
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cork-derived hierarchically porous hydroxyapatite with different stoichiometries for biomedical and environmental applications†
Francesca
Scalera
 a,
Alessandra
Quarta
a,
David M.
Tobaldi
a,
Robert C.
Pullar
bc and
Clara
Piccirillo
*a
a,
Alessandra
Quarta
a,
David M.
Tobaldi
a,
Robert C.
Pullar
bc and
Clara
Piccirillo
*a
aCNR NANOTEC, Institute of Nanotechnology, Campus Ecotekne, 73047 Lecce, Italy. E-mail: clara.piccirillo@nanotec.cnr.it
bDepartment of Materials and Ceramic Engineering/CICECO – Aveiro Institute of Materials, University of Aveiro, Campus Universitário de Santiago, 3810-193 Aveiro, Portugal
cDipartimento di Scienze Molecolari e Nanosistemi (DSMN), Ca’ Foscari University of Venice, Via Torino 155, 30172 Venezia Mestre, Italy
First published on 18th May 2021
Abstract
Hydroxyapatite (HA, Ca10(PO4)6(OH)2) is the major mineral component of human bone, but has a wide range of interesting and useful properties, and many applications beyond biomedicine. Here we produce HA-based materials from a naturally templated precursor which can have a variety of tailored microstructures and hierarchical porosities, and we investigate their use as designed functional materials for several applications. HA-based materials with different stoichiometries of Ca/P = 1.67 (HA) and 1.5 (Ca-deficient HA) were successfully synthesised from a previously prepared and reported cork-derived CaCO3. The CaCO3 was reacted with a phosphorus containing precursor solution at 60 °C, and also successively recalcined at 700 °C. The samples prepared at 60 °C maintained the 3D honeycomb structure of ∼20 μm hexagonal cells typical of cork, and showed high specific surface areas (SSA > 85 m2 g−1) and a high mesoporosity (average mesopores ∼10–15 nm). These features made them suitable for environmental remediation applications such as heavy metals (i.e. lead) removal from water, with calcium-deficient HA (higher degree of mesoporosity) adsorbing >99% of Pb(II) ions from solutions containing up to 1.5 g of lead per L. Recalcined samples (SSA = 29 m2 g−1), on the other hand, had very low cytotoxicity towards MG-63 osteoblasts, showing them to be biocompatible for biomaterials. All had cell viabilities of over 70% against a control, and the calcium-deficient HA proved to be even less cytotoxic than the control (>100% viability after 24 and 48 h). Therefore, cork was shown to be an excellent template agent for the design of HA materials with varied properties, tailored for different applications in the environmental and biomedical fields.
Introduction
Calcium phosphates (CaP) are a very important class of materials due to their many interesting properties and technological applications, particularly in the biomedical field.1 Hydroxyapatite (HA, Ca10(PO4)6(OH)2) is the most used CaP compound in biomedicine, as a bone substitute. HA is in fact the main component of human bone. Because of this, synthetic HA is employed when parts of bones need to be replaced, due to traumas and/or pathologies.2 In addition to biocompatibility, HA is capable of inducing bone regrowth (osteoconductivity). Stoichiometric HA has a Ca/P ratio of 1.67, while bone mineral typically has a slightly smaller ratio and contains some carbonate ions;3 because of this, calcium-deficient hydroxyapatite (CDHA) has also been considered for biomedical applications, along with mixtures of 75% HA and 25% β-tricalcium phosphate (β-TCP, Ca3(PO4)2, Ca![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P ratio = 1.5). The use of these in place of stoichiometric HA has led to improved bioactivity and resorbability of bone scaffolds.4
:P ratio = 1.5). The use of these in place of stoichiometric HA has led to improved bioactivity and resorbability of bone scaffolds.4
In addition to biomedicine, HA can also be used for environmental remediation, as it can remove heavy metals from contaminated soils or wastewaters.5,6 Generally, the main mechanisms involved in the removal are ion exchange between the calcium in the HA lattice and the heavy metal ions in solution,7 as well as a dissolution–precipitation process.8
The performance of a material can be affected by different parameters; in the case of cellular growth, for instance, literature shows that morphology and porosity play a key role for HA-based compounds.9,10 For both cellular proliferation and heavy metal removal, on the other hand, hierarchical porosity – pores with dimensions in different ranges on the nano- and micron scale – can lead to enhanced performance.11–13
In recent years, there has been a growing interest in developing materials inspired by nature; this is because often natural materials have a particular structure and/or morphology which can lead to enhanced performance.14 Plant- or wood-based compounds, for instance, were considered as template agents to prepare ceramics: the prepared ceramics (ecoceramics) replicate the 3D structure of the wood template.14 Literature reports various ceramics synthesised using wood template agents, including SiC and oxides.15–17
Cork, the bark of the oak tree Quercus suber L., is a promising wood material to be used as template agent, due to its interesting 3D structure. It consists of cells arranged in a hexagonal honeycomb shape in the radial direction, and rectangular in the transverse direction. The average dimensions of the cells are 40–45 μm and 15–20 μm in the radial and transverse directions, respectively. The walls of the cells are about 1–1.5 μm thick, and are crossed by channels with an average diameter smaller than 100 nm.18 Cork is a very sustainable material, as the tree is not harmed when the bark is harvested every 9–13 years; moreover, after harvesting, the tree acts as a carbon sink as it continues to grow a new layer of bark.19
Regarding its composition, the main components of cork are suberin and lignins, about 45 and 22% respectively. These organic molecules can be converted into inorganic graphitic carbon with a thermal treatment in an inert atmosphere (i.e. N2).20 If the process is performed with a slow heating ramp, the 3D structure of the cork can be preserved, leading to a porous carbon skeleton, which can be infiltrated with precursor solutions and successively converted into different ceramic materials upon subsequent heating. Literature reports several cork-derived ceramics, such as silicon carbide, hexagonal ferrites, zinc oxide and cerium oxide.20–23
In previous work carried out by our group, CaCO3 was prepared with cork as template agent.24 By carefully selecting the preparation conditions, a porous material was prepared, which maintained the 3D structure of cork and showed high surface area. In the present work, cork-derived CaCO3 is used as starting material (i.e. calcium source) for the synthesis of HA-based compounds; to achieve this, cork-derived CaCO3 was reacted with a phosphorus source – a (NH4)2HPO4 solution – in a solid–liquid reaction.25 Different reaction conditions (time and stoichiometric ratios) were considered, and the obtained materials were fully characterised. The obtained HA-based materials could have a variety of microstructures and hierarchical porosities depending upon processing, and were investigated as designed functional materials for a number of different applications. Functional properties were tested for diverse applications in biomedicine or for environmental remediation. The cytotoxicity of the materials was assessed with cellular growth experiments, and the adsorption capacity for heavy metals (i.e. lead) was also studied. Results showed that synthesis conditions could be tailored to make materials more suitable for use in either the biomedical or environmental fields.
Materials and methods
HA and CDHA preparation
The preparation of cork-derived CaCO3 was described in detail in previous work;24 a brief summary is given. Pyrolysed cork powder (average dimensions 200–400 μm) was infiltrated with calcium acetate and dried overnight. The powder was subsequently treated at 700 °C in an inert atmosphere (N2), the thermal treatment leading to the conversion of the acetate into carbonate, although some residual graphitic carbon was detected. As prepared CaCO3 was converted into CaP materials using a known protocol25 with some modifications.A weighed amount of CaCO3 was placed in distilled water, and a stoichiometric amount of (NH4)2HPO4 was added and pH was adjusted to 9. Two Ca/P ratios were considered, 1.5 and 1.67, for the synthesis of CDHA and HA, respectively. The reaction was carried out at 60 °C and pH = 9, and for different durations (0–16 h). After the reaction, the powder was separated from the solution by centrifugation; it was then washed with distilled water several times, to reach neutral pH, and successively dried at 60 °C.
Some selected samples were then calcined at 700 °C (1 °C min−1, 3 hours).
Characterisation
The phase composition of the samples was determined by X-ray diffraction (XRD), using an X’Pert PRO MRD diffractometer, with a CuKα radiation; the diffraction patterns were acquired with a step size of 0.005° 2θ and a counting time of 100 seconds, and the interval between 2θ = 20° and 60° was considered. Phase identification was performed compared to the standard diffraction patterns 00-005-0586 for CaCO3 (calcite) and 01-072-1243 for HA. Absolute unit cell parameters of the prepared samples were determined using LaB6 (NIST 660b) as an internal standard. Rietveld refinements were attained by means of the GSAS software package,26 and its graphical interface EXPGUI.27 The starting atomic parameters for hydroxyapatite, described in the space group P63/m, were taken from the literature.28 These parameters were refined: scale-factors and zero point; the background profile was modelled using a 10th order shifted Chebyshev polynomial function. Unit cell parameters and profile coefficients were modelled with two Lorentzian terms (LX and LY), whilst the angle independent Gaussian term (GW) was constrained to the value obtained for LaB6, with peak correction for asymmetry and sample displacement effects.Thermogravimetric analysis (TGA) was performed on as synthesised hydroxyapatite powders using SDT Q600 apparatus (TA Instruments). The calcination treatment was replicated to identify consequential weight losses during the different thermal treatment phases.
The morphology of the samples was analysed by scanning electron microscopy (SEM) using a Carl Zeiss Merlin instrument, equipped with a Gemini II column and an integrated high efficiency In-lens for secondary electrons. Samples were sputtered with gold before the analysis.
Surface area was determined by a Micrometric Gemini VII Surface Area Analyzer from Micrometric Instrument Corp.; N2 was employed as the adsorbate gas, and samples were degassed at 120 °C. Specific surface area (SSA) was calculated with the BET method.
Adsorption experiments
For some selected samples, adsorption experiments with Pb(II) ions were performed. A weighed amount of powder (0.016 g) was placed into a Pb(II) containing solution (8 ml) in a 5.5 cm Petri dish; the dishes were gently stirred (90 rpm) at 20 °C. At the end of the experiments, the solid powder was separated from the solution through centrifugation, and the supernatant was analysed to determine the residual Pb(II) concentration. The analysis were performed using an inductively coupled plasma – atomic emission spectrometer (ICP-AES, Varian 720-ES). As a first screening, different adsorption times were considered, to study the kinetics of the process. Successively different Pb(II) concentrations were tested for an adsorption time of 6 hours.Data from the kinetics study were fitted with the Lagergren and Crank models, according to eqn (1) and (2) respectively:
| ln(qt − qe) = lnqe − k × t | (1) |
| qt = K × t0.5 | (2) |
For the experiments performed with different Pb(II) concentrations, the removal efficiency was calculated with the formula:
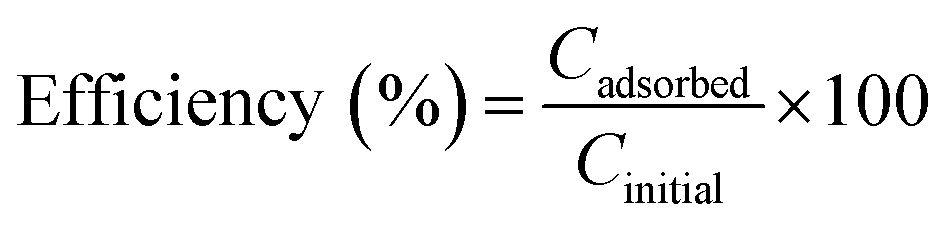 | (3) |
Biocompatibility
The biocompatibility of the samples was evaluated with the MTT assay performed on the MG-63 (osteoblast) cell line. The cells were grown in DMEM medium supplemented with 10% of foetal bovine serum (FBS), 2 mM glutamine, 100 IU mL−1 of penicillin and 100 μg mL−1 of streptomycin, and were cultured in an incubator at 37 °C in a humidified atmosphere with 5% CO2.The cells (2.5 × 104), suspended in 500 μL of culture medium, were seeded into each well of a 48 multiwell plate. After 24 h incubation, the sterilised powders were added to the wells at different concentrations (100, 250 and 500 μg mL−1, each point in triplicate); the cells were kept under incubation for three different times, 24, 48 and 96 h, respectively. At the end of the incubation time, the medium was removed, the cells were washed twice with PBS, and 200 μL of fresh serum-free medium containing 2 mg mL−1 MTT were added to each well. After 3 h of incubation at 37 °C, the medium was discarded from the wells and 500 μL of DMSO were added to dissolve the formazan salts. The plate was stirred for a few minutes at ambient temperature, and the absorbance of the solution at 570 nm was measured on a microplate reader (CLARIOstar Plus, BMG Labtech). To determine the percentage of cell viability, the treated samples were compared to the control samples according to eqn (4):
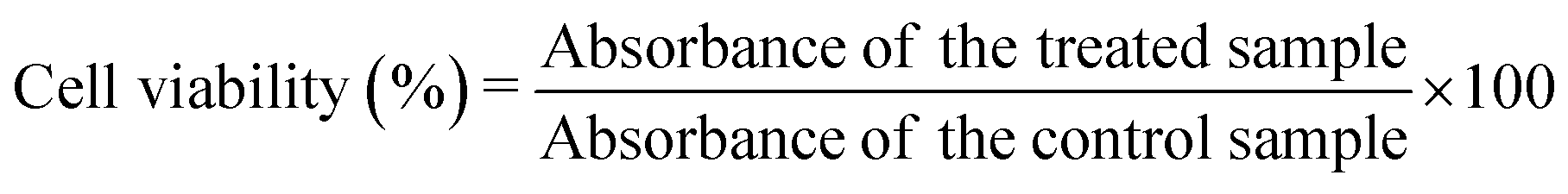 | (4) |
Results and discussion
Preparation of HA and CDHA
As stated in the previous section, HA with two different stoichiometries was prepared. In some preliminary experiments different reaction times were considered, to monitor the conversion of CaCO3 into HA; these first tests were all performed with pH adjusted to 9, to avoid the formation of different CaP phases.29 At this first stage only a stoichiometric HA Ca/P molar ratio was considered (1.67). Table 1 reports all the reaction conditions employed, while Fig. 1 shows the XRD patterns for the starting material, as well as the products for 4, 8 and 16 hours of reaction (samples HA_4, HA_8 and HA_16 respectively).| Reaction conditions | Composition | |
|---|---|---|
| HA_0 | Ca/P = 1.67, t = 0 h | CaCO3 |
| HA_4 | Ca/P = 1.67, t = 4 h | CaCO3–HA |
| HA_8 | Ca/P = 1.67, t = 8 h | CaCO3–HA |
| HA_16 | Ca/P = 1.67, t = 16 h | HA |
| CDHA_16 | Ca/P = 1.5, t = 16 h | HA |
| Thermal treatment | ||
| HA_16_700 | 700 °C, 3 h | HA |
| CDHA_16_700 | 700 °C, 3 h | HA |
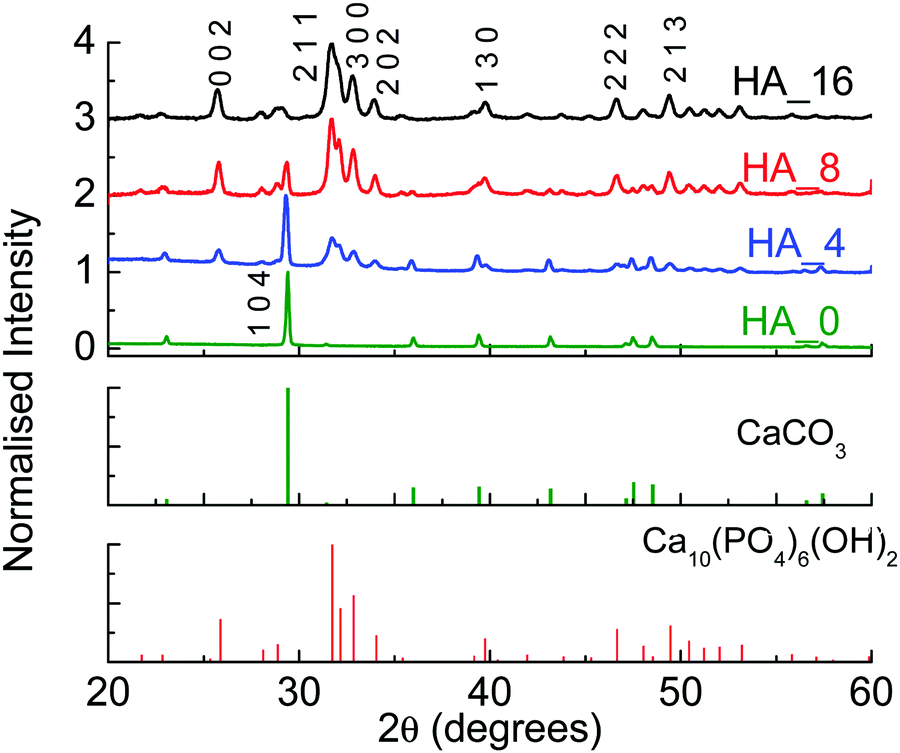 | ||
| Fig. 1 XRD patterns for the HA samples prepared for different reaction times. | ||
It can be seen that after 4 hours (HA_4), calcite is still the main component in the material, but some peaks belonging to HA are present (2θ = 31.7°). After 8 hours (HA_8) HA becomes the main component, but a significant amount of calcite is still detected. For 16 hours (HA_16), on the other hand, only traces of CaCO3 can be seen, indicating a clear conversion to HA. The peaks are not very sharp, but broad – this is reasonable, as the synthesis was performed at relatively low temperature (60 °C), and the powders are still relatively amorphous.
The morphology of the samples was studied by SEM, and the images are shown in Fig. 2. As previously reported, cork-derived CaCO3 maintained the characteristic cell structure (see Fig. 2(a)).24 Micrographs taken after 4, 8 and 16 hours of reaction show that the shape of the cells is also present in these HA-based materials, with clearly apparent hexagonal cells of around 20 μm diameter. This indicates that the mild reaction conditions are appropriate for the synthesis of biomorphic HA. With increasing reaction times, for instance after 8 hours (HA_8), the formation of crystals can be observed on the cell walls (see the red circles in Fig. 2(c)); this feature is even more obvious for HA_16 (16 hours), as can be seen in Fig. 2(d)–(f) – the latter two are micrographs with higher magnification. Flower-like HA crystals with needle shaped grains are formed, the individual needles having dimensions of about 500 nm in length and 40–50 nm in width. It is interesting to note that generally the formation of crystals with this shape has been reported to take place in the presence of surfactants, which determine and orient the crystal growth.30,31 In this work, however, no surfactant was employed.
 | ||
| Fig. 2 SEM micrographs of HA samples prepared for different reaction times. (a) HA_0: t = 0 h; (b) HA_4: t = 4 h; (c) HA_8: t = 8 h; (d) HA_16: t = 16 h; (e and f) HA_16 at higher magnification. | ||
Therefore, other elements affected the mechanisms of the crystal growth. Our hypothesis is that the residual carbonaceous fragments present in the starting material24 play a role in the orientation of the crystals. Indeed, literature reports HA crystal growth being oriented into small nanorods by the presence of graphitic material.32
It is also interesting to note that, in comparison with the CaCO3 starting material, HA_16 shows a more open structure. The cells in the original cork structure are mainly closed with a rear wall, but in HA_16 SEM shows that in the majority of cells the rear wall is missing. This indicates that the reaction to form the ceramic took place mainly on the side walls of the cells, whilst the rear wall was removed. The mechanisms allowing this to happen are not clear, but it was previously observed in other work on cork-based ceramic materials.22,23 A material with a more open and interconnected structure is very suitable for biomedicine applications, as open porosity will facilitate nutrient and oxygen diffusion, fundamental for cell proliferation and growth.33
The measured Ca/P molar ratio of HA_16 (see Table 2) shows that the value is slightly higher than the stoichiometric one – 1.73 vs. 1.67; literature already reports that for solid–liquid reactions between calcium and phosphorus, the Ca/P ratio in the final material is different from that in solution.34,35
| Sample | Ca (wt%) | P (wt%) | Ca/P (mol mol−1) |
|---|---|---|---|
| HA_16 | 30.98 ± 1.4 | 13.90 ± 0.6 | 1.73 ± 0.02 |
| CDHA_16 | 29.39 ± 0.9 | 14.54 ± 0.6 | 1.57 ± 0.02 |
| HA_16_700 | 37.01 ± 1.0 | 16.95 ± 0.5 | 1.69 ± 0.03 |
| CDHA_16_700 | 36.21 ± 1.6 | 18.34 ± 0.7 | 1.53 ± 0.03 |
Considering the results for HA preparation, CDHA was prepared using the same conditions of pH and 16 h reaction time, but a different stoichiometry, i.e. Ca:P = 1.5 (CDHA_16). As can be seen in Table 2, the measured Ca/P ratio of this sample is also slightly higher than that of the precursor solution, i.e. 1.57, similar to the observations for HA_16. Fig. 3 shows the XRD pattern for CDHA_16, which shows that HA is formed along with a small amount of CaCO3, and this sample appears more crystalline than HA_16.
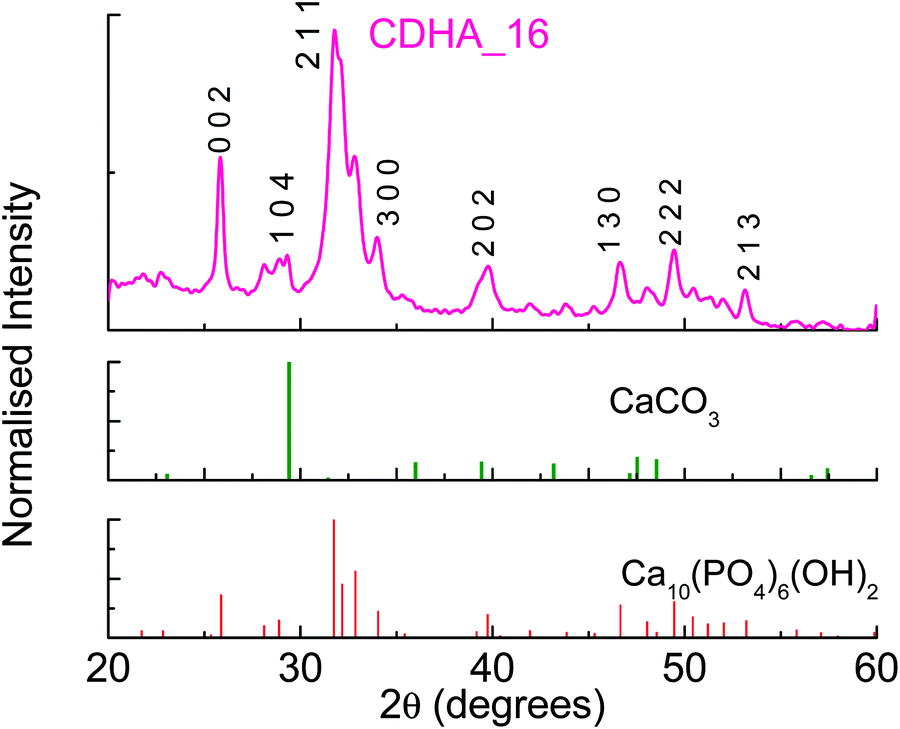 | ||
| Fig. 3 XRD pattern for sample CDHA_16. | ||
To have a proper comparison between the two forms of HA, stoichiometric and calcium-deficient, crystallographic data were obtained and they are reported in Table 3. It can be seen that the calcium-deficient sample has a shorter cell in the a-axis, while the c-axis dimension virtually does not change; this corresponds to a slightly smaller cell volume for CDHA_16 – 531.08 vs. 530.13 A3.
| Sample | Agreement factors | Unit cell parameters (Å) | Unit cell volume (Å3) | |||
|---|---|---|---|---|---|---|
| R F 2 (%) | R wp (%) | χ 2 | a = b | c | ||
| HA_16 | 6.00 | 4.08 | 5.98 | 9.4347(9) | 6.8893(10) | 531.08(10) |
| CDHA_16 | 6.86 | 3.11 | 3.56 | 9.4268(9) | 6.8885(9) | 530.13(9) |
| HA_16_700 | 7.96 | 6.71 | 14.03 | 9.4019(12) | 6.8800(12) | 526.68(13) |
| CDHA_16_700 | 9.68 | 3.59 | 4.49 | 9.4224(12) | 6.8821(13) | 529.14(12) |
Calcination of the powders
Samples HA_16 and CDHA_16 were recalcined at 700 °C in air; this thermal treatment was performed to remove the residual carbon present in the powders, and to increase the crystallinity of the materials. From Table 2 it can be seen that the Ca/P ratio slightly decreases with the temperature, becoming slightly closer to the nominal stoichiometric values, but with no significant difference to the non-calcined samples.
Fig. 4(a) shows the XRD patterns for both samples. It can be seen that hydroxyapatite is the only phase detected, indicating that the thermal treatment did not lead to the formation of other CaP-based phases, even in CDHA_16_700, which nominally had the β-tricalcium phosphate Ca:P ratio of 1.5. There is no evidence of CaCO3 in either sample. The samples shows a higher level of crystallinity, as the peaks are sharper. Crystallographic data of these samples (see Fig. S1, ESI† for an example of Rietveld refinement) show that the a-axis in the Ca-deficient HA (CDHA_16_700) was expanded compared to that of the stoichiometric HA_16_700, in order to accommodate this calcium deficiency in the HA structure (see Table 3). On the other hand, the c-axis only shows a tiny expansion compared to that observed in HA_16_700. The unit cell volume of HA_16_700 was found to be 526.68 Å3vs. 529.14 Å3 in CDHA_16_700. This is consistent with previous literature reports. Indeed, an expansion in the a-axis has been systematically reported in Ca-deficient HA, both in prepared samples36,37 and in simulations.38 The change in the c-axis seems to be related to the pH of formation: an alkaline environment generally leads to a slight expansion in the c-axis in comparison to stoichiometric HA,37 consistent with our experimental results.
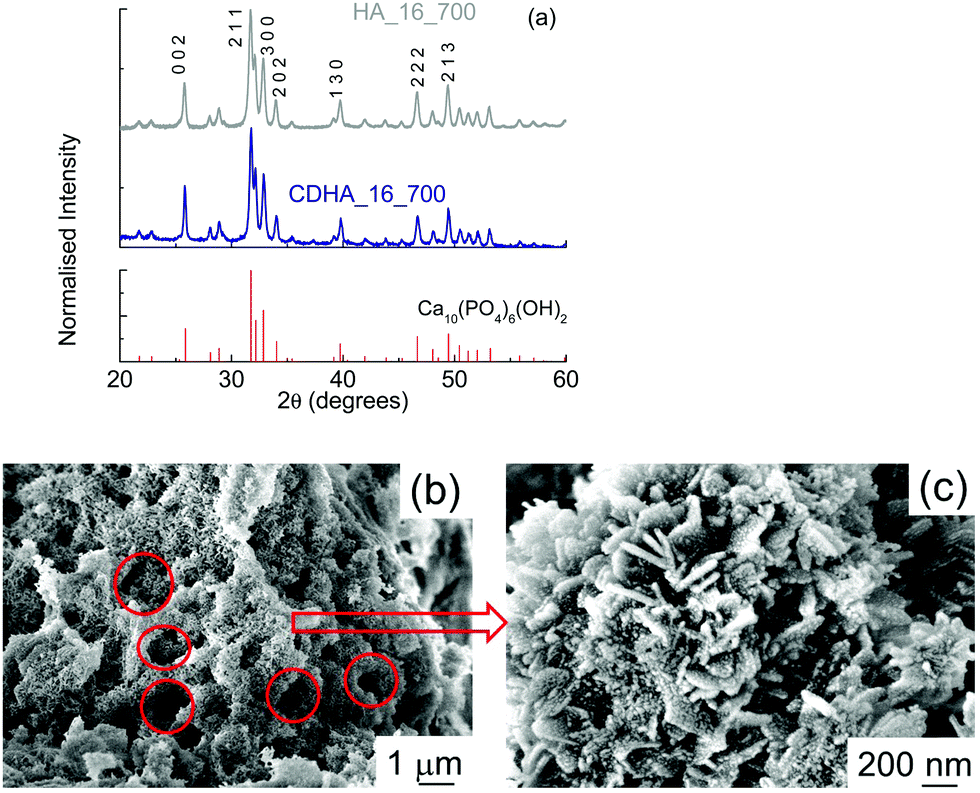 | ||
| Fig. 4 (a) XRD patterns for HA_16_700 and CDHA_16_700; (b and c) SEM micrographs of HA_16_700. | ||
TGA analysis for HA_16 (see Fig. S2, ESI†) shows a weight loss of slightly less than 20%, due to the combustion of the residual carbon; a first loss was observed for T < 400 °C (about 8%) while the rest was seen for 400 < T < 550 °C. The presence of two distinct steps indicates that residual carbon was present in different forms; it is likely that the first step is related to the combustion of residual fragments (i.e. unreacted salt(s) from the starting material), while the second is due to the residual graphite. Indeed, previous studies showed that graphite combustion takes place for T < 500 °C, although differences in temperature can be observed due to the chemical surroundings.20 The curve for CDHA_16 shows similar features, for both the behaviour of the curve and the residual weight at 700 °C (data not shown). These measurements indicate that for both syntheses, the CaP proportion is >80%.
SEM micrographs of HA_16_700 show a much more compact material (Fig. 4(b)); in fact, the structure of cork, with cells tens of microns in diameter, is not present anymore. Some smaller voids resembling cork geometry, however, can be observed (see the red circles), with diameters of about 1–2 μm. This indicates that, during the calcination, the thermal treatment led to an almost complete loss of the cork structure. This could be due to the heat released during the treatment from the combustion of the residual carbon material. In the micrograph with higher magnification (Fig. 4(c)) it can be seen that HA_16_700 still has a nanocrystalline structure, even if more irregular than that of HA_16; in some areas, in fact, the presence of some lamellae a few tens of nanometers thick and up to 250 nm long can be observed. Some lamellae are vertically aligned while other are positioned at an angle. These are similar to the nanoneedles which formed the flower-like crystals observed in HA_16. On their surface, some small rounded particles are present (10–20 nm); these are the crystallites formed during the recalcination, which progressively grow into larger structures. The mechanism of formation may be affected by the presence of the residual carbon, which is then eliminated during the process. CDHA_16_700 had a similar morphology (data not shown).
Surface area and porosity
Fig. 5 shows the BET adsorption and desorption curves for the samples as prepared with 16 h of reaction time (Fig. 5(a)) and calcined at 700 °C (Fig. 5(b)). | ||
| Fig. 5 BET adsorption and desorption isotherms for (a) HA_16 and CDHA_16; (b) HA_16_700 and CDHA_16_700. The numbers on the graphics indicate the values of the specific surface area of each sample. | ||
The values of the surface area are also indicated on the graphs. Considering the samples as prepared before calcination, their surface areas are about 87 and 85 m2 g−1 for HA_16 and CDHA_16, respectively. These values are more than three times higher than the starting CaCO3-based material used for the reaction, which showed a value of about 23 m2 g−1.24 This indicates that the reaction with the ammonium phosphate leads to a more porous structure (as well as increased macroporosity from the loss of the rear cell walls), and the growth of the flower-like crystals consisting of nanoscale needles will also contribute to this greatly increased surface area compared to the CaCO3 precursor. Moreover they have higher surface area also in comparison to other materials derived from natural templates.39,40
Despite the similar values of the surface area, the adsorption–desorption curves of the two samples show some marked differences: CDHA_16 has a much wider hysteresis loop. Both have Type IV isotherms typical of mesoporous solids, with multilayer adsorption followed by capillary condensation on desorption, which results in the hysteresis. Indeed, it can be seen that the adsorption curves overlap for the two samples indicating similar multilayer adsorption processes occur in them. However, the desorption process in CDHA_16 results in a significantly wider and longer hysteresis loop, indicating that capillary condensation is a more dominant process in this material compared to HA_16, despite it having a slightly smaller BET surface area. This shows that CDHA_16 has a greater mesoporous (2–50 nm pores) surface area than HA_16. The lack of steepness of both isotherms is between types H3 and H4, indicating the presence of narrow slit-like pores and internal voids of irregular shape and broad size distribution of this mesoporosity.41
BJH (Barrett, Joyner and Halenda) analysis provides approximate pore volumes of pores with diameters between 2 nm mesopores 300 nm macropores. This gives a mesopore volume between 0.284–0.295 cm3 g−1, and average pore diameters of 13.0 nm and 12.2 nm, from adsorption and desorption isotherms, respectively, for HA_16. However, BJH data for CDHA_16 gives a much larger mesopore volume of 0.328–0.371 cm3 g−1 for average pore diameters of 15.3 nm and 9.6 nm, providing further evidence of a much greater degree of mesoporosity being present in CDHA_16, despite both samples having almost equivalent specific surface areas. T-plot data indicated that HA_16 had greater microporosity (pores under 2 nm diameter) than CDHA_16, with microporosity contributions to the BET surface area of 11.5 m2 g−1 and 2.3 m2 g−1, respectively. Therefore, both samples clearly displayed hierarchical levels of porosity, combining the macroporous cellular structure of the cork template (20 μm cells) combined with their highly mesoporous and microporous nature.
For the calcined samples, on the other hand, the surface area is much smaller – about 29 m2 g−1 for both materials; this is in agreement with the more compact structure observed in the SEM micrographs (see Fig. 4). Despite the thermal treatment, however, the surface area of HA_16_700 and CDHA_16_700 is still higher than the original CaCO3 starting material (29 vs. 23 m2 g−1). Both still have Type IV isotherms, but with greatly reduced hysteresis loops, and less difference between the desorption processes. This indicates that a significant part of the reduction in surface area is due to the loss of mesoporosity within the material, as well as the more apparent loss of the macroporosity of the cellular cork microstructure and grain growth due to heating to 700 °C.
Tests of the functional properties
As a first screening, the kinetics of the adsorption was studied; Fig. 6(a) shows as an example the data as a function of time for HA_16 with an initial Pb(II) concentration of 2000 mg l−1. It can be seen that the amount increases progressively, reaching a plateau after about 5 hours. Data were fitted with Lagergren and Crank models, as reported in Fig. 6(a) and (c), respectively. It can be seen that, although both models give good fitting, the correlation coefficients indicate that the Lagergren model (i.e. kinetics of the first order) is the closest one (R2 = 0.969). Such kinetic behaviour was already observed for Pb(II) adsorption by HA-based materials,42,43 although other studies report different kinetic behaviour.6,44 Indeed, the kinetics of the adsorption can be affected by several factors, including the adsorption mechanism, the temperature and the possible presence of other ions/compounds, which are co-adsorbed.42 Similar experiments, performed with CDHA_16 and for other Pb(II) concentrations, showed the same result (data not shown), confirming the first order kinetics.
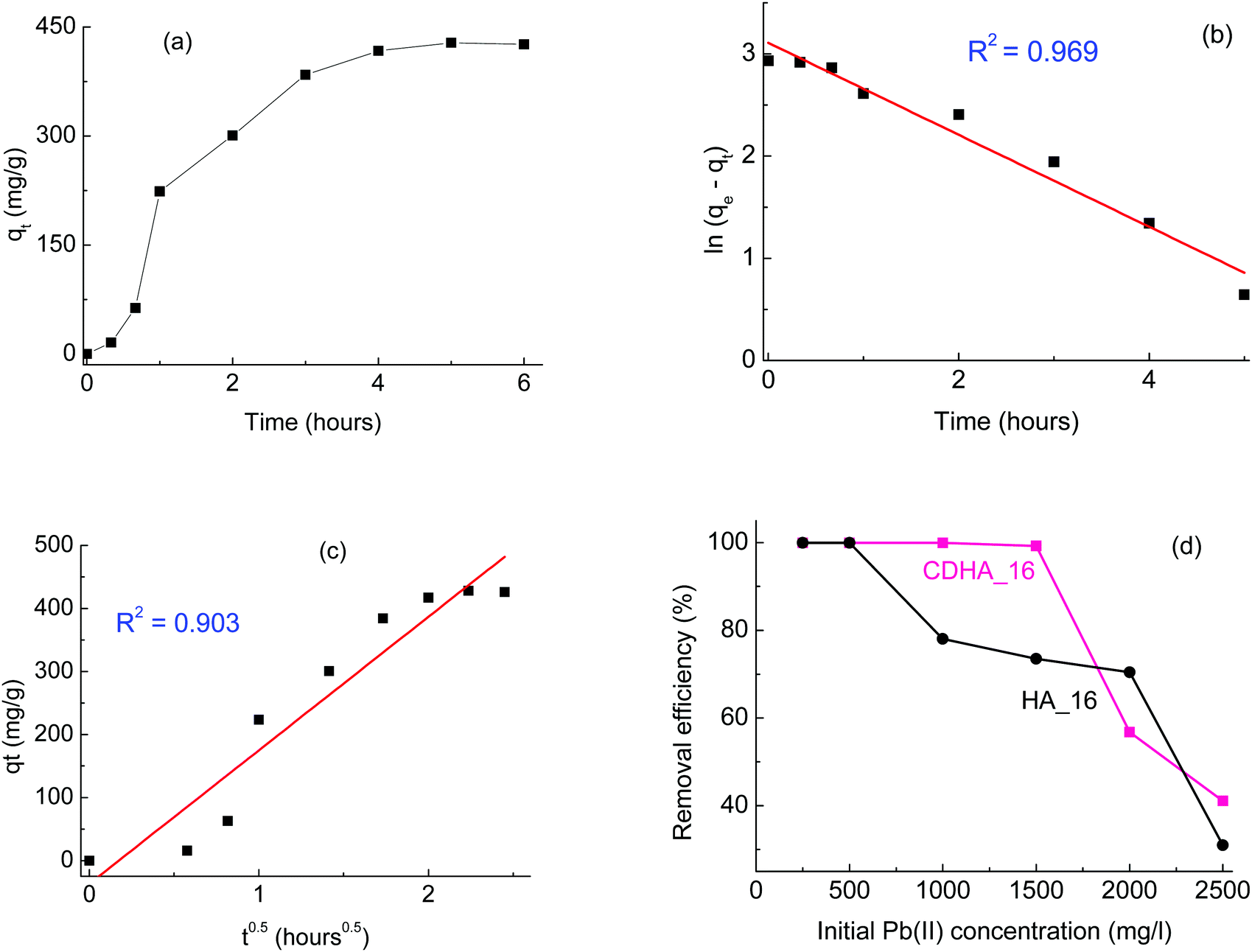 | ||
| Fig. 6 (a) Kinetic study of Pb(II) adsorption by HA_16 with initial Pb(II) concentration of 2000 mg l−1; (b) and (c) fitting of the data with the Lagergren and Crank models, respectively; (d) removal efficiency for HA_16 and CDHA_16 with different initial Pb(II) concentrations. | ||
After the kinetics study, more experiments were performed with different Pb(II) concentrations, in a range between 250 and 2500 mg l−1; in all cases a contact time of 6 hours was considered, based on the results of Fig. 6(a). The removal efficiency for both HA_16 and CDHA_16 at the different concentrations is shown in Fig. 6(d). It can be seen that both samples perform a complete Pb(II) removal for concentrations up to 500 mg l−1; for higher initial Pb(II) content, however, some differences can be observed. CDHA_16 shows an efficiency higher than 99% for Pb(II) values up to 1500 mg l−1; for HA_16, on the contrary, a decrease can be observed. This different behaviour cannot be attributed to the difference in surface area, since the values for the two samples are comparable, with HA_16 showing a slightly higher one (87 vs. 85 m2 g−1). However, the much greater degree of mesoporosity evidenced in CDHA_16 from the isotherm hysteresis loop could be the essential factor in this difference in Pb(II) absorption. The efficiency for HA_16, however, is still relatively high (>70%) for Pb(II) concentrations up to 2000 mg l−1.
Literature reports heavy metal removal performed with calcium-deficient hydroxyapatite,45 although only one study was performed comparing directly stoichiometric and non-stoichiometric materials.46 This investigation showed that calcium-deficient hydroxyapatite can have enhanced performance in the adsorption of metals; the authors explained this behaviour as being due to fact that the calcium deficient material was B-type carbonate HA (carbonate groups substituting phosphate ones), and the presence of carbonate ions favoured the metal immobilisation. Indeed, other studies report that carbonate ions can react with lead to form hydrated lead carbonate, hence showing higher removal.43 Carbonate ions substituting for phosphates were detected for both HA_16 and CDHA_16, as can be seen from their FTIR spectra (Fig. S3, ESI†), which show the peaks associated with B-type HA at 874, 1418 and 1455 cm−1.46 However, the spectra show very similar features between the two samples. Therefore, the presence of carbonate cannot explain the difference in the lead removal performances in our case, and the difference in mesoporosity remains the most likely explanation.
Previous work done on Pb(II) removal by HA-based material showed that a small fraction of the metal is released after the adsorption, indicating a strong between the material and the lead ion;6 a similar behaviour can be expected with the material of this study. For the material to be reused, therefore, an acid treatment would be necessary.
Overall, these results show that these HA-based materials derived from cork have very good performance for environment remediation, with Ca-deficient HA having particularly high efficiency (>99%) for Pb(II) removal for concentrations up to 1500 mg l−1.
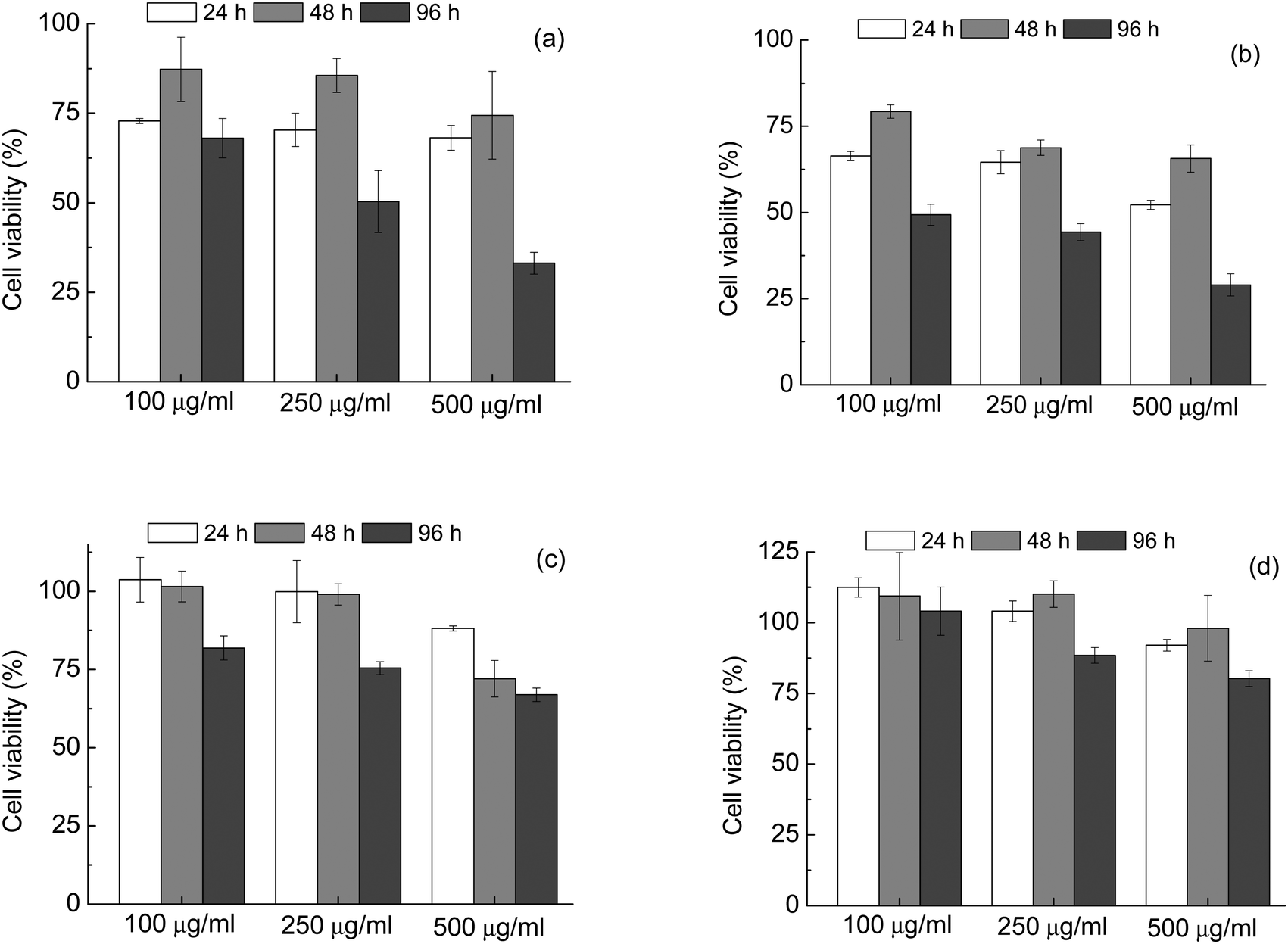 | ||
| Fig. 7 MTT cell viability assays for 100, 250 and 500 μg mL−1 of powdered samples of (a) HA_16, (b) CDHA_16, (c) HA_16_700, (d) CDHA_16_700, incubated with MG63 cells for 24, 48 and 96 h. All values relative to 100% for the control sample. | ||
Tests were carried out on both the samples prepared with 16 h reaction time (HA_16 and CDHA_16), and those calcined at 700 °C (HA_16_700 and CDHA_16_700).
It can be seen that in both as-prepared samples – HA_16 and CDHA_16, Fig. 7(a) and (b) respectively – there was an impact on the viability of MG63 cells, leading to a final decrease of the number of viable cells. For HA_16 values of around 70% were observed after 24 h and above 75% after 48 hours incubation times for all the concentrations considered. However, for the longest time of 96 hours the proportion of viable cells decreased to below 50% for the cells incubated with 250 and 500 μg mL−1 powders, indicating cytotoxicity. The same trend is observed for CDHA_16; in this case, however, the values for cell viability are even lower. These results indicate that these materials, in this form, are not ideal for biomedical applications. Although there is no data for this reported in literature, it is likely that the presence of residual carbon contamination has a negative effect on the cytotoxicity.
The thermal treatment of the samples had a beneficial effect on the biocompatibility, as the cell viability increased for both powders treated at 700 °C. For HA_16_700 (Fig. 7(c)) values of about 100% were seen for both 24 and 48 hours testing with concentrations of 100 and 250 μg mL−1. For 96 hours, a slight decrease was observed with the value still higher than 75%. Slightly lower cell viability (close to 70%) was observed for the highest powder concentration, i.e. 500 μg mL−1, likely due to high release of Ca ions and consequent cellular stress. Indeed, it has been reported that high concentrations of HA affect the viability of osteosarcoma cells, leading to cell vacuolation, organelles alteration and finally apoptosis.49–51 Despite this, all values for HA_16_700 were above the limit of 70% considered to indicate cytotoxicity. CDHA_16_700 (Fig. 7(d)) showed even lower cytotoxicity – in fact, for the lower concentrations, cell viability was even higher than the positive control, indicating an enhanced cell proliferation. Moreover, with the highest powder concentration the number of viable cells was still more than 75%. These data seem to confirm that calcium deficiency in the HA lattice is beneficial for biomedical applications.4,50
Based on these results, it seems that two aspects contribute to the overall biocompatibility of the HA powders: on one hand, the calcination and thus the removal of carbon residues, and on the others the Ca:P ratio, the samples with lower Ca levels (calcium deficient HA) being more biocompatible. It can be concluded that synthesis in calcium deficient conditions followed by a thermal treatment led to a material with the best performance in terms of biomedicine.
Conclusions
HA-based materials with different stoichiometries (Ca/P = 1.67 and 1.5) were successfully prepared from cork-derived CaCO3. The samples were prepared at 60 °C, and successively recalcined at 700 °C. The different Ca/P ratios affected the lattice cell paíeters for both as-prepared and recalcined samples.The as-prepared samples maintained the 3D honeycomb structure typical of cork, along with increased surface areas (>85 m2 g−1) and a high mesoporosity. This hierarchical porosity made the materials suitable for environmental remediation applications. Indeed, tests on the adsorption of Pb(II) proved these materials to have very good performance, especially in the calcium-deficient HA, which had a higher degree of mesoporosity. Recalcined samples, on the other hand, showed very low cytotoxicity; again, the calcium-deficient HA was the one giving the best results, as it was less cytotoxic than the control.
Therefore, cork was shown to be an excellent template agent for the preparation of designed materials with varied properties, tailored for different applications in the environmental and biomedical fields.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Clara Piccirillo and Francecsa Scalera would like to thank Fondazione con il Sud for funding the HApECOrk project (project 2015-0243). Robert Pullar would like to thank the FCT (Fundação para a Ciência e a Tecnologia, Portugal) Grant IF/00681/2015 for supporting this work, along with FCT project H2CORK (PTDC/CTM-ENE/6762/2014), and this work was developed within the scope of the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020 & UIDP/50011/2020, financed by national funds through the FCT/MEC and when appropriate co-financed by FEDER under the PT2020 Partnership Agreement. Alessandra Quarta would like to thank “Tecnopolo per la medicina di precisione” (TecnoMed Puglia) – Regione Puglia: DGR no. 2117 del 21/11/2018, CUP: B84I18000540002 and “Tecnopolo di Nanotecnologia e Fotonica per la medicina di precisione” (TECNOMED) – FISR/MIUR-CNR: delibera CIPE no. 3449 del 7-08-2017, CUP: B83B17000010001. David M. Tobaldi was funded by the project EleGaNTe – PON ARS01_01007. The authors would like to thank Dr. I. Vilarinho for her help with the surface area measurements.References
- S. V. Dorozhkin, Calcium orthophosphate bioceramics, Ceram. Int., 2015, 41, 13913 CrossRef CAS.
- R. Bosco, M. Iafisco, A. Tampieri, J. A. Jansen, S. C. G. Leeuwenburgh and J. J. J. P. van den Beucken, Hydroxyapatite nanocrystals functionalized with alendronate as bioactive components for bone implant coatings to decrease osteoclastic activity, Appl. Surf. Sci., 2015, 328, 516 CrossRef CAS.
- S. V. Dorozhkin and S. V. Amorphous, calcium (ortho)phosphates, Acta Biomater., 2010, 6, 4457 CrossRef CAS PubMed.
- X. Li, Y. Deng, M. Wang, X. Chen, Y. Xiao and X. Zhang, Stabilization of Ca-deficient hydroxyapatite in biphasic calcium phosphate ceramics by adding alginate to enhance their biological performances, J. Mater. Chem. B, 2018, 6, 84 RSC.
- Y. Yan, Q. Li, J. Yang, S. Zhou, L. Wang and N. Bolan, Evaluation of hydroxyapatite derived from flue gas desulphurization gypsum on simultaneous immobilization of lead and cadmium in contaminated soil, J. Hazard Mater., 2020, 400, 123038 CrossRef CAS PubMed.
- C. Piccirillo, I. S. Moreira, R. M. Novais, A. J. S. Fernandes, R. C. Pullar and P. M. L. Castro, Biphasic apatite-carbon materials derived from pyrolysed fish bones for effective adsorption of persistent pollutants and heavy metals, J. Environ. Chem. Eng., 2017, 5, 4884 CrossRef CAS.
- B. S. Liaw, T. T. Chang, H. K. Chang and W. K. Liu, Fish scale-extracted hydroxyapatite/chitosan composite scaffolds fabricated by freeze casting – An innovative strategy for water treatment, J. Hazard. Mater., 2020, 382, 121082 CrossRef CAS PubMed.
- A. Nzihou and P. Sharrock, Role of phosphate in the remediation and reuse of heavy metal polluted wastes and sites, Waste Biomass Valorization, 2010, 1, 163 CrossRef CAS.
- A. J. Nathanel, Y. M. Im and T. H. Oh, Intrinsic effect of anionic surfactant on the morphology of hydroxyapatite nanoparticles and its structural and biological properties, Adv. Powder Technol., 2020, 31, 234 CrossRef.
- M. Y. Ma, Y. J. Zhu, L. Li and S. W. Cao, Nanostructured porous hollow ellipsoidal capsules of hydroxyapatite and calcium silicate: preparation and application in drug delivery, J. Mater. Chem., 2008, 18, 2722–2727 RSC.
- Y. Hui, Z. Dong, P. Wenkun, D. Yao, G. Huichang and L. Tongxiang, Facile synthesis of copper doping hierarchical hollow porous hydroxyapatite beads by rapid gelling strategy, Mater. Sci. Eng., C, 2020, 109, 110531 CrossRef CAS PubMed.
- Y. Liu, J. Huang and H. Li, Synthesis of hydroxyapatite-reduced graphite oxide nanocomposites for biomedical applications: oriented nucleation and epitaxial growth of hydroxyapatite, J. Mater. Chem. B, 2013, 1, 1826 RSC.
- Y. C. Qi, J. Shen, Q. Y. Jiang, B. Jin, J. W. Chen, X. Zhang and J. L. Su, Hierarchical porous hydroxyapatite microspheres: synthesis and application in water treatment, J. Mater. Sci., 2016, 51, 2598 CrossRef CAS.
- N. S. Kumar, R. P. Suvarna, K. C. B. Naidu, P. Banerjee, A. Ratnamala and H. Manjunatha, A review on biological and biomimetic materials and their applications, Appl. Phys. A: Mater. Sci. Process., 2020, 126, 445 CrossRef.
- M. Singh, J. Martínez-Fernández and A. R. de Arellano-López, Environmentally conscious ceramics (ecoceramics) from natural wood precursors, Curr. Opin. Solid State Mater. Sci., 2003, 7, 247 CrossRef CAS.
- O. Gryshkov, N. Klyui, V. P. Temchenko, V. S. Kyselov, A. Chatterjee, A. E. Belyaev, L. Lauterboeck, D. Iarmolenko and B. Glasmacher, Porous biomorphic silicon carbide ceramics coated with hydroxyapatite as prospective materials for bone implants, Mater. Sci. Eng., C, 2016, 68, 143 CrossRef CAS PubMed.
- R. C. Pullar and R. M. Novais, Ecoceramics - Cork-based Biomimetic Ceramic 3-DOM Foams, Mater. Today, 2017, 20, 45 CrossRef.
- C. Brazinha, A. P. Fonseca, H. Pereira, O. M. N. D. Teodoro and J. G. Crespo, Gas transport through cork: Modelling gas permeation based on the morphology of a natural polymer material, J. Membr. Sci., 2013, 428, 52 CrossRef CAS.
- M. Demertzi, J. Amaral, P. L. Arroja and A. C. Dias, A carbon footprint simulation model for the cork oak sector, Sci. Total Environ., 2016, 566–567, 499 CrossRef CAS PubMed.
- A. Quarta, R. M. Novais, S. Bettini, M. Iafisco, R. C. Pullar and C. Piccirillo, A sustainable multi-function biomorphic material for pollution remediation or UV absorption: aerosol assisted preparation of highly porous ZnO-based materials from cork templates, J. Environ. Chem. Eng., 2019, 7, 102936 CrossRef CAS.
- V. O. Yukhymchuk, V. S. Kieslow, M. Y. Valakh, M. A. Skoryk, A. G. Rozhin, S. A. Kulinich and A. E. Belyaev, Biomorphic SiC ceramics prepared from cork oak as precursor, J. Phys. Chem. Solids, 2016, 91, 145 CrossRef CAS.
- R. C. Pullar, P. Marques, J. Amaral and J. A. Labrincha, Magnetic wood-based biomorphic Sr3Co2Fe24O41 Z-type hexaferrite ecoceramics made from cork templates, Mater. Des., 2015, 82, 297 CrossRef CAS.
- R. M. Novais and R. C. Pullar, Comparison of low and high pressure infiltration regimes on the density and highly porous microstructure of ceria ecoceramics made from sustainable cork templates, J. Eur. Ceram. Soc., 2019, 39, 1287 CrossRef CAS.
- F. Scalera, L. Carbone, S. Bettini, R. C. Pullar and C. Piccirillo, Biomimetic calcium carbonate with hierarchical porosity produced using cork as sustainable template agent, J. Environ. Chem. Eng., 2020, 8, 103594 CrossRef CAS.
- D. Phan Minh, N. Lyczko, H. Sebei, A. Nzihou and P. Sharrock, Synthesis of calcium hydroxyapatite from calcium carbonate and different hydroxyapatite sources: A comparative study, Mater. Sci. Eng., B, 2012, 177, 1080 CrossRef.
- A. C. Larson and R. B. Von Dreele, General structure analysis system (GSAS), Los Alamos National Laboratory Report LAUR, 2004.
- B. H. Toby, EXPGUI, a graphical use interface for GSAS, J. Appl. Crystallogr., 2004, 34, 210 CrossRef.
- R. M. Wilson, J. C. Elliott and S. E. P. Dowker, Rietveld refinement of the crystallographic structure of human dental enamel apatites, Am. Mineral., 1999, 84, 1406 CrossRef CAS.
- M. V. Nikolenko, K. V. Vasylenko, V. D. Myrhorodska, A. Kostyniuk and B. Likozar, Sythesis of calcium orthophosphates by Chemical Precipitation in aqueous solutions: the effect of the acidity, Ca/P molar ratio, and temperature on the phase composition and solubility of precipitates, Processes, 2020, 8, 1009 CrossRef CAS.
- D. Timpu, L. Sacarescu, T. Vasiliu, M. V. Dinu and G. David, Surface cation functionalized nano-hydroxyapatite – Preparation, characterization, effect of coverage on properties and related applications, Eur. Polym. J., 2020, 132, 109759 CrossRef CAS.
- T. P. J. Reardon, A. D. Handoko, L. Li, J. Huang and J. Tang, Dimensionally and compositionally controlled growth of calcium phosphate nanowires for bone tissue regeneration, J. Mater. Chem. B, 2013, 1, 6170 RSC.
- J. Liu, C. Xu, Y. Xu, Q. Ye, S. Fu, N. Hu and C. Yan, Bioinspired scaffolds with hierarchical structures for tailored mechanical behaviour and cell migration, Ceram. Int., 2020, 46, 24102 CrossRef CAS.
- M. Bohner, B. Le Gars Sontoni and N. Döbelin, β-tricalcium phosphate for bone substitution: synthesis and properties, Acta Biomater., 2020, 113, 23 CrossRef CAS PubMed.
- R. Wati and Y. Yusuf, Carbonated hydroxyapatite derived from Cerastoderma edule, Paphia undulata, and Meretrix meretrix shells, IOP Conf. Ser., 2019, 546, 042049 CAS.
- A. Turlybekuly, et al., Synthesis, characterization, in vitro biocompatibility and antibacterial properties study of nanocomposite materials based on hydroxyapatite-biphasic ZnO micro- and nanoparticles embedded in alginate matrix, Mater. Sci. Eng., C, 2019, 104, 109965 CrossRef CAS PubMed.
- R. M. Wilson, J. C. Elliott and S. E. P. Dowker, Rietveld refinements and spectroscopic studies of the structure of Ca-deficient apatite, Biomaterials, 2005, 26, 1317 CrossRef CAS PubMed.
- J. C. Elliott, Hydroxyapatite and Nonstoichiometric apatites, Stud. Inorg. Chem., 1994, 18, 11–189 Search PubMed.
- C. L. M. Camargo, N. S. Resende, C. A. C. Perez, C. R. A. Abreu, V. M. M. Salim and F. W. Tavares, Molecular dynamics simulation and experimental validation by X-ray data of hydroxyapatite crystalline structures, Fluid Phase Equilib., 2018, 470, 60 CrossRef CAS.
- X. Peng, T. Cheng, M. Cheng, J. Wang and X. Zhang, Hierarchically porous CaCO3 monoliths prepared from leaf templates, Mater. Lett., 2014, 129, 198–201 CrossRef CAS.
- J. Ramírez-Rico, J. Martínez-Fernandez and M. Singh, Biomorphic ceramics from wood-derived precursors, Int. Mater. Rev., 2017, 62, 465–485 CrossRef.
- K. S. W. Sing, Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984), Pure Appl. Chem., 1985, 57, 603 CAS.
- T. Kaludjerovic-Radoicic and S. Raicevic, Aqueous Pb sorption by synthetic and natural apatite: Kinetics, equilibrium and thermodynamic studies, Chem. Eng. J., 2010, 160, 503 CrossRef CAS.
- W. Anutrasakda, A. Phasuk and C. Tangku, Effect of different CO32− to PO43− molar ratios on the properties, morphology, and Pb(II) removal performances of carbonated hydroxyapatite, J. Environ. Chem. Eng., 2021, 9, 104658 CrossRef CAS.
- E. Bernalte, J. Kamieniak, E. P. Randviir and C. E. Banks, The preparation of hydroxyapatite from unrefined calcite residues and its application for lead removal from aqueous solution, RSC Adv., 2019, 9, 4054 RSC.
- Z. Zhu, L. Li, H. Zhang, Y. Qiu and J. Zhao, Adsorption of lead and cadmium on Ca-deficient hydroxyapatite, Sep. Sci. Technol., 2010, 45, 262 CrossRef CAS.
- S. Campisi, C. Castellano and A. Gervasini, Tailoring structural and morphological properties of hydroxyapatite materials to enhance the capture efficiency towards copper(II) and lead(II) ions, New J. Chem., 2018, 42, 4520 RSC.
- G. Thrivikraman, G. Madrasb and B. Basu, In vitro/In vivo assessment and mechanisms of toxicity of bioceramic materials and its wear particulates, RSC Adv., 2014, 4, 127632014 RSC.
- G. N. Attik, C. Villat, F. Hallay, N. Pradelle-Plasse, H. Bonnet, K. Moreau, P. Colon and B. Grosgogeat, In vitro biocompatibility of a dentine substitute cement on human MG63 osteoblasts cells: BiodentineTM versus MTA, Int. Endod. J., 2014, 47, 1133 CrossRef CAS PubMed.
- F. Qing, Z. Wang, Y. Hong, M. Liu, B. Guo, H. Luo and X. Zhang, Selective effects of hydroxyapatite nanoparticles on osteosarcoma cells and osteoblasts, J. Mater. Sci.: Mater. Med., 2012, 23, 2245 CrossRef CAS PubMed.
- A. Hoppe, N. S. Güldal and A. R. Boccaccini, A review of the biological response to ionic dissolution products from bioactive glasses and glass-ceramics, Biomaterials, 2011, 32, 2757 CrossRef CAS PubMed.
- E. Filová, T. Suchý, Z. Sucharda, M. Šupová, M. Žaloudková, K. Balík, V. Lisá, M. Šlouf and L. Bačáková, Support for the initial attachment, growth and differentiation of MG-63 cells: a comparison between nano-size hydroxyapatite and micro-size hydroxyapatite in composites, Int. J. Nanomed., 2014, 9, 3687 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1qm00584g |
| This journal is © the Partner Organisations 2021 |