
Biomimetic cytomembrane nanovaccines prevent breast cancer development in the long term†
Long
Xiao‡
 a,
Yu
Huang‡
b,
Yuhe
Yang‡
c,
Zhiwei
Miao‡
a,
Jie
Zhu
a,
Mengdan
Zhong
a,
Chencheng
Feng
a,
Wenkai
Tang
a,
Jinhua
Zhou
*b,
Lihong
Wang
*a,
Xin
Zhao
*c and
Zhirong
Wang
*a
a,
Yu
Huang‡
b,
Yuhe
Yang‡
c,
Zhiwei
Miao‡
a,
Jie
Zhu
a,
Mengdan
Zhong
a,
Chencheng
Feng
a,
Wenkai
Tang
a,
Jinhua
Zhou
*b,
Lihong
Wang
*a,
Xin
Zhao
*c and
Zhirong
Wang
*a
aCenter Laboratory, Zhangjiagang Traditional Chinese Medicine Hospital Affiliated to Nanjing University of Chinese Medicine, Zhangjiagang, Jiangsu 215600, China. E-mail: zjgzy001@njucm.edu.cn; zjgfy_spine_wzr@njucm.edu.cn
bDepartment of Obstetrics and Gynecology, The First Affiliated Hospital of Soochow University, Suzhou, Jiangsu 215006, China
cDepartment of Biomedical Engineering, The Hong Kong Polytechnic University, Hung Hom, Hong Kong, China. E-mail: xin.zhao@polyu.edu.hk
First published on 28th January 2021
Abstract
Cytomembrane cancer nanovaccines are considered a promising approach to induce tumor-specific immunity. Most of the currently developed nanovaccines, unfortunately, fail to study the underlying mechanism for cancer prevention and therapy, as well as immune memory establishment, with their long-term anti-tumor immunity remaining unknown. Here, we present a strategy to prepare biomimetic cytomembrane nanovaccines (named CCMP@R837) consisting of antigenic cancer cell membrane (CCM)-capped poly(lactic-co-glycolic acid) (PLGA) nanoparticles loaded with imiquimod (R@837) as an adjuvant to activate the immune system. We found that our CCMP@R837 system enhanced bone-marrow-derived dendritic cell uptake and maturation, as well as increased anti-tumor response against breast cancer 4T1 cells in vitro. Moreover, an immune memory was established after three-time immunization with CCMP@R837 in BALB/c mice. The CCMP@R837-immunized BALB/c mice exhibited suppressed tumor growth and a long survival period (75% of mice lived longer than 50 days after tumor formation). This long-term anti-tumor immunity was achieved by increasing CD8+ T cells and decreasing regulatory T cells in the tumor while increasing effector memory T cells in the spleen. Overall, our platform demonstrates that CCMP@R837 can be a potential candidate for preventive cancer vaccines in the clinic.
Introduction
Training the autologous immune system using cancer cell membrane (CCM) vaccines to recognize and fight cancers is a promising modality for cancer prevention and therapy.1–7 This strategy takes advantage of the natural capability of cytotoxic T lymphocytes (CTLs) to recognize the diverse receptors on the CCM, increasing their specificity to fight against tumor cells and reducing side effects.6,8–12 Moreover, it has a relatively low cost as fashioning the CCM directly into the nanoparticle (NP) format bypasses the heavy workload of proteomics13 and the engineering hurdles behind functionalizing NPs.8 Recently, several works have encapsulated NPs (e.g., metal–organic framework (MOF) NPs,14 gold NPs,15 poly(lactic-co-glycolic acid) (PLGA) NPs)16 with various CCMs (e.g., 4T1 cells, dendritic cells (DCs), and B16–F10 cells) for the purposes of cancer prevention and therapy. Most of the above-mentioned studies, however, did not examine the DC activation and the immune memory establishment using the nanovaccine strategies and the underlying mechanism of these approaches for cancer prevention and therapy, as well as their long-term anti-tumor immunity, remain unknown.6,14–17In this study, we encapsulate biodegradable PLGA NPs loading R837 (an agonist against toll-like receptor 7)18 as an adjuvant with 4T1 CCM as antigens. We demonstrate that the resulting NPs (denoted as CCMP@R837) possess an antigenic shell closely resembling the source cancer cells. Compared to its naked counterparts, our CCMP@R837 shows enhanced bone marrow-derived dendritic cell (BMDC) uptake and stronger stimulation effect to trigger the secretion of interleukin-12 (IL-12p70), which further stimulates the tissue-resident memory T (Trm) cells to secrete the effector molecules interferon (IFN)-γ and tumor necrosis factor (TNF)-α to achieve the anti-tumor effects (Scheme 1). The mature DCs are found to suppress Treg differentiation from naïve CD4+ T cells while activating the naïve CD8+ T cells, making them massively proliferate and differentiate into central-memory (TCM) and effector-memory (TEM) CD8+ T cells. When the immunized mice (treated with CCMP@R837) are challenged with 4T1 cancer cells, the pre-trained immune system recognizes and destroys cancer cells, with long-lasting anti-tumor immunity. Our work highlights the great potential of CCMP@R837 to develop nanovaccines for cancer immunotherapy.
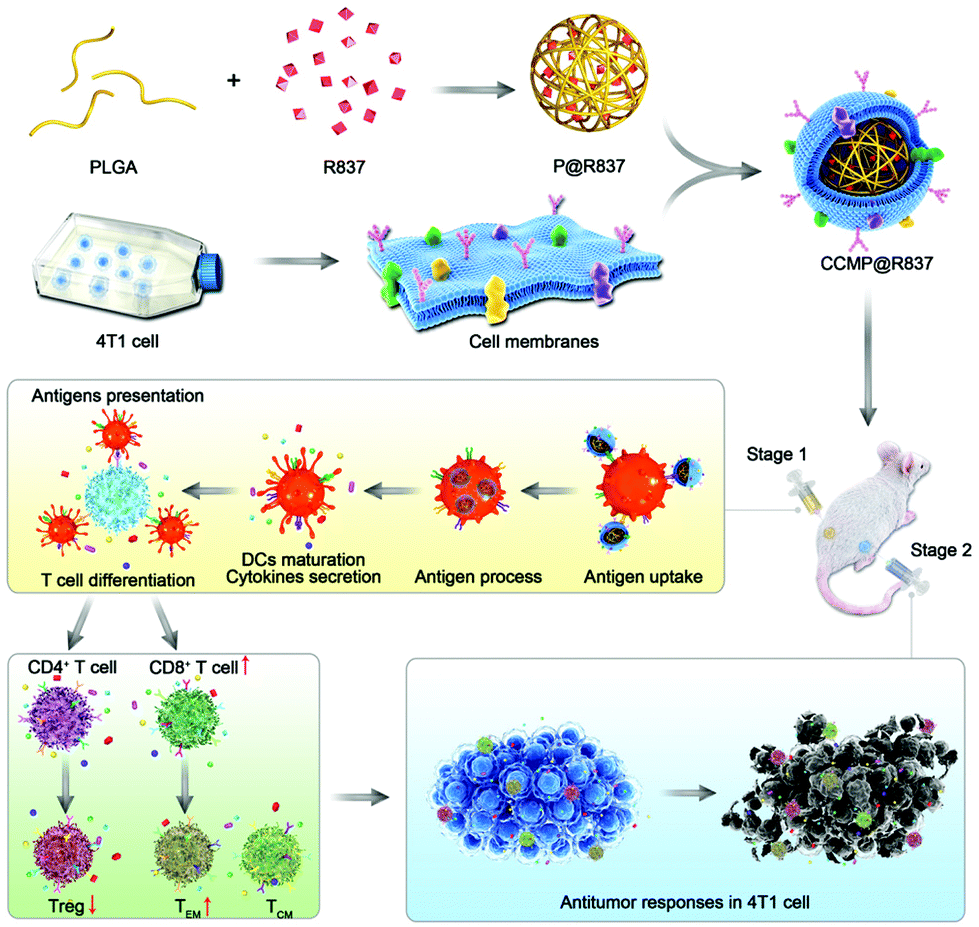 | ||
| Scheme 1 Fabrication and mechanism of cancer immunotherapy of CCMP@R837. CCMP@R837 NPs are fabricated by encapsulating biodegradable poly(lactic-co-glycolic acid) (PLGA) nanoparticles (NPs) loading R837 (an agonist against toll-like receptor 7) as an adjuvant with 4T1 cancer cell membrane as antigens. When the nanovaccines are injected into a mouse model (stage 1), they will be taken up by dendritic cells (DCs) and trigger the DCs to secrete cytokines (e.g., interleukin-12), which will then suppress Treg differentiation from naïve CD4+ T cells and activate the naïve CD8+ T cells to differentiate into central-memory (TCM) and effector-memory (TEM) CD8+ T cells. When the immunized mouse is challenged with 4T1 cancer cells (stage 2), the TCM and TEM cells will differentiate into cytotoxic T lymphocytes (CTLs) to recognize and destroy cancer cells, with long-lasting anti-tumor immunity. | ||
Materials and methods
Materials
PLGA, dimethyl sulfoxide (DMSO) and dichloromethane (CH2Cl2) were purchased from Sigma-Aldrich; R837 (imiquimod) was obtained from InvivoGen; all the antibodies for flow cytometry and the Zombie aqua fixable viability kit were obtained from BioLegend; IL-12p70, IFN-γ and TNF-α ELISA kits were purchased from Thermo Fisher.Cell line
The 4T1 mouse breast cancer cell line was acquired from the American Type Culture Collection (ATCC). 4T1 cells were cultured in the Roswell Park Memorial Institute (RPMI) 1640 medium (HyClone) supplemented with 10% fetal bovine serum (FBS, Gibco), penicillin (100 U mL−1, HyClone), and streptomycin (100 U mL−1, HyClone). BMDCs were isolated from the tibia and femur of the BLAB/c mice (4–6 weeks) according to an established method19 and cultured under the same conditions as 4T1.Animal
Female BALB/c mice (4–6 weeks) were purchased from Shanghai Biomodel Organism Science & Technology Development Co., Ltd and treated under protocols approved by the Institute of Animal Care Committee of Zhangjiagang Traditional Chinese Medicine Hospital (approval number: 2019A011), in accordance with international ethics guidelines and the National Institutes of Health of China, concerning the Care and Use of Laboratory Animals.Cell membrane isolation
4T1 cells were cultured in T75 flasks (Thermo Fisher) until the cells reached 80% confluence. The cells were detached with PBS-EDTA (Gibco, Thermo Fisher) followed by washing with 1× PBS (HyClone) three times. Afterwards, the obtained pellet was resuspended in a hypotonic lysing buffer with 1 tablet of the protease inhibitor (Thermo Fisher). Finally, gradient centrifugation (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 30 min and 100000g for 20 min) was used to obtain the 4T1 cell membrane pellet for further use.20
000g for 30 min and 100000g for 20 min) was used to obtain the 4T1 cell membrane pellet for further use.20
Synthesis of CCMP@R837
P@R837 NPs were fabricated as previously reported.5 Briefly, PLGA (25 mg mL−1) was dissolved in CH2Cl2 with 200 μg of R837 (in DMSO at 5 mg mL−1); then 0.5 mL of polyvinylpyrrolidone (PVP) solution (5% w/v) was added into the PLGA mixture followed by tip-sonication (40% power for 3 minutes, 3 seconds on and 1 second off). After that, 2.5 mL of PVP solution (5% w/v) was added dropwise into the obtained solution under constant stirring overnight at room temperature. The P@R837 NPs were obtained after centrifugation at 3500g for 25 min. The naked PLGA NPs (denoted as P) were fabricated using the same protocol, without adding R837. To prepare CCMP@R837, the 4T1 cell membrane pellet was bath-sonicated and physically co-extruded with P@R837 through a 200 nm polycarbonate membrane (Avanti Lipids) using a mini extruder (Avanti Lipids) for 11 passes. Fluorescence-labelled P, P@R837 and CCMP@R837 were named F#P, F#P@R837 and F#CCMP@R837, respectively. These NPs were prepared by adding coumarin 6 into the solution of PLGA.Characterization of the physical properties of CCMP@R837
The hydrodynamic sizes and zeta potentials of CCM, P@R837, and CCMP@R837 were explored using a Zetasizer Nano-ZS (Malvern Instruments) with dynamic light scattering (DLS) and electrophoretic light scattering (ELS), respectively. The protein content of CCMP@R837 was investigated by the micro (bicinchoninic acid) BCA assay kit. Additionally, the long-term stability of CCMP@R837 (stored at 4 °C without stirring in 1× PBS) was investigated using DLS. The release behaviour of CCMP@R837 was explored by high-performance liquid chromatography (HPLC) with an ultraviolet-visible detector at 325 nm. The morphology of CCMP@R837 was visualized using a transmission electron microscope (TEM, FEI).Characterization of the in vitro biological properties of CCMP@R837
The biocompatibility of CCMP@R837 was investigated in both BMDCs and 4T1 cells. Briefly, BMDCs (1 × 104 cells per well in a 96 well plate) and 4T1 cells (1 × 104 cells per well in a 96 well plate) were respectively treated with different formulas (CCM, P@R837, and CCMP@R837; P@R837 and CCMP@R837 contained the same amount of R837 per well) for 1, 2 and 3 days. NC means cells cultured without any NPs. Cell viability was assessed using a CellTiter-Glo reagent assay (Promega Corporation) according to the manufacturer's instructions.To quantify BMDCs’ uptake, F#P, F#P@R837 and F#CCMP@R837 were co-cultured with BMDCs (3 × 105 cells per well in a 24 well plate) for 1, 6 and 24 hours. After incubation, the BMDCs were detached and washed with 1× PBS, and then, trypan blue was used to eliminate any NPs bound onto the surface of BMDCs before flow cytometry analysis (BD, FACS Celesta).
The ability of CCMP@R837 in inducing BMDC activation was further explored by treating BMDCs with different formulations (R837, CCM, P@R837 and CCMP@R837, respectively) for 1, 2 and 3 days in vitro. After incubation, the BMDCs were detached and washed with 1× PBS, and then stained with CD11c-FITC, CD80-PE, and CD86-APC for further flow cytometry analysis. To further explore the immune activation ability of CCMP@R837, BMDCs were firstly treated with different formulations (R837, CCM, P@R837, R837 + CCM, and CCMP@R837). After 3 days of treatment, BMDCs were detached and washed with 1× PBS. Then, these BMDCs were co-cultured with splenocytes for another 3 days; this treatment made BMDCs process and present the related antigens to splenocytes. Finally, both BMDCs and splenocytes were collected and co-cultured with 4T1 cells for 24 hours. After that, the culture medium suspension was collected to evaluate the concentration of IFN-γ and IL-12p70 using an ELISA kit, and the 4T1 cells were gently washed three times using 1× PBS, and the cell viability of 4T1 was evaluated by the CellTiter-Glo reagent assay.
Immunogenicity of CCMP@R837
Female BALB/c mice (randomly divided into four groups) were subcutaneously treated with different formulations (saline, CCM, P@R837 and CCMP@R837) on day 0, 14, and 28. The body weight of mice was recorded every two days. Three days after pre-immunization, 4T1 cells were subcutaneously injected into the right flank of these mice. Then, the tumor size and the survival rate of each mouse were recorded every two days. 14 days post 4T1 cell injection, five mice were randomly selected from each group to analyze the immunological profile of the tumor tissue, inguinal lymph nodes and the concentration of TNF-α in the blood. Specifically, the peripheral blood was collected from each mouse and serum was isolated to analyze the concentration of TNF-α by ELISA. Afterwards, the mice were euthanized, and the tumor tissue and inguinal lymph nodes were collected respectively to prepare the single-cell suspension for flow cytometry analysis as described elsewhere.5 Firstly, the Zombie aqua fixable viability kit was used to gate the living cells, and then, the differentiated hematopoietic cells were gated in the CD45+ population (as a marker for the leukocytes). On the one hand, cytotoxic T lymphocytes (CTLs) were gated in CD45+, CD3+, and CD8+ (CD3 as a marker for both cytotoxic T cell and T helper cells and CD8 as a marker for cytotoxic T cells).21 The subpopulation of CTLs was further analyzed in central memory T cells (TCM, CD62L+CD44+) and effector memory T cells (TEM, CD62L−CD44+) to explore the main subpopulation in inhibiting cancer cells. On the other hand, helper T cells were gated in CD45+, CD3+, and CD4+, while the regulatory T cells (Treg) were gated in CD3+, CD4+, and Foxp3+ (CD4 as a marker for T helper cells and Foxp3+ as a marker for the regulatory T cells). Immunohistochemistry was used to evaluate the T cell subpopulation (CD3+, CD4+ and CD8+) and distribution of these subpopulations in the tumor area of mice treated with CCMP@R837. 21 days after the 4T1 injection, three mice were randomly selected from each group, and the tumor size was qualitatively investigated using small animal in vivo optical imaging.22Statistical analysis
To compare differences between groups, one-way ANOVA with Tukey's multiple comparison test was used (IBM SPSS Statistics 25). *p < 0.05, **p < 0.01 and ***p < 0.001 indicate statistical significance. Values are expressed as mean ± SD.Results and discussion
Physical characteristics of CCMP@R837
In this study, we fabricated PLGA NPs loading R837 as an adjuvant with 4T1 CCM as antigens, denoted as CCMP@R837. PLGA NPs loading R837 (denoted as P@R837) and naked PLGA NPs (denoted as P) were used as the control. To investigate whether the CCM coating was successful, we first characterized the morphology of the prepared NPs. CCMP@R837 showed a core–shell structure (Fig. 1a), where the dark part was the P@R837 core and the light coating was the CCM. We then examined the diameter and zeta potential of different NPs.23 The DLS (Fig. 1b) and ELS (Fig. 1c) results indicated that the average diameter of CCMP@R837 (164.2 ± 5.9 nm) was larger than that of P@R837 (122.4 ± 4.2 nm) due to the presence of CCM coating. In addition, the zeta potential increased from −40.6 ± 1.1 mV for P@R837 to −20.4 ± 2.6 mV for CCMP@R837 due to the presence of the CCM (−19.5 ± 1.1 mV) on the surface of P@R837. Furthermore, we checked the protein content of CCMP@R837 in comparison with the cell membrane by the micro BCA protein assay, and we found that every 1 mg of CCMP@R837 included 0.6 mg of proteins. In addition, it was found that after 28 days of storage at 4 °C, the diameter of CCMP@R837 exhibited negligible change, indicating the long-term stability of these NPs (Fig. 1d). We then investigated the drug release from CCMP@R837 (Fig. 1e). We first investigated the loading efficacy of R837 in CCMP@R837 and it was found to be 7.3 ± 1.1%. We then found that after the first 24 hours of immersion in phosphate-buffered saline (PBS), over 50% of loaded R837 was released, and after 72 hours, almost all R837 (93.2 ± 3.45%) was released. The above data indicated that CCMP@R837 was successfully synthesized.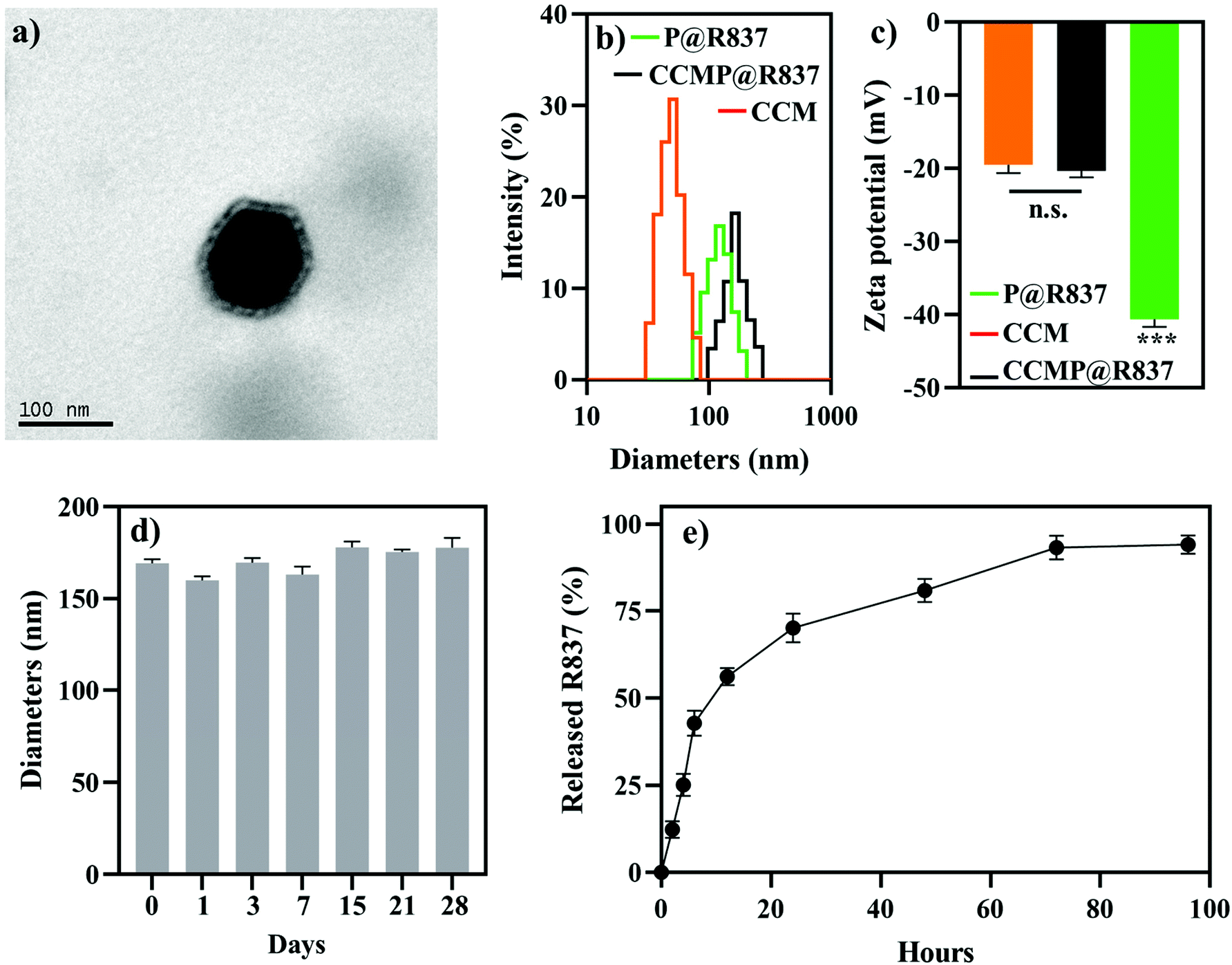 | ||
| Fig. 1 Physical characteristics of CCMP@R837. (a) Morphology of the resultant CCMP@R837 NPs. (b) Hydrodynamic diameter of CCM, P@R837, and CCMP@R837. (c) Surface zeta potential of CCM, P@R837, and CCMP@R837 (d) Hydrodynamic diameter of CCMP@R837 at 4 °C in 1× PBS for 28 days. (e) R837 release from CCMP@R837 in vitro. Data represent mean ± SD (n = 3). Please note that there is no significant difference between P@R837 and CCM, but a significant difference between CCMP@R837 and P@R837/CCM groups was observed (***p < 0.001). | ||
Biocompatibility and cellular uptake of CCMP@R837
After the successful synthesis of CCMP@R837, we examined the biocompatibility of these NPs using BMDCs and 4T1 cells. BMDC was selected as a model immune cell to evaluate the biocompatibility of the system, while 4T1 cells were used to investigate whether the system was cytotoxic to cancer cells. As shown in Fig. S-1a,† BMDCs treated with various formulations exhibited similar cell viability (around 100% compared with the negative control groups, NC, culture medium) except for the positive control (PC) groups treated with 1% Triton X-100 (the cell viability in the PC groups was around 10%). A similar trend was observed in the 4T1 cells treated with different formulations (Fig. S-1b†). These results suggested that the CCM, P@R837, and CCMP@R837 were not cytotoxic for both BMDCs and 4T1 cells in vitro. We further evaluated the BMDCs’ uptake of these NPs using flow cytometry (Fig. S-1c†). From 1 to 24 hours, an enhanced cellular uptake was found in the BMDCs treated with CCMP@R837 compared with P@R837, suggesting that the presence of the CCM can enhance the BMDC uptake. Additionally, comparing the uptake percentage of P and P@R837, more uptake percentage could be found in P@R837, which could be attributed to the immunogenic properties of R837.Immunogenicity of CCMP@R837 in vitro
After the cellular uptake investigations, the efficacy of CCMP@R837 boosting the CD86+ and CD80+ expression in BMDCs (indicating BMDC activation) was evaluated in vitro using flow cytometry. As shown in Fig. 2a, after 1 day of incubation, there was no significant maturation difference between the BMDCs treated with formulations containing R837 or not, whereas, after 2 days of incubation, BMDCs treated with CCMP@R837 exhibited a higher maturation percentage (81.7 ± 2.2% in CD86+, 78.9 ± 2.5% in CD80+) than those treated with R@837 (67.8 ± 5.5% in CD86+, 52.7 ± 3.7% in CD80+) and P@R837 (64.2 ± 4.0% in CD86+, 55.4 ± 4.8% in CD80+). Similarly, after three days of incubation, BMDCs treated with CCMP@R837 showed a much higher maturation percentage (88.9 ± 2.5% in CD86+, 80.0 ± 4.5% in CD80+) than those treated with R@837 (69.5 ± 4.2% in CD86+, 67.9 ± 6.7% in CD80+) and P@R837 (75.2 ± 4.1% in CD86+, 68.7 ± 3.9% in CD80+). The increased maturation percentage in BMDCs treated with CCMP@R837 indicated that the co-delivery of antigens and the adjuvant within the same nanocarriers could significantly boost BMDC activation, which is vital for immunogenicity.16,24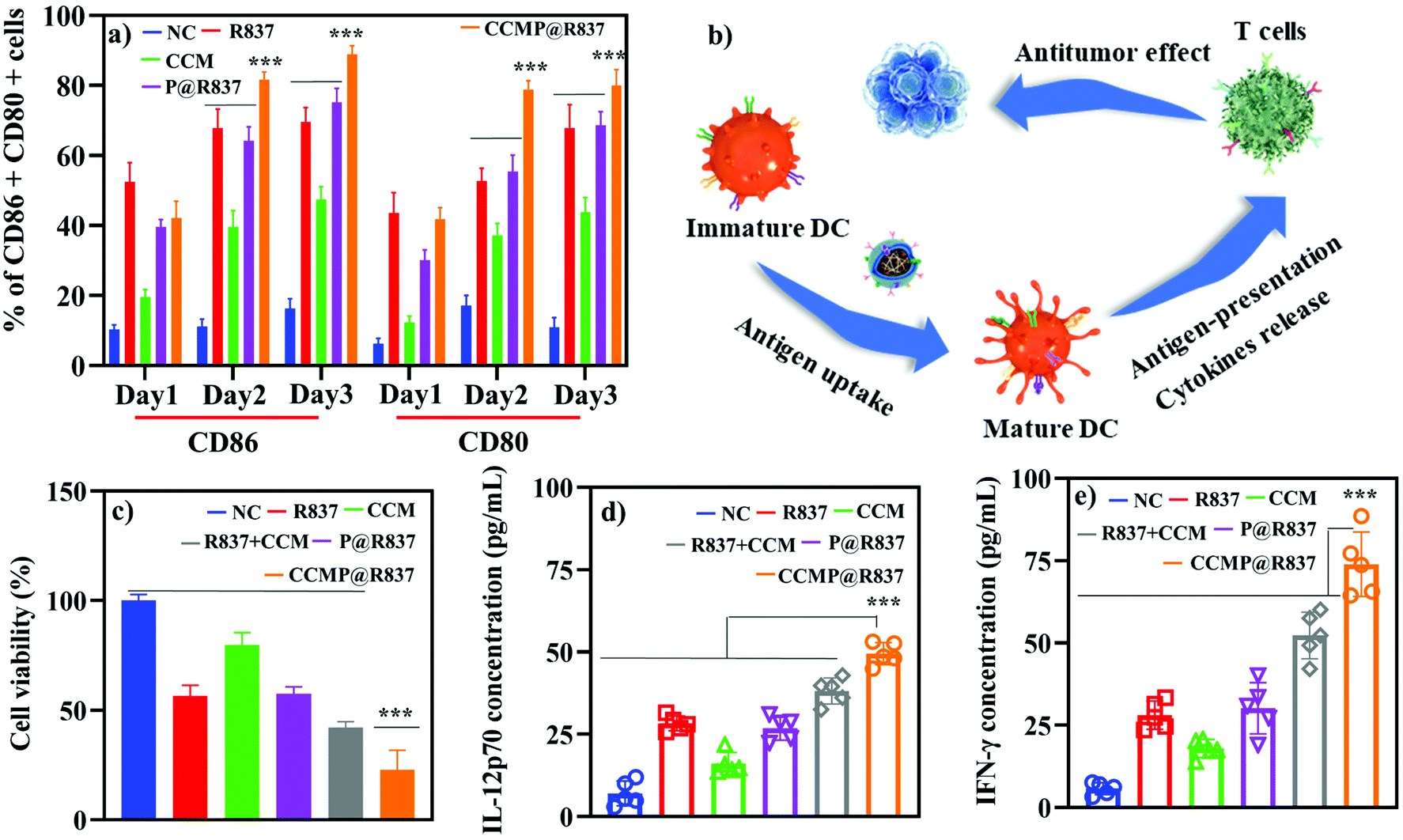 | ||
| Fig. 2 In vitro immunogenicity of CCMP@R837. (a) Maturation (CD86+ and CD80+ positive BMDCs) of BMDCs treated with different formulations. (b) Mechanism of in vitro antitumor effect of CCMP@R837. (c) Effect of CCMP@R837 on 4T1 cell viability. (d and e) Effect of CCMP@R837 on IL-12p70 (d) and IFN-γ (e) secretion. Data represent mean ± SD (n = 5; ***p < 0.001). | ||
To verify whether CCMP@R837 could trigger enhanced immune response, the BMDCs were treated with different formulations for inducing BMDC activation and antigen processing. As demonstrated in Fig. 2b, specifically, BMDCs were treated with the mixture of CCM and R837 as a control to explore whether CCMP@R837 would induce a stronger immune response than the control. BMDCs cultured only with the medium were used as NC. After three days of treatment, BMDCs were co-cultured with splenocytes for another three days to activate the T cells in the splenocytes; then both the BMDCs and the splenocytes were further co-cultured with 4T1 cells to investigate whether the induced T cells can kill 4T1 cancer cells. We then collected the suspension (medium of 4T1 cultured with BMDCs and splenocytes) to explore the mechanism underlying the inhibition of cancer cell growth. As shown in Fig. 2c, the viability of 4T1 cells treated with NC groups was regarded as 100%. We found that the 4T1 cells treated with CCMP@R837 exhibited the lowest cell viability compared to R837 + CCM groups (mixture of R837 and CCM), indicating that the co-delivery of antigens (from the cell membrane) and the adjuvant can enhance the immune response, with a higher concentration of the secreted cytokines IL-12p70 and IFN-γ (Fig. 2d and e). As reported, IL-12p70 activates CD8+ T cells to generate IFN-γ to induce apoptosis in cancer cells.25 According to the promising results in vitro, we further investigated the immunogenicity of CCMP@R837 in vivo.
Immunogenicity of CCMP@R837 in vivo
For the in vivo experiments, we first investigated the biocompatibility of CCMP@R837 in healthy BALB/c mice. We vaccinated the BALB/c mice three times on day 0, 14 and 28 and recorded their body weight (Fig. S-2†). Mice treated with different formulations exhibited a similar trend in body weight (increasing body weight with time), indicating the long-term biocompatibility of CCMP@R837. After 31 days of immunization, 4T1 cells were subcutaneously injected into the right flank of these mice (Fig. 3a). We found that the tumor size in mice treated with saline, CCM and P@R837 dramatically increased after 14 days of injection, whereas for the mice treated with CCMP@R837, a much less increase in the tumor size was observed, indicating the significant suppression effect of CCMP@R837 on tumor progression (Fig. 3b–e). The average tumor sizes of the mice injected with saline, CCM and P@R837 were 6, 4.6 and 3 times higher than those of the CCMP@R837-immunized mice (Fig. 3f). Additionally, we can clearly see from the survival rate results that after 23 days of treatment, there was the first death in saline, CCM and P@R837 groups due to the aggressive tumor progression; the first death in the CCMP@R837 group, however, happened 27 days after the 4T1 tumor cell injection. Additionally, the majority of mice in saline, CCM and P@R837 groups died between 20 days and 40 days after the treatment. Moreover, for the CCMP@R837 group, a 70% survival rate was observed after 50 days of treatment, with an average tumor size of 379 ± 103 mm3. The above results have demonstrated the strong inhibition effect on tumor progression in the mice immunized with CCMP@R837.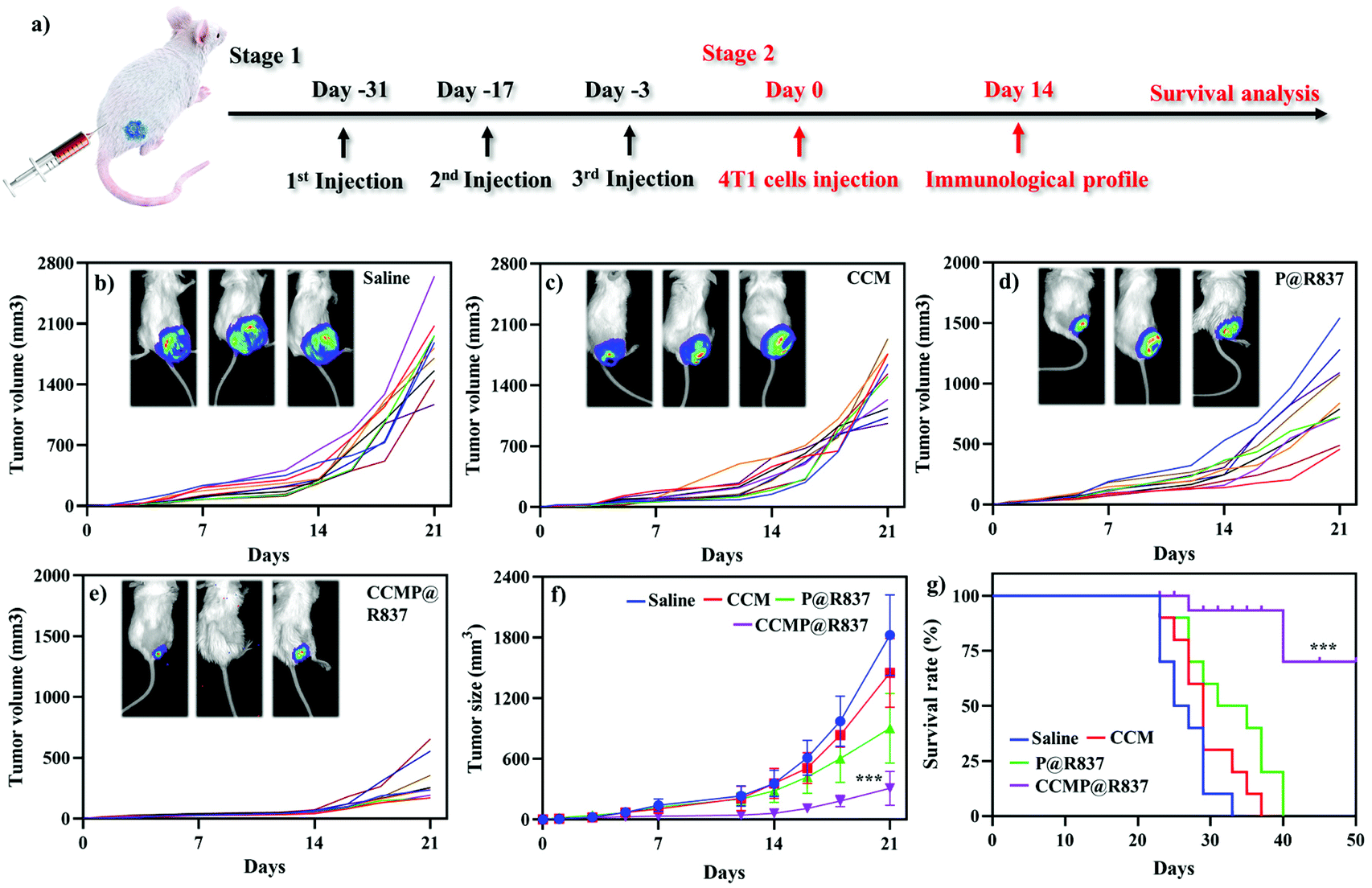 | ||
| Fig. 3 Antitumor effect of CCMP@R837 in vivo. (a) Treatment plan for BALB/c mice. Individual tumor size in mice treated with (b) saline, (c) CCM, (d) P@R837, and (e) CCMP@R837. Every single line indicated individual BALB/c mice. (f) Average tumor size of mice treated with various treatments. *** means the difference between CCMP@R836 groups and other groups at 21 days. (g) Survival rate of BALB/c treated with different formulations. *** means the difference between CCMP@R836 groups and other groups at 50 days. Data represent mean ± SD (n = 5; ***p < 0.001). | ||
We further explored the underlying mechanism of the increasing survival rate and the inhibition of tumor growth in mice treated with CCMP@R837. Five mice were randomly selected from each group; the spleen and tumor were collected for immunological proliferation and investigation at 14 days post 4T1 cell injection. In this experiment, the serum was isolated from mice, and the concentration of TNF-α in the serum was investigated. TNF-α produced by CD8+ CTLs is responsible for cancer cell elimination.26 As shown in Fig. S-3,† the TNF-α concentration in mice treated with CCMP@R837 was found to be 3, 1.5, and 4 times higher than those treated with CCM, P@R837 and silane. As known, CTLs play an important role in cleaning tumor cells by triggering the lysis of tumor cells. Thus, in the tumor tissue, the high percentage of CTLs indicates a strong anti-tumor immune response for inhibition of tumor growth.27 As shown in Fig. 4a, a significant population shift can be observed in the CTLs (positive expression of CD3+ and CD8+; CD3+ is involved in activating both the cytotoxic T cells and T helper cells; CD8+ is involved in the cytotoxic T cells) of the CCMP@R837 group. The percentage of CTLs in the CCMP@R837 group was found to be 1.9, 1.4, and 1.7 times higher than those in saline, P@R837 and CCM groups (Fig. 4b). In the tumor area, regulatory T cells (Tregs) are immunosuppressive and generally downregulate the proliferation of effector T cells.28 Therefore, the Tregs (CD4+, Foxp3+) were investigated to evaluate the immunosuppression in BALB/c mice immunized with different formulations (Fig. 4c). Saline, CCM and P@R837 groups revealed 1.7, 1.5, and 1.2 times higher percentage of Tregs in the tumor than the CCMP@R837 group, respectively (Fig. 4d), which indicated that the BALB/c mice pre-immunized with CCMP@R837 have created a less immunosuppressive microenvironment than those treated with saline, CCM and P@R837 groups.29 We additionally examined the T cells’ distribution in the tumor. As shown in Fig. 4e–g, plenty of CD3+, CD4+, and CD8+ T cells were found in the tumor of BALB/c mice treated with CCMP@R837, revealing the strong cell-mediated immune responses.
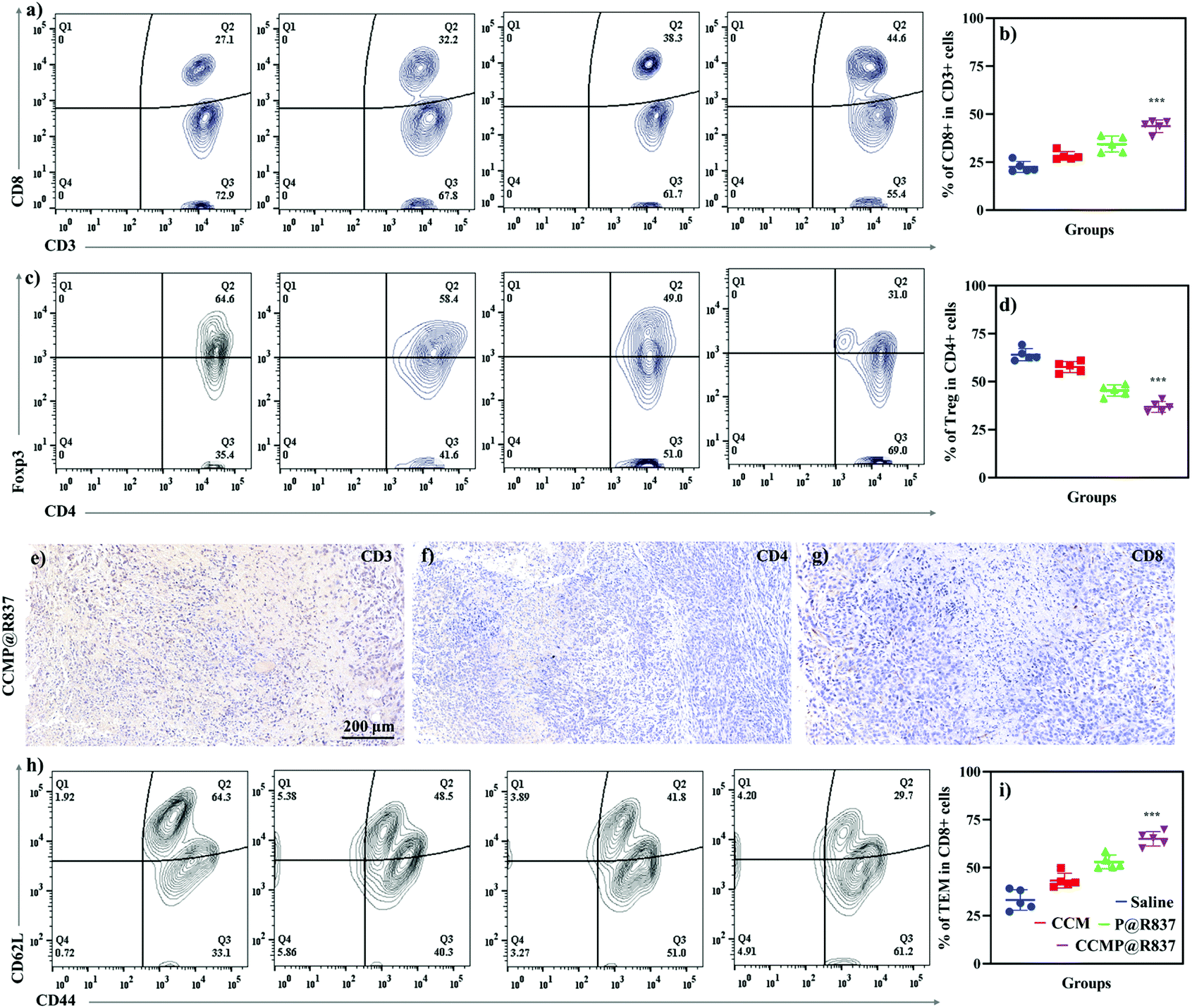 | ||
| Fig. 4 Immunogenicity of CCMP@R837 in vivo. (a) Flow cytometry results of the tumor cell suspension in CD3+CD8+ T cells. (b) Quantitative analysis of CD3+CD8+ T cells. (c) Flow cytometry results of the tumor cell suspension in CD4+Foxp3+ T cells. (d) Quantitative analysis of CD4+Foxp3+ T cells. The presence of (e) CD3+ T cells, (f) CD4+ T cells, and (g) CD8+ T cells in tumor tissue in mice treated with CCMP@R837. (h) Flow cytometry results of the spleen cell suspension in CD44+CD62L− T cells. (i) Quantitative analysis of CD44+CD62L− T cells. Data represent mean ± SD (n = 5; ***p < 0.001). | ||
The ability of CCMP@R837 to induce immune memory was further evaluated. After activation and antigen processing, CD8+ T cells partially differentiate into memory T cells, which can be further divided into central memory T cells (TCM) and effector T cells (TEM). In terms of functionality, TEM can quickly play the role of fighting against tumor cells. As for the TCM, it will be expanded when it is re-exposed to the cognate antigen.30 In this study, the spleen was collected to investigate the immunological profile in immunized BALB/c mice (Fig. 4h). We can see that the mice treated with CCMP@R837 showed a more significant shift of population from TCM to TEM compared to the mice treated with saline, CCM and P@R837, indicating activation of the anti-tumor effect. The higher number of TEM in the spleen of CCMP@R837-immunized mice demonstrated that the majority of T cells were activated to fight against tumor cells (Fig. 4i). Overall, the increased survival rate and the inhibited tumor growth in mice treated with CCMP@R837 could mostly be attributed to the presence of a higher percentage of CD8+ T cells and a lower percentage of Tregs in the tumor area. In addition, the presence of TEM in the spleen of mice treated with CCMP@R837 also contributes to the inhibition of tumor growth. That is, our CCMP@R837 has enhanced the differentiation of CD8+ T cells and TEM and inhibited the differentiation of Tregs, which ultimately facilitated the fight of immunized mice against tumors.
Conclusions
In this work, we fabricated nanovaccines by combining PLGA, R837, and CCM. We demonstrated that our CCMP@R837 can effectively enhance immunity and induce the generation of immune response to fight against breast cancer challenge. Compared with other pre-immunization treatments, such as saline, CCM and P@R837, our CCMP@R837 successfully trained the immune system of mice to be prepared for the 4T1 tumor challenge by boosting the maturation of antigen-presenting cells, promoting the differentiation of CTLs and downregulating the precentage of Tregs in the tumor. The upregulated percentage of TEM also plays a vital role in inhibiting tumor progression thanks to the full immune activation and resultant immune memory using our platform. Additionally, when the pre-immunized animals are challenged with 4T1 cancer cells, the TCM and TEM cells can quickly differentiate into CTLs to recognize and destroy cancer cells, with long-lasting anti-tumor immunity. For future study, it would be interesting to combine our platform with other therapy methods, such as photothermal therapy and irreversible electroporation, to further enhance the therapeutic efficency.Author contributions
L. Xiao, Y. Huang: methodology; Y. Yang: writing – original draft; Z. Miao: validation; J. Zhu, W. Tang: data curation; M. Zhong, C. Feng: formal analysis; J. Zhou: resources; L. Wang: writing – reviewing and editing; X. Zhao, Z. Wang: supervision.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
X. Zhao would like to acknowledge the General Research Fund (Grant no. 15202119) from the Research Grants Council of Hong Kong and the intra-faculty fund (P0013913) from the Hong Kong Polytechnic University. Z. R. Wang would like to acknowledge the financial support from the National Nature Science Foundation of China (82074473) and the Natural Science Foundation of Jiangsu Province (BK20191201).References
- J. Y. Zhu, D. W. Zheng, M. K. Zhang, W. Y. Yu, W. X. Qiu, J. J. Hu, J. Feng and X. Z. Zhang, Nano Lett., 2016, 16, 5895–5901 CrossRef CAS.
- Y. Min, K. C. Roche, S. Tian, M. J. Eblan, K. P. McKinnon, J. M. Caster, S. Chai, L. E. Herring, L. Zhang and T. Zhang, Nat. Nanotechnol., 2017, 12, 877–882 CrossRef CAS.
- A. V. Kroll, R. H. Fang, Y. Jiang, J. Zhou, X. Wei, C. L. Yu, J. Gao, B. T. Luk, D. Dehaini and W. Gao, Adv. Mater., 2017, 29, 1703969 CrossRef.
- D. J. Schwartzentruber, D. H. Lawson, J. M. Richards, R. M. Conry, D. M. Miller, J. Treisman, F. Gailani, L. Riley, K. Conlon and B. Pockaj, N. Engl. J. Med., 2011, 364, 2119–2127 CrossRef CAS.
- Q. Chen, L. Xu, C. Liang, C. Wang, R. Peng and Z. Liu, Nat. Commun., 2016, 7, 1–13 Search PubMed.
- R. H. Fang, C. M. J. Hu, B. T. Luk, W. Gao, J. A. Copp, Y. Tai, D. E. O'Connor and L. Zhang, Nano Lett., 2014, 14, 2181–2188 CrossRef CAS.
- Y. Guo, D. Wang, Q. Song, T. Wu, X. Zhuang, Y. Bao, M. Kong, Y. Qi, S. Tan and Z. Zhang, ACS Nano, 2015, 9, 6918–6933 CrossRef CAS.
- R. Kuai, L. J. Ochyl, K. S. Bahjat, A. Schwendeman and J. J. Moon, Nat. Mater., 2017, 16, 489–496 CrossRef CAS.
- Y. Ye, C. Wang, X. Zhang, Q. Hu, Y. Zhang, Q. Liu, D. Wen, J. Milligan, A. Bellotti and L. Huang, Sci. Immunol., 2017, 2, eaan5692 CrossRef.
- C. Wang, Y. Ye, Q. Hu, A. Bellotti and Z. Gu, Adv. Mater., 2017, 29, 1606036 CrossRef.
- C. M. J. Hu, L. Zhang, S. Aryal, C. Cheung, R. H. Fang and L. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10980–10985 CrossRef CAS.
- C. M. J. Hu, R. H. Fang, B. T. Luk, K. N. Chen, C. Carpenter, W. Gao, K. Zhang and L. Zhang, Nanoscale, 2013, 5, 2664–2668 RSC.
- J. Blonder, K. C. Chan, H. J. Issaq and T. D. Veenstra, Nat. Protoc., 2006, 1, 2784–2790 CrossRef CAS.
- W. L. Liu, M. Z. Zou, T. Liu, J. Y. Zeng, X. Li, W. Y. Yu, C. X. Li, J. J. Ye, W. Song and J. Feng, Nat. Commun., 2019, 10, 1–12 CrossRef.
- H. Sun, J. Su, Q. Meng, Q. Yin, L. Chen, W. Gu, Z. Zhang, H. Yu, P. Zhang and S. Wang, Adv. Funct. Mater., 2017, 27, 1604300 CrossRef.
- Y. Jiang, N. Krishnan, J. Zhou, S. Chekuri, X. Wei, A. V. Kroll, C. L. Yu, Y. Duan, W. Gao, R. H. Fang and L. Zhang, Adv. Mater., 2020, 32, 2001808 CrossRef CAS.
- R. Yang, J. Xu, L. Xu, X. Sun, Q. Chen, Y. Zhao, R. Peng and Z. Liu, ACS Nano, 2018, 12, 5121–5129 CrossRef CAS.
- S. P. Kasturi, I. Skountzou, R. A. Albrecht, D. Koutsonanos, T. Hua, H. I. Nakaya, R. Ravindran, S. Stewart, M. Alam and M. Kwissa, Nature, 2011, 470, 543–547 CrossRef CAS.
- A. Madaan, R. Verma, A. T. Singh, S. K. Jain and M. Jaggi, J. Biol. Methods, 2014, 1, e1 CrossRef.
- S. Y. Li, H. Cheng, B. R. Xie, W. X. Qiu, J. Y. Zeng, C. X. Li, S. S. Wan, L. Zhang, W. L. Liu and X. Z. Zhang, ACS Nano, 2017, 11, 7006–7018 CrossRef CAS.
- J. A. Ledbetter, J. P. Deans, A. Aruffo, L. S. Grosmaire, S. B. Kanner, J. B. Bolen and G. L. Schieven, Curr. Opin. Immunol., 1993, 5, 334–340 CrossRef CAS.
- L. Miao, J. Li, Q. Liu, R. Feng, M. Das, C. M. Lin, T. J. Goodwin, O. Dorosheva, R. Liu and L. Huang, ACS Nano, 2017, 11, 8690–8706 CrossRef CAS.
- F. Fontana, M. Fusciello, C. Groeneveldt, C. Capasso, J. Chiaro, S. Feola, Z. Liu, E. M. Mäkilä, J. J. Salonen and J. T. Hirvonen, ACS Nano, 2019, 13, 6477–6490 CrossRef CAS.
- A. S. Cheung, S. T. Koshy, A. G. Stafford, M. M. Bastings and D. J. Mooney, Small, 2016, 12, 2321–2333 CrossRef CAS.
- L. Escribà-Garcia, C. Alvarez-Fernández, M. Tellez-Gabriel, J. Sierra and J. Briones, J. Transl. Med., 2017, 15, 115 CrossRef.
- A. Ratner and W. R. Clark, J. Immunol., 1993, 150, 4303–4314 CAS.
- Y. Cao, Y. H. Feng, L. W. Gao, X. Y. Li, Q. X. Jin, Y. Y. Wang, Y. Y. Xu, F. Jin, S. L. Lu and M. J. Wei, Int. Immunopharmacol., 2019, 70, 110–116 CrossRef CAS.
- T. J. Curiel, J. Clin. Invest., 2007, 117, 1167–1174 CrossRef CAS.
- D. H. Munn and V. Bronte, Curr. Opin. Immunol., 2016, 39, 1–6 CrossRef CAS.
- M. Pepper and M. K. Jenkins, Nat. Immunol., 2011, 12, 467–471 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr08978h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |