
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolysaccharide-reinforced amyloid fibril hydrogels and aerogels†
Mattia
Usuelli
 a,
Till
Germerdonk
a,
Yiping
Cao‡
a,
Mohammad
Peydayesh
a,
Massimo
Bagnani
a,
Stephan
Handschin
b,
Gustav
Nyström
ac and
Raffaele
Mezzenga
*ad
a,
Till
Germerdonk
a,
Yiping
Cao‡
a,
Mohammad
Peydayesh
a,
Massimo
Bagnani
a,
Stephan
Handschin
b,
Gustav
Nyström
ac and
Raffaele
Mezzenga
*ad
aETH Zürich, Department of Health Sciences and Technology, Schmelzbergstrasse 9, 8092 Zürich, Switzerland. E-mail: raffaele.mezzenga@hest.ethz.ch
bETH Zürich, Scientific Center for Optical and Electron Microscopy (ScopeM), 8093 Zürich, Switzerland
cEMPA, Laboratory for Cellulose & Wood Materials, Überlandstrasse 129, 8600 Dübendorf, Switzerland
dETH Zürich, Department of Materials, Wolfgang-Pauli-Strasse 10, 8093 Zürich, Switzerland
First published on 25th June 2021
Abstract
β-Lactoglobulin amyloid fibrils are bio-colloids of high interest in many fields (e.g. water purification, cell growth, drug delivery and sensing). While the mechanical properties of pure amyloid fibril gels meet the needs of some applications, mechanical fragility often hinders a wider usage basin. In this work, we present a simple and sustainable approach for reinforcing amyloid fibril hydrogels and aerogels, upon the diffusion of polysaccharides (low-acetylated Gellan Gum and κ-carrageenan) inside their mesh. The formed hybrid materials show enhanced resistance upon compression, without any loss of the exquisite surface reactivity of the amyloid fibrils. The proposed approach can pave the way for designing composite materials that are both highly functional and environmentally friendly.
Hydrogels are versatile materials, comprising a solid skeleton interpenetrated by water. When the liquid phase is replaced by air, while minimizing structural damage, hydrogels transform into aerogels.1,2 The resulting arrested structures exhibit a large variety of functionalities and can be used for a wide range of applications that encompass drug delivery,3 bone regeneration,4 dehumidification5 and electrochemistry.6 Among the many potential building blocks of hydrogels and aerogels, the ones that respect sustainability criteria are more often being brought under the spotlight.7,8 In fact, increasing effort is put in pursuing a more equitable global society while operating within Planetary Boundaries (PB), which define a safe space for using the resources of Earth without affecting its resilience.9,10 Thus, functional properties and sustainable design should be increasingly blended, aiming at materials that positively impact human activities while safeguarding our planet.11
One promising answer to promote both requirements lies in synthesising hydrogels and aerogels out of under-exploited food sources;12,13 changes in temperature, pH or ionic strength allow to generate networks of proteins and polysaccharides with a broad range of functionalities. As a remarkable example, amyloid fibrillar structures (formed upon the self-assembly of hydrolyzed peptides) are used in a wide spectrum of applications, including water purification,14–16 iron fortification,17 regulation of gut microbial dysbiosis18 and creation of conductive materials.19,20 Thus, in vitro prepared amyloid fibrils are a promising candidate for designing eco-friendly, functional materials. However, the poor mechanical properties of pure amyloid gels are an often-encountered limitation. While this is not a drawback for some specific applications, where the mechanical strength is of secondary importance (e.g. drug delivery), enhancement of mechanical properties is desired for those contexts where gels are subjected to significant strains and stresses. Therefore, the functional amyloid fibril community is highly interested in ways to strengthen the fibrillar networks while preserving their surface functionality.21
From a historical perspective, biological networks have been combined with inorganic components (e.g. silica) to create synergistic effects. For example, there are reports of silica gels strengthened through the incorporation of a cellulose scaffold,22 pectin–silica hybrid materials23 and fibrillar networks with an amyloid core and a silicious shell.24 Although these strategies were proven to be effective in enhancing the properties of the materials, they might reduce their surface activity; moreover, the abovementioned sustainability concerns encourage the exploration of alternative routes based on renewable secondary components. Such exploration has been attempted with some classes of biopolymer gels (like those based on alginate,25 lentil protein26 and myofibrillar protein27) but not with gels containing amyloid fibrils as the main, functional component.
In this work, we present a simple approach for reinforcing β-lactoglobulin (βLG) amyloid fibril gels through the inclusion of polysaccharides (namely, low-acetylated Gellan Gum and κ-carrageenan). The protocol we follow nicely complies with the sustainability criteria, by being entirely based on largely available sources and salt solutions. In fact, βLG, Gellan Gum (GG) and κ-carrageenan (κ-carr) are obtained from whey proteins28 (a side stream of cheese production), bacterial fermentation29 and marine algae,30 respectively. All of them have, in the current global food scenario, a marginal role in nutrient and calorie intake. The risk of competition between their usage as food and their usage as harbingers of functional materials is therefore minimized.
We report a comprehensive analysis of the morphology of the network-forming units through a statistical evaluation of Atomic Force Microscopy (AFM) images, showing the potential of the polysaccharide chains to diffuse inside a mesh of amyloid fibrils. The macroscopic appearance (hydrogels and aerogels) and the microscopic structure (aerogels) of the synthesised materials are then analysed, and the mechanical properties of the hydrogels and aerogels are presented; such analyses confirm the diffusion of the polysaccharide units and show their ability to mechanically reinforce the materials. Finally, through adsorption experiments, we show that the mechanical enhancement does not come at the expense of the surface functionality of the amyloid component. The performed analyses corroborate the knowledge on synthesising materials that are both functional and renewable.
1. Materials and methods
1.1. βLG monomer purification
The purification of βLG monomers was carried out using Whey Protein Isolate (WPI; Fonterra, New Zealand) based on a simplified version of the protocol reported by Vigolo and co-workers.31 10 g of WPI was dissolved in 90 g of Milli-Q water, and the pH of the solution was adjusted to 4.2 with a concentrated HCl solution. A Schott bottle containing the solution was incubated in a shaking water bath (VWR, Switzerland) for more than 2 hours at 60 °C, until the solution became turbid suggesting the agglomeration of α-lactalbumin due to the proximity to its isoelectric point. The turbid solution was then transferred into plastic Falcon tubes and centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 074g for 20 minutes to precipitate the separated α-lactalbumin fraction. The obtained transparent solution, containing mainly βLG monomers and residual salts and sugars, was transferred into a Schott bottle and its weight fraction was determined through gravimetric analysis.
074g for 20 minutes to precipitate the separated α-lactalbumin fraction. The obtained transparent solution, containing mainly βLG monomers and residual salts and sugars, was transferred into a Schott bottle and its weight fraction was determined through gravimetric analysis.
1.2. βLG amyloid fibril synthesis
The pH of the βLG monomer solution generated as described above was adjusted to 2 using a concentrated HCl solution, and the solution was further diluted to 2 wt% using Milli-Q water at pH 2. The formation of amyloid fibrils was driven by heating at 90 °C for 5 hours, while stirring (at 120 rpm for the first 3 hours and at 170 rpm for the last 2 hours) with a 3 cm long Teflon magnetic stirrer. Afterwards, the Schott bottle containing the solution was cooled on ice and stored at 4 °C.1.3. Preparation of polysaccharides and salt solutions
κ-Carrageenan (Sigma Life Science, lot. 1432063) and low-acetylated Gellan Gum (Opal Biotech, lot. 18100601) solutions were prepared following the same protocol. Briefly, 1 g of polysaccharide was dissolved in 199 g of Milli-Q water by stirring for 1 hour at 90 °C. The resulting solutions were then cooled and stored at 4 °C. We decided to form 0.5 wt% solutions of both polysaccharides as this concentration corresponds to their reported maximum solubility in hot water (at the mentioned temperature).Salt (KCl, NaCl and CaCl2) solutions of different molarities were prepared using the following reagents: calcium chloride dihydrate for analysis (Merck kGaA, lot. A0263182 203), sodium chloride for analysis (Merck kGaA, lot. K43132604 207) and potassium chloride BioXtra ≥ 99.0% (Sigma, lot. BCBP9964 V). In the case of NaCl, the final solutions were prepared with Milli-Q water, the pH of which had been previously adjusted to 2 with a concentrated HCl solution. In the case of the other two salts, Milli-Q water at neutral pH was used.
1.4. AFM characterisation
The samples for AFM characterization were prepared following the protocols described below.For βLG amyloid fibrils, an aliquot of the 2 wt% dispersion was diluted to a final concentration of 0.01 wt%, using Milli-Q water at pH 2. Twenty μL of the abovementioned solution was then deposited on freshly cleaved mica and, after incubating for 2 minutes, the mica surface was rinsed with 1 mL of Milli-Q water at pH 2. Finally, the surface of the mica substrate was gently dried under a compressed air flow.
For κ-carrageenan and Gellan Gum, the initial 0.5 wt% dispersions were diluted with neutral Milli-Q water to a final concentration of 1 μg mL−1. (3-Aminopropyl)triethoxysilane (APTES, CAS 919-30-2, 99.9%, Sigma-Aldrich) was used to modify the surface charge of the mica substrate. After dilution with Milli-Q water, 20 μL of a 0.05 v/v % APTES solution was deposited on freshly cleaved mica and incubated for 1 minute. The surface was then rinsed with 3 mL of Milli-Q water and dried under compressed air flow. Subsequently, 20 μL of each of the diluted polysaccharide solutions was deposited on the surface-modified mica and incubated for 30 seconds. Finally, the deposited solutions were rinsed with 1 mL of Milli-Q water and dried under compressed air flow. AFM characterization was performed with a commercial cantilever (Bruker, USA) installed onto a Dimension FastScan Bio probe microscope (Bruker, USA), run in the tapping mode. The analysis of the colloidal units was performed using the open-source software FiberApp.32
1.5. Amyloid fibril hydrogel preparation
For the gelation of the amyloid fibril dispersion, 1 mL of the 2 wt% solution was transferred into a plastic syringe, the top part of which had been previously cut. In order to allow ions to diffuse in, but fibrils not to diffuse out, the plastic syringe was covered with a cut section of a dialysis tube (Spectra/Por®, MWCO 6–8 kDa, Spectrum Laboratories Inc.) and sealed with parafilm. Before usage, the dialysis tube was rinsed with deionized water. The tip of the syringe was then placed in contact with an excess of a 300 mM NaCl solution at pH 2 for at least 48 h to allow for the gelation of the entire sample. For a schematic representation of the process, we refer to the upper-left corner of Fig. 1.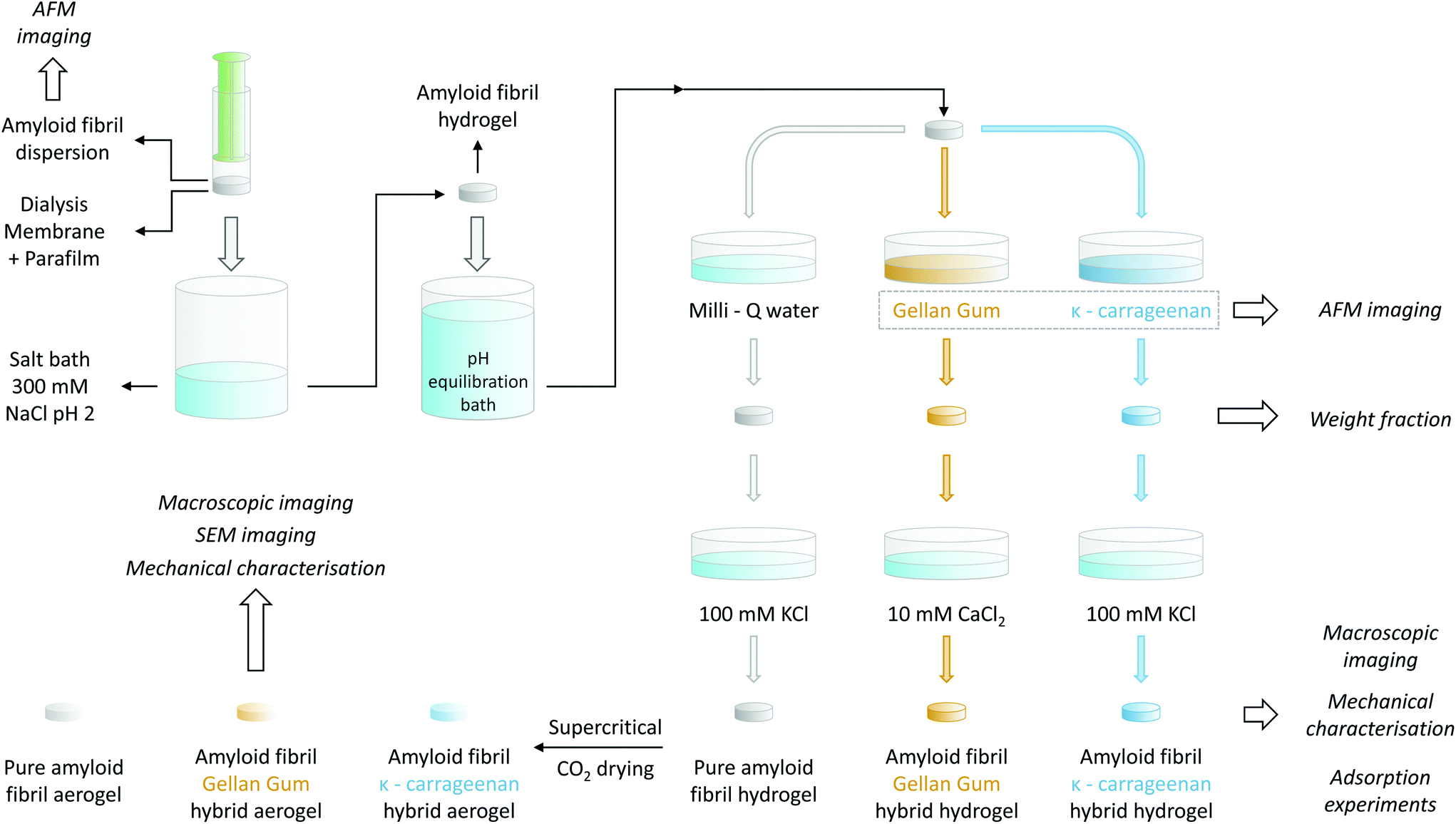 | ||
| Fig. 1 Schematic representing the different steps for the preparation of the amyloid fibril/polysaccharide hybrid hydrogels and aerogels. The details of the used protocol are explained in the Materials and methods section. | ||
1.6. Amyloid–polysaccharide hybrid hydrogel preparation
To allow for the diffusion of polysaccharides into the preformed βLG amyloid fibril hydrogels, it was necessary to first adjust their pH. As a calibration experiment, the preformed gels were placed into different 100 mL Milli-Q water solutions, the pH of which had been adjusted to a value ranging from 7 to 11 using a concentrated NaOH solution. After an equilibration time of at least 48 h, the final pH was measured again with a commercial pH meter (pHenomenal®, VWR, 1000 L or 1100 L). In this way, it was possible to construct an equilibration curve that allowed the determination of the final pH as a function of the initial one: the data points and the used fitting function are reported in Fig. S1.† Accordingly, the gels used for preparing pure βLG hydrogels/aerogels or the βLG/Gellan Gum hybrid ones were put in contact with a bath at pH 10.42 to reach a final pH value of ∼ 6.5, whereas the pH of the equilibration bath used for the preparation of βLG/κ-carrageenan hybrid hydrogels/aerogels was adjusted to 10.54 to reach a final pH of approximately 8.3.To mechanically reinforce the pure amyloid fibril gels, polysaccharides were allowed to diffuse inside their mesh for six days, according to the following protocol. The βLG hydrogels were transferred into plastic Petri dishes and were completely submerged in 5 mL of the polysaccharide solutions (either κ-carrageenan or low-acetylated Gellan Gum at a concentration of 0.5 wt%). Pure amyloid networks used as control were instead immersed in 5 mL of Milli-Q water for the same period of time.
Following the incubation in the polysaccharide solution, the hydrogels were removed from the Petri dishes using a metal spatula and were immersed in different salt solutions in new Petri dishes. In the case of the pure amyloid networks and the βLG/κ-carrageenan hybrid hydrogels, the salt bath consisted of 5 mL of a 100 mM KCl solution. In the case of the βLG/GG hybrid hydrogels, a 10 mM CaCl2 solution was used instead. To facilitate the formation of salt-induced intra- and inter-chain modifications of the polysaccharide units, the hydrogels remained immersed in the corresponding salt solutions for two days. All the mentioned steps are illustrated in Fig. 1.
1.7. Weight fraction determination
For weight fraction determination, the hydrogels were placed in aluminum disks and dried at 60 °C in an oven until the weight became stable. Their weight fraction was calculated by measuring the initial weight of the hydrogels and that of their dry mass, after evaporation, using a precision balance with an accuracy of 2 μg (Mettler AT20, Mettler Toledo). To avoid the influence of the presence of salt on weight fraction characterisation, the measurements were performed directly after the diffusion of the polysaccharides (without the gels being immersed in the salt baths mentioned above).1.8. Aerogel preparation
Pure amyloid hydrogels and polysaccharide-reinforced amyloid hydrogels, prepared as described above, were transformed into aerogels using the following protocol. The hydrogels were transformed into alcogels by solvent exchange through immersion in ethanol baths with increasing volume fractions of the alcoholic component (20%, 50%, 94% and 99.8% v/v ethanol). The alcogels were then transferred into home-made aluminum cages and the alcoholic medium was replaced with a gaseous environment through CO2 supercritical drying (Autosamdri®-931, Tousimis, USA).1.9. Mechanical characterisation
A stress-controlled rheometer (AR2000, TA Instruments) was used to characterize the hydrogels through oscillatory strain and frequency sweeps. The used geometry consisted of a plate–plate arrangement with a diameter of 40 mm, which was covered with a solvent trap during the measurements to prevent solvent loss, and was maintained at a constant temperature of 25 °C. To perform the measurements, the hydrogels were carefully transferred and centered in the above-mentioned geometry with a spatula and the upper plate was lowered to a gap value of 300 μm (with a concomitant gentle rotation in one direction to promote a nice spread of the gel under the plate). For the frequency sweeps (between the values of 0.01 Hz and 100 Hz), the elastic (G′) and the viscous (G′′) moduli were determined by oscillations at a strain equal to 0.1%; the strain sweeps (between 0.01% and 1000%) were instead performed at the constant frequency of 1 Hz.Compression tests on hydrogels and aerogels were conducted with a Zwick Z010 (ZwickRoell GmbH & Co. KG, Germany) with a 100 N cell and a 75 mm diameter plate. Measurements were set to start (0% strain) when a force value of 0.05 N was reached: the applied force was then measured every 0.01 mm while the plate was descending at a constant speed of 0.5 mm min−1, until a distance equal to 90% of the geometry gap at the beginning of the measurement was reached. For determining the intrinsic properties of the probed materials, the data needed to be normalized. The compressive strain was computed as:
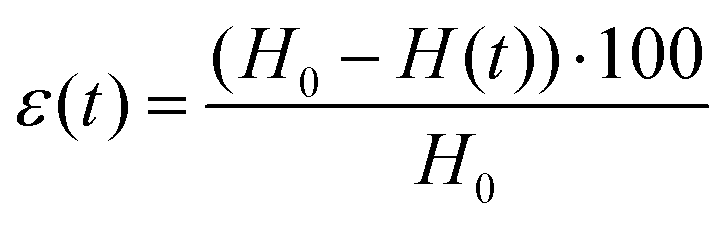 | (1) |
In the reported formula, H0 is the height of the geometry at the beginning of the measurement and H(t) is the height at a specific instant t. The pressure applied by the plate was determined instead by dividing the instantaneous force measured by the equipment by the initial cross-section of the probed hydrogel or aerogel:
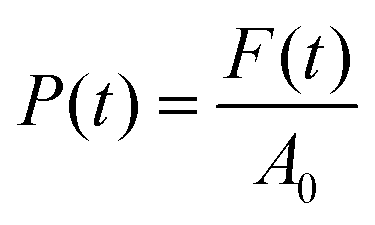 | (2) |
We are aware that, due to the incompressibility of water in the experimental pressure range, the assumption of the constant cross-section of hydrogels over the course of the experiment might be inaccurate. Nonetheless, we followed this procedure in order to better compare experiments between hydrogels and aerogels, and we present an alternative normalisation in the ESI, which takes into account the constancy of the water volume (Fig. S5†).
Before performing any statistical analysis (experiments for each type of hydrogel/aerogel were performed at least in triplicate), the curves were further re-normalized to take into account the potential imprecisions of the starting point of the measurements.
1.10. Pictures of hydrogels/aerogels and SEM imaging
Pictures of the hydrogels and of the aerogels were acquired with a Canon EOS 550D camera. For SEM-imaging, small pieces of the aerogels were mounted on SEM aluminium pin stubs with double adhesive carbon tape. The samples were additionally fixed with conductive silver paint (Plano, DE) by coating all around them. After drying, the samples were carefully cut with a razor blade and sputter-coated with 4 nm of platinum/palladium (CCU-10, Safematic, CH). SE in-lens images were obtained at a working distance of 4–5 mm using a scanning electron microscope (Merlin FE-SEM, Zeiss, DE), operated at an accelerating voltage of 1.5 kV.2. Results & discussion
2.1. Characterisation and analysis of the gel building blocks
Since the diffusion of the polysaccharides in the network depends on the geometry of the polymers, we first performed an AFM characterisation and analysis at the individual molecule/fibril level. In Fig. 2, the morphological properties of β-lactoglobulin amyloid fibrils (panel A), low-acetylated Gellan Gum (panel B) and κ-carrageenan (panel C) are presented.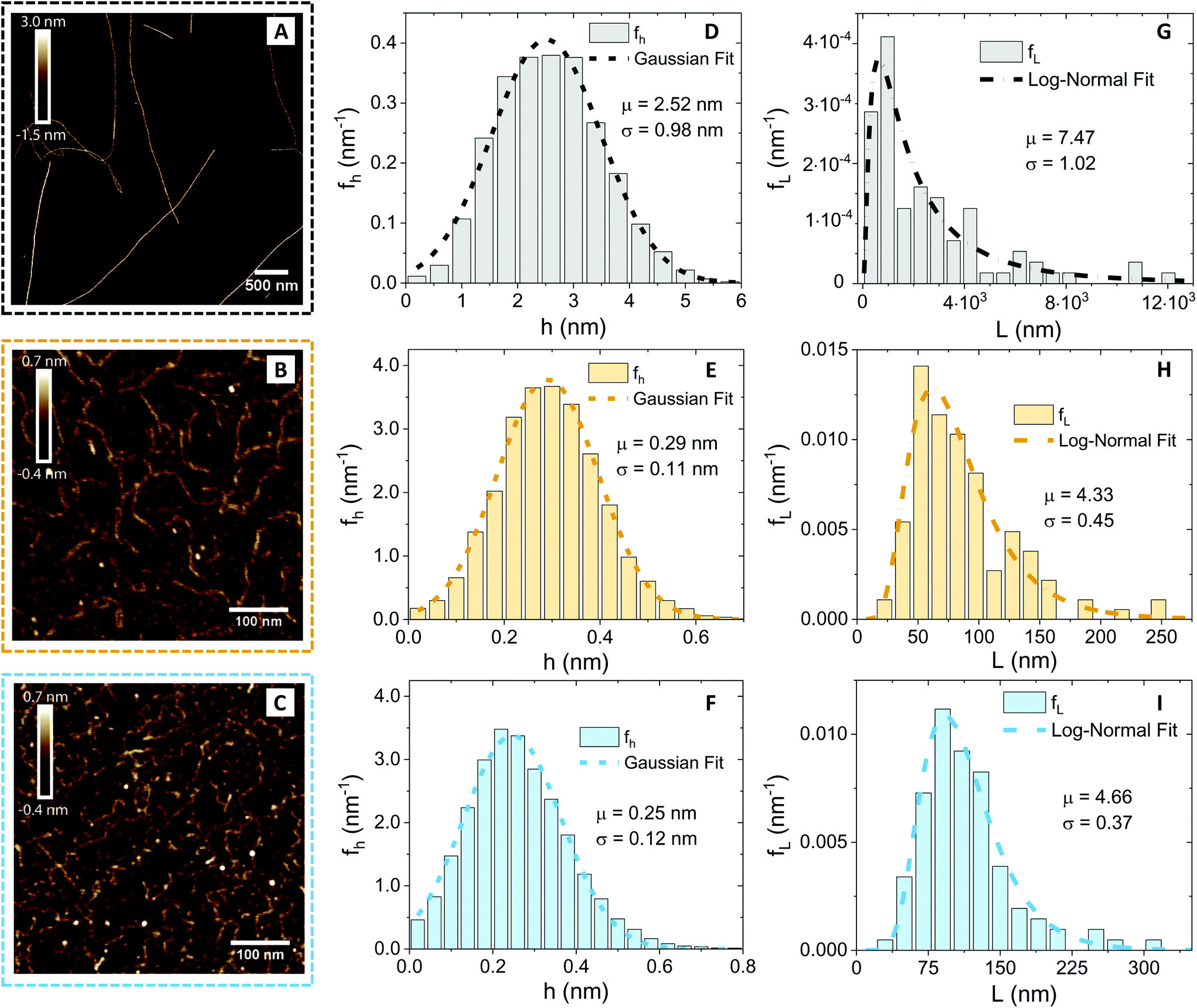 | ||
Fig. 2 Atomic force microscopy characterisation of the prepared amyloid fibrils and of the used polysaccharide units (κ-carrageenan and low-acetylated Gellan Gum). (A–C) Cropped section of AFM images obtained for β-lactoglobulin amyloid fibrils, low-acetylated Gellan Gum and κ-carrageenan, respectively. The color code associated with the three food units (gray/black for βLG, ochre for Gellan Gum and light blue for κ-carrageenan) is kept coherent in the other panels as well. (D–F) Height distributions, computed from the software FiberApp using the tracked points as raw data, together with a Gaussian fit 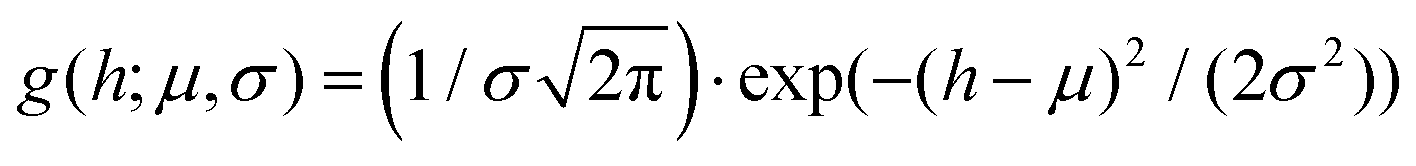 . The parameters extracted from the fitting (μ and σ) are reported in the panels, with the units of nm. (G–I) Contour length distributions, computed from the software FiberApp using the tracked objects as raw data, together with a log–normal fit . The parameters extracted from the fitting (μ and σ) are reported in the panels, with the units of nm. (G–I) Contour length distributions, computed from the software FiberApp using the tracked objects as raw data, together with a log–normal fit 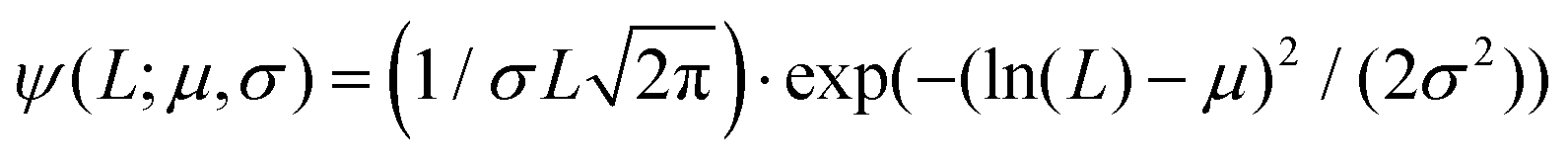 . The parameters extracted from the fitting (μ and σ) are reported in the panels, with a dimensional units (due to the fact that the natural logarithm is applied to the contour length). The contour length data and the probability densities were fed in the fitting algorithm with the units of nm and nm−1, respectively, for all the three considered polymers. . The parameters extracted from the fitting (μ and σ) are reported in the panels, with a dimensional units (due to the fact that the natural logarithm is applied to the contour length). The contour length data and the probability densities were fed in the fitting algorithm with the units of nm and nm−1, respectively, for all the three considered polymers. | ||
Visual inspection of the AFM images reveals profound differences, in morphology and shape, between the βLG amyloid fibrils and the polysaccharide units. A quantitative analysis (performed using the open-source software FiberApp32), together with further calculations, confirmed the visual impression (Table 1).
| Food unit | 〈h〉 (nm) | 〈L〉 (nm) | l p (nm) | R g (nm) | ξ m (nm) |
|---|---|---|---|---|---|
| βLG | 2.52 | 2950 | 5150 | 806 | 50 |
| Gellan Gum | 0.29 | 84 | 33 | 20 | — |
| κ-Carrageenan | 0.25 | 115 | 25 | 23 | — |
In panels D, E and F of Fig. 2, the height (h) distributions of the tracked points are reported, along with the performed Gaussian fits. In panels G, H and I, the length distributions of the tracked objects, along with log–normal fits, are shown. The average length of the semi-flexible polymers was computed from the extracted parameters (μ and σ) through the formula 〈L〉 = e(μ+σ2/2). Despite the possible inaccuracies in tracking (due to difficulties in properly following the contour of not-well-resolvable objects), the performed analysis shows how both the characteristic length and height of the polysaccharide units are one order of magnitude smaller than those of the amyloid fibrils. It is important to consider that not only the contour length L but also the persistence length (lp) defines the size of the semi-flexible polymers. For this reason, we report in the ESI (Fig. S2†) the computation of lp for the three filamentous colloids, extracted through fitting the mean-squared end-to-end distance of the polymers as a function of their internal contour length (l). Knowing both the contour and the persistence lengths, the three-dimensional radius of gyration (Rg) of the semi-flexible polymers can be computed as:33,34
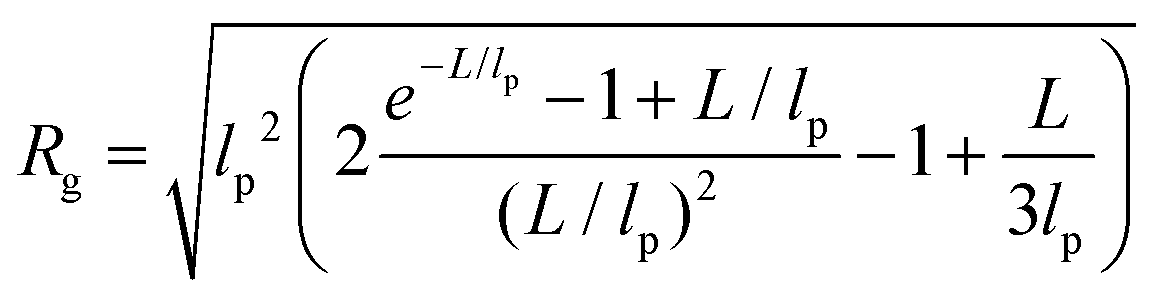 | (3) |
The characteristic dimensions of the polysaccharide units should not be compared with the single-filament properties of the amyloid fibrils but with the mesh size (ξm) of the network they form. Through the cubic lattice (lc) model, it is possible to estimate ξm at the studied protein concentration (c ∼ 2 wt%–20 mg mL−1). Assuming a 40% conversion (ϕ) of monomers into amyloids and a fibril radius (a) of 1.26 nm (Fig. 2D), and considering the density of the fibrils (ρ) equal to 1300 mg mL−1, we obtain:35
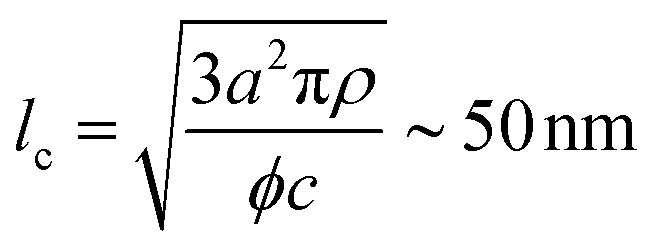 | (4) |
The radius of gyration of the polysaccharide units and the estimated mesh size of the amyloid network are similar, which makes the diffusion of the small chains inside the protein gels possible. This concept is schematically represented in Fig. 3A.
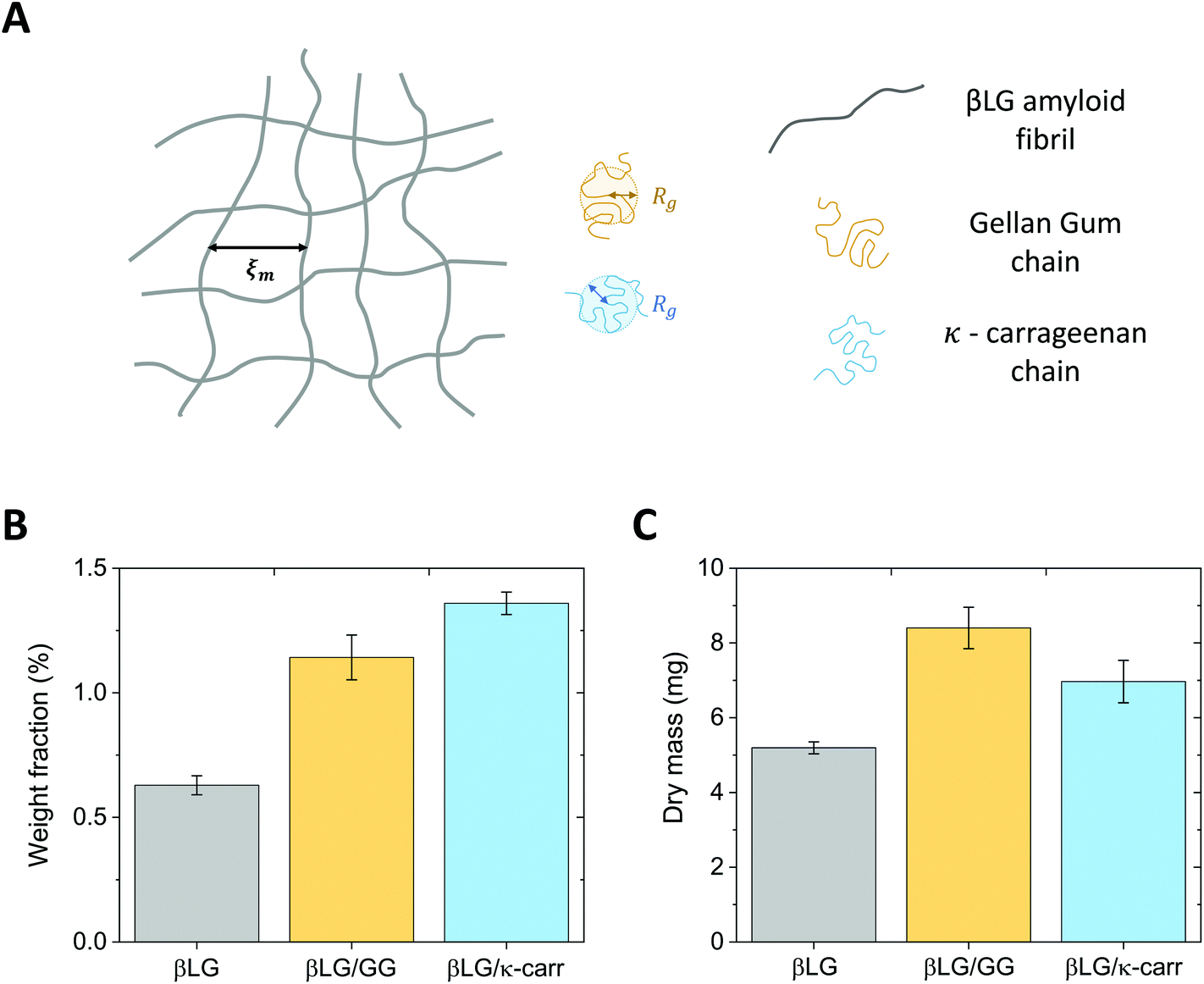 | ||
| Fig. 3 (A) Schematic representing the amyloid network together with the polysaccharide units. The characteristic dimensions that tune the possibility for the sugar chains, to diffuse inside the gel, are the mesh size of the amyloid network (ξm) and the radius of gyration (Rg) of low-acetylated Gellan Gum and κ-carrageenan. (B) Weight fraction characterisation of the pure βLG hydrogels and of the hybrid ones that clearly shows higher values for the protein–polysaccharide hybrid materials, confirming the diffusion of the chains inside the amyloid network. (C) Dry mass analysis of the pure βLG hydrogels and of the hybrid ones, which is aligned with the content of panel B when taking into account the shrinkage observed for the βLG/κ-carrageenan hybrid systems. | ||
2.2. Considerations of polysaccharide diffusion in amyloid hydrogels
Our βLG amyloid fibril hydrogels were prepared by the diffusion of ionic species (Na+ and Cl−) into amyloid fibril semi-diluted solutions with the aim of transforming entanglements into long-living physical crosslinks.36 The inclusion of the polysaccharides inside the mesh occurs then in a separate step, which allows for the diffusion of the chains into the formed hydrogels. Two conditions need to be fulfilled for this phenomenon to occur.The first condition, discussed above, is related to the relative size of the polysaccharide chains, compared to the average mesh size of the network; the AFM analysis shows that the κ-carrageenan and the Gellan Gum chains are small enough to be able to diffuse into the mesh. The second condition consists of the bio-colloids bearing surface charges of the same sign and is achieved by bringing the pH of the βLG hydrogels towards neutral values (upon immersion in alkaline baths). In fact, at the synthesis conditions (pH = 2), the amyloid fibrils are positively charged; if the used polysaccharides were brought directly in contact with the formed hydrogels at the mentioned pH conditions, we would have observed different phenomena as a function of the used polysaccharide (Fig. S3†). κ-Carrageenan is a linear sulphated galactan, the repeating disaccharide of which contains one sulfate group;30 the low pKa of this unit makes κ-carrageenan negatively charged at both neutral and acidic (pH ∼ 2) conditions, and consequently promotes its dissolution in both scenarios (Fig. S3A†). However, despite the fact that κ-carrageenan dissolves at both conditions, at acidic conditions it was shown to interact with βLG amyloid fibrils by decorating their surface through the formation of globular aggregates.37 This phenomenon is undesired as it would inhibit the efficient strengthening of the network. Gellan Gum is composed of a linear tetrasaccharide repeating unit that contains one carboxylic group.29 The pKa of this group is larger than the pH at which the βLG amyloid fibrils are usually synthesized; as a consequence, GG is expected to have a close-to-neutral surface charge at pH ∼ 2. The low surface charge drives, at such pH conditions, the flocculation and sedimentation of the carboxylated polysaccharide, inhibiting its solubility (Fig. S3B†). The reported analysis suggests the importance, for an effective diffusion of the polysaccharides, to shift the pH of the amyloid fibril hydrogels to values that are larger than the isoelectric point of βLG (IEP ∼ 5) to avoid both electrostatic complexation and limited solubility of the sugar chains.
Although the analysis described above supports the possibility that the polysaccharide units can diffuse inside the protein network, additional factors may have a negative effect. In particular, if the food units bear the same surface charge, electrostatic repulsion might hinder the diffusive phenomena. Furthermore, it has to be considered that the measured radius of gyration (Rg) does not reflect the real hydrodynamic dimensions of the units, which can actually be larger than the reported values. These concerns prompted the design of an experimental protocol whereby the amyloid hydrogels remained immersed in polysaccharide solutions for six days, to allow the dynamic phenomena to reach completion. The experimental confirmation of chain diffusion in the mesh is presented in Fig. 3B, which reports the weight fraction of the dry component of the gels, upon evaporation of the wet component in an oven at 60 °C. Although the initial βLG amyloid gels were synthesised from a 2 wt% protein solution, their effective solid content after pH equilibration was much lower (smaller than 0.75 wt%). This may appear surprising, but it is important to consider that the percentage of single protein molecules converted into amyloid fibrils depends on the used protocol and is still an open point in the current literature.38 As reported above, in the case of βLG, a recently published study reported a conversion of around 40% in weight,35 which is compatible with our observed reduction in weight fraction. In fact, during the pH equilibration towards close-to-neutral values, the isoelectric point of βLG is crossed, which may lead to the coagulation and sedimentation of unreacted peptide and protein units. Indeed, such a phenomenon was visually observed as white sedimented aggregates in the proximity of the gels (data not shown).
After the gels remained for six days in 0.5 wt% polysaccharide solutions, the weight fraction of the dry component increased to values larger than 1 wt%. While the average value of the weight fraction of βLG/GG hybrid hydrogels strongly reflects diffusive dynamics (due to limited shrinkage), we believe that the value reported for the βLG/κ-carrageenan hybrid hydrogels overestimates the truly diffused polysaccharide component (as a consequence of visible shrinkage that lowered the initial wet weight of the material). Such a phenomenon can give a misleading interpretation of the dry content of the hydrogels (due to different water content in the wet state). Fig. 3C shows the plot of the measured dry mass of the pure βLG and hybrid gels. It is evident that, while the βLG/GG system shows an appreciable degree of diffused polysaccharides (compatible with the weight fraction analysis shown in panel B), the βLG/κ-carrageenan one shows hints of less-effective diffusion (if one considers the shrinkage-induced water loss, the apparent contradiction of this observation with the weight fraction analysis disappears).
The different behaviours observed for the two polysaccharides, considering the morphological similarities of the single chains, probably stem from their different chemical compositions. The different pKa values of the charged groups of Gellan Gum and κ-carrageenan (carboxylic and sulfate, respectively), together with their diverse recurrency (one group per tetra-saccharide and one group per di-saccharide, respectively), make κ-carrageenan more charged than Gellan Gum, with two consequences that we experimentally observed. Firstly, κ-carrageenan diffused less effectively in the βLG amyloid fibril gels due to enhanced electrostatic repulsion. Secondly, protein gels immersed in κ-carrageenan exhibited a more pronounced shrinkage due to osmotic pressure imbalance between the arrested structure and the surrounding solution: the water phase was driven by thermodynamics to dilute the highly charged polysaccharide dispersion.
2.3. Macro- and microscopic appearances of the pure amyloid and hybrid hydrogels and aerogels
After having embedded the polysaccharides inside the proteinaceous matrix, we further promoted their network strengthening role by leaving the gels immersed in salt solutions (10 mM CaCl2 and 100 mM KCl, respectively for the βLG/GG and βLG/κ-carrageenan hybrid systems). In fact, Diener and co-workers recently showed how the cations of the used salts trigger drastic intra- and inter-chain modifications in both polysaccharides39,40by driving a coil-to-helix morphological change and further bundling of the resulting structures. To investigate how far such morphological rearrangements are possible within already formed arrested structures, we analysed the macro-, meso- and microstructural properties of the synthesized hydrogels and aerogels (Fig. 4). | ||
| Fig. 4 Macro- and microscopic appearances of the synthesised hydrogels and aerogels. The images surrounded by black, ochre and light blue dashed frames are representative of hydrogels and aerogels made out of pure βLG amyloid fibrils, of βLG/Gellan Gum hybrid networks and of βLG/κ-carrageenan hybrid materials, respectively. (A–C) Pictures of the pure/hybrid hydrogels. (D–F) Pictures of the pure/hybrid aerogels. (G–I) Microscopic characterization of the formed aerogels through Scanning Electron Microscopy (SEM) imaging. In panels H and I, the coloured arrows highlight local bundles. | ||
Fig. 4A shows the macroscopic appearance of a pure βLG hydrogel: its transparency can be immediately appreciated, together with its self-standing properties. Upon diffusion and reconfiguration of the Gellan Gum units (Fig. 4B) the hydrogel still shows remarkable transparency and has a size that is comparable to the initial size. In the case of κ-carrageenan (Fig. 4C), instead, we can observe a certain degree of hydrogel shrinkage, both at global (overall size) and local (morphology) scales. We also noticed that the degree of shrinkage depended on the pH of the system (data not shown). However, due to complexities in fine tuning the final pH without a buffer system (the reader is addressed to the caption of Fig. S1†), we decided not to pursue such an analysis further but to leave it open for future studies. As mentioned above, the observed shrinkage can be attributed to the osmotic pressure imbalance between the amyloid fibril gel and the surrounding κ-carrageenan dispersion.
Gentle removal of the liquid component allowed the transformation of the synthesized hydrogels into aerogels. In order to preserve the state of the network as close as possible to its initial wet state, we used supercritical CO2 drying, employing ethanol as an intermediate solvent.7 Despite the lack of capillary forces that could drastically destroy the structure, the solvent exchange steps can have a large influence on the gels. In fact, a recent perspective article showed how a change in the dielectric properties of the continuous phase (together with changes in its ability to provide hydrogen bonding) can drive the shrinkage phenomena in biopolymer gels, especially relevant at small mass fractions of the solid component.41 In our case, although we used a protocol with a step-wise solvent exchange, we observed marked shrinkage effects on the materials containing polysaccharides (and limited ones in pure amyloid networks).
Fig. 4D shows the appearance of a pure βLG amyloid fibril aerogel. Only a slight shrinkage was observed and the material showed high transparency (the ETH Zürich logo behind it can still be easily read). We hypothesize that enhanced turbidity compared to the liquid state is not caused by a change in the morphology of the network-forming units but instead by a higher refractive index difference between the amyloid fibrils and air (compared to the one between the bio-fibrillar colloids and water). This result is compatible with the considerations discussed above, because βLG amyloid fibrils have both hydrophilic and hydrophobic properties;15 as a consequence, solvent exchange steps are not drastically modifying the aggregation state of the formed network, as will be confirmed later on in the analysis of the acquired SEM images. However, in the case of hybrid aerogels containing Gellan Gum and κ-carrageenan (Fig. 4E and F, respectively), a certain degree of shrinkage (more accentuated for the κ-carrageenan case) and much higher turbidity (the ETH Zürich logo cannot be read anymore through the aerogels) were observed. This significant change in optical properties, different from what was observed with the pure βLG case, can be attributed to an aggregation-driven morphological change of the network forming units. In fact, shrinkage-induced bundling of the bio-polymeric components can lead to the formation of aggregates, the size of which becomes comparable with the wavelength of light in the visible range (in the order of hundreds of nm), leading to enhanced scattering phenomena and a more turbid appearance of the final materials.42 In this case, we hypothesize that the presence of the polysaccharides (for whom ethanol is a bad/non-solvent) makes the material more sensitive to the solvent exchange procedure, influencing the properties of the network. This is evidenced by the fact that pure polysaccharide hydrogels usually showed a large shrinkage when transformed into aerogels (at mass fractions compatible with the ones used in our study).41
To test the expected correlation between the diverse macroscopic appearances of the synthesised materials with the underlying microscopic structures, we performed Scanning Electron Microscopy imaging of the aerogels (Fig. 4G–I). Although the overall appearance looks similar for all the three materials, a higher degree of local aggregation is observed in the case of the protein–polysaccharide hybrid aerogels: brighter spots in the images (highlighted by means of coloured arrows) can be interpreted as fractures of local bundles and strands during crossfracturing of the aerogels. It is not clear whether such bundles stem from the protein–polysaccharide interactions, from a secondary polysaccharide network or from the aggregation phenomena during the shrinkage process. In addition, it is not possible to clearly identify, from these images, the location of the polysaccharide units upon being embedded in the network. To gain further insights indirectly, we analysed the effect of the diffused polysaccharides on the mechanical properties of the materials.
2.4. Network-strengthening role of the polysaccharide component
The question that we would now like to address is whether the diffused polysaccharide units can efficiently strengthen the mechanical properties of the final material. To gain experimental evidence on this point, we performed oscillatory sweeps (both frequency and amplitude ones, only for hydrogels) and mechanical compression tests (for both hydrogels and aerogels). The results of the oscillatory rheology characterization are shown in Fig. 5A. The first point that clearly emerges is the solid-like nature of the materials: for all the three types of networks, the elastic modulus (G′) dominates over the viscous one (G′′). At the same time, it can be observed that the loss tangent (tanδ, defined as the ratio between G′′ and G′), for the βLG/GG case, shifts towards higher values as the oscillatory frequency decreases (Fig. S4†). This can be attributed to Gellan Gum chains being not fully embedded in the network structure. We hypothesized that such units contribute to storing mechanical energy at fast deformation rates but increasingly relax and contribute to viscous dissipation as the probation frequency decreases.
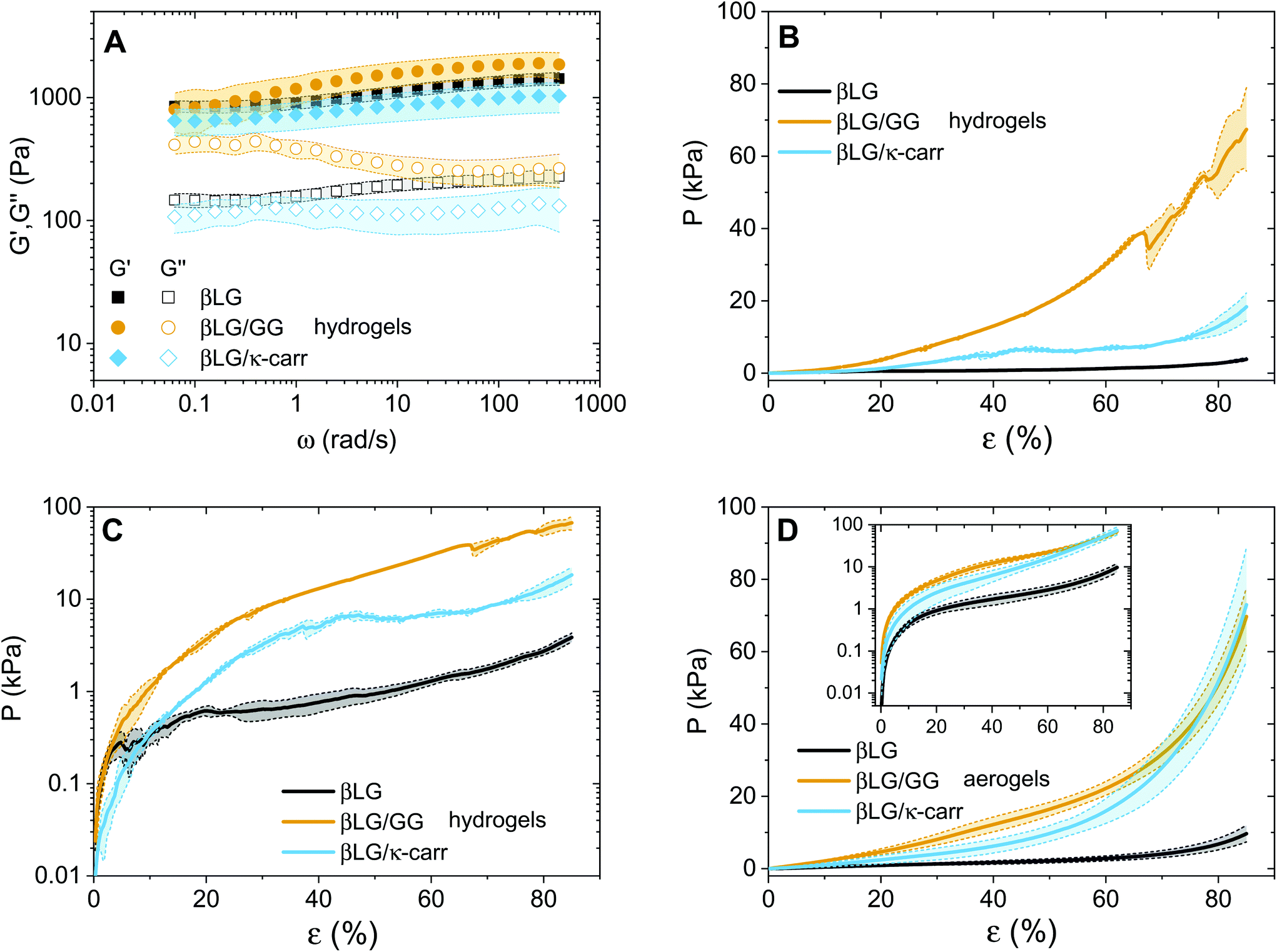 | ||
| Fig. 5 Mechanical characterisation of hydrogels and aerogels. In all the panels, light colors indicate error bars over multiple individual measurements. (A) Oscillatory frequency sweeps applied to the three typologies of hydrogels. (B and C) Mechanical compression experiments on pure and hybrid hydrogels. The same information is respectively plotted in a linear plot (B) and in a semi-logarithmic plot (C). (D) Mechanical compression experiments on pure and hybrid aerogels. Inset: The same information is plotted in a semi-logarithmic plot. | ||
A second point, that is more surprising, is how the absolute values of the elastic moduli are close to each other. In the case of the hybrid hydrogels, the networks are both denser (exhibiting an almost double weight fraction) and are supposedly further strengthened by the interactions between the protein and the polysaccharide fractions, leading to an expected enhanced elastic component.24 We believe that this point can be explained by considering the compression properties of the materials that are presented in Fig. 5B. In fact, while the shear elastic moduli of the materials look similar, the compression of the materials shows astonishingly different features. A clear difference can be seen among the three curves, both in absolute values and in overall behaviour. Regarding the values, the three materials need three different orders of magnitude of pressures to be compressed down to more than 80% of their initial height. In particular, the βLG/GG hydrogels require pressures two orders of magnitude larger than those needed for the pure βLG hydogels, while the βLG/κ-carrageenan ones need pressure one order of magnitude larger than those of the pure amyloid fibril networks. This comparison can be better appreciated in Fig. 5C, where the same information as in panel B is shown in a semi-logarithmic plot. It can be seen that the pure amyloid fibril network starts showing fluctuations in the measured pressure after a deformation rate of a few percentage points (less than 10%). For both hybrid gels, this behavior is observed instead much later (approximately 40% for the κ-carrageenan case and more than 60% for the Gellan Gum one). These observations suggest that the polysaccharide component has two drastic effects: it reinforces the materials and makes them withstand more severe deformations before being structurally affected. As reported in the Materials and methods section, the presented analysis is performed by normalizing the compressing force (F(t)) by the initial cross-section of the samples (A0). Although this approximation is not fully met in reality, it allows comparing the extracted values with the ones of the aerogels discussed below and for which the constant cross-section approximation is more valid. For completeness, in the ESI (Fig. S5†) we report an analysis based on the assumption of the conservation of the total volume of the gels being compressed, due to the incompressibility of the water phase. Despite the fact that the absolute values are inevitably different, the relative differences are very similar to the ones reported in Fig. 5B and C, thereby confirming the different mechanical responses of the three materials.
The observations on the compression behaviour of the pure amyloid and hybrid hydrogels can help better understand the oscillatory rheology properties shown in Fig. 5A. The plate–plate geometry setup used for the oscillatory tests had a diameter of 4 cm and a gap of 300 μm; therefore, the initial materials were subjected to considerable compression to fill the probation volume. It is probable that the composite networks did not spread under the plate as homogeneously as the pure βLG ones due to the highlighted difference in their compression properties. The consequent presence of potential in-homogeneous zones could influence the absolute values of the measured elastic and viscous moduli, making the comparison between the different materials less straightforward.
After having discussed the properties of the hydrogels, we can now focus on the mechanical properties of the aerogels (Fig. 5D). A similar trend as in panels B and C can be observed with two main differences. The first is the smoother behaviour of the curves, which can be attributed to the absence of the water phase. Water, in fact, tends to escape from the hydrogels while the compression measurements are performed, due to its incompressibility in the pressure range of interest. This further affects the networks and introduces fluctuations in the reported pressure values. The second difference is that the hybrid material curves are closer to each other due to the enhancement of the mechanical properties of the βLG/κ-carrageenan aerogels. A direct explanation of this experimental observation lies in the more pronounced shrinkage of these samples compared to that of the βLG/Gellan Gum ones; their denser networks can be assumed to be responsible for the observed phenomenon.
The experimental evidences presented so far show clearly the reinforcing role of the included polysaccharides. Moreover, in the ESI (section 6 and Fig. S6†), we share experimental evidence (through adsorption of water pollutants) that the strengthening of the materials does not negatively influence the surface reactivity of the amyloid fibrils. This observation, together with the other ones discussed in this work, makes us formulate the hypothesis that the polysaccharide component forms either local bundles or an independent network within the already-formed amyloid gels (section 7 in the ESI and Fig. S7†).
3. Conclusions
In this study we presented a sustainable pathway for reinforcing βLG amyloid fibril gels with polysaccharide chains (namely low-acetylated Gellan Gum and κ-carrageenan). The sugar chains were shown to effectively diffuse inside the initial networks and to reinforce their mechanical properties, while still allowing amyloid fibrils to express their surface functionality. The combination of the performed experiments allowed us to draw preliminary conclusions on how the polysaccharide component is embedded inside the network: the creation of local, thicker bundles with high bending rigidity or the creation of an independent network is the hypothesis that mostly aligns with the collected evidence.The developed approach shows a simple and bioresource-based route for synthesizing functional, innovative hydrogels and aerogels with a broad range of potential applications and paves the way for further studies aimed at up-scaling the production protocol (through direct mixing of the protein and polysaccharide components) and at better elucidating the role of the synthesis and final pH in the properties of the prepared materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr. Michael Diener, Dr. Milad Radiom and Dr. Maria Mitsi for insightful discussions. The help of Horst Adelmann and Xiangze Jia in performing the compression and AAS/UV-Vis experiments, respectively, is also greatly acknowledged.References
- S. S. Kistler, Nature, 1931, 127, 741–741 CrossRef CAS.
- S. S. Kistler, J. Phys. Chem., 1932, 36, 52–64 CrossRef CAS.
- T. Coviello, P. Matricardi and F. Alhaique, Expert Opin. Drug Delivery, 2006, 3, 395–404 CrossRef CAS PubMed.
- J. J. Chung, S. Li, M. M. Stevens, T. K. Georgiou and J. R. Jones, Chem. Mater., 2016, 28, 6127–6135 CrossRef CAS.
- X. Zheng, K. Chen and Z. Lin, Ind. Eng. Chem. Res., 2020, 59, 5760–5767 CrossRef CAS.
- O. Lev, Z. Wu, S. Bharathi, V. Glezer, A. Modestov, J. Gun, L. Rabinovich and S. Sampath, Chem. Mater., 1997, 9, 2354–2375 CrossRef CAS.
- S. Zhao, W. J. Malfait, N. Guerrero-Alburquerque, M. M. Koebel and G. Nyström, Angew. Chem., Int. Ed., 2018, 57, 7580–7608 CrossRef CAS PubMed.
- I. Smirnova and P. Gurikov, J. Supercrit. Fluids, 2018, 134, 228–233 CrossRef CAS.
- W. Steffen, K. Richardson, J. Rockström, S. E. Cornell, I. Fetzer, E. M. Bennett, R. Biggs, S. R. Carpenter, W. de Vries, C. A. de Wit, C. Folke, D. Gerten, J. Heinke, G. M. Mace, L. M. Persson, V. Ramanathan, B. Reyers and S. Sörlin, Science, 2015, 347, 1259855 CrossRef PubMed.
- J. D. Sachs, G. Schmidt-Traub, M. Mazzucato, D. Messner, N. Nakicenovic and J. Rockström, Nat. Sustain., 2019, 2, 805–814 CrossRef.
- E. A. Olivetti and J. M. Cullen, Science, 2018, 360, 1396–1398 CrossRef CAS PubMed.
- Y. Cao and R. Mezzenga, Nat. Food, 2020, 1, 106–118 CrossRef.
- S. Assenza and R. Mezzenga, Nat. Rev. Phys., 2019, 1, 551–566 CrossRef.
- S. Bolisetty and R. Mezzenga, Nat. Nanotechnol., 2016, 11, 365–371 CrossRef CAS PubMed.
- M. Peydayesh, M. K. Suter, S. Bolisetty, S. Boulos, S. Handschin, L. Nyström and R. Mezzenga, Adv. Mater., 2020, 32, 1907932 CrossRef CAS PubMed.
- M. Peydayesh and R. Mezzenga, Nat. Commun., 2021, 12, 1–17 CrossRef PubMed.
- Y. Shen, L. Posavec, S. Bolisetty, F. M. Hilty, G. Nyström, J. Kohlbrecher, M. Hilbe, A. Rossi, J. Baumgartner and M. B. Zimmermann, et al. , Nat. Nanotechnol., 2017, 12, 642 CrossRef CAS PubMed.
- B. Hu, S. Yu, C. Shi, J. Gu, Y. Shao, Q. Chen, Y. Li and R. Mezzenga, ACS Nano, 2020, 14, 2760–2776 CrossRef CAS PubMed.
- Y. Han, Y. Cao, S. Bolisetty, T. Tian, S. Handschin, C. Lu and R. Mezzenga, Small, 2020, 16, 2004932 CrossRef CAS PubMed.
- G. Nyström, M. P. Fernández-Ronco, S. Bolisetty, M. Mazzotti and R. Mezzenga, Adv. Mater., 2016, 28, 472–478 CrossRef PubMed.
- G. Wei, Z. Su, N. P. Reynolds, P. Arosio, I. W. Hamley, E. Gazit and R. Mezzenga, Chem. Soc. Rev., 2017, 46, 4661–4708 RSC.
- J. Fu, S. Wang, C. He, Z. Lu, J. Huang and Z. Chen, Carbohydr. Polym., 2016, 147, 89–96 CrossRef CAS PubMed.
- W. G. Willats, J. P. Knox and J. D. Mikkelsen, Trends Food Sci. Technol., 2006, 17, 97–104 CrossRef CAS.
- Y. Cao, S. Bolisetty, G. Wolfisberg, J. Adamcik and R. Mezzenga, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4012–4017 CrossRef CAS PubMed.
- T. Ramdhan, S. H. Ching, S. Prakash and B. Bhandari, Trends Food Sci. Technol., 2020, 106, 150–159 CrossRef CAS.
- M. Joshi, P. Aldred, J. Panozzo, S. Kasapis and B. Adhikari, Food Hydrocolloids, 2014, 35, 226–237 CrossRef CAS.
- Y. L. Xiong and S. P. Blanchard, J. Food Sci., 1993, 58, 164–167 CrossRef CAS.
- A. Kilara and M. N. Vaghela, in Proteins in food processing, ed. R. Y. Yada, Elsevier, 2nd edn, 2018, ch. 4, pp. 93–126 Search PubMed.
- G. Sworn and L. Stouby, in Handbook of hydrocolloids, ed. G. O. Phillips and P. A. Williams, Elsevier, 3rd edn, 2021, ch. 28, pp. 855–885 Search PubMed.
- V. L. Campo, D. F. Kawano, D. B. da Silva Jr. and I. Carvalho, Carbohydr. Polym., 2009, 77, 167–180 CrossRef CAS.
- D. Vigolo, J. Zhao, S. Handschin, X. Cao, A. J. deMello and R. Mezzenga, Sci. Rep., 2017, 7, 1211 CrossRef PubMed.
- I. Usov and R. Mezzenga, Macromolecules, 2015, 48, 1269–1280 CrossRef CAS.
- K. Kroy and E. Frey, in Scattering in polymeric and colloidal systems, ed. W. Brown and K. Mortensen, Taylor & Francis Group, London, 1st edn, 2000, ch. 5, pp. 197–248 Search PubMed.
- H. Benoit and P. Doty, J. Phys. Chem., 1953, 57, 958–963 CrossRef CAS.
- M. Usuelli, Y. Cao, M. Bagnani, S. Handschin, G. Nyström and R. Mezzenga, Macromolecules, 2020, 53, 5950–5956 CrossRef CAS.
- Y. Cao, S. Bolisetty, J. Adamcik and R. Mezzenga, Phys. Rev. Lett., 2018, 120, 158103 CrossRef CAS PubMed.
- O. G. Jones, S. Handschin, J. Adamcik, L. Harnau, S. Bolisetty and R. Mezzenga, Biomacromolecules, 2011, 12, 3056–3065 CrossRef CAS PubMed.
- Y. Cao and R. Mezzenga, Adv. Colloid Interface Sci., 2019, 269, 334–356 CrossRef CAS PubMed.
- M. Diener, J. Adamcik, A. Sánchez-Ferrer, F. Jaedig, L. Schefer and R. Mezzenga, Biomacromolecules, 2019, 20, 1731–1739 CrossRef CAS PubMed.
- M. Diener, J. Adamcik, J. Bergfreund, S. Catalini, P. Fischer and R. Mezzenga, ACS Macro Lett., 2020, 9, 115–121 CrossRef CAS.
- P. Gurikov, R. SP, J. S. Griffin, S. A. Steiner III and I. Smirnova, Ind. Eng. Chem. Res., 2019, 58, 18590–18600 CrossRef CAS.
- H. C. van de Hulst, Light scattering by small particles, Dover Publications, 1981 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: pH equilibration of the amyloid fibril hydrogels, computation of the persistence length of the polymeric units, dissolution of κ-carrageenan and low-acetylated Gellan Gum at pH 2, loss tangent (rheological data), alternative normalization of the compression experiments on the hydrogels, AuCl4− and methylene blue adsorption, hypotheses on the network structure. See DOI: 10.1039/D1NR03133C |
| ‡ Present address: Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. |
| This journal is © The Royal Society of Chemistry 2021 |