
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesymmetrised pentaporphyrinic gears mounted on metallo-organic anchors†
Seifallah
Abid
 a,
Yohan
Gisbert
a,
Mitsuru
Kojima
b,
Nathalie
Saffon-Merceron
c,
Jérôme
Cuny
d,
Claire
Kammerer
*a and
Gwénaël
Rapenne
*ab
a,
Yohan
Gisbert
a,
Mitsuru
Kojima
b,
Nathalie
Saffon-Merceron
c,
Jérôme
Cuny
d,
Claire
Kammerer
*a and
Gwénaël
Rapenne
*ab
aCEMES, Université de Toulouse, CNRS, 29 Rue Marvig, F-31055 Toulouse Cedex 4, France. E-mail: rapenne@cemes.fr
bDivision of Materials Science, Nara Institute of Science and Technology, NAIST, 8916-5 Takayama-cho, Ikoma, Nara 630-0192, Japan
cUniversité de Toulouse, UPS, Institut de Chimie de Toulouse, ICT FR 2599, 118 Route de Narbonne, 31062 Toulouse, France
dLCPQ, Université de Toulouse, CNRS, 118 Route de Narbonne, F-31062 Toulouse Cedex 9, France
First published on 18th February 2021
Abstract
Mastering intermolecular gearing is crucial for the emergence of complex functional nanoscale machineries. However, achieving correlated motion within trains of molecular gears remains highly challenging, due to the multiple degrees of freedom of each cogwheel. In this context, we designed and synthesised a series of star-shaped organometallic molecular gears incorporating a hydrotris(indazolyl)borate anchor to prevent diffusion on the surface, a central ruthenium atom as a fixed rotation axis, and an azimuthal pentaporphyrinic cyclopentadienyl cogwheel specifically labelled to monitor its motion by non-time-resolved Scanning Tunneling Microscopy (STM). Desymmetrisation of the cogwheels was first achieved sterically, i.e. by introducing one tooth longer than the other four. For optimal mechanical interactions, chemical labelling was also investigated as a preferential way to induce local contrast in STM images, and the electronic properties of one single paddle were modulated by varying the porphyrinic scaffold or the nature of the central metal. To reach such a structural diversity, our modular synthetic approach relied on sequential cross-coupling reactions on a penta(p-halogenophenyl)cyclopentadienyl ruthenium(II) key building block, bearing a single pre-activated p-iodophenyl group. Chemoselective Sonogashira or more challenging Suzuki–Miyaura reactions allowed the controlled introduction of the tagged porphyrinic tooth, and the subsequent four-fold cross-couplings yielded the prototypes of pentaporphyrinic molecular gears for on-surface studies, incorporating desymmetrised cogwheels over 5 nm in diameter.
1. Introduction
Prompted by the pioneering studies of Sauvage, Stoddart and Feringa,1 the field of artificial molecular machines has witnessed an impressive development over the last decades and different types of machines have been reported,2 some of them being technomimetic3 such as syringes,4 wheelbarrows,5 vehicles,6 scissors,7 and elevators.8 In addition, control over directionality of motion gave rise to artificial rotary motors9 responding to chemical,10 light,11 or electric stimuli.12 A vast majority of these machines have been studied collectively in solution or at interfaces, leading to average behaviour over populations of molecules. However, with the advent of scanning probe microscopy, resolution down to the atomic scale has been reached, thus allowing for in-depth investigations into nanoscale mechanics on single molecules adsorbed on surfaces.13 Even a race of nanocars was proposed14 and organised in 2017,15 showing that these nanomachines are now studied at the single-molecule level by many research teams worldwide.With this large array of artificial molecular machines at hand, the next step is now to master the assembly of several of such individual modules into complex functional nanoscale machineries. In this regard, gear systems appear as essential elementary units allowing the mechanical transmission of information over (long) distances and the conversion of the motive power of a motor through synchronised disrotatory motion of the cogwheels. Ancient machineries such as the Antikythera mechanism, which dates back to the Hellenistic period (150–100 BC), already incorporated decimeter-scale gear components in their structure.16 Over the centuries, miniaturisation of solid state cogwheels has been pushed forward as a function of available technology, with the smallest reported cogwheels having a diameter of 70 nm, obtained by e-beam lithography.17
As opposed to this top-down approach that is close to reaching its physical limits in terms of object size, molecular gear systems have alternatively been obtained by a monumentalisation strategy via the controlled assembly of specifically designed molecular cogwheels. In this regard, the C3-symmetric triptycene fragment has been extensively investigated as a three-blade cogwheel archetype in intramolecular gearing systems studied in solution. Pioneering examples of covalent bis-triptycenyl structures with intersecting rotation axes (i.e. bevel gear prototypes, Fig. 1a) were independently reported in 1980 by Mislow18 and Iwamura,19 who unambiguously demonstrated the disrotatory correlated motion of the meshed cogwheels by inserting a tag on one blade of each cogwheel.
 | ||
| Fig. 1 Selection of pioneering examples of molecular cogwheel systems exhibiting intramolecular gearing in solution (a and b) or intermolecular gearing when anchored on a surface (c and d). These selected examples have been reported by (a) Iwamura,20 (b) Siegel,21 (c) Moresco22 and (d) Soe.23 | ||
Analogous bis-triptycenyl systems bearing different linkers were reported over the years,24 some of which exhibited very elegant switch function in response to a chemical25 or photochemical26 stimulus. In a similar approach, prototypes of spur gears, involving parallel rotation axes, were developed more recently (Fig. 1b) and the impact of the inter-triptycene distance on the gearing fidelity (i.e. proportion of geared rotations over gear slippage) was highlighted.21,27 The complexity of intramolecular gearing systems has been further enhanced by varying the nature of the cogwheels28 or by multiplying the number of meshed cogwheels.29 However, in all these systems designed for in-solution studies, gearing mechanisms are inherently limited to intramolecular correlated motion of submolecular fragments bound to a rigid scaffold, which is required to maintain the distance and relative orientation of the rotation axes. To achieve intermolecular gearing and thus allow correlated motion over longer distances, pre-organisation of the assembly of cogwheels is required (Fig. 2), with (i) a linear arrangement leading to a train of gears, (ii) a parallel orientation of the rotation axes (depicted in red), and (iii) appropriate and locked intermolecular distances leading to proper intertwining of the teeth.
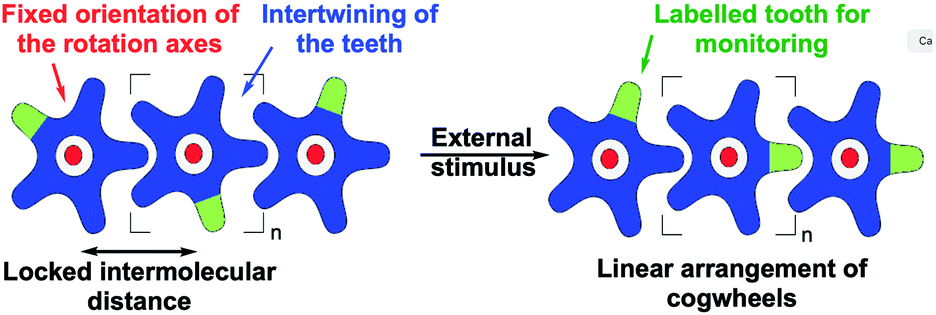 | ||
| Fig. 2 Schematic representation of the key parameters to be considered in the design of trains of molecular gears to achieve intermolecular correlated rotation in response to an external stimulus: top views of a train of hypothetical star-shaped cogwheels (in blue) incorporating a labelled tooth (in green) and mounted on rotation axes (in red) at fixed intermolecular distances, before and after an external stimulus leading to correlated motion. | ||
With this aim in view, specifically designed molecular cogwheels have been investigated as solid state self-assemblies,30 or alternatively deposited on surfaces and studied at the single molecule scale by means of Scanning Tunneling Microscopy (STM).31 To date, the smallest known functional train of gears deposited on a surface has been constructed on Au(111) using star-shaped pentaarylcyclopentadienyl derivatives as cogwheels, taking advantage of their radical state to promote chemical anchoring on the metallic surface (Fig. 1c).22 This anchoring locks the inter-cogwheel distance and favours intermolecular correlated motion over diffusion of the neighbouring species. Under these conditions, rotation of the driver cogwheel, induced by mechanical manipulation using the STM tip, was, for the first time, successfully propagated to two further molecules. The disrotatory correlated motion of these three cogwheels was evidenced by comparison of STM images, exploiting the presence of one tert-butyl substituent per molecule as a steric tag. As an alternative to chemical anchoring to fix the multimolecular gearing system, single metallic atoms have been deposited on a Pb(111) surface and hexaarylbenzene-based cogwheels (Fig. 1d) have been successfully mounted on top of such ad-atoms thus acting as the rotation axis.23,32 These approaches, which gave rise to the first functional trains of molecular gears on a surface, fulfill several of the criteria detailed in Fig. 2. However, they rely on the in situ generation of the rotation axes after the deposition of the molecular cogwheels on the surface.
As an alternative, it also appears possible to incorporate a metallic rotation axis in a molecular structure prior to deposition by means of coordination chemistry, and bring the pre-assembled functional metallo-organic species on the surface. This approach was exploited in the design, synthesis and on-surface study of a family of ruthenium-based rotary molecular machines,33 and in particular of an electron-fueled molecular motor.12b The central ruthenium atom acts as a ball bearing between an azimuthal pentaarylcyclopentadienyl rotating subunit and a hydrotris(indazolyl)borate stator, specifically functionalised with thioether groups. This scorpionate ligand lifts away from the surface the ruthenium rotation axis and thus the rotor, while affording tight anchoring on noble metal surfaces at low temperature.34 It is important to note here that translation of such complexes by STM lateral manipulation is still possible at 77 K while diffusion is totally frozen at 4 K, which allows a dedicated spatial molecular arrangement to be set up by STM manipulation and subsequently frozen. This ruthenium-based scaffold seemed particularly well adapted for the construction of trains of molecular gears on surfaces, with control over the rotation axis orientation, the intermolecular distance and the height of the cogwheels, decoupled from the surface. The cyclopentadienyl core was thus functionalised to mimic star-shaped cogwheels, with teeth ranging from pseudo-monodimensional biaryl fragments to bidimensional porphyrinic paddles for an optimal intermolecular transmission of motion.35 The resulting family of prototypes incorporate rotor subunits with a C5 symmetry (i.e. carrying five identical teeth), which will prevent direct monitoring of motions on a surface based on static STM images.
In order to get a deeper understanding of the single-molecule mechanics of such ruthenium-based cogwheel prototypes, it appears crucial for on-surface studies to lower the symmetry of the upper rotating subunit. This may be achieved by varying the shape of one of the five teeth (i.e. introducing a steric tag) or by chemically inducing a change in the STM contrast of one of the teeth (i.e. introducing a chemical tag). This labelling strategy is commonly applied to monitor molecular motions by STM, on the single molecule scale or within self-assemblies, and discrimination is usually performed by adding or removing a bulky group (steric tag)12b,22 or by inserting nitrogen atoms in the molecular backbone (chemical tag).31
The desymmetrisation of extended ruthenium-based porphyrinic cogwheels was thus tackled, exploiting the length of the paddles and the electronic features of the porphyrins as steric and chemical tags, respectively. Herein, we report the design supported by DFT calculations, the modular synthesis and the characterisation of a series of desymmetrised cogwheel prototypes bearing one labelled porphyrinic paddle.
2. Results and discussion
Lowering the symmetry of the star-shaped ruthenium-based cogwheels while maintaining their structural diversity implies the development of a modular synthetic approach, allowing for a controlled introduction of a sterically- or chemically-tagged porphyrinic tooth.2.1. Synthetic strategies towards desymmetrised pentaporphyrinic cyclopentadienyl cogwheels
Our general strategy for the synthesis of ruthenium-based rotary machines relies on a post-functionalisation of a piano-stool ruthenium complex as the key intermediate, incorporating the thioether-functionalised hydrotris(indazolyl)borate tripod in combination with a pentaarylcyclopentadienyl ligand. In anticipation of post-functionalisation via multiple transition metal-catalysed cross-coupling reactions, the cyclopentadienyl rotor part is pre-activated and thus it carries five p-halogenophenyl groups. Starting from the corresponding pentabrominated36 or pentaiodinated35 precursor, five-fold cross-coupling reactions gave rise to a variety of cogwheel prototypes of increasing diameter, with five identical teeth such as 4,4′-biphenyl fragments, carbazoles, BODIPYs and porphyrins. It is, however, important to mention here that the reactivity of the bromophenyl groups in the key precursor is dramatically reduced by the proximity of the electron-rich cyclopentadienyl platform, compared to bromobenzene as the reference, thus requiring highly active catalysts for such transformations. It has also been observed that the five halogenated positions react independently. A purely statistical approach for the desymmetrisation of the cyclopentadienyl cogwheel, with a concomitant introduction of both coupling partners in a 4 to 1 ratio, is thus doomed to failure and controlled synthetic routes were devised.From a retrosynthetic point of view, desymmetrised cyclopentadienyl cogwheels with one discriminated tooth may stem from a synthetic intermediate carrying this distinct tooth, along with four halogenophenyl moieties still available for a final four-fold cross-coupling (Scheme 1). The mono-functionalised intermediate may in turn be obtained according to two distinct strategies, relying on statistical (Scheme 1, bottom left) vs. chemoselective (bottom right) approaches. The statistical strategy leads back to the symmetrical penta(p-bromophenyl)-appended precursor 2, and implies the introduction of the distinct tooth via a Suzuki–Miyaura or Sonogashira cross-coupling under statistical conditions, presumably with moderate efficiency due to the formation of various by-products. As an alternative, a selective pathway has been envisioned, relying on the discrimination of aryl halides in cross-coupling reactions leading to ethynylaryl37 or biaryl38 patterns. A universal unsymmetrical 1,2,3,4,5-penta(p-halogenophenyl)cyclopentadienyl ruthenium(II) precursor with a single pre-activated p-iodophenyl group (1) was thus designed and synthesised,39 to allow for a chemoselective monofunctionalisation as a starting point for the synthesis of pentaporphyrinic cogwheels incorporating one sterically- or chemically-tagged tooth.
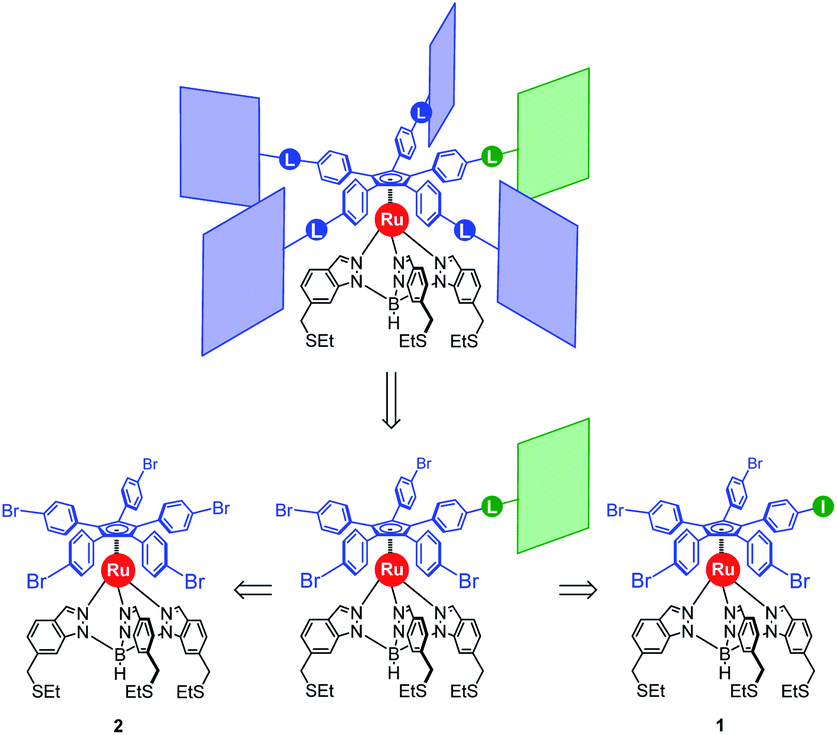 | ||
| Scheme 1 Retrosynthetic approaches towards desymmetrised cyclopentadienyl cogwheels, via statistical (left) vs. chemoselective (right) routes (L = linker). | ||
2.2. Steric desymmetrisation
As opposed to translation motion, the STM characterisation of rotary motion of Cn-symmetric objects on surfaces is not straightforward due to the absence of time resolution. This issue is commonly overcome by sterically lowering the symmetry of the objects, thus allowing the motion to be monitored by comparing static images recorded before and after the stimulus. This strategy was in particular applied to star-shaped cyclopentadienyl derivatives, bearing one truncated tolyl arm among ferrocenylphenyl arms12b or one extended p-tert-butylphenyl tooth compared to the remaining phenyl teeth.22 In the case of ruthenium-based cogwheels, the length of the linker between the cyclopentadienyl core and the tetrapyrrole macrocyle was chosen as the key parameter for steric labelling, while keeping the structure of the porphyrin constant. Indeed, in our previous work towards symmetric cogwheels,35 the diameter of the rotating platform could be successfully modulated by changing the structure of the linker. While a five-fold Suzuki–Miyaura coupling applied to the symmetrical pentabrominated precursor 2 delivered 4,4′-biphenyl patterns, a five-fold Sonogashira reaction afforded phenylethynylphenyl spacers leading to a cogwheel diameter up to 5 nm. Interestingly, the Sonogashira coupling was successfully carried out under very mild conditions (45 °C, 24 h, in triethylamine) starting from the symmetrical pentaiodinated precursor, while the standard pentabrominated counterpart 2 required much harsher conditions (95 °C, 48 h, in DMF/diisopropylamine). This dramatic change was expected in view of the higher reactivity of aryl iodides than that of aryl bromides in Sonogashira reactions,37 but most importantly it opens the way for chemoselective monofunctionalisation of the unsymmetrical mono-iodinated key precursor 1. This will thus be exploited to produce sterically tagged cogwheels, incorporating one tooth longer than the other four, while keeping the structure of the external porphyrins constant. According to our strategy, a chemoselective Sonogashira coupling reaction on the pre-activated precursor 1 will provide a monofunctionalised intermediate with a single long porphyrinic tooth, and the four remaining p-bromophenyl moieties will then undergo a four-fold Suzuki–Miyaura coupling to introduce shorter biphenyl-based teeth.For the series of ruthenium-based cogwheels, nickel(II) porphyrins have been selected as rigid and flat paddles, in view of the high stability of the nickel centre which avoids any metal exchange during studies on metallic surfaces. Such A3B-type macrocycles carry 3,5-di-tert-butylphenyl substituents on three meso positions which increase solubility during in-solution synthesis and limit π–π stacking on the surface among neighbouring cogwheels, which may hamper transmission of motion. The fourth meso position is functionalised in anticipation of Sonogashira and Suzuki–Miyaura reactions with a phenylethynyl group (3) or a phenylboronic acid pinacol ester moiety (4), respectively (Fig. 3). A3B-type porphyrins 3 and 4 were synthesised under Lindsey statistical conditions starting from pyrrole and appropriate benzaldehyde derivatives, and metalation was next performed in the presence of nickel(II) acetylacetonate (see the ESI section†). With both porphyrinic coupling partners in hand, the synthesis of sterically tagged cogwheel 9via a selective approach was tackled (Scheme 2). The higher reactivity of aryl iodides than that of their bromide counterparts in Sonogashira couplings was exploited for the monofunctionalisation of key precursor 1. The latter reacted under mild conditions with 5-(ethynylphenyl)porphyrin 3 in the presence of PdCl2(PPh3)2 and CuI as cocatalysts to afford the expected monoporphyrinic complex 8 in 51% yield. The four remaining p-bromophenylene groups were subsequently submitted to Suzuki–Miyaura coupling conditions in the presence of an excess of porphyrin 4 to give the pentaporphyrinic complex 9 in 36% yield. The structure of this sterically tagged cogwheel prototype incorporating five identical porphyrinic paddles but one longer linker was confirmed by combined characterisation techniques, unambiguously showing the presence of four 4,4′-biphenyl patterns and a single phenylethynylphenyl moiety acting as a steric tag.
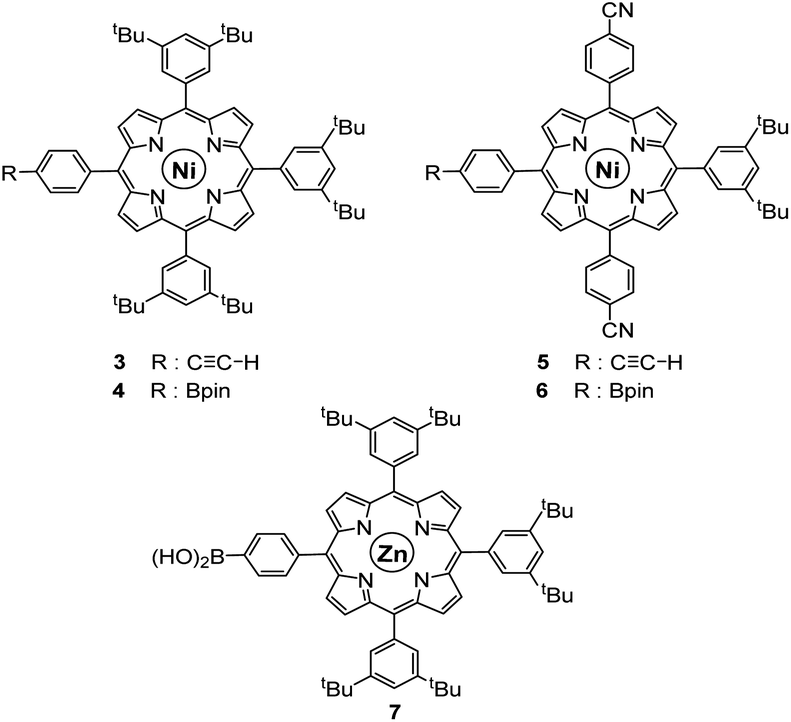 | ||
| Fig. 3 Structure of porphyrins 3–7 used as coupling partners in the synthesis of desymmetrised cogwheel prototypes. | ||
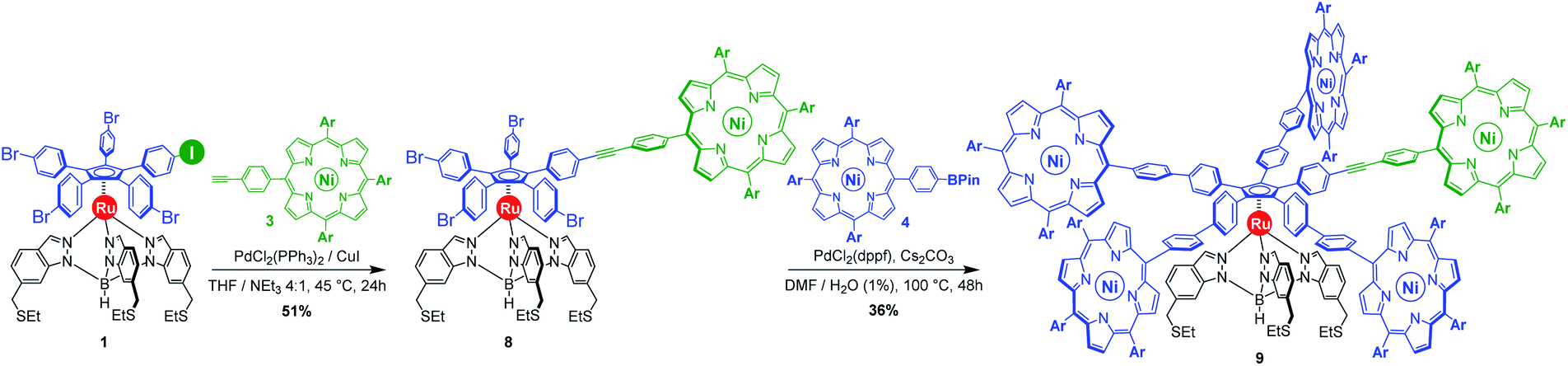 | ||
| Scheme 2 Synthesis of the sterically tagged cogwheel 9, incorporating one longer tooth (in green), via sequential chemoselective Sonogashira and four-fold Suzuki–Miyaura cross-coupling reactions. Ar = 3,5-di-tert-butylphenyl. | ||
Incorporation of a steric tag in molecular scaffolds is a standard method to evidence rotary motion in STM studies. However, in gearing systems this structural variation might modify the interlocking of teeth and thus intermolecular interactions, which may prevent the correlated motion of neighbouring cogwheels.22 As an alternative, we envisioned to induce a change in the STM contrast of one single tooth while keeping the shape of all cogwheels identical.
2.3. Chemical desymmetrisation
Contrast in STM images is correlated to local variations of tunneling current intensity at given tip height and bias voltage when scanning the surface. Submolecular contrast thus depends on the distribution of electronic density within a molecular object adsorbed on a surface. Such a submolecular contrast is routinely exploited to evidence the rotary motion of highly symmetric molecules, as in the case of the synchronised rotation of double-decker self-assemblies unambiguously evidenced thanks to the dipolar character of the upper porphyrinic deck.40 Chemical desymmetrisation has also been performed by intentionally substituting one submolecular fragment with an isosteric group featuring distinct electronic properties. By way of example, the hexaarylbenzene cogwheel used in the first example of intermolecular gearing on the surface31a incorporates five tert-butylphenyl teeth and a single tert-butylpyrimidyl fragment as the N-tagged tooth, inducing a contrast on a Cu(111) surface.For the ruthenium-based molecular gears, an optimal design favouring interlocking and gearing motion relies on cogwheels bearing teeth of identical length and with homogeneous peripheral shapes for the paddles. We thus aim at a chemical desymmetrisation involving a variation of the electronic properties of a single porphyrin, while keeping an identical substitution pattern at the 5,15 positions. In the present case, a bright contrast located on the tagged paddle will be obtained in STM images if the LUMO is centred on this discriminated porphyrin.
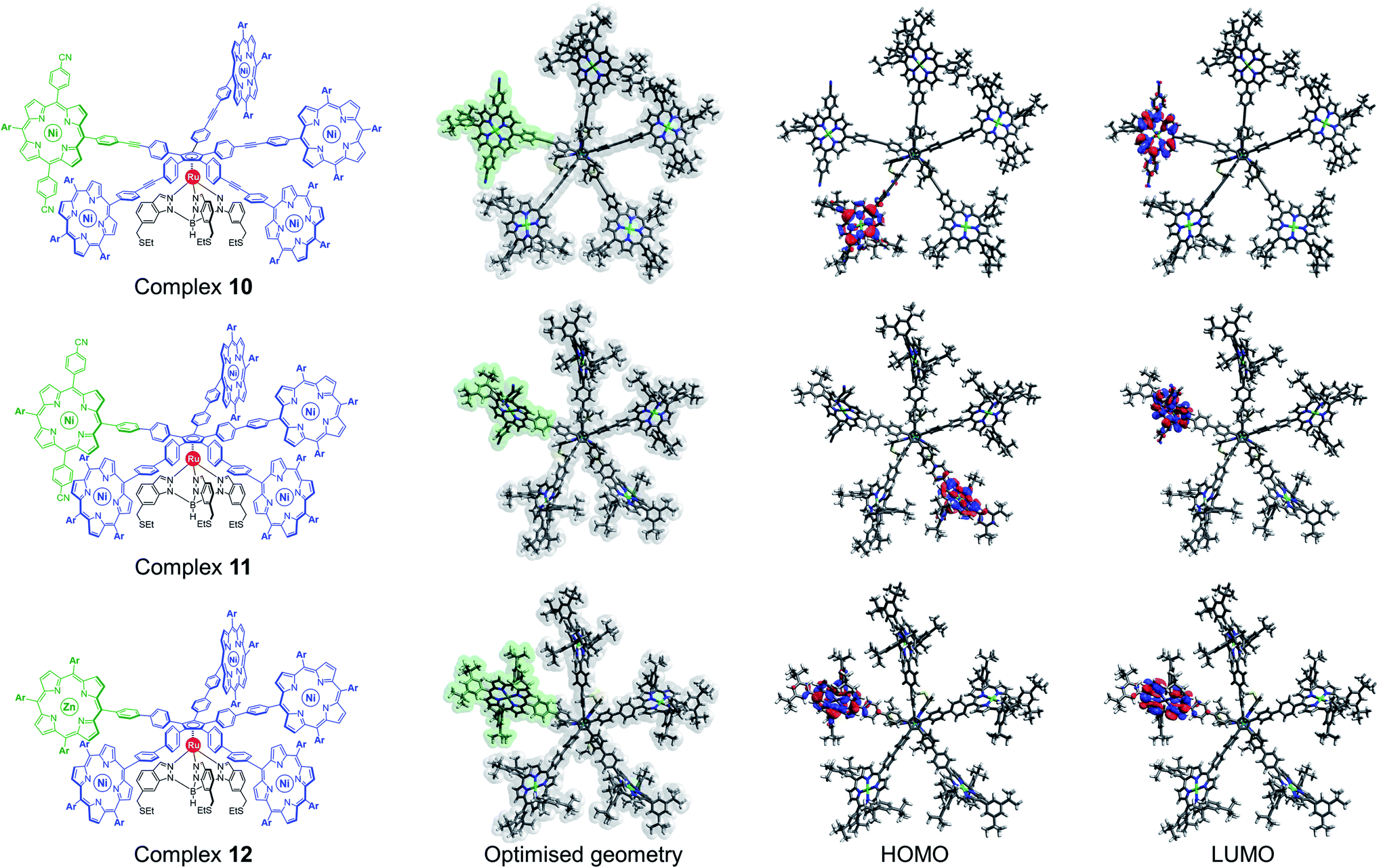 | ||
| Fig. 4 Chemical structure, DFT-optimised geometry (with the distinct porphyrin unit highlighted in green) and HOMO–LUMO maps of desymmetrised pentaporphyrinic ruthenium complexes 10, 11 and 12 (Ar = 3,5-di-tert-butylphenyl). | ||
In order to assess the possibility of forming a train of gears with this cogwheel design, further calculations were performed using the GFN2-xTB semi-empirical tight-binding model41b specifically dedicated to describe organometallic compounds containing a few thousand atoms. It also includes an accurate description of noncovalent interactions as required in the present study. The geometry of two interacting cogwheels 11 was optimised for three different distances between the rotation axes (Ru–Ru distances): 35.3, 37.5 and 41.2 Å. The final optimised structures are presented in Fig. S12–S14 in the ESI section.† According to these structures, even with the smallest distance (35.3 Å), the space between two porphyrinic paddles is sufficient to accommodate a paddle from the neighbouring cogwheel, thus demonstrating that this architecture leads to a proper intertwining of the teeth, which is a prerequisite for correlated rotation.
However, these results obtained in the gas phase have to be confirmed by further calculations considering the molecule adsorbed via its thioether-functionalised tripod on a metallic surface. Even if the cyclopentadienyl platform carrying the five porphyrins is lifted up and thus strongly decoupled from the surface, important changes in paddle conformations might result from interactions between porphyrinic meso 10,20-substituents and the metallic surface, especially in the case of 4-cyanophenyl moieties inserted on the tagged paddle. Moreover, it has been shown that the tripodal ligand undergoes a twist of the indazole fragments when adsorbed on a metal surface, leading to helical chirality of the hydrotris(indazolyl)borate subunit as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of right- and left-handed propellers.12b Owing to important steric interactions, tilting of the indazoles is expected to affect the orientation of the phenyl moieties directly connected to the cyclopentadienyl ring,13e which may thus induce helical chirality on the rotating cogwheel. In such a case, it could be envisioned to build trains of gears with alternating right- and left-handed cogwheels to allow propagation of disrotatory motion.
:1 ratio of right- and left-handed propellers.12b Owing to important steric interactions, tilting of the indazoles is expected to affect the orientation of the phenyl moieties directly connected to the cyclopentadienyl ring,13e which may thus induce helical chirality on the rotating cogwheel. In such a case, it could be envisioned to build trains of gears with alternating right- and left-handed cogwheels to allow propagation of disrotatory motion.
From an electronic point of view, regardless of the nature of the linker, the frontier orbitals of desymmetrised ruthenium complexes 10 and 11 are centred exclusively on the porphyrinic units (Fig. 4). As expected from the incorporation of electron-withdrawing substituents, the LUMO is located on the discriminated porphyrin carrying the 4-cyanophenyl substituents, which is promising to obtain a bright contrast on this paddle in STM studies.
Alternatively, the electronic properties of porphyrins can also be modulated by the presence or not of a coordinated metal, and by its nature. In the case of molecular gears to be actuated on a metallic surface, it appears highly desirable to use metal porphyrins as paddles since such free-base macrocycles are known to undergo metal insertion on noble metal surfaces.42 In addition, having a single free-base porphyrin as a discriminated paddle may lead to intermolecular scrambling in solution, which might in fine produce anisotropic population of cogwheel prototypes. DFT calculations were thus undertaken to probe the electronic features of a cogwheel prototype carrying five identical A3B-type tetrapyrrolic scaffolds, with four nickel(II) and one distinct zinc(II) metallic centres. The optimised geometry of the star-shaped complex 12 involving 4,4′-biphenyl linkers around the cyclopentadienyl core and the corresponding frontier orbitals are presented in Fig. 4 (bottom row). Interestingly, both the HOMO and LUMO orbitals are centred on the distinct zinc(II) porphyrin, which is again likely to produce contrast in STM images.
This preliminary study based on DFT calculations highlighted two promising ways of inducing a chemical desymmetrisation of pentaporphyrinic cogwheel prototypes, while keeping their shape unchanged. The synthesis of the three candidates was then tackled.
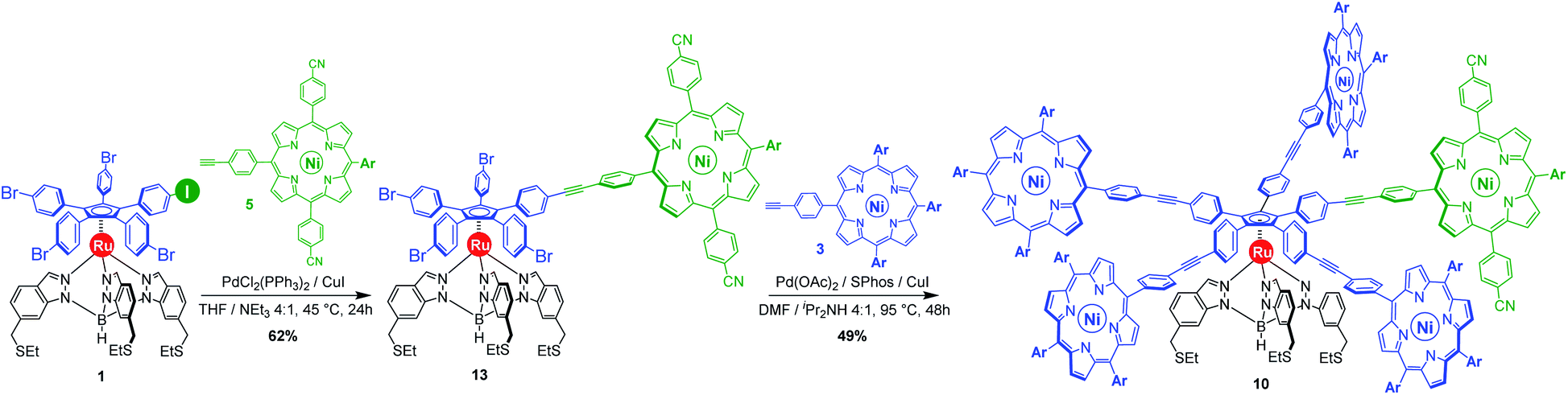 | ||
| Scheme 3 Synthesis of the chemically-tagged cogwheel 10, incorporating one distinct electron-deficient porphyrinic macrocycle (in green), via sequential Sonogashira cross-coupling reactions. Ar = 3,5-di-tert-butylphenyl. | ||
With those two distinct nickel(II) porphyrinic macrocycles as coupling partners, the synthesis of cogwheels incorporating one chemically-discriminated porphyrin paddle was tackled. Single functionalisation of the mono(p-iodophenyl) key precursor 1 was carried out with 2 equiv. of trans A2BC porphyrin 5 (Scheme 3). The reaction occurred at 45 °C in the presence of PdCl2(PPh3)2 (10 mol%) and CuI (6 mol%) as a catalytic system in a mixture of tetrahydrofuran and triethylamine (4:1). At this temperature, almost no side product resulting from the competitive oxidative addition of bromophenyl moieties was observed. After 24 h, ruthenium complex 13 bearing a single porphyrin as a result of the chemoselective Sonogashira coupling was isolated in 62% yield as a red solid.
Single crystals suitable for X-ray diffraction were successfully obtained by slow evaporation of a solution of 13 in a mixture of benzene and methanol (Fig. 5). The structure confirms the presence of the phenylethynylphenyl pattern as a linker between the cyclopentadienyl and porphyrin cores, with a torsion angle of 20° between both phenylene units. In the solid state, it appears that the tetrapyrrole macrocycle lies almost parallel to the central cyclopentadienyl ring, with a distance of 2.6 nm between the centre of the Cp ring and the outer tert-butyl carbons located on the porphyrin trans meso substituent. The structure also confirms the presence of four remaining p-bromophenyl moieties available for further cross-couplings.
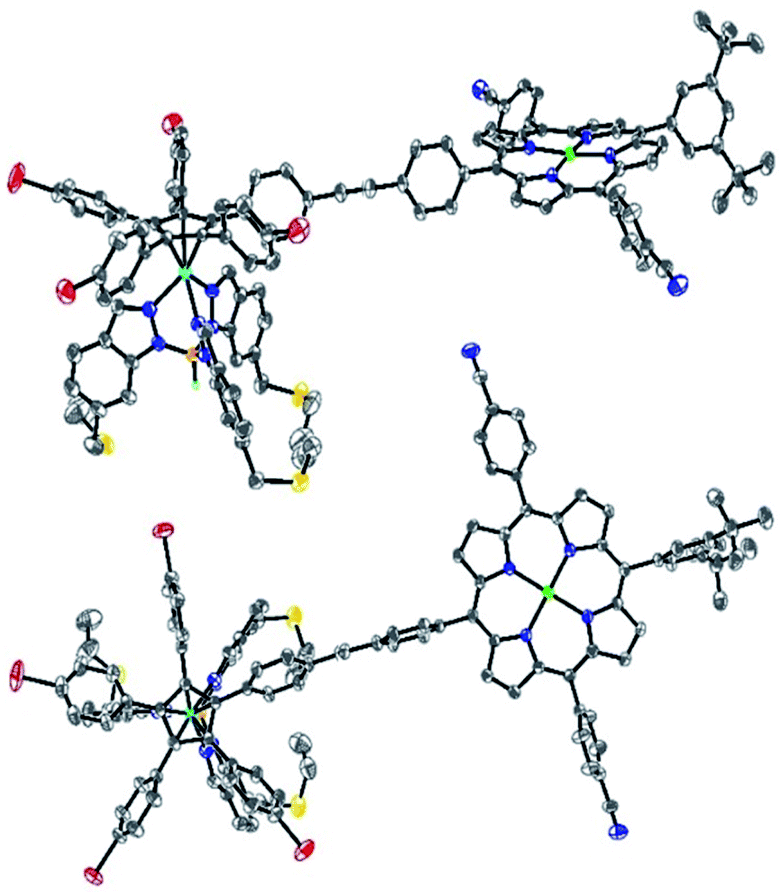 | ||
| Fig. 5 Side view (top) and top view (bottom) of the molecular structure of monoporphyrinic ruthenium complex 13. Thermal ellipsoids are drawn at 30% probability, and hydrogen atoms (except for B–H), solvent molecules and disordered atoms are omitted for clarity. | ||
Insertion of four identical porphyrinic paddles was next attempted with A3B-type ethynylphenylporphyrin 3 (6 equiv., i.e. 1.5 equiv./Br) as the coupling partner. After optimisation of the reaction conditions, it appeared that the Buchwald catalyst showed high efficiency in promoting the Sonogashira coupling of the very poorly reactive bromophenylenes. Using a combination of Pd(OAc)2 (40 mol%, i.e. 10 mol%/Br) and SPhos ligand (80 mol%) as the precatalyst, with CuI (30 mol%) as the second catalyst, the desired unsymmetrical pentaporphyrinic cogwheel 10 was isolated in 49% yield after 48 h at 95 °C in a DMF/diisopropylamine (4:1) mixture. This first example of a chemically-tagged ruthenium-based cogwheel was fully characterised (vide infra), thereby unambiguously confirming the presence of the five identical phenylethynylphenyl spacers carrying distinct porphyrinic units.
Next, the synthesis of smaller chemically-tagged cogwheels involving 4,4′-biphenyl spacers was tackled. In a similar way, it was envisioned to introduce first the distinct paddle via a chemoselective aryl–aryl bond formation starting from monoiodinated key precursor 1, and subsequently couple the four remaining bromophenylene moieties.
A huge variety of catalytic systems are known to achieve biaryl couplings, many of them involving palladium species.38 Based on the higher reactivity of aryl iodides than that of aryl bromides in the oxidative addition elementary step, successful examples of chemoselective monocoupling of bromoiodobenzene derivatives have been reported, but the extent of the selectivity relies on a subtle interplay between the nature of the coupling partner (and thus the cross-coupling type), the choice of the pre-catalyst and ligand system, the steric and electronic bias of the substrate, and the reaction conditions. In addition, our star-shaped ruthenium-based system includes four bromophenyl and one single iodophenyl positions that are spatially and electronically decoupled, and thus they react fully independently. As mentioned earlier, they are highly deactivated by the central electron-rich cyclopentadienyl ring, and the choice of catalyst is restricted to highly active systems, which is not in favour of a Br vs. I discrimination. An extensive optimisation was not desirable at that stage of a multi-step synthesis, involving costly and structurally complex coupling partners. It was thus decided to carry on with Suzuki–Miyaura cross-couplings for the creation of the aryl–aryl bond, even though very few examples of successful iodo- vs. bromoaryl discrimination have been reported in unbiased substrates.43 Interestingly, it has been inferred that substituting aryl bromide with aryl iodide leads to a change from oxidative addition to transmetalation in the rate-determining elementary step of such a cross-coupling.44 Given the success encountered in Sonogashira monocoupling on the monoiodinated ruthenium complex, it appears that temperature-controlled selective oxidative addition of the iodophenyl fragment is possible in our system, and we thus intended to facilitate the transmetalation step while maintaining mild temperature conditions as a way to ensure chemoselectivity. Our attention was drawn by the copper-mediated Suzuki–Miyaura coupling conditions reported by Savarin and Liebeskind, exhibiting high efficiency with aryl iodides as substrates in a nonbasic medium at room temperature.45 This palladium-catalysed process involves the copper(I) thiophene-2-carboxylate (CuTC) complex as a stoichiometric additive, which has been shown to facilitate the B to Pd transmetalation step when using arylboronic acids as coupling partners. Under such mild conditions, high I vs. Br selectivity has been reported on a simple bromoiodobenzene substrate, which prompted us to test these conditions on our more structurally complex system. A trans A2BC-type porphyrin carrying two electron-withdrawing 4-cyanophenyl substituents on the 10- and 20-positions as well as a phenylboronic acid moiety was thus required. Unfortunately, we failed to obtain this coupling partner, presumably due to nickel(II)-mediated homocoupling side reactions during the metalation step.
By default, a standard Suzuki–Miyaura cross-coupling was attempted, involving equimolar amounts of unsymmetrical monoiodinated complex 1 and nickel(II) porphyrin 6 incorporating a boronic acid pinacol ester moiety (Scheme 4). In the presence of PdCl2(dppf) (10 mol%) as a catalyst and 2 equiv. of Cs2CO3 as a base in a DMF/water (99:1) system, no conversion was observed at room temperature. Heating to 100 °C led to the formation of the desired monocoupled product, along with poly-substituted compounds. After tedious chromatographic separation, the desired monoporphyrinic ruthenium complex 14 was finally isolated in 8% yield, which is comparable to the result of a statistical reaction on symmetrical pentabrominated complex 2 (6% yield). Such similar yields obtained with unsymmetrical and symmetrical penta(p-halogenophenyl) precursors 1 and 2 confirm that no significant chemoselectivity is achieved in our system with a Suzuki–Miyaura coupling solely catalysed by palladium. Moreover, formation of polyporphyrinic side products leads to a challenging purification, which explains the very low yields obtained in this step.
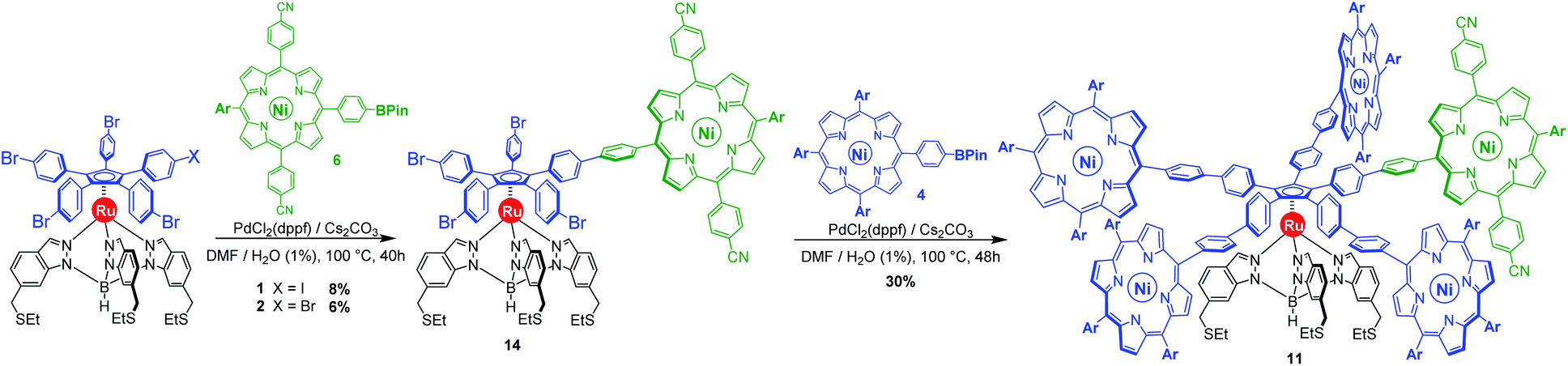 | ||
| Scheme 4 Synthesis of the chemically-tagged cogwheel 11, incorporating one distinct electron-deficient porphyrinic macrocycle (in green), via sequential Suzuki–Miyaura cross-coupling reactions. Ar = 3,5-di-tert-butylphenyl. | ||
Subsequent four-fold biaryl coupling was carried out on monofunctionalised complex 14 with A3B-type porphyrin 4 as the coupling partner (8 equiv.) to afford in 30% yield the chemically-tagged cogwheel 11 possessing five shorter identical 4,4′-biphenyl linkers and distinct porphyrin paddles.
Two desymmetrised cogwheels of increasing diameter 11 and 10 were thus obtained via sequential reactions, with the first single coupling being either chemoselective or statistical. In these rotors, the contrast in STM images will stem from the electronic differentiation of one macrocyclic scaffold, bearing electron-withdrawing cyanophenyl substituents. As an alternative, it was also envisioned to change the nature of one metallic centre while keeping the macrocycle constant, and the corresponding cogwheel prototype involving one zinc(II) and four nickel(II) centres was next synthesised.
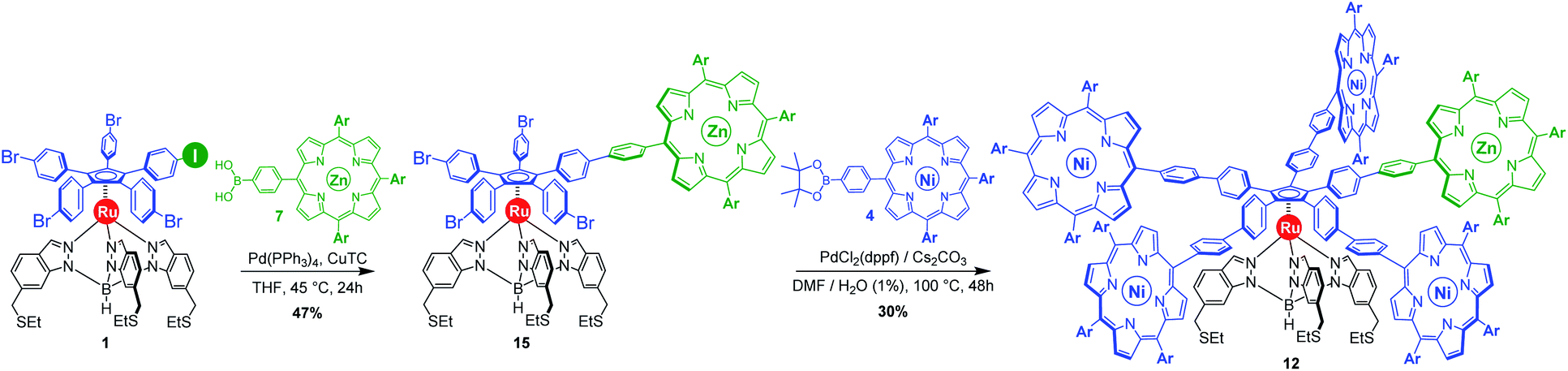 | ||
| Scheme 5 Synthesis of the chemically-tagged cogwheel 12, incorporating one distinct zinc(II) porphyrin (in green), via sequential chemoselective Suzuki–Miyaura cross-coupling reactions. Ar = 3,5-di-tert-butylphenyl. | ||
The monoporphyrinic ruthenium complex 15 was finally submitted to a four-fold Suzuki–Miyaura coupling under standard conditions with nickel(II) porphyrin 4 bearing a phenylboronic acid pinacol ester substituent. The desired desymmetrised cogwheel 12 possessing four nickel(II) A3B-type porphyrins and a single zinc(II) porphyrin, connected by five identical 4,4′-biphenyl linkers, was obtained in 30% yield. Thanks to Liebeskind copper-mediated conditions for the selective monofunctionalisation of the pentahalogenated precursor, the overall yield for the synthesis of desymmetrised pentaporphyrinic cogwheels 11 and 12, bearing 4,4′-biphenyl spacers, has undergone a six-fold increase and has now reached 3% over seven steps, starting from 3-amino-4-methylbenzoic acid.
Remarkably, the new cogwheel prototype 12 incorporates three distinct metal types shared on six different positions, with respective roles ranging from a mechanical ball bearing to electronic contrast agent.
2.4. Spectroscopic and electrochemical studies of chemically-tagged cogwheels
The influence of the chemically-distinct porphyrinic paddle on the spectroscopic and electrochemical properties of the larger ethynyl-containing cogwheel 10 and smaller biphenylene-derived cogwheels 11 and 12 was investigated.:4 of the signals of the chemically-tagged porphyrin (as identified in the spectra of intermediates 13, 14 and 15). This implies that the incorporation of a distinct porphyrin has a minor impact on the environment of the ruthenium-based cogwheel. In 1H and 13C NMR, the signals related to the tripodal ligand, the phenylethynylphenyl or 4,4′-biphenyl spacers and the four A3B-type nickel(II) porphyrins remain unchanged, as exemplified in Fig. 6 (signals depicted in blue and black) in the case of cogwheel 12. In contrast, specific patterns for the distinct porphyrin (A2BC-type nickel(II) porphyrin for 10 and 11, or A3B-type zinc(II) porphyrin for 12) are observed, in particular for the β-pyrrole protons (8.5–9.0 ppm region) and for the aromatic protons of the 3,5-bis-tert-butylphenyl moiety. This has been highlighted in green in Fig. 6 in the case of compound 12, for which the nature of the metal was expected to behave as a chemical tag. Indeed, all the signals belonging to the zinc(II) porphyrin and its meso substituents are clearly shifted downfield under the influence of the zinc centre, compared to the nickel(II) counterparts. This electron-deficient character of the zinc(II) porphyrinic fragment is very promising to generate spatial contrast in on-surface STM studies of single-molecules.
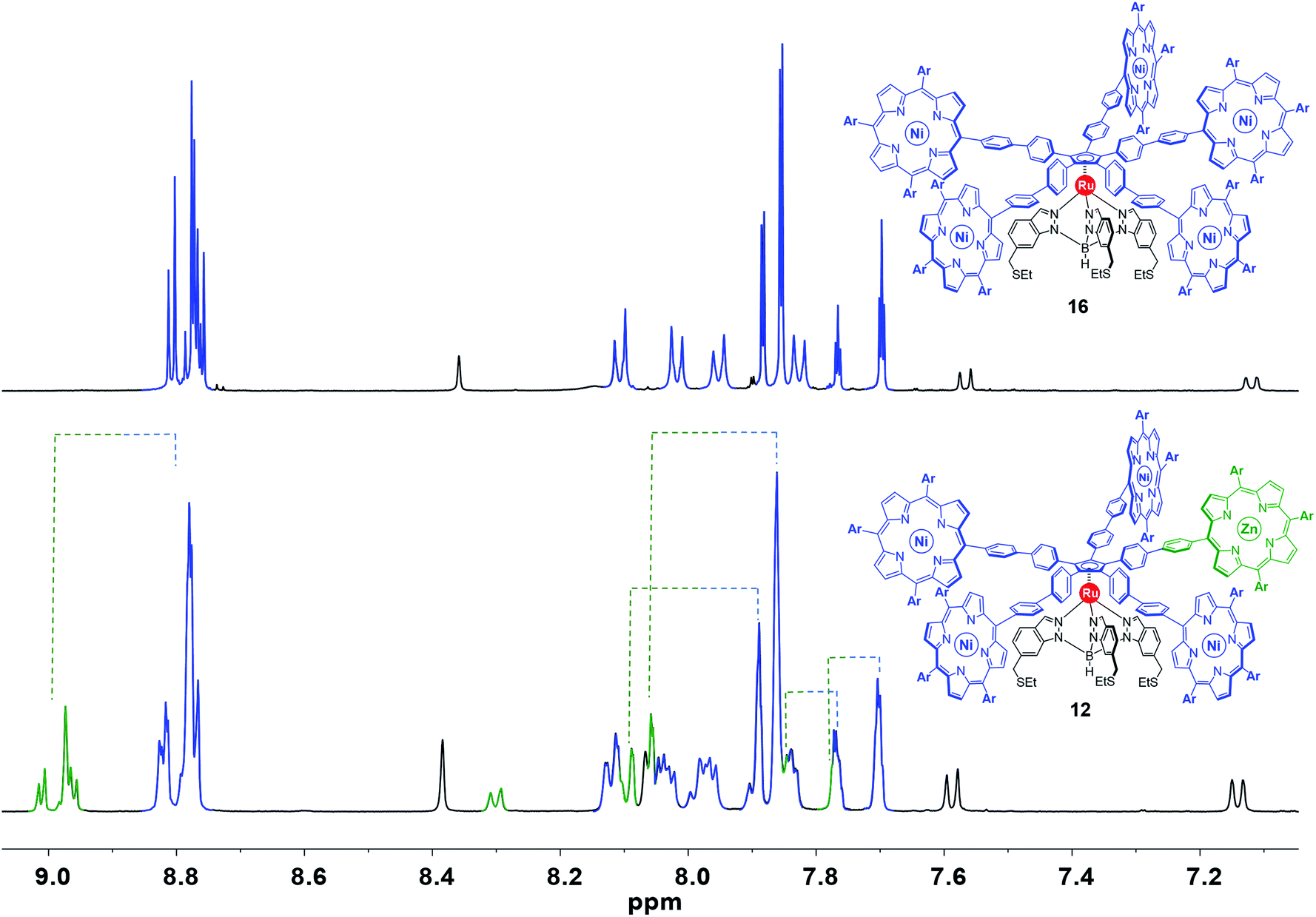 | ||
| Fig. 6 Aromatic region of the 1H NMR spectrum (500 MHz, CD2Cl2) of the symmetric cogwheel prototype 16 bearing 4,4′-biphenyl spacers and A3B-type nickel(II) 10,15,20-tris(3,5-di-tert-butylphenyl)porphyrins35 (top) and of the desymmetrised cogwheel prototype 12 incorporating an A3B-type zinc(II) porphyrin as the chemical tag. Spectra are coloured in black, green and blue according to each fragment of the molecules (Ar = 3,5-di-tert-butylphenyl). | ||
In addition, it is important to mention that porphyrins undergo unrestricted rotation around the phenylethynylphenyl or 4,4′-biphenyl axes, and that free rotation around the Cp–Ru–B axis takes place at room temperature. Variable-temperature 1H NMR experiments in CD2Cl2 down to −60 °C did not reveal any restricted rotation.
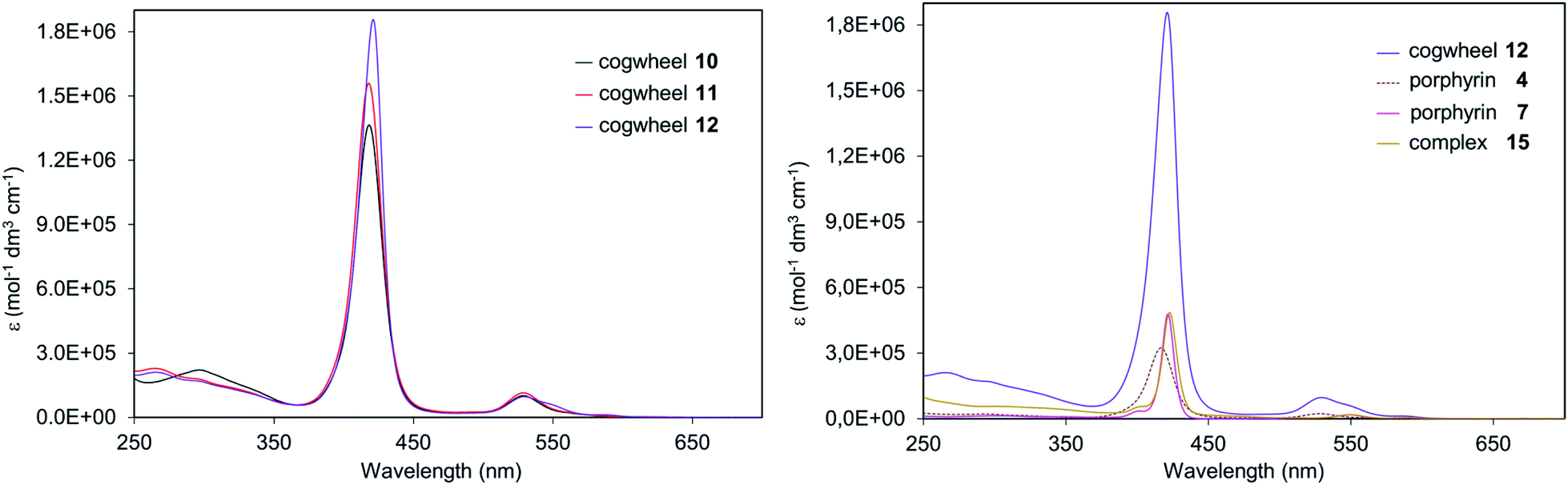 | ||
| Fig. 7 UV/Vis absorption spectra of desymmetrised pentaporphyrinic cogwheels 10, 11 and 12 (left) and of the series of compounds including porphyrins 4 and 7, complex 15 and cogwheel 12 (right) in CH2Cl2. | ||
In Fig. 7 (right), comparison of porphyrinic precursors 4 (dashed curve) and 7 (pink curve) highlights the known stronger absorption of zinc(II) porphyrins than that of nickel(II) analogues, which is further evidenced by the left spectrum with the zinc-containing cogwheel 12 having the highest molar extinction coefficient in the Soret band. Interestingly, sequential addition of porphyrin units on the ruthenium-based scaffold does not modify the absorption properties of each subunit, thus showing that the pentaarylcyclopentadienyl ruthenium(II) centre and the porphyrins are electronically decoupled (Fig. 7, right). Moreover, the intensity of the Soret and Q bands in such porphyrinic complexes (e.g.15vs.12) is directly proportional to the number of porphyrins present in the molecular structure, thus revealing the additivity of chromophores’ absorption properties in ruthenium complexes 10–12.
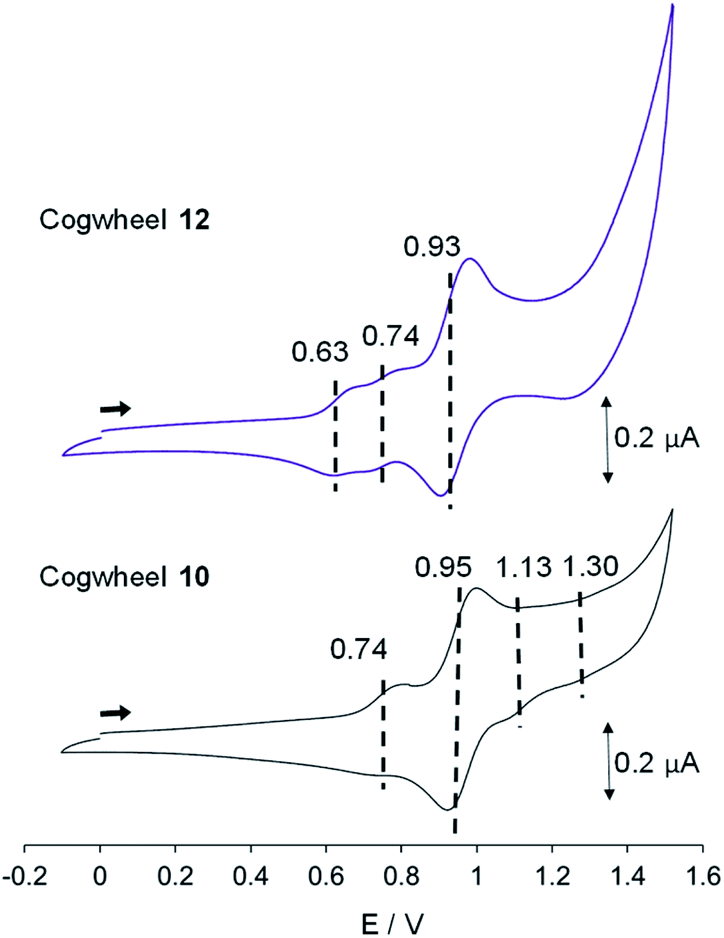 | ||
| Fig. 8 Cyclic voltammograms of cogwheels 10 and 12versus SCE (CH2Cl2, 0.1 M nBu4NPF6, 25 °C, scan rate: 0.1 V s−1). | ||
In the reduction region of the voltammogram (Fig. S4 and S5 in the ESI section†), no reversible wave is observed for ruthenium-based cogwheels 10 and 12 down to the voltage limit of −1.5 V. However, the first reduction of the porphyrin precursor 5 carrying two 4-cyanophenyl substituents occurs reversibly at E1/2 = −1.25 V (vs. SCE) and this redox behaviour is still observed in the corresponding monoporphyrinic ruthenium complex 13. It can thus be inferred that the first reduction in pentaporphyrinic complex 10 will take place on the tagged A2BC nickel(II) porphyrin, in line with its electron-deficient character compared to the remaining A3B nickel(II) porphyrins. This localised redox behaviour will be exploited in on-surface STM studies to induce contrast within the single molecular cogwheel.
3. Conclusion
Four desymmetrised pentaporphyrinic molecular gears were carefully designed and synthesised, with the aim of assembling them into trains of gears on a surface and ultimately achieving controlled intermolecular gearing. These star-shaped organometallic structures incorporate a hydrotris(indazolyl)borate ligand as an anchor to prevent diffusion on the surface, a central ruthenium atom as a fixed rotation axis, and an azimuthal pentaporphyrinic cyclopentadienyl cogwheel specifically labelled for STM studies.Desymmetrisation of the cogwheel architecture was first achieved by introducing one tooth longer than the other four, using a phenylethynylphenyl spacer instead of a 4,4′-biphenylene between the porphyrinic macrocycle and the cyclopentadienyl core. To allow for optimal mechanical interactions within the trains of gears, chemical labelling was next investigated as a way to induce local submolecular contrast in STM images while maintaining identical lengths and homogeneous peripheral shapes for the paddles. The electronic properties of one single porphyrinic paddle were successfully modulated either by introducing electron-withdrawing p-cyanophenyl substituents on the tetrapyrrole core or by changing the nature of the central metal from nickel(II) to zinc(II), as evidenced by spectroscopic and electrochemical studies.
To reach the required structural diversity, a modular synthetic approach was followed, relying on a penta(p-halogenophenyl)cyclopentadienyl ruthenium(II) key building block, bearing a single pre-activated p-iodophenyl group. Post-functionalisation was achieved via sequential palladium-catalysed cross-coupling reactions, starting with a chemoselective coupling of the iodophenylene moiety under Sonogashira or more challenging Suzuki–Miyaura conditions, requiring the assistance of copper(I) additives. This controlled introduction of the sterically- or chemically-tagged porphyrinic tooth followed by four-fold cross-couplings on the remaining p-bromophenyl positions yielded four prototypes of desymmetrised pentaporphyrinic molecular gears with diameters over 5 nm. Mechanical studies on surfaces and on the single molecule scale are currently underway.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Paul Sabatier University (Toulouse) and CNRS. It has received funding from the ANR (ACTION project ANR-15-CE29-0005), the European Union's Horizon 2020 research and innovation programme under the project MEMO, grant agreement no. 766864, the JSPS KAKENHI grant in aid for Scientific Research on Innovative Areas ‘‘Molecular Engine (no. 8006)’’ 18H05419 and the KAKENHI Grant-in-Aid for Challenging Research (20K21131). DFT calculations were performed using HPC resources from CALMIP (grant 2020-p20041). Y. G. thanks the French Ministry of National Education for a PhD Fellowship. Dr Jacques Bonvoisin is gratefully acknowledged for his help with cyclic voltammetry experiments.Notes and references
- (a) J.-P. Sauvage, Angew. Chem., Int. Ed., 2017, 56, 11080–11093 CrossRef CAS; (b) J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS; (c) B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 11060–11078 CrossRef CAS.
- (a) V. Balzani, A. Credi and M. Venturi, Molecular Devices and Machines. Concepts and Perspectives for the Nanoworld, Wiley-VCH, Weinheim, 2nd edn, 2008 CrossRef; (b) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS.
- (a) G. Rapenne, Org. Biomol. Chem., 2005, 3, 1165–1169 RSC; (b) A. A. Gakh, Molecular Devices: An Introduction to Technomimetics and Its Biological Applications, John Wiley & Sons, Hoboken, NJ, 2018 CrossRef; (c) C. Kammerer, G. Erbland, Y. Gisbert, T. Nishino, K. Yasuhara and G. Rapenne, Chem. Lett., 2019, 48, 299–308 CrossRef CAS.
- A. Ikeda, T. Tsudera and S. Shinkai, J. Org. Chem., 1997, 62, 3568–3574 CrossRef CAS.
- G. Rapenne and G. Jimenez-Bueno, Tetrahedron, 2007, 63, 7018–7026 CrossRef CAS.
- (a) Y. Shirai, A. J. Osgood, Y. Zhao, K. F. Kelly and J. M. Tour, Nano Lett., 2005, 5, 2330–2334 CrossRef CAS; (b) T. Kudernac, N. Ruangsupapichat, M. Parschau, B. Maciá, N. Katsonis, S. R. Harutyunyan, K.-H. Ernst and B. L. Feringa, Nature, 2011, 479, 208–211 CrossRef CAS; (c) T. Nishino, C. J. Martin, H. Takeuchi, F. Lim, K. Yasuhara, Y. Gisbert, S. Abid, N. Saffon-Merceron, C. Kammerer and G. Rapenne, Chem.–Eur. J., 2020, 26, 12010–12018 CrossRef CAS.
- T. Muraoka, K. Kinbara, Y. Kobayashi and T. Aida, J. Am. Chem. Soc., 2003, 125, 5612–5613 CrossRef CAS.
- J. D. Badjic, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science, 2004, 303, 1845–1849 CrossRef CAS.
- (a) S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46, 2592–2621 RSC; (b) M. Baroncini, S. Silvi and A. Credi, Chem. Rev., 2020, 120, 200–268 CrossRef CAS; (c) D. Dattler, G. Fuks, J. Heiser, E. Moulin, A. Perrot, X. Yao and N. Giuseppone, Chem. Rev., 2020, 120, 310–433 CrossRef CAS; (d) V. García-López, D. Liu and J. M. Tour, Chem. Rev., 2020, 120, 79–124 CrossRef; (e) A. Goswami, S. Saha, P. K. Biswas and M. Schmittel, Chem. Rev., 2020, 120, 125–199 CrossRef CAS.
- T. R. Kelly, H. De Silva and R. A. Silva, Nature, 1999, 401, 150–152 CrossRef CAS.
- N. Koumura, R. W. J. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS.
- (a) H. L. Tierney, C. J. Murphy, A. D. Jewell, A. E. Baber, E. V. Iski, H. Y. Khodaverdian, A. F. McGuire, N. Klebanov and E. C. H. Sykes, Nat. Nanotechnol., 2011, 6, 625–629 CrossRef CAS; (b) U. G. E. Perera, F. Ample, H. Kersell, Y. Zhang, G. Vives, J. Echeverria, M. Grisolia, G. Rapenne, C. Joachim and S.-W. Hla, Nat. Nanotechnol., 2013, 8, 46–51 CrossRef CAS.
- (a) L. Grill, K.-H. Rieder, F. Moresco, G. Rapenne, S. Stojkovic, X. Bouju and C. Joachim, Nat. Nanotechnol., 2007, 2, 95–98 CrossRef CAS; (b) W.-H. Soe, Y. Shirai, C. Durand, Y. Yonamine, K. Minami, X. Bouju, M. Kolmer, K. Ariga, C. Joachim and W. Nakanishi, ACS Nano, 2017, 11, 10357–10365 CrossRef CAS; (c) W.-H. Soe, C. Durand, O. Guillermet, S. Gauthier, H.-P. Jacquot de Rouville, S. Srivastava, C. Kammerer, G. Rapenne and C. Joachim, Nanotechnology, 2018, 29, 495401 CrossRef; (d) F. Eisenhut, J. Meyer, J. Krüger, R. Ohmann, G. Cuniberti and F. Moresco, Surf. Sci., 2018, 678, 177–182 CrossRef CAS; (e) Y. Zhang, J. P. Calupitan, T. Rojas, R. Tumbleson, G. Erbland, C. Kammerer, T. M. Ajayi, S.-W. Wang, L. A. Curtiss, A. T. Ngo, S. E. Ulloa, G. Rapenne and S.-W. Hla, Nat. Commun., 2019, 10, 3742 CrossRef; (f) G. J. Simpson, V. García-López, A. D. Boese, J. M. Tour and L. Grill, Nat. Commun., 2019, 10, 4631 CrossRef.
- C. Joachim and G. Rapenne, ACS Nano, 2013, 7, 11–14 CrossRef CAS.
- G. Rapenne and C. Joachim, Nat. Rev. Mater., 2017, 2, 17040–17042 CrossRef.
- T. Freeth, A. Jones, J. M. Steele and Y. Bitsakis, Nature, 2008, 454, 614–617 CrossRef CAS.
- J. Yang, J. Deng, C. Troadec, T. Ondarçuhu and C. Joachim, Nanotechnology, 2014, 25, 465305 CrossRef.
- W. D. Hounshell, C. A. Johnson, A. Guenzi, F. Cozzi and K. Mislow, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 6961–6964 CrossRef CAS.
- Y. Kawada and H. Iwamura, J. Org. Chem., 1980, 45, 2547–2548 CrossRef CAS.
- H. Iwamura, T. Ito, H. Ito, K. Toriumi, Y. Kawada, E. Osawa, T. Fujiyoshi and C. Jaime, J. Am. Chem. Soc., 1984, 106, 4712–4717 CrossRef CAS.
- D. K. Frantz, A. Linden, K. K. Baldridge and J. S. Siegel, J. Am. Chem. Soc., 2012, 134, 1528–1535 CrossRef CAS.
- K. H. Au Yeung, T. Kühne, F. Eisenhut, M. Kleinwächter, Y. Gisbert, R. Robles, N. Lorente, G. Cuniberti, C. Joachim, G. Rapenne, C. Kammerer and F. Moresco, J. Phys. Chem. Lett., 2020, 11, 6892–6899 CrossRef CAS.
- W.-H. Soe, S. Srivastava and C. Joachim, J. Phys. Chem. Lett., 2019, 10, 6462–6467 CrossRef CAS.
- (a) H. Iwamura and K. Mislow, Acc. Chem. Res., 1988, 21, 175–182 CrossRef CAS; (b) K. Okamura, Y. Inagaki, H. Momma, E. Kwon and W. Setaka, J. Org. Chem., 2019, 84, 14636–14643 CrossRef CAS.
- W. Setaka, T. Nirengi, C. Kabuto and M. Kira, J. Am. Chem. Soc., 2008, 130, 15762–15763 CrossRef CAS.
- H. Ube, Y. Yasuda, H. Sato and M. Shionoya, Nat. Commun., 2017, 8, 14296 CrossRef CAS.
- (a) F. Huang, G. Wang, L. Ma, Y. Wang, X. Chen, Y. Che and H. Jiang, J. Org. Chem., 2017, 82, 12106–12111 CrossRef CAS; (b) X. Jiang, S. Yang, M. J. Jellen, K. N. Houk and M. Garcia-Garibay, Org. Lett., 2020, 22, 4049–4052 CrossRef CAS.
- (a) A. M. Stevens and C. J. Richards, Tetrahedron Lett., 1997, 38, 7805–7808 CrossRef CAS; (b) S. Ogi, T. Ikeda, R. Wakabayashi, S. Shinkai and M. Takeuchi, Chem.–Eur. J., 2010, 16, 8285–8290 CrossRef CAS.
- (a) M. Nakamura, K. Kishimoto, Y. Kobori, T. Abe, K. Yoza and K. Kobayashi, J. Am. Chem. Soc., 2016, 138, 12564–12577 CrossRef CAS; (b) H. Ube, R. Yamada, J. Ishida, H. Sato, M. Shiro and M. Shionoya, J. Am. Chem. Soc., 2017, 139, 16470–16473 CrossRef CAS; (c) S. Toyota, K. Kawahata, K. Sugahara, T. Oki and T. Iwanaga, Asian J. Org. Chem., 2019, 8, 1919–1923 CrossRef CAS.
- (a) N. Tanaka, Y. Inagaki, K. Yamaguchi and W. Setaka, Cryst. Growth Des., 2020, 20, 1097–1102 CrossRef CAS; (b) I. Liepuoniute, M. J. Jellen and M. A. Garcia-Garibay, Chem. Sci., 2020, 11, 12994–13007 RSC.
- (a) F. Chiaravalloti, L. Gross, K.-H. Rieder, S. M. Stojkovic, A. Gourdon, C. Joachim and F. Moresco, Nat. Mater., 2007, 6, 30–33 CrossRef CAS; (b) C. Manzano, W.-H. Soe, H. S. Wong, F. Ample, A. Gourdon, N. Chandrasekhar and C. Joachim, Nat. Mater., 2009, 8, 576–579 CrossRef CAS.
- W.-H. Soe, M. Kleinwächter, C. Kammerer, G. Rapenne and C. Joachim, J. Phys. Chem. C, 2020, 124, 22625–22630 CrossRef CAS.
- G. Vives, H.-P. Jacquot de Rouville, A. Carella, J.-P. Launay and G. Rapenne, Chem. Soc. Rev., 2009, 38, 1551–1561 RSC.
- C. Kammerer and G. Rapenne, Eur. J. Inorg. Chem., 2016, 2214–2226 CrossRef CAS.
- G. Erbland, S. Abid, Y. Gisbert, N. Saffon-Merceron, Y. Hashimoto, L. Andreoni, T. Guérin, C. Kammerer and G. Rapenne, Chem.–Eur. J., 2019, 25, 16328–16339 CrossRef CAS.
- G. Erbland, Y. Gisbert, G. Rapenne and C. Kammerer, Eur. J. Org. Chem., 2018, 4731–4739 CrossRef CAS.
- R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
- J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1470 CrossRef CAS.
- Y. Gisbert, S. Abid, G. Bertrand, N. Saffon-Merceron, C. Kammerer and G. Rapenne, Chem. Commun., 2019, 55, 14689–14692 RSC.
- Y. Zhang, H. Kersell, R. Stefak, J. Echeverria, V. Iancu, U. G. E. Perera, Y. Li, A. Deshpande, K.-F. Braun, C. Joachim, G. Rapenne and S.-W. Hla, Nat. Nanotechnol., 2016, 11, 706–712 CrossRef CAS.
- (a) M. J. Frisch, et al., Gaussian 09 Revision D.01, Gaussian Inc., Wallingford CT, 2013, (see ESI† for full author list) Search PubMed; (b) C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS.
- H. Marbach, Acc. Chem. Res., 2015, 48, 2649–2658 CrossRef CAS.
- Stille cross-couplings have been far more documented for aryl bromide vs. aryl iodide discrimination but preliminary studies involving monoiodinated key precursor 1 and a tributyl(p-tolyl)stannane as a model coupling partner failed to give the desired monocoupled product in satisfactory yield.
- (a) G. B. Smith, G. C. Dezeny, D. L. Hughes, A. O. King and T. R. Verhoeven, J. Org. Chem., 1994, 59, 8151–8156 CrossRef CAS; (b) A. F. Littke, C. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020–4028 CrossRef CAS.
- C. Savarin and L. S. Liebeskind, Org. Lett., 2001, 3, 2149–2152 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2039772 and 2039773. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06379g |
| This journal is © The Royal Society of Chemistry 2021 |