
Conformationally rigid molecular and polymeric naphthalene-diimides containing C6H6N2 constitutional isomers†
Vincenzo Mirco
Abbinante
 a,
Gonzalo
García-Espejo
a,
Gabriele
Calabrese
b,
Silvia
Milita
*b,
Luisa
Barba
c,
Diego
Marini
b,
Candida
Pipitone
d,
Francesco
Giannici
d,
Antonietta
Guagliardi
*e and
Norberto
Masciocchi
*a
a,
Gonzalo
García-Espejo
a,
Gabriele
Calabrese
b,
Silvia
Milita
*b,
Luisa
Barba
c,
Diego
Marini
b,
Candida
Pipitone
d,
Francesco
Giannici
d,
Antonietta
Guagliardi
*e and
Norberto
Masciocchi
*a
aDipartimento di Scienza e Alta Tecnologia & INSTM, Università dell’Insubria, via Valleggio 11, 22100 Como, Italy. E-mail: norberto.masciocchi@uninsubria.it
bIstituto per la Microelettronica e Microsistemi, Consiglio Nazionale delle Ricerche, via Gobetti 101, 40129 Bologna, Italy. E-mail: milita@bo.imm.cnr.it
cIstituto di Cristallografia @ Elettra Synchrotron, Consiglio Nazionale delle Ricerche, c/o Area Science Park, 34139 Basovizza, Trieste, Italy
dDipartimento di Fisica e Chimica, Università di Palermo, viale delle Scienze, Ed. 17, 90128 Palermo, Italy
eIstituto di Cristallografia & To.Sca.Lab., Consiglio Nazionale delle Ricerche, via Valleggio 11, 22100 Como, Italy
First published on 16th June 2021
Abstract
Organic thin films based on naphthalenediimides (NDIs) bearing alkyl substituents have shown interesting properties for application in OLEDs, thermoelectrics, solar cells, sensors and organic electronics. However, the polymorphic versatility attributed to the flexibility of alkyl chains remains a challenging issue, with detrimental implications on the performances. Aryl analogues containing C6H6N2 constitutional isomers are herein investigated as one of the possible way-out strategies. The synthesis of molecular and polymeric species is described, starting from naphthaleneteracarboxyldianhydride with isomeric aromatic amines and hydrazine. The materials are fully characterized by spectroscopy, thermal and structural X-ray diffraction methods, both as bulk powders and thin films, revealing a rich structural landscape. Depending on the stereochemistry of the branching aryls, the compounds show a variety of parallel stacking of the NDI cores, and high structural stability upon heating, up to 560 °C in the polymeric form. Thin films prepared by spin coating from organic solvent solutions and studied by grazing-incidence X-ray diffraction exhibit a high degree of crystallinity indicating the intrinsic tendency of these molecules to self-assemble in an ordered fashion without the need for any post-processing technique. In line with other NDI-based diimides, UV-vis spectroscopy indicates the optical band gaps falling in the visible region (2.87–3.02 eV). DFT calculations reveal a significant lowering of the frontier orbital energies of the hydrazido derivative. Beyond solution processing, the high thermal stability and the absence of polymorphic forms of these materials suggest that sublimation-based routes for films and device preparation can also be followed.
1. Introduction
Organic molecular semiconductors are a class of compounds that have attracted a great deal of attention, in the form of thin films, for appealing applications in photovoltaics, optoelectronics, computing and sensing1 and, more recently, low-power thermoelectric generators.2 These compounds greatly benefit from easy chemical functionalization, rapid solution processability of pristine molecular entities, simple deposition of thin films and superb flexibility.3 After several years of fundamental studies, they have recently been incorporated into large commodities,4 and rapidly filled the market as flexible light panels, rollable solar cells, cell phone displays and battery-free artificial retina, to mention a few.5Among the most investigated species, a number of studies focused on symmetrically disubstituted naphthalene diimide (NDI)6 and perylene diimide (PDI)7 molecules, where the presence of π–π stacked aromatic cores, in optimized geometrical arrangements, enhances important functional properties, such as electron conductivity above all.8 In particular, NDI compounds (having a smaller aromatic core than the PDI congeners) possess two strong electron-withdrawing imide groups enabling several NDI-based systems to achieve relatively low LUMO energies, excellent ambient stability, and promising charge-transport properties. Nonetheless, very slight changes in the nature or position of the side groups may result in a big difference in molecular packing and in material properties, as observed, for example, in many of the NDI-based species discussed in a previous paper.9 Indeed, once isolated as powders and films, alkyl chain derivatives show a remarkable structural flexibility, witnessed by a rich polymorphic behavior and reversible crystal-to-crystal and crystal-to-mesophase transformations, with detrimental implications for the applications/devices.10 On the other hand, while the performances of these materials are heavily dependent on the supramolecular assembly of the constituent molecules in the solid state (mostly in thin films),11 predicting/devising the best performing geometry presently remains a challenging task. Stimulated by these considerations, we extended our interest to less flexible residues. Inspired by the earlier reports on amino-bearing residues in fullerenes, polyfluorenes and PDI derivatives,12 where the presence of the amino group was not found prejudicial to materials properties, here we focus our attention on NDI-based solids bearing amino- (or hydrazido)-substituted benzenes, where the presence of aromatic rings is expected to block, to some extent, the mobility of the N-linked moieties, preventing “easy” polymorphic transformations under working conditions.
The choice of aromatic residues (here, in the form of amino-substituted phenyls) is also encouraged by the increased conjugation which can extend off the NDI core, enhancing the electron mobility. To the best of our knowledge, studies on pendant amino groups for NDI are scarce and mostly limited to their use as fluorescent probes13 and in cancer therapy.14 Although, admittedly, NH2 groups are prone to oxidation and molecular degradation (more so in solution than in the solid phase), our studies show that the species presented here are stable in air up to (at least) 280 °C. Thus, amino-group reactivity seems not to be a worrisome issue.
One appealing, and so far underexplored, complementary aspect is the impact of the constitutional isomers considered in the present work on the NDI structural landscape, i.e. that of molecular entities possessing the same chemical formula but with a slightly different atom connectivity in their periphery. It is noteworthy that these NDI-materials sharing constitutional isomers are not the result of non-stereo-/regio-selective syntheses in which it is difficult to separate mixtures (as in the commercially available PDI-Br215 and N140016 dyes); instead, by using predefined aminoaromatic residues, they are prepared as pure chemical entities with unique crystallographic phases.
While having the same chemical composition (as polymorphs do), crystal forms determined by the presence of constitutional isomers possess distinct physical and chemical properties from those one would expect in (conformationally different) polymorphic forms, with additional upstream control and greater stability. Similar considerations would apply as well to the case of configurational stereoisomers, pointing to the importance of understanding and addressing the selective formation of solid organic materials of isomeric species in organic electronics,17 as much as it is considered in pharmaceutical and pigment industries.18 Notable examples in these industrially relevant fields are the polymorphism of rubrene for efficient OLEDs,19 stereoisomerism of amphetamine20 and, for what matters here, the constitutionally different Pigments Red 194 and Orange 43 molecules.21 It is noteworthy that an additional degree of freedom, that is the occurrence of structurally induced mesomeric forms in organic semiconductors, has also been recently unveiled.22
In the present work, the synthesis of a number of NDIs is presented, based on C6H6N2 isomers (see Chart 1). Their rich structural landscape is unveiled and deeply discussed based on the combination of multiple structural characterization methods including X-ray powder diffraction, spectroscopic and thermal analyses, DFT calculations, and variable temperature diffraction methods. UV-Vis reflectance spectra on powders and grazing-incidence X-ray diffraction analysis of spin-coated films are presented with a general discussion of the properties and the possible processability routes of these materials.
 | ||
| Chart 1 Molecular sketches and labeling of the species discussed in this work. | ||
2. Experimental section
(a) Materials
Reagents and solvents were purchased from Merck (p-phenylenediamine, >98%; m-phenylenediamine, 99%; o-phenylenediamine, >98%; phenylhydrazine, >97%; benzoic acid, >99%; methanol, 99.8%; dimethylformamide, 99.8%; toluene, 99,8%; dimethylacetamide, >99%; m-cresol, 99%), BLD (1,4,5,8-naphthalentetracarboxylic acid monoanhydride, 98%) and Riedel-de-Haën (triethylamine, 99%) and used without further purification. All reactions were performed under a N2 atmosphere.(b) Syntheses of the materials
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :299.2.
:447.4.
:447.4.
:447.4.
:299.2.
:447.4.
:447.4.
:447.4.
(c) Physico-chemical characterization
:θ geometry, equipped with a Lynxeye position sensitive detector. DS: 0.5°; generator setting: 40 kV, 40 mA; Ni-filtered Cu-Kα radiation, λ = 1.5418 Å. PXRD data for structure solution were collected in the 3–105° 2θ range for all samples (3–75° for poly-NppDI), sampling at 0.02°, with scan time lasting approximately 16 h.
(d) Preparation of thin films
The NDI derivatives were used as obtained from the washing and purification processes after their respective synthesis. For each compound, the solvent and the concentration of the solution were chosen close to the saturation limit. The least soluble NppDI derivative was dissolved in N-methyl-2-pyrrolidinone (NMP) and acetone (volume ratio 3:7) at a concentration of 1 mg mL−1, while the NmpDI and NphDI solids were dissolved in N,N-dimethylformamide (DMF), with concentrations of 17 and 30 mg mL−1, respectively. Thin films were prepared by spin-coating 20 μL of solution onto 1 × 1 cm2 silicon substrates covered with a 300 nm-thick thermal silicon oxide layer. Before spin coating, the SiO2/Si substrates were cleaned sequentially for 5 minutes in acetone, ethanol, isopropyl alcohol and deionized water, and blow-dried with nitrogen gas. Finally, the substrates were etched by Ar/O2 plasma for 15 minutes (flux ratio of 3:1), aiming at improving their surface wettability.
Though NMP and DMF are solvents not commonly used for the deposition of thin organic films for device fabrication (chloroform and chlorobenzene being much more widely employed), reports on thin film processing with less volatile solvents (surprisingly including also methanesulfonic acid) have appeared.27 Additionally, the high thermal stability and the absence of polymorphic phases of these molecular solids, later discussed, suggest that sublimation processing of thin films and devices is a further viable option.
For each film, the spin coating conditions were tuned to optimize the film homogeneity and reproducibility. The NppDI solution was stirred and heated up to 170 °C for 48 h before being centrifuged at 104 rpm for 5 minutes to separate the suspended powders from the solution. The NppDI films were spin-coated at 1000 rpm for 30 s followed by a treatment at 5500 rpm for 30 s. For variance, the NmpDI and NphDI solutions were heated up to ca. 130 °C for 10 minutes until a complete dissolution of the powders was achieved. The NmpDI films were spin-coated at 1500 rpm for 30 s, while the NphDI films were spin-coated at 1000 rpm for 30 s, followed by 2500 rpm for 30s. All films were left to dry at room temperature. Each sample has been investigated both as fabricated and after thermal annealing in air at 180 °C for 10 minutes.
(e) X-ray structural studies
The paucity of the diffraction peaks in the poly-NppDI PXRD trace, their breadth and severe overlap, did not enable (semi)automatic structure solution. A few competitive structural models were built through sensible stereochemical considerations and tested against the experimental pattern, eventually validating a set of collinear alternate copolymers of NDI/C6H4 fragments as the best model. The accuracy of the refined metrical and structural descriptors is less than ideal. Nevertheless, the material periodicity and its main stereochemical features are (reasonably) well addressed by the present model.
The final refinements were eventually carried out by the Rietveld method, maintaining the rigid bodies introduced at the structure solution stage (see Fig. 1 and Fig. S11, ESI†) The background was modelled by a polynomial function of the Chebyshev type and the peak profiles were described by the Fundamental Parameters Approach30 and a common (refinable) isotropic thermal factor was attributed to all atoms. March–Dollase correction for preferred orientation31 was applied in the form of g(hkl) and quoted below. Fractional atomic coordinates and crystal structure details were deposited with the CCDC (CSD Codes 2059769–2059772).† Specific sample-dependent issues are discussed in the Results section.
 | ||
| Fig. 1 Final Rietveld refinement plots, with difference plot and peak markers at the bottom. The observed and calculated curves (blue and red traces, respectively) have been vertically shifted for clarity. | ||
(f) Grazing incidence X-ray diffraction (GIXD) studies
GIXD measurements were performed at the XRD1 beamline of the ELETTRA synchrotron radiation facility in Trieste (Italy). λ = 1.00Å; beam size: 200 × 200 μm2. 2D-GIXD patterns were collected by using a 2 M Pilatus silicon pixel X-ray detector (DECTRIS Ltd) positioned perpendicular to the incident beam, 350mm away from the sample. Incidence angles of the X-ray beam with the film surface of αi = 0.05° and 0.1° were chosen to probe the uppermost film layers (the X-ray penetration depths are ca. 4 and 8 nm, for αi = 0.05° and 0.1° respectively), as well as αi = 0.2° to probe the full film thickness (penetration depth: ca. 30 μm). During the measurements, the samples were slowly rotated around the substrate normal (180° in 60 s), to average signals originating from laterally inhomogeneous film regions. Both as-prepared and annealed films were investigated to study the effect of thermal annealing on film crystallinity and mosaicity.
The visualization, analysis of the 2D-GIXD patterns and intensity profile extraction were performed using GIDVis software.32
(g) Quantum-chemical calculations
Ground state geometry optimization and HOMO–LUMO energy calculations for NphDI, NmpDI and NppDI were performed with the Gaussian16 package,33 using the nonlocal hybrid Becke three-parameter Lee–Yang–Parr (B3LYP) functional34,35 and the 6-31G(p,d) basis set. The experimentally determined crystal structure of each compound was used as a starting point for geometry optimization. The method and the chosen basis set were validated against the already published LUMO value of NphDI and HOMO/LUMO values of fluorinated analogues,36 with absolute differences of less than 0.1 eV.3. Results and discussion
(a) Synthesis
The syntheses of the NppDI24,25 and NphDI23 molecules were already reported, though their characterization was limited to standard spectroscopic and (for NphDI) electrochemical methods. In the present work, we have adopted the synthetic approach of ref. 24 for what concerns NppDI, whereas some modifications of the procedure reported in ref. 26 were applied to NphDI, regarding changes in the work-up procedure (see details in the Experimental section). In particular, instead of dissolving the extracted crude material in THF and subsequently adding water to crystallize the product at 4 °C overnight, the pure product was obtained upon washing the extracted reaction crude material with water, acetone and then CH2Cl2, thus improving the product yield from 43%26 to 65%.The syntheses of the NDI cores branched with m- and o-substituted diaminobenzenes (NmpDI and NopDI), not present in the scientific literature, were performed in strict similarity with the protocol used for the p-regioisomer. NmpDI was easily recovered as a pure solid in high yields (>80%). The synthesis of the NopDI molecule, attempted in several ways, mostly modifying temperature solvents and reaction times, proved to be very difficult. Indeed, two ring-closure adducts, known as Pigment Orange 4337 and Pigment Red 194,38 were systematically detected in the reaction mixture (see Chart S1, ESI†). Not being possible to isolate a single (pure) crystal phase of NopDI, its full structural and analytical characterization was abandoned. It is worth noting that such o-aminophenylene condensation has been used in the synthesis of BBL, or poly(benzimidazobenzophenanthroline), a soluble polymer somewhat reminiscent of both NopDI and poly-NppDI), which has recently attracted the attention of many for its excellent performances in n-channel OFETs.27c
Finally, aiming at enhancing the thermal stability and the conductive properties of this class of materials, the polymerization capability of the NppDI molecule was explored through the synthesis of a polymeric species, poly-NppDI. A one-pot reaction using a 1:0.8 NDA:p-diamino-benzene molar ratio proved to be unsuccessful (a complex mixture of products was obtained). Co-polymerization with NDA was also tried by reacting NDA and NppDI monomers overnight in 1:0.8 ratio in refluxing toluene, but this attempt also failed.
However, taking inspiration from polymerization reactions of NDA with a series of aromatic diamines,39 suitable conditions for the obtention of the sought copolymer were unearthed. Employing m-cresol as the solvent (a liquid with a high boiling point and solvent power) and with the crucial usage of two additives (excess benzoic acid – 4 eq. – and TEA – 1.5 eq.), poly-NppDI was synthesized with a 93% yield. The role of these additives can be explained by benzoic acid coordination to the NDA oxygen atoms, making carbonyl groups more susceptible to the nucleophilic attack, and by TEA-induced avoidance of proton transfer from the benzoic acid to p-diaminobenzene. Quantitative yields were obtained when employing 1 eq. of p-diaminobenzene as the unique nucleophile and heating at 175 °C for 15 h and then at 195 °C for an additional 3 hours. poly-NppDI appeared to be a nanosized material by X-ray diffraction analysis. Its structural analysis (later discussed) confirmed the formation of the sought material, though the average chain length is limited to a few (alternating) NDA/p-diaminobenzene monomers.
(b) Crystal structures
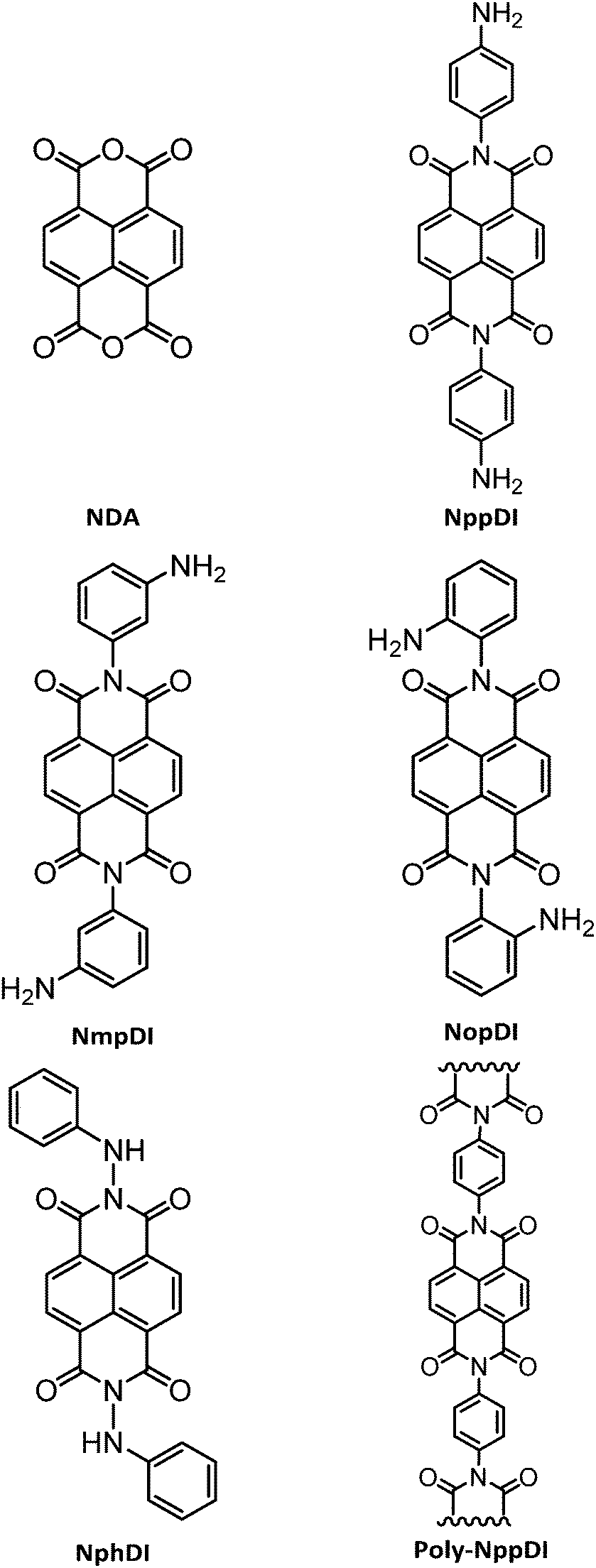
The crystal structures of the NppDI, NmpDI and NphDI molecular phases contain symmetrically disubstituted NDI cores, lying about inversion centers in monoclinic or orthorhombic space groups, with Z′ = 0.5. Their molecular structures and crystal packings are shown in Fig. 2, and the relevant stereochemical features are shown in Table 1, where the structural analogues 1,4,5,8-naphthalenetetracarboxyl-p-CH3-benzyldimide (NhbDI) and -p-CF3-benzyldimide (NfbDI) are also included.40 As detailed in the Experimental Section, the structural models presented here make use of rigid body descriptions of the stiff portions of the molecules. Accordingly, reliable structural information is mostly confined in the conformational freedom (the orientation of the branching residuals), in the intermolecular effects (the stacking periodicity) and in the degree of overlap of parallel, adjacent, NDI cores. With reference to the stacking periodicity, while for aliphatic substituents the π–π stacking distances were typically around 3.4 Å or lower9 (very similar to the interlayer interaction in graphite, 3.35 Å),41 in the present case this value increases to 3.5 Å or above for NppDI monomeric and polymeric derivatives, whereas parallel stacking does not occur in the monoclinic crystal structure of NmpDI. In the latter case, the aromatic cores, stretching out in the approximate [102] direction, stack in herringbone geometry, tilted away one from the other by ca. 48° (see Fig. S12, ESI†). Further comparative geometrical descriptors of NppDI vs. NmpDI are the tilt angles of the aromatic cores vs. the lamellar stacking (100) [63.2° vs. 29.7°] and the thickness of the molecular slabs [<11.5 vs. 16.0 Å], witnessing the more sprawled disposition of the NDI cores in NppDI. Only the phenylhydrazine derivative (NphDI) shows a short(er) NDI–core interaction. | ||
| Fig. 2 Schematic drawings of the molecular structure and crystal packings of (a and b) NppDI, (c and d) NmpDI and (e and f) NphDI, together with a note of their orientations. In (g), the polymeric poly-NppDI structure is shown. Color codes: C in grey, N in blue, O in red and H in white. Graphics realized by Schakal42 (a, c and e) and Mercury43 (b, d, f and g). | ||
| Species | τ, ° | d, Å | χ, ° | ψ, ° | H-Bonds, Å | DoO (eqn (1)) | E gap, eV |
|---|---|---|---|---|---|---|---|
| NppDI | 82.6 | 3.63 | 46.7 | 55.9 | 3.11 (N⋯N) 3.22 (N⋯O) | 0.46 | 2.87 |
| NmpDI | 78.4 | no π–π | — | — | None | — | 3.01 |
| NphDI | 84.7 | 3.38 | 49.2 | 64.9 | 3.06 (N⋯N) | 0.63 | 2.94 |
| NhbDI40 | 80.0 | 3.54 | 67.1 | 46.2 | None | 0.40 | n.a. |
| NfbDI40 | 78.6 | 3.40 | 73.7 | 39.8 | None | 0.58 | n.a. |
| Poly-NppDI | 63.0 | 3.50 | 90.0 | 44.0 | None | 0.69 | 2.90 |
| Aliphatic NDI | — | 3.27–3.42 | 68.4–80.5 | 42.2–53.4 | — | 0.62–0.72 |
As per the relative slip of the aromatic core, for all crystal phases studied here, the stereochemical descriptors (which in aliphatic systems and in NhbDI and NfbDI as well, have found to cluster into two specific portions of the χ, ψ plot)9 are more widely spread here (see Fig. 3). These findings indicate that the most relevant interactions driving the formation of the stacks are not anymore assignable to the NDI cores; instead, they must be attributed to the interlocking of the rigid (aromatic) branches. The geometrical degree of overlap (not the orbital one) defined by:
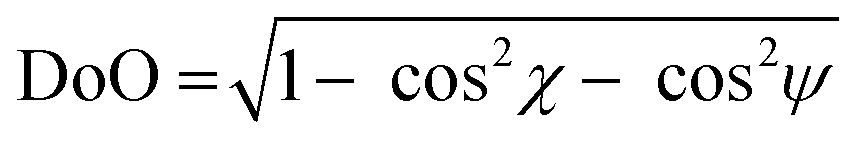 | (1) |
 | ||
| Fig. 3 Stereochemical descriptors χ,ψ for the π–π stacking of the crystal phases of this work (red dots, values from Table 2) and the aliphatic analogues (black dots) reported in ref. 9. | ||
Two of these materials, NmpDI and poly-NppDI, require a special set of comments. NmpDI crystallizes forming extended slabs running in the bc plane (thus normal to a), weakly bound by hydrogen bonds of the N–H⋯N type. Given that the m-positioned −NH2 residue was found to be crystallographically disordered in two positions roughly with a 1:3 ratio, this brings about a lack of ideal periodicity and the occurrence of a (possibly conditional) stereochemical local preference, with localized defects of the 2D type. This hypothesis is also partially confirmed by the less-than-ideal Rietveld fit shown in Fig. 1, possibly manifesting the slight misorientation of each molecule in the crystal from the average one, i.e. smoothing out the structural misfits.
Finally, poly-NppDI is a rare example of a crystalline polymer of NDI-based materials. Its extreme insolubility probably limits the growth of microstructured materials, and very broad peaks are observed in the PXRD trace. Thanks to the knowledge of the stereochemical feature of the NppDI molecular counterpart, it was possible to define a sensible structural model using symmetry restraints and geometrical considerations, later refined against the PXRD data using the standard Rietveld technique.
Our result revealed the occurrence of ca. 10 nm chains of elliptical section (suggesting a sequence of 8 NDI/1,4-phenylenes along the c axis and an average molecular weight of 2700 Dalton), packed in the crystal in a herringbone motif and a pseudo-hexagonal lattice (Fig. 2d), reminiscent of smectic E liquid crystalline phases. The fact that these chains are not extremely long is also witnessed by the less-than-ideal elemental analysis values reported in the Experimental section, where the C/N molar ratio is slightly above 11 (and should be 10 in an infinite chain), as if the chains were terminated by NDA, and not by p-phenylenediamine residues.
(c) Thermal stability
The thermal stabilities of the di- and poly-imide species were studied using the TG plots (Fig. 4) and the structural variations occurring upon heating were followed by variable-temperature X-ray experiments (Fig. 5). For NppDI, NmpDI, NphDI and poly-NppDI bulk samples, isolated in powdered form, the PXRD patterns were acquired from room temperature up to the decomposition temperature, measured by TG to occur near 300 °C for NphDI and at much higher temperatures for the other materials. Comparing TG and PXRD data, no polymorphic changes upon heating were observed. This is in striking difference with many aliphatic congeners, where the occurrence of several crystalline and liquid crystalline phases was the rule more than the exception.9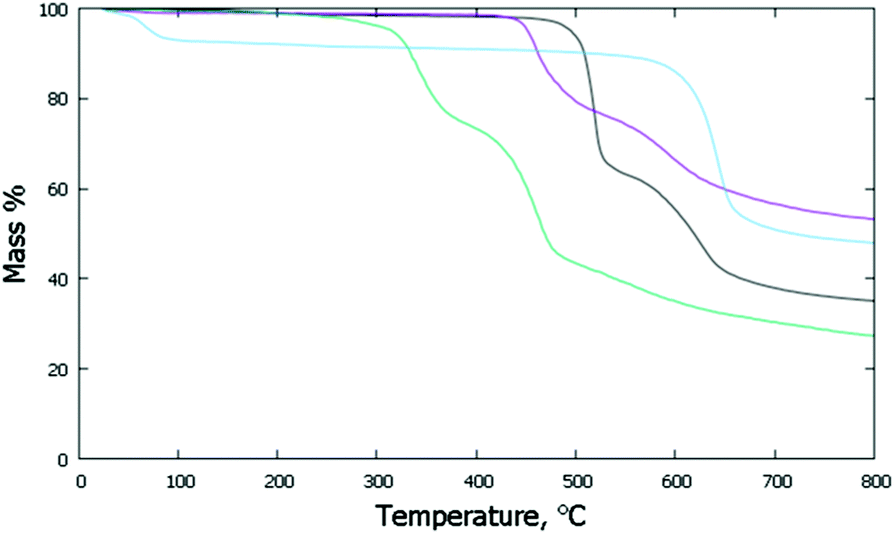 | ||
| Fig. 4 TG traces collected under N2 at a scan rate of 10° min−1: NppDI in black; NmpDI in magenta; NphDI in green; poly-NppDI in blue. | ||
 | ||
| Fig. 5 Variable temperature XRD scans (2θ in the 3–30° range), from RT (bottom) to 420, 420, 300, and 460 °C, respectively. | ||
The observed mass variations are not easy to interpret. In all three molecular compounds a double decomposition step is observed, accounting for a total of 52 to 61 w% loss. These values are in line with the elimination of the imidic branches and partial (and differential) decomposition (to CO or CO2) of the carbonyl residues. Of much simpler description is the TG curve of the polymeric poly-NppDI phase: the observed 41.2 w% loss is easily reproduced by the elimination of two C2O2N fragments (40.9%), leaving behind only condensed polyaromatic hydrocarbons.
The coefficients of thermal expansion were determined using the cell parameter variations derived from the structureless Le Bail fitting using the linear approximation:
| p(T) = p(To)[1 + κp (T − To)] | (2) |
| Species | TG inflection, ° | Mass loss, % | κ a | κ b | κ c | κ β | κ V | DLDa |
|---|---|---|---|---|---|---|---|---|
| a Note: DLD is the degree of lattice distortion, or the spontaneous strain, that is the square root of the sum of squared eigenvalues of strain tensor calculated for the 30–300° range, divided by 3. b Second inflection point and the total mass change after the second event. | ||||||||
| NppDI | 520.0 (624.2b) | 35.2 (60.9b) | 51(4) | −34(1) | 51(2) | 79(5) | 88(7) | 0.0126 |
| NmpDI | 460.3 (594.2b) | 22.0 (51.9b) | 37(3) | 19(1) | 55(2) | — | 112(3) | 0.0068 |
| NphDI | 334.8 (464.9b) | 25.4 (57.4b) | 26(2) | 104(4) | 11(2) | −46(1) | 150(2) | 0.0123 |
| Poly-NppDI | 643.0 | 41.2 | 109(3) | −4(3) | −1(3) | — | 104(6) | 0.0113 |
The κp values, spatially visualized in Fig. 6, are highly anisotropic, in line with the crystal symmetries of these materials. This is particularly true for poly-NppDI, where two nearly null coefficients are observed along the b and c axes, at the expenses of a very large value measured along a.
 | ||
| Fig. 6 Visualization of the thermal expansion tensors, calculated by comparing the lattice metrics at RT and at 300 °C. Positive lobes in green, (tiny) negative ones in red. | ||
Additionally, the very high decomposition temperature of poly-NppDI powders (onset near 615 °C) closely matches that of films of the 2,3,6,7 regioisomer44 (measured by Td5, the 5% weight loss temperature – 590 °C in N2 and 580 °C in air), confirming the extreme thermal stability of aromatic polyimide (co)polymers.45 The oxidative stability of all species was also established by performing TG/DSC measurements in air (see Fig. S9, ESI†).
(d) Spectroscopic analysis
The powdered materials appear light brown in color and, as expected, show “absorbance” spectra, spanning the wide 400–600 nm interval (Fig. 7). Such behavior is in line with other NDI-based diimides, though a wider band is observed here. Since most of the studied species (NphDI excluded, for the presence of an sp3 hybridized N atom) may manifest extended charge delocalization onto the aromatic branching residues, the occurrence of several accessible states with low-energy separation is in agreement with the observed broad absorption band falling in the visible region.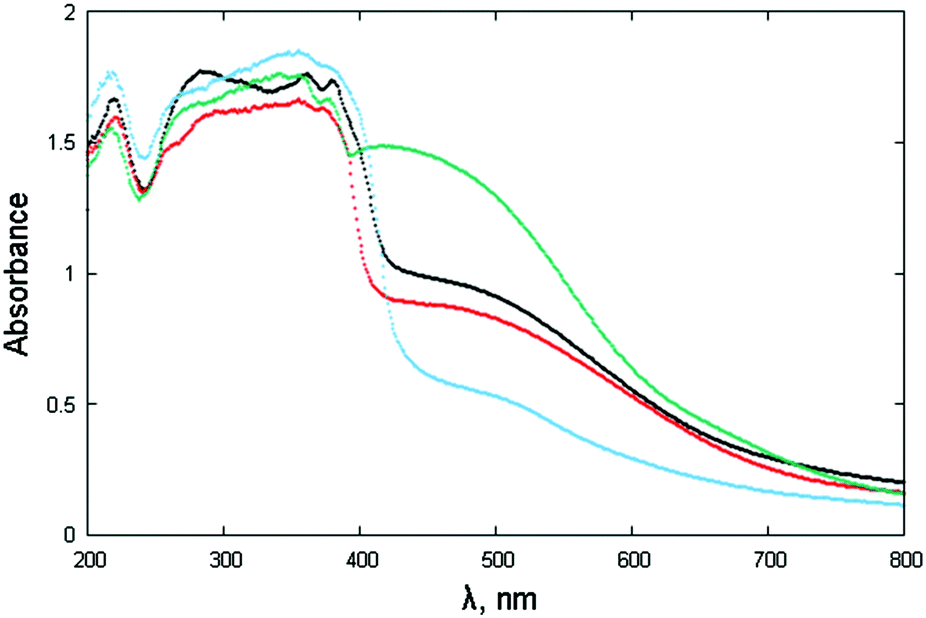 | ||
| Fig. 7 Absorbance spectra (in the form of F[R(E)] through Kubelka–Munk transformation of the diffuse reflectance spectra) of NppDI (black), NmpDI (red), NphDI (green) and poly-NppDI (blue). | ||
The steep increase observed in proximity to 400 nm, on the verge of the violet-UV range, has been interpreted by the occurrence of a direct bandgap, the actual values of which, falling in the narrow 2.87–3.01 eV range and derived as illustrated in Fig. S13 and S14 (ESI†), are reported in Table 1. The lowest values (2.87 and 2.90 eV) are found for the p-diaminophenylene derivatives, the molecular (NppDI) and polymeric (poly-NppDI) ones, respectively. Such observations, which cannot be explained by the crystal packing features discussed above and quantified in Table 1, speak for a slightly higher extent of conjugated bond overlap in these molecules. Slightly larger direct band gap energies are found for NmpDI (not manifesting π–π stacking in the solid) and NphDI (where significant bond conjugation through the N–N bond can easily be excluded). As reference values, for markedly unconjugated residues, the typical optical bandgap falls near 3.0 eV.46
(e) Electronic structure
As reported in Fig. 8, the HOMO and LUMO, derived from DFT calculations, are mostly localized on the side aromatic rings and on the naphthalene-diimide aromatic core, respectively. In NppDI and NmpDI, the HOMO levels are aligned (at ca. −5.6 eV); since they differ only for the (remote enough) position of the amino group on the side rings, this has negligible influence on the HOMO energy. Since the polyaromatic core is the same in both compounds (see the optimized geometries in Fig. S15 (ESI†), coordinates are provided in Tables S2–S4, ESI†), the energies of the LUMO differ by less than 0.1 eV (−3.15 and −3.23 eV). This finding can likely be ascribed to the difference in dihedral angles (reported in Table S5, ESI†), affecting a partial, though minor, electronic conjugation. On the other hand, in NphDI, the energies of both HOMO/LUMO are lowered (−5.95/−3.63 eV, in line with those reported in ref. 26) and favor 1e− reduction to the corresponding radical anion, useful for sensing and for the generation of n-type organic semiconductors. These results suggest that the HOMO/LUMO energy is sensitive to nitrogen substituents only when they are directly linked to the core rings. Experimentally measured optical band gaps (measured in the solid) deviate by ca. 0.4–0.6 eV from the DFT-calculated molecular ones (known to vary depending on the basis-set used47), an effect which was already observed in a large variety of organic semiconductors,48 and also in strictly related NDI-based molecules and congeners (up to ca. 0.7 eV).49 | ||
| Fig. 8 Energy scheme and the HOMO and LUMO isosurfaces for NphDI, NppDI, and NmpDI (left to right). | ||
To experimentally corroborate our DFT simulations, cyclic voltammetric measurements of NppDI, NmpDI and NphDI (in DMSO) were performed (see Fig. S10, ESI†) and the data were analyzed following the procedure used for similar compounds (n-dopable conjugated NDI-based polymers).50 Similarly to these polymers, our molecular compounds show two separate reduction steps (ca. 0.5 eV apart, see Table S6, ESI†), but with slightly lower electrochemical band gaps (ΔEgel, ca. 1.4 eV vs. ca. 2.0 eV in ref. 50). Such lowering is mostly attributed to the changes of the absolute energy values of the HOMOs, while those of the LUMOs are still located near −4 eV. Thus, LUMO-dependent reduction or partial n-doping is a favored process also in our molecular species synthesis.
(f) Thin film analysis
For the three molecular NDI derivatives, both the as-deposited and thermally annealed thin films have been investigated. The thin film structures, in terms of crystal phase, degree of crystallinity and texture (mosaicity) have been determined by the analysis of the 2D-GIXD measurements.51 As anticipated, these images have been collected just below and slightly above the critical angle for total reflection, and their mutual comparison enables the retrieval of information on the variation of the film structure along its thickness.52Due to the extremely poor solubility of NppDI (providing approximately 10× more dilute solutions than NmpDI and NphDI), the as-deposited (too thin) films did not provide measurable diffracted signals, and only few and weak signals appeared in the 2D-GIXD images after annealing (see Fig. S16, ESI†). Although the paucity of information does not allow a complete description of the NppDI film structure, the observed signals can be ascribed to the strongest reflections of the bulk phase, suggesting that the molecular arrangement in the film remains unchanged and that no distinct thin-film structure is formed.
Fig. 9 shows the 2D-GIXD images for the annealed NmpDI (a) and NphDI (b) thin films, together with the corresponding integrated intensity profiles along the qz (Fig. 9b and e) and qxy (Fig. 9c and f) directions. The 2D-GIXRD images of both films exhibit several intense Bragg spots having a perfect symmetry with respect to the central vertical line, overlapped with minor arc-shaped intensity contributions, the latter being more extended and pronounced for the NmpDI film than for NphDI (Fig. 9a and d). These features indicate the coexistence of a majority of crystallites/domains highly oriented along one specific direction with respect to the substrate surface (sharper h00 signals), and a minority of less, or even randomly, oriented crystallites (arc-shaped signals). For both films, all the reflections have been indexed by using the monoclinic crystallographic structure of the RT-stable bulk phases and by assuming the (100) texturing (Fig. S17 and S18a, ESI†), as confirmed by the qz integrated intensity profiles (Fig. 9b and e), where only h00 reflections are observed. The molecular orientations associated with this texturing are schematically reported in Fig. 9a and b. Interestingly, though sharing the same texturing, dictated by the stacking of molecular layers (arranged in slabs), the molecular contacts on the substrate surface distinctly differ with the angle between the aromatic cores and the surface being 83.7 and 40.8° for NmpDI and NphDI, respectively. However, in addition to peaks of the h00 class, the qxy integrated intensity profiles (Fig. 9c and f) show the presence of further peaks, witnessing the presence of largely misoriented crystallites.
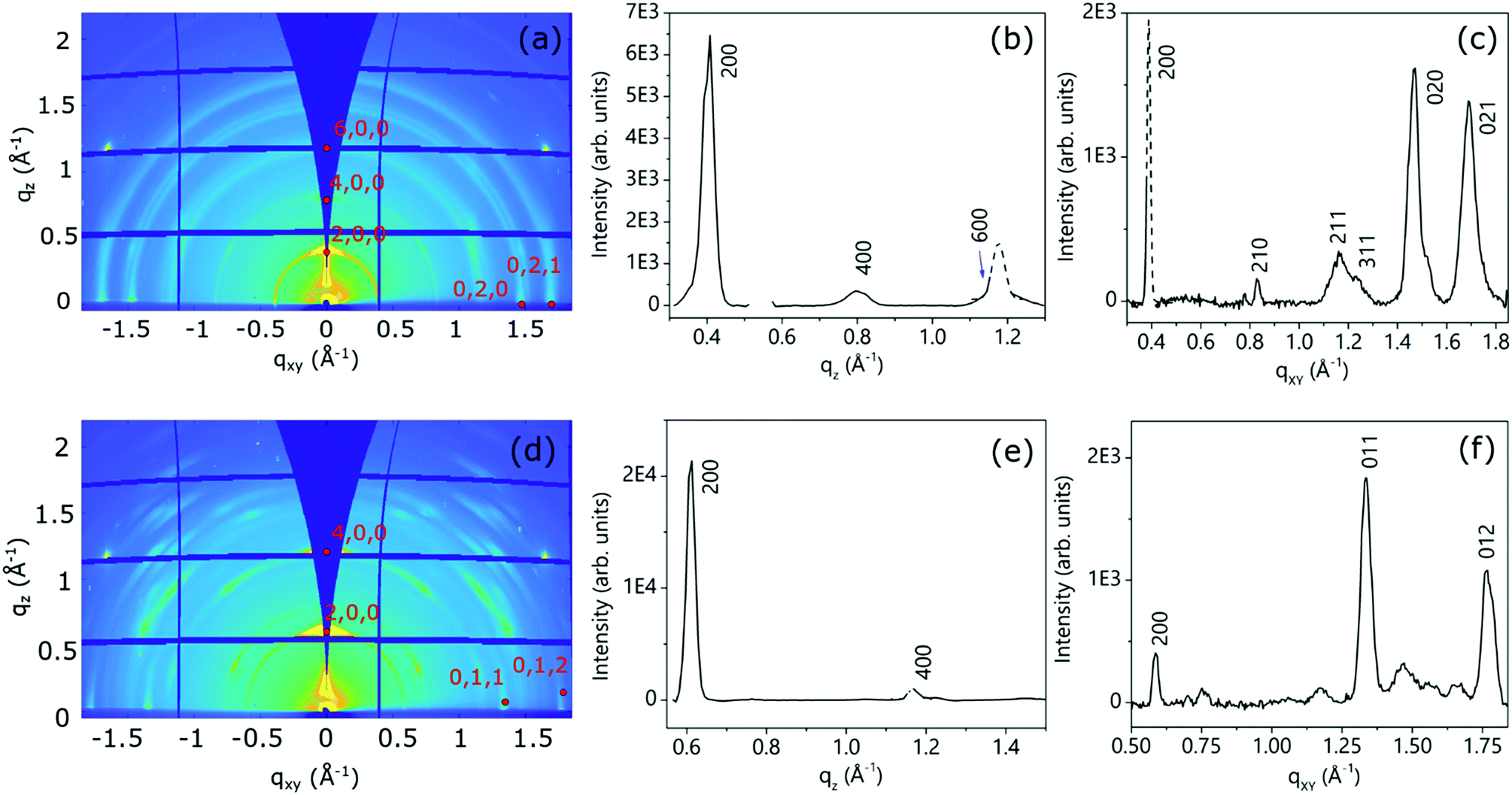 | ||
| Fig. 9 2D-GIXD images of annealed (a) NmpDI and (d) NphDI thin films recorded for αi = 0.1°. The corresponding integrated intensity profiles along qz, [(b and e)], and along qxy, [(c and f)] are reported. The dashed lines in b, c and e are Gaussian fits to the experimental data at the positions where the detector cuts hamper collection of the diffracted intensity. | ||
A closer look into the 2D-GIXD images of the NphDI film shows that all reflections are azimuthally misplaced with respect to those expected for an exact h00 texturing (Figure S18a, ESI†), as already observed in the 2D GIXD images of other highly ordered organic films53 (Fig. 10).
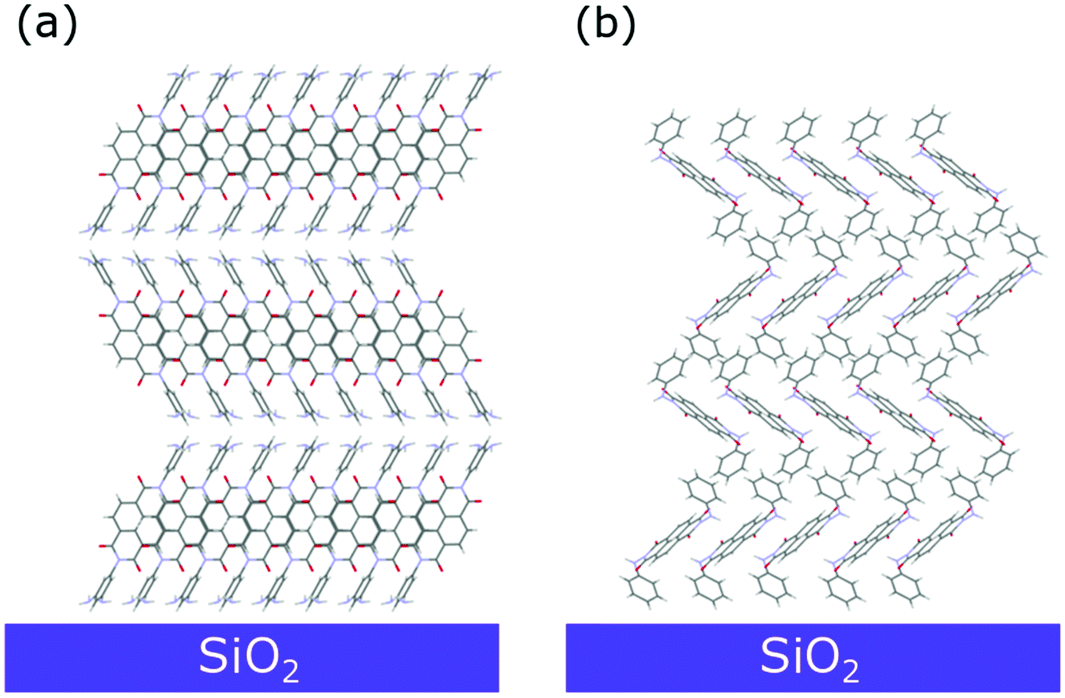 | ||
| Fig. 10 Sketch of the dominant molecular arrangement of the (a) NmpDI and (b) NphDI thin films on the substrate surface, as retrieved from GIXD analysis. | ||
However, the experimental positions of the reflection maxima can be properly reproduced by tilting by ca. 3° the (100) crystal planes with respect to the substrate surface (Fig. S18b, ESI1). This tilting only marginally affects the angle between the aromatic cores and the substrate surface (lowered to ca. 40.1°).
The mosaicity of the oriented crystallites/domains, i.e. their angular deviation with respect to the main orientation, is estimated from the analysis of the experimental azimuthal profile of the strong 200 reflection (see Fig. S19, ESI†). The very small value of the determined mosaicity, ca. ± 2° and ± 3.5° for NmpDI and NphDI, respectively, is a direct measure of the strong orientation of the domains (i.e., of the associated molecules).
The 2D GIXD patterns collected below and above the critical angle for total reflection (Fig. S20 and S21, ESI† for NmpDI and NphDI, respectively), probing only the first 4 nm close to the air interface or the entire film thickness, allowed us to establish the homogeneity of the molecular arrangement across the film thickness. Regardless of the probed depth, the films show the same texturing, as evidenced by consistence of the extracted qz profiles, and the same degree of mosaicity, given by the almost invariance of the width of azimuthal profiles. Interestingly, the as-deposited films exhibit a high degree of crystallinity (slightly lowered upon annealing), indicating the extreme propensity of these molecules to self-assemble in an ordered fashion on the substrate surface without the need for any postprocessing technique (Fig. S22 and S23, ESI†).
4. Conclusions
In this paper we presented the isolation and the thorough characterization of powders and thin films of new molecular crystals containing the NDI cores, substituted by different isomeric, constitutionally different, N-containing aromatic residues. The complete structural analysis demonstrated the similarity of the bulk and film structures, and the morphological texturing of the latter. In search for NDI-based organic solids not showing the polymorphic variability of the alkyl-chains analogues, in the present work we report species which exhibit high thermal stability and absence of polymorphic transitions up to melting. Both properties make the sublimation-route as a viable option for films and device preparation, in addition to solution-based methods, thus compensating the limited solubility properties of our systems. The structural, spectroscopic and thermal analyses presented in this work, together with the determination of the anisotropy of thermal expansion coefficients, add further knowledge on how NDI cores approach, and overlap, in the search for optimized performances (e.g., electron and thermal conductivity, and Seebeck coefficient), which are at the heart of thermoelectric generators based on aromatic-polyimides.54DFT calculations indicated a significant lowering of the frontier orbital energies of the hydrazido derivative (NphDI), fostering its use as an electronic acceptor. Such an effect can be further, and easily, enhanced by adding electron-withdrawing groups (such F or CF3 residues) on the aromatic rings in the molecular periphery. Work can be anticipated in the direction of exploring the synthesis and structure of NphDI fluorinated congeners, in search for soluble, filmable and dopable systems with improved electronic performances, electron mobility and rich(er) carrier concentrations. Indeed, the preliminary results indicate that polyfluorinated NDIs possess significantly enhanced solubilities in organic solvents commonly used for film deposition (acetonitrile, DMF and THF – but not chlorinated ones) and energy levels and redox properties suitable for n-doping.
Finally, we have also prepared and characterized an organic nanosized material, stable well above 500 °C and containing a 100% ordered 1:1 copolymer (of admittedly short chain length), together with its structure determination from the severely broadened XRD trace.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was partially supported by MIUR (PRIN-2017, Project 2017L8WW48, HY-TEC). We thank G. Chita for assistance during the synchrotron X-measurements at Elettra. The courtesy of G.B. Giovenzana (Università del Piemonte Orientale) for providing MS data is also acknowledged.Notes and references
- W. Brütting and C. Adachi, Physics of Organic Semiconductors, Wiley, NY (USA), 2012 Search PubMed.
- (a) Y. Chen, Y. Zhao and Z. Liang, Energy Environ. Sci., 2015, 8, 401–422 RSC; (b) M. Culebras, C. M. Gómez and A. Cantarero, Materials, 2014, 7, 6701–6732 CrossRef PubMed; (c) L. M. Cowen, J. Atoyo, M. J. Carnie, D. Baran and B. C. Schroeder, ECS J. Solid State Sci. Technnol., 2017, 6, N3080–N3088 CrossRef CAS.
- M. M. Hussain and N. El-Atab, Handbook of Flexible and Stretchable Electronics, CRC Press, Boca Raton, FL (USA), 2019 Search PubMed.
- M. Caironi and Y.-Y. Noh, Large Area and Flexible Electronics, Wiley VCH, Weinheim (Germany), 2015 Search PubMed.
- L. A. Francis and K. Iniewski, Novel Advances in Microsystems Technologies and Their Applications, CRC Press, Boca Raton, FL (USA), 2014 Search PubMed.
- M. Al Kobaisi, S. V. Bhosale, K. Latam, A. M. Raynor and S. V. Bhosale, Chem. Rev., 2016, 116, 1685–11796 CrossRef PubMed.
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed.
- (a) H. Dong, C. Wang and W. Hu, Chem. Commun., 2010, 46, 5211–5222 RSC; (b) A. Nowak-Król, K. Shoyama, M. Stolte and F. Würthner, Chem. Commun., 2018, 54, 13763–13772 RSC.
- S. Milita, F. Liscio, L. Cowen, M. Cavallini, B. A. Drain, T. Degousée, S. Luong, O. Fenwick, A. Guagliardi, B. C. Schroeder and N. Masciocchi, J. Mater. Chem. C, 2020, 8, 3097–3112 RSC.
- (a) M. Dharmarwardana, R. P. Welch, S. Kwon, V. K. Nguyen, G. T. McCandless, M. A. Omary and J. J. Gassensmith, Chem. Commun., 2017, 53, 9890–9893 RSC; (b) P. M. Alvey, J. J. Reczek, V. Lynch and B. L. Iverson, J. Org. Chem., 2010, 75, 7682–7690 CrossRef CAS PubMed; (c) J. J. Reczek, K. R. Villazor, V. Lynch, T. M. Swager and B. L. Iverson, J. Am. Chem. Soc., 2006, 128, 7995–8002 CrossRef CAS PubMed; (d) T. He, M. Stolte, C. Burschka, N. H. Hansen, T. Musiol, D.l. Kalblein, J. Pflaum, X. Tao, J. Brill and F. Würthner, Nat. Commun., 2015, 6, 5954 CrossRef CAS PubMed; (e) M. Diebold, E. Christ, L. Biniek, L. Karmazin, B. Heinrich, C. Contal, S. Ghosh, P. J. Mesini and M. Brinkmann, J. Mater. Chem. C, 2019, 7, 13120–13129 RSC; (f) X. Jiao, S. Maniam, S. J. Langford and C. R. Mc Neill, Phys. Rev. Mater., 2019, 3, 013606 CrossRef CAS.
- (a) R. Pfattner, M. Mas-Torrent, I. Bilotti, A. Brillante, S. Milita, F. Liscio, F. Biscarini, T. Marszalek, J. Ulanski, A. Nosal, M. Gazicki-Lipman, M. Leufgen, G. Schmidt, L. W. Molenkamp, V. Laukhin, J. Veciana and C. Rovira, Adv. Mater., 2010, 22, 4198–4203 CrossRef CAS PubMed; (b) S. Galindo, A. Tamayo, F. Leonardi and M. Mas-Torrent, Adv. Funct. Mater., 2017, 27, 1700526 CrossRef; (c) D. Gentili, M. Gazzano, M. Melucci, D. Jones and M. Cavallini, Chem. Soc. Rev., 2019, 48, 2502–2517 RSC.
- (a) Z.-G. Zhang, H. Li, B. Qi, D. Chi, Z. Jin, Z. Qi, J. Hou, Y. Li and J. Wang, J. Mater. Chem. A, 2013, 1, 9624–9629 RSC; (b) T. Akiyama, Bull. Chem. Soc. Jpn., 2019, 92, 1181–1199 CrossRef CAS; (c) T. de Roo, J. Haase, J. Keller, Ch. Hinz, M. Schmid, D. V. Seletskiy, H. Cölfen, A. Leitenstorfer and S. Mecking, Adv. Funct. Mater., 2018, 14, 2714–2719 Search PubMed; (d) B. Russ, M. J. Robb, B. C. Popere, E. E. Perry, C.-K. Mai, S. L. Fronk, S. N. Patel, T. E. Mates, G. C. Bazan, J. J. Urban, M. L. Chabinyc, C. J. Hawkerand and R. A. Segalman, Chem. Sci., 2016, 7, 1914–1919 RSC.
- F. Doria, A. Oppi, F. Manoli, S. Botti, N. Kandoth, V. Grande, I. Manet and M. Freccero, Chem. Commun., 2015, 51, 9105–9108 RSC.
- H. Wei, M. Lv, X. Duan, S. Li, Y. Yao, K. Wang, P. Zhang, X. Li and H. Chen, Med. Chem. Res., 2014, 23, 2277–2286 CrossRef CAS.
- F. Würthner, V. Stepanenko, Z. Chen, C. R. Saha-Möller, N. Kocher and D. Stalke, J. Org. Chem., 2004, 69, 7933–7939 CrossRef PubMed.
- L. Ferlauto, F. Liscio, E. Orgiu, N. Masciocchi, A. Guagliardi, F. Biscarini, P. Samorì and S. Milita, Adv. Funct. Mater., 2014, 24, 1090–1099 CrossRef.
- O. D. Jurchescu, D. A. Mourey, S. Subramanian, S. R. Parkin, B. M. Vogel, J. E. Anthony, T. N. Jackson and D. J. Gundlach, Phys. Rev., 2009, 80, 0852012009 Search PubMed.
- (a) A. S. Raw, M. S. Furness, D. S. Gill, R. C. Adams, F. O. Holcombe and L. X. Yu, Adv. Drug Delivery Rev., 2004, 56, 397–414 CrossRef CAS PubMed; (b) G. Goldbeck, E. Pidcock and C. Groom, Solid Form Informatics for Pharmaceuticals and Agrochemicals: Knowledge-Based Substance Development and Risk Assessment, CCDC, 2012 http://www.ccdc.cam.ac.uk/support-and-resources/ccdcresources/Solid_Form_Informatics.pdf; (c) J. Bernstein, Polymorphism in Molecular Crystals, Clarendon Press, Oxford, 2002 Search PubMed.
- D. W. Armstrong, K. R. Rund, U. B. Nair and G. L. Raid III, Current, 1996, 15, 57–61 CAS.
- S. Bergantin and M. Moret, Cryst. Growth Des., 2012, 12, 6035–6041 CrossRef CAS.
- J. Mizugichi, J. Phys. Chem. B, 2004, 108, 8926–8930 CrossRef.
- K. C. Lee, G. S. Ryu, S. Chen, G. Kim, Y. Y. Noh and C. Yang, Org. Electron., 2016, 37, 402–410 CrossRef CAS.
- K. Tagami. A. Saitoh, K. Sekido, M. Sekiya and M. Nishi, US Pat., US10065969B2, 2018 Search PubMed.
- C. R. DeBlase, K. Hernández-Burgos, J. M. Totter, D. J. Fortman, D. dos, S. Abreu, R. A. Timm, I. C. N. Diógenes, L. T. Kubota, H. D. Abruña and W. R. Dichtel, Angew. Chem., Int. Ed., 2015, 54, 13225–13229 CrossRef CAS PubMed.
- S. A. Bhat, C. Das and T. K. Maji, J. Mater. Chem. A, 2018, 6, 19834–19842 RSC.
- M. R. Ajayakumar, P. Mukhopadhyay, S. Yadav and D. Ghosh, Org. Lett., 2010, 12, 2646–2649 CrossRef CAS PubMed.
- See for example: (a) Do-K Kim, H. Lee, X. Zhang, J.-H. Bae and J. Park, Micromachines, 2019, 10, 727–736 CrossRef PubMed; (b) L. Ruan, D. Zhang, J. Tong, J. Kang, Y. Chang, L. Zhou, G. Qin and X. Zhang, in Ferroelectrics and Their Applications, ed. H. Irzaman, Intechopen, London, UK, 2018, ch. 8 Search PubMed; (c) F. S. Kim, C. H. Park, Y. Na and S. A. Jeneke, Org. Electr., 2019, 69, 301–317 CrossRef CAS.
- TOPAS-R, V. 3.0, 2005, Bruker AXS, Karlsruhe, Germany.
- A. A. Coelho, J. Appl. Crystallogr., 2003, 36, 86–95 CrossRef CAS.
- R. W. Cheary and A. Coelho, J. Appl. Crystallogr., 1992, 25, 109–121 CrossRef CAS.
- W. A. Dollase, J. Appl. Crystallogr., 1986, 19, 267–272 CrossRef CAS.
- B. Schrode, S. Pachmajer, M. Dohr, C. Röthel, J. Domke, T. Fritz, R. Resel and O. Werzer, J. Appl. Crystallogr., 2019, 52, 683–689 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Chem. Phys., 1994, 98, 11623–11627 CrossRef CAS.
- M. R. Ajayakumar and P. Mukhopadhyay, Chem. Commun., 2009, 3702–3704 RSC.
- J. Mizuguchi, Zeit. Kristallogr. NCS, 2002, 218, 137–138 Search PubMed.
- J. Mizuguchi, Zeit. Kristallogr. NCS, 2002, 218, 139–140 Search PubMed.
- K. Miyatake, H. Zhou, T. Matsuo, H. Uchida and M. Watanabe, Macromolcules, 2004, 37, 4961–4966 CrossRef CAS.
- H. E. Katz, T. Siegrist, J. H. Schön, C. Kloc, B. Batlogg, A. J. Lovinger and J. Johnson, ChemPhysChem, 2001, 3, 167–172 CrossRef.
- P. Trucano and R. Chen, Nature, 1975, 258, 136–137 CrossRef CAS.
- E. Keller, Chem. Zeit, 1980, 14, 56–60 CAS.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- M. Hasegawa and S. Horii, Polym. J., 2007, 39, 610–621 CrossRef CAS.
- See, e.g., (a) A. S. Mathews, H. Kim and C.-S. Ha, Macromol. Res., 2007, 15, 114–128 CrossRef CAS; (b) ed. K. L. Mittal, Polyimides and other high temperature polymers: synthesis, characterization, and applications, CRC Press, Boca Raton, FL, 2009, vol. 5 Search PubMed.
- N. B. Kolhe, R. Nandini Devi, S. P. Senanayak, B. Jancy, K. S. Narayan and S. K. Asha, J. Mater. Chem., 2012, 22, 15235–15246 RSC.
- A. Dittmer, R. Izsák, F. Neese and D. Maganas, Inorg. Chem., 2019, 58, 9303–9315 CrossRef CAS PubMed.
- J. C. S. Costa, R. J. S. Taveira, C. F. R. A. C. Lima, A. Mendes and L. M. N. B. F. Santos, Opt. Mater., 2016, 58, 51–60 CrossRef CAS.
- A. Insuasty, S. Carrara, L. Tang, C. Forsyth, C. F. Hogan, C. R. McNeill and S. J. Langford, ChemPlusChem, 2019, 84, 1638–1642 CrossRef CAS PubMed.
- P. Ledwon, D. Ovsiannikova, T. Jarosz, S. Gogoc, P. Nitschke and W. Domagala, Electrochim. Acta, 2019, 307, 525–535 CrossRef CAS.
- J. Lee, J. L. Hernandez, I. Pelse, J. R. Reynolds and C. Yang, J. Mater. Chem. C, 2018, 6, 10532–10537 RSC.
- W. Porzio, C. Scavia, L. Barba, G. Arrighetti and S. Milita, Eur. Polym. J., 2011, 47, 273–283 CrossRef CAS.
- (a) S. Milita, M. Servidori, F. Cicoira, C. Santato and A. Pifferi, Nucl. Instr. Meth. B, 2006, 246, 101–105 CrossRef CAS; (b) S. Milita, C. Santato and F. Cicoira, Appl. Surf. Sci., 2006, 252, 8022–8027 CrossRef CAS.
- K. Feng, H. Guo, J. Wang, Y. Shi, Z. Wu, M. Su, Z. Zhang, J. H. Son, H. Y. Woo and X. Guo, J. Am. Chem. Soc., 2021, 143, 1539–1552 CrossRef CAS PubMed , and references therein.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the 2D-GIXD, structural and spectroscopic characterization. See DOI: 10.1039/d1tc00564b |
| This journal is © The Royal Society of Chemistry 2021 |