
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigations into the structure, reactivity, and AACVD of aluminium and gallium amidoenoate complexes†
Kristian L.
Mears
 ,
Malavika A.
Bhide
,
Caroline E.
Knapp
* and
Claire J.
Carmalt
*
,
Malavika A.
Bhide
,
Caroline E.
Knapp
* and
Claire J.
Carmalt
*
Department of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: c.j.carmalt@ucl.ac.uk
First published on 1st December 2021
Abstract
Amidoenoate (AME = {ethyl-3-(R-amido)but-2-enoate}) complexes of aluminium and gallium, of the type: [AlCl2(AMER)] R = iPr (1-Al); [AlCl(AMER)2] R = iPr (2-Al), Dip (3-Al); [GaCl2(AMER)] R = iPr (1-Ga) and [GaCl(AMER)2] R = iPr (2-Ga), Dip (3-Ga), have been synthesised (iPr = isopropyl, Dip = 2,6-diisopropylphenyl). The coordination chemistry of these complexes has been studied in relation to precursor suitability. Investigations into the reactivity of the aluminium and gallium amidoenoate complexes involved reactions with hydride sources including alkali metal hydride salts, alkylsilanes, and magnesium hydride species and magnesium(I) dimers. The isolation of alkyl metal amidoenoate precursors including an aluminium hydride amidoenoate, [AlH(AMEDip)2] (4-Al) and dimethyl gallium amidoenoates [GaMe2(AMEDip)] (4-Ga), [GaMe2(AMEiPr)] (5-Ga) concluded the synthetic studies. A selection of the isolated complexes were used as precursors for aerosol assisted chemical vapour deposition (AACVD) at 500 °C. Thin films of either amorphous Al2O3 or Ga2O3 were deposited and subsequently annealed at 1000 °C to improve the materials’ crystallinity. The films were characterised by X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM), UV-visible (UV-vis) spectroscopy and energy dispersive X-ray analysis (EDXA).
Introduction
In recent decades, precursor design and synthesis has become increasingly important for the deposition of functional materials.1–4 Specifically, the customization of precursors which deliver specific properties demanded by the deposition technique such as volatility or solubility have placed ‘designer’ precursors center-stage for a wide-range of material deposition techniques, including aerosol-assisted chemical vapor deposition (AACVD),4–8 spin-coating,9–11 or by ink deposition.12–14 Recently, investigations into the structural parameters of metal-containing molecular precursors have been carried out in order to probe relationships between a precursor's molecular geometry (dictated by the bonding modes of the ligands bound to the metal) and their suitability for deposition.15 For example, recent studies have shown that molecular complexes to be employed as precursors which crystallize with a distorted geometry are able to be deposited at lower temperatures due to their inherent molecular strain,16,17 which can be quantified using the degree of distortion denoted as a ‘τ value’.18,19Methods employed by synthetic chemists to control such parameters include varying the chemical nature of the complex by choosing specific ligands to perform certain functions. For example, incorporating per-fluorinated ligands at metal centres in order to achieve a precursor with high volatility,20 or choosing fluxional ligands, such as donor-functionalised alkoxides with hemi-labile properties to encourage gas-phase decomposition during CVD.21–23 Ligands which aid metal–oxide formation often include a source of oxygen such as diketonates and β-ketoiminates (BKIs), as shown in Fig. 1 (A and B respectively), have become well established ligands in the literature for metal complexes used to deposit oxide materials.24–29 One advantage the BKI ligand has over the nitrogen free β-diketonates is the option to incorporate various steric profiles at the N-position.
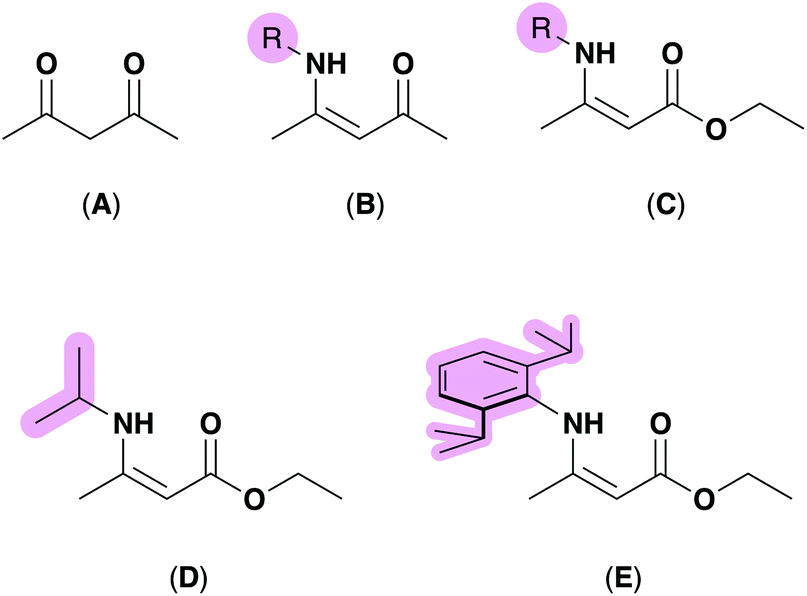 | ||
| Fig. 1 Top: previously reported ligand systems for oxide formation include β-diketones (A), β-diketoimines (B) and aminoenoates (C). Bottom: the aminoenoate pro-ligands employed in this work with -iPr (D) and -Dip (2,6-diisopropylphenyl) (E) at the N-position. R-groups that can be tuned to vary precursor geometry, highlighted in pink. | ||
This ability to tune the steric profile within the metal's coordination centre is almost solely considered for stabilising complexes that require significant stabilisation due to an unusual oxidation state or kinetic instability,30,31 however these design principles are becoming more common in precursor synthesis. For example, the aminoenoate (HAME), (Fig. 1, C) includes a nitrogen donor that can be functionalised to incorporate varying steric bulk, as well as two oxygen atoms to afford a high inherent oxygen content, but amidoenoate (AME) ligands have yet to receive the same attention as other (O,N)-coordinating ligands, such as BKI motifs.
Previously, we have shown that AME ligands can act as oxygen saturated systems at zinc centres (Fig. 2, A–D) to form precursors which successfully converted to zinc oxide (ZnO) via AACVD.32,33 McElwee-White and co-workers have employed AMEs at tungsten centres (Fig. 2, E and F), generating a range of tungsten oxide precursors with interesting molecular geometries which afforded W![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O ratio control in the resulting thin films depending on the deposition temperature used.34
:O ratio control in the resulting thin films depending on the deposition temperature used.34
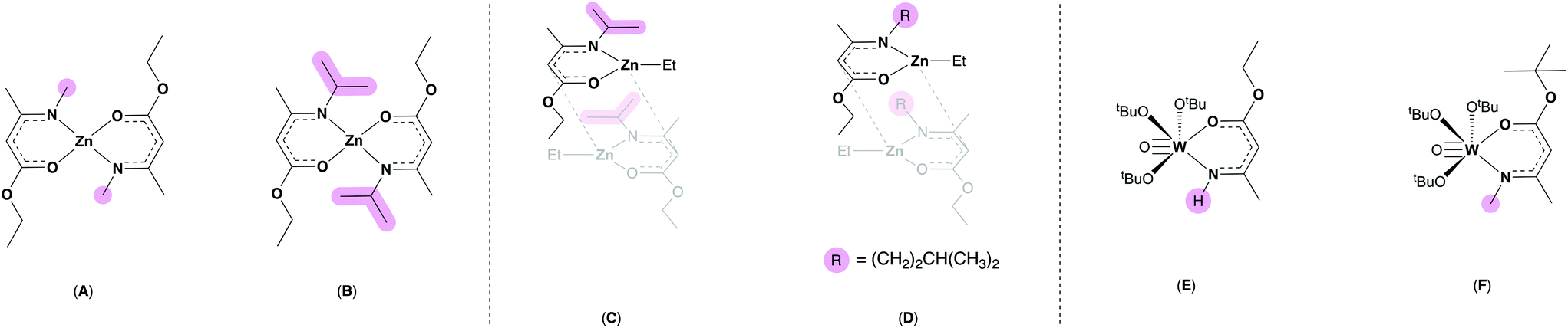 | ||
| Fig. 2 Previously reported zinc bis-amidoenoates (A and B), ethyl zinc amidoenoate dimers (C and D) and tungsten amidoenoate complexes (E and F).32–34 Differing steric profiles are highlighted in pink. | ||
As zinc and tungsten AME systems have been investigated previously and afforded viable oxide precursors, AME ligands at group 13 centres, such as aluminium and gallium may result in similarly successful precursors to these oxides, which have a range of applications including solar cells, abrasives and sensors.35,36 Here, we investigate AME ligands at aluminium and gallium centres as precursors towards aluminium oxide (α-Al2O3) or gallium oxide (β-Ga2O3) respectively.
As very few solution-based precursors towards aluminium oxide have been previously reported,37 we focused particularly on the synthesis of [Al(AME)] systems. For comparison with another important oxide material (β-Ga2O3), the analogous gallium complexes were also prepared, noting that precursors to gallium oxide often rely on alkoxides, diketonates and BKI systems.38,39
Results and discussion
Single crystals suitable for X-ray crystallographic analysis were obtained from saturated solutions of their respective mixtures of hexane and ether due to the oily nature of the isolated material. Obtained powder and crystalline samples of (1–2)-(Al,Ga) melted quickly at room temperature. Analysis of the crystalline material was therefore carried out using 1H and 13C-NMR spectroscopy (and in most cases corroborated with {1H–1H} COSY or NOESY, {1H–13C} HSQC and HMBC multinuclear NMR experiments (ESI†), as well as on-site positive-mode electrospray ionisation (ESI+) mass spectroscopy.The aminoenoate pro-ligand bearing an iPr-group at the N-position (HAMEiPr) was synthesised according to previously reported literature,32 and the bulkier analogue bearing a Dip-substituent, Dip = 2,6-diisopropylphenyl (HAMEDip) was isolated following a similar procedure (ESI). Initially, the lithium salts of these compounds were generated from a 1:1 metathesis reaction with lithium silylamide etherate (Scheme 1, left). The resulting [Li(AMEiPr)] and [Li(AMEDip)] salts which were both isolated as white crystalline material in excellent yields (80% and 86% respectively) were initially reacted with aluminium trichloride. In order to probe the stability offered by the amidoenoate ligand with the least-bulky R-group (iPr), a 1:1 reaction of [Li(AMEiPr)] with AlCl3 was carried out (Scheme 1, route A) in diethyl ether and afforded [AlCl2(AMEiPr)] (1-Al) as a pale oil that was titurated in hexane to afford a white powder in good yield (69%). The molecular structures of complexes 1-Al–3-Al are shown in Fig. 3 and selected bond lengths and angles are shown in Table 1. The geometry of [AlCl2(AMEiPr)] at the aluminium centre was found to be tetrahedral (τ4 = 0.92).
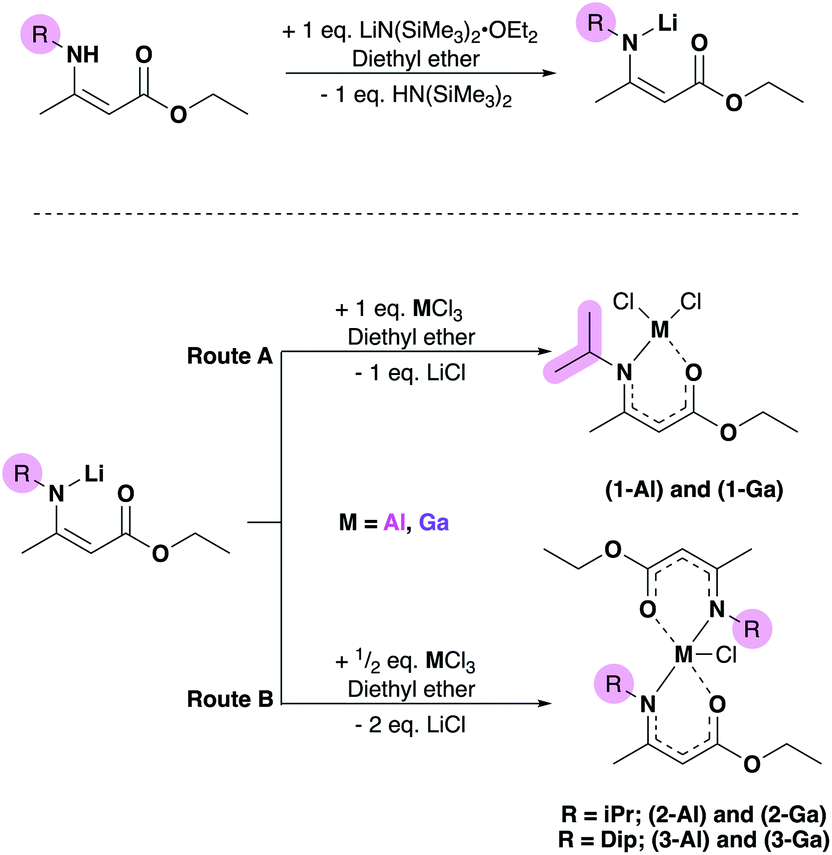 | ||
| Scheme 1 Left: lithium amidoenoate formation via reaction of pro-ligand with lithium silylamide etherate. Right: salt metathesis routes used to synthesize mono- and bis-aminoenoate metal complexes. R = iPr or Dip (highlighted). | ||
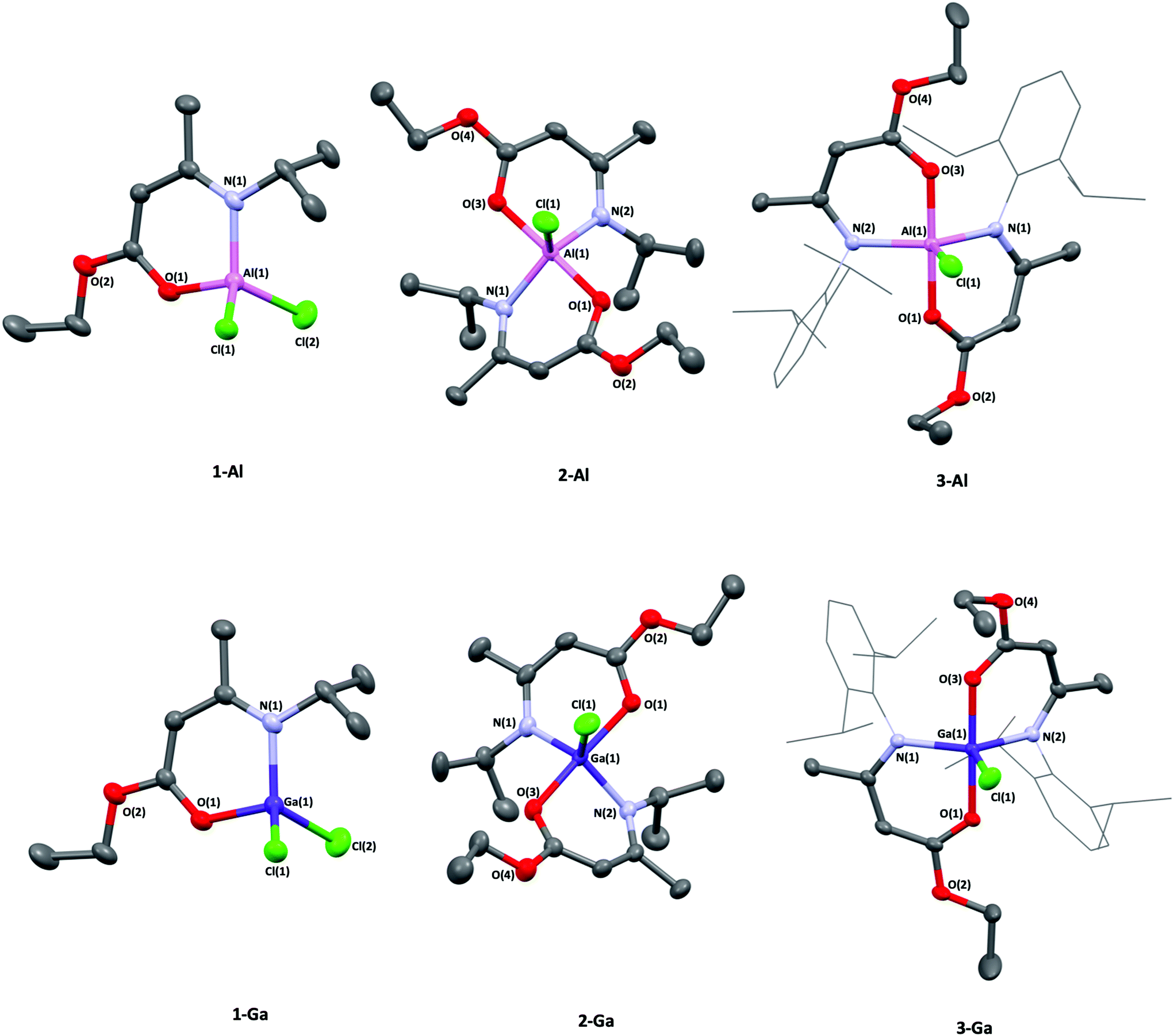 | ||
| Fig. 3 Molecular structures of aluminium chlorido amidoenoates, [AlCl2(AMEiPr)] (1-Al), [AlCl(AMEiPr)2] (2-Al), [AlCl(AMEDip)2] (3-Al) and analogous gallium chlorido amidoenoates, [GaCl2(AMEiPr)] (1-Ga), [GaCl(AMEiPr)2] (2-Ga), [GaCl(AMEDip)2] (3-Ga). Thermal ellipsoids shown at 30% probability. Dip-substituents are shown in wireframe format and hydrogens have been omitted for clarity. | ||
| 1-Al | 2-Al | 3-Al | 1-Ga | 2-Ga | 3-Ga | |
|---|---|---|---|---|---|---|
| Bond lengths (Å) | ||||||
| M–Cl(1) | 2.1287(5) | 2.2362(5) | 2.1969(5) | 2.1553(5) | 2.2443(5) | 2.2177(4) |
| M–Cl(2) | 2.1178(5) | — | — | 2.1661(5) | — | — |
| M–N(1) | 1.8721(11) | 1.9356(12) | 1.9746(12) | 1.9050(14) | 1.9506(16) | 1.9888(11) |
| M–N(2) | — | 1.9338(12) | 1.9782(11) | — | 1.9510(16) | 1.9861(12) |
| M–O(1) | 1.7689(9) | 1.8608(11) | 1.8477(10) | 1.8664(11) | 1.9960(14) | 1.9734(10) |
| M–O(3) | — | 1.8545(11) | 1.8541(10) | — | 1.9807(14) | 1.9687(10) |
| Bond angles (°) | ||||||
| O(1)–M–N(1) | 100.43(5) | 91.15(5) | 90.20(5) | 100.92(5) | 91.03(6) | 91.41(4) |
| N(1)–M–Cl(1) | 115.08(4) | 117.58 | 115.27(4) | 116.11(5) | 117.16(5) | 114.77(3) |
| O(1)–M–O(3) | — | 174.14(5) | 176.34(5) | — | 172.85(6) | 178.95(5) |
| N(1)–M–N(2) | — | 121.52(6) | 130.91(5) | — | 122.42(7) | 129.34(5) |
| Cl(1)–M–Cl(2) | 109.48(2) | — | — | 108.96(2) | _ | — |
| τ value | 0.92 | 0.87 | 0.76 | 0.96 | 0.84 | 0.83 |
Complex 1-Al represents the first example of a tetrahedral aluminium dichloride unit featuring a ligand that binds through both oxygen and nitrogen and was synthesised from the salt metathesis reaction of [Li(AMEiPr)] and AlCl3 (Scheme 1).
Two previous examples of aluminium dichlorides featuring (O,N)-binding ligands have been reported by Richards40 and Schulz,41 with the formulae [AlCl2{(OC(Me)CHC(Me)NC2H4NEt2)}] and [AlCl2{(OC(Me)C(H)C(Me)NC2H4NMe2)}] respectively, but are 5-coordinate complexes due to the extra donor functionality incorporated into the ligand.
All Al-heteroatom bonds in 1-Al were found to be significantly shorter than the 5-coordinate complexes reported by Richards and Schulz previously,40,41 which is likely a consequence of the comparatively electron deficient nature of the aluminum center in 1-Al. Complexes 2-Al and 3-Al both displayed a distorted trigonal bipyramidal geometry with a τ4 value of 0.87 for 2-Al and 0.76 for 3-Al, suggesting that the Dip-substituted amidoenoate ligands in 3-Al distort the geometry of the aluminium center significantly more than the iPr-substituents in 2-Al. The Al–Cl bond lengths (2.2362(5) Å and 2.1969(5) Å in 2-Al and 3-Al respectively) were similar to other 5-coordinate {Al–Cl} containing complexes bearing (O,N)-ligands.42–44 Following the successful isolation of (1–3)-Al, the heavier gallium analogues were targeted. Using similar routes (Scheme 1), the 1:1 and 1:2 reaction of GaCl3 and [Li(AMEiPr)] yielded complexes [GaCl2(AMEiPr)] (1-Ga) and [GaCl(AMEiPr)2] (2-Ga) as pale-yellow oils which could be titurated in hexane to give powders in good yields (62% and 63% respectively). [GaCl(AMEDip)2] (3-Ga), was isolated as a white powder in excellent yield (86%) from the salt metathesis of two equivalents of [Li(AMEDip)] and one equivalent of GaCl3. The geometries of complexes (1–3)-Ga were almost identical to those found for their aluminium analogues. Complex 1-Ga had a geometry similar to that of 1-Al, displaying a tetrahedral geometry with a τ4 value of 0.96 – only 4% distorted from a perfect tetrahedral geometry, whereas 1-Al had a distortion of 8%. Interestingly, the Ga–Cl(2) bond length (2.1661(5) Å) in 1-Ga was identical to that of a previously reported gallium bis-chlorido BKI complex with the formula [GaCl2(Me(O)C(H)C(NiPr)Me)].45
Complex 2-Ga featured a trigonal bipyramidal geometry, though slightly more distorted than the aluminium analogue with a τ5 value of 0.84 (3% more distorted than 2-Al). This complex also had the largest Ga–Cl(1) bond length of the set of {Ga–Cl} containing complexes presented in this study. The complex 3-Ga also displayed a trigonal bipyramidal geometry, though was less distorted than 3-Al. A further trend observed was that the complexes with the formula [MCl(AMEiPr)2], M = Al, Ga (2-Al and 2-Ga) featured the longest M–Cl(1) bond lengths, rather than the bulkier Dip-substituent containing system with the formula [MCl(AMEDip)2], M = Al, Ga (3-Al and 3-Ga). The result of this observation is proposed to be electronic, rather than steric in nature. The donating effect from the iPr group in 2-Al and 2-Ga likely affords a more basic amide and hence a stronger N-donor to the metal. The M–Cl bond lengthens as a result to stabilize the quantity of electron density at the metal center. For complexes 3-Al and 3-Ga, the bulkier Dip-substituents at the N-position are electron withdrawing, and so shorter M–Cl(1) bond lengths were observed, despite these complexes having to accommodate larger ligands in their coordination spheres.
In most cases, the M–O(1) bond lengths are shorter than the M–N(1) bond lengths in both the (1–3)-Al and (1–3)-Ga systems and is likely due to delocalisation in the ligand backbone (Table 1). Complex 2-Ga was the only case (for the complexes presented) where the M–O(1) bond length was longer than the M–N(1) bond length.45 This may be a consequence of gallium's slightly shorter covalent radius compared to aluminium, resulting in the lengthening of the M–O(1) bond to accommodate the two strongly donating NiPr groups, despite the oxophilic nature of the metal.
Reactivity studies
Chemical transformations between different functional groups are ubiquitous in organic chemistry and yet, often not considered for applications in precursor synthesis. An example where synthetic transformations in materials chemistry may be beneficial, is the abstraction of a chlorine atom in a precursor to a different “reaction handle” which could allow further functionalisation of a precursor. As metal amidoenoate systems have received little attention previously, we decided to conduct a reactivity study of some of the complexes presented. There were many synthetic possibilities, so to limit the scope we attempted to transform the Cl atom in complexes 3-Al and 3-Ga to their respective hydrides. The routes attempted are illustrated in Scheme 2.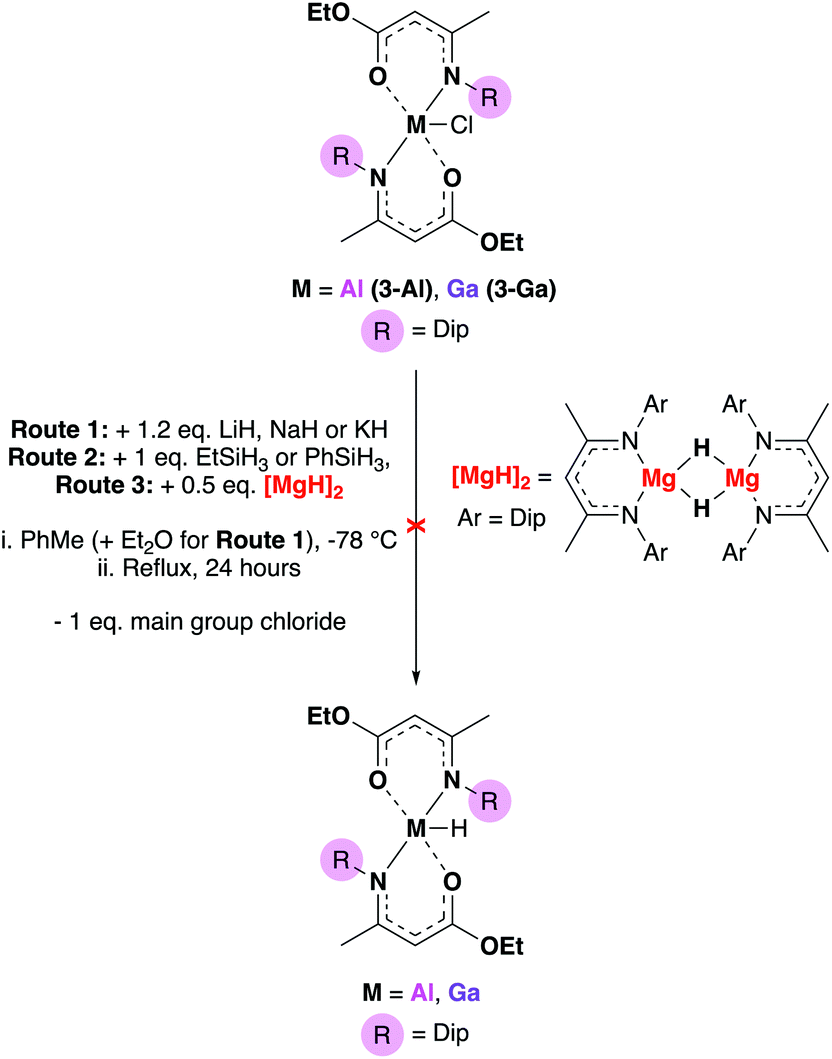 | ||
| Scheme 2 Attempted formation of group 13 amidoenoate hydrides using alkali metal hydrides, silanes and magnesium hydride dimers. | ||
Complexes 3-Al and 3-Ga were chosen for the reactivity study as these complexes had the most distorted structures of the set of amidoenoate complexes synthesised (τ5 values of 0.76 and 0.83 for 3-Al and 3-Ga respectively) and hence, it was hypothesized that replacing the chloride for a smaller hydride in these systems may be an energetically favourable process and result in the desired {M–H} product.
Conventional hydride sources including metal hydride salts and silanes with varying acidity were attempted but did not result in the conversion to the group 13 hydride. Jones’ accessible magnesium hydride dimer46 was then employed in order to probe if the formation of an [MgCl(BDIDip)] complex would drive the metathesis, but this route also resulted in no conversion (either by NMR or product isolation). For the cases of routes 1 and 2 (Scheme 2), crystalline starting material of 3-Al or 3-Ga could be obtained. These results suggested that the (O,N)-coordinating bulky Dip-substituted amidoenoate ligands provided significant stability at the group 13 centers. Final attempts at abstracting the Cl-atom from the main group centres involved employing Jones’ low-valent Mg(I) complex,47 [Mg(BDIMes)]2 (Mes = mesityl), in order to either reduce the complexes to form [(AME)2M–M(AME)2] dimers or by installing [M–Mg] (M = Al, Ga) bonds. At elevated temperatures, these reactions resulted in some AME-complex decomposition (as evidenced by 1H NMR experiments – see ESI†). As functionalisation of the chloride to a hydride was not achieved, the reactivity study proceeded to investigate whether a group 13-hydride synthon could be used to synthesize a metal hydride bis-amidoenoate complex (Scheme 3).
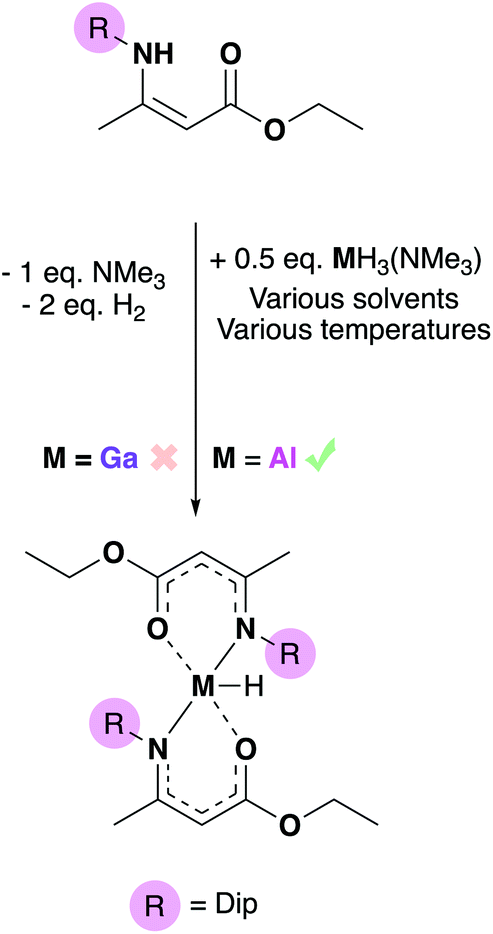 | ||
| Scheme 3 Attempted formation of aluminium and gallium bis-amidoenoate hydrides using amine stabilized alane or gallane synthons respectively with the Dip-substituted aminoenoate (R = Dip, highlighted). | ||
One equivalent of HAMEDip dissolved in toluene and was added dropwise to half an equivalent of [GaH3(NMe3)] in hexane at −78 °C. However, the acid–base reaction between HAMEDip and [GaH3(NMe3)] resulted in no observable hydrogen evolution and eventually, decomposition of the gallium hydride to gallium metal occurred. Despite [GaH3(NMe3)] being a successful synthon for the isolation of other {Ga–H} containing precursors we have reported previously, such as [GaH(NMe3−x{CH2CH2O}x)2],48,49 the amine-stabilized gallane employed was not found to react with the aminoenoate pro-ligand. Despite the various temperature and solvent conditions attempted (which included keeping the reaction mixture at 0 °C for 12 hours, or using various solvents including toluene, hexane and diethyl ether), a gallium hydride bis-amidoenoate complex could not be isolated via this route.
In contrast, when one equivalent of HAMEDip was added to a cooled (−78 °C) solution of half an equivalent of [AlH3(NMe3)] in toluene, vigorous bubbling consistent with the formation of dihydrogen was observed instantly. This mixture was allowed to stir for 4 hours, and subsequent removal of all volatiles yielded a white powder. The powder was redissolved in warm hexane and after 24 hours, yielded the complex [AlH(AMEDip)2] (4-Al) as block-like colourless crystals (Fig. 4). 4-Al was found to adopt a trigonal bipyramidal geometry akin to that of its chloride analogue 3-Al, though as expected the τ5 value of the hydride had increased to 0.79, suggesting that the smaller H-group afforded a geometrically more stable complex. Selected bond lengths and angles for 4-Al are shown in Table 2.
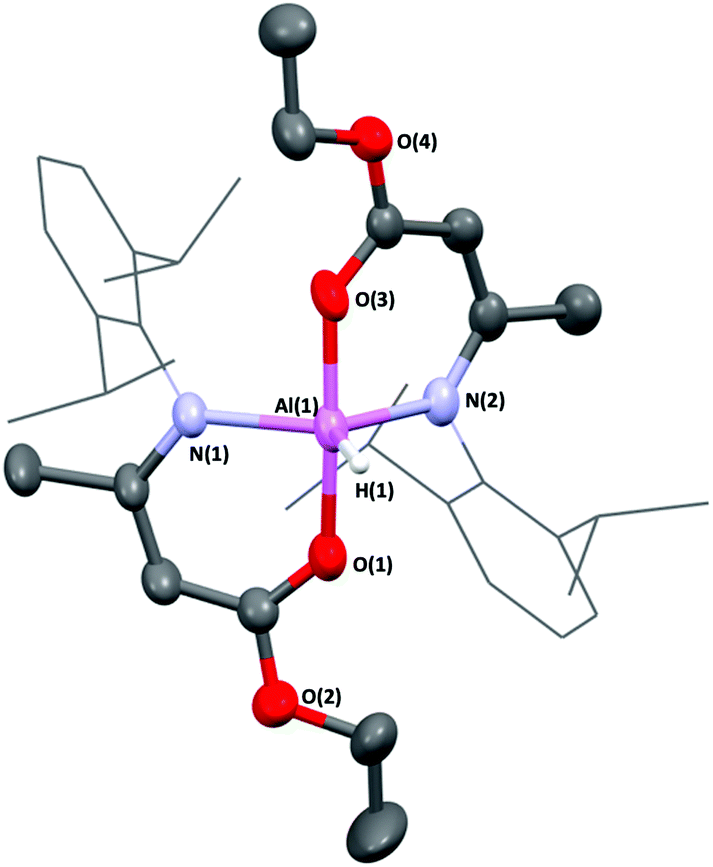 | ||
| Fig. 4 Molecular structure of [AlH(AMEDip)2] (4-Al). Thermal ellipsoids shown at 30%. Dip-substituents are in wireframe for clarity. | ||
| Bond lengths (Å) | |
| Al(1)–N(1) | 1.985(2) |
| Al(1)–N(2) | 1.984(2) |
| Al(1)–O(1) | 1.877(2) |
| Al(1)–O(2) | 1.879(2) |
| Bond angles (°) | |
| O(1)–Al(1)–O(3) | 173.79(9) |
| N(1)–Al(1)–N(2) | 126.48(9) |
| O(1)–Al(1)–N(1) | 89.30(8) |
| O(1)–Al(1)–N(2) | 87.55(8) |
| O(3)–Al(1)–N(1) | 88.34(9) |
| O(3)–Al(1)–N(2) | 89.24(8) |
Huang and co-workers have reported the only other {Al–H} containing complex stabilized by (O,N)-containing ligands, which featured β-ketoiminate (BKI) ligands and the same Dip-substituent at the N-position (formula [AlH(BKIDip)2]).44 Comparing the bond lengths in 4-Al and Huang's complex, both Al(1)–O(1) and Al(1)–O(3) bond lengths were significantly longer at 1.877(2) Å and 1.879(2) Å respectively, compared to 1.763(3) Å and 1.757(3) Å in [AlH(BKIDip)2]. However, the opposite was true for the Al(1)–N(1) and Al(1)–N(2) bond lengths (1.985(2) Å and 1.984(2) Å respectively in complex 4-Al compared to 2.079(3) Å and 2.064(3) Å in [AlH(BKIDip)2]).
This finding could potentially be a consequence of the extra oxygen atoms (O(2) and O(4)) within the enoate backbone of 4-Al. This may afford a resonance stabilization through inductive withdrawal of electron density from O(1) and O(3) respectively, causing a slight lengthening of the Al(1)–O(1) and Al(1)–O(3) bond lengths, concomitantly allowing the electron density of the N-groups to be more strongly donated to the Al center, resulting in slightly shorter Al–N bonds.
After the successful isolation of 4-Al, further attempts at synthesizing reactive gallium amidoenoate complexes were attempted. As hydride derivatives proved difficult to isolate previously, alkyl-substituents were targeted. By successfully obtaining [AlH(AMEDip)2] from an aluminium hydride adduct, we hypothesized that using a gallium alkyl species would provide a better route to the desired product, rather than trying to transform the chloride-substituent of complexes (1–3)-Ga to a methyl group. Scheme 4 shows the synthetic routes carried out towards the methyl gallium amidoenoate derivatives [GaMe2(AMEDip)] (4-Ga) and [GaMe2(AMEiPr)] (5-Ga). As neat trimethyl gallium is highly reactive, we employed the larger Dip-substituted aminoenoate pro-ligand in order to provide extra stabilization to the product formed. The 1:1 reaction of HAMEDip with [GaMe3] cooled to −78 °C in toluene was seen to evolve methane instantly upon addition. After allowing the reaction mixture to slowly warm to ambient temperature, all volatiles were removed yielding a viscous yellow oil. As tituration in hexane did not afford a solid, a range of NMR experiments and mass spectrometry confirmed that the oil was the pure 4-Ga (ESI†).
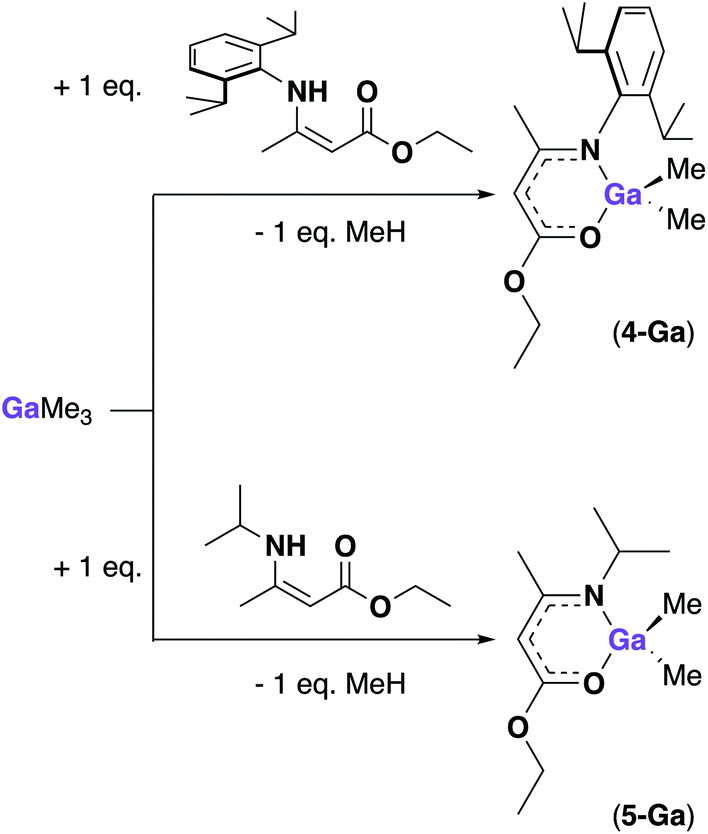 | ||
| Scheme 4 Synthetic routes to complexes 4-Ga [GaMe2(AMEDip)] and 5-Ga [GaMe2(AMEiPr)]. | ||
These included a singlet at δ = 0.04 ppm corresponding to the Ga(CH3)2 environment, a shift of 0.09 ppm when compared to free [GaMe3] (Ga(CH3)3 found at δ = −0.15 ppm) and two distinguished sets of doublets corresponding to ArCH(CH3)2 protons of the Dip-substituents at δ = 1.21 and 1.09 ppm with coupling values of 3JHH = 6.8 and 6.9 Hz respectively. The methine proton of the amidoenoate backbone was a singlet at δ = 4.90 ppm followed by the methine protons of the Dip-substituents (ArCH(CH3)2) at δ = 3.24 ppm.
The viscous nature of 4-Ga is proposed to be a consequence of the two remaining methyl groups bound to the gallium centre. Previous reports of methyl gallium derivatives also describe viscous products,28 however for solution-based applications such as AACVD, precursor viscosity does not present an issue. Following the successful isolation of 4-Ga, the smaller aminoenoate analogue (HAMEiPr) was employed in a similar reaction and afforded [GaMe2(AMEiPr)] (5-Ga) as a viscous pale oil. Multiple NMR experiments were again used to confirm that the oil was the pure complex [GaMe2(AMEiPr)] (ESI†). The peaks found included a singlet at δ = 0.11 ppm corresponding to the Ga(CH3)2 environment (downfield when compared to complex 4-Ga). A set of doublets corresponding to the isopropyl NCH(CH3)2 protons was observed at δ = 0.99 ppm (3JHH = 6.6 Hz) and overlapped with the terminal methyl group of the enoate tail (a triplet) at δ = 0.97 ppm (3JHH = 7.1 Hz). The methine proton of the amidoenoate backbone was a singlet at δ = 4.52 ppm, shifted up-field by 0.38 ppm when compared to complex 4-Ga. These differences suggest that the nature of the alkyl/aryl group at the N-position affects the acidity of the methine backbone proton in these complexes.
Aerosol-assisted chemical vapour deposition studies
Following the successful isolation of a range of aluminium and gallium amidoenoate complexes (1–4)-Al, (1–5)-Ga we performed a proof-of-concept study with a selection of these complexes (1-Ga, 3-Ga, 3-Al and 4-Al) in order to probe their merit as precursors towards their respective oxide (either α-Al2O3 or β-Ga2O3). This selection also provided a comparison between chemo (3-Al and 4-Al) and steric (1-Ga and 3-Ga) effects on deposition. Standard conditions for the depositions carried out included precursor (0.2 g) mixtures with toluene as the solvent (15 mL), an argon flow rate of 1 L min−1 and deposition temperatures of 500 °C. Films grown on silica coated float glass from 1-Ga, 3-Ga, 3-Al and 4-Al were amorphous as expected for the deposition of Al2O3 and Ga2O3 at 500 °C, and subsequent annealing at 1000 °C for 12 hours in air on quartz samples in order to improve the materials’ crystallinity was carried out and was evidenced by SEM analysis. Energy dispersive X-ray analysis (EDXA) confirmed negligible carbon contamination of all films and the expected elemental ratios (M2O3). Interestingly, the increased oxygen content installed in precursor 3-Ga (with 4 oxygen atoms in the precursor) did not afford a higher oxygen content when compared to precursor 1-Ga (with 2 oxygen atoms). This is likely due to the AACVD rig having an open-to-atmosphere exhaust and therefore diffusion of oxygen gas into the rig was a possibility.The chloride containing complex [AlCl(AMEDip)2] (3-Al) afforded ratios of Al2.2O2.9, whereas [AlH(AMEDip)2] (4-Al) resulted in the ratios of Al2.0O3.0. Similar results were observed for the gallium complexes with weight% ratios averaging 70:30 (Ga:O) for both complexes 1-Ga and 3-Ga, suggesting that film composition was not affected by the difference in steric bulk of these precursors. Furthermore, despite comparing the Cl-containing and Cl-absent precursors (3-Al and 4-Al respectively), X-ray photoelectron spectroscopy (XPS) survey analyses of all films suggested that no Cl was present in the deposited materials. This result implies that low atomic quantities of chlorine present in a precursor does not present a risk for chloride contamination during the AACVD process.
XPS analysis for films of α-Al2O3 deposited from 3-Al and 4-Al showed one distinct Al environment at an etch of 300 seconds. A representative example of XPS analysis from a film deposited from 3-Al is shown in Fig. 5.
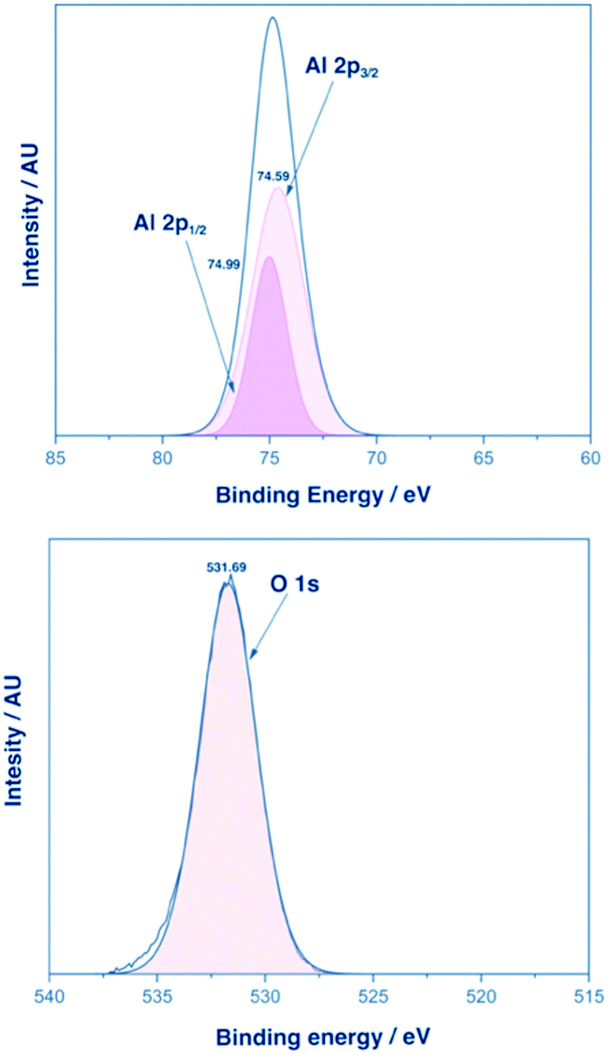 | ||
| Fig. 5 XPS analysis of an α-Al2O3 film deposited from complex 3-Al. Top: XPS of the Al 2p1/2 and 2p3/2 states. Bottom: XPS of the O 1s state. | ||
Peaks were found at 74.59 eV and 74.99 eV corresponding to the Al 2p3/2 and 2p1/2 environments respectively (separated by an energy gap of 0.40 eV) and with an intensity ratio of 1:2 (consistent with previously reported XPS analysis of Al3+).50–52 The O 1s binding energy was found at 531.39 eV and again confirmed the presence of an α-Al2O3 phase corresponding to the single environment observed. XPS analysis of thin films of β-Ga2O3 deposited from 1-Ga and 3-Ga showed the expected peaks for the Ga 2p1/2, Ga 2p3/2 as well as Ga 3d5/2 and 3d3/2 (ESI†) and were similar to previously reported gallium oxide XPS analyses.21,53
Scanning electron microscopy (SEM) was used to investigate the morphology of the deposited films, both on glass and quartz after annealing. Films deposited on glass with no annealing treatment showed a distribution of poorly connected, large particulates, consistent with the X-ray diffraction (XRD) patterns obtained for the samples deposited on glass, which were entirely amorphous and XRD patterns exhibited no conclusive information. Post-treatment annealing of the films was shown to improve the connectivity and definition of the surface particulates of all deposited material and improved the phase slightly as evidenced by XRD (ESI†). Example SEMs comparing the effects of annealing on films deposited from 3-Al are shown in Fig. 6. Similar SEMs were obtained for β-Ga2O3 deposited from 1-Ga and 3-Ga (ESI†).
 | ||
| Fig. 6 SEM images of the surface of a film deposited from 3-Al on glass (top) and on quartz after being annealed at 1000 °C for 12 hours in air (bottom). 1 μm scale bar inset. | ||
Ultraviolet–visible (UV-vis) spectroscopy was used to determine the transmittance of the films deposited on quartz. Table 3 shows the transmittance of the annealed thin films on quartz (ESI†). Transmittance measurements revealed that the annealed α-Al2O3 and β-Ga2O3 thin films transmitted 79–99% of visible light. Films deposited on glass were visibly coloured, however annealing the films on quartz afforded highly transparent and colourless films, which was reflected in the transmittance data.54,55
| Precursor | Transmittance (%) |
|---|---|
| [GaCl2(AMEiPr)] (1-Ga) | 90 |
| [GaCl(AMEDip)2] (3-Ga) | 79 |
| [AlCl(AMEDip)2] (3-Al) | 95 |
| [AlH(AMEDip)2] (4-Al) | 99 |
Conclusions
A variety of complexes containing the group 13 metals aluminium and gallium have been prepared using aminoenoate compounds as ligands to form the respective chlorido amidoenoate group 13 complexes, (1–4)-Al and (1–5)-Ga. This yielded the first presented example of a bis-chlorido aluminium species coordinated by an (O,N)-ligand. Small (iPr) and sterically demanding (Dip) R-groups at the N-position of the amidoenoate acted as a method to compare the coordination chemistry of these complexes, with degrees of distortion calculated for each complex that was isolated in the solid state.In most cases, the chlorido amidoenoate complexes were extremely stable to a variety of transformation reactions, such as hydride sources (including alkali metal hydride salts, alkyl silanes and magnesium hydride dimers) which were used in attempts to transform the {M–Cl} moiety to {M–H}. However, by varying the group 13 source, different functionalities could be installed as evidenced by the synthesis of amidoenoate complexes exhibiting {Al–H} (4-Al) and {Ga–C} (4-Ga and 5-Ga) bonds.
A proof-of-concept study using a selection of these complexes (1-Ga, 3-Ga, 3-Al and 4-Al) in AACVD experiments was carried out. These initial studies present the precursors as suitable options for routes to the deposition of the corresponding group 13 oxide. Standard deposition conditions for the films included a deposition temperature of 500 °C from toluene mixtures of the precursors, which deposited the expected amorphous material on glass. Subsequent annealing of the material on quartz substrates at 1000 °C in air for 12 hours afforded more crystalline oxide material. The films deposited were analysed using standard techniques (XRD, XPS, EDAX, SEM and UV-Vis) and confirmed the composition of the α-Al2O3 and β-Ga2O3 films, their elemental stoichiometry, transparency, and morphological characteristics. The geometries of the complexes, quantified using the τ-parameter, provide an indication of the precursors potential deposition profile. This shows that, at least for the system of the group 13 oxides Al2O3 and Ga2O3, precursors that are not sterically strained (τ-value closer to 0 or 1) afford materials that exhibit improved transmittances after deposition. In-depth AACVD investigations related to these systems are currently underway.
Experimental
All preparations were performed under an inert argon atmosphere using standard Schlenk techniques or using an MBraun nitrogen-filled glovebox. All chemicals were obtained from commercial sources. All solvents were obtained from a solvent purification system and stored over molecular sieves under an inert (Ar) environment. HAMEiPr, [AlH3(NMe3)] and [GaH3(NMe3)] were synthesised according to literature procedures.32,56 The syntheses of HAMEDip, [Li(AMEiPr)], [Li(AMEDip)] and complexes (1–4)-Al and (1–5)-Ga are detailed in the Experimental section. The oils obtained were titurated with hexane (combined with solvent, shaken vigorously, volatiles removed). This process was repeated until powders of the samples could be obtained. [GaMe3] (extremely pyrophoric!) was distilled prior to use.Single crystal X-ray diffraction (XRD) data were collected using a SuperNova Atlas (Dual) diffractometer using Cu Kα1 radiation of wavelength 1.54184 Å. Suitable crystals were selected and mounted on a nylon loop and the crystal kept at 150 K during data collection. Using Olex2,57 we solved the structure with the olex2.solve58 structure solution program using Charge Flipping and refined with the ShelXL59 refinement package using Least Squares minimization. Nuclear magnetic resonance (NMR) data were recorded in either CDCl3 or C6D6 (obtained from Sigma, dried on sieves and stored under an inert atmosphere) using a Bruker Avance 300, Bruker Avance Neo 500 or Bruker Avance Neo 700 MHz instrument at ambient temperature. 1H and 13C{1H} NMR assignments were confirmed by 1H–1H (COSY and NOESY) and 1H–13C (HSQC and HMBC) experiments where necessary. Mass spectra were obtained using a Micromass 70-SE spectrometer using electrospray chemical ionization in the positive mode (ESI+). XRD for thin films was carried out with a Bruker D8 Discover X-ray diffractometer using monochromatic Cu Kα1 and Cu Kα2 radiation of wavelengths 1.54056 and 1.54439 Å, respectively, emitted in an intensity ratio of 2:1, voltage = 40 kV; current = 40 mA. SEM/EDXA was performed using a Philips XL30 FEG with an electron beam accelerating energy of 30 kV. XPS profiling was performed using a Thermo Scientific K-Alpha XPS system using monochromatic Al Kα radiation at 1486.6 eV X-ray source. CasaXPS software was used to analyse the binding energy of the data, referenced to a C 1s peak at 284.8 eV. UV/vis/NIR transmission spectra were recorded using a PerkinElmer Lambda 950 spectrometer in the range of 300–1400 nm with a background of air, and Tauc plots from this data were used to calculate the band gaps.
Depositions were carried out under argon (99.99% from BOC). The synthesized precursors were placed into an inert AACVD glass bubbler and dissolved in an appropriate dry solvent. Two glass substrates were placed into a horizontal bed cold-wall reactor chamber, one resting on top of the graphite heating block, the second resting 0.5 cm above, ensuring a laminar flow of gas. The glass substrates were cleaned prior to deposition with isopropyl alcohol, water, and acetone and allowed to dry in air before the deposition took place. The temperature of the graphite block was controlled with a thermocouple composed of platinum-rhodium rods. An ultrasonic nebulizer containing a piezoelectric device (functioning at 20 kHz) was used to generate an aerosol mist of the precursor. The mist was carried into the reaction chamber via a flow of ultra-pure argon gas, which was optimized at a flow rate of 1 L min−1.
Synthesis of HAMEDip
2,6-Diisopropylaniline (149 mmol, 28.0 mL) was added dropwise to ethyl acetoacetate (75 mmol, 9.49 mL) dispersed over K-10 montmorillonite clay (40.0 g) in a 3-necked round-bottom flask fitted with an overhead mechanical stirrer. The reaction slurry was stirred at room temperature for 72 hours. The product was extracted via filtration by washing the clay with dichloromethane (50 mL × 3). The filtrate was concentrated under reduced pressure yielding the product as a white solid. This was purified by recrystallisation in hot methanol. Yield: 16.2 g, 77%. 1H NMR δ/ppm (500 MHz, CDCl3): 9.86 (s, 1H, NH), 7.29 (t, 1H, 3JHH = 7.7 Hz, Ar–Hpara), 7.17 (d, 2H, 3JHH = 7.6 Hz, Ar–Hmeta), 4.68 (s, 1H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHC), 4.17 (q, 2H, 3JHH = 7.1 Hz, OCH2CH3), 3.10 (sept, 2H, 3JHH = 6.9 Hz, CH(CH3)2), 1.63 (s, 3H, CH3CC), 1.31 (t, 3H, 3JHH = 7.1 Hz, OCH2CH3), 1.22 (d, 6H, 3JHH = 6.9 Hz, CH(CH3)2), 1.15 (d, 6H, 3JHH = 6.8 Hz, CH(CH3)2). 13C{1H} δ/ppm (75 MHz, C6D6): 170.7 (CO), 147.0 (N–CC), 133.0 (Ar–Cortho), 128.4 (Ar–Cpara), 123.7 (Ar–Cmeta), 77.2 (CC–CO), 58.9 (O–CH2CH3), 28.5 (CH(CH3)2), 24.8 (CH(CH3)2), 22.8 (CH(CH3)2), 19.9 (CH3CC), 14.7 (OCH2CH3).
CHC), 4.17 (q, 2H, 3JHH = 7.1 Hz, OCH2CH3), 3.10 (sept, 2H, 3JHH = 6.9 Hz, CH(CH3)2), 1.63 (s, 3H, CH3CC), 1.31 (t, 3H, 3JHH = 7.1 Hz, OCH2CH3), 1.22 (d, 6H, 3JHH = 6.9 Hz, CH(CH3)2), 1.15 (d, 6H, 3JHH = 6.8 Hz, CH(CH3)2). 13C{1H} δ/ppm (75 MHz, C6D6): 170.7 (CO), 147.0 (N–CC), 133.0 (Ar–Cortho), 128.4 (Ar–Cpara), 123.7 (Ar–Cmeta), 77.2 (CC–CO), 58.9 (O–CH2CH3), 28.5 (CH(CH3)2), 24.8 (CH(CH3)2), 22.8 (CH(CH3)2), 19.9 (CH3CC), 14.7 (OCH2CH3).
Synthesis of [Li(AMEiPr)]
HAMEiPr (75.33 mmol, 12.90 g) was dissolved in dry hexane (ca. 50 mL) and was cooled (−78 °C) using a dry ice and acetone bath. A solution of [LiHMDS·OEt2] (75.33 mmol, 18.20 g) in dry diethyl ether (ca. 200 mL) was added dropwise to the cooled mixture over a period of 30 minutes. The resulting mixture was allowed to stir overnight and slowly come to room-temperature. All volatiles were removed under reduced pressure yielding the product as a white/yellow powder. This powder was subsequently washed with cold hexane (10 mL × 2) and dried under vacuum. Yield: 10.64 g, 80%. 1H NMR δ/ppm (300 MHz, C6D6): 4.08 (s, 1H, CCHC), 3.78 (q, 2H, 3JHH = 7.0 Hz, OCH2CH3), 3.55 (hept, 1H, 3JHH = 6.3 Hz, NCH(CH3)2), 1.68 (s, 3H, CH3CN), 1.26 (d, 6H, 3JHH = 6.3 Hz, NCH(CH3)2), 1.22 (t, 3H, 3JHH = 7.0 Hz, OCH2CH3). 13C{1H} δ/ppm (75 MHz, C6D6): 168.3 (CO), 167.0 (N–CC), 75.5 (CC–CO), 61.3 (O–CH2CH3), 49.8 ((CH3)2CHN), 24.4 ((CH3)2CHN), 20.8 (CH3CCH), 14.9 (OCH2CH3).
Synthesis of [Li(AMEDip)]
HAMEDip (34.55 mmol, 10.00 g) was dissolved in dry hexane (ca. 60 mL) and cooled to and was cooled to −78 °C using a dry ice and ace-tone bath. A solution of [LiHMDS·OEt2] (34.55 mmol, 8.34 g) in dry diethyl ether (ca. 100 mL) was added dropwise to the cooled mixture over a period of 15 minutes. Solvent and volatiles were subsequently removed under reduced pressure yielding the product as a white/yellow powder. This powder was further washed with cold hexane (10 mL × 2) until the washings were colourless. The remaining white solid was dried under vacuum. Yield: 8.73 g, 86%. 1H NMR δ/ppm (300 MHz, C6D6): 7.14–7.08 (m, 3H, Ar–Hortho, para, meta), 4.32 (s, 1H, CCHC), 3.39 (br, 2H, OCH2CH3), 3.11 (br, 2H, CH(CH3)2), 1.59 (s, 3H, CH3CC), 1.22–1.20 (multiplet, 12H, CH2(CH3)2), 0.82 (t, 3H, 3JHH = 7.0 Hz, OCH2CH3). 13C{1H} δ/ppm (75 MHz, C6D6): 204.6 (CO), 170.9 (N–CC), 170.0 (Ar–Cipso) 140.3 (Ar–Cortho), 123.9 (Ar–Cpara), 123.4 (Ar–Cmeta), 75.5 (CC–CO), 61.3 (OCH2CH3), 28.3 (CH(CH3)2), 24.2 (CH(CH3)2), 23.7 (CH3CC), 14.4 (OCH2CH3).
Synthesis of [AlCl2(AMEiPr)] (1-Al)
[Li(AMEiPr)] (2.82 mmol, 0.50 g) was dissolved in dry hexane and added dropwise to a cooled (−78 °C) solution of AlCl3 (2.82 mmol, 0.38 g) in diethyl ether. The mixture was allowed to stir for 12 hours. The resulting colourless solution was filtered, and all volatiles removed yielding a pale oil which was titurated with hexane. Volatiles were removed under reduced pressure yielding a white powder. This powder was subsequently redissolved in warm hexane/diethyl ether (90:10) and stored at −20 °C overnight affording the product as colourless block-like crystals suitable for X-ray analysis. Yield: 0.52 g, 69%. 1H NMR δ/ppm (300 MHz, C6D6): 4.49 (s, 1H, CCHC), 3.68 (q, 2H, 3JHH = 7.1 Hz, OCH2CH3), 3.52 (m, 1H, NCH(CH3)2), 1.52 (s, 1H, CH2CN), 1.28 (d, 6H, 3JHH = 6.6 Hz, NCH(CH3)2, 0.86 (t, 3H, 3JHH = 7.1 Hz, OCH2CH3). 13C{1H} δ/ppm (75 MHz, C6D6): 177.3 (CO), 170.8 (N–CC), 83.4 (CC–CO), 68.2 (O–C H2CH3), 50.1 ((CH3)2CHN), 25.8 (CH3CCH), 23.5 ((CH3)2CHN, 14.4 (OCH2CH3).
Synthesis of [AlCl(AMEiPr)2] (2-Al)
[Li(AMEiPr)] (7.50 mmol, 1.32 g) was dissolved in diethyl ether (30 mL) and added dropwise to a cooled (−78 °C) diethyl ether (20 mL) solution of aluminium trichloride (3.75 mmol, 0.5 g). This mixture was allowed to stir for 12 hours and warm to room temperature. All volatiles were removed in under reduced pressure yielding a pale oil. The oil was titurated with hexane affording a powder, which was taken up in warm hexane/diethyl ether (30 mL, 90:10), filtered and cooled, yielding transparent block-like crystals suitable for X-ray analysis. Yield: 1.057 g, 69%. 1H NMR δ/ppm (300 MHz, C6D6): 4.70 (s, 2H, CCHC), 4.13 (sept, 2H, 3JHH = 6.5 Hz, NCH(CH3)2), 3.96 (q, 4H, J = 7.1 Hz, OCCH2CH3), 1.74 (s, 6H, CH3CN), 1.35 (d, 12H, 3JHH = 7.0 Hz, NCH(CH3)2, 1.06 (t, 6H, 3JHH = 7.1 Hz, OCH2CH3), 13C{1H} δ/ppm (75 MHz, C6D6): 209.9 (CO), 204.1 (CO), 174.3 (N–CC), 170.6 (N–CC), 82.0 (CC–CO), 61.7 (O–CH2CH3), 50.4 ((CH3)2CHN), 24.0 (CH3CCH), 23.1 ((CH3)2CHN), 14.8 (OCH2CH3). ESI+ Theoretical mass = 403.19; measured = 403.19.
Synthesis of [AlCl(AMEDip)2] (3-Al)
[Li(AMEDip)] (7.50 mmol, 2.22 g) was dissolved in diethyl ether (20 mL) and added dropwise to a cooled (−78 °C) diethyl ether (20 mL) solution of aluminium trichloride (3.75 mmol, 0.5 g). All volatiles were removed in vacuo and the product was taken up in hexane (20 mL) and filtered. Slow cooling of the filtrate yielded transparent block-like crystals suitable for X-ray analysis. Yield: 2.023 g, 84%. 1H NMR δ/ppm (300 MHz, C6D6): 7.04 (s, 6H, ArH), 4.87 (s, 2H, CCHC), 3.61 (p, 4H, 3JHH = 6.8 Hz, OCCH2CH3), 3.32–3.25 (m, 4H, Ar(CH)(CH3)2), 2.62–2.56 (p, 4H, 3JHH = 6.8 Hz, OCCH2CH3), 1.54 (d, 6H, 3JHH = 6.6 Hz, Ar(CH)(CH3)2), 1.40 (s, 6H, CH3CNC), 1.28 (d, 6H, 3JHH = 6.8 Hz, Ar(CH)(CH3)2), 1.11 (dd, 12H, 3JHH = 6.8, 2.0 Hz, Ar(CH)(CH3)2), 0.87 (t, 3JHH = 7.0 Hz, 6H, OCH2CH3). 13C{1H} δ/ppm (75 MHz, C6D6): 170.9 (CO), 160.2 (N–CC), 83.1 (CC–CO), 58.3 (O–CH2CH3), 44.2 ((CH3)2CHN), 23.9 ((CH3)2CHN), 18.9 (CH3CCH), 14.9 (OCH2CH3). ESI+ Theoretical mass = 639.34; mass measured = 639.35.
Synthesis of [AlH(AMEDip)2] (4-Al)
HAMEDip (4.48 mmol, 1.30 g) was dissolved in hexane (20 mL) and added dropwise to a cooled (−78 °C) hexane (20 mL) solution of [AlH3(NMe3)] (2.24 mmol, 0.2 g). Vigorous bubbling corresponding to dihydrogen gas liberation was observed. This mixture was allowed to stir overnight and slowly warm to room temperature. The resulting transparent solution was concentrated under reduced pressure. Slow cooling yielded transparent block-like crystals suitable for X-ray analysis. Yield: 1.090 g, 80%. 1H NMR δ/ppm (300 MHz, C6D6): 7.08–6.96 (m, 6H, ArH), 4.80 (s, 2H, CCHC), 3.42 (dsept, 4H, 3JHH = 27.4, 6.7 Hz, Ar(CH)(CH3)2), 3.18 (dq, 2H, 3JHH = 10.5, 7.1 Hz, OCH2CH3), 2.56 (dq, 2H, 3JHH = 10.7, 7.2 Hz, OCH2CH3), 1.37 (s, 6H, CH3C(N)C), 1.33 (s, 6H, Ar(CH)(CH3)2), 1.32 (d, 6H, 3JHH = 6.8 Hz, Ar(CH)(CH3)2), 1.20–1.09 (m, 12H, Ar(CH)(CH3)2), 0.83 (t, 6H, 3JHH = 7.1 Hz, OCH2CH3). 13C{1H} δ/ppm (75 MHz, C6D6): 175.7 (CO), 170.6 (N–CC), 147.2 (ArC), 144.6 (ArC), 143.4 (ArC), 125.7 (ArC), 124.3 (ArC), 123.3 (ArC), 82.1 (CC–CO), 60.1 (O–CH2CH3), 29.0 (Ar(CH)(CH3)2), 27.9 (Ar(CH)(CH3)2), 25.9 (Ar(CH)(CH3)2), 25.7 (Ar(CH)(CH3)2), 24.8 (CH3C(N)C), 24.2 (Ar(CH)(CH3)2), 24.0 (Ar(CH)(CH3)2), 14.5 (OCH2CH3). ESI+ Theoretical mass = 603.37; mass measured = 603.37.
Synthesis of [GaCl2(AMEiPr)] (1-Ga)
[Li(AMEiPr)] (3.75 mmol, 0.66 g) was dissolved in hexane (30 mL) and added dropwise to a cooled (−78 °C) hexane (20 mL) solution of gallium trichloride (3.75 mmol, 0.66 g). This mixture was allowed to stir for 12 hours and come to room temperature. The mixture was filtered, and the filtrate concentrated, yielding a pale oil. The oil was titurated with hexane affording a powder, which was taken up in warm hexane/diethyl ether (30 mL, 90:10), filtered and cooled. Slow cooling of the filtrate yielded transparent block-like crystals suitable for X-ray analysis. Yield: 0.724 g, 62%. 1H NMR δ/ppm (300 MHz, C6D6): 4.42 (s, 1H, CCHC), 3.65 (q, 2H, 3JHH = 7.1 Hz, OCH2CH3), 3.31 (sept, 1H, 3JHH = 6.1 Hz, NCH(CH3)2), 1.23–1.17 (m, 9H, CH3CNC; NCH(CH3)2), 0.79 (t, 3H, 3JHH = 7.1 Hz, OCH2CH3). 13C{1H} δ/ppm (75 MHz, C6D6): 174.1 (CO), 172.3 (N–CC), 82.3 (CC–CO), 62.6 (O–CH2CH3), 50.9 ((CH3)2CHN), 23.7 ((CH3)2CHN), 22.5 (CH3CNC), 14.1 (OCH2CH3).
Synthesis of [GaCl(AMEiPr)2] (2-Ga)
[Li(AMEiPr)] (7.50 mmol, 1.32 g) was dissolved in hexane (30 mL) and added dropwise to a cooled (−78 °C) hexane (20 mL) solution of gallium trichloride (3.75 mmol, 0.66 g). This mixture was allowed to stir for 12 hours and come to room temperature. The mixture was filtered and concentrated, yielding a pale oil. The oil was titurated with hexane affording a powder, which was taken up in warm hexane/diethyl ether (30 mL, 90:10) filtered and cooled. Slow cooling yielded transparent block-like crystals suitable for X-ray analysis. Yield: 1.039 g, 63%. 1H NMR δ/ppm (300MHz, C6D6): 4.58 (s, 2H, CCHC), 4.09 (q, 4H, 3JHH = 7.3 Hz, OCCH2CH3), 3.18 (dsept, 2H, 3JHH = 9.4, 6.6 Hz, NCH(CH3)2), 1.52 (s, 9H, CH3C(N)C; NCH(CH3)2), 1.08 (t, 6H, 3JHH = 7.1 Hz, OCH2CH3), 0.83 (d, 12H, 3JHH = 6.5 Hz, OCH2CH3). 13C{1H} δ/ppm (75 MHz, C6D6): 170.7 (CO), 160.3 (N–CC), 82.9 (CC–CO), 58.2 (O–CH2CH3), 44.3 ((CH3)2CHN), 24.0 (CH3C(N)C), 18.9 ((CH3)2CHN), 15.0 (OCH2CH3). ESI+ Theoretical mass, 445.64, mass measures = 431.18 ([M+] − CH3).
Synthesis of [GaCl(AMEDip)2] (3-Ga)
[Li(AMEDip)] (7.50 mmol, 2.22 g) was dissolved in diethyl ether (20 mL) and added dropwise to a cooled (−78 °C) hexane (20 mL) solution of [GaCl3] (1.14 mmol, 0.20 g). The resulting transparent solution was stirred for 12 hours, filtered and all volatiles were removed under reduced pressure. The remaining white solid was taken up in hexane (20 mL). Slow cooling of this solution yielded transparent block-like crystals suitable for X-ray analysis. Yield: 2.023 g, 84%. 1H NMR δ/ppm (300 MHz, C6D6): 7.08–6.99 (m, 6H, ArH), 4.88 (s, 2H, CCHC), 3.70 (p, 2H, 3JHH = 6.7 Hz, Ar(CH)(CH3)2), 3.44–3.35 (m, 2H, OCH2CH3), 3.33–3.25 (m, 2H, Ar(CH)(CH3)2), 2.77–2.71 (m, 4H, OCH2CH3), 1.55 (d, 6H, 3JHH = 6.6 Hz, Ar(CH)(CH3)2), 1.42 (s, 6H, CH3C(N)C), 1.29 (d, 6H, 3JHH = 6.8 Hz, Ar(CH)(CH3)2), 1.14 (dd, 12H, 3JHH = 8.2, 6.8 Hz, Ar(CH)(CH3)2), 0.87 (t, 6H, 3JHH = 7.1 Hz, OCH2CH3). 13C{1H} δ/ppm (75 MHz, C6D6): 174.5 (CO), 171.3 (N–CC), 146.2 (ArC), 145.0 (ArC), 143.9 (ArC), 126.4 (ArC), 124.9 (ArC), 123.3 (ArC), 82.6 (CC–CO), 60.4 (O–CH2CH3), 29.0 (Ar(CH)(CH3)2), 27.9 (Ar(CH)(CH3)2), 26.4 (Ar(CH)(CH3)2), 26.4 (Ar(CH)(CH3)2), 26.3 (CH3C(N)C), 25.2 (Ar(CH)(CH3)2), 24.7 (Ar(CH)(CH3)2), 24.1 (Ar(CH)(CH3)2), 14.5 (OCH2CH3). ESI+ Theoretical mass = 681.29; mass measured = 681.29.
Synthesis of [GaMe2(AMEDip)] (4-Ga)
HAMEDip (2.24 mmol, 0.65 g) was dissolved in toluene (20 mL) and added dropwise to a cooled (−78 °C) toluene (20 mL) solution of [GaMe3] (2.24 mmol, 0.26 g). Vigorous bubbling caused by methane evolution was observed. This mixture was allowed to stir for 12 hours and slowly warm to room temperature. The resulting yellow solution was concentrated under reduced pressure, yielding a yellow oil. Yield: 0.69 g, 91%. 1H NMR δ/ppm (300 MHz, C6D6): 7.05 (m, 3H, ArH), 4.90 (s, 1H, CCHC), 3.94 (q, 2H, 3JHH = 7.1 Hz, OCH2CH3), 3.24 (sept, 2H, 3JHH = 6.8 Hz, Ar(CH)(CH3)2), 1.45 (s, 3H, CH3C(N)C), 1.21 (d, 6H, 3JHH = 6.8 Hz, Ar(CH)(CH3)2), 1.09 (d, 6H, 3JHH = 6.9 Hz, Ar(CH)(CH3)2), 0.96 (t, 3H, 3JHH = 7.1 Hz, OCH2CH3), 0.04 (s, 6H, Ga(CH3)2). 13C{1H} δ/ppm (75 MHz, C6D6): 173.7 (CO), 173.5 (N–CC), 144.3 (ArC), 140.9 (ArC), 126.9 (ArC), 124.3 (ArC), 80.9 (CC–CO), 60.9 (O–CH2CH3), 28.1 (Ar(CH)(CH3)2), 24.9 (Ar(CH)(CH3)2), 24.5 (Ar(CH)(CH3)2), 23.3 (CH3C(N)C), 14.6 (OCH2CH3), −7.5 (Ga(CH3)2). ESI+ theoretical mass = 388.20; mass measured = 352.12 ([M+] − 3CH3).
Synthesis of [GaMe2(AMEiPr)] (5-Ga)
HAMEiPr (1.17 mmol, 0.200 g) was dissolved in toluene (30 mL) and added dropwise to a cooled (−78 °C) toluene (20 mL) solution of trimethyl gallium (1.17 mmol, 0.134 g). Vigorous bubbling associated with methane evolution. This mixture was allowed to stir overnight and slowly warm to room temperature. All volatiles were removed under reduced pressure yielding a pale-yellow oil. Yield: 0.248 g, 79%. 1H NMR δ/ppm (300MHz, C6D6): 4.52 (s, 1H, CCHC), 3.91 (q, 2H, 3JHH = 7.1 Hz, OCH2CH3), 3.45 (sept, 1H, 3JHH = 6.6 Hz, NCH(CH3)2), 1.49 (s, 3H, CH3C(N)C) 0.99 (d, 6H, 3JHH = 6.6 Hz, NCH(CH3)2), 0.97 (t, 3H, 3JHH = 7.1 Hz, OCH2CH3), 0.11 (s, 6H, Ga(CH3)2). 13C{1H} δ/ppm (75 MHz, C6D6): 171.6 (CO), 170.4 (N–CC), 79.6 (CC–CO), 60.3 (O–CH2CH3), 49.8 ((CH3)2CHN), 23.6 ((CH3)2CHN), 22.2 (CH3C(N)C), 14.7 (OCH2CH3), −3.6 (Ga(CH3)2). ESI+ Theoretical mass = 270.09; mass measured = 270.09.
Author contributions
KLM carried out all synthetic procedures. Crystallographic analysis was conducted by CEK. AACVD experiments were carried out by MAB and KLM. Thin film analysis was carried out by MAB. KLM wrote the manuscript. CEK and CJC supervised KLM and MAB and were involved in the design of the experiments, editing of the manuscript and discussions throughout the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the EPSRC (EP/N509577/1, EP/P020410/1 and EP/R513143). The authors also thank Dr Abil Aliev for help with NMR, Dr Sanjay Sathasivam for XPS, Dr Kersti Karu for mass spec measurements and Pilkington NSG for supplying the glass substrates.Notes and references
- S. Liu, Z. Zhang, D. Gray, L. Zhu, J. R. Abelson and G. S. Girolami, Chem. Mater., 2020, 32, 9316–9334 CrossRef CAS.
- J. C. Sarker and G. Hogarth, Chem. Rev., 2021, 121, 6057–6123 CrossRef CAS PubMed.
- C. E. Knapp and C. J. Carmalt, Chem. Soc. Rev., 2016, 45, 1036–1064 RSC.
- K. I. Y. Ketchemen, S. Mlowe, L. D. Nyamen, P. T. Ndifon, N. Revaprasadu and P. O'Brien, J. Mater. Sci. Mater. Electron., 2018, 29, 14462–14470 CrossRef CAS.
- S. Mlowe, D. J. Lewis, M. A. Malik, J. Raftery, E. B. Mubofu, P. O'Brien and N. Revaprasadu, Dalton Trans., 2016, 45, 2647–2655 RSC.
- H. Lu, D. S. Wright and S. D. Pike, Chem. Commun., 2020, 56, 854–871 RSC.
- F. Alam and D. J. Lewis, Sci. Rep., 2020, 10, 1–7 CrossRef PubMed.
- T. Wildsmith, M. S. Hill, A. L. Johnson, A. J. Kingsley and K. C. Molloy, Chem. Commun., 2013, 49, 8773–8775 RSC.
- T. Řičica, T. Světlík, L. Dostál, A. Růžička, K. Růžička, L. Beneš, P. Němec, M. Bouška and R. Jambor, Chem. – Eur. J., 2016, 22, 18817–18823 CrossRef.
- M. Tsaroucha, Y. Aksu, E. Irran and M. Driess, Chem. Mater., 2011, 23, 2428–2438 CrossRef CAS.
- K. Samedov, Y. Aksu and M. Driess, Chem. – Eur. J., 2012, 18, 7766–7779 CrossRef CAS.
- Y. Jo, S. J. Oh, S. S. Lee, Y. H. Seo, B. H. Ryu, J. Moon, Y. Choi and S. Jeong, J. Mater. Chem. C, 2014, 2, 9746–9753 RSC.
- M. Hu, X. Cai, Q. Guo, B. Bian, T. Zhang and J. Yang, ACS Nano, 2016, 10, 396–404 CrossRef CAS.
- K. Ankireddy, S. Vunnam, J. Kellar and W. Cross, J. Mater. Chem. C, 2013, 1, 572–579 RSC.
- N. Boysen, B. Misimi, A. Muriqi, J. L. Wree, T. Hasselmann, D. Rogalla, T. Haeger, D. Theirich, M. Nolan, T. Riedl and A. Devi, Chem. Commun., 2020, 56, 13752–13755 RSC.
- M. M. Stalzer, T. L. Lohr and T. J. Marks, Inorg. Chem., 2018, 57, 3017–3024 CrossRef CAS PubMed.
- S. Lee, G. Y. Lee, C. G. Kim, T. M. Chung and B. K. Park, RSC Adv., 2020, 10, 29659–29667 RSC.
- D. Rosiak, A. Okuniewski and J. Chojnacki, Polyhedron, 2018, 146, 35–41 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- A. Navulla, A. A. Tsirlin, A. M. Abakumov, R. V. Shpanchenko, H. Zhang and E. V. Dikarev, J. Am. Chem. Soc., 2011, 133, 692–694 CrossRef CAS.
- K. L. Mears, L. G. Bloor, D. Pugh, A. E. Aliev, C. E. Knapp and C. J. Carmalt, Inorg. Chem., 2019, 58, 10346–10356 CrossRef CAS PubMed.
- M. A. Bhide, K. L. Mears, C. J. Carmalt and C. E. Knapp, Chem. Sci., 2021, 12, 8822–8831 RSC.
- C. E. Knapp, D. A. Wann, A. Bil, J. T. Schirlin, H. E. Robertson, P. F. McMillan, D. W. H. Rankin and C. J. Carmalt, Inorg. Chem., 2012, 51, 3324–3331 CrossRef CAS.
- J. Hämäläinen, M. Ritala and M. Leskelä, Chem. Mater., 2014, 26, 786–801 CrossRef.
- G. G. Condorelli, G. Malandrino and I. L. Fragalà, Coord. Chem. Rev., 2007, 251, 1931–1950 CrossRef CAS.
- T. Blanquart, J. Niinistö, M. Ritala and M. Leskelä, Chem. Vap. Deposition, 2014, 20, 189–208 CrossRef CAS.
- D. L. Schulz, B. J. Hinds, D. A. Neumayer, C. L. Stern and T. J. Marks, Chem. Mater., 1993, 5, 1605–1617 CrossRef CAS.
- C. E. Knapp, P. Marchand, C. Dyer, I. P. Parkin and C. J. Carmalt, New J. Chem., 2015, 39, 6585–6592 RSC.
- J. S. Matthews, O. Just, B. Obi-Johnson and W. S. Rees, Chem. Vap. Deposition, 2000, 6, 129–132 CrossRef CAS.
- J. D. Queen, A. Lehmann, J. C. Fettinger, H. M. Tuononen and P. P. Power, J. Am. Chem. Soc., 2020, 142, 20554–20559 CrossRef CAS.
- R. Nakano, R. Jazzar and G. Bertrand, Nat. Chem., 2018, 10, 1196–1200 CrossRef CAS.
- J. A. Manzi, C. E. Knapp, I. P. Parkin and C. J. Carmalt, Eur. J. Inorg. Chem., 2015, 2015, 3658–3665 CrossRef CAS.
- M. A. Bhide, J. A. Manzi, C. E. Knapp and C. J. Carmalt, Molecules, 2021, 26, 3165 CrossRef CAS.
- X. Su, T. Kim, K. A. Abboud and L. McElwee-White, Polyhedron, 2019, 157, 548–557 CrossRef CAS.
- X. Shi-Gang, S. Li-Xin, Z. Rong-Gen and H. Xing-Fang, Surf. Coat. Technol., 2005, 199, 184–188 CrossRef.
- Z. Galazka, Semicond. Sci. Technol., 2018, 33, 113001 CrossRef.
- R. Branquinho, D. Salgueiro, L. Santos, P. Barquinha, L. Pereira, R. Martins and E. Fortunato, ACS Appl. Mater. Interfaces, 2014, 6, 19592–19599 CrossRef CAS.
- C. E. Knapp, D. Pugh, P. F. McMillan, I. P. Parkin and C. J. Carmalt, Inorg. Chem., 2011, 50, 9491–9498 CrossRef CAS PubMed.
- C. E. Knapp, I. D. Prassides, S. Sathasivam, I. P. Parkin and C. J. Carmalt, ChemPlusChem, 2014, 79, 122–127 CrossRef CAS PubMed.
- C. M. Beavers, G. H. Talbo and A. F. Richards, J. Organomet. Chem., 2011, 696, 2507–2511 CrossRef CAS.
- P. Steiniger, D. Dittrich, C. Scheiper, C. John, C. Wölper and S. Schulz, Z. Anorg. Allg. Chem., 2018, 644, 1367–1375 CrossRef CAS.
- A. Alaaeddine, C. M. Thomas, T. Roisnel and J. F. Carpentier, Organometallics, 2009, 28, 1469–1475 CrossRef CAS.
- P. Shukla, J. C. Gordon, A. H. Cowley and J. N. Jones, J. Organomet. Chem., 2005, 690, 1366–1371 CrossRef CAS.
- R. C. Yu, C. H. Hung, J. H. Huang, H. Y. Lee and J. T. Chen, Inorg. Chem., 2002, 41, 6450–6455 CrossRef CAS.
- R. O'Donoghue, S. Rahman, B. Mallick, M. Winter, D. Rogalla, H. W. Becker and A. Devi, New J. Chem., 2018, 42, 3196–3210 RSC.
- S. J. Bonyhady, C. Jones, S. Nembenna, A. Stasch, A. J. Edwards and G. J. McIntyre, Chem. – Eur. J., 2010, 16, 938–955 CrossRef CAS.
- S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754–1757 CrossRef CAS.
- D. Pugh, P. Marchand, I. P. Parkin and C. J. Carmalt, Inorg. Chem., 2012, 51, 6385–6395 CrossRef CAS PubMed.
- D. Pugh, L. G. Bloor, I. P. Parkin and C. J. Carmalt, Chem. – Eur. J., 2012, 18, 6079–6087 CrossRef CAS.
- L. Hiltunen, H. Kattelus, M. Leskelä, M. Mäkelä, L. Niinistö, E. Nykänen, P. Soininen and M. Tiittad, Mater. Chem. Phys., 1991, 28, 379–388 CrossRef CAS.
- A. Lale, E. Scheid, F. Cristiano, L. Datas, B. Reig, J. Launay and P. Temple-Boyer, Thin Solid Films, 2018, 666, 20–27 CrossRef CAS.
- R. H. Temperton, A. Gibson and J. N. O'Shea, Phys. Chem. Chem. Phys., 2019, 21, 1393–1398 RSC.
- D. Pugh, L. G. Bloor, S. Sathasivam, I. P. Parkin and C. J. Carmalt, Eur. J. Inorg. Chem., 2011, 2011, 1953–1960 CrossRef.
- M. Aguilar-Frutis, M. Garcia and C. Falcony, Appl. Phys. Lett., 1998, 72, 1700–1702 CrossRef CAS.
- M. G. Ju, X. Wang, W. Liang, Y. Zhao and C. Li, J. Mater. Chem. A, 2014, 2, 17005–17014 RSC.
- M. G. Gardiner and C. L. Raston, Coord. Chem. Rev., 1997, 166, 1–34 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr. Sect. A Found. Crystallogr., 2015, 71, 59–75 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Homonuclear and heteronuclear NMR data, X-ray crystallography collection data and XPS, SEM and UV-Vis data. CCDC 2101966, 2101967, 2101971, 2101972, 2101973, 2101974 and 2101975. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03365d |
| This journal is © The Royal Society of Chemistry 2022 |