
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpontaneous and induced chiral symmetry breaking of stereolabile pillar[5]arene derivatives upon crystallisation†
Haiying
Wang
 a,
Weiwei
Yang
a,
Kim K.
Baldridge
a,
Cai-Hong
Zhan
*b,
Tushar Ulhas
Thikekar
*ac and
Andrew C.-H.
Sue
*c
a,
Weiwei
Yang
a,
Kim K.
Baldridge
a,
Cai-Hong
Zhan
*b,
Tushar Ulhas
Thikekar
*ac and
Andrew C.-H.
Sue
*c
aSchool of Pharmaceutical Science & Technology, Tianjin University, 92 Weijin Road, Nankai District, Tianjin 300072, P. R. China. E-mail: tusthikekar@tju.edu.cn
bCollege of Chemistry and Life Sciences, Zhejiang Normal University, 688 Yingbin Road, Jinhua, Zhejiang Province 321004, P. R. China. E-mail: chzhan@zjnu.cn
cCollege of Chemistry and Chemical Engineering, Xiamen University, 422 Siming S Rd, Siming District, Xiamen, Fujian Province 361005, P. R. China. E-mail: andrewsue@xmu.edu.cn
First published on 27th July 2021
Abstract
Stereolabile pillar[5]arene (P[5]) derivatives, which are dynamic racemic mixtures in solution on account of their low inversion barriers, were employed as platforms to study chiral symmetry breaking during crystallisation. In the solid state, we showed that crystal enantiomeric excess of a conglomerate-forming P[5] derivative can be obtained by handpicking and Viedma ripening without the intervention of external chiral entities. On the other hand, in the presence of ethyl D/L-lactate as both optically-active solvents and chiral guests, the handedness of P[5] derivative crystals, either forming racemic compounds or conglomerates upon condensation, can be directed and subsequently inverted in a highly controllable manner.
Introduction
Chirality, the geometric property of a rigid object of being non-superimposable on its mirror image, is omnipresent in nature.1 Since the days of Biot and Pasteur, crystallography and chirality have been two deeply intertwined scientific disciplines.2 Resolution by crystallisation, originating from the manual separation of hemihedral crystals of sodium ammonium tartrate,3 has also evolved into a variety of techniques that are of both significant academic and industrial interest.4 In principle, the yield of isolating an enantiomer by resolving the corresponding racemate is 50% at best. Nonetheless, compounds that undergo rapid racemisation in solution,5 when coupled with the occurrence of conglomerate formation (vide infra), offer the opportunity to achieve chiral symmetry breaking, or total spontaneous resolution, deviating from the racemic equilibrium in favour of one handedness.6Pillar[5]arenes7 (P[5]s) are a class of macrocyclic compounds exhibiting rich conformational isomerism and dynamic stereoisomerism. Enantiomeric P[5] conformers8 are interconvertible to each other through a series of oxygen-through-the-annulus flipping motions of their aromatic units.9 By introducing either bulky substituents on the rims,10 or mechanically-interlocked structures using P[5]s as the ring component,11 the inversion processes of P[5]s can be completely inhibited, therefore enabling the isolation of two types of atropisomers by classical resolution methods or with the help of HPLC with chiral stationary phases. On the other hand, stereolabile P[5]s with low-to-moderate inversion barriers exist in solution as optically-inactive ensembles of fluxional conformers. The chiroptical characterisation and resolution of these P[5] derivatives showing rapid enantiomerisation12 can be non-trivial.13
A general method to transform stereolabile P[5]s into enantiopure compounds of either handedness is still lacking. We envisioned that chiral crystal formation followed by topochemical reactions14 might be a viable approach to achieve absolute asymmetric synthesis of chiral P[5]s. Although the spontaneous resolution phenomenon of P[5]s, i.e. the formation of mechanical mixtures of enantiopure conglomerate crystals, was already observed,15 no further exploration beyond X-ray crystallography has been described to date. Herein, we report our investigation into P[5] conglomerates including conventional triage and Viedma ripening.16 Chiral induction through co-crystallization with chiral guest molecules was also attempted on P[5]-based conglomerates and racemic compounds. In both cases, chiroptical characterisation and X-ray crystallography confirmed that crystal enantiomeric excess (CEE) was successfully obtained.
Results and discussion
Resolution by triage
It is well-known that racemic mixtures can be condensed (Fig. 1) in the solid state as racemic compounds, racemic conglomerates, and occasionally pseudoracemates.17 Structural data in the Cambridge Crystallographic Data Centre reveals that P[5] derivatives are indeed mostly racemic compounds, and a small fraction of P[5] derivatives crystallises as conglomerates in one of the 65 Sohncke space groups.18 Among P[5] derivatives crystallising in non-centrosymmetric space groups reported in the literature, P[5]HQ,19 featuring one hydroquinone and four 1,4,-dimethoxybenzene units, was chosen due to its ease of synthesis. Upon crystallisation in CH2Cl2, P[5]HQ (monoclinic, space group C2) spontaneously resolves (Fig. 2a) into racemic conglomerates, whose unit cell features four P[5]HQ macrocycles with CH2Cl2 solvent molecules in their cavities. Individual rhombohedral single crystals obtained by the vapour diffusion method (Et2O into CH2Cl2) were handpicked (Fig. S1, ESI†) and crushed into finely ground microcrystalline powder. Their KBr pellets were subjected to solid-state electronic circular dichroism (ECD) measurements.20 After repeated attempts of triage,3,21 several samples, especially smaller single crystals (≈1 mm), gave rise to (Fig. 2b) clearly distinguishable ECD spectra22 (Fig. S2 and S3, ESI†) Based on the comparison with the calculated ECD spectra (see the ESI† for computational details), the absolute configurations of these crystals were determined (Fig. 2b and S4, ESI†). | ||
| Fig. 1 The crystallisation of stereolabile pillar[5]arene macrocycles can result in (a) racemic compounds or (b) racemic conglomerates in the solid state. Solid solutions, or pseudoracemates, are another possibility, which happens much less frequently. | ||
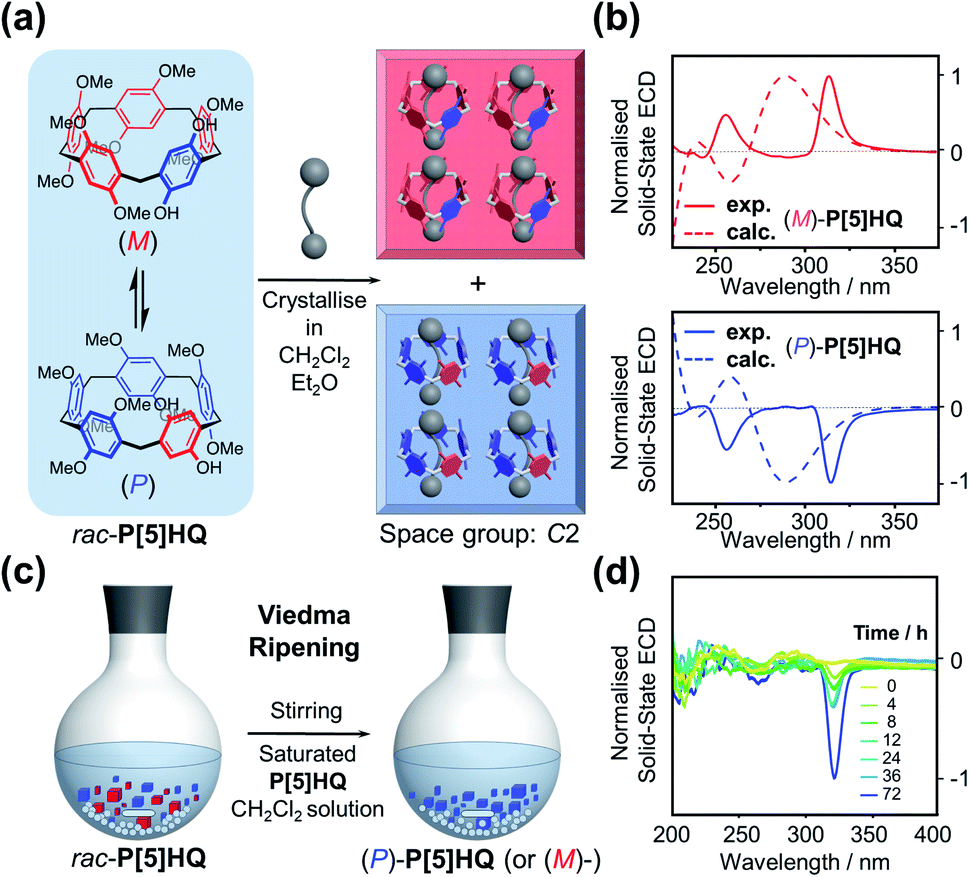 | ||
| Fig. 2 (a) Schematic representation of the spontaneous resolution of rac-P[5]HQ. The mechanical mixture of enantiomeric crystals can be resolved by triage. (b) Normalised solid-state ECD spectra of randomly handpicked (M)- and (P)-P[5]HQ single crystals (solid lines) and stimulated ECD spectra of (M)- and (P)-P[5]HQ (dashed lines). (c) Schematic representation of Viedma ripening. (d) Normalised solid-state ECD spectra of a near-racemic P[5]HQ sample gradually showing crystal enantiomeric excess during the process of Viedma ripening. | ||
Deracemisation by Viedma ripening
The fact that P[5]HQ crystallises as racemic conglomerates, coupled with its relatively low stereochemical inversion barrier (11.9 kcal mol−1 in CDCl3),9b sets the stage for conducting the so-called Viedma ripening (Fig. 2c), which was discovered by Cristóbal Viedma in 2005.16a In his seminal publication, racemic mixtures of NaClO3 conglomerate crystals in a saturated aqueous solution were converted to a single chiral phase by continuous abrasive grinding with glass beads. This attrition-enhanced deracemization process has proven to be an effective approach to obtain homochiral populations of conglomerate-forming crystals from an initially racemic state. Bulk crystalline P[5]HQ powder samples occasionally showed weak ECD signals, which can be presumably attributed to slight preferential crystallisation in the condensation process during synthesis. Continuous ball-milling of a near-racemic or rac-P[5]HQ slurry under an Ar atmosphere in its saturated CH2Cl2 solution, which promotes the continuous dissolution–crystallisation (secondary nucleation), resulted in solid samples showing (Fig. 2d, S5 and S6, ESI†) distinctive ECD signals in several days. The emergence of intense chiroptical signals from these P[5]HQ solids proves that scalemic mixtures, and eventually enantiopure compounds, could be obtained in the solid state for stereolabile P[5] derivatives whose resolution is not feasible in solution. This finding also expands the scope of Viedma ripening, which was applied to inorganic salts,16a intrinsically chiral organic molecules,23 achiral organic compounds,24 and metal complexes,25 and further to inherently chiral and conformationally labile macrocyclic compounds. With many applications reported, in particular the preparation of drug intermediates,26 we expect that the method of Viedma ripening will further promote the research and development of chiral P[5]s.Enantiopure crystallisation induced by optically active solvents
Inspired by the chiral induction effect on P[5] derivatives by assorted chiral guest molecules in solution,27 we also explored various chiral media for co-crystallisation, and eventually chose a pair of cheap commercially-available optically active solvents, ethyl D/L-lactate ((D)/(L)-EL), in which P[5]HQ shows moderate solubility. Crystalline P[5]HQ samples can be readily obtained in (L)-EL upon heating and slow cooling (Fig. 3a and S7, ESI†). Solid-state chiroptical spectral analysis shows (Fig. 3b and S8, ESI†) an ECD spectrum with positive Cotton effects at circa 255 and 315 nm. In contrast, crystallisation of P[5]HQ in ethyl D-lactate ((D)-EL) yielded solid samples exhibiting (Fig. 3b and S9, ESI†) inverted ECD signals.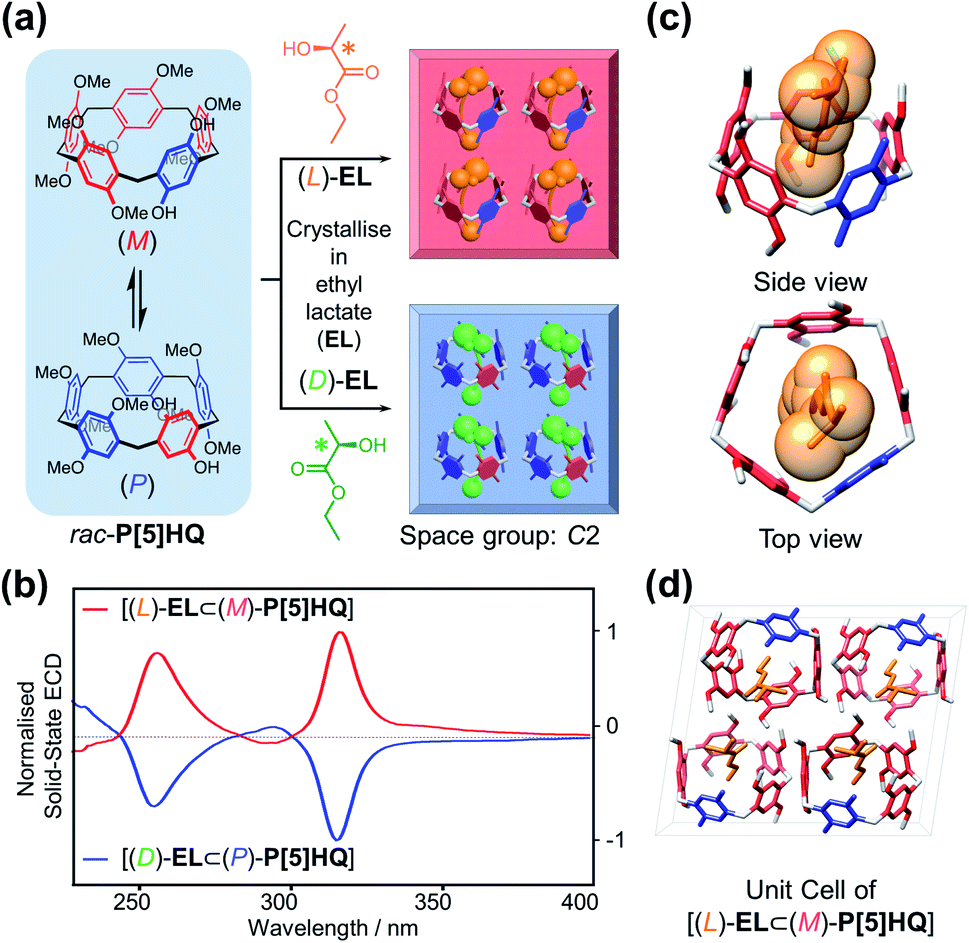 | ||
| Fig. 3 (a) Schematic representation of the crystallisation of P[5]HQ in (D)- and (L)-EL, respectively, forming homochiral crystals of opposite handedness. (b) Normalised solid-state ECD spectra of [(L)-EL⊂(M)-P[5]HQ] and [(D)-EL⊂(P)-P[5]HQ] crystals. Single crystal structures of (c) [(L)-EL⊂(M)-P[5]HQ] inclusion complex. (d) Unit cell of [(L)-EL⊂(M)-P[5]HQ]. | ||
X-ray crystallographic analysis was performed (Fig. 3c) for P[5]HQ single crystals obtained in (L)-EL to further reveal the absolute configuration, host–guest interactions, and packing modes in the solid state. It turned out that crystallisation of P[5]HQ in (L)-EL exclusively forms the [(L)-EL⊂(M)-P[5]HQ] inclusion complex. The chiral solvents sit in the cavities of the P[5]HQ macrocycles, forming 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes on account of several C–H⋯π interactions between the ethyl groups of EL and the aromatic units of P[5]HQ. Intermolecular hydrogen bonds can be found between the hydroxyl group of (L)-EL and a methoxy group on one of the nearby P[5]HQ rings (OH⋯O distance = 2.43 Å, Fig. S11, ESI†), providing additional stabilisation for the overall superstructure. The inclusion of guest molecules does not change the overall packing, as the (L)-EL solvomorph features an identical unit cell in the C2 space group with lattice constants similar to that of the original P[5]HQ crystal structure crystallised in CH2Cl2 (Fig. 3d). In the 1H NMR spectrum of the dissolved [(L)-EL⊂(M)-P[5]HQ] crystal sample, proton signals of the (L)-EL ethyl moiety show (Fig. S12, ESI†) minor upfield shifts (Δδ = −0.13 and −0.10 ppm for the methyl and methylene protons, respectively), indicating that the host–guest interaction between P[5]HQ and EL occurs in solution. The chirality transfer from EL presumably starts from the formation of diastereomeric complexes with P[5]HQ in solution, which subsequently leads to enantioselective crystallisation.
:1 complexes on account of several C–H⋯π interactions between the ethyl groups of EL and the aromatic units of P[5]HQ. Intermolecular hydrogen bonds can be found between the hydroxyl group of (L)-EL and a methoxy group on one of the nearby P[5]HQ rings (OH⋯O distance = 2.43 Å, Fig. S11, ESI†), providing additional stabilisation for the overall superstructure. The inclusion of guest molecules does not change the overall packing, as the (L)-EL solvomorph features an identical unit cell in the C2 space group with lattice constants similar to that of the original P[5]HQ crystal structure crystallised in CH2Cl2 (Fig. 3d). In the 1H NMR spectrum of the dissolved [(L)-EL⊂(M)-P[5]HQ] crystal sample, proton signals of the (L)-EL ethyl moiety show (Fig. S12, ESI†) minor upfield shifts (Δδ = −0.13 and −0.10 ppm for the methyl and methylene protons, respectively), indicating that the host–guest interaction between P[5]HQ and EL occurs in solution. The chirality transfer from EL presumably starts from the formation of diastereomeric complexes with P[5]HQ in solution, which subsequently leads to enantioselective crystallisation.
After discovering that P[5]HQ conglomerates can be turned into homochiral crystals of either handedness, depending on the choice of the crystallisation solvent, the same approach was applied to a structurally similar compound P[5]Q,19 which contains a quinone unit instead of hydroquinone in the scaffold but instead crystallises as a racemic compound in the P21/c space group (Fig. 4a). X-ray crystallographic analysis of a single crystal sample carefully cured in (L)-EL revealed (Fig. 4b) that the [(L)-EL⊂(M)-P[5]Q] inclusion complex was successfully obtained. The ethyl group of (L)-EL is also stabilised in the middle of the P[5]Q macrocycle by C–H⋯π interactions. It is noteworthy that when P[5]Q crystallised in EL, the solid-state packing changed completely to a non-centrosymmetric structure. Coincidentally, the [(L)-EL⊂(M)-P[5]Q] complex crystallises (Fig. 4c and S13, ESI†) in the C2 space group with almost identical unit cell parameters, packing, and hydrogen bonding patterns (OH⋯O distance = 2.13 Å, Fig. S13, ESI†) to those of P[5]HQ. The homochirality of both [(L)-EL⊂(M)-P[5]Q] and [(D)-EL⊂(P)-P[5]Q] was confirmed by the solid-state ECD spectral analysis. Typical antipodal ECD curves were observed (Fig. 4d) with Cotton effects at 321, 370, and 520 nm, respectively.
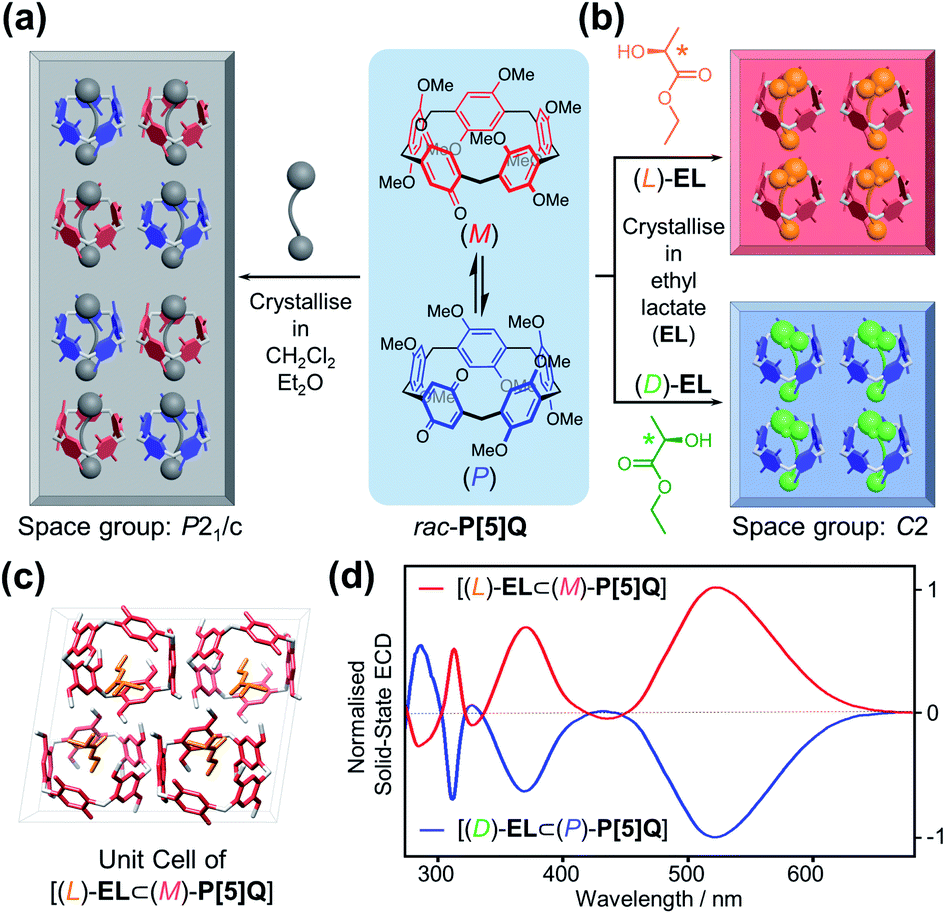 | ||
| Fig. 4 Schematic representation of rac-P[5]Q crystallising in (a) achiral solvents, forming a racemic compound, and (b) chiral solvents, leading to the exclusive formation of homochiral crystals. (c) Unit cell of [(L)-EL⊂(M)-P[5]Q]. (d) Normalised solid-state ECD spectra of [(L)-EL⊂(M)-P[5]Q] and [(D)-EL⊂(P)-P[5]Q] crystals. | ||
In order to further explore the chiral induction effects of (D)/(L)-EL as both crystallisation solvents and chiral guests, as well as devising a scalable method to produce P[5]-based homochiral crystalline materials, rac-P[5]HQ solids were stirred in (D)/(L)-EL aqueous solutions with different concentrations ranging from 0.1 to 4.5 M under ambient conditions for 24 h. The crystalline samples were tested by solid-state ECD (Fig. S16, ESI†). The ECD signal intensity increased with the concentration of the EL solution up to 2.6 M. Beyond this, the magnitude of the Cotton effects plateaued, indicating that the maximum (≈100%) CEE value was already reached. Analogous experiments were also conducted for P[5]Q, which was stirred in (D)/(L)-EL aqueous solutions with different concentrations ranging from 0.5 to 5.0 M. The ECD signal intensity increased with the concentration of the EL solution up to 4.0 M (L)-EL aqueous solutions for 24 h. Interestingly, all samples showed obvious ECD signals with consistent Cotton effect signs as those crystalline samples directly crystallised in pure (D)/(L)-EL, confirming the formation of [(L)-EL⊂(M)-P[5]HQ] and [(D)-EL⊂(P)-P[5]HQ] complexes. Taking advantage of this approach, both rac-P[5]Q and rac-P[5]HQ can be selectively turned into homochiral crystals of either handedness depending on the choice of the (D)/(L)-EL aqueous solutions (Fig. 5). These facile and green processes can be repeated for many rounds without loss of fidelity.
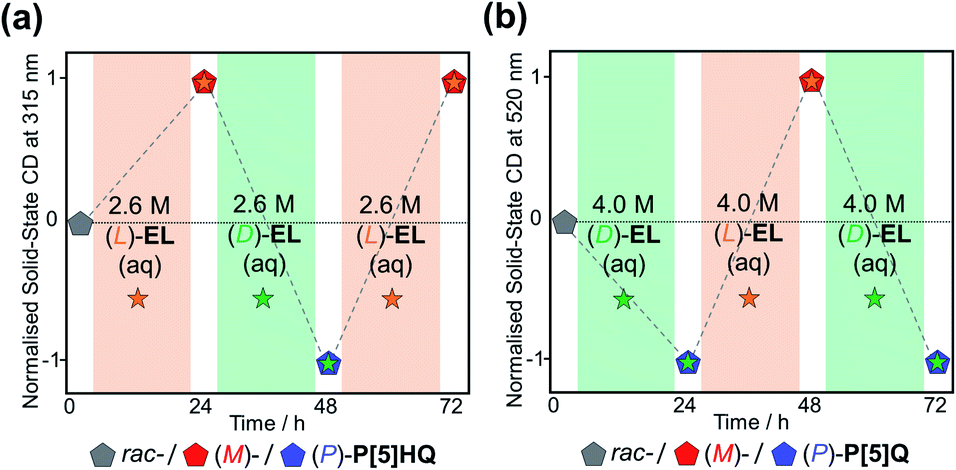 | ||
| Fig. 5 Chiral induction and inversion of (a) P[5]HQ and (b) P[5]Q in (D)- and /(L)-EL aqueous solutions, monitored by the changes of ECD signals at 315 and 520 nm, respectively. | ||
Conclusions
In summary, we have successfully demonstrated that scalemic mixtures or enantiopure compounds of stereolabile pillar[5]arene derivatives could be obtained through crystallisation, with and without the aid of chiral substances. A stereolabile pillar[5]arene derivative P[5]HQ that forms conglomerates in the solid state was resolved by triage, and its crystal enantiomeric excess could be enhanced through attrition-enhanced chiral symmetry breaking. Chiroptical properties of the resulting crystalline solids were characterised qualitatively by solid-state electronic circular dichroism spectroscopy. A versatile chiral induction approach, employing ethyl D/L-lactate as the chiral source, was also proved to be highly effective. Both X-ray crystallography and 1H NMR spectroscopy confirmed that these optically-active solvents transfer chiral information to the pillar[5]arene-based macrocycles through host–guest interactions. This method was not only applied to P[5]HQ, which forms conglomerates in the solid state, but also to a racemic compound P[5]Q. Overall, this work provides new strategies to manipulate the handedness of inherently chiral pillar[5]arene macrocycles. These pillararene-based chiral crystalline materials will be of interest for further studies of symmetry breaking during crystallisation,28 new routes to absolute asymmetric synthesis,14 and their chiroptical, pyroelectric, or piezoelectric properties.29Author contributions
A. C. H. S and C. Z. identified the pillararene-based conglomerates. T. U. T. and H. W. synthesised all compounds employed in this study and performed the experiments. W. Y. and C. Z. solved the crystal structures. K. K. B. performed the simulation of ECD spectra. A. C. H. S. conceptualised the study. A. C. H. S., H. W., C. Z. and T. U. T wrote the manuscript. All authors read and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the 973 National Basic Research Program of China (2015CB856500) and the National Science Foundation of China (21871208). The authors thank Professor Jay Siegel of Tianjin University for helpful discussions.Notes and references
- IUPAC Compendium of Chemical Terminology, http://goldbook.iupac.org, accessed April 2021 Search PubMed.
- H. D. Flack, Acta Crystallogr., 2009, 65, 371–389 CrossRef CAS PubMed.
- (a) L. Pasteur, Ann. Chim. Phys., 1848, 24, 442–459 Search PubMed; (b) G. B. Kauffman and R. D. Myers, J. Chem. Educ., 1975, 52, 777 CrossRef CAS; (c) Y. Tobe, Mendeleev Commun., 2003, 13, 93–94 CrossRef.
- J. Jacques, A. Collet and S. H. Wilen, Enantiomers, Racemates, and Resolutions, John Wiley & Sons, New York, 1981 Search PubMed.
- C. Wolf, Dynamic Stereochemistry of Chiral Compounds, The Royal Society of Chemistry, Cambridge, 2007 Search PubMed.
- D. K. Kondepudi, R. J. Kaufman and N. Singh, Science, 1990, 250, 975–976 CrossRef CAS PubMed.
- (a) T. Ogoshi, S. Kanai, S. Fujinami, T.-a. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed; (b) T. Ogoshi, Pillararene, The Royal Society of Chemistry, Cambridge, 2016 Search PubMed.
- Although planar chirality stereochemical descriptors pR/pS are commonly used in the pillararene chemistry community, here we adopted the P/M notations for inherently chiral macrocycles. See A. Szumna, Chem. Soc. Rev., 2010, 39, 4274–4285 RSC.
- (a) T. Ogoshi, K. Kitajima, T. Aoki, S. Fujinami, T.-a. Yamagishi and Y. Nakamoto, J. Org. Chem., 2010, 75, 3268–3273 CrossRef CAS PubMed; (b) N. L. Strutt, S. T. Schneebeli and J. F. Stoddart, Supramol. Chem., 2013, 25, 596–608 CrossRef CAS; (c) K. Du, P. Demay-Drouhard, K. Samanta, S. Li, T. U. Thikekar, H. Wang, M. Guo, B. van Lagen, H. Zuilhof and A. C.-H. Sue, J. Org. Chem., 2020, 85, 11368–11374 CrossRef CAS PubMed.
- (a) T. Ogoshi, K. Masaki, R. Shiga, K. Kitajima and T.-a. Yamagishi, Org. Lett., 2011, 13, 1264–1266 CrossRef CAS PubMed; (b) N. L. Strutt, D. Fairen-Jimenez, J. Iehl, M. B. Lalonde, R. Q. Snurr, O. K. Farha, J. T. Hupp and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 17436–17439 CrossRef CAS PubMed; (c) T. F. Al-Azemi, M. Vinodh, F. H. Alipour and A. A. Mohamod, Org. Chem. Front., 2019, 6, 603–610 RSC.
- (a) T. Ogoshi, D. Yamafuji, T. Aoki, K. Kitajima, T.-a. Yamagishi, Y. Hayashi and S. Kawauchi, Chem.–Eur. J., 2012, 18, 7493–7500 CrossRef CAS PubMed; (b) S.-H. Li, H.-Y. Zhang, X. Xu and Y. Liu, Nat. Commun., 2015, 6, 7590–7596 CrossRef PubMed; (c) W.-J. Li, Q. Gu, X.-Q. Wang, D.-Y. Zhang, Y.-T. Wang, X. He, W. Wang and H.-B. Yang, Angew. Chem., Int. Ed., 2021, 60, 9507–9515 CrossRef CAS PubMed.
- M. Reist, B. Testa, P.-A. Carrupt, M. Jung and V. Schurig, Chirality, 1995, 7, 396–400 CrossRef CAS.
- The racemisation half-lives of pillar[5]arene derivatives studied in ref. 9 are all in the range of milliseconds. Therefore, their chiral resolution is not feasible through conventional chromatographic techniques. An example of successful resolution of a A1/A2-disubstituted pillar[5]arene derivative and its detailed racemisation kinetic analysis can be found here: C. Xiao, W. Liang, W. Wu, K. Kanagaraj, Y. Yang, K. Wen and C. Yang, Symmetry, 2019, 11, 773–786 CrossRef CAS.
- M. Sakamoto and H. Uekusa, Advances in Organic Crystal Chemistry, Springer, Singapore, 2020 Search PubMed.
- I. Nierengarten, S. Guerra, H. Ben Aziza, M. Holler, R. Abidi, J. Barberá, R. Deschenaux and J.-F. Nierengarten, Chem.–Eur. J., 2016, 22, 6185–6189 CrossRef CAS PubMed.
- (a) C. Viedma, Phys. Rev. Lett., 2005, 94, 065504.1–065504.4 CrossRef PubMed; (b) L.-C. Sögütoglu, R. R. E. Steendam, H. Meekes, E. Vlieg and F. P. J. T. Rutjes, Chem. Soc. Rev., 2015, 44, 6723–6732 RSC; (c) C. Viedma, Cryst. Growth Des., 2007, 7, 553–556 CrossRef CAS; (d) R. Sivakumar, J. Kwiatoszynski, A. Fouret, T. P. T. Nguyen, P. Ramrup, P. S. M. Cheung, P. Cintas, C. Viedma and L. A. Cuccia, Cryst. Growth Des., 2016, 16, 3573–3576 CrossRef CAS; (e) J. E. Hein, B. H. Cao, C. Viedma, R. M. Kellogg and D. G. Blackmond, J. Am. Chem. Soc., 2012, 134, 12629–12636 CrossRef CAS PubMed.
- (a) E. L. Eliel and S. H. Wilen, Stereochemistry of Organic Compounds, John Wiley & Sons, New York, 1994 Search PubMed; (b) L. Pérez-García and D. B. Amabilino, Chem. Soc. Rev., 2007, 36, 941–967 RSC; (c) L. Pérez-García and D. B. Amabilino, Chem. Soc. Rev., 2002, 31, 342–356 RSC.
- (a) H. D. Flack, Helv. Chim. Acta, 2003, 86, 905–921 CrossRef CAS; (b) C. Dryzun and D. Avnir, Chem. Commun., 2012, 48, 5874–5876 RSC.
- C. Han, Z. Zhang, G. Yu and F. Huang, Chem. Commun., 2012, 48, 9876–9878 RSC.
- (a) R. Kuroda and T. J. C. Honma, Chirality, 2000, 12, 269–277 CrossRef CAS PubMed; (b) L. Ding, L. Lin, C. Liu, H. Li, A. Qin, Y. Liu, L. Song, H. Zhang, B. Z. Tang and Y. Zhao, New J. Chem., 2011, 35, 1781–1786 RSC; (c) E. Castiglioni, S. Abbate, G. Longhi and R. Gangemi, Chirality, 2007, 19, 491–496 CrossRef CAS PubMed; (d) M. Minguet, D. B. Amabilino, K. Wurst and J. Veciana, J. Chem. Soc., Perkin Trans. 2, 2001, 2, 670–676 RSC.
- J. Gal, Nat. Chem., 2017, 9, 604–605 CrossRef CAS PubMed.
- As a combined result of the difference in optical purity of the crystalline samples, the homogeneity of the KBr pellets, and the error in the weighing process, each sample measured showed different ECD signal levels. For better representation, the ECD spectra shown in the main text were linearly normalised by setting the most intense peaks to 1 or −1.
- C. Viedma, J. E. Ortiz, T. de Torres, T. Izumi and D. G. Blackmond, J. Am. Chem. Soc., 2008, 130, 15274–15275 CrossRef CAS PubMed.
- D. T. McLaughlin, T. P. T. Nguyen, L. Mengnjo, C. Bian, Y. H. Leung, E. Goodfellow, P. Ramrup, S. Woo and L. A. Cuccia, Cryst. Growth Des., 2014, 14, 1067–1076 CrossRef CAS.
- (a) M. Björemark, J. Jönsson and M. Håkansson, Chem.–Eur. J., 2015, 21, 10630–10633 CrossRef PubMed; (b) A. Lennartson, S. Olsson, J. Sundberg and M. Håkansson, Angew. Chem., Int. Ed., 2009, 48, 3137–3140 CrossRef CAS PubMed; (c) A. Lennartson and M. Håkansson, Angew. Chem., Int. Ed., 2009, 48, 5869–5871 CrossRef CAS PubMed.
- (a) W. L. Noorduin, P. van der Asdonk, A. A. Bode, H. Meekes, W. J. Van Enckevort, E. Vlieg, B. Kaptein, M. van der Meijden, R. M. Kellogg and G. Deroover, Org. Process Res. Dev., 2010, 14, 908–911 CrossRef CAS; (b) W. Maarten, M. van der Meijden, M. Leeman, E. Gelens, W. L. Noorduin, H. Meekes, W. J. van der Enckevort, B. Kaptein, E. Vlieg and R. M. Kellogg, Org. Process Res. Dev., 2009, 13, 1195–1198 CrossRef.
- (a) H. Zhu, Q. Li, Z. Gao, H. Wang, B. Shi, Y. Wu, L. Shangguan, X. Hong, F. Wang and F. Huang, Angew. Chem., Int. Ed., 2020, 59, 10868–10872 CrossRef CAS PubMed; (b) Y. Chen, L. Fu, B. Sun, C. Qian, R. Wang, J. Jiang, C. Lin, J. Ma and L. Wang, Org. Lett., 2020, 22, 2266–2270 CrossRef CAS PubMed; (c) J. Ji, Y. Li, C. Xiao, G. Cheng, K. Luo, Q. Gong, D. Zhou, J. J. Chruma, W. Wu and C. Yang, Chem. Commun., 2020, 56, 161–164 RSC; (d) Y. Chen, L. Fu, B. Sun, C. Qian, S. Pangannaya, H. Zhu, J. Ma, J. Jiang, Z. Ni, R. Wang, X. Lu and L. Wang, Chem.–Eur. J., 2021, 27, 5890–5896 CrossRef CAS PubMed.
- G. Coquerel and M. Hoquante, Symmetry, 2020, 12, 1796–1812 CrossRef CAS.
- P. Yuriy, Polar Crystals, LAP Lambert Academic Publishing, Germany, 2014 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and additional data of the synthesised compounds. CCDC 2073655, and 2073656. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02560k |
| This journal is © The Royal Society of Chemistry 2021 |