
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular strategies in artificial photosynthesis
Tom
Keijer
,
Tessel
Bouwens
,
Joeri
Hessels
and
Joost N. H.
Reek
 *
*
Homogeneous Supramolecular and Bio-inspired Catalysis, Van't Hoff Institute for Molecular Sciences (HIMS), University of Amsterdam (UvA), Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: j.n.h.reek@uva.nl
First published on 16th November 2020
Abstract
Artificial photosynthesis is a major scientific endeavor aimed at converting solar power into a chemical fuel as a viable approach to sustainable energy production and storage. Photosynthesis requires three fundamental actions performed in order; light harvesting, charge-separation and redox catalysis. These actions span different timescales and require the integration of functional architectures developed in different fields of study. The development of artificial photosynthetic devices is therefore inherently complex and requires an interdisciplinary approach. Supramolecular chemistry has evolved to a mature scientific field in which programmed molecular components form larger functional structures by self-assembly processes. Supramolecular chemistry could provide important tools in preparing, integrating and optimizing artificial photosynthetic devices as it allows precise control over molecular components within such a device. This is illustrated in this perspective by discussing state-of-the-art devices and the current limiting factors – such as recombination and low stability of reactive intermediates – and providing exemplary supramolecular approaches to alleviate some of those problems. Inspiring supramolecular solutions such as those discussed herein will incite expansion of the supramolecular toolbox, which eventually may be needed for the development of applied artificial photosynthesis.
1 Introduction
Humanity has a solar-power debt as we have been using fossil energy, which was collected by photosynthetic organisms over the course of millions of years, in a faster rate than is currently generated, leading to high CO2 emissions and climate change as a result. Sustainable energy is sufficiently available; one hour of incident solar power is enough to account for the yearly worldwide energy consumption.1 Decades of research into crystalline silicon solar cells resulted in the production of sustainable electricity on industrial scale for commercially competitive prices. However, 80% of the energy is consumed in the form of fuel, and it is likely that fuels will remain important in the future. Moreover, electrical power is not easily stored and transported.2 Combined with the mismatch in energy demand and solar energy supply due to the day/night cycle and the seasonal fluctuations, various energy storage strategies should be developed to allow the transition to a society that runs on sustainable energy.3 In this context, the production of a chemical fuel by converting solar-energy to fuel (a solar-fuel), by direct or indirect means, is crucial. Plants and cyanobacteria have been converting solar energy to fossil fuel for millions of years already.4 As such they form an inspiring example, or even a blue print, to generate man-made devices that directly produce fuels from solar energy, often referred to as artificial photosynthesis.2,5–71.1 The fundamental actions of photosynthesis
For both natural and artificial photosynthesis, the same three fundamental actions are required to convert solar energy into a fuel. (i) Light-harvesting, (ii) charge-separation and (iii) redox catalysis (Fig. 1).8,9 Nature has evolved to highly dedicated and ordered, but complex supramolecular architectures, that can capture the light, and transmit the energy with high efficiency. Also the catalytic processes carried out by complex enzymes are highly efficient.10 The excellent properties of biological systems are a result of fine-tuned molecular components for the various tasks, that are well-organized in space by the use of dynamic non-covalent bonds. As this organization is important, the efficiency in photosynthesis may be deduced from, but not reduced to, the properties of components at a molecular level.11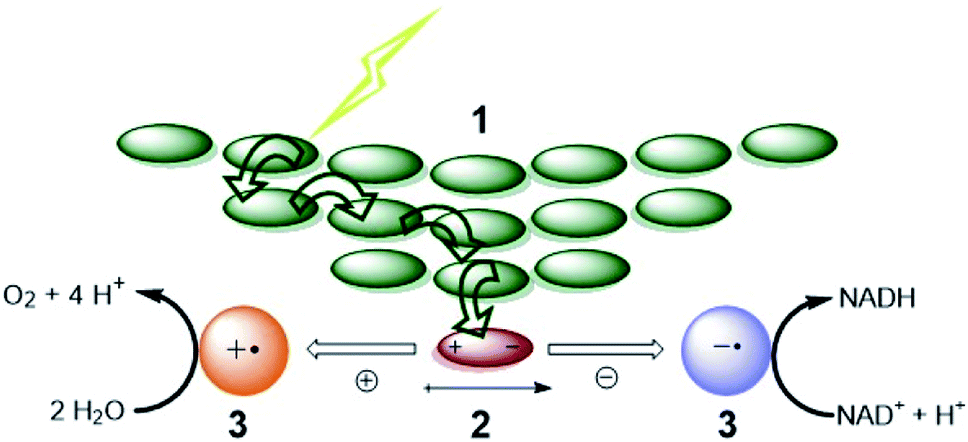 | ||
| Fig. 1 The fundamental actions of photosynthesis. (1) Light harvesting. (2) Charge-separation. (3) Redox catalysis. Notice that energy transfer occurs between chromophores (green arrows) after excitation to funnel the energy to the special pair where it leads to charge separation. Some energy losses are required to drive the charge separation forward. Charges are moved to catalysts that drive uphill chemical reactions. Adapted with permission from ref. 8. | ||
Supramolecular chemistry based on well-defined non-covalent interactions between molecules is a powerful tool to achieve larger, organized molecular structures with an increased level of complexity through self-assembly. As such, it provides a tool to optimize properties of structures required for artificial photosynthesis in a bio-inspired fashion. Like in natural systems, preorganization leads to improved energy transfer processes, charge-separation and redox catalysis. In this perspective we will focus on supramolecular strategies to perform the fundamental actions of photosynthesis in artificial systems. Such supramolecular strategies are underexplored, but have potential to circumvent the problems in state-of-the-art artificial photosynthesis. For those new to this interdisciplinary field, a brief background on solar to fuel devices will be presented. After exploring the limiting factors in present-day devices, exemplary supramolecular tools will be discussed in order to perform the three fundamental actions of photosynthesis more efficiently, taking all current and future bottlenecks into account. Firstly, we will focus on supramolecular light harvesting, charge-separation and the combination thereof. This is followed by supramolecular catalysis for artificial photosynthesis. Strategies in water oxidation and proton reduction are highlighted. A discussion of homogeneous artificial supramolecular photosynthesis will show the potential of combined supramolecular strategies. Finally, devices already implementing supramolecular strategies are presented in the last section. This perspective will demonstrate the power of supramolecular chemistry and attempts to inspire readers to apply supramolecular thinking to overcome problems associated with artificial photosynthesis, hopefully adding to the expanding supramolecular toolbox.
2 Artificial photosynthesis
2.1 From electricity to fuels
Any man-made system that performs the three fundamental actions of photosynthesis can be considered an artificial photosynthesis device. Three prominent device designs are PV-electrocatalysis, particulate semiconductor photocatalysts (PSP) and photoelectrochemical cells (PEC).12,13 Dye-sensitized solar cells (DSSC) are solar cells based on molecular components. For the purpose of fuel production, the molecular strategy ties dyes to (molecular) catalysts that perform photoredox reactions. This type of PEC is called a dye-sensitized photoelectrochemical cell (DS-PEC).14 The supramolecular solutions presented in this perspective may serve each of the designs, but the DS-PEC will profit the most from supramolecular strategies as they are based on molecular components. Catalysis is currently the bottleneck in DS-PEC performance, but some efficiency limiting processes in DSSC (e.g. charge recombination) are similar to processes that may limit the efficiency of DS-PEC. Studying and stretching the limits of DSSC with clever supramolecular strategies potentially translate to DS-PEC as well.2.2 Device designs
A schematic picture of the n-type and p-type DSSC is depicted in Fig. 2. Both types consist of a semiconductor, most often TiO2 for n-type or NiO for p-type, on a conducting substrate (FTO). A molecular dye with the proper energy levels is anchored onto the semiconductor, usually by covalent bonds. Photoexcitation of the dye results in rapid injection into the semiconductor, and the ionized molecular dye is neutralized by the redox mediator (or the catalyst in DS-PEC). The ionized redox mediator gets regenerated at the counter electrode. Forward electron propagation is depicted in Fig. 2 with green arrows and this pathway leads to photocurrent. The recombination pathways (red arrows), are non-productive pathways responsible for efficiency losses in these devices. These processes are mainly the neutralization of the ionized dye by charges from the semiconductor (Fig. 2, path 6), regeneration of the redox mediator by the semiconductor of the working electrode (Fig. 2, path 7), and the excited electron decaying to the ground state before injection (Fig. 2, path 5). Recombination pathway 7 is readily minimized by passivation of the surface via Al2O3 deposition for example. Similar processes occur for both n-type and p-type DSSC, but because the charge mobility in NiO is much lower the charge recombination events are more dominant in p-type cells. Attempts to circumvent charge recombination have mainly focused on finetuning the properties of the dyes.32–35 So called push–pull dyes prevent charge-recombination, since it leads naturally to an increased distance between the oxidized (for n-type) part of the dye and the surface, typically, minimizing recombination pathway 5. This has led to huge efficiency gains for n-type up to 14.7%.36 Currently, the p-type electrodes suffer most from recombination, the record for p-type solar cells is 2.51%.37 In order to have a working DS-PEC, analogous n-/p-type electrodes need to be coupled to catalysts that oxidize water at the anode (n-type) and produce fuel at the cathode (p-type). The low efficiencies observed for p-type DSSC will limit p-type electrodes and hence the overall efficiency of the tandem DS-PEC design. Interestingly, supramolecular strategies are emerging as a viable option to inhibit charge recombination in these devices. In this perspective we will highlight supramolecular strategies to reduce charge recombination in DSSC that act as stepping stones to efficient supramolecular artificial photosynthesis using the tandem DS-PEC design.
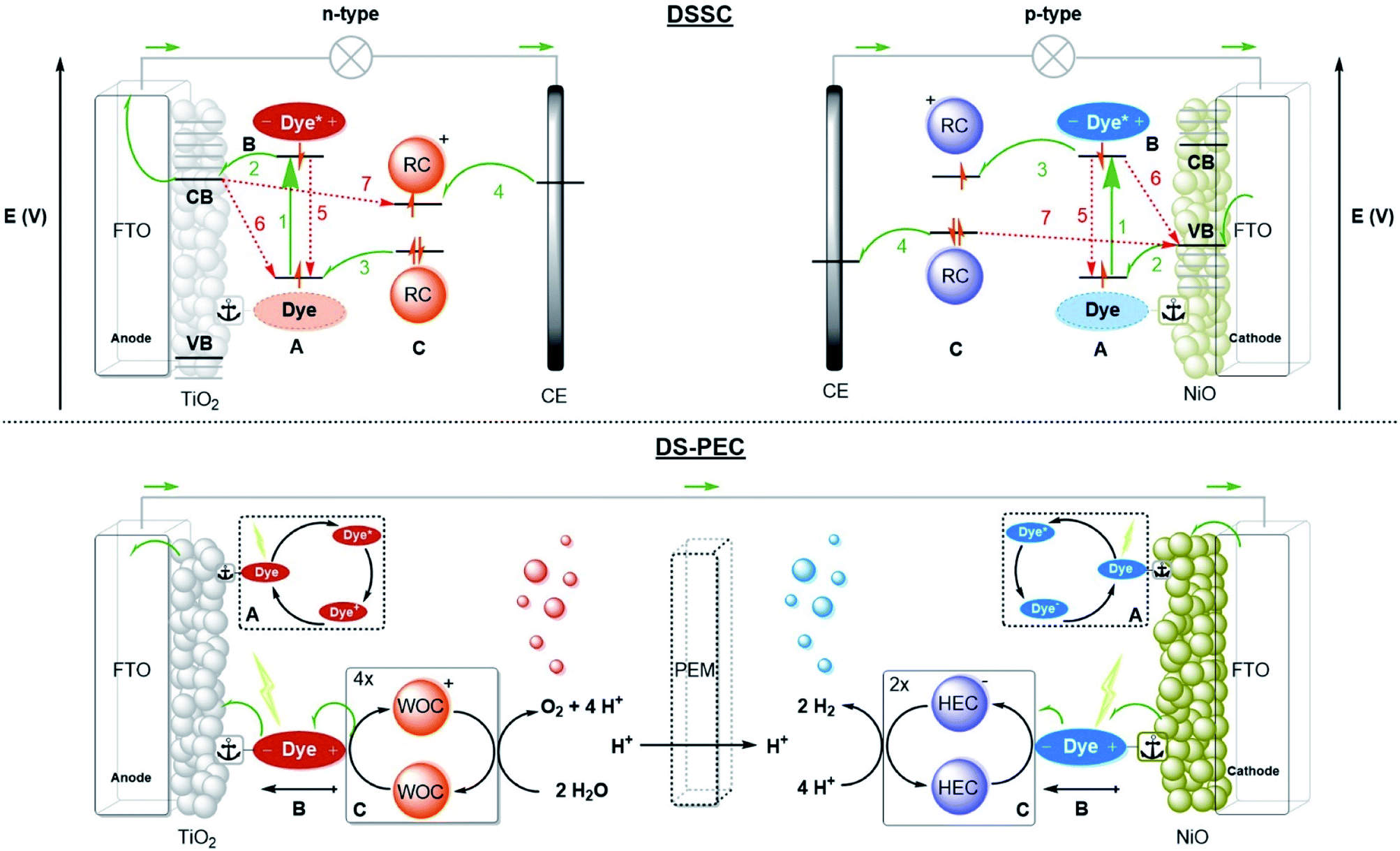 | ||
| Fig. 2 Operational mode of the DSSC (top) and schematic diagram of a DS-PEC (bottom). (A) Dyes anchored on a semiconductor deposited on conducting FTO glass are excited by light. (B) The dye performs charge-separation and (C) oxidizes or reduces the redox couple (RC) in the electrolyte that delivers the charge to the counter electrode (CE) closing the circuit. For n-type the dye is excited (process 1) followed by fast electron injection into the conduction band (CB) (process 2). The dark colour represents the dyes in the depicted electron configuration. The dye is oxidatively quenched by the redox couple (RC) (process 3). The RC is then regenerated at the counter electrode (CE) (process 4). In p-type the electrons move the other way around, but the processes are equivalent to n-type. The dye is excited (process 1) after which it is reductively quenched by an electron in the valence band (VB) of the NiO. The electron of the reduced dye is transferred to the RC (process 3) after which the redox mediator is regenerated at the CE. The possible recombination pathways are depicted with red arrows. A DS-PEC performs processes (A) & (B) in a similar fashion. Process (C) is altered by substituting the RC for a redox catalyst performing reactions such as water splitting. The catalyst may be anchored to the semiconductor surface or dye as diffusion is not required. Water oxidation catalysts (WOC) oxidize water to oxygen and protons. Four oxidations are required to generate a single oxygen molecule. The hydrogen evolution catalyst (HEC) combines the electrons and protons liberated at the anode to form hydrogen gas at the cathode. Every hydrogen molecule requires two electrons from the HEC. The half reactions are often separated by a proton exchange membrane (PEM). | ||
The operation of a DS-PEC is similar to that of a DSSC. The charge recombination pathways discussed for DSSC also apply here and result in loss of efficiency. Next to this, the catalyst properties play a role. The overpotential required for the catalytic reactions, that is the extra potential with respect to the thermodynamic potential, results in losses and as such catalysts with sufficient rates at low overpotential are needed. Also, processes span hugely different timescales, and the catalyst should be compatible with that. Based on the photon flux and absorption cross-section of the dye, the estimated photon absorption rate is 1–10 photons per s.39 This value increases considerably when light-harvesting antennae funnel energy towards the catalyst. The photophysics and electron transfer processes typically take place within picoseconds to nanoseconds, while the catalysis takes place at the millisecond timescale. This means that overall photosynthesis involves processes that differ orders of magnitude in timescale. Because the catalytic reactions are multi-electron processes, but the electron transfer are single electron transfer processes with time-delays related to the photon flux, catalysts should operate by a mechanism in which the intermediates are sufficiently stable, both in terms of decomposition and in terms of charge recombination.
With all these challenges in mind one can design supramolecular strategies to address these issues. Although such strategies are just being developed, it already provided interesting examples which will be discussed in this perspective.
3 Supramolecular light harvesting and charge separation
3.1 Inspiration from nature
Solar light is a dilute source of energy and efficient artificial photosynthesis requires the conversion of every photon. Molecular photosensitizers with long lived triplet excited-state lifetimes are frequently used in photocatalytic experiments to buy time for diffusion controlled forward reactions. This approach has several potential downsides as discussed by Würthner et al.40 (i) The long-lived triplets formed increase both the likelihood of productive pathways as well as competitive back-reactions. (ii) Singlet oxygen can be produced that damages organic ligands. (iii) Triplet sensitizers often use precious metals (e.g. Ru). (iv) Absorption coefficients are generally lower than organic dyes. (v) In addition, triplet formation dissipates photon energy being invariably lower in potential energy than the singlet. The application of supramolecular preorganization reduces the influence of diffusion and thus the need for triplet lifetimes.In natural photosystems the lifetime of the excited state is achieved by proper spatial organization of chromophores in huge protein structures. The organization of chromophores needs to be precise as otherwise quenching would lead to exciton (electron–hole pair) trapping and dissipation of the photon energy.41 Despite the very high local concentration in light-harvesting proteins (10−1 M), the chromophores are not quenching, because orbital overlap is prevented. The supramolecular organization of chromophores inside the protein matrix yields a so-called antenna system that collects photon energy and directs this towards the reactive center where charge-separation (CS) happens. These natural supramolecular structures lead to very high quantum yields (close to unity) and are able to deliver excitons and redox equivalents at a sufficient rate to drive multielectron catalysis, which is fast with regards to the number of photons available per second. Thus, the lesson learned from these natural structures is that chromophore organization can be beneficial for two reasons. (1) Close packing of light absorbing molecules results in delocalization of excitons across several of the same chromophores or rapid energy transfer among them via homo-FRET (Förster resonance energy transfer). Therefore, distant chromophores can deliver the photon energy they harvest to a chromophore close to a redox partner (e.g. a catalyst). Thus, an increased fraction of the incident solar energy is delivered at the reactive center. (2) Organizing chromophores in an energy cascade transfers the harvested energy towards a reactive center at the expense of small energy losses. Back-transfer is subsequently inhibited since that process has become energetically uphill. This energetic funneling can be used to deliver an exciton closer to a redox partner via hetero-FRET. In addition, the downward energy cascade for electron transfer (i.e. redox reactions) as observed in natural photosynthesis leads to long-lived charge-separated states. Due to microscopic reversibility of both the electron transfer and catalytic steps, the rate limiting steps are in equilibrium with the initial excited state. Therefore, long CS lifetime is required to drive the slow catalysis and prevent back-reactions.
Inspired by the natural system, scientists have prepared many different types of light-harvesting antenna systems and charge-separation architectures.40,42–45 Most were studied in solution using transient absorption techniques, assayed for their energy or electron transfer rates and the lifetimes of the exciton and charge-separated state was determined. Whereas the properties of these beautiful structures are well documented, implementation into artificial photosynthesis has yet to be developed. Systems that display the photophysical properties required for photosynthesis can be prepared via self-assembly. Also, the integration of light-harvesting, charge-separation and catalysis into a working device can be performed in a supramolecular manner. Towards this goal, it is important to understand the influence of the supramolecular bond on photophysical properties of assemblies. In the next section, we shall highlight several examples of supramolecular light harvesting antennae, CS assemblies and their integration into efficient light harvesting architectures.
3.2 Supramolecular chromophore organization
Porphyrins are synthetically accessible and stable analogues of nature's chlorophylls and therefore thoroughly studied chromophores. When porphyrins are metallated, coordination of axial ligands is possible. Several strategies employ this axial binding to produce discrete supramolecular assemblies. Zn-porphyrins are well explored, where coordination occurs at only one axial position. These mainly consist of pyridinyl and imidazolyl metal coordination.Pyridine–Zn interactions show binding constants of 103–104 M in apolar solvents and therefore supramolecular assemblies use multiple interactions to produce more stable structures under dilute conditions. Often the porphyrins have a double function, acting as the structural backbone and light-harvesting functionality. For example, ZnII-porphyrins can be combined with pyridine substituted free base porphyrins. The fluorescence of ZnII-porphyrins overlaps with the Q-band absorption of the free base porphyrins. The excited ZnII-porphyrin molecule transfers energy to the freebase porphyrin. Complex architectures, like supramolecular porphyrin cages, boxes, ladders and rings could be formed this way. All these assemblies show fast (picosecond) energetically downhill energy transfer processes enhanced by supramolecular preorganization. Supramolecular assemblies have been thoroughly studied for the purpose of PET and CS,45 such as self-assembled dimers of porphyrins and fullerenes as well as trimers and porphyrin squares.46 Another strategy is to use a rigid pyridine functionalized template to organize porphyrins in close proximity, resulting in rapid energy transfer among the chromophores.47 In addition to axial binding, functionalization of porphyrin macrocycles with specific binding motifs can result in supramolecular organization of chromophore arrays using metal–ligand, hydrogen bonding, ion-dipole and π–π stacking interactions.48 When these interactions are adequately chosen, extensive arrays (J-aggregates) can be formed via self-assembly, which show energy transfer over large distances.49 A complementary approach comprises the use of a scaffold such as a polymer or dendrimer, and by means of its functionalities induce supramolecular organization. A plethora of scaffolds has been explored, including (viral) proteins, DNA, and lipid membranes, (non-natural) peptides and dendrimers.43
The supramolecular interactions used to properly preorganize chromophores, can also alter the photophysical properties. For example, axial coordination of nitrogen ligands to porphyrin metal centers desymmetrizes covalent, cyclic porphyrin arrays.50 This desymmetrization decreases excitonic coupling in the antenna system (Fig. 3). Supramolecular binding can therefore partially localize the exciton in a supramolecular antenna, leading to increased fluorescence of an acceptor upon selective excitation of the donors. Desymmetrization is crucial for the purpose of energy funneling and CS as these are inherently asymmetric processes.51 Supramolecular interactions aid in this regard and supramolecular donor–acceptor complexes yield superior CS lifetimes compared to covalent analogues.52
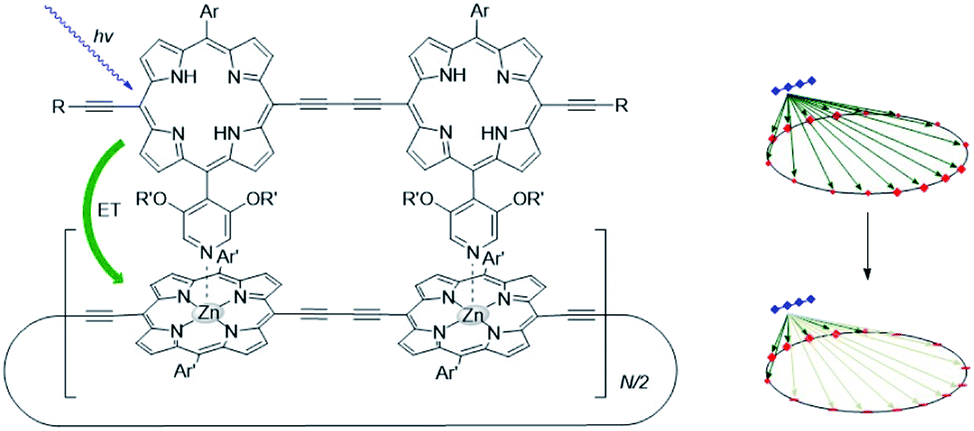 | ||
| Fig. 3 As the porphyrin dimer becomes excited, energy transfer occurs to the ring with the same rate (∼1.3 ps) no matter its size (N = 6, 8, 10, 12 or 30). A representation of the line-dipole model on the right shows the effect of strain induced by coordination. The transferred excitation is delocalized over 6 porphyrins. Adapted with permission from ref. 50. | ||
When heavy atoms are used in the self-assembly of supramolecular antennae they can quench exited states and indeed decreased fluorescence intensity was first reported by Drain and Lehn.53 This phenomenon can be attributed to singlet to triplet intersystem crossing due to the heavy-atom effect.54 Elliott et al. attempted to resolve this problem by electronically decoupling (using insulating triazole linkers) both singlet and triplet forming chromophores to heavy-atom coordinating pyridines.55 Since all photochemistry is determined by kinetics, metal–ligand based supramolecular assemblies can still be used, for example as the rate for singlet energy transfer competes with the rate for intersystem crossing.56 Thus metal–ligand interactions are an interesting strategy to produce chromophore assemblies, but care has to be taken that heavy-atoms can induce triplet formation at the expense of energy efficiency.
Hydrogen bonds are regularly employed for the supramolecular organization of chromophores. Triplet energy transfer,57 singlet energy transfer58 and electron transfer59 are all facilitated by hydrogen-bonding interactions, leading to increased rates of transfer due to D–A orbital overlap (following the Dexter mechanism of energy transfer or via PET).
Clearly, supramolecular bonding interactions are not innocent and their use complicates the study of light-harvesting assemblies. However, careful use of self-assembly by supramolecular interactions can provide directionality of energy and electron transfer and can lead to increased CS state lifetime as well. Therefore, they are useful in the development of artificial photosynthesis. The influence of supramolecular bonds on photophysics is well explored. Now is the time to use these bonds for the integration of the fundamental actions of photosynthesis. Below examples will illustrate that otherwise inaccessible structures are obtained using self-assembly strategies, that above all show enhanced light-harvesting functionality over covalent analogues.
3.3 Selected examples of supramolecular strategies to construct light harvesting and charge separation systems
The group of Kuroda attempted to generate large size antenna systems using non-covalent strategies.60 Hydrogen-bonding interactions have been used in conjunction with metal-porphyrin coordination, all based on multiple interactions to reach high binding constants such that these dyads are formed efficiently under dilute conditions. A free-base porphyrin was symmetrically substituted with spacers at the 4′-phenyl positions linked to pyrazine units. These ditopic nitrogen ligands act as a guest by coordinating Zn-porphyrin cages that in turn are formed via multiple hydrogen bonds. As such, a single H2-porphyrin binds 8 light-harvesting Zn-porphyrins (Fig. 4). The energy transfer efficiency from a single Zn-porphyrin cage to the free base was found to be 82%, somewhat lower than previously reported related covalent systems. However, the fluorescence of the free base in the self-assembled structure is enhanced 18-fold demonstrating the antenna effect. More elaborate H2-porphyrins with increasing pyrazine functionalities linked in a linear or branching topology revealed that the enhanced light-absorption could be further increased (77-fold) when the ratio of 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for Zn:H2 porphyrin was reached.61
:1 for Zn:H2 porphyrin was reached.61
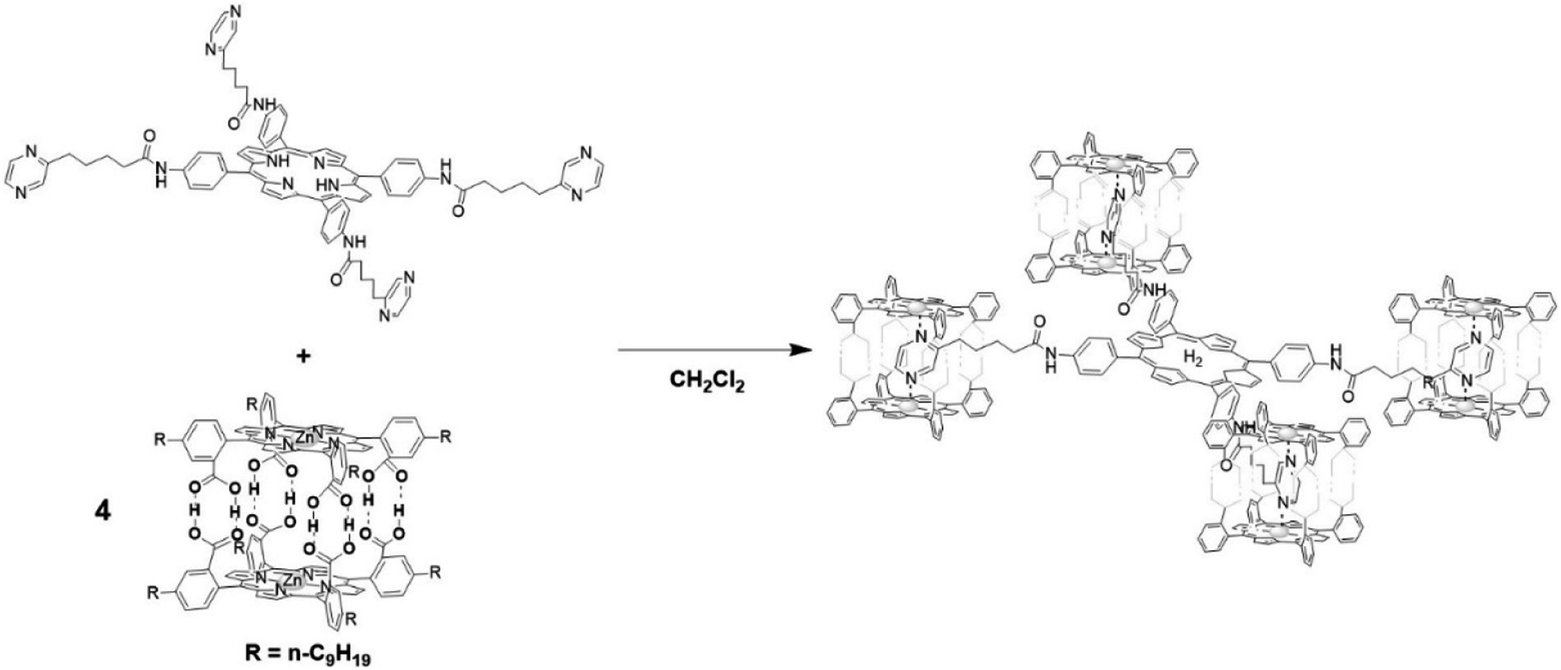 | ||
| Fig. 4 A porphyrin acceptor functionalized with ditopic pyrazine units (top) that act as guests to the donor Zn-porphyrin cage assembled with hydrogen bonds. A 8:1 donor:acceptor ratio is achieved here. Some atoms in the antenna system are omitted for clarity. | ||
Dendrimers are hyperbranched well-defined macromolecular structures that are prepared in iterative stepwise synthetic procedures. These structures are interesting scaffolds for applications as light-harvesting antennae, for example by the functionalization of the end groups with porphyrin dyes. Even more interesting is that simple addition of a coordinating C60 acceptor, turns a porphyrin dendrimer into an integrated light-harvesting assembly (Fig. 5).62 The authors observed excited-state energy migration across the porphyrins, as is also typically observed in the natural LH system, and an impressively long charge-separated lifetime after PET to the C60 (0.25 ms).
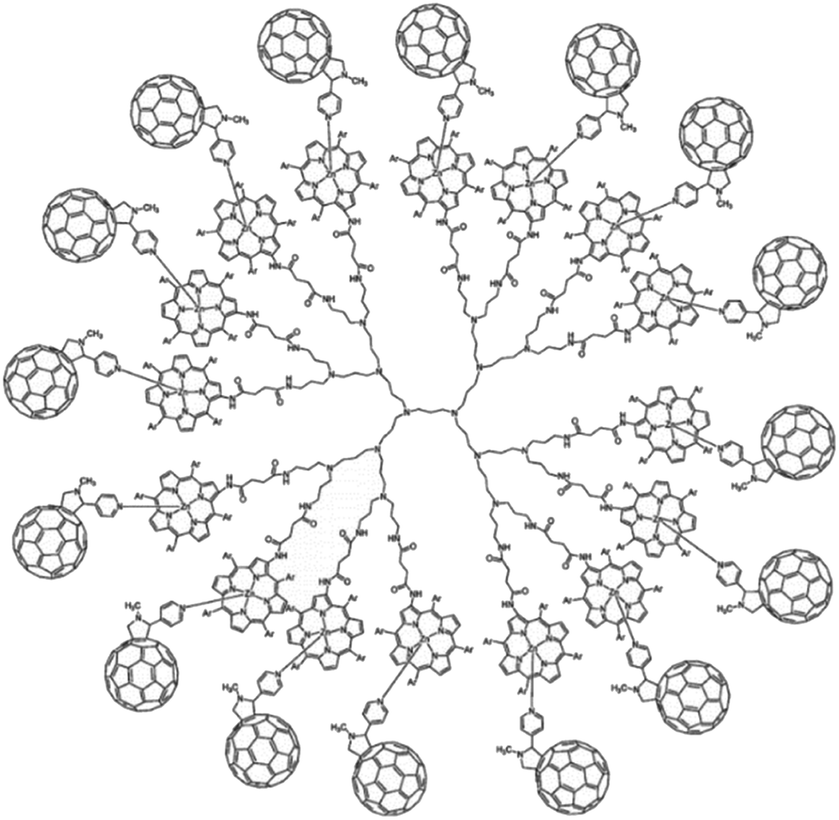 | ||
| Fig. 5 Dendrimer with 16 porphyrin units act as a light-harvesting antenna, and achieves long lasting charge separation when electron accepting fullerenes bind the porphyrin metal atom. Used with permission from ref. 62. | ||
In a supramolecular tour-de-force Kobuke's group developed an integrated light harvesting system that could be extended to CS assembly.63,64 The supramolecular antenna was formed by using imidazolyl substituted Zn(II)-porphyrin building blocks. The nonameric porphyrin antenna possessed three Zn-porphyrins that did not partake in the assembly. Thus, simply mixing in a pyridinyl tripodal ligand containing an energy accepting porphyrin and covalently attached fullerene electron acceptor provided an integrated assembly (Fig. 6). The supramolecular structure attained a total energy conversion efficiency of 85% where the charge-separated state had a half-life of 0.2 μs. The authors took their approach even further when they incorporated supramolecular antennae into lipid bilayers (known as dynamic self-assembled interfaces, vide infra).65 Transient experiments were not performed, but the authors deduced inter antenna energy transfer between the different antenna systems assembled within the membrane.
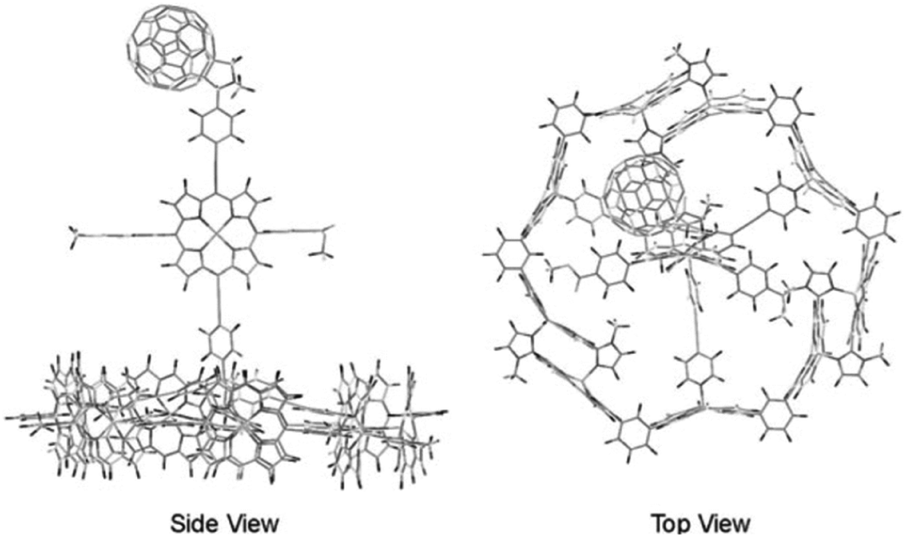 | ||
| Fig. 6 Atomistic model of an integrated antenna/charge-separation assembly. Adapted with permission from ref. 64. | ||
Above examples have illustrated that light harvesting architectures can be built using supramolecular strategies that collect photons and transfer their energy over considerable distances. Though supramolecular interactions add to overall complexity of the system, they are a vital tool for assembling multi-chromophoric antennae and integrating them with charge-separation functionality. Such well-organized multi-chromophoric systems often yield enhanced properties such as increased absorption, panchromaticity and charge-separation lifetimes (i.e. lowered recombination rates) vital to efficient artificial photosynthesis devices. Meanwhile, these complex structures can be constructed from simple components with high-absorption coefficients that assemble with little synthetic effort and when properly designed, conserve energy and prevent degradative processes by circumventing triplet formation. The ability to direct energy through a supramolecular assembly may prove crucial to collect, transfer and funnel energy for the purpose of artificial photosynthesis. Proper connection to redox catalysis is yet to be demonstrated, however, the implementation of integrated supramolecular antennae/CS assemblies in an artificial photosynthetic device warrants reduced recombination and increased overall photon efficiency. Indeed, when supramolecular interactions are used to preorganize dyes for light-harvesting and CS onto conducting solid materials enhanced device performance is often reported (see Section 6.1).
4 Supramolecular catalysis
For the efficient conversion of solar energy into fuel, redox catalysis is essential. In artificial photosynthetic devices water oxidation provides electrons and protons, whereas proton reduction (or CO2 reduction) delivers the fuel in an overall thermodynamically uphill reaction. The rate of these multi-electron processes is limited by the photon flux, which provides the redox equivalents with a certain speed. This means that reactive intermediates are produced that should be sufficiently stable to prevent decomposition or recombination. The redox catalysis is mostly performed by transition metals since they can access several oxidation states. Important properties for the catalysts used in artificial photosynthetic devices are (1) catalysis at low overpotential as overpotential leads to losses (2) stability of the catalyst (3) the rate of the catalyst such that the amount of catalyst can be optimized. Supramolecular strategies provide handles to optimize catalysts with respect to these properties. This section focusses on catalysis for artificial photosynthesis using the supramolecular approach. Whereas eventually these properties should be evaluated in a light driven device, these basic properties are easily measured using electrocatalysis or catalysis driven by chemical oxidants/reductants.4.1 Chemical and electrochemical water oxidation
A key reaction in natural photosynthesis is the oxidation of water, which is benign and abundant, providing electrons and protons for the production of the fuel. Water oxidation catalysis provides four protons and four electrons from two water molecules leading to the formation of dioxygen. Interestingly, PSII operates using a CaMn4O5-cluster. Therefore, chemists have developed analogous clusters that mimic the activity of PSII, but these show low stability and activity at least partly because these synthetic clusters lack the protein environment.66 The best synthetic catalyst for water oxidation are currently based on ruthenium and iridium, and in this section we will describe how supramolecular strategies can be used to further increase the performance of these catalysts. The power of supramolecular strategies is illustrated by simple water oxidation catalyst (WOC) that operate via a dinuclear mechanism, facilitated by π–π stacking interactions,67,68 as well as more complex multi-component systems. To explain this supramolecular effect rationally, the catalytic cycle for water oxidation is explained first.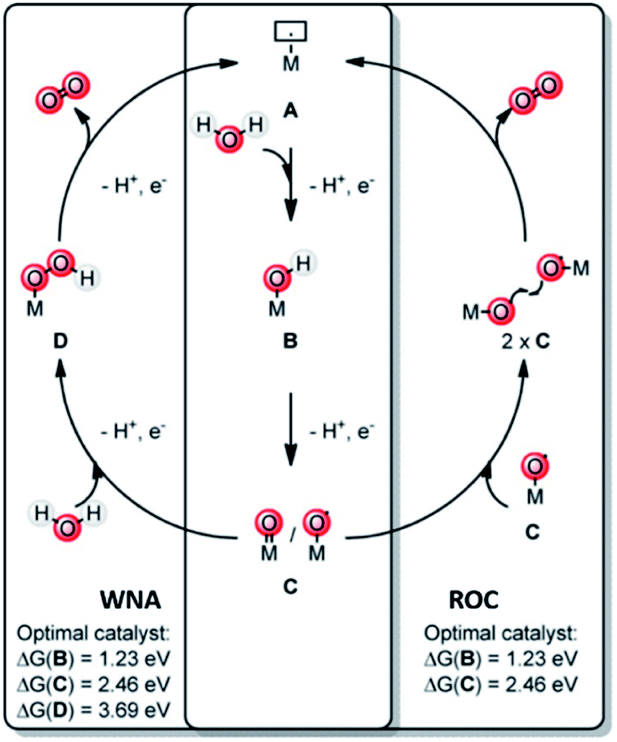 | ||
| Fig. 7 Water oxidation catalysis follows either of two mechanisms. A mononuclear WNA mechanism or the binuclear ROC mechanism. Used with permission from ref. 69. | ||
Catalysts that operate via the ROC mechanism could also be preorganized using supramolecular strategies, because supramolecular chemistry can aid in the spatial organization of two metallo-oxo species without exerting a fixed M–M distance.
000 respectively.72
Besides π–π stacking to bring catalysts together, concentrating catalysts by putting them in a confined space is beneficial. In 2012 the groups of Yang and Li showed that confining Ru(bda)(pic)2 (pic = picoline) WOCs in mesoporous silica led to improved TOFs (Fig. 8).73 Interestingly, the use of different sizes of amphiphilic RuWOCs can lead to selection between the mononuclear WNA mechanism and the dinuclear ROC mechanism (Fig. 9).74 Yang et al. showed that the use of amphiphilic groups on the axial ligands of Ru(bda) systems led to self-assembly of liposome-like structures. A large amphiphilic substituent increased the Ru–Ru distance which inhibited the ROC mechanism, but a small amphiphilic group favored the ROC mechanism resulting in faster catalysis.
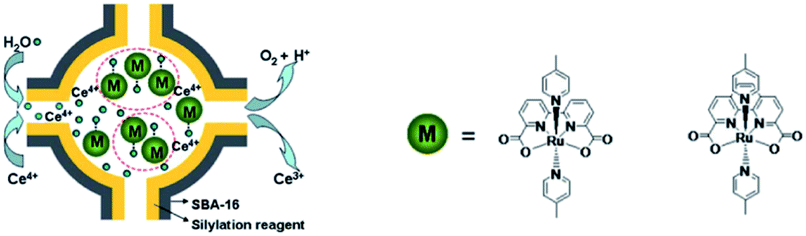 | ||
| Fig. 8 WOC encapsulated in mesoporous silica. Used with permission from ref. 73. | ||
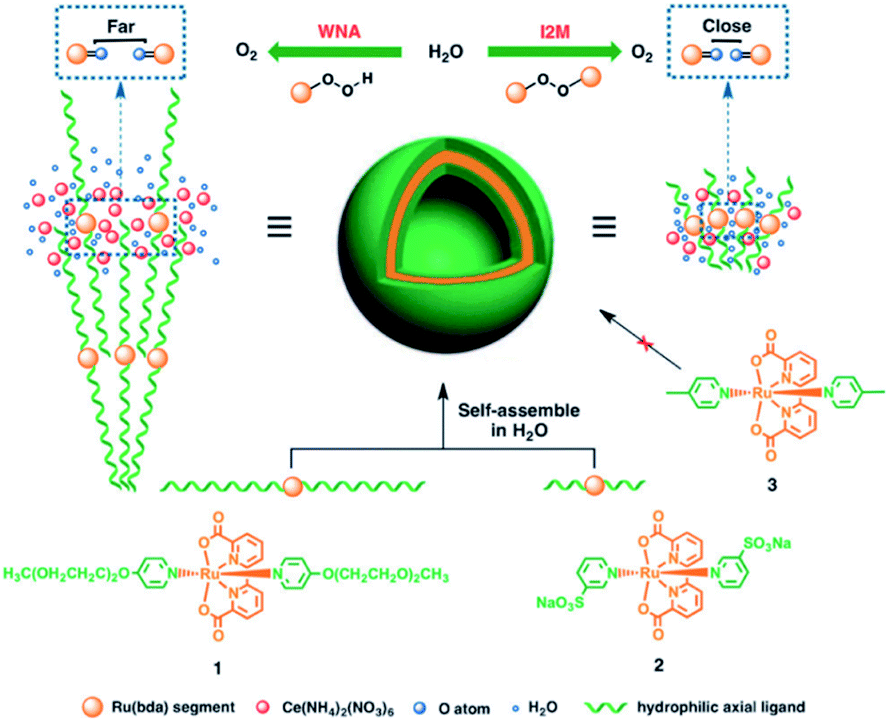 | ||
| Fig. 9 Alkyl chain substituted WOC self-assemble into micellar vesicles in water, preorganizing the WOC for the ROC mechanism. Used with permission from ref. 74. | ||
Supramolecular interactions are also exploited to increase local concentrations facilitating electrochemical water oxidation, as showcased by our group in 2018.75 We incorporated increasing equivalents of sulfonated Ru-WOC in a guanidinium functionalized M12L24 supramolecular cage (Fig. 10). Interestingly, the rate of catalysts that follow the dinuclear ROC mechanism was enhanced up to 130 times through encapsulation, while the rate of a Ru-WOC that operates via the mononuclear WNA mechanism was unchanged by increasing the local concentration. This example shows that great rate enhancements can be achieved by spatial organization of catalytically active species following the dinuclear mechanism.
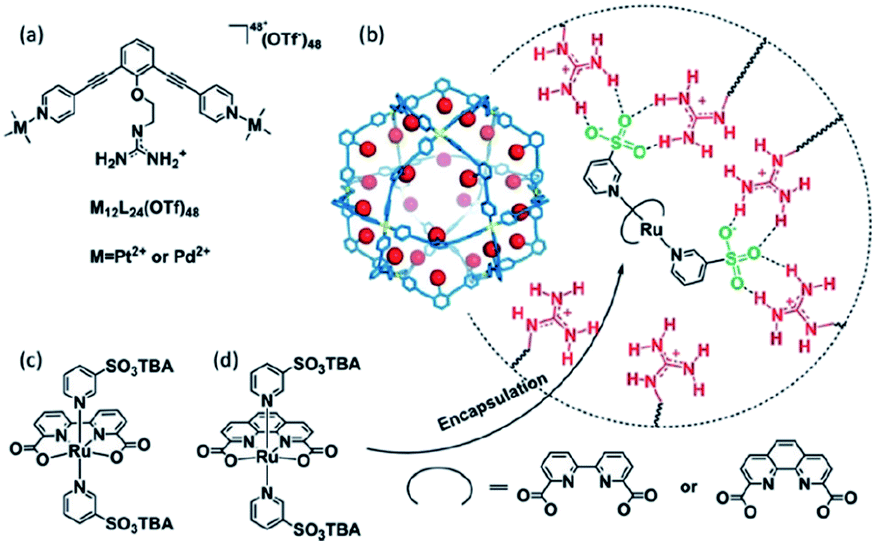 | ||
| Fig. 10 Sulfonated WOC entrapped in a M12L24 supramolecular sphere. Used with permission from ref. 75. | ||
The group of Würthner showed that ditopic pyridinyl ligands bind the axial positions of Ru(bda) complexes resulting in the formation of a supramolecular macrocycle catalyst trimer, [Ru(bda)bpb]3.76 They showed that supramolecular interactions spread out the catalytic sites while capable of oxidizing water using the WNA mechanism. These trimers showed exceptionally stable, due to limited accessibility of the active sites by steric repulsion of the organic scaffolds. A follow-up study investigated the effect of the ring size and the role of the hydrogen bonding network between water molecules that reside within the trimeric structure (Fig. 11).77 Higher rates were obtained when all reactive sites point towards the interior of the macrocycle, because the favorable hydrogen bonding network stabilizes the generated protons.
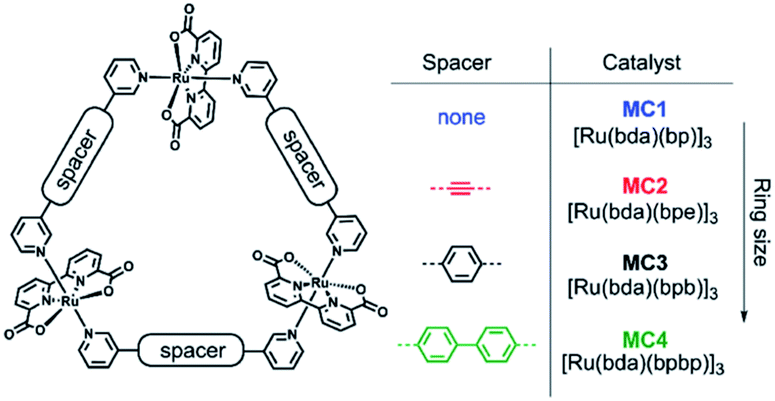 | ||
| Fig. 11 A supramolecular macrocyclic WOC trimer performs better with increased ring size. Used with permission from ref. 76. | ||
4.2 Chemical and electrochemical proton reduction
Water oxidation catalysis provides the electrons and protons that are needed to generate the fuel by a reduction reaction. The simplest reaction is the direct combination of protons and electrons to form molecular hydrogen. Again, critical properties of the catalyst include rate, stability and overpotential at which proton reduction catalysis is achieved. The 2nd and 3rd row transition metals Rh and Pt are effective hydrogen evolving catalysts (HEC), both as metal–ligand complexes and colloidal nanoparticles. In fact, Pt electrodes can do proton reduction catalysis at virtually no overpotential. However, hydrogen evolving enzymes utilize abundant 1st row transition metals such as Ni and Fe to catalyse proton reduction (Fig. 12), also at the thermodynamic potential. This has inspired chemists to produce structural and functional mimics of the hydrogenase active site (H-cluster) using an expanded set of abundant transition metals including Fe, Co, Ni and Mo. Though there has been a rise in efficiency and stability of molecular catalysts and lowering of overpotentials, further development has stagnated. Meanwhile, the [FeFe]-hydrogenase active site works at almost no overpotential and has TOF ∼ 10000 s−1. The apo-enzyme is inactive, and many H-cluster mimics that have been synthesized are active, but require considerable overpotential. Interestingly, when the inactive laboratory synthesized H-cluster mimic [Fe2(pdt)(CO)4(CN)2] is reconstituted in its native protein environment the activity is fully restored.78 This is a clear demonstration that the catalysts' reactivity is dictated by an interplay between the active site and its protein environment utilizing supramolecular interactions. Computational79,80 and bioengineering10 studies have pinpointed the relevant proton transfer pathways and those residues crucial to the catalytic activity.
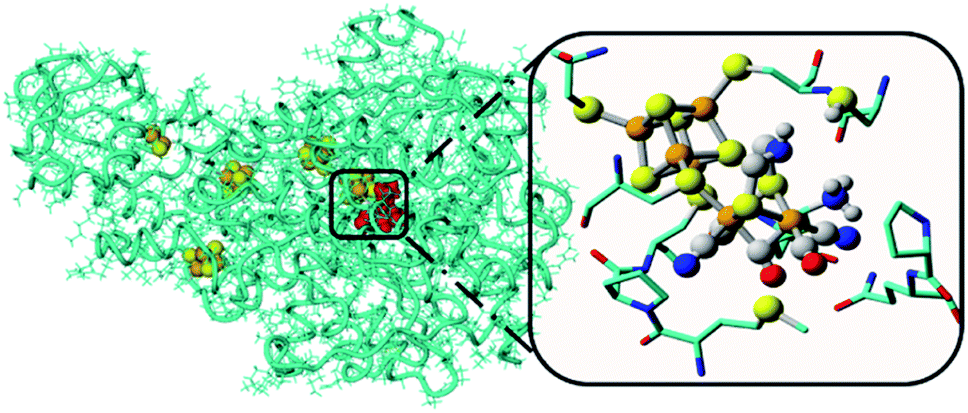 | ||
| Fig. 12 The hydrogenase enzyme with the active site depicted in red and other FeS-clusters in yellow/orange. The highlighted area shows a close-up of the H-cluster and the tight embedding in the surrounding protein environment. Adapted with permission from ref. 10. | ||
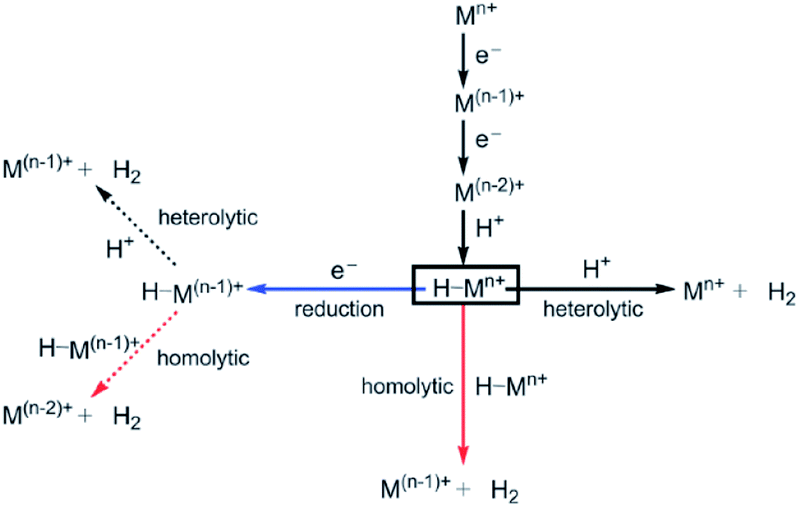 | ||
| Fig. 13 Possible mechanisms for the hydrogen evolution reaction. Used with permission from ref. 2. | ||
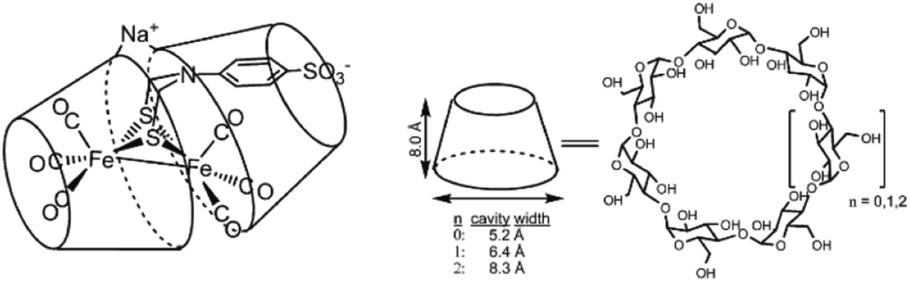 | ||
| Fig. 14 A sulfonated [FeFe]-hydrogenase mimic is stabilized in a cyclodextrin, but catalytically inactive. Used with permission from ref. 88. | ||
The assembly of supramolecular cages by metal ligand interactions provides a universal strategy to generate a diverse set of cage compounds.89 Cages of various shapes and sizes can be produced that are highly modular in character and can bind a diverse set of guest molecules under various conditions. We have introduced a template ligand approach as a general strategy to encapsulate catalysts in molecular cages. Along these lines, we recently used zinc(II)porphyrin based supramolecular cages to encapsulate an [FeFe]-hydrogenase mimic with an appended pyridine containing phosphine ligand (Fig. 15).90 The catalyst encapsulation is based on coordination of the pyridyl phosphine ligand to the Zn-porphyrin building blocks of the cage. The phosphine containing complex is an active proton reduction catalyst, but disproportionates upon electrochemical reduction in solution, which was established by IR-spectroelectrochemistry. However, no disproportionation is observed when the catalyst is bound in the supramolecular cage, leading to a more stable catalyst upon encapsulation. Most interestingly, the overpotential for proton reduction is lowered with 150 mV when the hydrogenase mimic is in the cage compared to the catalyst free in solution. The cationic nature of the cage apparently lowers the barrier for proton reduction catalysis. The same effect is observed when [FeFe]-hydrogenase mimics are encapsulated in a Pd12L24 supramolecular sphere, which is also highly cationic in nature, decreasing the overpotential by 250 mV.91 Moreover, these larger spheres also allow for co-encapsulation and thus preorganization of acid substrate in close proximity to the catalyst, leading to a rate enhancement of two orders of magnitude compared to the system were only external acid is present. Thus, supramolecular encapsulation strategies provide additional tools to control the overpotential and activity of catalysts. In addition, the ET rate from the electrode to electroactive components in these type of larger spheres can be controlled over two orders of magnitude by changing the connectivity, providing new tools yet to be explored to match the reaction rate of catalysts.92
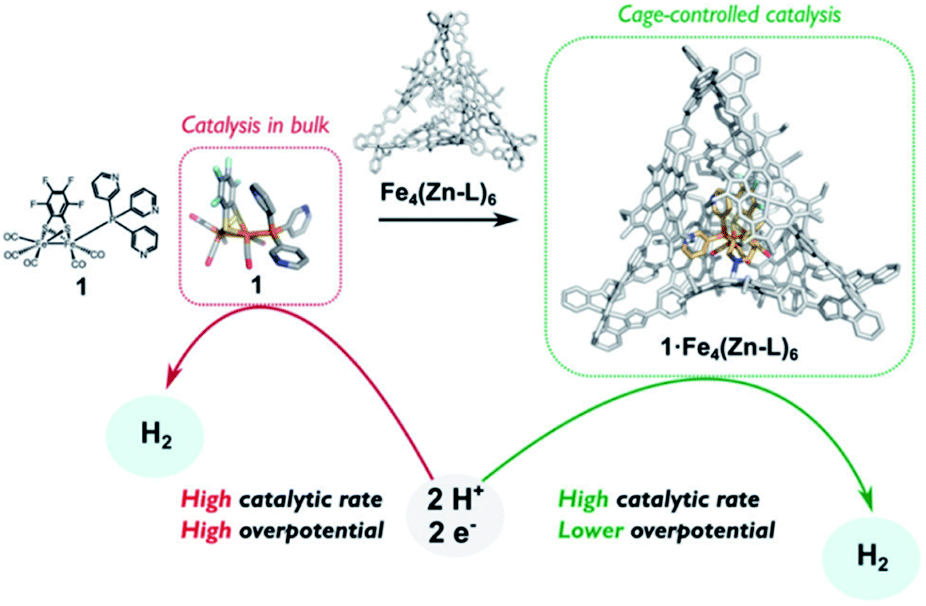 | ||
| Fig. 15 Comparison between catalyst in homogeneous solution and the catalyst controlled by a tetrahedral supramolecular cage. Used with permission from ref. 90. | ||
Next to creation of well-defined second coordination spheres by encapsulation, HEC can be embedded in polymer matrices that act as second coordination sphere. The potential of this approach was illustrated by Brezinski et al., who reported a molecular hydrogenase mimic in a polymer framework that folds around the metal cluster (Fig. 16).93 They used atom-transfer radical polymerization to form this metallopolymer. Interestingly, the embedded hydrogenase mimic showed enhanced catalytic properties when compared to the natural [FeFe]-hydrogenase enzyme. The rate increased 25 times to an incredible kobs = 250000 s−1 and an overpotential of a mere 0.3 V (compared to Pt). Moreover, these metallopolymers operated in air at 200 nM concentrations. By copolymerizing pyridine and pyrene functionalities Reisner et al. were able to attach cobaloxime HECs to a polymer matrix that showed enhanced catalytic stability as a result.94 Additionally, the polymer assembled onto multi walled carbon nanotubes (MWCN) as the pyrene functionalities connected via π–π interactions. When deposited onto a MWCN modified glassy carbon electrode catalyst loadings comparable to metal oxide sensitization were reported, denoting the potential of supramolecular functionalization of electrodes. Besides enhanced stability, polymers facilitating a second coordination may influence reaction selectivity. This was demonstrated by Liu and McCrory, who embedded Co-phtalocyanine CO2 reduction catalysts in poly-vinylpyridines.95 They postulated that high CO product selectivity is obtained by sluggish proton transfer through the polymer, while protonated pyridine was sufficiently acidic to act as proton donor to CO2.
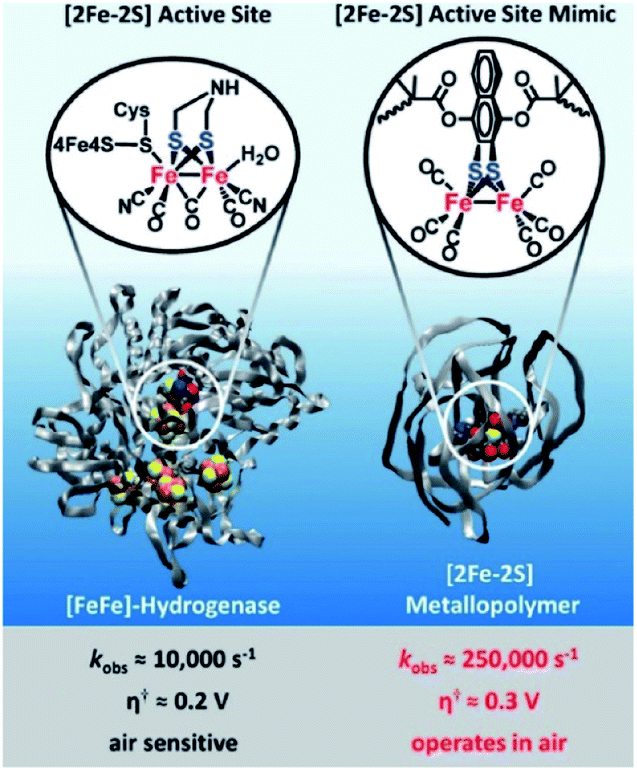 | ||
| Fig. 16 Comparison of catalytic parameters of a natural [FeFe]-hydrogenase enzyme and a metallopolymer [FeFe]-hydrogenase mimic. Used with permission from ref. 93. | ||
In the above examples the catalytic reactions were evaluated by electrochemical methods, providing insight in the important properties of the catalysts and highlighting the effects of supramolecular encapsulation. Electrochemical methods can also be implemented in artificial photosynthesis using PV-electrolysis set-ups. For the preparation of integrated systems in which light is directly used to generate fuel, supramolecular approaches can also be fruitful and this will be discussed in the next section.
5 Supramolecular artificial photosynthesis
Supramolecular assemblies are mainly explored for light-driven water oxidation, proton reduction and to a lesser extent for CO2 reduction. The catalytic systems can be divided in two categories. (i) Dye-catalyst assemblies where a light absorbing functionality is preorganized to a molecular catalyst using non-covalent bonds, and (ii) dynamic self-assembled interfaces where dye and catalyst are co-embedded in a self-assembled membrane. These strategies are discussed below.5.1 Supramolecular photochemical water oxidation
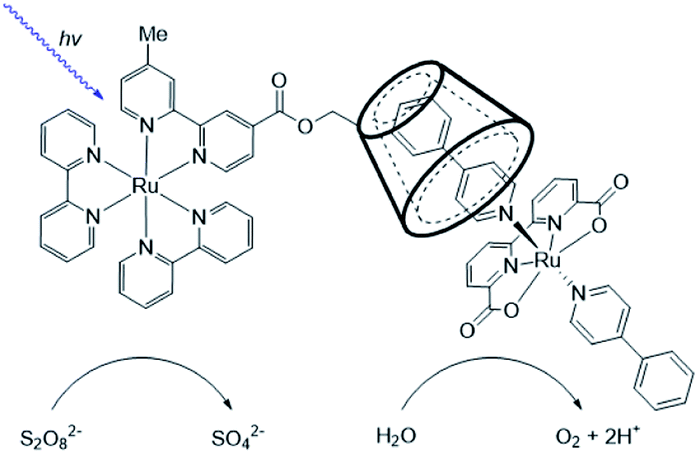 | ||
| Fig. 17 Supramolecular dye-WOC assemblies are formed by cyclodextrin host-guest chemistry. This results in enhanced stability in photocatalytic water oxidation in presence of a sacrificial oxidant. Cl− ions are omitted. | ||
Cobalt based polyoxometalates (POMs) are water oxidation catalysts and were preorganized to light absorbing carbon nanodots using electrostatic interactions.98 The supramolecular construct was used in light driven water oxidation, resulting in TONs up to 552. Paille and co-workers investigated metal organic frameworks (MOF), such as MOF-545 as a light-absorbing supramolecular host for water oxidation catalysts.99 They chose a POM [(PW9O34)2Co4(H2O)2]10− as the WOC, which was immobilized in the MOF by mild aqueous impregnation (Fig. 18). This system reached a TON of 70 per POM after one hour of photocatalysis and could be recovered after catalysis by filtration, retaining most of its activity. The authors mentioned that the sacrificial oxidant was limiting the conversion. All control experiments in which one of the components was absent, showed low conversion, denoting the importance of both the catalyst-sensitizer preorganization and enhanced POM stability when encapsulated. When the supramolecular assembly is loaded on thin films, the TON is increased 23-fold.100
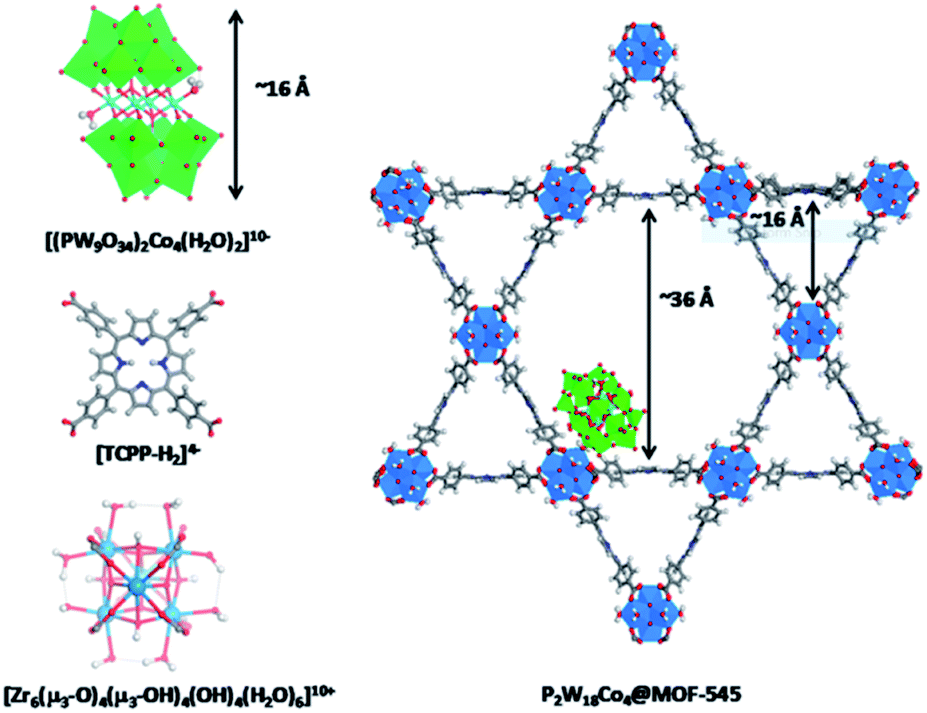 | ||
| Fig. 18 The components (left) are the catalytic POM (top) light-harvesting porphyrin strut (middle) and structural Zr-cluster (bottom) that lead to a supramolecular artificial photosynthetic assembly (right). Used with permission from ref. 99. | ||
Bonchio et al. wanted to increase the dye-catalyst ratio, as this is also found in natural photosynthesis. By using a Ru-POM structure as water oxidation catalyst, a dye-WOC assembly was formed in 5:1 ratio.101 The catalyst intercalated into π-stacked perylene bisimides (PBI) chromophores and the structure was likely supported by hydrogen bonds (Fig. 19). The assembly aggregated into clusters bearing a resemblance to the PS II architecture as shown by electron microscopy. Transient absorption revealed very rapid electron transfer (25 ps) to the catalyst, though the TON was still rather low (∼3).
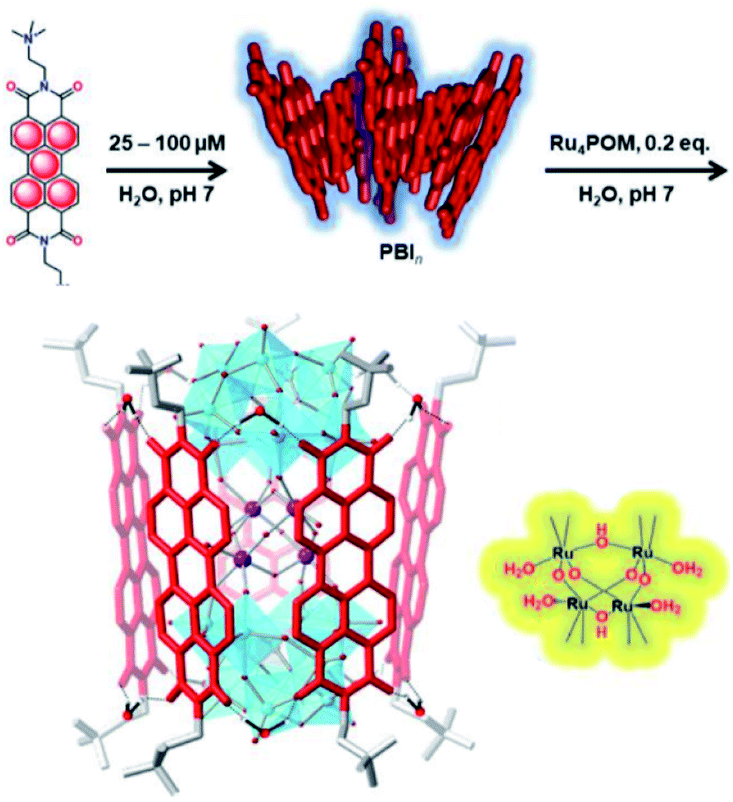 | ||
| Fig. 19 A supramolecular dye-WOC assembly is formed in a 5:1 ratio when Ru-POM is added to π-stacked PBI in water. The Ru-oxo core of the POM is depicted besides the assembly. Used with permission from ref. 101. | ||
The most studied reaction for solar fuels is water splitting, with water oxidation the most difficult reaction. A related reaction to overall water splitting that requires less energy and is more easily performed is the conversion of H2S to elemental S and H2 gas. Although H2S is not as abundant as water, it is sufficiently available as a toxic and unwanted side product of the petrochemical industry. Jing et al. developed a supramolecular Ni-cage that encapsulates the dye fluorescein by hydrophobic effects in aqueous solution (Fig. 20).102 Upon excitation of the encapsulated dye, the cage framework quenches the excited state and the cornerstones facilitate proton reduction with a TON of 433 per Ni atom after 24 hours of irradiation. Interestingly, the oxidized dye is ejected from the capsule, preventing recombination, and oxidizing the S2− ions in the basic solution to solid elemental sulfur. As such, overall H2S splitting is performed by spatial control over redox events. Control experiments with a similar mononuclear Ni catalyst and cage occupying inactive guests both gave approximately 3 times lower H2 formation. More importantly, no elemental S was observed in those experiments.
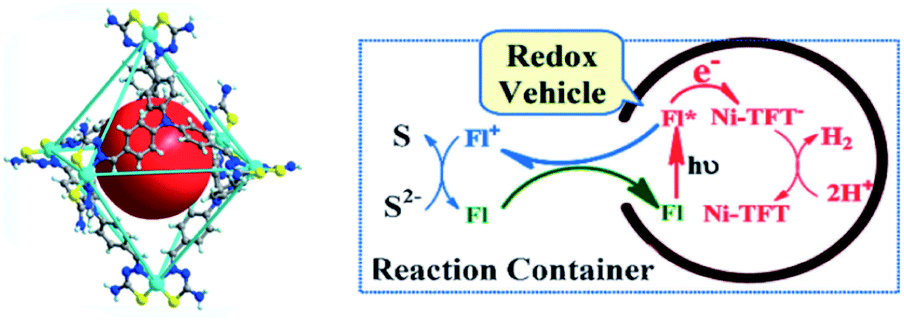 | ||
| Fig. 20 The supramolecular Ni-TFT cage (left) has ample space to encapsulate fluorescein (red ball). A scheme of the overall H2S splitting at pH 11–13 is depicted (right). The cage encapsulates fluorescein (FL) and expels the dye as it is oxidized. Used with permission from ref. 102. | ||
5.2 Supramolecular photochemical proton reduction
A thorough review of Co based H2 generation is available.104 Metallosupramolecular cobaloximes were first investigated by Fihri et al. in the group of Fontecave.105 They appended a pyridinyl functionalized dye to one axial position of the cobaloxime (Fig. 21). This led to TON of 103 after 15 hours of photoirradiation.
 | ||
| Fig. 21 This dye-HEC assembly sparked interest into supramolecular assemblies for proton reduction. | ||
These metallosupramolecular complexes used comparable conjugated dye-catalyst linking ligands, but the question remained what the influence of the bridging ligand was on catalytic properties. Therefore, amide coupled analogues were prepared in the group of Sun, showing that dye-catalyst assemblies with a CH2-spacer that breaks the conjugation showed enhanced photocatalytic properties.106 Most likely, the conjugated bridge leads to both increased electron transfer rate as well as unwanted recombination rates.
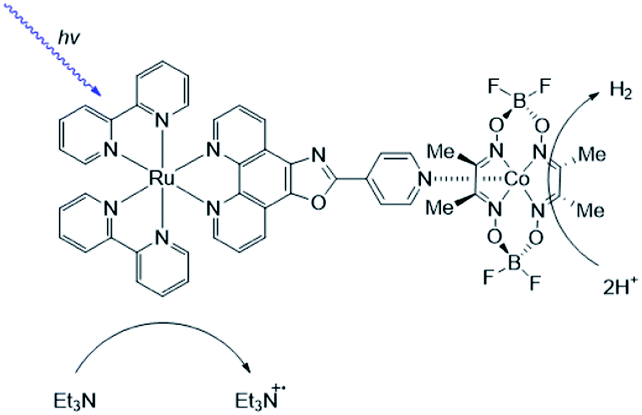
As [FeFe]-hydrogenase are very efficient and the active site is based on abundant metal catalysts their mimics are frequently studied for (light driven) hydrogen production.107,108 Our group investigated pyridinyl phosphine appended [FeFe]-hydrogenase mimics.109 In the presence of Zn-porphyrins, a dye-catalyst assembly forms which shows hydrogen formation (TON = 5) under illumination (Fig. 22) in the presence of a tertiary amine as sacrificial electron donor. Interestingly, only experiments containing a mixture of porphyrins yielded hydrogen. This suggest that the active species has two different chromophores, highlighting that asymmetry in the structure plays an important role for photocatalysis.
 | ||
| Fig. 22 The photo active species of an [FeFe]-hydrogenase mimic bears two different chromophores. Used with permission from ref. 109. | ||
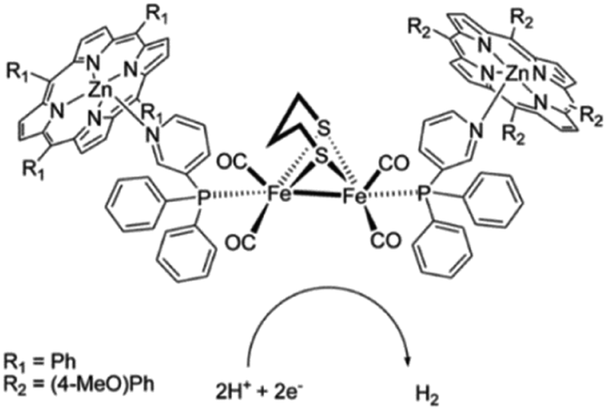
As was shown above, encapsulation can lead to stabilization of hydrogenase mimics. MOFs serve as interesting materials to encapsulate these complexes in a supramolecular fashion. Using a well-known MOF (UiO-66) Pullen and Fei et al. applied post-synthetic exchange (PSE) to partly replace the struts in a MOF with [FeFe]-hydrogenase mimics (Fig. 23).110 In the presence of a Ru photosensitizer, light-driven hydrogen production was observed, which increased three-fold to a TON of 3 compared to the free catalyst in absence of MOF material. MOFs based on porphyrins such as ZrPF (e.g. in Fig. 18) are able to coordinate [FeFe]-hydrogenase mimics in the pores without PSE.111 Again a significant improvement in catalytic activity was seen up to ∼3.6 over 120 minutes compared to similar homogeneous complexes with substoichiometric TON (∼0.1).
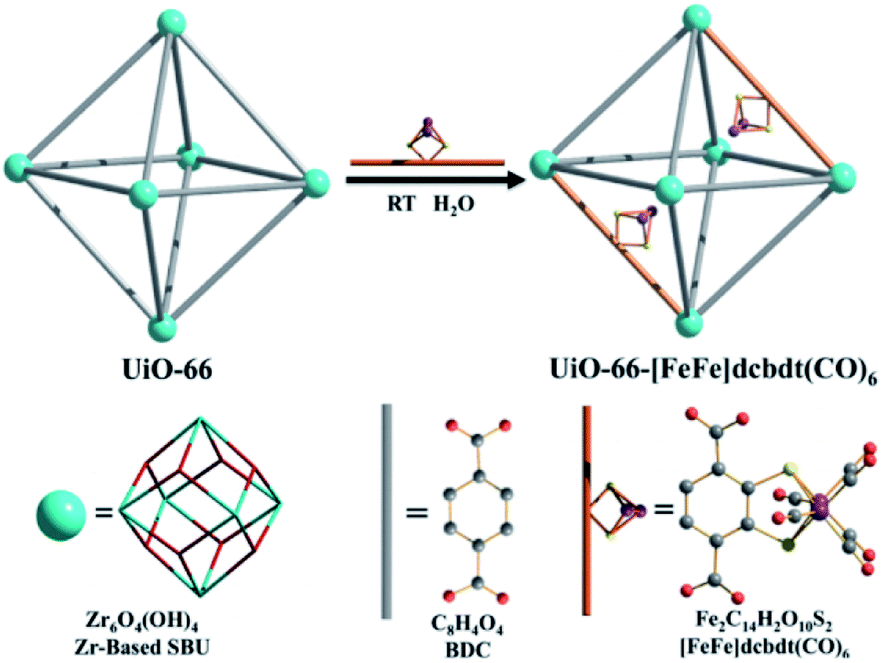 | ||
| Fig. 23 The [FeFe]-hydrogenase mimic is incorporated into the MOF structure by PSE. Used with permission from ref. 110. | ||
In analogy to MOFs, homogeneous metallo-organic cages serve as multi-nuclear self-assembled structures that can be used for light-driven proton reduction. For example, Duan et al. assembled a tetrahedral supramolecular cage that encapsulates a fluorescein photosensitizer in the cavity.112 The cage is held together by four CoIII-ions that act as structural cornerstones and catalytic sites for proton reduction (Fig. 24). Upon irradiation a PET event leads to reductions of the metal nodes, which in turn leads to proton reduction with a TON of 11000 per cage. The host-guest assembly enhances the catalysis as corroborated by the fact that cage analogues that are too small for dye encapsulation, operate at lower efficiency. Moreover, competitive binding of equimolar adenosine triphosphate (ATP) (expelling the dye from the cage) decreased the TON by a factor 6. Clearly the preorganization of sensitizers in a multinuclear catalytic cage is beneficial to photocatalytic proton reduction.
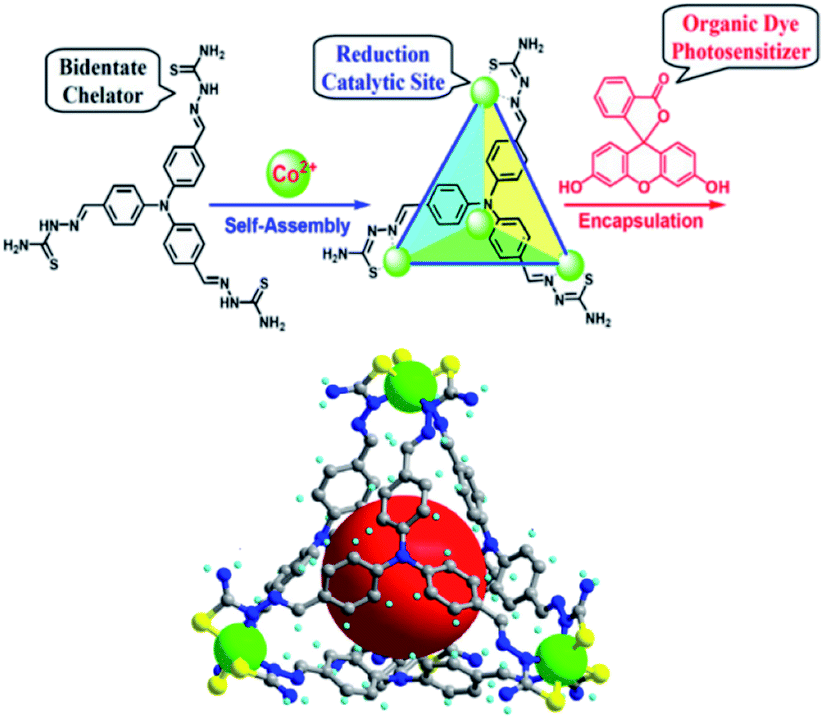 | ||
| Fig. 24 Supramolecular cages self-assemble when a ligand and Co2+ ions are combined in a 1:1 stoichiometry. The cage encapsulates an organic fluorescein photosensitizer in a following step. The assembly is an efficient light drive proton reduction catalyst. Used with permission from ref. 112. | ||
 | ||
| Fig. 25 Schematic representation of the influence of cone-angle on self-assembled lipid topology. | ||
Bilayers allow for both compartmentalization and organization. CS across the membrane can be achieved and with this also the back electron transfer can be slowed down, as was shown by Murata and co-workers.115 They demonstrated electron transfer from a sacrificial electron donor present in the interior of the vesicle to a photosensitizer embedded in the bilayer. The photoexcited electron is then donated to an acceptor in the exterior aqueous layer of the vesicle to evolve hydrogen. The charge of the amphiphilic head group can be changed to study the effect on catalysis.116 Wu and co-workers performed the encapsulation of both the water insoluble chromophore, an [FeFe]-hydrogenase mimic and the sacrificial electron donor ascorbate (indicated H2A in Fig. 26).117 With these components in close proximity H2 can be produced. Although TON was low (0.13) the photostability of the catalyst was enhanced.
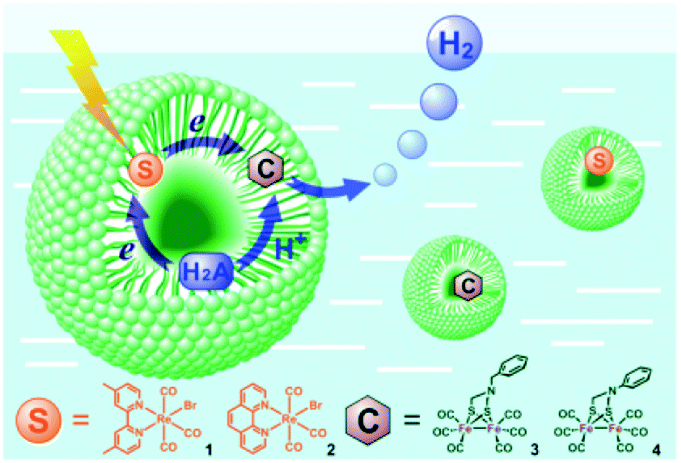 | ||
| Fig. 26 A schematic representation of Re sensitizers co-embedded with [FeFe]-hydrogenase mimics in SDS micelles using ascorbic acid (H2A in the figure) as a sacrificial proton and electron donor. Used with permission from ref. 117. | ||
Troppmann and König studied dye-HEC assemblies preorganized onto membrane bilayers, producing hydrogen with a TON of 165 (Fig. 27).118 Moreover, self-reorganization within this system is studied by varying the membrane fluidity. More fluidic membranes result in higher mobility of the embedded complexes. The use of membranes with higher mobility showed better performance of the system in terms of H2 evolution, allowing sensitizers and HECs to meet transiently at the membrane periphery.
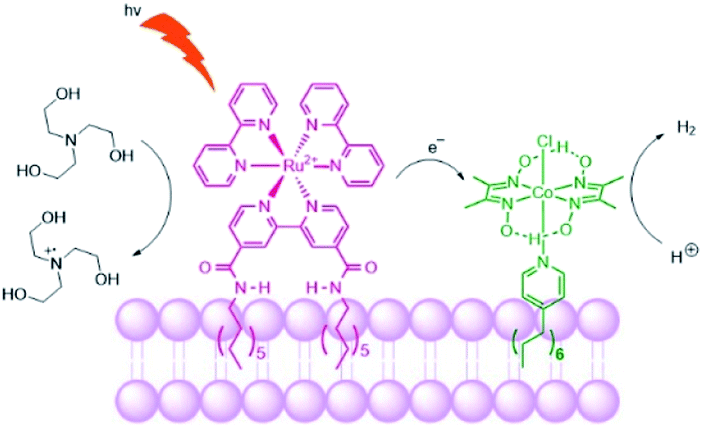 | ||
| Fig. 27 Schematic representation of the photosensitizer embedded and the cobaloxime catalyst in the liposome membrane. The sacrificial electron donor is triethanolamine. | ||
These effects were further investigated in the group of Woutersen and Reek.119 In their system the hydrophobic hydrogenase mimic as HEC was fully embedded within the membrane layer (Fig. 28). This system evolved hydrogen with a TON of 67. The authors found that the membrane effect is two-fold. In the first place the high local concentration of catalyst in the vesicles, changes the reaction pathway. Instead of protonation after the first reduction event, two of such complexes lead to disproportionation providing the di-reduced and nonreduced complex. Secondly, the membrane provides a constant pH for the HEC as there is no shift in reduction potential of HEC while varying the pH of the aqueous solution. This implies that the bilayer provides an environment with a certain pH that is independent of the exterior.
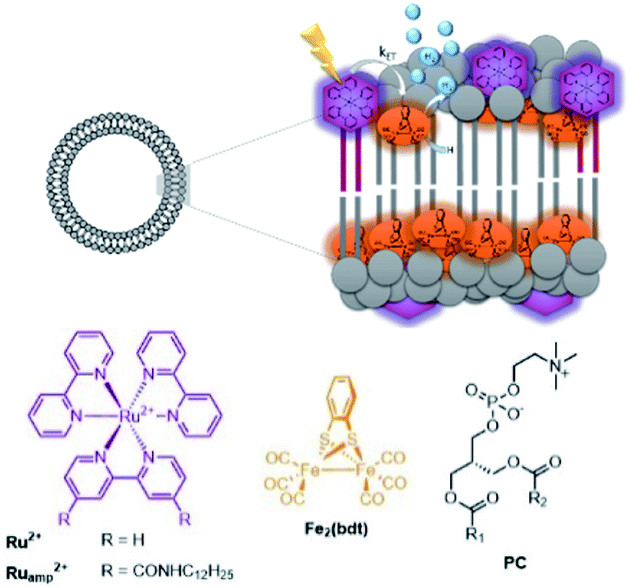 | ||
| Fig. 28 A self-assembled bilayer preorganizes a Ru-photosensitizer and [FeFe]-hydrogenase mimic for light driven proton reduction. Used with permission from ref. 119. | ||
A seminal paper by Moore, Moore and Gust, describes a membrane bound CS triad which was used to reduce quinones at the periphery of the membrane (Fig. 29).120 The reduced quinone was protonated at the bulk water interface after which it diffused to the other side of the membrane, where it delivered the proton to the interior. This process results in a proton gradient. Interestingly, CFOF1-ATP synthase, an enzyme that converts adenosine diphosphate to ATP using a proton flow, was co-embedded into the membrane. The photogenerated proton gradient resulted in a proton-motive force, that allowed the synthesis of adenosine triphosphate (ATP) with a quantum efficiency of 7%. This supramolecular assembly mimics light-harvesting as observed in photosynthetic bacteria, using an ATP-synthase to generate ATP as the fuel. It should be noted that in the natural system light energy is converted into chemical fuel both by the proton gradient across the membrane, as well as light driven proton reduction to form protonated nicotinamide adenine dinucleotide (NADH), and that this system is one rare mimic of the former.
 | ||
| Fig. 29 Covalent artificial reaction center (1) is embedded in a lipid bilayer, where it reduces quinones (Qs) under illumination. The quinone imports protons into the vesical formed by the bilayer. The arising proton gradient is used to drive the CFOF1-ATP synthase enzyme for the production of ATP. Used with permission from ref. 120. | ||
A similar system was reported in the group of Matile.121 Self-assembled naphthalene diimide (NDI) rigid-rods breached unilamellar vesicles to allow photosynthetic energy conversion. EDTA was oxidized at the exterior, while internal quinones could be reduced at the interior of the vesicle.
Besides vesicles other soft materials can be considered as exemplified by the supramolecular hydrogel scaffolds reported Wasielewski and Stupp.122 Perylene monoimide amphiphiles acted as chromophores and self-assembled into a 3D scaffold, that preorganized HEC using electrostatic interactions.
6 Supramolecular photosynthetic devices
The state-of-the-art applications for artificial photosynthesis focused on DSSC and DS-PEC devices, as discussed in Section 2, suffer from recombination processes lowering the efficiency. Moreover, the catalytic processes need optimization as they should require minimal overpotential, are very active and have sufficient stability. The supramolecular strategies discussed in Sections 3–5 address some of these issues, but mostly describe systems that are in homogeneous solutions and are aimed at understanding the role of supramolecular organization on (photo)chemical and (photo)catalytic properties of assemblies. In the next section, we will focus on a few studies that have actually employed supramolecular strategies in functional devices.6.1 Supramolecular DSSC
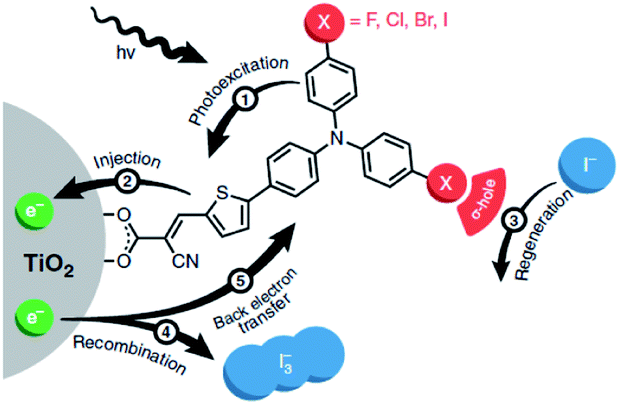 | ||
| Fig. 30 The redox mediator binds the dye after electron injection due to halogen-bonding substituents on the dye. It is then quickly regenerated and as a result back electron transfer is inhibited. Used with permission from ref. 123. | ||
Supramolecular antennae as discussed in Section 3 are applied in n-type DSSC. J-aggregates have been assembled by spin-coating Zn-porphyrin solutions onto TiO2.125 Interestingly, free base porphyrins heterogenized on TiO2 could be self-assembled with spherical agglomerates of Zn-porphyrins via H-bonding diphenylalanine substitutions (Fig. 31).126 The implementation of such antenna led to a doubling of the DSSC efficiency showing that implementation of supramolecular antennae can considerably boost the efficiency in devices.
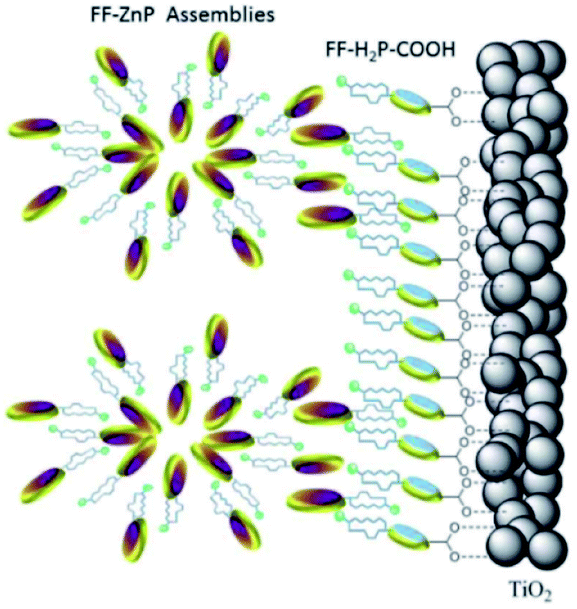 | ||
| Fig. 31 Supramolecular antenna complexes are assembled onto n-type semi-conductors (TiO2) using hydrogen-bonding interactions. H2-porphyrin acceptor (blue discs) act as the seed for the growth of Zn-porphyrin antennae acting as energy donor. Used with permission from ref. 126. | ||
Additional strategies reported in this area are a TiO2 functionalized material with coordinating ligands that bind porphyrins in multi-chromophoric assemblies.127 It also has been demonstrated that supramolecular porphyrin assemblies on TiO2 inhibit charge-recombination by stepwise charge separation.128
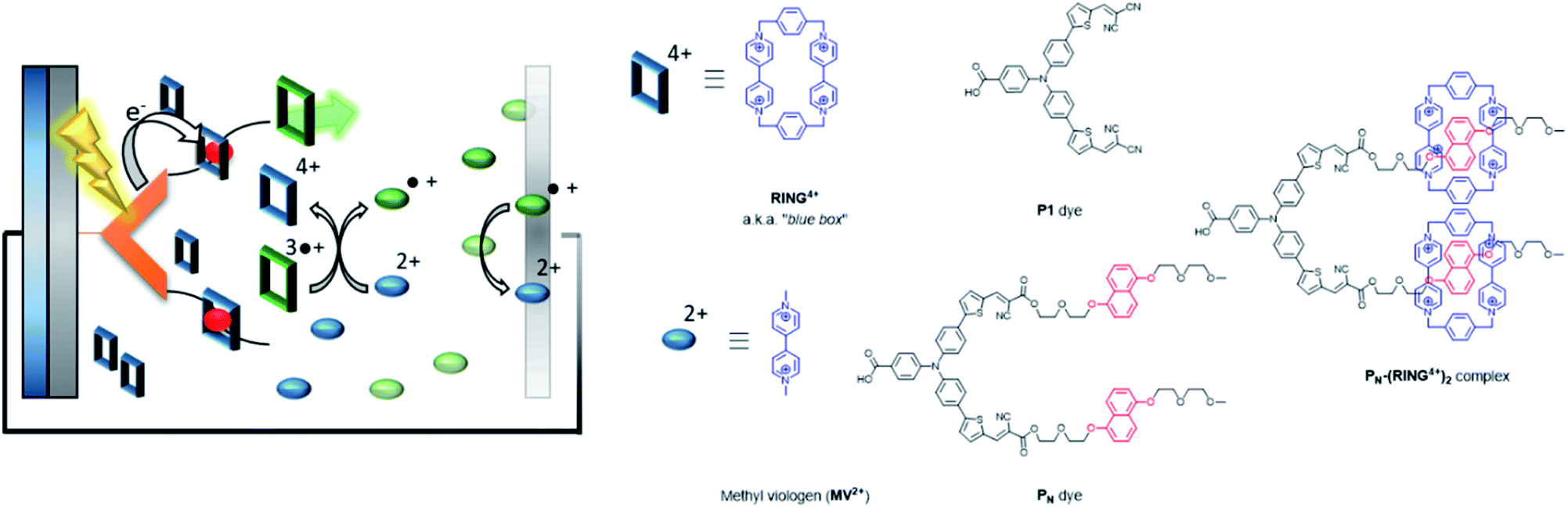 | ||
| Fig. 32 Supramolecular interactions are implemented in p-type DSSC using the “blue box” as redox shuttle. The PN dye is heterogenized onto NiO, and binds the ring prior to excitation. As soon as the ring is reduced, it loses affinity for the chromophore, dethreads and moves away while carrying the charge. Used with permission from ref. 129. | ||
Implementation of supramolecular ZrIV-phosphonate chemistry developed in the group of Mallouk provides a tool to create layer-by-layer assemblies.130 By sequentially dipping a metal-oxide (ITO, TiO2, NiO) coated glass slide in solution of phosphonic acid functionalized components alternated by a ZrIV solution, Meyer and co-workers synthesized an assembly purely based on molecular components with long lived photoinduced charge separated states that function as p/n-junctions (Fig. 33).131 Electron recombination occurred after 5.6 s for the photoanode and an impressive 26 ms for the photocathode. This bottom-up approach is very promising for future hybrid technologies, and has already been implemented in the preparation of supramolecular DS-PEC (vide infra).
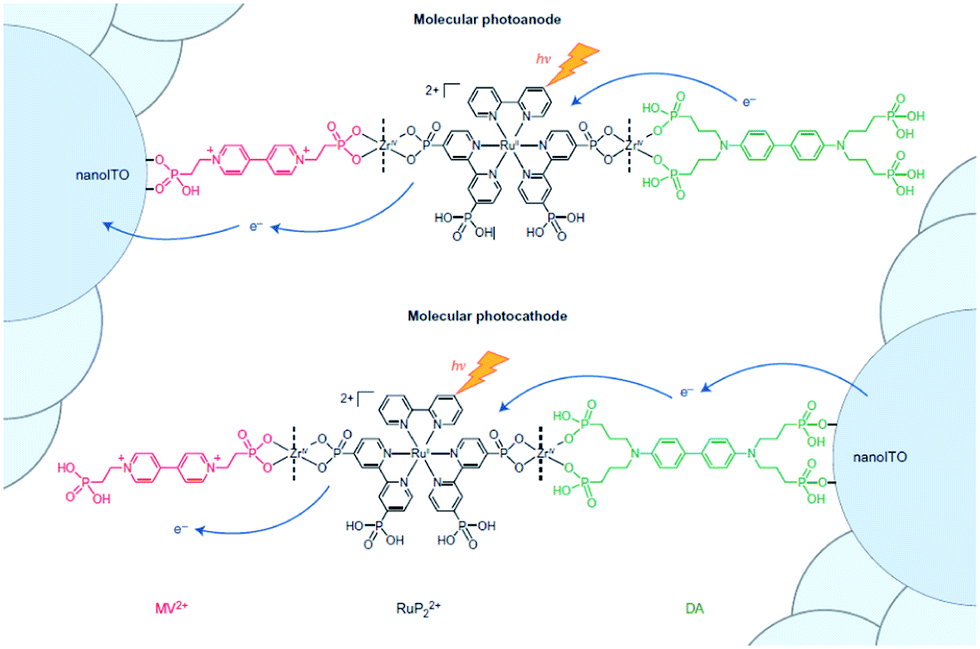 | ||
| Fig. 33 Molecular p/n-junctions are accessible by supramolecular ZrIV-phosphonate chemistry. Used with permission from ref. 131. | ||
Supramolecular p/n junctions can also be obtained via π–π stacking, NDI's based, zippers. By electronically tuning the NDI's via substitutions, a redox gradient can be prepared that enhances fill factors in optoelectronic devices.132,133
6.2 Supramolecular DS-PEC
Although supramolecular DS-PEC development is at a very early stage, these seminal devices show potential and as such are a source of inspiration. The first example of a DS-PEC using supramolecular strategies reported by the group of Wu was based on pyridine functionalized Ru-dye that was attached via a carboxylate linker to the NiO semiconductor (Fig. 34). The pyridine function was used to bind to a cobaloxime HEC.134 This dye-HEC design was already reported by Fihri et al. for solution based experiments and discussed above (Section 5.2.1). The supramolecular strategy to make this device resulted in a system that produced steady photocurrent suggesting a stable assembly on NiO. This was verified by absorption studies prior and after the photocatalysis experiments. The authors reported 68% faradaic efficiency for the production of H2. This work depicts that even a single dative bond can be sufficient to prepare dye-HEC assemblies for proton reduction.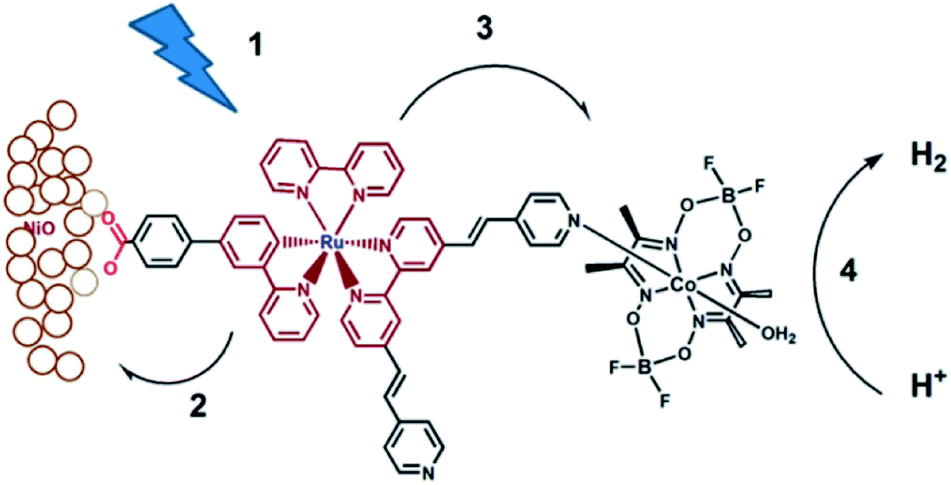 | ||
| Fig. 34 The dye-HEC assembly is anchored to NiO to produce a functional DS-PEC producing molecular hydrogen under illumination. The catalyst is preorganized to the dye by a dative, stable during operation. Used with permission from ref. 134. | ||
Another strategy—for water oxidation this time—was reported by the group of Sun.135 A Ru-photosensitizer was heterogenized on a TiO2 electrode using phosphate anchoring groups. Both the Ru-photosensitizer and the WOC were functionalized with long aliphatic chains leading to preorganization when the WOC was co-deposited with poly(methyl methacrylate) as supporting polymer material. The aliphatic chain was crucial to obtain stable light-driven water oxidation catalysis keeping the catalyst in the polymer matrix. Later, the group of Concepcion took the same catalyst and combined it with aliphatic chain functionalized chromophores already heterogenized on a metal oxide.136 The catalyst self-assembled onto the hydrophobic layer formed by the chromophores. A follow-up paper used this strategy to arrive at photoanodes for water oxidation operating at a current density of ∼2.2 mA cm−2 (Fig. 35).137 Clearly, hydrophobic interactions are suitable to preorganize chromophores and catalysts via self-assembly on electrode surfaces.
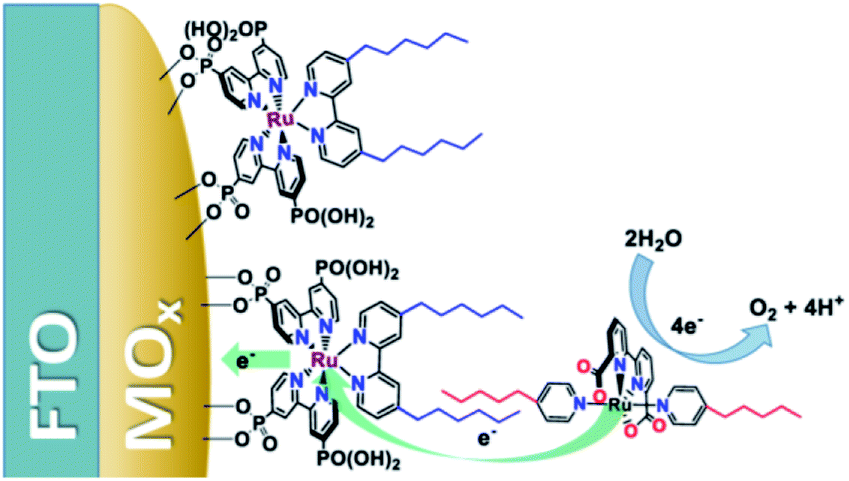 | ||
| Fig. 35 Heterogenized dyes bear aliphatic tails, allowing for supramolecular preorganization of Ru-WOC in DS-PEC devices. Used with permission from ref. 137. | ||
Lastly, Zr-phosphonate chemistry has been used to prepare dye-catalyst assemblies using a similar layer-by-layer deposition strategy as described above (Section 6.1.3). This was applied to water oxidation catalysis138,139 and proton reduction catalysis.140 In both examples the supramolecular strategy was of added value as it induced directionality in electron transfer, while inhibiting recombination from the surface to the catalyst. In an attempt to prepare an artificial analogue of PS II in terms of component numbers and their spatial separation, Wang et al. prepared a multi-component assembly containing acceptors, donors, chromophores and WOC (Fig. 36).141 Charge-separation occurred over a distance of 70 Å. The layer-by-layer strategy has recently been extended to supramolecular DS-PEC for CO2 reduction.142
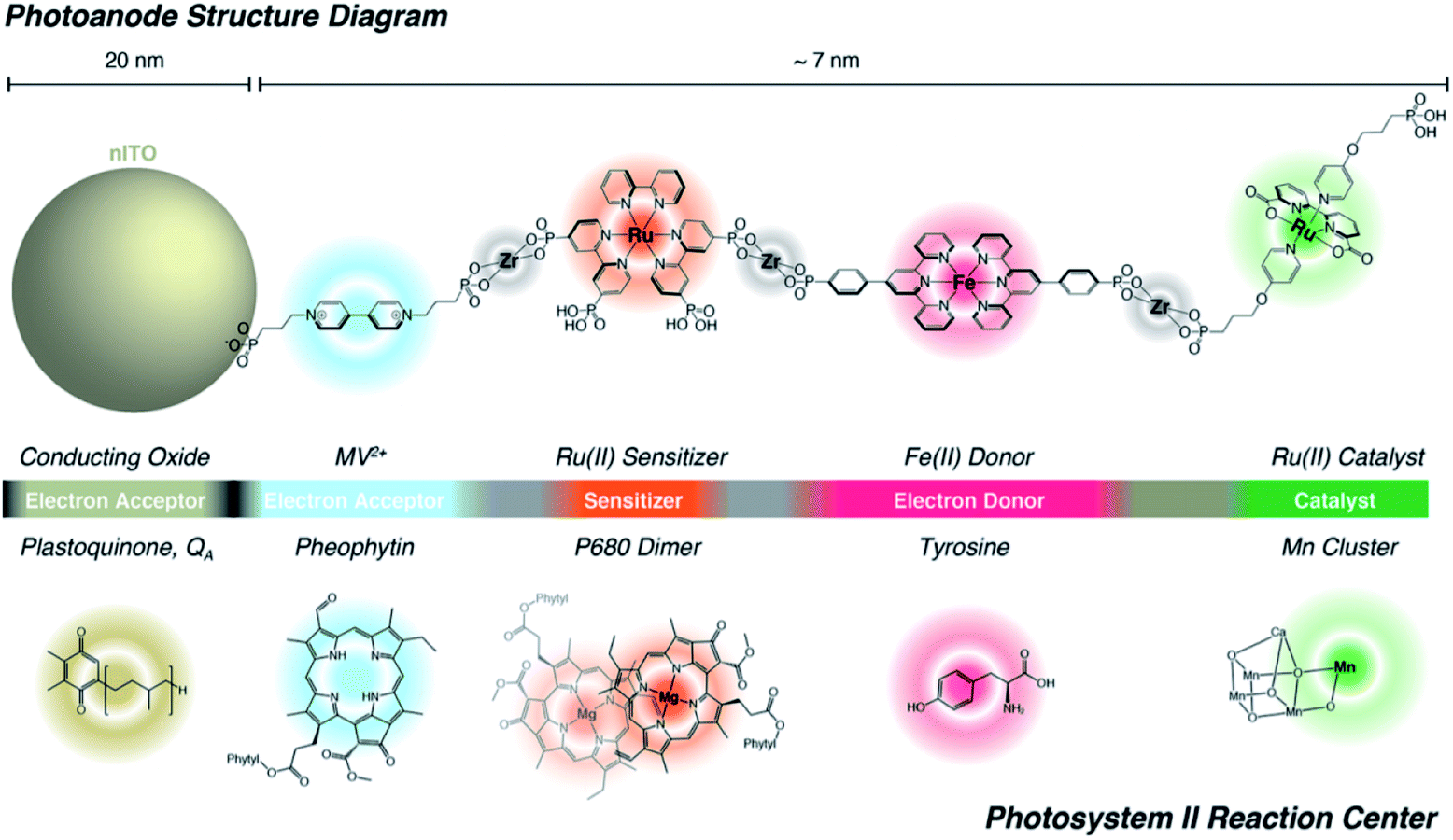 | ||
| Fig. 36 The supramolecular layer-by-layer deposition using Zr-phosphonate chemistry preorganizes chromophores reminding of the PS II. The top system is fully artificial, but the components serve the same purpose those in the PS II Reaction Centre and transfer electrons over a large distance. Used with permission from ref. 141. | ||
7 Conclusions
In this perspective we have discussed the use of supramolecular approaches in the field of solar to fuel devices. Though this emerging field is still under development, it has already proven to provide additional tools to control key aspects of artificial photosynthesis (1) light absorption (2) charge separation (3) redox catalysis. In addition, such tools can also be used in the construction of devices based on molecular components. For the light absorption, supramolecular organization of chromophores leads to the antenna effect and fast directional energy funnelling and charge-separation in light harvesting systems. Various supramolecular strategies are available to improve catalyst properties. For water oxidation catalysis, two reaction pathways are known, and the one that proceeds via a dinuclear mechanism can be stimulated and enhanced by supramolecular preorganization. In addition, supramolecular preorganization of the catalyst and the substrate, leading to high local concentrations, also enhances catalysis as is demonstrated for proton reduction catalysis. Catalyst stability is one of the key parameters to optimize, and encapsulation of catalysts in supramolecular cages can prevent degradation of catalysts, for example by preventing (dinuclear) disproportionation of reactive intermediates. Supramolecular preorganization can also be used to construct devices and electrodes, and preorganization of redox mediators and chromophores in DSSC have resulted in higher efficiencies.So far mostly components for artificial photosynthetic devices have been explored. The next challenge is to implement the components responsible for the different actions of artificial photosynthesis in working devices. Next to using components that use some of the discussed supramolecular strategies, self-assembly could also be to generate the highly organized, complex molecular architectures within the device. Supramolecular chemistry does not only enable facile synthesis of assemblies such as antennae, charge-separating and catalytic components, it may also allow for their integration into a fully artificial photosynthetic device. Such an assembly will be heavily inspired by natural photosynthesis, but may be simplified and more robust. We are looking forward to the days that these assemblies are coupled to inorganic materials, showing high efficiency and stability as operative DS-PEC.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by the Netherlands Organisation for Scientific Research (NWO) and National Science Foundation China (NSFC) as part of the program Cooperation China (NSFC) – Supramolecular Chemistry & Catalysis and was supported by the Holland Research School of Molecular Chemistry (HRSMC) and Foundation for Fundamental Research on Matter (FOM), which is part of NWO.Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS.
- S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- M. Roeb and H. Müller-Steinhagen, Science, 2010, 329, 773–774 CrossRef CAS.
- N. Armaroli and V. Balzani, Chem.–Eur. J., 2016, 22, 32–57 CrossRef CAS.
- R. J. Detz, K. Sakai, L. Spiccia, G. W. Brudvig, L. Sun and J. N. H. Reek, ChemPlusChem, 2016, 81, 1024–1027 CrossRef CAS.
- M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS.
- V. Balzani, A. Credi and M. Venturi, Curr. Opin. Chem. Biol., 1997, 1, 506–513 CrossRef CAS.
- R. Croce and H. Van Amerongen, Nat. Chem. Biol., 2014, 10, 492–501 CrossRef CAS.
- P. Knörzer, A. Silakov, C. E. Foster, F. A. Armstrong, W. Lubitz and T. Happe, J. Biol. Chem., 2012, 287, 1489–1499 CrossRef.
- J. M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 2836–2850 CrossRef CAS.
- T. J. Jacobsson, V. Fjällström, M. Edoff and T. Edvinsson, Energy Environ. Sci., 2014, 7, 2056–2070 RSC.
- A. V. Akimov, A. J. Neukirch and O. V. Prezhdo, Chem. Rev., 2013, 113, 4496–4565 CrossRef CAS.
- F. Li, K. Fan, B. Xu, E. Gabrielsson, Q. Daniel, L. Li and L. Sun, J. Am. Chem. Soc., 2015, 137, 9153–9159 CrossRef CAS.
- G. Peharz, F. Dimroth and U. Wittstadt, Int. J. Hydrogen Energy, 2007, 32, 3248–3252 CrossRef CAS.
- J. Jia, L. C. Seitz, J. D. Benck, Y. Huo, Y. Chen, J. W. D. Ng, T. Bilir, J. S. Harris and T. F. Jaramillo, Nat. Commun., 2016, 7, 13237 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- T. Hisatomi and K. Domen, Nat. Catal., 2019, 2, 387–399 CrossRef CAS.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS.
- M. Pazoki, U. B. Cappel, E. M. J. Johansson, A. Hagfeldt and G. Boschloo, Energy Environ. Sci., 2017, 10, 672–709 RSC.
- E. Benazzi, J. Mallows, G. H. Summers, F. A. Black and E. A. Gibson, J. Mater. Chem. C, 2019, 7, 10409–10445 RSC.
- H. J. Snaith, Nat. Photonics, 2012, 6, 337–340 CrossRef CAS.
- X. Yang, M. Yanagida and L. Han, Energy Environ. Sci., 2013, 6, 54–66 RSC.
- J. Halme, P. Vahermaa, K. Miettunen and P. Lund, Adv. Mater., 2010, 22, E210–E234 CrossRef CAS.
- K. Takagi, S. Magaino, H. Saito, T. Aoki and D. Aoki, J. Photochem. Photobiol., C, 2013, 14, 1–12 CrossRef CAS.
- P. R. F. Barnes, K. Miettunen, X. Li, A. Y. Anderson, M. Bessho, T. Grazel and B. C. O'Regan, Adv. Mater., 2013, 25, 1881–1922 CrossRef CAS.
- R. Katoh and A. Furube, J. Photochem. Photobiol., C, 2014, 20, 1–16 CrossRef CAS.
- F. Fabregat-Santiago, G. Garcia-Belmonte, I. Mora-Sero and J. Bisquert, Phys. Chem. Chem. Phys., 2011, 13, 9083–9118 RSC.
- E. M. J. Johansson, R. Lindblad, H. Siegbahn and A. Hagfeldt, ChemPhysChem, 2014, 15, 1006–1017 CrossRef CAS.
- H. N. Tsao, C. Yi, T. Moehl, J. H. Yum, S. M. Zakeeruddin, M. K. Nazeeruddin and M. Grätzel, ChemSusChem, 2011, 4, 591–594 CrossRef CAS.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS.
- C. J. Wood, G. H. Summers and E. A. Gibson, Chem. Commun., 2015, 51, 3915–3918 RSC.
- A. Nattestad, A. J. Mozer, M. K. R. Fischer, Y. B. Cheng, A. Mishra, P. Bäuerle and U. Bach, Nat. Mater., 2010, 9, 31–35 CrossRef CAS.
- K. Kakiage, Y. Aoyama, T. Yano, K. Oya, J. I. Fujisawa and M. Hanaya, Chem. Commun., 2015, 51, 15894–15897 RSC.
- I. R. Perera, T. Daeneke, S. Makuta, Z. Yu, Y. Tachibana, A. Mishra, P. Bäuerle, C. A. Ohlin, U. Bach and L. Spiccia, Angew. Chem., Int. Ed., 2015, 54, 3758–3762 CrossRef CAS.
- J. R. Swierk, D. D. Méndez-Hernández, N. S. McCool, P. Liddell, Y. Terazono, I. Pahk, J. J. Tomlin, N. V Oster, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1681–1686 CrossRef CAS.
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, 2008 Search PubMed.
- P. D. Frischmann, K. Mahata and F. Würthner, Chem. Soc. Rev., 2013, 42, 1847–1870 RSC.
- D. L. Dexter and J. H. Schulman, J. Chem. Phys., 1954, 22, 1063–1070 CrossRef CAS.
- J. Otsuki, J. Mater. Chem. A, 2018, 6, 6710–6753 RSC.
- H. Q. Peng, L. Y. Niu, Y. Z. Chen, L. Z. Wu, C. H. Tung and Q. Z. Yang, Chem. Rev., 2015, 115, 7502–7542 CrossRef CAS.
- G. De La Torre, G. Bottari, M. Sekita, A. Hausmann, D. M. Guldi and T. Torres, Chem. Soc. Rev., 2013, 42, 8049–8105 RSC.
- G. Bottari, O. Trukhina, M. Ince and T. Torres, Coord. Chem. Rev., 2012, 256, 2453–2477 CrossRef CAS.
- Y. Shinozaki, K. Ohkubo, S. Fukuzumi, K. Sugawa and J. Otsuki, Chem.–Eur. J., 2016, 22, 1165–1176 CrossRef CAS.
- R. F. Kelley, J. L. Suk, T. M. Wilson, Y. Nakamura, D. M. Tiede, A. Osuka, J. T. Hupp and M. R. Wasielewski, J. Am. Chem. Soc., 2008, 130, 4277–4284 CrossRef CAS.
- M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910–1921 CrossRef CAS.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef.
- P. Parkinson, N. Kamonsutthipaijit, H. L. Anderson and L. M. Herz, ACS Nano, 2016, 10, 5933–5940 CrossRef CAS.
- E. Iengo, G. D. Pantosx, J. K. M. Sanders, M. Orlandi, C. Chiorboli, S. Fracassoc and F. Scandola, Chem. Sci., 2011, 2, 676–685 RSC.
- P. Li, S. Amirjalayer, F. Hartl, M. Lutz, B. de Bruin, R. Becker, S. Woutersen and J. N. H. Reek, Inorg. Chem., 2014, 53, 5373–5383 CrossRef CAS.
- C. M. Drain and J. M. Lehn, J. Chem. Soc., Chem. Commun., 1994, 2313–2315 RSC.
- C. M. Drain, F. Nifiatis, A. Vasenko and J. D. Batteas, Angew. Chem., Int. Ed., 1998, 37, 2344–2347 CrossRef CAS.
- A. B. S. Elliott, J. E. M. Lewis, H. Van Der Salm, C. J. McAdam, J. D. Crowley and K. C. Gordon, Inorg. Chem., 2016, 55, 3440–3447 CrossRef CAS.
- H. Dekkiche, A. Buisson, A. Langlois, P. L. Karsenti, L. Ruhlmann, P. D. Harvey and R. Ruppert, Inorg. Chem., 2016, 55, 10329–10336 CrossRef CAS.
- J. L. Sessler, B. Wang and A. Harriman, J. Am. Chem. Soc., 1995, 117, 704–714 CrossRef CAS.
- J. Otsuki, K. Iwasaki, Y. Nakano, M. Itou, Y. Araki and O. Ito, Chem.–Eur. J., 2004, 10, 3461–3466 CrossRef CAS.
- J. L. Sessler, A. Hardman and B. Wang, J. Am. Chem. Soc., 1993, 115, 10418–10419 CrossRef CAS.
- Y. Kuroda, K. Sugou and K. Sasaki, J. Am. Chem. Soc., 2000, 122, 7833–7834 CrossRef CAS.
- K. Sugou, K. Sasaki, K. Kitajima, T. Iwaki and Y. Kuroda, J. Am. Chem. Soc., 2002, 124, 1182–1183 CrossRef CAS.
- S. Fukuzumi, K. Saito, K. Ohkubo, T. Khoury, Y. Kashiwagi, M. A. Absalom, S. Gadde, F. D'Souza, Y. Araki, O. Ito and M. J. Crossley, Chem. Commun., 2011, 47, 7980–7982 RSC.
- Y. Kuramochi, A. Satake and Y. Kobuke, J. Am. Chem. Soc., 2004, 126, 8668–8669 CrossRef CAS.
- Y. Kuramochi, A. S. D. Sandanayaka, A. Satake, Y. Araki, K. Ogawa, O. Ito and Y. Kobuke, Chem.–Eur. J., 2009, 15, 2317–2327 CrossRef CAS.
- N. Nagata, Y. Kuramochi and Y. Kobuke, J. Am. Chem. Soc., 2009, 131, 10–11 CrossRef CAS.
- I. Zaharieva, M. M. Najafpour, M. Wiechen, M. Haumann, P. Kurz and H. Dau, Energy Environ. Sci., 2011, 4, 2400–2408 RSC.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS.
- C. J. Richmond, R. Matheu, A. Poater, L. Falivene, J. Benet-Buchholz, X. Sala, L. Cavallo and A. Llobet, Chem.–Eur. J., 2014, 20, 17282–17286 CrossRef CAS.
- J. Hessels, R. J. Detz, M. T. M. Koper and J. N. H. Reek, Chem.–Eur. J., 2017, 23, 16413–16418 CrossRef CAS.
- L. Duan, A. Fischer, Y. Xu and L. Sun, J. Am. Chem. Soc., 2009, 131, 10397–10399 CrossRef CAS.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS.
- Y. Xie, D. W. Shaffer and J. J. Concepcion, Inorg. Chem., 2018, 57, 10533–10542 CrossRef CAS.
- B. Li, F. Li, S. Bai, Z. Wang, L. Sun, Q. Yang and C. Li, Energy Environ. Sci., 2012, 5, 8229 RSC.
- B. Yang, X. Jiang, Q. Guo, T. Lei, L. P. Zhang, B. Chen, C. H. Tung and L. Z. Wu, Angew. Chem., Int. Ed., 2016, 55, 6229–6234 CrossRef CAS.
- F. Yu, D. Poole, S. Mathew, N. Yan, J. Hessels, N. Orth, I. Ivanović-Burmazović and J. N. H. Reek, Angew. Chem., Int. Ed., 2018, 57, 11247–11251 CrossRef CAS.
- M. Schulze, V. Kunz, P. D. Frischmann and F. Würthner, Nat. Chem., 2016, 8, 576–583 CrossRef CAS.
- V. Kunz, J. O. Lindner, M. Schulze, M. I. S. Röhr, D. Schmidt, R. Mitrić and F. Würthner, Energy Environ. Sci., 2017, 10, 2137–2153 RSC.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66–69 CrossRef CAS.
- B. Ginovska, S. Raugei and W. J. Shaw, Molecular Dynamics Studies of Proton Transport in Hydrogenase and Hydrogenase Mimics, Elsevier Inc., 1st edn, 2016, vol. 578 Search PubMed.
- S. Qiu, L. M. Azofra, D. R. MacFarlane and C. Sun, Phys. Chem. Chem. Phys., 2018, 20, 6735–6743 RSC.
- S. Dementin, V. Belle, P. Bertrand, B. Guigliarelli, G. Adryanczyk-Perrier, A. L. De Lacey, V. M. Fernandez, M. Rousset and C. Léger, J. Am. Chem. Soc., 2006, 128, 5209–5218 CrossRef CAS.
- B. Ginovska-Pangovska, A. Dutta, M. L. Reback, J. C. Linehan and W. J. Shaw, Acc. Chem. Res., 2014, 47, 55 CrossRef.
- A. Pannwitz and O. S. Wenger, Chem. Commun., 2019, 55, 4004–4014 RSC.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS.
- J. Duan, S. Mebs, K. Laun, F. Wittkamp, J. Heberle, T. Happe, E. Hofmann, U. P. Apfel, M. Winkler, M. Senger, M. Haumann and S. T. Stripp, ACS Catal., 2019, 9, 9140–9149 CrossRef CAS.
- A. Dutta, A. M. Appel and W. J. Shaw, Nat. Rev. Chem., 2018, 2, 244–252 CrossRef CAS.
- M. L. Singleton, J. H. Reibenspies and M. Y. Darensbourg, J. Am. Chem. Soc., 2010, 132, 8870–8871 CrossRef CAS.
- M. L. Singleton, D. J. Crouthers, R. P. Duttweiler, J. H. Reibenspies and M. Y. Darensbourg, Inorg. Chem., 2011, 50, 5015–5026 CrossRef CAS.
- F. J. Rizzuto, L. K. S. von Krbek and J. R. Nitschke, Nat. Rev. Chem., 2019, 3, 204–222 CrossRef.
- S. S. Nurttila, R. Zaffaroni, S. Mathew and J. N. H. Reek, Chem. Commun., 2019, 55, 3081–3084 RSC.
- R. Zaffaroni, N. Orth, I. Ivanović-Burmazović and J. N. H. Reek, Angew. Chem. Int. Ed., 2020, 59, 18485–18489 CrossRef CAS.
- R. Zaffaroni, E. O. Bobylev, R. Plessius, J. I. van der Vlugt and J. N. H. Reek, J. Am. Chem. Soc., 2020, 142, 8837–8847 CrossRef.
- W. P. Brezinski, M. Karayilan, K. E. Clary, N. G. Pavlopoulos, S. Li, L. Fu, K. Matyjaszewski, D. H. Evans, R. S. Glass, D. L. Lichtenberger and J. Pyun, Angew. Chem., Int. Ed., 2018, 57, 11898–11902 CrossRef CAS.
- B. Reuillard, J. Warnan, J. J. Leung, D. W. Wakerley and E. Reisner, Angew. Chem., 2016, 128, 4020–4025 CrossRef.
- Y. Liu and C. C. L. McCrory, Nat. Commun., 2019, 10, 1–10 CrossRef.
- F. Li, Y. Jiang, B. Zhang, F. Huang, Y. Gao and L. Sun, Angew. Chem., Int. Ed., 2012, 51, 2417–2420 CrossRef CAS.
- H. Li, F. Li, B. Zhang, X. Zhou, F. Yu and L. Sun, J. Am. Chem. Soc., 2015, 137, 4332–4335 CrossRef CAS.
- Y. Choi, D. Jeon, Y. Choi, J. Ryu and B. S. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 13434–13441 CrossRef CAS.
- G. Paille, M. Gomez-Mingot, C. Roch-Marchal, B. Lassalle-Kaiser, P. Mialane, M. Fontecave, C. Mellot-Draznieks and A. Dolbecq, J. Am. Chem. Soc., 2018, 140, 3613–3618 CrossRef CAS.
- G. Paille, M. Gomez-Mingot, C. Roch-Marchal, M. Haouas, Y. Benseghir, T. Pino, M. H. Ha-Thi, G. Landrot, P. Mialane, M. Fontecave, A. Dolbecq and C. Mellot-Draznieks, ACS Appl. Mater. Interfaces, 2019, 11, 47837–47845 CrossRef CAS.
- M. Bonchio, Z. Syrgiannis, M. Burian, N. Marino, E. Pizzolato, K. Dirian, F. Rigodanza, G. A. Volpato, G. La Ganga, N. Demitri, S. Berardi, H. Amenitsch, D. M. Guldi, S. Caramori, C. A. Bignozzi, A. Sartorel and M. Prato, Nat. Chem., 2019, 11, 146–153 CrossRef CAS.
- X. Jing, Y. Yang, C. He, Z. Chang, J. N. H. Reek and C. Duan, Angew. Chem., Int. Ed., 2017, 56, 11759–11763 CrossRef CAS.
- A. F. Heyduk and D. G. Nocera, Science, 2001, 293, 1639–1641 CrossRef CAS.
- V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238–7266 CrossRef CAS.
- A. Fihri, V. Artero, M. Razavet, C. Baffert, W. Leibl and M. Fontecave, Angew. Chem., Int. Ed., 2008, 47, 564–567 CrossRef CAS.
- C. Li, M. Wang, J. Pan, P. Zhang, R. Zhang and L. Sun, J. Organomet. Chem., 2009, 649, 2814–2819 CrossRef.
- W. Lubitz, E. J. Reijerse and J. Messinger, Energy Environ. Sci., 2008, 1, 15–31 RSC.
- F. Gloaguen and T. B. Rauchfuss, Chem. Soc. Rev., 2009, 38, 100–108 RSC.
- A. M. Kluwer, R. Kapre, F. Hartl, M. Lutz, A. L. Spek, A. M. Brouwer, P. W. N. M. Van Leeuwen and J. N. H. Reek, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10460–10465 CrossRef CAS.
- S. Pullen, H. Fei, A. Orthaber, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2013, 135, 16997–17003 CrossRef CAS.
- K. Sasan, Q. Lin, C. Mao and P. Feng, Chem. Commun., 2014, 50, 10390–10393 RSC.
- X. Jing, C. He, Y. Yang and C. Duan, J. Am. Chem. Soc., 2015, 137, 3967–3974 CrossRef CAS.
- C. Orain, F. Quentel and F. Gloaguen, ChemSusChem, 2014, 7, 638–643 CrossRef CAS.
- M. Hansen, S. Troppmann and B. König, Chem.–Eur. J., 2016, 22, 58–72 CrossRef CAS.
- K. Watanabe, K. Moriya, T. Kouyama, A. Onoda, T. Minatani, S. Y. Takizawa and S. Murata, J. Photochem. Photobiol., A, 2011, 221, 113–122 CrossRef CAS.
- L. He, C. Luo, Y. Hou, C. Li, Q. Zhou, Y. Sun, W. Wang, B. Zhang and X. Wang, Int. J. Hydrogen Energy, 2011, 36, 10593–10599 CrossRef CAS.
- H.-Y. Wang, W.-G. Wang, G. Si, F. Wang, C.-H. Tung and L.-Z. Wu, Langmuir, 2010, 26, 9766–9771 CrossRef CAS.
- S. Troppmann and B. König, Chem.–Eur. J., 2014, 20, 14570–14574 CrossRef CAS.
- R. Becker, T. Bouwens, E. C. F. Schippers, T. Gelderen, M. Hilbers, S. Woutersen and J. N. H. Reek, Chem.–Eur. J., 2019, 25, 13921–13929 CrossRef CAS.
- G. Steinberg-Yfrach, J. L. Rigaud, E. N. Durantini, A. L. Moore, D. Gust and T. A. Moore, Nature, 1998, 392, 479–482 CrossRef CAS.
- S. Bhosale, A. L. Sisson, P. Talukdar, A. Fürstanberg, N. Banetji, E. Vauthey, G. Bollot, J. Mareda, C. Röger, F. Würthner, N. Sakai and S. Matile, Science, 2006, 313, 84–86 CrossRef CAS.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, M. McClendon, A. R. Koltonow, A. P. S. Samuel, D. J. Kiebala, M. R. Wasielewski and S. I. Stupp, Nat. Chem., 2014, 6, 964–970 CrossRef CAS.
- F. G. L. Parlane, C. Mustoe, C. W. Kellett, S. J. Simon, W. B. Swords, G. J. Meyer, P. Kennepohl and C. P. Berlinguette, Nat. Commun., 2017, 8, 1761 CrossRef.
- Y. Uemura, T. N. Murakami and N. Koumura, J. Phys. Chem. C, 2014, 118, 16749–16759 CrossRef CAS.
- A. Huijser, B. M. J. M. Suijkerbuijk, R. J. M. K. Gebbink, T. J. Savenije and L. D. A. Siebbeles, J. Am. Chem. Soc., 2008, 130, 2485–2492 CrossRef CAS.
- G. Charalambidis, K. Karikis, E. Georgilis, B. L. M'Sabah, Y. Pellegrin, A. Planchat, B. Lucas, A. Mitraki, J. Bouclé, F. Odobel and A. G. Coutsolelos, Sustainable Energy Fuels, 2017, 1, 387–395 RSC.
- N. K. Subbaiyan, C. A. Wijesinghe and F. D'Souza, J. Am. Chem. Soc., 2009, 131, 14646–14647 CrossRef CAS.
- M. Planells, L. Pellejà, P. Ballester and E. Palomares, Energy Environ. Sci., 2011, 4, 528–534 RSC.
- T. Bouwens, S. Mathew and J. N. H. Reek, Faraday Discuss., 2019, 215, 393–406 RSC.
- G. Cao, H. G. Hong and T. E. Mallouk, Acc. Chem. Res., 1992, 25, 420–427 CrossRef CAS.
- B. H. Farnum, K.-R. Wee and T. J. Meyer, Nat. Chem., 2016, 8, 845–852 CrossRef CAS.
- R. S. K. Kishore, O. Kel, N. Banerji, D. Emery, G. Bollot, J. Mareda, A. Gomez-Casado, P. Jonkheijm, J. Huskens, P. Maroni, M. Borkovec, E. Vauthey, N. Sakai and S. Matile, J. Am. Chem. Soc., 2009, 131, 11106–11116 CrossRef CAS.
- R. Bhosale, J. Míśek, N. Sakai and S. Matile, Chem. Soc. Rev., 2010, 39, 138–149 RSC.
- Z. Ji, M. He, Z. Huang, U. Ozkan and Y. Wu, J. Am. Chem. Soc., 2013, 135, 11696–11699 CrossRef CAS.
- X. Ding, Y. Gao, L. Ye, L. Zhang and L. Sun, ChemSusChem, 2015, 8, 3992–3995 CrossRef CAS.
- L. Wang, D. E. Polyansky and J. J. Concepcion, J. Am. Chem. Soc., 2019, 141, 8020–8024 CAS.
- D. Wang, L. Wang, M. D. Brady, C. J. Dares, G. J. Meyer, T. J. Meyer and J. J. Concepcion, J. Phys. Chem. C, 2019, 123, 30039–30045 CrossRef CAS.
- K. Hanson, D. A. Torelli, A. K. Vannucci, M. K. Brennaman, H. Luo, L. Alibabaei, W. Song, D. L. Ashford, M. R. Norris, C. R. K. Glasson, J. J. Concepcion and T. J. Meyer, Angew. Chem., Int. Ed., 2012, 51, 12782–12785 CrossRef CAS.
- X. Ding, Y. Gao, L. Zhang, Z. Yu, J. Liu and L. Sun, ACS Catal., 2014, 4, 2347–2350 CrossRef CAS.
- M. A. Gross, C. E. Creissen, K. L. Orchard and E. Reisner, Chem. Sci., 2016, 7, 5537–5546 RSC.
- D. Wang, R. N. Sampaio, L. Troian-Gautier, S. L. Marquard, B. H. Farnum, B. D. Sherman, M. V. Sheridan, C. J. Dares, G. J. Meyer and T. J. Meyer, J. Am. Chem. Soc., 2019, 141, 7926–7933 CrossRef CAS.
- D. Wang, Y. Wang, M. D. Brady, M. V. Sheridan, B. D. Sherman, B. H. Farnum, Y. Liu, S. L. Marquard, G. J. Meyer, C. J. Dares and T. J. Meyer, Chem. Sci., 2019, 10, 4436–4444 RSC.
| This journal is © The Royal Society of Chemistry 2021 |