
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle-atom nickel terminating sp2 and sp3 nitride in polymeric carbon nitride for visible-light photocatalytic overall water splitting†
Yanrui
Li
a,
Yiqing
Wang
a,
Chung-Li
Dong
 b,
Yu-Cheng
Huang
b,
Jie
Chen
c,
Zhen
Zhang
a,
Fanqi
Meng
d,
Qinghua
Zhang
d,
Yiliang
Huangfu
a,
Daming
Zhao
a,
Lin
Gu
d and
Shaohua
Shen
*a
b,
Yu-Cheng
Huang
b,
Jie
Chen
c,
Zhen
Zhang
a,
Fanqi
Meng
d,
Qinghua
Zhang
d,
Yiliang
Huangfu
a,
Daming
Zhao
a,
Lin
Gu
d and
Shaohua
Shen
*a
aInternational Research Center for Renewable Energy, State Key Laboratory of Multiphase Flow in Power Engineering, Xi'an Jiaotong University, Xi'an 710049, China. E-mail: shshen_xjtu@mail.xjtu.edu.cn
bDepartment of Physics, Tamkang University, 151 Yingzhuan Rd, New Taipei City 25137, Taiwan
cDivision of Physical Science and Engineering, King Abdullah University of Science and Technology, Thuwal, 23955-6900, Saudi Arabia
dBeijing National Laboratory for Condensed Matter Physics, Institute of Physics, Chinese Academy of Sciences, Beijing, 100190, China
First published on 26th January 2021
Abstract
Polymeric carbon nitride (PCN) has been widely used as a metal-free photocatalyst for solar hydrogen generation from water. However, rapid charge carrier recombination and sluggish water catalysis kinetics have greatly limited its photocatalytic performance for overall water splitting. Herein, a single-atom Ni terminating agent was introduced to coordinate with the heptazine units of PCN to create new hybrid orbitals. Both theoretical calculation and experimental evidence revealed that the new hybrid orbitals synergistically broadened visible light absorption via a metal-to-ligand charge transfer (MLCT) process, and accelerated the separation and transfer of photoexcited electrons and holes. The obtained single-atom Ni terminated PCN (PCNNi), without an additional cocatalyst loading, realized efficient photocatalytic overall water splitting into easily-separated gas-product H2 and liquid-product H2O2 under visible light, with evolution rates reaching 26.6 and 24.0 μmol g−1 h−1, respectively. It was indicated that single-atom Ni and the neighboring C atom served as water oxidation and reduction active sites, respectively, for overall water splitting via a two-electron reaction pathway.
Introduction
Solar-driven water splitting based on semiconductors is a fascinating technology for converting earth-abundant solar radiation energy into clean and renewable hydrogen fuel.1,2 In the past few decades, numerous semiconductors including metal oxides, sulfides and nitrides have been developed for efficient photocatalytic hydrogen evolution in the presence of sacrificial reagents as hole scavengers.3–5 Photocatalytic overall water splitting using a particulate photocatalyst in the absence of sacrificial reagents serves as a simpler, more cost-effective approach for large-scale solar-fuel conversion, but still suffers from low efficiency. Up to now, some metal-based semiconductor particulates have been developed for photocatalytic overall water splitting.6–8 For example, Takata et al. achieved a quantum efficiency of almost unity for overall water splitting over SrTiO3,9 which is the highest value of photocatalytic water splitting reported so far, but is still limited in the ultraviolet region. Other than these well-investigated metal-based semiconductors, significant progress has also been achieved in metal-free polymeric semiconductors.10 Among them, polymeric carbon nitride (PCN) has attracted the greatest attention because of its low cost, facile preparation and environmental benignity,11,12 which could be activated for overall water splitting via visible light photocatalysis.13,14 However, PCN still suffers from fast charge carrier recombination and insufficient driving force for photoexcited electron–hole separation, resulting in relatively poor photocatalytic activity for overall water splitting. It is therefore important to explore strategies and methodologies to prompt the charge carrier kinetics of PCN photocatalysts.PCN possesses a molecular structure of 1D heptazine-based polymer chains conjugated by hydrogen bonding in the planar arrangement, with 2D melon-based sheets stacked along the c-direction with weak interlayer van der Waals forces.15 Upon photoexcitation, the electrons generated in the heptazine units could migrate to the extended heptazine rings in three ways including, interplanar migration along the π-stacking direction, transporting from one heptazine unit to another with different symmetric planes in the same polymer chains, or intrachain migration along the polymer chains via bridging N atoms (Fig. S1 in the ESI†).16,17 During migration, the photocarriers would be dissipated by electron–hole recombination during intrachain migration along the polymer chains, or the fast recombination of photoexcited electron–hole pairs confined in the heptazine units, or the interrupted charge transport by the hydrogen bonds. To overcome these limitations, representative solutions include the design of plane heterostructural carbon ring (Cring)–C3N4 nanosheets to build an in-built electric field for fast spatial transfer of photoexcited electrons18 and the integration of dual co-catalysts to spatially separate H2 and O2 evolution reactions.19
Recently, constructing atomically dispersed active sites on PCN has been considered as an ideal modification strategy by bringing together the fine-tunable molecular structure and the single-atom active sites of PCN to promote charge transfer and accelerate surface reactions.20,21 For instance, Wei et al. confined single Co1–P4 sites on the PCN matrix by a facile phosphidation method.22 The photoexcited charges generated in the conjugate ring could be rapidly separated by the neighboring Co1–P4 active sites and the carrier lifetime was prolonged by about 20 times relative to pristine PCN. Xiong et al. designed PCN–Pt2+ coordination by mixing dicyandiamide and H2PtCl6via one-step thermal polymerization. This coordination unit would act as a new light absorption center through a metal-to-ligand charge transfer (MLCT) process, resulting in the improved visible-near-infrared photocatalytic performance.23 Wu et al. anchored single-atom Pt onto PCN via a mild liquid-phase route by mixing dicyandiamide and H2PtCl6 in solution. It was demonstrated that the intrinsic change of the surface trap states of PCN induced by the single Pt atoms enhanced the photocatalytic water reduction performance.24 Note that the thermal polymerization of nitrogen-rich precursors for the synthesis of PCN will produce intermediates such as the heptazine ring,25 melem26 and low-dimensional C–N polymers,27 which all possess nitrogen with lone-pair electrons (see the polymerization pathway in Fig. S2 in the ESI†). During the thermal polymerization process, metal atoms could coordinate with these intermediates containing sp2 or sp3 hybridized N atoms,25,28,29 which may alter the stacking mode of conjugate rings in PCN and manipulate the molecular and electronic structures.23,30,31 Therefore, it is essential to investigate the chemical coordination between single metal atoms and intermediates during the thermal polymerization process, which could regulate the fine structure of the as-synthesized PCN, with an engineered band structure for extended light absorption and coordinated active sites for promoted charge transfer and catalytic reactions. High photocatalytic performances can thus be anticipated.
Bearing the favorable coordination between metal atoms and polymerization intermediates, herein, we utilized single-atom Ni as a terminating agent to coordinate with triangular edge (sp2) nitrogen and amino groups/bridge (sp3) nitrogen in heptazine units during the polymerization reaction for PCN synthesis. The single-atom Ni terminated PCN (PCNNi), without an additional cocatalyst loading, exhibited much increased photocatalytic activity for overall water splitting, with gas-product H2 and liquid-product H2O2 generation rates reaching 26.6 and 24.0 μmol g−1 h−1, respectively, under visible light irradiation (λ > 420 nm). It was demonstrated that the hybridization of single-atom Ni with the aromatic conjugated ring created a new highest occupied molecular orbital (HOMO) and new lowest unoccupied molecular orbital (LUMO) in PCNNi, synergistically broadening the optical absorption range via the MLCT process and accelerating the separation and transfer of photoexcited electrons and holes generated in the heptazine units. Theoretical calculations and electrochemical experiments further revealed that the single-atom Ni and the neighboring C atom served as oxidation and reduction active sites for overall water splitting into H2O2 and H2, respectively, through a two-electron reaction pathway. This study presents an alternative approach to engineer the atomic and electronic structures of PCN for great performance enhancement in photocatalytic overall water splitting, with liquid H2O2 and gaseous H2 generated as easily-separated products, rather than the explosive gaseous mixture of O2 and H2.
Results and discussion
Morphological and structural properties of PCNNi
It has been well documented that three kinds of nitrogen atom exist in the heptazine units of PCN, i.e., triangular edge nitrogen (NA), amino groups/bridge nitrogen (NB) and central tertiary nitrogen (NC) as described in Scheme 1a. Both sp2 NA and sp3 NB possess lone-pair electrons, providing coordination sites to confine metal atoms.32 It could be proposed that NA atoms in the aromatic conjugate rings are coordinated by Ni atoms (Scheme 1b, coordination structure I). These Ni atoms exist between the melon polymer chains, breaking the hydrogen bonding (N–H⋯N) formed by the terminal –NH2 or –NH– groups (Scheme 1c and d). Meanwhile, Ni atoms also coordinate with NB atoms in the heptazine units (Scheme 1b, coordination structure II), terminating amidogen polycondensation in the polymerization process thus interrupting the heptazine chains, which brings about numerous defective structures. To conclude, single-atom Ni could act as a terminating agent that coordinates with NA atoms to break the hydrogen bonding and with NB atoms to interrupt the polymerization process, which would trigger remarkable alterations in the molecular structure of PCN. In this study, a series of single-atom Ni coordinated PCN was synthesized by thermal polymerization of melamine and nickel chloride as precursors under a nitrogen atmosphere (see the Experimental section in the ESI†). The samples are named PCNNi-1, PCNNi-2, PCNNi-3, and PCNNi-4 with Ni contents determined to be 0.3 wt%, 0.5 wt%, 1.0 wt%, and 1.9 wt%, respectively, by inductively coupled plasma mass spectrometry. As shown in Table S1,† the Ni contents could be well controlled in PCNNi synthesized in different batches. Pristine PCN presents a bulky morphology (Fig. S3a†), similar to the previous reported PCN obtained from the thermal polymerization process.33 In comparison, all the PCNNi samples display nanosheet-like structures (Fig. 1a and S3b–d†), which should be due to the single-atom Ni interrupting the polymerization process to produce small and thin pieces of PCN. One can hardly observe any Ni-based nanoparticles in these PCNNi samples, even in PCNNi-4 with the maximum content of Ni (Fig. S4†), implying the possibility of single-atom Ni existing in the PCN matrix, which will be confirmed in the following sections.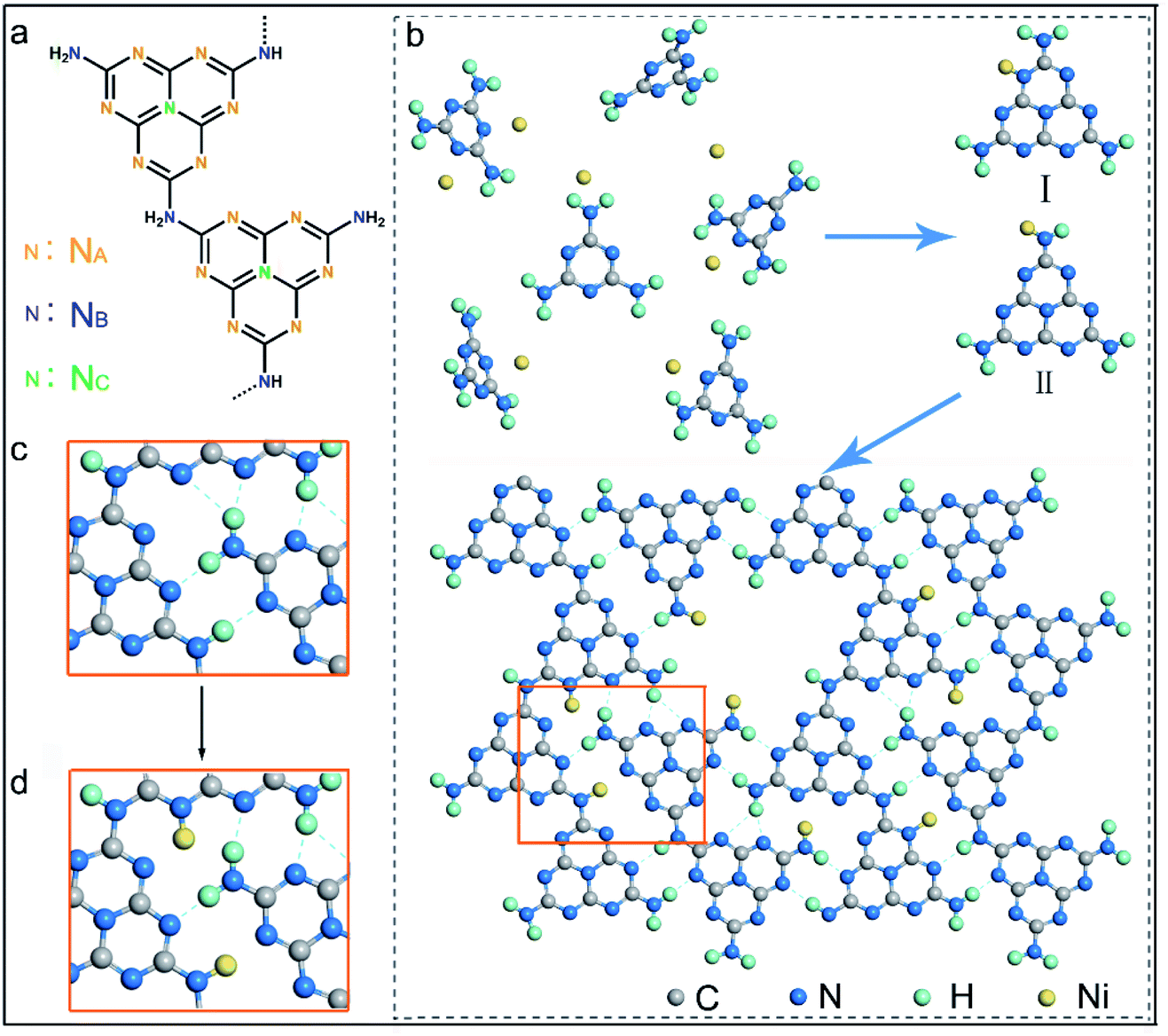 | ||
| Scheme 1 (a) Molecular structure of heptazine-based PCN. (b) Proposed schemes of thermal polymerization of heptazine units with Ni–N coordination. Regional schemes of (c) PCN with hydrogen bonding between terminal –NH2 and –NH– groups, and (d) PCN with broken hydrogen bonding with single-atom Ni as the terminating agent. | ||
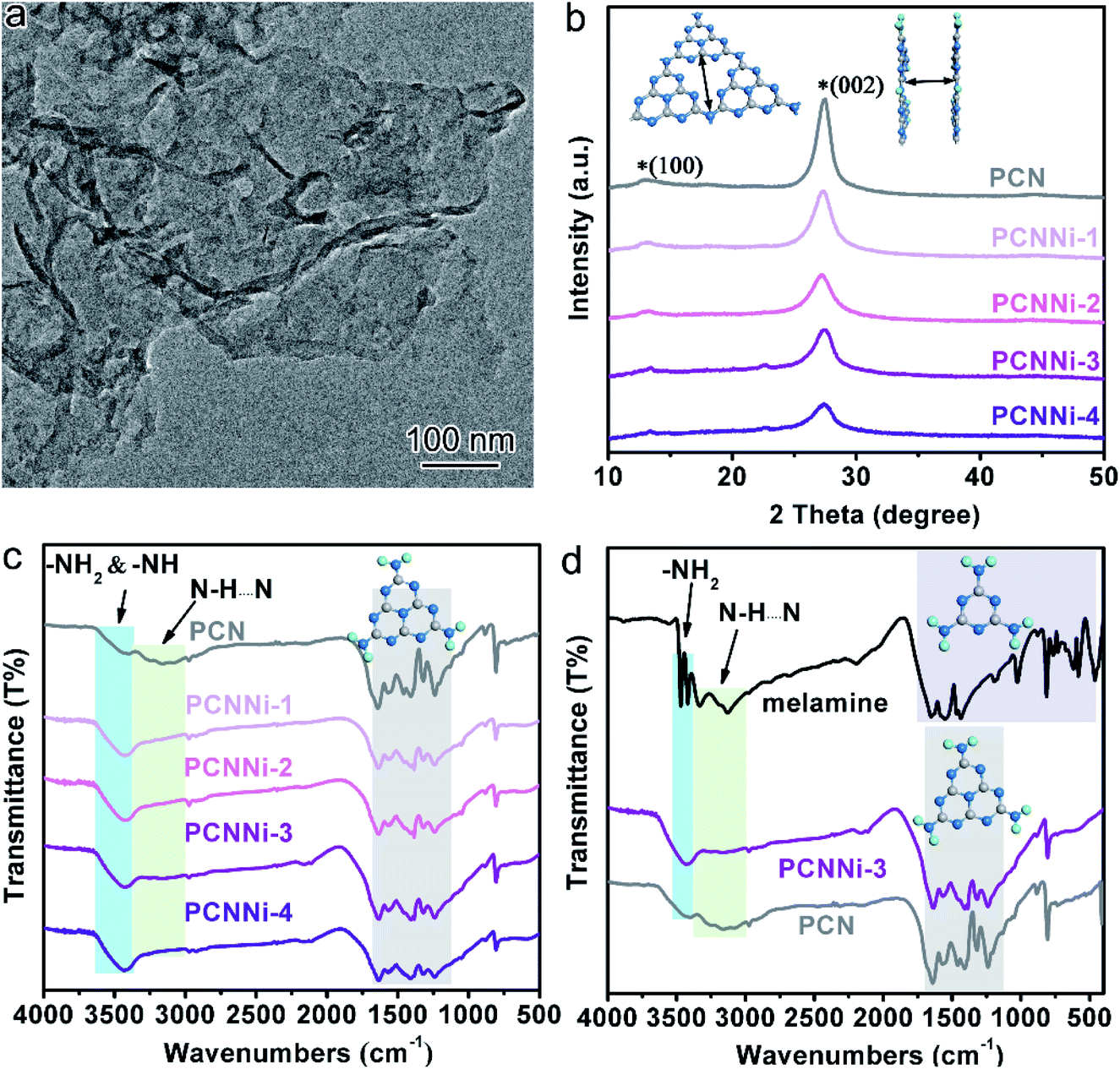 | ||
| Fig. 1 (a) TEM image of PCNNi-3. (b) XRD patterns of PCN and PCNNi with different Ni contents. (c) FTIR spectra of PCN and PCNNi with different Ni contents. (d) FTIR spectra of melamine, PCNNi-3 and PCN. | ||
The crystalline phases of the PCNNi samples were determined from the X-ray powder diffraction (XRD) pattern. As shown in Fig. 1b, both pristine PCN and PCNNi display two diffraction peaks at 2θ = 13.1° and 27.3°, corresponding to the (100) and (002) planes of PCN, which should be related to the in-plane structural repeating units of heptazine and the interlayer stacking of the conjugated aromatic rings for the graphitic-like structure of PCN, respectively.34 Noteworthily, both the diffraction peaks show gradually decreased intensity with the increasing Ni contents in PCNNi. Furthermore, as presented in Table S2,† the full width at half maximum (FWHM) of the peak at 2θ = 27.3° increases from 1.86° for PCN to 2.36° for PCNNi-4, indicating the loss of the ordered structure in PCN. No peak assigned to Ni-based species is observed in PCNNi, again supporting the possible existence of single-atom Ni. The thermal stability of PCNNi was then examined by thermogravimetric analysis (TGA) under nitrogen (Fig. S5†). It is observed that the onset temperature of mass loss decreases from 480 °C for pristine PCN to 380 °C for PCNNi-4. Interestingly, the onset temperature is proportional to the increasing Ni contents in PCNNi. This reduced thermal stability should be related to the increasing ratio of small and thin pieces of PCN with a disordered structure, which were created during the polymerization process interrupted by the single-atom Ni terminating agent. Fourier transform infrared (FT-IR) spectroscopy was further conducted to investigate the influence of the single-atom Ni terminating agent on the thermal polymerization process and on the molecular structure of PCN. As shown in Fig. 1c, the characteristic bands in the region of 1100–1600 cm−1 belonging to the fingerprint of heptazine rings are very similar for PCN and PCNNi,13 suggesting that the single-atom Ni as a terminating agent does not change the basic structure of the heptazine units during polymerization. However, the bands in the region of 3000–3400 cm−1 typical for hydrogen bonding display a progressive decrease in intensity, whilst the characteristic bands of NH2– or –NH– in the region of 3400–3600 cm−1 are gradually strengthened, with the increasing Ni contents in PCNNi. For more detailed analyzation, the FTIR spectrum of the melamine precursor is shown as reference (Fig. 1d).27 The bands in the region of 500–1800 cm−1 correspond to the fingerprint of melamine. The two sharp bands featuring in the region of 3400–3500 cm−1 (centered at 3428 cm−1 and 3482 cm−1) are assigned to the symmetric and asymmetric stretching of NH2–, respectively, which shows that plenty of amidogens exist in the melamine precursor. One will observe that these bands are stronger for PCNNi than PCN, with intensities increasing depending on the increasing Ni contents in PCNNi (Fig. 1c and d). This confirms our hypothesis that single-atom Ni coordinates with NB in heptazine units, interrupting the polymerization reaction with more NH2– or –NH– groups created in PCNNi. Hence, the N to C ratio of PCNNi was increased compared with that of PCN (Table S3†). Furthermore, the melamine precursor displays two obvious peaks in the region of 3000–3400 cm−1 (centered at 3134 cm−1 and 3327 cm−1), which is the typical indicator of hydrogen bonding (N–H⋯N) formed between melamine molecules. However, this fingerprint of hydrogen bonding in PCNNi is found to be much weaker than that of PCN (Fig. 1c and d), revealing that the hydrogen bonding is broken by the coordination between single-atom Ni and NA/NB atoms in heptazine units. To conclude, FTIR analysis provides direct evidence from the viewpoint of the molecular structure that single-atom Ni terminates sp2 and sp3 nitrogen with lone-pair electrons in the heptazine units during the thermal polymerization of the melamine precursor, leading to diminishing of hydrogen bonds between the neighboring melon chain and increasing residual NH2– or –NH– groups in PCN. As calculated from the nitrogen adsorption and desorption isotherms (Fig. S6†), the Brunauer–Emmett–Teller (BET) specific surface areas (SBET) were determined to be 5.1, 6.1, 7.3, 7.5 and 8.0 m2 g−1 for PCN, PCNNi-1, PCNNi-2, PCNNi-3 and PCNNi-4, respectively. This slight increase in SBET should be due to the small and thin pieces of PCN produced via interrupting the polymerization process by the single-atom Ni, which should not be responsible for the great improvement in the photocatalytic activity of PCNNi.
To evidence the atomic dispersion of Ni as speculated in previous analysis, atomic-resolution high-angle-annular-dark-field scanning transmission electron microscopy (HAADF-STEM) was conducted on PCNNi. The HAADF-STEM image clearly displays the isolated bright dots of atomic size in white as highlighted by the red circles (Fig. 2a), which can unambiguously determine the atomic distribution of Ni in PCN. A quantitative analysis of the Ni atom positions in the HAADF-STEM image indicates that the measured nearest-neighbor distances of the bright dots are in accordance with the Rayleigh distribution for a random distribution of single-atom Ni over the support of PCN. X-ray absorption structure spectroscopy (XAS) is sensitive to local atomic/electronic structures and provides atomic-level structural information. For more insights into the chemical and bonding situations of the atom-distributed Ni in PCNNi, X-ray absorption near-edge structure (XANES) spectra at the Ni K-edge were monitored over PCNNi along with Ni foil and NiO as references. As shown in Fig. 2b, the spectral profile of the Ni K-edge of PCNNi is apparently different from that of Ni or NiO. The shoulder-like feature at the K-edge has been interpreted as 1s to d transition, which is usually forbidden according to the Fermi golden rule. However, this transition will be allowed, if Ni 2p–3d rehybridization happens with energy band formation in Ni foil. Thus, the absence of the shoulder-like feature in PCNNi suggests that the band structure of single-atom Ni is different from that of Ni foil. Additionally, the intensity of the Ni K-edge of PCNNi is higher (lower) than that of Ni foil (NiO), revealing that the single-atom Ni possesses an oxidation state higher (lower) than that of metallic Ni (fully oxidized NiO) and holds more (less) unoccupied states than metallic Ni (fully oxidized NiO). Considering that Ni2+ in the NiCl2 precursor possesses a 3d8 electron configuration, which will coordinate with N atoms in heptazine units, electron transfer from N atoms to the unoccupied orbitals of Ni atoms19,20 gives rise to the reduced unoccupied orbitals of Ni in PCNNi, in comparison to NiO with 3d8 (4s2 electron transfers to oxygen sites) electron configuration. Note that metallic Ni has 3d84s2 electron configuration while NiO has 3d8 electron configuration; the single-atom Ni should possess a partially filled 4s orbital (Fig. 2b, inset), attributed to the electron transfer from neighboring N to Ni atoms. Fig. 2c shows the Fourier Transformed (FTs) amplitude of the extended X-ray absorption fine structure (EXAFS) of Ni K-edge spectra. Ni foil and NiO display strong peaks at 2.2 Å and 2.4 Å, attributed to the Ni–Ni bond and Ni–O bond, respectively. In comparison, both the peaks are invisible for PCNNi, implying the absence of Ni–O and Ni–Ni bonds. Yet, a peak located at about 1.6 Å assigned to the Ni–N bond is detected,35,36 again suggesting that the Ni species are well isolated as single-atom sites and coordinated with N atoms in the framework of PCN.
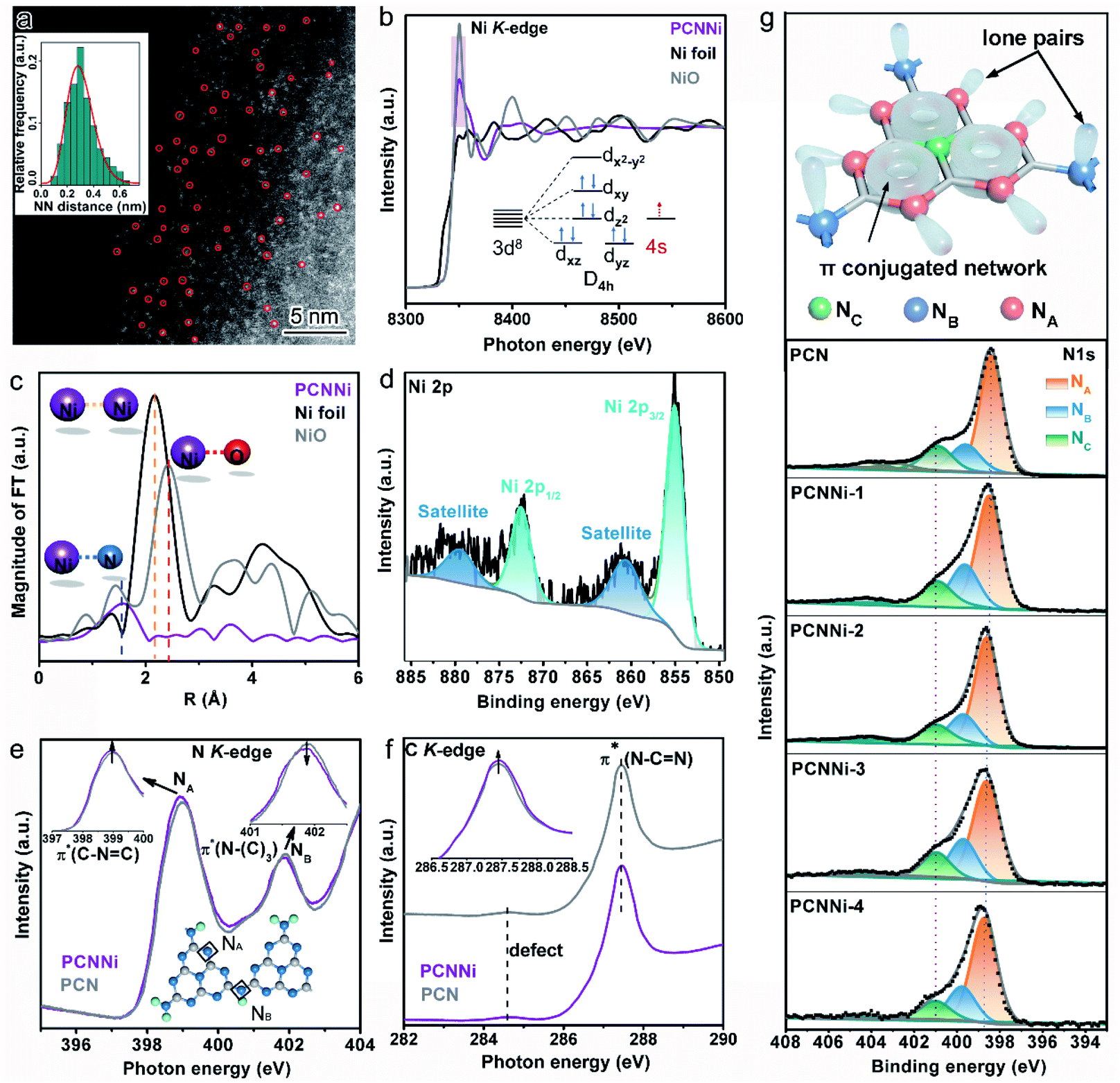 | ||
| Fig. 2 (a) HAADF-STEM image of PCNNi-3. (b) Ni K-edge XANES spectra of PCNNi-3 and PCN. The inset in (b) shows the outer electron configuration of single-atom Ni in PCNNi-3. (c) Fourier Transformed (FTs) amplitude of the extended X-ray absorption fine structure (EXAFS) of the Ni K-edge spectra for PCNNi-3, Ni foil and NiO. (d) XPS Ni 2p spectrum of PCNNi-3. (e) N K-edge and (f) C K-edge XANES spectra of PCNNi-3 and PCN. (g) Atomic model of the heptazine units in PCN, and N 1s XPS spectra of PCN and PCNNi. | ||
X-ray photoelectron spectroscopy (XPS) measurements were also conducted to investigate the valence state of single-atom Ni coordinated with PCN. It was previously recorded that the Ni 2p XPS peaks for Ni2+ in NiO are located at 855.5 eV and 873.6 eV.37,38 In comparison, these two Ni 2p XPS peaks identified for PCNNi (Fig. 2d and S7a†) are shifted to lower binding energies of 855.0 eV and 872.4 eV,39,40 respectively. Note that no XPS peak indexed to metallic Ni0 (853.2 eV) and NiN3 (852.7 eV) is observed.41 These comparative results confirm that the oxidation state of single-atom Ni sites in PCNNi is lower than that of Ni2+ in the fully oxidized NiO but higher than that of metallic Ni0, which well agrees with the Ni K-edge XANES analysis.
In addition to the characterization of the metal Ni site, XANES measurements on the non-metal N and C K-edge were carried out to monitor the local atomic and electronic structure changes of PCNNi vs. PCN. In the N K-edge XANES spectra (Fig. 2e), both PCNNi and PCN display two typical π* resonances at 399 eV and 402 eV, associated with the triangular edge nitrogen (NA, see the inset of Fig. 2e) and the bridge nitrogen (NB, see the inset of Fig. 2e), respectively.42 The NA peak intensity of PCNNi is stronger than that of PCN, due to the coordination between single-atom Ni and NA atoms.19 Thereafter, the more unoccupied 2p states at NA atoms and the greater π* characteristics, which could generally accelerate the electron transfer from NA to single-atom Ni and strengthen the hybridization of triangular C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and single-atom Ni in PCNNi, respectively, benefit photocatalysis. Meanwhile, the intensity of peak NB is decreased, suggesting the structure breaking of bridge nitrogen, caused by the introduction of the single-atom Ni terminating agent during the polymerization reaction.41 As the N 2p orbital is strongly hybridized with the C 2p orbital in the heptazine unit, complementary C K-edge spectra are provided to illustrate the hybridization of single-atom Ni with heptazine units (Fig. 2f). The overall spectral profile of PCNNi resembles that of PCN, indicating that the C atoms in the heptazine units are not disturbed by the single-atom Ni, which is consistent with the unchanged C 1s XPS spectra and (see the detailed analysis in Fig. S7b in the ESI†). Interestingly, the peak related to π*C–N–C at ca. 287.5 eV is higher in PCNNi than in PCN, indicating that the heptazine unit anchored with single-atom Ni has a greater π* characteristic, as evidenced in the N K-edge spectra. These analyses well support that single-atom Ni acts as a terminating agent to coordinate with N atoms with lone pair electrons in the heptazine units during the thermal polymerization of PCN, which agrees well with the FTIR results.
C and single-atom Ni in PCNNi, respectively, benefit photocatalysis. Meanwhile, the intensity of peak NB is decreased, suggesting the structure breaking of bridge nitrogen, caused by the introduction of the single-atom Ni terminating agent during the polymerization reaction.41 As the N 2p orbital is strongly hybridized with the C 2p orbital in the heptazine unit, complementary C K-edge spectra are provided to illustrate the hybridization of single-atom Ni with heptazine units (Fig. 2f). The overall spectral profile of PCNNi resembles that of PCN, indicating that the C atoms in the heptazine units are not disturbed by the single-atom Ni, which is consistent with the unchanged C 1s XPS spectra and (see the detailed analysis in Fig. S7b in the ESI†). Interestingly, the peak related to π*C–N–C at ca. 287.5 eV is higher in PCNNi than in PCN, indicating that the heptazine unit anchored with single-atom Ni has a greater π* characteristic, as evidenced in the N K-edge spectra. These analyses well support that single-atom Ni acts as a terminating agent to coordinate with N atoms with lone pair electrons in the heptazine units during the thermal polymerization of PCN, which agrees well with the FTIR results.
We further carried out N 1s XPS measurements to detect the local chemical environment of single-atom Ni and N atoms. The N 1s spectrum of PCN could be deconvoluted into three peaks at 398.4 eV, 399.6 eV, and 400.89 eV (Fig. 2g), which are assigned to triangular edge nitrogen (CN–C) (NA, see the inset of Fig. 2g), amino groups (C–NHx) (NB, see the inset of Fig. 2g) and central tertiary nitrogen (N–(C)3) (NC, see the inset of Fig. 2g), respectively.43,44 The N 1s spectra of PCNNi could be fitted with three peaks analogous to PCN, suggesting the well-preserved heptazine-based structure in PCNNi. According to the detailed assignment of the N 1s peak (Table S4†), both peak NA and peak NB shift toward higher binding energies, which indicates that the electron densities at NA and NB are reduced, likely due to charge transfer from N atoms to single-atom Ni.30 This result also confirms the coordination between single-atom Ni and N atoms (NA and NB) by Ni–N chemical bonding, in good agreement with the FTIR and XAS results. Furthermore, in the Solid-state 13C CP-MAS NMR spectra (Fig. S8†), the lower chemical shifts of PCNNi than PCN indicate the reduced electron densities of aromatic conjugated rings, which could again evidence the coordination between single-atom Ni and the heptazine network.
Detailed investigation into the chemical interaction between single-atom Ni and coordinated N sites in intermediate products obtained at different temperatures indicates that single-atom Ni coordinates with N sites in heptazine formed at the early stage of thermal polymerization (see Fig. S9 and S10 and related discussion in the ESI†).
Optical properties and band structure of PCNNi
The optical properties of the as-obtained PCNNi samples were determined using UV-visible diffuse reflectance spectra (DRS). As compared to pure PCN, the optical absorption of PCNNi redshifts to longer wavelengths with increasing Ni contents (Fig. 3a). The Tauc plots translated from UV-vis spectra by the Kubelka–Munk equation showed two band gaps at 2.73 and 2.3 eV for PCNNi, respectively (Fig. 3b). The bandgap of 2.73 eV is attributed to π → π* transition, i.e., band-to-band transition from the HOMO to LUMO in the conjugated electron system of PCN.45 The bandgap of 2.3 eV in PCNNi should be related to the Ni–N coordination structure, in which electron transition happens between the metal and ligand, i.e., the metal-to-ligand charge transfer (MLCT) process, with transition energy lower than that of band-to-band transition,46 contributing to optical absorption in visible light.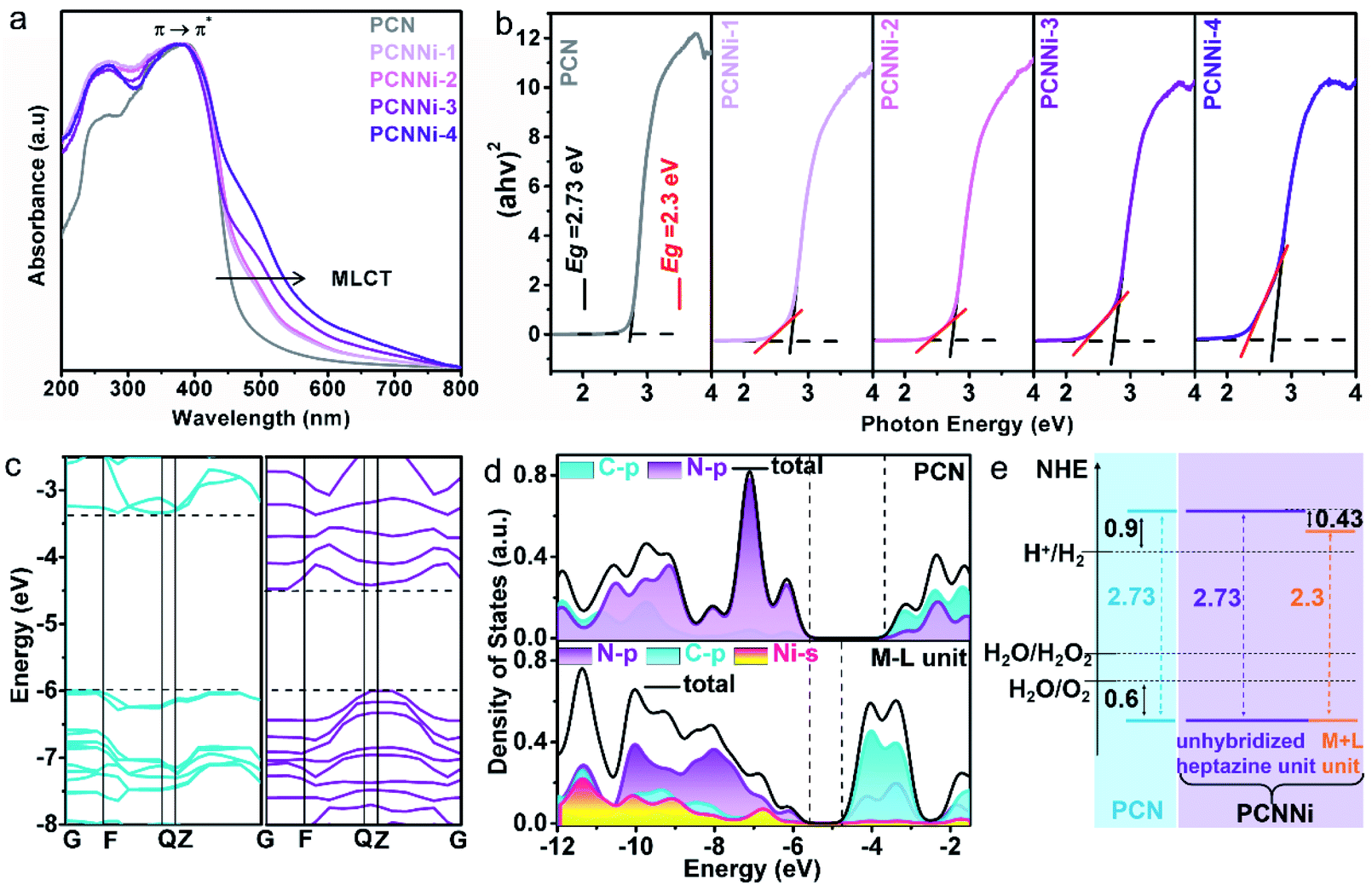 | ||
| Fig. 3 (a) UV-vis diffuse reflectance spectra of PCN and PCNNi. (b) Tauc plots of PCN and PCNNi calculated by the Kubelka–Munk method. DFT calculations of the (c) band structures of PCN (left) and the M–L unit (right) and the (d) projected density of states of PCN (top) and the M–L unit (down). (e) Band structure diagrams of PCN and PCNNi. | ||
The band structures and electronic structures of PCNNi were further explored using XPS VB spectra and density-functional theory (DFT) calculations. The HOMO tails of PCNNi ends at 0.8 eV (Fig. S11†), similar to that of pure PCN, indicating that the HOMO maximum remains unchanged with the Ni–N coordination. This result can be proved by the DFT calculation results. As shown in Fig. 3c, the new HOMO in PCNNi is at the same energy level as the HOMO of the pristine PCN. Interestingly, a new LUMO state in PCNNi is created below the original LUMO of the pristine PCN, which should be ascribed to the hybridization of single-atom Ni with the heptazine unit (namely, M–L unit). The densities of states (DOS) were calculated for the pure PCN and the M–L unit, as displayed in Fig. 3d. The LUMO of pure PCN is composed of the 2p hybridization states of C and N atoms, while the HOMO is dominated by the 2p states of N atoms. In contrast, the orbital distributions of PCNNi are significantly changed upon the coordination of single-atom Ni with the heptazine unit. For the M–L unit, the 4s orbitals of single-atom Ni and the 2p orbitals of N atoms are hybridized, which contributes to the new HOMO. The LUMO of the M–L unit is mainly composed of the C 2p state, while N 2p and Ni 4s states contribute very slightly. Based on the above analysis, the band structures of PCN and PCNNi could be schematically proposed in Fig. 3e. It was reported that PCN possesses the LUMO at a potential of ca. 0.9 eV higher than the H+/H2 redox potential, and the HOMO at a potential of ca. 0.6 eV lower than the H2O/O2 redox potential,47 giving rise to the bandgap of ∼2.73 eV. In comparison, a narrower bandgap of 2.3 eV was observed for PCNNi, implying that electron transition occurs between the new HOMO and LUMO of the M–L unit. Considering that the HOMO position of PCNNi was unchanged relative to PCN, the narrowed bandgap should have resulted from the downward shift of the LUMO by 0.43 eV. Although shifted, the LUMO and HOMO positions of PCNNi still guarantee enough potential to drive water reduction and oxidation reactions, respectively. Encouragingly, the built-in electric field created by the new LUMO of the M–L unit and the original LUMO of PCN would provide a driving force to steer the migration of photoexcited electrons from the unhybridized heptazine unit to the M–L unit.
Photocatalytic performance for overall water splitting into H2 and H2O2
The photocatalytic water splitting performances of the as-synthesized PCNNi samples were evaluated under visible light (λ > 420 nm) without any sacrificial reagents. As shown in Fig. 4a, the pristine PCN can hardly produce H2 from pure water under visible light. In comparison, PCNNi steadily produces H2 from pure water without loading additional cocatalysts. With the increasing Ni contents in PCNNi, the photocatalytic activity is gradually increased, with a highest H2 evolution rate of 26.65 μmol g−1 h−1 achieved over PCNNi-3. However, further increase in Ni contents would decrease the photocatalytic activity of PCNNi-4. The apparent quantum yields (AQY) of PCNNi-3 at 380 nm, 420 nm and 500 nm were examined to be 1.53%, 1.12% and 0.05%, respectively, which depends on the optical absorption profile of PCNNi (Fig. S12†). It is clear that PCNNi could photocatalyze overall water splitting at a longer wavelength of 500 nm, which indicates that the extended optical absorption should contribute to the improved photocatalytic activity. Interestingly, rather than O2, H2O2 was detected as the water oxidation product over PCNNi (see raw GC data in Fig. S13†), indicating that a two-electron process is responsible for pure water splitting (Fig. 4b). For all the PCNNi photocatalysts, the H2O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 molar ratios are measured to be ∼0.9:1, slightly smaller than the stoichiometric ratio of 1:1 for overall water splitting into H2O2 and H2, possibly due to the decomposition of H2O2 during the reaction.48 Moreover, PCNNi shows quite good photocatalytic stability for overall water splitting under visible light, with H2 and H2O2 evolution rates remaining almost unchanged during the 3-cycle (24 h) reaction (Fig. S14a and b†), which exhibits very good reproducibility as confirmed in different batches of PCNNi (Fig. 4b). The well maintained single-atom Ni–N coordination structure after the photocatalytic reaction (Fig. S14c and d†) further confirms the good chemical stability of PCNNi for photocatalytic overall water splitting. By carrying out rotating Pt disk–Pt ring electrodes (RRDE) measurements, the electron transfer number is calculated to be 2 for water oxidation (Fig. 4c), which well explains the production of H2O2 rather than O2 during photocatalytic water splitting over PCNNi. To reveal the great enhancement in photocatalytic performances for overall water splitting, the charge transfer processes are systematically investigated on PCNNi in the following sections.
:H2 molar ratios are measured to be ∼0.9:1, slightly smaller than the stoichiometric ratio of 1:1 for overall water splitting into H2O2 and H2, possibly due to the decomposition of H2O2 during the reaction.48 Moreover, PCNNi shows quite good photocatalytic stability for overall water splitting under visible light, with H2 and H2O2 evolution rates remaining almost unchanged during the 3-cycle (24 h) reaction (Fig. S14a and b†), which exhibits very good reproducibility as confirmed in different batches of PCNNi (Fig. 4b). The well maintained single-atom Ni–N coordination structure after the photocatalytic reaction (Fig. S14c and d†) further confirms the good chemical stability of PCNNi for photocatalytic overall water splitting. By carrying out rotating Pt disk–Pt ring electrodes (RRDE) measurements, the electron transfer number is calculated to be 2 for water oxidation (Fig. 4c), which well explains the production of H2O2 rather than O2 during photocatalytic water splitting over PCNNi. To reveal the great enhancement in photocatalytic performances for overall water splitting, the charge transfer processes are systematically investigated on PCNNi in the following sections.
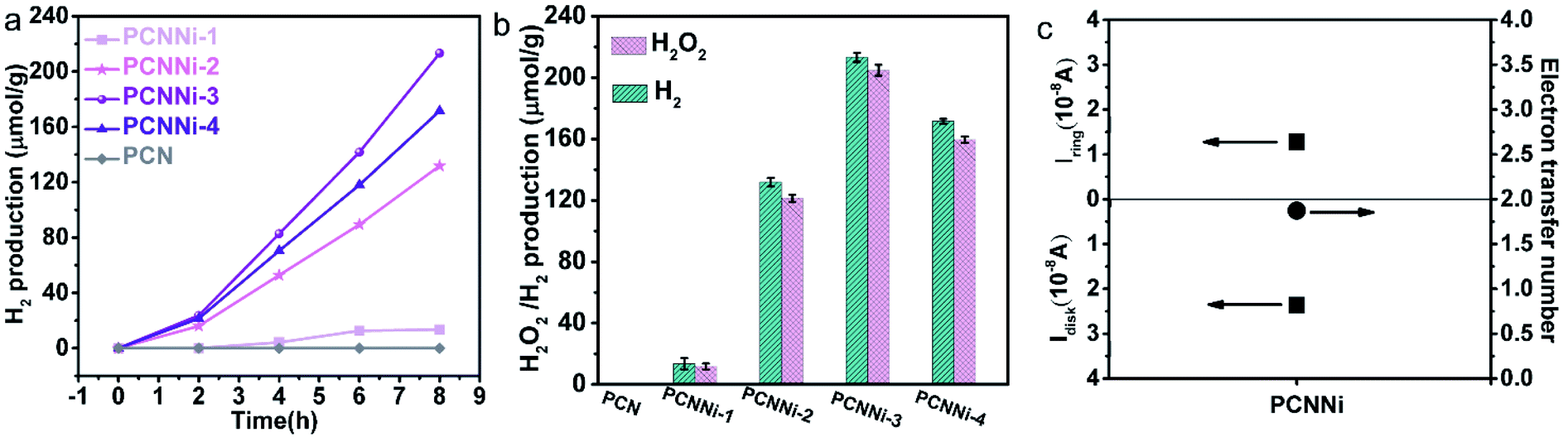 | ||
| Fig. 4 (a) Photocatalytic hydrogen generation from pure water under visible light (λ > 420 nm), (b) photocatalytic overall water splitting into hydrogen and hydrogen peroxide without any sacrificial reagent under visible light (λ > 420 nm) with activity marked by the error bar, (c) the measured Pt disk current and Pt exchange current and corresponding electron transfer number. | ||
Mechanism analysis of photocatalytic overall water splitting
As previously reported for the band structure of PCN,49 C–N sp3 hybridization gives rise to high energy σ and σ* molecular orbitals, while C–N sp2 hybridization constitutes a conjugated network, forming low energy π bonding and π* antibonding orbitals (Fig. 5a, left). Besides, the unbonded lone pairs (LP) of electrons on pyridinic N atoms create a LP orbital above the π bonding orbital (Fig. 5a, left). As shown in Fig. S15,† PCN shows strong photoluminescence (PL) emission at ca. 460 nm, which is related to the electron transition between the LP orbital and π* antibonding orbital. In comparison, all the PCNNi samples show a significant decrease in PL emission intensity, which implies the inhibited charge carrier recombination in PCNNi with the introduction of single-atom Ni. Interestingly, PCNNi has PL emissions located at almost the constant position (Fig. 5b, taking PCCNi-3 as an example), when excited by photons with different energies higher than the measured bandgap of PCN (e.g., light with wavelengths varying from 310 nm to 370 nm). This observation suggests that excited electrons in PCNNi would relax from σ* orbitals to the same π* antibonding orbital through intersystem crossing (ISC).49 Hence, similar to PCN, PCNNi has PL emission originating from the radiative recombination of electrons in the π* antibonding orbital and holes in the LP orbital (Fig. 5a, left). As previously analyzed (Fig. 3c–e), the M–L unit creates a new LUMO in the band structure of PCNNi with the introduction of the single-atom Ni terminating agent (Fig. 5a, right). It could then be deduced that the new LUMO could act as the trap site to capture electrons from the unhybridized heptazine unit, resulting in inhibited charge carrier recombination, which could be well evidenced by the decreased PL emission of PCNNi (Fig. S15†). As further evidenced in electrochemical impedance spectroscopy (EIS), PCNNi exhibits much decreased charge-transfer resistances, as compared with PCN, suggesting that the single-atom Ni could greatly improve charge transfer ability for the surface water redox reaction, especially for PCNNi-3 (Fig. S16†). Furthermore, the time-resolved PL spectra (Fig. 5c) demonstrate that PCNNi-3 possesses an average carrier lifetime (5.922 ns) much longer than that of PCN (1.759 ns). This again proves the inhibited charge recombination and then the improved charge transfer ability, due to the migration of electrons from σ* and π* antibonding orbitals of the unhybridized heptazine unit to the new LUMO created in the M–L unit. In addition, a low-energy tail was observed in the PL curve of PCNNi, and the peak can be fitted with three Gaussian peaks centered at 460 nm, 480 nm, and 525 nm (Fig. 5b, inset). Considering the absorption spectrum with photoexcitations of 2.73 eV and 2.3 eV, the emission peaks at 460 nm and 525 nm should originate from the electron transitions of the π* → LP orbital in the heptazine unit and the LUMO → HOMO in the M–L unit, respectively. The emission peak at 480 nm may be related to other mid-states due to the structural defect in PCNNi.47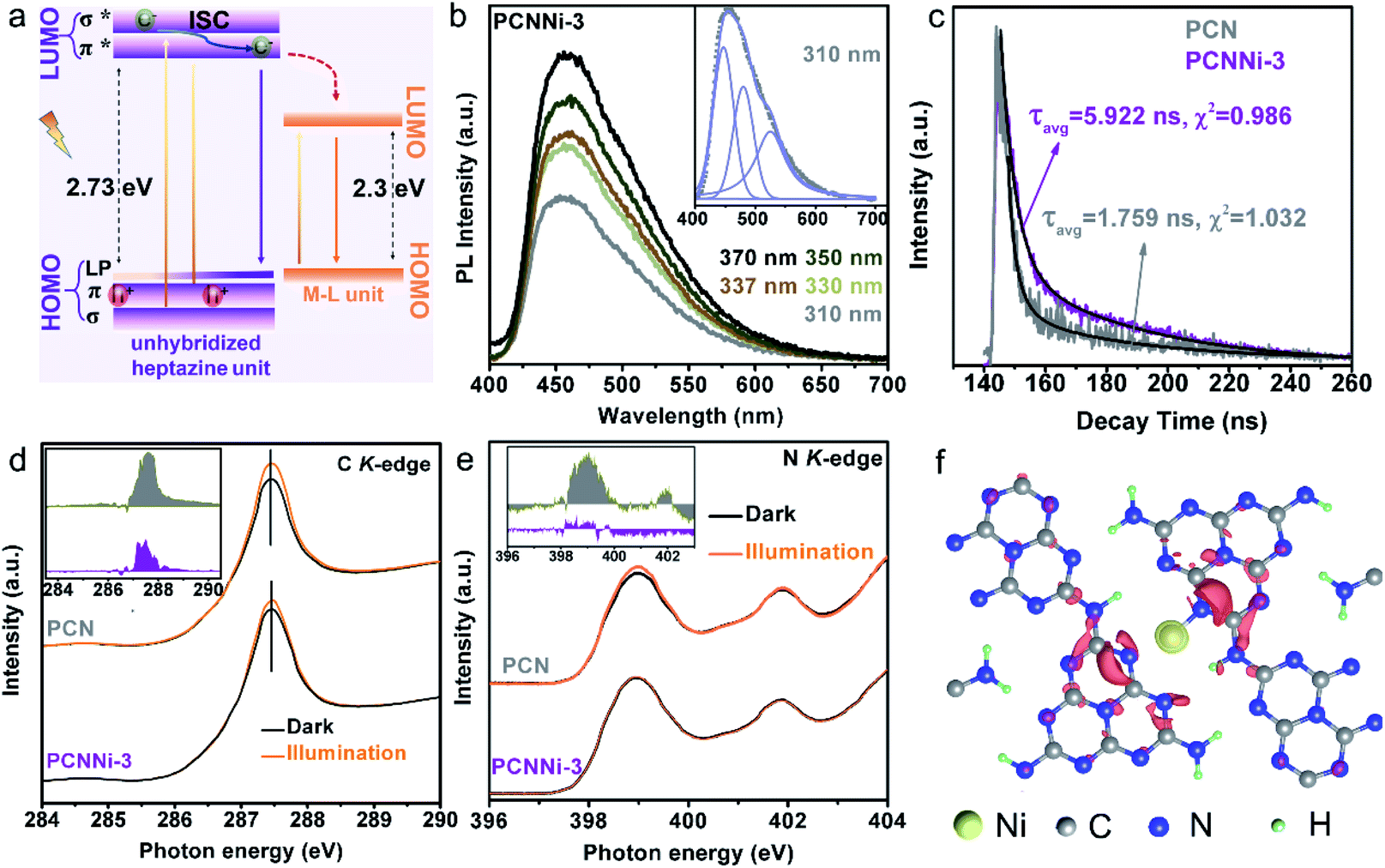 | ||
| Fig. 5 (a) Illustration of the charge transfer processes in the unhybridized heptazine unit (left) and the M–L unit (right). (b) Steady-state fluorescence spectra of PCNNi-3 at different excitation wavelengths. (c) Time-resolved transient PL decay spectra of PCN and PCNNi-3 with the excitation wavelength of 337 nm. (d) C K-edge and (e) N K-edge XANES spectra of PCN and PCNNi-3 with or without illumination. The insets in (d) and (e) show the spectral difference between in the dark and under illumination, respectively. (f) Differential charge density mapping of the M–L unit (single-atom Ni coordinated with sp2 NA atoms). Red and yellow represent electron accumulation and electron depletion regions, respectively. | ||
C K-edge and N K-edge XANES spectra were collected in the dark and under illumination to further investigate the charge transfer processes in PCNNi upon photoexcitation. As mentioned in DFT calculations (Fig. 3d), the HOMO of the M–L unit is derived from the hybridization of Ni 4s and N 2p orbitals, while the LUMO mainly consists of C 2p orbitals. It is well known that the electrons are excited and transferred from the LUMO to HOMO, contributing to the emerging peaks in C and N K-edge spectra. As shown in Fig. 5d and e, the C K-edge and N K-edge spectra of both PCN and PCNNi exhibit higher peak intensities under illumination than in the dark. Moreover, the spectral deviation between illuminated and dark conditions (the inset in Fig. 5d and e) is less significant in PCNNi, as compared to PCN, which should be related to the transfer of photoexcited electrons in M–L units via the MLCT process. In detail, since the single-atom Ni has its non-vacant 4s orbital hybridized with the N 2p orbital, the photoexcited electrons tend to transfer from the Ni 4s state to the 2p states of neighboring N or even C atoms through the MLCT process.9
The differential charge density was calculated to further confirm the MLCT process in the M–L unit under illumination. As shown in Fig. 5f and S17,† upon photoexcitation, the charge density in the M–L unit is redistributed, with electron transfer from single-atom Ni to the adjacent C and N atoms, resulting in electron depletion at single-atom Ni and electron accumulation at C and N atoms. Therefore, the single-atom Ni builds a bridge for photoexcited electrons to transfer between the adjacent heptazine-based polymer chains, accelerating the separation and transfer of photoexcited electrons and holes generated in PCNNi. In contrast, such a charge transfer process is always interrupted by the hydrogen bonds in the pristine PCN (see charge migration pathways in PCN in Fig. S1 in the ESI†).
Based on the above characterization and analysis, new HOMO and LUMO are created by the M–L unit in the band structure of PCNNi with the introduction of the single-atom Ni terminating agent. Electron transition happens via the MLCT process in the M–L unit, broadening the optical absorption in visible light. Meanwhile, the downshifted LUMO of the M–L unit can steer the migration of photoexcited electrons from the unhybridized heptazine unit to the M–L unit, resulting in the inhibited charge carrier recombination. As a result, the broadened optical absorption and the improved charge transfer behaviors synergistically contribute to great enhancement in photocatalytic performances for overall water splitting into H2 and H2O2. As proposed in the DFT calculations (Fig. 5f and S17†), holes are enriched at the single-atom Ni and electrons are aggregated at the adjacent C and N atoms. DOS results (Fig. 3d) indicate that the 2p orbital of the C atom is the major contributor to the HOMO states of the M–L unit to accept the photoexcited electrons from LUMO states. Thus, single-atom Ni and the adjacent C atoms are favorable to act as water oxidation and reduction active sites, respectively. Then, the reaction pathways for water reduction and oxidation were further theoretically calculated for PCNNi with single-atom Ni coordinated with sp2 NA and sp3 NB atoms, with free energy diagrams shown in Fig. 6 and S18,† respectively. For pure PCN, a free energy difference of 0.0665 eV is required for the formation of the transition state (TS) for hydrogen evolution (Fig. 6a). In comparison, PCNNi holds a much lower free energy difference of 0.0015 eV (Fig. 6a), due to electron accumulation at the reductive active sites of C atoms via the MLCT process, leading to the reduced barrier energy for TS formation. For easy understanding, the water reduction reaction pathway is schematically depicted in Fig. 6c–f. According to the principle of minimum energy, with an initial model of the photocatalytic water splitting reaction constructed at a reasonable charge distribution site,48 single-atom Ni firstly absorbs two H2O molecules (Fig. 6c) and breaks the H–O–H bond into H and OH. Then, two H atoms tend to adsorb at the neighboring C atoms of the single-atom Ni (Fig. 6d), and subsequently the H–H bond is formed as the intermediate between two H adatoms (Fig. 6e), with H2 releasing from PCNNi finally (Fig. 6f). In this study, H2O2 rather than O2 is produced over PCNNi as the water oxidation product during photocatalytic water splitting. Herein, the reaction pathways of water oxidation into H2O2 (pathway 1) and O2 (pathway 2) were theoretically investigated for PCNNi (Fig. 6b). The energy calculations show that the free energy difference of 0.5554 eV for TS2.1 is lower than that of 0.8330 eV for TS2.2, indicating that HO–OH species rather than O–O species are formed as the intermediates during the water oxidation reaction. Furthermore, the absorption energy of H2O2 (0.1911 eV) is lower than that of O2 (0.2115 eV), resulting in the easier desorption of H2O2 from the surface of PCNNi. Hence, the oxidation reaction is likely to generate H2O2 rather than O2. As shown in Fig. 6c–f, with the H2O molecules absorbed at single-atom Ni and broken into H and OH (Fig. 6c), two OH remaining at the single-atom Ni (Fig. 6d) tend to form the TS of HO–OH species (Fig. 6e). Subsequently, H2O2 is formed and released from the single-atom Ni (Fig. 6f). For PCNNi with single-atom Ni coordinated with sp3 NB atoms (see the reaction pathway and detailed analysis in Fig. S18 in the ESI†), the calculated results also show that the barrier energy of PCNNi is lower than that of pure PCN for the water reduction reaction. And, the water oxidation reaction prefers to generate H2O2 rather than O2 over PCNNi.
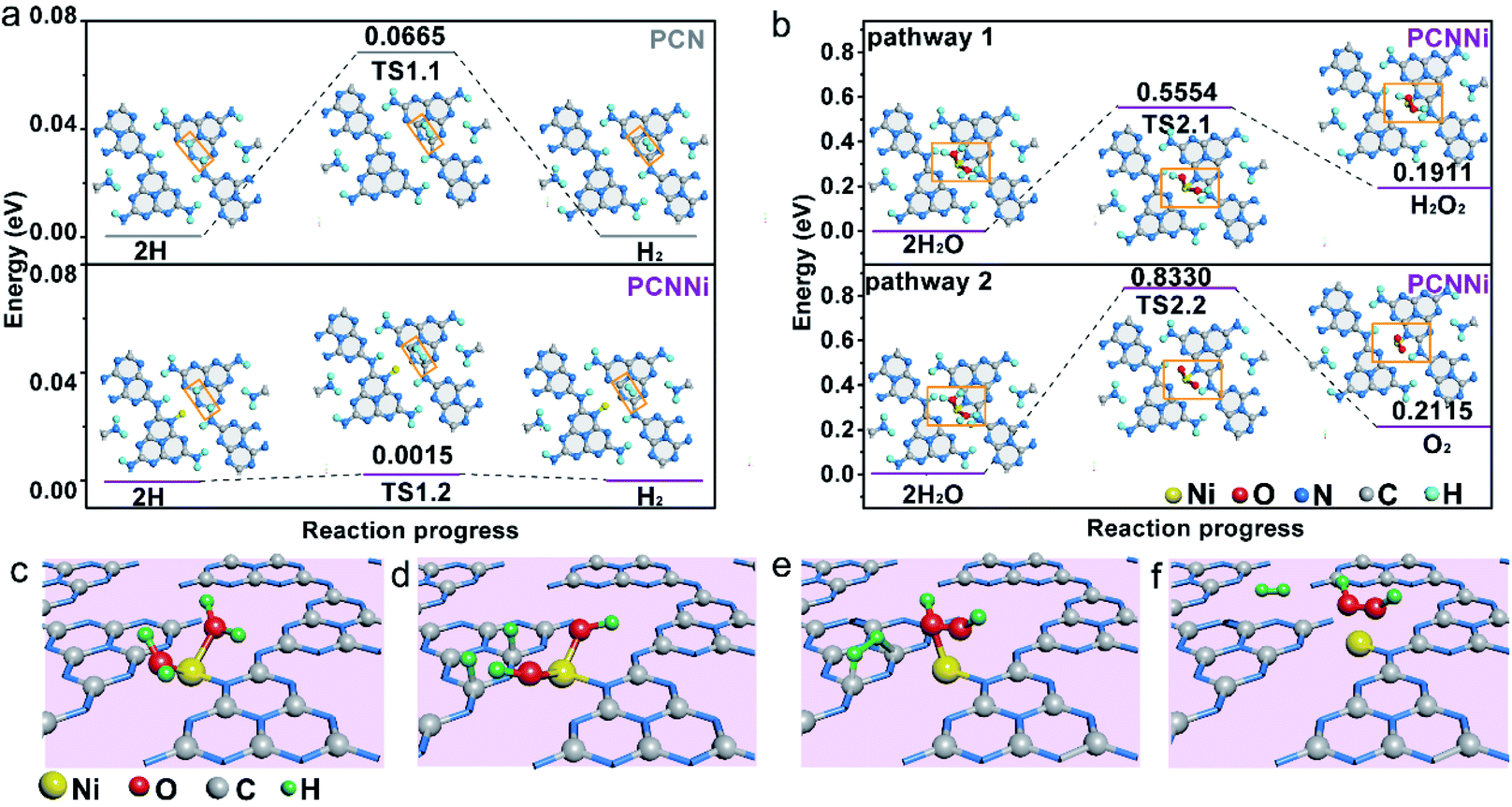 | ||
| Fig. 6 Reaction pathways of (a) water reduction to H2 and (b) water oxidation to H2O2 (top) and O2 (down) for PCNNi (single-atom Ni coordinated with sp2 NA atoms). (c–f) Proposed mechanisms of photocatalytic pure water splitting into H2 and H2O2 for PCNNi (single-atom Ni coordinated with sp2 NA atoms). | ||
Conclusion
We successfully introduced single-atom Ni as a terminating agent to coordinate with sp2 or sp3 N atoms in heptazine units in PCN (PCNNi) to realize efficient photocatalytic overall water splitting into easily-separated gas-product H2 and liquid-product H2O2 under visible light, without an additional cocatalyst loading. It was experimentally revealed that new HOMO and LUMO states were created in PCNNi by the hybridization of single-atom Ni with N atoms in the heptazine ring, resulting in an altered band structure and electronic structure. The superior overall water splitting activity achieved over PCNNi could be attributed to the synergy of the efficient visible light absorption and steered charge migration to the surface-active sites. Theoretical calculations further displayed that the single-atom Ni and the neighboring C atom acted as oxidation and reduction active sites for overall water splitting into H2O2 and H2, respectively, through a two-electron reaction pathway. This work demonstrates great potential and promising prospects to use single atoms for molecular engineering of the organic matrix of PCN to modulate the band structure and active sites for efficient solar-driven overall water splitting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51961165103 and 21875183), the National Key Research and Development Program of China (2017YFE0193900), the National Program for Support of Top-notch Young Professionals, the “Fundamental Research Funds for the Central Universities”, the Project Supported by the Natural Science Basic Research Plan in Shaanxi Province of China (2018JQ2028), the China Postdoctoral Science Foundation (2018M640981), and “The Youth Innovation Team of Shaanxi Universities”. C.L.D is grateful to MoST for financially supporting this work under contracts MoST 107-2112-M-032-004-MY3 and 108-2218-E-032-003-MY3.References
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S. T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS.
- L. Cheng, Q. Xiang, Y. Liao and H. Zhang, Energy Environ. Sci., 2018, 11, 1362 RSC.
- S. Chandrasekaran, L. Yao, L. Deng, C. Bowen, Y. Zhang, S. Chen, Z. Lin, F. Peng and P. Zhang, Chem. Soc. Rev., 2019, 48, 4178 RSC.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120(2), 919 CrossRef CAS.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109 RSC.
- L. Wang, X. Zheng, L. Chen, Y. Xiong and H. Xu, Angew. Chem., Int. Ed., 2018, 57, 3454 ( Angew. Chem. , 2018 , 130 , 3512 ) CrossRef CAS.
- Z. Li, L. Zhang, Y. Liu, C. Shao, Y. Gao, F. Fan, J. Wang, J. Li, J. Yan, R. Li and C. Li, Angew. Chem., Int. Ed., 2020, 59, 935 ( Angew. Chem. , 2020 , 132 , 945 ) CrossRef CAS.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seke, T. Hisatomi and K. Domen, Nature, 2020, 581, 411 CrossRef CAS.
- L. Wang, Y. Zhang, L. Chen, H. Xu and Y. Xiong, Adv. Mater., 2018, 30, 1801955 CrossRef.
- V. W. Lau, I. Moudrakovski, T. Botari, S. Weinberger, M. B. Mesch, V. Duppel, J. Senker, V. Blum and B. V. Lotsch, Nat. Commun., 2016, 7, 12165 CrossRef CAS.
- Y. Zheng, L. Lin, B. Wang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868 CrossRef CAS.
- Y. Fu, C. Liu, M. Zhang, C. Zhu, H. Li, H. Wang, Y. Song, H. Huang, Y. Liu and Z. Kang, Adv. Energy Mater., 2018, 8, 1802525 CrossRef.
- H. Su, M. Liu, W. Cheng, X. Zhao, F. Hu and Q. Liu, J. Mater. Chem. A, 2019, 7, 11170 RSC.
- Y. Kang, Y. Yang, L. Yin, X. Kang, G. Liu and H. M. Cheng, Adv. Mater., 2015, 27, 4572 CrossRef CAS.
- C. Merschjann, S. Tschierlei, T. Tyborski, K. Kailasam, S. Orthmann, D. Hollmann, T. Schedel-Niedrig, A. Thomas and S. Lochbrunner, Adv. Mater., 2015, 27, 7993 CrossRef CAS.
- B. Choudhury, K. K. Paul, D. Sanyal, A. Hazarika and P. K. Giri, J. Phys. Chem. C, 2018, 122, 9209 CrossRef CAS.
- W. Che, W. Cheng, T. Yao, F. Tang, W. Liu, H. Su, Y. Huang, Q. Liu, J. Liu, F. Hu, Z. Pan, Z. Sun and S. Wei, J. Am. Chem. Soc., 2017, 139, 2468 CrossRef.
- G. Zhang, Z. Lan, L. Lin, S. Lin and X. Wang, Chem. Sci., 2016, 7, 3062 RSC.
- Y. Zhang, A. Thomas, M. Antonietti and X. Wang, J. Am. Chem. Soc., 2009, 131, 50 CrossRef CAS.
- G. Vilé, D. Albani, M. Nachtegaal, Z. Chen, D. Dontsova, M. Antonietti, N. López and J. P. Ramírez, Angew. Chem., Int. Ed., 2015, 54, 11265 CrossRef.
- W. Liu, L. Cao, W. Cheng, Y. Cao, X. Liu, W. Zhang, X. Mou, L. Jin, X. Zheng, W. Che, Q. Liu, T. Yao and S. Wei, Angew. Chem., Int. Ed., 2017, 56, 9312 CrossRef CAS.
- Y. Li, Z. Wang, T. Xia, H. Ju, K. Zhang, R. Long, Q. Xu, C. Wang, L. Song, J. Zhu, J. Jing and Y. Xiong, Adv. Mater., 2016, 28, 6959 CrossRef CAS.
- X. Li, W. Bi, L. Zhang, S. Tao, W. Chu, Q. Zhang, Y. Luo, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 2427 CrossRef CAS.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159 CrossRef CAS.
- C. Y. Hsu and K. S. Chang, J. Phys. Chem. C, 2018, 122, 3506 CrossRef CAS.
- V. W. Lau, M. B. Mesch, V. Duppel, V. Blum, J. Senker and B. V. Lotsch, J. Am. Chem. Soc., 2015, 137, 1064 CrossRef CAS.
- X. Wang, X. Chen, A. Thomas, X. Fu and M. Antonietti, Adv. Mater., 2009, 21, 1609 CrossRef CAS.
- K. Iwase, T. Yoshioka, S. Nakanishi, K. Hashimoto and K. Kamiya, Angew. Chem., Int. Ed., 2015, 54, 11068 ( Angew. Chem. , 2015 , 127 , 11220 ) CrossRef CAS.
- H. Li, Y. Xia, T. Hu, Q. Deng, N. Du and W. Hou, J. Mater. Chem. A, 2018, 6, 6238 RSC.
- G. Wu, S. Hu, Z. Han, C. Liu and Q. Li, New J. Chem., 2017, 41, 15289 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. A. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS.
- D. Zhao, C. Dong, B. Wang, C. Chen, Y. Huang, Z. Diao, S. Li, L. Guo and S. Shen, Adv. Mater., 2019, 31, 1903545 CrossRef CAS.
- G. Weng, Y. Xie, H. Wang, C. Karpovich, J. Lipton, J. Zhu, J. Kong, L. D. Pfefferle and A. D. Taylor, Angew. Chem., Int. Ed., 2019, 58, 13727 CrossRef CAS.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty, D. Su, K. Attenkofer and H. Wang, Energy Environ. Sci., 2018, 11, 893 RSC.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078 CrossRef CAS.
- Y. Li, X. Li, Z. Wang, H. Guo and T. Li, Ceram. Int., 2016, 42, 14565 CrossRef CAS.
- Z. Gao, J. Liu, X. Chen, X. Zheng, J. Mao, H. Liu, T. Ma, L. Li, W. C. Wang and X. W. Du, Adv. Mater., 2019, 31, 1804769 CrossRef.
- Z. Zhao, H. Wu, H. He, X. Xu and Y. Jin, J. Mater. Chem. A, 2015, 3, 7179 RSC.
- D. Liu, S. Ding, C. Wu, W. Gan, C. Wang, D. Cao, Z. U. Rehman, Y. Sang, S. Chen, X. Zheng, Y. Wang, B. Ge and L. Song, J. Mater. Chem. A, 2018, 6, 6840 RSC.
- W. Ni, A. Krammer, C. Hsu, H. Chen, A. Schüler and X. Hu, Angew. Chem., Int. Ed., 2019, 58, 7445 CrossRef CAS.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783 CrossRef.
- K. Akaike, K. Aoyama, S. Dekubo, A. Onishi and K. Kanai, Chem. Mater., 2018, 30, 2341 CrossRef CAS.
- Y. Kang, Y. Yang, L. C. Yin, X. Kang, G. Liu and H. M. Cheng, Adv. Mater., 2015, 27, 4572 CrossRef CAS.
- G. Zhang, A. Savateev, Y. Zhao, L. Li and M. Antonietti, J. Mater. Chem. A, 2017, 5, 12723 RSC.
- H. Xiang, J. Cheng, X. Ma, X. Zhou and J. J. Chruma, Chem. Soc. Rev., 2013, 42, 6128 RSC.
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Fu, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 441 ( Angew. Chem. , 2010 , 122 , 451 ) CrossRef CAS.
- P. Kumar, E. Vahidzadeh, U. K. Thakur, P. Kar, K. M. Alam, A. Goswami, N. Mahdi, K. Cui, G. M. Bernard, V. K. Michaelis and K. Shankar, J. Am. Chem. Soc., 2019, 141, 5415 CrossRef CAS.
- L. Lin, X. Ye, X. Gao and X. J. Huang, J. Mol. Catal. A: Chem., 2015, 406, 137 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, additional structural characterization, photocatalytic measurements and DFT calculations for reaction pathways. See DOI: 10.1039/d0sc07093a |
| This journal is © The Royal Society of Chemistry 2021 |