
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceClimate change influence on the levels and trends of persistent organic pollutants (POPs) and chemicals of emerging Arctic concern (CEACs) in the Arctic physical environment – a review†
Hayley
Hung
 *a,
Crispin
Halsall
b,
Hollie
Ball
b,
Terry
Bidleman
c,
Jordi
Dachs
d,
Amila
De Silva
e,
Mark
Hermanson
f,
Roland
Kallenborn
gh,
Derek
Muir
e,
Roxana
Sühring
ij,
Xiaoping
Wang
k and
Simon
Wilson
l
*a,
Crispin
Halsall
b,
Hollie
Ball
b,
Terry
Bidleman
c,
Jordi
Dachs
d,
Amila
De Silva
e,
Mark
Hermanson
f,
Roland
Kallenborn
gh,
Derek
Muir
e,
Roxana
Sühring
ij,
Xiaoping
Wang
k and
Simon
Wilson
l
aAir Quality Processes Research Section, Environment and Climate Change Canada, 4905 Dufferin Street, Toronto, Ontario M5P 1W4, Canada. E-mail: hayley.hung@ec.gc.ca
bLancaster Environment Centre, Lancaster University, Lancaster LA1 4YQ, UK
cDepartment of Chemistry, Umeå University, Umeå, SE-901 87, Sweden
dInstitute of Environmental Assessment and Water Research, Spanish National Research Council (IDAEA-CSIC), Barcelona, Catalonia 08034, Spain
eAquatic Contaminants Research Division, Environment and Climate Change Canada, Burlington, Ontario L7S 1A1, Canada
fHermanson & Associates LLC, 2000 W 53rd Street, Minneapolis, Minnesota 55419, USA
gDepartment of Arctic Technology, University Centre in Svalbard (UNIS), Longyearbyen, 9171, Norway
hFaculty of Chemistry, Biotechnology and Food Sciences, Norwegian University of Life Sciences (NMBU), Ås, 1432, Norway
iDepartment for Environmental Science, Stockholm University, 114 19 Stockholm, Sweden
jDepartment of Chemistry and Biology, Ryerson University, Toronto, Ontario M5B 2K3, Canada
kKey Laboratory of Tibetan Environment Changes and Land Surface Processes, Institute of Tibetan Plateau Research, Chinese Academy of Sciences, Beijing 100101, China
lArctic Monitoring and Assessment Programme Secretariat, The Fram Centre, 9296 Tromsø, Norway
First published on 25th February 2022
Abstract
Climate change brings about significant changes in the physical environment in the Arctic. Increasing temperatures, sea ice retreat, slumping permafrost, changing sea ice regimes, glacial loss and changes in precipitation patterns can all affect how contaminants distribute within the Arctic environment and subsequently impact the Arctic ecosystems. In this review, we summarized observed evidence of the influence of climate change on contaminant circulation and transport among various Arctic environment media, including air, ice, snow, permafrost, fresh water and the marine environment. We have also drawn on parallel examples observed in Antarctica and the Tibetan Plateau, to broaden the discussion on how climate change may influence contaminant fate in similar cold-climate ecosystems. Significant knowledge gaps on indirect effects of climate change on contaminants in the Arctic environment, including those of extreme weather events, increase in forests fires, and enhanced human activities leading to new local contaminant emissions, have been identified. Enhanced mobilization of contaminants to marine and freshwater ecosystems has been observed as a result of climate change, but better linkages need to be made between these observed effects with subsequent exposure and accumulation of contaminants in biota. Emerging issues include those of Arctic contamination by microplastics and higher molecular weight halogenated natural products (hHNPs) and the implications of such contamination in a changing Arctic environment is explored.
Environmental significanceSignificant changes in the physical environment of the Arctic have been observed under a rapidly changing climate. Associated with such changes, much evidence of climate change-induced impacts on new emission sources, remobilization, transport and circulation of persistent organic pollutants (POPs) and Chemicals of Emerging Arctic Concern (CEACs) has also been observed. This review compiled these observed changes in the Arctic and similar findings in cold-climate environments in Antarctica and on the Tibetan Plateau, to curate the current knowledge on this issue and to identify knowledge gaps in order to guide future research to better understand their implications on Arctic contamination, quantify their impacts and investigate their linkages to contaminant exposures and observed effects on the Arctic ecosystems. |
A. Introduction
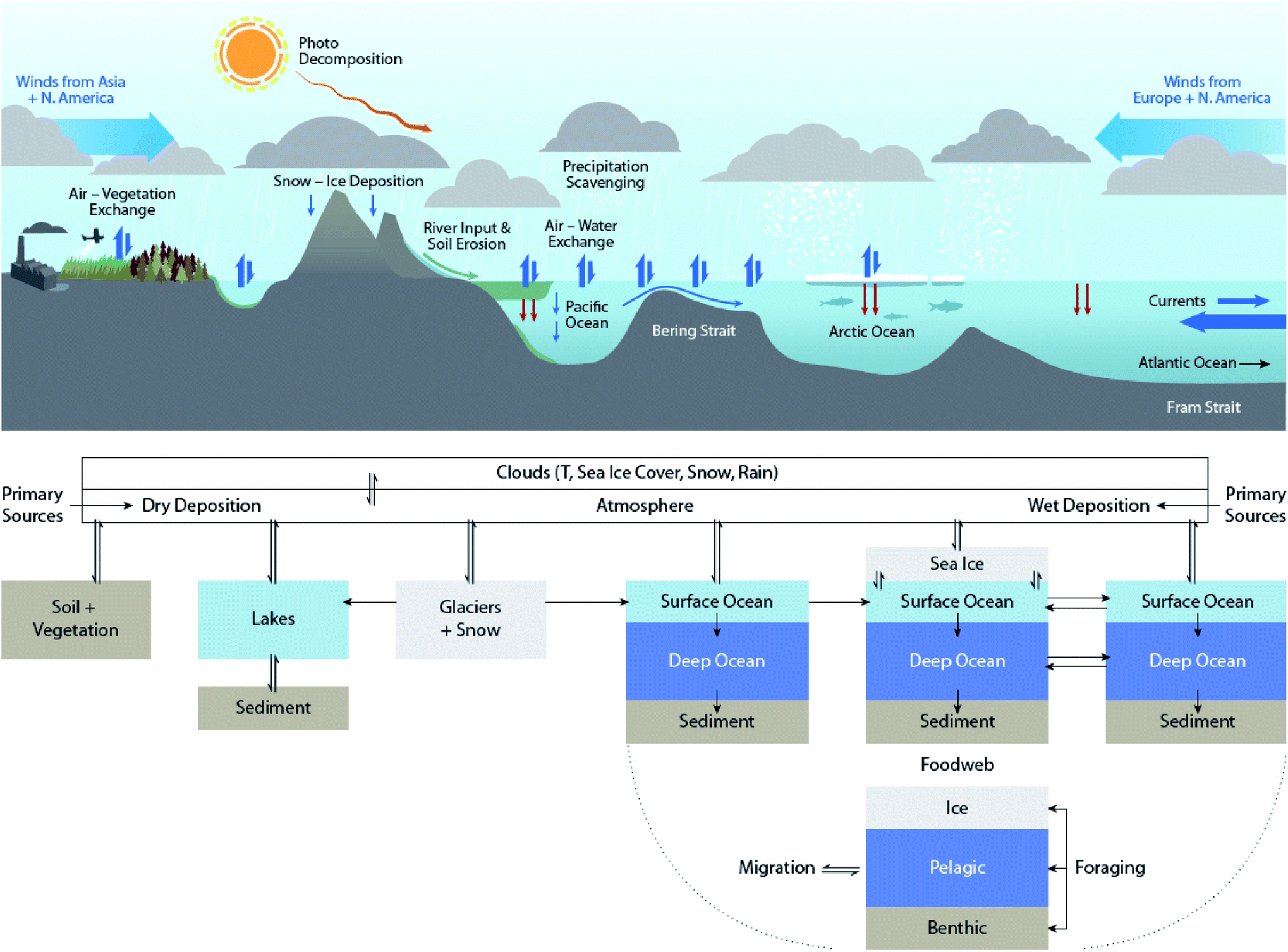
How can climate change influence the distribution of contaminants in the Arctic, as well as globally?The Arctic environment is vulnerable to global environmental changes. The Arctic cryosphere (marine and terrestrial) is rapidly declining with observable and documented impacts on Arctic ecosystems.1–6 According to the most recent assessment by the intergovernmental panel on climate change,7 warming occurs faster and with greater magnitude in the Arctic compared to other parts of the world (see Fig. 1A in de Wit et al. (2022)8 in this special issue). Climate change in the Arctic is resulting in extreme sea ice retreat (see Fig. 1C in de Wit et al. (2022)8), melting glaciers, thawing permafrosts and a greater frequency of extreme weather events.9 Such changes also affect the environmental distribution and fate of contaminants such as persistent organic pollutants (POPs). Effects on contaminants occur not only because of changes in Arctic surface conditions (e.g. increased open water area, loss of glaciers, perturbation of snow deposition patterns) and physical processes (e.g. air and water circulation patterns, precipitation rates), but also because the most important drivers of the POP transport, partitioning, and transformation are their physical–chemical properties, many of which are temperature-dependent. Temperature-dependent properties of POPs include their vapor pressure, Henry's law constant, partitioning coefficients, and rates of degradation or transformation (including photolysis, hydrolysis, etc.).10 Lamon et al. (2009)11 estimated that a 1 °C increase as a result of climate change would increase the volatility of POPs such as polychlorinated biphenyls (PCBs) by 10 to 15% and thus increase their mobility. Other biogeochemical processes affecting POPs, such as the ‘biological pump’ and degradation in marine waters,12,13 also show a climate sensitivity.
 | ||
| Fig. 1 Simplified summary of transport and transformation processes of POPs in the abiotic environment in the northern hemisphere that can be influenced by climate change. Modified from Ma et al. (2016).10 | ||
As a result of climate teleconnections between the Arctic and other parts of the world, the amplified effects of climate change in the Arctic will contribute to higher sea levels and more intensive and frequent extreme climate events (e.g. heavy rainfalls, droughts, severe storms) globally.9,14,15 Together with generally increasing global surface temperatures and decreasing ice and snow cover, these climate-related changes will also serve to mobilize and redistribute POPs in the environment of more southerly latitudes. Thus, the amplified effects of climate change on contaminants in the Arctic can provide early warning signals for similar effects globally.
Many of the processes responsible for the distribution and transformation of POPs can be influenced by climate change. Fig. 1 summarizes the abiotic processes that drive the environmental distribution of POPs in the northern hemisphere. These processes are subject to climate change influence to various extents. The impacts of climate change on biogeochemical processes can often have opposing effects on POPs. For example, ambient temperature increases favor the volatilization of chemicals, thus enhancing their mobility in the atmosphere and potential for long-range atmospheric transport (LRAT). On the other hand, increases in precipitation enhance the scavenging, deposition, and removal of chemicals from the atmosphere.
Climate change occurs over long periods of time (i.e. decades), therefore its effects on POPs need to be evaluated over similar time scales. Here we attempt to make use of available temporal and spatial measurements of POPs in air, water, oceanic- and lake-sediment cores, and ice- and snow-cores to provide insight into whether the cumulative impacts of climate change within the Arctic exacerbate or diminish contaminant transport and accumulation patterns in different abiotic media. Fig. 2 shows the locations of key studies on the physical environment reviewed here. In addition to POPs, we will also explore the effects of climate change on chemicals of emerging Arctic concern (CEACs), identified by the Arctic Monitoring and Assessment Programme (AMAP) as chemicals newly detected in the Arctic environment.16 Given the relatively recent discovery of CEACs, less is known about their sources and environmental fate; however, it is expected that climate change will influence the presence and fate of CEACs similarly as for POPs.
 | ||
| Fig. 2 Locations of key studies on the Arctic physical environment referenced in this review. | ||
B. Direct and indirect effects of climate change on contaminants in the Arctic
What are the influencing factors associated with climate change that affect contaminant transport, accumulation, and occurrence in different abiotic media (i.e. physical environmental compartments)?Changes in ecosystem processes and structure (e.g. food webs, deposition pathways, cryosphere, etc.), as well as thermodynamically-driven interactions between chemicals and their surrounding environment, will inevitably influence the presence, lifetime, and mobility of chemicals in Arctic ecosystems – either directly or indirectly. The direct effects of climate change cause changes to the physical environment, such as ambient temperature changes in air and water, and changes to physical processes in the Arctic (e.g. retreating ice, melting permafrost, increasing precipitation, erratic warming events, changing river flows, etc.), whereas indirect effects are the secondary changes resulting from the consequences of primary changes to the physical environment. Examples of indirect effects include new or different pollutant sources associated with changes or shifts in anthropogenic activities within the Arctic (e.g. shipping, mineral exploitation), or variations in biological processes (e.g. changes in the magnitude and extent of the oceanic biological pump driving contaminants to deeper waters). Direct and indirect effects of climate change relevant to the presence and distribution of contaminants in the Arctic are summarized in Table 1.
| Influence of climate change | Contaminant-related processes potentially affected |
|---|---|
| Direct influences | |
| Increased ambient temperatures (sea, land, atmosphere) | • Pollutant re-mobilization |
| • Long-range transport | |
| • Transformation pathways | |
| Ocean acidification | • Transformation conditions |
| Changing weather patterns (e.g. precipitation, seasonal characteristics, frequency of extreme events) | • Long-range atmospheric transport |
| • Deposition and precipitation event frequency | |
| Sea level rise | • Transfer between terrestrial and marine environments |
| • Marine transport pathways | |
| Loss of cryosphere | • Translocation, re-mobilization, and redistribution of contaminants |
| Different radiation characteristics [black carbon (BC), cloud condensation nuclei (CCN)] | • Photochemistry and transformation pathways |
| Altered carbon cycling and sequestration | • Biotic and abiotic transformation |
| Water mass transport changes (e.g. changing ocean currents) | • Long-range oceanic transport |
| Increased dust aerosol loadings | • Additional advective transport and particle-mediated transport |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|
| Indirect influences | |
| Food web composition change (e.g. invading species) | • Bioaccumulation and transformation |
| Re-mobilization of pollutants | • Re-emission from sediment, ice surfaces, and soils |
| Land degradation (e.g. increased erosion) and flooding | • Re-mobilization and re-emission |
| • Transfer between terrestrial and marine environments | |
| Biodiversity loss | • Bioaccumulation and transformation |
| Behavioral pattern changes (e.g. animal migration) | • Contaminant exposure |
| • Bioaccumulation | |
| • Biovector-based transport of pollutants | |
| Human socio-economic development | • New pollutant sources |
| • New exposure routes | |
| New economic opportunities in the Arctic | • New pollutant sources |
| • Re-mobilization | |
| Increased agricultural disease and pests | • Pesticide and pest control agent use |
| Increased frequency of boreal forest fires | • Pollutant re-mobilization and redistribution |
| Effects on the oceanic biological pump | • Ocean sequestration of pollutants |
The direct effects of climate change on the presence and profiles of various organic (and inorganic) pollutants have been previously documented.17–20 However, under the current, most accepted climate change scenarios forecast for the Arctic, it is expected that the indirect effects of climate change will have a significantly stronger effect on the environmental fate of contaminants compared to direct influences.4,10,21–24 Compound-specific environmental factors like environmental stability, transformation, bioavailability, and environmental mobility can be monitored in future studies aimed at characterizing and summarizing the effect of climate change on the presence and behavior of chemical contaminants in the Arctic, bearing in mind that such factors may vary geographically across the region.4,21,24,25
Many CEACs are directly associated with human activities, and usually those occurring within the Arctic region itself. AMAP (2017)16 identified 17 CEAC groups, of which, approximately 11 may originate from local sources in addition to long-range transport (LRT) (Table 2). Additionally, Röhler et al. (2020)26 developed a sample clean-up and analytical method to screen air samples for new chemicals of concern in Arctic air. In air collected from the Zeppelin station on Svalbard (Fig. 2) they found 700+ and 1200+ chemicals of interest in the particle and gas phase fractions, respectively, including 73 new potential CEACs (i.e. compounds previously not reported in Arctic environments). There is very little information about the occurrence and transport pathways of many CEACs; thus, it is difficult to determine how climate change would affect their distributions in the Arctic environment. Better knowledge of the sources, environmental fate, and impacts of climate change on CEACs is required to coordinate pollutant regulations on a circum-Arctic scale.16
| CEAC group | Abbreviation | Characteristic compounds | Main pathways to the Arctic | ||
|---|---|---|---|---|---|
| Long-range transport | Local sources | Notes | |||
| Per- and polyfluoroalkyl substances | PFAS | Perfluoroalkyl acids (PFAA) including perfluorooctanesulfonate (PFOS) | X | X | Local communities and airports may act as local sources |
| Perfluorooctanoic acid (PFOA), perfluorohexane sulfonate (PFHxS) | |||||
| Volatile neutral PFAA precursors including fluorotelomer alcohol (FTOH) and perfluoroalkylsulfonamide-based substances | |||||
| Brominated flame retardants | BFR | Decabromodiphenylether (PBDE-209) | X | X | Dump sites may act as a local source |
| Chlorinated flame retardants | CFR | Dechlorane plus | X | X | Dump sites may act as a local source |
| Organophosphate ester-based flame retardants and plasticizers | OPE | Chlorinated OPE such as tris(2-chloroethyl) phosphate (TCEP) | X | X | Airports may act as local sources |
| Alkylated OPE such as tri-n-butyl phosphate (TnBP), tris meta-(cresyl) phosphate (TmCP) | |||||
| Aryl-OPE such as 2-ethylhexyl diphenyl phosphate (EHDPP) | |||||
| Phthalates | Diethylphthalate | X | X | ||
| Short-chain chlorinated paraffins | SCCP | C10H17Cl5 | X | X | |
| Siloxanes | Hexamethyldisiloxane (HMDS) decamethylcyclopentasiloxane (D5) | X | X | Atmospheric transport is a major source to the Arctic, however, local sources (e.g. personal care product use) also exist | |
| Pharmaceuticals and personal care products | PPCP | Ibuprofen, caffeine | X | Sewage outflows are primary local sources | |
| Polychlorinated naphthalenes | PCN | 75 Congeners with 1–8 Cl, in technical mixtures (e.g., Halowaxes, Nibren waxes, Seekay waxes, Clonacire waxes), and formed by combustion and other high temperature processes | X | X | |
| Hexachlorobutadiene | HCBD | HCBD | X | ||
| Current-use pesticides | CUP | Chlorpyrifos, chlorothalonil, Dacthal | X | X | Agricultural applications are primary sources |
| Pentachlorophenol and pentachloroanisole | PCP & PCA | PCP & PCA | X | X | PCP is a wood preservative, PCA is a metabolite and possibly a natural product |
| Organotins | RnSnX4−n, where R represents an alkyl or aryl group and X is represented by an anion such as chloride, oxide, hydroxide, acetate, or other functional group, e.g. tributyltin (TBT) | X | Use associated with population and shipping densities. Harbors are major local sources | ||
| Polycyclic aromatic hydrocarbons | PAH | Naphthalene, anthracene | X | X | Summer wildfires in sub-Arctic regions act as episodic sources |
| Unintentionally produced PCBs | uPCB | PCB-11 | X | X | |
| Halogenated natural products | HNP | Brominated phenols, mixed halogenated compounds | X | X | Phytoplankton blooms, macroalgae are primary sources |
| Marine plastics and microplastics | μPlastic | Polyethylene (PE), polypropylene (PP), polyvinyl chloride (PVC), including monomers and additives synthetic fibers, indigo denim, tire particles | X | X | |
C. Within-Arctic redistribution of POPs and CEACs
What are the most sensitive transfer processes that affect the movement of contaminants between polar reservoirs?Given the semi-volatile nature of POPs and many CEACs, transfer of contaminants between environmental compartments (e.g. air to surfaces, water to sediments, ice to water, etc.) will be affected by changes in ambient temperature and Arctic processes, such as snow, rainfall, and the changing cryosphere. Re-mobilization of legacy POPs present in Arctic surface seawater back to the atmosphere due to the decline in sea ice cover was examined by Ma et al. (2011).27 By combining empirical monitoring data and model-based scenario assessments, this study showed that the physical–chemical properties of the respective substances combined with the diminishing cryosphere is resulting in the re-emission of contaminants from Arctic surface compartments (i.e. ‘reservoirs’ such as snow, ice, soil, sea water) and influencing longer-term time trends of selected POPs in the Arctic atmosphere.
The Arctic marine environment (e.g. marine sediments) is considered an important depositional sink for legacy POPs in the Arctic, largely due to the settling of organic matter-bound POPs through the biological pump operating in oceanic surface waters.12 For most of the substances addressed in this report, the Arctic marine environment and associated food webs are the largest environmental reservoirs due to the areal extent of the Arctic Ocean and coastal seas relative to terrestrial areas. Therefore, the loss of the terrestrial cryosphere is not expected to significantly contribute to the diffusive re-mobilization of legacy POPs on an Arctic-wide basis.28 Wöhrnschimmel et al. (2013)29 examined the temporal profiles of PCBs and their responses to Arctic environmental changes. This comprehensive study combined a chemical fate model, scenario assessments, and empirical data evaluations in the context of a changing Arctic. Based on these evaluations, the authors concluded that long-range atmospheric transport from potential primary sources (e.g. fresh usage) and secondary sources (e.g. volatilization from soil), rather than secondary re-emissions from Arctic compartments, will continue to be the major inputs of PCBs and other POPs into the Arctic environment in the future.
More recently, Ubl et al. (2017)30 used a cluster analysis to study the relationship between atmospheric transport regimes and PCB concentrations in air measured at Zeppelin station on Svalbard. They found that the concentrations of less-volatile PCBs, e.g. PCB-101, were higher during the winter with land-derived air masses originating from Europe. However, air concentrations of lighter PCBs (represented by PCB-28) were higher during the summer, and were associated with air masses of an oceanic origin and longer residence times closer to the station (Fig. 3). They attributed this observation to the increased emissions from local PCB sources (e.g. PCBs in paints, mines, and heavily contaminated soil) during the warm season, which masked the effect of LRT. In a warming Arctic, emissions from local environmental reservoirs will likely become more important for more volatile chemicals (e.g. PCB-28).
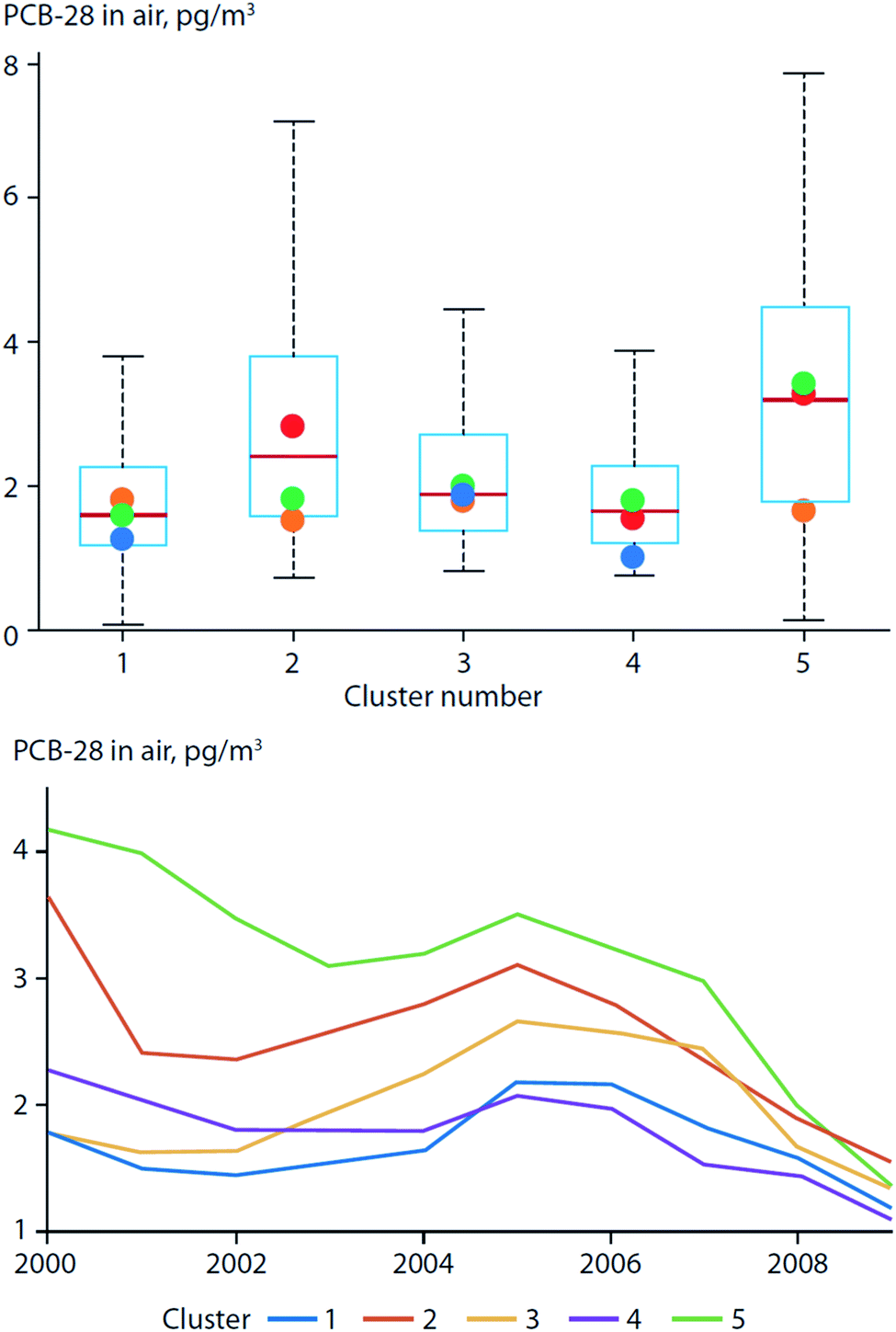 | ||
| Fig. 3 PCB-28 concentrations in air from Zeppelin station on Svalbard classified by origin of air masses (land-derived: clusters 1, 3, and 4; ocean-derived: clusters 2 and 5). (top) Box plot of PCB-28 concentrations in air. Boxes show interquartile range with whiskers that extend to the most-extreme values; colored dots indicate median concentrations per season. Concentration variability and seasonal variation were highest for ocean-derived transport regimes compared to land-derived transport regimes originating from continents. PCB-28 concentrations in cluster 2 were highest during the summer (red dot). (bottom) Running median (4 years) of PCB-28 concentrations by cluster between 2000 and 2009. Highest concentrations of PCB-28 were observed when air masses originated from oceans. Modified from Ubl et al. (2017).30 | ||
In the light of a rapidly changing environment, internal Arctic redistribution and transformation processes will be more prominent in shaping the environmental fate of legacy POPs, such as PCBs, through the following processes:
(1) Increases in temperature in all Arctic media will generally increase transformation rates of contaminants.
(2) Wet deposition will occur more often in the form of rain instead of snow, which will have effects on the scavenging effectiveness and deposition profile of POPs present in the atmosphere.
(3) Microbial transformation will play a more prominent role in the degradation of POPs. Climate change will modify the structure of microbial communities and the rate of microbial degradation.
(4) Transarctic ice transport will be less important for the marine redistribution processes of POPs, particularly after riverine release of POPs.
(5) Changes in food web composition will contribute to changes in environmental partitioning and accumulation of POPs, as well settling fluxes of organic matter-bound chemicals.
(6) Increasing ablation and melt of ice caps and glaciers will continue to be a source for POP re-mobilization in the Arctic environment.31,32 Legacy POPs with physical chemical properties similar to PCBs may be expected to exhibit similar fate characteristics.
Increased re-mobilization of legacy POPs from marine reservoirs due to accelerating Arctic environmental change and marine cryosphere loss, in combination with complex inner-Arctic redistribution processes, may add to the overall POP levels and potential exposure risk for marine food webs and indigenous populations.
C.1. Effects of warming and biogeochemical change on air–water-sediment exchange
The decrease in sea ice coverage appears to be enhancing primary production in the Arctic Ocean due to the increased light entering the sea, longer growing season, and increased nutrient availability from marginal sources. Climate change will also modify the seasonal timing of phytoplankton blooms in the Arctic.33 Massive phytoplankton blooms underneath regions fully covered by sea ice have been observed, suggesting blooms may be initiated whenever light and nutrients are sufficient for photosynthesis, independent of the timing of ice retreat.34,35 Since many POPs and CEACs are hydrophobic with a tendency to associate with organic matter, after entering surface seawater, these chemicals sorb to particles and are transported to the deep ocean via vertical sinking, resulting in increased sedimentation of chemical contaminants in a process driven mainly by the biological pump operating in near-surface waters.12 This process also draws dissolved chemicals out of the seawater, inducing an air–water fugacity gradient that enhances their flux from air to water.12,36–38 In addition, the ocean sequestration of atmospheric POPs also modifies or can modulate the transport of POPs to the Arctic. Under a climate change scenario, the activity of the biological pump will change in magnitude and geographic extent. High-productivity regions and their large settling fluxes of organic matter are expected to shift northward to Arctic regions currently covered by sea ice,33 thus leading to a larger sequestration of POPs in deep Arctic seawater and sediments. Liu et al. (2020)39 reported the full-depth profiles of polycyclic aromatic hydrocarbon (PAH) concentrations in sea water from the Pacific to the Arctic and Atlantic Oceans at 44 sites measured on-board the RV Xuelong (Snow Dragon) research expedition from July to September 2012. PAHs in the water columns generally showed a “surface enrichment and depth-depletion” pattern. Using particle-reactive radioisotopes, Liu et al. (2020)39 showed high scavenging efficiency, with PAH sinking fluxes of 2773 ± 1259 ng m−2 d−1. Lateral transport, traced by transient tracers, did not show a significant contribution to the PAH inventories in the Arctic surface ocean. Their findings indicate the importance of biogeochemical-modulated contaminant transport, which would be enhanced due to climate change.Another important biogeochemical process influencing contaminant exchange is the degradation of organic pollutants by microorganisms. For example, microbial degradation of hexachlorocyclohexanes (HCHs) has been reported for Arctic waters,13,40 and occurs at a higher magnitude during bloom events that is sufficient to induce a depletion of dissolved phase HCH concentrations and drive atmospheric deposition of HCHs. Other organic pollutants besides HCHs could be microbially degraded in the Arctic Ocean, a process that could be favored under a climate change scenario with higher temperatures. On the other hand, organic pollutants exert an influence on microbial community structures and functions that could impact degradation. For example, Cerro-Gálvez (2019)41 has shown that inputs of organic pollutant mixtures to coastal waters from Svalbard increase growth of the microbial biosphere and induce a transcriptomic response related to the degradation of organic pollutants and other detoxification processes.
C.2. Gas–particle partitioning and climate change
Volatilization rates as well as gas–particle partitioning are two of the key factors that determine the environmental fate of semi-volatile organic chemicals (SVOCs), including POPs and many CEACs. Gas–particle partitioning and volatilization rates of SVOCs can be estimated from their octanol–air partitioning coefficients (KOA) and vapor pressures,42–45 both of which are temperature-dependent, and therefore likely to be impacted by climate change.In addition to temperature-driven changes to gas–particle partitioning,46,47 particle concentrations in air seem to be an important driver for partitioning of organic contaminants onto particles.42,46–48 Elevated coarse particle loads at lower temperatures, e.g. in the Arctic49 were found to lead to higher OPE fractions on particles.46,48 Consequently, the higher average temperatures due to climate change could result in a higher fraction of OPEs being found in the gas-phase.
It is not clear whether an increased gas-phase concentration of OPEs (and other SVOCs) would lead to an increase or decrease in atmospheric transport compared to particle-associated long-range transport. An increase could occur, because larger particles (and the contaminants sorbed to them) are scavenged by wet and dry deposition, which reduces the overall travel distance of the particle and any sorbed contaminants.50 Higher gas-phase concentrations could therefore reduce the removal of contaminants from the atmosphere, allowing them to travel further. This hypothesis has been supported by model predictions of PAH concentrations in the Arctic under future climate change scenarios. Using a chemical transport model (GEOS-Chem) Friedman et al. (2014)51 predicted increasing concentrations of volatile PAHs and decreasing concentrations of particle-bound PAH concentrations under climate change conditions. For soil/snow–air partitioning, Casal et al. (2018)52 demonstrated that PCB and HCH concentrations show temperature-dependent variation; however, such seasonality is not observed for high molecular weight PAHs. On the other hand, particle adsorption can reduce degradation of contaminants.53,54 Therefore, higher gas-phase concentrations could also lead to increased degradation (e.g. photochemical oxidation) and, thereby reduce the potential for long-range atmospheric transport, which has been discussed for decabromodiphenylether (PBDE-209).55
C.3. Sea-spray aerosols and their influence on contaminant transport
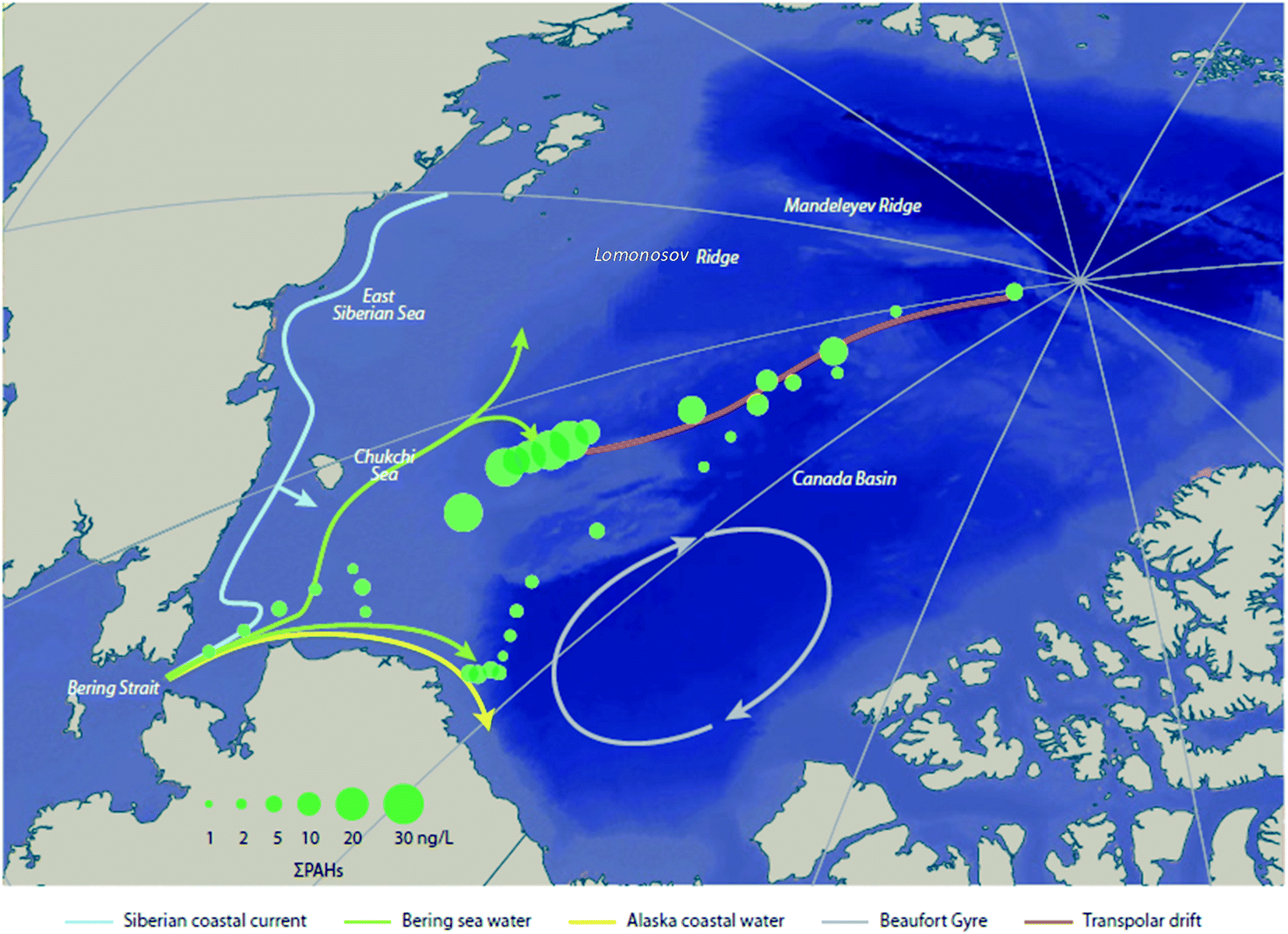
Sea spray aerosols (SSAs) are droplets of sea water that are ejected into the atmosphere by the bursting of bubbles on the sea surface. The bubbles, that are mostly formed by breaking waves, fragment into small “film droplets” (SSAs < 1 μm) and/or burst, releasing larger “jet droplets” (SSAs > 1 μm).56,57 The formation of SSAs is a complex process that is highly dependent on environmental parameters such as air temperature, water temperature, and wind speed.58 The bubbles that ultimately lead to the formation of SSAs have been shown to enrich organic matter, and with it, persistent and mobile chemicals such as per- and polyfluoroalkyl substances (PFASs), OPEs, and other organic compounds that are present in the water column.59Johansson et al. (2019)60 reported a 62000-fold enrichment of PFAS concentrations in SSAs compared to bulk water in a sea-spray chamber experiment. They concluded that SSAs were a significant source of PFASs to the atmosphere. Using the Norwegian Earth System Model (NorESM), Johansson et al. (2019)60 estimated the global emissions and transport of PFASs due to SSAs. They concluded that PFAS emissions through SSAs would be highest in latitudes between 45°N and 60°N, and 45°S and 60°S, respectively (Fig. 4). The apparently higher modeled PFOA emissions by SSA in the Southern Hemisphere compared to the Northern Hemisphere (Fig. 4) is due to the assumptions that the enrichment factors were not affected by either the seawater concentration of PFAS or seawater chemistry, and PFAS seawater concentrations were uniform. Thus, the modelled emissions correspond to SSA production which has higher fluxes between 45°S and 60°S.61 Spatially resolved and reliable seawater concentrations for PFAS are needed to better estimate PFAS emissions via SSA. Casal et al. (2017)62 have shown that SSAs can be an important source of PFASs to Antarctic waters when scavenged during snow deposition events. Such a process could be important for the Arctic Ocean as well.
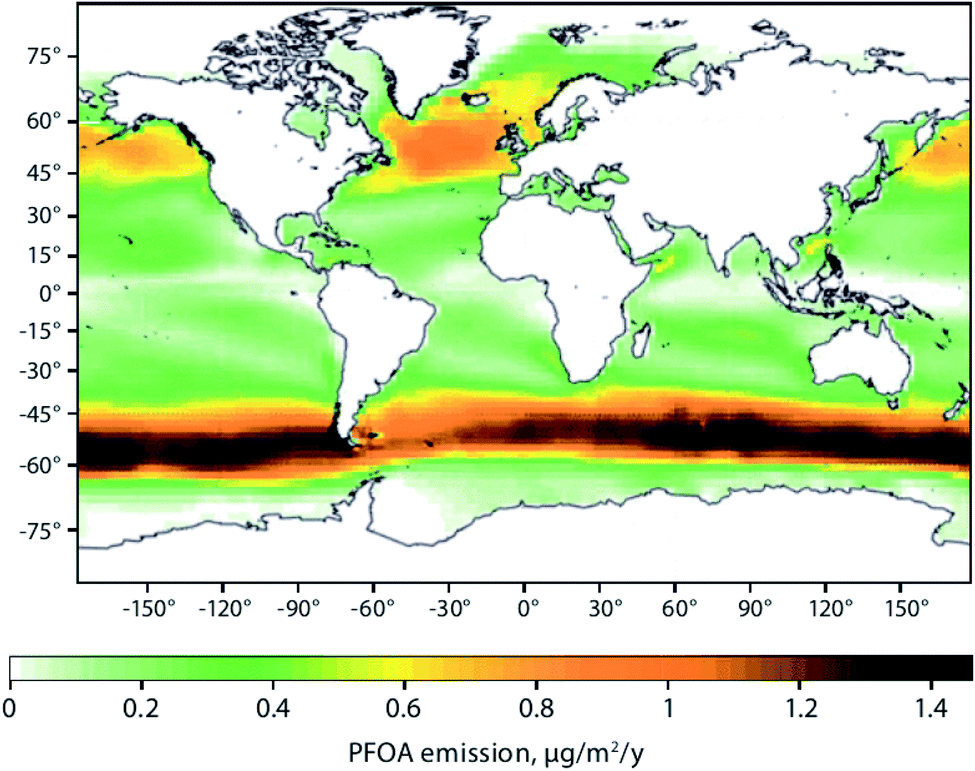 | ||
| Fig. 4 Spatial depiction of total predicted yearly emissions of perfluorooctanoic acid (PFOA) via sea spray aerosols. Source: Johansson et al. (2019).60 Note that the higher PFOA emissions between 45–60°S are driven by higher SSA formation in this region rather than elevated PFOA concentrations in the surface seawater. | ||
Since the formation and size of SSAs are highly dependent on environmental factors, climate change could have a substantial impact on their role as vectors for PFASs and other organic chemicals to the atmosphere. However, the complex interactions of water temperature, air temperature, wind speed, and salinity in SSA formation mean it is not possible to draw global conclusions regarding the impact of climate change on SSAs and their role in contaminant transport.
The reduction of sea ice due to climate change is expected to lead to a general increase in SSAs within the Arctic.63 However, laboratory studies by Mårtensson et al. (2003)64 and in situ measurements by Nilsson et al.65 showed that increasing water temperatures could lead to a decrease of nano-sized SSAs, which have been shown to have the highest enrichment potential for organic matter66 and, by extension, organic contaminants potentially as well. For larger SSAs (>100 nm), the same experiments showed an increase of the number of SSAs with increasing seawater temperature.64,65
Model predictions for Europe by Soares et al. (2016)67 suggest an increase of sea salt emissions into the air north of Iceland and around the United Kingdom under climate change scenarios. These findings are congruent with the regions predicted to have elevated PFAS emissions associated with SSAs60 (Fig. 4). Climate change impact on SSA-related emissions of organic chemicals, such as PFAS, in the Arctic is uncertain and warrants further investigation.
D. Influence of climate patterns on the behavior of POPs
How do large-scale climate variation patterns influence POP transport to, and distribution within, the Arctic?Climate variation patterns, such as the Arctic Oscillation (AO), North Atlantic Oscillation (NAO) and El Nino Southern Oscillation (ENSO) (ESI Text S1 and Fig. S1† for definitions), not only affect volatilization of POPs from secondary sources, they can also influence the atmospheric circulation patterns which distribute chemicals around the globe (Fig. 1). LRAT follows patterns defined by the physics of atmospheric circulation and physical–chemical properties of the chemicals.10 For instance, the trans-Pacific transport of POPs follows west to east storm tracks across the Pacific Ocean.68,69 Storm tracks are climatological winds that occur repeatedly, but their positions and strength may change with climate variabilities. Hung et al. (2005)70 reported that the gas-phase concentrations of PCB-31, PCB-101, PCB-153 and γ-HCH measured in air at the Canadian Arctic station of Alert (1993–1999) during the spring months showed statistically significant (p < 0.1) correlations with the Pacific North American (PNA) index (Fig. 5). In addition, concentrations of PCB-31, PCB-44 and PCB-138 correlated with the NAO index. Although the underlying mechanisms responsible for these correlations are unknown, such relationships suggest climate variation patterns are capable of influencing the distribution and levels of POPs spatially and temporally within the Arctic. Temperature anomalies associated with climate variation patterns can enhance the volatilization of POPs and affect gas–particle partitioning of atmospheric contaminants, which, in turn, affects their transport mechanisms to the Arctic. Climate variation patterns, such as the NAO and AO, can also alter moisture fluxes,71 which will affect upstream scavenging of POPs, influencing their transport potential.
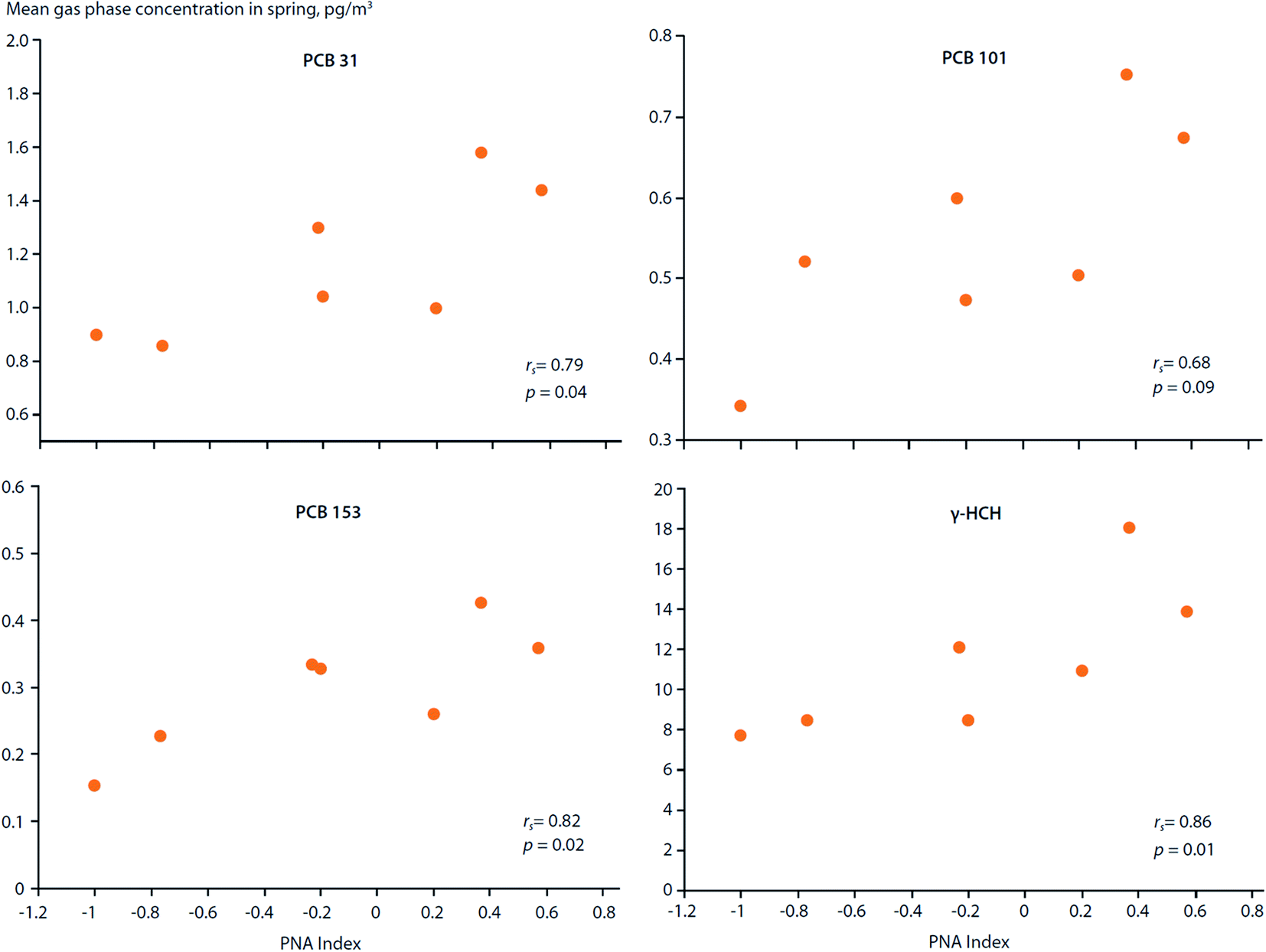 | ||
| Fig. 5 Correlations between the Pacific North American (PNA) climate index and spring air concentrations of PCBs and γ-HCH measured at Alert, Canada between 1993–1999. Source: Hung et al. (2005, 2022).70,72 | ||
Becker et al. (2008)73 studied twelve years of α-HCH and γ-HCH measurements in air collected from Alert (Canada) and Zeppelin Mountain (Svalbard). It was found that AO fluctuations influenced the α-HCH time-series at Zeppelin, but not at Alert, and not for γ-HCH at either location (Fig. 6, top panel). During the 1990s, the AO was predominantly in a positive phase; after 2000, the AO was mainly in a negative phase. A change in trends after 2000 is apparent for α-HCH measured at Zeppelin, which was supported by a statistically significant relationship (p < 0.05) between the summer AO index (June–August) and α-HCH concentrations (Fig. 6). The authors have also found that there were much greater variations in concentrations between summer and winter months when AO was in the negative phase (post-2000) (Fig. 6, bottom panel).
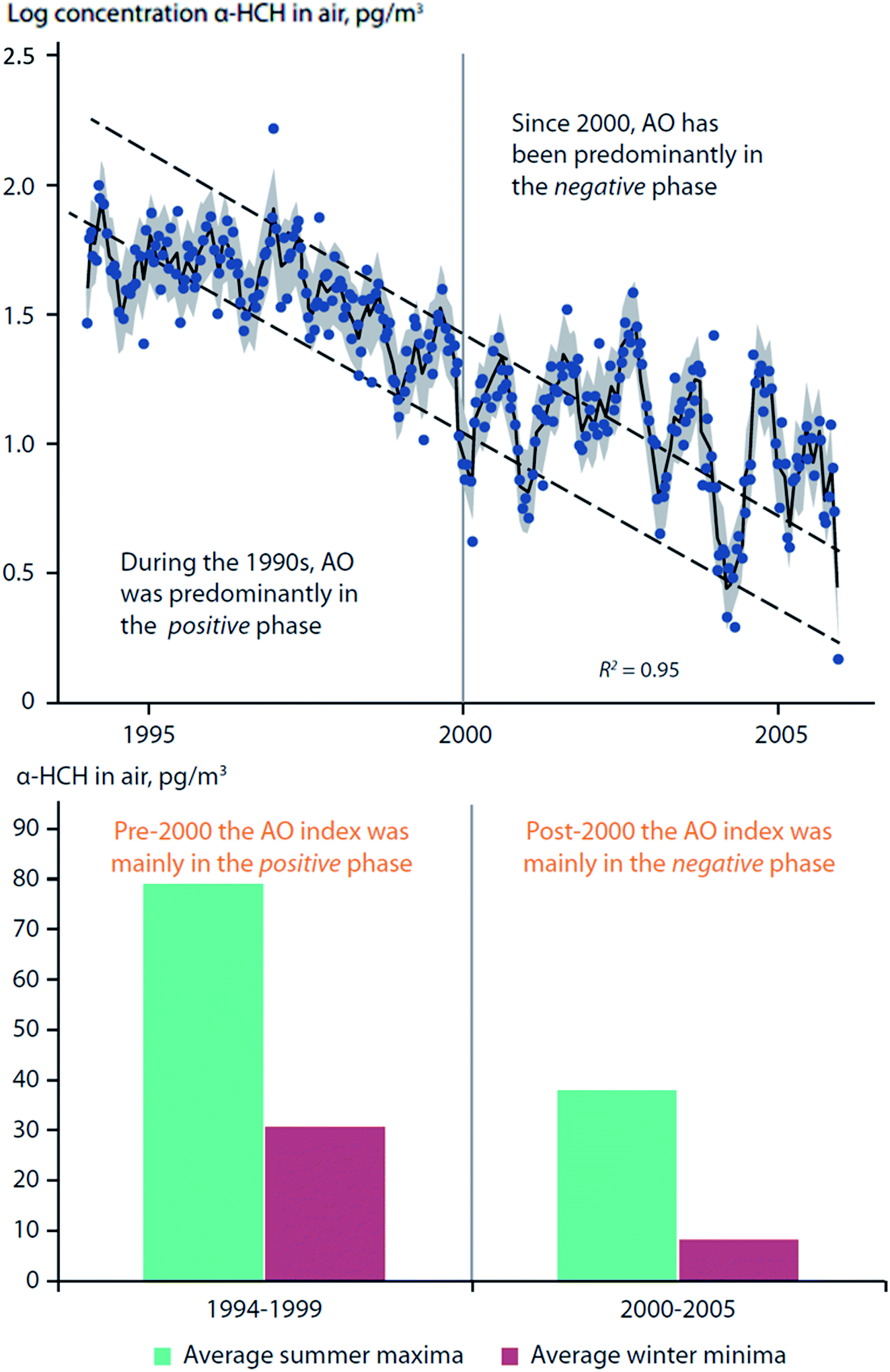 | ||
| Fig. 6 Arctic oscillation (AO) influence on α-HCH air concentrations measured at Zeppelin Mountain, Svalbard. (top) Bi-weekly measurements of α-HCH in air between 1994–2006. Dashed lines indicate expected differences between summer and winter concentrations, assuming constant decline. (bottom) Difference in average summer- and winter- α-HCH air concentrations pre- and post-2000. Adopted from Becker et al. (2008).73 | ||
Climatic oscillations can also influence the transport of contaminants in Arctic Ocean and coastal seas. During the July to September 2010 R/V Xuelong (Snow Dragon) ship-based expedition, Lin et al. (2019)74 found that the PAH concentrations in surface seawater from the Eurasian margin of the Chukchi Plateau were nearly an order of magnitude higher than those in the North American margin (Fig. 7). By examining sea ice back trajectories and stable oxygen isotope data, the authors concluded that the observed concentration difference was driven by the Transpolar Drift and Beaufort Gyre under an enhanced AO; pointing towards significant PAH input from river runoff and ice-melt originating from the Eurasian margin under a warming Arctic. A mass balance model showed that 83% of the PAH input to the Chukchi Plateau was from marginal sources (i.e. river discharge, shelf input, and sediment-laden meltwater), with nearly 100% of PAHs associated with river discharge coming from Eurasia. The model also suggested that 64% of PAHs would be removed by volatilization, indicating the Arctic Ocean has shifted from a receptor to a strong secondary source for PAHs to air. This study demonstrated that a warming Arctic will likely remobilize PAHs from environmental sinks in marginal areas to surface seawater and the atmosphere, thus influencing the global distribution of these compounds.
 | ||
| Fig. 7 Concentrations of total dissolved PAHs (ng L−1) in surface waters of the Arctic Ocean. Modified from Lin et al. (2019).74 | ||
E. Changing long-range transport, secondary emissions, human activities, and local sources
How do contaminant pathways into- and within-the Arctic change due to climate change, and how do local sources compare to long-range transport?Long-range atmospheric transport continues to be a source of POPs and other contaminants (e.g. CEACs) to the Arctic. Oceanic transport is also relevant for more water-soluble contaminants (e.g. perfluoroalkyl acids (PFAAs)) released into coastal seas in temperate regions and then transported northwards. The declining concentrations observed for many POPs in the Arctic atmosphere over the last 15 years or so, is to some extent, being affected by re-mobilization (e.g. revolatilization) of previously deposited stocks of certain chemicals present in Arctic seawater, meltwater, and other compartments. In addition, climate change effects on the Arctic and sub-Arctic, such as an increased frequency and extent of forest fires, changing permafrost, and local pollution sources influencing CEAC levels, in particular, have the potential to influence contaminant levels in the region, and are addressed in the following sections.
E.1. Changes in long-range transport and secondary emissions – air
Climate change has brought about an ablation of Arctic sea ice, permanent ice, and glaciers. Reductions in the areal extent of sea ice, or earlier seasonal break-up and thaw of ice floes, can affect the water to air transfer of chemicals that have accumulated in surface seawater. For instance, an abrupt increase in air concentrations of α-HCH in the Canadian Arctic coincided with ice breakup in the central Canadian Archipelago.75 Hung et al. (2016)4 reported increasing air concentrations of PCB-52 and PCB-101 at the coastal site of Stórhöfði, Iceland in close proximity to retreating sea ice and the deglaciating, retreating ice caps of Mýrdalsjökull and Eyjafjallajökull (Fig. 8, top and second panels). After consistently declining trends from the 1990s to the early 2000's, β-HCH measured in air at Alert, Canada and Stórhöfði, Iceland show complex trends (Fig. 8, third and fourth panels).4 At Alert, β-HCH concentrations increased from 2003 to 2007, and declined thereafter to 2012. In 2007, episodes marked by high concentrations of β-HCH at Alert corresponded with a record low in Arctic sea ice extent. This observation is consistent with the hypotheses of Li et al. (2002)76 that the dominant transport pathway for β-HCH is via the ocean, and re-emissions from open waters sustain levels in Arctic air.77 In contrast, at Stórhöfði the seasonal summer maxima was consistently apparent in β-HCH concentration variability (Fig. 8, fourth panel). The different β-HCH concentration patterns observed at Alert and Stórhöfði could be explained by differences in their surrounding environments. In the early 2000s, Alert was mostly ice-bound throughout the year, whereas Stórhöfði is close to open ocean year-round. Therefore, the high concentration episodes of β-HCH observed at Alert were probably due to LRT, while the consistent β-HCH summer peak at Stórhöfði was probably due to increased volatilization from nearby open waters.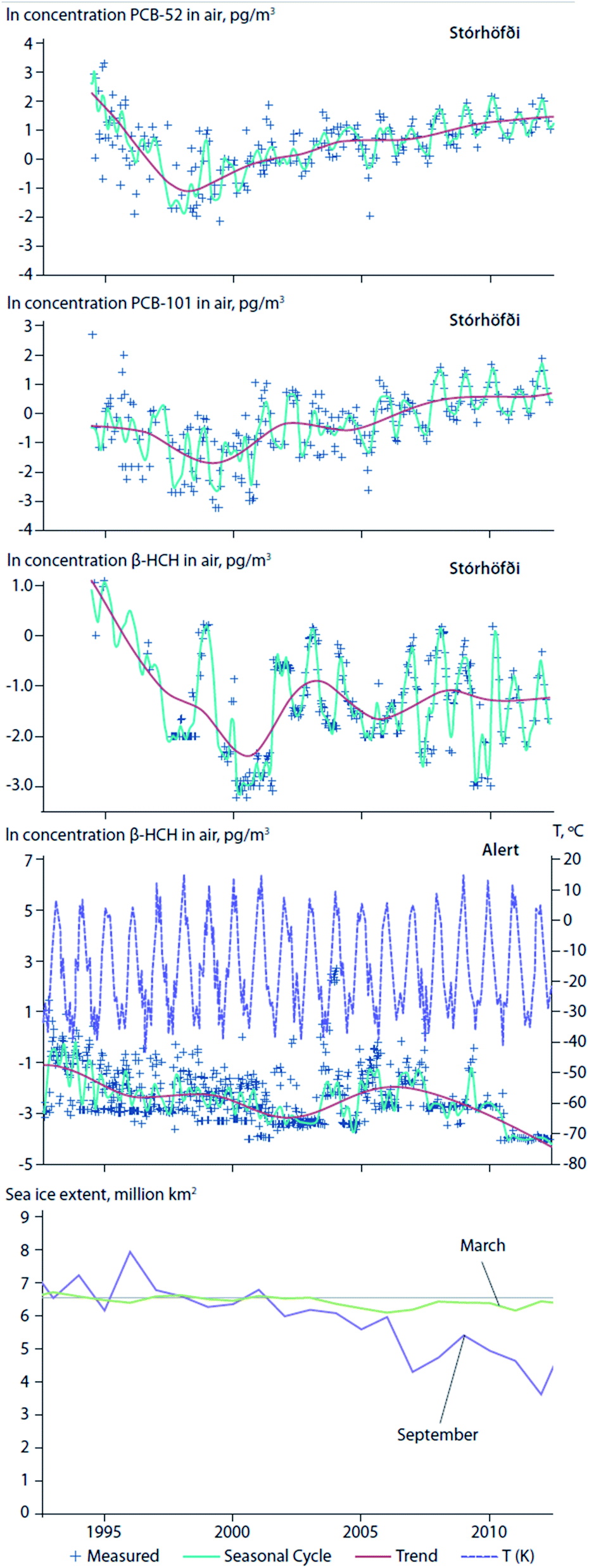 | ||
| Fig. 8 Temporal trends of contaminant concentrations in air at Alert, Canada and Stórhöfði, Iceland in relation to sea ice extent. Temporal trends of PCB-52 (top panel) and PCB-101 (second panel) concentrations in air at Stórhöfði, Iceland. Temporal trends of β-HCH at Alert (third panel) and Stórhöfði (fourth panel). Air temperatures at Alert are shown in purple dashed line in panel (third panel). September Average Sea Ice Extents (2002–2012) (million km2) given in italics red fonts under each year on the x-axes79 (adopted from Hung et al., 2016).4 September is usually the month when sea ice reaches its annual minimum extent. For comparison, the 1981 to 2010 average is 6.52 million km2. (bottom) Average sea ice extent in March and September. Source: NSIDC (2015).79 | ||
Anttila et al. (2016)78 examined the potential influence of secondary emissions of PAHs and some POPs on future trends by comparing air measurements and time series at the remote Arctic site of Pallas to a site in southern Scandinavia located closer to primary sources between 1994 and 2011. Assuming that future emissions of legacy POPs remain similar to the time periods during which the atmospheric trends were developed (1994–2011), their projections indicated PCBs will be depleted sooner, and chlordanes later, at the Arctic site of Pallas compared to southern Scandinavia, while the most long-lived chemicals will remain in the atmosphere for another couple of decades.
POPs may also be directly released into the sea as a result of melting snow and ice.80 Pućko et al.81–85 have shown that sea ice brines can concentrate POPs relative to sea ice and deliver them efficiently to lower trophic levels in the food chain (e.g. phytoplankton, zooplankton). Gioia et al. (2008)86 showed that air concentrations of PCBs in the Atlantic sector of the Arctic Ocean were higher in the sea ice marginal zone, probably related to their volatilization from melting ice. Recently, at coastal Antarctica, it has been shown that snow melting drives coastal seawater levels of POPs and a net volatilization of PCBs and other POPs, regardless of their physical–chemical properties.87
It is predicted that climate change will increase the frequency and intensity of wildfires which release contaminants, particularly PAHs, but also previously deposited PCBs, into the atmosphere. Yu et al. (2019)88 found elevated air concentrations of phenanthrene, pyrene, and retene at the Canadian High Arctic station of Alert from 2001–2005 and in 2015 which coincided with more frequent summer forest fire events in Canada, Alaska, and Greenland in these years. In this study, a global 3-D transport model also predicted that warming would result in higher air concentrations of lighter PAHs due to revolatilization from environmental sinks, while particle-bound PAHs (e.g. benzo[a]pyrene), were less affected by temperature. Recently, Luo et al.89 reconstructed PAH emissions from forest fires in the northern boreal forest using forest carbon stocks and the Moderate Resolution Imaging Spectroradiometer (MODIS) satellite images. They have shown that wildfires from northern Russia were major sources of benzo[a]pyrene to the Arctic. Also, increasing PAHs from forest fire plays a major role in offsetting the declining trends of PAHs in the Arctic. In an earlier study, using a combination of statistical fingerprinting techniques and MODIS satellite images, Sofowote et al. (2011)90 attributed significant contributions of retene in air at the Canadian sub-Arctic site of Little Fox Lake in autumn 2008 to wildfires occurring in British Columbia, the western United States, and north-eastern Asia during this time. Lin et al. (2020)91 used an integrated source apportionment technique to reveal five potential sources of PAHs found in 34 surface sediments from the northern Bering–Chukchi margin. The northeast Chukchi Sea exclusively had PAH profiles indicative of contributions from softwood combustion (characterized by retene), a likely result of the increasing wildfires in Alaska related to climate change. Eckhardt et al. (2007)92 reported extremely high concentrations of PCBs measured at Zeppelin, Svalbard, in spring 2006 and July 2004 as a result of biomass burning in Eastern Europe and boreal forest fires in North America, respectively. Together with warming, an increase in wildfires within and outside the Arctic would release POPs and CEACs (such as PAHs) to the atmosphere and enhance their mobility in the environment.
E.2. Changes in long-range transport and secondary emissions – ice/snow, surface water and land
POPs that were previously deposited from the atmosphere and subsequently incorporated into ice ‘reservoirs’ such as snow and glaciers (including ice sheets, ice fields, and ice caps), are being released and remobilized in meltwater due to climate change-induced glacier ablation and melting. These phenomena have been studied outside of the Arctic in alpine systems such as the European Alps93–96 and in Tibetan Plateau glaciers. For example, dichlorodiphenyltrichloroethane (DDT) peaked in the sediment of glacier-fed Lake Oberaar, Switzerland, around 1970 then re-increased in the 1990s, coinciding with the accelerated melting of the adjacent glacier.95 In lake sediment from the central Tibetan Plateau (Fig. 9), two concentration peaks of DDTs and HCHs were observed; one in the 1970s, corresponding to their heavy usage, and the second one between 1990–2000, possibly indicating a significant release from melting glaciers, as the production and usage of DDTs and HCHs has declined significantly since the 1970s.97,98 This phenomenon is broadly consistent with observations in the European Alps. In addition, flood waters from melting glaciers have influenced POP profiles in receiving lake sediments. Sun et al. (2018)98 found that there was no difference in the (DDD + DDE)/ΣDDT ratio measured throughout the depth of sediment cores collected from a Tibetian Plateau lake receiving glacial meltwater. In other words, the glacial meltwater retained the DDT signature emitted and deposited in the glacier during the peak usage period in the 1970s. The environment of the Tibetan Plateau, being subject to climatic conditions similar to polar regions, exhibits weak microbiological activity due to low temperatures, thus limiting the transformation of DDT to DDD and DDE. Similarly, a constant β-HCH/ΣHCHs ratio was also observed throughout the lake sediment core depth, however, the ratio was much higher than that of the technical mixture range. The authors attribute this observation to the weathering of HCHs during the process of long-range atmospheric transport, and thus the unchanging HCH signature reaffirms that there is limited degradation of HCHs in the Tibetan Plateau environment after deposition. These studies provide some insights into the influence of melting glaciers on the fate of POPs and confirm that melting glaciers can modify, and even amplify, the organochlorine pesticide (OCP) burden of the contemporary Tibetan environment.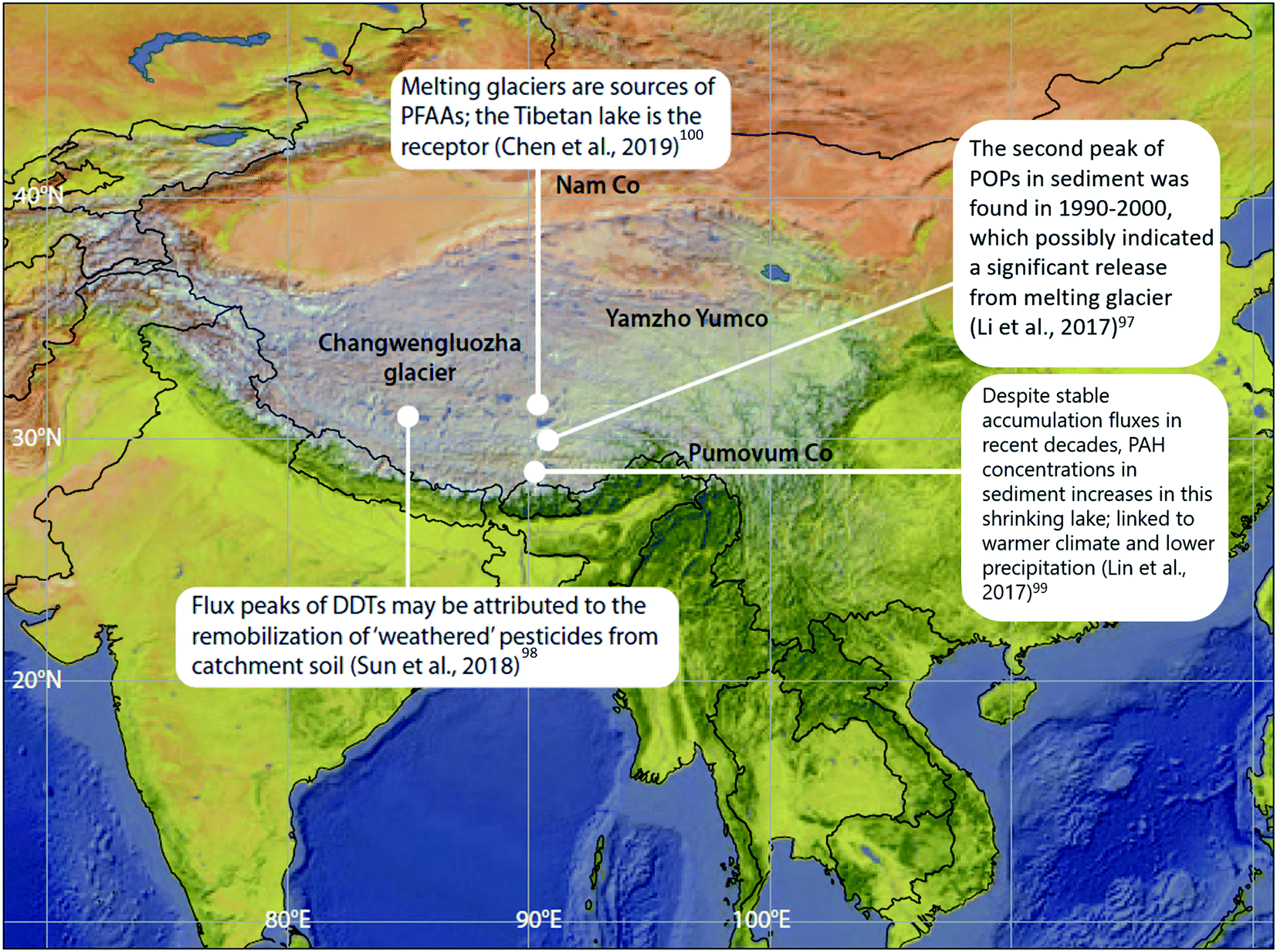 | ||
| Fig. 9 Climate change-related effects on the distribution of POPs, PAHs and PFAAs observed on the Tibetan Plateau. | ||
The situation in Tibet is complicated however, because lakes in the Tibetan Plateau are experiencing different environmental changes. In addition to potentially receiving glacial meltwater, some lakes are experiencing greater precipitation, while others are shrinking as a result of the warming climate and reduced precipitation. In the shrinking lake Pumoyum Co located in the southern Tibetan Plateau (Fig. 9), sediment PAH concentrations increased over the past decade, but accumulation fluxes remained stable,99 as opposed to the increasing fluxes of OCPs observed in lakes in the northern Tibetan Plateau.97,98 This observation is attributable to the recent low precipitation rates and lower catchment erosion within the watershed of Pumoyum Co.99 This indicates that the impact of climate change and warming is complex, whereby pollutant migration can be influenced significantly by catchment erosion (via sedimentation rates), which is dependent on the meteorological conditions (dry or wet) of the lake's catchment.
Chen et al. (2019)100 studied the release of water soluble PFASs from a Tibetan glacier. The PFAS composition in Lake Nam Co (Fig. 9) was similar to that observed in glacier ice. Additionally, during the melt season, release fluxes of PFAAs were strongly related to glacial melt intensity (i.e. river flow rate). Moreover, at the end of the melt season, when melt intensity is very low, PFAA release fluxes were found to be positively correlated with PFAA concentrations in glacier ice. This evidence suggests that melting glaciers are sources of PFAAs, while the Tibetan lakes serve as the receptors. Such effects will increase the risks of emerging pollutants to freshwater sources and is of great concern.
Similar to these studies, it may be possible to relate temporal contaminant deposition in Arctic systems receiving enhanced contaminant loads due to climate-induced melting glaciers. Pouch et al.101,102 reported contaminant deposition in sediment cores collected in 2013 along a transect between the inner and outer areas of two fjords, Hornsund and Kongsfjorden, on the Spitsbergen coast of Svalbard. Approximately 67% and 77% of the drainage basins are covered by glaciers in the Hornsund and Kongsjorden fiords, respectively. Inner parts of the fjords receive enhanced inputs of glacier meltwater103,104 and these inner parts (i.e. glacial bays) are characterized by high sediment accumulation rates (0.23–0.39 cm per year) and low organic matter content.101,102 Pouch et al.101,102 observed sediment cores sampled from the innermost glacial bays of the fjords contained greater concentrations of ∑PCB7 and ∑PAH12 (Fig. 10, top and middle panels), and hexachlorobenzene (HCB). In the glacial bays of Hornsund and Kongsfjorden, ∑PCB7 were as high as 1.47 and 1.41 ng g−1 dw, respectively, compared to the outer fjord maximas of 0.25 and 0.62 ng g−1 dw. Similarly, the Hornsund glacial bay sediment in site H5 contained maxima of 1974 ng g−1 dw ∑PAH12 and 144 pg g−1 dw HCB compared to 209 ng g−1 dw ∑PAH12 and 53.6 pg g−1 dw HCB from the corresponding outer fjord area in H1. The congener profile in the inner fjord areas was dominated by light chlorinated PCBs, (PCB-28 and PCB-52), which tend to prevail in atmospheric samples, providing further evidence that they originate from direct inputs of glacier meltwater. The exponentially higher rate of deposition of contaminants in the glacier bays of Svalbard post-1990 were attributed to the corollary increase in glacier runoff due to climate change. Van Pelt et al. (2019)105 applied a multilayer subsurface model combined with a surface energy balance to estimate glacier run off for all of Svalbard using high resolution climate data (i.e. air temperature, precipitation, relative humidity, cloud cover, and air pressure), glacier coverage, elevation parameters, and snow/firn conditions. The multi-decadal runoff simulation (Fig. 10, bottom panel) indicates a gradual increase in annual run off from 11 Gigaton per annum (Gt/a) to 34 Gt/a, primarily driven by projected temperature increases during the summer melt period.
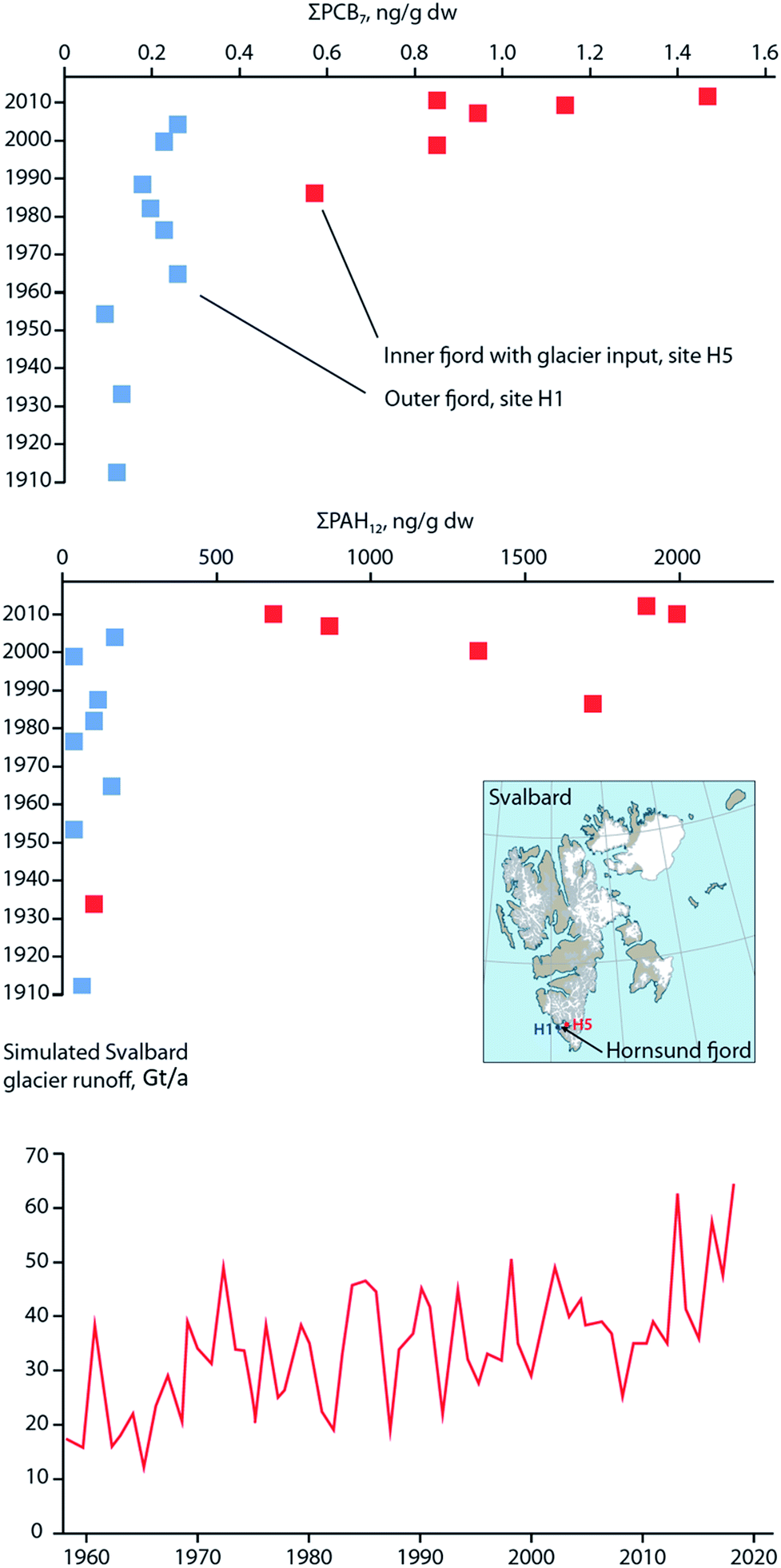 | ||
| Fig. 10 Sediment contaminant concentrations and glacial run-off in Svalbard. (top and middle) concentrations of ∑PCB7 and ∑PAH12, respectively, in dated sediment cores from two sites located in the outer fjord (H1), and inner glacial bay (H5), in Hornsund fjord, southern Svalbard. Data source: Pouch et al. (2017). (bottom panel) Simulated glacier runoff (Gigaton per annum) for all of Svalbard from 1957 to 2018. Data source: van Pelt et al. (2019).105 | ||
In the post-2007 glacial-mass loss era in Lake Hazen, in which the runoff rate reached 1–1.8 Gt/a (Fig. 12) and sediment accumulation rates increased by a factor of eight, analyses of dated sediment cores indicated enhanced delivery of PFAS to the lake.106 The peak PFAS sedimentary fluxes in Lake Hazen are coincident with the glacier discharge volumes (Fig. 12).
Fluxes of POPs and CEACs have also been shown to increase in Lake Hazen, the largest lake by volume north of the Arctic Circle, based on a study using dated sediment cores.107 Surface temperatures of glacier-covered regions of the Lake Hazen watershed experienced a 2.6 °C warming over the period 2000–2012, with the greatest change occurring from 2007 to 2012. The increasing air temperatures and decreasing surface albedo, resulted in major increases in glacial runoff inputs to the lake. Glacier runoff increased lake outflow by 370% for the period 2007–2012 relative to 1996–2006. Sediment accumulation rates post-2007 were on average eight times higher relative to the pre-1948 baseline period. Legacy OCPs were determined in a sediment core from a deep point (267 m) in the lake. OCP fluxes peaked in the 1970–80s, consistent with the known uses of DDT and other OCPs, and showed rising concentrations post-2000 (Fig. 11) reflecting re-mobilization of OCPs previously deposited and stored in glaciers. Previous studies based on a core collected in 1990 at the same location had shown low fluxes of OCPs.108
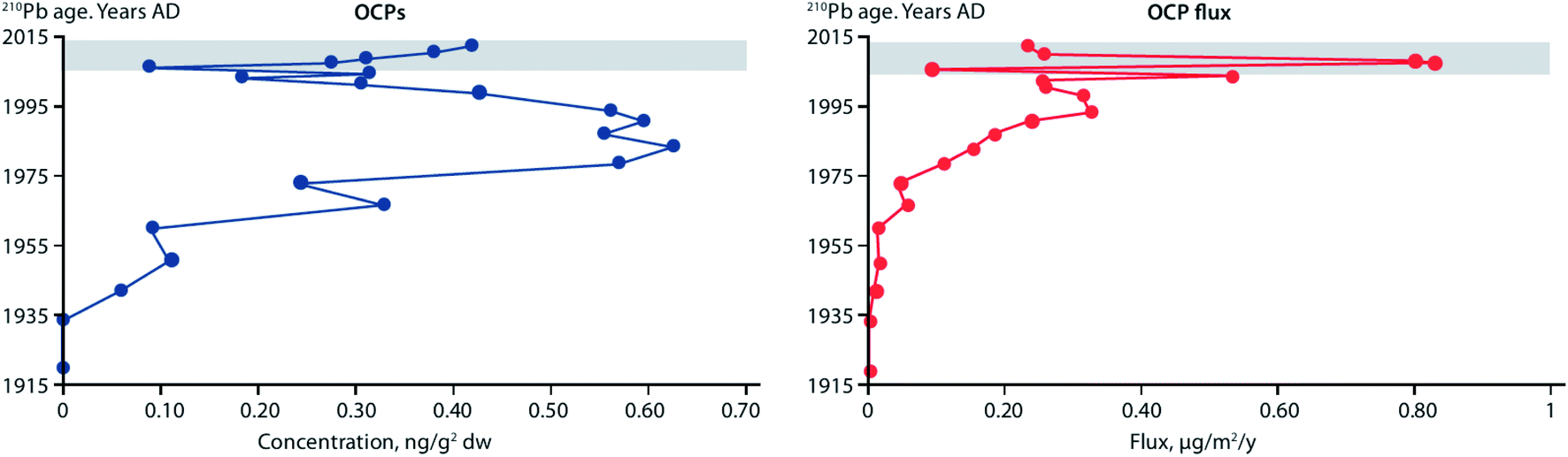 | ||
| Fig. 11 Concentrations and fluxes of total organochlorine pesticides (OCPs), including DDT, HCH, dieldrin, endrin, and chlordane-related compounds, in a dated sediment core from Lake Hazen, Canada. Grey area denotes period of elevated glacial runoff and higher sedimentation rates. Redrawn from Lehnherr et al. (2018).107 | ||
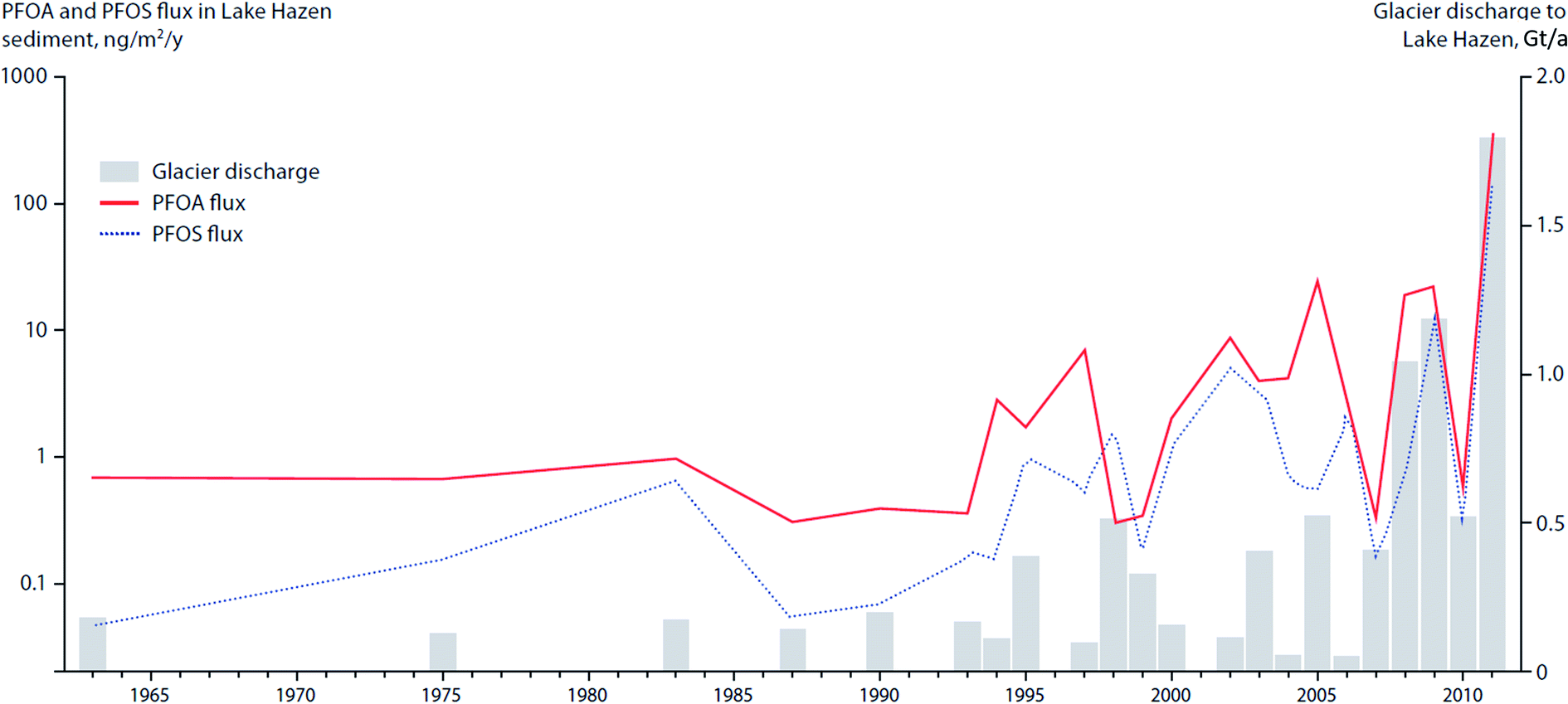 | ||
| Fig. 12 Deposition of PFAA into Lake Hazen sediments in relation to climate-driven variability in glacier discharge rates. Data sources: MacInnis et al. (2019)106 and Lehnherr et al. (2018).107 | ||
Climate change has been linked to increasing meltwater discharges, shorter periods of ice-coverage, increased particle loads, and generally higher flow rates in rivers discharging into the Arctic Ocean.109,110 The increased discharge volume and flow velocity from rivers is expected to bring with it an increased contaminant load of both dissolved and particle-bound contaminants.111 The increased contaminant load has been hypothesized to include remobilized legacy POPs retained in sediments,111 PAHs from sediment-laden ice and coal deposits eroded by deglaciation;91 as well as POPs and CEACs with sources close to settlements.109,111–113
Taken together, these studies reporting increased contaminant deposition via glacier melt in alpine and Arctic regions, provide insights into the influence of melting glaciers on the fate of POPs, and confirm that melting glaciers can amplify the contaminant burden of receiving waters globally. As such, further characterization of contaminants in the cryosphere is warranted to estimate potential releases through meltwaters into Arctic rivers, lakes, and ultimately the ocean. This is particularly germane for those substances known to biomagnify in Arctic marine food webs.
Some CEACs, such as PFASs, especially the anionic PFAAs and halogenated OPEs, have appreciable water solubility, low Henry's law constants, and slow degradation rates, which presents concerns regarding their susceptibility to long-range oceanic and riverine transport.113–115 In both of these substance groups, concentrations similar to, or exceeding, those of traditional POPs have been reported in Arctic environmental media, including air, snow, water, and biota.16,106,116–118
Investigating potential LRT pathways of selected CEAC, Sühring et al. (2016b),46 McDonough et al. (2018),116 and Schmidt et al. (2019)119 found strong indications for oceanic- and riverine-based LRT of chlorinated organophosphate esters (Cl-OPEs) and PFASs. Spatial analysis of Cl-OPEs in the Arctic indicated higher concentrations in near-shore areas, suggesting input from snow melt runoff and riverine sources.46,116 Moreover, model estimates further supported the hypothesis that oceanic (including riverine) transport of Cl-OPEs could explain some of the high concentrations reported in the Arctic, despite the lack of predicted atmospheric LRT for most OPEs,120 with an estimated median inventory of ∑11OPE mass of 4100 tons in the Canadian Arctic Ocean based on measurements across the Canadian Arctic.121 However, sampling of glacier-fed rivers and lake water in the Canadian Lake Hazen watershed has revealed the presence of Cl-OPEs, implying atmospheric transport and subsequent deposition and accumulation in snow and glacier ice.118 Cl-OPEs were the most prevalent OPEs in six glacier-fed tributaries sampled in 2015 and 2018. The estimated loading into Lake Hazen by glacier-fed rivers in 2015 was 2.62 ± 1.3 kg Cl-OPEs, and 7.04 ± 3.24 kg total OPEs (∑OPE14 comprised of Cl-OPEs, alkyl-OPEs, and aryl-OPEs). Sun et al. (2020)118 also reported relatively lower OPE concentrations in non-glacier impacted Arctic lakes. ∑OPE14 concentrations in East and West Lakes on Melville Island, Canada corresponded to 4.7–5.3 ng L−1 and in 6.5 ng L−1 in small lake on Cornwallis Island, Canada. These levels are approximately half the concentration of Lake Hazen (10–15 ng L−1 ∑OPE14) despite being further north and more remote from anthropogenic activity. These results highlight the delivery of OPEs to Arctic lakes via annual atmospheric deposition, with release of accumulated OPEs in glaciers acting as an additional significant source in specific watersheds.
PFAAs can also be deposited from the atmosphere via snowfall. Recently, MacInnis et al. (2019b)106 investigated the delivery of PFASs from overlying snow and ice into Lake Hazen during the short and pronounced summer-melt period. ∑PFAA were found to be well mixed throughout the 250 m water column during the ice-covered period (September to May, ∑PFAA <0.5 ng L−1), elevated in the top 5 m during the melting period (late May, ∑PFAA up to about 3.1 ng L−1), and were well-mixed during the ice-free open water period (July–August, ∑PFAA <0.5 ng L−1). Many Arctic lakes, like Lake Hazen, are covered by ice and snow for many months of the year resulting in a sudden surface pulse of contaminants during spring melt. Depth-concentration profiling of contaminants in Arctic lake waters during melting events can provide pertinent data for understanding the contaminant-specific flux into underlying waters, as well as transfer further downstream with exiting river or meltwater flow.
E.3. Indirect effects of a changing Arctic on contaminant sources
It is well known that PCBs from domestic,122–124 industrial123,125 and military sources126–131 contribute to local Arctic pollution. PCBs are also leaking from building materials and electrical installations in major settlements in the Arctic, e.g. Longyearbyen and Barentsburg on Svalbard.132 Many of these sources were registered and, where possible, treated, sealed or removed.132 Unfortunately, emissions from diffusive sources continue even 40 years after the global ban of PCBs. However, less is known about the emissions of CEACs associated with local domestic and/or industrial sources,16,25 which are expected to increase as the indirect result of increasing population and human activities due to Arctic warming. For example, many of CEACs are released into the environment through sewage. Many sewage systems are inadequate, deteriorating133–135 and difficult to upgrade in the Arctic.136,137 Untreated sewage is either directly released into estuarine, coastal, and freshwater systems, or stored for long periods in sewage lagoons. In a warming Arctic, the permafrost underneath such lagoons is melting, and can no longer prevent contaminants from seeping into the adjacent soil and aquifers.135,138Sühring et al. (2016b)46 reported tri-n-butyl phosphate (TnBP), an OPE which is a major component in aircraft fire-resistant hydraulic fluid, Skydrol 500B-4,139 in air samples near a military airport at Resolute Bay in the Canadian Arctic. Consistent with these results, Sun et al. (2020),118 reported TnBP in two lakes, Resolute Lake and Meretta Lake, at concentrations ranging from 20–40 ng L−1. These lakes are in closer proximity to the airport and waste disposal sites in the Resolute Bay community, compared to four other lakes that had TnBP at much lower concentrations, 1–3 ng L−1.118 High local concentrations of TnBP have previously been linked to emissions from airports.140 Additional OPEs with apparent local sources (i.e. high concentrations and detection frequencies at land-based stations, and low detection rates from ship-based sampling) include 2-ethylhexyl diphenyl phosphate (EHDPP)46,141 and tris meta-(cresyl) phosphate (TmCP).46 EHDPP contamination has been linked to the use of hydraulic fluids in fishing boat motors from the higher concentrations detected close to the coast or around harbors compared to those measured in open ocean waters.119 An increase in fishing activities in the Arctic due to reduced sea ice, as well as an increase of industrial activities, military activities, shipping, and tourism (increasing frequency of flights) are likely to result in an increase of these types of contaminants (e.g. TnBP and EHDPP) in local environments.
Furthermore, the retreat of land-fast ice (glaciers) and permafrost is expected to allow the exploitation of previously unavailable resources (minerals, petroleum, gas etc.). For mining and refining, new settlements and infrastructure will be established or expanded, leading to an increased potential for local pollution.137,142–144 Pollution can also be related to water discharges from offshore installations, as well as from unplanned discharges. Unplanned discharges include leaks of petrochemicals, but also discharge of PFAS-containing fire-fighting foams in the case of incidents, training, or testing of firefighting equipment as reported for Longyearbyen and Ny-Ålesund (Svalbard).145 Sühring et al. (2017)146 reported the potential significance of offshore oil and gas installations as sources for marine PFAS contamination based on a model simulation using use and discharge data of offshore fire-fighting foams. Lescord et al. (2015)147 also noted highly elevated perfluorooctane sulfonic acid (PFOS) concentrations (30–40 ng L−1) in water from lakes downstream of the Resolute Bay airport, and also reported levels of other PFAS substances associated with fire-fighting activity, including fluorotelomer sulfonates and other perfluoroalkyl sulfonates.
The development of economically-feasible technology for fossil fuel production from shale gas (i.e. fracking technologies) and oil sands in the North American Arctic has led to increased land-based exploitation of petroleum resources.148–150 Such production technologies are associated with considerable environmental consequences, including large pollutant burdens and destruction of habitats in oil sands regions.151–154 Recent reports confirm an increase in local contamination of PAHs and POPs in the context of oil sands and shale gas production sites.155–158 Furthermore, the reclamation of previously degraded sites is currently progressing very slowly in Canada and the US, leaving large areas without the expected conservational or recreational value.155,159–162
The increased probability of oil spill incidents associated with oil and gas production and refinement are expected to add to the pollutant loads in the Arctic. Experiences from earlier oil spill incidents in the Arctic (both on land and offshore) revealed that such events have severe consequences both for humans and wildlife in the region.163–166 Furthermore, as a consequence of reduced summer sea ice, new shipping routes along the northeast and the northwestern Arctic coasts are currently being explored as potential international shipping routes167 that will serve as viable alternatives for the major Asia–Europe/America shipping routes.168–170 Such Arctic shipping routes will shorten transportation times considerably, and thus are economically beneficial.171 However, Arctic transit routes will inevitably lead to local pollution in the region. Four major environment threats need to be considered as a result of increases in Arctic shipping in the regulatory context: accidents resulting in related oil/chemical spills, emissions from combustion engines, release of wastes into Arctic waters (e.g. bilge release and ballast tank maintenance), and emissions of chemicals used as surface coatings on ship hulls.172
F. Changing Arctic cryosphere (snow, permafrost, sea ice and glacial melts) – contaminant amplification
How does cryospheric change affect fate of contaminants in the Arctic?While climate change is driving a strong declining trend of areal sea ice cover during the Arctic summer, the nature of sea ice is also changing. The Arctic Ocean is now dominated by first-year sea ice (FYI) with a substantial decline in older, multi-year ice (MYI); in other words, there has been a shift from a perennial icescape to an annual one.173 Climate warming is resulting in the thawing of permafrost in lake catchments and shorelines with a subsequent alteration of lake and river water chemistry. The thawing of ice-rich permafrost ground is resulting in the formation of thermokarst, the detached layers of land, shallow lakes, and wetlands created from land subsidence. In turn, this can influence watershed hydrology and delivery of dissolved organic carbon (DOC) and particulate organic carbon (POC) to lakes and to coastal seas.174,175 Furthermore, sea-level air temperatures are increasing176 and, as an apparent consequence, precipitation type (snow vs. rain) and amounts are changing, and possibly increasing across large parts of the Arctic.177 The influence of these cryosphere changes on contaminant behavior is likely to be pronounced and is explored further in the following sections.
F.1. Contaminant fate in changing sea ice
MYI used to cover ∼60% of the Arctic Ocean, but over the last few decades has declined to <30%, with the oldest ice (4+ years old) comprising only 3–5% of the ice cover in the Arctic.173 With regards to the entry and fate of organic contaminants, sea ice has previously been viewed simply as a seasonal barrier limiting the transfer of contaminants between the atmosphere and the surface ocean.84 However, field studies have demonstrated that the sea ice snowpack is a notable repository of atmospherically-derived chemical pollutants, including PFASs178 and OCPs.85 In addition, sea ice itself also contains these chemicals, with α- and γ-HCH present in brine-rich FYI at concentrations much higher than the underlying sea water and other Arctic media.82 This implies that single-season ice or FYI can accumulate organic contaminants to such an extent that concentrations of these chemicals are amplified relative to the underlying sea water or surface meltwater.84 This process appears to be controlled by brine, the saline water present in young ice. As sea ice forms, thickens and then ages most of the salts present in the freezing seawater are rejected into the underlying ocean, leaving only a small amount trapped within a network of highly saline brine pockets. As ice continues to grow, more salts are expelled, and seasonal meltwater at the surface ‘flushes’ the sea ice, reducing its bulk salinity further. Seasonal or FYI has a higher bulk salinity179 and contains more brine per unit volume than MYI, and this brine appears to influence the behavior and fate of organic micropollutants present in the sea ice system, which is dominated by FYI.82,180Garnett et al. (2019)180 conducted experiments in a controlled sea ice facility to understand the uptake and accumulation of a range of POPs in young, growing sea ice. Sea ice formation was shown to result in the entrainment of the chemicals from seawater. The subsequent contaminant concentration profiles in bulk ice showed the highest levels in both the upper (ice–atmosphere interface) and lower (ice–ocean interface) layers, indicating that the incorporation and distribution of POPs is strongly influenced by brine advection within young ice. Fig. 13 shows the similarity in the vertical profiles of contaminants and NaCl in ice, with both displaying a characteristic ‘C’-shape driven by brine dynamics in the ice with the dissolved contaminants behaving akin to the salt ions. Application of a one-dimensional sea ice brine dynamics model was able to accurately replicate the organic contaminant profile in the ice. The concentrations of seven POPs (α- HCH, γ-HCH, PCB-28, PCB-52, chlorpyrifos, PBDE-47, PBDE-99) were higher in the ice brine than the bulk ice (i.e. frozen water, trapped air, brine pockets) demonstrating that the contaminant solutes are behaving similarly to the salt ions in the brine (a solution within the ice matrix). The chemical concentrations in ice brine were similar to, or higher than, their concentrations in the beneath-ice seawater, indicating an enrichment of semi-volatile contaminants. This enrichment occurs in the same way as the ice brine becomes more concentrated due to the ‘freezing out’ of water molecules and the lack of incorporation of solutes into or between ice crystals. Enrichment factors (EF), the ratio of the chemical concentration in ice brine to that in the underlying seawater, exceeded one for some of the chemicals in the study (e.g. α-HCH, chlorpyrifos, and PCB congeners).
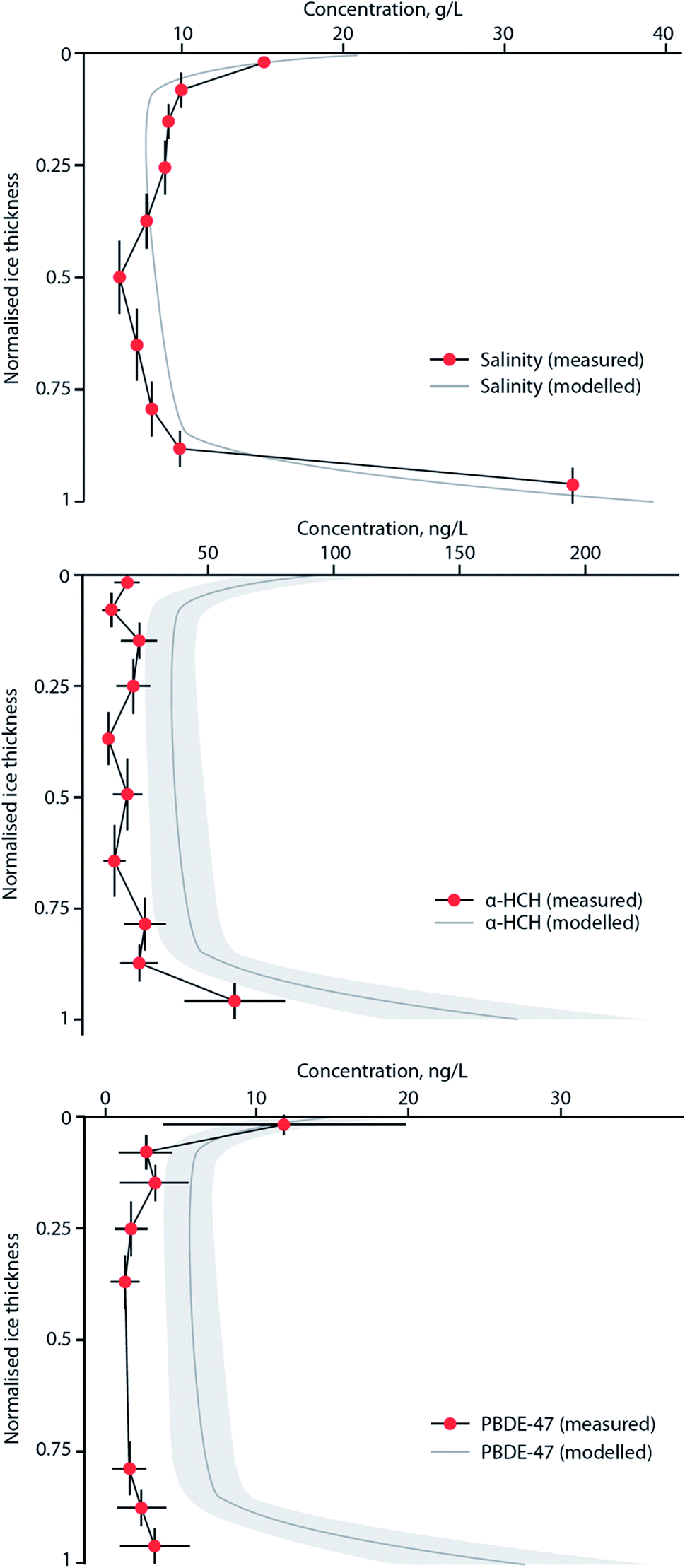 | ||
| Fig. 13 Modelled and measured concentration profiles for salinity, α-HCH, and PBDE-47 in artificial sea ice grown at −18 °C to a depth of 26 cm in a specialized sea ice facility. Vertical bars indicate ice layer thickness. Variability (±2SD) is indicated for both measured data (horizontal bars) and results of a one-dimensional sea ice brine dynamics model (grey shade). Source: Garnett et al., (2019).180 | ||
Table 3 highlights EFs calculated from the experimental ice chamber work of Garnett et al. (2019),180 as well as those derived from several Arctic sea ice studies. The highest EFs were observed in the saltiest brine located in the upper ice sections (closest to the atmosphere) similar to what was also observed in FYI in the Amundsen Gulf of the Canadian Arctic. EFs up to four were observed for α- and γ-HCH,82,85 with concentrations that exceeded any other abiotic media in the Arctic. A common feature on the surface of new and young ice is frost flowers. These are clusters of dendritic ice crystals that have a very high salt content and form at the interface between a warm ice surface and a cold atmosphere, particularly on new ice which possesses a thin liquid brine layer on the surface (see ref. 181). Contaminant enrichment in frost flowers is significant, with EFs >10 for PBDE-47 and PBDE-99 observed in the experimental sea ice facility.180 Importantly, this phenomenon was also observed in frost flowers sampled on coastal sea ice near Barrow, Alaska,182 with EFs approaching 40 for an array of organic contaminants. In a warmer Arctic, the frequency of frost flower occurrence is likely to increase over much wider areas of the Arctic Ocean. The implications of this enrichment of contaminants in frost flowers is unclear at present, but is likely to result in enhanced surface to air transfer, and ‘re-cycling’ of contaminants in the Arctic marine environment over a large area.
| Enrichment factor | Bulk ice depth, cm | Chemical | Data sourceb | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| NaCl | α-HCH | γ-HCH | Chlorpyrifos | PCB-28 | PCB-52 | PBDE-47 | PBDE-99 | |||
| a na: not applicable, nm: not measured. b Data sources: (1) Garnett et al. (2019);180 (2) Pućko et al. (2010);82 (3) Pućko et al. (2011).85 | ||||||||||
| EFbulk ice/seawater | 17 ± 1 | 0.4 ± <0.1 | 0.1 ± <0.1 | 0.1 ± 0.1 | 0.2 ± 0.1 | 0.2 ± 0.1 | 0.2 ± <0.1 | 0.1 ± 0.1 | 0.4 ± 0.2 | (1) |
| 26 ± 1 | 0.3 ± <0.1 | 0.2 ± 0.1 | 0.3 ± 0.2 | 0.3 ± 0.2 | 0.2 ± 0.1 | 0.1 ± <0.1 | 0.2 ± 0.1 | 0.4 ± 0.2 | (1) | |
| 30 | 0.4 | 0.4 | 0.5 | nm | nm | nm | nm | nm | (2) | |
| 90 | 0.2 | 0.3 | 0.3 | nm | nm | nm | nm | nm | (3) | |
| 5 | 0.3 | 0.3 | 0.4 | nm | nm | nm | nm | nm | (3) | |
| EFbrine/seawater | 26 ± 1 | 1.4 ± <0.1 | 0.6 ± 0.2 | 1.0 ± 0.8 | 1.2 ± 0.7 | 1.3 ± 0.5 | 1.2 ± 0.3 | 0.7 ± 0.5 | 0.9 ± 0.6 | (1) |
| 90 | 4.4 | 3.9 | 4 | nma | nm | nm | nm | nm | (2) | |
| EFfrost flower/seawater | naa | 2.4 ± <0.1 | 0.2 ± 0.1 | 0.2 ± 0.2 | 0.3 ± 0.2 | 0.2 ± 0.1 | 0.4 ± 0.1 | 6.6 ± 4.4 | 24 ± 15 | (1) |
| EFfrost flower/sea ice layer | na | 5.0 ± <0.1 | 1.5 ± 0.5 | 2.0 ± 1.5 | 2.4 ± 1.4 | 2.5 ± 0.9 | 3.0 ± 0.7 | 30 ± 20 | 50 ± 31 | (1) |
F.2. Role of the marine snowpack and seasonal thaw
The transfer of organic contaminants between ice-rafted snowpack and sea ice is facilitated by the ingress of the ice brine into the base of the snowpack during the winter. Observations conducted on FYI in the Amundsen Gulf in the Canadian Arctic estimated that upward percolating sea ice brine provides as much as 50% of the α- and γ-HCH burden present in the overlying snowpack.85 This process is clearly facilitated by the movement of brine (i.e. ‘brine engine’) present in young and FYI and is greatly diminished or absent in MYI.84 Snow meltwater can percolate into ice during the onset of thaw and was observed to result in an increase of 2 to 32% of the α- and γ-HCH burden in the sea ice.As the melt season progresses, the thawing snowpack gives rise to melt ponds which are a dominant feature on ice floes during the early summer.183 Organic contaminants present in the melting snowpack may either volatilize back to the atmosphere, depending on their volatility and aqueous solubility, and/or be supplied to melt-ponds. Gas exchange between the atmosphere and the melt-pond surface may serve to increase levels in the pond water, particularly for those contaminants initially found at very low levels in either the snowpack or melt-pond. For example, current use pesticides (CUPs), advected into the Arctic through LRAT during the early summer will undergo air–surface exchange with net loading into melt-pond water. In addition, wet deposition through late season snowfall and rainfall events can significantly enhance contaminant loading to melt-ponds.83Fig. 14 illustrates a time-series of CUP concentrations in the snowpack and melt-ponds over the May/June melt season on landfast FYI between Griffith and Cornwallis Island in the Canadian Arctic Archipelago. Of the five CUPs investigated, chlorpyrifos (insecticide) and Dacthal (herbicide) showed substantially higher concentrations in the melt-pond water compared to predicted concentrations based solely on gas-exchange with the atmosphere. This indicates the important role of the melting snowpack in releasing contaminant residues, while also pointing to the significance of precipitation events which serve to add these chemicals to the melt-pond directly. Precipitation in the form of snowfall and, importantly, rain events in late winter, are predicted to increase over large parts of the Arctic Ocean,184 enhancing the transfer of atmospheric contaminants to sea ice surfaces and melt-ponds. Melt-pond enrichment factors (MEF) (the ratio of contaminant concentrations in the melt-pond to those in the underlying seawater) of ∼2–10 were calculated for the pesticides endosulfan I, chlorothalonil, and chlorpyrifos. This was based only on net loading through gaseous transfer from the atmosphere using concentrations measured in air during the Arctic summer period. MEFs for Dacthal, based on direct measurements in melt-pond water, were found to be much higher, at ∼10–16. As a consequence of melt pond enrichment, the exposure of ice algae and phytoplankton to contaminants could significantly increase following late-season melt-pond drainage, either via water percolating through the remaining ice, or drainage directly into beneath-ice seawater.
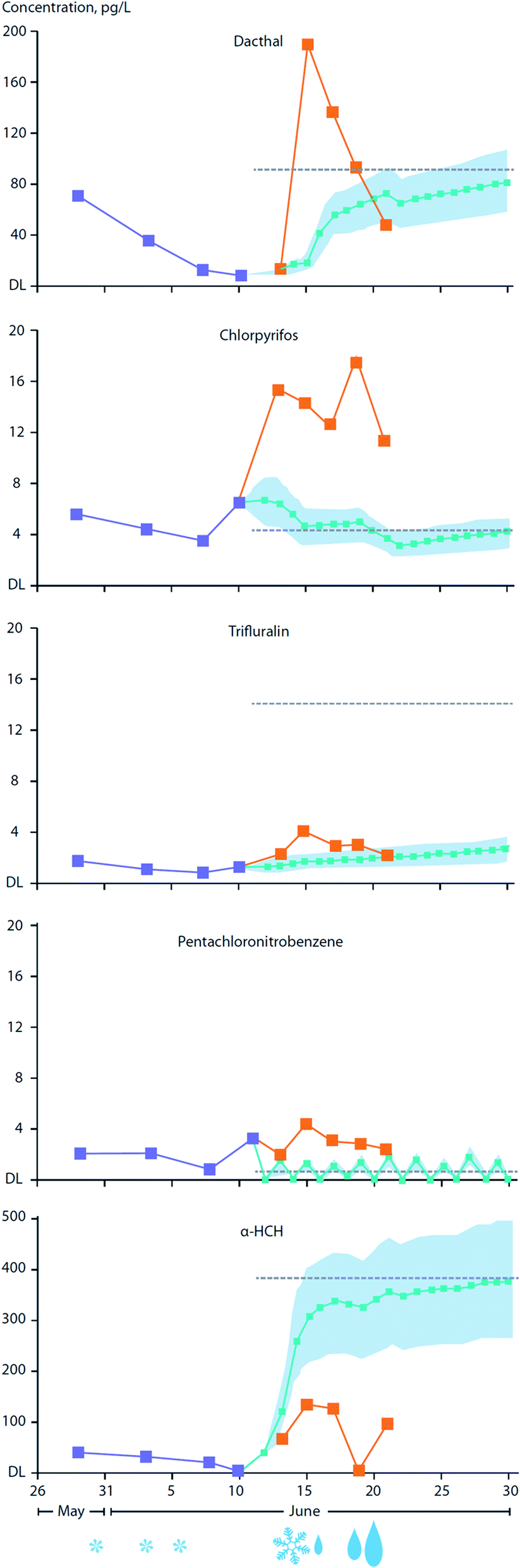 | ||
| Fig. 14 Concentrations of Dacthal, chlorpyrifos, trifluralin, pentachloronitrobenzene, and α-HCH in snow (Csnow) and in melt ponds as measured (Cmp/Mes), modeled (Cmp/Mod) and calculated at equilibrium partitioning with atmosphere (Cmp/Eq), along with the relative types and amounts of precipitation (denoted as size and type of symbols underneath graphs) as a function of season progression. Light blue shadows show uncertainty range for CMP/Mod; concentration scales not uniform between graphs. Measurements were conducted on melting landfast ice in the Canadian Archipelago. Source: Pućko et al. (2017).83 | ||
Contaminant enrichment in sea ice is driven mainly by brine processes in young FYI. As FYI is prevalent over a much wider area of the Arctic Ocean following the decline in the extent of MYI, then seasonal enrichment and subsequent release of contaminants from melting ice is likely to provide a significant route of contaminant exposure to the lower trophic levels of the marine food web. Importantly, many organisms situated at the base of the pelagic food web are abundant in sea ice and inhabit the network of brine inclusions and channels located towards the base of the ice. The exposure of sympagic biota in particular, such as ice algae and the associated zooplankton present in brine channels, will be significant over a much wider area in a warming Arctic Ocean. There is some uncertainty over the release of organic contaminants during ice melting, with less soluble contaminants retained in the ice matrix until final melt.180 Furthermore, the various stages of melt pond evolution, and subsequent meltwater drainage, could give rise to meltwater percolating into the ice pack appreciably. Alternatively, and in addition to percolation, meltwater may drain abruptly into the underlying seawater depending on ice breakup and the nature of the melt season (e.g. rapid thaw vs. slow thaw).83 This will have implications for the delivery and exposure of organic contaminants to ice-associated biota. For example, the earlier onset of seasonal thaw, formation of melt ponds, and final drainage following ice floe breakup, could coincide with the widespread under-ice phytoplankton spring bloom.34 However, this will vary spatially across the Arctic Ocean and coastal seas, as well as temporally, depending on the ice type and the nature of seasonal warming each year.
F.3. Permafrost degradation and mobilization of POPs
Climate warming is resulting in the thawing of permafrost in lake catchments and shorelines with resulting alterations in lake and river water chemistry. The thawing of ice-rich permafrost ground causes the formation of thermokarsts, detached layers of land, shallow lakes, and wetlands formed from land subsidence, that can influence watershed hydrology and delivery of DOC and POC to lakes and the ocean.174,175Permafrost thawing and formation of thermokarsts has implications for the partitioning and transport of POPs that may be deposited to terrestrial environments via atmospheric deposition and then mobilized with DOC/POC runoff. Eickmeyer et al. (2016)185 found that slump-affected lakes contained higher total organic carbon (TOC)-normalized concentrations of ΣPCBs, HCB and ΣDDT than nearby reference lakes that were unaffected by thaw slumps (Fig. 15). ΣPCBs, HCB and ΣDDT concentrations were positively correlated with mean total sedimentation rate for each lake (Fig. 15). The higher TOC-normalized concentrations in slump-affected lakes were explained by the reduced availability of organic matter for adsorption in the water column, so that the POPs were associated with a smaller pool of organic carbon. Slump-affected lakes are generally observed with lower DOC concentrations than unaffected lakes, which is thought to be related to the low DOC in runoff from recently disturbed areas of exposed mineral soils.174,186
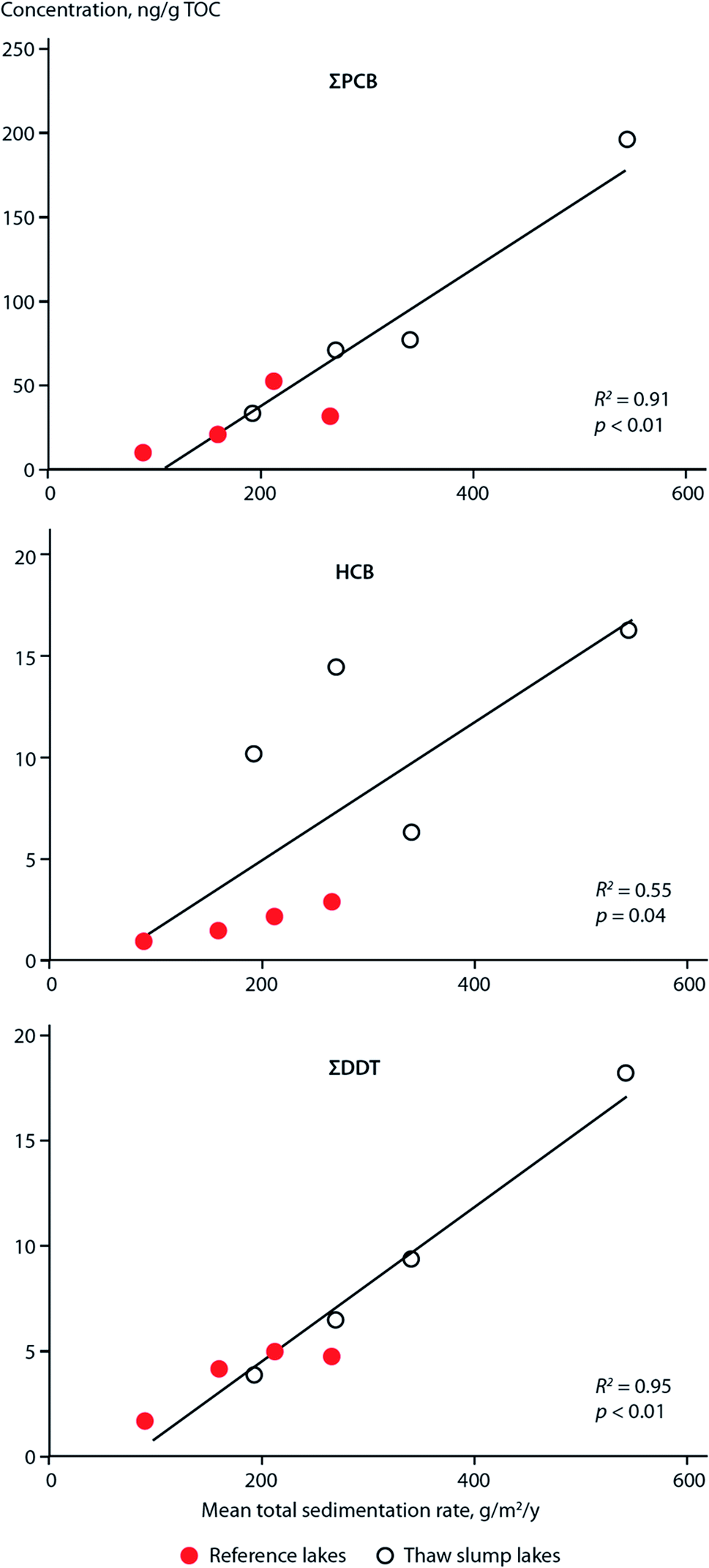 | ||
| Fig. 15 Concentrations (ng g−1 TOC) of ΣPCBs, HCB and ΣDDT in sediments from eight study lakes, including reference lakes (red points) and lakes affected by thaw slumps (white points) in tundra uplands adjacent to the Mackenzie Delta, Canada, plotted versus the mean total sedimentation rate (g per m2 per year) in each lake. Redrawn from Eickmeyer et al. (2016).185 | ||
Fluxes of POPs (concentrations × sedimentation rates) were generally higher and more variable in slump-affected lakes than the reference lakes in the tundra uplands adjacent to the Mackenzie Delta.185 Although temporal resolution was limited due to low sedimentation rates, the historical profiles generally had sub-surface maxima in both reference and impacted lakes. Subsurface maxima have generally been observed in arctic lake sediment cores because of the greater emissions of POPs in previous decades.187,188 Thus, it appears that current permafrost degradation and slumping at these sites increased inputs of POPs but not sufficiently to cause a major change in inputs relative to past deposition in the 1960–80s. Fluxes of POPs have also been shown to increase in Lake Hazen in the Canadian Arctic based on a studies using dated sediment cores (see Section E.2).
Cabrerizo et al. (2019)189 compared concentrations of PCBs and OCPs in river water from the West Lake catchment with the nearby East Lake river on Melville Island in the Canadian Arctic archipelago. The West Lake catchment experienced impacts from permafrost degradation associated with significant warming during the period 2007 to 2012, while the catchment of East Lake has undergone little change.190 PCB concentrations and homolog profiles were similar in both rivers during the snowmelt period in mid-June (Fig. 16), suggesting a similar source such as snow melt runoff. However, in the brief snow-free summer period during July, concentrations and PCB homolog profiles differed between the two rivers. Much higher concentrations of dissolved and particulate PCBs and OCPs were observed in West River, and lower proportions of di-, octa- and nona-chlorinated PCB congeners were seen the East River water, suggesting differing sources. It should be noted that atmospheric deposition is the only source of POPs to these very remote catchments.
 | ||
| Fig. 16 Concentrations of ΣPCBs70 and homolog groups based on degree of chlorination (CL). In suspended particulate matter during melt run off (mid-June) and ice-free (mid-July) conditions in West and East Rivers on Melville Island, Canada in 2016. Redrawn from Cabrerizo et al. (2019).189 | ||
Higher concentrations of PCBs on suspended particulate matter (SPM) were also detected in West Lake (ΣPCB70 = 75 pg L−1) in comparison to East Lake (ΣPCB70 = 1 pg L−1). West Lake has been impacted by several subaqueous slumps since 2007 which may be related to permafrost degradation, although this is unconfirmed.191 West Lake is very turbid, with 50- to 100-fold greater turbidity compared to East Lake. Thus, the source of higher PCBs in West Lake is likely a combination of greater catchment inputs and higher SPM content. West Lake's elevated levels of SPM-associated PCBs are also reflected in the higher PCB levels detected in its biota, including zooplankton, fish stomach contents, and landlocked char.189,192 Whether elevated SPM-associated POPs in lake water is also observed in other turbid lakes or thermokarst-impacted lakes is not known at present. Using satellite imagery, one study found that 288 lakes on Banks Island in the western Canadian Arctic archipelago had been impacted by retrogressive thaw slumps, with the majority of the changes occurring between 1999 and 2013, a period of significant warming.193
F.4. Deposition of POPs to Svalbard glaciers and ice caps in a changing climate
The deposition of various contaminants, particularly POPs, has been identified in ice cores, surface snows, or both from four glaciers located on Svalbard.125,194–198 The four glacier sites are shown in Fig. 17. One goal of these studies was to identify potential POP source regions, which are generally understood to be the northern areas of Europe and Russia. Being that the glaciers and ice caps sampled in these studies are >500 meters above sea level (MASL), they are likely receptors of pollution from LRAT, although it is recognized that the coastal communities in the southern part of Spitzbergen, the main island of Svalbard, are also a potential source of POPs.199 | ||
| Fig. 17 Four glacial sites on Svalbard where ice core and/or surface snow have been analyzed for POPs deposition. | ||
Contaminants, including PCBs, brominated flame retardants (BFRs), and pesticides (both legacy and current-use) are well characterized in these Svalbard glaciers and/or the surface snow at the sites. Historical data from ice cores indicates that in some cases, the discontinued use of pesticides has resulted in lower inputs to the glaciers. However, for PCBs, inputs appear to vary across the years, indicating significant recycling in the global environment despite the cessation of new production. Cross-Svalbard results are available for some pesticides195 and show that the eastern site at Austfonna receives inputs of a larger number of pesticides and greater amounts of some, than Holtedahlfonna, suggesting that the dominance of southeast winds on Svalbard favors deposition to Austfonna. This was also observed in elemental carbon deposition work by Forsström et al. (2009).200 More recently, a cross-Svalbard investigation of PCB deposition to surface snow at the four sites in Fig. 17 shows greater inputs to the western Kongsvegen site than the eastern sites at Lomonosovfonna and Austfonna.198 It was concluded that the Kongsvegen site was affected by local sources of PCB from the western communities on Svalbard, in addition to an apparent overlay of a commercial PCB mixture from Europe observed at all sites. The differences in elevations of the four sites are key to this. Kongsvegen (700 MASL) and Austfonna (740 MASL) are within the tropospheric boundary layer (TBL) during the winter, therefore, both may be affected by local sources, although Austfonna is more remotely located from the western settlements. Holtedahlfonna (1150 MASL) and Lomonosovfonna (1202 MASL) are above the TBL all year201 and as a result, local emissions are less likely to accumulate there.
Air temperatures in Svalbard are increasing, and as an apparent consequence, precipitation amounts are also increasing. The influence of these changes on POP deposition could be significant because higher air temperatures will result in higher vapor pressures that retain highly volatile contaminants in the gas phase, but also shift traditionally less-volatile contaminants towards the gas phase.
Characterization of the effects of climate change on transport and accumulation of POPs in the Svalbard cryosphere and other glaciated areas in the Arctic requires four types of reliable information:1 LRAT sources;2 the climate change forcing or warming at those sources;3 present and historical deposition of contaminants to ice caps and glaciers;4 the current climate-change influences on those ice caps and glaciers. Of these four, the only reliable existing data for Svalbard glaciers are for contaminant deposition.3 Investigators have hypotheses about source regions,1 as noted earlier as being in northern Europe and Russia (i.e. northern Eurasia), however the exact source locations within these areas are not known. Additionally, there are some data suggesting positive temperature anomalies in these regions that may change the emissions of POPs.2 The most significant missing information regards the current climate change impact on Svalbard glaciers.4
The effect of temperature increases on the glaciers themselves is particularly important because on glaciers, contaminants interact only with air and the snow surface, as there is no canopy, soil or meltwater.202 The temperature at glacier surfaces is therefore critical. It is known that air temperatures will affect surface snow temperatures, however and these can vary widely from year to year independent of the effects of climate change. For example, Erath (2005)203 found that temperatures in the upper 0.5 m of seasonal snow at Kongsvegen. Svalbard varied from −18 °C in April 2000 to −5 °C in April 2001. Although both temperatures are well-below freezing, the striking difference shows that the unusually cold, late winter in 2000 influenced these snow temperatures.
Current conditions of air temperatures and precipitation amounts on Svalbard glaciers and ice caps are less well understood than those at sea level because of the absence of long-term data, characteristic of glaciers throughout the Arctic.204 Due to this data gap, the effects of climate change on glaciers are typically described on large spatial scales, while the contaminant results are reported on smaller spatial scales, such as single sampling sites. For example, Claremar et al. (2012)204 mention that the glacial mass on all of Svalbard has been falling for a century as a result of warming following the end of the Little Ice Age in about 1870.
Glacial recession is another large-scale measurement, and Erath (2005)203 shows that the Kongsvegen glacier has receded several km since 1960. Aas et al. (2015)205 mention using the placement of stakes to measure net snow accumulation during the previous winter on several Svalbard glaciers. However, these stake measurements are read only once each year, which loses finer-scale seasonal information. While each of these approaches is an indication of the effects of temperature and precipitation changes on glaciers over time, they do not describe the more localized or short-term effects, which may vary among sites because of differences in elevation. A potential solution to account for elevation differences is to use existing long-term data from one elevation to estimate the temperature at another elevation using the lapse rate (i.e. the rate at which temperature falls with altitude). However, Erath (2005)203 notes that the lapse rates at Kongsvegen differ from typical averages and are different between seasons. Automatic weather stations (AWSs) offer another solution. AWSs can operate unattended for extended periods of time, but with varying levels of success.203 On Svalbard, there are numerous AWSs, but only two AWSs have been installed and working on glaciers since the 2000s. Continuous data from AWSs would better clarify warming since the 1990s caused by increasing greenhouse gas forcing.204
The two operating AWS systems on Svalbard are at Kongsvegen site 6 (used by Erath, 2005203) at 537 MASL, and the other at Austfonna (370 MASL).205 Both of these AWSs measure air temperature and precipitation, two vital variables for understanding how the climate at these sites may be changing. However, both of these are well below the peak altitudes on both glaciers where samples for POPs measurement have been collected (700 MASL at Kongsvegen and 740 MASL at Austfonna), and there are no AWSs at Holtedahlfonna and Lomonosovfonna. Both existing AWSs have provided data to verify results of climate models,205 in this case the Weather Research & Forecasting (WRF) model combined with the Glacier Climatic Mass Balance (CMB) model. The results of a 10 year study by Aas et al. (2015)205 show that the model overestimated the measured temperature at Kongsvegen by 0.2 °C and underestimated the temperature at Austfonna by 1.9 °C, but that the variances between measured and modeled temperatures were consistent. Erath (2005)203 showed that the AWS at Kongsvegen provided some useful snow accumulation data, but AWS maintenance problems caused a loss of data from time to time. In the WRF-CMB model results, snow accumulation data tended to be variable, and it was concluded that the model was insufficient to resolve a number of small-scale processes (i.e. those on the order of less than one km because of the topography on Svalbard), which could include processes that might affect contaminant deposition.
While these model results are encouraging, the indication is that more AWSs would help understand the conditions at higher elevation glaciers where contaminant samples are collected. Without more AWS data, the understanding of how glacier climatic processes are changing will be very uncertain. Continued investigation of POPs deposition may need to rely on other information or estimates to describe climate-related changes over time.
The deposition of contaminants, such as BFRs, CUPs, and PFASs, have been previously reported in the Devon Island ice cap, located at 2000 MASL, Canada.206–209 In these studies, temporal trends in depositional fluxes were related to changes in emission inventories. Climate change was not explored as a significant influence in contaminant deposition in these studies. However, the utility of ice cores has long been demonstrated as a proxy for paleoclimate records, with much of that research focusing on climate reconstruction dating thousands of years ago. Thus, there is untapped potential for using ice core analyses to relate contaminant trends to more recent climate data in order to understand the influence of climate change on contaminant dynamics. New tracers for environmental processes are emerging that could be used to examine sources and mechanisms of contaminant transport (i.e. advective vs. diffusive transport) to the Arctic. For example, a recent study presented analysis of four aromatic acids in ice cores that have potential to serve as tracers for biomass burning events and biogenic aerosols210 and another report developed a method for using short chain fatty acids in ice cores as a proxy for sea surface microlayer aerosol transport.211
G. Climate change and emerging contaminant issues
G.1. Halogenated natural products (HNPs) and the influence of climate change
Halogenated natural products (HNPs) are organic compounds containing bromine, chlorine, iodine, and sometimes fluorine, that are mostly biosynthesized by a variety of marine (mainly) and terrestrial organisms. HNPs are considered CEACs16,212 as many have persistent, bioaccumulative, and toxic properties similar to those of anthropogenic contaminants, and in some cases, have been found at levels in the environment that equal or exceed those of POPs.16 This section distinguishes between two groups of HNPs: light halomethanes and haloethanes, referred to as natural halocarbons (nHCs), and higher-molecular weight HNPs (hHNPs).HNPs are produced through biosynthesis by marine bacteria, phytoplankton, macroalgae and some invertebrate animals. Hydrogen peroxide, released during photosynthesis and photorespiration, oxidizes seawater halides under catalysis by vanadium peroxidase. Oxidized halogen species then react with organic substrates within the species or with dissolved organic matter.213–218 Two subclasses of HNPs exist:
• Volatile, low molecular weight natural halocarbons (nHCs) are mainly methanes and ethanes containing chlorine, bromine and/or iodine; mixed substitutions are common. They play a key role in regulating tropospheric and stratospheric ozone. A large database exists for nHCs in the atmosphere and ocean surface water.219
• Higher molecular weight halogenated natural products (hHNPs) have similar physicochemical properties (e.g. partitioning coefficients such as KOW, KAW, KOA) as many POPs and CEACs,212,215 therefore food web bioaccumulation and toxic properties should be similar, recognizing potential differences in metabolism.220 Far less information exists for hHNPs in abiotic media, though the database is larger for fish, birds, and marine mammals.215,221 Bromoanisoles (BAs), which originate from bromophenols (BPs), are the most frequently reported hHNPs in air and water, however, there are a number of other hHNPs measured in the environment (Table 4). See Bidleman et al.212,215,216 for more information.
| hHNP group | Abbreviation |
|---|---|
| Bromoanisoles | BAs |
| Bromophenols | BPs |
| Hydroxylated polybrominated diphenyl ethers | OH-PBDEs |
| Methoxylated polybrominated diphenyl ethers | MeO-PBDEs |
| Mixed halogen compound (sesquiterpene) | MHC-1 |
| Polybrominated dibenzo-p-dioxins | PBDDs |
| Polybrominated hexahydroxanthene derivatives | PBHDs |
| Polyhalogenated carbazoles | PHCs |
| polyhalogenated 1,1′-dimethyl-2,2′-dimethylbipyrroles | PDBPs |
| Polyhalogenated 1′-methyl-1,2′-bipyrroles | PMBPs |
| Polyhalogenated N-methyl indoles | PMIs |
| Polyhalogenated N-methylpyrroles | PMPs |
Approximately half of the total bromine reaching the stratosphere in 2016 consisted of natural tribromomethane (CHBr3) and other brominated nHCs, however, there is no indication of long-term trends so far.223 Most models predict an increase in nHC emissions over this century, but the impacts of this increase are uncertain. Increased nHC emissions may lead to a reduction in tropospheric ozone; this effect would then be carried over to the stratosphere due to an increase in convective transport, particularly in the tropics.224 Stratospheric ozone loss from short-lived nHCs has nearly twice the radiative effect of long-lived (mostly anthropogenic) halocarbons, and therefore an increase in nHCs could have important impacts on future climate.225 On the other hand, offsetting factors such as increased tropospheric height, and greater chemical degradation and removal of particulate bromine, could mitigate the effects of higher ocean flux; in this case, the bromine from nHCs would not be a major source of stratospheric ozone depletion and climate forcing in the future.226
Halocarbons are produced by an array of organisms including marine bacteria, phytoplankton, macroalgae and some invertebrate animals. In the Arctic, ice algae and microorganisms present in sea ice and frost flowers can release nHCs to the atmosphere.227–229 In addition, climate change may impact areas where macroalgae are, or could be, commercially farmed; these farming operations and drying of the harvested crop may increase nHC emissions.217 Most commercial macroalgae farming occurs in tropical-subtropical regions,230 with recent expansion to the Nordic region.231
hHNPs have been reported in air, water and sediments from locations around the globe, including the Arctic (Fig. 18). Bromoanisoles (BAs) are the most frequently reported hHNPs in air and water. Fig. 19 summarizes the spatial distributions of two BAs, 2,4-dibromoanisole (2,4-diBA) and 2,4,6-tribromoanisole (2,4,6-triBA) in air and seawater, and demonstrates their presence at Arctic latitudes. BAs originate from bacterial O-methylation of bromophenols (BPs) that are produced by phytoplankton and macroalgae but are also released from anthropogenic activities including wastewater and seawater chlorination, and various industrial processes. BPs are ionized at seawater pH, but neutral BAs are volatile and found in air worldwide.
 | ||
| Fig. 18 Locations where hHNPs have been reported quantitatively in air, water and sediments. See Table 4 for expansion of chemical abbreviations. Location abbreviations: BAI = Banks Island, Canada; KOO = Kootany River, B.C., Canada; HUB = Hudson Bay, Canada; LAB = Labrador Sea, Canada; NOW = Northwater Polynya, Canada; LAM = Lake Michigan, U.S.A.; LAH = Lake Huron, Canada-U.S.A.; LAS = Lake Superior, Canada-U.S.A.; WLM, MLM = White and Muskegon Lakes, Michigan, U.S.A.; BIR = Birkenes, Norway; LIS = Lista, Norway; AND = Andøya, Norway; ZEP = Zeppelin Mountain (Ny Ålesund), Norway; BAL = Baltic Sea; ABI = Abisko, Sweden; PAL = Pallas, Finland; RÅÖ = Råö, Sweden; SWC = Swedish West Coast; GEB = German Bight (North Sea); REU = Réunion; LIA = Liaodong Bay (Bohai Sea); ECS = East China Sea, YES = Yellow Sea; SKO = coastal South Korea (including Busan); GBR = Great Barrier Reef, Australia; AUP = Aupouri Peninsula, New Zealand; AMS = American Samoa; SIA = Signey Island, Antarctica. References available in Bidleman et al. (2020).216 | ||
 | ||
| Fig. 19 Distribution of 2,4-dibromoanisole (2,4-DiBA) and 2,4,6-tribromoanisole (2,4,6-TriBA) in air (top) and surface water (bottom) at indicated latitudes. See Fig. 18 for locations and Bidleman et al. (2020)216 for data summary and references. Note the occurrence of these compounds at high latitude sites close to and within the Arctic. | ||
The few co-located measurements that exist for BAs in air and water indicate net sea-to-air exchange that occasionally approaches near equilibrium.232–234 Nearly all the data in Fig. 19 stem from single measurements or short-term campaigns. The only current air monitoring of 2,4,6-triBA is being done at stations in temperate and Arctic Norway, where annual means showed no trends between 2007 and 2018.235 A set of air measurements at the Arctic station Pallas, Finland showed a significant increase in 2,4-diBA between 2006 and 2012 (p < 0.05); an increase in 2,4,6-triBA was also suggested, but was not significant (p = 0.064).212
Since hHNPs are produced in the ocean, transport via ocean currents seems likely (e.g. movement from productive coastal/shelf areas to the open ocean and tropical/temperate to polar regions). hHNPs are pseudo-persistent; always there, albeit with seasonal cycles. Continuous production implies continuous destruction, therefore, there must be efficient pathways for the breakdown of these compounds in the environment and metabolism by organisms.220 However, information on removal rates and processes within the Arctic are currently unknown, and ocean transport of hHNPs has scarcely been investigated.
The same organisms (bacteria, phytoplankton, and macroalgae) generate both nHCs and hHNPs. Production of these two compound classes is expected to respond similarly to climate-induced shifts in macroalgae range, phytoplankton and bacteria growth, and changes in chemical and physical stressors.216 How these changes will affect the levels and impact of hHNPs is unknown. Few temporal trends records for hHNPs exist which might be useful to infer climatic influence. So far, evidence for changing BA concentrations in air are weak or lacking (see above). There is some evidence for increasing methoxylated polybrominated diphenyl ethers (MeO-PBDEs) in recent times derived from the sediment record in the East China Sea, where MeO-PBDEs were correlated with phytoplankton lipid biomarkers.236
Both nHC and hHNP emissions may also be affected by commercial macroalgae operations, with potential increased exposure to biota and humans.237 Macroalgae are used for direct human consumption, in diets for farmed fish, in pharmaceuticals and personal care products, as stabilizers and emulsifiers, as “functional foods” that provide health benefits to people and animals, and as raw materials for biofuels.230,238 The macroalgae market is rapidly expanding, with a doubling of output between 2005–2015, and over 90% resulting from commercial operations.230 Such expansion may affect HNP emissions.
As climate change is expected to affect the production and emissions of hHNPs, baseline and temporal trend measurements in biotic and abiotic media are needed to evaluate future changes. Synergies can be sought through the analysis of hHNPs in air and water samples collected under long-term monitoring programs for POPs, and the use of archived biotic samples and sediment/ice cores to rebuild trends. While the nHCs scientific community is advanced in investigating sources, atmospheric and oceanic transport, and forecasting climate change impacts through modeling, these activities are nascent or non-existent for hHNPs. Collaborations between hHNPs and nHCs communities should be established, with joint measurement campaigns and an evaluation of hHNP transport, bioaccumulation, and fate in chemistry-climate models used for nHCs. Monitoring of hHNPs in producing species (i.e. macroalgae, phytoplankton, invertebrates) is also necessary to estimate existing stocks and temporal trends to better understand climate change impacts.
• In the Arctic, microplastic pollution has been discovered in sea ice,246–248 snow,249 surface and sub-surface ocean waters250,251 and deep-sea sediment,240 as well as Arctic biota.252,253
• A wide range of polymers, with many particles being in the lower size ranges (diameters <500 μm), have been identified in Arctic media, including in Arctic snow,249 suggesting multiple sources of microplastics, both local and long-range, exist.245
• Due to their presence in marine biota, their ability to sorb POPs and other pollutants, and potential to leach chemical additives and CEACs,254,255 microplastics present a risk to Arctic ecosystems.
• Microplastics can accumulate in sea ice,250,256 but particle concentrations show a very wide range in values, in part attributable to different measurement methodologies. For example, Obbard et al. (2014)246 observed particle concentrations of 0.04–0.24 particles per L (meltwater) in central Arctic Ocean sea ice, whereas much higher values of 1145–4270 particles per L were reported in sea ice from the Fram Strait.248
• Mesocosm studies have provided evidence that microplastic particles present at very high concentrations in sea ice and snow can alter the ice albedo and promote surface melt.256 However, there is a general lack of knowledge concerning the processes and rates of microplastic incorporation within sea ice, and any resulting effect on sea ice properties.
• Fig. 20 illustrates a likely feedback loop of the effect of continued input of microplastics to the Arctic environment. It probably represents a worse-case scenario, but highlights how increasing anthropogenic activities in the Arctic due to climate change could give rise to increasing plastic pollution within the Arctic and enhance ice melt through decreasing albedo; unless restrictions on plastic use and disposal are put into place.
 | ||
| Fig. 20 Feedback loop of climate change and microplastic pollution in the Arctic. | ||
H. Conclusions and future outlook
Contaminant pathways in the physical environment are changing; most notably the intra-Arctic transfer of POPs marked by re-mobilization of previously deposited ‘reservoirs’, including release from melting glaciers and permafrost (through slumping and erosion), as well as changing source types within and outside the Arctic. It is now apparent that the rate of decline of certain legacy POPs (particularly OC pesticides) in the Arctic atmosphere is slowing and can be attributed to revolatilization of ‘old stocks’ present in seawater, particularly with the decline in areal sea ice extent and changes in the cryosphere in general. This phenomenon has been observed at several of the air monitoring stations, but not all. It is important to recognize that climatic oscillations (e.g. AO) also affect the temporal trends of POPs in the Arctic atmosphere, as well as transport patterns with surface ocean currents. These oscillations provide wider teleconnections to temperate regions, and in a warming Arctic their influence on contaminant behavior and patterns of entry into the Arctic can be pronounced.There are a number of contaminant transfer processes that are sensitive to a warming Arctic. These include the gas/particle distribution of semi-volatile compounds in the atmosphere, with atmospheric transport models simulating enhanced mobility for those chemicals shifted more towards the gas phase with warmer air temperatures. Enhanced deposition from the atmosphere (air-to-surface transfer) through increased scavenging of both gas and particle-bound chemicals brought about through higher rates of precipitation across large parts of the Arctic is likely, although empirical monitoring studies are limited. For the Arctic Ocean, increased primary productivity under climate change is enhancing the drawdown and transfer of contaminants from surface waters to deeper waters, through the process known as the ‘biological pump’. The best evidence for this is from ship-based cruises in the western Arctic examining the atmospheric drawdown and transfer of PAHs from the surface ocean to deeper waters. A key feature of the changing cryosphere across the Arctic Ocean is the replacement of MYI by brine-rich FYI. This in turn is affecting contaminant dynamics in the sea ice system, as POPs and CEACs have been observed to accumulate, and even enrich in the brine fraction of FYI. This has implications for the subsequent exposure to sympagic organisms occupying brine channels and the lower parts of the ice in contact with the underlying seawater. As brine-rich FYI dominates ice coverage over large parts of the Arctic, the additional impacts of climate change, such as earlier or erratic thawing, could result in the more efficient delivery of contaminants to organisms at the base of the marine food web.
Climate change in the Arctic is clearly exacerbating intra-Arctic contaminant mobility and transfer between physical environmental compartments. There are now clear examples of contaminant release from glacier melt in the coastal fjords of Svalbard, as well as freshwater systems in the Canadian Arctic, particularly for watersheds dominated by glacier cover and melt (e.g. Lake Hazen, Ellesmere Island). These observations mirror the contaminant transfer observed with melting glaciers in mountainous areas in temperate regions. However, in the Arctic, re-mobilization of POPs in watersheds is also occurring due to permafrost degradation and erosion, resulting in generally higher and more variable contaminant concentrations in ‘slump -affected’ lakes in the western Canadian Arctic (where observational studies have been conducted). The relevance of such processes to biotic exposures in freshwater systems may be site- or regionally-specific, for example differing between high Arctic tundra and sub-Arctic boreal forest biomes, and therefore difficult to assess in other circumpolar regions. Site-specific processes in terrestrial and freshwater environments may introduce a stronger local or regional component into long-term time trends of POPs in the Arctic. Indeed, elevated concentrations of POPs in freshwater and marine systems as a consequence of this would then result in increased exposure to biota and respective food webs. Subsequent increases in POP concentrations in higher trophic-level organisms are likely to be specific to certain geographic regions (where re-mobilization or altered contaminant pathways are clearly apparent). While increasing contaminant levels in these organisms may be a result of re-mobilization of contaminants, they may also be coincidently influenced by other ecological changes, including altered feeding habits from changes in prey abundance and/or behavior. These biological phenomena are explored in further detail in Borgå et al.257
There is now evidence of indirect effects of climate change on Arctic and sub-Arctic systems, such as the increased frequency and magnitude of wildfires. Fires in the boreal forests of Canada, Alaska and Eastern Europe have resulted in short-term elevated air concentrations of PAHs and PCBs in the high Arctic. It is foreseeable that extreme weather events (e.g. severe rain events, snowstorms, and unseasonal warming in parts of the Arctic, that in turn can lead to forest fires or unusual melt events) will become more frequent due to climate change. Yet, knowledge of the role of such events on the overall transport and distribution pathways of POPs to Arctic terrestrial and marine environments is very limited.
There are additional emerging issues that are affecting contaminant dynamics in a warming Arctic, such as microplastics. However, microplastic particles can transport POPs and CEACs, and thus serve as a likely source of these chemicals to Arctic systems. With regard to climate change-related effects, there is a lack of knowledge concerning the processes and rates of microplastic incorporation within sea ice, the resulting effect on sea ice properties and the potential release of microplastics and associated contaminants entrained in the ice. Additionally, many hHNPs biosynthesized by bacteria, phytoplankton, and microalgae, possess POP-like properties and are present in the Arctic marine environment. Globally their production is likely to increase with climate change and hence may provide an additional stress on higher trophic level organisms, alongside synthetic POPs/CEACs.
Increased human activities as an indirect effect of climate change (shipping, tourism, oil and gas development, fisheries) have the potential to increase local emissions within the Arctic. New primary sources (industries, communities, tourism) will contribute to a potential increase of CEACs, which clearly affect local areas in the vicinity of these sources. Elevated levels of some CEACs (PFASs, BFRs, OPEs, PAHs) have now been shown near Arctic communities. The relative proportion of contaminant input to the Arctic environment due to long-range atmospheric and/or oceanic transport versus local emissions may change for certain chemicals due to significant changes in human activities as a result of warming, although further assessments are required to understand the contributions from these different sources. New CEACs are being identified in the Arctic atmosphere using cutting-edge analytical methodologies, although whether these chemicals arise through long-range transport into the Arctic or from local emissions (or a combination thereof) needs further investigation.
H.1. Recommendations and future research needs
Major observations of climate change influences on POPs and CEACs in the physical environment are summarized in Fig. 21.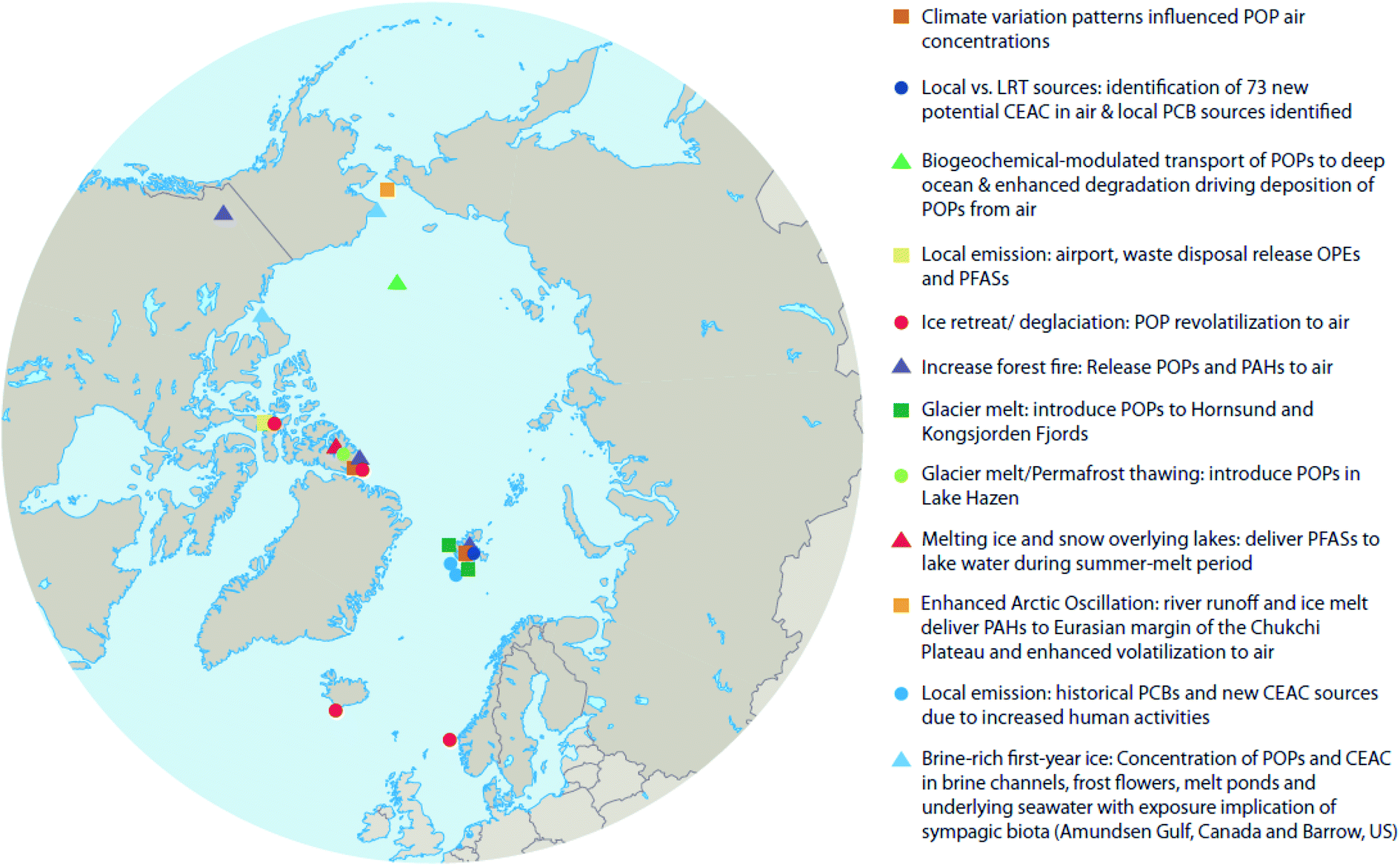 | ||
| Fig. 21 Locations of major observations of direct- and indirect-climate change influences on POPs and CEACs in physical environmental media of the Arctic. | ||
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
HH is grateful to the support of the Northern Contaminants Program (Crown-Indigenous Relations and Northern Affairs Canada) and the Government of Canada Chemicals Management Plan for funding most of her work on Arctic contaminant fate and climate change influence over the last 20 years. CH is grateful for the EISPAC project (NE/R012857/1), part of the Changing Arctic Ocean programme, jointly funded by the UKRI Natural Environment Research Council (NERC) and the German Federal Ministry of Education and Research (BMBF). HB's PhD (NE/L002604/1) is funded through UK NERC's ENVISION Doctoral Training Centre. TB's contribution is supported by the Swedish Research Environment Ecochange. The authors would like to acknowledge Jennifer Balmer for her help with text and figure editing.References
- J. C. Comiso, Large Decadal Decline of the Arctic Multiyear Ice Cover, J. Clim., 2012, 25(4), 1176–1193 CrossRef.
- J. C. Comiso and D. K. Hall, Climate trends in the Arctic as observed from space, Wiley Interdiscip. Rev. Clim. Change, 2014, 5(3), 389–409 CrossRef PubMed.
- D. Grémillet, J. Fort, F. Amélineau, E. Zakharova, T. Le Bot and E. Sala, et al., Arctic warming: nonlinear impacts of sea–ice and glacier melt on seabird foraging, Glob. Change Biol., 2015, 21(3), 1116–1123 CrossRef PubMed.
- H. Hung, A. A. Katsoyiannis, E. Brorström-Lundén, K. Olafsdottir, W. Aas and K. Breivik, et al., Temporal trends of Persistent Organic Pollutants (POPs) in arctic air: 20 years of monitoring under the Arctic Monitoring and Assessment Programme (AMAP), Environ. Pollut., 2016, 217, 52–61 CrossRef CAS PubMed.
- B. K. Biskaborn, S. L. Smith, J. Noetzli, H. Matthes, G. Vieira and D. A. Streletskiy, et al., Permafrost is warming at a global scale, Nat. Commun., 2019, 10(1), 264 CrossRef PubMed.
- D. Yumashev, C. Hope, K. Schaefer, K. Riemann-Campe, F. Iglesias-Suarez and E. Jafarov, et al., Climate policy implications of nonlinear decline of Arctic land permafrost and other cryosphere elements, Nat. Commun., 2019, 10, 1900 CrossRef PubMed.
- H. O. Pörtner, D. C. Roberts, V. Masson-Delmotte, P. Zhai, M. Tignor and E. Poloczanska, et al., IPCC Special Report on the Ocean and Cryosphere in a Changing Climate. Intergovernmental Panel on Climate Change (IPCC), Geneva, Switzerland. 70 2019, p. 77 Search PubMed.
- C. de Wit, K. Vorkamp and D. Muir, Influence of climate change on Persistent Organic Pollutants and Chemicals of Emerging Concern in the Arctic: State of knowledge and recommendations for future research, Environ. Sci.: Processes Impacts, 2022 Search PubMed.
- J. Cohen, J. A. Screen, J. C. Furtado, M. Barlow, D. Whittleston and D. Coumou, et al., Recent Arctic amplification and extreme mid-latitude weather, Nat. Geosci., 2014, 7(9), 627–637 CrossRef CAS.
- J. Ma, H. Hung and R. W. Macdonald, The influence of global climate change on the environmental fate of persistent organic pollutants: A review with emphasis on the Northern Hemisphere and the Arctic as a receptor, Glob. Planet. Change, 2016, 146, 89–108 CrossRef.
- L. Lamon, H. von Waldow, M. MacLeod, M. Scheringer, A. Marcomini and K. Hungerbühler, Modeling the Global Levels and Distribution of Polychlorinated Biphenyls in Air under a Climate Change Scenario, Environ. Sci. Technol., 2009, 43(15), 5818–5824 CrossRef CAS PubMed.
- C. Galbán-Malagón, N. Berrojalbiz, M.-J. Ojeda and J. Dachs, The oceanic biological pump modulates the atmospheric transport of persistent organic pollutants to the Arctic, Nat. Commun., 2012, 3(1), 862 CrossRef PubMed.
- C. J. Galbán-Malagón, N. Berrojalbiz, R. Gioia and J. Dachs, The “Degradative” and “Biological” Pumps Controls on the Atmospheric Deposition and Sequestration of Hexachlorocyclohexanes and Hexachlorobenzene in the North Atlantic and Arctic Oceans, Environ. Sci. Technol., 2013, 47(13), 7195–7203 CrossRef PubMed.
- S. Kusangaya, M. L. Warburton, E. Archer van Garderen and G. P. W. Jewitt, Impacts of climate change on water resources in southern Africa: A review, Phys. Chem. Earth, 2014, 67–69, 47–54 CrossRef.
- G. McBean and I. Ajibade, Climate change, related hazards and human settlements, Curr. Opin. Environ. Sustain., 2009, 1(2), 179–186 CrossRef.
- AMAP, AMAP Assessment 2016, Chemicals of Emerging Arctic Concern, Arctic Monitoring and Assessment Programme (AMAP), Oslo, Norway, 2017a, p. xvi+353 Search PubMed.
- M. Dalla Valle, E. Codato and A. Marcomini, Climate change influence on POPs distribution and fate: A case study, Chemosphere, 2007, 67(7), 1287–1295 CrossRef CAS PubMed.
- M. Rajkumar, M. N. V. Prasad, S. Swaminathan and H. Freitas, Climate change driven plant–metal–microbe interactions, Environ. Int., 2013, 53, 74–86 CrossRef CAS PubMed.
- D. Vione and A. Scozzaro, Photochemistry of surface fresh waters in the framework of climate change, Environ. Sci. Technol., 2019, 53(14), 7945–7963 CrossRef CAS PubMed.
- S. R. Wilson, S. Madronich, J. D. Longstreth and K. R. Solomon, Interactive effects of changing stratospheric ozone and climate on tropospheric composition and air quality, and the consequences for human and ecosystem health, Photochem. Photobiol. Sci., 2019, 18, 775–803 CrossRef CAS PubMed.
- AMAP, ed. Kallenborn R., Borgå K., Christensen J. H., Dowdall M., Evenset A. and Odland J. O., et al. Combined Effects of Selected Pollutants and Climate Change in the Arctic Environment, Arctic Monitoring and Assessment Programme (AMAP), AMAP Technical Report No. 5, Oslo, 2011, p. 108 Search PubMed.
- R. Kallenborn, C. Halsall, M. Dellong and P. Carlsson, The influence of climate change on the global distribution and fate processes of anthropogenic persistent organic pollutants, J. Environ. Monit., 2012, 14(11), 2854 RSC.
- J. M. Pacyna, I. T. Cousins, C. Halsall, A. Rautio, J. Pawlak and E. G. Pacyna, et al., Impacts on human health in the Arctic owing to climate-induced changes in contaminant cycling – The EU ArcRisk project policy outcome, Environ. Sci. Policy, 2015, 50, 200–213 CrossRef CAS.
- P. Carlsson, K. Breivik, E. Brorström-Lundén, I. Cousins, J. Christensen and J. O. Grimalt, et al., Polychlorinated biphenyls (PCBs) as sentinels for the elucidation of Arctic environmental change processes: a comprehensive review combined with ArcRisk project results, Environ. Sci. Pollut. Res., 2018, 25(23), 22499–22528 CrossRef CAS PubMed.
- R. Kallenborn, E. Brorström-Lundén, L.-O. Reiersen and S. Wilson, Pharmaceuticals and personal care products (PPCPs) in Arctic environments: indicator contaminants for assessing local and remote anthropogenic sources in a pristine ecosystem in change, Environ. Sci. Pollut. Res., 2018, 25(33), 33001–33013 CrossRef CAS PubMed.
- L. Röhler, M. Schlabach, P. Haglund, K. Breivik, R. Kallenborn and P. Bohlin-Nizzetto, Non-target and suspect characterisation of organic contaminants in Arctic air, Part II: Application of a new tool for identification and prioritisation of chemicals of emerging Arctic concern in air, Atmos. Chem. Phys., 2020, 20, 9031–9049 CrossRef.
- J. Ma, H. Hung, C. Tian and R. Kallenborn, Revolatilization of persistent organic pollutants in the Arctic induced by climate change, Nat. Clim. Change, 2011, 1(5), 255–260 CrossRef CAS.
- F. Rigét, A. Bignert, B. Braune, M. Dam, R. Dietz and M. Evans, et al., Temporal trends of persistent organic pollutants in Arctic marine and freshwater biota, Sci. Total Environ., 2019, 649, 99–110 CrossRef PubMed.
- H. Wöhrnschimmel, M. MacLeod and K. Hungerbühler, Emissions, fate and transport of persistent organic pollutants to the Arctic in a changing global climate, Environ. Sci. Technol., 2013, 47, 2323–2330 CrossRef PubMed.
- S. Ubl, M. Scheringer and K. Hungerbühler, Relationships between atmospheric transport regimes and PCB concentrations in the air at Zeppelin, Spitsbergen, Environ. Sci. Technol., 2017, 51, 9784–9791 CrossRef CAS PubMed.
- C. Steinlin, C. Bogdal, M. Scheringer, P. A. Pavlova, M. Schwikowski and P. Schmid, et al., Polychlorinated biphenyls in glaciers. 2. Model results of deposition and incorporation processes, Environ. Sci. Technol., 2014, 48, 7849–7857 CrossRef CAS PubMed.
- X. Wang, C. Wang, T. Zhu, P. Gong, J. Fu and Z. Cong, Persistent organic pollutants in the polar regions and the Tibetan Plateau: A review of current knowledge and future prospects, Environ. Pollut., 2019, 248, 191–208 CrossRef CAS PubMed.
- P. Wassmann, Arctic marine ecosystems in an era of rapid climate change, Prog. Oceanogr., 2011, 90, 1–17 CrossRef.
- K. R. Arrigo, D. K. Perovich, R. S. Pickart, Z. W. Brown, G. L. van Dijken and K. E. Lowry, et al., Massive phytoplankton blooms under Arctic sea ice, Science, 2012, 336(6087), 1408 CrossRef CAS PubMed.
- K. E. Lowry, G. L. van Dijken and K. R. Arrigo, Evidence of under-ice phytoplankton blooms in the Chukchi Sea from 1998 to 2012, Top. Stud. Oceanogr., Deep-Sea Res., Part II, 2014, 105, 105–117 CrossRef CAS.
- E. Jurado and J. Dachs, Seasonality in the “grasshopping” and atmospheric residence times of persistent organic pollutants over the oceans, Geophys. Res. Lett., 2008, 35(17), L17805 CrossRef.
- Z. Z. A. Kuzyk, R. W. Macdonald, S. C. Johannessen and G. A. Stern, Biogeochemical controls on PCB deposition in Hudson Bay, Environ. Sci. Technol., 2010, 44(9), 3280–3285 CrossRef CAS PubMed.
- N. Berrojalbiz, J. Dachs, M. J. Ojeda, M. C. Valle, J. Castro-Jiménez and J. Wollgast, et al., Biogeochemical and physical controls on concentrations of polycyclic aromatic hydrocarbons in water and plankton of the Mediterranean and Black Seas, Global Biogeochem. Cycles, 2011, 25(4), GB4003 CrossRef.
- M. Liu, M. Chen, M. Duan, Y. Lin, X. Liu, J. Liang, H. Ke, H. Li, M. Chen and M. Cai, Particulate export of PAHs firstly traced by 210Po/210Pb disequilibrium: Implication on the “shelf sink effect” in the Arctic Ocean, J. Geophys. Res.: Oceans, 2021, 126(8), e17384 Search PubMed.
- T. Harner, L. M. M. Jantunen, T. F. Bidleman, L. A. Barrie, H. Kylin and W. M. J. Strachan, et al., Microbial degradation is a key elimination pathway of hexachlorocyclohexanes from the Arctic Ocean, Geophys. Res. Lett., 2000, 27(8), 1155–1158 CrossRef CAS.
- E. Cerro-Gálvez, P. Casal, D. Lundin, B. Piña, J. Pinhassi and J. Dachs, et al., Microbial responses to anthropogenic dissolved organic carbon in the Arctic and Antarctic coastal seawaters, Environ. Microbiol., 2019, 21(4), 1466–1481 CrossRef PubMed.
- T. F. Bidleman, Atmospheric processes, Environ. Sci. Technol., 1988, 22(4), 361–367 CrossRef CAS.
- J. F. Pankow, An absorption model of gas/particle partitioning of organic compounds in the atmosphere, Atmos. Environ., 1994, 28(2), 185–188 CrossRef CAS.
- Y.-F. Li, M. Qin, P.-F. Yang, L.-Y. Liu, L.-J. Zhou and J.-N. Liu, et al., Treatment of particle/gas partitioning using level III fugacity models in a six-compartment system, Chemosphere, 2021, 271, 129580 CrossRef CAS PubMed.
- Y.-F. Li, L.-N. Qiao, N.-Q. Ren, R. W. Macdonald and K. Kannan, Gas/particle partitioning of semi-volatile organic compounds in the atmosphere: Transition from unsteady to steady state, Sci. Total Environ., 2020, 710, 136394 CrossRef CAS PubMed.
- R. Sühring, M. L. Diamond, M. Scheringer, F. Wong, M. Pućko and G. Stern, et al., Organophosphate esters in Canadian Arctic air: Occurrence, levels and trends, Environ. Sci. Technol., 2016, 50(14), 7409–7415 CrossRef PubMed.
- K. Kondo, N. Kagi and N. Namiki, Study on the mechanism of SVOC adsorption onto airborne particles in indoor air, Japan Archit. Rev., 2018, 1(4), 528–537 CrossRef.
- R. Sühring, H. Wolschke, M. L. Diamond, L. M. Jantunen and M. Scheringer, Distribution of organophosphate esters between the gas and particle phase–model predictions vs. measured data, Environ. Sci. Technol., 2016, 50, 6644–6651 CrossRef PubMed.
- C. W. Götz, M. Scheringer, M. MacLeod, F. Wegmann and K. Hungerbühler, Regional differences in gas–particle partitioning and deposition of semivolatile organic compounds on a global scale, Atmos. Environ., 2008, 42(3), 554–567 CrossRef.
- S. Thuens, C. Blodau, F. Wania and M. Radke, Comparison of atmospheric travel distances of several PAHs calculated by two fate and transport models (the tool and ELPOS) with experimental values derived from a peat bog transect, Atmosphere, 2014, 5(2), 324–341 CrossRef.
- C. L. Friedman, Y. Zhang and N. E. Selin, Climate change and emissions impacts on atmospheric PAH transport to the Arctic, Environ. Sci. Technol., 2014, 48(1), 429–437 CrossRef CAS PubMed.
- P. Casal, J. Castro-Jiménez, M. Pizarro, A. Katsoyiannis and J. Dachs, Seasonal soil/snow-air exchange of semivolatile organic pollutants at a coastal arctic site (Tromsø, 69°N), Sci. Total Environ., 2018, 636, 1109–1116 CrossRef CAS PubMed.
- S. Zhou, A. K. Y. Lee, R. D. McWhinney and J. P. D. Abbatt, Burial effects of organic coatings on the heterogeneous reactivity of particle-borne benzo[a]pyrene (BaP) toward ozone, J. Phys. Chem. A, 2012, 116, 7050–7056 CrossRef CAS PubMed.
- Q. Liu, J. Liggio, D. Wu, A. Saini, S. Halappanavar and J. J. B. Wentzell, et al., Experimental Study of OH-Initiated Heterogeneous Oxidation of Organophosphate Flame Retardants: Kinetics, Mechanism, and Toxicity, Environ. Sci. Technol., 2019, 53(24), 14398–14408 CrossRef CAS PubMed.
- A. Möller, Z. Xie, M. Cai, G. Zhong, P. Huang and M. Cai, et al., Polybrominated diphenyl ethers vs. alternate brominated flame retardants and dechloranes from east Asia to the Arctic, Environ. Sci. Technol., 2011, 45(16), 6793–6799 CrossRef PubMed.
- D. E. Spiel, The sizes of the jet drops produced by air bubbles bursting on sea- and fresh-water surfaces, Tellus B, 1994, 46(4), 325–338 CrossRef.
- X. Wang, G. B. Deane, K. A. Moore, O. S. Ryder, M. D. Stokes, C. M. Beall, D. B. Collins, M. V. Santander, S. M. Burrows, C. M. Sultana and K. A. Prather, The role of jet and film drops in controlling the mixing state of submicron sea spray aerosol particles, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(27), 6978–6983 CrossRef CAS PubMed.
- R. Lewis and E. Schwartz, Sea Salt Aerosol Production: Mechanisms, Methods, Measurements and Models—A Critical Review, Washington, D. C., American Geophysical Union, 2004 Search PubMed.
- C. J. McMurdo, D. A. Ellis, E. Webster, J. Butler, R. D. Christensen and L. K. Reid, Aerosol enrichment of the surfactant PFO and mediation of the water–air transport of gaseous PFOA, Environ. Sci. Technol., 2008, 42(11), 3969–3974 CrossRef CAS PubMed.
- J. H. Johansson, M. E. Salter, J. C. Acosta Navarro, C. Leck, E. D. Nilsson and I. T. Cousins, Global transport of perfluoroalkyl acids via sea spray aerosol, Environ. Sci.: Processes Impacts, 2019, 21(4), 635–649 RSC.
- M. E. Salter, P. Zieger, J. C. Acosta Navarro, H. Grythe, A. Kirkevåg and B. Rosati, et al., An empirically derived inorganic sea spray source function incorporating sea surface temperature, Atmos. Chem. Phys., 2015, 15(19), 11047–11066 CrossRef CAS.
- P. Casal, Y. Zhang, J. W. Martin, M. Pizarro, B. Jiménez and J. Dachs, Role of snow deposition of perfluoroalkylated substances at coastal Livingston Island (Maritime Antarctica), Environ. Sci. Technol., 2017, 51(15), 8460–8470 CrossRef CAS PubMed.
- H. Struthers, A. M. L. Ekman, P. Glantz, T. Iversen, A. Kirkevåg and E. M. Mårtensson, et al., The effect of sea ice loss on sea salt aerosol concentrations and the radiative balance in the Arctic, Atmos. Chem. Phys., 2011, 11, 3459–3477 CrossRef CAS.
- E. M. Mårtensson, E. D. Nilsson, G. de Leeuw, L. H. Cohen and H.-C. Hansson, Laboratory simulations and parameterization of the primary marine aerosol production, J. Geophys. Res.: Atmos., 2003, 108(D9), 4297 CrossRef.
- E. D. Nilsson, E. M. Mårtensson, J. S. van Ekeren and C. O'Dowd, Primary marine aerosol emissions: Size resolved eddy covariance measurements with estimates of the sea salt and organic carbon fractions, preprint, 2007, DOI:10.5194/acpd-7-13345-2007. https://acp.copernicus.org/preprints/acpd-2007-0355/.
- W. C. Keene, H. Maring, J. R. Maben, D. J. Kieber, A. A. P. Pszenny and E. E. Dahl, et al., Chemical and physical characteristics of nascent aerosols produced by bursting bubbles at a model air–sea interface, J. Geophys. Res., 2007, 112(D21), D21202 CrossRef.
- J. Soares, M. Sofiev, C. Geels, J. H. Christensen, C. Andersson, S. Tsyro and J. Langner, Impact of climate change on the production and transport of sea salt aerosol on European seas, Atmos. Chem. Phys., 2016, 16, 13081–13104 CrossRef CAS.
- R. Bailey, L. A. Barrie, C. J. Halsall, P. Fellin and D. C. G. Muir, Atmospheric organochlorine pesticides in the western Canadian Arctic: Evidence of transpacific transport, J. Geophys. Res.: Atmos., 2000, 105(D9), 11805–11811 CrossRef CAS.
- L. Zhang, J. Ma, S. Venkatesh, Y. Li and P. Cheung, Modeling evidence of episodic intercontinental long-range transport of lindane, Environ. Sci. Technol., 2008, 42, 8791–8797 CrossRef CAS PubMed.
- H. Hung, P. Blanchard, C. J. Halsall, T. F. Bidleman, G. A. Stern and P. Fellin, et al., Temporal and spatial variabilities of atmospheric polychlorinated biphenyls (PCBs), organochlorine (OC) pesticides and polycyclic aromatic hydrocarbons (PAHs) in the Canadian Arctic: Results from a decade of monitoring, Sci. Total Environ., 2005, 342(1–3), 119–144 CrossRef CAS PubMed.
- R. W. Macdonald, T. Harner and J. Fyfe, Recent climate change in the Arctic and its impact on contaminant pathways and interpretation of temporal trend data, Sci. Total Environ., 2005, 342(1–3), 5–86 CrossRef CAS PubMed.
- H. Hung, P. Blanchard, C. J. Halsall, T. F. Bidleman, G. A. Stern, P. Fellin, D. C. G. Muir and L. A. Barrie, et al., Corrigendum to “Temporal and Spatial Variabilities of Atmospheric Polychlorinated Biphenyls (PCBs), Organochlorine (OC) Pesticides and Polycyclic Aromatic Hydrocarbons (PAHs) in the Canadian Arctic: Results from a Decade of Monitoring”, Sci Total Environ, 2005, 342, 119–144 ( Sci. Total Environ. , 2022 , 150150 ) CrossRef CAS PubMed.
- S. Becker, C. J. Halsall, W. Tych, R. Kallenborn, Y. Su and H. Hung, Long-term trends in atmospheric concentrations of α- and γ-HCH in the Arctic provide insight into the effects of legislation and climatic fluctuations on contaminant levels, Atmos. Environ., 2008, 42(35), 8225–8233 CrossRef CAS.
- Y. Lin, M. Cai, M. Chen, P. Huang, R. Lei, M. Chen, D. Gui and H. Ke, Strong Signals of Remobilized Margin PAHs to the Basin in a Warming Arctic. Abstract #242, SETAC North America 40th Annual Meeting, Toronto, Ontario, Canada, 2019, 72, ISSN: 1087–8939 Search PubMed.
- L. M. Jantunen, P. A. Helm, H. Kylin and T. F. Bidleman, Hexachlorocyclohexanes (HCHs) in the Canadian Archipelago. 2. Air–water gas exchange of α- and γ-HCH, Environ. Sci. Technol., 2008, 42(2), 465–470 CrossRef CAS PubMed.
- Y.-F. Li, R. W. Macdonald, L. M. M. Jantunen, T. Harner, T. F. Bidleman and W. M. J. Strachan, The transport of β-hexachlorocyclohexane to the western Arctic Ocean: a contrast to α-HCH, Sci. Total Environ., 2002, 291(1–3), 229–246 CrossRef CAS.
- H. Wöhrnschimmel, P. Tay, H. von Waldow, H. Hung, Y. F. Li and M. MacLeod, et al., Comparative assessment of the global fate of α- and β-hexachlorocyclohexane before and after phase-out, Environ. Sci. Technol., 2012, 46(4), 2047–2054 CrossRef PubMed.
- P. Anttila, E. Brorström-Lundén, K. Hansson, H. Hakola and M. Vestenius, Assessment of the spatial and temporal distribution of persistent organic pollutants (POPs) in the Nordic atmosphere, Atmos. Environ., 2016, 140, 22–33 CrossRef CAS.
- NSIDC, State of the Cryosphere: Is the Cryosphere Sending Signals About Climate Change? National Snow and Ice Data Center (NSIDC). https://nsidcorg/cryosphere/sotc/sea_icehtml, accessed January 2015.
- H. N. Geisz, R. M. Dickhut, M. A. Cochran, W. R. Fraser and H. W. Ducklow, Melting glaciers: A probable source of DDT to the Antarctic marine ecosystem, Environ. Sci. Technol., 2008, 42(11), 3958–3962 CrossRef CAS PubMed.
- M. Pućko, R. W. Macdonald, D. G. Barber, B. Rosenberg, Y. Gratton and G. A. Stern, α-HCH enantiomer fraction (EF): A novel approach to calculate the ventilation age of water in the Arctic Ocean?, J. Geophys. Res.: Atmos., 2012, 117(C8), 8038 CrossRef.
- M. Pućko, G. A. Stern, R. W. Macdonald and D. G. Barber, α- and γ-hexachlorocyclohexane measurements in the brine fraction of sea ice in the Canadian High Arctic using a sump-hole technique, Environ. Sci. Technol., 2010, 44(24), 9258–9264 CrossRef PubMed.
- M. Pućko, G. A. Stern, A. E. Burt, L. M. Jantunen, T. F. Bidleman and R. W. Macdonald, et al., Current use pesticide and legacy organochlorine pesticide dynamics at the ocean-sea ice-atmosphere interface in resolute passage, Canadian Arctic, during winter-summer transition, Sci. Total Environ., 2017, 580, 1460–1469 CrossRef PubMed.
- M. Pućko, G. A. Stern, R. W. Macdonald, L. M. Jantunen, T. F. Bidleman and F. Wong, et al., The delivery of organic contaminants to the Arctic food web: Why sea ice matters, Sci. Total Environ., 2015, 506–507, 444–452 CrossRef PubMed.
- M. Pućko, G. A. Stern, R. W. Macdonald, B. Rosenberg and D. G. Barber, The influence of the atmosphere–snow–ice–ocean interactions on the levels of hexachlorocyclohexanes in the Arctic cryosphere, J. Geophys. Res., 2011, 116(C2), C02035 CrossRef.
- R. Gioia, R. Lohmann, J. Dachs, C. Temme, S. Lakaschus and D. Schulz-Bull, et al., Polychlorinated biphenyls in air and water of the North Atlantic and Arctic Ocean, J. Geophys. Res., 2008, 113(D19), D19302 CrossRef.
- P. Casal, G. Casas, M. Vila-Costa, A. Cabrerizo, M. Pizarro and B. Jiménez, et al., Snow Amplification of Persistent Organic Pollutants at Coastal Antarctica, Environ. Sci. Technol., 2019, 53(15), 8872–8882 CrossRef CAS PubMed.
- Y. Yu, A. Katsoyiannis, P. Bohlin-Nizzetto, E. Brorström-Lundén, J. Ma and Y. Zhao, et al., Polycyclic aromatic hydrocarbons not declining in arctic air despite global emission reduction, Environ. Sci. Technol., 2019, 53, 2375–2382 CrossRef CAS PubMed.
- J. Luo, Y. Han, Y. Zhao, Y. Huang, X. Liu and S. Tao, et al., Effect of northern boreal forest fires on PAH fluctuations across the arctic, Environ. Pollut., 2020, 261, 114186 CrossRef CAS PubMed.
- U. M. Sofowote, H. Hung, A. K. Rastogi, J. M. Westgate, Y. Su and P. DeLuca, et al., Assessing the long-range transport of PAH to a sub-arctic site using positive matrix factorization and potential source contribution function, Atmos. Environ., 2011, 45(4), 967–976 CrossRef CAS.
- Y. Lin, L. Liu, M. Cai, L. A. Rodenburg, M. Chitsaz and Y. Liu, et al., Isolating different natural and anthropogenic PAHs in the sediments from the northern Bering–Chukchi margin: Implications for transport processes in a warming Arctic, Sci. Total Environ., 2020, 736, 139608 CrossRef CAS PubMed.
- S. Eckhardt, K. Breivik, S. Manø and A. Stohl, Record high peaks in PCB concentrations in the Arctic atmosphere due to long-range transport of biomass burning emissions, Atmos. Chem. Phys., 2007, 7(17), 4527–4536 CrossRef CAS.
- E. C. Bizzotto, S. Villa, C. Vaj and M. Vighi, Comparison of glacial and non-glacial-fed streams to evaluate the loading of persistent organic pollutants through seasonal snow/ice melt, Chemosphere, 2009, 74(7), 924–930 CrossRef CAS PubMed.
- C. Bogdal, P. Schmid, M. Zennegg, F. S. Anselmetti, M. Scheringer and K. Hungerbühler, Blast from the past: Melting glaciers as a relevant source for persistent organic pollutants, Environ. Sci. Technol., 2009, 43(21), 8173–8177 CrossRef CAS PubMed.
- C. Bogdal, D. Nikolic, M. P. Lüthi, U. Schenker, M. Scheringer and K. Hungerbühler, Release of legacy pollutants from melting glaciers: Model evidence and conceptual understanding, Environ. Sci. Technol., 2010, 44(11), 4063–4069 CrossRef CAS PubMed.
- K. R. Miner, J. Blais, C. Bogdal, S. Villa, M. Schwikowski and P. Pavlova, et al., Legacy organochlorine pollutants in glacial watersheds: a review, Environ. Sci.: Processes Impacts, 2017, 19(12), 1474–1483 RSC.
- J. Li, G.-L. Yuan, M.-Z. Wu, Y. Sun, P. Han and G.-H. Wang, Evidence for persistent organic pollutants released from melting glacier in the central Tibetan Plateau, China, Environ. Pollut., 2017, 220, 178–185 CrossRef CAS PubMed.
- Y. Sun, G. L. Yuan, J. Li, J. Tang and G. H. Wang, High-resolution sedimentary records of some organochlorine pesticides in Yamzho Yumco Lake of the Tibetan Plateau: Concentration and composition, Sci. Total Environ., 2018, 615, 469–475 CrossRef CAS PubMed.
- H. Lin, X. Wang, P. Gong, J. Ren, C. Wang and X. Yuan, et al., The influence of climate change on the accumulation of polycyclic aromatic hydrocarbons, black carbon and mercury in a shrinking remote lake of the southern Tibetan Plateau, Sci. Total Environ., 2017, 601–602, 1814–1823 CrossRef CAS PubMed.
- M. Chen, C. Wang, X. Wang, J. Fu, P. Gong and J. Yan, et al., Release of perfluoroalkyl substances from melting glacier of the Tibetan Plateau: Insights into the impact of global warming on the cycling of emerging pollutants, J. Geophys. Res.: Atmos., 2019, 7442–7456, DOI:10.1029/2019JD030566.
- A. Pouch, A. Zaborska and K. Pazdro, Concentrations and origin of polychlorinated biphenyls (PCBs) and polycyclic aromatic hydrocarbons (PAHs) in sediments of western Spitsbergen fjords (Kongsfjorden, Hornsund, and Adventfjorden), Environ. Monit. Assess., 2017, 189(4), 175 CrossRef PubMed.
- A. Pouch, A. Zaborska and K. Pazdro, The history of hexachlorobenzene accumulation in Svalbard fjords, Environ. Monit. Assess., 2018, 190(6), 360 CrossRef CAS PubMed.
- K. Husum, J. A. Howe, A. Baltzer, M. Forwick, M. Jensen and P. Jernas, et al., The marine sedimentary environments of Kongsfjorden, Svalbard: an archive of polar environmental change, Polar Res., 2019, 38, 3380 CrossRef CAS.
- M. Błaszczyk, D. Ignatiuk, A. Uszczyk, K. Cielecka-Nowak, M. Grabiec and J. A. Jania, et al., Freshwater input to the Arctic fjord Hornsund (Svalbard), Polar Res., 2019, 38, 3506 CrossRef.
- W. van Pelt, V. Pohjola, R. Pettersson, S. Marchenko, J. Kohler and B. Luks, et al., A long-term dataset of climatic mass balance, snow conditions, and runoff in Svalbard (1957–2018), Cryosphere, 2019, 13, 2259–2280 CrossRef.
- J. J. MacInnis, I. Lehnherr, D. C. G. Muir, R. Quinlan and A. O. De Silva, Characterization of perfluoroalkyl substances in sediment cores from High and Low Arctic lakes in Canada, Sci. Total Environ., 2019, 666, 414–422 CrossRef CAS PubMed.
- I. Lehnherr, V. L. St. Louis, M. Sharp, A. S. Gardner, J. P. Smol and S. L. Schiff, et al., The world's largest High Arctic lake responds rapidly to climate warming, Nat. Commun., 2018, 9(1), 1290 CrossRef PubMed.
- D. C. G. Muir, N. P. Grift, W. L. Lockhart, P. Wilkinson, B. N. Billeck and G. J. Brunskill, Spatial trends and historical profiles of organochlorine pesticides in Arctic lake sediments, Sci. Total Environ., 1995, 160–161, 447–457 CrossRef.
- I. C. Burkow and R. Kallenborn, Sources and transport of persistent pollutants to the Arctic, Toxicol. Lett., 2000, 112–113, 87–92 CrossRef CAS PubMed.
- S. St. George, Hydrological dynamics in the Winnipeg River basin, Manitoba, in, Report of Activities 2006, Manitoba Science, Technology, Energy and Mines, Manitoba Geological Survey, Winnipeg, Manitoba, 2006, pp. 226–230 Search PubMed.
- T. Teran, LLaAM. Climate change effects on POPs' environmental behaviour: A scientific perspective for future regulatory actions, Atmos. Pollut. Res., 2012, 3(4), 466–476 CrossRef CAS.
- Environment Canada, Fisheries and Oceans Canada, Indian and Northern Affairs Canada. Land-based pollution in the Arctic ocean: Canadian actions in a regional and global context, Arctic, 2008, 61(S1), 111–121 Search PubMed.
- T. F. M. Rodgers, J. W. Truong, L. M. Jantunen, P. A. Helm and M. L. Diamond, Organophosphate Ester Transport, Fate, and Emissions in Toronto, Canada, Estimated Using an Updated Multimedia Urban Model, Environ. Sci. Technol., 2018, 52(21), 12465–12474 CrossRef CAS PubMed.
- T. Reemtsma, U. Berger, H. P. H. Arp, H. Gallard, T. P. Knepper and M. Neumann, et al., Mind the gap: Persistent and mobile organic compounds—water contaminants that slip through, Environ. Sci. Technol., 2016, 50(19), 10308–10315 CrossRef CAS PubMed.
- M. Neumann and I. Schliebner, Protecting the Sources of Our Drinking Water from Mobile Chemicals. A Revised Proposal for Implementing Criteria and an Assessment Procedure to Identify Persistent, Mobile and Toxic (PMT) and Very Persistent, Very Mobile (vPvM) Substances Registered under REACH, German Environmental Agency, ISSN: 2363-8273 20, https://wwwumweltbundesamtde/en/publikationen/protecting-the-sources-of-our-drinking-water-from, 2017 Search PubMed.
- C. A. McDonough, A. O. De Silva, C. Sun, A. Cabrerizo, D. Adelman and T. Soltwedel, et al., Dissolved organophosphate esters and polybrominated diphenyl ethers in remote marine environments: arctic surface water distributions and net transport through Fram Strait, Environ. Sci. Technol., 2018, 52(11), 6208–6216 CrossRef CAS PubMed.
- D. Muir, R. Bossi, P. Carlsson, M. Evans, A. De Silva and C. Halsall, et al., Levels and trends of poly- and perfluoroalkyl substances in the Arctic environment – An update, Emerging Contam., 2019, 5, 240–271 CrossRef.
- Y. Sun, A. O. De Silva, K. A. St Pierre, D. C. G. Muir, C. Spencer and I. Lehnherr, et al., Glacial melt inputs of organophosphate ester flame retardants to the largest high Arctic lake, Environ. Sci. Technol., 2020, 54(5), 2734–2743 CrossRef CAS PubMed.
- N. Schmidt, V. Fauvelle, A. Ody, J. Castro-Jiménez, J. Jouanno and T. Changeux, et al., The Amazon river: A major source of organic plastic additives to the tropical North Atlantic?, Environ. Sci. Technol., 2019, 53(13), 7513–7521, DOI:10.1021/acsest9b01585.
- R. Sühring, M. Scheringer, T. F. M. Rodgers, L. M. Jantunen and M. L. Diamond, Evaluation of the OECD POV and LRTP screening tool for estimating the long-range transport of organophosphate esters, Environ. Sci.: Processes Impacts, 2020, 22(1), 207–216 RSC.
- R. Sühring, M. L. Diamond, S. Bernstein, J. K. Adams, J. K. Schuster and K. Fernie, et al., Organophosphate esters in the Canadian Arctic Ocean, Environ. Sci. Technol., 2021, 55(1), 304–312 CrossRef PubMed.
- T. M. Sandanger, J. O. Odland, A. Tkachev and I. C. Burkow, Persistent organic pollutants in plasma of delivering women from Arkhangelsk, Sci. Total Environ., 2003, 306(1–3), 171–178 CrossRef CAS PubMed.
- M. Jartun, R. T. Ottesen, T. Volden and Q. Lundkvist, Local sources of polychlorinated biphenyls (PCB) in Russian and Norwegian settlements on Spitsbergen Island, Norway, J. Toxicol. Environ. Health, Part A, 2009, 72(3–4), 284–294 CrossRef CAS PubMed.
- A. Dudarev, V. Chupakhin, S. Vlasov and S. Yamin-Pasternak, Traditional diet and environmental contaminants in coastal Chukotka II: Legacy POPs, Int. J. Environ. Res. Public Health, 2019, 16(5), 695 CrossRef CAS PubMed.
- O. Garmash, M. H. Hermanson, E. Isaksson, M. Schwikowski, D. Divine and C. Teixeira, et al., Deposition history of polychlorinated biphenyls to the Lomonosovfonna Glacier, Svalbard: A 209 congener analysis, Environ. Sci. Technol., 2013, 47(21), 12064–12072 CrossRef CAS PubMed.
- Z. A. Kuzyk, J. P. Stow, N. M. Burgess, S. M. Solomon and K. J. Reimer, PCBs in sediments and the coastal food web near a local contaminant source in Saglek Bay, Labrador, Sci. Total Environ., 2005, 351–352, 264–284 CrossRef CAS PubMed.
- I. Kalinovich, A. Rutter, J. Poland, G. Cairns and R. Rowe, Remediation of PCB contaminated soils in the Canadian Arctic: Excavation and surface PRB technology, Sci. Total Environ., 2008, 407(1), 53–66 CrossRef CAS PubMed.
- I. K. Kalinovich, A. Rutter, R. K. Rowe and J. S. Poland, Design and application of surface PRBs for PCB remediation in the Canadian Arctic, J. Environ. Manage., 2012, 101, 124–133 CrossRef CAS PubMed.
- C. Luttmer, S. Ficko, K. Reimer and B. Zeeb, Deciduous vegetation (Betula glandulosa) as a biomonitor of airborne PCB contamination from a local source in the Arctic, Sci. Total Environ., 2013, 445–446, 314–320 CrossRef CAS PubMed.
- T. M. Brown, S. J. Iverson, A. T. Fisk, R. W. Macdonald, C. C. Helbing and K. J. Reimer, Local contamination, and not feeding preferences, explains elevated PCB concentrations in Labrador ringed seals (Pusa hispida), Sci. Total Environ., 2015, 515–516, 188–197 CrossRef CAS PubMed.
- T. M. Brown, Z. Z. A. Kuzyk, J. P. Stow, N. M. Burgess, S. M. Solomon and T. A. Sheldon, et al., Effects-based marine ecological risk assessment at a polychlorinated biphenyl-contaminated site in Saglek, Labrador, Canada, Environ. Toxicol. Chem., 2013, 32(2), 453–467 CrossRef CAS PubMed.
- H. R. Pedersen, R. T. Ottesen, G. W. Gabrielsen, C. Schrum, A. Evenset and A. Ruus, et al., PCBs on Svalbard, Norwegian Environment Agency and Governor of Svalbard, Longyearbyen, 2011, ISBN: 978-82-91850-36-8 https://wwwsysselmannenno/contentassets/9a8cf80ee4094bfd8c515631bb9191fe/pcb-on-svalbard---report-2011pdf.%202011 Search PubMed.
- K. Daley, H. Castleden, R. Jamieson, C. Furgal and L. Ell, Municipal water quantities and health in Nunavut households: an exploratory case study in Coral Harbour, Nunavut, Canada, Int. J. Circumpolar Health, 2014, 73(1), 23843 CrossRef PubMed.
- A. Sarkar, M. Hanrahan and A. Hudson, Water insecurity in Canadian Indigenous communities: Some inconvenient truths, Rural Remote Health, 2015, 15(4), 3354 Search PubMed.
- P. E. Jensen, T. W. Hennessy and R. Kallenborn, Water, sanitation, pollution, and health in the Arctic, Environ. Sci. Pollut. Res., 2018, 25(33), 32827–32830 CrossRef PubMed.
- J. A. Warren, J. E. Berner and T. Curtis, Climate change and human health: infrastructure impacts to small remote communities in the north, Int. J. Circumpolar Health, 2005, 64(5), 487–497 CrossRef PubMed.
- T. D. Prowse, C. Furgal, R. Chouinard, H. Melling, D. Milburn and S. L. Smith, Implications of climate change for economic development in northern Canada: Energy, resource, and transportation sectors, Ambio, 2009, 38(5), 272–281 CrossRef PubMed.
- R. Gunnarsdóttir, P. D. Jenssen, P. Erland Jensen, A. Villumsen and R. Kallenborn, A review of wastewater handling in the Arctic with special reference to pharmaceuticals and personal care products (PPCPs) and microbial pollution, Ecol. Eng., 2013, 50, 76–85 CrossRef.
- C. E. Healy, R. S. Nair, W. E. Ribelin and C. L. Bechtel, Subchronic rat inhalation study with Skydrol 500B-4 fire resistant hydraulic fluid, Am. Ind. Hyg. Assoc. J., 1992, 53(3), 175–180 CrossRef CAS PubMed.
- A. Marklund, B. Andersson and P. Haglund, Organophosphorus flame retardants and plasticizers in Swedish sewage treatment plants, Environ. Sci. Technol., 2005, 39(19), 7423–7429 CrossRef CAS PubMed.
- A. Salamova, M. H. Hermanson and R. A. Hites, Organophosphate and halogenated flame retardants in atmospheric particles from a European Arctic site, Environ. Sci. Technol., 2014, 48(11), 6133–6140 CrossRef CAS PubMed.
- B. C. Forbes, N. Fresco, A. Shvidenko, K. Danell and F. S. Chapin, Geographic variations in anthropogenic drivers that influence the vulnerability and resilience of social-ecological systems, Ambio, 2004, 33(6), 377–382 CrossRef PubMed.
- D. G. Clark, J. D. Ford, T. Pearce and L. Berrang-Ford, Vulnerability to unintentional injuries associated with land-use activities and search and rescue in Nunavut, Canada, Soc. Sci. Med., 2016, 169, 18–26 CrossRef PubMed.
- A.-S. Crépin, M. Karcher and J.-C. Gascard, Arctic climate change, economy and society (ACCESS): Integrated perspectives, Ambio, 2017, 46(S3), 341–354 CrossRef PubMed.
- J. S. Skaar, E. M. Ræder, J. L. Lyche, L. Ahrens and R. Kallenborn, Elucidation of contamination sources for poly- and perfluoroalkyl substances (PFASs) on Svalbard (Norwegian Arctic), Environ. Sci. Pollut. Res., 2019, 26(8), 7356–7363 CrossRef CAS PubMed.
- R. Sühring, C. Phillips, R. Gioia, R. Rowles and I. Abdoellakhan, Perfluorinated compounds in offshore fire-fighting foams – a source for marine contamination?, Organohalogen Compd., 2017, 79, 667–669 Search PubMed.
- G. L. Lescord, K. A. Kidd, A. O. De Silva, M. Williamson, C. Spencer and X. Wang, et al., Perfluorinated and polyfluorinated compounds in lake food webs from the Canadian high Arctic, Environ. Sci. Technol., 2015, 49(5), 2694–2702 CrossRef CAS PubMed.
- Fracking, ed. R. E. Hester and R. M. Harrison, Cambridge, Royal Society of Chemistry, 2014 Search PubMed.
- J. O. Robertson and G. V. Chilingar, Environmental Aspects of Oil and Gas Production, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2017, ISBN: 978-1-119-11737-7 Search PubMed.
- J. A. Jacobs and S. M. Testa, Environmental Considerations Associated with Hydraulic Fracturing Operations: Adjusting to the Shale Revolution in a Green World, Wiley, 1st edn, 2019 Search PubMed.
- J. E. Lindstrom and J. F. Braddock, Biodegradation of petroleum hydrocarbons at low temperature in the presence of the dispersant Corexit 9500, Mar. Pollut. Bull., 2002, 44(8), 739–747 CrossRef CAS PubMed.
- J. Jautzy, J. M. E. Ahad, C. Gobeil and M. M. Savard, Century-long source apportionment of PAHs in Athabasca oil sands region lakes using diagnostic ratios and compound-specific carbon isotope signatures, Environ. Sci. Technol., 2013, 47(12), 6155–6163 CrossRef CAS PubMed.
- A. E. Bauer, L. M. Hewitt, J. L. Parrott, A. J. Bartlett, P. L. Gillis and L. E. Deeth, et al., The toxicity of organic fractions from aged oil sands process-affected water to aquatic species, Sci. Total Environ., 2019, 669, 702–710 CrossRef CAS PubMed.
- D. Zhao, C. Su, G. Liu, Y. Zhu and Z. Gu, Performance and autopsy of nanofiltration membranes at an oil-field wastewater desalination plant, Environ. Sci. Pollut. Res. Int., 2019, 26(3), 2681–2690 CrossRef CAS PubMed.
- M. Noah, M. Lappé, B. Schneider, A. Vieth-Hillebrand, H. Wilkes and J. Kallmeyer, Tracing biogeochemical and microbial variability over a complete oil sand mining and recultivation process, Sci. Total Environ., 2014, 499, 297–310 CrossRef CAS PubMed.
- J. R. Graney, M. S. Landis, K. J. Puckett, W. B. Studabaker, E. S. Edgerton and A. H. Legge, et al., Differential accumulation of PAHs, elements, and Pb isotopes by five lichen species from the Athabasca Oil Sands Region in Alberta, Canada, Chemosphere, 2017, 184, 700–710 CrossRef CAS PubMed.
- C. Trellu, A. Miltner, R. Gallo, D. Huguenot, E. D. van Hullebusch and G. Esposito, et al., Characteristics of PAH tar oil contaminated soils-black particles, resins and implications for treatment strategies, J. Hazard. Mater., 2017, 327, 206–215 CrossRef CAS PubMed.
- M. S. Landis, W. B. Studabaker, J. P. Pancras, J. R. Graney, E. M. White and E. S. Edgerton, Source apportionment of ambient fine and coarse particulate matter polycyclic aromatic hydrocarbons at the Bertha Ganter-Fort McKay community site in the Oil Sands Region of Alberta, Canada, Sci. Total Environ., 2019, 666, 540–558 CrossRef CAS PubMed.
- RRTAC, Woody plant establishment and management program for oil sands mine reclamation, Reclamation Research Technical Advisory Committee (RRTAC), Report # OSESG-RRTAC 83-5, Techman Engineers Ltd, Edmonton, Alberta, Canada, 1983, p. 124, DOI:10.7939/R36N21.
- R. C. Rooney, S. E. Bayley and D. W. Schindler, Oil sands mining and reclamation cause massive loss of peatland and stored carbon, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(13), 4933–4937 CrossRef CAS PubMed.
- K. E. Kovalenko, J. J. H. Ciborowski, C. Daly, D. G. Dixon, A. J. Farwell and A. L. Foote, et al., Food web structure in oil sands reclaimed wetlands, Ecol. Appl., 2013, 23(5), 1048–1060 CrossRef CAS PubMed.
- M. Noah, S. Poetz, A. Vieth-Hillebrand and H. Wilkes, Detection of residual oil-sand-derived organic material in developing soils of reclamation sites by ultra-high-resolution mass spectrometry, Environ. Sci. Technol., 2015, 49(11), 6466–6473 CrossRef CAS PubMed.
- A. Jernelöv, The threats from oil spills: Now, then, and in the future, Ambio, 2010, 39(5–6), 353–366 CrossRef PubMed.
- M. Aune, A. S. Aniceto, M. Biuw, M. Daase, S. Falk-Petersen and E. Leu, et al., Seasonal ecology in ice-covered Arctic seas – Considerations for spill response decision making, Mar. Environ. Res., 2018, 141, 275–288 CrossRef CAS PubMed.
- J. Carroll, F. Vikebø, D. Howell, O. J. Broch, R. Nepstad and S. Augustine, et al., Assessing impacts of simulated oil spills on the Northeast Arctic cod fishery, Mar. Pollut. Bull., 2018, 126, 63–73 CrossRef CAS PubMed.
- L. Vergeynst, J. H. Christensen, K. U. Kjeldsen, L. Meire, W. Boone and L. M. V. Malmquist, et al., In situ biodegradation, photooxidation and dissolution of petroleum compounds in Arctic seawater and sea ice, Water Res., 2019, 148, 459–468 CrossRef CAS PubMed.
- E. Harrould-Kolieb, Shipping Impacts on Climate: A Source with Solutions, Oceana, Washington, DC, USA, 2008, p. 17 Search PubMed.
- Wilson J. M. and Rodriguez S. M., Arctic Assessments: U.S. Plans for Future Presence, Nova Science Publishers, 2012; p. 135 Search PubMed.
- E. Tedsen, S. Cavalieri and R. A. Kraemer, Arctic Marine Governance: Opportunities for Transatlantic Cooperation, Springer, 2014, p. 267 Search PubMed.
- F. Lasserre and O. Faury, Arctic Shipping: Climate Change, Commercial Traffic and Port Development, Routledge, 2020, p. 234 Search PubMed.
- L. C. Smith and S. R. Stephenson, New trans-Arctic shipping routes navigable by midcentury, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(13), 4871–4872 CrossRef CAS PubMed.
- Caddell R., and D. R. Thomas, Shipping, Law and the Marine Environment in the 21st Century: Emerging Challenges for the Law of the Sea: Legal Implications and Liabilities, Lawtext Publishing Limited, 2013, p. 327 Search PubMed.
- NSIDC, All about Sea Ice, National Snow and Ice Data Center (NSIDC), http://nsidcorg/cryosphere/seaice/indexhtml, accessed January 2019 Search PubMed.
- S. V. Kokelj, R. E. Jenkins, D. Milburn, C. R. Burn and N. Snow, The influence of thermokarst disturbance on the water quality of small upland lakes, Mackenzie Delta region, Northwest Territories, Canada, Permafr. Periglac. Process, 2005, 16(4), 343–353 CrossRef.
- A. Bring, I. Fedorova, Y. Dibike, L. Hinzman, J. Mård and S. H. Mernild, et al., Arctic terrestrial hydrology: A synthesis of processes, regional effects, and research challenges, J. Geophys. Res.: Biogeosci., 2016, 121(3), 621–649 CrossRef.
- J. E. Overland and M. Wang, Increased variability in the early winter subarctic North American atmospheric circulation, J. Clim., 2015, 28(18), 7297–7305 CrossRef.
- AMAP, Snow, water, ice and permafrost in the Arctic (SWIPA), Arctic Monitoring and Assessment Programme (AMAP), Oslo, Norway, 2017, xiv+269 Search PubMed.
- M. Cai, Z. Zhao, Z. Yin, L. Ahrens, P. Huang and M. Cai, et al., Occurrence of Perfluoroalkyl Compounds in Surface Waters from the North Pacific to the Arctic Ocean, Environ. Sci. Technol., 2012, 46(2), 661–668 CrossRef CAS PubMed.
- D. Notz and M. G. Worster, Desalination processes of sea ice revisited, J. Geophys. Res., 2009, 114(C5), C05006 CrossRef.
- J. Garnett, C. Halsall, M. Thomas, J. France, J. Kaiser and C. Graf, et al., Mechanistic Insight into the Uptake and Fate of Persistent Organic Pollutants in Sea Ice, Environ. Sci. Technol., 2019, 53(12), 6757–6764 CrossRef CAS PubMed.
- D. G. Barber, J. K. Ehn, M. Pućko, S. Rysgaard, J. W. Deming and J. S. Bowman, et al., Frost flowers on young Arctic sea ice: The climatic, chemical, and microbial significance of an emerging ice type: Frost flowers on young Arctic sea ice, J. Geophys. Res.: Atmos., 2014, 119(20), 593–611 Search PubMed.
- T. A. Douglas, F. Domine, M. Barret, C. Anastasio, H. J. Beine and J. Bottenheim, et al., Frost flowers growing in the Arctic ocean-atmosphere-sea ice-snow interface: 1. Chemical composition, J. Geophys. Res.: Atmos., 2012, 117(D14), D00R09 CrossRef.
- F. Fetterer and N. Untersteiner, Observations of melt ponds on Arctic sea ice, J. Geophys. Res.: Oceans, 1998, 103(C11), 24821–24835 CrossRef.
- AMAP, Snow, water, ice and permafrost in the Arctic (SWIPA): Climate change and the cryosphere. Arctic Monitoring and Assessment Programme (AMAP), Oslo, Norway. 2011; xii + p. 538 Search PubMed.
- D. C. Eickmeyer, L. E. Kimpe, S. V. Kokelj, M. F. J. Pisaric, J. P. Smol and H. Sanei, et al., Interactions of polychlorinated biphenyls and organochlorine pesticides with sedimentary organic matter of retrogressive thaw slump-affected lakes in the tundra uplands adjacent to the Mackenzie Delta, NT, Canada: Contaminants in Thermokarst Areas, J. Geophys. Res.: Biogeosci., 2016, 121(2), 411–421 CrossRef CAS.
- M. S. Thompson, F. J. Wrona and T. D. Prowse, Shifts in plankton, nutrient and light relationships in small tundra lakes caused by localized permafrost thaw, Arctic, 2012, 65(4), 367–376 CrossRef.
- D. C. G. Muir, A. Omelchenko, N. P. Grift, D. A. Savoie, W. L. Lockhart and P. Wilkinson, et al., Spatial trends and historical deposition of polychlorinated biphenyls in Canadian midlatitude and Arctic lake sediments, Environ. Sci. Technol., 1996, 30(12), 3609–3617 CrossRef CAS.
- G. A. Stern, E. Braekevelt, P. A. Helm, T. F. Bidleman, P. M. Outridge and W. L. Lockhart, et al., Modern and historical fluxes of halogenated organic contaminants to a lake in the Canadian arctic, as determined from annually laminated sediment cores, Sci. Total Environ., 2005, 342(1–3), 223–243 CrossRef CAS PubMed.
- A. Cabrerizo, D. C. G. Muir, C. Teixeira, S. F. Lamoureux and M. J. Lafreniere, Snow deposition and melting as drivers of polychlorinated biphenyls and organochlorine pesticides in Arctic rivers, lakes, and ocean, Environ. Sci. Technol., 2019, 53(24), 14377–14386 CrossRef CAS PubMed.
- S. F. Lamoureux and M. J. Lafrenière, Seasonal fluxes and age of particulate organic carbon exported from Arctic catchments impacted by localized permafrost slope disturbances, Environ. Res. Lett., 2014, 9(4), 045002 CrossRef CAS.
- K. E. Roberts, S. F. Lamoureux, T. K. Kyser, D. C. G. Muir, M. J. Lafrenière and D. Iqaluk, et al., Climate and permafrost effects on the chemistry and ecosystems of High Arctic Lakes, Sci. Rep., 2017, 7(1), 13292 CrossRef CAS PubMed.
- A. Cabrerizo, A. De Silva and D. Muir, Impact of climate change on the mobilization and bioaccumulation of persistent organic pollutants in arctic freshwater systems, in, Synopsis of Research Conducted under the 2017-2018 Northern Contaminants Program, Aboriginal Affairs and Northern Development Canada, Ottawa, Ontario, 2018 Search PubMed.
- A. G. Lewkowicz and R. G. Way, Extremes of summer climate trigger thousands of thermokarst landslides in a High Arctic environment, Nat. Commun., 2019, 10(1), 1329 CrossRef PubMed.
- M. H. Hermanson, E. Isaksson, C. Teixeira, D. C. G. Muir, K. M. Compher and Y.-F. Li, et al., Current-use and legacy pesticide history in the Austfonna Ice Cap, Svalbard, Norway, Environ. Sci. Technol., 2005, 39(21), 8163–8169 CrossRef CAS PubMed.
- R. M. Ruggirello, M. H. Hermanson, E. Isaksson, C. Teixeira, S. Forsström and D. C. G. Muir, et al., Current use and legacy pesticide deposition to ice caps on Svalbard, Norway, J. Geophys. Res., 2010, 115, D18308, DOI:10.1029/2010D014005.
- M. H. Hermanson, E. Isaksson, S. Forsström, C. Teixeira, D. C. G. Muir and V. A. Pohjola, et al., Deposition history of brominated flame retardant compounds in an ice core from Holtedahlfonna, Svalbard, Norway, Environ. Sci. Technol., 2010, 44(19), 7405–7410 CrossRef CAS PubMed.
- K. Y. Kwok, E. Yamazaki, N. Yamashita, S. Taniyasu, M. B. Murphy and Y. Horii, et al., Transport of perfluoroalkyl substances (PFAS) from an arctic glacier to downstream locations: Implications for sources, Sci. Total Environ., 2013, 447, 46–55 CrossRef CAS PubMed.
- M. H. Hermanson, E. Isaksson, D. Divine, C. Teixeira and D. C. G. Muir, Atmospheric deposition of polychlorinated biphenyls to seasonal surface snow at four glacier sites on Svalbard, 2013–2014, Chemosphere, 2020, 243, 125324 CrossRef CAS PubMed.
- Q. Lundkvist, H. R. Pedersen, R. T. Ottesen, T. Volden, M. Jartun, G. W. Gabrielsen, J. U. Skåre, R. Kallenborn, A. Ruus, S. Dahle, A. Evenset, D. Vongraven, B. M. Jenssen, M. Ekker and R. Hindrum, PCBs in Svalbard: Status of knowledge and management, Report No 1/2008 Governor of Svalbard, 2008, p. 38, https://coreacuk/display/30804432 Search PubMed.
- S. Forsström, J. Ström, C. A. Pedersen, E. Isaksson and S. Gerland, Elemental carbon distribution in Svalbard snow, J. Geophys. Res., 2009, 114(D19), D19112 CrossRef.
- I. Esau and I. Repina, Wind climate in Kongsfjorden, Svalbard, and attribution of leading wind driving mechanisms through turbulence-resolving simulations, Adv. Meteorol, 2012, 568454, 1–16 Search PubMed.
- G. L. Daly and F. Wania, Simulating the influence of snow on the fate of organic compounds, Environ. Sci. Technol., 2004, 38(15), 4176–4186 CrossRef CAS PubMed.
- S. Erath, Simulation of the Mass and Energy Balance at Kongsvegen 2001-2003, MSc Thesis, Dept of Meteorology and Geophysics, Innsbruk University, Tyrol, Austria, 2005 Search PubMed.
- B. Claremar, F. Obleitner, C. Reijmer, V. Pohjola, A. Waxegård and F. Karner, et al., Applying a mesoscale atmospheric model to Svalbard glaciers, Adv. Meteorol, 2012, 321649, 1–22 Search PubMed.
- K. S. Aas, T. Dunse, E. Collier, T. V. Schuler, T. K. Berntsen and J. Kohler, et al., Simulating the climatic mass balance of Svalbard glaciers from 2003 to 2013 with a high-resolution coupled atmosphere-glacier model, Cryosphere, 2016, 10, 1089–1104 CrossRef.
- T. Meyer, D. C. G. Muir, C. Teixeira, X. Wang, T. Young and F. Wania, Deposition of brominated flame retardants to the Devon Ice Cap, Nunavut, Canada, Environ. Sci. Technol., 2012, 46(2), 826–833 CrossRef CAS PubMed.
- X. Zhang, T. Meyer, D. C. G. Muir, C. Teixeira, X. Wang and F. Wania, Atmospheric deposition of current use pesticides in the Arctic: Snow core records from the Devon Island Ice Cap, Nunavut, Canada, Environ. Sci.: Processes Impacts, 2013, 15, 2304–2311 RSC.
- J. J. MacInnis, K. French, D. C. G. Muir, C. Spencer, A. Criscitiello and A. O. De Silva, et al., Emerging investigator series: a 14-year depositional ice record of perfluoroalkyl substances in the High Arctic, Environ. Sci.: Processes Impacts, 2017, 19(1), 22–30 RSC.
- H. M. Pickard, A. S. Criscitiello, C. Spencer, M. J. Sharp, D. C. G. Muir and A. O. De Silva, et al., Continuous non-marine inputs of per- and polyfluoroalkyl substances to the High Arctic: a multi-decadal temporal record, Atmos. Chem. Phys., 2018, 18(7), 5045–5058 CrossRef CAS.
- X. Wan, K. Kawamura, K. Ram, S. Kang, M. Loewen and S. Gao, et al., Aromatic acids as biomass-burning tracers in atmospheric aerosols and ice cores: A review, Environ. Pollut., 2019, 247, 216–218 CrossRef CAS PubMed.
- A. Pokhrel, K. Kawamura, O. Seki, S. Matoba and T. Shiraiwa, Ice core profiles of saturated fatty acids (C 12:0 –C 30:0 ) and oleic acid (C 18:1 ) from southern Alaska since 1734 AD: A link to climate change in the Northern Hemisphere, Atmos. Environ., 2015, 100, 202–209 CrossRef CAS.
- T. F. Bidleman, E. Brorström-Lundén, K. Hansson, H. Laudon, O. Nygren and M. Tysklind, Atmospheric transport and deposition of bromoanisoles along a temperate to Arctic gradient, Environ. Sci. Technol., 2017, 51(19), 10974–10982 CrossRef CAS PubMed.
- Y.-K. Lim, S.-M. Phang, N. Abdul Rahman, W. T. Sturges and G. Malin, Halocarbon emissions from marine phytoplankton and climate change, Int. J. Environ. Sci. Technol., 2017, 14(6), 1355–1370 CrossRef CAS.
- R. Wever, B. E. Krenn and R. Renirie, Marine vanadium-dependent haloperoxidases, their isolation, characterization, and application, Mar. Enzymes Specialized Metab., Part B Methods in Enzymology, 2018, 605, 141–201 CAS.
- T. F. Bidleman, A. Andersson, L. M. Jantunen, J. R. Kucklick, H. Kylin and R. J. Letcher, et al., A review of halogenated natural products in Arctic, subarctic and Nordic ecosystems, Emerging Contam., 2019, 5, 89–115 CrossRef.
- T. F. Bidleman, A. Andersson, P. Haglund and M. Tysklind, Will climate change influence production and environmental pathways of halogenated natural products?, Environ. Sci. Technol., 2020, 54(11), 6468–6485 CrossRef CAS PubMed.
- F. S.-L. Keng, S.-M. Phang, N. Abd Rahman, E. C. Leedham Elvidge, G. Malin and W. T. Sturges, The emission of volatile halocarbons by seaweeds and their response towards environmental changes, J. Appl. Phycol., 2020, 32(2), 1377–1394 CrossRef CAS.
- T. F. Bidleman, J. Kucklick, H. Kylin, R. Letcher, L. M. Jantunen and F. Wong, Halogenated Natural Products. AMAP Assessment 2016: Chemicals of Emerging Arctic Concern, Oslo, Norway, Arctic Monitoring and Assessment Programme, 2017, pp. 243–267 Search PubMed.
- F. Ziska, B. Quack, K. Abrahamsson, S. D. Archer, E. Atlas and T. Bell, et al., Global sea-to-air flux climatology for bromoform, dibromomethane and methyl iodide, Atmos. Chem. Phys., 2013, 13, 8915–8934 CrossRef.
- W. Vetter and G. W. Gribble, Anthropogenic persistent organic pollutants—lessons to be learned from natural halogenated products, Environ. Toxicol. Chem., 2007, 26, 2249–2252 CrossRef CAS PubMed.
- M. B. Alonso, A. Azevedo, J. P. M. Torres, P. R. Dorneles, E. Eljarrat and D. Barceló, et al., Anthropogenic (PBDE) and naturally-produced (MeO-PBDE) brominated compounds in cetaceans — A review, Sci. Total Environ., 2014, 481, 619–634 CrossRef CAS PubMed.
- R. Hossaini, P. K. Patra, A. A. Leeson, G. Krysztofiak, N. L. Abraham and S. J. Andrews, et al., A multi-model intercomparison of halogenated very short-lived substances (TransCom-VSLS):linking oceanic emissions and tropospheric transport for a reconciled estimate of the stratospheric source gas injection of bromine, Atmos. Chem. Phys., 2016, 16(14), 9163–9187 CrossRef CAS.
- W. M. O. Scientific Assessment of Ozone Depletion: 2018. Global Ozone Research and Monitoring Project, Report No. 58, World Meteorological Organization (WMO), Geneva, Switzerland, vol. 588, 2018 Search PubMed.
- F. Ziska, B. Quack, S. Tegtmeier, I. Stemmler and K. Krüger, Future emissions of marine halogenated very-short lived substances under climate change, J. Atmos. Chem., 2017, 74, 245–260 CrossRef CAS.
- R. Hossaini, M. P. Chipperfield, S. A. Montzka, A. Rap, S. Dhomse and W. Feng, Efficiency of short-lived halogens at influencing climate through depletion of stratospheric ozone, Nat. Geosci., 2015, 8(3), 186–190 CrossRef CAS.
- S. Falk, B.-M. Sinnhuber, G. Krysztofiak, P. Jöckel, P. Graf and S. T. Lennartz, Brominated VSLS and their influence on ozone under a changing climate, Atmos. Chem. Phys., 2017, 17(18), 11313–11329 CrossRef CAS.
- W. T. Sturges, G. F. Cota and P. T. Buckley, Bromoform emission from arctic ice algae, Nature, 1992, 358, 660–662 CrossRef CAS.
- A. Granfors, M. Ahnoff, M. M. Mills and K. Abrahamsson, Organic iodine in Antarctic sea ice: A comparison between winter in the Weddell Sea and summer in the Amundsen Sea, J. Geophys. Res.: Biogeosci., 2014, 119(12), 2276–2291 CrossRef CAS.
- A. Granfors, M. Andersson, M. Chierici, A. Fransson, K. Gårdfeldt and A. Torstensson, et al., Biogenic halocarbons in young Arctic sea ice and frost flowers, Mar. Chem., 2013, 155, 124–134 CrossRef CAS.
- FAO, The global status of seaweed production, trade and utilization, Globefish Research Programme, vol. 124Food and Agricultural Organization of the United Nations (FAO), Rome, Italy, 2018, p. 120 Search PubMed.
- L. Hasselström, J.-B. Thomas, J. Nordström, G. Cervin, G. M. Nylund and H. Pavia, et al., Socioeconomic prospects of a seaweed bioeconomy in Sweden, Sci. Rep., 2020, 10(1), 1610 CrossRef PubMed.
- O. Pfeifer and K. Ballschmiter, Halogenated methyl-phenyl ethers (HMPE; halogenated anisoles) in the marine troposphere and in the surface water of the Atlantic Ocean: An indicator of the global load of anthropogenic and biogenic halophenols, Organohalogen Compd., 2002, 57, 483–486 CAS.
- F. Wong, L. M. Jantunen, M. Pućko, T. Papakyriakou, G. A. Stern and T. F. Bidleman, Air–water exchange of anthropogenic and natural organohalogens on International Polar Year (IPY) expeditions in the Canadian Arctic, Environ. Sci. Technol., 2011, 45, 876–881 CrossRef CAS PubMed.
- T. F. Bidleman, K. Agosta, A. Andersson, P. Haglund, P. Liljelind and A. Hegmans, et al., Sea–air exchange of bromoanisoles and methoxylated bromodiphenyl ethers in the Northern Baltic, Mar. Pollut. Bull., 2016, 112(1–2), 58–64 CrossRef CAS PubMed.
- P. Bohlin-Nizzetto, W. Aas and V. Nikiforov, Monitoring of Environmental Contaminants in Air and Precipitation, Annual Report 2018. Report 11/2019, Norwegian Institute for Air Research (NILU), Kjeller, Norway, 2019 Search PubMed.
- Y. Fan, C.-A. Huh, J. Lan, M. Zhao, Z. Zhao and G. Li, et al., Major sources of MeO/OH-BDEs in the East China Sea elucidated from their records and phytoplankton biomarkers, Environ. Pollut., 2014, 192, 1–8 CrossRef PubMed.
- K. Haraguchi, Y. Ito, M. Takagi, Y. Fujii, K. H. Harada and A. Koizumi, Levels, profiles and dietary sources of hydroxylated PCBs and hydroxylated and methoxylated PBDEs in Japanese women serum samples, Environ. Int., 2016, 97, 155–162 CrossRef CAS PubMed.
- S. L. Holdt and S. Kraan, Bioactive compounds in seaweed: functional food applications and legislation, J. Appl. Phycol., 2011, 23(3), 543–597 CrossRef CAS.
- L. Van Cauwenberghe, A. Vanreusel, J. Mees and C. R. Janssen, Microplastic pollution in deep-sea sediments, Environ. Pollut., 2013, 182, 495–499 CrossRef CAS PubMed.
- M. Bergmann, V. Wirzberger, T. Krumpen, C. Lorenz, S. Primpke and M. B. Tekman, et al., High quantities of microplastic in Arctic deep-sea sediments from the HAUSGARTEN Observatory, Environ. Sci. Technol., 2017, 51(19), 11000–11010 CrossRef CAS PubMed.
- J. Brahney, M. Hallerud, E. Heim, M. Hahnenberger and S. Sukumaran, Plastic rain in protected areas of the United States, Science, 2020, 368(6496), 1257–1260 CrossRef CAS PubMed.
- C. M. Free, O. P. Jensen, S. A. Mason, M. Eriksen, N. J. Williamson and B. Boldgiv, High-levels of microplastic pollution in a large, remote, mountain lake, Mar. Pollut. Bull., 2014, 85(1), 156–163 CrossRef CAS PubMed.
- K. Zhang, J. Su, X. Xiong, X. Wu, C. Wu and J. Liu, Microplastic pollution of lakeshore sediments from remote lakes in Tibet plateau, China, Environ. Pollut., 2016, 219, 450–455 CrossRef CAS PubMed.
- C. L. Waller, H. J. Griffiths, C. M. Waluda, S. E. Thorpe, I. Loaiza and B. Moreno, et al., Microplastics in the Antarctic marine system: An emerging area of research, Sci. Total Environ., 2017, 598, 220–227 CrossRef CAS PubMed.
- N. Evangeliou, H. Grythe, Z. Klimont, C. Heyes, S. Eckhardt and S. Lopez-Aparicio, et al., Atmospheric transport is a major pathway of microplastics to remote regions, Nat. Commun., 2020, 11(1), 3381 CrossRef CAS PubMed.
- R. W. Obbard, S. Sadri, Y. Q. Wong, A. A. Khitun, I. Baker and R. C. Thompson, Global warming releases microplastic legacy frozen in Arctic Sea ice, Earth's Future, 2014, 2(6), 315–320 CrossRef.
- M. Bergmann, I. Peeken, B. Beyer, T. Krumpen, S. Primpke and M. B. Tekman, et al., Vast quantities of microplastics in Arctic sea ice—A prime temporary sink for plastic litter and a medium of transport, Fate and Impact of Microplastics in Marine Ecosystems, Elsevier, 2017, pp. 75–76 Search PubMed.
- I. Peeken, S. Primpke, B. Beyer, J. Gütermann, C. Katlein and T. Krumpen, et al., Arctic sea ice is an important temporal sink and means of transport for microplastic, Nat. Commun., 2018, 9(1), 1505 CrossRef PubMed.
- M. Bergmann, S. Mützel, S. Primpke, M. B. Tekman, J. Trachsel and G. Gerdts, White and wonderful? Microplastics prevail in snow from the Alps to the Arctic, Sci. Adv., 2019, 5(8), eaax1157 CrossRef CAS PubMed.
- A. L. Lusher, V. Tirelli, I. O'Connor and R. Officer, Microplastics in Arctic polar waters: the first reported values of particles in surface and sub-surface samples, Sci. Rep., 2015, 5(1), 14947 CrossRef CAS PubMed.
- L. D. K. Kanhai, K. Gårdfeldt, O. Lyashevska, M. Hassellöv, R. C. Thompson and I. O'Connor, Microplastics in sub-surface waters of the Arctic Central Basin, Mar. Pollut. Bull., 2018, 130, 8–18 CrossRef CAS PubMed.
- S. Kühn, F. L. Schaafsma, B. van Werven, H. Flores, M. Bergmann and M. Egelkraut-Holtus, et al., Plastic ingestion by juvenile polar cod (Boreogadus saida) in the Arctic Ocean, Polar Biol., 2018, 41(6), 1269–1278 CrossRef PubMed.
- S. Morgana, L. Ghigliotti, N. Estévez-Calvar, R. Stifanese, A. Wieckzorek and T. Doyle, et al., Microplastics in the Arctic: A case study with sub-surface water and fish samples off Northeast Greenland, Environ. Pollut., 2018, 242, 1078–1086 CrossRef CAS PubMed.
- M. Cole, P. Lindeque, C. Halsband and T. S. Galloway, Microplastics as contaminants in the marine environment: A review, Mar. Pollut. Bull., 2011, 62(12), 2588–2597 CrossRef CAS PubMed.
- A. Bakir, I. A. O'Connor, S. J. Rowland, A. J. Hendriks and R. C. Thompson, Relative importance of microplastics as a pathway for the transfer of hydrophobic organic chemicals to marine life, Environ. Pollut., 2016, 219, 56–65 CrossRef CAS PubMed.
- N.-X. Geilfus, K. M. Munson, J. Sousa, Y. Germanov, S. Bhugaloo and D. Babb, et al., Distribution and impacts of microplastic incorporation within sea ice, Mar. Pollut. Bull., 2019, 145, 463–473 CrossRef CAS PubMed.
- K. M. Borgå, A. Melissa, H. Routti, K. Fernie, J. Giebichenstein and D. Muir, et al., The influence of global climate change on accumulation and toxicity of persistent organic pollutants and chemicals of emerging Arctic concern in Arctic food webs, Environ. Sci.: Processes Impacts, 2022 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1em00485a |
| This journal is © The Royal Society of Chemistry 2022 |