
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWhich factors govern the adsorption of peptides to Zr(IV)-based metal–organic frameworks?†
Alexandra
Loosen
,
Francisco
de Azambuja
 and
Tatjana N.
Parac-Vogt
*
and
Tatjana N.
Parac-Vogt
*
Department of Chemistry, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium. E-mail: tatjana.vogt@kuleuven.be
First published on 2nd February 2022
Abstract
Fundamental insight into the interactions between biological molecules and materials is essential for the design and development of adsorbents for many applications. Herein, we use a range of peptides to study the nature of these interactions and identify parameters pertinent to their adsorption onto Zr-based metal–organic framework (MOF) materials differing in structure and surface properties. A combination of experimental techniques revealed that MOF characteristics such as available Zr(IV) sites, nature of the linker, and hydrophilicity had a large influence on peptide adsorption. Experiments with peptides protected at N- and C-terminals indicated that adsorption to more hydrophilic MOFs MOF-808 and UiO-66 is governed by C-terminal carboxylate coordination to the Zr6 cluster, while adsorption onto the more hydrophobic NU-1000 is driven by electrostatic repulsion of the N-terminal group and hydrophobic interactions with the MOF surface/linker. Systematic variation in the amino acid's side chain suggests that other interactions like hydrophobic, aromatic, sulfur– or cation–π, and carboxylate interactions also influence adsorption, mostly arising from the hydrophobic/aromatic nature of the linkers and Zr6 cluster connectivity. Furthermore, the adsorption of peptides was strongly dependent on the pH of the solution, which could be used to modulate the affinity of the MOF material towards peptides. These results provide unique molecular insight towards understanding adsorption of biomolecules onto MOF materials and their design as nanozymes for catalytic reactions with peptides and proteins.
Introduction
Ubiquitous to all forms of life, amino acids (AA) are also largely used in various areas of science such as in solid-phase peptide synthesis,1,2 pharmaceutics,3 biomedical sensors4 and food supplements.5 In many of these applications, the different affinity of AAs towards solid surfaces is an important feature for purification, synthesis and/or detection steps. Common adsorbents for AAs are generally based on zeolites,6,7 silica,8,9 molecular sieves,10 metals and oxides.11 However, these materials feature limitations related to stability,12 spatial limitations,12 low pore volume or narrow pores,7,13 and grafting of adsorbent functionalities onto the solid,14 making the design of new adsorbent materials a milestone for the development of new technologies. To this end, fundamental understanding of AA's adsorption onto solid surfaces is key to designing novel adsorbents/applications, providing guidelines to (improve) processes, and extending adsorption technology to more complex systems such as peptides, proteins and other biomolecules.Metal–organic frameworks (MOFs) have the potential to become excellent adsorbents for AA and peptide adsorption, overcoming some of the problems currently faced by other materials. MOFs consist of inorganic nodes joined together by organic linkers. Clusters and linkers can vary widely in nature, which allows for tuning the structure's stability and characteristics towards specific applications. The highly porous structure and high internal surface areas of MOFs have been transformative to the adsorption field.15–18 Many reports have showcased MOFs’ potential as adsorbents for a broad range of substrates like gases,19 antibiotics,20 organic contaminants in water,21–23 organophosphorus pesticides,24 and enzyme immobilization.25 Moreover, they have been used to separate AA enantiomers via capillary electrophoresis,26 and their use as biosensors is receiving increasing attention due to their potential to selectively interact with specific biomolecules even in complex mixtures.27–29 Thus, engineering MOF structures with specific features could generate enhanced adsorbents for different classes of biomolecules.30
MOFs’ emerging catalytic applications towards biomolecules are also dependent of substrate-solid surface interaction, and deeper molecular insights into the adsorption phenomena are essential for the development of their catalytic reactivity.31–34 In this area, we have been particularly interested in the development of Zr6O8-based MOFs (Zr-MOFs) as nanozymes for the formation35 and cleavage36–38 of peptide bonds, including the selective hydrolysis of proteins which generates fragments in the optimal range for the emerging area of middle-down proteomics.39–41 However, the use of MOFs as a new generation of nanozymes has been hampered by the strong MOF–protein (fragments) interactions, which precludes further analysis of the protein digest.36 Interestingly, Zr-MOFs and a mixed Ce/Zr-UiO-66 MOF have shown distinct rates of peptide bond hydrolysis upon variation of the side chain in dipeptide substrates, which suggests different affinities of the peptide substrates towards the MOF catalyst.37,38,42 Preliminary adsorption studies have reinforced this hypothesis, but driving forces which influence interaction of biomolecules onto MOF materials are still poorly understood.42 Therefore, unraveling the factors affecting the adsorption of proteins’ building blocks such as amino acids and dipeptides could provide valuable information about the MOF reactivity towards proteins and further inspire their development as nanozymes.
In this context, we report a detailed investigation of adsorption properties of three Zr-MOFs towards a range of amino acids and peptides differing in nature of their side chains.36–38 The structures of these Zr-MOFs vary in cluster connectivity, type of linker and surface characteristics, which allowed us to evaluate key factors influencing the adsorption of biomolecules (Fig. 1 and Table 1). Additionally, the effect of amino acid structure and solution parameters which are pertinent for hydrolytic reactions, such as the pH and temperature have also been investigated. In the future, these parameters may be leveraged to design MOF materials with specific reactivity or adsorption characteristics.
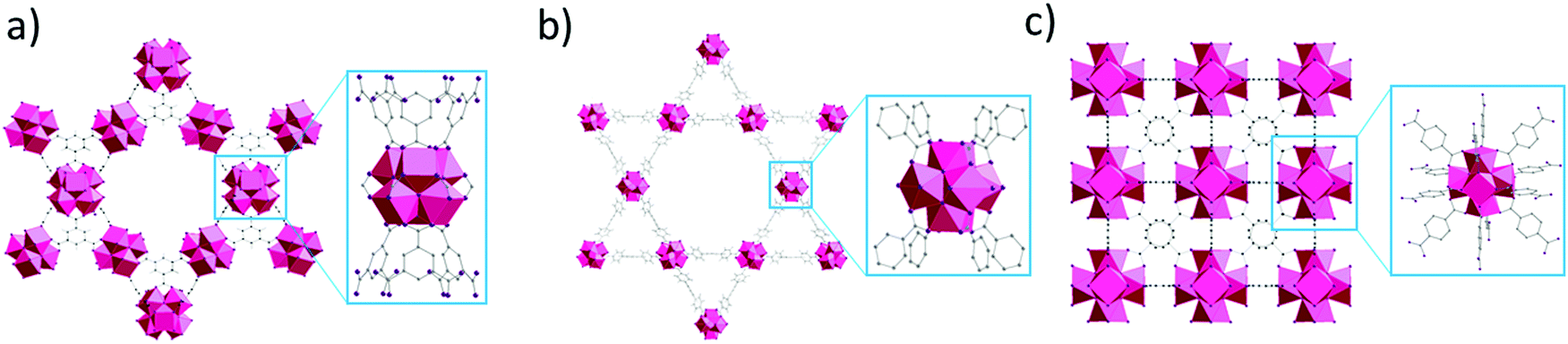 | ||
| Fig. 1 MOF structures used in this study, and magnification of their clusters with specific linker connectivity. (a) MOF-808, (b) NU-1000 and (c) UiO-66. [Zr6O8] = pink polyhedron, O = purple, C = gray. | ||
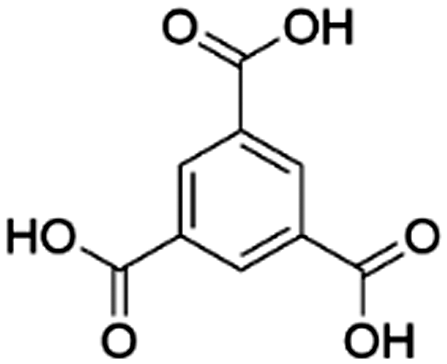
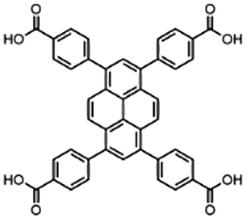
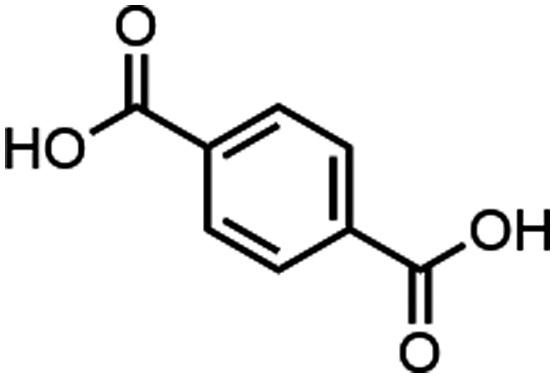
| Entry | MOF | Connectivity Zr6 cluster | Particle size (μm) | BET surface aread (m2 g−1) | Linker | log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P linkera P linkera |
pI |
|---|---|---|---|---|---|---|---|
|
a logP of linkers determined with ChemDraw. A higher logP corresponds to a higher hydrophobicity.
b Measured by SEM (Fig. S2, ESI).
c Determined by TGA.35,45
d From N2 physisorption.
|
|||||||
| 1 | MOF-808 | 6 | 237 | 1264 |
|
0.70 | 4.043 |
| 2 | NU-1000 | 8 | 3b | 219236 |
|
9.28 | 4.325 |
| 3 | UiO-66 | 9c | 0.335 | 1405 |
|
1.15 | 5.544 |
Experimental
Materials and methods
Unless otherwise noted, reactions were performed without any precautions against air and moisture. Adsorption reactions were performed in 1.5 mL vials sealed with a PTFE/silicone-lined screw cap with center hole. The pD was determined using a conventional pH meter by applying the relationship pD = pHread + 0.41.46 Unless otherwise noted, results reported are the average of three experiments. Data analysis performed using Microsoft Excel 365. MOFs were synthesized according to literature procedures with slight modifications of the modulator and the washing method in the case of MOF-808 and UiO-66 (see ESI†). 1H NMR spectra were recorded with a Bruker Avance 400 spectrometer in D2O (NS = 16, D1 = 14) and with 0.1 M TMSPA-d4 as an internal reference. Spectra were processed using Topspin software (Bruker). Powder X-ray Diffraction (PXRD) patterns was performed on a Malvern PANalytical Empyrean diffractometer (in transmission mode) over a 1.3–45° 2θ range, using a PIXcel3D solid state detector and Cu anode (Cu Kα1:1.5406 Å; Cu Kα2:1.5444 Å). N2 physisorption isotherms were measured on a Micromeritics 3Flex surface analyzer at −196 °C. Prior to measurements, samples were evacuated at 120 °C under vacuum for 12 h. Surface areas were calculated using the multi-point BET method applied to the isotherm adsorption branch taking into account the Rouquerol consistency criteria and the micropore volume was calculated at P/P0 = 0.5.47
Terephthalic acid (Fluka), formic acid (≥99%, VWR), N,N-dimethylformamide (Fischer Scientific), benzene-1,3,5-tricarboxylic acid (J&K Scientific), HCl (37%, ChemLab), ZrOCl2·8H2O (98%, Alfa Aesar), ZrCl4 (Thermo Fisher), benzoic acid (Sigma-Aldrich), 4,4′,4′′,4′′′-(1,3,6,8-pyrenetetrayl)tetrakis-benzoic acid (synthesized according to literature),48 acetone (Fisher Scientific), methanol (Fisher Scientific), ethanol (Fisher Scientific), NaOD (40 wt%, Sigma-Aldrich), DCl (35 wt%, Sigma-Aldrich), TMSPA-d4 (Sigma-Aldrich), glycylglycine (TCI), glycylalanine (Sigma-Aldrich), alanylglycine (Sigma-Aldrich), glycylleucine (TCI), glycyllysine (Bachem), glycylmethionine (Bio Connect BV), glycylaspartic acid (Sigma-Aldrich), glycylserine (Fluorochem), glycylthreonine (Sigma-Aldrich), glycylphenylalanine (Fluka), glycyltryptophan (TCI), glycylhistidine (Sigma-Aldrich), glycylasparagine (Sigma-Aldrich), glycylglutamic acid (Sigma-Aldrich), glycylisoleucine (Sigma-Aldrich), glycylarginine (Bachem), glycyltyrosine (Fluka), glycylglycylglycine (TCI Europe), glycylglycine methyl ester hydrochloride (AK Scientific), 2-(2-acetamidoacetamido)acetic acid (95%, Fluorochem). All chemicals were commercially obtained and used without further purification.
Adsorption study of glycylglycine
800 μL of D2O was mixed with 2 μmol MOF and stirred for 10 min. 200 μL of a 10 mM glycylglycine (GG) solution was added and pD was adjusted with NaOD or DCl. Reactions were carried out at pD 3.4, 5.4, 7.4 and 8.4, each reaction in threefold. Reactions were done in individual vials at room temperature and 500 rpm and the supernatant was collected after 6 h by 2 times 10 min centrifugation at 14000 rpm. Adsorption was determined based on 1H NMR spectroscopy using 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt (TMSPA-d4) as internal standard. Adsorption of GG was determined by quantification of GG in solution, and subtraction from initial concentration present before reaction. When hydrolysis side reaction was observed, the amount of G was determined by quantification of G in solution and divided by 2 to calculate backwards the amount of GG in solution it should originate from, and subsequently subtracted from the initial concentration GG present before reaction.
Adsorption study of glycylglycine with varying temperature
800 μL of D2O was mixed with 2 μmol MOF and stirred for 10 min. 200 μL of a 10 mM glycylglycine (GG) solution was added and pD was adjusted with NaOD to pD 7.4. Reactions were carried out at 25, 40 and 60 °C, each reaction in two- or threefold. Reactions were done in individual vials at room temperature and 500 rpm and the supernatant was collected after 6 h by 2 times 10 min centrifugation at 14000 rpm. Adsorption was determined based on 1H NMR spectroscopy using 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt (TMSPA-d4) as internal standard. Adsorption was determined by quantification of GG and G in solution, and subtraction from initial concentration of GG present before reaction.
Digestion of MOF samples
MOF samples were digested after reaction with 2 mM GG or 2 mM G as described above to see what is left on the MOF. The samples were collected by 2 times 10 min centrifugation at 14000 rpm and air-dried overnight. NaOD (1 M, 600 μL for 10 mg MOF, Table S1, ESI†) was added to 2 μmol MOF, sonicated and briefly stirred. The samples were left for 24 h after which D2O was added until a total volume of 540 μL. The supernatant was collected by 10 min centrifugation. GG or G in supernatant after digestion was determined based on 1H NMR spectroscopy using TMSPA-d4 as internal standard.
Adsorption study of C- and N-blocked GG
800 μL of D2O was added to 2 μmol MOF and mixed with 200 μL of glycylglycine methyl ester hydrochloride or 2-(2-acetamidoacetamido)acetic acid solution (GGOMe or N-acetylGG, 10 mM) and pD was adjusted to 3.4 and 7.4 with DCl or NaOD, each reaction in twofold. After 6 h of stirring at room temperature and 500 rpm, the supernatant was collected and adsorption was determined based on 1H NMR using TMSPA-d4 as internal standard. Adsorption of GGOMe or N-acetylGG was determined by quantification of GGOMe or N-acetylGG in solution, and subtraction from initial concentration present before reaction.Adsorption study of glycine and glycylglycylglycine
800 μL of D2O was mixed with 2 μmol MOF and stirred for 10 min. 200 μL of a 10 mM glycine (G) or glycylglycylglycine (GGG) solution was added and pD was adjusted with NaOD or DCl. Reactions were carried out at pD 7.4, each reaction in twofold. Reactions were done in individual vials at room temperature and 500 rpm and the supernatant was collected after 6 h by 2 times 10 min centrifugation at 14000 rpm. Adsorption was determined based on 1H NMR spectroscopy using TMSPA-d4 as internal standard. Adsorption of G was determined by quantification of G in solution, and subtraction from initial concentration present before reaction.
Adsorption study of dipeptides
The adsorption of different dipeptides was followed. 900 μL of D2O was added to 2 μmol MOF and mixed with 100 μL of dipeptide solution (20 mM) and pD was adjusted to 7.4 with NaOD, each reaction in twofold. After 6 h of stirring at room temperature and 500 rpm, the supernatant was collected and adsorption was determined based on 1H NMR using TMSPA-d4 as internal standard. Adsorption of peptides was determined by quantification of peptides in solution, and subtraction from initial concentration present before reaction.Results
Based on our previous work on developing artificial peptidases,36–38 three well-known Zr-MOFs have been selected for this work: (1) MOF-808, a 6-connected MOF with 1,3,5-benzenetricarboxylate (BTC) linkers;49 (2) NU-1000, a 8-connected MOF with 1,3,6,8-(p-benzoate)pyrene (TBAPy) linkers;48 and (3) UiO-66, a 12-connected MOF with 1,4-benzenedicarboxylate (BDC) linkers (Fig. 1 and Table 1).50 These MOFs feature Zr6O8 clusters with a different coordination environment depending on the connectivity (Fig. 1), which imparts key differences in availability of metal sites and their overall physicochemical properties (Table 1). To evaluate the adsorption behavior of these MOFs, we have considered three major factors:1. Effect of pH and temperature conditions of substrate solution on adsorption;
2. Impact of substrates’ structure on adsorption, namely AA's side chain nature and position, as well as the length of the peptide;
3. Influence of MOF characteristics such as the connectivity of the Zr6 cluster, surface charge, type of linker and surface hydrophobicity on the adsorption.
The amount of peptide adsorption onto the MOF was monitored by 1H NMR spectroscopy through comparison of the initial peptide concentration with the peptide concentration in the supernatant after incubation with the MOF. This method's suitability was confirmed by analysis of the recovered MOF material. Using adsorption experiments conducted at room temperature and pH 7.0 after 6 h of incubation as a representative example, we could prove that all GG that was removed from solution was actually adsorbed on the MOF. The MOF material recovered from the incubation with GG was digested, and the amount of recovered peptide was quantified by 1H NMR spectroscopy (Table S4, ESI†).
To ensure that the stability of MOF structure was being preserved during experiments, PXRD analyses were done before and after the experiments. In general, the structure of all MOFs remained well preserved after adsorption experiments at various pH and temperatures, and with the diverse set of substrates probed (Fig. S7–S15, ESI†). This ensured that the stability and heterogeneous character of MOFs were preserved during experiments, and the results did not relate to structural changes MOFs might have undergone in the course of incubations.
Effect of pH on adsorption of glycylglycine onto Zr-MOFs
In a first step, the effect of pH of the dipeptide solution on adsorption was examined, as the degree of AA protonation is pH dependent (Fig. 2a). The simplest dipeptide, glycylglycine (GG), was used as the model dipeptide for this study. The adsorption at room temperature was measured at various pH values by quantifying the decrease of GG's 1H NMR resonance at 3.77–3.95 ppm (Fig. S5, ESI†). For each MOF, the change of GG concentration in solution was plotted as a function of pH (Fig. 2b). Under the conditions used in this study, very limited hydrolysis of GG was observed in the case of MOF-808 at pH 7.0 and 8.0, and for the UiO-66 MOF at pH 8.0.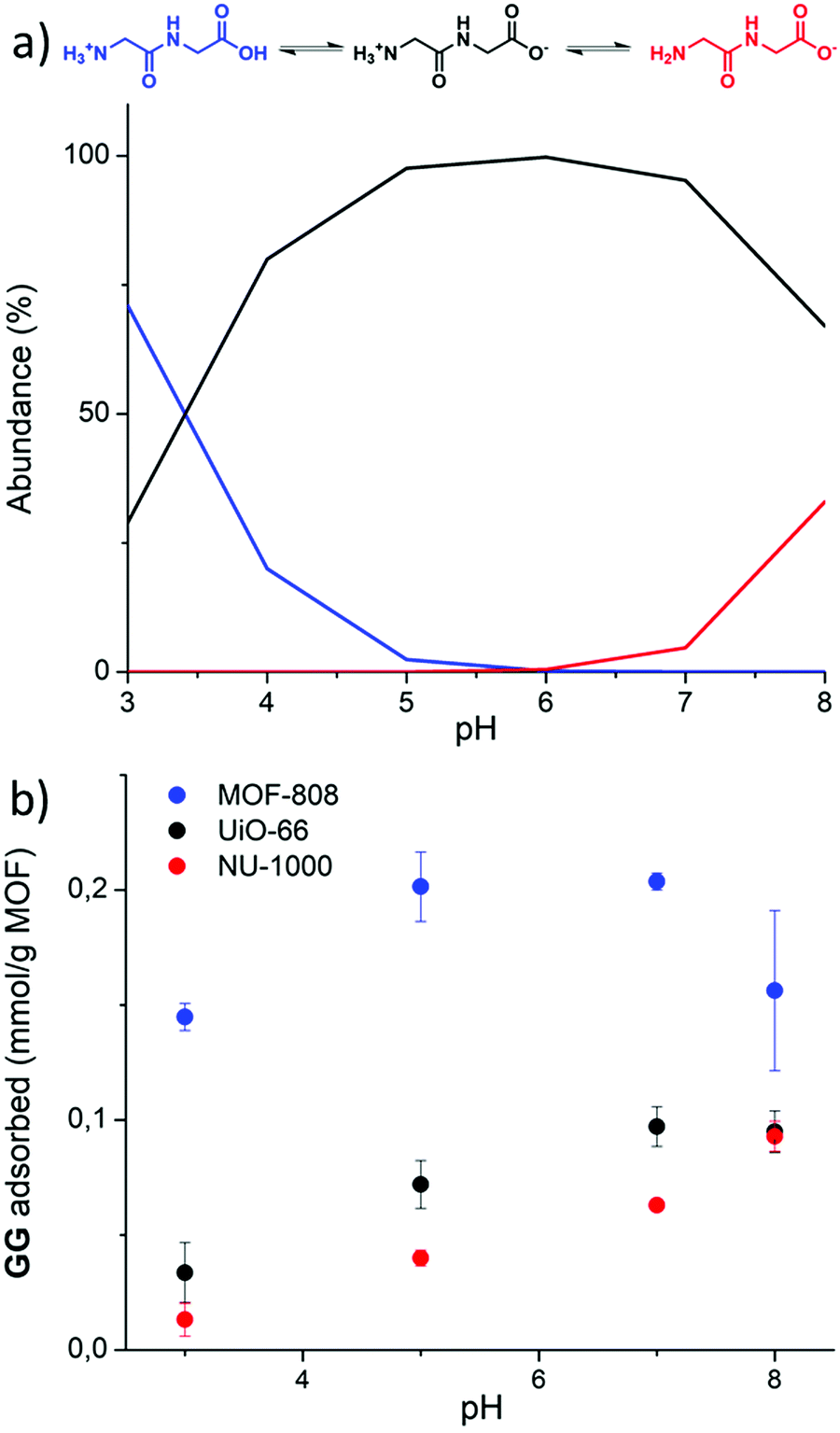 | ||
| Fig. 2 Acidity of the solution affects glycylglycine (GG) adsorption. (a) Abundance of the different forms of GG depends on the solution acidity, (b) effect of pH on adsorption of GG onto Zr-MOFs. 100% adsorption of GG = 0.600 mmol g−1 UiO-66, 0.463 mmol g−1 NU-1000, 0.741 mmol g−1 MOF-808. | ||
In general, upon increasing the pH in the range of 3.0 to 7.0 more GG was adsorbed onto MOF material; at pH 8.0, GG adsorption was higher only for NU-1000 (Fig. 2b). The decrease in GG adsorption observed for MOF 808 and UiO-66 at pH values above 7.0 might be related to GG hydrolysis, or loss of MOF 3D structure. At pH 7.0 and 8.0, ca. 4 and 7% of GG was hydrolyzed to glycine (G) by MOF 808, respectively, while for UiO-66 about 5% of hydrolysis was observed at pH 8.0. As adsorption of G onto Zr-MOFs was lower than GG (see results below), hydrolysis could be causing the decrease in adsorption shown in Fig. 2b. In addition, the larger drop observed for MOF-808 at pH 8.0 suggested a potential loss of MOF structure, as reported previously under similar conditions (60 °C, pH 8.0).37 Although PXRD patterns of the MOF series showed that all MOFs were stable under adsorption conditions at various pH values (Fig. S7–S9, ESI†), the persistent larger error bar obtained at pH 8.0, even after multiple replicates, hints that additional factors might be playing a role. It is plausible that some change in MOF structure not easily detected through PXRD takes place, which could lead to the drop in adsorption.
In a second step, to probe if the carboxylic acid and/or the amino group of GG were involved in adsorption, and whether the strength of interaction depends on their protonation state, C- and N-terminal protected GG dipeptides were used as control substrates to measure adsorption at pH 3.0 and pH 7.0 (Fig. 3). Esterification of GG's C-terminal group prevents its effective coordination to Zr(IV) centra, and makes it more hydrophobic (Table S5, ESI† entry 1 vs. 19). Higher hydrophobicity of glycylglycine methyl ester (GGOMe) compared to GG could disfavor its adsorption onto more hydrophilic UiO-66 and MOF-808 materials. Thus, GGOMe was used as a control substrate with protected C-terminal. At pH 3.0, adsorption decreased for all MOFs when GGOMe was used as a substrate (Fig. 3a). Curiously, the decrease in adsorption in the presence of NU-1000 was much lower at pH 3.0 compared to the other MOFs, and similar adsorptions were observed for GGOMe and GG at pH 7.0 (Fig. 3b). This might be due to the greater surface hydrophobicity of NU-1000 compared to other MOFs used in the study, which originates from its linker structure (Table 1). In agreement with this trend, a large decrease in adsorption of GGOMe compared to GG was observed for MOF-808, with the larger drop detected at pH 7.0. The adsorption of GGOMe to UiO-66 followed the same trend observed for MOF-808, showing lower adsorption of protected dipeptide compared to free GG, both at pH 3.0 and pH 7.0. However, the effect was smaller than in the case of MOF-808.
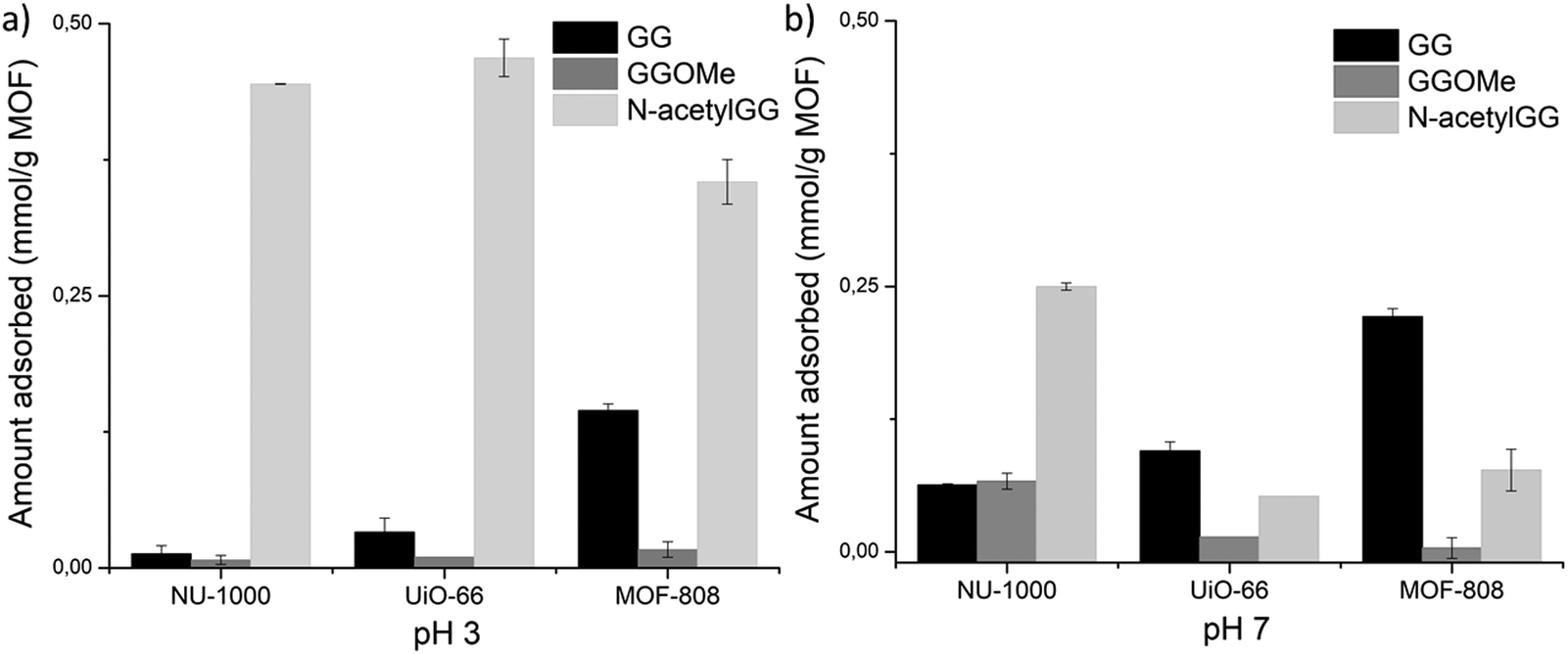 | ||
| Fig. 3 Blocking C- and N-terminal groups of GG perturbs adsorption in different ways depending on pH: (a) pH 3.0, (b) pH 7.0. | ||
Similar experiments were used to probe the influence of the N-terminal group on adsorption (Fig. 3). N-Acetyl glycylglycine (N-acetylGG) was used as a control N-terminal protected substrate since the acetyl group eliminates the positively charged ammonium terminus, in the pH range 3.0 to 7.0 (Fig. 2a), without significantly affecting the nature of the substrate. Overall, the results indicated that the N-terminal group of GG impacts the adsorption differently depending on the MOF and the pH of solution. At pH 3.0, a large increase of adsorption was observed for all three MOFs, being proportionally much higher for NU-1000 and UiO-66 than for MOF-808 (Fig. 3a). At pH 7.0, changes of overall lower magnitude were observed; however, two patterns emerged. A 4-fold increase in adsorption compared to GG was observed for NU-1000, while decreases in adsorption were observed for UiO-66 (∼35%) and MOF-808 (∼50%) (Fig. 3b).
Effect of distance between the C- and N-terminal of peptides on adsorption
Initial results pointed that the adsorption of GG onto Zr-MOFs most likely occurs via carboxylate–Zr interactions, and that the positively charged N-terminal had a considerable contribution on adsorption. Therefore, the effect of distance between the C- and N-terminal of peptides on adsorption was examined by comparing the amino acid G, dipeptide GG and tripeptide GGG at pH 7.0. Based on previous findings, we assumed all substrates preferentially coordinate the Zr6 clusters via the carboxylate group.51 For all MOFs, adsorption of G was the lowest among the three substrates (Fig. 4). On the other hand, upon increasing the distance of NH3+ with respect to the coordinating COO− group in GGG, adsorption onto NU-1000 increased (Fig. 4), while similar adsorptions were observed for GG and GGG onto UiO-66 and MOF-808. Together these results indicate that the distance between N- and C-terminal groups may have a significant influence on the adsorption, and that this effect is different for hydrophobic versus hydrophilic MOFs.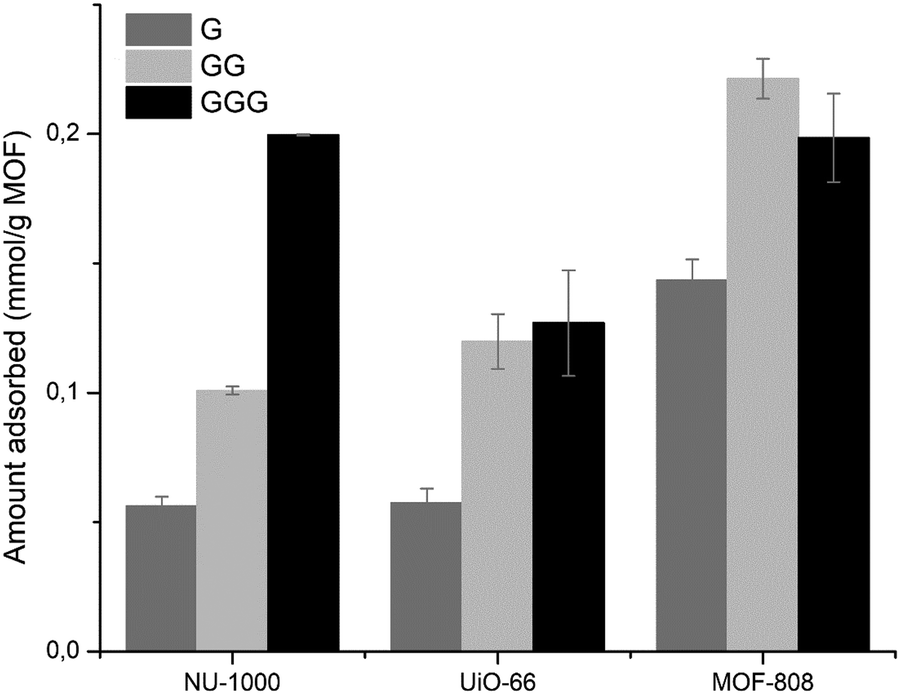 | ||
| Fig. 4 Effect of distance between C- and N-terminal on adsorption of peptides onto Zr-MOFs. | ||
As the Zr-MOFs in the series can readily hydrolyze the dipeptide GG to G,36–38 even more so at higher temperatures, the adsorption of GG was studied at different temperatures. Increasing the temperature results in faster reaction rates of GG hydrolysis by Zr-MOFs and thus more hydrolysis product G, which adsorbs comparatively less than the initial GG substrate (Fig. 4). Consequently, upon increasing the temperature from 25 to 60 °C, a lower total adsorption was observed (Fig. S6, ESI†). Remarkably, at 60 °C the adsorption of UiO-66 was lower compared to NU-1000, while the reversed trend was observed at 25 and 40 °C. This scenario matched the greater hydrolysis observed for UiO-66 at 60 °C in this work, since the decrease in adsorption from GG to G was bigger for UiO-66 compared to NU-1000.
Effect of dipeptides’ side chain on adsorption
To study the effect of AA's side chain in the adsorption of peptides, we have carried out an elaborate study using NU-1000 as a representative MOF structure, and several Gly–X dipeptides as probing substrates (Fig. 5, 6, and Table S5, ESI†) at room temperature and pH 7.0. Then, we used a smaller set of dipeptides to probe the behavior of MOF-808 and UiO-66 (Fig. 7 and Table S5, ESI†). In general, the type of side chain has a major effect on the amount of adsorption, resulting in different adsorption profiles, and trends depending on the functional groups present in the substrates.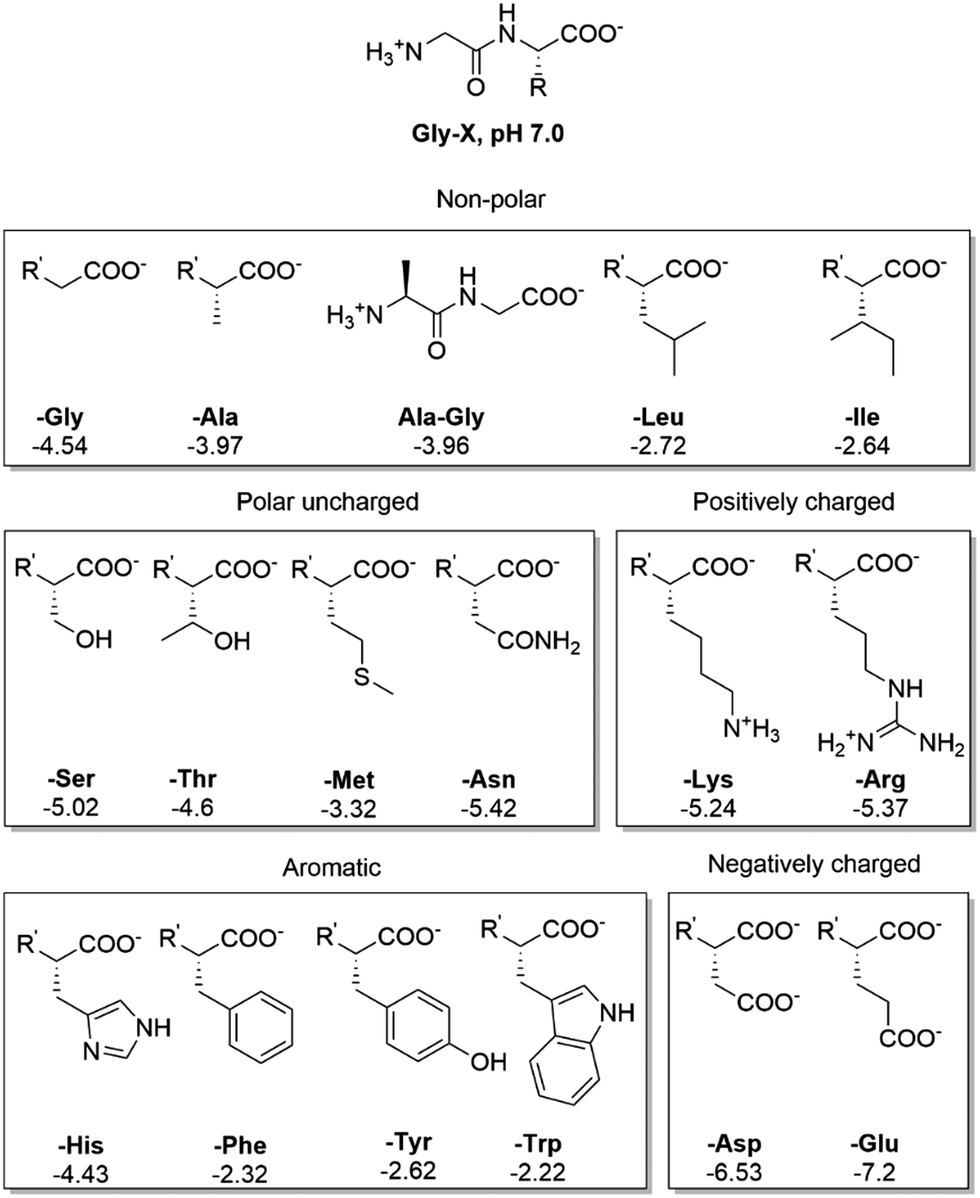 | ||
Fig. 5 Structure of side chains of Gly–X dipeptides and their logD values (more negative logD values correspond to a higher hydrophilicity). R′ = H3N+ –CH2–CONH– . . | ||
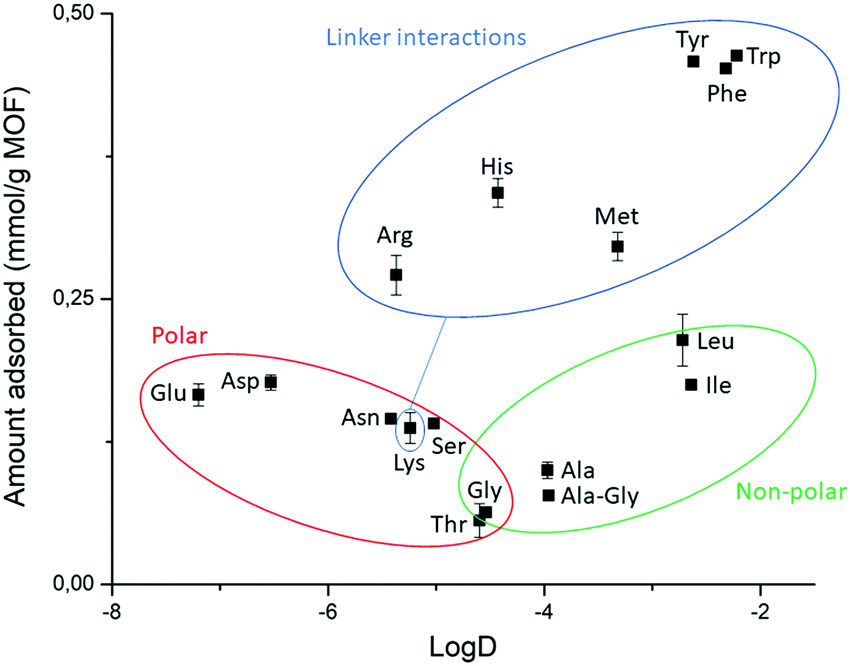 | ||
| Fig. 6 Effect of dipeptide's side chain nature on adsorption shows that different types of interactions govern adsorption onto NU-1000. Polar dipeptides (red), non-polar dipeptides (green), dipeptides involved in interactions with linker (blue). Dipeptide GG belongs to the non-polar group (green). A more negative logD value corresponds to a higher hydrophilicity. | ||
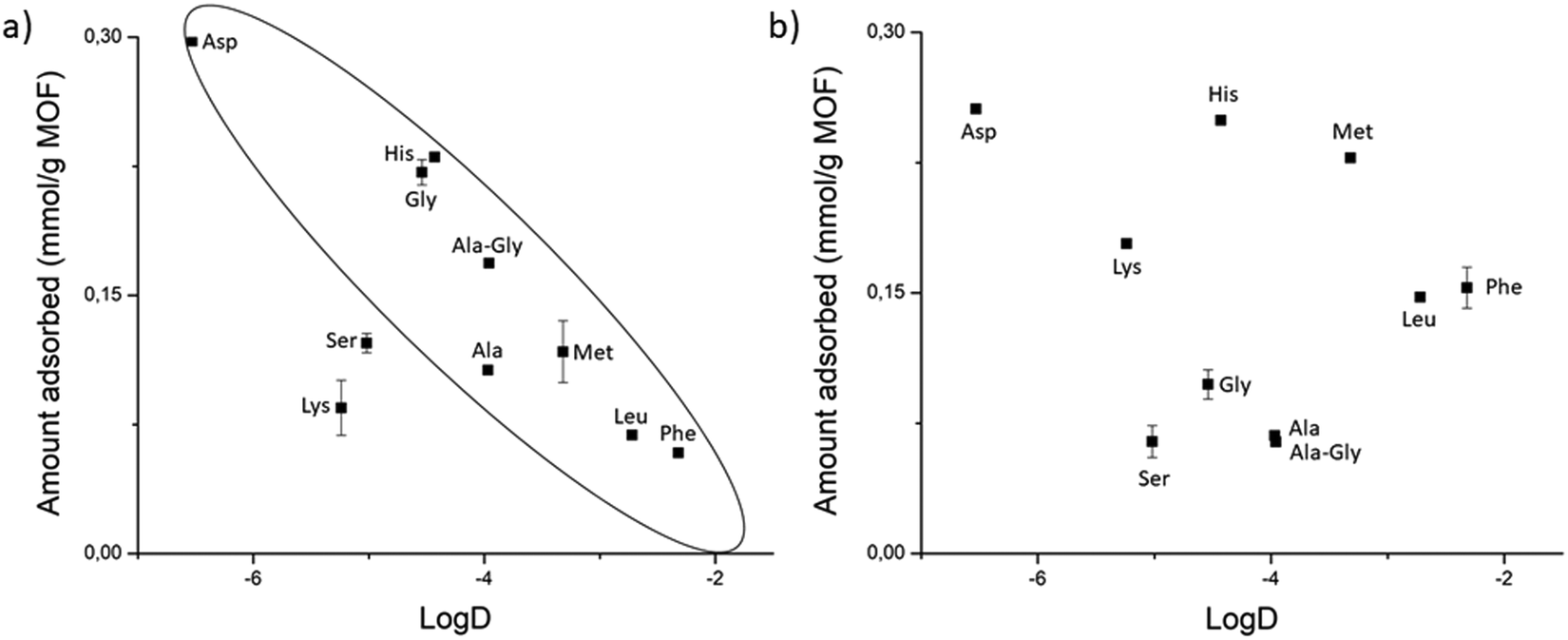 | ||
| Fig. 7 Effect of the type of side chain X in Gly–X or Ala–Gly on the amount of adsorption onto (a) MOF-808, and (b) UiO-66. A more negative logD value corresponds to a higher hydrophilicity. | ||
In general, the same hydrophobicity-dependent trend was observed for the set of aromatic dipeptides, i.e., adsorption increases with increasing hydrophobicity of the side chain (Fig. 6 blue group). The presence of aromatic groups in Gly–X peptide (X = His, Phe, Tyr and Trp) resulted in the highest adsorption in the dipeptide series; nearly 100% for X = Phe, Tyr and Trp (0.463 mmol g−1 NU-1000, Table S5, ESI†), while the more hydrophilic Gly–His was the least adsorbed. The sulfur containing Gly–Met dipeptide adsorption was higher than expected from its logD value. Based on the initial results with GG, the positively charged side chain in Gly–Lys and Gly–Arg should decrease the adsorption relatively to GG. However, a higher than expected adsorption was observed for these dipeptides, with an apparent greater beneficial effect of the guanidinium group of Arg in comparison to the Lys side chain's ammonium group.54 These results suggest that, besides hydrophobicity, peptide-linker interactions might be playing a role, given that different types of interactions with aromatic systems like the TBAPy linkers of NU-1000 are known for dipeptides.54–58
Dipeptides bearing side chains with functional groups that can coordinate to the Zr6 cluster37,59,60 such as carboxylates, amides, and hydroxyl groups, were generally more adsorbed than GG, with an increase in hydrophilicity (lower logD) leading to an increase in adsorption onto NU-1000. This trend might be related to the potential coordination of side chain functional groups to the Zr6 cluster, with a relative strength of coordination of carboxylate > amide > hydroxyl. Gly–Ser featuring a hydroxyl side chain was the least adsorbed, and additional steric hindrance at this side chain further decreased adsorption, as seen for Gly–Thr. Gly–Asp and Gly–Glu had the highest adsorption in the polar series, hinting again to the key role of carboxylate group in the adsorption, as observed above for the dipeptide C-terminal nature. Interestingly, adsorption decreased when glycine was moved to the N-terminal position of the dipeptide (X–Gly peptides), as 22% of Gly–Ala (0.100 mmol g−1 NU-1000) was adsorbed onto NU-1000 compared to 17% of Ala–Gly (0.078 mmol g−1 NU-1000) (Table S5 entries 2–3, ESI†).
D values. These outlying dipeptides showed adsorption levels roughly comparable to non-polar substrates like Gly–Ala and Gly–Met, indicating that productive dipeptide-linker interactions, which were presumably responsible for their adsorption levels with NU-1000, are not as effective in MOF-808.
Discussion
Relevance of C- and N-terminal groups to the adsorption of dipeptides onto Zr-MOFs
Carboxylate group coordination to Zr(IV) sites is of great importance to the adsorption of dipeptides, as predicted from previous DFT calculations showing that the strongest mode of interaction between GG and the Zr6 cluster is via a bidentate binding of the terminal carboxylate group.51 In our results, MOFs with more available Zr(IV) sites for the coordination of the carboxylate group resulted in greater adsorption of dipeptides. MOF-808, which has the lowest connectivity, and thus the largest number of available coordination sites on Zr(IV), showed the highest amount of adsorption in the series (Fig. 2b). This MOF was also the most affected by preventing interactions through esterification of the carboxylate group (Fig. 3), in striking agreement with the lower peptide bond hydrolysis efficiency upon blocking GG at the C-terminal reported previously for MOF-808.37 Further, removing the steric hindrance nearby the carboxylate by moving the glycine to the N-terminal (Gly–Ala vs. Ala–Gly, Fig. 7a) increased the adsorption for MOF-808. Conversely, MOF-808 is the least affected by blocking the N-terminal at low pH (Fig. 3a), and increasing the distance of N-terminal with respect to the COO− group from G to GGG is of limited benefit for this MOF (Fig. 4), supporting that carboxylate binding is the strongest and probably the most prevalent binding interaction when Zr(IV) sites are accessible.In contrast, several results indicated that electrostatic repulsion of the N-terminal ammonium group counterbalances carboxylate coordination, and may play a key role in dipeptide adsorption, even though carboxylate coordination likely remains the strongest binding interaction. This explains the increase of adsorption upon increasing the pH from 3.0 to 7.0 (Fig. 2b). At low pH, a combined effect of the fully protonated form of GG, the intrinsic positive charge of Zr(IV) centra and the overall positive charge of MOF surface entailed from its protonated oxygens (see discussion below) disfavors GG adsorption (Fig. 2) likely due to the electrostatic repulsion between MOF and dipeptide. Consistent with this interplay between carboxylate coordination and electrostatic repulsion, removal of the electrostatic repulsion between the overall positively charged MOF surface and NH3+ group by acetylation of the N-terminal greatly increases adsorption at low pH (Fig. 3a). Following this trend, counterbalancing repulsion forces diminish at higher pH values (mostly because of the MOF structure, see discussion below), and facilitate coordination of GG's carboxylate group to the Zr(IV), thereby increasing the adsorption.51
The contribution of carboxylate coordination and N-terminal electrostatic repulsion in the adsorption of dipeptides correlates with MOF structure, pH of solution and substrate structure. For example, the unexpected lower adsorption of N-acetylGG in comparison with GG at pH 7.0 suggests that carboxylate coordination is more relevant for more hydrophilic MOF-808 and UiO-66 (Fig. 3b). In this pH, only hydroxyl groups are bound to the Zr(IV) centers, which are more difficult to be replaced by an incoming ligand (i.e., a poor leaving group) (entry 3, Table S3 see discussion on section ‘Main aspects of MOF structure affecting adsorption of glycylglycine’, ESI†). Thus, ligand exchange at pH 7.0 is less favored, which decreases adsorption onto MOF-808 and UiO-66 despite absence of electrostatic repulsions. In this sense, the divergent trend observed for the adsorption of N-acetylGG at pH 3.0, in which easy to be displaced aqua ligands are bound to the Zr6 cluster (entry 1, Table S3, ESI†), and at pH 7.0, in which Zr–OH moieties are prevalent, suggests that unfavorable repulsion effects play a bigger role at pH 3.0, counterbalancing more efficiently the beneficial presence of easier to displace aqua ligands. The slower ligand exchange at pH 7.0 might be related to the absence of a NH3+ acidic hydrogen, underlining the relevance of the Zr–OH group protonation event for the ligand exchange. However, more results are needed to confirm this hypothesis.
In contrast to the relevance of carboxylate coordination for MOF-808 and UiO-66, the more hydrophobic NU-1000 MOF is largely affected by modulation of electrostatic repulsion. Acetylation of the N-terminal group of GG, which eliminates electrostatic repulsion of the N-terminal ammonium group, caused a great increase in adsorption onto NU-1000 both at pH 3.0 and 7.0 (N-acetylGG vs. GG in Fig. 3). This trend is also in agreement with higher adsorption observed upon increasing the distance between C- and N-terminal groups from G to GGG (Fig. 4), as repulsive coulombic forces are inversely related to the distance between the charges. Interestingly, similar interplay between C- and N-terminal groups’ distance was observed in the adsorption of G. This amino acid was the least adsorbed compared to other glycine-only peptides for all MOFs (Fig. 4), pointing out that the substrate structure also has an effect in the balance between carboxylate coordination and electrostatic repulsion, and that the latter sharply increases by bringing the ammonium group closer to the cluster.
The lower adsorption of G compared to GG also allowed us to infer that temperature plays only a minor role in the adsorption of GG as its effect in the hydrolytic activity, resulting in formation of G, seems to supersede those in the adsorption itself.
Main aspects of MOF structure affecting adsorption of glycylglycine
The type of MOF clearly had a strong influence on the amount of GG adsorbed (Fig. 2b and 3), and some clear trends could be observed in this work depending on the MOF characteristics. Interestingly, no major relationship between adsorption and MOF materials’ porosity or particle size was observed, even though we cannot rule out an influence of these structural variables. If diffusion of dipeptides inside the material were limiting the overall adsorption process, smaller substrates should have been the ones mostly adsorbed. However, several results evidenced an opposite trend, such as: (1) the lower adsorption of G compared to GG for all MOFs; (2) adsorption of dipeptides with different side chains is largely controlled by the side chain nature, e.g., the greater adsorption of Gly–Asp to MOF-808 and UiO-66 compared to NU-1000, even though NU-1000's pores are the biggest among the MOFs probed.36 Likewise, the smaller particle size of UiO-66 compared to MOF-808 did not impart a higher GG adsorption onto UiO-66. On the contrary, MOF-808 adsorbed 2–3 times more GG than UiO-66 depending on the pH. Therefore, the primary factors influencing adsorption of dipeptides are likely not related to the architecture Zr-MOFs tested.The availability of Zr(IV) sites for the coordination of carboxylate largely influenced the adsorption observed. MOF-808 has the most available Zr(IV) sites due to its low connectivity (6-connected), which combined with the more hydrophilic linker (Table 1), makes this MOF the most hydrophilic in the series. These features resulted in the highest adsorption of the hydrophilic GG dipeptide in the MOF series (Fig. 2b and 3). On the other hand, NU-1000's 8-connected structure has intrinsically less available Zr(IV) sites, which together with its highly hydrophobic linker, provides the MOF with the most hydrophobic surface in the series, and resulted in the lowest adsorption of GG among the MOFs probed. Further, halfway between MOF-808 and NU-1000 MOFs, UiO-66 showed an intermediate adsorption of GG. This is consistent with the overall hydrophilic nature of this MOF, presumably arising from the BDC linkers’ low logP value (more hydrophilic, Table 1) but compensated by the lower availability of hydrophilic Zr(IV) sites (in our case, a 9-connected structure).35,50 UiO-66's lower availability of Zr(IV) explains the lower GG adsorption compared to MOF-808, though both MOFs have similar hydrophilic nature. Conversely, UiO-66's higher hydrophilicity prompts a higher adsorption of hydrophilic GG compared to NU-1000, despite the similar connectivity of both structures in this work (9 versus 8, respectively, Table 1). These results suggest that the hydrophilic/hydrophobic character of the MOF plays a key role in the adsorption of peptides.61 Moreover, the highest adsorption by MOF-808 underline the preponderance of available Zr(IV) sites in adsorption, which is line with the prominent role of carboxylate coordination discussed above. The role of available metal sites is also coherent with previous reports showing that more available coordination sites in MOFs resulted in higher adsorption of substrates,62–65 and in an enhanced catalytic activity.66–69
Acidity of the substrate solution modulates the MOF surface charge, and subsequently impacts dipeptide adsorption given that the N-terminal group remains largely protonated until pH 8.0. Besides influencing the protonation states of GG (Fig. 2a), the pH also affects the MOF surface charge since Zr(IV) centers are, in addition to structural linkers, also connected to several oxygen based ligands in the Zr6 node. Three types of oxygen ligands may be present, and they exist in different protonation states: μ3-OH, –OH2 and –OH depending on pH (Fig. 8),70 influencing also an isoelectric point (Table 1). At pH 3.0, all oxygen ligands are protonated (Table S3, ESI†), and considering the iso-electric points range between 4 to 5.5 (Table 1), all MOFs bear an overall positive charge in this case. With increasing pH, the overall MOF surface charge becomes neutral (pH ≈ pI) and eventually negative (pH > pI). In this scenario, the μ3-OH group is initially deprotonated to μ3-O−, and a further increase of pH causes the –OH2 group to lose one proton. These variations on MOF surface charge correlate well with the observed effect of pH on adsorption (Fig. 2b). The positively charged MOF surface at low pH enhances the electrostatic repulsion with the NH3+ group of GG (Fig. 2a), and likely contributes to the lower adsorption in more acidic solutions. The repulsion forces diminish at higher pH as the MOF surface is progressively deprotonated, thus contributing to a higher adsorption. This hypothesis is further supported by the higher adsorption of N-acetylGG compared to GG at pH 3.0 for all MOFs, as N-acetylation of GG prevents protonation of the NH2 group at lower pH values (see discussion above and Fig. 3).
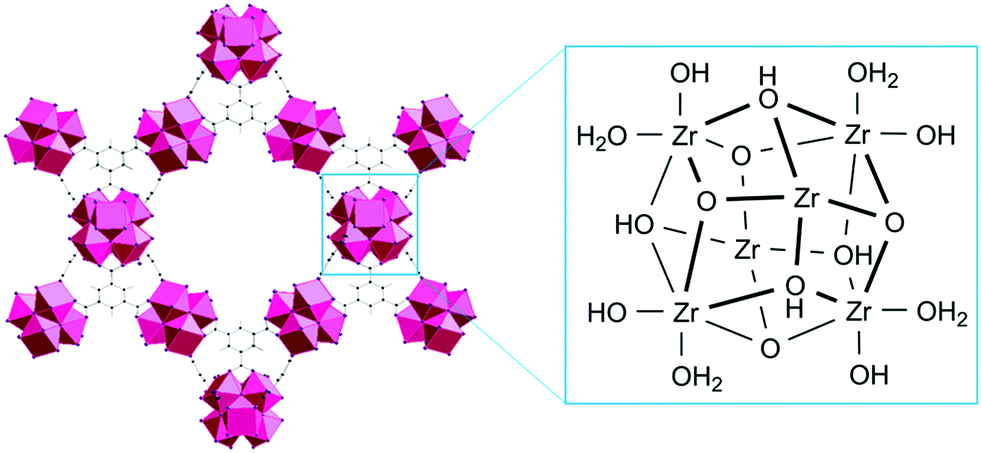 | ||
| Fig. 8 Illustrative depiction of the Zr6 node as secondary building unit of Zr-MOF with μ3-O, μ3-OH, –OH2 and –OH ligands. One –OH and one –OH2 group are replaced upon coordination of a carboxylate linker to Zr. Linkers are omitted for clarity. | ||
Together, the correlation of adsorption trends with the MOF structure underline the potential of these materials to be designed into selective adsorbents for biomolecules like peptides and proteins since the cluster connectivity, hydrophilic/-phobic and surface charge (isoelectric point) are all properties intrinsically linked to the MOF structure, and can be tuned by a proper combination of linkers and metal nodes.
Effect of dipeptides’ side chain on adsorption
Introduction of side chains to the dipeptide substrates revealed a new set of binding interactions that play a role in the adsorption of dipeptides, apart from those observed for the C- and N-terminal groups of GG discussed above. More specifically, hydrophobic interactions, potential cation–π or π–π interactions involving the aromatic linkers, and coordination of side chain's functional groups to the cluster could be inferred from the trends observed in the adsorption of various dipeptides to the three MOFs investigated. In some cases, the new interactions are effective in increasing the adsorption related to GG, which was generally attributed to factors discussed earlier (hydrophilicity of MOF/substrate, electrostatic repulsion,…).Hydrophobic interactions play a key role in the adsorption of hydrophobic dipeptides onto a hydrophobic MOF surface, as clearly observed by the affinity of non-polar dipeptides towards the hydrophobic MOF NU-1000 (Fig. 6). Likewise, the aromaticity of the linkers provides the MOF with a π-system able to interact through π–π stacking with aromatic side chains of Gly–His, –Phe, –Tyr and –Trp dipeptides, which were nearly fully adsorbed onto NU-1000 (Fig. 6). Interactions with the linker's π-system are likely also present for Gly–Met as methionine-aromatics interactions play an important role as stabilizer of protein structures.56,57 Similarly, cation–π interactions might arise between Gly–Lys/Arg and NU-1000's linker.54 Finally, the equivalent adsorption of nonpolar hydrophobic dipeptides (Gly–Leu/Ile) and polar carboxylate bearing dipeptides (Gly–Asp/Glu) onto NU-1000 shows that the hydrophobic interactions may enhance adsorption as efficiently as strong carboxylate–Zr coordination in hydrophobic MOFs, underlining the importance of hydrophobic interactions for the trends observed with NU-1000.
The high affinity of Gly–Asp for MOF-808 and UiO-66, and the higher adsorption of Gly–X peptides (X = Asp, Asn, Glu, Ser, and His) compared to GG onto NU-1000 evidence that functional groups with affinity for Zr(IV) like carboxylate, amide and hydroxyl groups can enhance adsorption by coordinating to the Zr6 cluster. This trend is obviously more relevant for MOFs with more available metal sites like MOF-808; however, adsorption of various peptides onto MOF-808 also correlated well with their hydrophilicity (Fig. 7a), suggesting that both properties are related when it comes to influence the adsorption.
All these different types of interaction that are linked to the type of side chain seem to be playing a role in the adsorption onto UiO-66. However, adsorption to this MOF changes almost on a case-by-case basis, which may be related to the low availability of Zr(IV) sites and the less pronounced hydrophilic/hydrophobic character of UiO-66. Similarly, the outlying adsorption behavior of some dipeptides cannot be entirely rationalized by the interactions discussed above. For example, Gly–Lys adsorption to MOF-808 is lower than expected from its hydrophilicity. This might have been caused by weak cation–π interactions with the poor π system of the BTC linker, and/or an electrostatic repulsion of the positively charged side chain with the positive character of available Zr(IV) coordination sites. Nevertheless, for all MOFs, Gly–Lys and Gly–Ser are not in line with the trends expected or related to any other substrates, which showcases many factors are at play in cases where several types of interactions are possible.
MOF characteristics vs. dipeptide's side chain
A comparative analysis cross-examining the substrate nature and MOF characteristics confirms some of the findings discussed above, and highlights the potential of MOFs to be developed into selective adsorbents or catalysts for peptides, and eventually proteins (Fig. 9). For example, NU-1000 has the superior ability to adsorb dipeptides able to engage in hydrophobic and linker interactions compared to MOF-808 and UiO-66. Similarly, MOF-808 has the highest affinity towards dipeptides that can coordinate to the Zr6 cluster due to its several available metal sites. Nevertheless, a more interesting aspect of this comparison is the distinct intrinsic selectivity of these MOFs towards different dipeptides. For example, NU-1000 shows great affinity for Gly–Phe, Gly–Leu, Gly–Met, and Gly–His, while UiO-66 favors the adsorption of Gly–Asp, Gly–His, Gly–Met, and the highest affinity for Gly–Lys among all MOFs. However, both show low affinity for Gly–Gly, Gly–Ala, Ala–Gly, and Gly–Ser. In turn, MOF-808's high adsorption for Gly–Asp, Gly–Gly, and Gly–His contrasts its low affinity for Gly–Leu and Gly–Phe. These trends arguably arise from the several factors and interaction types outlined in this work that may influence adsorption, and illustrate how properties like cluster connectivity and hydrophobicity of MOF surface can be tuned to selectively favor the adsorption of dipeptides with affinity for Zr(IV) or those of hydrophobic nature, respectively, thereby paving a direct link between structure and function.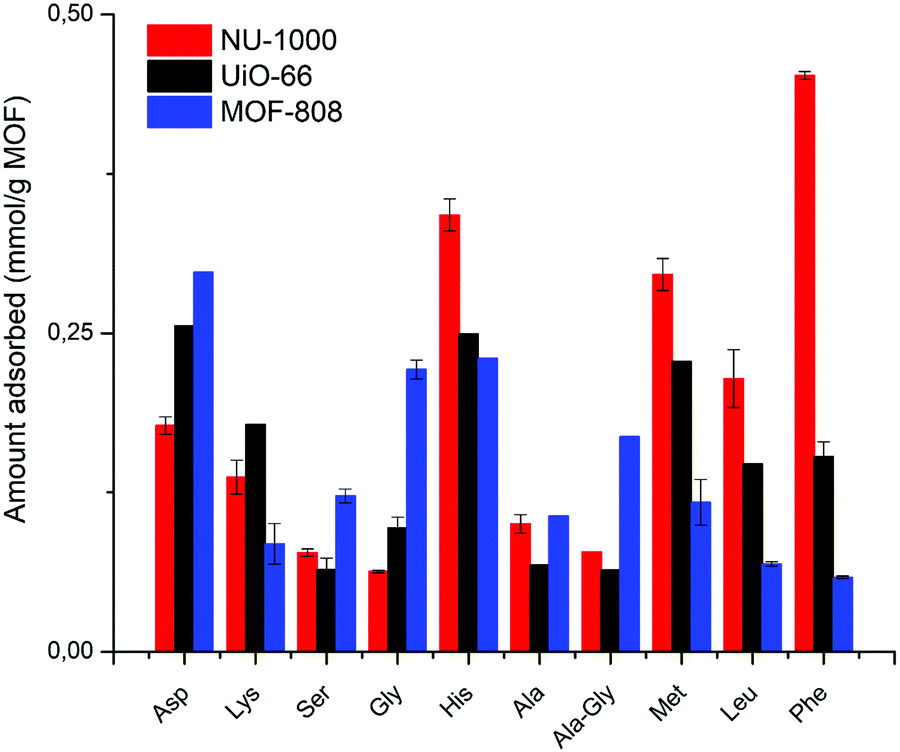 | ||
| Fig. 9 Comparison of the effect of different Zr-MOFs on adsorption of several dipeptides. X-Axis: increasing hydrophobicity (less negative logD) from left to right. Results reported without an error bar refer to a single measurement. | ||
Conclusion
In summary, through a systematic adsorption study using several dipeptides and three distinct Zr-MOFs, we determined which types of binding interaction and experimental factors influence the affinity of peptides towards Zr-MOFs. Our results show that MOF materials’ porosity or particle size are not the major factor influencing adsorption, but that adsorption is largely favored by C-terminal carboxylate coordination to available Zr(IV) sites, and disfavored by electrostatic repulsion of the N-terminal ammonium group. The governing balance between these two forces depends on the interplay between solution conditions (pH), MOF structure, and substrate nature. Moreover, a detailed screening with a range of dipeptides bearing diverse side chains revealed that a number of additional interactions (hydrophobic, aromatic, cation–π, sulfur–π, coordination), mostly arising from the hydrophobic/aromatic nature of the linkers and cluster connectivity, may be used to modulate the affinity of the MOF material towards a specific type of dipeptide. These findings arguably offer unique guidelines for future studies aiming at designing and controlling the adsorption of biomolecules onto MOF materials, as well as at the development of MOF based heterogeneous nanozymes towards peptides and proteins.Author contributions
All experiments were performed by A. L. The manuscript was written by A. L., F. d. A. and T. P. V. The project was supervised by T. P. V.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank K. U. Leuven and Research Foundation Flanders (FWO) for financial support. A. L. (48730/1S10318N) and F. d. A. (195931/1281921N) thank the FWO Flanders (Belgium) for fellowships. We thank Charlotte Simms, and Vincent Lemmens for the nitrogen physisorption measurements, and Dr Simon Smolders for helpful discussions.Notes and references
- R. B. Merrifield, Solid phase peptide synthesis. I. The synthesis of a tetrapeptide, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- K. G. Varnava and V. Sarojini, Making solid-phase peptide synthesis greener: a review of the literature, Chem. – Asian J., 2019, 14, 1088–1097 CrossRef CAS PubMed.
- K. Nakanishi, T. Sakiyama and K. Imamura, On the adsorption of proteins on solid surfaces, a common but very complicated phenomenon, J. Biosci. Bioeng., 2001, 91, 233–244 CrossRef CAS.
- R. Thull, Surface processes in artificial organs. An overview, Med. Prog. Technol., 1982, 9, 119–128 CAS.
- S. Adachi, T. Yamanaka and R. Matsuno, Adsorption of dipeptides on activated carbon, Food Sci. Technol. Int., Tokyo, 1998, 4, 269–273 CAS.
- J. E. Krohn and M. Tsapatsis, Amino acid adsorption on zeolite β, Langmuir, 2005, 21, 8743–8750 CrossRef CAS PubMed.
- S. Munsch, M. Hartmann and S. Ernst, Adsorption and separation of amino acids from aqueous solutions on zeolites, Chem. Commun., 2001, 1978–1979 RSC.
- M. Meng, L. Stievano and J.-F. Lambert, Adsorption and thermal condensation mechanisms of amino acids on oxide supports. 1. Glycine on silica, Langmuir, 2004, 20, 914–923 CrossRef CAS PubMed.
- Q. Gao, W. Xu, Y. Xu, D. Wu, Y. Sun, F. Deng and W. Shen, Amino acid adsorption on mesoporous materials: influence of types of amino acids, modification of mesoporous materials, and solution conditions, J. Phys. Chem. B, 2008, 112, 2261–2267 CrossRef CAS PubMed.
- A. J. O’Connor, A. Hokura, J. M. Kisler, S. Shimadzu, G. W. Stevens and Y. Komatsu, Amino acid adsorption onto mesoporous silica molecular sieves, Sep. Purif. Technol., 2006, 48, 197–201 CrossRef.
- D. Costa, L. Savio and C.-M. Pradier, Adsorption of amino acids and peptides on metal and oxide surfaces in water environment: a synthetic and prospective review, J. Phys. Chem. B, 2016, 120, 7039–7052 CrossRef CAS PubMed.
- J. Goscianska, A. Olejnik and R. Pietrzak, Adsorption of L-phenylalanine onto mesoporous silica, Mater. Chem. Phys., 2013, 142, 586–593 CrossRef CAS.
- D. Palit and S. P. Moulik, Adsorption behaviors of L-histidine and DL-tryptophan on cholesterol, silica, alumina, and graphite, J. Colloid Interface Sci., 2001, 239, 20–26 CrossRef CAS PubMed.
- V. Hiremath, A. H. Jadhav, H. Lee, S. Kwon and J. G. Seo, Highly reversible CO2 capture using amino acid functionalized ionic liquids immobilized on mesoporous silica, Chem. Eng. J., 2016, 287, 602–617 CrossRef CAS.
- J. Canivet, A. Fateeva, Y. Guo, B. Coasne and D. Farrusseng, Water adsorption in MOFs: fundamentals and applications, Chem. Soc. Rev., 2014, 43, 5594–5617 RSC.
- X. Liu, X. Wang and F. Kapteijn, Water and metal–organic frameworks: from interaction toward utilization, Chem. Rev., 2020, 120, 8303–8377 CrossRef CAS PubMed.
- R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dincă, A. Bavykina, J. Gascon, A. Ejsmont and J. Gościańska, The Current Status of MOF and COF Applications, Angew. Chem., Int. Ed., 2021, 60, 23975–24001 CrossRef CAS PubMed.
- L. Feng, J. Pang, P. She, J. L. Li, J. S. Qin, D. Y. Du and H. C. Zhou, Metal–organic frameworks based on group 3 and 4 metals, Adv. Mater., 2020, 32, 2004414 CrossRef PubMed.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Selective gas adsorption and separation in metal–organic frameworks, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- M. K. Alsaedi, G. K. Alothman, M. N. Alnajrani, O. A. Alsager, S. A. Alshmimri, M. A. Alharbi, M. O. Alawad, S. Alhadlaq and S. Alharbi, Antibiotic Adsorption by Metal-Organic Framework (UiO-66): A Comprehensive Kinetic, Thermodynamic, and Mechanistic Study, Antibiotics, 2020, 9, 722 CrossRef CAS PubMed.
- I. Akpinar and A. O. Yazaydin, Adsorption of atrazine from water in metal–organic framework materials, J. Chem. Eng. Data, 2018, 63, 2368–2375 CrossRef CAS.
- I. Taima-Mancera, P. Rocío-Bautista, J. Pasán, J. H. Ayala, C. Ruiz-Pérez, A. M. Afonso, A. B. Lago and V. Pino, Influence of ligand functionalization of UiO-66-based metal-organic frameworks when used as sorbents in dispersive solid-phase analytical microextraction for different aqueous organic pollutants, Molecules, 2018, 23, 2869 CrossRef PubMed.
- S. Rojas and P. Horcajada, Metal–organic frameworks for the removal of emerging organic contaminants in water, Chem. Rev., 2020, 120, 8378–8415 CrossRef CAS PubMed.
- Q. Yang, J. Wang, X. Chen, W. Yang, H. Pei, N. Hu, Z. Li, Y. Suo, T. Li and J. Wang, The simultaneous detection and removal of organophosphorus pesticides by a novel Zr-MOF based smart adsorbent, J. Mater. Chem. A, 2018, 6, 2184–2192 RSC.
- P. Li, J. A. Modica, A. J. Howarth, E. Vargas, P. Z. Moghadam, R. Q. Snurr, M. Mrksich, J. T. Hupp and O. K. Farha, Toward design rules for enzyme immobilization in hierarchical mesoporous metal-organic frameworks, Chem, 2016, 1, 154–169 CAS.
- N. Ye, J. Ma, J. An, J. Li, Z. Cai and H. Zong, Separation of amino acid enantiomers by a capillary modified with a metal–organic framework, RSC Adv., 2016, 6, 41587–41593 RSC.
- L. Wang, Z. Hu, S. Wu, J. Pan, X. Xu and X. Niu, A peroxidase-mimicking Zr-based MOF colorimetric sensing array to quantify and discriminate phosphorylated proteins, Anal. Chim. Acta, 2020, 1121, 26–34 CrossRef CAS PubMed.
- Y.-W. Zhao, Y. Wang and X.-M. Zhang, Homochiral MOF as circular dichroism sensor for enantioselective recognition on nature and chirality of unmodified amino acids, ACS Appl. Mater. Interfaces, 2017, 9, 20991–20999 CrossRef CAS PubMed.
- P. Chandrasekhar, A. Mukhopadhyay, G. Savitha and J. N. Moorthy, Remarkably selective and enantiodifferentiating sensing of histidine by a fluorescent homochiral Zn-MOF based on pyrene-tetralactic acid, Chem. Sci., 2016, 7, 3085–3091 RSC.
- M. Bazargan, F. Ghaemi, A. Amiri and M. Mirzaei, Metal–organic framework-based sorbents in analytical sample preparation, Coord. Chem. Rev., 2021, 445, 214107 CrossRef CAS.
- J. R. Bour, A. M. Wright, X. He and M. Dincă, Bioinspired chemistry at MOF secondary building units, Chem. Sci., 2020, 11, 1728–1737 RSC.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Metal–Organic Frameworks in Heterogeneous Catalysis: Recent Progress, New Trends, and Future Perspectives, Chem. Rev., 2020, 120, 8468–8535 CrossRef CAS PubMed.
- K. Hemmer, M. Cokoja and R. A. Fischer, Exploitation of Intrinsic Confinement Effects of MOFs in Catalysis, ChemCatChem, 2021, 13, 1683–1691 CrossRef CAS.
- F. Chen, H. F. Drake, L. Feng, J. A. Powell, K.-Y. Wang, T.-H. Yan and H.-C. Zhou, Metal–Organic Frameworks as Versatile Platforms for Organometallic Chemistry, Inorganics, 2021, 9, 27 CrossRef CAS.
- F. de Azambuja, A. Loosen, D. Conic, M. van den Besselaar, J. N. Harvey and T. N. Parac-Vogt, En Route to a Heterogeneous Catalytic Direct Peptide Bond Formation by Zr-Based Metal–Organic Framework Catalysts, ACS Catal., 2021, 11, 7647–7658 CrossRef CAS.
- A. Loosen, F. de Azambuja, S. Smolders, J. Moons, C. Simms, D. De Vos and T. N. Parac-Vogt, Interplay between structural parameters and reactivity of Zr6-based MOFs as artificial proteases, Chem. Sci., 2020, 11, 6662–6669 RSC.
- H. G. T. Ly, G. Fu, A. Kondinski, B. Bueken, D. De Vos and T. N. Parac-Vogt, Superactivity of MOF-808 toward Peptide Bond Hydrolysis, J. Am. Chem. Soc., 2018, 140, 6325–6335 CrossRef CAS PubMed.
- H. G. T. Ly, G. Fu, F. de Azambuja, D. De Vos and T. N. Parac-Vogt, Nanozymatic Activity of UiO-66 Metal–Organic Frameworks: Tuning the Nanopore Environment Enhances Hydrolytic Activity toward Peptide Bonds, ACS Appl. Nano Mater., 2020, 3, 8931–8938 CrossRef CAS.
- F. de Azambuja, J. Moons and T. N. Parac-Vogt, The Dawn of Metal-Oxo Clusters as Artificial Proteases: From Discovery to the Present and Beyond, Acc. Chem. Res., 2021, 54, 1673–1684 CrossRef PubMed.
- P. B. Pandeswari and V. Sabareesh, Middle-down approach: a choice to sequence and characterize proteins/proteomes by mass spectrometry, RSC Adv., 2019, 9, 313–344 RSC.
- H. Zhong, Y. Li, Y. Huang and R. Zhao, Metal–organic frameworks as advanced materials for sample preparation of bioactive peptides, Anal. Methods, 2021, 13, 862–873 RSC.
- A. Loosen, C. Simms, S. Smolders, D. E. De Vos and T. N. Parac-Vogt, Bimetallic Ce/Zr UiO-66 Metal–Organic Framework Nanostructures as Peptidase and Oxidase Nanozymes, ACS Appl. Nano Mater., 2021, 4, 5748–5757 CrossRef CAS.
- Z.-J. Lin, H.-Q. Zheng, Y.-N. Zeng, Y.-L. Wang, J. Chen, G.-J. Cao, J.-F. Gu and B. Chen, Effective and selective adsorption of organoarsenic acids from water over a Zr-based metal-organic framework, Chem. Eng. J., 2019, 378, 122196 CrossRef CAS.
- M. R. Azhar, H. R. Abid, V. Periasamy, H. Sun, M. O. Tade and S. Wang, Adsorptive removal of antibiotic sulfonamide by UiO-66 and ZIF-67 for wastewater treatment, J. Colloid Interface Sci., 2017, 500, 88–95 CrossRef CAS PubMed.
- G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Defect engineering: tuning the porosity and composition of the metal–organic framework UiO-66 via modulated synthesis, Chem. Mater., 2016, 28, 3749–3761 CrossRef CAS.
- P. K. Glasoe and F. A. Long, Use of glass electrodes to measure acidities in deuterium oxide, J. Phys. Chem., 1960, 64, 188–190, DOI:10.1021/j100830a521.
- J. Rouquerol, F. Rouquerol and K. S. W. Sing, Absorption by Powders and Porous Solids, Academic Press, London, 1999.
- T. C. Wang, N. A. Vermeulen, I. S. Kim, A. B. Martinson, J. F. Stoddart, J. T. Hupp and O. K. Farha, Scalable synthesis and post-modification of a mesoporous metal-organic framework called NU-1000, Nat. Protoc., 2016, 11, 149–162 CrossRef CAS PubMed.
- H. Reinsch, S. Waitschat, S. M. Chavan, K. P. Lillerud and N. Stock, A facile “green” route for scalable batch production and continuous synthesis of zirconium MOFs, Eur. J. Inorg. Chem., 2016, 4490–4498 CrossRef CAS.
- M. J. Katz, Z. J. Brown, Y. J. Colón, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, A facile synthesis of UiO-66, UiO-67 and their derivatives, Chem. Commun., 2013, 49, 9449–9451 RSC.
- D. Conic, K. Pierloot, T. Parac-Vogt and J. Harvey, Mechanism of the Highly Effective Peptide Bond Hydrolysis by MOF-808 Catalyst Under Biologically Relevant Conditions, Phys. Chem. Chem. Phys., 2020, 22, 25136–25145 RSC.
- A. P. Brogan, T. J. Dickerson and K. D. Janda, Enamine-Based Aldol Organocatalysis in Water: Are They Really “All Wet”?, Angew. Chem., 2006, 118, 8278–8280 CrossRef.
- N. Mase, Y. Nakai, N. Ohara, H. Yoda, K. Takabe, F. Tanaka and C. F. Barbas, Organocatalytic direct asymmetric aldol reactions in water, J. Am. Chem. Soc., 2006, 128, 734–735 CrossRef CAS PubMed.
- K. Kumar, S. M. Woo, T. Siu, W. A. Cortopassi, F. Duarte and R. S. Paton, Cation–π interactions in protein–ligand binding: Theory and data-mining reveal different roles for lysine and arginine, Chem. Sci., 2018, 9, 2655–2665 RSC.
- R. Loewenthal, J. Sancho and A. R. Fersht, Histidine-aromatic interactions in barnase: Elevation of histidine pKa and contribution to protein stability, J. Mol. Biol., 1992, 224, 759–770 CrossRef CAS.
- D. S. Weber and J. J. Warren, The interaction between methionine and two aromatic amino acids is an abundant and multifunctional motif in proteins, Arch. Biochem. Biophys., 2019, 672, 108053 CrossRef CAS PubMed.
- C. C. Valley, A. Cembran, J. D. Perlmutter, A. K. Lewis, N. P. Labello, J. Gao and J. N. Sachs, The methionine-aromatic motif plays a unique role in stabilizing protein structure, J. Biol. Chem., 2012, 287, 34979–34991 Search PubMed.
- G. B. McGaughey, M. Gagné and A. K. Rappé, π-stacking interactions: alive and well in proteins, J. Biol. Chem., 1998, 273, 15458–15463 CrossRef CAS PubMed.
- H.-H. Mautschke, F. Drache, I. Senkovska, S. Kaskel and F. L. I. Xamena, Catalytic properties of pristine and defect-engineered Zr-MOF-808 metal organic frameworks, Catal. Sci. Technol., 2018, 8, 3610–3616 RSC.
- B. Villoria-del-Álamo, S. Rojas-Buzo, P. García García and A. Corma, Zr-MOF-808 as Catalyst for Amide Esterification, Chem. – Eur. J., 2020, 27, 4588–4598 CrossRef PubMed.
- Q. Zha, X. Sang, D. Liu, D. Wang, G. Shi and C. Ni, Modification of hydrophilic amine-functionalized metal-organic frameworks to hydrophobic for dye adsorption, J. Solid State Chem., 2019, 275, 23–29 CrossRef CAS.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, Unusual and highly tunable missing-linker defects in zirconium metal–organic framework UiO-66 and their important effects on gas adsorption, J. Am. Chem. Soc., 2013, 135, 10525–10532 CrossRef CAS PubMed.
- X. Zhang, Y. Yang, L. Song, J. Chen, Y. Yang and Y. Wang, Enhanced adsorption performance of gaseous toluene on defective UiO-66 metal organic framework: equilibrium and kinetic studies, J. Hazard. Mater., 2019, 365, 597–605 CrossRef CAS PubMed.
- C. A. Clark, K. N. Heck, C. D. Powell and M. S. Wong, Highly defective UiO-66 materials for the adsorptive removal of perfluorooctanesulfonate, ACS Sustainable Chem. Eng., 2019, 7, 6619–6628 CrossRef CAS.
- P. Ghosh, Y. J. Colón and R. Q. Snurr, Water adsorption in UiO-66: the importance of defects, Chem. Commun., 2014, 50, 11329–11331 RSC.
- K. Epp, A. L. Semrau, M. Cokoja and R. A. Fischer, Dual Site Lewis-Acid Metal-Organic Framework Catalysts for CO2 Fixation: Counteracting Effects of Node Connectivity, Defects and Linker Metalation, ChemCatChem, 2018, 10, 3506–3512 CrossRef CAS.
- F. Vermoortele, B. Bueken, G. L. Le Bars, B. Van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier and V. Van Speybroeck, Synthesis modulation as a tool to increase the catalytic activity of metal–organic frameworks: the unique case of UiO-66 (Zr), J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
- Y. Liu, R. C. Klet, J. T. Hupp and O. Farha, Probing the correlations between the defects in metal–organic frameworks and their catalytic activity by an epoxide ring-opening reaction, Chem. Commun., 2016, 52, 7806–7809 RSC.
- J. Jin, Porphyrin-based metal–organic framework catalysts for photoreduction of CO2: understanding the effect of node connectivity and linker metalation on activity, New J. Chem., 2020, 44, 15362–15368 RSC.
- R. C. Klet, Y. Liu, T. C. Wang, J. T. Hupp and O. K. Farha, Evaluation of Brønsted acidity and proton topology in Zr-and Hf-based metal–organic frameworks using potentiometric acid–base titration, J. Mater. Chem. A, 2016, 4, 1479–1485 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary experiments, key literature data and MOF characterization before/after experiments. See DOI: 10.1039/d1ma01027a |
| This journal is © The Royal Society of Chemistry 2022 |