
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Toward the creation of high-performance heterogeneous catalysts by controlled ligand desorption from atomically precise metal nanoclusters
Tokuhisa
Kawawaki
 abc,
Yuki
Kataoka
a,
Momoko
Hirata
a,
Yuki
Iwamatsu
a,
Sakiat
Hossain
a and
Yuichi
Negishi
*abc
abc,
Yuki
Kataoka
a,
Momoko
Hirata
a,
Yuki
Iwamatsu
a,
Sakiat
Hossain
a and
Yuichi
Negishi
*abc
aDepartment of Applied Chemistry, Faculty of Science, Tokyo University of Science, Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: negishi@rs.tus.ac.jp
bPhotocatalysis International Research Center, Tokyo University of Science, 2641 Yamazaki, Noda, Chiba 278-8510, Japan
cResearch Institute for Science and Technology, Tokyo University of Science, Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan
First published on 12th April 2021
Abstract
Ligand-protected metal nanoclusters controlled by atomic accuracy (i. e. atomically precise metal NCs) have recently attracted considerable attention as active sites in heterogeneous catalysts. Using these atomically precise metal NCs, it becomes possible to create novel heterogeneous catalysts based on a size-specific electronic/geometrical structure of metal NCs and understand the mechanism of the catalytic reaction easily. However, to create high-performance heterogeneous catalysts using atomically precise metal NCs, it is often necessary to remove the ligands from the metal NCs. This review summarizes previous studies on the creation of heterogeneous catalysts using atomically precise metal NCs while focusing on the calcination as a ligand-elimination method. Through this summary, we intend to share state-of-art techniques and knowledge on (1) experimental conditions suitable for creating high-performance heterogeneous catalysts (e.g., support type, metal NC type, ligand type, and calcination temperature), (2) the mechanism of calcination, and (3) the mechanism of catalytic reaction over the created heterogeneous catalyst. We also discuss (4) issues that should be addressed in the future toward the creation of high-performance heterogeneous catalysts using atomically precise metal NCs. The knowledge and issues described in this review are expected to lead to clear design guidelines for the creation of novel heterogeneous catalysts.
 Tokuhisa Kawawaki | Tokuhisa Kawawaki Assistant Professor of the Department of Applied Chemistry at Tokyo University of Science. He received a PhD degree (2015) in Applied Chemistry from the University of Tokyo. In 2016–2017, he worked as a Japan Society for the Promotion of Science (JSPS) Postdoctoral fellow (PD) at the University of Melbourne. In 2017–2018, he worked as a JSPS super PD (SPD) at Kyoto University. In 2019, he moved to his current position. His current research topics include the synthesis of metal nanoparticles and nanoclusters in solution and their applications for photoelectrochemistry and photocatalysts. |
 Yuki Kataoka | Yuki Kataoka Master course student in the Negishi group at Tokyo University of Science. He received a MSc (2021) in Chemistry from Tokyo University of Science. His research interests include the activation of water-splitting photocatalysts by heteroatom doping of cocatalysts and elucidation of calcination process for loading precise metal NCs on photocatalysts. |
 Momoko Hirata | Momoko Hirata Master course student in the Negishi group at Tokyo University of Science. She received a BSc (2020) in Chemistry from Tokyo University of Science. Her research interests include the elucidation of calcination process for loading precise metal NCs on photocatalysts. |
 Yuki Iwamatsu | Yuki Iwamatsu Master course student in the Negishi group at Tokyo University of Science. He received a BSc (2020) in Chemistry from Tokyo University of Science. His research interests include the electrochemical application of precise metal NCs. |
 Sakiat Hossain | Sakiat Hossain Assistant Professor at Tokyo University of Science. He obtained his BSc (2005) from Ramakrishna Mission Residential College, Narendrapur, Calcutta University, MSc (2007) from the Indian Institute of Technology Delhi, and PhD (2013) from the Indian Institute of Technology Kanpur under the supervision of Prof. V. Chandrasekhar. He joined Tokyo University of Science in 2015. His research interests include the synthesis of novel metal clusters and study of their properties. |
 Yuichi Negishi | Yuichi Negishi Professor of the Department of Applied Chemistry at Tokyo University of Science. He received a PhD degree in Chemistry in 2001 under the supervision of Prof. Atsushi Nakajima from Keio University. Before joining Tokyo University of Science in 2008, he was employed as an Assistant Professor at Keio University and at the Institute for Molecular Science. His current research interests include the precise synthesis of stable and functionalized metal nanoclusters and their applications in energy and environmental materials. |
1. Introduction
1.1. Catalysts
Modern affluent society is supported by a wide variety of substances. Most of those are produced by chemical reactions, and most of the chemical reactions are promoted by catalysts. Although the number of elements that can be handled stably on the earth is limited, catalysts can produce a wide variety of useful chemical substances from this limited number of elements and therefore are indispensable materials in our society. Researchers have developed various catalysts one after another to meet the demands of society, leading to the establishment of a fulfilling life and helping to overcome food shortages and reduce environmental pollution.1 However, our society still has many problems related to resources, energy, and the environment. Therefore, future studies are expected to develop novel catalysts that will address these problems facing modern society.The concept of catalysis was first presented in 1836. Berzelius described in his paper that “catalysis is the acceleration of a slow chemical reaction by the presence of a foreign substance”.2 Then, in 1901, Ostwald defined a catalyst as “any substance which alters the rate of a chemical reaction without appearing in the final product”.3 This definition also clearly distinguished between “chemical equilibrium” and “reaction rate”. Thus, a catalyst is a substance that can change a chemical reaction without being consumed. In reality, although the catalyst does not change before and after the chemical reaction, it forms a reaction intermediate with the reactant during catalysis. Therefore, in modern chemistry, the catalyst is defined as “a substance that changes the reaction rate in a chemical reaction but does not appear in the general reaction formula and does not change the equilibrium constant, reduces the apparent activation energy by creating a new reaction pathway, and allows a specific reaction among several possible reactions to proceed selectively”.4
A catalyst contained in the same phase as the reactant is called a homogeneous catalyst. For example, molecular catalysts,5 such as metal complexes used when synthesizing pharmaceuticals and organic compounds in a solvent, are classified as homogeneous catalysts. These catalysts can generally be designed at the molecular level, which allows the desired reaction to proceed with high selectivity. However, because all of the substances related to the reaction are dissolved in a solvent, it is often difficult to separate products and recover the catalyst. Furthermore, in such a reaction system, the activity is generally not so high because the reaction temperature is limited to below the boiling point of the solvent.
A catalyst with a phase different from that of the reactant and product, such as a solid-state catalyst6,7 (metal/metal oxide nanoparticles loaded on a support), is called a heterogeneous catalyst.8 Generally, heterogeneous catalysts are used in large-scale industrial processes. In this reaction system, the reaction temperature can be raised to the heat-resistant temperature of solid-state catalysts such that high activity is often obtained. In addition, it is relatively easy to separate products and recycle the catalysts in this reaction system, which is a great advantage in industrial processes. However, the surfaces of heterogeneous catalysts are not always homogeneous, and it is difficult to design and control them at the molecular level. For this reason, reactions using heterogeneous catalysts often have low selectivity.
In the development of catalysts, it is very important to account for the activity, selectivity, durability/stability, and environmental burden.9 Understanding the reaction mechanism is also essential for improving the activity and selectivity. Thus, it is necessary to develop many catalysts for which these factors are improved.
1.2. Atomically precise metal nanoclusters as catalysts
Ligand-protected metal nanoclusters controlled by atomic accuracy (i. e. atomically precise metal NCs)10–46 have attracted considerable attention as novel homogeneous and heterogeneous catalysts. Research on the synthesis and determination of geometrical structure of these atomically precise metal NCs began in the 1970s, and initially gold (Au) NCs protected by phosphine (PR3) and halogen (X) (Aun(PR3)mXl NCs; n = number of Au atoms; m and l = number of PR3 and X, respectively)47–58 and platinum (Pt) and palladium (Pd) NCs protected by PR3 and/or carbon monoxide (CO) (Mn(PR3)m(CO)l; M = Pt or Pd)59–67 were studied as the main target. However, since the precise synthesis of thiolate (SR)-protected Au NCs (Aun(SR)m NCs),68 which show higher stability than the above-described metal NCs, was achieved in 2005, SR-protected Au,10–17,19,21,26,33,36,41,42 silver (Ag),10,18,20,22,43 copper (Cu)31,32 and their alloy25,27–30,34,35,37–40,44 NCs have become the main research focus. In addition, numerous studies have been conducted on metal NCs with alkyne (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR),73–76 selenolate,77–84 and tellurolate33 as ligands in recent years. For the synthesis and electronic/geometrical structure of these metal NCs, we point the interested reader to a number of reviews published in the last few years by several groups, including ours.23,24,85–95
CR),73–76 selenolate,77–84 and tellurolate33 as ligands in recent years. For the synthesis and electronic/geometrical structure of these metal NCs, we point the interested reader to a number of reviews published in the last few years by several groups, including ours.23,24,85–95
Regarding these atomically precise metal NCs, in 2010, Jin et al. found that Au25(PET)18 (PET = phenylethanethiolate) can function as either a homogeneous catalyst (sometimes called quasi-homogeneous; Fig. 1A) or a heterogeneous catalyst (Fig. 1B–D) for the selective hydrogenation of benzalacetone by hydrogen (H2).96 Since then, the catalytic reaction using atomically precise metal NCs has attracted much attention. As described in Section 1.1, heterogeneous catalysts have an advantage in the recycling. In addition, the use of support typically results in the improvements of the catalytic activity; e.g. the catalytic activity of Au99(SPh)42 (SPh = phenylthiolate) for the hydrogenation of 4-nitrobenzaldehyde to 4-nitrobenzyl alcohol was reported to be improved tenfold by supporting them on cerium(IV) oxide (CeO2).97 Thus, the recent studies have particularly focused on the use of atomically precise metal NCs in a heterogeneous catalyst.
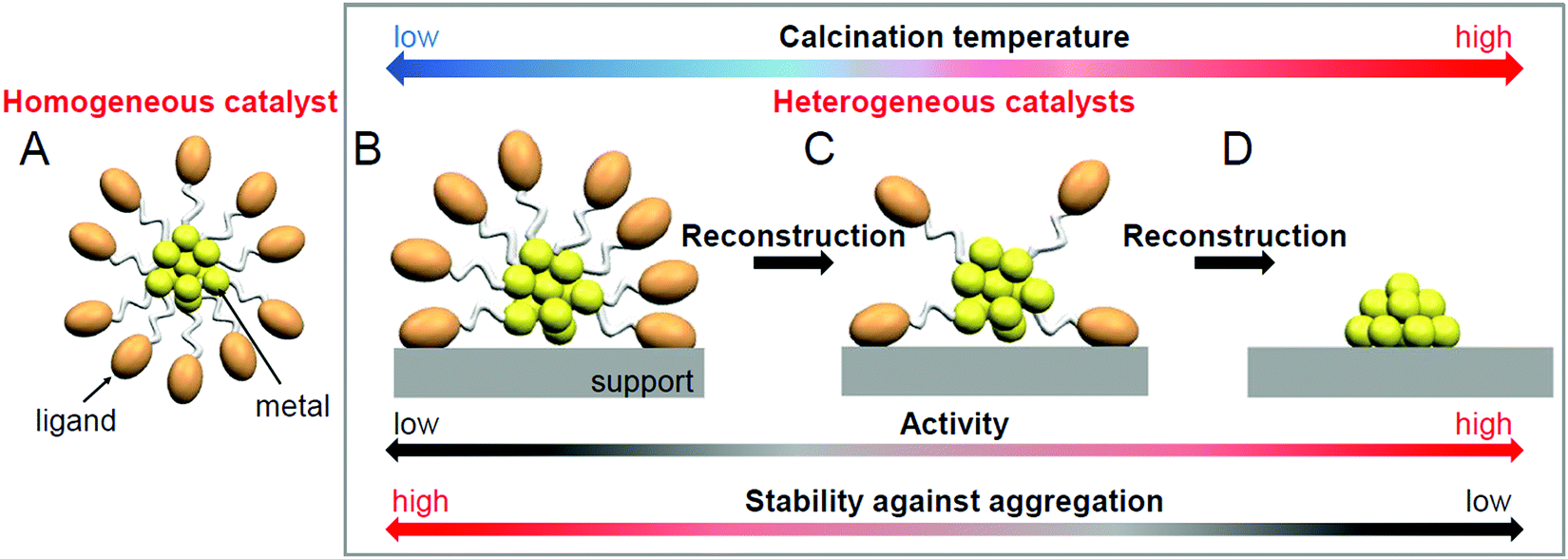 | ||
| Fig. 1 Categorization of catalysts: (A) homogeneous catalyst (sometimes called quasi-homogeneous catalyst) and (B–D) heterogeneous catalysts. This review discusses heterogeneous catalysts prepared using atomically precise, ligand-protected metal NCs (B–D). Note that calcination induces not only the ligand elimination but also the reconstruction of the metal-core structure. | ||
Atomically precise metal NCs are attracting attention as heterogeneous catalysts for the following reasons:
(1) In a conventional heterogeneous catalyst, metal nanoparticles (NPs) on a support have a relatively large size distribution. For such a system, it is difficult to identify highly active particles and selectively load them. In contrast, for heterogeneous catalysts loaded with atomically precise metal NCs, metal NCs with an atomically defined chemical composition are loaded such that highly active metal NCs can be identified and thereby selectively loaded onto supports.
(2) Ligand-protected metal NCs exhibit different physicochemical properties and functions from bulk metals, and these properties and functions change depending on the number of constituent atoms and the type of ligand.86,98–120 Therefore, it is possible to create a new heterogeneous catalyst with a unique catalytic activity when using these metal NCs.
(3) In recent years, it has become possible to synthesize various types of atomically precise metal NCs.18,22–24,31–33,69–84 Therefore, it is possible to create a heterogeneous catalyst suitable for the desired reaction based on the design, and it is expected that high reaction selectivity could be achieved.
(4) For atomically precise metal NCs, it is often possible to determine the geometrical structure through single-crystal X-ray diffraction (SC-XRD) analysis.26,28–30,37,39,40,98–100,121–180 Therefore, for heterogeneous catalysts using atomically precise metal NCs, it is possible to obtain a deep understanding of the correlation between the geometrical/interface structure of catalysts and catalytic activity and the reaction mechanism.
As described above, the use of atomically precise metal NCs as catalysts has many advantages for both the development of practical catalysts and the understanding of the mechanism of the catalytic reaction. Therefore, it seems possible to create a heterogeneous catalyst, in which the above-described four factors (activity, selectivity, durability/stability, and environmental burden) are improved using this catalyst system. However, the presence of ligands on the surface of the metal NCs interrupts the approach of the reactant to the surface of the metal NCs, which often leads to a decrease in catalytic activity (Fig. 1B). Therefore, many attempts have been made to create heterogeneous catalysts with even higher activity by removing part (Fig. 1C) or all (Fig. 1D) of the ligands.24,181,182 For example, in 2009, Tsukuda et al. succeeded in preparing a heterogeneous catalyst with both high activity and selectivity by removing all the ligands of triphenylphosphine (PPh3)-protected Au11 NCs.183 In this way, it is expected that a heterogeneous catalyst having even higher activity than that containing the ligands will be created when an appropriate amount of ligands is removed from the metal NCs.
1.3. Purpose and contents of this review
As mentioned in Section 1.2, ligand elimination induces activation of catalysts. However, ligand elimination also induces reconstruction and aggregation of metal NCs (Fig. 1B–D). When aggregation occurs, the catalytic activity decreases as the proportion of surface atoms in the loaded metal NCs decreases. Therefore, in a ligand elimination, it is extremely important to select suitable conditions. The conditions appear to vary depending on the type of support, the metal NCs, the ligands, and the catalytic reaction. In addition, if the desorbed ligands from metal NCs are adsorbed on the support, they will also affect the catalytic activity. Indeed, Burns et al. reported that nickel-oxide supported-Au NPs decreased the catalytic activity when the sulfate ions existed on the support.184 Thus, to create a highly active heterogeneous catalyst, it is necessary to deeply understand the mechanism of the ligand elimination. However, as the number of studies on the creation of highly functional heterogeneous catalysts based on such a ligand elimination has increased rapidly in recent years, it is difficult for researchers (especially beginners) to extract all of the knowledge gained from previous studies.This review summarizes representative previous studies on the preparation of heterogeneous catalysts using atomically precise metal NCs while focusing on calcination as a ligand-elimination method. The purpose of this review is to discuss existing knowledge on (1) the experimental conditions suitable for the creation of highly functional heterogeneous catalysts (types of support, metal NCs, and ligands and calcination temperature), (2) the calcination mechanism, and (3) the mechanism of catalytic reaction over the created heterogeneous catalyst.
In the next section (Section 2), we describe previous studies while classifying them by the catalytic reaction. Then, we describe the experimental conditions suitable for the creation of high-performance heterogeneous catalysts (Section 3.1), calcination mechanisms (Section 3.2), and the mechanism of the catalytic reaction over the prepared heterogeneous catalyst (Section 3.3). After a brief summary in Section 4, the future outlook is described in Section 5.
For metal NCs whose constituent atoms are not controlled with atomic precision, the creation of heterogeneous catalysts using calcination has been well studied over many years.185–200 However, this review summarizes the previous studies while focusing on examples using atomically precise metal NCs, the study of which only started in recent years. In addition, the ligands of metal NCs can be removed not only by calcination but also by light irradiation,201–203 oxidation,204–207 or reduction208 treatment. However, we do not touch on such methods because of space limitations. For readers interested in the elimination of ligands by methods other than calcination, it is better to read other reviews or original papers.
2. Examples of activation of atomically precise metal nanoclusters
In this section, we summarize representative studies on the functionalization of heterogeneous catalysts based on ligand elimination while classifying the studies by the catalytic reaction. The geometrical structures of the atomically precise metal NCs used in these studies are shown in Fig. 2. In addition, the metal NCs and supports used in each study are summarized in Table 1,128,178,180,183,200,209–247 and the characteristics of each support are summarized in Table 2.183,184,198,209,211,212,214,220,234,248–258 For readers interested in the details of the experimental conditions in each catalytic reaction, it is better to read the original paper. In the following, for example, Aun NCs loaded on a metal oxide are represented as Aun/metal oxide.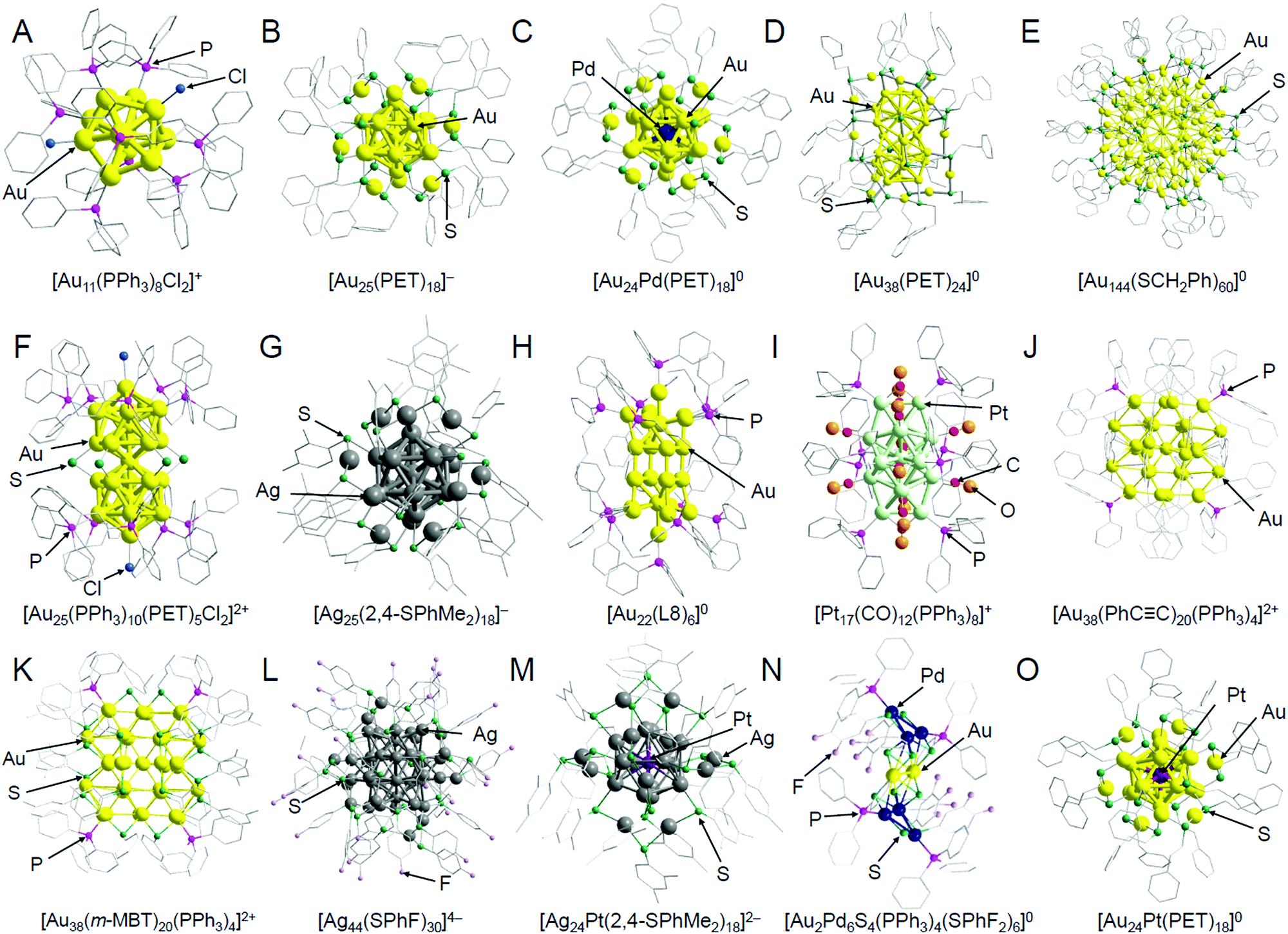 | ||
| Fig. 2 Geometrical structures of atomically precise, ligand-protected metal NCs: (A) [Au11(PPh3)8Cl2]+,171 (B) [Au25(PET)18]−,42 (C) [Au24Pd(PET)18]0,172 (D) [Au38(PET)24]0,173 (E) [Au144(SCH2Ph)60]0,174 (F) [Au25(PPh3)10(PET)5Cl2]2+,175 (G) [Ag25(2,4-SPhMe2)18]−,43 (H) [Au22(L8)6]0,176 (I) [Pt17(CO)12(PPh3)8]+,177 (J) [Au38(PhCC)20(PPh3)4]2+,178 (K) [Au38(m-MBT)20(PPh3)4]2+,178 (L) [Ag44(SPhF)30]4−,44 (M) [Ag24Pt(2,4-SPhMe2)18]2−,179 (N) [Au2Pd6S4(PPh3)4(SPhF2)6]0,180 and (O) [Au24Pt(PET)18]0.26 The gray points indicate carbon atoms, and hydrogen atoms are not shown. Au25(SR)18 (SR = SC12 or SG) discussed in Sections 2.1.1, 2.1.2, and 2.4 is considered to have a similar geometrical structure as [Au25(PET)18]− (B). Au24Pd(SC12)18 discussed in Section 2.1.1 is considered to have a similar geometrical structure as [Au24Pd(PET)18]0 (C). Au38(SC12)24 discussed in Section 2.1.4 is considered to have a similar geometrical structure as [Au38(PET)24]0 (D). Au144(PET)60 discussed in Sections 2.1.1–2.1.4 is considered to have a similar geometrical structure as [Au144(SCH2Ph)60]0 (E). These graphics were reproduced with permission from ref. 26, 42–44 and 171–180. Copyright 2016 Royal Society of Chemistry, 2008 American Chemical Society, 2015 American Chemical Society, 2013 Springer-Nature, 2014 American Chemical Society, 2016 American Chemical Society, 2010 American Chemical Society, 2018 AAAS, 2007 American Chemical Society, 2014 American Chemical Society, 2017 American Chemical Society, 2017 AAAS, 2017 American Chemical Society, 2018 Royal Society of Chemistry, respectively. | ||
| Reaction | NCsa | Support | Metal weight | Calc. atmosphere | Calc. temp./°C | Particle size/nm | Ref.b |
|---|---|---|---|---|---|---|---|
| a Counter ion is omitted. b Reference number. c No information. | |||||||
| Oxidation of benzyl alcohol | Au11(PPh3)8Cl2 | SBA-15 | 0.4 wt% | Vacuum | 200 | 0.8 ± 0.3 | 183 |
| Au25(SC12)18 | MWCNTs | 0.05–1.0 wt% | Vacuum | 300 | 1.1 ± 0.2 | 209 | |
| 450 | 1.2 ± 0.2 | ||||||
| Au24Pd(SC12)18 | 300 | 1.2 ± 0.3 | |||||
| 450 | 1.2 ± 0.3 | ||||||
| Au25(SC12)18 | HPCSs | 1.0 wt% | Vacuum | 500 | 1.2 ± 0.3 | 210 | |
| Au25(PET)18 | HPCSs | 1.0 wt% | Vacuum | 550 | 1.0 ± 0.2 | 211 | |
| Au38(PET)24 | 1.2 ± 0.3 | ||||||
| Au144(PET)60 | 1.6 ± 0.1 | ||||||
| Au∼330(PET)∼80 | 2.0 ± 0.2 | ||||||
| Au13Cu8(PPh2Py)12 | CNTs | —c | Vacuum | 370 | —c | 128 | |
| Au25(SPh-pNH2)17 | SBA-15 | 0.04 wt% | Air | 400 | 1.9 ± 0.6 | 212 | |
| 1.07 wt% | 1.8 ± 0.5 | ||||||
| Oxidation of glucose | Au38(PET)24 | AC | 1.38 wt% | Air | 120 | ∼1.6 | 213 |
| Aerobic oxidation of cyclohexane | Au10(SG)10 | HAP | 0.2 wt% | Vacuum | 300 | 1.0 ± 0.4 | 214 |
| Au18(SG)14 | 1.0 ± 0.4 | ||||||
| Au25(SG)18 | 1.1 ± 0.5 | ||||||
| Au39(SG)24 | 1.1 ± 0.5 | ||||||
| Au38(PET)24 | CeO2 or Al2O3 | 2 wt% | Air | 250 or 350 | —c | 215 | |
| Au38(PET)24 | CeO2 | 0.5 wt% | Air | 150 | ∼1 | 216 | |
| 250 | |||||||
| Al2O3 | 150 | 2–3 | |||||
| 250 | |||||||
| Au38(PET)24 | CeO2 | 0.5 wt% | Air | 250 | ∼1 | 217 | |
| Al2O3 | 2–3 | ||||||
| Au25(PET)18 | TiO2 | 0.54 wt% | Air | 150 | 1.1 ± 0.3 | 218 | |
| 250 | —c | ||||||
| SiO2 | 0.34 wt% | 150 | 1.2 ± 0.6 | ||||
| 250 | —c | ||||||
| Au144(PET)60 | TiO2 | 0.36 wt% | 150 | 1.6 ± 0.6 | |||
| 250 | —c | ||||||
| SiO2 | 0.25 wt% | 150 | 1.7 ± 0.4 | ||||
| 250 | —c | ||||||
| Oxidation of styrene | Au25(SG)18 | HAP | 0.5 wt% | Vacuum | 300 | 1.4 ± 0.6 | 219 |
| Au25(PET)18 | SiO2 or HAP | 1 wt% | Vacuum | 200 | —c | 220 | |
| Au38(PET)24 | |||||||
| Au144(PET)60 | |||||||
| Au25(PET)18 | CeO2 | 1 wt% | Vacuum | 260 | —c | 221 | |
| Au25(PPh3)10(PET)5Cl2 | |||||||
| Au25(PET)18 | SiO2 | ∼1 wt% | Vacuum | 200 | 3–4 | 222 | |
| Au25(PPh3)10(PET)5Cl2 | |||||||
| Au25(MHA)18 | HAP | 1 wt% | N2 | 300 | 2.4 ± 1.3 | 223 | |
| TiO2 | 2.0 ± 0.8 | ||||||
| AC | 4.0 ± 5.0 | ||||||
| PGO | 3.8 ± 3.1 | ||||||
| SiO2 | 5.0 ± 3.9 | ||||||
| Ag25(2,4-SPhMe2)18 | AC | 4 wt% | Air | 250 | 1.2 ± 0.3 | 224 | |
| 350 | 2.6 ± 0.4 | ||||||
| 450 | 2.8 ± 0.6 | ||||||
| Oxidation of CO | Au38(SC12)24 | TiO2 | 0.7 wt% | H2/He | 400 | 3.9 ± 1.0 | 225 |
| Au25(PET)18 | TiO2 | 1 wt% | O2 | 150 | —c | 226 | |
| Fe2O3 | 2 wt% | ||||||
| CeO2 | |||||||
| Au38(PET)24 | CeO2 | 1 wt% | O2 | 100–250 | —c | 227 | |
| Au144(PET)60 | CeO2 | 1 wt% | O2/CO or H2 | 80 | —c | 228 | |
| Au25(PET)18 | CeO2 | 0.66 wt% | O2 | 150 | —c | 229 | |
| Au25(1SNap)18 | 0.63 wt% | ||||||
| Au25(2SNap)18 | 0.63 wt% | ||||||
| Au36(S-c-C5H9)24 | 0.74 wt% | ||||||
| Au36(SPh)24 | 0.72 wt% | ||||||
| Au36(SPh-tBu)24 | 0.64 wt% | ||||||
| Au36(2SNap)24 | 0.64 wt% | ||||||
| Au38(PET)24 | 0.69 wt% | ||||||
| Au38(SPh)24 | 0.73 wt% | ||||||
| Au38(o-MBT)24 | 0.71 wt% | ||||||
| Au28(S-c-C6H11)20 | CeO2 | 0.32 μmol | O2 | 150 or 300 | —c | 230 | |
| Au28(TBBT)20 | —c | ||||||
| Au25(PET)18 | CeO2 | ∼1 wt% | O2 | 150–350 | —c | 231 | |
| Au22(L8)6 | TiO2, SiO2, or CeO2 | ∼0.5 wt% | O2 | 22–400 | —c | 232 | |
| Au38(PET)24 | CeO2 | 2 wt% | O2 | 150 or 250 | 2–4 | 233 | |
| Au25(PET)18 | Co3O4-EP-FDU-12 | 1.1–2 wt% | Air | 300 | —c | 234 | |
| Au144(PET)60 | CuO-EP-FDU-12 | 1.1 wt% | 1.7 ± 0.2 | ||||
| Pt17(CO)12(PPh3)8 | γ-Al2O3 | 0.15 wt% | Vacuum | 500 | 1.1 ± 0.2 | 235 | |
| Semihydrogenation of alkynes | Au25(PET)18 | TiO2, CeO2, SiO2, or Al2O3 | 1 wt% | Vacuum | 300 | —c | 236 |
| Au38(PhCC)20(Ph3P)4 |
TiO2 | 0.4 wt% | Vacuum | 130 | 1.4 | 178 | |
| Au38(m-MBT)20(Ph3P)4 | |||||||
| Reduction of 4-nitrophenol | Au25(PET)18 | CB | 0.3 wt% | Air | 125 | 1.5 ± 0.2 | 237 |
| 200 | 1.7 ± 0.8 | ||||||
| 250 | 1.9 ± 1.1 | ||||||
| 350 | 2.1 ± 0.9 | ||||||
| Reduction of nitrobenzene | Au25(MHA)18 | HAP | 1 wt% | N2 | 300 | 2.4 ± 1.3 | 223 |
| TiO2 | 2.0 ± 0.8 | ||||||
| AC | 4.0 ± 5.0 | ||||||
| PGO | 3.8 ± 3.1 | ||||||
| SiO2 | 5.0 ± 3.9 | ||||||
| Hydrogenation of 3-nitrostyren | Au25(Cys)18 | ZnAl-HT | 1.1 wt% | Air | 300 | 1.7 ± 0.5 | 238 |
| H2 generation | Ag44(SPhF)30 | MPC | 0.2 wt% | Vacuum | 150 and 300 | 1–2 | 239 |
| Photocatalytic HER and OER | Au25(SG)18 | BaLa4Ti4O15 | 0.1 wt% | Reduced pressure | 300 | 1.2 ± 0.3 | 240 |
| Au10(SG)10 | BaLa4Ti4O15 | 0.1 wt% | Reduced pressure | 300 | 0.9 ± 0.2 | 241 | |
| Au15(SG)13 | 1.0 ± 0.2 | ||||||
| Au18(SG)14 | 1.1 ± 0.2 | ||||||
| Au22(SG)16 | 1.5 ± 0.4 | ||||||
| Au25(SG)18 | 1.2 ± 0.3 | ||||||
| Au29(SG)20 | 1.6 ± 0.4 | ||||||
| Au33(SG)22 | 1.7 ± 0.5 | ||||||
| Au39(SG)24 | 1.5 ± 0.3 | ||||||
| Au25(SG)18 | SrTiO3 | 0.1 wt% | Reduced pressure | 300 | 1.0 ± 0.2 | 242 | |
| Au25(SG)18 | Cr2O3-BaLa4Ti4O15 | 0.1 wt% | Reduced pressure | 300 | 1.1 ± 0.2 | 243 | |
| Au25(PET)18 | BaLa4Ti4O15 | ∼0.1 wt% | Reduced pressure | 300 | —c | 244 | |
| Au24Pt(PET)18 | |||||||
| Rh2(SG)2 | BaLa4Ti4O15 | 0.09 wt% | Reduced pressure | 300 | 1.3 ± 0.3 | 245 | |
| Ag25(2,4-SPhMe2)18 | g-C3N4 | 0.2 wt% | Ar | 150 | 1.5 ± 0.3 | 200 | |
| Ag24Pt(2,4-SPhMe2)18 | |||||||
| Electrocatalytic HER and OER | Pd6(SC12)12 | AC | —c | Vacuum | 200 | —c | 246 |
| Au2Pd6S4(PPh3)4(SPhF2)6 | MoS2 | 0.05 wt% | Ar | 100 | —c | 180 | |
| Au25(PET)18 | CoSe2 | ∼0.2 mg cm−2 | N2 | 300 | —c | 247 | |
| Support | Surface area/m2 g−1 | Lewis acid site | Brønsted acid site |
|---|---|---|---|
| a Ref. 183. b Ref. 212. c Ref. 234. d Ref. 209. e Ref. 211. f Ref. 214. g Ref. 198. h Ref. 184. i Ref. 310. j Ref. 220. k No information. l Ref. 248. m Ref. 249. n Ref. 250. o Ref. 251. p Ref. 252. q Ref. 253. r Ref. 254. s Ref. 255. t Ref. 256. u Ref. 257. v Ref. 258. w General value. | |||
| SBA-15 (m-SiO2) | 866a | —k | —k |
| 570 ± 5b | |||
| 395.2c | |||
| MWCNTs | 250–300d | —k | Existr |
| HPCSs | 2300e | —k | —k |
| HAP | ∼100f | —k | —k |
| CeO2 | 65g | Exists | A little exists |
| Al2O3 | 172g | Exists | Absents |
| TiO2 | 48 (P-25)g | Exists | A little exists |
| 9.6 (anatase)h | |||
| 50 (anatase)i | |||
| 6.7 (rutile)h | |||
| SiO2 | 200 ± 25j | Exists | Absents |
| PGO | —k | —k | —k |
| AC | 250–2410lw | —k | —k |
| CB | —k | —k | —k |
| Fe2O3 | 10mw | A little existt | A little existt |
| γ-Al2O3 | 403nw | Existu | Absentu |
| ZnAl-HT | —k | —k | —k |
| BaLa4Ti4O15 | 6.1ow | —k | —k |
| SrTiO3 | 3–27pw | Existv | Absentv |
| g-C3N4 | 43–288qw | —k | —k |
| MoS2 | —k | —k | —k |
| CoSe2 | —k | —k | —k |
2.1. Oxidation reaction
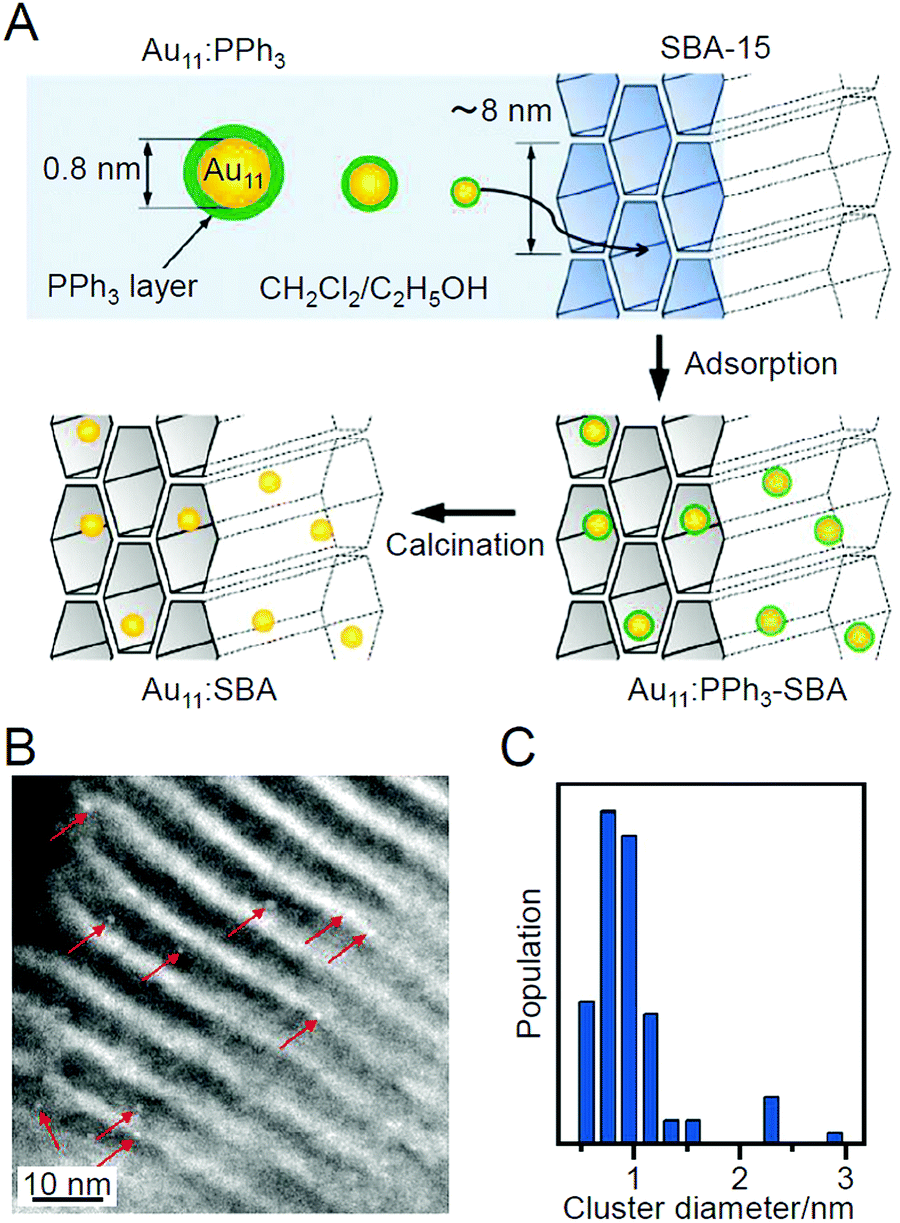 | ||
| Fig. 3 (A) Synthesis procedure of sub-nanometer-sized Au NCs within SBA-15 using PPh3-protected Au11 NCs as a precursor. (B) Representative HAADF-STEM image and (C) size distribution of Au NCs prepared by adsorption method. Reproduced with permission from ref. 183. Copyright 2009 American Chemical Society. | ||
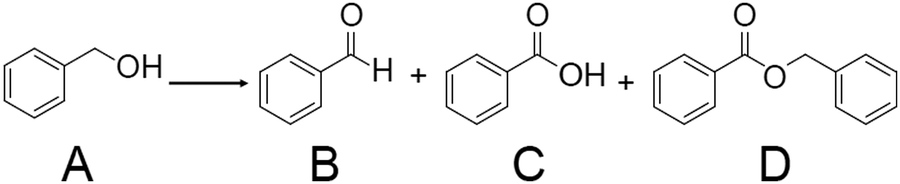 | ||
| Scheme 1 Benzyl alcohol oxidation using metal NC catalysts. Generally, the reaction is conducted in the presence of a base such as cesium carbonate (Cs2CO3) or potassium carbonate (K2CO3). In the literature, the reaction was conducted with H2O or toluene as a solvent under O2 or air atmosphere. In ref. 183, H2O2 was used as an oxidant. A = benzyl alcohol, B = benzaldehyde, C = benzoic acid, D = benzyl benzoate, respectively. | ||
In 2012, the same group studied the use of Au25(SC12)18 (Fig. 2B) and Au24Pd(SC12)18 (Fig. 2C; SC12 = 1-dodecanethiolate) as metal NCs with multi-walled carbon nanotubes (MWCNTs) as a support.209 In this study, the ligands were removed by calcination at 300 or 450 °C for 2 h under vacuum. In both cases, no aggregation was observed when the Au loading on the MWCNTs was less than 0.2 wt% (0.2 wt% Au). However, aggregation was clearly observed when the loading was greater than 0.2 wt% (Fig. 4A). This finding indicates that the optimization of the loading amount is very important to suppress the aggregation of the loaded metal NCs. The catalytic activity of the obtained catalysts (Au24M/MWCNTs; M = Au or Pd) for the oxidation of benzyl alcohol was investigated, and the results showed that the catalytic activity appeared only after ligand elimination for these catalysts. Moreover, in this study, it was shown that the oxidation reaction proceeds using atmospheric molecular O2 as an oxidant over these catalysts and that the catalytic activity of Au25/MWCNTs can be improved by nearly three times by simply replacing one Au atom with Pd (Fig. 4B).
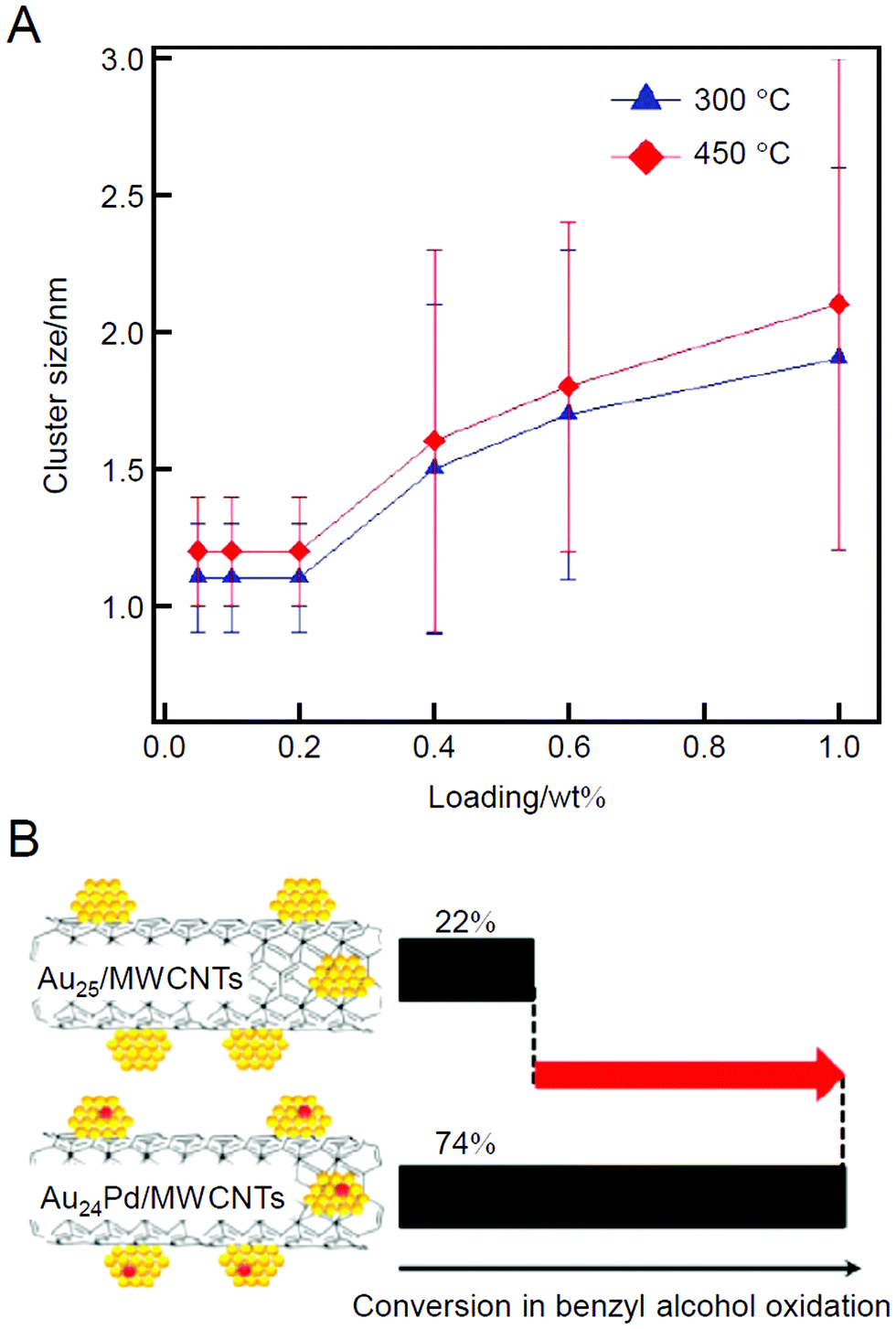 | ||
| Fig. 4 (A) Dependence of the loading amounts of Au25 NCs on the Au particle size in Au25/MWCNTs. The error bars indicate the size distribution determined by TEM. (B) Catalytic performances of Au25/MWCNTs and Au24Pd/MWCNTs for benzyl alcohol oxidation. Reproduced with permission from ref. 209. Copyright 2012 American Chemical Society. | ||
As described above, ligand elimination induces an increase in catalytic activity. In 2014, the same group reported that moderate amounts of the remaining ligands led to control of the reaction selectivity.210 In that study, Au25(SC12)18 (Fig. 2B) was used as the metal NCs and hierarchically porous carbon nanosheets (HPCSs; Fig. 5A) were used as the support. HPCSs have a larger specific surface area than MWCNTs (Table 2) and have more defects on their surface because of their porous structure. Therefore, almost no aggregation of Au25 was observed for Au25/HPCSs with 1.0 wt% Au even after calcination at 500 °C for 2 h. The study using the obtained catalysts revealed that (1) a part of the SRs could not be removed from the Au25 surface by the calcination below 500 °C and (2) the alkyl chains, once desorbed from the Au25 NC surfaces by S–C dissociation at low temperature, were adsorbed on the surfaces of the HPCSs. A study on the oxidation ability of benzyl alcohol for a series of catalysts prepared at different calcination temperatures revealed that (1) ligand elimination is necessary for the emergence of the catalytic activity of this catalyst, (2) the catalytic activity increases with the amount of ligand elimination, and (3) the catalyst with remaining partial ligands had the highest selectivity for the conversion to benzaldehyde (Scheme 1B) (Fig. 5B). This study was of great interest as it showed that the reaction selectivity can be controlled by controlling the amount of remaining ligands. In 2016, the same group used a series of Aun NCs (Au25, Au38, Au144, or Au∼330) to study the size dependence of Au NCs on this catalytic reaction, and the results revealed that Au144 shows the highest conversion efficiency.211
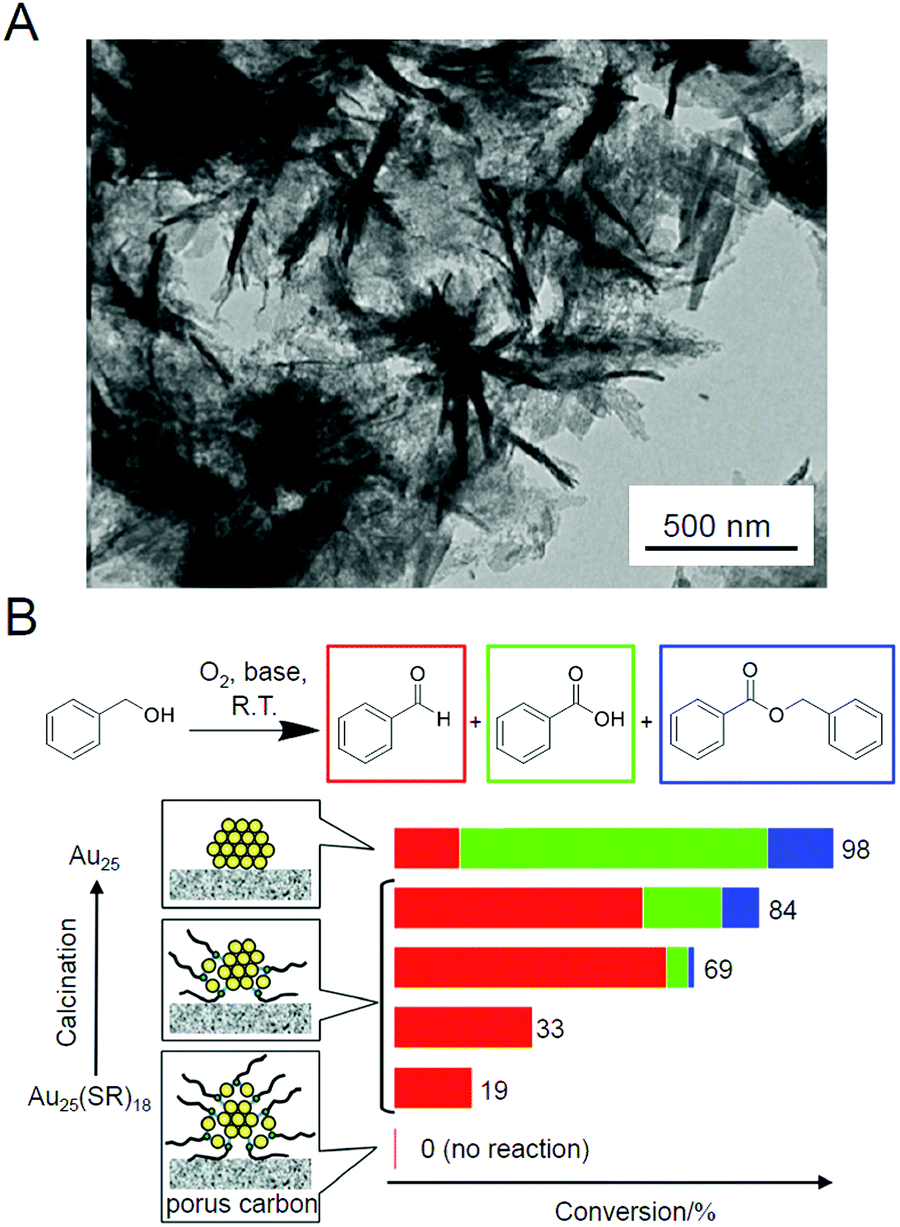 | ||
| Fig. 5 (A) TEM image of HPCSs. (B) Catalytic performance of Au25(SC12)18−x/HPCSs for aerobic oxidation of benzyl alcohol. Reproduced with permission from ref. 210. Copyright 2014 American Chemical Society. | ||
Regarding benzyl alcohol oxidation, Zheng et al. also reported results in 2013.128 In their study, the newly synthesized Au13Cu8(PPh2Py)12 (PPh2Py = diphenyl-2-pyridylphosphine) was loaded on CNTs, and then the catalysts were calcined at 370 °C under vacuum to prepare Au13Cu8/CNTs without ligand. The Au13Cu8/CNTs showed 46.9% conversion efficiency and 61.0% selectivity to benzaldehyde under O2 atmosphere at 80 °C. This catalyst resulted in a conversion efficiency of benzyl alcohol that was 33.2% higher than that for Au25/CNTs under the same reaction conditions, demonstrating the effectiveness of alloying in improving the conversion.
In addition to these studies, in 2015, Tuel et al. conducted a study using Au25(SPh-pNH2)17 (SPh-pNH2 = 4-aminothiophenolate)259 as a metal NC, whose geometrical structure has not been revealed to date, and SBA-15 as a support.212 On the basis of the TGA results of unsupported Au25(SPh-pNH2)17, they judged that calcination for 12 h at 400 °C was necessary to completely remove the ligands from this catalyst. In the TEM images of the catalysts with 0.04 or 1.07 wt% Au, particles with an average size of 1.8–1.9 nm were observed. Because this particle size is larger than that observed in the TEM image of Au25(SPh-pNH2)17 (0.8 ± 0.2 nm), it is assumed that the aggregation of Au NCs was not completely suppressed in this experiment. The authors investigated the catalytic activity of the catalysts for the conversion of benzyl alcohol to benzaldehyde (Scheme 1B). The results revealed that uncalcined Au25(SPh-pNH2)17/SBA-15 exhibited little catalytic activity for the oxidative dehydrogenation reaction using a strong base and that the catalytic activity increased continuously with the calcination temperature (Fig. 6). These findings indicate that it is crucial to eliminate ligand for proceeding this reaction (oxidative dehydrogenation reaction using a strong base). In contrast, for radical reactions using strong oxidants, uncalcined Au25(SPh-pNH2)17/SBA-15 showed catalytic activity; however, calcination led to a decrease in the catalytic activity. This result indicates that the effectiveness of ligand elimination depends on the proceeding reaction mechanism.
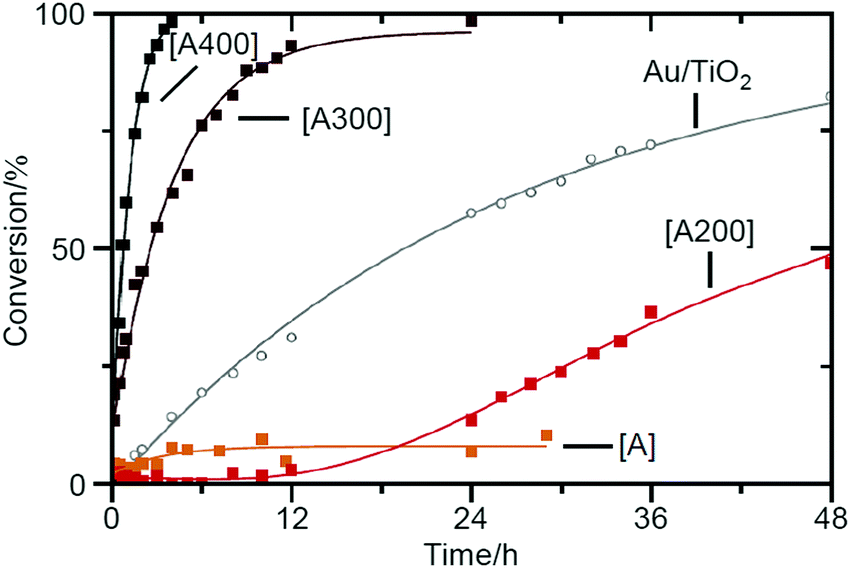 | ||
| Fig. 6 Kinetic survey of benzyl alcohol dehydrogenation over Au25/SBA-15 materials (1.07 wt% Au) as-synthesized [A] (orange) and calcined at 200 °C [A200] (red), 300 °C [A300] (brown), and 400 °C [A400] (black) and comparison data obtained for Au/TiO2 (gray) (reference from the World Gold Council). Reproduced with permission from ref. 212. Copyright 2015 Elsevier. | ||
As described above, benzyl alcohol is often used as a reactant for alcohol oxidation. Li and Huang et al. investigated glucose oxidation (Scheme 2) in 2017.213 The products, gluconic acid and its salts, are widely used as additives in foods, beverages, and pharmaceuticals. Currently, microbiota and Pd-bismuth (Bi) alloys are used as catalysts for the glucose oxidation; however, the catalysts must be produced with higher activity and fewer purification processes to save resources and energy and achieve low environmental impact. The authors found that calcination of Au38(PET)24/AC (Fig. 2D; AC = activated carbon) at 120 °C removes a part of the ligands, and the obtained catalyst (Au38/AC-120) showed higher catalytic activity than conventional catalysts such as Pd/AC, Pd-Bi/AC, and Au/AC (Fig. 7). For this reaction, the conversion efficiency decreased when all the ligands were removed from Au38(PET)24 by calcination at high temperature. In general, ligand removal induces structural change of metal NCs.244 This result implies that it is very important to maintain the geometrical and/or electronic structure of the precursor Au38(PET)24 for this catalytic reaction to proceed.
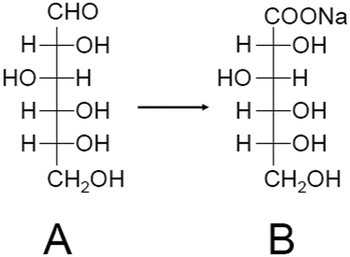 | ||
| Scheme 2 Glucose oxidation to gluconic acid using metal NC catalysts. The reaction was conducted under O2 atmosphere in NaOH solution kept at pH 9.5 in ref. 213. A = glucose, B = gluconic acid. | ||
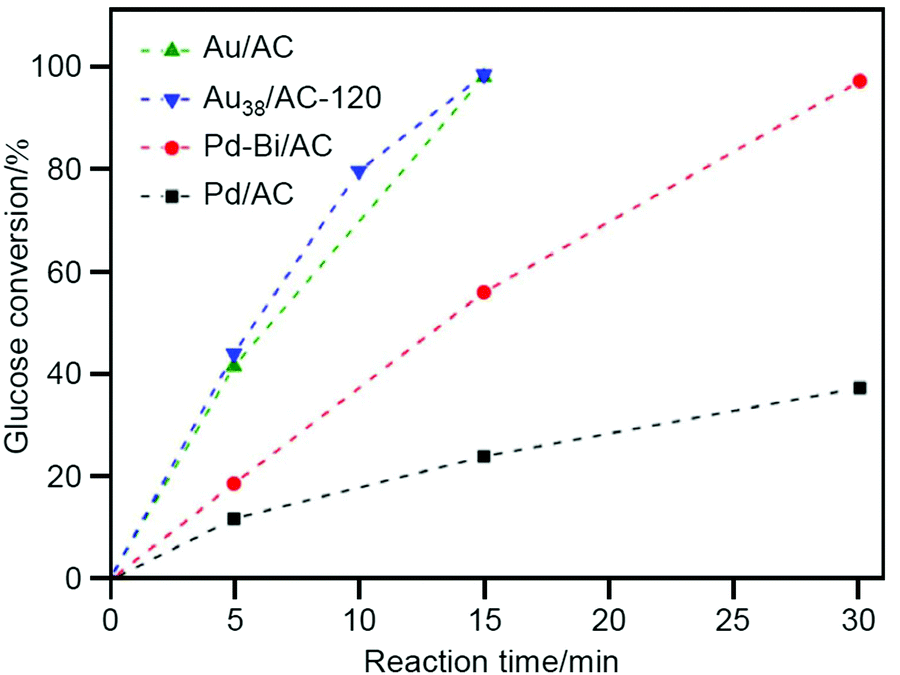 | ||
| Fig. 7 Glucose conversion vs. time over Pd/AC, Pd-Bi/AC, Au/AC, and Au38/AC-120. Reproduced with permission from ref. 213. Copyright 2017 Wiley-VCH. | ||
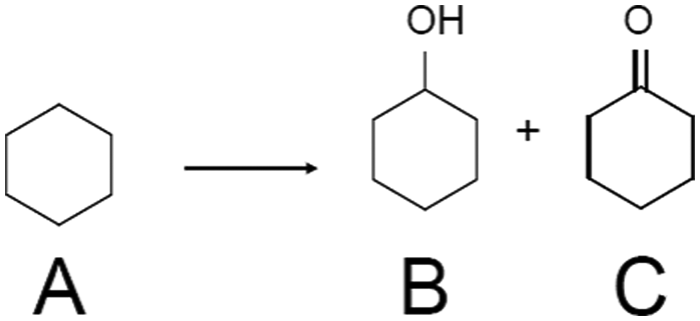 | ||
| Scheme 3 Cyclohexane oxidation using metal NC catalysts. In this reaction, TBHP was essential to initiate the reaction,259 suggesting that this oxidation proceeds via a complex radical chain mechanism.260,261 The reaction was conducted without solvent under O2 atmosphere in the literature.214,215,217,218 A = cyclohexane, B = cyclohexanol, C = cyclohexanone. Products B and C are called KA-oil. In ref. 214, cyclohexyl-t-butyl peroxide, which is an intermediate, and cyclohexanethiol were also produced (the latter was produced by the reaction between the reactant and the sulfur remaining on the support). | ||
In 2011, Tsukuda et al. prepared a Aun/HAP catalyst using Aun(SG)m (SG = glutathionate; n = 10, 18, 25, or 39) as metal NCs and hydroxyapatite (HAP) as a support.214 In this experiment, the ligands were removed by calcination under vacuum at 300 °C for 2 h. The complete elimination of the ligands was confirmed by inductively coupled plasma (ICP) analysis. In this study, the aggregation of Aun NCs on the support was suppressed by the following factors: (1) HAP with a large specific surface area (Table 2) was used as a support; (2) the loading amount was suppressed to 0.2 wt% Au; and (3) Aun(SG)m NCs were adsorbed in solution at pH where charge repulsion between the metal NCs was likely to occur. Each of the obtained Aun/HAP proceeded the oxidation reaction from cyclohexane to KA oil with a selectivity of more than 99%. A comparison of the catalytic activity of a series of the catalysts including Au∼85/HAP prepared by another method showed that Aun/HAP shows the highest conversion efficiency for cyclohexane oxidation in the n range of 40–80 (Fig. 8).
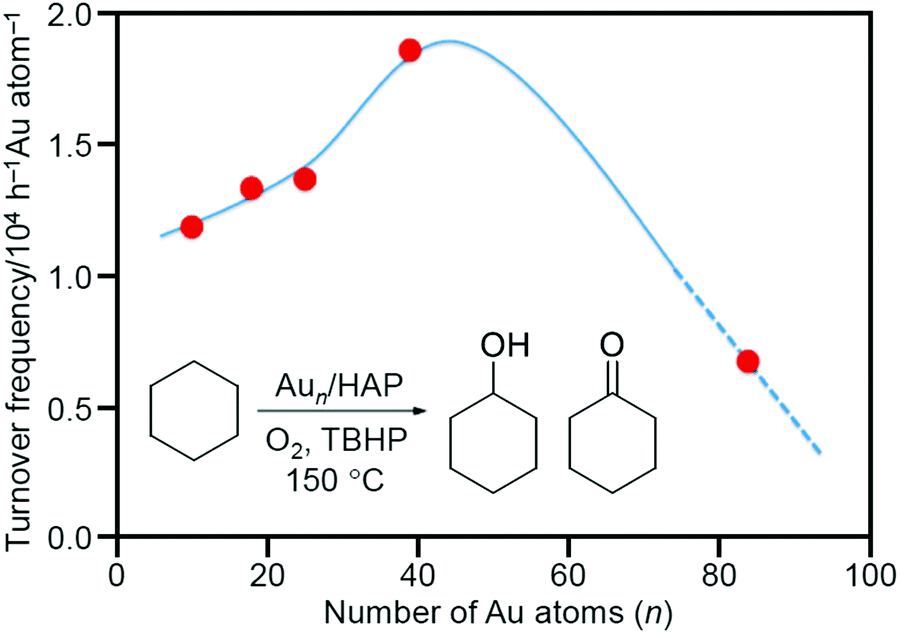 | ||
| Fig. 8 Turnover frequency values as a function of the number of Au atoms, n. The curve is a guide for the eye. Reproduced with permission from ref. 214. Copyright 2011 American Chemical Society. | ||
Based on the results obtained by Tsukuda et al., Barrabés and Bürgi et al. prepared Au38/CeO2 and Au38/Al2O3 (Al2O3 = aluminum oxide) using Au38(PET)24 (Fig. 2D) as a metal NC in 2015.215 They tracked the details of the calcination mechanism using TGA and temperature-programmed desorption/mass spectrometry (TPD/MS). Their results clarified that the SR-desorption process differed depending on the calcination atmosphere and the support. It was also shown that SR desorption occurred with two steps for both catalysts (Fig. 9A). X-ray absorption fine structure (XAFS) analysis revealed that ligands were removed at 250 °C for both catalysts. Both catalysts showed catalytic activity for the cyclohexane oxidation. Because the catalysts after calcination exhibited higher catalytic activity than those before calcination, the removal of the ligands was considered effective for improvement of the catalytic activity in these catalysts. Between the two, Au38/CeO2 showed higher activity. In Au38/CeO2, Au became cationic, which seems to induce high activity. In this study, cyclohexanethiol was also produced (Fig. 9B), suggesting that the loaded Au NCs also promoted the reaction between the cyclohexane and residual SR (or sulfur (S)).
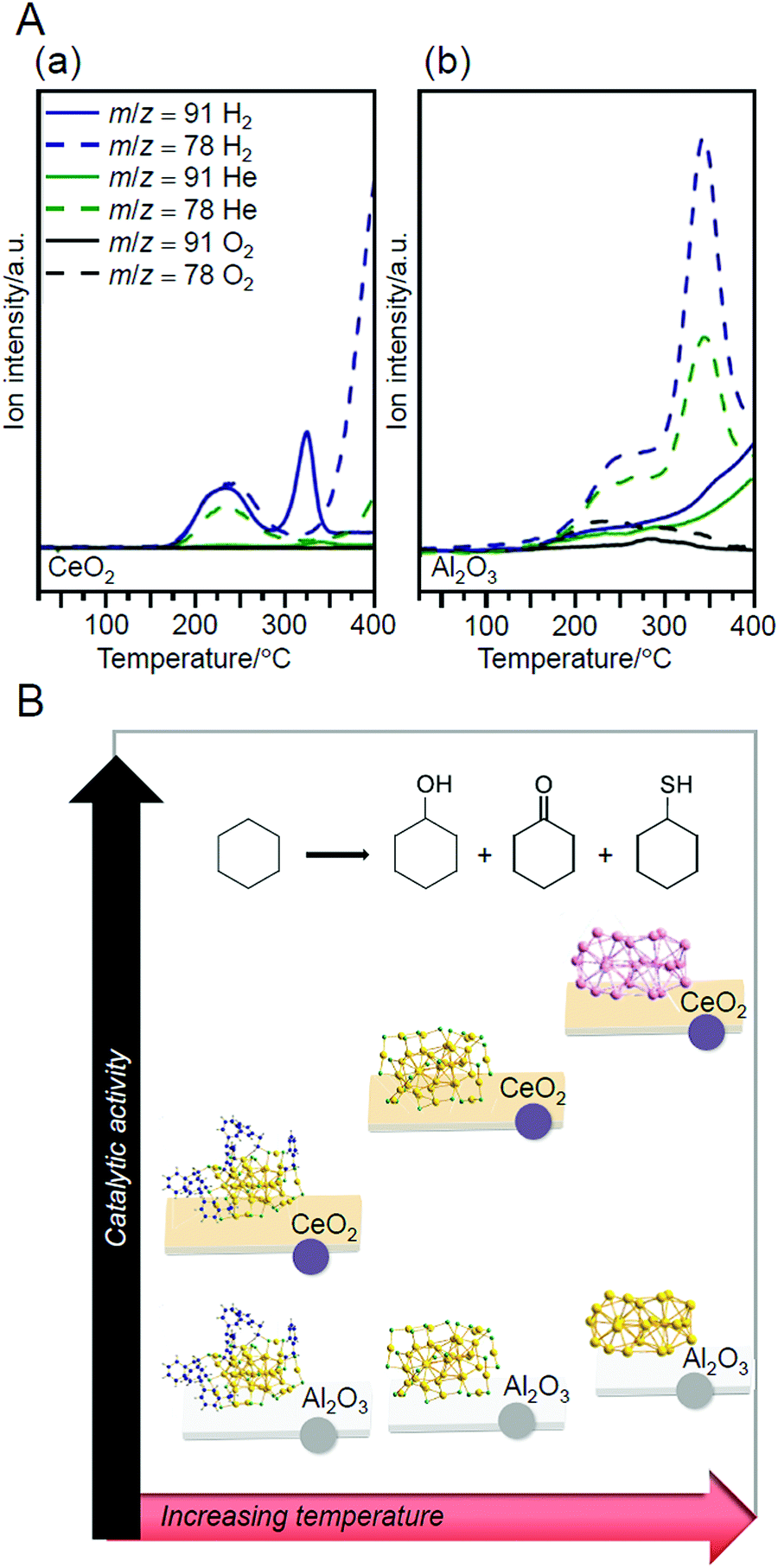 | ||
| Fig. 9 (A) Desorption of each compound from (a) Au38(PET)24/CeO2 and (b) Au38(PET)24/Al2O3 under different atmospheres at a heating rate of 10 °C min−1, obtained by TPD/MS. The compounds with m/z = 91 and 78 are originated from SC8H9 and C6H5, respectively. (B) Relation between the catalytic activity of each catalyst for aerobic oxidation of cyclohexane and the treatment temperature. Reproduced with permission from ref. 215. Copyright 2015 American Chemical Society. | ||
Barrabés and Bürgi et al. tracked the SR-desorption process during calcination under O2 atmosphere in more detail using X-ray absorption near edge structure (XANES; Fig. 10A) analysis and X-ray photoelectron spectroscopy (XPS) in 2018.216 They observed that the S, which was desorbed from Au38 NCs, was adsorbed on a support (CeO2 or Al2O3) in the vicinity of the Au38 NCs (Fig. 10B), and the desorbed S was oxidized in the order of SR, SO32−, and SO42− with increasing temperature. The S and oxidized S on the support have the following possibilities: (1) react directly with the substrate to produce by-products; (2) become a poison that suppresses the charge transfer between the support and metal; or (3) change the electron density of the support and thereby affect the catalytic properties. This study is significant because it clearly demonstrates that these possibilities should also be considered in the creation of heterogeneous catalysts based on the calcination of Aun(SR)m NCs/metal oxides. In their study in 2019, they revealed by multiple spectroscopies that S on these supports was involved in the reaction to produce the aforementioned cyclohexanethiol.217
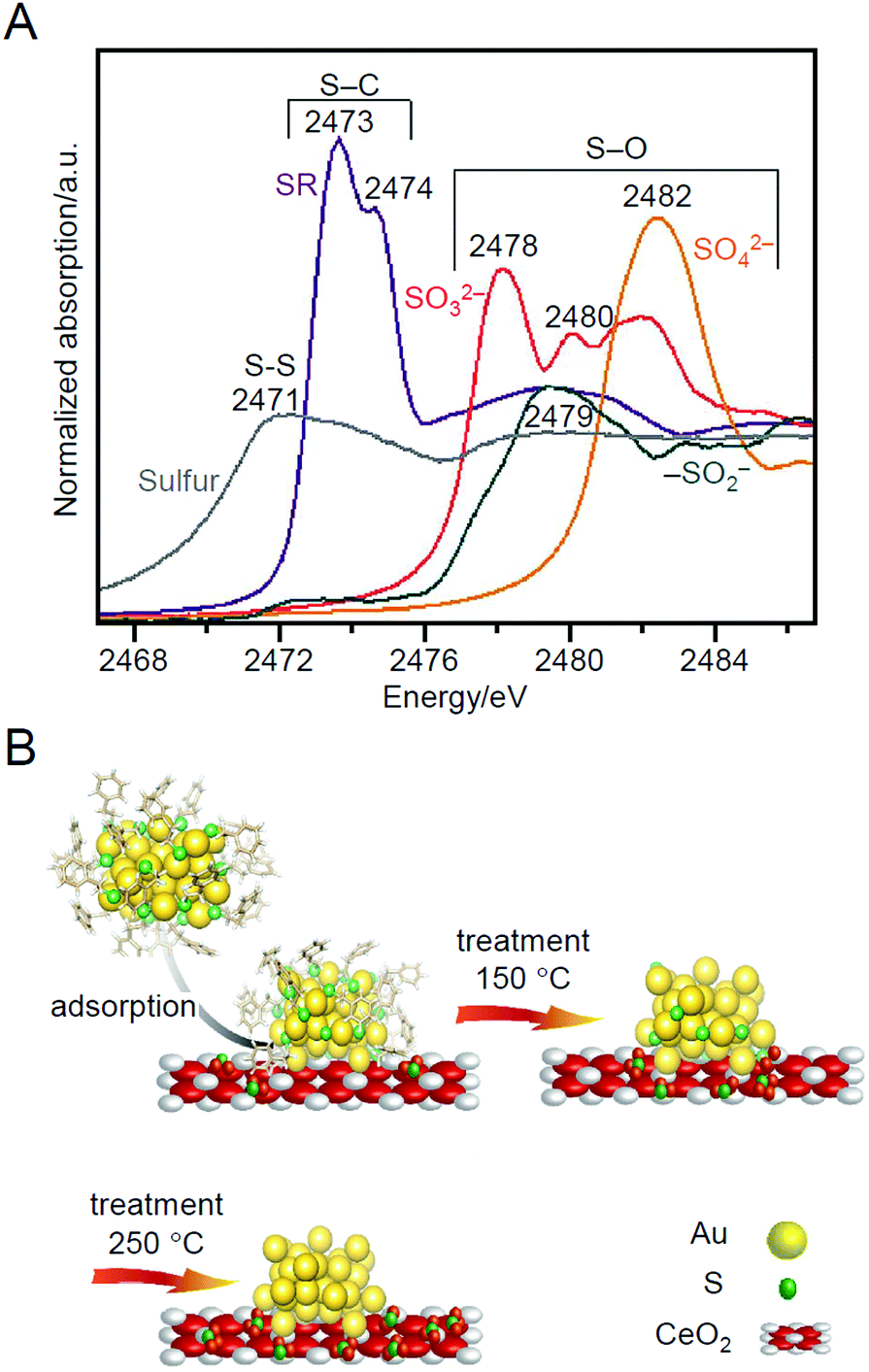 | ||
| Fig. 10 (A) XANES spectra at S K-edge of different reference materials. (B) Schematic of thiolate ligands evolution upon NC deposition on CeO2 and oxidative treatments. Reproduced from ref. 216. Copyright 2018 Wiley-VCH. | ||
In 2019, Barrabés et al. used two types of metal NCs, Au25(PET)18 and Au144(PET)60 (Fig. 2E), and two types of supports, titanium dioxide (TiO2) and silicon dioxide (SiO2), to study the effect of the size of Aun NCs and the type of the support on the catalytic activity.218 In this study, first, it was revealed by TGA that the ligand elimination starts at 150 °C and almost all the ligands could be removed at 250 °C for the catalysts prepared using all the combinations. However, calcination at 250 °C resulted in significant aggregation of metal NCs. Therefore, Au25(PET)18−x/TiO2, Au144(PET)60−x/TiO2, Au25(PET)18−x/SiO2, and Au144(PET)60−x/SiO2 prepared by the partial ligand removal by calcination at 150 °C under air were used as catalysts for the catalysis test. As for the size effect, it was clarified that Au144(PET)60−x showed about two times higher conversion efficiency than Au25(PET)18−x for both supports (Fig. 11). The difference in the conversion efficiency was interpreted to be caused by the difference in the adsorption efficiency of the substrate, activation of molecular O2, and other factors. For the support effect, SiO2 showed higher activity for the conversion efficiency of cyclohexane, whereas TiO2 showed higher values for the selectivity of KA oil production (Fig. 11). It was interpreted that this difference was caused by the different reaction mechanism depending on the support.
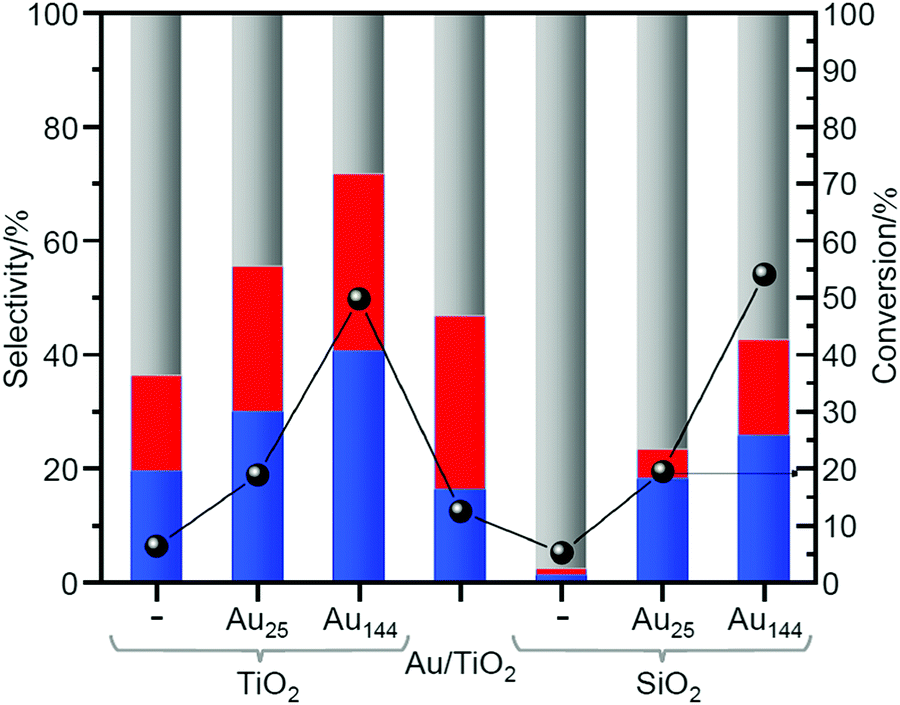 | ||
| Fig. 11 Catalytic activity of loaded Au25 and Au144 NCs (pre-treated at 150 °C) in cyclohexane oxidation. Cyclohexane conversion normalized to the Au loading (except for the blank measurements) (right axis); product distribution (left axis): red, blue, and gray represent cyclohexanol, cyclohexanone, and by-products, respectively. Reproduced with permission from ref. 218. Copyright 2019 Elsevier. | ||
 | ||
| Scheme 4 Styrene oxidation using metal NC catalysts. In this reaction, typically, TBHP is used as an oxidant because it is known to be more effective than O2 for alkene oxidations.219 The reaction was conducted in toluene or acetonitrile in the literature.219–224 A = styrene, B = styrene oxide, C = acetophenone, D = benzaldehyde, E = 3-phenylpropionic acid. In ref. 219, by-products such as benzoic acid and benzyl alcohol are also produced. | ||
In 2010, Tsukuda et al. prepared catalysts using Au25(SG)18 as the metal NCs and HAP as the support.219 The ligands were completely removed by calcination at 300 °C for 2 h under vacuum. This calcination slightly increased the particle size on the support from 1.0 ± 0.4 nm to 1.4 ± 0.6 nm (Fig. 12A). In the catalytic reaction, styrene oxidation was investigated using tert-butyl hydroperoxide (TBHP) as an oxidant, and the obtained catalyst showed 100% conversion and 92% styrene oxide (Scheme 4B) selectivity. This selectivity of the styrene oxides was higher than that of AuNP/HAP loaded with larger AuNP by adsorption or impregnation methods (Fig. 12B). Although the Au25/HAP converted styrene to styrene oxide, the conversion ability to further oxidize the styrene oxide was low, leading to the high selectivity of styrene oxide. This study also reported preliminary results that the catalytic reaction proceeds with 22% conversion and 36% styrene oxide selectivity even using molecular O2 as an oxidant when the reaction system was heated by microwave.
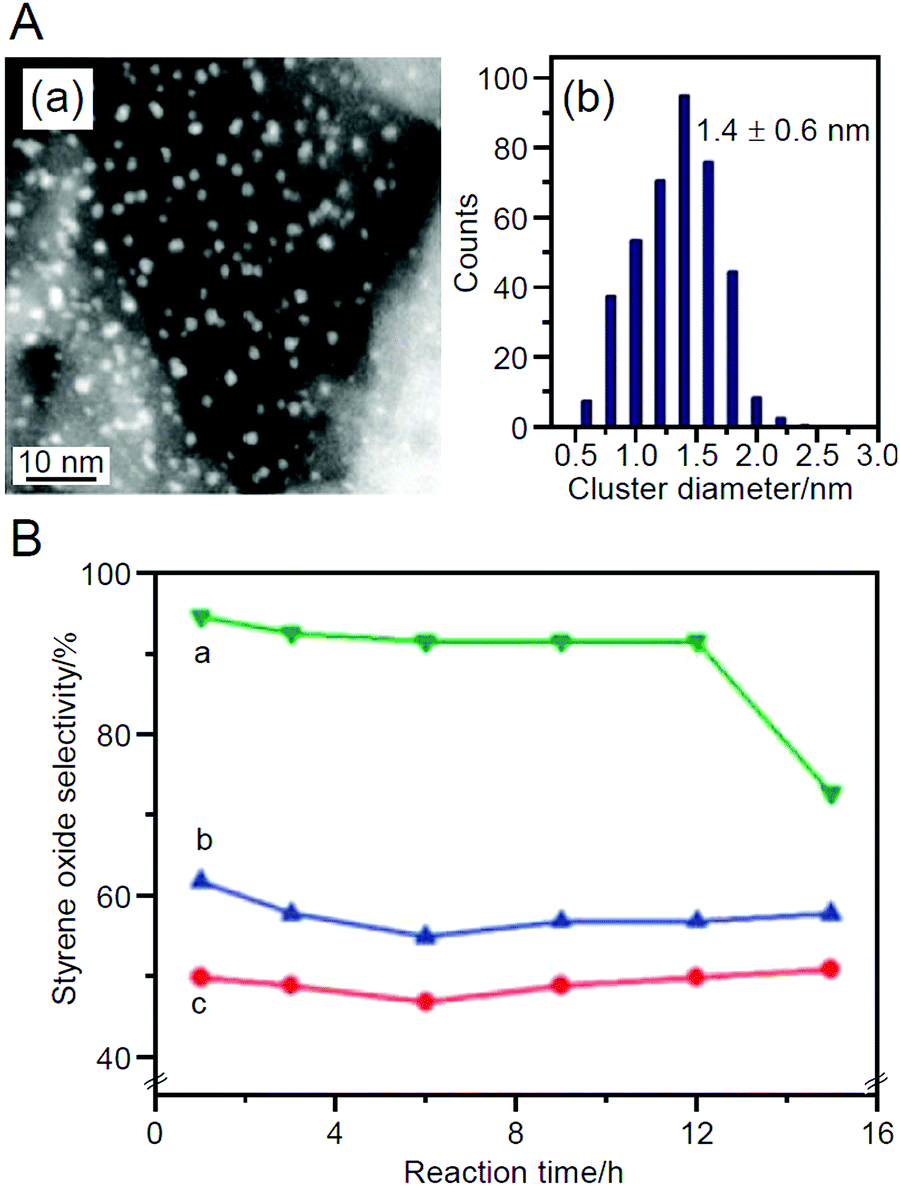 | ||
| Fig. 12 (A) (a) Representative HAADF-STEM image of Au25/HAP (0.5 wt% Au) and (b) size distribution of the Au NCs. (B) Selectivity of styrene oxide for (a) Au25/HAP (0.5 wt% Au), (b) AuNP/HAP (0.5 wt% Au) prepared by adsorption of HAuCl4, and (c) AuNP/HAP (0.5 wt% Au) prepared by impregnation method. Reproduced with permission from ref. 219. Copyright 2010 Royal Society of Chemistry. | ||
In 2010, Jin et al. studied the size and the support effects of catalysts using three different sizes of metal NCs (Au25(PET)18, Au38(PET)24, and Au144(PET)60) and two different supports (SiO2 and HAP).220 In this research, unlike in the work of Tsukuda et al., only a part of the ligands was removed by calcination at 200 °C under vacuum. Such an elimination of a part of the ligands increased the conversion efficiency (the main product was benzaldehyde; Scheme 4D) for both combinations of catalysts (Fig. 13A). For example, in the reaction using TBHP as the initiator, molecular O2 as the oxidant, and toluene as the solvent, the conversion efficiency increased with decreasing metal core size (Fig. 13B). It was interpreted that this phenomenon occurred because of the efficient activation of molecular O2 by small Au NCs. As for the support, HAP was shown to be a more suitable support for high activation than SiO2 (Fig. 13B). Unlike SiO2, HAP had electronic interactions with the loaded Au NCs. In addition, HAP suppressed the aggregation of loaded Au NCs more than SiO2. The higher conversion efficiency achieved when using HAP as the support rather than SiO2 was attributed to these factors. Because the ligand was only partially removed in this study, the details of the reaction mechanism were discussed based on the geometrical structure of Au25(PET)1842,262 which was determined by SC-XRD (Fig. 13C).
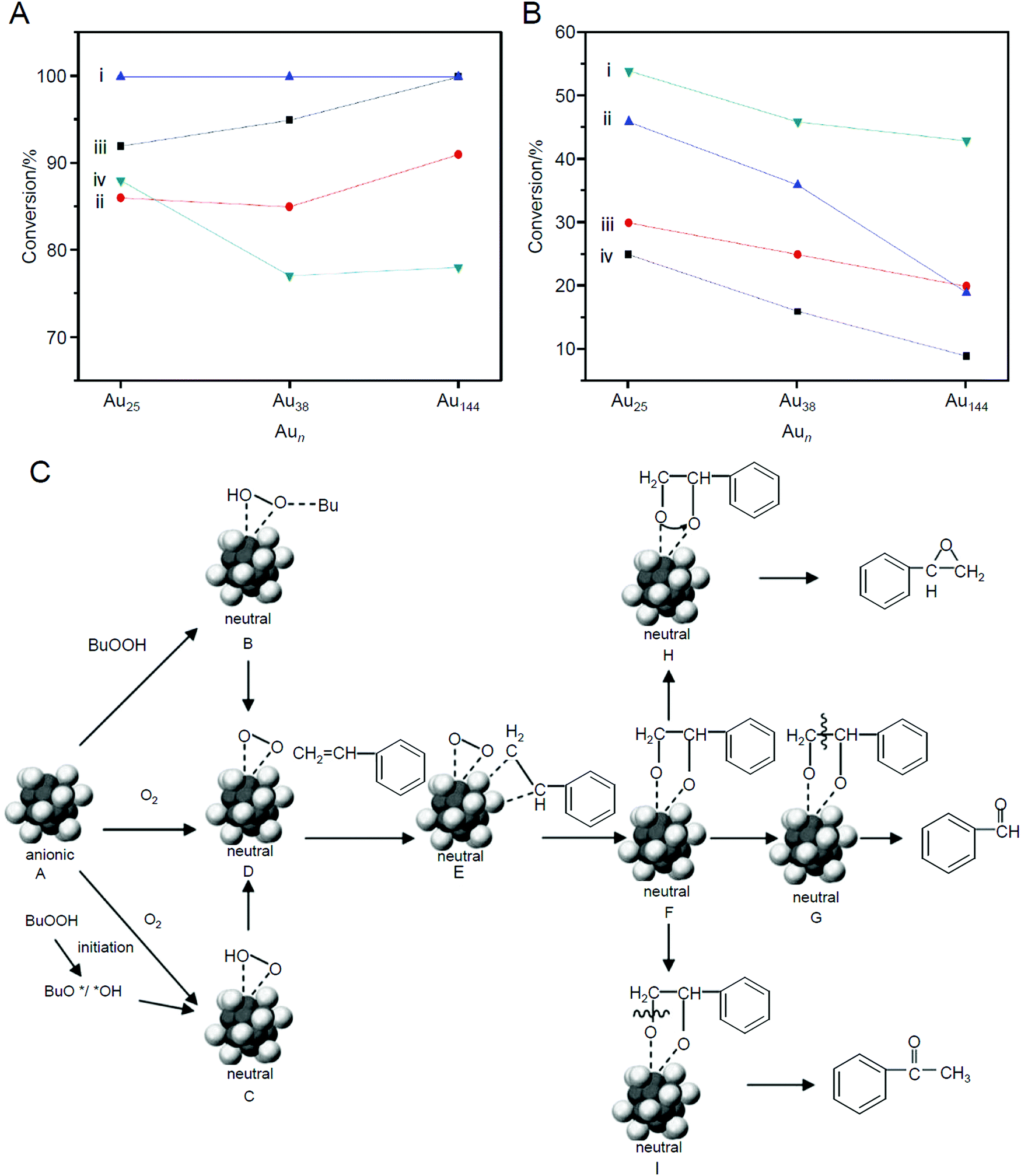 | ||
| Fig. 13 (A) Catalytic result of styrene oxidation with TBHP as the oxidant on (i) calcined Aun/HAP, (ii) uncalcined Aun/HAP, (iii) calcined Aun/SiO2, and (iv) uncalcined Aun/SiO2 catalysts. (B) Catalytic result of styrene oxidation with TBHP as an initiator and O2 as the main oxidant on (i) calcined Aun/HAP, (ii) uncalcined Aun/HAP, (iii) calcined Aun/SiO2, and (iv) uncalcined Aun/SiO2 catalysts. (C) Proposed mechanism of selective oxidation of styrene catalyzed by Au25(PET)18 NCs. For clarity, the PET ligands are not shown. Dark gray: Au atoms of the core, light gray: Au atoms of the shell. Reproduced with permission from ref. 220. Copyright 2010 Wiley-VCH. | ||
In 2011, Jin et al. also conducted a study using Au25(PET)18 and [Au25(PPh3)10(PET)5Cl2]2+ (Fig. 2F) as metal NCs and CeO2 as the support.221 TBHP was used as the oxidant, and acetonitrile was used as the solvent, which is different from their study in 2010. As a result, styrene oxide (Scheme 4B) was obtained with high selectivity (≥93%) using both catalysts. This result indicates that the type of solvent also greatly affects the selectivity of the product. Au25(PET)18/CeO2 showed a higher conversion efficiency than Au25(PPh3)10(PET)5Cl2/CeO2, indicating that the metal core structure also has a significant effect on the activity. Calcination at 260 °C under vacuum resulted in a significant decrease in the activity, especially for Au25(PET)18/CeO2. At this calcination temperature, all the ligands could be removed. In this case, the aggregation of metal NCs is likely to occur, and the cationic Au sites, which are easily attacked by the nucleophilic –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 group, disappear. It was interpreted that the activity significantly decreased in the calcination at 260 °C because of these factors. The authors speculated that the latter factor is also largely involved in the higher conversion of Au25(PET)18/CeO2 than Au25(PPh3)10(PET)5Cl2/CeO2. These results support their proposed reaction mechanism for this reaction (Fig. 13C).
CH2 group, disappear. It was interpreted that the activity significantly decreased in the calcination at 260 °C because of these factors. The authors speculated that the latter factor is also largely involved in the higher conversion of Au25(PET)18/CeO2 than Au25(PPh3)10(PET)5Cl2/CeO2. These results support their proposed reaction mechanism for this reaction (Fig. 13C).
Kumar et al. also studied a system similar to that of Jin et al. in 2013.222 In their study, Au25(PET)18 or Au25(PPh3)10(PET)5Cl2 was used as the metal NCs and SiO2 was used as the support. TBHP and acetonitrile were used as the oxidant and solvent, respectively. In the catalysts before calcination, Au25(PET)18/SiO2 showed high conversion efficiency, and Au25(PPh3)10(PET)5Cl2/SiO2 showed high selectivity for benzaldehyde (Scheme 4D). XANES (Fig. 14) and ultraviolet photoemission spectroscopy (UPS) demonstrated that Au in Au25(PPh3)10(PET)5Cl2/SiO2 is more cationic than Au in Au25(PET)18/SiO2, which is interpreted to lead to the easy oxidation of styrene into benzaldehyde when using Au25(PPh3)10(PET)5Cl2/SiO2. In this study, the ligand removal was also conducted by calcination. This improved the conversion efficiency; however, the particle size of the metal NCs after calcination increased to ∼3–4 nm.
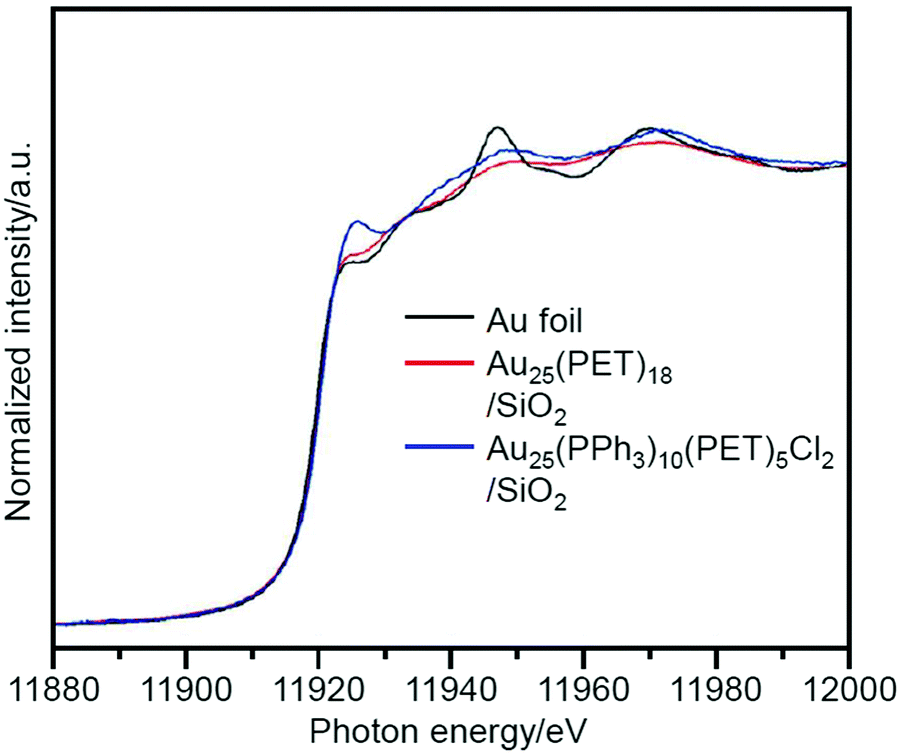 | ||
| Fig. 14 Normalized Au L3-edge XANES spectra of Au25(PET)18/SiO2, Au25(PPh3)10(PET)5Cl2/SiO2, and Au foil. These spectra indicate that Au in Au25(PPh3)10(PET)5Cl2/SiO2 is more cationic than that in Au25(PET)18/SiO2. Reproduced with permission from ref. 222. Copyright 2013 Wiley-VCH. | ||
Yan et al. conducted a more extensive study on the effect of the type of support on the particle size and catalytic activity of loaded metal NCs using Au25(MHA)18 (MHA = 6-mercaptohexanoic acid) and five different supports (HAP, TiO2 (P-25), AC, pyrolyzed graphene oxide (PGO), and fused SiO2) in 2015.223 This work revealed that only the use of HAP and TiO2 relatively suppressed the aggregation of Au25. In Au25/HAP, a strong interaction occurred between the phosphate ion (PO43−) of HAP and Au. A strong interaction also occurred in Au25/TiO2. It was interpreted that the aggregation of Au25 was effectively suppressed on HAP and TiO2 surfaces, unlike the surfaces on other supports, for these reasons. This study also suggested that ∼50% of the S remained bound to Au when calcined at 300 °C under nitrogen (N2) atmosphere. The obtained catalysts showed catalytic activity for styrene oxidation using TBHP as the oxidant; however, the conversion efficiency and the selectivity of styrene oxide (Scheme 4B) were comparable to those of the catalyst before calcination. In this study, the particle size slightly increased by calcination (from 1.5 to 2.4 nm).
In addition to these studies on Au NCs, Scott et al. conducted a study using [Ag25(2,4-SPhMe2)18]− (2,4-SPhMe2 = 2,4-dimethylbenzenethiolate; Fig. 2G) as a metal NC and AC as a support in 2019.224 They prepared Ag25/AC containing 4 wt% Ag by calcination at 250 °C under air. The TEM and XAFS (Fig. 15) measurements revealed that the Ag25 hardly aggregated and that most of the ligands were removed from the Ag25. Quantitative analysis of the XPS spectrum suggested a residual of 0.55% S, which was interpreted to be transferred from the Ag25 surface onto the AC. When the calcination temperature was increased to 350 and 450 °C, the amount of S continuously decreased (0.14% at 350 °C and ∼0.01% at 450 °C); however, there was significant aggregation of Ag25 in the catalysts. The catalysts prepared by calcination at 250 °C showed a conversion efficiency of 61.4% and 94% selectivity of styrene oxide (Scheme 4B) when TBHP was used as the oxidant. These values were higher than those of the catalyst before calcination, indicating that removal of the ligands by calcination led to improvement of the activity of the catalyst. Unfortunately, both the conversion efficiency and selectivity decreased when TBHP was used as the initiator and molecular O2 was used as the oxidant. These issues are expected to be overcome in the future by improving the activity through modifications such as heteroatom substitutions.
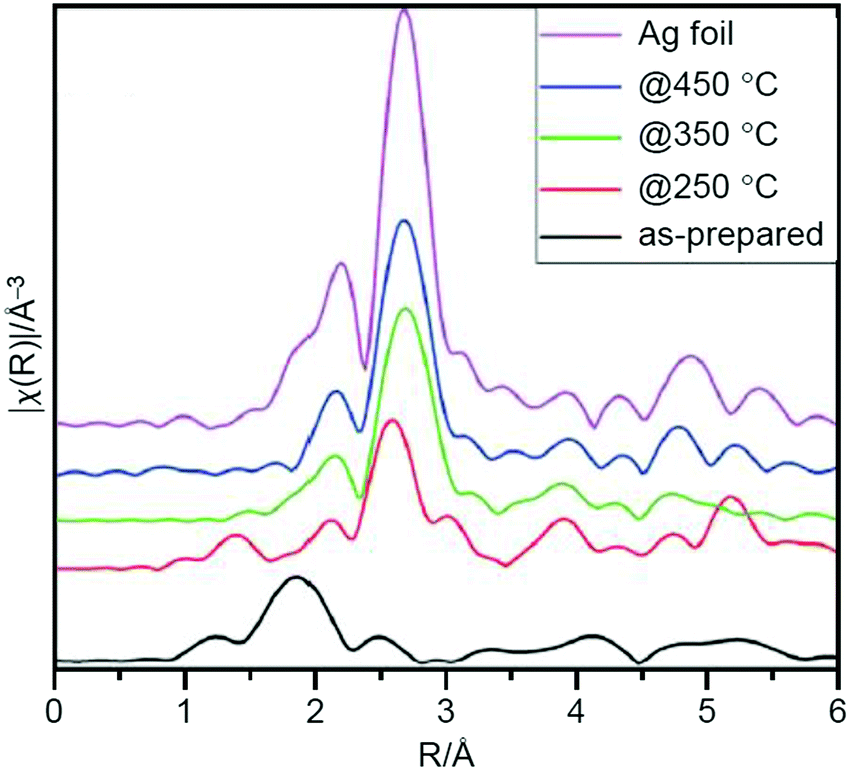 | ||
| Fig. 15 Ag K-edge FT-EXAFS spectra in R-space for as-prepared and activated Ag25/AC catalysts and Ag foil reference. Reproduced with permission from ref. 224. Copyright 2019 Royal Society of Chemistry. | ||
 | ||
| Scheme 5 CO oxidation using metal NC catalysts. The reaction was conducted while supplying reactant gas mixture containing of CO, O2, and inert gas. Inclusion of water vapor in the feed gas has been shown to benefit the CO conversion.226 | ||
In 2012, Spivey et al. conducted a study using Au38(SC12)24 as a metal NC and TiO2 as a support.225 In this study, the ligands were removed by calcination at 400 °C for 1 h under a mixture gas of H2–helium (He). The catalytic activity for CO oxidation started to appear by removing ligands. However, unfortunately, remarkable aggregation of Au38(SC12)24 had already occurred when Au38(SC12)24 were adsorbed on the TiO2 surface. Therefore, it was difficult to elucidate the catalytic activity of Au38/TiO2 for CO oxidation in this study.
In contrast, in the study by Jin et al. in 2012, the aggregation of metal NCs during adsorption was almost completely suppressed.226 In this study, Au25(PET)18 was used as the metal NC and TiO2, iron(III) oxide (Fe2O3), or CeO2 was used as the support. Among these combinations, Au25(PET)18/CeO2 showed the highest conversion efficiency. For this catalyst, high activity was induced by pre-treatment at 150 °C for 1.5 h under O2 atmosphere (Fig. 16A). This temperature is lower than the temperature at which the ligand was removed (190 °C). Therefore, the authors described that high activity can be induced without the ligand elimination for this catalyst. On the basis of several experiments conducted under various conditions, it was suggested that (1) O2 adsorbed onto CeO2 could be activated by the pre-treatment and (2) the reaction occurred at the interface between Au25(PET)18 and CeO2 (perimeter; Fig. 16B).
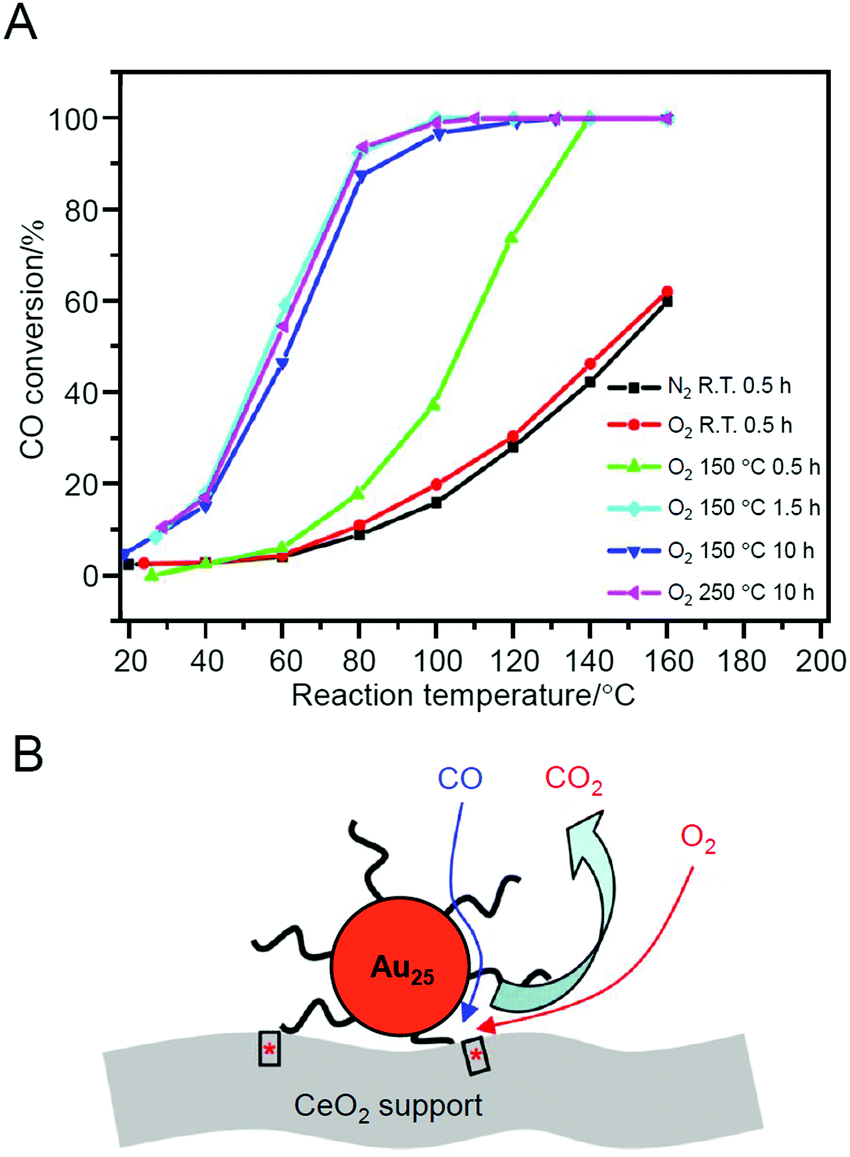 | ||
| Fig. 16 (A) Reaction temperature dependence of CO conversion over Au25(PET)18/CeO2 catalyst after different pre-treatments. Gas hourly space velocity (GHSV) = 7500 mL g−1 h−1. (B) Proposed model for CO oxidation at the perimeter sites of Au25(PET)18/CeO2 catalyst. Reproduced with permission from ref. 226. Copyright 2012 American Chemical Society. | ||
Jin et al. also studied Au38(PET)24/CeO2 in 2013227 and demonstrated that Au38(PET)24/CeO2 shows the highest activity when calcined at 175 °C (Fig. 17A). According to the TGA results, part of the ligands was interpreted to be removed in Au38(PET)24/CeO2 during the calcination at this temperature. However, the conversion efficiency of this sample after 20 h of reaction was inverted by the sample prepared by calcination at 130 °C (namely, without the ligand elimination) (Fig. 17B). This finding indicates that the protection by ligands is very important for the preparation of highly stable catalysts. Thus, this study revealed the following two factors for the preparation of Au38(PET)24/CeO2 catalyst that promote CO oxidation: (1) pre-treatment (calcination) under O2 atmosphere is necessary for the activation; and (2) however, it is more appropriate not to completely remove the ligands from the viewpoint of stability. In their study in 2016, it was shown that when Au144(PET)60/CeO2 was used as a catalyst, the inclusion of a reductive gas (H2 or CO) along with O2 further improved the effect of the pre-treatment.228 Furthermore, their study in 2018 revealed that the catalytic activity of Aun(SR)m/CeO2 (n = 25, 36, or 38) decreases overall with the bulkiness of the functional groups directly attached to S.229 In addition to these facts, Jin et al. also demonstrated that both the ligand and metal core structures affect the catalytic activity by comparing the catalytic activity of Au28(S-c-C6H11)20 (S-c-C6H11 = cyclohexanethiolate) and Au28(TBBT)20 (TBBT = 4-tert-butylbenzenethiolate).230
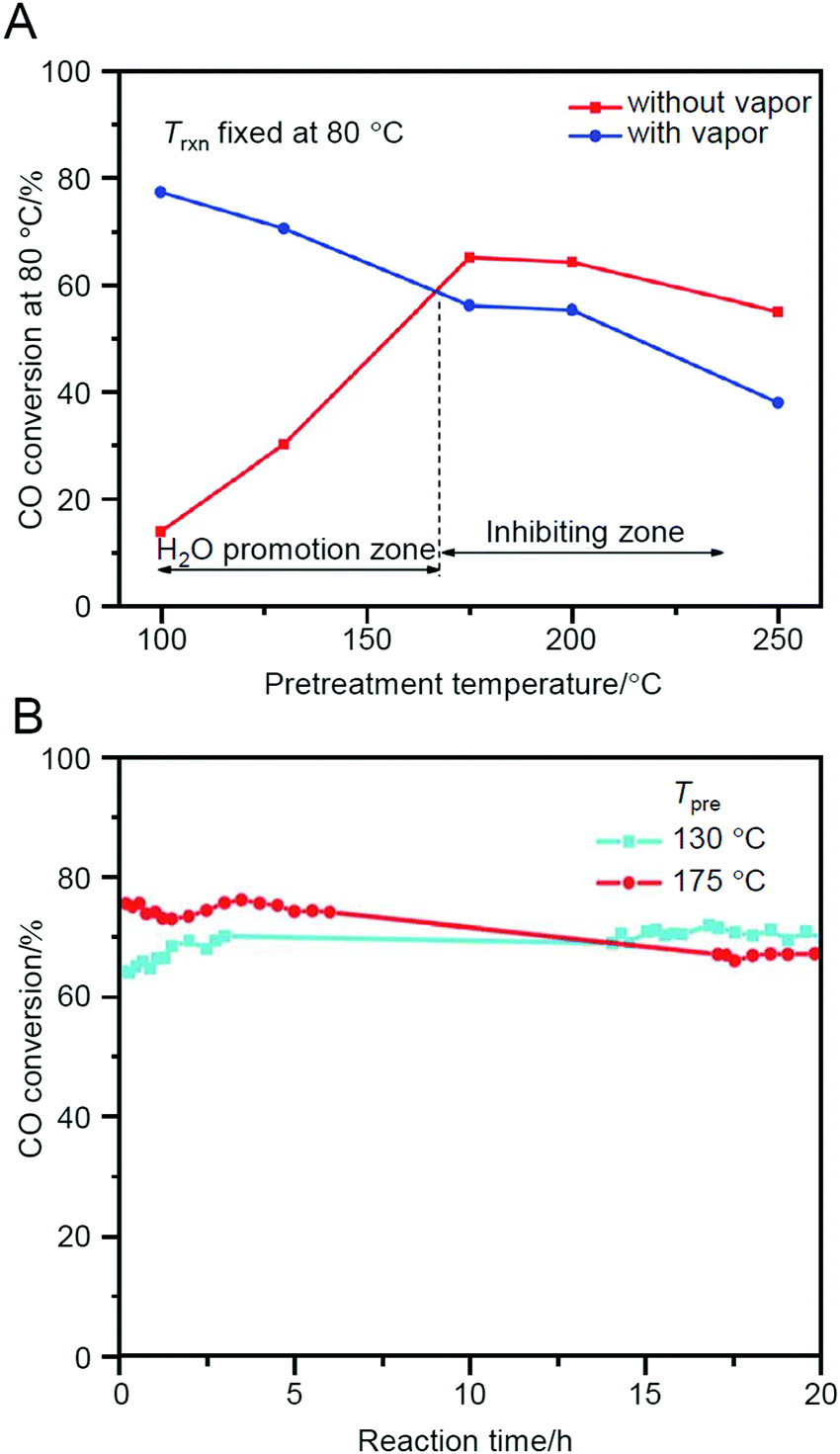 | ||
Fig. 17 (A) CO conversion at fixed reaction temperature (Trxn) of 80 °C as a function of catalyst pre-treatment temperature for Au38(PET)24/CeO2 catalyst. GHSV = 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL g−1 h−1. (B) CO conversion at Trxn = 100 °C over Au38(SR)24/CeO2 catalyst as a function of reaction time. Pre-treatment conditions: O2 for 2 h and pre-treatment temperature (Tpre) as shown in the figure. GHSV: 30000 mL g−1 h−1. Feed gas: without water vapor. Reproduced with permission from ref. 227. Copyright 2013 Royal Society of Chemistry. 000 mL g−1 h−1. (B) CO conversion at Trxn = 100 °C over Au38(SR)24/CeO2 catalyst as a function of reaction time. Pre-treatment conditions: O2 for 2 h and pre-treatment temperature (Tpre) as shown in the figure. GHSV: 30000 mL g−1 h−1. Feed gas: without water vapor. Reproduced with permission from ref. 227. Copyright 2013 Royal Society of Chemistry. | ||
Wu and Overbury et al. performed a detailed investigation of the reaction mechanism for Au25(PET)18/CeO2 in 2014 (Fig. 18).231 They showed that (1) CO could not be adsorbed on the catalyst without pre-treatment and (2) after pre-treatment at 150 °C or higher under O2 atmosphere, the PET existed at the interface between Au25(PET)18 and CeO2 started to desorb (Au25(PET)18−x/CeO2), and CO could be adsorbed on the exposed Au. The presence of such exposed Au was interpreted to begin to generate catalytic activity for CO oxidation at low temperature. Regarding the mechanism, it was revealed that when Au25(PET)18−x/CeO2 was used as a catalyst, the reaction proceeds mainly by the Mars–van Krevelen mechanism (reduction mechanism), in which CO activated by Au25(PET)18−x reacts with the oxygen in CeO2 to produce CO2, and O2 in the gas phase compensates for the oxygen in CeO2 (Fig. 18B). In addition, it was suggested that when the catalyst with the ligands further removed (Au25/CeO2) is used, the Langmuir–Hinshelwood mechanism, in which CO and O2 react on the Au25 surface to produce CO2, also occurred in parallel to the Mars–Van Krevelen mechanism (Fig. 18C). To further reinforce this interpretation, in 2016, Wu et al. conducted a study using Au22(L8)6 (L8 = 1,8-bis(diphenylphosphino)octane; Fig. 2H),176 which contains coordinatively unsaturated Au, as a metal NC. As a result, it was confirmed that Au22(L8)6/metal oxide (TiO2, SiO2, or CeO2) promotes CO oxidation at room temperature without pre-treatment.232
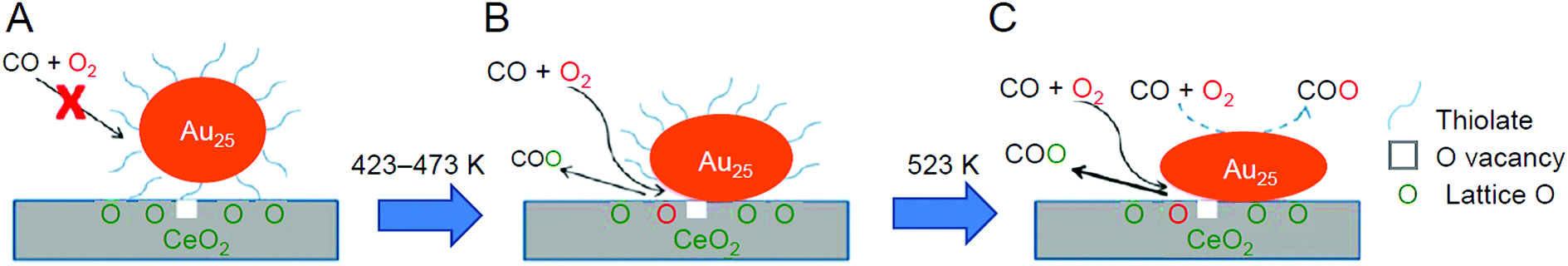 | ||
| Fig. 18 CO oxidation mechanism on (A) intact, (B) partially dethiolated, and (C) fully dethiolated Au25(PET)18/CeO2 catalysts. Reproduced with permission from ref. 231. Copyright 2014 American Chemical Society. | ||
Thus, many catalysts require pre-treatment at an appropriate temperature. In 2020, Barrabés et al. tracked the phenomena occurring during the pre-treatment of Au38(PET)24/CeO2 at 150 °C using in situ XAFS.233 The results suggested that although S remained on Au, the staple structure (Aun(SR)n+1; n = 1 or 2) surrounding the Au23 core (Fig. 2D) collapses with pre-treatment at 150 °C (Fig. 19A). This study also showed that Au NCs on the support change during CO oxidation reaction, which led to the adsorption of compounds with Au–S bonds on CeO2, which are eventually oxidized to SO32− and SO42−. They also reported that increasing the pre-treatment temperature to 250 °C causes the complete removal of the ligands from the Au NCs, resulting in higher activity than that of the catalyst pre-treated at 150 °C (Fig. 19B). These results have led to new information on the structural changes induced by pre-treatment. However, the effect of the pre-treatment on the catalytic activity was not completely consistent with that reported by Jin et al. and Wu et al.; a deeper understanding of this difference is expected to be attained in future study.
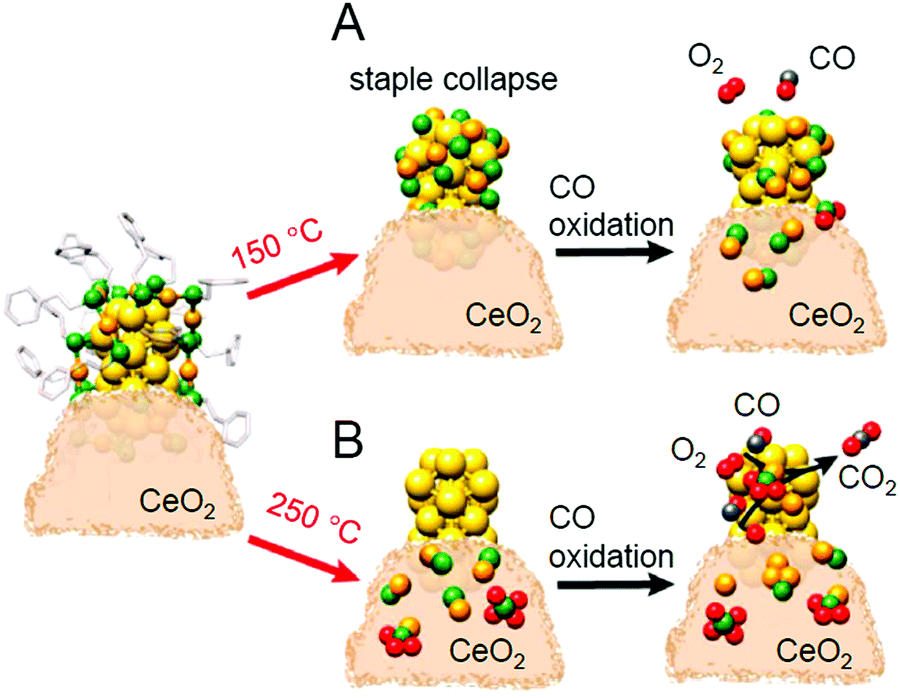 | ||
| Fig. 19 Evolution of Au38(PET)24/CeO2 catalyst structure estimated from EXAFS analyses: catalysts after pre-treatment at (A) 150 °C and (B) 250 °C and subsequent CO oxidation. Reproduced with permission from ref. 233. Copyright 2020 American Chemical Society. | ||
Thus, when CeO2 is used as a support, the aggregation of metal NCs is suppressed and high activation is induced. In 2012, Dai and Fan et al. reported the use of CuO-EP-FDU-12 or Co3O4-EP-FDU-12, in which the surface of mesoporous silica (EP-FDU-12) is modified with nanostructures of copper oxide (CuO) or cobalt oxide (Co3O4), as a support, resulting in the production of catalysts with very high stability.234 They prepared Au144/CuO-EP-FDU-12 or Au25/Co3O4-EP-FDU-12 by adsorbing Au144(PET)60 onto CuO-EP-FDU-12 or Au25(PET)18 onto Co3O4-EP-FDU-12, followed by calcination at 300 °C under air. It was shown that such calcination removed the ligands; however, only slight aggregation of Au144 (Fig. 20) or Au25 occurred on the support. Au NCs on silica tend to aggregate because Au NCs and silica have a weak interaction with each other. However, in their structures, the transition metal nanostructures (CuO or Co3O4) are strongly immobilized on the silica pore surface. It was interpreted that the aggregation of Au NCs was suppressed in Au144/CuO-EP-FDU-12 and Au25/Co3O4-EP-FDU-12 because these transition metal nanostructures strongly fix Au NCs on their surfaces. Both of the obtained catalysts exhibited high activity for CO oxidation. Because the catalyst without calcination showed little activity, it was concluded that calcination is essential for inducing catalytic activity for these catalysts.
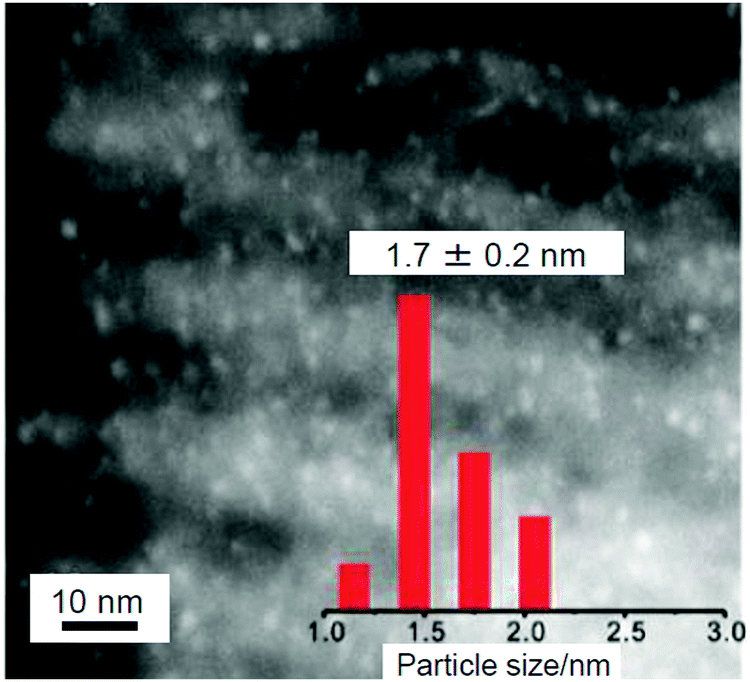 | ||
| Fig. 20 STEM image of Au144 NCs (1.1 wt% Au) loaded on CuO-EP-FDU-12 after calcination at 300 °C and size distribution of Au NCs. Reproduced with permission from ref. 234. Copyright 2012 Royal Society of Chemistry. | ||
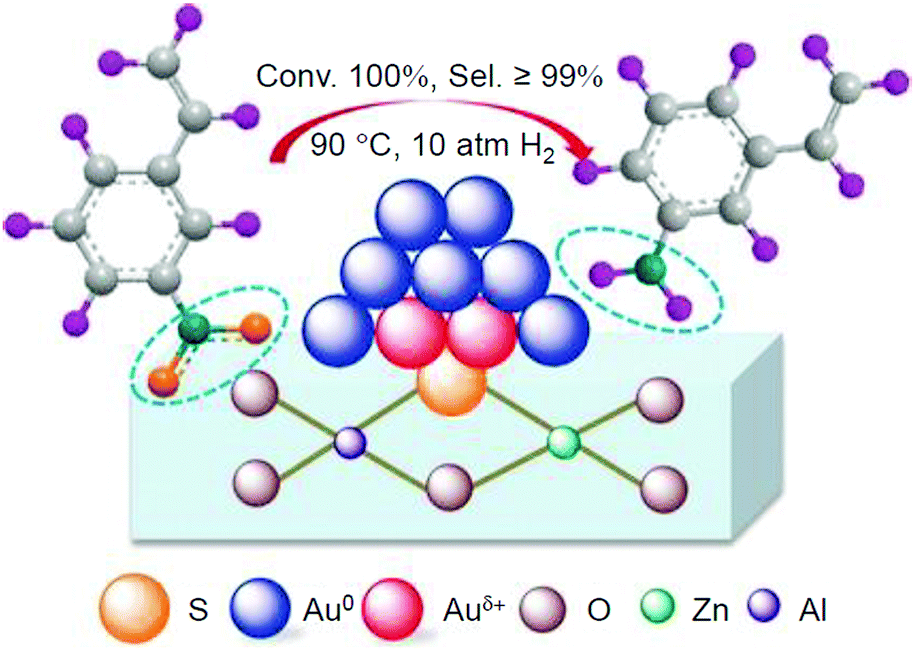
In addition to these studies using atomically precise Au NCs, there have been examples using atomically precise Pt NCs. Negishi and Nagaoka et al. reported on the use of Pt17/γ-Al2O3 as a catalyst for CO oxidation in 2020.235 Pt is widely used as a catalyst for exhaust-gas purification; however, because Pt is a rare and expensive metal, reduction of its use through miniaturization is required. Nair and Negishi et al. found an easy method to synthesize Pt17 NCs protected by CO and PPh3 ([Pt17(CO)12(PPh3)8]Cln; n = 1, 2; Fig. 2I) in 2017.177 In their synthesis method, Ptn(CO)m(PPh3)l NCs, in which [Pt17(CO)12(PPh3)8]Cln was the main product, were first prepared by mixing the reagents and heating the solvent under air atmosphere. [Pt17(CO)12(PPh3)8]Cln was isolated from the obtained mixture in high purity using the difference in solubility in the solvent. This method has the advantage that precise Pt NCs can be synthesized without any special synthesis equipment264 or ligands.265,266 In their study in 2020, [Pt17(CO)12(PPh3)8]Cln was first adsorbed on γ-Al2O3 (Pt17(CO)12(PPh3)8/γ-Al2O3), which was then calcined at 500 °C to obtain Pt17/γ-Al2O3 (Fig. 21A). Pt17 NCs hardly aggregated in the series of operations (Fig. 21B). Compared with other noble metals (e.g., Au), Pt binds strongly to O (318.4 ± 6.7 kJ mol−1 for Pt–O vs. 223 ± 21.1 kJ mol−1 for Au–O). Furthermore, because γ-Al2O3 has a complicated structure in which Al atoms are arranged octahedrally or tetrahedrally, cationic sites are present owing to the surface defects in γ-Al2O3.267 Pt atoms interacted with these cation sites and, as a result, Pt clusters hardly aggregated on γ-Al2O3.268 According to TGA analysis, several CO appeared to be coordinated to the surface of Pt17/γ-Al2O3 (Fig. 21A(c)). The HAADF-STEM images suggested that the loaded Pt17 NCs had bi-layer or tri-layer structures (Fig. 21C). Previous studies have suggested that CO and O2 are activated on the terrace Pt and step Pt, respectively, during the oxidation reaction of CO.269–271 In the structure of Fig. 21C, most of the terrace Pt atoms are located near the step Pt atoms. Thus, the reaction of CO and O2, i.e., the oxidation of CO, was expected to proceed effectively over Pt17/γ-Al2O3.
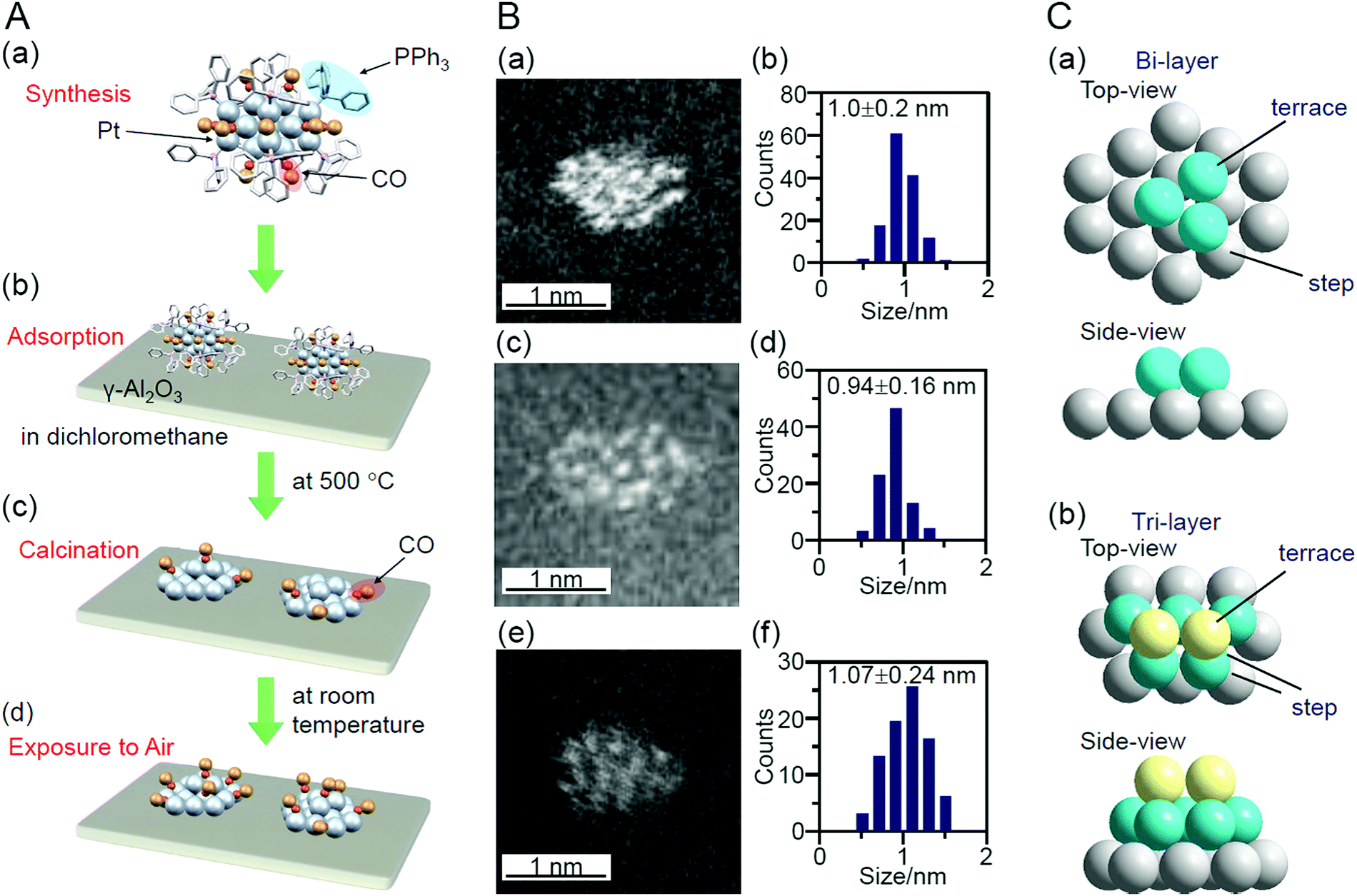 | ||
| Fig. 21 (A) Schematic of preparation of Pt17/γ-Al2O3: (a) precise synthesis of [Pt17(CO)12(PPh3)8]Cln, (b) adsorption of [Pt17(CO)12(PPh3)8]Cln on γ-Al2O3 (Pt17(CO)12(PPh3)8/γ-Al2O3), (c) calcination of ligands while maintaining the NC size (Pt17/γ-Al2O3), and (d) exposure of Pt17/γ-Al2O3 to air. (B) HAADF-STEM images (a, c, e) and size distribution (b, d, f) of (a, b) [Pt17(CO)12(PPh3)8]Cln, (c, d) Pt17(CO)12(PPh3)8/γ-Al2O3, and (e and f) Pt17/γ-Al2O3. (C) Two proposed structures for Pt17 on γ-Al2O3, which were estimated based on the HAADF-STEM images of Pt17/γ-Al2O3: (a) bi-layer and (b) tri-layer structure. In (a) and (b), both top and side views are shown. Reproduced from ref. 235. Copyright 2020 Royal Society of Chemistry. | ||
In fact, Pt17/γ-Al2O3, loaded with 0.15 wt% Pt17, showed a higher CO oxidation activity than PtNP/γ-Al2O3 (an industrially used Pt catalyst), in which 0.15 wt% PtNP was loaded by the impregnation method. In this experiment, Pt17/γ-Al2O3 and PtNP/γ-Al2O3 were coated on a honeycomb support to evaluate their catalytic performance in a state similar to actual vehicle mounting conditions. A gas mixture consisting of 1% CO, 0.5% O2, and 98.5% N2 was circulated over the honeycomb support at a space velocity of 50000 h−1 while the temperature of the honeycomb support was increased to 400 °C at a rate of 20 °C min−1. The conversion ratio of CO over the catalyst was estimated by evaluating the components of the mixed gas before and after circulation using an exhaust gas analyzer (Fig. 22A). When PtNP/γ-Al2O3 was used as the catalyst, the catalytic activity started to appear at approximately 270 °C, and the conversion reached 50% at approximately 350 °C (light-off temperature) and nearly 100% at approximately 370 °C (Fig. 22B). In contrast, when Pt17/γ-Al2O3 was used as the catalyst, the catalytic activity started from approximately 240 °C, and the conversion reached 50% at approximately 330 °C and almost 100% at approximately 350 °C (Fig. 22B). These results indicate that Pt17/γ-Al2O3 exhibits higher catalytic activity at each temperature than PtNP/γ-Al2O3; namely, Pt17/γ-Al2O3 can convert CO at lower temperatures than PtNP/γ-Al2O3. Similar experiments showed that Pt17/γ-Al2O3 also has a higher catalytic activity for propylene oxidation than PtNP/γ-Al2O3. Currently, the catalytic activity of exhaust-gas-treating catalysts at low temperatures is expected to be improved with the spread of vehicles that frequently stop and restart their engines (e.g., hybrid vehicles).272 These results strongly suggest that Pt17/γ-Al2O3 could be used as an exhaust-gas-treating catalyst to overcome this issue.
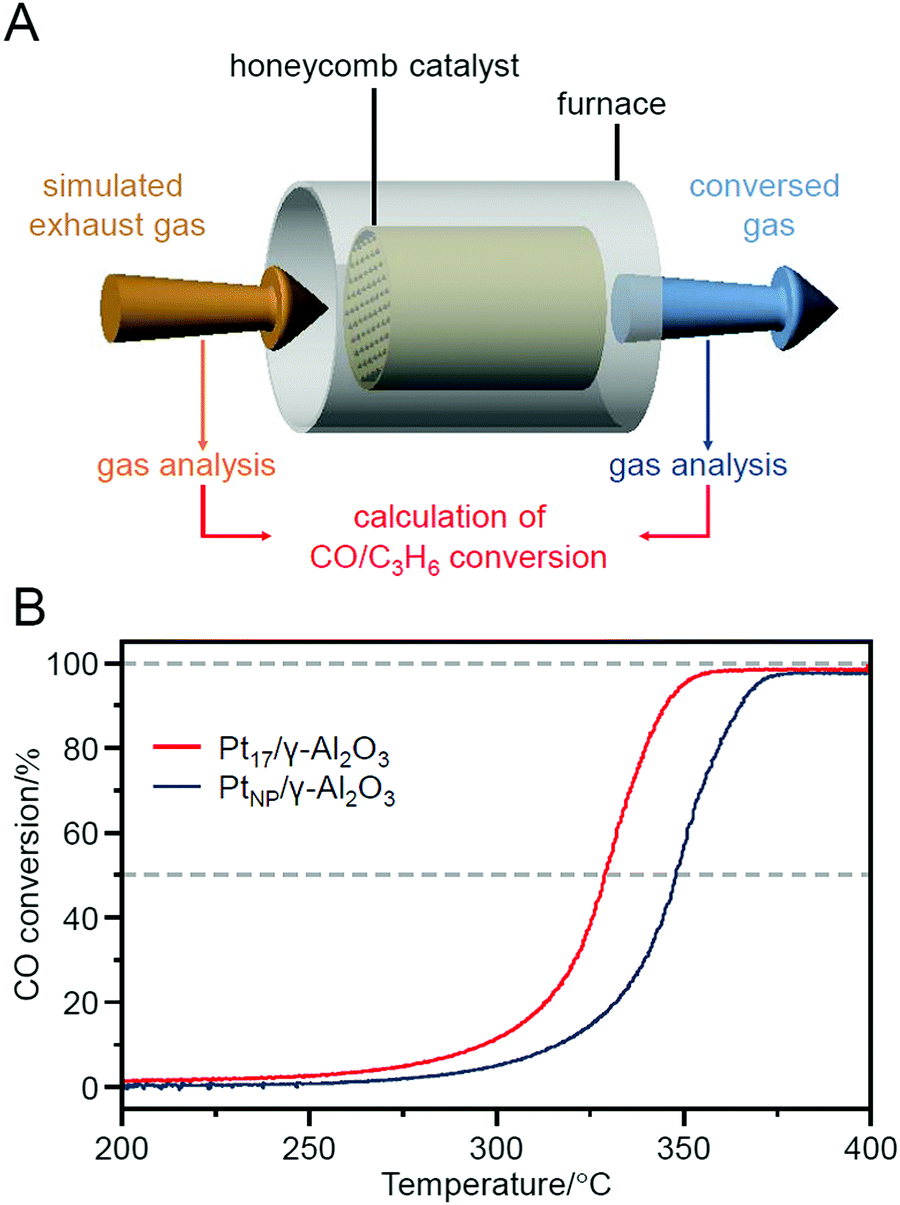 | ||
| Fig. 22 (A) Schematic of the estimation of CO conversions over Pt17/γ-Al2O3 or PtNP/γ-Al2O3 coated on a cordierite honeycomb substrate. (B) CO conversions over Pt17/γ-Al2O3 or PtNP/γ-Al2O3. Reproduced from ref. 235. Copyright 2020 Royal Society of Chemistry. | ||
As mentioned above, the precursor [Pt17(CO)12(PPh3)8]Cln can be obtained with atomic precision only by mixing the reagents under air atmosphere, heating the solvent, and using a simple separation procedure. The loaded Pt17 is a Ptn cluster within the size range showing high catalytic activity.271 Thus, it is expected that by using the method established in this study, many research groups can conduct further investigations on Pt17/γ-Al2O3 to obtain a deeper understanding of this catalyst and will find new ways of using Pt17/γ-Al2O3 catalysts and Pt17 loaded on the other oxide supports.
2.2. Reduction reaction
 | ||
| Scheme 6 Semihydrogenation of terminal alkyne using metal NC catalysts. Generally, the reaction is conducted in the presence of a base, such as pyridine, because amine additives can largely promote the semihydrogenation reaction,273 and pyridine gave the best catalytic result among them.236 The reaction was conducted under H2 atmosphere in ethanol or/and H2O. A = terminal alkyne, B = alkene. | ||
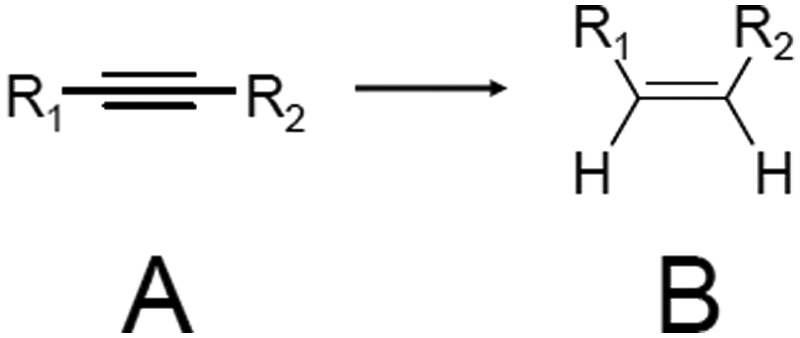
In 2014, Jin et al. used Au25(PET)18/oxide (TiO2, CeO2, SiO2, or Al2O3) for the semihydrogenation reaction of various terminal alkyne compounds (the products are the terminal alkenes; Scheme 6).236,273 In this study, the ligands were not removed because the catalyst was calcined at 150 °C. Au25(PET)18/TiO2 obtained under such conditions showed, for example, a conversion efficiency and selectivity of almost 100% for the semihydrogenation reaction of 4-phenyl-1-butyne. For the mechanism, it was speculated that the reaction started by adsorption of the terminal alkyne to the plane triangular Au3 site in Au25(PET)18 (Fig. 23). Au25(PET)18/TiO2 showed little activity against semihydrogenation of internal alkynes (Scheme 7),236,273 which do not contain the terminal alkyne. In contrast, a sample prepared by the calcination of Au25(PET)18/TiO2 at 300 °C for 1 h under vacuum showed a conversion efficiency in the range of 52.6–59.7% and selectivity of more than 97% for such semihydrogenation of internal alkynes. This finding indicates that ligand removal is essential for the progress of semihydrogenation of internal alkynes.
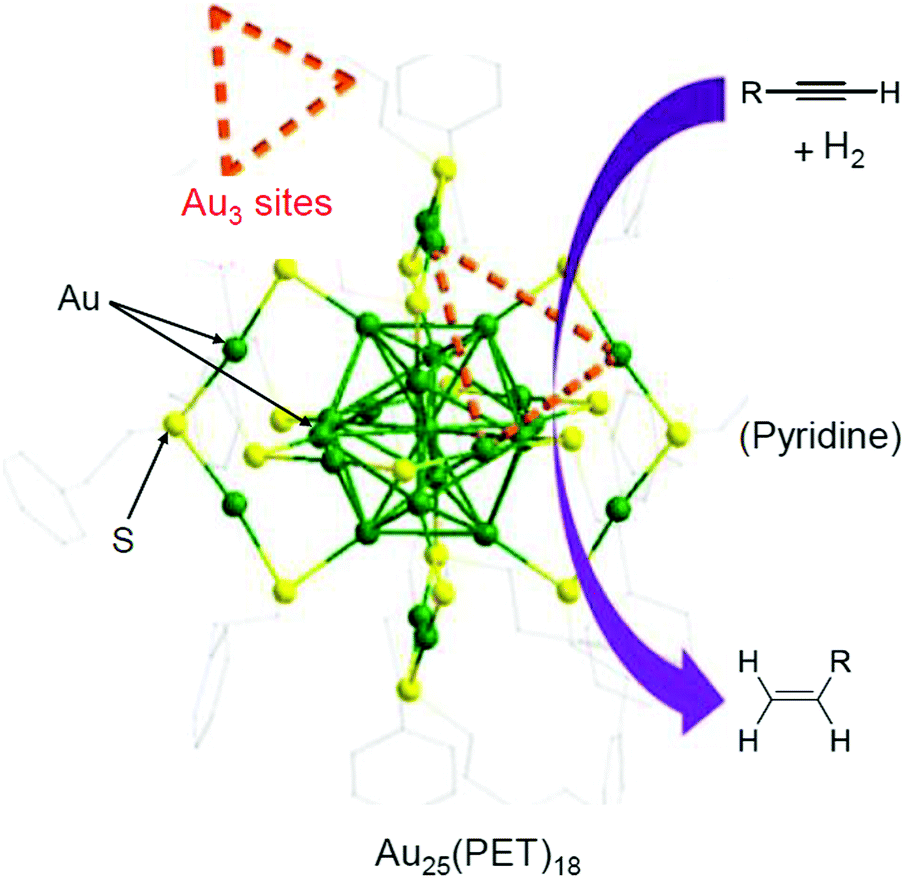 | ||
| Fig. 23 Schematic illustration of semihydrogenation of Au25(PET)18/TiO2. Reproduced with permission from ref. 236. Copyright 2014 American Chemical Society. | ||
 | ||
| Scheme 7 Semihydrogenation of internal alkyne using metal NC catalysts. Generally, the reaction is conducted in the presence of a base, such as pyridine, because amine additives can largely promote the semihydrogenation reaction,273 and pyridine gave the best catalytic result among them.236 The reaction was conducted under H2 atmosphere in ethanol or/and H2O. A = internal alkyne, B = alkene. | ||
In 2017, Wang et al. succeeded in the precise synthesis of [Au38(PhCC)20(PPh3)4]2+ (PhCC = phenylacetylene; Fig. 2J) and [Au38(m-MBT)20(PPh3)4]2+ (m-MBT = 3-methylbenzenethiolate; Fig. 2K). These two Au NCs have the same number of Au atoms and ligands. The authors conducted a catalytic study using these two NCs.178 Au38(PhCC)20(PPh3)4/TiO2 showed a conversion efficiency of more than 97% for semihydrogenation of both the terminal alkyne and internal alkyne. This result differs from the above result for Au25(PET)18/TiO2: for Au25(PET)18/TiO2, semihydrogenation of the internal alkyne proceeded only after ligand removal. This difference was interpreted to be caused by the differences in the mechanisms of the semi-hydrogenation reactions for the two systems. For Au38(m-MBT)20(PPh3)4/TiO2, only a very low conversion efficiency was observed. The authors also investigated the effect of calcination of Au38(PhCC)20(PPh3)4/TiO2 on the semihydrogenation reaction of diphenylacetylene. For calcination at 130–340 °C, the selectivity of trans-stilbene improved with increasing temperature. It was interpreted that the elimination of part of the ligands formed a larger space on the surface of the metal NCs, which worked effectively for the formation of these bulky trans-stilbene.
 | ||
| Scheme 8 Hydrogenation of nitrobenzene using metal NC catalysts. The reaction was conducted under a flow of H2 in toluene. A = nitrobenzene, B = aniline. | ||
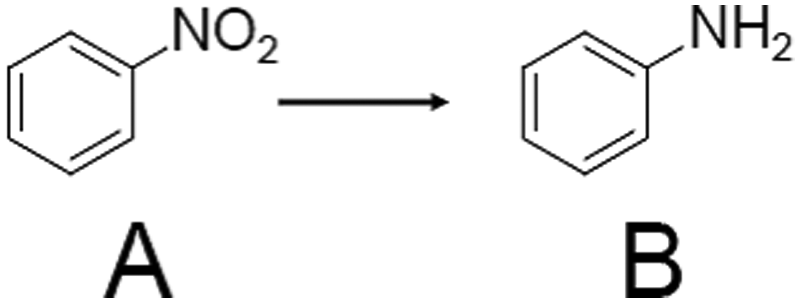
In 2015, Yan et al. studied the catalysis for hydrogenation of nitrobenzene to aniline (Scheme 9).223 Aniline is treated as an intermediate in the manufacture of medical drugs, polymers, herbicides, and other substances. Aniline is often synthesized from the corresponding aromatic nitro compound by catalytic hydrogenation using a commercially available catalyst. Pt, Pd, and iridium (Ir) are mainly used as catalysts. However, in these catalytic reactions, not only the nitro group but also the carbonyl and other double bonds are reduced. Therefore, the development of a catalyst that selectively reduces nitro groups is required. In the study by Yan et al. described in Section 2.1.3, Au25/HAP (more exactly, Au25 aggregates to a particle size of 2.4 nm on HAP in this study) showed a conversion efficiency and selectivity of more than 99% for the hydrogenation reaction from nitrobenzene to aniline. Almost no activity was observed for Au25(MHA)18/HAP without calcination. From this result, it was concluded that ligand removal is necessary for this reaction as well.
 | ||
| Scheme 9 Reduction of 4-nitrophenol using metal NC catalysts. Generally, the reaction proceeds in the presence of NaBH4. The reaction was conducted in a mixture of H2O and tetrahydrofuran. A = 4-nitrophenol, B = 4-aminophenol. | ||
In 2017, Zhang and Liu et al. prepared the catalyst Au25/ZnAl-HT (ZnAl-HT = hydrotalcite) using Au25(Cys)18 (Cys = cysteine) and ZnAl-HT. They found that it selectively reduces the nitro group of 3-nitrostyrene (Fig. 24 and Scheme 10).238 This catalyst was prepared by calcination at a temperature above 300 °C. Based on TEM images, it was interpreted that calcination caused a slight increase in particle size from 1.4 to 1.7 nm. The obtained Au25/ZnAl-HT showed a conversion efficiency of 100% and 3-vinylaniline selectivity of 99% at a reaction temperature of 90 °C (Fig. 24). 3-Nitrostyrene has two functional groups, the nitro group and the alkene (CC). It was interpreted that Au25/ZnAl-HT selectively reduced the nitro group because the former was easier to approach to ZnAl-HT than the latter. Because this catalyst showed no catalytic activity when uncalcined, it was interpreted that this catalytic reaction could only proceed by removing the ligand of Au25 NCs.
 | ||
| Fig. 24 Schematic of hydrogenation of 3-nitrostyrene using Au25/ZnAl-HT. Reproduced with permission from ref. 238. Copyright 2017 Wiley-VCH. | ||
 | ||
| Scheme 10 Hydrogenation of 3-nitrostyrene using metal catalysts. The reaction was conducted under H2 atmosphere in toluene. A = 3-nitrostyrene, B = 3-vinylaniline, C = 3-ethylaniline, D = 3-ethylnitrobenzene. | ||
2.3. H2 generation
Ammonia borane is attracting attention as a lightweight, highly stable, and highly efficient hydrogen storage agent. In 2015, Tsukuda et al. reported that Ag44/MPC (MPC = mesoporous carbon) functions as a catalyst for dehydrogenation of ammonia borane (NH3BH3; Scheme 11).239 In catalyst preparation, first, [Ag44(SPhF)30](PPh4)4 (SPhF = 4-(fluorophenyl)thiolate; Fig. 2L) was adsorbed on MPC, and then the obtained sample was calcined at 150 °C for 30 min and then at 300 °C for 2 h under vacuum. It was revealed that the Ag–S bond dissociated and the ligand was desorbed as SPhF or (SPhF)2 by such calcination (Fig. 25A). This ligand desorption mechanism was largely different from that observed for SC12-protected Ag NCs (Agn(SC12)m NCs), in which S–C dissociation also proceeds at the same time. The S–C bond in [Ag44(SPhF)30](PPh4)4 is stronger than that in Agn(SC12)m NCs. This difference was interpreted to be related to the selective dissociation of the Ag–S bond and the complete desorption of S from the Ag44 surface. The obtained Ag44/MPC showed high catalytic activity for the dehydrogenation reaction from ammonia borane under oxygen or air atmosphere. Some comparison experiments revealed that the dehydrogenation reaction using Ag44/MPC as a catalyst proceeded via a mechanism different from that proceeding when NPs composed of other metals such as Pt and Pd were used as catalysts (Fig. 25B). | ||
| Scheme 11 Reaction of dehydrogenation of ammonia diborane using metal NC catalysts. The reaction was conducted in H2O under air atmosphere. | ||
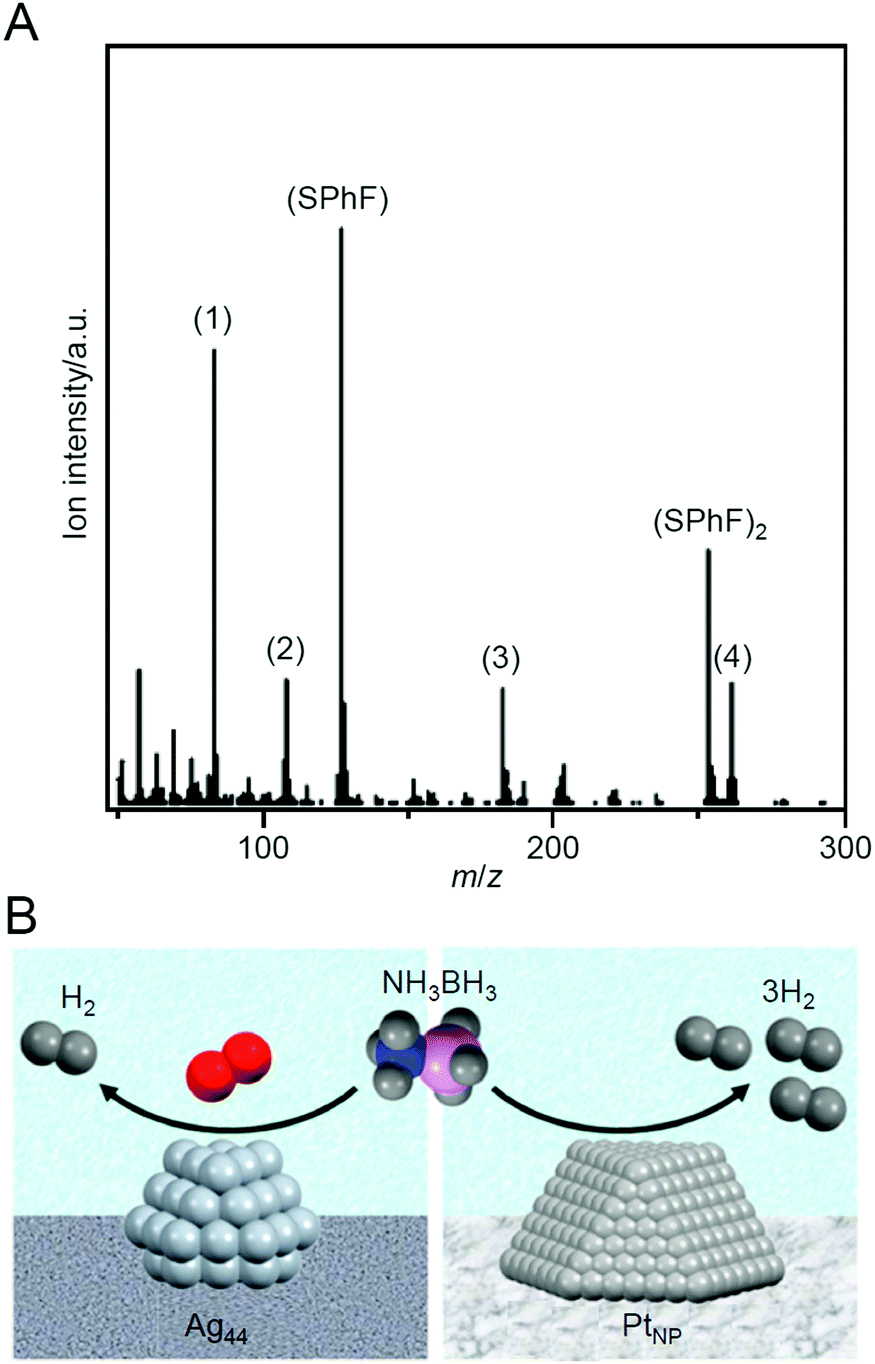 | ||
| Fig. 25 (A) Typical mass spectrum of the desorbed species from [Ag44(SPhF)30](PPh4)4 at 297 °C. The mass peaks of (1) PhF, (2) PPh, (3) PPh2, and (4) PPh3 are fragments of PPh4 and SPhF, produced by electron-impact ionization. (B) Comparison of H2 generation from NH3BH3 in the presence of Ag44 NCs/MPC and PtNP/MPC. Reproduced with permission from ref. 239. Copyright 2015 American Chemical Society. | ||
2.4. Photocatalytic water splitting
With global warming and the depletion of fossil resources, our fossil-fuel-dependent society is expected to shift to one that instead uses H2 as a clean and renewable energy source. Water-splitting photocatalysts (Fig. 26A) can produce H2 from water using sunlight, both of which are almost infinite on the earth.274 Therefore, the photocatalytic water-splitting reaction is considered one of the cleanest energy-production reactions for humankind. However, further improvement of the activity is necessary to enable their practical application.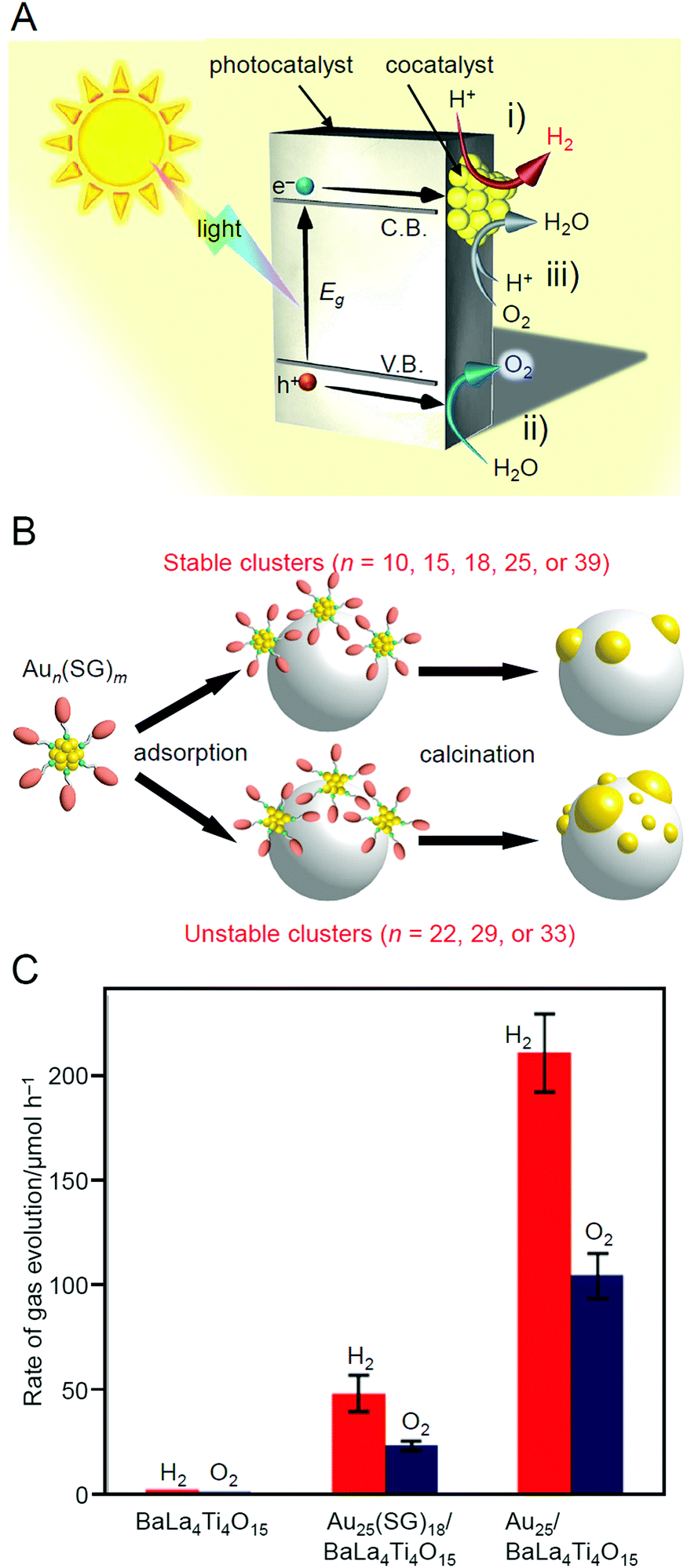 | ||
| Fig. 26 (A) Schematic of photocatalytic water splitting using a one-step photoexcitation system (C.B., conduction band; V.B., valence band; Eg, band gap) showing the processes of (i) hydrogen evolution, (ii) oxygen evolution, and (iii) oxygen photoreduction. (B) Schematic of the aggregation of Aun(SG)m on the photocatalyst depending on the cluster size (0.1 wt% Au). (C) Comparison of the photocatalytic activities of BaLa4Ti4O15, Au25(SG)18/BaLa4Ti4O15, and Au25/BaLa4Ti4O15. Reproduced with permission from ref. 241 and ref. 244. Copyright 2015 American Chemical Society and 2019 American Chemical Society. | ||
In many cases, water-splitting photocatalysts need to load metal NPs/NCs (cocatalysts) as active sites (Fig. 26A),275 and their particle-size control is an extremely effective means for improving the water-splitting activity.276,277 Several previous studies have shown that the use of atomically precise metal NCs as precursors could load metal NCs with controlled particle size on photocatalysts, which could lead to improvement of the water-splitting activity.200,239–245,278–280
In 2013, Negishi and Kudo et al. succeeded in loading Au25 on BaLa4Ti4O15, which is one of the highly active photocatalysts. In the experiment, Au25(SG)18 was first adsorbed on BaLa4Ti4O15 with high efficiency (>99%) by stirring them in water.240 Glutathione has two carboxyl groups and one amino group. It can be considered that Au25(SG)18 was efficiently adsorbed on the photocatalyst because the polar functional groups of Au25(SG)18 formed hydrogen bonds with the –OH groups on the surface of BaLa4Ti4O15. Then, the obtained Au25(SG)18/BaLa4Ti4O15 was calcined at 300 °C for 2 h to remove the ligands from Au25(SG)18. TEM images and XPS spectra revealed that such calcination resulted in (1) removal of ligands from Au NCs and (2) little aggregation of the loaded Au NCs. The obtained Au25(SG)18/BaLa4Ti4O15 exhibited higher water-splitting activity than AuNP/BaLa4Ti4O15 in which a large AuNP was loaded by the impregnation method. In 2015, the same group clarified that (1) this loading method is also effective for loading other sizes of Aun NCs; (2) however, to load Aun NCs on BaLa4Ti4O15 while preventing the increase in particle size, it is necessary to use stable Aun(SG)m NCs as a precursor (Fig. 26B), and (3) it is essential to remove the ligands by calcination to obtain higher activity (Fig. 26C).241 Furthermore, in 2018, the same group clarified that the precise loading of Au25 is also possible in the same way when SrTiO3 is used as a photocatalyst.242
Because such an overall water splitting is an uphill reaction (Fig. 27A),281 the reverse reaction proceeds easily.243 Therefore, to create a highly active photocatalyst in such a reaction system, it is necessary to efficiently suppress the reverse reaction. In addition, light irradiation of the photocatalyst promotes aggregation of the cocatalyst.240 Therefore, to create a highly stable and active photocatalyst, it is necessary to take some measures to suppress the aggregation of the cocatalyst. One effective means to suppress the reverse reaction of water splitting is to form a chromium oxide (Cr2O3) shell on the surface of the loaded cocatalyst. This Cr2O3 shell is permeable to H+ but not to O2 approaching from the outside (Fig. 27B). Domen et al. reported that when the cocatalyst surface is covered by the Cr2O3 shell, it is possible to suppress only the progress of the reverse reaction while maintaining the H2 evolution activity.186,188–190,282–288 In their studies, they first loaded the cocatalysts and then formed the Cr2O3 shell on the cocatalysts using the photodeposition method (Fig. 27C(a)). However, when Au25/BaLa4Ti4O15 was irradiated with light, the loaded metal NCs aggregated.240 Thus, when the method reported by Domen et al. is used as is, it is difficult to form the Cr2O3 shell on the surface of the loaded metal NCs while maintaining the number of atoms of the metal NCs. Meanwhile, studies in the field of surface science have revealed that when a metal oxide support loaded with metal NCs is heated under H2 or O2 atmosphere, a strong metal–support interaction (SMSI) is induced, thereby leading to the formation of an oxide film on the metal NCs.289–296 Therefore, Kurashige et al. attempted to form a Cr2O3 shell on the surface of the cocatalyst using such a SMSI effect (Fig. 27C(b)).243 As a result, they created a highly functional water-splitting photocatalyst that addresses both the reverse reaction and the aggregation of Au25.
 | ||
| Fig. 27 (A) Activation energy of water-splitting reaction.281 (B) Schematic model of H2 evolution reaction on core/shell noble-metal/Cr2O3 particulate system as a cocatalyst for photocatalytic overall water splitting.287 (C) Comparison of the procedures of Cr2O3 shell formation in (a) the literature286 and (b) our work.243 (D) TEM images (a, c and e) and HR-TEM images (b, d and f) of (a and b) Cr2O3/BaLa4Ti4O15 (0.5 wt% Cr), (c and d) Au25(SG)18/Cr2O3/BaLa4Ti4O15 (0.1 wt% Au; 0.5 wt% Cr), and (e and f) Cr2Oy/Au25/BaLa4Ti4O15 (0.1 wt% Au; 0.5 wt% Cr). Cr2Oy indicates the chromium oxide in which part of the chromium was oxidized to a highly oxidized state (>3+). In (c and e), the insets show the core-size distributions of the particles. In (b and f), the thicknesses of the Cr2O3 or Cr2Oy shells are indicated by yellow double-pointed arrows. In (f), the particle size is also indicated by a white double-pointed arrow. Reproduced with permission from ref. 243 and ref. 287. Copyright 2018 American Chemical Society and 2009 American Chemical Society. | ||
In the Cr2O3-shell-formation process used in their study, the Cr2O3 layer was first formed on BaLa4Ti4O15 using the photodeposition method before loading Au25 (Fig. 27D(a) and (b)). Then, Au25(SG)18 was adsorbed on the surface of the obtained Cr2O3/BaLa4Ti4O15 (Fig. 27D(c) and (d)). The photocatalyst after the adsorption of Au25(SG)18 (Au25(SG)18/Cr2O3/BaLa4Ti4O15) was calcined at 300 °C for 2 h under a reduced pressure. The Au25 was covered with a Cr2O3 shell during calcination via the SMSI effect (Fig. 27D(e) and (f)). Part of the chromium was oxidized to a highly oxidized state (>3+) by calcination; however, it was reduced to Cr2O3 by irradiation with UV light. The obtained Cr2O3/Au25/BaLa4Ti4O15 showed nearly 20 times higher water-splitting activity than Au25/BaLa4Ti4O15. Furthermore, even when the light irradiation was continued for 10 h, almost no aggregation of Au25 occurred.243 In 2019, Kurashige et al. reported that substituting one Au atom of Cr2O3/Au25/BaLa4Ti4O15 with a Pt atom could induce even higher water-splitting activity.244 In 2020, they succeeded in obtaining the highest quantum yield (16% at 270 nm) for BaLa4Ti4O15 catalysts by using Rh instead of Au as the base element of the cocatalysts (Fig. 28).245 For Rh2−xCrxO3/BaLa4Ti4O15, the cocatalyst is not currently controlled with atomic precision. However, if a precise synthesis method is established for Rhn NCs in the future, it may become possible to control cocatalysts with atomic precision for Rh2−xCrxO3/BaLa4Ti4O15. It is expected that the establishment of such precise synthesis methods will enable us to create even more highly active Rh2−xCrxO3/BaLa4Ti4O15.
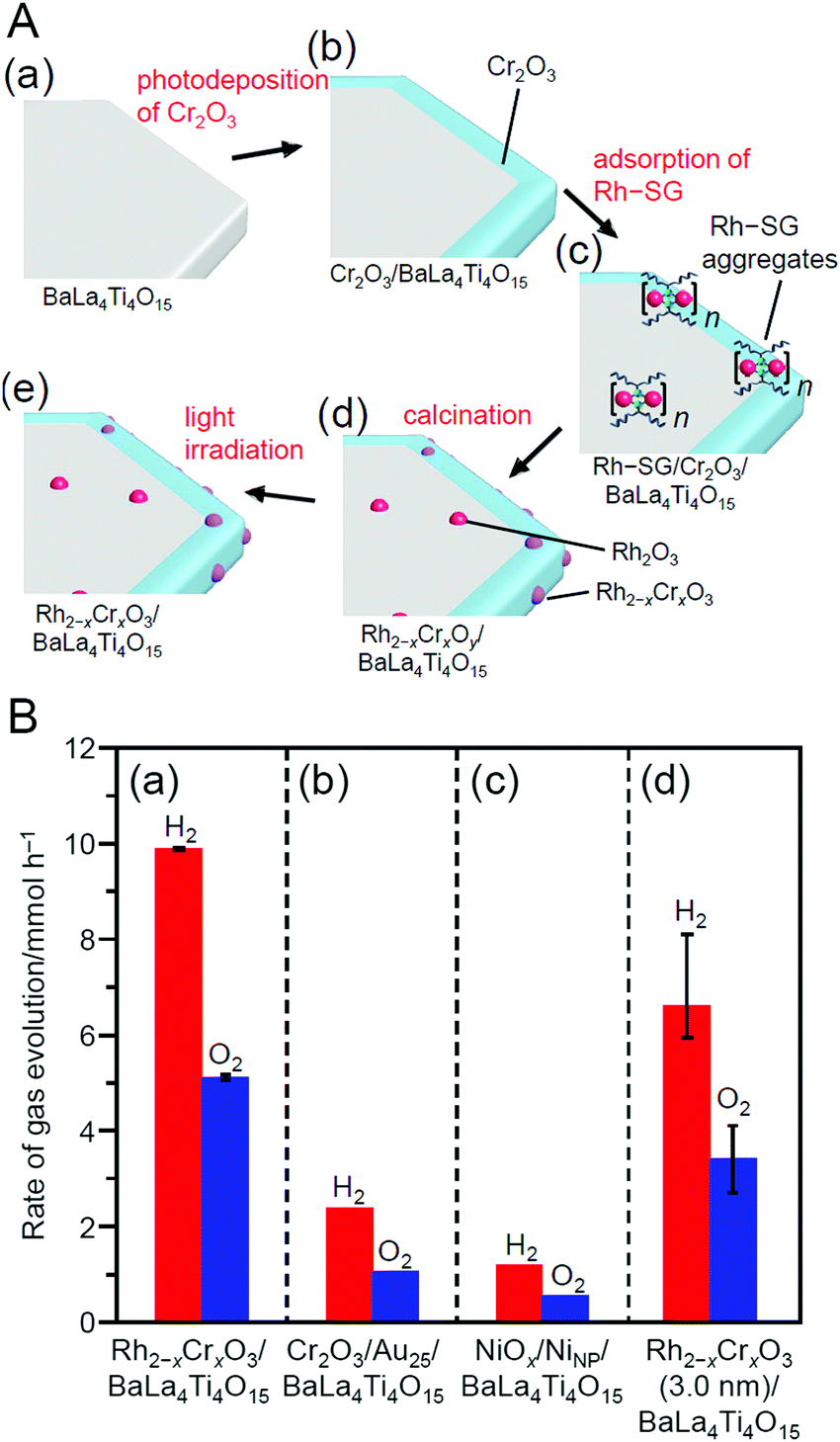 | ||
| Fig. 28 (A) Schematic of the experimental procedure for the formation of Rh2−xCrxO3/BaLa4Ti4O15. (a) BaLa4Ti4O15, (b) Cr2O3/BaLa4Ti4O15, (c) Rh–SG/Cr2O3/BaLa4Ti4O15, (d) Rh2−xCrxOy/BaLa4Ti4O15, and (e) Rh2−xCrxO3/BaLa4Ti4O15. Rh2−xCrxOy indicates Rh2−xCrxO3 including highly oxidized Cr (>3+). (B) Comparison of rates of H2 and O2 evolution by photocatalytic water-splitting over different photocatalysts (a) Rh2−xCrxO3/BaLa4Ti4O15 (0.09 wt% Rh and 0.10 wt% Cr), (b) Cr2O3/Au25/BaLa4Ti4O15 (0.10 wt% Au and 0.50 wt% Cr), (c) NiOx/NiNP/BaLa4Ti4O15 (0.50 wt% Ni), and (d) Rh2−xCrxO3 (3.0 nm)/BaLa4Ti4O15 (0.10 wt% Rh and 0.15 wt% Cr). In this study, NiOx/NiNP/BaLa4Ti4O15 and Rh2−xCrxO3 (3.0 nm)/BaLa4Ti4O15 loaded with Rh2−xCrxO3 NPs of ∼3.0 nm were prepared by the impregnation method. Reproduced with permission from ref. 245. Copyright 2020 Wiley-VCH. | ||
The above studies used photocatalysts that can achieve the overall water-splitting reaction using UV light. However, to enable the practical application of water-splitting photocatalysts in the future, it is necessary to use visible light, which accounts for ∼40% of solar energy, for the water-splitting reaction.297 To completely decompose water, the positions of its conduction band (C. B.) and valence band (V. B.) must have sufficient energy for the H2 evolution reaction (HER) and O2 evolution reaction (OER) to proceed, respectively (Fig. 26A). Furthermore, to suppress the recombination of excited electrons and holes, the photocatalyst needs to have both high crystallinity and high specific surface area. Because of these requirements, there are currently only a few semiconductor photocatalysts that can completely split water in one step using visible light.298 Nevertheless, overall water splitting can also be achieved by combining a semiconductor causing its half-reaction (HER and OER) and a redox couple responsible for the transfer of charges (electrons and holes) between them.299 Such a system, which imitates the photosynthesis occurring in plants, is called the Z-scheme system (Fig. 29A).
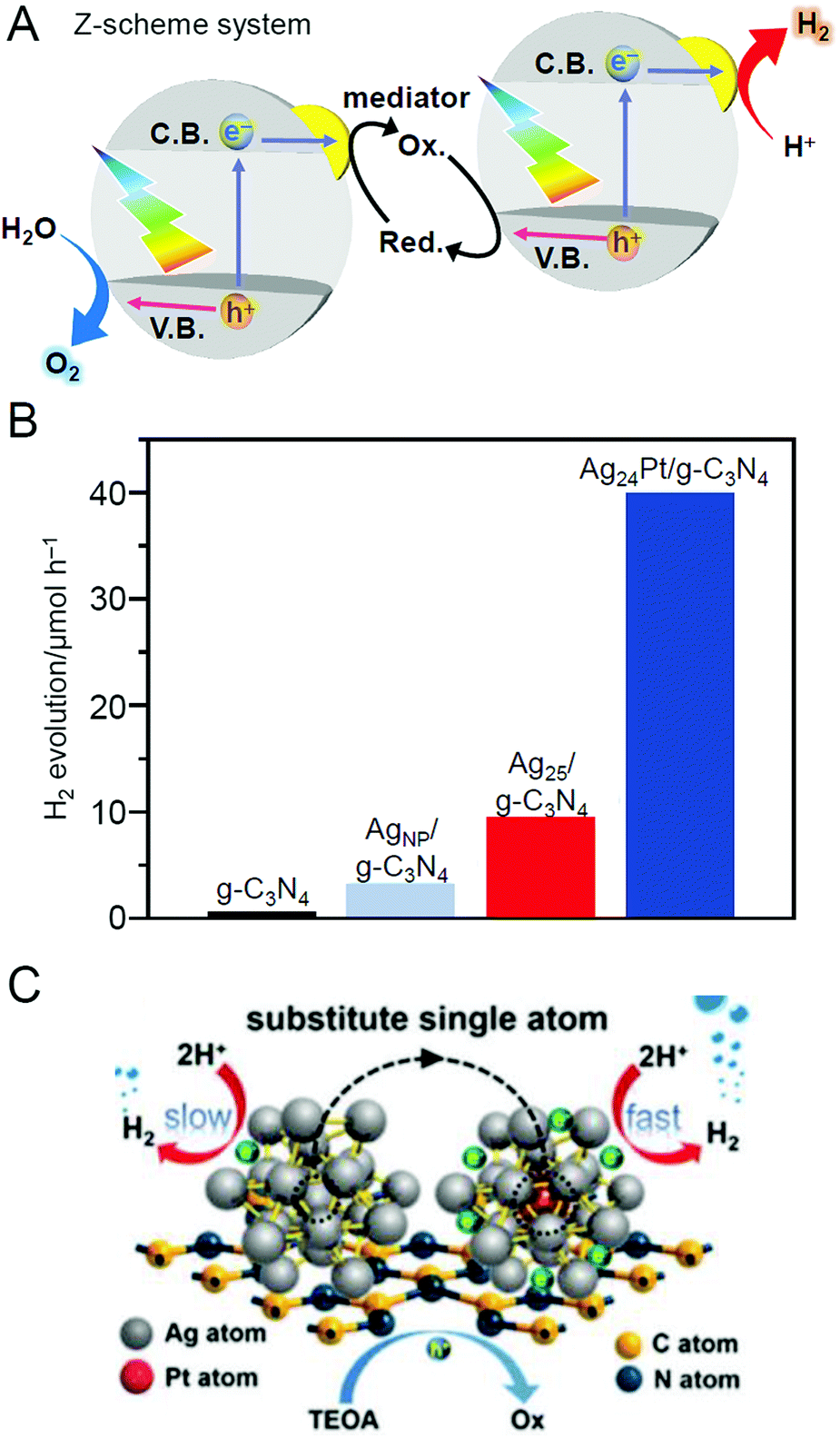 | ||
| Fig. 29 (A) Schematics of Z-scheme system for overall water splitting. (B) Comparison of the photocatalytic H2-evolution activities of g-C3N4, AgNP/g-C3N4, Ag25/g-C3N4, and Ag24Pt/g-C3N4. (C) Proposed photocatalytic H2-evolution mechanism. Reproduced with permission from ref. 200 and ref. 297. Copyright 2017 Royal Society of Chemistry and 2020 Royal Society of Chemistry. | ||
In 2017, Yang et al. succeeded in highly activating the graphitic carbon nitride (g-C3N4), which functions as a HER photocatalyst in the Z-scheme system, by using atomically precise Ag25(2,4-SPhMe2)18PPh4 (Fig. 2G) and [Ag24Pt(2,4-SPhMe2)18](PPh4)2 (Fig. 2M) as precursors of cocatalysts.200 In the experiment, each NC was first adsorbed on g-C3N4 by stirring them in mixed solvent of toluene and dichloromethane for 12 h. Then, the ligand was removed from Ag25 or Ag24Pt by calcination at 150 °C under argon (Ar) atmosphere for 2 h. These photocatalysts were dispersed in an aqueous solution containing triethanolamine (TEOA; sacrificial agent), and the HER was proceeded by irradiating the aqueous solution with visible light (>420 nm). As a result, Ag25/g-C3N4 and Ag24Pt/g-C3N4 generated 76 and 330 times more H2 than g-C3N4 without the cocatalyst, respectively (Fig. 29B). The authors reported that Ag24Pt/g-C3N4 showed higher activity than Ag25/g-C3N4 because Pt substitution facilitated the transfer of photoexcited electrons from g-C3N4 to the cocatalyst (Fig. 29C). Thus, it was shown that cocatalyst control using atomically precise metal NCs is also effective for high activation of the g-C3N4 HER photocatalyst.
2.5. Electrocatalytic reaction
In the photocatalytic water-splitting reaction described in Section 2.4, H2 and O2 are directly generated from water by sunlight. These reactions convert light into chemical energy and are therefore also called artificial photosynthesis. In contrast, the use of a solar cell300 enables us to obtain electrical energy from sunlight. When water is electrolyzed using the obtained electrical energy,301–304 it is possible to obtain chemical energy from sunlight and water, similar to the case of a photocatalytic reaction.305,306The electrolysis of water consists of two half-reactions, the HER and OER. When a voltage is applied on the metal electrode, a reduction reaction proceeds at the cathode and an oxidation reaction proceeds at the anode, and water molecules are electrolyzed into H2 and O2. However, in reality, the reaction does not proceed even if a higher potential than the redox potential (HER: 0 V vs. SHE, OER: 1.23 V vs. SHE) in each reaction is applied onto the metal electrode. This is because the activation energy in each reaction is high. Therefore, precious-metal particles are loaded on the electrode as catalysts.307 Previous studies have revealed that atomically precise metal NCs are also extremely promising as such electrode catalysts.180,246,247
In 2017, Chen et al. conducted a study using Pd6(SC12)12 as a precursor of an electrode catalyst (Fig. 30A).246 In their experiment, Pd6(SC12)12 was first adsorbed on AC, and then the catalysts were calcined at 200 °C under reduced pressure. During adsorption and calcination (Fig. 30B), almost no aggregation of metal NCs occurred. In the HER measurement, 0.5 M sulfuric acid (H2SO4) was used as the electrolyte. The Pd6/AC showed high HER activity and durability (Fig. 30C). For example, Pd6/AC has 2.86 and 10.22 times higher current densities than Pd6(SC12)12/AC and commercially available Pt/carbon (C) catalysts, respectively, when comparing the HER activity at an applied voltage of −0.423 V. The removal of the ligand formed a wider space around Pd and reduced the charge density of Pd. It was interpreted that because the removal of the ligand made it easier for the reaction substrate (H+) to approach Pd, it improved the HER activity. In contrast, Pd6(SC12)12/AC showed higher activity than Pd6/AC and Pt/C for the OER. These results indicate that the presence or absence of ligand removal should be determined while taking the reaction mechanism into consideration.
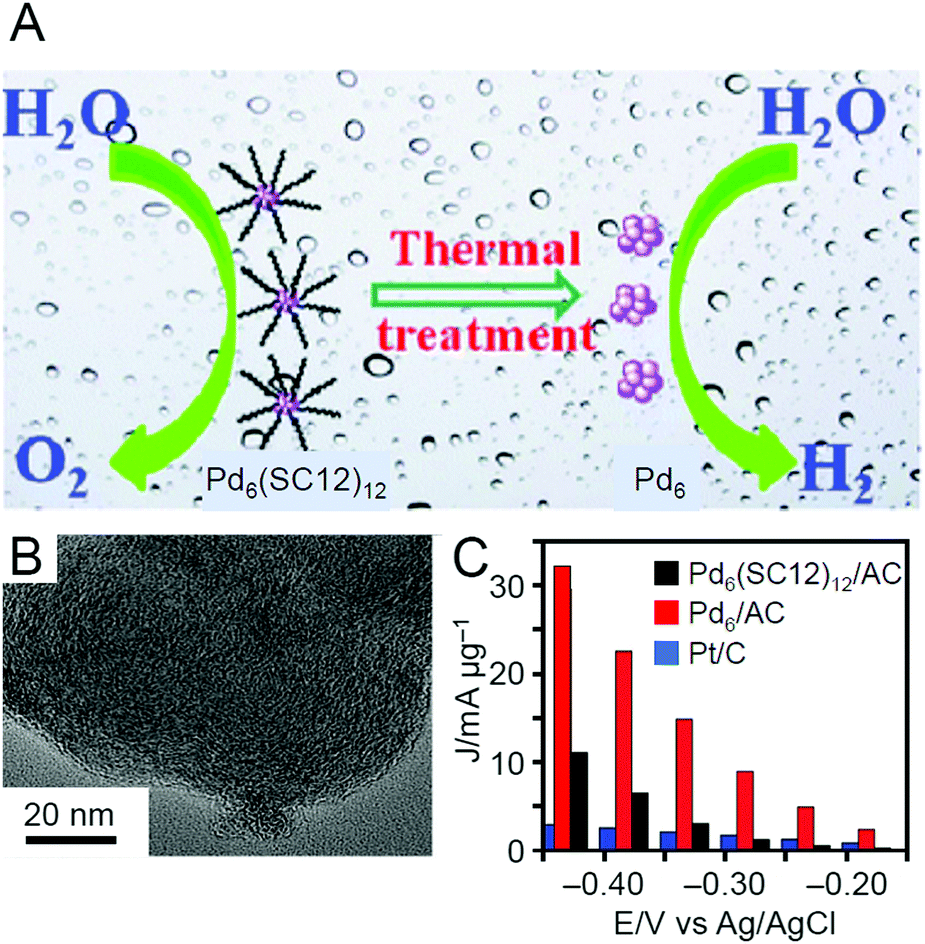 | ||
| Fig. 30 (A) Schematic of difference in electrochemical reaction before and after thermal treatment of Pd6(SC12)12/AC (B) HR-TEM images of Pd6/AC. (C) Comparison of the current densities of the HER on the samples at different potentials. Reproduced with permission from ref. 246. Copyright 2017 Royal Society of Chemistry. | ||
In 2018, Zhu et al. used Au2Pd6S4(PPh3)4(SPhF2)6 (SPhF2 = 3,4-difluorobenzenethiolate; Fig. 2N), which was newly synthesized in their study, as a metal NC and molybdenum disulfide (MoS2) having high HER activity as a support.180 The catalyst was obtained by first adsorbing Au2Pd6S4(PPh3)4(SPhF2)6 on MoS2 and then calcining the catalysts at 100 °C under Ar atmosphere (Fig. 31A and B). The paper did not refer to whether the ligand was removed by such calcination. The resulting catalyst (hereafter referred to as Au2Pd6/MoS2 regardless of the existence of the ligand) showed higher HER activity than MoS2, Au2/MoS2, Pd3/MoS2, and (Au2 + Pd3)/MoS2 (Fig. 31C). Several experiments and density functional theory (DFT) calculation revealed that Au2Pd6/MoS2 exhibited high HER activity because the adsorption site for H atom increased by the charge transfer from Au2Pd6 to MoS2 (Fig. 31D). These results indicate that the loading (or adsorption) of atomically precise metal NCs is also effective for enhancing the functionality of existing HER catalysts.
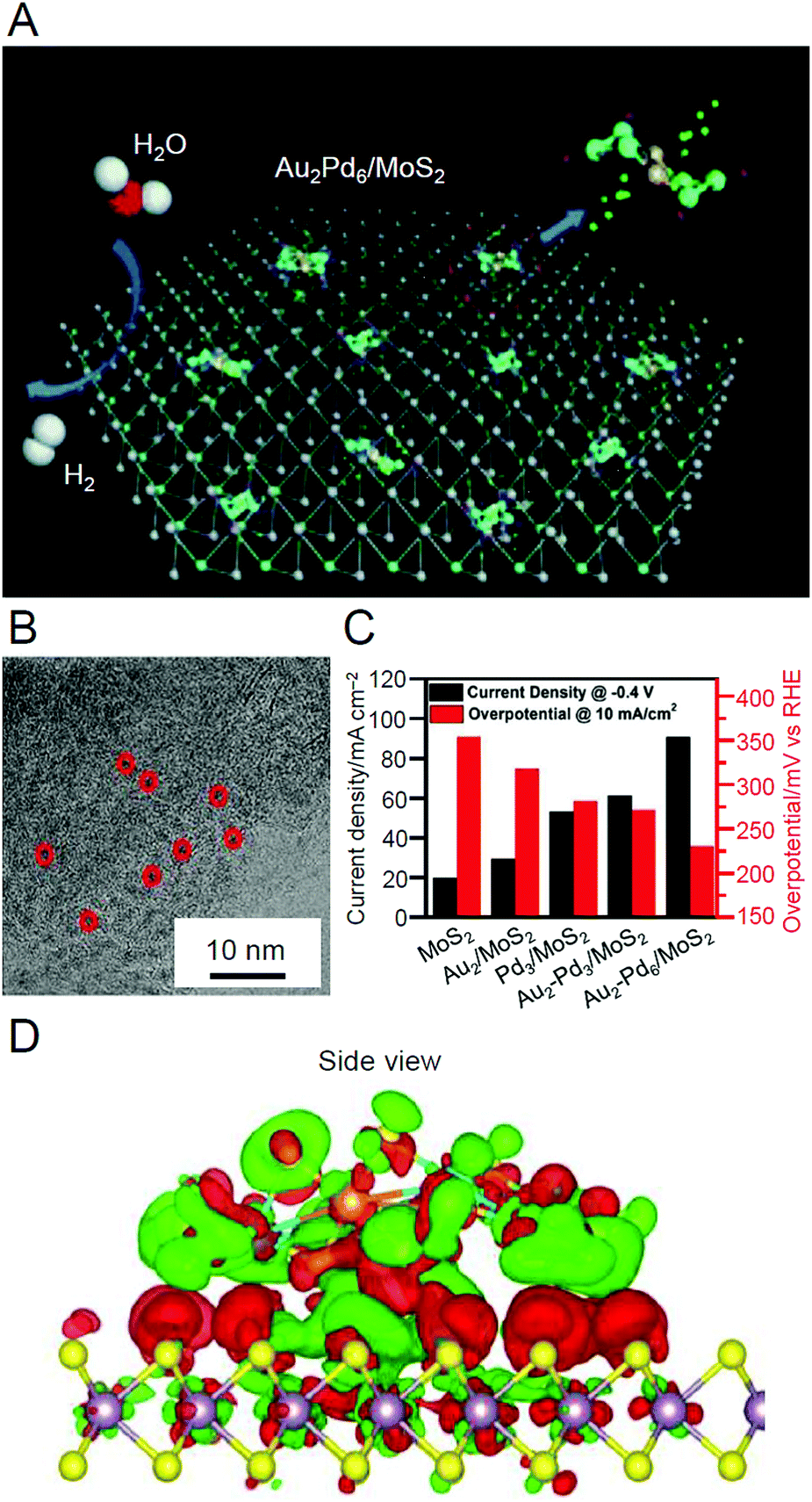 | ||
| Fig. 31 (A) Schematic of HER on Au2Pd6/MoS2 system. (B) HR-TEM image of Au2Pd6/MoS2 system (Au2Pd6 NCs marked by red circles). (C) Comparison of the overpotential at a current density of 10 mA cm−2 (red column on the right) and the current density at −0.4 V vs. RHE (black column on the left). (D) Charge deformation density of Au2Pd6/MoS2 system. The charge density of Au2Pd6 NC decreased while the charge density of MoS2 increased, indicating the charge transfer from NC to MoS2. Reproduced with permission from ref. 180. Copyright 2018 Royal Society of Chemistry. | ||
On the other hand, in 2017, Jin et al. succeeded in promoting the OER, which is often a bottleneck in the electrolysis of water, using atomically precise metal NCs (Fig. 32A).247 In this study, Au25(PET)18/diselenium cobalt nanosheets (CoSe2) were first prepared. The obtained Au25(PET)18/CoSe2 showed higher OER activity than catalysts such as CoSe2 and Pt/C. Then, the authors showed that even higher OER activity could be achieved by calcining Au25(PET)18/CoSe2 at 300 °C under N2 atmosphere (Fig. 32B and C). They interpreted that the OER activity was improved because the electronic interaction between Au NCs (slightly increased in size by calcination) and CoSe2 was increased by calcination of the ligand. They also investigated why the OER activity of the CoSe2 catalyst was improved by loading Au NCs by conducting additional experiments and DFT calculation (Fig. 32D). In addition, this study revealed that the OER activity increases with increasing size of Au NCs.
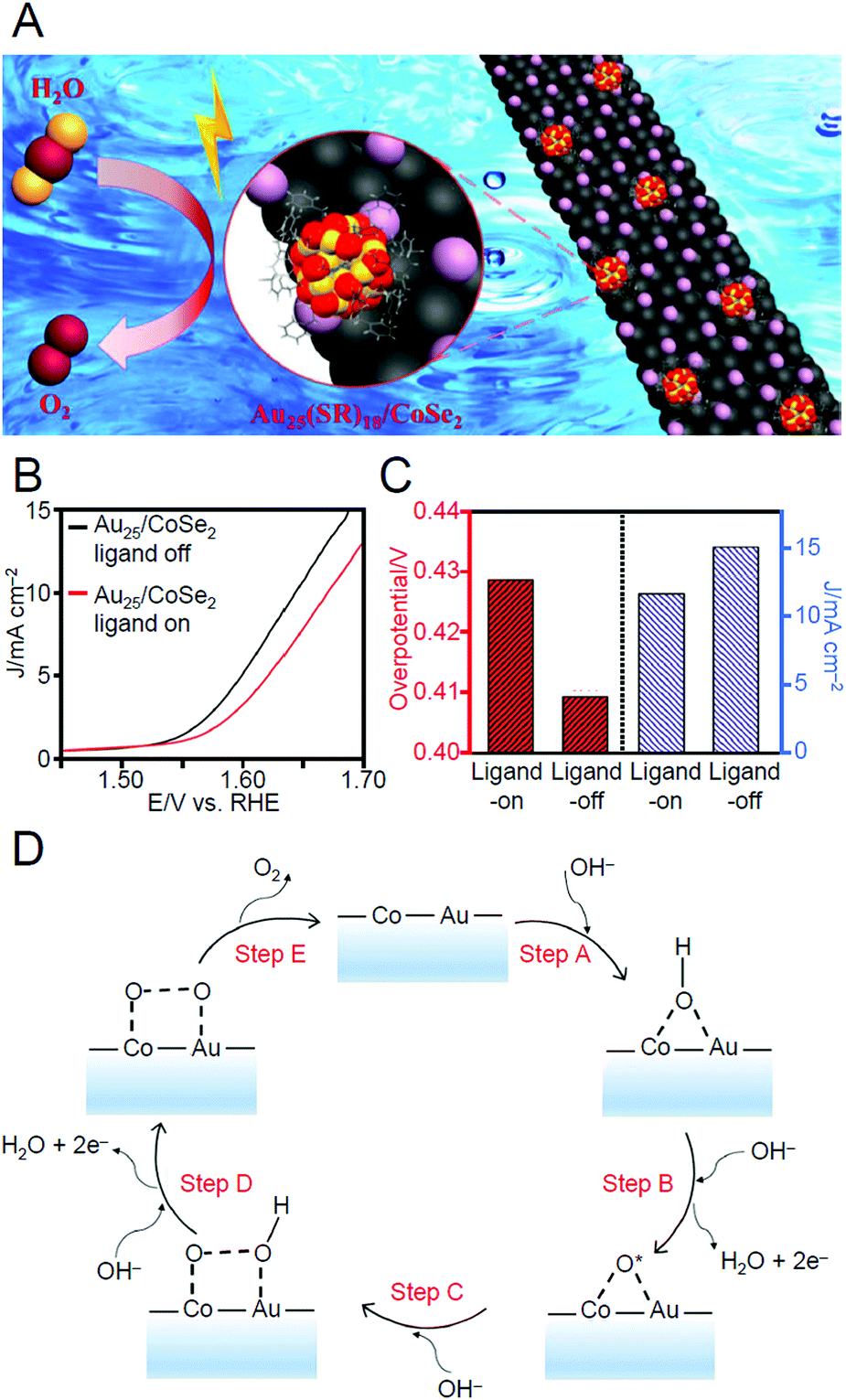 | ||
| Fig. 32 (A) Schematic of OER on Au25/CoSe2 catalysts. (B) OER polarization curves for ligand-on and ligand-off Au25/CoSe2 catalysts. (C) Comparison of the overpotential (at 10 mA cm−2) and the current density at the overpotential of 0.45 V for ligand-on and ligand-off Au25/CoSe2 catalysts. (D) Proposed mechanisms for OER in the presence of a Au atom on the CoSe2 catalyst surface. Reproduced with permission from ref. 247. Copyright 2017 American Chemical Society. | ||
In addition to these photocatalytic water-splitting reactions, Crudden, Tsukuda, and Häkkinen reported the study on the electrocatalytic reduction of CO2 to CO using N-heterocyclic carbene (NHC)-fuctionalized Au11 NCs (Fig. 33A), which was obtained by exchanging the ligand of [Au11(PPh3)8Cl2]+ with N-heterocyclic carbene.308 This reaction is an important one because CO is the key component in many high-volume carbon–carbon bond-formation reactions. Although the Au NCs did not show the catalytic activity before the calcination, when the phosphine was removed by the calcination at 180 °C for 2 h, the Au NCs showed the catalytic activity for this reaction (Fig. 33A). In this study, they found that Au11 NCs bearing a methylated NHC shows an especially high activity (Fig. 33B) and that this high activity is caused by high stability of this NC.
 | ||
| Fig. 33 (A) Geometrical structure of NHC-functionalized Au11 NCs. (B) Electrocatalytic activity for the reduction of CO2 to CO; Au11 NCs bearing a methylated NHC (red), Au11 NCs without NHC (blue), and Au11 NCs bearing an isopropylated NHC (green). Reproduced with permission from ref. 308. Copyright 2019 Springer-Nature. | ||
3. Toward the creation of functional heterogeneous catalysts
In this section, we discuss the suitable conditions for catalyst preparation (Section 3.1), the reaction mechanism during the preparation of catalysts (Section 3.2), and the mechanism of catalysis (Section 3.3), which have been clarified in previous studies.3.1. Suitable experimental conditions
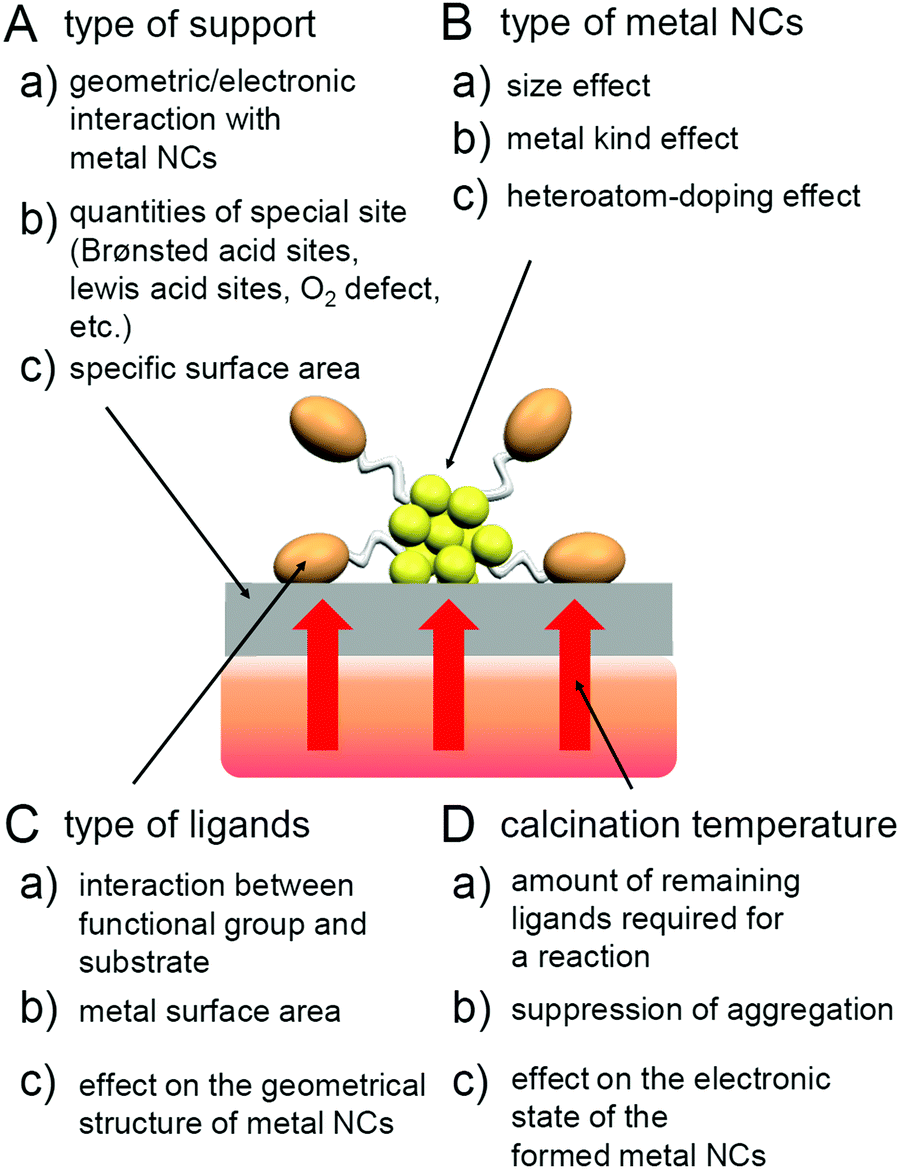 | ||
| Fig. 34 Important factors to consider in creation of highly functional catalysts. | ||
In most of the previous studies, Au has been used as the base element of the loaded metal NCs. For such NCs, the catalytic activity for oxidation reactions is often improved when CeO2 (Table 2) is used as a support.215,217,221,226–233,255 CeO2 changes the charge state of Au in metal NCs to be cationic and induces O2 activation.231,233 Because of these factors, the oxidation reaction is accelerated when CeO2 is used as a support. In addition, the aggregation of Au NCs is suppressed on CeO2 because of the strong interaction between Au NCs and CeO2.217,255 Through this means, the highly active Au NCs/CeO2 can be created without decreasing the unique catalytic activity of small Au NCs. Similar effects were observed when HAP and TiO2 were used as supports (Table 2).178,214,216,218–220,223,225,232,236 In contrast, there is very little information on the selection of the support for the reduction reaction because of the limited number of study examples. However, because suppression of aggregation of Au NCs is extremely important in this case as well, a highly active catalyst is often obtained when HAP or TiO2 is used as the support.219,223
Regarding the correlation between the support and the aggregation of Au NCs, Donoeva and Häkkinen et al. studied how the behavior of PPh3-protected Au NCs during calcination changes depending on the type of the support in 2020.255 In this study, they revealed the following about the behavior of Au9(PPh3)8: (1) when SiO2 containing a Brønsted acid site was used as the support, Au9(PPh3)8 dissociated into Au–PPh3 complexes during calcination because the interaction between Au9(PPh3)8 and SiO2 is weak; and (2) when CeO2 containing Lewis acid sites was used at the support, the ligand could be removed by calcination from Au9(PPh3)8 while maintaining the Au NC size because the interaction between Au9(PPh3)8 and CeO2 is strong. This interpretation was well supported by DFT calculations (Fig. 35A). This study also showed that when TiO2 containing some Brønsted acid sites and small Lewis acid sites was used as a support, little aggregation of Au NCs occurred. This result is generally in good agreement with the series of experimental results (Fig. 35B) reported by Anderson and Nakayama et al. from 2013 to 2016.309–311
 | ||
| Fig. 35 (A) Born–Haber cycle for (a) oxidative fragmentation and (b) ligand migration for the two supports, HO–SiO2 and HO–CeO2. The numbers in pink correspond to values for the hydroxylated amorphous SiO2 surface, and the numbers in blue correspond to energy values for the hydroxylated CeO2(111) surface. ΔHtot is the total reaction enthalpy for the processes, ΔHrd is the energy to reduce and disintegrate the cluster into AuL species in the gas phase, ΔHrp is the energy to reduce and peel the ligands from the cluster forming Au9 and L species in the gas phase, ΔHrh is the energy to remove a H atom from the surface, and ΔHint is the energy due to the interaction between the end species and the surface. Note that the Born–Haber cycle implicitly takes into account the charge imbalance in the total reaction enthalpy ΔHtot. (B) Schematic illustration of gold clusters deposited on titania nanosheet: (a) before annealing and (b) after annealing. The green and orange lines represent the results that can be obtained from atomic force microscopy and scanning tunnel microscopy, respectively. The lines indicating the emission of electrons represent the XPS results. Reproduced with permission from ref. 255 and ref. 311. Copyright 2020 Wiley-VCH and 2016 Royal Society of Chemistry. | ||
To suppress the aggregation of metal NCs on the support, increasing the specific surface area of the support is also effective (Fig. 34A). As described in Section 2.1.1, Tsukuda et al. succeeded in loading ∼1 nm Au NCs on SBA-15 without any aggregation.183 SiO2 has only weak interaction with Au. However, the SBA-15 used in this study had a high specific surface area (866 m2 g−1; Table 2); therefore, Au NCs hardly aggregated on the support. The same authors have also successfully suppressed the aggregation of metal NCs on HPCSs (2300 m2 g−1; Section 2.1.1) and MPC (2300 m2 g−1; Section 2.1.1).209,210 It is expected that increase of a high specific surface area leads to the increase of distance between Au NCs on the substrate and thereby the increase of the number of the defects, which well fix the Au NCs on the substrate, between Au NCs. These factors seem to be related to the suppression of the aggregation of the Au NCs on the substrate. Furthermore, Dai et al. succeeded in suppressing the aggregation of Au NCs on the support by loading CuO or Co3O4 as anchor particles on SBA-15 (395 m2 g−1).234 Because the metal NCs do not easily aggregate on these supports, it is possible to increase the amount of metal NCs loaded on them (Table 2). In the future, it is expected that techniques to create supports with high specific surface area will also be established for supports that show the strong interaction with metal NCs. In addition, recent studies have demonstrated that loading metal NCs on layered double hydroxides;312–314 protecting each metal NC by a silica cage,315–320 zinc oxide hollow cage,321 or atomic-layer-deposition of alumina overlayers;322 and including metal NCs in metal organic frameworks323–328 are also effective for suppressing the aggregation of metal NCs. Therefore, these techniques are expected to be more widely employed for creating highly functional heterogeneous catalysts using atomically precise metal NCs in the future.
In terms of the Au NC size that is effective for creating a highly active catalyst (Fig. 34B), previous studies have demonstrated that, overall, small Au NCs show high activity for catalytic reactions.220,241 However, for benzyl alcohol oxidation, Au144 NCs showed highest conversion efficiency211 and for cyclohexane oxidation, Aun NCs in the n range of 40–80 showed the highest conversion efficiency.212
In terms of metal elements, there are only a few studies on metal NCs consisting of metal elements other than Au. However, for Ag222,239 and Pt,235 it has also been shown that when the number of constituent atoms of NCs is reduced to fewer than 50, higher activity can be achieved for some reactions than for NPs with large particle size. In addition, highly active catalysts have been created by heteroatom doping.209,244 However, to discuss the generality of these effects, it is necessary to conduct studies on metal NCs of various sizes and metal elements in the future.
In some studies, hydrophilic SRs, such as SG, have been used as a ligand of metal NCs.32,96,240–243 When these ligands are used, metal NCs can adsorb on the metal-oxide support with high efficiency via hydrogen bond formation in solution. Even for the hydrophobic SR-protected metal NC, when part of the ligands is replaced by hydrophilic ligands, they can be adsorbed and loaded on the metal-oxide support with high efficiency.244 The catalysts obtained using these methods have the following advantages: (1) it is easy to estimate the quantities of the loaded metal atoms; (2) there is high reproducibility of the metal loading; and (3) it is possible to improve the quantity of the loaded metal atoms.
Meanwhile, when metal NCs are used as catalysts by allowing some or all of the ligands to remain as opposed to removing all of them, the design and selection of the functional group structure of the ligands is more important. In this case, it is necessary to design and select a ligand that can create a suitable structure of the metal NCs178,221,222,229–231,236 and suitable vacancy size on the surface of metal NCs231,236 for the desired reaction to proceed (Fig. 34C).
Previous studies have also shown that the temperature required for the ligand elimination varies depending on the type of ligand and support. Thus, in catalyst preparation, it is necessary to know the start temperature of the ligand elimination and the temperature at which the ligand elimination is completed in the studied system in advance.178,183,209,212,215,218–222,224,226,227 Typically, TGA of unsupported metal NCs is used for this purpose. However, since the support might affect the ligand elimination temperature, it seems to be better to check the presence/absence of metal–ligand bonds by XAFS on the supported metal NCs obtained by the calcination at each temperature. In the calcination, the temperature is typically increased at a heating rate of 10 °C min−1 and kept at a certain temperature in the range of 150–500 °C for several hours. The finish temperature should be determined while checking the particle size at each calcination temperature using TEM, HR-TEM, HAADF-STEM, and XPS309,310 because too high temperature induces the aggregation of metal NCs (Table 3).178,183,209,210,212,214,216–219,223–225,233,234,236,237,239 It is expected that the elimination speed of the ligands and the electronic state of the formed metal NCs change depending on the calcination atmospheres.215,222,226,229 In the previous studies, calcination atmospheres such as vacuum, reduced pressure, air, O2, H2, and inert gas have been used. However, at present, the effects of the calcination atmosphere on the elimination speed of the ligands and the electronic state of the formed metal NCs remain unclear. Therefore, it is expected that more studies will be conducted to clarify these points in the future.
| Method | Representative purpose |
|---|---|
| TGA | Check of the temperature at which mass loss starts and finishes |
| TG/TPD-MS | Track of the desorbed species at each temperature |
| XPS | Check of the presence and elimination of ligands |
| Elucidation of the charge states of metal NCs | |
| Estimation of the occurring of the aggregation | |
| XAFS | Check of the presence and elimination of ligands |
| Elucidation of the charge states of metal NCs | |
| Elucidation of the interaction between metal NCs and substrate | |
| Estimation of the particle size and atomic packing structure of the metal NCs (from the average coordination number) | |
| STEM-EDX | Check of the presence and elimination of ligands |
| Check of the dispersion of metal NCs on the substrate | |
| TEM | Determination of particle size of metal NCs |
| HR-TEM/HAADF-STEM | Determination of particle size of metal NCs |
| Determination of atomic packing structure of metal NCs | |
| FT-IR/DRIFTS | Estimation of the charge states of each element |
| Confirmation of the presence of certain bonds | |
| Estimation of the specific surface area of metal NCs | |
| ICP-MS/AES/OES | Determination of the quantities of the ligands included in catalysts |
| Determination of chemical composition of alloy NCs |
3.2. Mechanism of calcination
As an example, herein, we focus on a system that uses SR-protected Au NCs as the metal NCs. In this case, two patterns of dissociation have been reported on the surface of the Au NCs upon calcination: dissociation of Au–S bonds209 and dissociation of both Au–S and S–C bonds210,216 (Fig. 36B). It has been reported in several papers that the ligand elimination occurs in two steps.209,210,215,216,330 In each paper, different factors have been proposed, such as the difference in the desorption temperature of the ligand between the Au NC–support interface and the surface of the Au NCs232 or the difference in the desorption temperature of the ligand depending on the S environment owing to the presence of two types of S atom in Au NCs.184,215 It was previously thought that the dissociated SR or S would be vaporized and eliminated from the system by further heating. However, based on recent results, it is interpreted that SR or S migrate from the surface of the Au NCs onto the support during calcination (Fig. 36B)216 and are then oxidized to SO3 or SO4 on the support (Fig. 36C).216–218,224 The generated sulfur oxides appear to adsorb on the support via dipole–dipole interaction.314 Tsukuda et al. pointed out that desorbed alkyl chains are also adsorbed on carbon supports (Fig. 36B).209 Barrabés et al. noted that Au+–S− migrates from Au NCs to the support during calcination (Fig. 36C).233 When the calcination temperature is further increased, the S oxides vaporize and are removed from the support (Fig. 36D).331 Although the results described above were obtained for Au NCs, similar adsorption of S oxides on the support has also been reported by Scott et al. during the calcination of Ag25(2,4-SPhMe2)18/AC.224 In addition, Adnan et al. reported NO3− counter anions were also adsorbed on the support when Au8(PPh3)(NO3)2 was used as a precursor.332
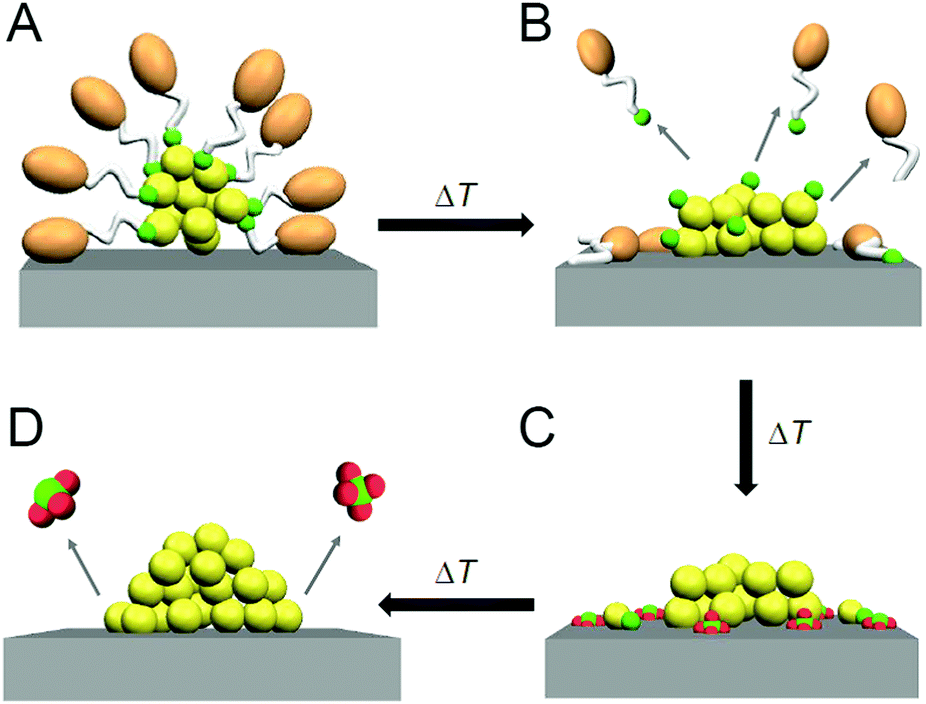 | ||
| Fig. 36 Schematic of proposed desorption mechanism of the ligand: (A) adsorption of metal NCs on a support, (B) S–C or metal–S dissociation and migration of organic and S-compounds from metal NCs to support, (C) oxidation of S on a support, and (D) desorption of S oxides from a support. | ||
As shown in Fig. 35A, the behavior of the ligand and metal core during calcination strongly depends on the nature of the support. Therefore, the ligand-elimination mechanism should vary depending on the system used. However, researchers working on other systems should know the above phenomena and the factors that contribute to their occurrence.
 | ||
| Fig. 37 Illustration of the structure evolution of (A) Au24Pd NCs and (B) Au24Pt NCs supported on BaLa4Ti4O15 upon calcination, (C) AuxPdy NCs supported on TiO2 upon calcination, and (D) Au24Pd NCs supported on MWCNTs upon calcination. Reproduced with permission from ref. 211, ref. 244 and ref. 335. Copyright 2019 American Chemical Society, 2020 American Chemical Society, and 2013 Royal Society of Chemistry. | ||
3.3. Mechanism in catalysis
 | ||
| Fig. 38 (A) Proposed structures of Au24M/BaLa4Ti4O15 for M = (a) Au, (b) Pd, and (c) Pt during the water-splitting reaction. (B) Resonance structures of carbocationic intermediate. Reproduced with permission from ref. 211 and ref. 244. Copyright 2016 Wiley-VCH and 2019 American Chemical Society. | ||
 | ||
| Fig. 39 Results of operando DRIFTS of Au24Pd/TiO2 catalysts during CO oxidation: (A) after the calcination under O2 atmosphere, followed under H2 atmosphere and (B) just after the calcination under O2 atmosphere. Spectra taken upon cooling are indicated by dotted lines. These spectra were acquired in helium at room temperature. Note that the absolute intensities of different samples cannot be compared. Reproduced with permission from ref. 335. Copyright 2020 American Chemical Society. | ||
4. Conclusion
It is essential to continue to develop high-performance catalysts to solve the problems that our society is currently facing and to enrich our lives in the future. From this point of view, atomically precise metal NCs have high potential as components of new functional catalysts. In this review, we have summarized previous studies on the creation of heterogeneous catalysts using atomically precise metal NCs. Through this summary, the current state of research on the development of catalysts using atomically precise metal NCs became clear, as described in the following.Validity of the use of atomically precise metal NCs
The use of atomically precise metal NCs is effective in elucidating the mechanism of catalytic reactions. Using this route, a deep understanding of the mechanism has been obtained for many reactions, such as CO oxidation by Aun(SR)m/CeO2, styrene oxidation by Aun(SR)m/HAP, semihydrogenation of terminal alkyne compounds by Au25(PET)18/TiO2, photocatalytic water splitting by Au25/BaLa4Ti4O15, and oxidation of benzyl alcohol by Au24Pd/MWCNTs.Necessity of ligand elimination
The effect of ligand elimination on the conversion efficiency depends on the type of catalytic reaction. However, overall, the ligand-desorbed catalyst exhibits a higher conversion efficiency than the ligand-remaining catalyst. Several recent DFT calculations have predicted that higher activity can be achieved through the elimination of ligands also in the application of metal NCs as electrocatalysts.337–342 However, there are also reactions in which the presence of ligands leads to an improvement of the conversion efficiency. In addition, the presence of ligands has advantages in changing the reaction selectivity and suppressing aggregation of metal NCs, thereby improving the catalyst durability. Even in these cases, calcination at a low temperature, which does not cause ligand elimination, often induces an improvement in catalytic activity.Selection of support
To remove the ligands while maintaining the size of the metal NCs, it is necessary to use a support that strongly interacts with the metal NCs. In addition, if a material with a high specific surface area and/or a porous material with an appropriate pore size is used as a support, it is possible to suppress the aggregation of metal NCs. Regarding the suppression of aggregation of metal NCs, the introduction of anchor nanomaterials or atoms,244 which strongly fix the loaded metal NCs on the support, is also an effective means.Selection of metal NCs
The appropriate size of Au NCs depends on the type of catalytic reaction. However, overall, a smaller size results in higher activity. Nevertheless, in reactions such as benzyl alcohol oxidation and cyclohexane oxidation, an appropriate Au NC size exists. It is difficult to discuss the size effects of metal NCs other than Au NCs, as well as the doping effects, because of the small number of study examples.Mechanism in calcination
The calcination process involves a complex process that was not initially predicted. Specifically, calcination first causes dissociation of Au–S bonds and/or S–C bonds on the surface of metal NCs. The dissociated SR or S moves from the Au NC surface to the support during calcination and is then oxidized to SO32− and SO42− on the support. In a specific system, not only SR and S but also the alkyl chain and Au+–S− are transferred to the support. When the calcination temperature is further increased, the sulfur oxide is vaporized and removed from the support. In addition to these processes, when the precursor contains NO3−, NO3− is also adsorbed on the support. Ligand calcination also induces structural deformation of metal NCs. Along with this, in alloy NCs, heteroatoms change the positions in metal NCs depending on the binding energy with the support.
The above insight is expected to lead to clear design guidelines for the creation of novel heterogeneous catalysts. However, the current knowledge is not presented as a unified view but as a collection of case studies. In the future, it is expected that researchers studying catalysts using metal NCs and/or metal NPs (such as thermal catalysts, photocatalysts, and electrocatalysts) will exchange more information. Such cooperation beyond the fields would enable us to obtain further knowledge on the creation of novel heterogeneous catalysts and make the practical application of heterogeneous catalysts using atomically precise metal NCs more realistic.
5. Outlook
It is expected that the following issues will be overcome in the future to enable the creation of various highly functional heterogeneous catalysts using atomically precise metal NCs.Use of various precise metal NCs
Most of the metal NCs used in the studies of catalysts are the metal NCs shown in Fig. 2. However, in recent years, it has become possible to precisely synthesize metal NCs of various sizes and chemical compositions for both pure metal and alloy NCs.24,25,87,88 In the future, it is expected that a deeper understanding will be obtained on (1) the correlations among the metal element, size, and catalytic activity, (2) the heteroatom substitution effect,343 (3) the mechanism in each reaction, (4) the correlations among the metal element, size, and durability by using various metal NCs from such a present “library” of atomically precise metal NCs.Improvement of durability
In most studies, metal NCs aggregate during calcination. In addition, even when the metal NCs can be loaded on the support with the same particle size, the particle size often increases after the catalytic reaction. As described above, increasing the specific surface area of the support is effective in suppressing the aggregation of metal NCs. In the future, it is expected that such ingenious attempts will be applied to various metal-oxide supports. For this purpose, it may be necessary to establish a new preparation method for the support. In addition, it has been reported that the formation of a protective shell on the surface of metal NCs suppresses the aggregation of metal NCs as well as the reverse reaction.243,275,297 In the future, many protective shells with high transparency of reactants and a small contact area between the metal NCs and films are expected to be newly produced. Also, it is expected that the information about whether further calcination/heat treatment is needed or not before recycling, which is not well understood at present, will be obtained in future study.Elucidation of geometrical structures of loaded metal NCs
To understand the structure–physical property relationship, it is essential to obtain a deeper understanding of the geometrical structure of the loaded metal NCs. Therefore, it is expected that the geometrical structures of loaded metal NCs will be elucidated by direct observation using spherical aberration-corrected (Cs-corrected) TEM344 and STEM. In addition, the geometrical structure determined using those methods is not necessarily the same as that present during the catalytic reaction. Therefore, it is expected that operand XAFS measurement345 will also be widely used for understanding the geometrical structure during catalytic reactions.Structural control of loaded metal NCs
Regarding the geometrical structure, even if atomically precise metal NCs having no variation in the geometrical structure are used as precursors, variation of the geometrical structure of the loaded metal NCs appears to exist. To identify the geometrical structure of metal NCs that create high activity and thereby selectively load such superior NCs as a catalyst on the support, it is necessary to establish a new method for controlling the geometrical structure of the loaded NCs. Previous studies have established many methods to control the size and geometrical structure of metal NCs dispersed in solution (after-treatment method for as-prepared metal NCs in solution).87,91,346,347 In the future, it is expected that such after-treatment methods will also be established for the loaded NCs, and thereby control of the geometrical structure could also be realized for the loaded metal NCs.Perfect elimination of ligands from catalysts
Recent studies have revealed that compounds produced by calcination are not easily removed through vaporization once adsorbed on the support. These compounds seem to (1) cause direct reaction with the reactants to produce by-products, (2) result in the catalyst poisoning that suppresses charge transfer between the support and metal, and (3) affect the catalyst properties. Previous studies have shown that further calcination leads to vaporization of the final products of these compounds (such as SO32− and SO42−) from supports;331 however, excessive heating promotes aggregation of metal NCs. Therefore, in the future, it is expected that new elimination methods will be established for compounds generated by calcination, such as chemical washing.Synthesis of platinum group metal NCs stable in air
In most previous studies, Au has been used as the base element for metal NCs. The superiority of using Au as a catalyst relates to the following factors: (1) Au NCs show high catalytic activity at low temperature; (2) Au NCs exhibit catalytic selectivity different from that of other precious metals; and (3) Au NCs have very low toxicity. However, Au NCs have a low melting point, and their catalytic activities do not improve dramatically even at high temperature. Therefore, it is not suitable to use Au NCs as catalysts at high temperature. For catalysts that must be used at high temperatures, such as catalysts for eliminating automobile exhaust gas, it will be necessary to use metal NCs consisting of elements such as Pt, Pd, and Rh. Although there are many reports of the synthesis of atomically precise metal NCs composed of these metal elements,53–59 they are difficult to synthesize in the atmosphere. In the future, it is expected that many metal NCs that are stable in the atmosphere will be found for metal NCs consisting of elements such as Pt, Pd, and Rh.Guidance by DFT calculation
As described above, it has become possible to control the chemical composition of loaded metal NCs with atomic accuracy in recent years. For such heterogeneous catalysts composed of atomically precise metal NCs, DFT calculations may be able to predict highly functional catalyst systems.337–342,348 Thus, in the future, it is expected that studies for improving the functionality of heterogeneous catalysts will shift from searching for the optimum system based on trial-and-error experiments to prediction by DFT calculation.
If the above problems are overcome, it will be possible to create a heterogeneous catalyst with high activity, selectivity, stability, and durability with a smaller amount of precious metal used, which would help address the resource, energy, and environmental problems currently facing modern society.
Author contributions
Y. N. constructed the structure of this review. T. K., Y. K., M. H., and Y. I. surveyed the literature, compiled figures and tables, and wrote the manuscript. Y. N. and S. H. revised the entire draft before submission. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (grant number 20H02698, 20H02552), Scientific Research on Innovative Areas “Coordination Asymmetry” (grant number 17H05385 and 19H04595), and Scientific Research on Innovative Areas “Innovations for Light-Energy Conversion” (grant number 18H05178 and 20H05115). Funding from Asahi Glass Foundation, TEPCO Memorial Foundation Research Grant (Basic Research), Kato Foundation for Promotion of Science (grant number KJ-2904), and Nissanken are gratefully acknowledged.References
- J. N. Galloway, F. J. Dentener, D. G. Capone, E. W. Boyer, R. W. Howarth, S. P. Seitzinger, G. P. Asner, C. C. Cleveland, P. A. Green, E. A. Holland, D. M. Karl, A. F. Michaels, J. H. Porter, A. R. Townsend and C. J. Vörösmarty, Biogeochemistry, 2004, 70, 153–226 CrossRef CAS.
- J. J. Berzelius, Ann. Chim., 1836, 61, 146–151 Search PubMed.
- W. Z. Ostwald, Phys. Chem., 1901, 37, 385 Search PubMed.
- J. T. Richardson, Principles of Catalyst Development, Springer, New York, 1989 Search PubMed.
- A. Miyashita, A. Yasuda, H. Takaya, K. Toriumi, T. Ito, T. Souchi and R. Noyori, J. Am. Chem. Soc., 1980, 102, 7932–7934 CrossRef CAS.
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301–309 CrossRef CAS.
- J. M. Thomas, R. Raja and D. W. Lewis, Angew. Chem., Int. Ed., 2005, 44, 6456–6482 CrossRef CAS PubMed.
- U. Hanefeld, Catalysis: An Integrated Textbook for Students, ed. L. Lefferts, 2018 Search PubMed.
- T. Tsukuda and H. Häkkinen, Protected Metal Clusters, From Fundamentals to Applications, Elsevier, Amsterdam, 2015 Search PubMed.
- M. Agrachev, M. Ruzzi, A. Venzo and F. Maran, Acc. Chem. Res., 2019, 52, 44–52 CrossRef CAS PubMed.
- K. Kwak and D. Lee, Acc. Chem. Res., 2019, 52, 12–22 CrossRef CAS PubMed.
- B. Nieto-Ortega and T. Bürgi, Acc. Chem. Res., 2018, 51, 2811–2819 CrossRef CAS PubMed.
- H. Qian, M. Zhu, Z. Wu and R. Jin, Acc. Chem. Res., 2012, 45, 1470–1479 CrossRef CAS PubMed.
- N. A. Sakthivel and A. Dass, Acc. Chem. Res., 2018, 51, 1774–1783 CrossRef CAS PubMed.
- R. L. Whetten, H.-C. Weissker, J. J. Pelayo, S. M. Mullins, X. López-Lozano and I. L. Garzón, Acc. Chem. Res., 2019, 52, 34–43 CrossRef CAS PubMed.
- T. Tsukuda, Bull. Chem. Soc. Jpn., 2012, 85, 151–168 CrossRef CAS.
- B. Bhattarai, Y. Zaker, A. Atnagulov, B. Yoon, U. Landman and T. P. Bigioni, Acc. Chem. Res., 2018, 51, 3104–3113 CrossRef CAS PubMed.
- Z. Gan, N. Xia and Z. Wu, Acc. Chem. Res., 2018, 51, 2774–2783 CrossRef CAS PubMed.
- A. Ghosh, O. F. Mohammed and O. M. Bakr, Acc. Chem. Res., 2018, 51, 3094–3103 CrossRef CAS PubMed.
- S. Hossain, Y. Niihori, L. V. Nair, B. Kumar, W. Kurashige and Y. Negishi, Acc. Chem. Res., 2018, 51, 3114–3124 CrossRef CAS PubMed.
- J. Yan, B. K. Teo and N. Zheng, Acc. Chem. Res., 2018, 51, 3084–3093 CrossRef CAS PubMed.
- T. Kawawaki, Y. Imai, D. Suzuki, S. Kato, I. Kobayashi, T. Suzuki, R. Kaneko, S. Hossain and Y. Negishi, Chem. – Eur. J., 2020, 26, 16150–16193 CrossRef CAS PubMed.
- X. Kang, Y. Li, M. Zhu and R. Jin, Chem. Soc. Rev., 2020, 49, 6443–6514 RSC.
- S. Bhat, A. Baksi, S. K. Mudedla, G. Natarajan, V. Subramanian and T. Pradeep, J. Phys. Chem. Lett., 2017, 8, 2787–2793 CrossRef CAS PubMed.
- S. Tian, L. Liao, J. Yuan, C. Yao, J. Chen, J. Yang and Z. Wu, Chem. Commun., 2016, 52, 9873–9876 RSC.
- S. Yamazoe, W. Kurashige, K. Nobusada, Y. Negishi and T. Tsukuda, J. Phys. Chem. C, 2014, 118, 25284–25290 CrossRef CAS.
- C. Kumara, C. M. Aikens and A. Dass, J. Phys. Chem. Lett., 2014, 5, 461–466 CrossRef CAS PubMed.
- C. Yao, Y.-j. Lin, J. Yuan, L. Liao, M. Zhu, L.-h. Weng, J. Yang and Z. Wu, J. Am. Chem. Soc., 2015, 137, 15350–15353 CrossRef CAS PubMed.
- W. Fei, S. Antonello, T. Dainese, A. Dolmella, M. Lahtinen, K. Rissanen, A. Venzo and F. Maran, J. Am. Chem. Soc., 2019, 141, 16033–16045 CrossRef CAS PubMed.
- C. Sun, N. Mammen, S. Kaappa, P. Yuan, G. Deng, C. Zhao, J. Yan, S. Malola, K. Honkala, H. Häkkinen, B. K. Teo and N. Zheng, ACS Nano, 2019, 13, 5975–5986 CrossRef CAS PubMed.
- S. Lee, M. S. Bootharaju, G. Deng, S. Malola, W. Baek, H. Häkkinen, N. Zheng and T. Hyeon, J. Am. Chem. Soc., 2020, 142, 13974–13981 CrossRef CAS PubMed.
- W. Kurashige, S. Yamazoe, M. Yamaguchi, K. Nishido, K. Nobusada, T. Tsukuda and Y. Negishi, J. Phys. Chem. Lett., 2014, 5, 2072–2076 CrossRef CAS PubMed.
- S. Sharma, W. Kurashige, K. Nobusada and Y. Negishi, Nanoscale, 2015, 7, 10606–10612 RSC.
- Y. Niihori, M. Eguro, A. Kato, S. Sharma, B. Kumar, W. Kurashige, K. Nobusada and Y. Negishi, J. Phys. Chem. C, 2016, 120, 14301–14309 CrossRef CAS.
- Y. Niihori, S. Hossain, B. Kumar, L. V. Nair, W. Kurashige and Y. Negishi, APL Mater., 2017, 5, 053201 CrossRef.
- L. V. Nair, S. Hossain, S. Takagi, Y. Imai, G. Hu, S. Wakayama, B. Kumar, W. Kurashige, D.-e. Jiang and Y. Negishi, Nanoscale, 2018, 10, 18969–18979 RSC.
- S. Hossain, Y. Imai, D. Suzuki, W. Choi, Z. Chen, T. Suzuki, M. Yoshioka, T. Kawawaki, D. Lee and Y. Negishi, Nanoscale, 2019, 11, 22089–22098 RSC.
- S. Hossain, Y. Imai, Y. Motohashi, Z. Chen, D. Suzuki, T. Suzuki, Y. Kataoka, M. Hirata, T. Ono, W. Kurashige, T. Kawawaki, T. Yamamoto and Y. Negishi, Mater. Horiz., 2020, 7, 796–803 RSC.
- S. Hossain, T. Ono, M. Yoshioka, G. Hu, M. Hosoi, Z. Chen, L. V. Nair, Y. Niihori, W. Kurashige, D.-e. Jiang and Y. Negishi, J. Phys. Chem. Lett., 2018, 9, 2590–2594 CrossRef CAS PubMed.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS PubMed.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS PubMed.
- C. P. Joshi, M. S. Bootharaju, M. J. Alhilaly and O. M. Bakr, J. Am. Chem. Soc., 2015, 137, 11578–11581 CrossRef CAS PubMed.
- H. Yang, Y. Wang, H. Huang, L. Gell, L. Lehtovaara, S. Malola, H. Häkkinen and N. Zheng, Nat. Commun., 2013, 4, 2422 CrossRef PubMed.
- B. Kumar, T. Kawawaki, N. Shimizu, Y. Imai, D. Suzuki, S. Hossain, L. V. Nair and Y. Negishi, Nanoscale, 2020, 12, 9969–9979 RSC.
- Y. Negishi, S. Hashimoto, A. Ebina, K. Hamada, S. Hossain and T. Kawawaki, Nanoscale, 2020, 12, 8017–8039 RSC.
- C. E. Briant, B. R. C. Theobald, J. W. White, L. K. Bell, D. M. P. Mingos and A. J. Welch, J. Chem. Soc., Chem. Commun., 1981, 201–202 RSC.
- M. McPartlin, R. Mason and L. Malatesta, J. Chem. Soc. D, 1969, 334 RSC.
- G. Schmid, Chem. Rev., 1992, 92, 1709–1727 CrossRef CAS.
- M. Schulz-Dobrick and M. Jansen, Z. Anorg. Allg. Chem., 2007, 633, 2326–2331 CrossRef CAS.
- B. K. Teo, X. Shi and H. Zhang, J. Am. Chem. Soc., 1992, 114, 2743–2745 CrossRef CAS.
- J. D. Roth, G. J. Lewis, L. K. Safford, X. Jiang, L. F. Dahl and M. J. Weaver, J. Am. Chem. Soc., 1992, 114, 6159–6169 CrossRef CAS.
- Y. Shichibu and K. Konishi, Small, 2010, 6, 1216–1220 CrossRef CAS PubMed.
- H. Hirai, S. Takano, T. Nakamura and T. Tsukuda, Inorg. Chem., 2020, 59, 17889–17895 CrossRef CAS PubMed.
- K. Konishi, M. Iwasaki and Y. Shichibu, Acc. Chem. Res., 2018, 51, 3125–3133 CrossRef CAS PubMed.
- B. K. Teo and H. Zhang, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 5067–5071 CrossRef CAS PubMed.
- V. G. Albano, P. L. Bellon, M. Manassero and M. Sansoni, J. Chem. Soc. D, 1970, 1210–1211 RSC.
- G. Schmid, R. Pfeil, R. Boese, F. Bandermann, S. Meyer, G. H. M. Calis and J. W. A. van der Velden, Chem. Ber., 1981, 114, 3634–3642 CrossRef CAS.
- E. G. Mednikov and L. F. Dahl, Philos. Trans. R. Soc., A, 2010, 368, 1301–1332 CrossRef CAS PubMed.
- A. Ceriotti, N. Masciocchi, P. Macchi and G. Longoni, Angew. Chem., Int. Ed., 1999, 38, 3724–3727 CrossRef CAS PubMed.
- I. Ciabatti, C. Femoni, M. C. Iapalucci, G. Longoni and S. Zacchini, J. Cluster Sci., 2014, 25, 115–146 CrossRef CAS.
- S. S. Kurasov, N. K. Eremenko, Y. L. Slovokhotov and Y. T. Struchkov, J. Organomet. Chem., 1989, 361, 405–408 CrossRef CAS.
- L. Hao, G. J. Spivak, J. Xiao, J. J. Vittal and R. J. Puddephatt, J. Am. Chem. Soc., 1995, 117, 7011–7012 CrossRef CAS.
- E. Cattabriga, I. Ciabatti, C. Femoni, T. Funaioli, M. C. Iapalucci and S. Zacchini, Inorg. Chem., 2016, 55, 6068–6079 CrossRef CAS PubMed.
- C. Cesari, I. Ciabatti, C. Femoni, M. C. Iapalucci, F. Mancini and S. Zacchini, Inorg. Chem., 2017, 56, 1655–1668 CrossRef CAS PubMed.
- P. Chini, J. Organomet. Chem., 1980, 200, 37–61 CrossRef CAS.
- M. Paolieri, I. Ciabatti and M. Fontani, J. Cluster Sci., 2019, 30, 1623–1631 CrossRef CAS.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS PubMed.
- A. Baksi, M. S. Bootharaju, X. Chen, H. Häkkinen and T. Pradeep, J. Phys. Chem. C, 2014, 118, 21722–21729 CrossRef CAS.
- I. Chakraborty, W. Kurashige, K. Kanehira, L. Gell, H. Häkkinen, Y. Negishi and T. Pradeep, J. Phys. Chem. Lett., 2013, 4, 3351–3355 CrossRef CAS PubMed.
- K. S. Sugi, I. Chakraborty, T. Udayabhaskararao, J. S. Mohanty and T. Pradeep, Part. Part. Syst. Charact., 2013, 30, 241–243 CrossRef CAS.
- Z. Lei, X.-K. Wan, S.-F. Yuan, Z.-J. Guan and Q.-M. Wang, Acc. Chem. Res., 2018, 51, 2465–2474 CrossRef CAS PubMed.
- J.-J. Li, Z.-J. Guan, Z. Lei, F. Hu and Q.-M. Wang, Angew. Chem., Int. Ed., 2019, 58, 1083–1087 CrossRef CAS PubMed.
- X.-K. Wan, Q. Tang, S.-F. Yuan, D.-e. Jiang and Q.-M. Wang, J. Am. Chem. Soc., 2015, 137, 652–655 CrossRef CAS.
- S. Ito, S. Takano and T. Tsukuda, J. Phys. Chem. Lett., 2019, 10, 6892–6896 CrossRef CAS PubMed.
- Y. Negishi, W. Kurashige and U. Kamimura, Langmuir, 2011, 27, 12289–12292 CrossRef CAS PubMed.
- W. Kurashige, M. Yamaguchi, K. Nobusada and Y. Negishi, J. Phys. Chem. Lett., 2012, 3, 2649–2652 CrossRef CAS.
- W. Kurashige, K. Munakata, K. Nobusada and Y. Negishi, Chem. Commun., 2013, 49, 5447–5449 RSC.
- W. Kurashige, S. Yamazoe, K. Kanehira, T. Tsukuda and Y. Negishi, J. Phys. Chem. Lett., 2013, 4, 3181–3185 CrossRef CAS.
- W. Kurashige, Y. Niihori, S. Sharma and Y. Negishi, J. Phys. Chem. Lett., 2014, 5, 4134–4142 CrossRef CAS PubMed.
- S. Hossain, W. Kurashige, S. Wakayama, B. Kumar, L. V. Nair, Y. Niihori and Y. Negishi, J. Phys. Chem. C, 2016, 120, 25861–25869 CrossRef CAS.
- Y. Niihori, S. Hossain, S. Sharma, B. Kumar, W. Kurashige and Y. Negishi, Chem. Rec., 2017, 17, 473–484 CrossRef CAS PubMed.
- X. Kang and M. Zhu, Small, 2019, 15, 1902703 CrossRef CAS.
- T. Kawawaki, A. Ebina, Y. Hosokawa, S. Ozaki, D. Suzuki, S. Hossain and Y. Negishi, Small, 2021, 2005328 CrossRef PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- A. Ebina, S. Hossain, H. Horihata, S. Ozaki, S. Kato, T. Kawawaki and Y. Negishi, Nanomaterials, 2020, 10, 1105 CrossRef CAS PubMed.
- X. Kang, H. Chong and M. Zhu, Nanoscale, 2018, 10, 10758–10834 RSC.
- X. Kang and M. Zhu, Chem. Mater., 2019, 31, 9939–9969 CrossRef CAS.
- X. Kang and M. Zhu, Coord. Chem. Rev., 2019, 394, 1–38 CrossRef CAS.
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC.
- H. Yu, B. Rao, W. Jiang, S. Yang and M. Zhu, Coord. Chem. Rev., 2019, 378, 595–617 CrossRef CAS.
- T. Omoda, S. Takano and T. Tsukuda, Small, 2020, 2001439 CrossRef.
- Y. Zhu, H. Qian, B. A. Drake and R. Jin, Angew. Chem., Int. Ed., 2010, 49, 1295–1298 CrossRef CAS.
- G. Li, C. Zeng and R. Jin, J. Am. Chem. Soc., 2014, 136, 3673–3679 CrossRef CAS PubMed.
- Y. Negishi, W. Kurashige, Y. Niihori, T. Iwasa and K. Nobusada, Phys. Chem. Chem. Phys., 2010, 12, 6219–6225 RSC.
- Y. Negishi, T. Iwai and M. Ide, Chem. Commun., 2010, 46, 4713–4715 RSC.
- Y. Negishi, R. Arai, Y. Niihori and T. Tsukuda, Chem. Commun., 2011, 47, 5693–5695 RSC.
- Y. Negishi, K. Igarashi, K. Munakata, W. Ohgake and K. Nobusada, Chem. Commun., 2012, 48, 660–662 RSC.
- W. Kurashige and Y. Negishi, J. Cluster Sci., 2012, 23, 365–374 CrossRef CAS.
- Y. Negishi, U. Kamimura, M. Ide and M. Hirayama, Nanoscale, 2012, 4, 4263–4268 RSC.
- Y. Negishi, K. Munakata, W. Ohgake and K. Nobusada, J. Phys. Chem. Lett., 2012, 3, 2209–2214 CrossRef CAS PubMed.
- Y. Niihori, W. Kurashige, M. Matsuzaki and Y. Negishi, Nanoscale, 2013, 5, 508–512 RSC.
- Y. Negishi, W. Kurashige, Y. Kobayashi, S. Yamazoe, N. Kojima, M. Seto and T. Tsukuda, J. Phys. Chem. Lett., 2013, 4, 3579–3583 CrossRef CAS.
- Y. Negishi, W. Kurashige, Y. Niihori and K. Nobusada, Phys. Chem. Chem. Phys., 2013, 15, 18736–18751 RSC.
- A. Puls, P. Jerabek, W. Kurashige, M. Förster, M. Molon, T. Bollermann, M. Winter, C. Gemel, Y. Negishi, G. Frenking and R. A. Fischer, Angew. Chem., Int. Ed., 2014, 53, 4327–4331 CrossRef CAS PubMed.
- D. Bahnemann, A. Henglein, J. Lilie and L. Spanhel, J. Phys. Chem., 1984, 88, 709–711 CrossRef CAS.
- H. N. Ghosh, J. B. Asbury and T. Lian, J. Phys. Chem. B, 1998, 102, 6482–6486 CrossRef CAS.
- B. Ohtani, R. M. Bowman, D. P. Colombo Jr., H. Kominami, H. Noguchi and K. Uosaki, Chem. Lett., 1998, 579–580 CrossRef CAS.
- A. Yamakata, T.-a. Ishibashi and H. Onishi, Chem. Phys. Lett., 2001, 333, 271–277 CrossRef CAS.
- T. Yoshihara, R. Katoh, A. Furube, Y. Tamaki, M. Murai, K. Hara, S. Murata, H. Arakawa and M. Tachiya, J. Phys. Chem. B, 2004, 108, 3817–3823 CrossRef CAS.
- J. Lee, H. S. Shim, M. Lee, J. K. Song and D. Lee, J. Phys. Chem. Lett., 2011, 2, 2840–2845 CrossRef CAS.
- X.-Q. Gong, A. Selloni, O. Dulub, P. Jacobson and U. Diebold, J. Am. Chem. Soc., 2008, 130, 370–381 CrossRef CAS.
- M. R. Nellist, F. A. L. Laskowski, J. Qiu, H. Hajibabaei, K. Sivula, T. W. Hamann and S. W. Boettcher, Nat. Energy, 2018, 3, 46–52 CrossRef CAS.
- M. R. Nellist, J. Qiu, F. A. L. Laskowski, F. M. Toma and S. W. Boettcher, ACS Energy Lett., 2018, 3, 2286–2291 CrossRef CAS.
- G. N. Vayssilov, Y. Lykhach, A. Migani, T. Staudt, G. P. Petrova, N. Tsud, T. Skála, A. Bruix, F. Illas, K. C. Prince, V. Matolin, K. M. Neyman and J. Libuda, Nat. Mater., 2011, 10, 310–315 CrossRef CAS PubMed.
- K. He, J. Xie, Z.-Q. Liu, N. Li, X. Chen, J. Hu and X. Li, J. Mater. Chem. A, 2018, 6, 13110–13122 RSC.
- Y. Niihori, K. Yoshida, S. Hossain, W. Kurashige and Y. Negishi, Bull. Chem. Soc. Jpn., 2019, 92, 664–695 CrossRef CAS.
- S. Wang, X. Meng, A. Das, T. Li, Y. Song, T. Cao, X. Zhu, M. Zhu and R. Jin, Angew. Chem., Int. Ed., 2014, 53, 2376–2380 CrossRef CAS PubMed.
- J. Chen, L. Liu, X. Liu, L. Liao, S. Zhuang, S. Zhou, J. Yang and Z. Wu, Chem. – Eur. J., 2017, 23, 18187–18192 CrossRef CAS PubMed.
- R. Anumula, P. Xiao, C. Cui, H. Wu, G. Cui, W.-H. Fang, Z. Luo and J. Yao, Nanoscale, 2020, 12, 7864–7869 RSC.
- S. R. Biltek, A. C. Reber, S. N. Khanna and A. Sen, J. Phys. Chem. A, 2017, 121, 5324–5331 CrossRef CAS PubMed.
- G. Sun, X. Kang, S. Jin, X. Li, D. Hu, S. Wang and M. Zhu, Acta Phys.-Chim. Sin., 2018, 34, 799–804 CAS.
- X. Liu, J. Yuan, J. Chen, J. Yang and Z. Wu, Part. Part. Syst. Charact., 2019, 36, 1900003 CrossRef.
- X. Kang, S. Wang, Y. Song, S. Jin, G. Sun, H. Yu and M. Zhu, Angew. Chem., Int. Ed., 2016, 55, 3611–3614 CrossRef CAS PubMed.
- H. Yang, Y. Wang, J. Lei, L. Shi, X. Wu, V. Mäkinen, S. Lin, Z. Tang, J. He, H. Häkkinen, L. Zheng and N. Zheng, J. Am. Chem. Soc., 2013, 135, 9568–9571 CrossRef CAS PubMed.
- M.-b. Li, S.-k. Tian and Z. Wu, Chin. J. Chem., 2017, 35, 567–571 CrossRef CAS.
- C. Zhou, H. Li, Y. Song, F. Ke, W. W. Xu and M. Zhu, Nanoscale, 2019, 11, 19393–19397 RSC.
- R. Anumula, A. C. Reber, P. An, C. Cui, M. Guo, H. Wu, Z. Luo and S. N. Khanna, Nanoscale, 2020, 12, 14801–14807 RSC.
- X. Kang, L. Xiong, S. Wang, Y. Pei and M. Zhu, Inorg. Chem., 2018, 57, 335–342 CrossRef CAS PubMed.
- J. Xiang, P. Li, Y. Song, X. Liu, H. Chong, S. Jin, Y. Pei, X. Yuan and M. Zhu, Nanoscale, 2015, 7, 18278–18283 RSC.
- X. Kang, C. Silalai, Y. Lv, G. Sun, S. Chen, H. Yu, F. Xu and M. Zhu, Eur. J. Inorg. Chem., 2017, 1414–1419 CrossRef CAS.
- Y. Song, S. Weng, H. Li, H. Yu and M. Zhu, Inorg. Chem., 2019, 58, 7136–7140 CrossRef CAS PubMed.
- Q. Li, K. J. Lambright, M. G. Taylor, K. Kirschbaum, T.-Y. Luo, J. Zhao, G. Mpourmpakis, S. Mokashi-Punekar, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2017, 139, 17779–17782 CrossRef CAS PubMed.
- S. Yang, J. Chai, Y. Song, J. Fan, T. Chen, S. Wang, H. Yu, X. Li and M. Zhu, J. Am. Chem. Soc., 2017, 139, 5668–5671 CrossRef CAS PubMed.
- T.-H. Chiu, J.-H. Liao, F. Gam, I. Chantrenne, S. Kahlal, J.-Y. Saillard and C. W. Liu, J. Am. Chem. Soc., 2019, 141, 12957–12961 CrossRef CAS PubMed.
- C. Yao, C.-Q. Xu, I.-H. Park, M. Zhao, Z. Zhu, J. Li, X. Hai, H. Fang, Y. Zhang, G. Macam, J. Teng, L. Li, Q.-H. Xu, F.-C. Chuang, J. Lu, C. Su, J. Li and J. Lu, Angew. Chem., Int. Ed., 2020, 59, 8270–8276 CrossRef CAS PubMed.
- Q. Li, T.-Y. Luo, M. G. Taylor, S. Wang, X. Zhu, Y. Song, G. Mpourmpakis, N. L. Rosi and R. Jin, Sci. Adv., 2017, 3, e1603193 CrossRef PubMed.
- Y. Li, M. J. Cowan, M. Zhou, M. G. Taylor, H. Wang, Y. Song, G. Mpourmpakis and R. Jin, ACS Nano, 2020, 14, 6599–6606 CrossRef CAS.
- Q. Li, M. G. Taylor, K. Kirschbaum, K. J. Lambright, X. Zhu, G. Mpourmpakis and R. Jin, J. Colloid Interface Sci., 2017, 505, 1202–1207 CrossRef CAS PubMed.
- S. Takano, S. Ito and T. Tsukuda, J. Am. Chem. Soc., 2019, 141, 15994–16002 CrossRef CAS PubMed.
- H. Yang, Y. Wang, J. Yan, X. Chen, X. Zhang, H. Häkkinen and N. Zheng, J. Am. Chem. Soc., 2014, 136, 7197–7200 CrossRef CAS PubMed.
- L. Liao, S. Zhou, Y. Dai, L. Liu, C. Yao, C. Fu, J. Yang and Z. Wu, J. Am. Chem. Soc., 2015, 137, 9511–9514 CrossRef CAS PubMed.
- J. Yan, H. Su, H. Yang, S. Malola, S. Lin, H. Häkkinen and N. Zheng, J. Am. Chem. Soc., 2015, 137, 11880–11883 CrossRef CAS.
- Q. Li, S. Wang, K. Kirschbaum, K. J. Lambright, A. Das and R. Jin, Chem. Commun., 2016, 52, 5194–5197 RSC.
- N. Yan, L. Liao, J. Yuan, Y.-j. Lin, L.-h. Weng, J. Yang and Z. Wu, Chem. Mater., 2016, 28, 8240–8247 CrossRef CAS.
- M. S. Bootharaju, C. P. Joshi, M. R. Parida, O. F. Mohammed and O. M. Bakr, Angew. Chem., Int. Ed., 2016, 55, 922–926 CrossRef CAS.
- S. Wang, H. Abroshan, C. Liu, T.-Y. Luo, M. Zhu, H. J. Kim, N. L. Rosi and R. Jin, Nat. Commun., 2017, 8, 848 CrossRef.
- S. Yang, J. Chai, T. Chen, B. Rao, Y. Pan, H. Yu and M. Zhu, Inorg. Chem., 2017, 56, 1771–1774 CrossRef CAS.
- L. He, J. Yuan, N. Xia, L. Liao, X. Liu, Z. Gan, C. Wang, J. Yang and Z. Wu, J. Am. Chem. Soc., 2018, 140, 3487–3490 CrossRef CAS PubMed.
- G. Soldan, M. A. Aljuhani, M. S. Bootharaju, L. G. AbdulHalim, M. R. Parida, A.-H. Emwas, O. F. Mohammed and O. M. Bakr, Angew. Chem., Int. Ed., 2016, 55, 5749–5753 CrossRef CAS.
- X. Kang, H. Abroshan, S. Wang and M. Zhu, Inorg. Chem., 2019, 58, 11000–11009 CrossRef CAS.
- X. Kang, X. Wei, S. Wang and M. Zhu, Inorg. Chem., 2020, 59, 8736–8743 CrossRef CAS.
- J. Chai, S. Yang, Y. Lv, H. Chong, H. Yu and M. Zhu, Angew. Chem., Int. Ed., 2019, 58, 15671–15674 CrossRef CAS.
- W. Zhang, S. Zhuang, L. Liao, H. Dong, N. Xia, J. Li, H. Deng and Z. Wu, Inorg. Chem., 2019, 58, 5388–5392 CrossRef CAS.
- X. Zou, S. Jin, S. Wang, M. Zhu and R. Jin, Nano Futures, 2018, 2, 045004 CrossRef CAS.
- J. Fan, Y. Song, J. Chai, S. Yang, T. Chen, B. Rao, H. Yu and M. Zhu, Nanoscale, 2016, 8, 15317–15322 RSC.
- B. Rao, T. Zhao, S. Yang, J. Chai, Y. Pan, S. Weng, H. Yu, X. Li and M. Zhu, Dalton Trans., 2018, 47, 475–480 RSC.
- N. A. Sakthivel, M. Stener, L. Sementa, M. Medves, G. Ramakrishna, A. Fortunelli, A. G. Oliver and A. Dass, J. Phys. Chem. C, 2019, 123, 29484–29494 CrossRef.
- C. Kumara, K. J. Gagnon and A. Dass, J. Phys. Chem. Lett., 2015, 6, 1223–1228 CrossRef CAS PubMed.
- J. Chai, Y. Lv, S. Yang, Y. Song, X. Zan, Q. Li, H. Yu, M. Wu and M. Zhu, J. Phys. Chem. C, 2017, 121, 21665–21669 CrossRef CAS.
- J. Yan, H. Su, H. Yang, C. Hu, S. Malola, S. Lin, B. K. Teo, H. Häkkinen and N. Zheng, J. Am. Chem. Soc., 2016, 138, 12751–12754 CrossRef CAS.
- L. Huang, J. Yan, L. Ren, B. K. Teo and N. Zheng, Dalton Trans., 2017, 46, 1757–1760 RSC.
- J. Yan, S. Malola, C. Hu, J. Peng, B. Dittrich, B. K. Teo, H. Häkkinen, L. Zheng and N. Zheng, Nat. Commun., 2018, 9, 3357 CrossRef PubMed.
- S. Zhuang, D. Chen, L. Liao, Y. Zhao, N. Xia, W. Zhang, C. Wang, J. Yang and Z. Wu, Angew. Chem., Int. Ed., 2020, 59, 3073–3077 CrossRef CAS PubMed.
- S. Jin, M. Zhou, X. Kang, X. Li, W. Du, X. Wei, S. Chen, S. Wang and M. Zhu, Angew. Chem., Int. Ed., 2020, 59, 3891–3895 CrossRef CAS.
- S. Wang, S. Jin, S. Yang, S. Chen, Y. Song, J. Zhang and M. Zhu, Sci. Adv., 2015, 1, e1500441 CrossRef PubMed.
- T. Higaki, C. Liu, D. J. Morris, G. He, T.-Y. Luo, M. Y. Sfeir, P. Zhang, N. L. Rosi and R. Jin, Angew. Chem., Int. Ed., 2019, 58, 18798–18802 CrossRef CAS PubMed.
- L. C. McKenzie, T. O. Zaikova and J. E. Hutchison, J. Am. Chem. Soc., 2014, 136, 13426–13435 CrossRef CAS.
- M. A. Tofanelli, T. W. Ni, B. D. Phillips and C. J. Ackerson, Inorg. Chem., 2016, 55, 999–1001 CrossRef CAS PubMed.
- H. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. Jin, J. Am. Chem. Soc., 2010, 132, 8280–8281 CrossRef CAS PubMed.
- N. Yan, N. Xia, L. Liao, M. Zhu, F. Jin, R. Jin and Z. Wu, Sci. Adv., 2018, 4, eaat7259 CrossRef CAS PubMed.
- Y. Shichibu, Y. Negishi, T. Watanabe, N. K. Chaki, H. Kawaguchi and T. Tsukuda, J. Phys. Chem. C, 2007, 111, 7845–7847 CrossRef CAS.
- J. Chen, Q.-F. Zhang, T. A. Bonaccorso, P. G. Williard and L.-S. Wang, J. Am. Chem. Soc., 2014, 136, 92–95 CrossRef CAS PubMed.
- L. V. Nair, S. Hossain, S. Wakayama, S. Takagi, M. Yoshioka, J. Maekawa, A. Harasawa, B. Kumar, Y. Niihori, W. Kurashige and Y. Negishi, J. Phys. Chem. C, 2017, 121, 11002–11009 CrossRef CAS.
- X.-K. Wan, J.-Q. Wang, Z.-A. Nan and Q.-M. Wang, Sci. Adv., 2017, 3, e1701823 CrossRef PubMed.
- X. Liu, J. Yuan, C. Yao, J. Chen, L. Li, X. Bao, J. Yang and Z. Wu, J. Phys. Chem. C, 2017, 121, 13848–13853 CrossRef CAS.
- Y. Du, J. Xiang, K. Ni, Y. Yun, G. Sun, X. Yuan, H. Sheng, Y. Zhu and M. Zhu, Inorg. Chem. Front., 2018, 5, 2948–2954 RSC.
- V. Sudheeshkumar, K. O. Sulaiman and R. W. J. Scott, Nanoscale Adv., 2020, 2, 55–69 RSC.
- R. Jin, G. Li, S. Sharma, Y. Li and X. Du, Chem. Rev., 2021, 121, 567–648 CrossRef CAS PubMed.
- Y. Liu, H. Tsunoyama, T. Akita and T. Tsukuda, J. Phys. Chem. C, 2009, 113, 13457–13461 CrossRef CAS.
- H. A. Almukhlifi and R. C. Burns, Appl. Catal., A, 2015, 502, 174–187 CrossRef CAS.
- M. Cargnello, C. Chen, B. T. Diroll, V. V. T. Doan-Nguyen, R. J. Gorte and C. B. Murray, J. Am. Chem. Soc., 2015, 137, 6906–6911 CrossRef CAS.
- N. Sakamoto, H. Ohtsuka, T. Ikeda, K. Maeda, D. Lu, M. Kanehara, K. Teramura, T. Teranishi and K. Domen, Nanoscale, 2009, 1, 106–109 RSC.
- K. Maeda, N. Sakamoto, T. Ikeda, H. Ohtsuka, A. Xiong, D. Lu, M. Kanehara, T. Teranishi and K. Domen, Chem. – Eur. J., 2010, 16, 7750–7759 CrossRef CAS PubMed.
- T. Ikeda, A. Xiong, T. Yoshinaga, K. Maeda, K. Domen and T. Teranishi, J. Phys. Chem. C, 2013, 117, 2467–2473 CrossRef CAS.
- K. Maeda, A. Xiong, T. Yoshinaga, T. Ikeda, N. Sakamoto, T. Hisatomi, M. Takashima, D. Lu, M. Kanehara, T. Setoyama, T. Teranishi and K. Domen, Angew. Chem., Int. Ed., 2010, 49, 4096–4099 CrossRef CAS PubMed.
- A. Xiong, T. Yoshinaga, T. Ikeda, M. Takashima, T. Hisatomi, K. Maeda, T. Setoyama, T. Teranishi and K. Domen, Eur. J. Inorg. Chem., 2014, 767–772 CrossRef CAS.
- T. Yoshinaga, M. Saruyama, A. Xiong, Y. Ham, Y. Kuang, R. Niishiro, S. Akiyama, M. Sakamoto, T. Hisatomi, K. Domen and T. Teranishi, Nanoscale, 2018, 10, 10420–10427 RSC.
- Y. H. Li, J. Xing, Z. J. Chen, Z. Li, F. Tian, L. R. Zheng, H. F. Wang, P. Hu, H. J. Zhao and H. G. Yang, Nat. Commun., 2013, 4, 2500 CrossRef PubMed.
- E. Cui and G. Lu, J. Phys. Chem. C, 2013, 117, 26415–26425 CrossRef CAS.
- E. Cui and G. Lu, Int. J. Hydrogen Energy, 2014, 39, 7672–7685 CrossRef CAS.
- Y. Zhang, D. A. J. M. Ligthart, X.-Y. Quek, L. Gao and E. J. M. Hensen, Int. J. Hydrogen Energy, 2014, 39, 11537–11546 CrossRef CAS.
- M. Hojamberdiev, M. M. Khan, Z. Kadirova, K. Kawashima, K. Yubuta, K. Teshima, R. Riedel and M. Hasegawa, Renewable Energy, 2019, 138, 434–444 CrossRef CAS.
- M. Luo, P. Lu, W. Yao, C. Huang, Q. Xu, Q. Wu, Y. Kuwahara and H. Yamashita, ACS Appl. Mater. Interfaces, 2016, 8, 20667–20674 CrossRef CAS PubMed.
- S. Xu, A. J. Du, J. Liu, J. Ng and D. D. Sun, Int. J. Hydrogen Energy, 2011, 36, 6560–6568 CrossRef CAS.
- Q. Wu, S. Xiong, P. Shen, S. Zhao, Y. Li, D. Su and A. Orlov, Catal. Sci. Technol., 2015, 5, 2059–2064 RSC.
- X. L. Du, X. L. Wang, Y. H. Li, Y. L. Wang, J. J. Zhao, L. J. Fang, L. R. Zheng, H. Tong and H. G. Yang, Chem. Commun., 2017, 53, 9402–9405 RSC.
- S. Liu and Y.-J. Xu, Sci. Rep., 2016, 6, 22742 CrossRef CAS PubMed.
- F.-X. Xiao, Z. Zeng, S.-H. Hsu, S.-F. Hung, H. M. Chen and B. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 28105–28109 CrossRef CAS PubMed.
- A. Kogo, N. Sakai and T. Tatsuma, Nanoscale, 2015, 7, 14237–14240 RSC.
- L. D. Menard, F. Xu, R. G. Nuzzo and J. C. Yang, J. Catal., 2006, 243, 64–73 CrossRef CAS.
- E. W. Elliott, R. D. Glover and J. E. Hutchison, ACS Nano, 2015, 9, 3050–3059 CrossRef CAS PubMed.
- J. Kilmartin, R. Sarip, R. Grau-Crespo, D. D. Tommaso, G. Hogarth, C. Prestipino and G. Sankar, ACS Catal., 2012, 2, 957–963 CrossRef CAS.
- B. Zhang, J. Fang, J. Li, J. J. Lau, D. Mattia, Z. Zhong, J. Xie and N. Yan, Chem. – Asian J., 2016, 11, 532–539 CrossRef CAS PubMed.
- S. Das, A. Goswami, M. Hesari, J. F. Al-Sharab, E. Mikmeková, F. Maran and T. Asefa, Small, 2014, 10, 1473–1478 CrossRef CAS PubMed.
- S. Xie, H. Tsunoyama, W. Kurashige, Y. Negishi and T. Tsukuda, ACS Catal., 2012, 2, 1519–1523 CrossRef CAS.
- T. Yoskamtorn, S. Yamazoe, R. Takahata, J.-i. Nishigaki, A. Thivasasith, J. Limtrakul and T. Tsukuda, ACS Catal., 2014, 4, 3696–3700 CrossRef CAS.
- S. Yamazoe, T. Yoskamtorn, S. Takano, S. Yadnum, J. Limtrakul and T. Tsukuda, Chem. Rec., 2016, 16, 2338–2348 CrossRef CAS PubMed.
- C. Lavenn, A. Demessence and A. Tuel, J. Catal., 2015, 322, 130–138 CrossRef CAS.
- C. Liu, J. Zhang, J. Huang, C. Zhang, F. Hong, Y. Zhou, G. Li and M. Haruta, ChemSusChem, 2017, 10, 1976–1980 CrossRef CAS PubMed.
- Y. Liu, H. Tsunoyama, T. Akita, S. Xie and T. Tsukuda, ACS Catal., 2011, 1, 2–6 CrossRef CAS.
- B. Zhang, S. Kaziz, H. Li, M. G. Hevia, D. Wodka, C. Mazet, T. Bürgi and N. Barrabés, J. Phys. Chem. C, 2015, 119, 11193–11199 CrossRef CAS.
- B. Zhang, A. Sels, G. Salassa, S. Pollitt, V. Truttmann, C. Rameshan, J. Llorca, W. Olszewski, G. Rupprechter, T. Bürgi and N. Barrabés, ChemCatChem, 2018, 10, 5372–5376 CrossRef CAS.
- B. Zhang, C. García, A. Sels, G. Salassa, C. Rameshan, J. Llorca, K. Hradil, G. Rupprechter, N. Barrabés and T. Bürgi, Catal. Commun., 2019, 130, 105768 CrossRef CAS.
- C. García, S. Pollitt, M. van der Linden, V. Truttmann, C. Rameshan, R. Rameshan, E. Pittenauer, G. Allmaier, P. Kregsamer, M. Stöger-Pollach, N. Barrabés and G. Rupprechter, Catal. Today, 2019, 336, 174–185 CrossRef.
- Y. Liu, H. Tsunoyama, T. Akita and T. Tsukuda, Chem. Commun., 2010, 46, 550–552 RSC.
- Y. Zhu, H. Qian and R. Jin, Chem. – Eur. J., 2010, 16, 11455–11462 CrossRef CAS PubMed.
- Y. Zhu, H. Qian, A. Das and R. Jin, Chin. J. Catal., 2011, 32, 1149–1155 CrossRef CAS.
- J. Liu, K. S. Krishna, Y. B. Losovyj, S. Chattopadhyay, N. Lozova, J. T. Miller, J. J. Spivey and C. S. S. R. Kumar, Chem. – Eur. J., 2013, 19, 10201–10208 CrossRef CAS PubMed.
- J. Fang, J. Li, B. Zhang, X. Yuan, H. Asakura, T. Tanaka, K. Teramura, J. Xie and N. Yan, Nanoscale, 2015, 7, 6325–6333 RSC.
- K. O. Sulaiman, V. Sudheeshkumar and R. W. J. Scott, RSC Adv., 2019, 9, 28019–28027 RSC.
- S. Gaur, J. T. Miller, D. Stellwagen, A. Sanampudi, C. S. S. R. Kumar and J. J. Spivey, Phys. Chem. Chem. Phys., 2012, 14, 1627–1634 RSC.
- X. Nie, H. Qian, Q. Ge, H. Xu and R. Jin, ACS Nano, 2012, 6, 6014–6022 CrossRef CAS PubMed.
- X. Nie, C. Zeng, X. Ma, H. Qian, Q. Ge, H. Xu and R. Jin, Nanoscale, 2013, 5, 5912–5918 RSC.
- W. Li, Q. Ge, X. Ma, Y. Chen, M. Zhu, H. Xu and R. Jin, Nanoscale, 2016, 8, 2378–2385 RSC.
- Y. Li, Y. Chen, S. D. House, S. Zhao, Z. Wahab, J. C. Yang and R. Jin, ACS Appl. Mater. Interfaces, 2018, 10, 29425–29434 CrossRef CAS PubMed.
- Y. Chen, C. Liu, Q. Tang, C. Zeng, T. Higaki, A. Das, D.-e. Jiang, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2016, 138, 1482–1485 CrossRef CAS PubMed.
- Z. Wu, D.-e. Jiang, A. K. P. Mann, D. R. Mullins, Z.-A. Qiao, L. F. Allard, C. Zeng, R. Jin and S. H. Overbury, J. Am. Chem. Soc., 2014, 136, 6111–6122 CrossRef CAS PubMed.
- Z. Wu, G. Hu, D.-e. Jiang, D. R. Mullins, Q.-F. Zhang, L. F. Allard, Jr., L.-S. Wang and S. H. Overbury, Nano Lett., 2016, 16, 6560–6567 CrossRef CAS.
- S. Pollitt, V. Truttmann, T. Haunold, C. Garcia, W. Olszewski, J. Llorca, N. Barrabés and G. Rupprechter, ACS Catal., 2020, 10, 6144–6148 CrossRef CAS PubMed.
- G. Ma, A. Binder, M. Chi, C. Liu, R. Jin, D.-e. Jiang, J. Fan and S. Dai, Chem. Commun., 2012, 48, 11413–11415 RSC.
- Y. Negishi, N. Shimizu, K. Funai, R. Kaneko, K. Wakamatsu, A. Harasawa, S. Hossain, M. E. Schuster, D. Ozkaya, W. Kurashige, T. Kawawaki, S. Yamazoe and S. Nagaoka, Nanoscale Adv., 2020, 2, 669–678 RSC.
- G. Li and R. Jin, J. Am. Chem. Soc., 2014, 136, 11347–11354 CrossRef CAS PubMed.
- A. Shivhare, D. M. Chevrier, R. W. Purves and R. W. J. Scott, J. Phys. Chem. C, 2013, 117, 20007–20016 CrossRef CAS.
- Y. Tan, X. Y. Liu, L. Zhang, A. Wang, L. Li, X. Pan, S. Miao, M. Haruta, H. Wei, H. Wang, F. Wang, X. Wang and T. Zhang, Angew. Chem., Int. Ed., 2017, 56, 2709–2713 CrossRef CAS PubMed.
- M. Urushizaki, H. Kitazawa, S. Takano, R. Takahata, S. Yamazoe and T. Tsukuda, J. Phys. Chem. C, 2015, 119, 27483–27488 CrossRef CAS.
- Y. Negishi, M. Mizuno, M. Hirayama, M. Omatoi, T. Takayama, A. Iwase and A. Kudo, Nanoscale, 2013, 5, 7188–7192 RSC.
- Y. Negishi, Y. Matsuura, R. Tomizawa, W. Kurashige, Y. Niihori, T. Takayama, A. Iwase and A. Kudo, J. Phys. Chem. C, 2015, 119, 11224–11232 CrossRef CAS.
- W. Kurashige, R. Kumazawa, Y. Mori and Y. Negishi, J. Mater. Appl., 2018, 7, 1–11 Search PubMed.
- W. Kurashige, R. Kumazawa, D. Ishii, R. Hayashi, Y. Niihori, S. Hossain, L. V. Nair, T. Takayama, A. Iwase, S. Yamazoe, T. Tsukuda, A. Kudo and Y. Negishi, J. Phys. Chem. C, 2018, 122, 13669–13681 CrossRef CAS.
- W. Kurashige, R. Hayashi, K. Wakamatsu, Y. Kataoka, S. Hossain, A. Iwase, A. Kudo, S. Yamazoe and Y. Negishi, ACS Appl. Energy Mater., 2019, 2, 4175–4187 CrossRef CAS.
- W. Kurashige, Y. Mori, S. Ozaki, M. Kawachi, S. Hossain, T. Kawawaki, C. J. Shearer, A. Iwase, G. F. Metha, S. Yamazoe, A. Kudo and Y. Negishi, Angew. Chem., Int. Ed., 2020, 59, 7076–7082 CrossRef CAS PubMed.
- X. Gao and W. Chen, Chem. Commun., 2017, 53, 9733–9736 RSC.
- S. Zhao, R. Jin, H. Abroshan, C. Zeng, H. Zhang, S. D. House, E. Gottlieb, H. J. Kim, J. C. Yang and R. Jin, J. Am. Chem. Soc., 2017, 139, 1077–1080 CrossRef CAS PubMed.
- O. Ioannidou and A. Zabaniotou, Renewable Sustainable Energy Rev., 2007, 11, 1966–2005 CrossRef CAS.
- H. Yin, Z. Ma, M. Chi and S. Dai, Catal. Today, 2011, 160, 87–95 CrossRef CAS.
- F. Rashidi, T. Sasaki, A. M. Rashidi, A. N. Kharat and K. J. Jozani, J. Catal., 2013, 299, 321–335 CrossRef CAS.
- Y. Miseki, H. Kato and A. Kudo, Energy Environ. Sci., 2009, 2, 306–314 RSC.
- A. Kudo, A. Tanaka, K. Domen and T. Onishi, J. Catal., 1988, 111, 296–301 CrossRef CAS.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS.
- W. Ren, L. Xiong, G. Nie, H. Zhang, X. Duan and S. Wang, Environ. Sci. Technol., 2020, 54, 1267–1275 CrossRef CAS PubMed.
- A. Longo, E. J. J. de Boed, N. Mammen, M. van der Linden, K. Honkala, H. Häkkinen, P. E. de Jongh and B. Donoeva, Chem. – Eur. J., 2020, 26, 7051–7058 CrossRef CAS PubMed.
- S. S. Karpova, V. A. Moshnikov, S. V. Mjakin and E. S. Kolovangina, Semiconductors, 2013, 47, 392–395 CrossRef CAS.
- J. Datka, A. M. Turek, J. M. Jehng and I. E. Wachs, J. Catal., 1992, 135, 186–199 CrossRef CAS.
- Z. Bao, V. Fung, F. Polo-Garzon, Z. D. Hood, S. Cao, M. Chi, L. Bai, D.-e. Jiang and Z. Wu, J. Catal., 2020, 384, 49–60 CrossRef CAS.
- C. Lavenn, F. Albrieux, G. Bergeret, R. Chiriac, P. Delichère, A. Tuel and A. Demessence, Nanoscale, 2012, 4, 7334–7337 RSC.
- Y.-J. Xu, P. Landon, D. Enache, A. F. Carley, M. W. Roberts and G. J. Hutchings, Catal. Lett., 2005, 101, 175–179 CrossRef CAS.
- B. P. C. Hereijgers and B. M. Weckhuysen, J. Catal., 2010, 270, 16–25 CrossRef CAS.
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS PubMed.
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 405–408 CrossRef CAS.
- I. Ciabatti, C. Femoni, M. C. Iapalucci, S. Ruggieri and S. Zacchini, Coord. Chem. Rev., 2018, 355, 27–38 CrossRef CAS.
- T. Imaoka, H. Kitazawa, W.-J. Chun, S. Omura, K. Albrecht and K. Yamamoto, J. Am. Chem. Soc., 2013, 135, 13089–13095 CrossRef CAS PubMed.
- K. Yamamoto, T. Imaoka, W.-J. Chun, O. Enoki, H. Katoh, M. Takenaga and A. Sonoi, Nat. Chem., 2009, 1, 397–402 CrossRef CAS PubMed.
- S. Blonski and S. H. Garofalini, Surf. Sci., 1993, 295, 263–274 CrossRef CAS.
- J. H. Kwak, J. Hu, D. Mei, C.-W. Yi, D. H. Kim, C. H. F. Peden, L. F. Allard and J. Szanyi, Science, 2009, 325, 1670–1673 CrossRef CAS PubMed.
- Y. Watanabe, X. Wu, H. Hirata and N. Isomura, Catal. Sci. Technol., 2011, 1, 1490–1495 RSC.
- C. Yin, F. R. Negreiros, G. Barcaro, A. Beniya, L. Sementa, E. C. Tyo, S. Bartling, K.-H. Meiwes-Broer, S. Seifert, H. Hirata, N. Isomura, S. Nigam, C. Majumder, Y. Watanabe, A. Fortunelli and S. Vajda, J. Mater. Chem. A, 2017, 5, 4923–4931 RSC.
- U. Heiz, A. Sanchez, S. Abbet and W.-D. Schneider, J. Am. Chem. Soc., 1999, 121, 3214–3217 CrossRef CAS.
- R. M. Heck, R. J. Farrauto and S. T. Gulati, Catalytic Air Pollution Control: Commercial Technology, John Wiley & Sons, America, 3rd edn, 2009 Search PubMed.
- D. Ren, L. He, L. Yu, R.-S. Ding, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, J. Am. Chem. Soc., 2012, 134, 17592–17598 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- K. Maeda, ACS Catal., 2013, 3, 1486–1503 CrossRef CAS.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- T. Kawawaki, Y. Kataoka, S. Ozaki, M. Kawachi, M. Hirata and Y. Negishi, Chem. Commun., 2021, 57, 417–440 RSC.
- Y. Negishi, Bull. Chem. Soc. Jpn., 2014, 87, 375–389 CrossRef CAS.
- W. Kurashige, Y. Niihori, S. Sharma and Y. Negishi, Coord. Chem. Rev., 2016, 320–321, 238–250 CrossRef CAS.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851–7861 CrossRef CAS.
- K. Maeda, J. Photochem. Photobiol. C, 2011, 12, 237–268 CrossRef CAS.
- K. Maeda, D. Lu and K. Domen, Chem. – Eur. J., 2013, 19, 4986–4991 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2006, 45, 7806–7809 CrossRef CAS PubMed.
- M. Yoshida, K. Takanabe, K. Maeda, A. Ishikawa, J. Kubota, Y. Sakata, Y. Ikezawa and K. Domen, J. Phys. Chem. C, 2009, 113, 10151–10157 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. C, 2007, 111, 7554–7560 CrossRef CAS.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS.
- S. J. Tauster, Acc. Chem. Res., 1987, 20, 389–394 CrossRef CAS.
- E. J. Braunschweig, A. D. Logan, A. K. Datye and D. J. Smith, J. Catal., 1989, 118, 227–237 CrossRef CAS.
- A. D. Logan, E. J. Braunschweig, A. K. Datye and D. J. Smith, Langmuir, 1988, 4, 827–830 CrossRef CAS.
- S. Labich, E. Taglauer and H. Knözinger, Top. Catal., 2000, 14, 153–161 CrossRef CAS.
- L. Liu, C. Ge, W. Zou, X. Gu, F. Gao and L. Dong, Phys. Chem. Chem. Phys., 2015, 17, 5133–5140 RSC.
- X. Liu, M.-H. Liu, Y.-C. Luo, C.-Y. Mou, S. D. Lin, H. Cheng, J.-M. Chen, J.-F. Lee and T.-S. Lin, J. Am. Chem. Soc., 2012, 134, 10251–10258 CrossRef CAS PubMed.
- H. Tang, J. Wei, F. Liu, B. Qiao, X. Pan, L. Li, J. Liu, J. Wang and T. Zhang, J. Am. Chem. Soc., 2016, 138, 56–59 CrossRef CAS PubMed.
- T. Kawawaki, Y. Mori, K. Wakamatsu, S. Ozaki, M. Kawachi, S. Hossain and Y. Negishi, J. Mater. Chem. A, 2020, 8, 16081–16113 RSC.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. J. Martin, M. Higashi, D. Kong, R. Abe and J. Tang, Chem. Rev., 2018, 118, 5201–5241 CrossRef CAS PubMed.
- D. E. Carlson and C. R. Wronski, Appl. Phys. Lett., 1976, 28, 671–673 CrossRef CAS.
- X.-T. Wang, T. Ouyang, L. Wang, J.-H. Zhong and Z.-Q. Liu, Angew. Chem., Int. Ed., 2020, 59, 6492–6499 CrossRef CAS PubMed.
- C. Huang, Y. Zou, Y.-Q. Ye, T. Ouyang, K. Xiao and Z.-Q. Liu, Chem. Commun., 2019, 55, 7687–7690 RSC.
- M. K. Sheehan, M. Rudden, H. Cai and C.-K. Tsung, Catal. Lett., 2016, 146, 309–318 CrossRef CAS.
- L. Liu, X. Zhang, L. Yang, L. Ren, D. Wang and J. Ye, Natl. Sci. Rev., 2017, 4, 761–780 CrossRef CAS.
- C. Jiang, S. J. A. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- M. R. Narouz, K. M. Osten, P. J. Unsworth, R. W. Y. Man, K. Salorinne, S. Takano, R. Tomihara, S. Kaappa, S. Malola, C.-T. Dinh, J. D. Padmos, K. Ayoo, P. J. Garrett, M. Nambo, J. H. Horton, E. H. Sargent, H. Häkkinen, T. Tsukuda and C. M. Crudden, Nat. Chem., 2019, 11, 419–425 CrossRef CAS PubMed.
- D. P. Anderson, J. F. Alvino, A. Gentleman, H. A. Qahtani, L. Thomsen, M. I. J. Polson, G. F. Metha, V. B. Golovko and G. G. Andersson, Phys. Chem. Chem. Phys., 2013, 15, 3917–3929 RSC.
- D. P. Anderson, R. H. Adnan, J. F. Alvino, O. Shipper, B. Donoeva, J.-Y. Ruzicka, H. Al Qahtani, H. H. Harris, B. Cowie, J. B. Aitken, V. B. Golovko, G. F. Metha and G. G. Andersson, Phys. Chem. Chem. Phys., 2013, 15, 14806–14813 RSC.
- H. S. Al Qahtani, R. Higuchi, T. Sasaki, J. F. Alvino, G. F. Metha, V. B. Golovko, R. Adnan, G. G. Andersson and T. Nakayama, RSC Adv., 2016, 6, 110765–110774 RSC.
- S. Wang, S. Yin, G. Chen, L. Li and H. Zhang, Catal. Sci. Technol., 2016, 6, 4090–4104 RSC.
- L. Li, L. Dou and H. Zhang, Nanoscale, 2014, 6, 3753–3763 RSC.
- S. Yin, J. Li and H. Zhang, Green Chem., 2016, 18, 5900–5914 RSC.
- S. H. Joo, J. Y. Park, C.-K. Tsung, Y. Yamada, P. Yang and G. A. Somorjai, Nat. Mater., 2009, 8, 126–131 CrossRef CAS PubMed.
- M. A. Habeeb Muhammed and T. Pradeep, Small, 2011, 7, 204–208 CrossRef CAS PubMed.
- A. Samanta, B. B. Dhar and R. N. Devi, J. Phys. Chem. C, 2012, 116, 1748–1754 CrossRef CAS.
- V. Sudheeshkumar, A. Shivhare and R. W. J. Scott, Catal. Sci. Technol., 2017, 7, 272–280 RSC.
- H. Chen, C. Liu, M. Wang, C. Zhang, G. Li and F. Wang, Chin. J. Catal., 2016, 37, 1787–1793 CrossRef CAS.
- Y. Zheng, X. Zhang, Y. Yao, X. Chen and Q. Yang, RSC Adv., 2015, 5, 105747–105752 RSC.
- P. M. Arnal, M. Comotti and F. Schüth, Angew. Chem., Int. Ed., 2006, 45, 8224–8227 CrossRef CAS PubMed.
- V. Sudheeshkumar, A. Lushington, X. Sun and R. W. J. Scott, ACS Appl. Nano Mater., 2018, 1, 6904–6911 CrossRef CAS.
- X. Cui, J. Wang, B. Liu, S. Ling, R. Long and Y. Xiong, J. Am. Chem. Soc., 2018, 140, 16514–16520 CrossRef CAS PubMed.
- X. Li, T. W. Goh, L. Li, C. Xiao, Z. Guo, X. C. Zeng and W. Huang, ACS Catal., 2016, 6, 3461–3468 CrossRef CAS.
- L. Liu, Y. Song, H. Chong, S. Yang, J. Xiang, S. Jin, X. Kang, J. Zhang, H. Yu and M. Zhu, Nanoscale, 2016, 8, 1407–1412 RSC.
- L. Sun, Y. Yun, H. Sheng, Y. Du, Y. Ding, P. Wu, P. Li and M. Zhu, J. Mater. Chem. A, 2018, 6, 15371–15376 RSC.
- C. Liu, C. Zeng, T.-Y. Luo, A. D. Merg, R. Jin and N. L. Rosi, J. Am. Chem. Soc., 2016, 138, 12045–12048 CrossRef CAS PubMed.
- Y. Luo, S. Fan, W. Yu, Z. Wu, D. A. Cullen, C. Liang, J. Shi and C. Su, Adv. Mater., 2018, 30, 1704576 CrossRef PubMed.
- L. M. Rossi, J. L. Fiorio, M. A. S. Garcia and C. P. Ferraz, Dalton Trans., 2018, 47, 5889–5915 RSC.
- Z. Wu and R. Jin, ACS Nano, 2009, 3, 2036–2042 CrossRef CAS PubMed.
- Y. Tai, W. Yamaguchi, M. Okada, F. Ohashi, K.-i. Shimizu, A. Satsuma, K. Tajiri and H. Kageyama, J. Catal., 2010, 270, 234–241 CrossRef CAS.
- R. H. Adnan and V. B. Golovko, Catal. Lett., 2019, 149, 449–455 CrossRef CAS.
- H. Häkkinen and U. Landman, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, R2287–R2290 CrossRef.
- L. Vitos, A. V. Ruban, H. L. Skriver and J. Kollár, Surf. Sci., 1998, 411, 186–202 CrossRef CAS.
- C. Garcia, V. Truttmann, I. Lopez, T. Haunold, C. Marini, C. Rameshan, E. Pittenauer, P. Kregsamer, K. Dobrezberger, M. Stöger-Pollach, N. Barrabés and G. Rupprechter, J. Phys. Chem. C, 2020, 124, 23626–23636 CrossRef CAS PubMed.
- K. T. Chan, J. B. Neaton and M. L. Cohen, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 235430 CrossRef.
- D. R. Kauffman, D. R. Alfonso, D. N. Tafen, C. Wang, Y. Zhou, Y. Yu, J. W. Lekse, X. Deng, V. Espinoza, J. Trindell, O. K. Ranasingha, A. Roy, J.-S. Lee and H. L. Xin, J. Phys. Chem. C, 2018, 122, 27991–28000 CrossRef CAS.
- D. R. Alfonso, D. Kauffman and C. Matranga, J. Chem. Phys., 2016, 144, 184705 CrossRef.
- N. Austin, S. Zhao, J. R. McKone, R. Jin and G. Mpourmpakis, Catal. Sci. Technol., 2018, 8, 3795–3805 RSC.
- A. V. Nagarajan, R. Juarez-Mosqueda, M. J. Cowan, R. Jin, D. R. Kauffman and G. Mpourmpakis, SN Appl. Sci., 2020, 2, 680 CrossRef CAS.
- S. Zhao, N. Austin, M. Li, Y. Song, S. D. House, S. Bernhard, J. C. Yang, G. Mpourmpakis and R. Jin, ACS Catal., 2018, 8, 4996–5001 CrossRef CAS.
- D. A. Pichugina, N. A. Nikitina and N. E. Kuz’menko, J. Phys. Chem. C, 2020, 124, 3080–3086 CrossRef CAS.
- J. Han, J. Lu, M. Wang, Y. Wang and F. Wang, Chin. J. Chem., 2019, 37, 977–988 CrossRef CAS.
- R. Takahata, S. Yamazoe, Y. Maehara, K. Yamazaki, S. Takano, W. Kurashige, Y. Negishi, K. Gohara and T. Tsukuda, J. Phys. Chem. C, 2020, 124, 6907–6912 CrossRef CAS.
- H. Asakura, S. Hosokawa, T. Ina, K. Kato, K. Nitta, K. Uera, T. Uruga, H. Miura, T. Shishido, J. Ohyama, A. Satsuma, K. Sato, A. Yamamoto, S. Hinokuma, H. Yoshida, M. Machida, S. Yamazoe, T. Tsukuda, K. Teramura and T. Tanaka, J. Am. Chem. Soc., 2018, 140, 176–184 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, A. Das and R. Jin, J. Phys. Chem. Lett., 2015, 6, 2976–2986 CrossRef CAS.
- S. Tian, Y. Cao, T. Chen, S. Zang and J. Xie, Chem. Commun., 2020, 56, 1163–1174 RSC.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |