
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fabrication and mechanical properties of knitted dissimilar polymeric materials with movable cross-links†
Ryohei
Ikura
 a,
Shunsuke
Murayama
b,
Junsu
Park
a,
Yuka
Ikemoto
c,
Motofumi
Osaki
d,
Hiroyasu
Yamaguchi
aef,
Akira
Harada
g,
Go
Matsuba
*b and
Yoshinori
Takashima
*adef
a,
Shunsuke
Murayama
b,
Junsu
Park
a,
Yuka
Ikemoto
c,
Motofumi
Osaki
d,
Hiroyasu
Yamaguchi
aef,
Akira
Harada
g,
Go
Matsuba
*b and
Yoshinori
Takashima
*adef
aDepartment of Macromolecular Science, Graduate School of Science, Osaka University, Toyonaka, Osaka 560-0043, Japan. E-mail: takasima@chem.sci.osaka-u.ac.jp
bGraduate School of Organic Material Engineering, Yamagata University, 4-3-16 Jonan, Yonezawa, Yamagata 992-8510, Japan. E-mail: gmatsuba @yz.yamagata-u.ac.jp
cJapan Synchrotron Radiation Research Institute, Sayo-gun, Hyogo 679-5198, Japan
dInstitute for Advanced Co-Creation Studies, Osaka University, Toyonaka, Osaka 560-0043, Japan
eProject Research Center for Fundamental Sciences, Graduate School of Science, Osaka University, Machikaneyama-cho, Toyonaka, Osaka 560-0043, Japan
fInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICS-OTRI), Osaka University, Suita, Osaka 565-0871, Japan
gSANKEN (The Institute of Scientific and Industrial Research), Osaka University, Ibaraki, Osaka 567-0047, Japan
First published on 24th March 2022
Abstract
Bulk polymerization of liquid main chain monomers in the presence of a linear polymer bearing acetylated γ-cyclodextrin (TAcγCD) (primary polymer) results in composite materials consisting of dissimilar polymer chains, hereinafter called the designs of movable cross-network elastomer knitting dissimilar polymers (KP elastomers). The post polymerized chains (secondary polymer) penetrate cavities in the TAcγCD units and form movable cross-links that connect between the primary and secondary polymers. The KP elastomers successfully improve the mechanical properties based on two key design components. One is a long movable range. The secondary polymers easily move in the TAcγCD cavities on the primary polymers due to the lack of a chemical cross-linker between the primary and secondary polymers. Thus, the KP elastomers exhibit high toughness because the movable cross-links are not fixed, which results in effective stress dispersion. The other is the synergistic effect of the multiphase structure. The network designs of the KP elastomers enable composite materials with movable cross-links that can even connect immiscible polymers, namely, poly(ethyl acrylate) (PEA) and polystyrene (PS). The obtained materials contain a three-phase structure: PEA, PS, and a mixed phase. The synergistic effect of the three-phase structure simultaneously improves the toughness and stiffness.
Design, System, ApplicationMovable cross-linked materials connecting between dissimilar polymers were designed by the methods for the formation of movable cross-links “polymer threading through polymerization”. We carried out the bulk polymerization of liquid main chain monomers in the presence of a linear polymer bearing TAcγCD units. The network designs of the obtained elastomers (KP elastomer) successfully improve the mechanical properties based on two key-systems. One is a long movable range. The KP elastomers exhibit high toughness because the movable cross-links are not interlocked, which results in effective stress dispersion. The other is the synergistic effect of the multiphase structure: PEA (contributing effective stress dispersion with high mobility), PS (contributing high stiffness and strength as hard domain), and a mixed phase (connecting each phase to work together). Blending of immiscible polymers cannot improve mechanical properties due to severe phase separation. However dissimilar knitting methods successfully improve toughness and stiffness of polymeric materials by the synergistic effect of the three-phase structure. These designs allow the application for extension of lifetime of products that can also promote the replacement of heavy materials (metals and ceramics) with lightweight polymeric materials. |
Introduction
With increasing interest in establishing a sustainable society, the demand for tougher polymeric materials is increasing daily. Polymers with improved toughness can reduce waste by extending the lifetime of products. This can also promote the replacement of heavy materials (metals and ceramics) with lightweight polymeric materials, leading to a reduction in fuel consumption. In recent decades, many efforts, such as the addition of filler,1–4 polymer blending5–7 and minimization of structural defects8,9 have been attempted to prepare tougher polymeric materials.Regarding polymers with enhanced toughness, three broad categories of polymer networks have been designed based on their stress dispersion properties. One is interpenetrating brittle and ductile networks.10–14 The brittle network acts as sacrificial bonds, which dissipate stress and prevent catastrophic crack propagation. Another is a reversible cross-linked network. The reversible cross-links are constructed by dynamic covalent bonds15–17 and noncovalent bonds.18–52 These bonds are usually weaker than covalent bonds and act as sacrificial bonds. The other type is a movable cross-linked network that disperses stress based on the mobility of cross-links along the axis of the polymers.53,54
The preparation methods for movable cross-linked materials fall into five broad categories. These methods are based on macrocyclic molecules, and these ring-shaped molecules enable us to construct supramolecular architectures, such as pseudo-rotaxane/rotaxane,55 pseudo-polyrotaxane/polyrotaxane,56–58 catenane/polycatenane,59,60 and [c2]-daisy chains.61–64 The first method is “connecting between the ring units of polyrotaxane”.54,65–73 The second method is “polymerization of the polymerizable groups on the ring units of polyrotaxane”.74–79 Reversible cross-linked materials prepared from polyrotaxane were also studied.80–86 The third method is “formation of a rotaxane/polyrotaxane structure in a side chain”.87–95 The fourth method is “the polycondensation of the inclusion complex”.96–110
Previously, we prepared elastomers with movable cross-links by copolymerization between CD monomers and main chain monomers (single movable cross-network (SC) elastomers)111,112 (Fig. 1), which is the fifth method “polymer threading through polymerization”.111–114 SC elastomers show a higher toughness and Young's modulus than covalently cross-linked elastomers.112 Inoue and his coworker revealed that improvements in the mechanical properties of SC elastomers were derived from the viscoelastic behavior and relaxation mode of the movable cross-links.115,116 Notably, the advantage of the fifth method is an easy preparation of movable cross-linked materials through the polymerization of vinyl monomers. By expanding the fifth method, the combination of two dissimilar polymers using movable cross-links will improve the toughness and Young's modulus of the material.
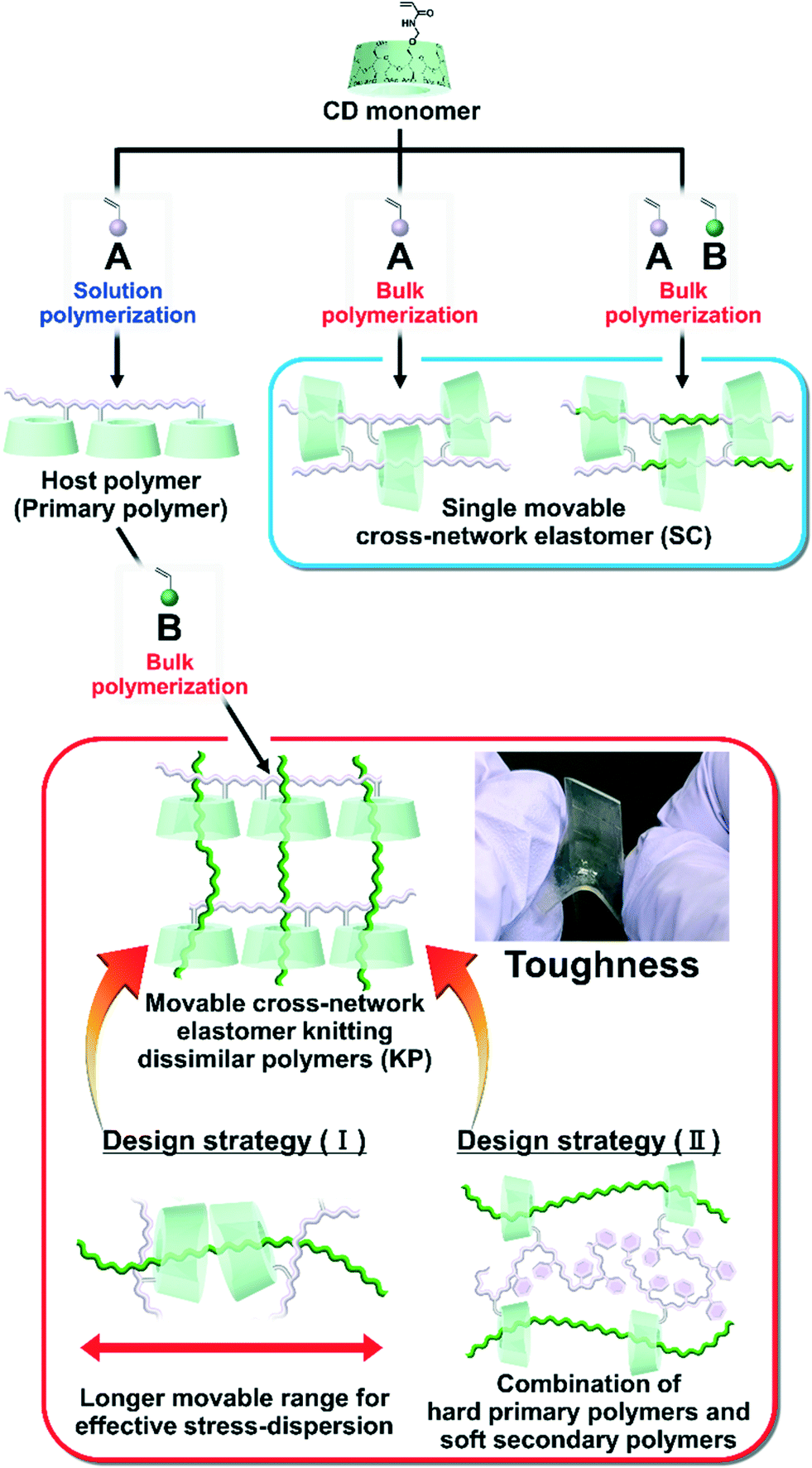 | ||
| Fig. 1 Routes toward two different material designs with movable cross-links: single movable cross-network elastomer (SC)115,116 and movable cross-network elastomer for knitting dissimilar polymers (KP). KP elastomers allow for the two design strategies: (I) and (II). | ||
Herein, we aimed to improve the mechanical properties by two design strategies: (I) a longer movable range for effective stress dispersion and (II) a combination of hard primary polymers with a high Young's modulus and soft secondary polymers for effective stress dispersion (Fig. 1). To realize these design strategies, we invented an elastomer with movable cross-links that connect dissimilar polymers in two steps. First, we copolymerized the peracetylated γCD (TAcγCD) monomer and main chain monomer in organic solution to obtain the host polymer (primary polymer). Next, we polymerized another liquid main chain monomer in the presence of a host polymer. The post polymerized chains (secondary polymer) penetrate the TAcγCD units and form movable cross-links that connect the dissimilar polymers. This knitting of primary and secondary polymers results in an elastomer (KP elastomer) that consists of host monomer-penetrated movable cross-links and dissimilar polymers. This report focuses on the mechanical properties of the KP elastomers with regard to the network designs, stress dispersion behavior, and domain structures.
Results and discussion
2-1. Preparation of the KP elastomers
Fig. 2 shows the chemical structures of the main chain monomers, primary polymers, KP elastomers, SC elastomers, and polymer blends. Ethyl acrylate (EA), butyl acrylate (BA), hexyl acrylate (HA), and styrene (St) were employed as the liquid main chain monomers (Fig. 2a). To solubilize in hydrophobic solvent, peracetylated CD monomer (mono-6O-acrylamidomethyl-triicosaacetyl-γ-cyclodextrin; abbreviated TAcγCDAAmMe) was prepared according to a previous report.43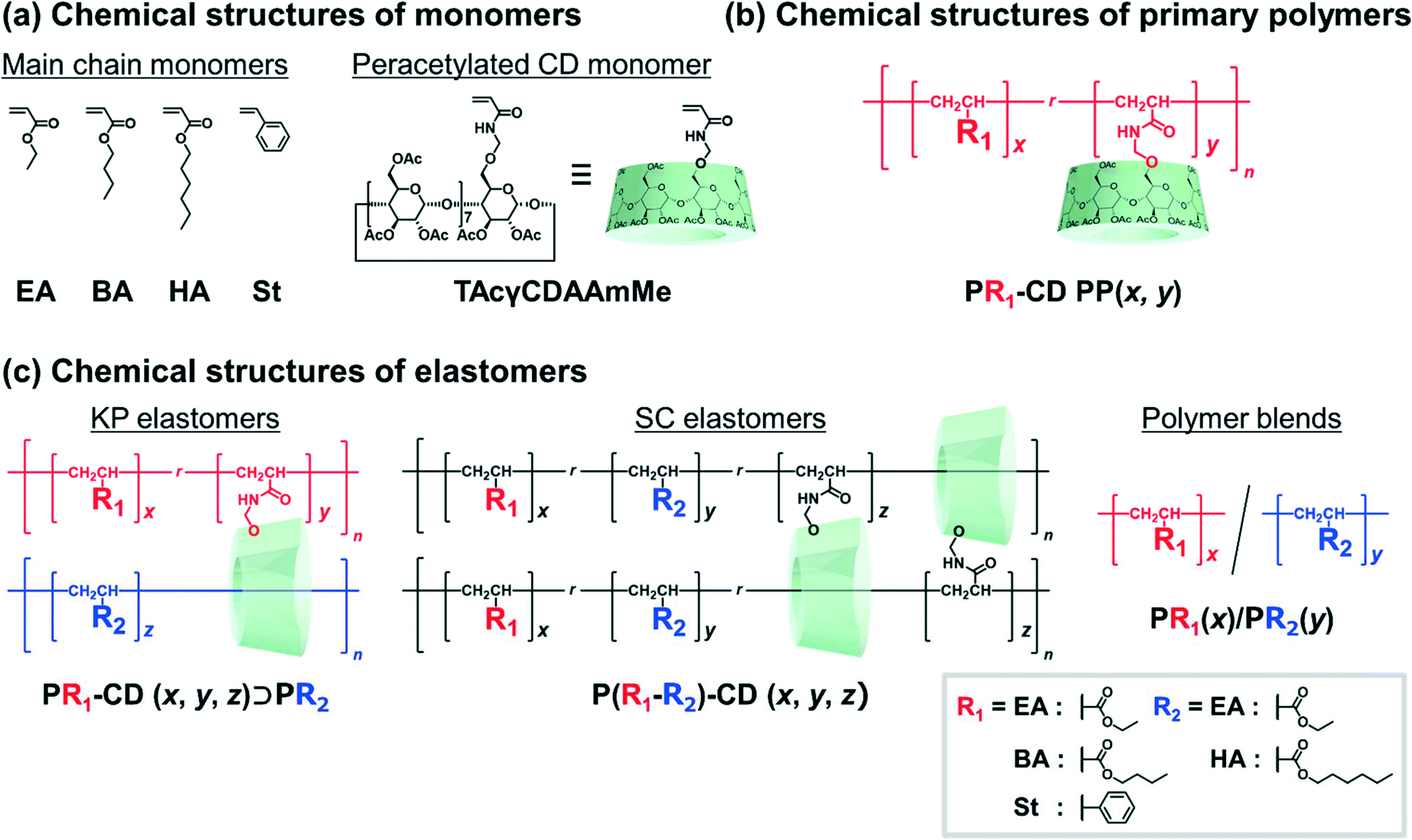 | ||
| Fig. 2 Chemical structures of (a) the main chain monomers (EA, BA, HA, and St), the peracetylated CD monomer (TAcγCDAAmMe), (b) the primary polymers (PR1–PAcγCD (x, y)), (c) the KP elastomers (PR1–CD (x, y)⊃PR2 (z)), the SC elastomers (P(R1–R2)–CD (x, y, z)), and the polymer blends (PR1(x)/PR2(y)). | ||
The primary polymers were obtained by solution copolymerization between main chain monomer R1 and TAcγCD monomer in hydrophobic solvents (chloroform or toluene) (Schemes S1–S3 and Tables S1–S3†). The solvents act as competitive guests to prevent the formation of movable cross-links in the primary polymers.111 The obtained primary polymers are abbreviated PR1–CD PP (x, y), where x and y are the mol% contents of the R1 and TAcγCD units, respectively. PR1 (poly(R1); a homopolymer without TAcγCD units) was also prepared by solution polymerization (Schemes S4 and S5†).
The KP elastomers were obtained by the bulk polymerization of another main chain monomer R2 in the presence of PR1–CD PP (x, y) (Schemes S6–S8 and Tables S4–S6†). The obtained KP elastomers were abbreviated PR1–CD (x, y)⊃PR2 (z), where x, y, and z are the mol% contents of the R1, TAcγCD, and R2 units, respectively. The TAcγCD units form movable cross-links between the PEA chains.111 Therefore, EA was mainly employed as the main chain monomer for the secondary polymer. As control samples to investigate the properties derived from network structures, P(R1–R2)–CD (x, y, z) (SC elastomers) were obtained from bulk polymerization (Scheme S9 and Table S7†). PR1(x)/PR2(y) (polymer blends) were obtained from bulk polymerization in the presence of the homopolymer (Schemes S10 and S11†). PEA–CD (x, y) and PEA were prepared according to a previous report111 Almost quantitively polymerization reactions were confirmed from integral values of 1H-nuclear magnetic resonance (NMR) spectra and absence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C peaks in the 13C-NMR and Fourier transform infrared (FT-IR) spectra (Fig. S3–S35†).
C peaks in the 13C-NMR and Fourier transform infrared (FT-IR) spectra (Fig. S3–S35†).
2-2. Mechanical properties of the KP elastomers
The mechanical properties of the KP elastomers were investigated by tensile tests (Fig. 3, S37, S38, and Table S10†). Prior to the KP elastomers with dissimilar polymers, tensile tests of PEA–CD (20, 1)⊃PEA (79) and control elastomers (PEA–CD (99, 1) and PEA) were carried out to reveal the effect of the structure itself. As a results, PEA–CD (20, 1)⊃PEA (79) showed the highest fracture stress and strain. Toughness was calculated from the integral of the stress–strain curve representing the tensile test results. Fig. 3a shows plots of the relation between the toughness and Young's moduli of PEA–CD (20, 1)⊃PEA (79). The toughness and Young's modulus of PEA–CD (20, 1)⊃PEA (79) were 16 ± 1 and 5.5 ± 2.2 times higher than those of PEA, indicating the presence of TAcγCD units as cross-linkers. Interestingly, the toughness of PEA–CD (20, 1)⊃PEA (79) was 3.5 ± 0.2 times higher than that of PEA–CD (99, 1), indicating that the network structure of PEA–CD (20, 1)⊃PEA (79) resulted in a higher toughness.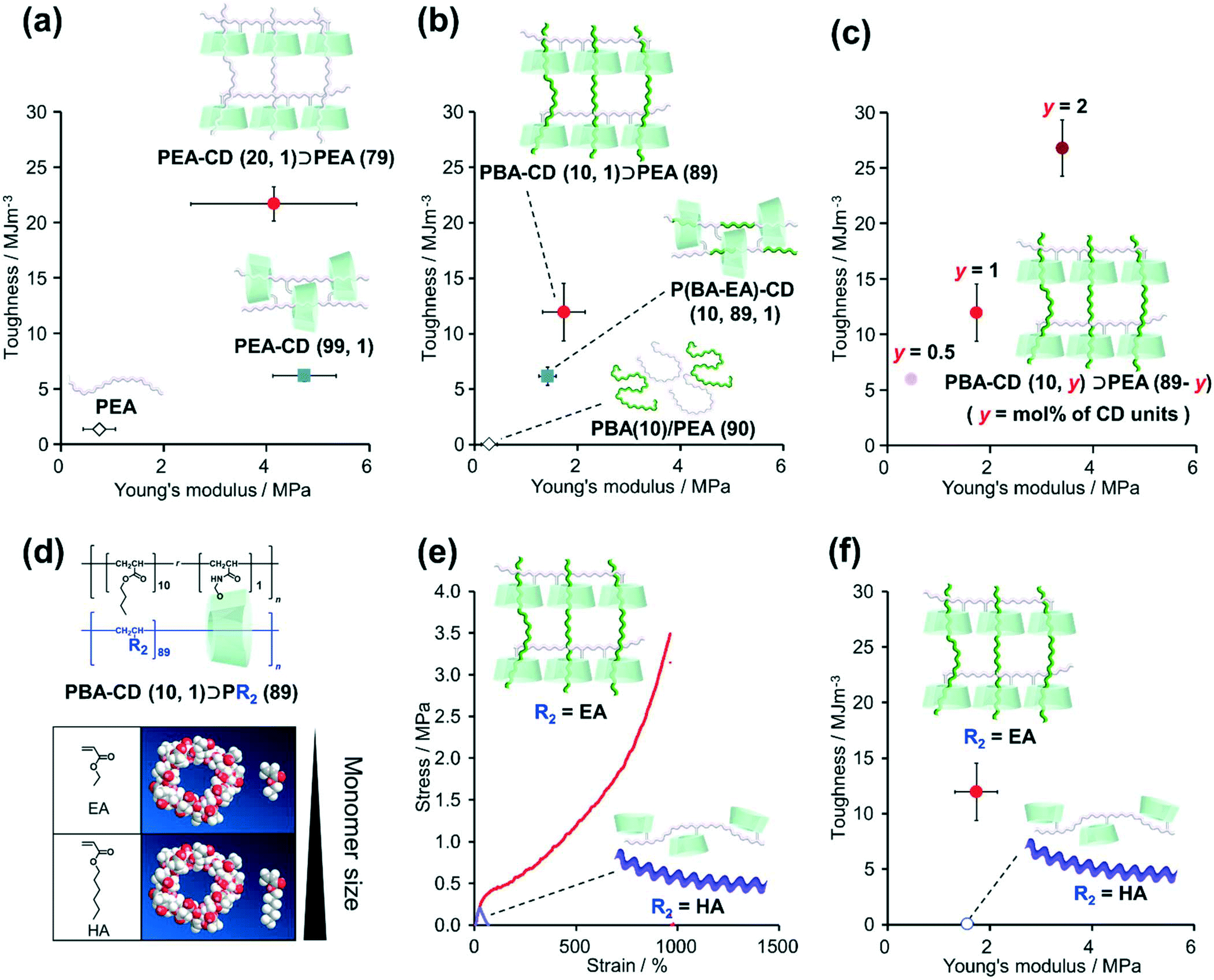 | ||
| Fig. 3 Plots of the toughness and Young's modulus of (a) PEA–CD (20, 1)⊃PEA (79) (red filled circle), PEA–CD (99, 1) (blue filled square), PEA (black open rhombus), (b) PBA–CD (10, 1)⊃PEA (89) (red filled circle), P(BA–EA)–CD (10, 89, 1) (blue filled square), PBA(10)/PEA(90) (black open rhombus), (c) PBA–CD (10, y)⊃PEA (10-y) (y = 0.5: pale red filled circle, y = 1: red filled circle, and y = 2: dark red filled circle). (d) 3D models (Chem3D® Ultra 16.0.1.4) of TAcγCD, EA, and HA. (e) Stress–strain curves of PBA–CD (10, 1)⊃PEA (89) (red line) and PBA–CD (10, 1)⊃PHA (89) (purple line). (f) Plots of the toughness and Young's modulus of PBA–CD (10, 1)⊃PEA (89) (red filled circle) and PBA–CD (10, 1)⊃PHA (89) (purple open circle). | ||
Similar results were obtained with the KP elastomers with dissimilar polymers and their control samples. PBA–CD (10, 1)⊃PEA (79) also showed the highest strength and stretchability (Fig. S37 and Table S10†). PBA–CD (10, 1)⊃PEA (79) exhibited clear fracture points and (6.4 ± 1.4) × 102 times and 6.2 ± 1.5 times higher toughness and Young's moduli compared with PBA(10)/PEA (90), confirming the formation of movable cross-links (Fig. 3b). PBA–CD (10, 1)⊃PEA (89) showed a 1.9 ± 0.4 times higher toughness than P(BA–EA)–CD (10, 89, 1). Fig. 3c shows the relation between the toughness and Young's modulus of PBA–CD (10, y)⊃PEA (10-y) (mol% of TAcγCD units: y = 0.5, 1, and 2). With increasing TAcγCD units, the toughness and Young's moduli of PBA–CD (10, y)⊃PEA (10-y) increased. These results confirmed that the TAcγCD units acted as cross-links.
2-3. Relation between cross-link formation and secondary polymer diameter
We investigated the relation between the mechanical properties of the materials and the size of the secondary monomers EA and HA (Scheme S12†). Fig. 3d shows the 3D models (Chem3D® Ultra 16.0.1.4) of TAcγCD, EA, and HA. The structures of the models were generated by minimizing the internal energy of each molecule by MM2 molecular dynamics. As HA is larger than the cavity diameter of TAcγCD, we assumed that the poly(hexyl acrylate) (PHA) chains rarely penetrated TAcγCD.Fig. 3e shows the stress–strain curves of PBA–CD (10, 1)⊃PEA (89) and PBA–CD (10, 1)⊃PHA (89). While PBA–CD (10, 1)⊃PEA (89) exhibited a clear fracture point, PBA–CD (10, 1)⊃PHA (89) showed plastic deformation. The toughness of PBA–CD (10, 1)⊃PHA (89) decreased to only 0.42% of that of PBA–CD (10, 1)⊃pEA (89) (Fig. 3f) despite having similar Young's moduli. This implied that temporary cross-links formed between the TAcγCD units and chain ends or side chains of PHA instead of stable movable cross-links. These results supported our assumption from the 3D models, which suggested that the high toughness was due to complexation between the TAcγCD units and secondary polymers. SC elastomers with PBA linear polymer (PBA(10)/PEA–CD(89, 1)) show lower toughness than PBA–CD (10, 1)⊃PEA (89), supporting connection between dissimilar polymers improved mechanical properties (Scheme S13 and Fig. S39†).
2-4. Investigation of the network structure by swelling tests
To characterize the network structures, we conducted swelling tests using chloroform as the swelling solvent. The polymers were immersed in excess chloroform. The swelling ratio (Q) was determined by the following equation:where W is the weight of the swollen polymer and W0 is the initial weight of the polymer before swelling. The Q of the P(BA–EA)–CD (10, 90-z, z) (z = 0.5, 1, and 2) polymers decreased with increasing mol% content of the TAcγCD unit, indicating the formation of interlocked cross-links (Fig. 4a). On the other hand, all PBA–CD (10, y)⊃PEA (90-y) (y = 0.5, 1, and 2) samples dissolved in chloroform (Fig. 4b). These solubilities allowed the GPC measurements of the KP elastomers (Fig. S36, Tables S8, and S9†). The GPC profiles of KP elastomers showed two peaks with the order of molecular weights comparable to (i) primary polymers and (ii) PEA obtained by bulk polymerization, respectively. These results suggest that the dethreading of the secondary polymer from the TAcγCD units caused the elastomers to dissolve.
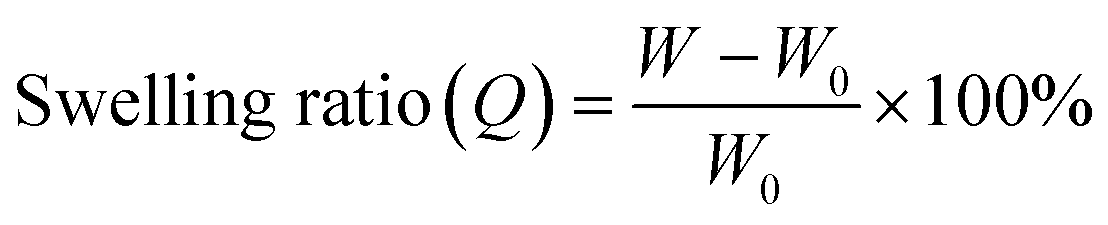
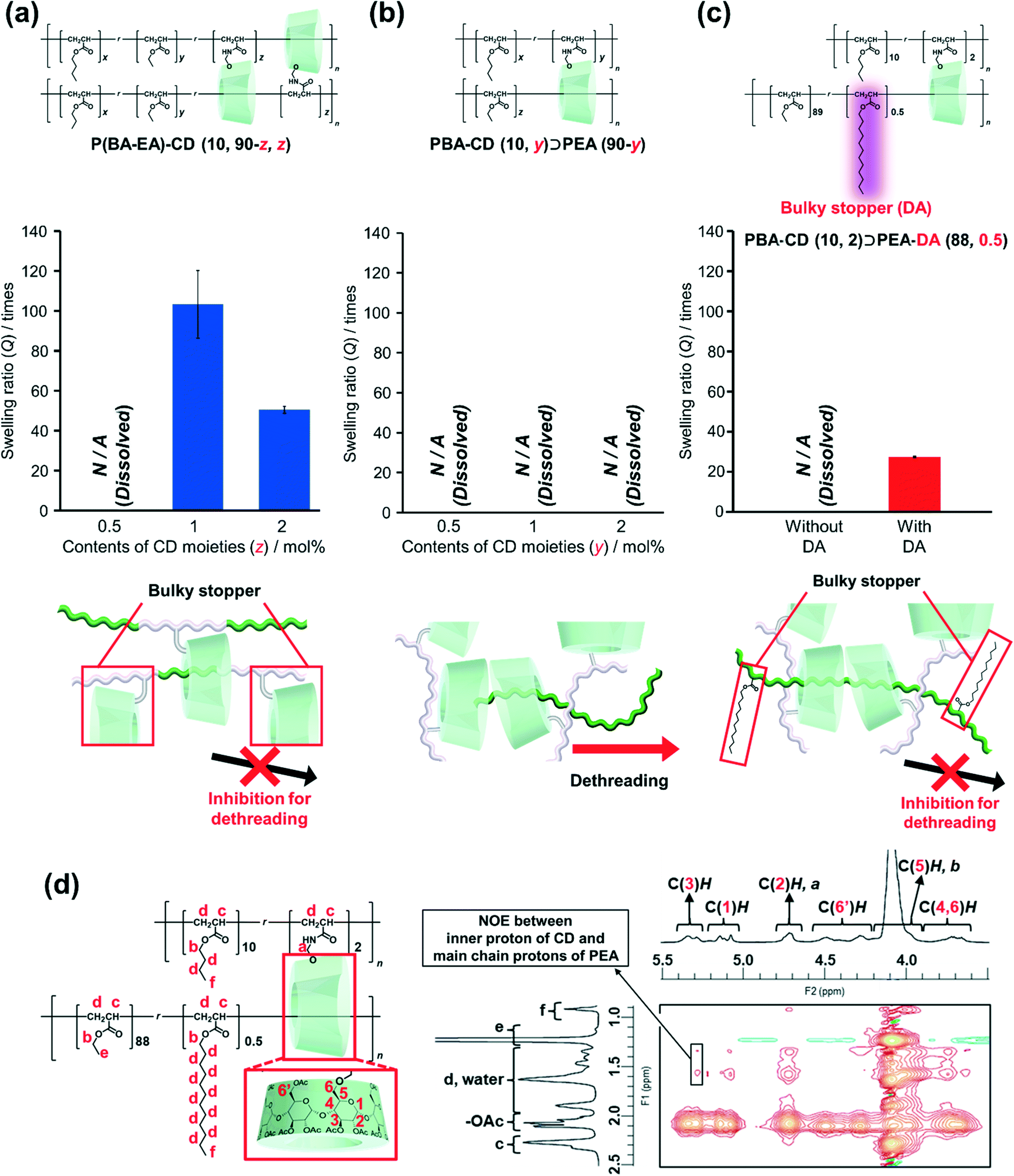 | ||
| Fig. 4 Swelling ratios and proposed structures of (a) P(BA–EA)–CD (10, 90-z, z), (b) PBA–CD (10, y)⊃PEA (90-y), (c) PBA–CD (10, 2)⊃PEA–DA (88) and PBA–CD (10, 2)⊃PEA–DA (88, 0.5). Contents of the CD moieties were 0.5, 1, and 2. (d) 600 MHz 2D 1H–1H NOESY NMR spectrum of the PBA–CD (10, 2)⊃PEA–DA (88, 0.5) slurries with chloroform-d. | ||
To confirm this presumption, we also prepared PBA–CD (10, 2)⊃PEA–DA (88, 0.5), where the secondary polymer contained bulky DA units to prevent the TAcγCD units from passing over (Scheme S14†). While PBA–CD (10, 2)⊃PEA (88) was dissolved, PBA–CD (10, 2)⊃PEA–DA (88, 0.5) was swollen to give a smaller Q than that of P(BA–EA)–CD (10, 88, 2) (Fig. 4c). These results indicated the presence of movable cross-links in PBA–CD (x, y)⊃PEA (z) between the TAcγCD units and secondary polymers.
2D nuclear Overhauser effect spectroscopy (NOESY) NMR spectra were obtained to confirm the complexation between the TAcγCD units and PEA. To prevent peak broadening, chloroform-d organogels of PBA–CD (10, 2)⊃PEA–DA (88, 0.5) were ball-milled into slurries before taking measurements. The spectrum of the ball-milled PBA–CD (10, 2)⊃PEA–DA (88, 0.5) slurries with chloroform-d exhibited NOE correlation signals between the protons in the TAcγCD units and PEA chains (Fig. 4d). Since C(3)H protons were located on the internal side of the TAcγCD rings, NOE signals of C(3)H indicated that the PEA main chains penetrated the TAcγCD units. On the other hand, the NOE signals of C(3)H were not observed in the spectrum of the reference sample: chloroform-d solutions of P(EA–DA) and PBA–CD PP(20, 1) (Fig. S40†). These results indicated that the TAcγCD units formed inclusion complexes with the PEA secondary polymer chains.
2-5. Investigation of the structural information by SAXS and DSC
To elucidate the deformation mechanism, we investigated nanoscale structures by small-angle X-ray scattering (SAXS) measurements upon tensile deformation. Fig. 5 shows the SAXS profiles of PBA(10)/PEA(90), P(BA–EA)–CD (10, 89, 1), and PBA–CD (10, 1)⊃PEA (89) with various λ strains (λ = 0%, 30%, 60%, 90%, and 120%) in the tensile and vertical directions. The intensities of the small scattering vector were in the order PBA(10)/PEA(90) > PBA–CD (10, 1)⊃PEA (89) > P(BA–EA)–CD (10, 89, 1) at λ = 0%. These results indicated that PBA–CD (10, 1)⊃PEA (89) was more homogenous than PBA(10)/PEA(90) but less homogeneous than P(BA–EA)–CD (10, 89, 1).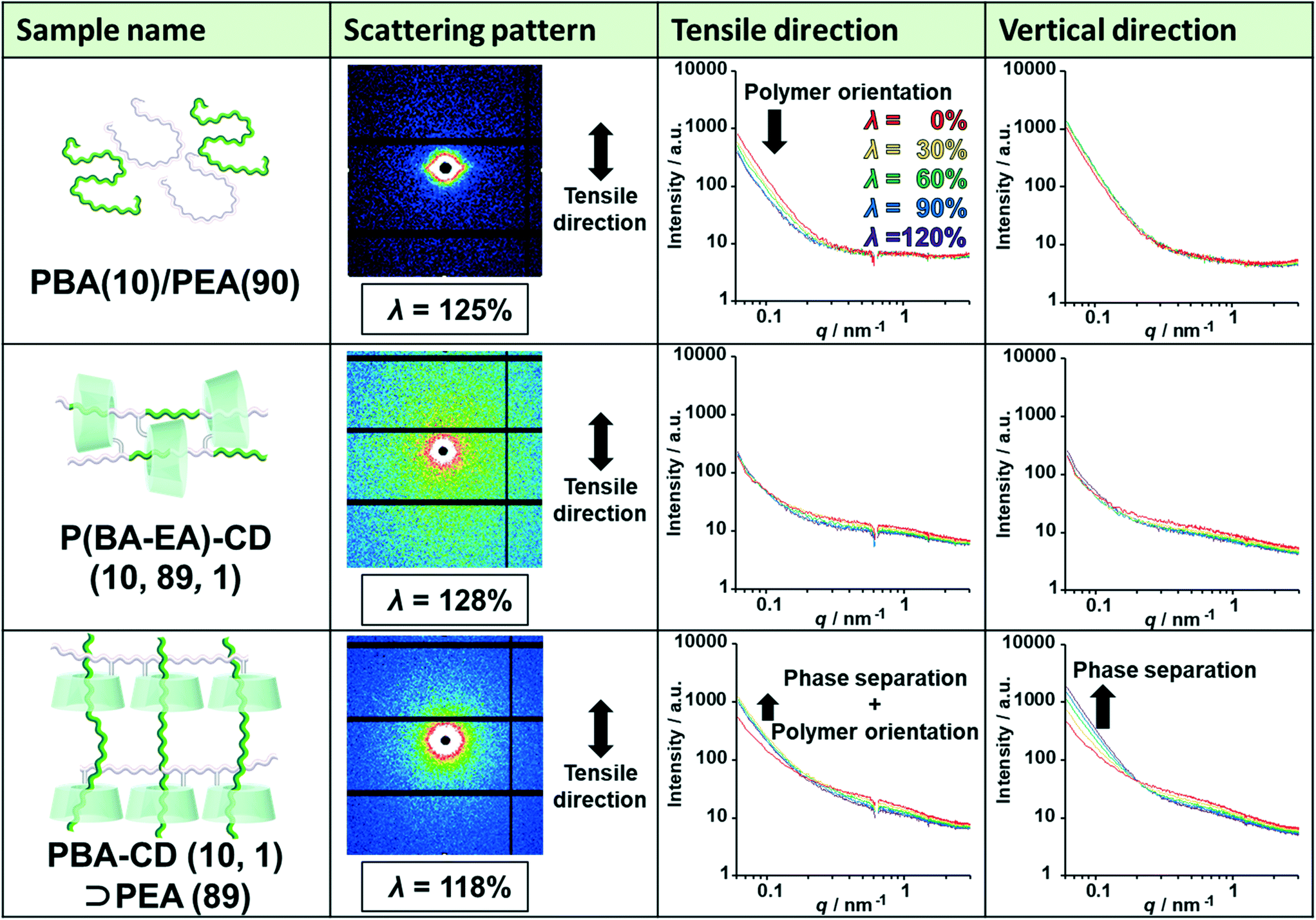 | ||
| Fig. 5 Scattering patterns of PBA(10)/PEA(90) with 125% strain, P(BA–EA)–CD (10, 89, 1) with 128% strain, and PBA–CD (10, 1)⊃PEA (89) with 118% strain. SAXS profiles of PBA(10)/PEA(90), P(BA–EA)–CD (10, 89, 1), and PBA–CD (10, 1)⊃PEA (89) with λ strains (λ = 0%: red line, λ = 30%: yellow line, λ = 60%: green line, λ = 90%: blue line, and λ = 120%: purple line) in the tensile and vertical directions. | ||
The intensities of PBA(10)/PEA(90) in the tensile direction decreased through deformation, indicating the orientation of the polymer chains. The streak-like scattering patterns of PBA(10)/PEA(90) supported the polymer orientation. Notably, P(BA–EA)–CD (10, 89, 1) did not show the orientation. The intensities of PBA–CD (10, 1)⊃PEA (89) in both directions increased through deformation, showing strain-induced phase separation between the primary and secondary polymers. The small orientation of PBA–CD (10, 1)⊃PEA (89) was also observed from the higher intensity in the vertical direction at a high value of λ. These results exhibited dethreading of the secondary polymer through deformation of the KP elastomers.
The thermal properties were also evaluated by differential scanning calorimetry (DSC) (Fig. S41†). PBA(10)/PEA(90) showed a characteristic endothermic peak after the endothermic transition step of glass transition temperature (Tg = −18 °C) of the PEA polymer. These peaks indicated that the PEA chains underwent enthalpy relaxation below Tg within the time scale of the cooling process in the measurements. Enthalpy relaxation is the structural relaxation of glass from nonequilibrium conformations toward an overall equilibrium state below Tg.117,118 It is known that an increased number of cross-links restricts enthalpy relaxation.119 The absence of the enthalpy relaxation peaks of PBA–CD (10, 1)⊃PEA (89) supported the formation of movable cross-links on the PEA secondary polymer. PEA–CD (20, 1)⊃PEA (79) and the reference samples also showed similar tendencies in their DSC results (Fig. S42†). In the thermal gravimetric analysis (TGA) results, PBA(10)/PEA(90) showed two steps of pyrolysis (below and above 360 °C). However, PBA–CD (10, 1)⊃PEA (89) showed one step of pyrolysis and higher thermostability at 350 °C than PBA(10)/PEA(90) (Fig. S43†). Those results could be contributed by the improvement of homogeneity by the formation of movable cross-links between primary and secondary polymers. Furthermore, compared to PEA, PEA–CD (20, 1)⊃PEA (79) also showed higher thermostability at 350 °C (Fig. S44†).
2-6. Evaluation of the stress dispersion property by stress-relaxation tests
To clarify relationship between the time scale of deformation and stress dispersion against strain, we performed tensile tests of two kinds of movable cross-linked materials with various tensile rates![[small lambda, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e152.gif) ( = 1.0, 0.30, 0.10, 0.030, and 0.010 mm s−1). While the stress of P(BA–EA)–CD (10, 89, 1) decreased with decreasing tensile rate, the fracture strain values are not significantly changed by the tensile rate (Fig. 6a). PBA–CD (10, 1)⊃PEA (89) exhibited elastic behavior at = 1.0 and 0.30 mm s−1 (Fig. 6b). The tensile strain drastically improved at = 0.10 and 0.030 based on the effective mobility of the cross-links and dethreading. At too slow of a tensile rate ( = 0.010), the movable cross-links in PBA–CD (10, 1)⊃PEA (89) lost elasticity to result in plastic deformation like polymeric materials without solid cross-link. While the slow deformation is supposed to be derived from effective mobility of the movable cross-links, the interlocked cross-links of P(BA–EA)–CD (10, 89, 1) prevent improved fracture strain. Dethreading of secondary polymers in PBA–CD (10, 1)⊃PEA (89) were also observed in cyclic tensile tests (Fig. S45†).
( = 1.0, 0.30, 0.10, 0.030, and 0.010 mm s−1). While the stress of P(BA–EA)–CD (10, 89, 1) decreased with decreasing tensile rate, the fracture strain values are not significantly changed by the tensile rate (Fig. 6a). PBA–CD (10, 1)⊃PEA (89) exhibited elastic behavior at = 1.0 and 0.30 mm s−1 (Fig. 6b). The tensile strain drastically improved at = 0.10 and 0.030 based on the effective mobility of the cross-links and dethreading. At too slow of a tensile rate ( = 0.010), the movable cross-links in PBA–CD (10, 1)⊃PEA (89) lost elasticity to result in plastic deformation like polymeric materials without solid cross-link. While the slow deformation is supposed to be derived from effective mobility of the movable cross-links, the interlocked cross-links of P(BA–EA)–CD (10, 89, 1) prevent improved fracture strain. Dethreading of secondary polymers in PBA–CD (10, 1)⊃PEA (89) were also observed in cyclic tensile tests (Fig. S45†).
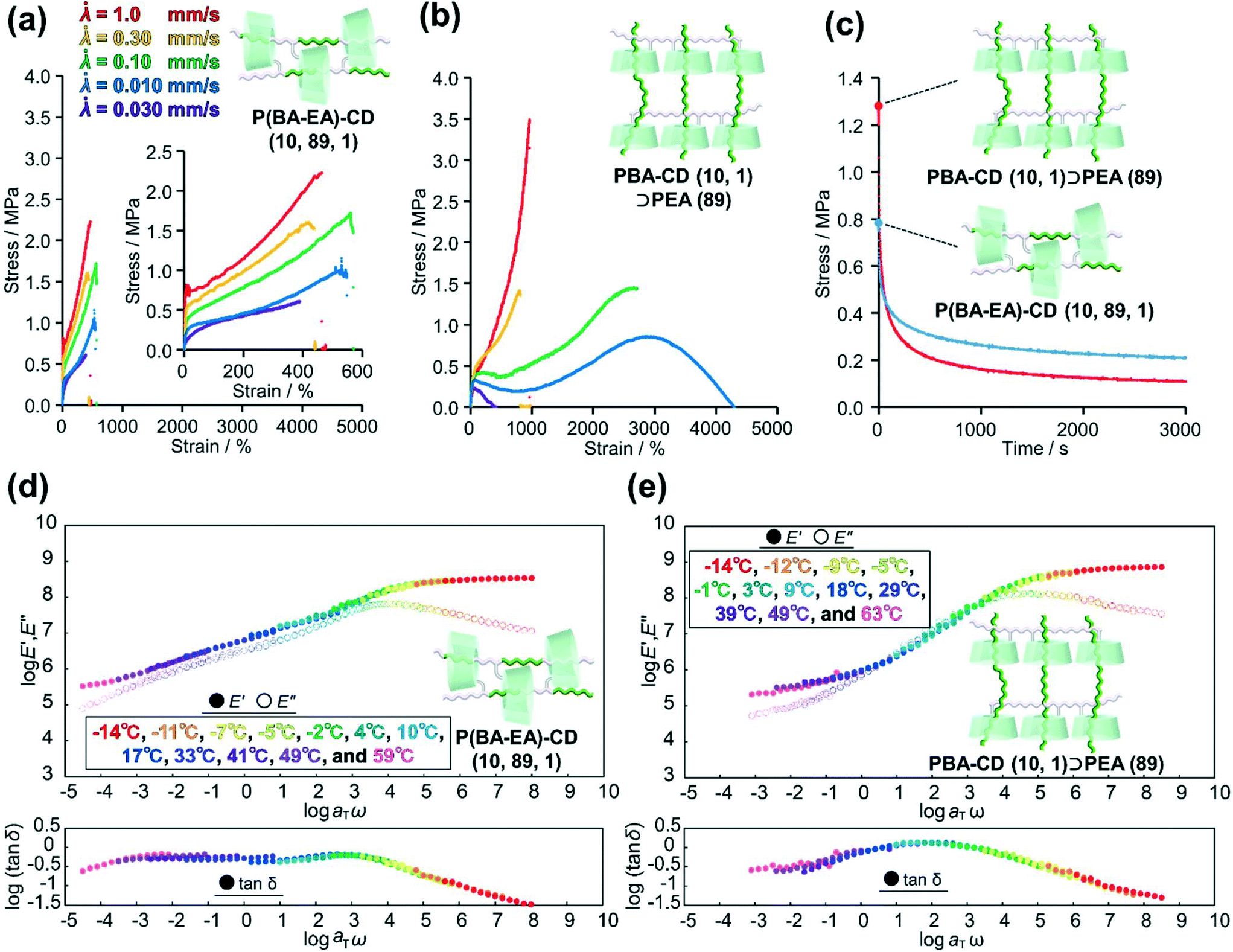 | ||
Fig. 6 Stress strain curves of (a) PBA–CD (10, 1)⊃PEA (89) and (b) P(BA–EA)–CD (10, 89, 1) with a tensile rate ( = 1.0 mm s−1: red line, = 0.30 mm s−1: yellow line, = 0.10 mm s−1: green line, = 0.030 mm s−1: blue line, and = 0.0010 mm s−1: purple line). (c) Stress relaxation of PBA–CD (10, 1)⊃PEA (89) and P(BA–EA)–CD (10, 89, 1) at 400% strain. Composite curves of the logarithmic moduli (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) E′ and logE′′) and log(tanδ) of (d) P(BA–EA)–CD (10, 89, 1) and (e) PBA–CD (10, 1)⊃PEA (89). E′ and logE′′) and log(tanδ) of (d) P(BA–EA)–CD (10, 89, 1) and (e) PBA–CD (10, 1)⊃PEA (89). | ||
Mentioned in Introduction, the high mechanical properties of the movable cross-linked materials were attributed to the stress dispersion properties at the cross-linking points. Herein, we carried out stress-relaxation tests to compare the stress dispersion properties between two kinds of movable cross-linked materials. The test pieces were continuously stretched until reaching 400%. Then, the test piece was held at the strain 400%, and the stress of the test piece was recorded for 3000 s (Fig. 6c). At the start of the stress-relaxation tests, the stress of PBA–CD (10, 1)⊃PEA (89) was higher than that of P(BA–EA)–CD (10, 89, 1). However, the stress of the former became lower than the latter in approximately 60 s. Consequently, PBA–CD (10, 1)⊃PEA (89) showed a smaller residual stress than P(BA–EA)–CD (10, 89, 1). These results suggested that PBA–CD (10, 1)⊃PEA (89) dispersed the internal stress more effectively.
To obtain further insight into the stress-relaxation behavior of these movable cross-linked materials, we carried out curve fitting using the Kohlrausch–Williams–Watts models, as described by the following equation. Notably, the stress σ versus relaxation time t curves were well fitted (R2 > 0.99) (Fig. S46†).
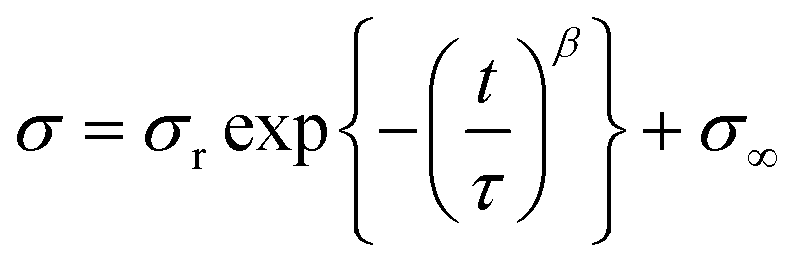
In the above equation, σr is the relaxable stress, σ∞ is the residual stress, τ is the time constant, and β is the stretching exponent. The obtained fitting parameters are summerized in Table 1. Consequently, PBA–CD (10, 1)⊃PEA (89) showed faster and larger stress dispersion, leading to its high toughness shown in the tensile test.
| Relaxable component | Residual components | |||
|---|---|---|---|---|
| σ r (MPa) | τ (s) | β | σ ∞ (MPa) | |
| P(BA–EA)–CD (10, 89, 1) | 0.67 | 140 | 0.29 | 0.15 |
| PBA–CD (10, 1)⊃PEA (89) | 1.33 | 30 | 0.29 | 0.07 |
Fig. 6d and e show the composite curves of the logarithmic storage and loss moduli (logE′ and logE′′) and log(tanδ) (tanδ = E′′/E′) of P(BA–EA)–CD (10, 89, 1) and PBA–CD (10, 1)⊃PEA (89) obtained from dynamic mechanical analysis (DMA; see ESI† for detail). The composite curves of the E′ and E′′ were constructed following the time (angular frequency)-temperature superposition principle: each modulus was a horizontally shifted modulus using the horizontal shift factor, αT, and a reference temperature (20 °C). Since the time–temperature superposition principle gave well-fitted composite curves, here we discuss the dynamics of polymer chain using the composite curves. Relaxations of E′ and E′′ for PBA–CD (10, 1)⊃PEA (89) were observed at higher frequency than those of P(BA–EA)–CD (10, 89, 1). In addition, PBA–CD (10, 1)⊃PEA (89) showed higher peaks of tanδ than P(BA–EA)–CD (10, 89, 1). These results showed a faster and larger relaxation mode of PBA–CD (10, 1)⊃PEA (89) at ω = 10−2–104 rad per s. In addition, PBA–CD (10, 1)⊃PEA (89) exhibited a lower activation energy than P(BA–EA)–CD (10, 89, 1) (Fig. S48†), suggesting lower energy barrier for the relaxation. These results supported the effective stress dispersion over a longer movable range to realize high toughness. PEA–CD (99, 1) and PEA–CD (10, 1)⊃PEA (89) also showed similar tendencies in their DMA results (Fig. S47 and S48†).
2-7. Preparation of the KP elastomers with immiscible polymers and their mechanical properties
As mentioned above, the homogeneity of PBA–CD (10, 1)⊃PEA (89) was improved by the movable cross-links that connected the primary and secondary polymers. Herein, PEA and PS were employed as primary and secondary polymers, respectively. The combination of these polymers was reported to have very low miscibility (<0.01 wt% of PEA in PS).120The polymer blend without cross-links PS (20)/PEA (80) showed phase separation, forming heterogeneous domains on the order of 0.1 mm (Fig. 7a). The KP elastomers PS–CD (20, 1)⊃PEA (79) with immiscible polymers were white elastomers without heterogeneous domains on the order of 0.1 mm. These results confirmed an (i) improved miscibility and (ii) phase separation in the obtained elastomer. PS–CD (20, 1)⊃PEA (79) showed a 170 ± 30 times increase in toughness and 1.6 ± 0.3 times increase in the Young's modulus of PS (20)/PEA (80), indicating the formation of movable cross-links between the primary and secondary polymers (Fig. 7b, S49, and Table S11†). Interestingly, PS–CD (20, 1)⊃PS (79) exhibited a 1.6 ± 0.3 times increase in toughness and a 8.2 ± 0.4 times increase in the Young's modulus of the KP elastomers with similar polymers PEA–CD (20, 1)⊃PEA (79).
 | ||
| Fig. 7 (a) Photographs and (b) plots of the toughness and Young's modulus of PS–CD (20, 1)⊃PEA (79) (red filled circle), PEA–CD (20, 1)⊃PEA (79) (blue filled circle), and PS(20)/PEA(80) (black open rhombus). (c) DSC curves of PS–CD (20, 1)⊃PEA (79) (red line), PEA–CD (20, 1)⊃PEA (79) (blue line), and PS(20)/PEA(80) (gray line). | ||
To obtain further insight into the phase separation, we performed DSC measurements. PS–CD (20, 1)⊃PEA (79) showed three Tg steps at −12, 40 and 101 °C (Fig. S50†). The transition at −12 °C represented the Tg of the PEA secondary polymer in PS–CD (20, 1)⊃PEA (79). The high mobility of the secondary polymer resulted in effective stress dispersion by the movable cross-links. The SAXS measurements of PS–CD (20, 1)⊃PEA (79) showed the dethreading of the secondary polymer upon deformation, supporting the high mobility of the secondary polymers and movable cross-links (Fig. S51†). The transition at 101 °C represented the Tg of the PS primary polymer, which acted as a hard domain to give a high fracture stress and Young's modulus. The transition at 40 °C represented a compatible phase between PEA and PS in PS–CD (20, 1)⊃PEA (79). In contrast, heterogeneous PS (20)/PEA (80) did not show this step. In DMA measurements of PS–CD (20, 1)⊃PEA (79), E′ and E′′ at 60 °C drastically decreased beyond to fit composite curves (Fig. S52†). These results supported that the movable cross-links improved the compatibility of the polymers, enabling the multiphase structure to work collaboratively. The improved compatibility was also supported by the one pyrolysis step of PS–CD (20, 1)⊃PEA (79) in the TGA results (Fig. S53†). The higher compatibilities allows the improvements of thermal stabilities of PEA chains by chain transfer, in which terminal radicals of depolymerizing PEA chains were trapped by tertiary protons in PS chains.121–123 Consequently, movable cross-links between immiscible polymers realized a three-phase structure to simultaneously achieve a high toughness and Young's modulus.
Conclusion
We conducted free-radical polymerization of a main chain monomer in the presence of a linear polymer with TAcγCD (primary polymer). The post polymerized chains (secondary polymer) penetrate the TAcγCD units and act as movable cross-links that connect the two dissimilar polymers. The obtained KP elastomers have a longer movable range than the one-pot fabricated materials with movable cross-links (Fig. 8). The secondary polymers have no bulky stopper, allowing dethreading upon tensile deformation. The larger movable range results in faster and more effective stress dispersion to achieve higher toughness. In addition, we successfully prepared even tougher and stiffer materials using movable cross-links to connect immiscible polymers (PEA and PS). These materials contain a three-phase structure: PEA, PS and a mixed phase. The PEA phase contributes to effective stress dispersion by the movable cross-links on account of its low Tg and high mobility. The PS phase contributes to a high fracture stress and Young's modulus as a hard domain with a high Tg (∼100 °C). The movable cross-links connect the two phases and result in a mixed phase, enabling each phase to work together. These findings should provide design strategies for simultaneously enhancing the toughness, stiffness, and compatibility of polymeric materials and their composites.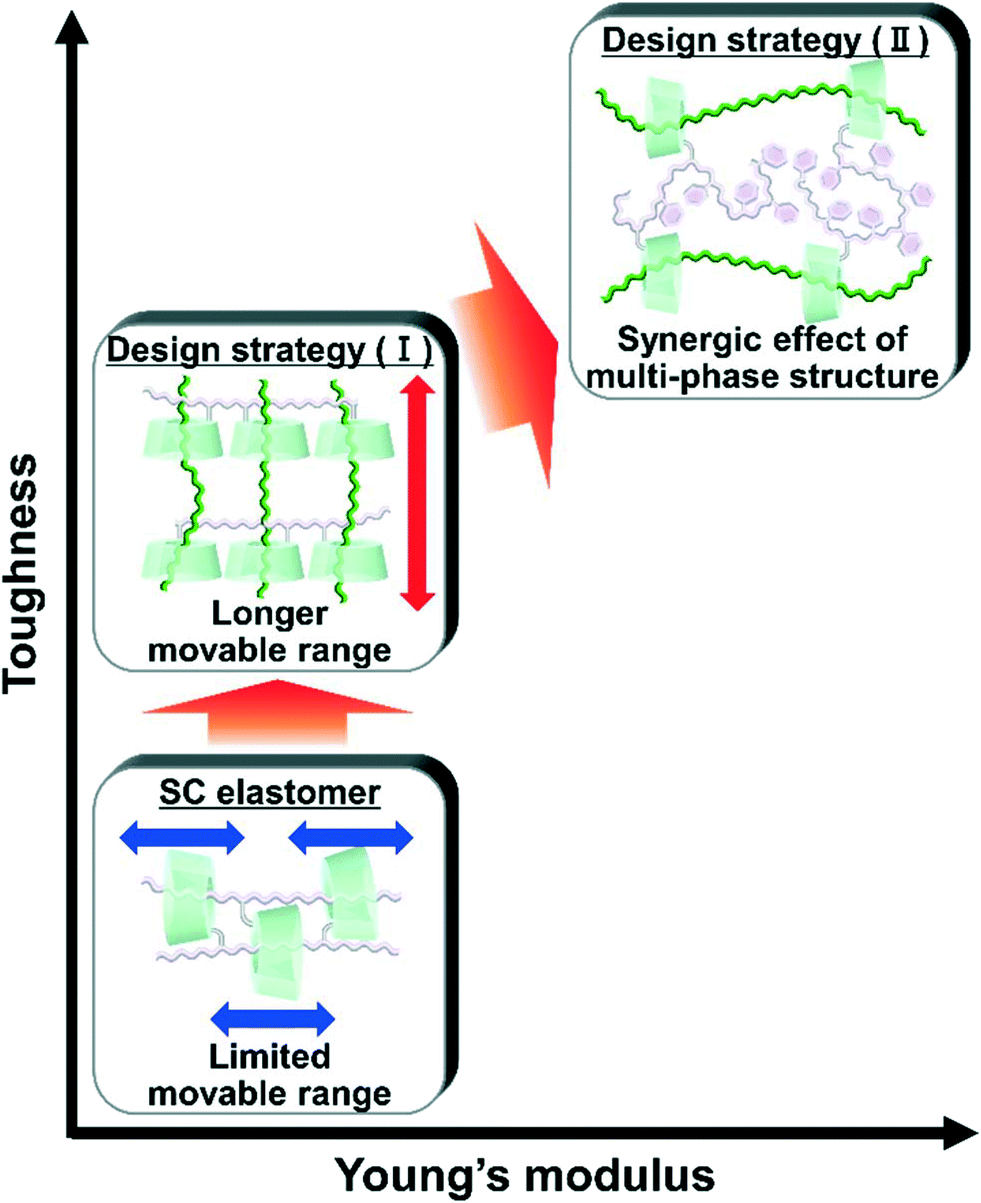 | ||
| Fig. 8 Achieving a higher toughness and Young's modulus by the two design strategies. | ||
Author contributions
Y. T. conceived and directed the study. R. I. performed syntheses and spectroscopic studies. All authors contributed to characterizations and discussion. Y. I. discussed and explained the results of the FT-IR spectroscopies. S. M. and G. M. discussed and explained the results of the SAXS measurements. M. O. wrote a section relating with DMA measurements. R. I., J. P., and Y. T. co-wrote the paper. A. H. and H. Y. gave valuable suggestions. Y. T. oversaw the project as well as contributed to the execution of the experiments and interpretation of the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by a Grant-in-Aid for Scientific Research (B) (No. JP26288062) from JSPS of Japan and Scientific Research on Innovative Area JP19H05721 from MEXT of Japan, JST-Mirai Program Grant Number JPMJMI18E3; the Kao Foundation for Arts and Sciences; and the Yazaki Memorial Foundation for Science. The authors appreciate Prof. Sadahito Aoshima and Prof. Arihiro Kanazawa (Graduate School of Science, Osaka University) for support with GPC measurements. The authors also appreciate Prof. Tadashi Inoue and Prof. Osamu Urakawa (Graduate School of Science, Osaka University) for support with DMA measurements. The authors also appreciate the Analytical Instrument Faculty of the Graduate School of Science, Osaka University, for support with the NMR, FT-IR, and TGA measurements. The authors also appreciate the Analytical Instrument Faculty of the Graduate School of Science, Osaka University, for support with the NMR, FT-IR, and TGA measurements. The authors would like to thank Dr. Noboru Ohta (SPring-8, JASRI) for the synchrotron radiation scattering measurements. The synchrotron radiation experiments were performed at BL40B2 (Proposal No. 2021A1593) and BL43 (Proposal No. 2020A1524) of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI).References
- J. N. Coleman, U. Khan, W. J. Blau and Y. K. Gun'ko, Carbon, 2006, 44, 1624–1652 CrossRef CAS.
- G. G. Tibbetts, M. L. Lake, K. L. Strong and B. P. Rice, Compos. Sci. Technol., 2007, 67, 1709–1718 CrossRef CAS.
- T. Ramanathan, A. A. Abdala, S. Stankovich, D. A. Dikin, M. Herrera-Alonso, R. D. Piner, D. H. Adamson, H. C. Schniepp, X. Chen, R. S. Ruoff, S. T. Nguyen, I. A. Aksay, R. K. Prud'Homme and L. C. Brinson, Nat. Nanotechnol., 2008, 3, 327–331 CrossRef CAS PubMed.
- M. Nogi, S. Iwamoto, A. N. Nakagaito and H. Yano, Adv. Mater., 2009, 21, 1595–1598 CrossRef CAS.
- S. Wu, J. Appl. Polym. Sci., 1988, 35, 549–561 CrossRef CAS.
- S. Wu, Polym. Eng. Sci., 1990, 30, 753–761 CrossRef CAS.
- T. H. Courtney, Mechanical Behavior of Materials, Waveland Press Inc., Illinois, 2016 Search PubMed.
- N. Sasaki, T. Matsunaga, M. Shibayama, S. Suzuki, Y. Yamamoto, R. Yoshida, C. Ito, T. Sakai and U. Chung, Macromolecules, 2008, 41, 5379–5384 CrossRef.
- S. Kondo, T. Hiroi, Y. S. Han, T. H. Kim, M. Shibayama, U. Il Chung and T. Sakai, Adv. Mater., 2015, 27, 7407–7411 CrossRef CAS PubMed.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- K. Singh, A. Ohlan, P. Saini and S. K. Dhawan, Polym. Adv. Technol., 2008, 19, 647–657 CrossRef.
- J. Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- E. Ducrot, Y. Chen, M. Bulters, R. P. Sijbesma and C. Creton, Science, 2014, 344, 186–190 CrossRef CAS PubMed.
- K. Sato, T. Nakajima, T. Hisamatsu, T. Nonoyama, T. Kurokawa and J. P. Gong, Adv. Mater., 2015, 27, 6990–6998 CrossRef CAS PubMed.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- K. Imato, M. Nishihara, T. Kanehara, Y. Amamoto, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2012, 51, 1138–1142 CrossRef CAS PubMed.
- A. Ciferri, Supramolecular Polymers, Marcel Dekker Inc., New York, 2000 Search PubMed.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS PubMed.
- Y. Gu, E. A. Alt, H. Wang, X. Li, A. P. Willard and J. A. Johnson, Nature, 2018, 560, 65–69 CrossRef CAS PubMed.
- S. Burattini, H. M. Colquhoun, J. D. Fox, D. Friedmann, B. W. Greenland, P. J. F. Harris, W. Hayes, M. E. MacKay and S. J. Rowan, Chem. Commun., 2009, 6717–6719 RSC.
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. MacKay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051–12058 CrossRef CAS PubMed.
- J. Fox, J. J. Wie, B. W. Greenland, S. Burattini, W. Hayes, H. M. Colquhoun, M. E. MacKay and S. J. Rowan, J. Am. Chem. Soc., 2012, 134, 5362–5368 CrossRef CAS PubMed.
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120–1124 CrossRef CAS.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS PubMed.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. Bin Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- J. C. Lai, L. Li, D. P. Wang, M. H. Zhang, S. R. Mo, X. Wang, K. Y. Zeng, C. H. Li, Q. Jiang, X. Z. You and J. L. Zuo, Nat. Commun., 2018, 9, 2725 CrossRef PubMed.
- Y. Miwa, K. Taira, J. Kurachi, T. Udagawa and S. Kutsumizu, Nat. Commun., 2019, 10, 1828 CrossRef PubMed.
- S. Wang and M. W. Urban, Nat. Rev. Mater., 2020, 5, 562–583 CrossRef CAS.
- M. W. Urban, D. Davydovich, Y. Yang, T. Demir, Y. Zhang and L. Casabianca, Science, 2018, 362, 220–225 CrossRef CAS PubMed.
- H. Wang, Y. Yang, M. Nishiura, Y. Higaki, A. Takahara and Z. Hou, J. Am. Chem. Soc., 2019, 141, 3249–3257 CrossRef CAS PubMed.
- D. C. Tuncaboylu, M. Sari, W. Oppermann and O. Okay, Macromolecules, 2011, 44, 4997–5005 CrossRef CAS.
- E. A. Appel, F. Biedermann, U. Rauwald, S. T. Jones, J. M. Zayed and O. A. Scherman, J. Am. Chem. Soc., 2010, 132, 14251–14260 CrossRef CAS PubMed.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- M. Zhang, D. Xu, X. Yan, J. Chen, S. Dong, B. Zheng and F. Huang, Angew. Chem., Int. Ed., 2012, 51, 7011–7015 CrossRef CAS PubMed.
- Y. Takashima, S. Hatanaka, M. Otsubo, M. Nakahata, T. Kakuta, A. Hashidzume, H. Yamaguchi and A. Harada, Nat. Commun., 2012, 3, 1270–1278 CrossRef PubMed.
- T. Kakuta, Y. Takashima, M. Nakahata, M. Otsubo, H. Yamaguchi and A. Harada, Adv. Mater., 2013, 25, 2849–2853 CrossRef CAS PubMed.
- K. Miyamae, M. Nakahata, Y. Takashima and A. Harada, Angew. Chem., Int. Ed., 2015, 54, 8984–8987 CrossRef CAS PubMed.
- V. Kardelis, K. Li, I. Nierengarten, M. Holler, J. F. Nierengarten and A. Adronov, Macromolecules, 2017, 50, 9144–9150 CrossRef CAS.
- X. Zhao, X. Chen, H. Yuk, S. Lin, X. Liu and G. Parada, Chem. Rev., 2021, 121, 4309–4372 CrossRef CAS PubMed.
- J. Liu, C. S. Y. Tan, Z. Yu, N. Li, C. Abell and O. A. Scherman, Adv. Mater., 2017, 29, 1–7 Search PubMed.
- S. Nomimura, M. Osaki, J. Park, R. Ikura, Y. Takashima, H. Yamaguchi and A. Harada, Macromolecules, 2019, 52, 2659–2668 CrossRef CAS.
- H. Aramoto, M. Osaki, S. Konishi, C. Ueda, Y. Kobayashi, Y. Takashima, A. Harada and H. Yamaguchi, Chem. Sci., 2020, 11, 4322–4331 RSC.
- J. Park, S. Murayama, M. Osaki, H. Yamaguchi, A. Harada, G. Matsuba and Y. Takashima, Adv. Mater., 2020, 32, 2002008 CrossRef CAS PubMed.
- B. V. K. J. Schmidt and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2017, 56, 8350–8369 CrossRef CAS PubMed.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- B. J. B. Folmer, R. P. Sijbesma, R. M. Versteegen, J. A. J. Van Der Rijt and E. W. Meijer, Adv. Mater., 2000, 12, 874–878 CrossRef CAS.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- Y. Yanagisawa, Y. Nan, K. Okuro and T. Aida, Science, 2018, 359, 72–76 CrossRef CAS PubMed.
- C.-F. Chow, S. Fujii and J.-M. Lehn, Angew. Chem., 2007, 119, 5095–5098 CrossRef.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed.
- Y. Koyama, Polym. J., 2014, 46, 315–322 CrossRef CAS.
- Y. Noda, Y. Hayashi and K. Ito, J. Appl. Polym. Sci., 2014, 131, 1–9 CrossRef.
- C. J. Bruson and J. F. Stoddart, The Nature of The Mechanical Bond: From Molecules to Machines, John Wiley & Sons, Inc., Hoboken, 2016 Search PubMed.
- A. Harada and M. Kamachi, Macromolecules, 1990, 23, 2823–2824 CrossRef.
- A. Harada, J. Li and M. Kamachi, Nature, 1992, 356, 325–327 CrossRef CAS.
- A. Harada, J. Li and M. Kamachi, Nature, 1994, 370, 1992–1994 CrossRef.
- M. Cesario, J. Guilhem, C. Pascard, C. O. Dietrich, A. Edel, J. P. Sauvage and J. P. Kintzinger, J. Am. Chem. Soc., 1986, 108, 6250–6254 CrossRef CAS.
- K. Endo, T. Shiroi, N. Murata, G. Kojima and T. Yamanaka, Macromolecules, 2004, 37, 3143–3150 CrossRef CAS.
- M. C. Jiménez, C. Dietrich-buchecker and J. Sauvage, Angew. Chem., Int. Ed., 2000, 1, 3284–3287 CrossRef.
- M. C. Jimenez-Molero, C. Dietrich-Buchecker and J. P. Sauvage, Chem. – Eur. J., 2002, 8, 1456–1466 CrossRef CAS.
- L. Fang, M. Hmadeh, J. Wu, M. A. Olson, J. M. Spruell, A. Trabolsi, Y. Yang, M. Elhabiri and J. F. Stoddart, J. Am. Chem. Soc., 2009, 131, 7126–7134 CrossRef CAS PubMed.
- P. G. Clark, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 13631–13633 CrossRef CAS PubMed.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef CAS.
- T. Karino, M. Shibayama, Y. Okumura and K. Ito, Phys. B, 2006, 385–386 I, 807–809 CrossRef.
- Y. Shinohara, K. Kayashima, Y. Okumura, C. Zhao, K. Ito and Y. Amemiya, Macromolecules, 2006, 39, 7386–7391 CrossRef CAS.
- T. Ogoshi, T. Aoki, S. Ueda, Y. Tamura and T. A. Yamagishi, Chem. Commun., 2014, 50, 6607–6609 RSC.
- X. Li, H. Kang, J. Shen, L. Zhang, T. Nishi, K. Ito, C. Zhao and P. Coates, Polymer, 2014, 55, 4313–4323 CrossRef CAS.
- H. Murakami, R. Baba, M. Fukushima and N. Nonaka, Polymer, 2015, 56, 368–374 CrossRef CAS.
- G. Kali, H. Eisenbarth and G. Wenz, Macromol. Rapid Commun., 2016, 37, 67–72 CrossRef CAS PubMed.
- J. Watanabe, T. Ooya, K. H. Nitta, K. D. Park, Y. H. Kim and N. Yui, Biomaterials, 2002, 23, 4041–4048 CrossRef CAS PubMed.
- T. Ooya, T. Ichi, T. Furubayashi, M. Katoh and N. Yui, React. Funct. Polym., 2007, 67, 1408–1417 CrossRef CAS.
- K. Koyanagi, Y. Takashima, H. Yamaguchi and A. Harada, Macromolecules, 2017, 50, 5695–5700 CrossRef CAS.
- A. Bin Imran, K. Esaki, H. Gotoh, T. Seki, K. Ito, Y. Sakai and Y. Takeoka, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- H. Gotoh, C. Liu, A. Bin Imran, M. Hara, T. Seki, K. Mayumi, K. Ito and Y. Takeoka, Sci. Adv., 2018, 4, 7629 CrossRef PubMed.
- J. H. Seo, M. Fushimi, N. Matsui, T. Takagaki, J. Tagami and N. Yui, ACS Macro Lett., 2015, 4, 1154–1157 CrossRef CAS.
- S. Choi, T. woo Kwon, A. Coskun and J. W. Choi, Science, 2017, 357, 279–283 CrossRef CAS PubMed.
- D. J. Yoo, A. Elabd, S. Choi, Y. Cho, J. Kim, S. J. Lee, S. H. Choi, T. woo Kwon, K. Char, K. J. Kim, A. Coskun and J. W. Choi, Adv. Mater., 2019, 31, 1–10 Search PubMed.
- C. Gong and H. W. Gibson, J. Am. Chem. Soc., 1997, 119, 8585–8589 CrossRef CAS.
- S. Li, G. H. Weng, W. Lin, Z. Bin Sun, M. Zhou, B. Zhu, Y. Ye and J. Wu, Polym. Chem., 2014, 5, 3994–4001 RSC.
- M. Nakahata, S. Mori, Y. Takashima, H. Yamaguchi and A. Harada, Chem, 2016, 1, 766–775 CAS.
- Y. Kobayashi, Y. Zheng, Y. Takashima, H. Yamaguchi and A. Harada, Chem. Lett., 2018, 47, 1387–1390 CrossRef CAS.
- S. Y. Zheng, C. Liu, L. Jiang, J. Lin, J. Qian, K. Mayumi, Z. L. Wu, K. Ito and Q. Zheng, Macromolecules, 2019, 52, 6748–6755 CrossRef CAS.
- H. Ke, L. P. Yang, M. Xie, Z. Chen, H. Yao and W. Jiang, Nat. Chem., 2019, 11, 470–477 CrossRef CAS PubMed.
- Q. Lin and C. Ke, Chem. Commun., 2022, 58, 250–253 RSC.
- T. Arai, K. Jang, Y. Koyama, S. Asai and T. Takata, Chem. – Eur. J., 2013, 19, 5917–5923 CrossRef CAS PubMed.
- C. Gong and H. W. Gibson, J. Am. Chem. Soc., 1997, 119, 5862–5866 CrossRef CAS.
- K. Iijima, Y. Kohsaka, Y. Koyama, K. Nakazono, S. Uchida, S. Asai and T. Takata, Polym. J., 2014, 46, 67–72 CrossRef CAS.
- J. Sawada, D. Aoki, S. Uchida, H. Otsuka and T. Takata, ACS Macro Lett., 2015, 4, 598–601 CrossRef CAS.
- J. Sawada, D. Aoki, M. Kuzume, K. Nakazono, H. Otsuka and T. Takata, Polym. Chem., 2017, 8, 1878–1881 RSC.
- K. Iijima, D. Aoki, H. Otsuka and T. Takata, Polymer, 2017, 128, 392–396 CrossRef CAS.
- T. Kureha, D. Aoki, S. Hiroshige, K. Iijima, D. Aoki, T. Takata and D. Suzuki, Angew. Chem., Int. Ed., 2017, 56, 15393–15396 CrossRef CAS PubMed.
- J. Sawada, D. Aoki, H. Otsuka and T. Takata, Angew. Chem., 2019, 131, 2791–2794 CrossRef.
- L. Li, Q. Lin, M. Tang, E. H. R. Tsai and C. Ke, Angew. Chem., 2021, 133, 10274–10281 CrossRef.
- N. Ogata, K. Sanui and J. Wada, J. Polym. Sci., Polym. Lett. Ed., 1976, 14, 459–462 CrossRef CAS.
- Y. Delaviz and H. W. Gibson, Macromolecules, 1992, 25, 4859–4862 CrossRef CAS.
- T. Murakami, B. V. K. J. Schmidt, H. R. Brown and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1156–1165 CrossRef CAS.
- S. Ikejiri, Y. Takashima, M. Osaki, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2018, 140, 17308–17315 CrossRef CAS PubMed.
- Y. Takashima, Y. Hayashi, M. Osaki, F. Kaneko, H. Yamaguchi and A. Harada, Macromolecules, 2018, 51, 4688–4693 CrossRef CAS.
- C. Y. Shi, Q. Zhang, C. Y. Yu, S. J. Rao, S. Yang, H. Tian and D. H. Qu, Adv. Mater., 2020, 32, 1–7 Search PubMed.
- D. Zhao, Z. Zhang, J. Zhao, K. Liu, Y. Liu, G. Li, X. Zhang, R. Bai, X. Yang and X. Yan, Angew. Chem., 2021, 60, 16224–16229 CrossRef CAS PubMed.
- H. W. Gibson, D. S. Nagvekar, J. Powell, C. Gong and W. S. Bryant, Tetrahedron, 1997, 53, 15197–15207 CrossRef CAS.
- H. W. Gibson, D. S. Nagvekar, N. Yamaguchi, S. Bhattacharjee, H. Wang, M. J. Vergue and D. M. Hercules, Macromolecules, 2004, 37, 7514–7529 CrossRef CAS.
- K. Yamabuki, Y. Isobe, K. Onimura and T. Oishi, Polym. J., 2008, 40, 205–211 CrossRef CAS.
- O. Jazkewitsch and H. Ritter, Macromolecules, 2011, 44, 375–382 CrossRef CAS.
- T. Cai, W. J. Yang, Z. Zhang, X. Zhu, K. G. Neoh and E. T. Kang, Soft Matter, 2012, 8, 5612–5620 RSC.
- T. Murakami, B. V. K. J. Schmidt, H. R. Brown and C. J. Hawker, Macromolecules, 2015, 48, 7774–7781 CrossRef CAS.
- K. Iwaso, Y. Takashima and A. Harada, Nat. Chem., 2016, 8, 625–632 CrossRef CAS PubMed.
- A. Goujon, T. Lang, G. Mariani, E. Moulin, G. Fuks, J. Raya, E. Buhler and N. Giuseppone, J. Am. Chem. Soc., 2017, 139, 14825–14828 CrossRef CAS PubMed.
- R. Ikura, J. Park, M. Osaki, H. Yamaguchi, A. Harada and Y. Takashima, Macromolecules, 2019, 52, 6953–6962 CrossRef CAS.
- R. Ikura, Y. Ikemoto, M. Osaki, H. Yamaguchi, A. Harada and Y. Takashima, Polymer, 2020, 196, 122465 CrossRef CAS.
- M. Ogawa, A. Kawasaki, Y. Koyama and T. Takata, Polym. J., 2011, 43, 909–915 CrossRef CAS.
- M. B. Yi, T. H. Lee, G. Y. Han, H. Kim, H. J. Kim, Y. Kim, H. S. Ryou and D. U. Jin, ACS Appl. Polym. Mater., 2021, 3, 2678–2686 CrossRef CAS.
- Y. Kashiwagi, T. Katashima, Y. Takashima, A. Harada and T. Inoue, Nihon Reoroji Gakkaishi, 2019, 47, 99–104 CrossRef CAS.
- Y. Kashiwagi, O. Urakawa, S. Zhao, Y. Takashima, A. Harada and T. Inoue, Macromolecules, 2021, 54, 3321–3333 CrossRef CAS.
- R. A. Fava, Polymer, 1968, 9, 137–151 CrossRef CAS.
- B. Wuxderlich and D. M. Bodily, J. Polym. Sci., Polym. Symp., 2007, 6, 137–148 Search PubMed.
- K. Jin, L. Li and J. M. Torkelson, Polymer, 2017, 115, 197–203 CrossRef CAS.
- R. H. Somani and M. T. Shaw, Macromolecules, 1981, 14, 1549–1554 CrossRef CAS.
- P. Gardner, R. Lehrle and D. Turner, J. Anal. Appl. Pyrolysis, 1993, 25, 11–24 CrossRef CAS.
- D. M. Bate and R. S. Lehrle, Polym. Degrad. Stab., 1998, 62, 57–66 CrossRef CAS.
- T. Uemura, T. Kaseda, Y. Sasaki, M. Inukai, T. Toriyama, A. Takahara, H. Jinnai and S. Kitagawa, Nat. Commun., 2015, 6, 1–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Preparative methods, NMR and FT-IR spectra, GPC, TGA, DMA, and SAXS measurements, mechanical properties and structures. See DOI: 10.1039/d2me00016d |
| This journal is © The Royal Society of Chemistry 2022 |