratio of CTA and monomer. Also here, PI–RAFT seems to be slightly advantageous. It should be noted that the theoretical Mn for thermal RAFT polymerization is, due to the presence of the initiator, slightly lower than displayed. For reasons of clarity values are not plotted but can be found in Table S4.† The dispersity of the polymers does not paint a clear picture. The absolute values are in either case too high to be considered a controlled radical polymerization (Đ > 1.3). No clear trend that would indicate an advantage or disadvantage of the PI–RAFT mechanism is visible. Also, SEC curves look similar for both methods with a slightly increased tendency of thermal RAFT polymerization for low molecular weight tailing, likely associated to accumulation of dead chains. In water, the highest targeted DP of 5000 seems to be too large for the used column system, as the unsymmetrical curve shape indicates a cut-off by the exclusion limit of the SEC system.
The conversion was probed by NMR spectroscopy and reveals interesting findings. For thermally induced polymerization, conversion was found to be high for most samples. Only in DMF at a targeted DP of 5000 a value below 90% was obtained, presumably due to the overall too low initiator concentration (which was linked to the CTA). For PI–RAFT polymerization radical concentration is expected to scale directly with the concentration of CTA. We performed measurements without CTA (Fig. S4†) which revealed that no auto-initiation is present in water while DMF is able to produce radicals under the used irradiation conditions. Still, the conversion after 1 h irradiation in DMF is limited (∼20%) compared to the obtained values in the presence of xanthate. Even at very low xanthate concentrations (dilution factor 500) reasonable conversions were obtained (∼65% in water and 96% in DMF). It should be emphasized that irradiation time was held constant in all cases at 1 h. The discrepancy between the solvents might be best explained by auto initiation of DMF providing additional radical. The conversions are of course also a function of the propagation rate of the monomer but still illustrates the high initiation efficiency of xanthates in PI–RAFT.
Interestingly, when PI–RAFT was conducted in DMF, a distinct decrease of the conversion at low CTA concentrations was detected also coinciding with a lowered dispersity of respective polymers. Reversible deactivation would be a valid explanation for this observation, as the probability of the back reaction is expected to increase with increasing CTA concentrations. In water the solubility of the xanthate prevented a test polymerization at the same concentration. However, a practical application of this effect seems out of scope as very high CTA concentrations limit the achievable polymer length drastically (DP < 10).
To obtain more insight by monitoring the influence of the absolute concentration of xanthate, a series of polymerizations with constant targeted DP (50) and varying monomer concentrations was performed (Fig. 2). Here, the expected outcome is identical (PNAM50) reducing the impact of the SEC calibration on the results. Water, which seems to be a better solvent for this polymerization, was used for these tests and thermal RAFT was compared to the PI-process.
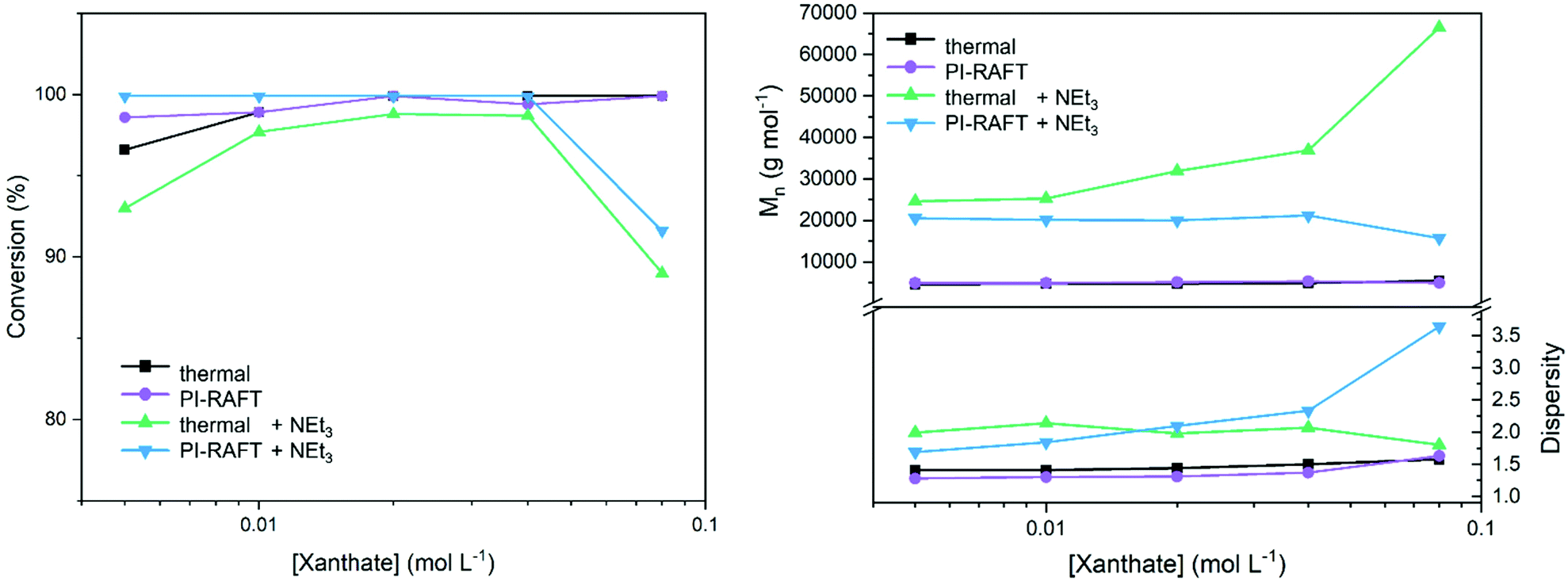 | ||
| Fig. 2 Variation of CTA concentration in the polymerization of NAM and Xan using a constant targeted DP (50). | ||
As previously found, the xanthate concentration had little effect on the conversion for photo-polymerizations. Also, dispersities were relatively unaffected by the nature of radical generation with slightly decreased values for the conventional process using an azo initiator. No distinct decrease in conversion or dispersities at high xanthate concentrations was detected. However, CTA concentrations where conversion hinted toward increased reversible deactivation (0.4 mol L−1, Fig. 1) were unachievable when maintaining a DP of 50.
To further probe the impact of reversible deactivation, a similar set of experiments with the addition of NEt3 was performed. As demonstrated by the groups of Boyer44 and Qiao,32 the addition of a tertiary amine to a PET–RAFT or PI–RAFT polymerization, respectively results in the formal reduction of the thiocarbonylthio radical to an anionic species (Scheme S1†). This has been shown to prevent degradation of the CTA end group when using methacrylic monomers. In the present case, the removal of thiocarbonylthio radical species from the equilibrium should lead to a decreased efficiency of reversible deactivation. In the absence of an active chain-transfer equilibrium, dispersities should increase if reversible deactivation plays a significant role in the process. Indeed, dispersities are markedly increased when compared to a process without addition of base. The effect is more pronounced at higher CTA concentrations, as here also the NEt3 concentration is higher here (3 equivalents per CTA molecule were used).
However, also in the case of thermal RAFT polymerization under the addition of NEt3 control over the polymerization is lost as indicted by increased values for Mn and Đ. It is likely that the base is able to degrade the CTA to an extend or interfers with the RAFT process in a different way than anticipated.
The essence of our initial findings is that reversible deactivation is unlikely play a role the control of the polymerization using the PI methodology at least for the monomer/CTA system described here. While reversible deactivation is likely a part of the mechanism, it is not able to produce defined polymers without an interlinked chain-transfer process. However, a significant advantage lies in the efficient re-initiation and high end group fidelity of the method, when compared to a conventional RAFT process. To probe these benefits and push the limits of what can be achieved by a controlled radical polymerization process, PI–RAFT was utilized to produce multiblock copolymers via repeated chain extension reactions. Multiblocks are an ideal tool to probe livingness as dead chains that can't be reactivated cease to grow upon chain extension, an effect that can be visualized via SEC. In detail, a decreased livingness would result in a low molecular weight tailing of the size distribution as for instance found in RAFT-made multiblocks with a high number of blocks.45
While it is possible to produce multiblock copolymers via conventional RAFT polymerization46–52 this strategy has certain limitations. In order to achieve near quantitative conversion for each chain extension, while not unnecessarily sacrificing livingness, the amount of initiator has to be optimized carefully.46 Still, a high number of blocks can only be achieved when the block length is kept relatively short (DP around 10) to maintain a relatively high monomer concentration. Otherwise, driving the reaction to quantitative conversion requires higher amounts of initiator, resulting in dead chains. On the other side, block length is also restricted on the lower end, as if the monomer to (macro)–CTA ratio is too low, the probability of every macromolecule re-initiating drops drastically.53 This then results in defects of the final structure in terms of missing blocks for a share of macromolecules. Some of these boundaries can be removed using sulfur-free RAFT in an emulsion process.54 Here, slow propagating monomers can be used and higher block length can be targeted, as irreversible termination is reduced. Similarly, PI–RAFT enables multiblock copolymers with high block length (DP = 100) as illustrated by Qiao and coworkers.24 Multiblocks have also been described in the context of PET–RAFT polymerization,22 but even here, low molecular weight tailing, associated to dead chains is detected.
In our case, initial experiments were performed using NAM as monomer producing pseudo-block copolymers. The aim was to produce multiblock copolymers in a one-pot process via consecutive monomer addition after the previous block was driven to high conversions. Thus, the monomer concentration was checked after each extension step and illumination time was adjusted to achieve high conversions (Tables S1–S3†) making in-between purification unnecessary. As we were able to show that with the herein established PI–RAFT protocol, high conversions can be achieved even at very low concentrations of xanthate it might be possible to achieve a high number of blocks while simultaneously targeting relatively high DPs for individual segments.
Our first goal was a multiblock copolymer with 20 consecutive chain extensions and a DP of 50 per block in water (Fig. 3). Conversion was probed via NMR and irradiation time was adjusted to achieve high conversions (Table S1†). The polymers were further subjected to SEC analysis to monitor the level of control. Once viscosity increased to a point where stirring was impossible, the reaction mixture was diluted with water.
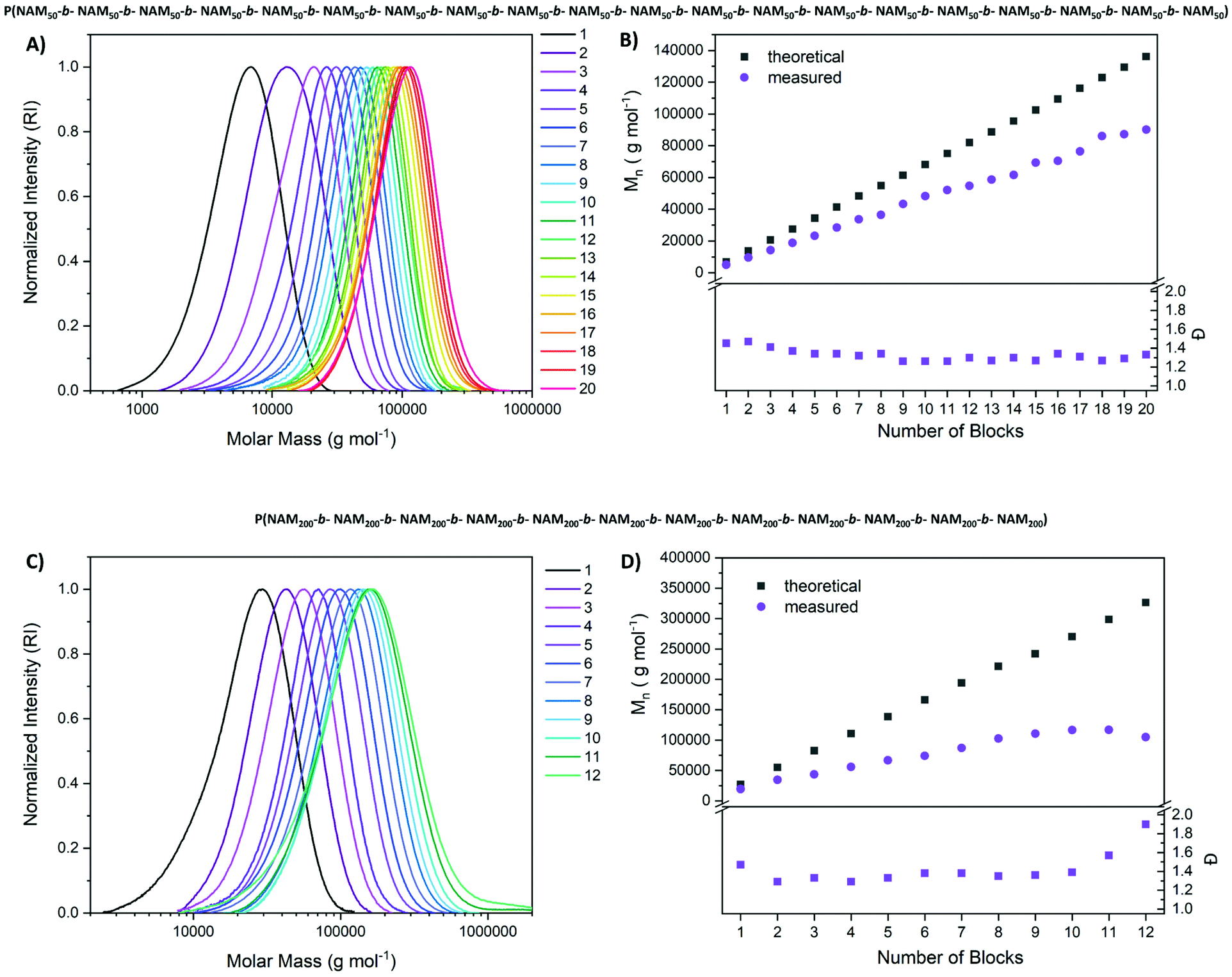 | ||
| Fig. 3 Synthesis of pseudo-multiblock copolymers from NAM with a DP per block of 50 and 200 respectively. SEC was measured in THF and calibrated with poly(styrene). (A and C) SEC traces of pseudo-multiblocks with DP 50 and 200 respectively, (B and D) Mn and Đ of pseudo-multiblocks with DP 50 and 200 respectively derived from SEC measurements. | ||
The chromatograms in Fig. 3 reveal a highly controlled process with a shift in size distribution upon each chain extension. These shifts become less obvious for high block numbers as here the addition of one block is a small change to the overall molecular weight. Still distinct chain extension is visible up to the ikosa block copolymer. More importantly, the low molecular weight tailing that would be expected for a process fuelled by an exogenous radical source is missing completely. Seemingly all chains are re-initiated, and the shape of the distribution remains highly symmetrical. When Mn is plotted as a function of the number of blocks, a linear increase of molecular weight upon chain extension is visible. One reason for the deviation from the theoretical values could be the relative SEC calibration (poly(styrene) (PS)). Impressively, the dispersity decreases to 1.3 over the first chain extensions and remains constant at this level, an effect which was also observed in sulfur-free RAFT polymerization.54 As after 20 blocks, irradiation times were markedly increased compared to the initial protocol and, also viscosity became very high, no further extensions were attempted. In addition, it was increasingly difficult to achieve high conversions as even after 6 h of irradiation only 95% of monomer conversion was reached. The cumulative DP calculated based on the signal of the end group (Fig. S10†) points toward a highly living polymerization as it fits well to the targeted values, even though the signal is relatively weak. The final product with a targeted DP of 1000 and 20 individual chain extensions possessed a molecular weight of 90100 g mol−1 and a dispersity of 1.33 (SEC in THF, PS calibration).
Intrigued by these results, a higher molecular weight pseudo-multiblock was attempted as well. Here, the targeted DP per block was set to 200 with otherwise same parameters and adjustments regarding viscosity and irradiation time. Also in this case, an efficient chain extension was observed up to block 10, after which the distribution broadened and dispersities increased. The cumulative irradiation time cannot be held responsible for this finding as this value is lower compared to the first multiblock with a DP of 50 per block (46.5 h for ikosablock (DP = 20 × 50) vs. 28.5 h for dodekablock (DP = 12 × 200)). As also the number of chain extensions is lower for the second multiblock, the overall molecular weight of the polymer seems to be the best possible explanation. Perhaps inaccessibility of the end group and a resulting slow chain transfer or activation is responsible for this effect.
To demonstrate the feasibility of the shown approach a real multiblock copolymer, using different monomers was produced (Fig. 4). Dimethyl acrylamide (DMA) and N-isopropyl acrylamide (NiPAAm) were chosen in addition to the already established NAM. Initial tests revealed that DMA is, in combination with Xan and UV-light able to produce polymers with a low dispersity (test reaction Mn = 2900 g mol−1Đ = 1.21 for a targeted DP of 50). This effect has already been described by Sumerlin and coworkers and was attributed to a positive influence of reversible deactivation.23 To reference this to our initial system, the Ctr was determined using the before described methods for DMA/Xan as well. The determined Ctr (0.89) is in the same range as for the NAM/Xan combinations which hints towards a similar chain transfer capability. Hence, control is likely provided by a different mechanism like reversible deactivation. It seems unintuitive that DMA in particular would enable a control via this mechanism while for similar monomers like NAM or NiPAAm this effect is at least strongly reduced.
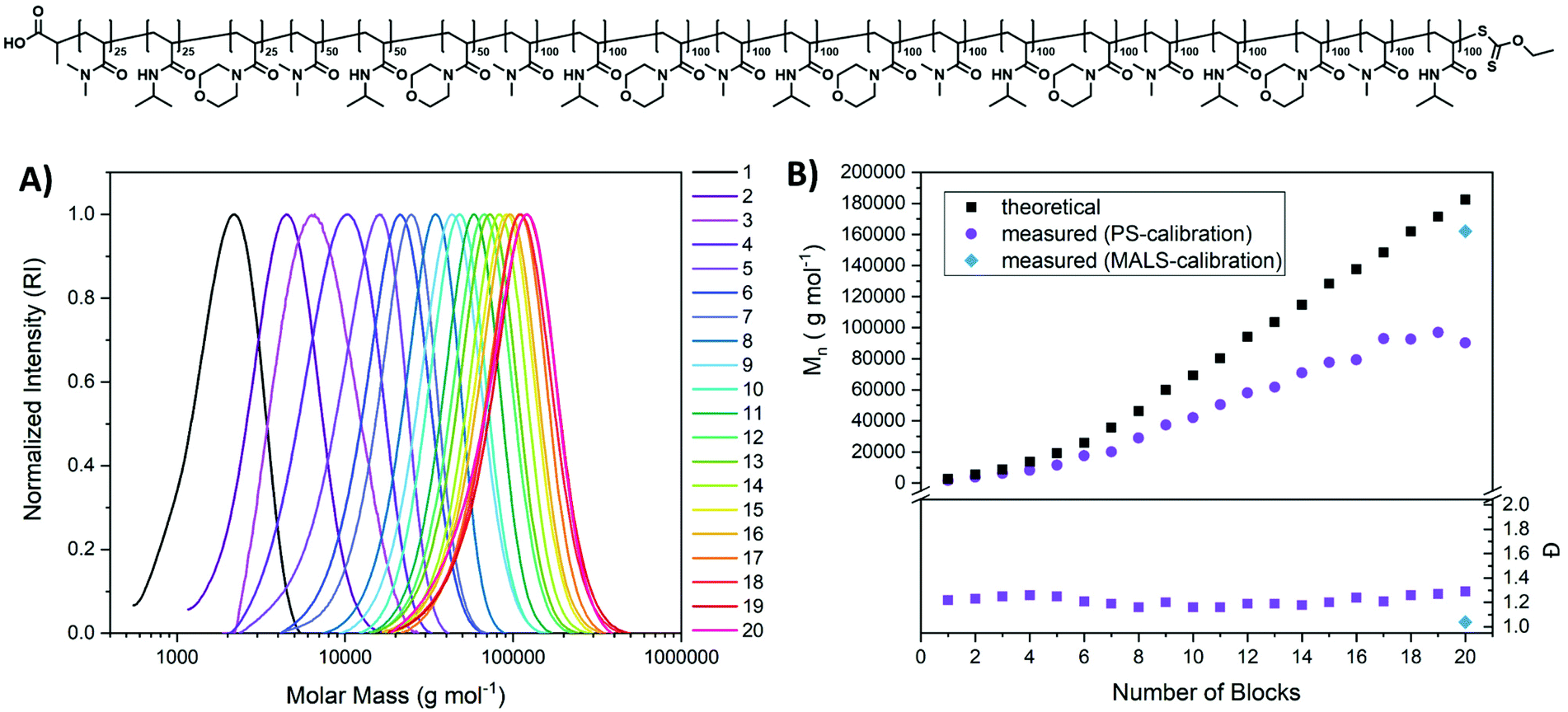 | ||
| Fig. 4 Synthesis of multiblock copolymers from DMA, NiPAAm, and NAM with varying DP per block (25/50/100). SEC measurements were performed in THF and calibrated with poly(styrene). SEC traces (A) were used to determine Mn and Đ (B). The final polymer was investigated via aqueous SEC (0.3% formic acid, 0.1 mol L−1 NaCl) using a MALS detector (blue diamonds). | ||
However, the used method of determining the Ctr only generates information about the CTA consumption in the beginning of the reaction and not directly about the RAFT main equilibrium. It is further possible that macro-CTAs possess a slightly increased chain transfer coefficient. However, it was not possible to investigate this for the present systems.
The multiblock were created by a repeating sequence of DMA, NiPAAM and NAM with varying chain length. For the first three blocks a DP of 25 per block was targeted and a DP of 50 for the subsequent three blocks, while after this, the targeted block length was 100 for all following blocks. This was done mainly to improve the tracking of multiblock copolymer growth by SEC as for later blocks larger chain extensions are beneficial for detection. But it also illustrates the control over polymer architecture and the potential length of multiblocks that can be created using this method.
The dispersity remains low throughout the process, and due to the involvement of DMA, the overall values were lower than for previous multiblocks (∼1.2–1.3). Also, the increase of molecular weight per block follows the trend observed before: a linear increase when referenced to the targeted block length but an offset to the expected molecular weight. This can again be a result of the unfitting SEC calibration which could be more intense here as the different blocks will display different hydrodynamic behaviour based on their solvatisation.
To probe the actual molecular weight of the final multiblock copolymer, it was investigated via aqueous SEC using a multi angle light scattering (MALS) detector (Fig. S9†). While conventional calibration using a poly(vinyl pyridine) standard results in an underestimation of the molecular weight, the value obtained via light scattering is with Mn = 162100 g mol−1 very close to the expected value based on the targeted DP. Also, dispersity is with Đ = 1.04 significantly lower compared to conventional calibration. While this value is likely underestimated in this measurement it still underlines the precision of the herein presented synthetic method.
Again, low molecular weight tailing, which would indicate formation of dead chains is virtually absent from SEC traces. However, also here necessary irradiation times increased with higher block numbers indicating either a slower activation, or a slower propagation, or a combination thereof at higher polymer lengths. Also decreasing concentration of Xan end group in the reaction volume could be held accountable for this observation.
Overall, a highly defined ikosablock copolymer could be produced rapidly (51.5 hours of total irradiation, only 10 h for the first 10 blocks) with impressive precision ((Mn = 90300 g mol−1, Đ = 1.29). It should be emphasized that the used method is simple, using only water as a solvent, a UV-lamp that can be found in most synthetic laboratories and a one necked flask with a rubber septum. Also, oxygen removal is performed in a straightforward way by purging with nitrogen after each new monomer addition. As conversion can be probed after each step with the possibility to continue irradiation if the result is not satisfying, no laborious optimizations are required beforehand. The apparent high livingness of the polymers could be a result of the persistent radical effect, but also associated to rapid deactivation by the xanthate radical.55
One finding that sticks out from the described experiments is that, apparently the definition of a polymer with a given targeted DP can be improved when multiple chain extensions are involved as opposed to one polymerization step. Indeed, when comparing polymers with an overall targeted DP of 1000, the molecular weight distribution of multiblock copolymers is much narrower (and also the overall molecular weight is lower, see Fig. S7†). This seems unintuitive but could be explained with reversible deactivation. Since for multiblock copolymers total irradiation times are much longer (>factor 20), much more activation and deactivation cycles are involved. Whereas speed of monomer addition for a single reaction step is mainly governed by the kp of the monomer, for multiblock synthesis monomer addition is segmented over the whole process. Thus, the number of reversible deactivation events per monomer addition steps is much higher throughout the whole polymerization for sequential addition of blocks.
To probe this hypothesis, we tested feeding the monomer to the reaction mixture. If our assumptions are correct, dispersity should scale with feeding rate. Indeed, as shown in Table 1 (SEC traces in Fig. S8†), with slower addition of monomer, dispersity values can be reduced significantly when reducing the addition of monomer to 6.67 μL min−1 resulting in an overall polymerization time of 5 h (as opposed to 1 h under non-feeding conditions). However, monomer addition can also be too slow as shown by increased dispersities for 10 h reaction time. Indeed, in the virtual absence of monomer the CTA will degrade. This is illustrated by the absence of conversion for a feeding rate of 1.11 μL min−1 (30 h of addition). In this case the signals of the CTA vanished from the NMR spectrum of the product, indicating degradation. Consequently, as the ratio between activation and monomer addition can be tuned up to a certain maximum of achievable control, this seems also to be a trade-off in livingness as end groups can potentially degrade in the absence of monomer as also found in other reports.56,57 The partial success of the feeding strategy could also be explained by another hypothesis: when performing the polymerization in one (quick) step, the low Ctr CTA might be consumed slowly leading to a certain continuing supply of small radicals throughout the process, as not all CTA is consumed at the beginning of the reactions. This is then resolved when the overall polymerization time is extended via feeding. This however does not sufficiently explain the success of the multiblock synthesis which should show a strong low molecular weight tailing of the size distribution, since re-initiation has to happen with each block anew.
| Feeding rate (μL min−1) | Feeding time (min) | Irradiation time (min) | Conversiona (%) |
M
nb (g mol−1) |
Đ |
|---|---|---|---|---|---|
| a Determined via1H-NMR spectroscopy in D2O. b Determined via SEC chromatography in THF using a PS calibration. | |||||
| — | 0 | 60 | 94.4 | 19300 |
1.53 |
| 36.36 | 55 | 60 | 84.7 | 17000 |
1.48 |
| 6.67 | 300 | 310 | 89.2 | 13400 |
1.30 |
| 3.33 | 600 | 610 | 84.9 | 10200 |
1.42 |
| 1.11 | 1800 | 1810 | 0.7 | — | — |
A different observation from feeding experiments is that the overall molecular weight seems to decrease with feeding time, which is also in accordance with observations from comparing pseudo-multiblocks and polymers that were produced in one step (Fig. S7†). While it doesn't seem to be a result of auto initiation by the solvent (water, Fig. S4†), we currently have no explanation for this behaviour.
Conclusions
To probe the impact of reversible deactivation in PI–RAFT polymerization we used the combination of an acrylamide (NAM) and a xanthate as a benchmark system, since chain transfer coefficients are relatively low (Ctr < 1) for this combination. Hence, a positive impact of the PI mechanism could lead to increased control over the polymerization. In addition, xanthates are well known for their efficient photochemistry. Our experiments did not show a significant increase in control over the molecular weight distribution of produced polymers. Even though PI–RAFT seems slightly superior in achieving targeted molecular weight at high DPs, it is still the RAFT chain-transfer equilibrium which determined whether a polymer distribution was narrow or not. In further experiments we could show that dispersity could be lowered (down to a minimum of 1.3) by a slow monomer addition, effectively increasing the number of activation–deactivation events per monomer addition, which seems still insufficient as basis for a controlled synthesis.However, a significant advantage is posed by the high livingness associated with PI–RAFT. Indeed, in chain extension experiments, dead chain usually associated with irreversible termination were virtually absent. Thus, it was possible to produce multiblock copolymers with very high molecular weight (per block and in total). For example, a repeating sequence of three acrylamides with block length up to 100 repeating units and a total number of 20 blocks was produced with impressive precision ((Mn = 90300 g mol−1, Đ = 1.29). It should be noted that the herein presented method is rapid (51.5 hours for the above mentioned ikosa block) and easy to use (standard laboratory UV lamp, reaction in water, degassing by N2-purging).
This study highlights the enormous potential of PI–RAFT in the synthesis of polymeric materials, in particular for the production of segmented macromolecules.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge funding by the DFG (Emmy-Noether-Program, HA 7725/2-1). The authors also thank the NMR core facility of the Institute of Chemistry (University of Potsdam) of Prof. Dr Heiko Möller, Dr Matthias Heydenreich, and Angela Krtitschka. Prof. Dr Helmut Schlaad and Sascha Prentzel from the Institute of Chemistry (University of Potsdam) are gratefully acknowledged for providing the facility to perform SEC measurements.Notes and references
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed.
- X. Pan, M. A. Tasdelen, J. Laun, T. Junkers, Y. Yagci and K. Matyjaszewski, Prog. Polym. Sci., 2016, 62, 73–125 CrossRef CAS.
- E. Skliutas, M. Lebedevaite, E. Kabouraki, T. Baldacchini, J. Ostrauskaite, M. Vamvakaki, M. Farsari, S. Juodkazis and M. Malinauskas, Nanophotonics, 2021, 10, 1211–1242 CrossRef.
- F. Lauterbach, M. Rubens, V. Abetz and T. Junkers, Angew. Chem., Int. Ed., 2018, 57, 14260–14264 CrossRef CAS PubMed.
- A. Anastasaki, V. Nikolaou, G. S. Pappas, Q. Zhang, C. Wan, P. Wilson, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Sci., 2014, 5, 3536–3542 RSC.
- Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245–6260 CrossRef CAS.
- X. Pan, N. Malhotra, A. Simakova, Z. Wang, D. Konkolewicz and K. Matyjaszewski, J. Am. Chem. Soc., 2015, 137, 15430–15433 CrossRef CAS PubMed.
- T. Zhang, T. Chen, I. Amin and R. Jordan, Polym. Chem., 2014, 5, 4790–4796 RSC.
- A. Anastasaki, V. Nikolaou, G. Nurumbetov, P. Wilson, K. Kempe, J. F. Quinn, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Rev., 2016, 116, 835–877 CrossRef CAS PubMed.
- M. Vorobii, O. Pop-Georgievski, A. de los Santos Pereira, N. Y. Kostina, R. Jezorek, Z. Sedláková, V. Percec and C. Rodriguez-Emmenegger, Polym. Chem., 2016, 7, 6934–6945 RSC.
- Y. Guillaneuf, D. Bertin, D. Gigmes, D.-L. Versace, J. Lalevée and J.-P. Fouassier, Macromolecules, 2010, 43, 2204–2212 CrossRef CAS.
- D.-L. Versace, J. Lalevée, J.-P. Fouassier, D. Gigmes, Y. Guillaneuf and D. Bertin, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2910–2915 CrossRef CAS.
- T. G. McKenzie, Q. Fu, M. Uchiyama, K. Satoh, J. Xu, C. Boyer, M. Kamigaito and G. G. Qiao, Adv. Sci., 2016, 3, 1500394 CrossRef PubMed.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- R. Chapman, K. Jung and C. Boyer, Photo RAFT Polymerization, in RAFT Polymerization, ed. G. Moad and E. Rizzardo, 2021, DOI:10.1002/9783527821358.ch12.
- S. Li, G. Han and W. Zhang, Polym. Chem., 2020, 11, 1830–1844 RSC.
- M. H. Stenzel and C. Barner-Kowollik, Mater. Horiz., 2016, 3, 471–477 RSC.
- N. Corrigan and C. Boyer, Trends Chem., 2020, 2, 689–706 CrossRef CAS.
- J. Phommalysack-Lovan, Y. Chu, C. Boyer and J. Xu, Chem. Commun., 2018, 54, 6591–6606 RSC.
- M. Hartlieb, Macromol. Rapid Commun., 2021, 2100514, DOI:10.1002/marc.202100514.
- M. L. Allegrezza and D. Konkolewicz, ACS Macro Lett., 2021, 10, 433–446 CrossRef CAS.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed.
- R. N. Carmean, T. E. Becker, M. B. Sims and B. S. Sumerlin, Chem, 2017, 2, 93–101 CAS.
- T. G. McKenzie, Q. Fu, E. H. H. Wong, D. E. Dunstan and G. G. Qiao, Macromolecules, 2015, 48, 3864–3872 CrossRef CAS.
- J. D. Coyle, Tetrahedron, 1985, 41, 5393–5425 CrossRef CAS.
- C. Ding, C. Fan, G. Jiang, X. Pan, Z. Zhang, J. Zhu and X. Zhu, Macromol. Rapid Commun., 2015, 36, 2181–2185 CrossRef CAS PubMed.
- J. Lalevée, N. Blanchard, M. El-Roz, X. Allonas and J. P. Fouassier, Macromolecules, 2008, 41, 2347–2352 CrossRef.
- M. L. Allegrezza, N. De Alwis Watuthanthrige, Y. Wang, G. A. Garcia, H. Ren and D. Konkolewicz, Polym. Chem., 2020, 11, 6129–6133 RSC.
- C. P. Easterling, Y. Xia, J. Zhao, G. E. Fanucci and B. S. Sumerlin, ACS Macro Lett., 2019, 8, 1461–1466 CrossRef CAS.
- T. Otsu, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2121–2136 CrossRef CAS.
- J. Xu, S. Shanmugam, N. A. Corrigan and C. Boyer, in Controlled Radical Polymerization: Mechanisms, American Chemical Society, 2015, vol. 1187, ch. 13, pp. 247–267 Search PubMed.
- Q. Fu, K. Xie, T. G. McKenzie and G. G. Qiao, Polym. Chem., 2017, 8, 1519–1526 RSC.
- J. R. Lamb, K. P. Qin and J. A. Johnson, Polym. Chem., 2019, 10, 1585–1590 RSC.
- J. Li, X. Pan, N. Li, J. Zhu and X. Zhu, Polym. Chem., 2018, 9, 2897–2904 RSC.
- Z. Zhang, N. Corrigan and C. Boyer, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202114111.
- X. Wu, B. Gross, B. Leuschel, K. Mougin, S. Dominici, S. Gree, M. Belqat, V. Tkachenko, B. Cabannes-Boué, A. Chemtob, J. Poly and A. Spangenberg, Adv. Funct. Mater., 2021, 2109446, DOI:10.1002/adfm.202109446.
- C. Bray, G. Li, A. Postma, L. T. Strover, J. Wang and G. Moad, Aust. J. Chem., 2021, 74, 56–64 CrossRef CAS.
- H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS PubMed.
- M. D. Thum, S. Wolf and D. E. Falvey, J. Phys. Chem. A, 2020, 124, 4211–4222 CrossRef CAS PubMed.
- Y. K. Chong, J. Krstina, T. P. T. Le, G. Moad, A. Postma, E. Rizzardo and S. H. Thang, Macromolecules, 2003, 36, 2256–2272 CrossRef CAS.
- J. Chiefari, R. T. A. Mayadunne, C. L. Moad, G. Moad, E. Rizzardo, A. Postma and S. H. Thang, Macromolecules, 2003, 36, 2273–2283 CrossRef CAS.
- G. Moad, J. Chiefari, Y. K. Chong, J. Krstina, R. T. A. Mayadunne, A. Postma, E. Rizzardo and S. H. Thang, Polym. Int., 2000, 49, 993–1001 CrossRef CAS.
- A. Valdebenito and M. V. Encinas, Polym. Int., 2010, 59, 1246–1251 CrossRef CAS.
- J. Xu, S. Shanmugam, H. T. Duong and C. Boyer, Polym. Chem., 2015, 6, 5615–5624 RSC.
- A. Kuroki, P. Sangwan, Y. Qu, R. Peltier, C. Sanchez-Cano, J. Moat, C. G. Dowson, E. G. L. Williams, K. E. S. Locock, M. Hartlieb and S. Perrier, ACS Appl. Mater. Interfaces, 2017, 9, 40117–40126 CrossRef CAS PubMed.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4 DOI:10.1038/ncomms3505.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Macromolecules, 2014, 47, 3451–3460 CrossRef CAS.
- G. Gody, R. Barbey, M. Danial and S. Perrier, Polym. Chem., 2015, 6, 1502–1511 RSC.
- L. Martin, G. Gody and S. Perrier, Polym. Chem., 2015, 6, 4875–4886 RSC.
- J. Zhang, R. Deubler, M. Hartlieb, L. Martin, J. Tanaka, E. Patyukova, P. D. Topham, F. H. Schacher and S. Perrier, Macromolecules, 2017, 50, 7380–7387 CrossRef CAS.
- A. Kerr, M. Hartlieb, J. Sanchis, T. Smith and S. Perrier, Chem. Commun., 2017, 53, 11901–11904 RSC.
- T. G. Floyd, S. Häkkinen, M. Hartlieb, A. Kerr and S. Perrier, in RAFT Polymerization, 2021, pp. 933–981, DOI:10.1002/9783527821358.ch20.
- G. Gody, P. B. Zetterlund, S. Perrier and S. Harrisson, Nat. Commun., 2016, 7, 10514 CrossRef CAS PubMed.
- N. G. Engelis, A. Anastasaki, G. Nurumbetov, N. P. Truong, V. Nikolaou, A. Shegiwal, M. R. Whittaker, T. P. Davis and D. M. Haddleton, Nat. Chem., 2017, 9, 171–178 CrossRef CAS PubMed.
- N. Ballard and J. M. Asua, ACS Macro Lett., 2020, 9, 190–196 CrossRef CAS.
- J. F. Quinn, L. Barner, C. Barner-Kowollik, E. Rizzardo and T. P. Davis, Macromolecules, 2002, 35, 7620–7627 CrossRef CAS.
- S. R. Turner and R. W. Blevins, Macromolecules, 1990, 23, 1856–1859 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and supplementary graphs and tables. See DOI: 10.1039/d1py01530c |
| This journal is © The Royal Society of Chemistry 2022 |