
Approaches towards biomaterial-mediated gene editing for cancer immunotherapy
Sydney R.
Shannon
a,
Elana
Ben-Akiva
a and
Jordan J.
Green
 *ab
*ab
aDepartment of Biomedical Engineering, Institute for NanoBioTechnology, and the Translational Tissue Engineering Center, Johns Hopkins University School of Medicine, Baltimore, MD 21231, USA. E-mail: green@jhu.edu
bDepartments of Ophthalmology, Oncology, Neurosurgery, Materials Science & Engineering, and Chemical & Biomolecular Engineering, and the Bloomberg∼Kimmel Institute for Cancer Immunotherapy, Johns Hopkins University School of Medicine, Baltimore, MD 21231, USA
First published on 13th July 2022
Abstract
Gene therapies are transforming treatment modalities for many human diseases and disorders, including those in ophthalmology, oncology, and nephrology. To maximize the clinical efficacy and safety of these treatments, consideration of both delivery materials and cargos is critical. In consideration of the former, a large effort has been placed on transitioning away from potentially immunoreactive and toxic viral delivery mechanisms towards safer and highly tunable nonviral delivery mechanisms, including polymeric, lipid-based, and inorganic carriers. This change of paradigm does not come without obstacles, as efficient non-viral delivery is challenging, particularly to immune cells, and has yet to see clinical translation breakthroughs for gene editing. This mini-review describes notable examples of biomaterial-based gene delivery to immune cells, with emphasis on recent in vivo successes. In consideration of delivery cargos, clustered regularly interspaced palindromic repeat (CRISPR) technology is reviewed and its great promise in the field of immune cell gene editing is described. This mini-review describes how leading non-viral delivery materials and CRISPR technology can be integrated together to advance its clinical potential for therapeutic gene transfer to immune cells to treat cancer.
1. Introduction
Since the first clinical trial was approved in 1990,1 the field has generated excitement about its promise in treating and preventing a variety of human diseases. Many research groups are now focused on developing gene transfer agents that are safe, effective, and specific, including for targeted gene delivery to immune cells. Successful delivery has the power to modulate the behavior of the immune system at the molecular level, with implications ranging from durable remission of cancer to novel approaches for the treatment of autoimmune diseases.In 2017, the Food and Drug Administration of the United States (FDA) approved the use of the first chimeric antigen receptor-T cell (CAR-T) therapy (tisagenlecleucel) to treat acute lymphoblastic leukemia, validating the clinical potential of genetic engineering for immunotherapy.2 This particular therapy relies on the ex vivo delivery of genes to modify T cells by lentivirus transduction. Immune cells are among the hardest cells to transfect with nucleic acids in vivo due to limited endocytosis and protein production, meager lymphocyte viability, and inherent immune protective functions.3–5 For instance, some T lymphocytes are programmed to act as anti-viral or anti-bacterial effectors and have been shown to eject danger-associated DNA in response to bacterially-associated CpG (cytosine–guanine oligonucleotides).6 Additionally, leuokocytes can have innate nucleic acid sensors, such as toll-like receptors (TLRs), that can become activated during delivery of exogenous genes and trigger reduced gene expression.4 Thus, next-generation delivery materials must assist in overcoming these obstacles.
Viral gene delivery approaches, including lentiviruses, other retroviruses, adenoviruses, and adeno-associated viruses, have largely dominated the field of immune cell gene delivery as they exhibit high cellular transduction efficiencies.7 However, viral approaches have also posed challenges regarding manufacturability, mutagenicity, immunoreactivity, and toxicity. Due to these inherent limitations, non-viral polymeric, lipid-based, and inorganic carriers, have been explored. In addition to an improved safety profile, they can allow for large nucleic acid cargo capacity, ease in scale-up for manufacture, tunability of physical and chemical properties, and the potential for tissue-specific targeting.
Biomaterial and particle properties can be designed to assist in attaining the desired non-viral nanocarrier features. For example, flexible nucleic acid cargo capacity can be attained by electrostatic binding between large anionic nucleic acids and cationic biomaterials without a cargo size limitation being imposed by a viral capsid.8 Particles can be designed to degrade over hours to days by hydrolysis of ester linkages in a biomaterial's structure.9,10 Further, through the use bioreducible disulfide linkages, gene delivery nanoparticles such as those composed of lipids11 or polymers,12 can biodegrade rapidly upon reaching the reducing environment of the cytosol, releasing biological cargo intracellularly in an environmentally-triggered fashion. Beyond reducing potential toxicities, these biodegradable functionalities also help ensure rapid release of nucleic acid cargo to the desired compartment, in contrast to non-degrading biomaterials. Using biomaterial library approaches, the physicochemical properties of engineered particles can be tuned as well, including particle size and surface properties, enabling tissue and immune cell-specific delivery following systemic administration for both lipid-based (such as selective organ targeting (SORT) technology)13 and polymer-based (such as poly(beta-amino ester) (PBAE))14 nanocarriers.
In order to further advance the therapeutic application of non-viral gene editing of immune cells towards the clinic, challenges associated with extracellular delivery, intracellular transport, and with gene editing need to be considered. Extracellular barriers include anatomical barriers, enzymatic degradation, particle instability, non-specific protein interactions, aggregation, and clearance (Fig. 1). These barriers can result in poor biodistribution and quick clearance of gene delivery nanocarriers, particularly charged carriers, before they can reach target cells. Biomaterial approaches can improve extracellular transport, such as PEGylation to reduce interactions of nanoparticle surfaces with serum proteins and cellular components.15,16 Intracellular transport barriers include challenges with cellular uptake, endosomal escape, and cytoplasmic trafficking (Fig. 1). Cellular uptake of nucleic acids is a challenge as it is difficult to transport large polyanions across a non-polar cell membrane.5 It is facilitated through use of a biomaterial nanocarrier and can occur through different types of cellular entry mechanisms including clathrin-mediated and caveolae-mediated endocytosis, macropinocytosis, and phagocytosis, which are dictated by the physicochemical properties of the nanoparticle, especially size.17 In particular, particle sizes ∼80–120 nm are advantageous for targeting a range of cells by caveolae-mediated and clathrin-mediated endocytosis and larger particle sizes ∼500 nm–10 μm are advantageous for targeting immune cells such as macrophages by phagocytosis.18 Once internalized into a vesicle, an even more challenging step is that the particle must escape to the cytosol, or else be degraded or recycled out of the cell. Endosomal escape can occur through multiple mechanisms such as proton buffering-mediated osmotic swelling (including with cationic polymers with pKas in the physiological range), biomaterials that can fuse with endosomal membranes (such lipid-based materials), and pore-forming materials (such as cell penetrating peptides).19 Further challenges can also be associated with the biological cargo and with gene editing, including release of encapsulated components from the nanocarrier, and as needed, nuclear translocation and/or assembly of editing components.
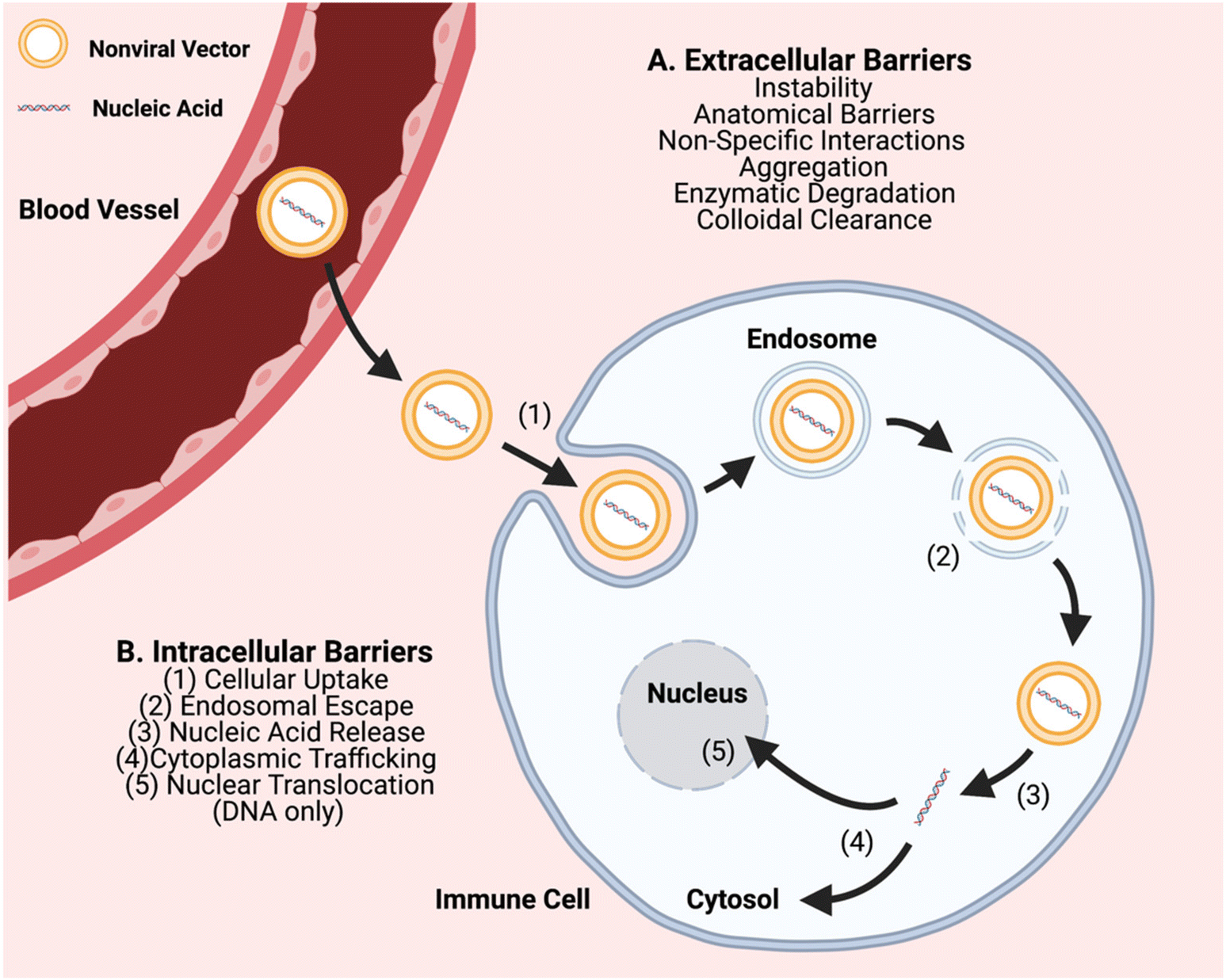 | ||
| Fig. 1 Schematic of the primary in vivo non-viral gene delivery barriers associated with common non-viral vehicles. As loaded delivery carriers travel through systemic circulation, they encounter many anatomical barriers, including epithelial/endothelial linings and the extracellular matrix (ECM). Additionally, professional phagocytes in the area are responsible for colloidal clearance, limiting a carrier's ability to act directly on target cells. Likewise, proteins (i.e. nucleases) exist in the blood and ECM to degrade exposed nucleic acids. Ultimately, crossing the cellular plasma membrane is a key rate limiting step for overall biomaterial gene transfection. Because nucleic acids cannot typically pass through the membrane unprotected, physical means or active cellular uptake mechanisms (endocytosis, pinocytosis, phagocytosis, fusion) are necessary. Intracellular steps of endosomal escape, nucleic acid release from the vehicle, and trafficking/translocation are then necessary for successful transfection. | ||
The development of clustered regularly interspaced short palindromic repeat (CRISPR)-associated protein (Cas) nucleases and related genome editing technology is revolutionizing the field of biology. CRISPR-associated protein 9 (Cas9) and related technologies enable precise editing of DNA in ways not previously possible and are useful for applications ranging from diagnostics to therapeutics. CRISPR has significant advantages over other gene-editing technologies, such as zinc finger nucleases (ZFNs) or transcription activator-like effector nucleases (TALENs), due to its simplicity, high efficiency, and broad applicability. Thus far, CRISPR technology has largely been effective for in vitro applications, with in vivo applications presenting significant additional challenges. In many ways, the general challenges of gene delivery are made even greater with CRISPR as there are more components to deliver to target cells with safety and efficacy. A target of increasing interest in the gene delivery and gene editing field are immune cells due to their key roles in critical diseases spanning from infectious diseases, to type 1 diabetes, to cancer. Non-viral biomaterials have great potential to deliver on the promise of CRISPR technology. This mini-review focuses on the recent advancements within these two fields, specifically in the context of immune cells.
2. Biomaterials for gene delivery to immune cells
Notable in vivo examples of biomaterial-based gene delivery vehicles for immune cell editing are collated in Table 1. While the overall focus of this mini-review is on cancer immunotherapy, a brief update on the use of these materials as nucleic acids carriers for specific applications to address the COVID-19 global pandemic is also discussed.| Material class | Biomaterial | Agent delivered | Immune cell target | Noted advantages | Primary outcome | Ref. |
|---|---|---|---|---|---|---|
| Polymer | Poly(β-amino ester) (PBAE) | In vitro-transcribed synthetic mRNA (IVT mRNA), DNA | T lymphocytes | (1) Biocompatible | For ref. 7: similar efficacy in murine cancer models in vivo when compared to ex vivo engineering T cell therapy | 20 and 21 |
| (2) Ease of manufacturing, distribution, and administration | For ref. 8: long term disease (cancer) remission through circulating T cell genetic modification | |||||
| (3) Low off-target binding (non-T cell) | ||||||
| (4) Low toxicity | ||||||
| Polymer | Poly(β-amino ester) (PBAE) | IVT mRNA | Macrophages | (1) Minimal toxicity with repeated dosing | (1) Delivers mRNA to produce anti-tumor effects without systemic injury | 22 |
| (2) Biodegradable | ||||||
| (3) Enhanced cytosolic nucleic acid persistence | ||||||
| Polymer | Polyethyleneimine (PEI-F25 LMW) | mRNA | Peripheral Mononuclear Blood Cells (PBMC) | (1) Minimal in vivo toxicity | (1) Prolonged survival and reduced pro-inflammatory markers within murine GvHD model | 23 |
| Polymer-lipid | Charge-altering releasable transporters (CART) | mRNA | T lymphocytes | (1) High mRNA encapsulation efficiency | (1) Relatively high CD4 and CD8 T cell transfection | 5 |
| (2) High endocytic rate due to biomaterial-based combination | ||||||
| Polymer-lipid | PBAE terpolymer-PEG lipid (A1-L3) | mRNA | Dendritic cells | (1) Highly tunable and simple synthesis | (1) High mRNA expression levels within multiple pulmonary cell types | 15 |
| (2) Tissue specificity (lung) | ||||||
| (3) Biodegradable | ||||||
| Lipid | DODAP-DOPE LNP 1,2-dioleoyl-3-dimethylammonium propane (DODAP); 1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) | DNA | Macrophages, dendritic cells, B lymphocytes | (1) Tissue selectivity (spleen) due to incorporation of helper lipid (DODAP) | (1) High murine splenic transfection post IV administration, yielding prophylactic and anti-tumor effects | 24 |
| (2) Minimal toxicity | ||||||
| Lipid | (R)-2-(2-((3R,5R,7R)-Adamantan-1-yl)acetoxy)-3-(dodecanoyloxy)propyl(2-(trimethylammonio)ethyl) phosphate (A-11) | mRNA | Macrophages (Kupffer cells) | (1) Colloidal stability with incorporation of adamantyl-based phospholipids | (1) Delivery of mRNA to non-hepatocytes without targeting ligand | 25 |
| Lipid | 306O i10 3,3′-diamino-N-methyldipropylamine (306); isodecyl acrylate (Oi10) | mRNA | Macrophages (Kupffer cells) | (1) Limited toxicity and immunogenicity with repeated dosing | (1) Delivery of three distinct mRNA sequences simultaneously | 26 |
| (2) High potency due to strong positive charge | (2) Transfection of multiple hepatocellular types simultaneously | |||||
| (3) Tissue specificity (liver) | ||||||
| Lipid | OF-Deg-Lin Ionizable Lipid | mRNA | B lymphocytes | (1) Increased cellular uptake | (1) Induce protein production in splenic B cells | 27 |
| (2) Increased protection/shielding of nucleic acid | ||||||
| (3) Induced biodegradable linkages | ||||||
| (4) Negligible toxicity | ||||||
| (5) Tissue specificity (spleen) | ||||||
| Lipid | (B-11) (3,6-bis(4-(bis (2-hydroxydodecyl) amino) butyl) piperazine-2,5-dione (CKK-E12); 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) | mRNA | Dendritic cells, macrophages, neutrophils, B lymphocytes | (1) Robust and easily variable synthesis | (1) Tumor shrinkage and extended murine survival through increase cytotoxic T cell activity | 28 |
2.1 Polymeric carriers
Polymeric carriers have been particularly useful in the realm of gene delivery as they have highly tunable chemical structures and compositions, including hydrophobicity, molecular weight, biodegradability, and charge density, that ultimately tune gene loading and gene transfection.29,30 Cationic polymeric carriers have also shown low immunogenicity and toxicity for many structures, as well as controllable degradation and elimination for high biocompatibility. Positively charged amine groups commonly found in cationic polyelectrolytes can electrostatically interact (aggregate) with negatively charged nucleic acids in aqueous conditions, forming stable polyplexes or hydrogels. Early carriers of this type included poly(ethylenimine) (PEI) and derivatives engineered to increase biocompatibility and stability during the transfection process.31,32 However, despite its expansive applications, PEI at clinically relevant concentrations and molecular weights has shown unwanted cytotoxicity.29,32,33 Therefore, biomaterials researchers have endeavored to take learnings from PEI (the importance of charged amine groups to bind nucleic acids and of titratable amine groups to aid in endosomal escape) to create more biocompatible and less cytotoxic material structures with other monomers and functional groups, especially those that include degradable linkages, including chitosan, poly(L-lysine) (PLL), poly (β-amino ester) (PBAE), charge-altering releasable transporters (CART), which are composed of poly(carbonate)-b-(α-amino ester)s, and poly(lactide-co-glycolide) (PLGA).29Thus far, polymer-mediated immunomodulatory gene delivery has largely exhibited ex vivo and in vitro successes for both innate and adaptive immune cells. Notably, Chakraborty et al. developed a variety of serum-independent acetylated-PEI/pDNA complexes coated with anionic poly(ethylene-alt-maleic-acid) (PEMA) to transfect immune cells.34 With diligent optimization, the group found that their Immunoplex formation with pDNA-polymer ratios of 30 and 10 wt% PEMA (IP30-10) reached a maximum of an eight-fold increase in GFP-reporter expression in RAW264.7 murine macrophage cells when compared to other polymer variants. While this work is influential, the group recognizes that further modifications must be made to apply the polymeric carrier to in situ situations.34 Additionally, Lou et al. developed an mRNA random copolymer p(HPMA-DMAE-co-PDTEMA-co-AzEMAm) (pHDPA) polyplex that effectively targets and transfects RAW264.7 murine macrophages and D1 dendritic cells.35 This previously developed polyplex optimized for targeting,36 was further modified with GALA peptides that were shown to enhance endosomal escape (coined PPx-GALA).37 In this study, the PPx-GALA polyplex showed 36% and 50% transfection efficiencies for murine macrophages and dendritic cells, respectively. The results of this work show promising applications for this polyplex as an in vivo mRNA vaccine.35
Other in vitro immunomodulatory successes focus on engineered T cells, specifically chimeric antigen receptor (CAR) T cells.38 To date, most CAR T cell therapies have employed viral vectors.38 However, to reduce viral-associated off target effects and permanent genetic changes, non-viral delivery methods have come to the forefront of the CAR T cell manufacturing.38–40 Thus far, few polymeric gene carriers have been developed to transfect primary human T cells.31 With that motivation, Olden et al., among other researchers, synthesized a library of pHEMA-g-pDMAEMA comb and sunflower polymers that could transfect mRNA and pDNA into CD4+/CD8+ primary human T cells with 25% and 18% transfection efficiencies, respectively. These results suggest this polymer's potential use in the realm of CAR-T cell manufacturing.31
Due to unwanted off-target effects, cytotoxicity, and limitations associated with complex serum, in vivo polymeric carriers in the realm of immunomodulation have been limited. These barriers were overcome by Smith et al., who successfully transfected DNA into lymphocytes in vivo using synthetic nanoparticles.21 This group synthesized poly (β-amino ester)-based (PBAE) nanoparticles coupled with T-cell-targeting anti-CD3e f(ab′)2 molecules, as well as peptides for microtubule-associated sequences (MTAS) and nuclear localization signals (NLS). The results showed that these nanocarriers transfected T cells with DNA encoding leukemia-specific CARs at 34% efficiency in vivo, programming functional antigen-recognition and anti-tumor effects (Fig. 2).21
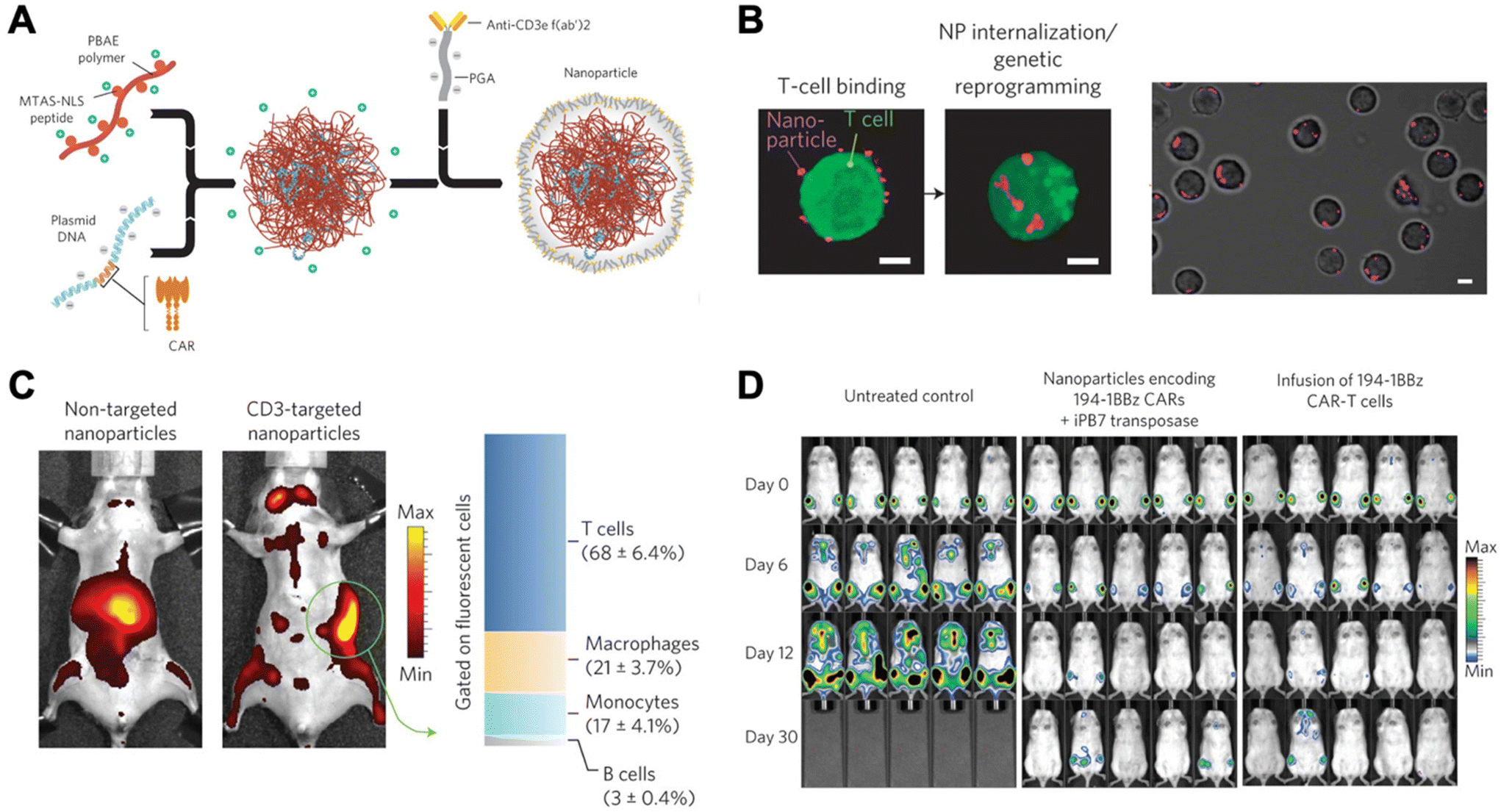 | ||
| Fig. 2 Design and in vivo functionality of lymphocyte-programming nanoparticles. (A) Schematic describing the fabrication of the poly(β-amino ester) nanoparticles. (B) Confocal microscopy indicates rapid internalization of particles from the cell surface of T cells within 120 min. (C) Biodistribution of nanoparticles. A bar graph on the right represents percentages of splenocytes positive for fluorescently labelled nanoparticles in animals treated with CD3-targeted nanoparticles. T cells (CD3+), macrophages (F4/80+, CD11b+, CD11c−), monocytes (CD11b+, Gr1+, F4/80low), and B cells (B220+) were measured using flow cytometry. (D) Bioimaging of firefly luciferase expressing leukemia cells systemically injected. Ex vivo CAR-T cells, a current standard, were used for comparison. Reproduced from ref. 21 with permission from Springer Nature, copyright 2017.21 | ||
Since their first pioneering publication, this group further developed their proof-of-concept to deliver mRNA, thereby avoiding the necessity for the genetic material to cross nuclear membranes.20 The nanocarrier used was composed of a PBAE variant previously developed by Tzeng et al.;41 this type of polymer is particularly useful in that it is biocompatible, enables efficient endosomal escape, and exhibits a relatively low toxicity, especially when compared to PEI.20 These PBAE nanocarriers that encapsulated CAR-specific mRNA, were conjugated with anti-CD8 antibodies and coated with polyglutamic acid (PGA) for optimized characteristics. Once injected in vivo, these nanocarriers transiently transfected T cells with 10% efficiency to express CARs. Although these in vivo methods have focused on T cells and CAR therapies, recent literature has also featured the genetic modulation of other immune cell types. For instance, Zhang et al. showed effective genetic reprogramming of tumor-associated macrophage cells (TAMs) in three separate in vivo experiments.22 This was done using PBAE-mRNA particles coated with PGA/Di-mannose for charge shielding purposes. These results exhibited promising applications in combinatory human cancer treatments for the future.
Further, Przybylski et al. developed and optimized a polycationic PEI-based mRNA nanoparticle that transduces bone marrow cells in a GvHD murine model to yield statistically significant cellular survival when compared to other treatment groups and untreated controls.23 Optimization of polymer synthesis and particle formulation also plays a role in Kaczmarek et al.'s development of polymer-lipid nanoparticles that functionally deliver mRNA to pulmonary immune cells (primarily dendritic cells).15 Although these in vivo transfection rates were low (2%), the optimized methodology used proved to yield more effective polymer formulations when compared to pre-optimized forms.
McKinlay et al. developed poly(carbonate)-b-(α-amino ester)-based nanoparticles named charge-altering releasable transporters (CARTs) and found that by tuning the hydrophobic lipid block of the amphipathic polymer, high mRNA transfection to Jurkat T cells (∼80%) in vitro was observed along with modest efficiency at transfecting T cells (∼1.5%) in vivo.5 In this delivery system, mixed hydrophobic blocks composed of both oleyl and nonenyl functionalities, showed significantly higher transfection rates at Jurkat T cell transfection when compared to either single hydrophobic lipid block CART or commercially-available Lipofectamine 2000. Although in vivo transfection of CD4 and CD8 T cells with the mixed-lipid block CARTs was relatively modest, it outperformed the transfection with single-lipid block CARTs (<1%).5 These experiments varying the hydrophobic nature of the polymer are just one of numerous approaches possible to yield an improved clinical candidate. Ultimately, with the increasing ability to tune characteristics, polymeric carriers may represent a clinically relevant group of non-viral delivery materials for translational immunomodulation in a variety of immune cell types.
2.2 Lipid-based carriers
As a result of the first therapeutic siRNA lipid-based delivery vehicle being globally approved in 2018, excitement surrounding the advancement of biomaterial-based gene delivery to immune cells drastically rose.42 Among biomaterials, ionizable lipid-based carriers in lipid nanoparticles (LNPs) have become the most thoroughly investigated and widely used non-viral nucleic acid delivery method.11 Their ease of manufacturability, diverse functionalization capacity, and ability to transfect multiple cell types has aided in their succces.24,38,43–45 The first ionizable lipids developed, including 1,2-dioleoyl-3-dimethylammonium propane (DODAP) and 1,2-dioleyoxy-N,N-dimethyl-3-aminopropane (DODMA), were useful in their potential to overcome issues with permanently cationic lipid systems. Since then, the chemical structure of these lipids has been altered, along with the synthesis of other adaptable ionizable compounds.24Despite advantages and favorable modifications, their applications are severely limited by the commonly observed weak correlation between ex vivo/in vitro and in vivo transfection performance.5,46–48 For instance, Paunovska et al. synthesized a library of hundreds of DNA-barcoded LNPs (lipid nanoparticles) that showed weak correlations in macrophage transfection efficiency when comparing in vivo and in vitro systems.48 Because of this discrepancy, many research groups are focusing on optimizing in vitro/ex vivo outcomes for more viable in vivo translation in the future. For instance, Billingsley et al. developed a library of ionizable lipid nanoparticles (LNPs) that were composed of various alkyl chains and polyamine cores to deliver mRNA to T lymphocytes.38 The experiment was optimized and showed that one specific LNP (C14-4) outperformed other LNPs in transfecting mRNA into the Jurkat cell line in vitro. This C14-4 LNP was able to successfully deliver CAR mRNA to primary human T cells ex vivo while reducing cytotoxicity and inducing similar potent cancer cell death when compared to the standard electroporation (EP) and EP/lentivirus, respectively.38 These results suggest the great potential in moving away from current costly ex vivo protocols. Additionally, Zukancic et al. analyzed differences associated with PEG in immunocellular transfection rates and organ-specific transfection for more effective in vivo translation.16 This group developed a PEGylated lipid nanoparticle (LNP) loaded with pDNA that showed relatively effective gene transfection in lymph nodes; the LNPs with 3% Tween 20 surfactant containing branched PEG (approximately 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 relative light units per mg tissue) showed greater than a five-fold increase in transfection rate within the lymph node when compared to formulations containing Tween 80 surfactant or the gold-standard PEG-DSPE (both approximately 15000 relative light units per mg tissue). In another instance, Harris et al. sought to optimize in vitro Jurkat and primary T cell gene transfection by comparing a variety of transfection variables and materials, including those that are both viral and nonviral.49 The group found that commercial Lipofectamine LTX, a non-viral formulation that combines negatively charged pDNA and positively charged liposomes, significantly outperformed other materials (including branched PEI, linear PEI, jetPEI, and TurboFect). More specifically, Lipofectamine exhibited a maximized Jurkat T cell transfection efficiency of 63% and a noticeably lower, but still maximized CD3+ T cell transfection efficiency of 8.1%. This group's further analysis suggests that the significantly lower primary T cell transfection efficiency may be due to delivery issues at the levels of cellular uptake or DNA cytoplasmic sensing.49 This comparative analysis is especially helpful in targeting locations for improvement within the delivery pathway in vitro, ultimately setting the stage in vivo translation of not only lipid-based, but also other non-viral gene delivery materials.
000 relative light units per mg tissue) showed greater than a five-fold increase in transfection rate within the lymph node when compared to formulations containing Tween 80 surfactant or the gold-standard PEG-DSPE (both approximately 15000 relative light units per mg tissue). In another instance, Harris et al. sought to optimize in vitro Jurkat and primary T cell gene transfection by comparing a variety of transfection variables and materials, including those that are both viral and nonviral.49 The group found that commercial Lipofectamine LTX, a non-viral formulation that combines negatively charged pDNA and positively charged liposomes, significantly outperformed other materials (including branched PEI, linear PEI, jetPEI, and TurboFect). More specifically, Lipofectamine exhibited a maximized Jurkat T cell transfection efficiency of 63% and a noticeably lower, but still maximized CD3+ T cell transfection efficiency of 8.1%. This group's further analysis suggests that the significantly lower primary T cell transfection efficiency may be due to delivery issues at the levels of cellular uptake or DNA cytoplasmic sensing.49 This comparative analysis is especially helpful in targeting locations for improvement within the delivery pathway in vitro, ultimately setting the stage in vivo translation of not only lipid-based, but also other non-viral gene delivery materials.
Despite the challenges discussed and outlined in Fig. 1, several groups have been able to show effective in vivo lipid-based gene transfection to immune cells. For example, Kimura et al. synthesized a novel lipid system to safely deliver antigen-encoding pDNA specifically to splenic immune cells in vivo.24 This lipid combination primarily featured 1,2-dioleoyl-3-dimethylammonium propane (DODAP), a material previously rendered ineffective in the realm of gene delivery. When combined with helper lipid 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), the DODAP-DOPE LNP achieved gene expression within antigen presenting cells, with emphasis on the statically significant gene expression in B lymphocytes.24 B cells also served as transfection targets for Fenton et al.'s work involving ionizable lipid mRNA carriers.27 These lipid-based nanoparticles, termed OF-Deg-Lin, were synthesized to include degradable linkages for increased delivery impact in vivo. Impressively, this group reported that 85% of LNP-induced protein production occurred within splenic immune cells. This marks an impressive feat, as many RNA therapies have limited systemic delivery outside of the liver.25
Of late, the creation of lipid libraries has proved to be a highly effective optimization strategy for particle synthesis and in vivo transfection. For instance, Oberli et al. utilized a two-phase optimization library, in which B-11 proved to be an effective mRNA carrier to a variety of immune cell types. Among those cells, 4.6% of DCs, 1.2% of macrophages, 3.3% of neutrophiles, and 0.06% of B cells were transfected with the LNP genetic reporter.28 Additionally, Gan et al. identified an ionizable adamantyl-containing LNP (constrained LNP) that successfully delivers mRNA to Kupffer cells, immune macrophages without targeting ligands.25 This LNP was developed as part of a vast library and was identified using FIND, a high throughput in vivo (in this case, in mice) assay.50 While these empirical library screening techniques have largely been effective, Zhao et al. developed a structure-based screening technique that minimizes the use of trial-and-error methods and therefore requires a lighter workload.51 The library selected two lipid-like materials (lipidoids) containing imidazole, named 93-O17S and 9322-O17S, that are most effective at delivering reporter and Cre-recombinase mRNA to T lymphocytes in vivo. Most notably, lipidoid 93-O17S showed 8.2% and 6.5% gene recombination efficiencies in splenic CD4+ and CD8+ T cells, respectively.51
Even more recently, Hajj et al. developed an in vivo branched tailed ionizable lipid-like (306i10) mRNA delivery system that has the ability to transfect Kupffer cells (86–88% efficacy) with three functionally distinct mRNAs simultaneously.26 While these recent experiments require further in vivo optimization, their clear successes open the door for advancements in the clinical translation of lipid-based gene delivery to immune cells, including for potential genetic editing of immune cells.
2.3 Inorganic carriers
Inorganic nonviral delivery vehicles, often containing metals, inorganic salts, or ceramics, are less established and of lesser prevalence for both in vitro and in vivo applications than are cationic lipid and polymeric carriers.7 These inorganic formulations can be beneficial in that they can be more stable when stored and often do not require dissociation of the nucleic acid from the carrier structure for effective gene activity in immune cells.3 For instance, Peng et al. developed a series of porous metal–organic frameworks (coined Ni-IRMOF-74-II to -V) that bind ssDNA and effectively deliver it to immune cells in vitro.3 This experiment showed that Ni-IRMOF-74-II and -III consistently outperformed commercial methods (Lipo and Neofect) in effective ssDNA transfection of RAW264.7 mouse macrophages, THP-1 human macrophages, CD4+ T cells, and B cells. Inorganic delivery vehicles represent an alternative class of immunomodulatory nonviral gene delivery that will continue to gain momentum with further engineering and chemical optimization.2.4 Biomaterial-based genetic vaccines for SARS-CoV2
Nucleic acid gene delivery for immunomodulation has played a large role in the context of the COVID-19 global pandemic. Two COVID-19 vaccines, released by Pfizer-BioNTech (Comirnaty) and Moderna (Spikevax), mark the first mRNA vaccines to be approved by the FDA. These approvals follow emergency use authorizations by the FDA for Pfizer-BioNTech's COVID-19 vaccine (BNT162b2) and Moderna's COVID-19 vaccine (mRNA-1273), both of which are lipid nanoparticle delivery vehicles containing mRNA that encodes the spike glycoprotein on the surface of the SARS-CoV-2 virus.52,53 These non-viral genetic vaccines represent a triumph in the realm of in vivo translation of biomaterial-based gene delivery. In terms of pipeline development, 33 out of 124 vaccines in clinical development are RNA or DNA based.54 Although most of these nucleic acid-based vaccines are intended for local intramuscular or intradermal delivery, their development marks an important milestone in the advancement of immunomodulatory non-viral gene delivery and are a harbinger of future, even more targeted, biomaterial-based systems to genetically modify immune cells.3. CRISPR-based gene editing of immune cells for cancer immunotherapy
CRISPR is currently at the forefront of gene editing technology, and has led to huge strides in the ability to utilize gene editing for therapeutic purposes. CRISPR utilizes a single guide RNA (sgRNA) that is designed to bind specifically to the DNA sequence that is to be edited, in combination with the Cas9 enzyme, which recognizes the sgRNA and cleaves the corresponding DNA at that site via a double-stranded break (DSB). The CRISPR/Cas9 system can be used to knock-out the target gene when the DSB is repaired by non-homologous end joining (NHEJ). NHEJ is highly error-prone, so it often introduces loss-of-function mutations into the gene. Alternatively, CRISPR/Cas9 can be used to knock-in a gene of interest using homology-directed repair (HDR), in which a DNA template containing the gene to be knocked-in is used to repair the DSB. The reader is directed to the cited articles for more detailed information on CRISPR mechanisms.55,56 In the context of editing immune cells for cancer immunotherapy applications, CRISPR/Cas9 has been most extensively utilized to edit CAR T cells.57 However, researchers are starting to investigate the use of CRISPR/Cas9 for other immune modulation applications, and there are many other potential applications that will likely make CRISPR an important tool in the future of immunotherapy.583.1 Considerations on the use of biomaterials to edit immune cells
Gene editing via CRISPR/Cas9 presents distinct encapsulation and delivery challenges with biomaterials as it is typically necessary to transport multiple types of biological cargo. Utilizing CRISPR/Cas9 gene editing requires the delivery of at least two components, the target-specific sgRNA and the Cas9 endonuclease, and there are multiple forms in which these components can be delivered. Three primary approaches are currently being investigated for biomaterial-mediated gene editing: (1) protein based: delivering Cas9 protein with sgRNA in a ribonucleoprotein (RNP) complex, (2) DNA based: delivering DNA plasmid(s) encoding for both Cas9 and the sgRNA, and (3) RNA based: delivering mRNA encoding for Cas9 with a separate sgRNA.59 Each of these approaches possess unique advantages and disadvantages and design constraints for delivery vehicles, and the cargo format needs to be selected for each gene editing application with consideration to these factors. Although RNP delivery results in the shortest-lasting presence of Cas9 in cells, the Cas9 is immediately available with no need for transcription or translation. Delivery of plasmid DNA is attractive as it is the simplest to manufacture and the least expensive to produce of the available options. However, DNA needs to be delivered to the nucleus, and this extra trafficking step can significantly hamper gene delivery efficiency, especially in difficult-to-transfect immune cells. On the other hand, delivery of mRNA eliminates the need for nuclear localization and there is no risk of insertional mutagenesis, but the difference in cargo lengths between Cas9 mRNA and the smaller sgRNA may pose an encapsulation challenge, the expression kinetics may lead to inefficiency, and RNA is challenging to manufacture at scale.59While non-viral delivery materials have been successfully employed for CRISPR/Cas9-based gene editing of non-immune cells, the application to immune cells is still emerging. As this field is growing rapidly, it is expected that many of the biomaterials developed for nucleic acid delivery to immune cells that were discussed in the previous section of this review will be useful and applied towards delivery of CRISPR/Cas9 components for gene editing. Despite the Cas9 protein being positively-charged, when complexed with sgRNA, the resulting RNPs are highly anionic.60 As a result, Cas9/sgRNA RNPs can be packaged using many of the same non-viral approaches as nucleic acids, such as cationic LNPs and polymers. LNPs have been employed for gene editing via delivery of RNPs,61,62 DNA,63 and mRNA,64,65 and some have been developed that can successfully deliver both the Cas9 mRNA and sgRNA in a single delivery vehicle despite their size difference.64 Cationic polymers have also been utilized for delivery of the CRISPR/Cas9 components, primarily in DNA,66–68 RNA,67 or DNA/RNA co-delivery69 formats, although delivery of RNPs with biodegradable polymers has also been demonstrated by balancing cationic polymer charge with anionic groups.70 While many polymer systems have been developed for delivery of mRNA and oligonucleotides separately, these previously used vehicles may not be optimal for both in combination. Therefore, new co-encapsulation formulation strategies may be needed for certain polymeric carriers. Nucleic acid encapsulation properties of a polymeric nanocarrier and delivery efficacy may also be further boosted by fine control of nucleic-acid loading via kinetic control of self-assembly.71 Thus, there are multiple strategies that non-viral biomaterials can provide to be able to deliver both gRNA and Cas9 endonuclease (or genetic information encoding it) in vivo. As immune cells are relatively resistant to transfection, the development of highly efficacious and safe non-viral delivery materials, such as those described in section 1, that can efficiently deliver biological components intracellularly to immune cells, will be critical in achieving CRISPR/Cas9-mediated gene editing of immune cells in situ. In particular, the extracellular delivery barriers and especially the ability to avoid the liver and clearance from the blood, will be critical for in vivo gene editing of immune cells. The recent research on advanced lipid-based materials such as lipidoids,51 biodegradable multi-component polymers such as poly(beta-amino ester)s,14,20,21 and novel nanocarrier constituents such as those with the SORT technology,13 all point to promising directions of using biomaterials to overcome the targeting challenges to genetically modulate immune cells in vivo. Further, given their flexibility for easy swapping of nucleic acid sequences to be encapsulated and modularity, biomaterial-based systems for gene editing can be an enabling technology to advance basic research in selecting the best nucleic acid cargos to help ensure gene editing fidelity.72 A final key consideration for the ideal biomaterials for genetic editing of immune cells is that the biomaterials themselves, as well as the subsequent nanocarriers that they form, can be manufactured scalably and inexpensively, and that the products can be stored stably over time. In this manner, biomaterial-based systems, unlike viral-based systems and cell-based systems, have great potential for the technology to be low cost and broadly accessible.
3.2 T cells
CRISPR has been researched extensively in the engineering of CAR T cells during the last decade.73 CAR T cells are traditionally cytotoxic T cells that are isolated from a patient, engineered ex vivo to introduce a CAR specific to the patient's tumor cells, expanded, and then re-introduced into the patient to recognize and kill the tumor cell targets.57,74 CAR T cells are typically transduced ex vivo with a gene encoding for the CAR. However, gene-editing technologies have shown the potential to significantly improve potency of CAR T cells.57,73,75 CRISPR/Cas9 has been used to disrupt the endogenous TCR and knock in a CD19 CAR at the T cell receptor α constant (TRAC) locus so that it is under its transcriptional control.76 The CRISPR-edited T cells showed significantly enhanced potency over conventionally engineered CAR T cells. Other researchers have performed similar successful studies utilizing CRISPR instead of traditional viral transduction methods to introduce CARs targeting tumor antigens into T cells.73,77,78In addition to using CRISPR to introduce the CAR into T cells, CRISPR technology has been utilized to edit CAR T cells in other ways to improve their functionality in vivo.73,75 There are a number of mechanisms of resistance to CAR T cells, such as PD-1 blockade and limited CAR T cell persistence in vivo. PD-1 blockade, in particular, can be a significant hurdle in effective CAR T cell treatment. As a result, there has been interest in knocking out the PD-1 gene in CAR T cells using gene editing technologies.73,79–82 Lu et al. reported on a clinical trial in which they tested the safety and efficacy of CRISPR/Cas9 edited T cells in patients with refractory non-small-cell lung cancer.83 The T cells were isolated from patients and expanded and edited ex vivo to disrupt the PD-1 gene and then reinfused into the patients. Plasmids encoding Cas9 and sgRNA were delivered into the T cells by electroporation, and the editing efficiency was fairly low at 5.81%, but using ribonucleoprotein (RNP) complexes significantly enhanced the editing efficiency. The cohort was too small to show significant therapeutic efficacy, but the study did confirm the safety and feasibility of CRISPR-engineered T cells as a cancer therapy. There was very limited off-target editing, and none of the patients had any severe treatment-related adverse events. Stadtmauer et al. similarly used CRISPR to knock-out PD-1 in CAR T cells, and also knocked out the endogenous T cell receptor (TCR) in an effort to increase engagement with the CAR (Fig. 3).84 CAR T cells have similarly been improved by targeting protein tyrosine phosphatase N2 (PTPN2), which enhanced T cell-mediated immunosurveillance, cytotoxicity, homing, and overall therapeutic efficacy. PTPN2-deficient HER-2 targeting CAR T cells were successful in eradicating HER-2 expressing breast cancer in a mouse model.85 Another technique has been to knock-out the TGF-β receptor in CAR T cells to reduce conversion to regulatory T cells and prevent CAR T cell exhaustion.86
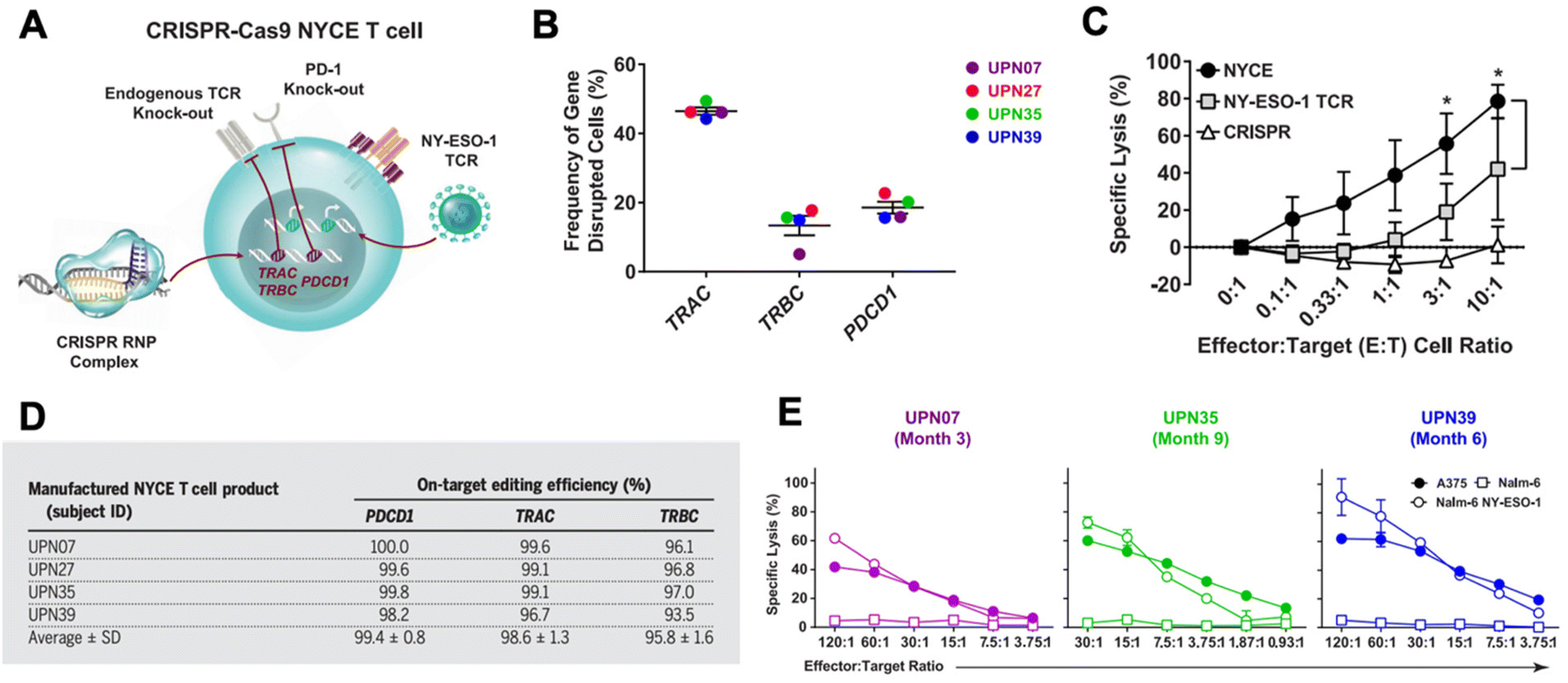 | ||
| Fig. 3 CRISPR/Cas9 engineering of T cells in patients with cancer. (A) T cells were isolated from four cancer patients (UPN07, UPN27, UPN35, and UPN39) and were loaded with CRISPR/Cas9 RNPs delivering three sgRNAs that resulted in disruption of the TRAC and TRBC (leading to endogenous TCR deletion) and the PDCD1 (leading to PD-1 deletion) loci. The cells were then transduced with a lentiviral vector to express a TCR specific for the cancer antigen NY-ESO-1. (B) The frequency of TRAC, TRBC, and PDCD1 editing in the total T cell population was determined. (C) Cytoxicity of NYCE T cells (T cells with both CRISPR knockouts and TCR transduction) against NY-ESO-1-expressing cancer cells was measured and compared to T cells with TCR transduction but without CRISPR editing (NY-ESO-1 TCR) and T cells with CRISPR editing but without TCR transduction (CRISPR). (D) The average on-target CRISPR/Cas9 editing efficiency for each target in each patient was measured. (E) To ensure that the NYCE T cells retain their antigen-specific cytolytic activity after infusion, the T cells were recovered from patients at the times specified post-infusion and expanded in the presence of the NY-ESO-1 antigen. The ability of the expanded effector cells to elicit a cytotoxic effect against NY-ESO-1-expressing cells A375 and Nalm-6 NY-ESO-1 compared to non-antigen-specific Nalm-6 cells was measured. Reproduced from ref. 84 with permission from American Association for the Advancement of Science, copyright 2020.84 | ||
Another shortcoming of CAR T cells is the high cost associated with isolating patient T cells and engineering and culturing them ex vivo. Universal CAR T cells provide an interesting alternative that could allow the therapy to be scaled up and administered more broadly, and there has been interest in using CRISPR technology to knock-out genes that could cause immune reactions between patients.87 Because of the polymorphic human leukocyte antigen (HLA) genes, CAR T cells generated from one patient cannot be used for another patient unless they are an HLA-match. Ren et al. attempted to create universal CAR T cells to address this problem by disrupting the HLA genes using CRISPR gene editing.88 The authors used lentiviral delivery of CAR and electro-transfer of Cas9 mRNA and sgRNAs targeting the endogenous TCR, β-2 microglobulin, and PD-1, thereby generating CAR T cells deficient in TCR, HLA class I, and PD-1. The edited T cells showed potent anti-tumor cytotoxicity in vitro and in animal models. Additionally, the TCR and HLA-deficient cells had reduced alloreactivity and did not cause graft-versus-host disease.
3.2 Macrophages and natural killer (NK) cells
Researchers have begun to investigate CRISPR/Cas9 for genetic engineering of NK cells and macrophages for cancer immunotherapy. NK cells are cytotoxic lymphocytes that play a key role in cancer immunity. NK cells become activated in the absence of MHC class I and, thereby, recognize tumor cells that have downregulated MHC class I expression in order to avoid CD8+ T cell recognition and killing.89 As a result, there has been interest in adoptive transfer of NK cells for cancer immunotherapy. However, NK cells often become exhausted in the tumor microenvironment, and they lack the antigen specificity of T cells. Thus, researchers have looked to genetic engineering in order to tackle these shortcomings of NK cells.90 However, progress has been slower compared to T cells, as NK cells are traditionally very difficult to transfect using viral vectors.91 Rautela et al. successfully used CRISPR-Cas 9 RNPs to genetically modify primary human NK cells.91 They disrupted CIS, a negative regulator for IL-15 signaling, which heightened NK cell responsiveness to IL-15 and, thereby, made them more potent. Pomeroy et al. used CRISPR/Cas9 to knock out ADAM17 and PDCD1, inhibitory signaling molecules in NK cells, to increase their potency for cancer immunotherapy.92 Huang et al. utilized CRISPR/Cas9 RNPs to knock-in CD16 and DNAM-1 into the NK-92 cell line.93 CD16 is critical for NK cell-mediated antibody dependent cellular cytotoxicity (ADCC), and DNAM-1 is a receptor for certain ligands on cancer cells. Thus, knocking-in both genes encoding for these proteins leads to significantly enhanced NK-92 cytotoxicity. There are many potential future applications for CRISPR/Cas9-mediated engineering of NK cells for cancer immunotherapy, including engineering CAR NK cells, promoting activating pathways, disrupting inhibitory pathways, and improving tumor infiltration of NK cells.90Macrophages have also been shown to play a critical role in cancer immunity. Tumor-associated macrophages (TAMs) typically possess a highly immunosuppressive phenotype that contributes to tumor progression and promotes further immune evasion by the tumor. As a result, there has been significant interest in targeting TAMs in the tumor microenvironment in order to kill them or repolarize them to a more immunostimulatory, anti-tumorigenic phenotype. Ray et al. engineered cationic arginine-coated gold nanoparticles that were able to deliver the complete CRISPR/Cas9 machinery to RAW264.7 murine macrophages (Fig. 4).94 They used this delivery platform to knock out SIRP-α in the macrophages. SIRP-α binds to CD47 on cancer cells and prevents phagocytosis of the cancer cells by the macrophages. They showed that CRISPR/Cas9-mediated disruption of SIRP-α led to enhanced phagocytosis of cancer cells by the macrophages. Through these approaches, non-viral biomaterials can be used to deliver either nucleic acids encoding Cas9 machinery or to deliver the combined RNP complexes to target immune cells for genetic editing.
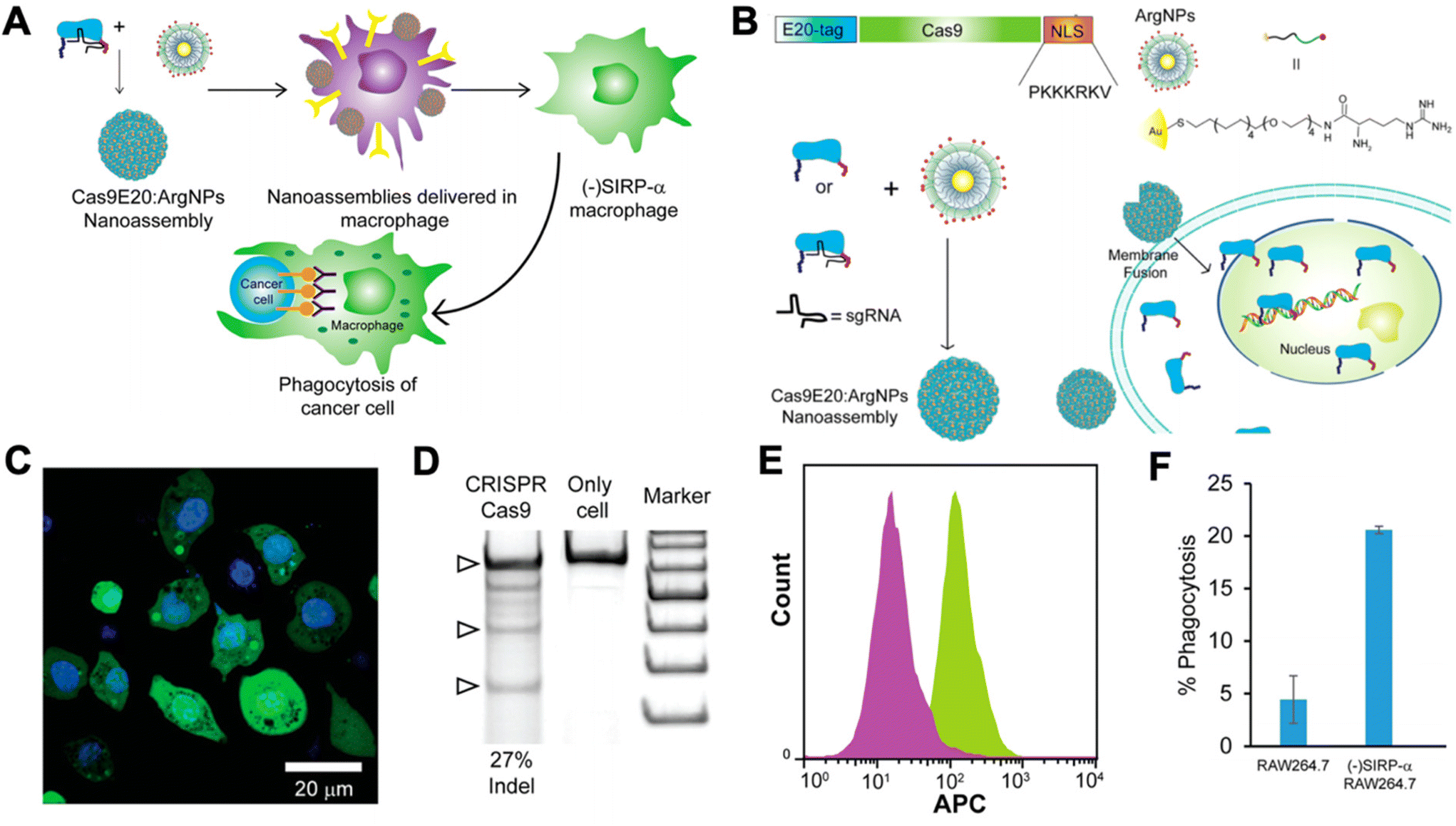 | ||
| Fig. 4 CRISPR/Cas9 nanoparticles for genetic engineering of macrophages for cancer immunotherapy. (A) Nanoparticle-mediated delivery of CRISPR/Cas9 machinery to macrophages to knock down SIRP-α, resulting in cancer cell phagocytosis. (B) Schematic of particle design. Cas9 was tagged with a nuclear localization signal (NLS) and an E20-tag. E20-tagged Cas9 and cationic arginine nanoparticles (ArgNPs) were mixed together and self-assembled into superstructures via carboxylate-guanidium binding. The particles are then delivered intracellularly via a membrane fusion mechanism that leads to direct payload release into the cytoplasm. (C) Delivery efficiency of Cas9 to the cytoplasm was assessed by delivering fluorescently labeled Cas9 to RAW264.7 macrophages in vitro (nuclei stained with Hoechst). (D) Delivery of Cas9 RNPs to target the SIRP-α gene in RAW264.7 cells resulted in efficient gene editing, as determined by an indel (insertion and deletion) assay. (E) Fluorescence histogram on Cas9-treated RAW264.7 cells (purple) and untreated cells (green) stained with APC anti-SIRP-α knockout indicates that the knockout of SIRP-α did occur. (F) Percentage of phagocytosis of U2OS cancer cells after co-culture with untreated RAW264.7 cells or SIRP-α knockout cells. Reproduced from ref. 94 with permission from American Chemical Society, copyright 2018.94 | ||
4. Conclusion
CRISPR/Cas9 has revolutionized the field of genetic engineering, enabling highly precise and efficient knock-out or knock-in of target genes. Similarly, an increased understanding of the importance of the immune system in cancer has ushered in a new era in cancer treatment with the successes of cancer immunotherapies. Researchers have begun to utilize CRISPR/Cas9 as a tool in engineering cancer immunotherapies, particularly with respect to CAR T cells. Simultaneously, great strides are being made in the development of non-viral biomaterials, in particular lipids and polymers, for the safe, efficient, and targeted delivery of nucleic acids and proteins to target immune cells, including T cells and macrophages. Although much of the early gene editing research has been performed thus far ex vivo utilizing viral vectors and/or electroporation, increasingly the advancements in the fields of gene editing and biomaterial-mediated delivery are being leveraged together. In ongoing research, the biomaterial approaches described here for gene and protein delivery to immune cells are being applied for delivery of CRISPR/Cas9. The development of biocompatible and biodegradable materials for efficient delivery of CRISPR/Cas9 to immune cells is advancing with a goal of enabling in situ genetic engineering of immune cells in a safe and highly controlled manner. This convergence of immunotherapy and gene therapy holds great promise not just for new paradigms of cancer treatment, but for treatment of infectious diseases, autoimmune diseases, and many other human diseases as well.Conflicts of interest
Patents related to technology discussed in the manuscript have been filed by Johns Hopkins University with co-inventors J. J. G. and E. B., J. J. G is a co-founder and CTO of Dome Therapeutics, co-founder of OncoSwitch Therapeutics, co-founder of WyveRNA Therapeutics, and was a scientific advisory board member of Tidal Therapeutics. Any potential conflicts of interest are managed by the Johns Hopkins University Committee on Outside Interests.Acknowledgements
SRS thanks the National Science Foundation (DGE-1746891) for fellowship support. EBA thanks the NIH NCI for fellowship support (F31CA250367). The authors thank the NIH for support of this work (R01EY031097, P41EB028239, and R01CA228133).References
- R. M. Blaese, K. W. Culver, A. D. Miller, C. S. Carter, T. Fleisher, M. Clerici, G. Shearer, L. Chang, Y. Chiang, P. Tolstoshev, J. J. Greenblatt, S. A. Rosenberg, H. Klein, M. Berger, C. A. Mullen, W. J. Ramsey, L. Muul, R. A. Morgan and W. F. Anderson, Science, 1995, 270, 475–480 CrossRef CAS PubMed.
- A. Mullard, Nat. Rev. Drug Discovery, 2017, 16, 669 Search PubMed.
- S. Peng, B. Bie, Y. Sun, M. Liu, H. Cong, W. Zhou, Y. Xia, H. Tang, H. Deng and X. Zhou, Nat. Commun., 2018, 9, 1293 CrossRef PubMed.
- D. Peer, Immunol. Rev., 2013, 253, 185–197 CrossRef PubMed.
- C. J. McKinlay, N. L. Benner, O. A. Haabeth, R. M. Waymouth and P. A. Wender, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5859–E5866 CrossRef PubMed.
- B. Ingelsson, D. Söderberg, T. Strid, A. Söderberg, A.-C. Bergh, V. Loitto, K. Lotfi, M. Segelmark, G. Spyrou and A. Rosén, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E478–E487 CrossRef CAS PubMed.
- M. S. Al-Dosari and X. Gao, AAPS J., 2009, 11, 671 CrossRef CAS PubMed.
- N. Montserrat, E. Garreta, F. Gonzalez, J. Gutierrez, C. Eguizabal, V. Ramos, S. Borros and J. C. Izpisua Belmonte, J. Biol. Chem., 2011, 286, 12417–12428 CrossRef CAS PubMed.
- J. C. Sunshine, D. Y. Peng and J. J. Green, Mol. Pharm., 2012, 9, 3375–3383 CrossRef CAS PubMed.
- C. Wang, Y. Zhang and Y. Dong, Acc. Chem. Res., 2021, 54, 4283–4293 CrossRef CAS PubMed.
- L. Yang, F. Ma, F. Liu, J. Chen, X. Zhao and Q. Xu, Mol. Ther.–Nucleic Acids, 2020, 19, 1357–1367 CrossRef CAS PubMed.
- J. Karlsson, S. Y. Tzeng, S. Hemmati, K. M. Luly, O. Choi, Y. Rui, D. R. Wilson, K. L. Kozielski, A. Quinones-Hinojosa and J. J. Green, Adv. Funct. Mater., 2021, 31(17), 2009768 CrossRef CAS PubMed.
- Q. Cheng, T. Wei, L. Farbiak, L. T. Johnson, S. A. Dilliard and D. J. Siegwart, Nat. Nanotechnol., 2020, 15, 313–320 CrossRef CAS PubMed.
- Y. Rui, D. R. Wilson, S. Y. Tzeng, H. M. Yamagata, D. Sudhakar, M. Conge, C. A. Berlinicke, D. J. Zack, A. Tuesca and J. J. Green, Sci. Adv., 2022, 8, eabk2855 CrossRef CAS PubMed.
- J. C. Kaczmarek, K. J. Kauffman, O. S. Fenton, K. Sadtler, A. K. Patel, M. W. Heartlein, F. DeRosa and D. G. Anderson, Nano Lett., 2018, 18, 6449–6454 CrossRef CAS PubMed.
- D. Zukancic, E. J. A. Suys, E. H. Pilkington, A. Algarni, H. Al-Wassiti and N. P. Truong, Pharmaceutics, 2020, 12, 1068 CrossRef CAS PubMed.
- S. Varma, S. Dey and D. Palanisamy, Curr. Pharm. Biotechnol., 2022, 23, 679–706 Search PubMed.
- K. Kettler, K. Veltman, D. van de Meent, A. van Wezel and A. J. Hendriks, Environ. Toxicol. Chem., 2014, 33, 481–492 CrossRef CAS PubMed.
- T. Van de Vyver, S. C. De Smedt and K. Raemdonck, Adv. Drug Delivery Rev., 2022, 181, 114041 CrossRef CAS PubMed.
- N. N. Parayath, S. B. Stephan, A. L. Koehne, P. S. Nelson and M. T. Stephan, Nat. Commun., 2020, 11, 6080–6080 CrossRef CAS PubMed.
- T. T. Smith, S. B. Stephan, H. F. Moffett, L. E. McKnight, W. Ji, D. Reiman, E. Bonagofski, M. E. Wohlfahrt, S. P. S. Pillai and M. T. Stephan, Nat. Nanotechnol., 2017, 12, 813–820 CrossRef CAS PubMed.
- F. Zhang, N. N. Parayath, C. I. Ene, S. B. Stephan, A. L. Koehne, M. E. Coon, E. C. Holland and M. T. Stephan, Nat. Commun., 2019, 10, 3974–3974 CrossRef CAS PubMed.
- S. Przybylski, M. Gasch, A. Marschner, M. Ebert, A. Ewe, G. Helmig, N. Hilger, S. Fricke, S. Rudzok, A. Aigner and J. Burkhardt, PLoS One, 2017, 12, e0176517 CrossRef PubMed.
- S. Kimura, I. A. Khalil, Y. H. A. Elewa and H. Harashima, J. Controlled Release, 2021, 330, 753–764 CrossRef CAS PubMed.
- Z. Gan, M. P. Lokugamage, M. Z. C. Hatit, D. Loughrey, K. Paunovska, M. Sato, A. Cristian and J. E. Dahlman, Bioeng. Transl. Med., 2020, 5, e10161 CAS.
- K. A. Hajj, J. R. Melamed, N. Chaudhary, N. G. Lamson, R. L. Ball, S. S. Yerneni and K. A. Whitehead, Nano Lett., 2020, 20, 5167–5175 CrossRef CAS PubMed.
- O. S. Fenton, K. J. Kauffman, J. C. Kaczmarek, R. L. McClellan, S. Jhunjhunwala, M. W. Tibbitt, M. D. Zeng, E. A. Appel, J. R. Dorkin, F. F. Mir, J. H. Yang, M. A. Oberli, M. W. Heartlein, F. DeRosa, R. Langer and D. G. Anderson, Adv. Mater., 2017, 29, 1606944 CrossRef PubMed.
- M. A. Oberli, A. M. Reichmuth, J. R. Dorkin, M. J. Mitchell, O. S. Fenton, A. Jaklenec, D. G. Anderson, R. Langer and D. Blankschtein, Nano Lett., 2017, 17, 1326–1335 CrossRef CAS PubMed.
- A. Aied, U. Greiser, A. Pandit and W. Wang, Drug Discovery Today, 2013, 18, 1090–1098 CrossRef CAS PubMed.
- M. Foldvari, D. W. Chen, N. Nafissi, D. Calderon, L. Narsineni and A. Rafiee, J. Controlled Release, 2016, 240, 165–190 CrossRef CAS PubMed.
- B. R. Olden, Y. Cheng, J. L. Yu and S. H. Pun, J. Controlled Release, 2018, 282, 140–147 CrossRef CAS PubMed.
- A. Hall, U. Lächelt, J. Bartek, E. Wagner and S. M. Moghimi, Mol. Ther., 2017, 25, 1476–1490 CrossRef CAS PubMed.
- G. Liu, Y. Li, L. Yang, Y. Wei, X. Wang, Z. Wang and L. Tao, RSC Adv., 2017, 7, 18252–18259 RSC.
- A. Chakraborty, J. J. Martín Lasola, N. Truong and R. M. Pearson, ACS Appl. Bio Mater., 2020, 3, 6263–6272 CrossRef CAS PubMed.
- B. Lou, S. De Koker, C. Y. J. Lau, W. E. Hennink and E. Mastrobattista, Bioconjugate Chem., 2019, 30, 461–475 CrossRef CAS PubMed.
- B. Lou, A. De Beuckelaer, G. R. Dakwar, K. Remaut, J. Grooten, K. Braeckmans, B. G. De Geest, E. Mastrobattista, S. De Koker and W. E. Hennink, J. Controlled Release, 2018, 284, 73–83 CrossRef CAS PubMed.
- H. Hatakeyama, E. Ito, H. Akita, M. Oishi, Y. Nagasaki, S. Futaki and H. Harashima, J. Controlled Release, 2009, 139, 127–132 CrossRef CAS PubMed.
- M. M. Billingsley, N. Singh, P. Ravikumar, R. Zhang, C. H. June and M. J. Mitchell, Nano Lett., 2020, 20, 1578–1589 CrossRef CAS PubMed.
- M. A. Dobrovolskaia and M. Bathe, WIREs Nanomed. Nanobiotechnol., 2021, 13, e1657 CAS.
- Y. Xiao, K. Shi, Y. Qu, B. Chu and Z. Qian, Mol. Ther.–Methods Clin. Dev., 2019, 12, 1–18 CrossRef CAS PubMed.
- S. Y. Tzeng, H. Guerrero-Cázares, E. E. Martinez, J. C. Sunshine, A. Quiñones-Hinojosa and J. J. Green, Biomaterials, 2011, 32, 5402–5410 CrossRef CAS PubMed.
- S. M. Hoy, Drugs, 2018, 78, 1625–1631 CrossRef CAS PubMed.
- Y. Zhao, H. Zheng, X. Wang, X. Zheng, Y. Zheng, Y. Chen, W. Fei, J. Zhu, W. Wang and C. Zheng, AAPS PharmSciTech, 2021, 22, 22 CrossRef CAS PubMed.
- C. Pennetta, N. Bono, F. Ponti, M. C. Bellucci, F. Viani, G. Candiani and A. Volonterio, Bioconjugate Chem., 2021, 32(4), 690–701 CrossRef CAS PubMed.
- F. Ponti, M. Campolungo, C. Melchiori, N. Bono and G. Candiani, Chem. Phys. Lipids, 2021, 235, 105032 CrossRef CAS PubMed.
- J. Buck, P. Grossen, P. R. Cullis, J. Huwyler and D. Witzigmann, ACS Nano, 2019, 13, 3754–3782 CrossRef CAS PubMed.
- N. Veiga, Y. Diesendruck and D. Peer, Adv. Drug Delivery Rev., 2020, 159, 364–376 CrossRef CAS PubMed.
- K. Paunovska, C. D. Sago, C. M. Monaco, W. H. Hudson, M. G. Castro, T. G. Rudoltz, S. Kalathoor, D. A. Vanover, P. J. Santangelo, R. Ahmed, A. V. Bryksin and J. E. Dahlman, Nano Lett., 2018, 18, 2148–2157 CrossRef CAS PubMed.
- E. Harris, D. Zimmerman, E. Warga, A. Bamezai and J. Elmer, Biotechnol. Bioeng., 2021, 118, 1674–1687 CrossRef PubMed.
- C. D. Sago, M. P. Lokugamage, K. Paunovska, D. A. Vanover, C. M. Monaco, N. N. Shah, M. Gamboa Castro, S. E. Anderson, T. G. Rudoltz, G. N. Lando, P. Munnilal Tiwari, J. L. Kirschman, N. Willett, Y. C. Jang, P. J. Santangelo, A. V. Bryksin and J. E. Dahlman, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E9944–E9952 CrossRef CAS PubMed.
- X. Zhao, J. Chen, M. Qiu, Y. Li, Z. Glass and Q. Xu, Angew. Chem., Int. Ed., 2020, 59, 20083–20089 CrossRef CAS PubMed.
- S. E. Oliver, J. W. Gargano, M. Marin, M. Wallace, K. G. Curran, M. Chamberland, N. McClung, D. Campos-Outcalt, R. L. Morgan, S. Mbaeyi, J. R. Romero, H. K. Talbot, G. M. Lee, B. P. Bell and K. Dooling, Morb. Mortal. Wkly. Rep., 2020, 69, 1922–1924 CrossRef CAS PubMed.
- Nat. Nanotechnol., 2020, 15, 963 Search PubMed.
- The COVID-19 candidate vaccine landscape and tracker, https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines, 2021.
- M. Adli, Nat. Commun., 2018, 9, 1911 CrossRef PubMed.
- A. Pickar-Oliver and C. A. Gersbach, Nat. Rev. Mol. Cell Biol., 2019, 20, 490–507 CrossRef CAS PubMed.
- G. I. Ellis, N. C. Sheppard and J. L. Riley, Nat. Rev. Genet., 2021, 22(7), 427–447 CrossRef CAS PubMed.
- S. R. Bailey and M. V. Maus, Nat. Biotechnol., 2019, 37, 1425–1434 CrossRef CAS PubMed.
- Y. Rui, D. R. Wilson and J. J. Green, Trends Biotechnol., 2019, 37, 281–293 CrossRef CAS PubMed.
- C. A. Lino, J. C. Harper, J. P. Carney and J. A. Timlin, Drug Delivery, 2018, 25, 1234–1257 CrossRef CAS PubMed.
- Y. A. Aksoy, B. Yang, W. Chen, T. Hung, R. P. Kuchel, N. W. Zammit, S. T. Grey, E. M. Goldys and W. Deng, ACS Appl. Mater. Interfaces, 2020, 12, 52433–52444 CrossRef CAS PubMed.
- J. A. Zuris, D. B. Thompson, Y. Shu, J. P. Guilinger, J. L. Bessen, J. H. Hu, M. L. Maeder, J. K. Joung, Z.-Y. Chen and D. R. Liu, Nat. Biotechnol., 2015, 33, 73–80 CrossRef CAS PubMed.
- C. Liang, F. Li, L. Wang, Z.-K. Zhang, C. Wang, B. He, J. Li, Z. Chen, A. B. Shaikh, J. Liu, X. Wu, S. Peng, L. Dang, B. Guo, X. He, D. W. T. Au, C. Lu, H. Zhu, B.-T. Zhang, A. Lu and G. Zhang, Biomaterials, 2017, 147, 68–85 CrossRef CAS PubMed.
- J. B. Miller, S. Zhang, P. Kos, H. Xiong, K. Zhou, S. S. Perelman, H. Zhu and D. J. Siegwart, Angew. Chem., Int. Ed., 2017, 56, 1059–1063 CrossRef CAS PubMed.
- J. D. Finn, A. R. Smith, M. C. Patel, L. Shaw, M. R. Youniss, J. van Heteren, T. Dirstine, C. Ciullo, R. Lescarbeau, J. Seitzer, R. R. Shah, A. Shah, D. Ling, J. Growe, M. Pink, E. Rohde, K. M. Wood, W. E. Salomon, W. F. Harrington, C. Dombrowski, W. R. Strapps, Y. Chang and D. V. Morrissey, Cell Rep., 2018, 22, 2227–2235 CrossRef CAS PubMed.
- H. Deng, S. Tan, X. Gao, C. Zou, C. Xu, K. Tu, Q. Song, F. Fan, W. Huang and Z. Zhang, Acta Pharm. Sin. B, 2020, 10, 358–373 CrossRef CAS PubMed.
- K. Tu, H. Deng, L. Kong, Y. Wang, T. Yang, Q. Hu, M. Hu, C. Yang and Z. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 16018–16030 CrossRef CAS PubMed.
- Y. Rui, M. Varanasi, S. Mendes, H. M. Yamagata, D. R. Wilson and J. J. Green, Mol. Ther.–Nucleic Acids, 2020, 20, 661–672 CrossRef CAS PubMed.
- Y. Rui, D. R. Wilson, K. Sanders and J. J. Green, ACS Appl. Mater. Interfaces, 2019, 11, 10472–10480 CrossRef CAS PubMed.
- Y. Rui, D. R. Wilson, J. Choi, M. Varanasi, K. Sanders, J. Karlsson, M. Lim and J. J. Green, Sci. Adv., 2019, 5, eaay3255 CrossRef CAS PubMed.
- Y. Hu, Y. Zhu, N. D. Sutherland, D. R. Wilson, M. Pang, E. Liu, J. R. Staub, C. A. Berlinicke, D. J. Zack, J. J. Green, S. K. Reddy and H. Q. Mao, Nano Lett., 2021, 21, 5697–5705 CrossRef CAS PubMed.
- L. Chechik, O. Martin and E. Soutoglou, Front. Cell Dev. Biol., 2020, 8, 319 CrossRef PubMed.
- H. Mollanoori, H. Shahraki, Y. Rahmati and S. Teimourian, Hum. Immunol., 2018, 79, 876–882 CrossRef CAS PubMed.
- R. G. Majzner and C. L. Mackall, Nat. Med., 2019, 25, 1341–1355 CrossRef CAS PubMed.
- N. N. Shah and T. J. Fry, Nat. Rev. Clin. Oncol., 2019, 16, 372–385 CAS.
- J. Eyquem, J. Mansilla-Soto, T. Giavridis, S. J. C. van der Stegen, M. Hamieh, K. M. Cunanan, A. Odak, M. Gönen and M. Sadelain, Nature, 2017, 543, 113–117 CrossRef CAS PubMed.
- T. L. Roth, C. Puig-Saus, R. Yu, E. Shifrut, J. Carnevale, P. J. Li, J. Hiatt, J. Saco, P. Krystofinski, H. Li, V. Tobin, D. N. Nguyen, M. R. Lee, A. L. Putnam, A. L. Ferris, J. W. Chen, J.-N. Schickel, L. Pellerin, D. Carmody, G. Alkorta-Aranburu, D. del Gaudio, H. Matsumoto, M. Morell, Y. Mao, M. Cho, R. M. Quadros, C. B. Gurumurthy, B. Smith, M. Haugwitz, S. H. Hughes, J. S. Weissman, K. Schumann, J. H. Esensten, A. P. May, A. Ashworth, G. M. Kupfer, S. A. W. Greeley, R. Bacchetta, E. Meffre, M. G. Roncarolo, N. Romberg, K. C. Herold, A. Ribas, M. D. Leonetti and A. Marson, Nature, 2018, 559, 405–409 CrossRef CAS PubMed.
- F. Mo, S. Duan, X. Jiang, X. Yang, X. Hou, W. Shi, C. J. J. Carlos, A. Liu, S. Yin, W. Wang, H. Yao, Z. Yu, Z. Tang, S. Xie, Z. Ding, X. Zhao, B. D. Hammock and X. Lu, Signal Transduction Targeted Ther., 2021, 6, 80 CrossRef CAS PubMed.
- L. J. Rupp, K. Schumann, K. T. Roybal, R. E. Gate, C. J. Ye, W. A. Lim and A. Marson, Sci. Rep., 2017, 7, 737 CrossRef PubMed.
- B. D. Choi, X. Yu, A. P. Castano, H. Darr, D. B. Henderson, A. A. Bouffard, R. C. Larson, I. Scarfò, S. R. Bailey, G. M. Gerhard, M. J. Frigault, M. B. Leick, A. Schmidts, J. G. Sagert, W. T. Curry, B. S. Carter and M. V. Maus, J. Immunother. Cancer, 2019, 7, 304 CrossRef PubMed.
- W. Hu, Z. Zi, Y. Jin, G. Li, K. Shao, Q. Cai, X. Ma and F. Wei, Cancer Immunol. Immunother., 2019, 68, 365–377 CrossRef CAS PubMed.
- S. Su, B. Hu, J. Shao, B. Shen, J. Du, Y. Du, J. Zhou, L. Yu, L. Zhang, F. Chen, H. Sha, L. Cheng, F. Meng, Z. Zou, X. Huang and B. Liu, Sci. Rep., 2016, 6, 20070 CrossRef CAS PubMed.
- Y. Lu, J. Xue, T. Deng, X. Zhou, K. Yu, L. Deng, M. Huang, X. Yi, M. Liang, Y. Wang, H. Shen, R. Tong, W. Wang, L. Li, J. Song, J. Li, X. Su, Z. Ding, Y. Gong, J. Zhu, Y. Wang, B. Zou, Y. Zhang, Y. Li, L. Zhou, Y. Liu, M. Yu, Y. Wang, X. Zhang, L. Yin, X. Xia, Y. Zeng, Q. Zhou, B. Ying, C. Chen, Y. Wei, W. Li and T. Mok, Nat. Med., 2020, 26, 732–740 CrossRef CAS PubMed.
- E. A. Stadtmauer, J. A. Fraietta, M. M. Davis, A. D. Cohen, K. L. Weber, E. Lancaster, P. A. Mangan, I. Kulikovskaya, M. Gupta, F. Chen, L. Tian, V. E. Gonzalez, J. Xu, I.-Y. Jung, J. J. Melenhorst, G. Plesa, J. Shea, T. Matlawski, A. Cervini, A. L. Gaymon, S. Desjardins, A. Lamontagne, J. Salas-Mckee, A. Fesnak, D. L. Siegel, B. L. Levine, J. K. Jadlowsky, R. M. Young, A. Chew, W.-T. Hwang, E. O. Hexner, B. M. Carreno, C. L. Nobles, F. D. Bushman, K. R. Parker, Y. Qi, A. T. Satpathy, H. Y. Chang, Y. Zhao, S. F. Lacey and C. H. June, Science, 2020, 367, eaba7365 CrossRef CAS PubMed.
- F. Wiede, K.-H. Lu, X. Du, S. Liang, K. Hochheiser, G. T. Dodd, P. K. Goh, C. Kearney, D. Meyran, P. A. Beavis, M. A. Henderson, S. L. Park, J. Waithman, S. Zhang, Z.-Y. Zhang, J. Oliaro, T. Gebhardt, P. K. Darcy and T. Tiganis, EMBO J., 2020, 39, e103637 CrossRef CAS PubMed.
- N. Tang, C. Cheng, X. Zhang, M. Qiao, N. Li, W. Mu, X.-F. Wei, W. Han and H. Wang, JCI Insight, 2020, 5(4), e133977 CrossRef PubMed.
- C. Georgiadis, R. Preece, L. Nickolay, A. Etuk, A. Petrova, D. Ladon, A. Danyi, N. Humphryes-Kirilov, A. Ajetunmobi, D. Kim, J.-S. Kim and W. Qasim, Mol. Ther., 2018, 26, 1215–1227 CrossRef CAS PubMed.
- J. Ren, X. Liu, C. Fang, S. Jiang, C. H. June and Y. Zhao, Clin. Cancer Res., 2017, 23, 2255–2266 CrossRef CAS PubMed.
- N. Shimasaki, A. Jain and D. Campana, Nat. Rev. Drug Discovery, 2020, 19, 200–218 CrossRef CAS PubMed.
- L. O. Afolabi, A. O. Adeshakin, M. M. Sani, J. Bi and X. Wan, Immunology, 2019, 158, 63–69 CrossRef CAS PubMed.
- J. Rautela, E. Surgenor and N. D. Huntington, J. Leukocyte Biol., 2020, 108, 1397–1408 CrossRef CAS PubMed.
- E. J. Pomeroy, J. T. Hunzeker, M. G. Kluesner, W. S. Lahr, B. A. Smeester, M. R. Crosby, C.-L. Lonetree, K. Yamamoto, L. Bendzick, J. S. Miller, M. A. Geller, B. Walcheck, M. Felices, B. R. Webber, T. K. Starr and B. S. Moriarity, Mol. Ther., 2020, 28, 52–63 CrossRef CAS PubMed.
- R.-S. Huang, H.-A. Shih, M.-C. Lai, Y.-J. Chang and S. Lin, Front. Immunol., 2020, 11, 1008 CrossRef CAS PubMed.
- M. Ray, Y.-W. Lee, J. Hardie, R. Mout, G. Y. Tonga, M. E. Farkas and V. M. Rotello, Bioconjugate Chem., 2018, 29, 445–450 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |