
Design of molecular water oxidation catalysts with earth-abundant metal ions
Mio
Kondo†
 abc,
Hayato
Tatewaki†
a and
Shigeyuki
Masaoka
*ab
abc,
Hayato
Tatewaki†
a and
Shigeyuki
Masaoka
*ab
aDivision of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: masaoka@chem.eng.osaka-u.ac.jp
bInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICS-OTRI), Osaka University, Suita, Osaka 565-0871, Japan
cJST, PRESTO, 4-1-8 Honcho, Kawaguchi, 332-0012, Japan
First published on 11th May 2021
Abstract
The four-electron oxidation of water (2H2O → O2 + 4H+ + 4e−) is considered the main bottleneck in artificial photosynthesis. In nature, this reaction is catalysed by a Mn4CaO5 cluster embedded in the oxygen-evolving complex of photosystem II. Ruthenium-based complexes have been successful artificial molecular catalysts for mimicking this reaction. However, for practical and large-scale applications in the future, molecular catalysts that contain earth-abundant first-row transition metal ions are preferred owing to their high natural abundance, low risk of depletion, and low costs. In this review, the frontier of water oxidation reactions mediated by first-row transition metal complexes is described. Special attention is paid towards the design of molecular structures of the catalysts and their reaction mechanisms, and these factors are expected to serve as guiding principles for creating efficient and robust molecular catalysts for water oxidation using ubiquitous elements.
 Mio Kondo | Mio Kondo is an associate professor in the Division of Applied Chemistry, Graduate School of Engineering, Osaka University. She earned her PhD from the University of Tokyo in 2008 under the supervision of Prof. Hiroshi Nishihara. After postdoctoral studies at Kyoto University with Prof. Susumu Kitagawa, she joined the laboratory of Prof. Shigeyuki Masaoka at the Institute for Molecular Science as a research associate in 2011. She was promoted to Associate Professor at Osaka University in 2019. Her current research goal is the development of function-integrated molecule-based catalytic systems for small molecule conversions. |
 Hayato Tatewaki | Hayato Tatewaki obtained his BSc in Chemistry in 2019 from Osaka University. He is currently pursuing his MSc at Osaka University, under the direct supervision of Prof. Shigeyuki Masaoka. His research focus is on the development of oxidation reactions using multinuclear metal complexes as catalysts. |
 Shigeyuki Masaoka | Shigeyuki Masaoka is a professor in Osaka University. He earned his PhD from Kyoto University in 2004 with Prof. Susumu Kitagawa. He then joined the laboratory of Prof. Matthew J. Rosseinsky at the University of Liverpool as a postdoctoral research fellow. In 2005, he joined the laboratory of Prof. Ken Sakai at Kyushu University as an assistant professor. He started his independent career at the Institute for Molecular Science as an associate professor in 2011. In 2019, he moved to the current position. His current research theme includes the development of molecular modules for artificial photosynthesis. |
1. Introduction
Energy shortage and global warming are two of the most serious global problems facing humanity. Artificial photosynthesis systems have attracted significant attention as promising solutions to these problems.1–8 Natural photosynthesis converts the sunlight energy to chemical energy in the form of carbohydrates. The artificial photosynthesis reaction mimics the natural photosynthesis and can produce valuable chemical fuels from earth-abundant sources, such as H2O and CO2, in an eco-friendly fashion. Artificial photosynthesis primarily consists of two half-reactions, i.e. reduction and oxidation reaction. The oxidation reaction comprises the four-electron oxidation of water (2H2O → O2 + 4H+ + 4e−) and generates energy-rich electrons, and the formed electrons are used for the production of chemical fuels. Therefore, water oxidation is indispensable for realising artificial photosynthesis. However, the water oxidation reaction requires the transfer of four electrons and the formation of a double bond between two oxygen atoms therefore, the reaction has been regarded as the main bottleneck in the artificial photosynthesis reaction sequence.Efficient catalysis of water oxidation has been studied extensively. The catalysts reported so far can be mainly classified as heterogeneous and homogeneous catalysts. While the former basically consists of metal oxides,9–11 the latter usually includes metal complexes. Among them, homogeneous catalysts are advantageous for probing reaction mechanisms at the molecular level with well-established spectroscopic techniques. Further, reaction mechanism-based design and high tunability of the catalysts are significant advantages of homogeneous catalysis systems. Therefore, the development of metal-complex-based homogeneous catalysts for water oxidation is a promising research prospect.
The first example of an artificial homogeneous catalyst for water oxidation is a μ-oxo-bridged dinuclear ruthenium complex, the so-called ‘blue dimer’, which was reported by Meyer et al. in 1982.12 Building on the pioneering discovery of the blue dimer, ruthenium-based complexes have mainly been studied as candidates for homogeneous water oxidation catalysts,13–17 and several examples of the ruthenium-based complexes with excellent catalytic activities have been reported.18–33 However, these complexes are not necessarily suitable for potential large-scale practical applications because of the limited supply and high cost of elemental ruthenium.
In this context, earth-abundant first-row transition metal ions such as manganese, iron, cobalt, nickel, and copper are suitable alternatives, as these metal ions are inexpensive and have a lesser risk of depletion in nature. However, complexes with these earth-abundant metal ions are much less explored as water oxidation catalysts compared to ruthenium-based complexes, and the development of earth-abundant metal-based molecular catalysts remains an emerging field.34 Therefore, establishing guiding principles for designing this catalyst class is highly important.
This paper reviews the frontier of water oxidation reactions mediated by the earth-abundant-first-row transition-metal complexes. Although several excellent and relevant reviews are available,35–43 this review aims to extract the principles from the literature for the design of molecular water oxidation catalysts based on the earth-abundant first-row-transition-metal complexes. The review begins with an introduction to the challenges in creating efficient and robust catalysts for the purpose. Significant examples of the catalysts with a focus on their molecular structures and reaction mechanisms are presented next, and finally, the vital aspects for obtaining excellent catalysis are discussed.
2. Challenges in water oxidation mediated by complexes of earth abundant first-row metal ions
The oxidation of water to produce dioxygen may seem a straightforward transformation because of the structural simplicity of the substrate and products. However, the reaction is highly challenging owing to the requirement of multiple (four) electron transfer, rearrangement of multiple bonds in the cleavage of four O–H bonds, and formation of an O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond. The Gibbs free energy (ΔG) for the water-splitting reaction (eqn (1)) is ∼237 kJ mol−1. Therefore, the standard redox potential (E°) for the water oxidation reaction (eqn (2)) is +1.229 V vs. the normal hydrogen electrode (NHE) according to the Nernst equation (eqn (3)), where n is the number of electrons transferred in the reaction and F is the Faraday constant.
O bond. The Gibbs free energy (ΔG) for the water-splitting reaction (eqn (1)) is ∼237 kJ mol−1. Therefore, the standard redox potential (E°) for the water oxidation reaction (eqn (2)) is +1.229 V vs. the normal hydrogen electrode (NHE) according to the Nernst equation (eqn (3)), where n is the number of electrons transferred in the reaction and F is the Faraday constant.| H2O(l) → 1/2O2(g) + H2(g) | (1) |
| 2H2O → O2 + 4H+ + 4e− | (2) |
| ΔG = −nFE° | (3) |
Therefore, gaining insights into the natural and artificial water oxidation catalysts will be highly beneficial for obtaining the keys to the development of novel molecular catalysts for water oxidation based on the earth-abundant first-row transition metal ions. In nature, the water oxidation reaction is catalysed by a Mn4CaO5 cluster in the oxygen-evolving complex (OEC) of photosystem II. The Mn4CaO5 cluster contains the earth-abundant manganese ions, which belong to the first row of transition metals. Mn4CaO5 is a highly active catalyst with an extraordinary turnover frequency (TOF: 100–400 s−1)44 for the water-oxidation reaction and possesses a substantially low overpotential (η < 0.3 V).45 However, the structure of Mn4CaO5 cluster is stable only under biological conditions, and the extraction of the cluster from the protein is highly challenging. Moreover, artificial replication of the cluster to exhibit similar reactivity is difficult, although there exist a large number of reports on the syntheses of the synthetic models of the cluster.46–49 While simple structural mimicry of the natural enzyme is quite difficult, these promising advances indicate that high-performance molecular water oxidation catalysts can be constructed using earth-abundant first-row transition metal ions.
Next, we focus on the artificial system for water oxidation. As described earlier, the development of molecular catalysts for water oxidation has been mainly studied on metal complexes containing the ruthenium ion. The reported investigations on ruthenium-based complexes provide important insights and principles for constructing artificial water oxidation catalysts.
Water oxidation can be broadly divided into three steps: (1) transfer of multiple electrons; (2) generation of the key active intermediate for catalysis; and (3) O–O bond formation by the reaction of the key intermediate(s). The multi-electron transfer reaction can be achieved mainly using the redox-active nature of metal centres. During this process, proton-coupled electron transfer (PCET) is also frequently used to avoid the formation of highly charged species. After the accumulation of the multiple charges, the formation of catalytically active key intermediate, in which an oxygen atom is bound to a high valent metal centre, takes place. For instance, in the case of ruthenium-based complexes, Ru(V)O, and Ru(IV)–O˙ species are widely known to be the key intermediates during water oxidation. For the final O–O bond formation step, two kinds of pathways exist. The first is the nucleophilic attack of a water molecule on the oxygen atom in the key intermediate, i.e. water nucleophilic attack (WNA). This mechanism generally operates in an intermolecular fashion. The other is the coupling reaction between two key M–O intermediates (I2M). This process can proceed both in an inter- and intra-molecular fashion. The manner of the O–O bond formation is governed by the structure, arrangement, and reactivity of the aforementioned intermediate. Collectively, the complexes that can accumulate multiple charges and generate a high valent key intermediate with an oxygen atom bound to the metal centre are regarded as potential candidates for water oxidation. In addition, first-row transition metal ions participate in many biological reactions which involve O2. For example, the Mn4CaO5 cluster in the OEC can serve as a water oxidation catalyst in the natural photosynthesis process.50,51 Iron-based metalloenzymes can catalyse several oxidation reactions and supply the O2 required for respiration.52–59 Copper-based complexes in metalloenzymes are known to transport or activate O2 for subsequent oxidation of substrates.60,61 Therefore, complexes of earth-abundant first-row transition metal ions should be potential candidates for water oxidation catalysis. However, the design of new water-oxidation catalysts by the simple replacement of the ruthenium ion within the same molecular framework with these metal ions is not useful, mainly because of the lability of the earth-abundant metal ion complexes towards substitution, which renders them less stable than their ruthenium-based counterparts. Because of the high lability of such metal ions, the incorporation of two multidentate ligands of different types for forming heteroleptic complexes, a frequently used strategy for the construction of ruthenium-based water oxidation catalysts, becomes difficult. Further, ligand-scrambling results in the formation of several other complexes during the syntheses. The lability also affects the stability of high-valent species required for the water-oxidation catalysis. In other words, ligand-leaching from the metal ions upon oxidation is a problem in the case of complexes with earth-abundant first-row transition metal ions. Thus, molecular designs for mitigating such decomposition pathways are critical.
Despite the various technical challenges, the development of molecular water oxidation catalysts using earth-abundant first-row transition metal ions derives inspiration from the excellent example of the natural system, which clearly indicates that the construction of such efficient catalysts is possible. In addition, the numerous studies on ruthenium-based artificial catalysts provided important guidelines to achieve the reaction. The business currently required in this field is the establishment of a focused catalyst design strategy for employing the earth-abundant first-row transition metal complexes (Scheme 1). In the following sections, we aim to compile the factors essential for designing active catalysts for water oxidation with metal complexes of the earth-abundant first-row transition metal ions. The reported water oxidation catalysts will be classified based on their ligand structures, and their structural features, activities, and their reaction mechanisms will be discussed.
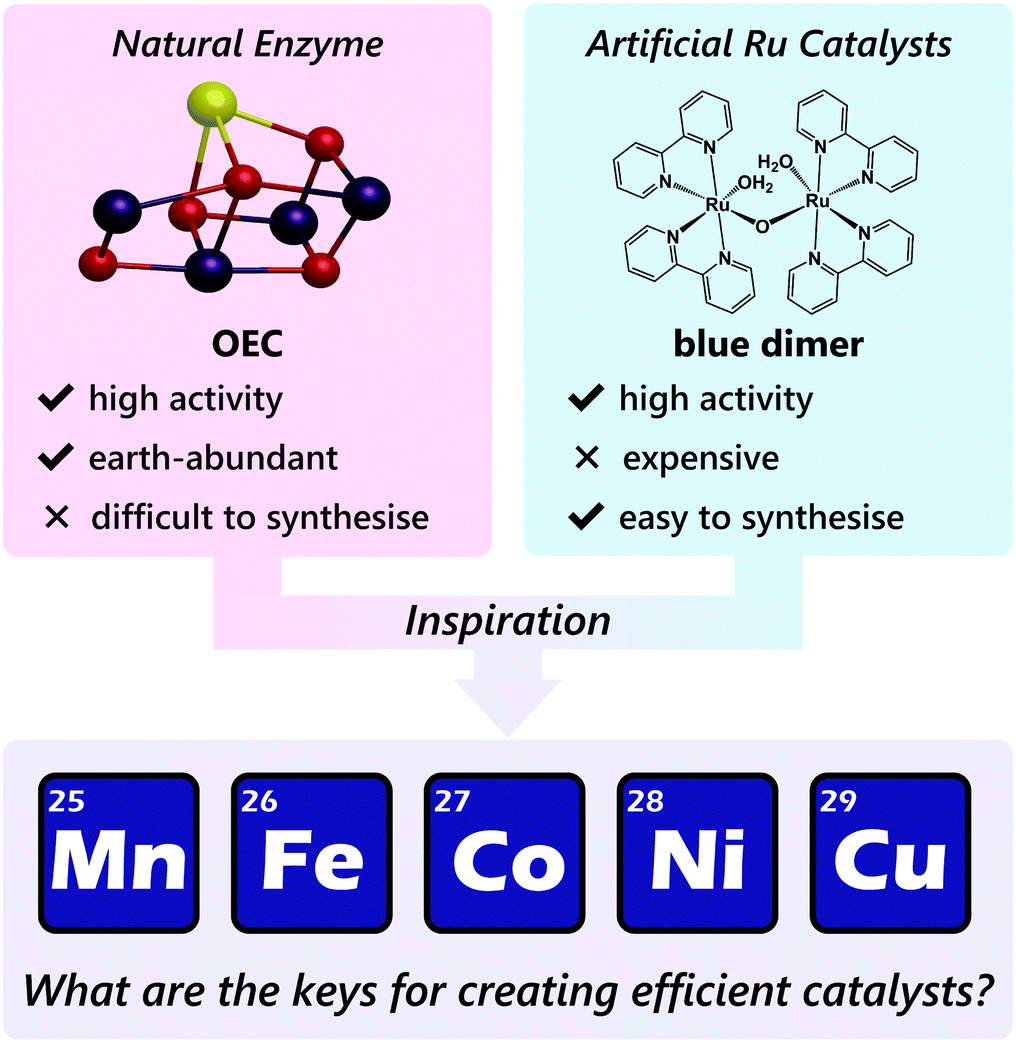 | ||
| Scheme 1 Features of the natural enzyme and artificial ruthenium-based catalysts for water oxidation (top), and the approach for the development of efficient molecular water oxidation catalysts containing earth-abundant first-row transition metal ions. | ||
3. Water oxidation catalysed by complexes of earth abundant first-row transition metal ions
Representative examples of the water oxidation catalysts are organised based on the nature of the constituent ligand(s). Chart 1 summarise the structures of the water oxidation catalysts described in this review. The purpose of this review is to unveil the effect of ligand structures on the catalytic activity and reaction mechanism of homogeneous catalysts containing earth-abundant first-row transition metal ions, and propose the rational strategy for designing catalysts with excellent performance. Therefore, in this review, following sections are grouped by ligand of water oxidation catalysts. In the beginning of each section, we will briefly introduce the structural features of the ligand focused on the section, which is then followed by the description on the examples of complexes bearing the corresponding ligand(s) mainly from the viewpoint of activity and catalytic mechanism. Finally, at the end of each section, we will provide the analysis on the properties of ligands based on the aforementioned discussion. | ||
| Chart 1 Structures of ligands or metal complexes introduced in this review. | ||
3.1 Mononuclear water oxidation catalysts with 2,2′-bipyridyl ligands and their derivatives
The strong electron-donation and chelation abilities of the nitrogen atoms render the 2,2′-bipyridyl moieties as excellent ligands for forming metal complexes with various metal ions. Notably, blue dimer, the first molecular water oxidation catalyst reported, comprises the 2,2′-bipyridine motif,12 and the ruthenium-based complexes with 2,2′-bipyridyl ligands are a prominent catalyst class for water oxidation.13–17 Inspired by their success in ruthenium-based water oxidation catalysts, the bipyridyl complexes of the earth-abundant first-row transition metal ions and their derivatives were studied.In 2012, Mayer et al. reported the first example of a copper-based molecular catalyst (Cu(bpy)(OH)2, 1 in Chart 2, bpy = 2,2′-bipyridine,) for water oxidation.62 This pioneering study demonstrated the capability of copper complexes as homogeneous electrocatalysts for water oxidation. The complex contains one bpy and two hydroxide anions as ligands and was prepared in situ by a straightforward process of mixing commercially available copper salts and bipyridine at high pH. Electrochemical studies confirmed the catalytic activity of 1 for water oxidation, and the cyclic voltammograms (CVs) of 1 under various pHs showed large irreversible peaks attributed to the water oxidation. The controlled potential electrolysis (CPE) of the complex at 1.35 V (vs. NHE) in a buffered aqueous solution (pH = 12) resulted in the formation of O2 with a faradaic efficiency of 90%. The overpotential (η) of the reaction was estimated to be 750 mV. EPR measurements of the complex revealed that 1 exists in equilibrium with the dimeric complex (Fig. 1). Further, the resting state of the complex varied with pH. Remarkably, 1 had very high reaction rates, and its TOF was estimated from CV using the following equation (eqn (4)).
 | (4) |
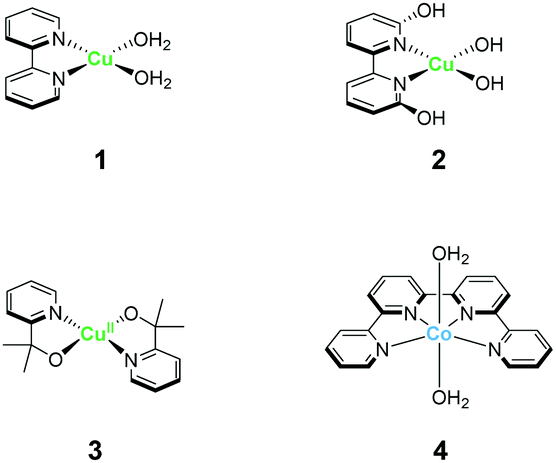 | ||
| Chart 2 Structure of mononuclear water oxidation catalysts with 2,2′-bipyridyl ligands and their derivatives. | ||
 | ||
| Fig. 1 The acid–base equilibrium of 1 in solution at different pH values. | ||
Inspired by the findings of Mayer et al., the catalytic activity of copper–bipyridine analogues was investigated. Lin and Papish independently reported the electrocatalytic activity of copper complexes containing the 6,6′-dihydroxy-2,2′-bipyridine ligands (6,6′-dhbpy).66,67 Lin investigated the electrochemical properties of a basic aqueous solution containing a Cu(II) salt and 6,6′-dhbpy. The key feature of their system is the presence of hydroxyl groups on the ligand. The ligand was designed to mimic the role of tyrosine Z or Yz in photosystem II for providing redox activity and enabling PCET during the catalysis. Importantly, such features should facilitate the access to high-oxidation-state copper species, which are required for the catalyst, lead to lower the overpotential. The complex was prepared by mixing a Cu(II) salt with 6,6′-dhbpy under basic pH and was obtained as a one-dimensional polymer with infinite structure. Electrochemical measurements of a solution containing the polymer-based complex indicated that the water oxidation reaction proceeds at η = 510–560 mV in the 12.0–14.0 pH range, which is substantially lower than η of 1. However, the TOF value of the system estimated based on eqn (4) was found to be lower (0.4 s−1) than that of 1. Papish et al. isolated the discrete complex [(6,6′-dhbpy)2Cu]n+ (2) and studied its catalytic activity. Similar to the approach of Lin et al., they aimed to lower the overpotential of the reaction by facilitating PCET and stabilising the key reaction intermediate. In their report, η and TOF were estimated to be 477 mV and 0.356 s−1, respectively. In both cases, it is indicated that the pendant hydroxyl groups on the ligand play an important role in the catalysis. The catalytic cycle reported by Lin et al. is shown in Fig. 2. In their proposed catalytic cycle, 6,6′-dhbpy serves as a non-innocent ligand and participates in electron transfer and PCET reactions. Due to such nature of the ligand, the catalytically-active high-oxidation-state copper species form more readily, which contributes to the lowering of η value of the system compared to that of 1. In this system, the Cu(III)–OH species (B in Fig. 2) act as a key intermediate for O–O bond formation and the O–O bond formation reaction proceeds via the WNA mechanism. Further investigations are required to clarify the nature of the catalytic intermediate and the catalytic mechanism.
 | ||
| Fig. 2 Plausible catalytic cycle for the water oxidation reaction mediated by copper–bipyridine complex 2. | ||
A copper complex with bidentate ligands containing a pyridyl moiety is also known to serve as a water oxidation catalyst. In 2017, Brudvig et al. reported the novel copper complex, Cu(pyalk)2 (pyalkH = 2-pyridinyl-2-propanol, 3).68 The analogous iridium complex with the pyalkH ligand was previously studied for water oxidation catalysis69 and demonstrated oxidation resistance. The group also showed that pyalkH undergoes facile deprotonation and forms the strongly electron-donating alkoxide anion, which can stabilise high oxidation states. Due to such reasons, pyalkH was expected to be a good ligand for designing water oxidation catalysts. In the electrochemical measurements of complex 3 at pH 13.3, a large reversible catalytic current was observed. Controlled-potential electrolysis of 3 was conducted at 1.1 V (vs. Ag/AgCl), and the formation of ∼600 μmol of dioxygen with a faradaic efficiency of ∼75%, which corresponds to a catalytic turnover of >30, was confirmed. The η and TOF of the catalytic reaction were 520–580 mV (pH 10.4–13.3) and ∼0.7 s−1 based on eqn (4), respectively. The group also investigated the stability of the catalyst by UV-vis absorption spectroscopy, SEM, EDX, and electrochemical measurements, and confirmed the robustness of the complex as a homogeneous catalyst. The control experiment using copper(II) picolinate indicated that the oxidation- and dissociation-resistant pyalkH ligand is likely the origin of the high stability of 3. In the subsequent report by the same group, a catalytic mechanism was proposed based on quantum chemical calculations, UV-vis absorption spectroscopy, kinetic isotope effect, and electrochemical measurements (Fig. 3). In the proposed mechanism, the complex 3 first isomerises to the cis-form (A), and the subsequent oxidation of the metal centre and the binding of the hydroxide anion generates the Cu(III)–OH species (C). The cis form alone is responsible for water oxidation. Next, the PCET reaction proceeds to generate the Cu(III)–O˙ species (D), which is the key intermediate. The intermediate reacts with water via WNA mechanism to form the O–O bond, which is the rate-determining step. The resultant peroxo species (E) is further oxidised via PCET and liberates dioxygen. Remarkably, the pyalk ligand remains bound even with a labile pyridine group that opens up a coordination site for water binding, which confirms the high stability of the complex.
 | ||
| Fig. 3 Catalytic cycle for water oxidation with the copper–PyalkH complex 3. | ||
In 2020, Zhang et al. reported a relevant mononuclear copper molecular catalyst for electrochemical water oxidation.70 The catalyst was simply generated in situ from copper salt and an organic ligand, 1-mesityl-1H-1,2-3-triazole-4-carboxylic acid, in a 0.1 M sodium acetate solution. The complex can catalyze electrochemical water oxidation at a very low overpotential (0.31 V) under neutral conditions. It was also revealed that the catalysis undergoes with first-order kinetics to the catalyst, suggesting a single-site mechanism for water oxidation.
In 2012, Lau et al. reported a cobalt complex Co(qpy)(OH2)2 bearing the tetradentate polypyridyl ligand 4 (qpy = 2,2′:6′,2′′:6′′,2′′-quaterpyridine).71 The structure of qpy comprises two 2,2′-bipyridine moieties connected with each other, and thus, the ligand is expected to have strong chelation and oxidation resistance. Further, 4 was the first reported example of a cobalt-based complex that can drive water oxidation photochemically. Upon irradiating visible light (457 nm) on a borate buffer solution containing catalytic amounts of 4, the photosensitiser [Ru(bpy)3]2+, and the sacrificial electron acceptor sodium peroxodisulfate, O2 evolution occurred with a turnover number (TON) of 335. The oxygen atom in this reaction was confirmed to originate from water by 18O-labelling experiments, and mass spectroscopy. The authors also performed several photochemical control experiments and dynamic light scattering measurements and confirmed the absence of cobalt oxide nanoparticle formation during the catalysis. These results highlight the use of multidentate ligands with oxidation-resistance as an effective strategy for stabilising high-valent cobalt–oxo species without forming heterogeneous metal oxide species.
As described below, 2,2′-bipyridyl ligand-based complexes with earth-abundant first-row transition metal ions can function as efficient catalysts for water oxidation. The inherent feature of this class of ligands is their high resistance towards oxidation reactions (Chart 3). The ease of preparation of the catalysts is another advantage. Additionally, the electronic structures and properties of the ligands can be tuned by introducing substituents at appropriate positions, which can stabilise the high-valent key reaction intermediates. Some reports disclosed that an increase in coordination positions on the ligand or the use of an anionic ligand enhance the stability of the resultant catalyst molecules. However, the reports on the 2,2′-bipyridyl ligand-based complexes with earth-abundant first-row transition metal ions are mainly limited to copper-based complexes with square planar geometry. The bidentate structure of 2,2′-bipyridyl type ligand often leads to the formation of tris(2,2′-bipyridyne)–metal complexes in the case of metal ions, which prefer hexacoordinated octahedral geometry. Therefore, a substitution-labile site on the complex, which functions as a water binding site, cannot be generated.
 | ||
| Chart 3 Summary of features of water oxidation catalysts with 2,2′-bipyridyl ligands and their derivatives. | ||
In this regard, the utilisation of a ligand with more coordination sites would be preferable for the construction of water oxidation catalysts based on metal complexes containing Mn, Fe, Co, and Ni ions.
3.2 Mononuclear water oxidation catalysts with PY5-type ligands
The development of first-row transition metal complexes that are stable during the catalysis reaction is key for obtaining efficient water oxidation catalysts. In this regard, the use of multidentate ligands is an important strategy. The derivatives of 2,6-(bis(bis-2-pyridyl)-methane)-pyridine (PY5) are excellent candidates owing to their pentadentate coordination and are suitable for constructing stable structures. Another important feature of these ligands is the open-site, which serves as a substrate binding site, can generate by the reaction with a metal ion in an octahedral geometry. Due to these reasons, several first-row transition metal complexes with PY5 ligands have been developed, and their catalytic activity for water oxidation was investigated.In 2012, Berlinguette et al. reported the catalytic activity of the PY5-based cobalt complex 5 (Chart 4).72 This is the first example of a cobalt complex that catalyses water oxidation. The authors used 2,6-(bis(bis-2-pyridyl)methoxy-methane)-pyridine as the ligand, wherein the methoxy groups were introduced to prevent ligand oxidation which causes catalyst deactivation. The metal centre of the complex has an octahedral geometry (Chart 4), and five out of the six coordinating atoms are nitrogen derived from the pyridine moieties of the ligand; a water molecule coordinates at the remaining site. Electrochemical measurements revealed that 5 undergoes PCET to furnish [CoIII–OH]2+. A sweep to the positive potential region indicated an irreversible catalytic current at ca. 1.45 V (vs. NHE), which indicates the formation of a [CoIV–OH]3+ species that are reactive towards OH−. It is noteworthy that the stabilisation of the metal ion within the pentadentate framework of PY5-type ligands allows access to this reactive intermediate. Building on this success, the electrocatalytic activity of 5 for water oxidation was intensively studied, and catalysis with η = 500 mV at pH = 9.2 was achieved. The authors performed several electrochemical measurements using [Co(OH2)6]2+ and excluded heterogeneous catalysis of water oxidation by the cobalt-oxide-based film formed on the electrode. Overall, the study confirmed the homogeneous water oxidation catalysis by 5 and demonstrated that the PY5-type are an important class of ligands for the design and construction of water oxidation catalysts based on earth-abundant transition metal ions. In a later study, the catalytic cycle of 5 was proposed based on electrochemical studies and quantum chemical calculations73 (Fig. 4). The electrochemical studies revealed that the two-step PCET reaction from the initial state of the complex affords the key Co(IV)–oxyl catalytic intermediate (C). This species has an intermediate-spin-quartet configuration of the formally CoIV-d5 centre. The structurally-rigid PY5-type ligand framework is responsible for generating the ligand field, in which this highly active state is almost isoenergetic to the low-spin doublet ground state. The O–O bond formation step was intensively investigated, and both intra- and inter-molecular pathways were considered. The results of the DFT calculations, together with electrochemical measurements, indicated that the nucleophilic attack of the hydroxide anion in an intermolecular fashion (C → D) is the most plausible pathway. By further oxidation of the complex via PCET mechanism, dioxygen is liberated, and the coordination of a water molecule regenerates the initial state. Thus, the structure of complex 5, which remains robust pentacoordinate structure during the catalytic process and also has an open site at the octahedral apex as a substrate binding site, is useful for water oxidation.
 | ||
| Chart 4 Structures of mononuclear water oxidation catalysts with PY5-type ligands. | ||
 | ||
| Fig. 4 Proposed water-oxidation mechanism using Co–PY5 complex 5 as a catalyst. | ||
Ott and Thapper et al. developed several complexes with the 2,6-(bis(bis-2-pyridyl)hydroxy-methane)-pyridine (PY5-OH) ligand. The hydroxy groups were introduced on the ligand to explore the possibilities for further functionalisation suitable for surface immobilisation. The first report by the group on the complex bearing a cobalt ion, [Co(PY5-OH)Cl](BF4) (6)74 investigated the photo-, electro-, and chemical-catalytic activities of the complex. The CVs of 6 in borate buffer (pH 8 and 9) showed a catalytic current at 1.2–1.3 V (vs. NHE), and the overpotentials for the reaction were 540 and 510 mV at pH = 8 and 9, respectively. Light-driven water oxidation using 6 as the catalyst was investigated in the borate buffer (pH 8) in the presence of [Ru(bpy)3]2+ as the photosensitiser and S2O82− as the sacrificial electron acceptor. Upon visible-light irradiation (λ = 470 ± 10 nm, 820 mE cm−2 s−1), O2 evolution with a TOF of 1.3 ± 0.2 s−1 and TON of 51 ± 3 was observed. The catalytic reaction with [Ru(bpy)3]3+ as the chemical oxidant proceeded with TOF of 15 ± 1. The removal of chloride ligand and further oxidation resulted in the formation of a μ-oxo bridged dimer complex, and this dimeric intermediate was found to be almost inactive for water oxidation. Therefore, the presence of chloride is essential for stabilisation of the complex. The authors also proposed the catalytic mechanism based on EPR measurements and UV-vis absorption spectroscopy (Fig. 5). In the mechanistic studies, the authors used m-chloroperoxybenzoic acid (m-CPBA) as the oxidant. The key Co(IV)O intermediate (B) is generated in the presence of m-CPBA, wherein the temporary detachment of a pyridine ligand provides the coordination site for substrate binding. Subsequently, the O–O bond formation occurs via the WNA pathway (B → C), and the liberation of the O2 from the complex closes the catalytic cycle. The authors also analysed the reaction mixture after the catalysis by DLS and UV-vis absorption spectroscopy and confirmed the molecular integrity of the catalyst during the turnover. Although the catalytic mechanism of 6 is different from that of 5, these studies disclosed that Co complexes bearing PY5-type ligands could serve as molecular catalysts for water oxidation.
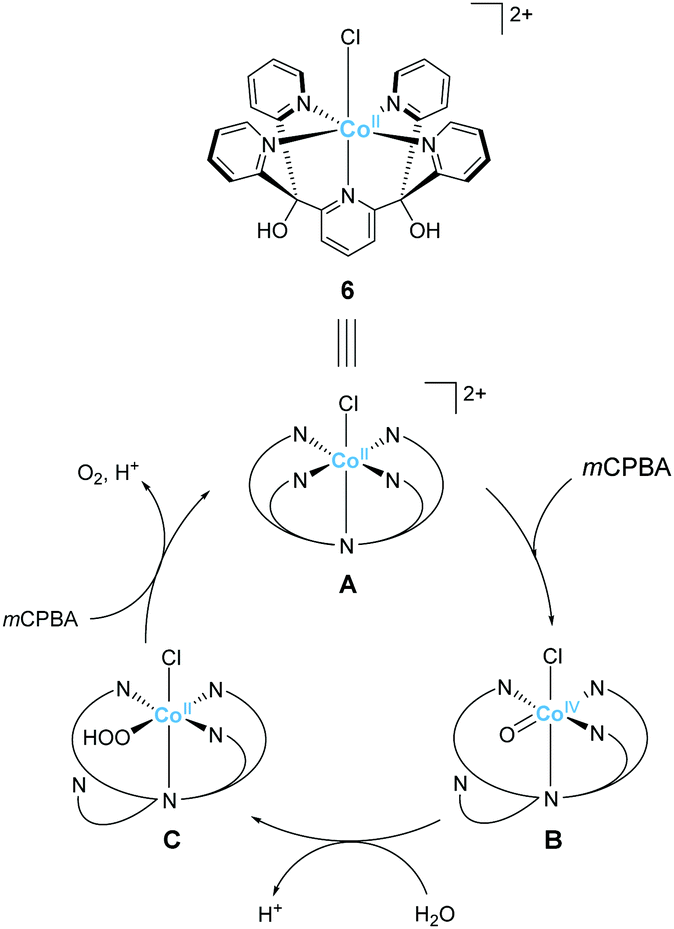 | ||
| Fig. 5 Postulated mechanism for water oxidation by Co–PY5-OH complex 6. | ||
In 2016, the same group reported two iron complexes bearing the PY5-OH ligand (7 and 8).75 In their report, cerium ammonium nitrate (CAN) and [Ru(bpy)3]3+ were used as the chemical oxidants. Interestingly, under acidic conditions (pH 1.5) and with CAN as the oxidant, 7 exhibited higher activity than 8. In contrast, the catalytic activity of 8 was better than that of 7 when the reaction was performed at pH 8 using [Ru(bpy)3]3+ as the oxidant. The TON and TOF of 7 at pH 1.5 were 16 and 0.75 s−1, respectively, and those of 8 at pH 8 were 26.5 and 2.2 s−1, respectively. Notably, 8 also catalyses light-driven water oxidation at pH 8 (TON: 43, TOF: 0.6 s−1). The authors carefully examined the reaction mechanism using EPR and quantum chemical calculations and proposed the mechanism shown in Fig. 6 and 7.
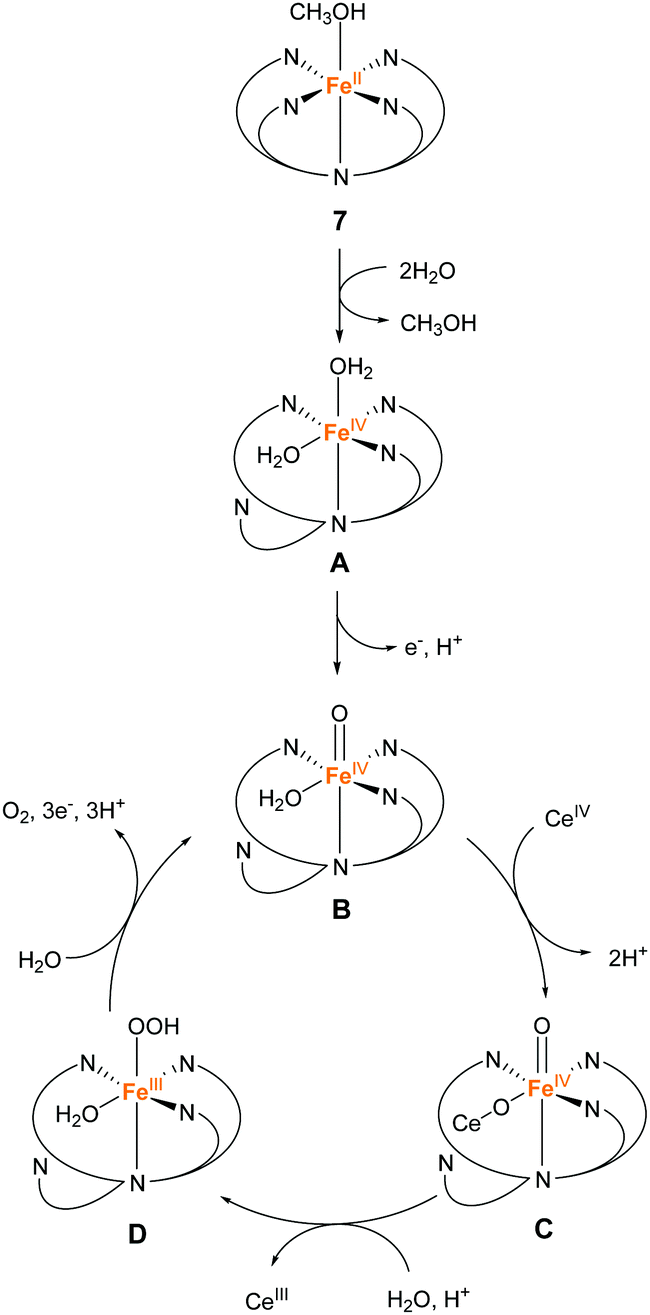 | ||
| Fig. 6 Proposed mechanism for water oxidation by Fe–PY5-OH complex 7 using CAN as a chemical oxidant. | ||
 | ||
| Fig. 7 Proposed mechanism for water oxidation by Fe–PY5-OH complex 8 using [Ru(bpy)3]3+ as a chemical oxidant. | ||
At pH 1.5, the two sites with the labile ligands in the cis configuration are necessary for efficient catalysis. The coordination of two water molecules to 7 and the subsequent PCET affords Fe(IV)O as the key intermediate (B in Fig. 6), and then Ce cation coordinates to the intermediate B, forming intermediate C. After that, the WNA reaction of this intermediate C leads to the bond formation between the two oxygen atoms (D in Fig. 6). Intermediate D releases oxygen due to further WNA and returns to intermediate C. It should be noted that 8 cannot catalyse the reaction under acidic conditions because one of the coordination sites is occupied permanently by the chloride ligand.
In contrast, the de-coordination of a pyridyl group and the reaction with water proceed initially in the catalysis mediated by 8 at pH 8 (Fig. 7). Subsequently, one- and two-electron oxidations involving the transfer of two protons proceed to afford the catalytic intermediate Fe(V)O (C). The Fe(V)O species reacts with water to form the O–O bond via WNA (D). Under these conditions, the stability of 7, which lacks a chloride ligand, is lower than that of 8. Intermediate D releases oxygen due to further PCET and returns to intermediate B. The results of the study showed the application of the PY5-type ligands for preparing molecular catalytic systems for water oxidation under various experimental conditions.
The most recent example of a PY5-based complex for water oxidation was reported by Sun et al. in 2016.76 They synthesised a nickel complex (9) bearing 2,6-(bis(bis-2-pyridyl)methyl-methane)-pyridine (PY5-Me) as the ligand and investigated its catalytic activity. From the results of electrochemical studies, the TON and TOF values of water oxidation mediated by the complex in a phosphate buffer were 19 and 145 s−1 (eqn (4) was used for the calculation of TOF), respectively. The robustness of the catalyst was confirmed by DLS, XPS, and electrochemical measurements. Further, the catalytic mechanism of 9 was recently suggested based on the quantum chemical calculations.77 The formation of the active species Ni(V)O, which was supposed to be the key intermediate from the original report, was difficult to access in this system due to its high redox potential (2.17 V), therefore Ni(IV)O was suggested to be the actual key intermediate involved in the O–O bond formation. An overview of the proposed catalytic cycle is shown in Fig. 8. In this catalytic cycle, two-step PCET reactions from the initial state afford the key Ni(IV)O intermediate (B).78,79 The intermediate then reacts with water via WNA, leading to O–O bond formation (B → C). The authors note that HPO42− plays a crucial role in this reaction by abstracting a proton from the water molecule during the attack. Quantum chemical calculations indicated that the presence of HPO42− lowered the activation energy for O–O bond formation to 11.5 kcal mol−1, which is in excellent agreement with the experimental kinetic data (barrier of 12.9 kcal mol−1). Further PCET (C → D), oxidation (D → E), and O2 dissociation (E → 9) regenerated the initial catalyst state. Additionally, the authors calculated the activation barriers for the O–O bond-formation process for various M-PY5-Me (M = Mn, Fe, Co, Ni, and Cu) complexes. The calculation results suggest that the Cu-analogue is expected to show a lower energy barrier compared to 9. Further, its redox potential is comparable to that of 9, whereas the complexes bearing Mn, Fe, and Co ions likely have a higher energy barrier for water oxidation compared to that of 9. They also calculated the ligand exchange reaction of PY5-MIVO species and found that the binding strength of PY5 to the metal ions follows the trend Co > Ni > Mn > Cu > Fe. Further, a difference of 6.4 kcal mol−1 was observed between Ni (56.4 kcal mol−1) and Cu (50.0 kcal mol−1). These results indicate that both the energy barrier of the catalysis and the stability of the catalysts should be considered for developing superior molecular water oxidation catalysts from earth-abundant transition metal complexes.
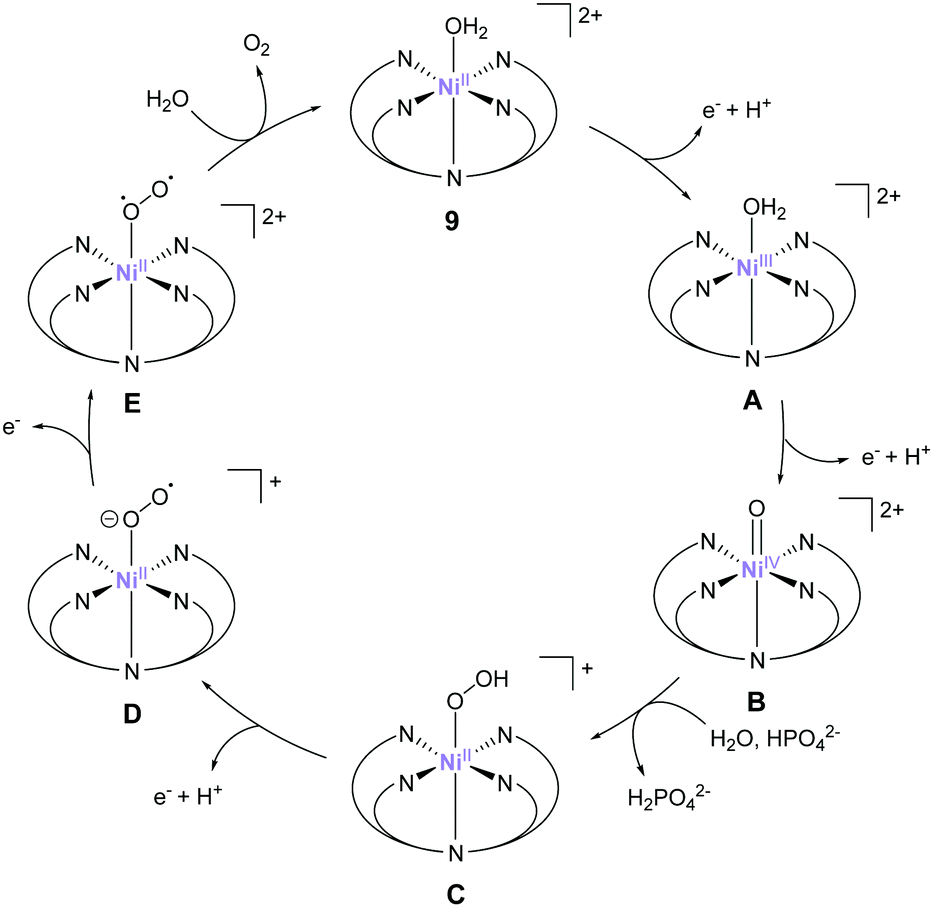 | ||
| Fig. 8 Proposed mechanism for electrocatalytic water oxidation by Ni–PY5-Me complex 9. | ||
The aforementioned examples of PY5-complexes indicate that the following features of PY5 are essential for catalysis (Chart 5). First, because PY5 is a pentadentate ligand, the dissociation of the entire ligand from the metal ion is not facile, which is an important attribute for mitigating catalyst decomposition. Second, the alkyl linker between the pyridyl moieties allows flexible ligand movement, which allows partial dissociation of the ligand for efficient catalysis. Third, the methoxy, hydroxy, or methyl protecting groups on the alkyl linker inhibit undesired pathways for ligand oxidation. Actually, the molecular integrity of the catalysts was proved by various studies. Therefore, this class of ligands are promising candidates for the development of water oxidation catalysts based on first-row transition metal ions. However, a recent report pointed out the possibility of heterogeneous nanoparticle formation during the catalysis, and that the resultant nanoparticles are the true species that catalyse water oxidation in iron-based PY5-complexes.80 Therefore, further in-depth mechanistic studies are required to elucidate the prevailing catalysis mechanism.
 | ||
| Chart 5 Summary of features of water oxidation catalysts with PY5-type ligands. | ||
3.3 Mononuclear water oxidation catalysts with alkylamine-pyridine complexes
The success of the PY5-type ligands in water oxidation catalysis with earth-abundant first-row transition metal ions highlights the utility of multidentate ligand systems. The intrinsic structural rigidity and oxidation resistance of these ligands confers high catalyst stability during the reaction. The use of alkylamine-pyridine type ligands is another important approach in the water-oxidation catalyst design owing to their capacity to stabilise high oxidation states and their ability to serve as highly-active oxidation catalysts.81–83 Also, the structural flexibility of the alkylamine moiety and coordination ability of pyridyl fragments are promising features for the ligand design. Moreover, the derivatisation of the structure is relatively straightforward, which is advantageous for fine-tuning the structural and electronic features of the complexes. Owing to several such beneficial attributes, this class of ligands were studied for the development of water oxidation catalysts.In 2011, Fillol and Costas et al. reported the catalytic activity of a series of iron complexes bearing the tetradentate alkylamine-pyridine type ligands (10–16 in Chart 6).84 Ligand availability, modularity, and versatility of these complexes allowed the systematic study of a large number of structures. The following are the key structural features of these complexes: 10–14 have two labile sites that are disposed in a cis configuration; 15 has two labile sites in a trans configuration; and 16 has only one labile site. The catalytic activity of the complexes was examined using CeIV as the chemical oxidant at pH 1. Although 10-14 exhibited catalytic activity, 15 and 16 were inactive for water oxidation, which strongly indicates that the presence of two labile sites in a cis configuration is essential for the desired reactivity. Among the complexes investigated, 11 showed the highest activity with a TOF of 0.23 s−1 and TON of 360. This result indicates that the use of lower coordination rigidity and lower ligand basicity are keys to improving the catalytic activity in this class of complexes. The authors also elucidated the plausible catalysis mechanism of the active complexes based on UV-vis absorption spectroscopy (Fig. 9). In this catalytic cycle, the iron centre is initially oxidised by the Ce(IV) oxidant to the Fe(IV)O species (A), the formation of which was confirmed by ESI-MS. The further oxidation of the species by Ce(IV) affords the Fe(V)O species (B), which serve as the key intermediate for O–O bond formation. Subsequently, the WNA reaction occurs, wherein the hydrogen bonding between a hydroxyl ligand and H2O molecule (C) assist in the formation of the Fe(III)–(OOH) species (D). Finally, the dissociation of O2 accompanied by the transfer of three electrons and three protons regenerates the Fe(IV)O catalytic resting state. Overall, this study succeeded in identifying the key features for the construction of water oxidation catalysts using alkylamine-pyridine complexes.
 | ||
| Chart 6 Structures of alkylamine-pyridine complexes. | ||
 | ||
| Fig. 9 Postulated mechanism for water oxidation by iron complexes 10–14 based on tetradentate nitrogen ligands. | ||
The complexes developed in the aforementioned report pursued a novel design of the iron-based complexes for water oxidation. However, the potential oxidation of the alkylamine ligand during the catalytic reaction leads to catalyst deactivation. To overcome this problem, the same group reported a novel complex similar to 11 in 2019.85 In this complex (17), the hydrogen atoms at the benzylic methylenic positions were replaced by deuterium, which imparted robustness against C–H oxidation. Therefore, the TOF of 17 reached 3800, which is substantially higher than that of 11. Notably, the initial reaction rates of these complex are identical, indicating that the deuteration does not affect the reaction rates. This study clearly demonstrates that catalytic activity can be improved by the identification of structural limitations at the molecular level and with remediation by rational design.
In 2014, Smith et al. reported the catalytic activity of a series of manganese-based complexes comprising the tetradentate pyridinophane ligand (18–20).86 Among them, 18 and 19 were found to catalytically disproportionate H2O2 in aqueous solution. In contrast, 20 exhibited water-oxidation catalysis. The CPE experiment of the complex at 1.23 V (vs. NHE) confirmed the formation of dioxygen with a faradaic efficiency of 74–81% and TON of 16–24. The unique water-oxidation reactivity of 20 largely relies on the bulkiness of the substituent on the pyridinophane scaffold. The installation of a bulky substituent (t-Bu) in 20 prevents the formation of the dimeric complex, which is inactive for water oxidation. It should also be noted that the electron-donating ability of the t-Bu groups enables facile access to higher oxidation states. The study highlights the importance of rational modification for achieving remarkable changes in catalytic activity. The mechanism for the catalysis with 20 was reported by the same group in 2017 (Fig. 10).87 The catalytic cycle was proposed based on DFT calculations. In its initial state, the complex undergoes deprotonation (20 → A), and the subsequent PCET reactions (A → B → C) generate the key catalytic intermediate C (Fig. 10). The next step involves the formation of O–O bond via intramolecular coupling between the two oxygen atoms (I2M mechanism). The O–O bond formation process proceeds via coupling between the two oxyl radical fragments (D → E). The energy barrier for the intramolecular O–O bond formation was estimated as 14.7 kcal mol−1. The authors also claimed two important features of the pyridinophane scaffold, which were its ability to (1) provide an appropriate environment for generating various oxidation and spin states and (2) its flexible nature that allowed the formation of the octahedral complex with the two labile sites in a cis configuration.
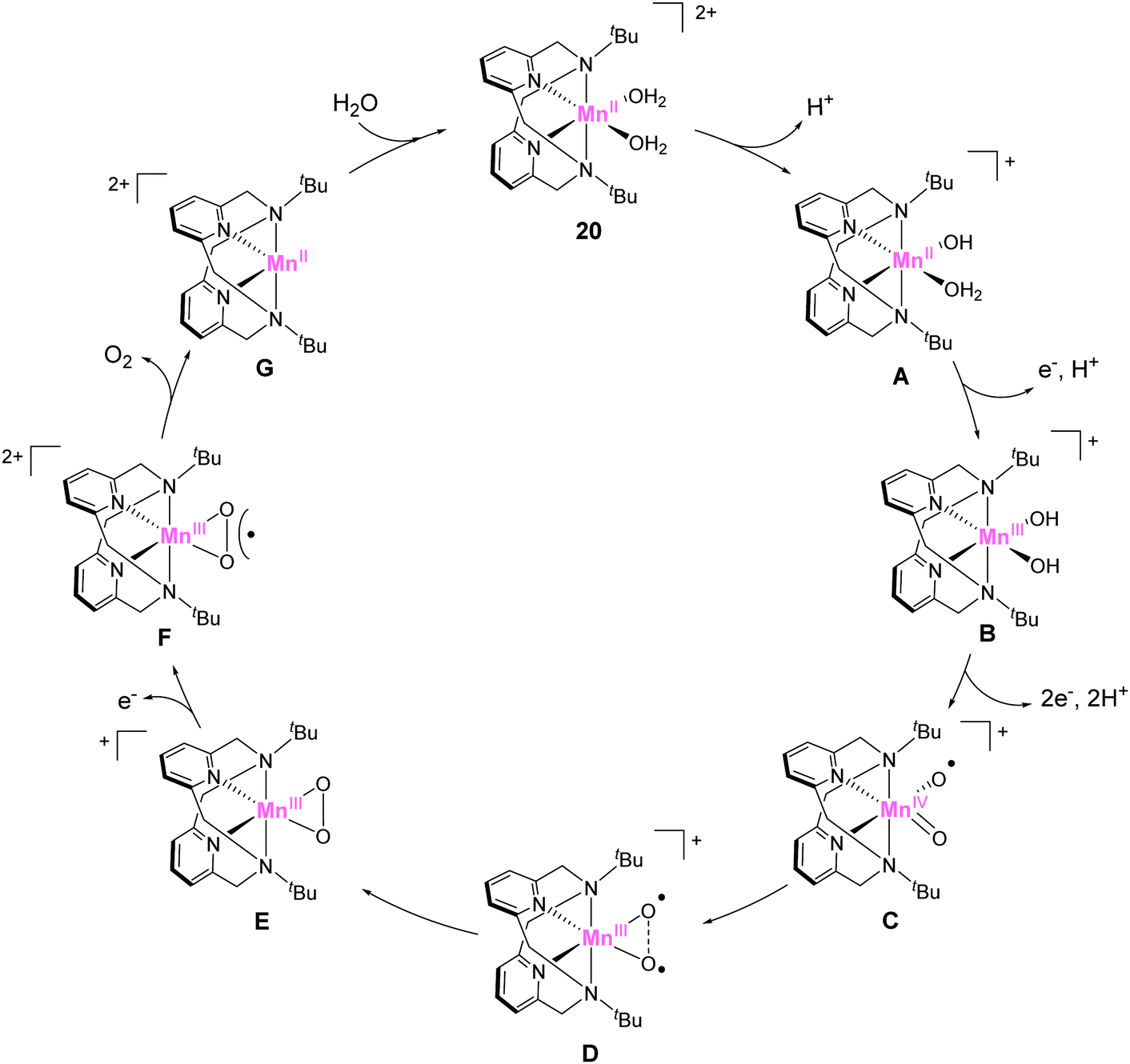 | ||
| Fig. 10 Proposed catalytic cycle for water oxidation mediated by manganese pyridinophane complex 20. | ||
Recently, Liao et al. proposed another catalytic pathway of 20 based on DFT calculations.88 They proposed two possible mechanisms for the O–O bond formation. The first involves the intramolecular coupling of the radical species similar to the mechanism proposed by Smith et al., and the other possible pathway is the WNA reaction of the Mn(IV)O. The activation energies for the intramolecular I2M reaction and intermolecular WNA reaction were 13.8 and 13.1 kcal mol−1, respectively. In the latter WNA mechanism, the oxo moiety that is not attacked by a water molecule serves as a base for deprotonating the water molecule bound to the Mn(IV)O moiety. Overall, they concluded that the O–O bond formation process likely proceeds both via the intramolecular I2M and intermolecular WNA processes.
In 2016, Lu reported that a nickel analogue of the complex 21 was active for water oxidation.89 Similar to the complexes developed by Costas et al. (10–14), this complex also has the two labile sites in the cis configuration. Electrochemical analysis of the complex (21) in acetate buffer showed an irreversible catalytic current at an onset potential of ca. 1.35 V (vs. NHE). Further, the CPE of the complex at 1.60 V (η = 750 mV) confirmed the formation of dioxygen, and the stability of the complex during the CPE was confirmed by UV-vis absorption spectroscopy, electrochemical measurements, and ESI-mass spectroscopy. The faradaic efficiency and TON of the reaction were ∼70% and 3.4 (33 h), respectively. The proposed mechanism of water oxidation mediated by 21 is shown in Fig. 11. The mechanism was proposed based on electrochemical measurements and DFT calculations. Initially, one of the water molecules in 21 is replaced by an acetate anion via ligand exchange. The species thus formed undergoes a 2e− per one H+ PCET process, and the subsequent ligand exchange between acetic acid and water molecules generates the bis- hydroxy-Ni(IV) species (A). The O–O bond formation step occurs via the HO–OH coupling of the two cis-OH groups with the assistance of the acetate (B) to generate the Ni(II)–(OOH) species (C). Notably, kinetic isotope effect studies indicated that the atom-proton transfer process for the O–O bond formation is the rate-determining step. Further, the calculated energy barrier of the O–O bond formation was 18.9 kcal mol−1. Finally, the Ni(II)–(OOH) intermediate underwent further oxidation to yield O2 and re-entered the catalytic cycle (C → 21). Although this study demonstrates that the nickel complex with alkylamine-pyridine type ligands and cis labile sites can serve as water oxidation catalysts, other studies demonstrated that the complex with the trans labile sites exhibits higher activity.90 Therefore, the strategy for designing highly active nickel-based catalysts should differ from that for iron-based complexes.
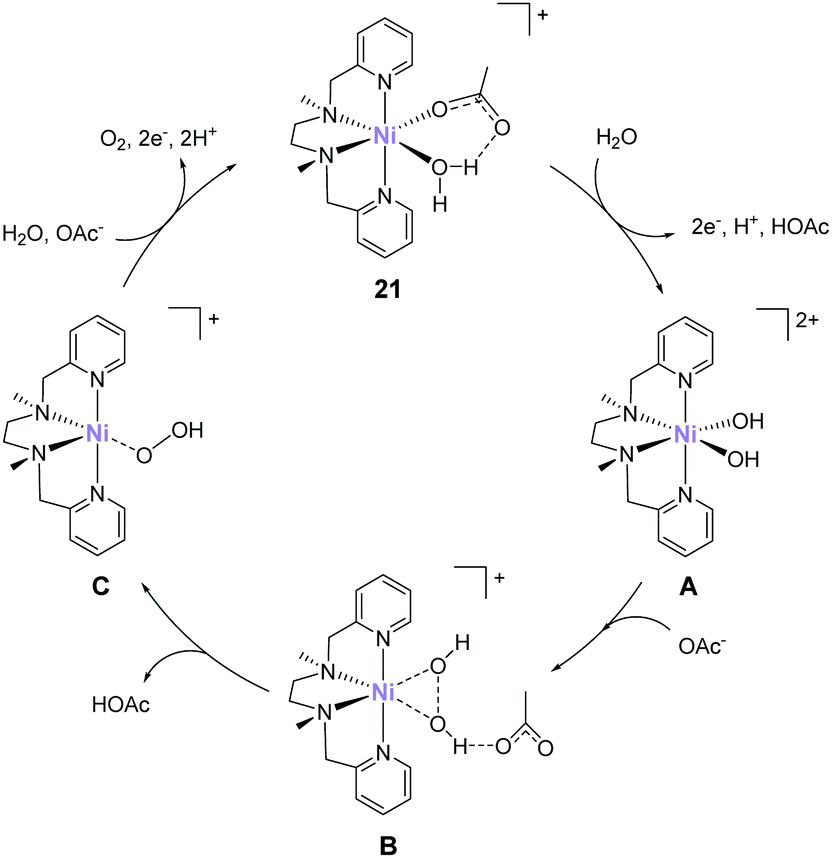 | ||
| Fig. 11 Proposed pathway for water oxidation by Ni–PY5 complex 21 in phosphate buffer solution. | ||
Pentadentate ligands were also utilised as alkylamine pyridyl-type ligands. Brudvig et al. reported a series of manganese complexes with pentadentate alkylamine pyridyl-type ligands (22 and 23) in 2013.91 They reported chemical oxidation reactions with Oxone or hydrogen peroxide as the oxidants, and 22 showed the evolution of O2 in the presence of both Oxone and hydrogen peroxide, whereas 23 acted as a water oxidation catalyst only in the presence of hydrogen peroxide. The difference between these two complexes is that 22 has a pentadentate anionic ligand, whereas 23 has a pentadentate neutral ligand. The high σ-donating ability of the anionic ligand should stabilise the high-valent manganese species in water. Indeed, the MnIV/MnIII redox potential of 22 is located in the more negative potential region compared to that of 23, which indicates that 22 is more easily oxidised to the high-valent state than 23. Although 22 exhibited better activity than 23, the TON (3–4) and TOF (0.01 s−1) of the complex are lower than those of reported manganese-based water oxidation catalysts. Further, the authors indicated that the low activity of these complexes is due to their decomposition during the catalysis. The mechanisms of degradation may include oxidation of the ligand to produce a different complex or an inactive manganese oxide material. However, this study demonstrates that the inclusion of an anionic carboxamido ligand trans to the labile coordination site promotes the formation of a high-valent Mn species which can facilitate oxygen formation. In other words, the use of an anionic ligand that remains unprotonated during the catalysis is an essential strategy for obtaining manganese-based water oxidation catalysts.
Meyer et al. explored the catalytic activity of a copper complex bearing the polypeptide ligand 24.92 Polypeptides are biologically-relevant molecules comprising several amino acids which are covalently-linked together. This class of ligands are particularly interesting because of their excellent ligand tunability via precise control of the chemical structures and pKa values of the constituent amino acids. Also, the metal complexes can be prepared from the ligands via the facile self-assembly processes. The electrochemical measurement of the complex was performed in 0.25 M phosphate buffer, and one pH-dependent reversible oxidation wave appeared at 0.58 V versus NHE (pH 11). The result indicated that the oxidation of the metal centre involves the PCET process. Upon sweeping the potential to the positive region, an irreversible oxidation wave was observed at ca. 1.32 V, suggesting the catalytic activity of the complex for water oxidation. Further, a CPE experiment was conducted to identify the product of the reaction, and the evolution of dioxygen with a faradaic efficiency of 99% was confirmed. The TON after 8 h reached ca. 13 and the onset potential for water oxidation was determined to be ca. 1.10 V, which corresponds to an overpotential of ca. 520 mV at pH 11. The authors also calculated the TOF to be 33 s−1 based on cyclic voltammetry (eqn (4)) and proposed a plausible cycle for the catalysis (Fig. 12) based on the electrochemical analysis. First, the proton-coupled oxidation of the CuII–aqua complex generated the corresponding CuIII–hydroxo complex (A), which was indicated by the pH-dependent behaviour of the first oxidation wave. A further oxidation coupled with the transfer of electron led to the formation of the high-valent CuIVO (B) or CuIII(O˙) (C) species, which were analogous to the highly reactive RuV–oxo (or RuIV–oxyl) intermediate. The pH-dependent behaviour of the potential of the catalytic current also supported their hypothesis.79,93 This high-valent intermediate then reacts with H2O to produce the [CuII–OOH]3− (D) or [CuII(HOOH)]2− (E) species. The study also revealed that the rate-determining step was the O–O bond formation process. Further oxidation of these species coupled with proton transfer resulted in the formation of [CuIII–OO˙]2− (H), from which, the liberation of dioxygen completed the catalytic cycle. This study demonstrated that bio-relevant polypeptides could also be employed as ligands for water oxidation catalysts containing an earth-abundant first-row transition metal ion.
 | ||
| Fig. 12 Proposed mechanism for water oxidation mediated by the copper complex bearing the polypeptide ligand 24. | ||
Meyer et al. also developed an iron complex with an anionic pentadentate alkylamine pyridyl-type ligand (25).94 This is the first example of an iron-complex electrocatalyst for water oxidation. The complex showed a quasi-reversible peak, which was attributed to the Fe(III)(OH2)/Fe(II)(OH2) process at 0.38 V vs. NHE in dry propylene carbonate (PC). Upon further sweep of the potential to the positive region, an irreversible oxidation peak assigned to the Fe(III)(OH2) → Fe(V)O oxidation process was observed at 1.58 V. In the presence of water, a catalytic current coupled with the Fe(III)(OH2) → Fe(V)O process emerged. The CPE of the complex was performed at 1.58 V in PC-8% H2O solution containing 0.5 M LiClO4. The evolution of dioxygen was confirmed after 15 h of electrolysis in spite that the faradaic efficiency of the catalysis is not so high (45%). TON and TOF of the reaction were 29 and 0.15 s−1 (TOF was determined based on eqn (4)), respectively. The catalysis mechanism of the complex was formulated based on the electrochemical studies using H2O and D2O as the substrates (Fig. 13). The mechanism was analogous to the previously reported ruthenium polypyridyl complex-based catalysts.13–17 The PCET reaction of the initial state affords the key Fe(V)O intermediate (A), and its reaction with a water molecule results in the O–O bond formation (A → B) via the WNA mechanism. The rate-determining step is the O–O bond formation process. The subsequent PCET reaction (B → C) and ligand exchange between the dioxygen and water molecules completed the cycle and regenerated the initial state (C → 25). It is noteworthy that the onset of catalytic current for the water oxidation with 25 occurs at an overpotential that is 200–250 mV lower than those of the relevant ruthenium-based complexes. The result indicates that the use of anionic ligands allows the formation of reactive high-valent species under mild conditions.
 | ||
| Fig. 13 Proposed catalytic cycle for the water oxidation reaction by the iron complex with an anionic pentadentate alkylamine pyridyl-type ligand 25. | ||
Note that some iron complexes bearing alkylamine-pyridine ligands can act as homogeneous catalysts under acidic conditions, although they can form iron hydroxide nanoparticles, which can act as a heterogeneous catalyst, in photocatalysis under basic conditions.95
The copper complex with an anionic pentadentate alkylamine pyridyl-type ligand (26) was also reported by Meyer et al. in 2014.96 The complex serves as a stable water oxidation catalyst in 0.1 M phosphate buffer solution. The results of electrochemical measurements and kinetic analysis revealed that the complex functions as a single-site catalyst and operates via the oxidative activation of the catalyst. The authors also reported that the HPO42− anion in the buffer works as a proton acceptor and is involved in the atom proton transfer process of the O–O bond formation step.
As described above, alkylamine-pyridine ligands can have diverse structures (Chart 7). Tetradentate and pentadentate ligands are available, and the structural differences in these ligands induce changes in the catalytic mechanism. In addition, anionic ligands can be prepared by introducing an amide moiety, which is different from the PY5-type ligands. Further, the anionic nature of the ligands can contribute to the stabilisation of high-valent species. It should also be noted that the high flexibility of the scaffold is advantageous for the stabilisation of reactive intermediates. However, the alkyl moieties of the ligands undergo facile oxidation during the catalytic reaction, which leads to the deactivation of the catalysts in certain cases. Therefore, modifying moieties by approaches such as deuteration are required for preventing ligand oxidation. Although further optimisation of the structure is required for obtaining highly-active catalysts, the alkylamine-pyridine ligands are a useful class of scaffolds for the development of various kinds of complexes containing earth-abundant metal ions and for enabling water oxidation.
 | ||
| Chart 7 Summary of features of water oxidation catalysts with alkylamine-pyridine ligands. | ||
3.4 Mononuclear water oxidation catalysts with macrocyclic ligands
The tetradentate macrocyclic ligands described in this review have planar structures with four coordinating nitrogen atoms. Therefore, the resultant complexes can exhibit a robust coordination environment, which is completely distinct from the tetradentate alkylamine pyridyl-type ligands described in the previous section. It is also expected that the complexes with this class of ligands possess labile sites at axial positions of the metal ion. For this reason, complexes bearing the tetradentate macrocyclic ligands are expected to be important candidates for the catalysis of oxidation reactions, including water oxidation.In 2010, Bernhard and Collins et al. reported a series of iron complexes bearing the tetra-amido macrocyclic ligand (TAML) as the first example of an iron-based molecular catalyst for water oxidation (27–31 in Chart 8).97 Simple structure with low molecular weight, use of iron (i.e. the most abundant and benign transition metal), and high water solubility of the complexes are the distinct advantages of these catalysts. Moreover, the ease of ligand modification afforded opportunities for accessing various kinds of ligands with electron-donating or withdrawing groups, which allowed the systematic investigation of the catalytic activity of the resultant complexes. The activity of these complexes was examined at pH 0.7 using CAN as the oxidant. Although 27 was not active for water oxidation, the complexes 28–31 catalysed the water oxidation reaction with relatively high reaction rates. The reaction rates increased depending on the electron-withdrawing ability of the substituents, and the complex with the most electron-deficient structure, 31, showed the highest TOF value (>1.3 s−1). However, the rapid liberation of O2 with first-order kinetics was terminated after 20 s, and a slower steady logarithmic evolution was observed in the second step, indicating the instability of the catalyst in acidic media. Although the stability of the catalysts was not so high, the study demonstrated for the first time that iron complexes can serve as water oxidation catalysts and that their catalytic activity can be precisely tuned by modification of the ligand structures.
 | ||
| Chart 8 Structures of mononuclear water oxidation catalysts with macrocyclic ligands. | ||
Subsequently, Liao and Sienbahn clarified the reaction mechanism for the catalysis by Fe–TAML complexes using quantum chemical calculations (Fig. 14),98 and explored the mechanism of 31. In their proposed mechanism, a two-step PCET reaction and one-electron oxidation afford the key Fe(V)O intermediate (C). WNA to this intermediate leads to the formation of the bond between the two oxygen atoms (D), and the subsequent deprotonation (D → E), PCET reaction (E → F), and liberation of dioxygen regenerates the initial catalyst state. The authors also discussed the origin of the low stability of the complex during the catalysis and proposed three possible pathways for the decomposition of the complex. The first was the possibility that the oxo group acts as a base and removes a proton from the water molecule. The generated hydroxide ion attacks one of the carbon atoms of the ligand and causes catalyst degradation. The second possibility is a nucleophilic attack of the nitrate anion originating from the Ce(IV) oxidant on the benzene ring of the ligand, which also causes catalyst degradation. The third likely pathway is the self-oxidation of the amide ligand by interaction with the Fe(V)O species to form an N-oxide. The authors undertook calculations, which demonstrated that the attack of water on the ligand is the main pathway for the degradation of the catalyst. Therefore, these degradation pathways must be considered during the ligand design to avoid undesired catalyst degradation.
 | ||
| Fig. 14 Proposed pathway for water oxidation with iron complexes bearing the tetra-amido macrocyclic ligands (TAMLs) 28–31. | ||
Gupta and Dhar reported the chemical and photochemical evolution of O2 catalysed by iron complexes with biuret-modified tetra-amidomacrocyclic ligands (32 and 33).99 They anticipated that the lone pair of the –NMe group can be effectively delocalised along the complex, which would alter the electron density of the iron atom. Indeed, the introduction of the –NMe moiety in the tail position increased the stability of the complex, which was a severe problem in the case of the Fe–TAML complexes developed by Bernhard and Collins et al. The introduction of an additional –NO2 moiety in the head aromatic ring further enhanced the stability of the complex. The electrochemical measurements of the complexes in aqueous media indicated the formation of high-valent Fe(V) species, which are expected to be important for the water oxidation reaction. The chemical reaction was performed with CAN as the oxidant at pH 1. The TON over a period of 300 s was calculated to be (10 ± 1) and (17 ± 1) for 32 and 33, respectively, and the TOF was 0.03 and 0.06 s−1 for 32 and 33, respectively. The photocatalytic reaction was performed by irradiating visible light (λmax = 440 nm) on the solutions containing the catalysts, with [Ru(bpy)3]2+ as the photosensitiser, and Na2S2O8 as a sacrificial electron acceptor, at pH 8.7 (basic condition). As a result, 32 was found to have better overall efficiency (TON = 220 ± 10, TOF = 0.67 s−1) than 33 (TON = 60 ± 4, TOF = 0.21 s−1). It should be noted that the UV-vis absorption spectroscopy, HRMS, and DLS measurements confirmed the molecular nature of the catalytic system and the formation of Fe2O3 nanoparticles was not detected. Another significant achievement of this study was the identification of the reaction intermediate during the photochemical reaction. The results of EPR, UV-vis absorption, and high-resolution mass spectroscopies demonstrated that a high valent Fe(V)O intermediate is photochemically generated as the active intermediate for water oxidation. This is the first example of an iron-based complex that promotes photocatalytic water oxidation through the formation of a Fe(V)O intermediate. The reaction mechanism of the light-driven water oxidation mediated by this complex is shown in Fig. 15. Photoirradiation of the reaction mixture leads to the formation of the FeIV–OH (A) first, followed by the formation of Fe(V)O (B) via a two-step PCET process. The Fe(V)O intermediate subsequently undergoes WNA to form the O–O bond (C). The generated species are oxidized further (2 eq.) and release dioxygen to complete the catalytic cycle by regenerating the initial FeIII–aqua complex (C → 32). The study successfully demonstrated that the appropriate modification of the TAML ligands can improve the stability of the corresponding catalyst and achieve water oxidation under both chemical (acidic medium) and photochemical (basic medium) conditions.
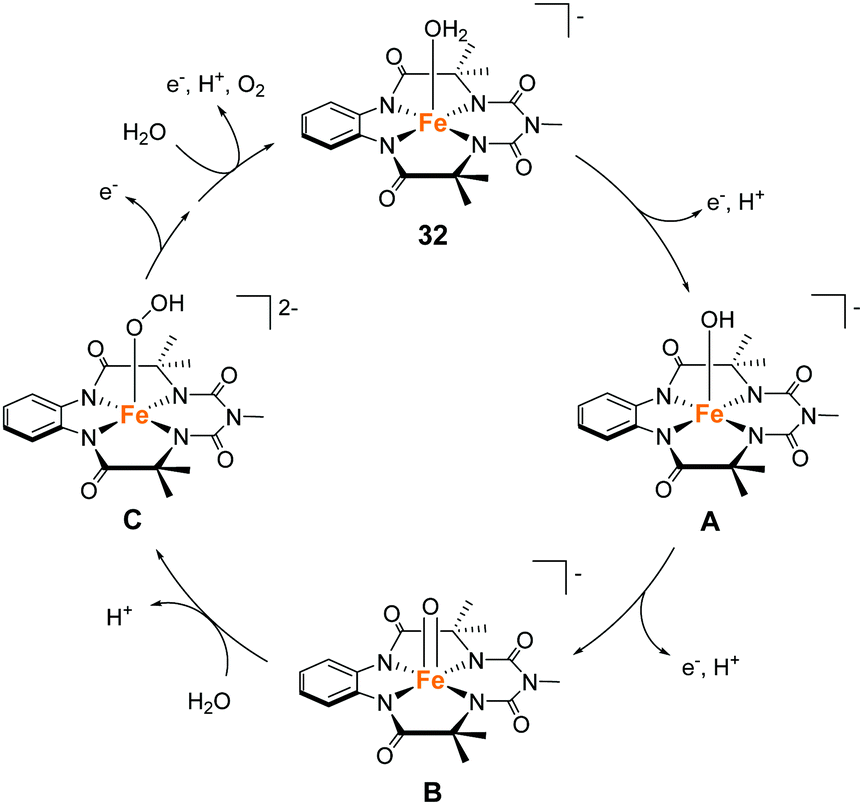 | ||
| Fig. 15 Proposed water oxidation mechanism of the iron complex with biuret-modified tetra-amidomacrocyclic ligand 32. | ||
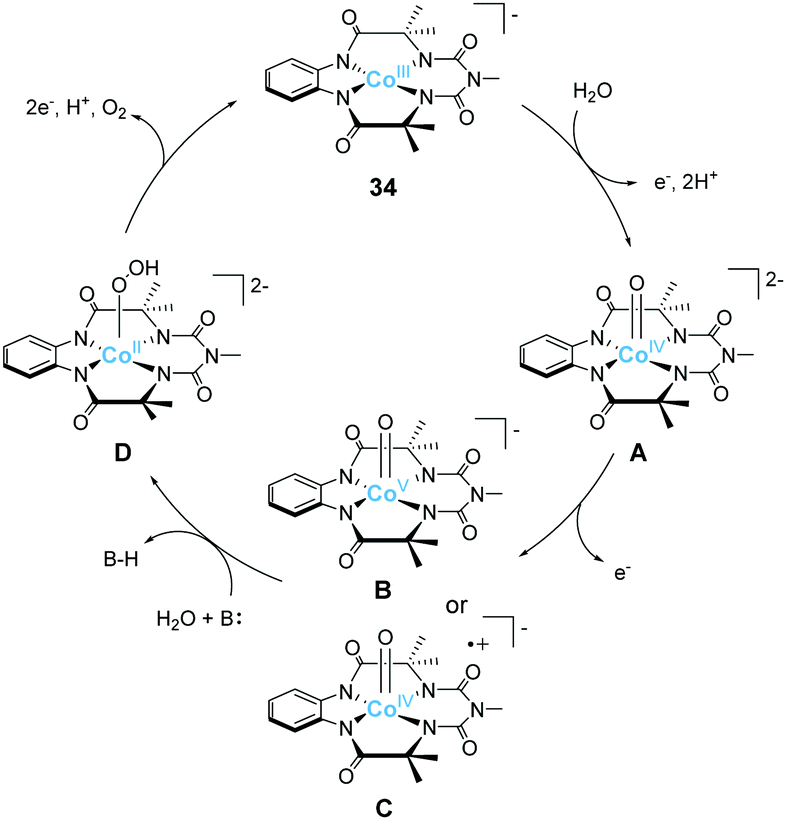
Gupta et al. also reported the catalytic activity of a cobalt complex bearing a biuret-modified TAML (bTAML) ligand (34).100 The complex displayed two redox waves in the cyclic voltammogram in the absence of water. The first wave was assigned to be the oxidation of Co3+ to Co4+. The electrochemical measurements in 0.1 M phosphate buffer (pH = 9.2) showed an irreversible wave at 1.1 V (vs. NHE), and a catalytic current was observed at 1.5 V. The first irreversible wave exhibited pH-dependent behaviour, and this process was stepwise or concerted two-proton and one electron transfer reaction. In contrast, the catalytic current showed pH-independent nature. The CPE experiment was then performed in 0.1 M phosphate buffer (pH = 9.2), and the formation of dioxygen was detected. The faradaic efficiency for the process was calculated to be 62%. UV-vis absorption spectroscopy, electrochemical measurements, XPS, SEM, and DLS ruled out the possibility of the formation of any heterogeneous cobalt oxide during the electrolysis. The TOF value calculated via cyclic voltammetry (eqn (4)) was 5.68 s−1. The proposed mechanism for water oxidation catalysed by 34 is as follows (Fig. 16). First, the one-electron oxidation of the complex with the loss of two protons affords [CoIV(O)bTAML]2− (A). [CoIV(O)bTAML]2− is then further oxidised to [Co(O)bTAML]− (C), which is isoelectronic with CoVO (B). The exact oxidation state of the cobalt ion could not be determined due to the redox non-innocent nature of the ligand, and thus the state was proposed to be either CoVO or CoIVO(bTAML radical cation), although the high-valent CoVO species is unstable in general.79 These species are the key intermediates responsible for the O–O bond formation. Further, the positive kinetic isotope effect observed for the catalytic reaction indicated the involvement of base-assisted nucleophilic attack of H2O on the key intermediate (B or C → D). The cobalt-hydroperoxo or peroxo species thus formed (D) subsequently underwent oxidation to liberate O2 and regenerated the initial catalyst state (D → 34). This study demonstrates that the redox non-innocent ligand plays a crucial role in the formation of the reactive key intermediate and promotes water oxidation.
Zhang et al. unveiled the catalytic activity of a series of Co–TAML complexes and their derivatives (35–38) for water oxidation.101 In their study, three complexes with redox active ligands (35–37) and the complex with a redox inactive ligand (38) were investigated. In the cyclic voltammograms of 35–37 in phosphate buffer (pH = 7), irreversible catalytic currents were observed, whereas 38 did not show a catalytic current. These results indicate the importance of redox activity of the ligands on the overall catalytic activity. To further confirm the activity, CPE experiments were performed on 35 and the formation of dioxygen with a faradaic efficiency of greater than 90% was detected. The overpotential of the reaction was estimated to be 380 mV (pH 7). The homogeneous nature of the catalytic system was indicated by electrochemical measurements, XPS, and SEM. The TOFs of the reaction were determined from the cyclic voltammetry results (eqn (4)) and the values were 7.53, 7.58, and 8.81 s−1 for 35, 36, and 37, respectively. The catalytic mechanism was examined in detail by the electrochemical approach in conjunction with quantum chemical calculations (Fig. 17). From its initial state, the [(L4−)CoIII]− complex undergoes ligand-based PCET (2e− + H+) to afford [(L2−)CoIII–OH] (A). The oxidation of metal centre coupled with the transfer of protons occurs next, leading to the formation of the catalytically-active [(L2−)CoIVO] (B) state. The attack of water on the intermediate generates the O–O bond, which is the rate-determining step. It should be noted that the O–O bond formation process proceeds with the help of a buffer anion. The peroxide intermediate (C) undergoes further oxidation via the PCET process to release dioxygen and thereby completes the catalytic cycle. This study highlights the relevance of the interplay between the ligand- and metal-centred redox activity for improving water oxidation catalysts.
 | ||
| Fig. 16 Proposed catalytic mechanism of the cobalt complex bearing the biuret-modified TAML (bTAML) ligand 34. | ||
 | ||
| Fig. 17 Proposed mechanism for water oxidation mediated by the Co-TAML complex 35. | ||
Llobet et al. developed the copper-based catalysts for water oxidation (39–42) that comprised ligands which were similar to TAMLs.102 The copper centres of the complexes showed a square planar geometry. In the complex 42, electrocatalytic water oxidation with a very small overpotential (η = 170 mV at pH 11.5) was achieved owing to the strong electron-donating ability of the aromatic ring on the ligands. Foot-of-the-wave analysis revealed that the catalytic rate constant of the reaction is 3.6 s−1 at pH 11.5. The stability of the catalyst was studied by the electrochemical analysis, UV-vis absorption spectroscopy, and EDX, which provided insights into the molecular nature of the catalyst. The results of the electrochemical experiments and quantum chemical calculations revealed the unique mechanism of the complex (Fig. 18). Initially, the complexes were oxidised from the Cu(II) to the Cu(III) state (A) at 0.48–0.56 V vs. NHE. Subsequently, the pH-dependent ligand-based oxidation occurred, which was followed by the coordination of the hydroxyl ligand to afford the L˙Cu(III)(OH) species (B). Electrochemical analysis and DFT calculations indicated that the rate-determining step involved the generation of the radical cation species and the reaction with a hydroxyl anion. The reaction of L˙Cu(III)(OH) with the additional hydroxy anion and the electron transfer from the hydroxy anion to the cyclic ligand generates the Cu(III)(HO⋯OH)˙− species (C), which comprises a radical anion fragment with a partial O–O bond. Then, the Cu(III)(HO···OH)˙− transforms to Cu(III)(HO–OH) (D) through a second intramolecular electron transfer, thus converting the HO–OH unit to a singlet state. It must be noted that the oxygen atoms are not directly bound to the copper centres in the Cu(III)(HO⋯OH)˙− and Cu(III)(HO–OH) intermediates. Instead, the (HO⋯OH)˙− and (HO–OH)2− moieties are bound to the complex via hydrogen bonding. Then, the Cu(III)(HO–OH) intermediate is further oxidised to the [(L)Cu(III)(HOO˙)] species by metal-based electron transfer and the subsequent PCET processes. Finally, O2 and a free proton are dissociated from the complex, which regenerates the initial state. The results of the DFT calculations also indicate that the ligand-based oxidation reaction to produce C from B is essential for determining the overpotential of the reaction. Therefore, it is expected that the electronic structure of the aromatic ring in the ligand can largely affect the overpotentials. Indeed, the overpotentials were largely reduced upon introduction of electron-donating groups on to the ligand (η = 700 (39), 400 (40), 270 (41), and 170 (42) mV at pH 11.5). However, the value of the TOFs determined by foot-of-the-wave analysis decreased with an increase in the electron-donating ability of the introduced substituents (TOF: 3.56 (39), 3.58 (40), 0.43 (41), 0.16 (42) s−1). The results suggest that using a redox non-innocent ligand can be an effective strategy for lowering the overpotential of the catalysts owing to the ability of the ligand to assist in the generation of the highly electrophilic OH-bound species without the formation of the Cu(IV)O or Cu(III)–oxyl species.
 | ||
| Fig. 18 Plausible catalytic pathway of water oxidation promoted by the copper-based complexes with TAML-like ligands 39–42. | ||
Llobet et al. further developed a copper-based TAML complex (43) for water oxidation which operates in neutral condition.103 They employed a highly π-extended TAML with two phenyl-bisamidate units for accessing to several oxidation states in the ligand at a lower potential range, which leads to the stabilization of the accumulated oxidative equivalents by charge delocalization. In the complex 43, the storage of oxidative equivalents (holes) both by the metal centre and the ligand allowed the catalytic water oxidation achieved under neutral conditions at a surprisingly low overpotential (200 mV). Foot-of-the-wave analysis revealed that the catalytic rate constant of the reaction is 140 s−1 at pH 7. The stability of the catalyst was also intensively studied by the electrochemical analysis, SEM, XANES, EXAFS and EDX, which confirms the molecular nature of the catalyst. The catalytic cycle of complex 43, proposed based on the electrochemical experiments, spectroscopic characterization and quantum chemical calculations, is shown in Fig. 19.
 | ||
| Fig. 19 Proposed mechanism for water oxidation promoted by the copper-based TAML complex 43. | ||
1,4,8,11-Tetramethyl-1,4,8,11-tetraazacyclotetradecane (TMC) ligands are another class of macrocycles used for the construction of complexes with square planar geometry. In contrast to the TAML-type, the TMC ligand is flexible, and the mechanism for the water oxidation can be expected to be different. Further, this class of ligands are capable of stabilising metal–oxo species in transition metal-catalysed oxidation reactions.104
In 2014, Lu and Zhang reported the first example of a Ni-based molecular catalyst for water oxidation.90 They employed the [NiII(meso-L)](ClO4)2 complex (44, L: 5,5,7,12,12,14-hexamethyl-1,4,8,11-tetra-azacyclotetradecane), which has a TMC-type ligand, as the catalyst. This complex was reported to serve as an electrocatalyst for water reduction.105 The CV of the complex in 0.1 M sodium phosphate buffer at pH 7.0 showed a small irreversible oxidation wave which was assigned to the NiIII/NiII process seen at 0.87 V (vs. NHE). Note that the redox potential of this oxidation process is pH-dependent, indicating that the process involves PCET. At more positive potentials, an additional irreversible current was observed at 1.41 V, which could be caused by water oxidation. The onset potential for water oxidation was observed at ca. 0.99 V vs. NHE, which corresponded to an overpotential of ca. 170 mV. Based on this result, the CPE of the complex at 1.55 V (vs. NHE) was carried out on an ITO electrode, and the evolution of O2 with a faradaic efficiency of 97.5% and TON of 15 was confirmed. Several control experiments were also performed, and the reaction mixture was analysed by electrochemical measurements, ESI-MS, and absorption spectroscopy. Notably, the catalyst was recovered almost quantitatively from the reaction mixture after the CPE experiment, and the structure of the recovered complex was confirmed by single-crystal X-ray structural analysis. The results of these experiments strongly indicate that the complex functions as a homogeneous catalyst. This high stability of the complex can be attributed to its low overpotential required for water oxidation. The reaction mechanism of the complex was proposed based on the results of a previous electrochemical study, and DFT calculations.106,107 The study revealed that the [NiII(meso-L)]2+ and [(meso-L)NiII(OH2)2]2+ species exist in equilibrium.108 In the catalytic cycle (Fig. 20), the complex in its initial state undergoes a PCET reaction to afford the hexacoordinated octahedral species, trans-Ni(III)–(OH2,OH) (A). A further PCET process generates the key intermediate state. In this key state, the three different species, trans-Ni(III)–(OH2,O˙) (B), the five coordinated Ni(III)–O˙ species (C), and cis-Ni(III)–(OH,OH) (D), are in an equilibrium, and the difference in the Gibbs free energy between them is within 1.6 kcal mol−1. Subsequently, the energetics of the O–O bond formation pathway from the three species were calculated. It should be noted that the reaction rate is first-order dependent on the concentration of nickel species. Therefore, O–O bond formation should be promoted by monometallic Ni species (B, C or D). The authors compared three different pathways for O–O bond formation; WNA, OH attack, and HO–OH coupling. Comparison of the Gibbs energies during the reaction suggested that HO–OH coupling from intermediate D was the most energetically stable and feasible pathway. The formed Ni(II)–HO–OH (F) is further oxidised and liberates dioxygen with the concomitant regeneration of the initial state of the catalyst. This study points to the advantages of the TMC-type ligand during water-oxidation catalysis, which includes flexible structures for minimising the thermodynamic energy of O–O bond formation.
 | ||
| Fig. 20 Proposed mechanism for water oxidation by the nickel complex with 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane (TMC)-type ligand 44. | ||
In 2016, Sun et al. reported a copper-based complex [Cu(TMC)(H2O)](NO3)2 (45) for water oxidation catalysis.104 The CV of the complex in 0.1 M phosphate buffer exhibited a quasi-reversible reduction wave due to the CuII/CuI redox process. In addition, a prominent catalytic current started at ca. 1.4 V (vs. NHE), which was indicative of the electrocatalytic water oxidation. From the electrochemical measurements (eqn (4)), the TOF was determined to be 30 s−1. The authors also performed the CPE experiment in a homogeneous solution; the evolution of dioxygen with the faradaic efficiency of 89% was confirmed. However, in this case, the deposition of the catalyst on the counter electrode was observed. Therefore, to prevent the catalyst decomposition and improve the durability the complex was immobilised on a Nafion-coated carbon cloth electrode. As a result, no current decay was observed during the electrolysis, and oxygen evolution with a faradaic efficiency of 98% was achieved. Further, the TON reached 362 in this experimental condition. The proposed reaction mechanism was established based on electrochemical and kinetic studies (Fig. 21). The mechanism is as follows. The initial state undergoes a PCET reaction to afford Cu(III)–OH state (A), and the further oxidation of the state generates Cu(III)(TMC·)OH3+ or Cu(IV)(TMC˙)(OH)3+ species (B). It should be noted, however, that the oxidation of the simple aliphatic ligand or the formation of high-valent Cu(IV) species does not seem to occur easily.79 The WNA reaction on this state generates the bond between the two oxygen atoms, which is regarded as the rate-determining step. As a result, the Cu(II)–HO–OH species (C) forms and a further two-electron oxidation of the state with the dissociation of two protons results in the evolution of O2 and the regeneration of the initial state. The study demonstrates that the TMC-type ligand can also be utilised for the development of copper-based catalysts for water oxidation.
 | ||
| Fig. 21 Plausible catalytic cycle for water oxidation mediated by the copper-based complex with TMC ligand 45. | ||
As shown above, macrocyclic ligands can provide square planar tetracoordinated environment, and thus simultaneously gave open labile sites, which is essential to drive the catalytic reaction, to the resultant complexes (Chart 9). Due to this inherent feature, the macrocyclic ligands are regarded to be fine candidates for oxidation reactions, and several catalysts have been reported. Notably, complexes bearing these ligands have been employed for water oxidation, and the first examples of iron- and nickel-based catalysts for the reaction were developed using these ligands. The ligands introduced in this section can be divided into two broad categories: (1) TAML-type ligands and (2) TMC-type ligands. The TAML-type ligands are anionic and thus produce a strong ligand field at the resultant complex. The structure of the ligand is rigid, and thus, it provides a robust coordination environment. Moreover, the facile modification of the ligand structures enables the addition of various features to the complexes, such as the tuning of redox properties and the creation of redox non-innocent ligands. All these features contribute to stabilising the high-valent key reaction intermediates, and in some cases, the lowering of overpotential. In contrast, TMC-type ligands are neutral and have structural flexibility. This structural flexibility contributes to the production of labile sites arranged in a cis fashion and the facile O–O bond formation proceeds in the case of the nickel-based complex. The copper analogue bearing the TMC-type ligand also serves as a water oxidation catalyst, in which the redox non-innocent nature of the ligand plays an important role.
 | ||
| Chart 9 Summary of features of water oxidation catalysts with macrocyclic ligands. | ||
3.5 Water oxidation catalysts with porphyrin/corrole ligands
Derivatives of porphyrin and corrole are vital constituents of natural enzymes and are known to exhibit unique photochemical and electrochemical properties and reactivities.109–116 Importantly, this class of compounds can be used for the construction of metal complexes with rigid structures. The resulting complexes are also typically stable against oxidation and catalyse various kinds of oxidation reactions both in the natural and artificial systems.55,117,118 Therefore, the porphyrin and corrole class of ligands are expected to be good candidates for the preparation of molecular catalysts for water oxidation.In 1994, Natura et al. reported a series of Mn-based complexes (46–48 in Chart 10)119 that electrochemically catalysed water oxidation. The complexes were the first Mn-based water-oxidation catalysts and were developed by bridging two triphenyleneporphyrin moieties with an o-phenylene linker, wherein two porphyrin rings were facing each other in the complex. All these complexes exhibited an irreversible catalytic current at ca. 1.2 V vs. Ag/Ag+ in the presence of water. Importantly, the CPE experiments of the complexes at 1.2–2.0 V (vs. Ag/Ag+) confirmed the evolution of dioxygen. Among the complexes investigated, 46 displayed the highest activity and a TON of 9.2. The use of 18O-labelled H2O as the substrate revealed that the oxygen atoms in the O2 evolved are derived from H2O. The detailed water oxidation catalysis mechanism of 46 was also investigated by the same group with mCPBA as the oxidant.120 The ESR and Raman spectroscopic analyses indicated the formation of the bis-Mn(V)–oxo-porphyrin species during the reaction. In their proposed mechanism, the two Mn(III)–OH units are oxidised initially to form bis-Mn(V)O species (A in Fig. 22). Subsequently, the intramolecular O–O bond formation between the oxo moieties occurs to afford the Mn(IV)–O–O–Mn(IV) intermediate. Finally, ligand exchange with the hydroxy ligands occurs and the bridged peroxy group liberates the dioxygen and regenerates the initial catalyst state. They compared the stability of the high-valent oxo species of mononuclear and dinuclear porphyrins. While the oxo species of the relevant mononuclear manganese porphyrin complex decomposed within several minutes, the dimer complexes retain their catalytic activities for several hours. Based on these results, they concluded that the higher stability of the high-valent oxo species of the dimer complex is due to the existence of an inner hydrophobic space created by the two porphyrin moieties and bulky meso mesityl groups. These results indicate that the creation of a reaction field that can stabilise the reactive high-valent oxo species is a promising strategy for obtaining efficient molecular catalysts for water oxidation.
 | ||
| Chart 10 Structures of mononuclear water oxidation catalysts with porphyrin/corrole ligands. | ||
 | ||
| Fig. 22 Proposed catalytic cycle of water oxidation promoted by Mn-based bis-porphyrin complexes 46–48. | ||
Inspired by the aforementioned findings, several porphyrin and corrole-based complexes began to attract considerable attention. In 2006, Sun et al. demonstrated that the manganese-based corrole complexes 49 and 50 could catalyse electrochemical water oxidation.121 Corrole derivatives function as trianionic ligands and thus are expected to stabilize high-valent metal species. In their later work, they extensively studied the mechanism of the water oxidation reaction with a manganese-based corrole complex (51).122 The authors specifically focused on the elucidation of the O–O bond formation process. Experimental results indicated that the Mn(V)O species forms upon oxidation with tert-butyl hydroperoxide (TBHP), and the subsequent reaction with n-Bu4NOH results in the formation of dioxygen. These processes were monitored by real-time mass spectroscopy, and the intermediates were characterised by UV-vis absorption and high-resolution mass spectroscopies. As shown in Fig. 23, the catalyst in its initial state undergoes oxidation to the key intermediate Mn(V)O species (A) by reaction with TBHP. Next, the nucleophilic attack by the hydroxyl anion on the intermediate leads to the formation of the hydroperoxide species (B). Further deprotonation and one-electron oxidation of the species causes the formation of peroxo radical species (C), and the initial state is recovered upon the release of dioxygen and one electron.
 | ||
| Fig. 23 Plausible catalytic pathway of water oxidation by the manganese-based corrole complex 51. | ||
Because the Co(II)–Hangman porphyrins are known to activate the O–O bond of dioxygen,123 Nocera et al. explored the catalytic activity of Co(III)–Hangman corrole complexes (52 and 53) for water oxidation.124 In these complexes, the macrocyclic frameworks were modified with electron-withdrawing groups to boost the oxidising power of the corrole subunit. The complexes were deposited on FTO-coated glass electrodes, and electrochemical measurements were performed. The measurements in 0.1 M phosphate buffer exhibited an increase of the irreversible current with an onset potential of 1.25 V (vs. Ag/AgCl), indicating the occurrence of water oxidation. It should also be noted that the intensities of the catalytic current were larger in these complexes compared to that for the corrole complex without the hanging group, and 53 exhibited higher activity than 52. This result indicates that the ability of the hanging group to preorganize water within the hangman cleft appears to be beneficial for the O–O bond formation step. The CPE experiments of the complexes were conducted at 1.4 V (vs. Ag/AgCl), and 53 was found to be the most efficient catalyst and evolved dioxygen with near quantitative faradaic efficiency. The authors also performed several experiments to confirm the molecular nature of the complexes during the catalysis. They found that the introduction of electron-withdrawing groups is an effective strategy for enhancing the catalytic activity. The role of the hanging groups was further studied in a later report.125,126 Quantum chemical modelling suggested that the carboxy moiety functions as the base to activate the water molecule towards the reaction with the metal oxo key intermediate. It was also disclosed that the high degree of peripheral fluorination contributes to the oxidation resistance and high reactivity of the metal–oxo species, which are the key catalytic intermediates. In other words, the introduction of electron-withdrawing groups on the corrole scaffold tunes the electrophilicity of the metal–oxo species and prevents decomposition of the corrole radical cation species. The study clearly demonstrated the advantages of this class of complexes, which included ease of structural modification and precise tunability of the properties based on structural modifications.
Sakai et al. reported the light-driven water oxidation reaction catalysed by water-soluble cobalt–porphyrin complexes (54–56).127 The catalytic activity of these complexes was investigated in a phosphate buffer solution (pH 11) using [Ru(bpy)3]2+ as the photosensitizer, and Na2S2O8 as the sacrificial electron acceptor. The photo-irradiation of the reaction mixture for 30 min resulted in the formation of O2 with a TOF of 0.118–0.170 s−1 and TON of 88–122. The DLS measurements revealed the absence of nanoparticle formation, which excludes the participation of cobalt oxide species in the catalysis. Further, the initial reaction rates showed a second-order dependence on the concentration of the catalyst, which indicates that the O–O bond formation process involves two molecules of the catalyst. Based on this result, the authors proposed two possible pathways for this step (Fig. 24). The first is the radical coupling between Co(III)–oxyl radical species (A), the other involves the radical coupling of the Co(IV)–oxyl radical species (B). In both cases, a bimolecular radical coupling process appears to be the rate-determining step. This work successfully confirmed the suitability of cobalt-porphyrin derivatives for photochemical water oxidation.
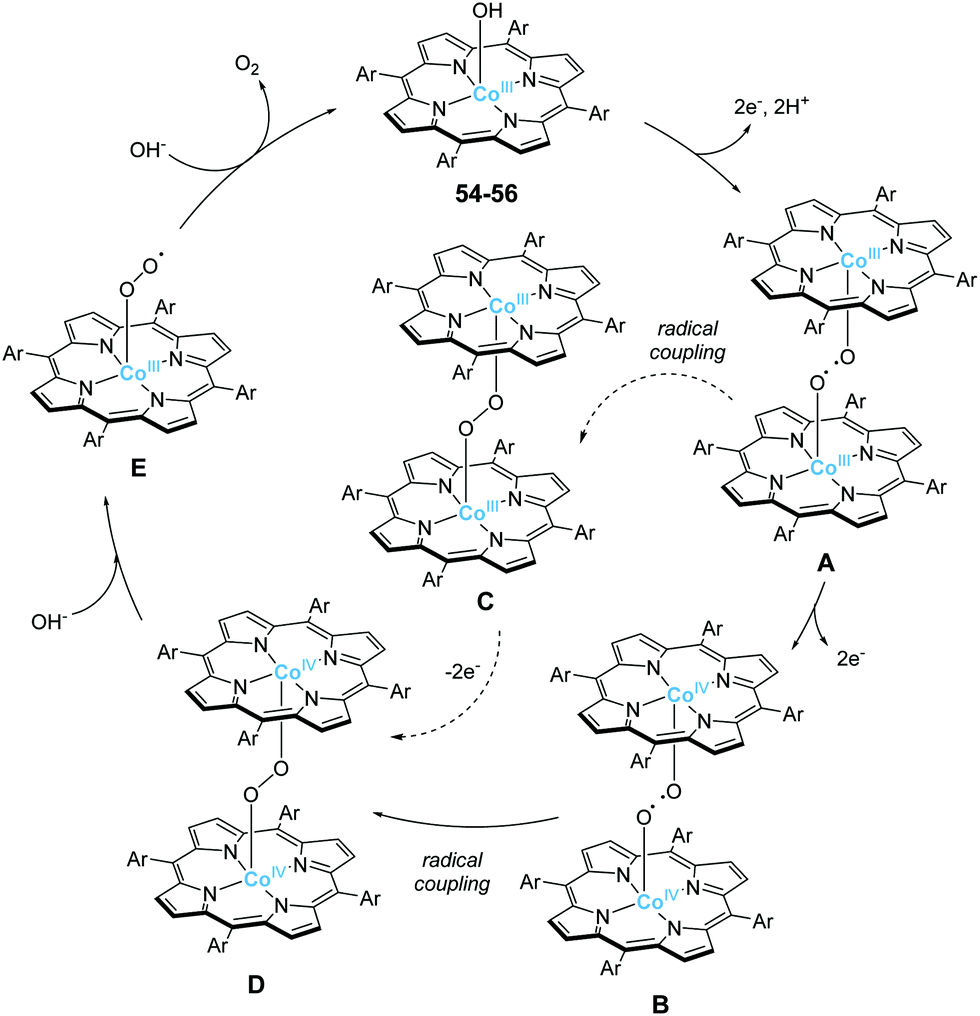 | ||
| Fig. 24 Plausible reaction mechanism of photocatalytic water oxidation by the water-soluble cobalt–porphyrin complexes 54–56. | ||
In 2013, Groves et al. reported a highly active electrocatalyst for water oxidation based on cobalt porphyrins.128 They developed a series of water-soluble cationic porphyrins (57–59), and investigated their catalytic activity in aqueous media at neutral pH (0.2 M aqueous sodium phosphate buffer). Among these complexes, 57, which contained the most electron-deficient groups at meso positions, exhibited the highest activity. The TOF of the reaction mediated by the complex was calculated based on electrochemical measurements (eqn (4)), and was remarkably high at 1400 s−1. Further, the onset potential was estimated to be ∼1.2 V (vs. Ag/AgCl), and CPE experiments confirmed the formation of dioxygen with 85–90% faradaic efficiency. During electrochemical analysis, 57 exhibited higher oxidation potentials than the other two complexes, indicating the stronger oxidation power of the formed species. In other words, the complex can provide stronger thermodynamic driving force for water oxidation than other complexes. The absence of cobalt nanoparticle formation during the catalysis is an evidence for the homogeneous nature of the catalytic system. The authors also proposed a plausible reaction mechanism (Fig. 25). In this mechanism, from the resting Por–Co(III)–OH species (A), one-electron oxidation of the porphyrin ring proceeds to produce porphyrin radical cation species, and +Por–Co(III)–OH (B) forms. Further one-electron oxidation coupled with the transfer of one proton affords the key reactive intermediate +Por–Co(III)–O (C). The O–O bond formation step then occurs via WNA on +Por–Co(III)–O, and this step is rate determining. Further ligand exchange with a water molecule generates dioxygen as the product, and the resting state is regenerated. Notably, the pKa of buffer drastically affects the onset potential in this catalytic system, which indicates that the assistance of an anion is essential for promoting the proton transfer reaction. The study clearly demonstrates that cobalt porphyrin bearing electron-deficient groups can achieve water oxidation with a remarkably high TOF and modest overpotentials.
 | ||
| Fig. 25 Plausible catalytic cycle of water oxidation promoted by the water-soluble cationic cobalt porphyrins 57–59. | ||
Cao et al. developed a cobalt-based corrole complex, [Co(tpfc)(py)2] (60, tpfc = 5,10,15-tris(pentafluoro-phenyl)corrole and py = pyridine), as an electrocatalyst for water oxidation.129 The catalytic activity of 60 was investigated by coating the complex on indium tin oxide (ITO) electrodes. The CPE experiments using the modified electrode at 1.4 V (vs. Ag/AgCl) confirmed the formation of dioxygen with a faradaic efficiency greater than 95% and a TOF of 0.2 s−1. The homogeneous nature of the catalyst was disclosed by several experiments, including electrochemical measurements, UV-vis absorption spectroscopy, and MALDI-TOF mass spectrometry. The reaction mechanism of the complex was proposed based on DFT calculations. The catalyst in its initial state exists as the Cor–Co(III)–(OH2,py) species. In the catalytic cycle of the Co–Hangman corrole complex reported by Nocera et al. (vide supra),124 the concerted transfer of the two electrons and two protons affords the key Co–oxo intermediate. Therefore, in this study, the possibility of a similar 2e−/2H+ oxidation process from the initial state was investigated. Proposed catalytic cycle based on the experimental results and DFT calculation, is shown in Fig. 26. The potentials required to form the intermediates, Cor˙+–Co(III)–(OH,py), and Cor˙+–Co(III)–(O˙−, py) via PCET process were calculated to be 0.45 and 1.37 V (vs. NHE), respectively. The authors claimed that these values were comparable to the experimentally-observed values (Cor˙+–Co(III)–(OH,py): 0.78 V, Cor˙+–Co(III)–(O˙−,py), 1.34 V (vs. NHE)), although the calculated and experimentally obtained potentials for the first oxidation step are different to some extent. The formed key active species, Cor˙+–Co(III)–(O˙−,py) (B), then undergoes the O–O bond formation via WNA mechanism (B → C). Next, the PCET process occurs at −0.56 V (vs. NHE), to generate the Cor–Co(III)–(OO˙−, py) species (D), which is further oxidised at −0.02 V (vs. NHE) to form Cor˙+–Co(III)–(OO˙,py) (E). As a result, the energy barrier for the formation of the dioxygen is as low at 3.6 kcal mol−1. Similar to the report by Groves et al.,128 the dependence of the reaction rate on phosphate concentration indicated that an additional, base-dependent atom-proton transfer (APT) process contributed to the rate-determining O–O bond formation process. The study disclosed that the cobalt corrole complex is also an interesting candidate for water oxidation catalysis.
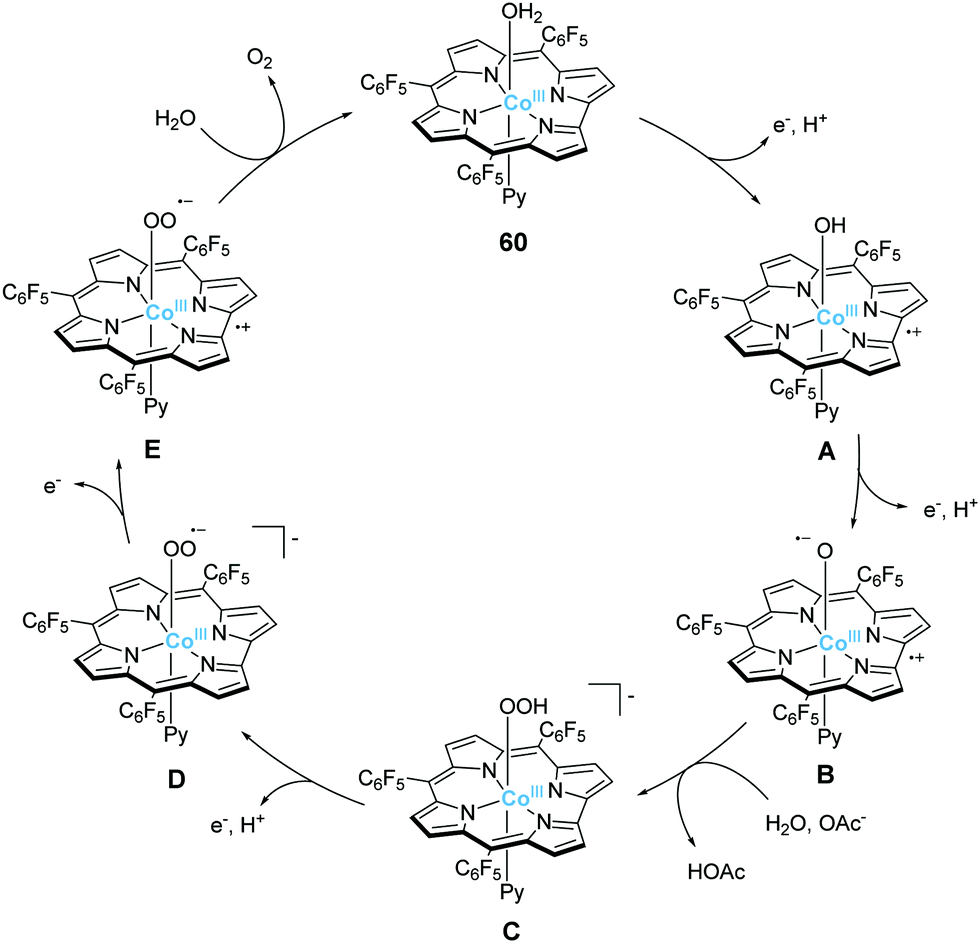 | ||
| Fig. 26 Proposed mechanism for water oxidation mediated by the cobalt-based corrole complex 60. | ||
Cao et al. reported in 2015 that Ni–porphyrin complexes can also catalyse water oxidation.130 They anticipated that Ni–porphyrin could serve as a water oxidation catalyst owing to the higher redox potential exhibited by the high-valent Ni atoms than that of Co in the same valence state. Indeed, the complex 61 was water-soluble and displayed a large irreversible catalytic current at 1.0 V (vs. NHE) in a phosphate buffer solution. This onset potential is substantially lower than the Co porphyrin analogue by ∼200 mV. The CPE experiments at 1.32 V (vs. NHE) in 0.1 M phosphate butter at pH 7.0 confirmed the evolution of dioxygen with >90% faradaic efficiency. The molecular nature of the catalyst was also confirmed by several experiments. The TOF was determined to be 0.67 s−1 based on the electrochemical analysis (eqn (4)). In the CV measurements of the complex, the decrease of the catalytic current with increasing scan rates indicated that the rate-determining step of the reaction involved the chemical reaction. The kinetic isotope effect of the reaction was 1.55, and the O–O bond formation was determined to be the rate-determining step. Electrochemical and spectroscopic measurements and DFT calculations indicated that the two-electron oxidised species of the complex is the key intermediate in the catalysis, and the proposed mechanism is shown in Fig. 27. In this catalysis mechanism, the oxidation of Por–Ni(II), i.e. the initial state of the catalyst, proceeds at the porphyrin ring to give Por˙+–Ni(II) (A). Subsequently, coordination of water occurs to afford Por˙+–Ni(II)–OH2 (B), and the PCET of Por˙+–Ni(II)–OH2 generates Por˙+–Ni(III)–OH (C). The further release of the proton from the species gives Por˙+–Ni(III)–O˙ (D) as the key intermediate of the reaction. The subsequent WNA reaction leads to the formation of the O–O bond, which can proceed in two possible pathways. In one pathway, the cluster constructed from four water molecules functions as a proton acceptor, and the energy barrier of this pathway was estimated to be 24.9 kcal mol−1. In another pathway, the acetate anion serves as a proton acceptor, and the energy barrier is 18.0 kcal mol−1. The authors also investigated the transition state formed between the key intermediate and the proton acceptors. The results of the investigation revealed that the acetate anion can easily form the transition state compared to the water cluster. Therefore, the latter pathway was found more plausible. The formed species (E) undergoes further oxidation via the PCET process (E → F), which leads to the release of O2 and the regeneration of the initial state of the catalyst. This study achieved the lowering of the porphyrin-based catalyst's overpotential by focusing on the nature of the central metal ion.
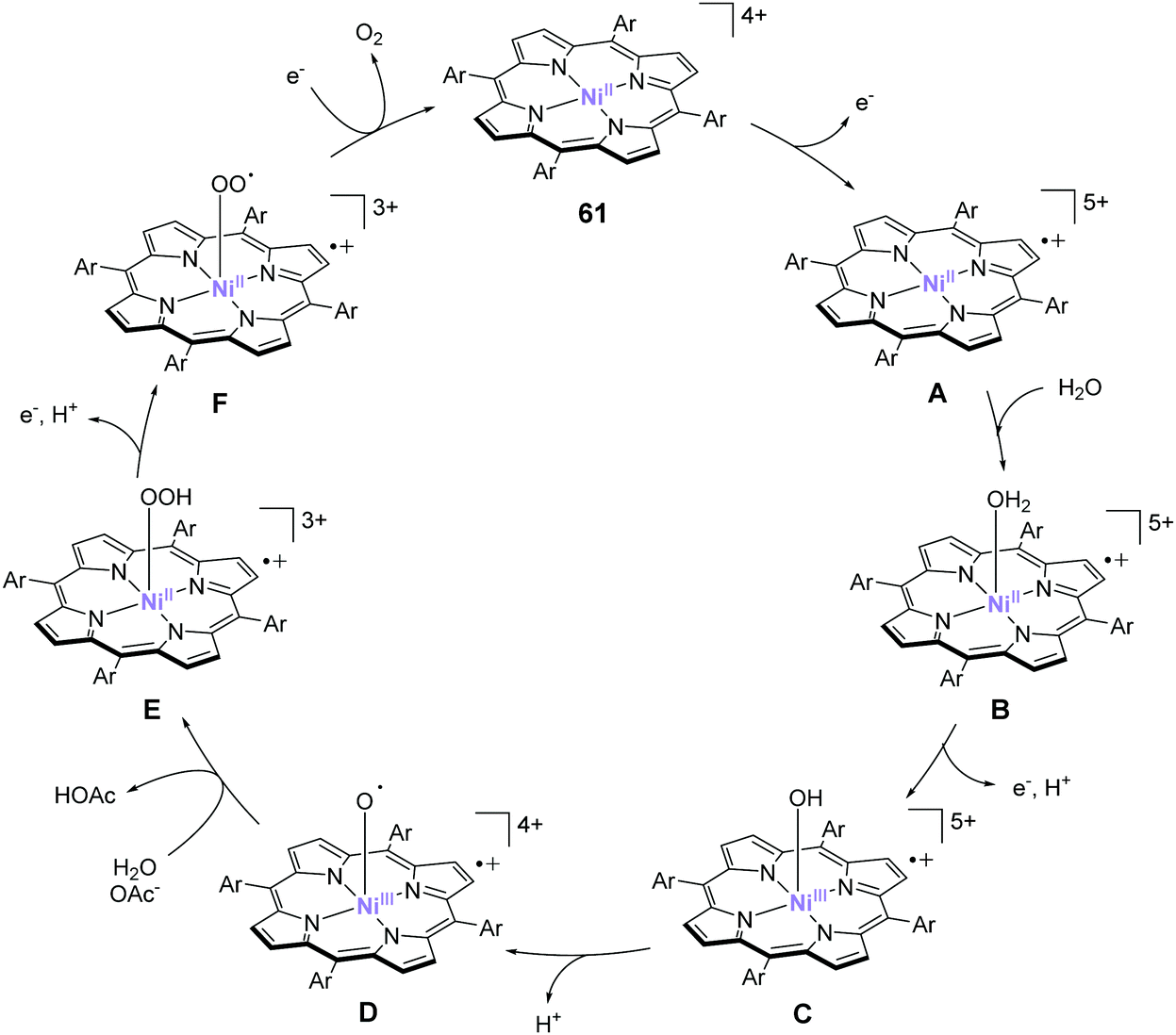 | ||
| Fig. 27 Plausible catalytic cycle for water oxidation facilitated by the water-soluble Ni–porphyrin complex 61. | ||
In 2019, Cao et al. reported the water oxidation reaction catalysed by a water-soluble copper–porphyrin complex (62).131 Electrochemical measurements of the complex in 0.1 M phosphate buffer (pH = 7.0) displayed a pronounced catalytic current. The onset of catalytic current occurred at 1.13 V (vs. NHE), which corresponds to the relatively low overpotential of 310 mV. The molecular nature and stability of the catalyst were confirmed by various techniques, including electrochemical measurements, UV-vis absorption spectroscopy, SEM, and EDX. The CPE conducted at 1.30 V (vs. NHE) detected the formation dioxygen with a faradaic efficiency of >93%. The TOF value of the catalysis was determined as 30 s−1 based on cyclic voltammetry (eqn (4)). This is a rare example of a copper-based catalyst for water oxidation which operates under neutral conditions. Notably, the complex catalyses the two-electron oxidation of water to form H2O2 under acidic conditions. Based on the result, the authors suggested that the one-electron oxidized species of the complex trigger the O–O bond formation, although the complete catalytic mechanism has not been proposed. In other words, the O–O bond formation without generating the high-valent CuIV–oxo/CuIII–oxyl radical species is the origin of the low overpotential, which promotes water oxidation in the case of 62.
To date, various kinds of complexes bearing porphyrin and corrole ligands have been reported. The redox-active and robust nature of the ligands enables the transfer of multiple electrons in the resultant complexes and assists the formation of the key reactive intermediates (Chart 11). The anionic nature of these complexes greatly contributes to the stabilisation of the high-valent oxo species. It should also be noted that the modification of the complexes by introducing substituents at the meso-positions and peripheral moieties is an important strategy for controlling the catalytic activities. Naruta et al. demonstrated the important strategy to stabilize high-valent metal–oxo species by introducing a hydrophobic group near the catalytic centres. Also, Groves et al. reported that the appropriate modification of porphyrin rings by the introduction of electron-deficient groups imparts remarkable activity to the catalysts. In other words, the fine-tuning of electronic structure by the chemical modification of the porphyrin and corrole derivatives affords catalysts with excellent performance. These findings highlight that the usefulness of this class of complexes as water oxidation catalysts.
 | ||
| Chart 11 Summary of features of water oxidation catalysts with porphyrin/corrole ligands. | ||
3.6 Dinuclear water oxidation catalysts
The first example of an artificial water oxidation catalyst, the blue dimer, is a dinuclear metal complex. Dinuclear complexes are regarded as promising candidates for water oxidation catalysis owing to their potential for enhanced electron transfer due to the presence of two metal centres in the structure. Further, the delocalization of the charge between the two metal centres is expected to stabilise the high-valent species. Therefore, dinuclear complexes are expected to be suitable scaffolds for constructing water oxidation catalysts based on earth-abundant first-row transition metal ions.Crabtree and Brudvig et al. developed the bis(μ-O)-bridged dinuclear manganese complex (63 in Chart 12) for water oxidation in 1999.132 The most significant feature of the complex is the labile aqua ligand that is located trans to the μ-oxo ligand. The activity of the complex was examined at pH 8.6 using NaClO as the chemical oxidant. The TON of the reaction was 4 for 6 h, and thereafter, the catalytic reaction terminated due to the formation of MnO4− as evidenced by UV-vis absorption spectroscopy. The proposed catalytic cycle of 63 is shown in Fig. 28. First, the complex is oxidised by NaClO to the Mn(IV)Mn(IV) state (A). The aqua ligand of the dimer is subsequently exchanged with the ClO− (A → B), and a further two-electron oxidation of the dimer leads to the formation of the Mn(V)Mn(V)O state (C), which acts as the key intermediate for O–O bond formation. The nucleophilic attack of the hydroxy anion on this oxo species generates O2 (C → D), with the concomitant regeneration of the Mn(IV)Mn(IV) state of the complex. When oxone was used as the chemical oxidant, the reaction followed a similar mechanism, and the reaction rate reached 2420 mol of O2 mol h−1. The major drawback of this catalyst is the need for the use of oxygen transfer reagents such as NaClO4 and oxone to drive the reaction. The reaction with CAN133 as the oxidant or under electrochemical conditions134 did not generate O2 owing to the instability of the complex under the experimental conditions. It should be also noted that there is a possibility of the formation of manganese oxide nanoparticles in this system which are the true catalytic species.135,136
 | ||
| Chart 12 Structures of dinuclear water oxidation catalysts. | ||
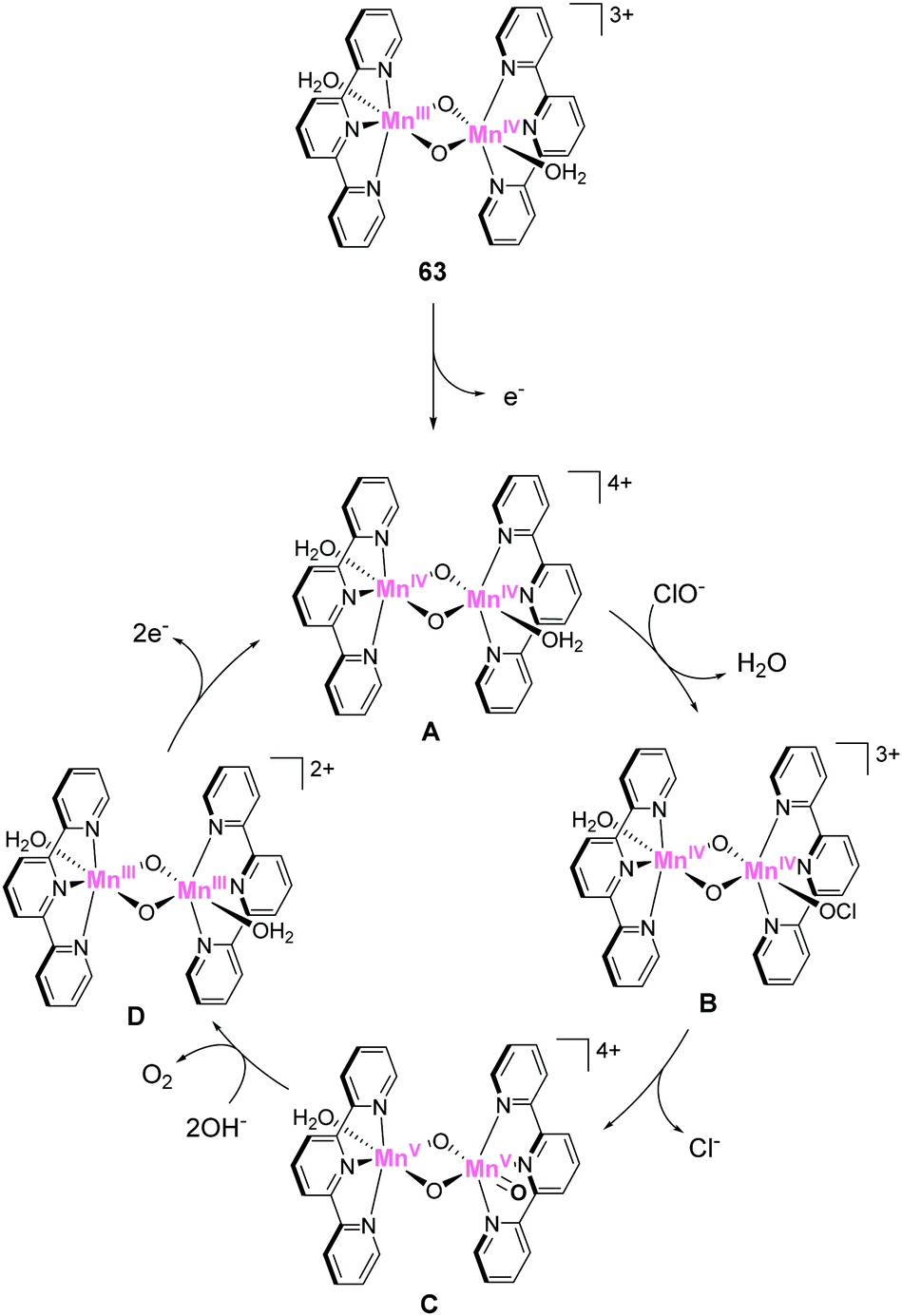 | ||
| Fig. 28 Plausible catalytic pathway for water oxidation mediated by the bis(μ-O)-bridged dinuclear manganese complex 63. | ||
In 2003, McKenzie reported the use of a manganese dimer complex bearing a monoanionic pentadentate ligand (64) as a water-oxidation catalyst. Originally, the authors intended to study these complexes as model systems of the manganese lipoxygenase.137 However, mechanistic investigations revealed the ability of 64 to function as a water oxidation catalyst.138 The complex has metal centres with a unique hepta-coordinated structure, wherein the manganese ions are present in various oxidation states. The carboxyl moiety of the ligands plays two roles. The first is as a bridging ligand to form the dimeric complex, and the second is for the formation of hydrogen bonds with the aqua ligands. The catalytic activity of the complex was investigated by adding the chemical oxidant TBHP to a solution containing the complex, which confirmed the generation of dioxygen. Further, periodic additions of TBHP afforded dioxygen generation repeatedly. They also explored the catalytic reaction using Ce(IV) as the chemical oxidant which also afforded dioxygen generation; however, the dioxygen generated was lower than that with TBHP. To gain insights into the nature of the active species of the reaction, electrochemical studies, ESI-MS, EPR, and UV-vis absorption spectroscopic investigations were carried out using TBHP as the chemical oxidant, and the formation of the high-valent Mn species was confirmed. Because O2 evolution can occur through disproportionation of TBHP, the authors performed the reaction using 18O-labelled water as the substrate to examine the source of the oxygen atoms in the generated O2. As a result, 18O–16O was mainly generated, indicating that both water and TBHP are the oxygen sources. The catalytic cycle of 64, proposed based on the experimental results, is shown in Fig. 29. The complex is initially oxidised by TBHP to afford the monomeric intermediate with an OH− ligand (A). The intermediate immediately dimerises to afford the μ-oxo bridged Mn(III)Mn(III) state (B). This intermediate is then oxidised by TBHP to the bis μ-oxo species with the Mn(IV)Mn(IV) oxidation states (C), wherein the source of the oxygen atom is TBHP. During this process, the carboxylate arms are concomitantly displaced to accommodate the two μ-oxo groups. The μ-oxo moieties of the species undergo intramolecular O–O bond formation (C → D). In this process, the coordination of the carboxylate arms to the metal centre assists the reaction. Subsequent ligand exchange with water molecules dissociates O2 from the complex and closes the catalytic cycle. It must be noted that this complex did not form the inactive MnO4− species observed in the catalysis mediated by 63 (vide supra).
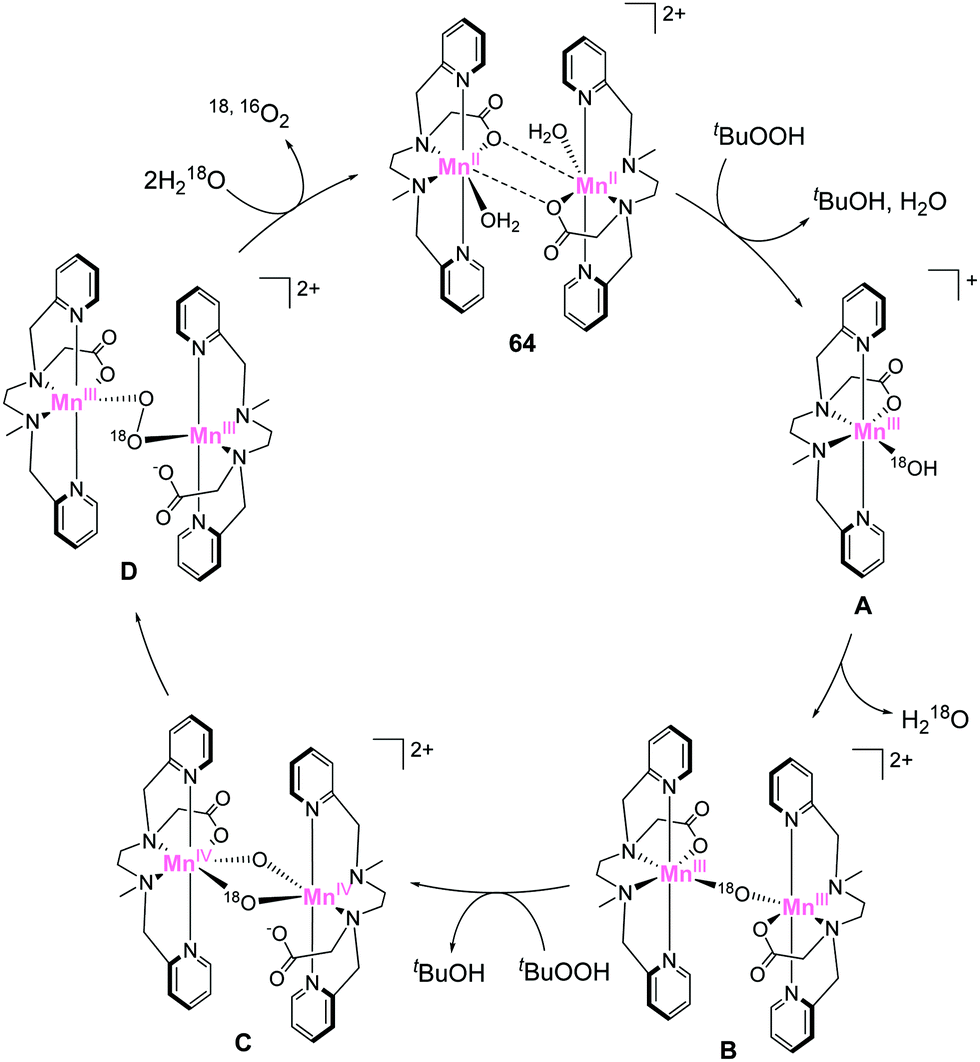 | ||
| Fig. 29 Catalytic cycle for water oxidation by the manganese dimer complex with a monoanionic pentadentate ligand 64 initially proposed by McKenzie et al. | ||
Further studies on the catalytic mechanism of 64 based on quantum chemical calculations139 indicated a different pathway. In the newly proposed mechanism (Fig. 30), the dimer is in equilibrium with the monomeric species (A) and the reaction is initiated from the monomeric state. First, the monomeric species is oxidised to the Mn(IV)O state (B), which then dimerises to form the Mn(IV)–(μ-O)2–Mn(IV) diamond core (D), in which pyridyl groups of the ligand are detached. This species exists in equilibrium with the Mn(III)–(μ-O)–Mn(IV)–O˙ key intermediate (C), which is formed by the coordination of a pyridyl moiety to the metal centre. This mixed-valence state is attacked by a water molecule and forms an O–O bond via the WNA mechanism. From the resultant Mn(III)–(μ-O)–Mn(III)–OOH species (E), O2 dissociates without any oxidation process, and the initial monomeric species is regenerated. In this step, the second manganese centre delivers the additional oxidising equivalent to release O2. As shown above, in both mechanisms, the flexible nature of the ligand and the dinuclear core of the complex play an important role in facilitating the O–O bond formation, which provides an intriguing insight into the development of the dinuclear complex-based water oxidation catalysts.
 | ||
| Fig. 30 Updated catalytic mechanism for water oxidation mediated by 64 reported in 2011. | ||
In 2011, Åkermark et al. reported a manganese dimer complex (65) as a water oxidation catalyst.140 Although several manganese-based complexes were developed for water oxidation (vide supra), many of the complexes require oxygen atom donors to drive the reaction, and cannot catalyse water oxidation when single-electron oxidants are used. The complex developed by the group is the first example of a manganese-based complex that promotes water oxidation in the presence of [Ru(bpy)3]3+ as the single-electron oxidant. The key to their success was the use of an oxidation-resistant negatively charged ligand. Such features of the ligand drastically reduce the redox potentials of the metal centre without causing catalyst decomposition. The catalytic activity of this complex was investigated in phosphate buffer. Upon addition of a single-electron oxidant [Ru(bpy)3]3+, oxygen evolution with a TOF of 0.027 s−1, and TON of ca. 25 was achieved. They also studied the light-driven reaction by using [Ru(bpy)2(deep)]2+ (deep = 4,4′-bis(ethoxycarbonyl)-2,2′-bipyridine) as the photosensitizer and Na2S2O8 as the sacrificial electron acceptor. Under these conditions, oxygen evolution occurred with a TON of approximately 4. Several control experiments revealed that the complex is essential for enabling the light-driven water oxidation. The results of the study clearly demonstrated that the manganese-based complex could catalyse water oxidation even in the absence of the oxygen donors when the complex was appropriately designed.
In 2012, Stahl and Llobet et al. reported two kinds of μ-peroxo cobalt dimeric complexes (66 and 67).141 These complexes are analogues of the ruthenium-based dimeric water oxidation complexes. The complex 66 was stable enough to remain active for several hours, and cobalt oxide particles were not observed during the reaction. This is particularly noteworthy considering that several cobalt-based dimeric complexes with bidentate bipyridyl ligands undergo facile decomposition under similar conditions.141 Electrochemical studies of the complexes revealed that these complexes could generate the Co(IV)Co(IV) states via the two-step one-electron oxidation reactions. The CV of 66 exhibited a quasi-reversible redox wave at 1.56 V (vs. NHE), and the wave was assigned to the Co(IV)Co(III)/Co(III)Co(III) process. In the differential pulse voltammetry, two oxidations were observed with approximately equal intensities at 1.56 and 1.82 V. The latter wave was assigned to the Co(IV)Co(IV)/Co(IV)Co(III) process. Upon further sweep of the potential to the positive region, the catalytic current was observed at 1.91 V, indicating the electrocatalytic activity of the complex for water oxidation. The group also performed the CPE of the complex at 2.0 V in phosphate buffer (pH 2.1) and detected the evolution of dioxygen with a faradaic efficiency of 77%. The CV of 67 displayed similar features as that of 66 with the shift of potentials to the negative region; the quasi-reversible wave and catalytic wave were observed at 1.35 and 1.84 V, respectively. In the CPE experiment, 67 exhibited higher current than 66; however, the faradaic efficiency of the reaction mediated by 67 was not reported. The authors also proposed a preliminary reaction mechanism based on the experimental results (Fig. 31). In this mechanism, Co(III)Co(III) is initially oxidised to the Co(IV)Co(III) state (A), which then undergoes PCET, as confirmed by differential pulse voltammetry (DPV). As a result, the Co(IV)Co(IV)–peroxo species forms, as observed in the case of the ruthenium-based complex, and the oxygen evolution reaction proceeds to close the catalytic cycle. The study highlights the importance of the non-labile ligand framework in minimizing ligand exchange and decomposition of the resultant complexes under catalytic conditions.
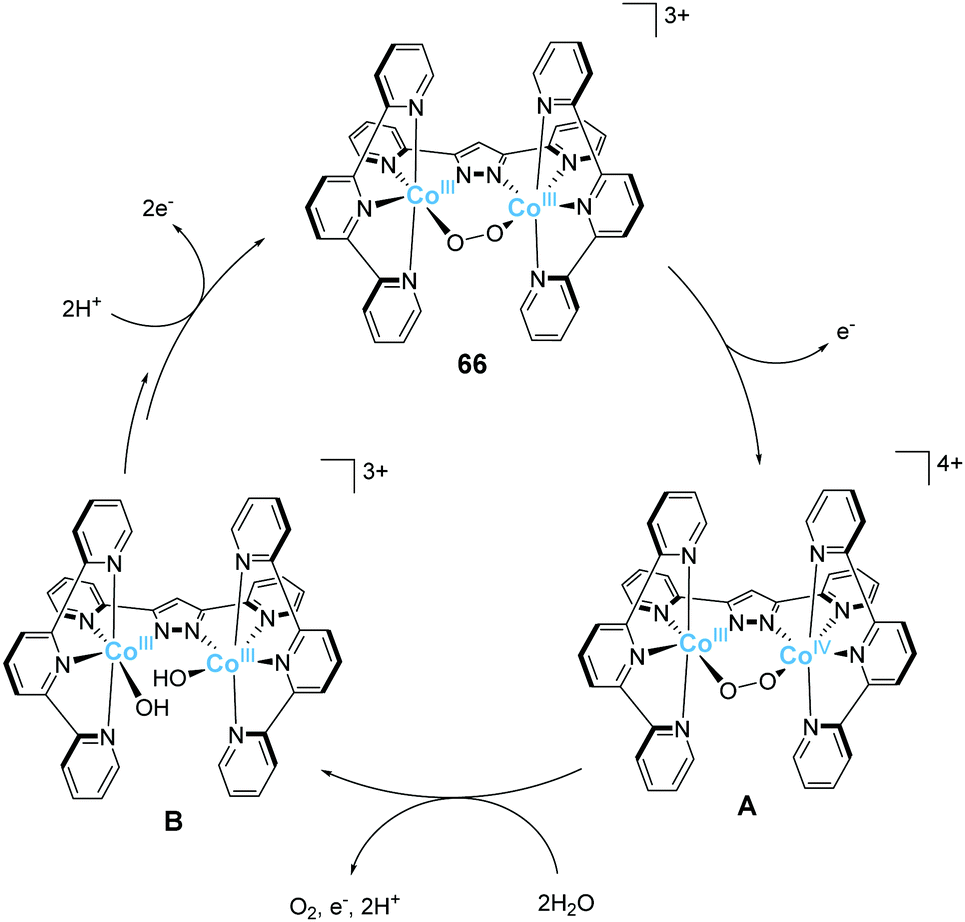 | ||
| Fig. 31 Plausible catalytic pathway of water oxidation mediated by the μ-peroxo cobalt dimeric complex 66. | ||
Thapper et al. reported the μ-O2 bridged cobalt dimer complex (68) as a catalyst for electrochemical and photochemical water oxidation.142 The key feature of the complex is its doubly-bridged μ-OH/μ-O2 motif, which enhances the kinetic stability of the complex. Electrochemical measurements of the complex in CH3CN displayed a reversible redox wave attributed to CoIVCoIII/CoIIICoIII at 1.34 V (vs. NHE). In the CV of the complex in borate buffer (pH 8), oxidation was observed at 1.05 V, and the further sweep of the potential exhibited a catalytic current for water oxidation. The onset of catalytic current was ca. 1.30 V which gave an overpotential of 540 mV. Encouraged by the result, the catalytic activity of the complex for light-driven water oxidation was then investigated. Experiments were performed by using [Ru(bpy)3]2+ as a photosensitizer and Na2S2O8 as the sacrificial electron acceptor. Under visible light irradiation of at pH 8, the TON and TOF values of the reaction reached 58 and 1.4 s−1, respectively; however, the catalysis terminated after 60 s. DLS measurements revealed that the absence of cobalt oxide formation during the catalytic reaction, which demonstrates the activity of 68 as a discrete catalyst. The reaction mechanism of 68 was proposed based on EPR spectroscopy and kinetic analyses and is shown in Fig. 32. The initial complex undergoes a two-step one-electron oxidation to form the Co(IV)–(μ-O2)–Co(IV) state (B). WNA of a water molecule on the peroxo moiety of the complex leads to O–O bond formation (B → C) and a further nucleophilic attack by another water molecule leads to the evolution of dioxygen and the (OH)CoIII–CoIII–(OH) species (D). Further oxidation and deprotonation results in (OH)CoIII–CoIVO species, and the second oxidation and deprotonation afford a reactive bis(Co(IV)O) species (E) that undergoes intramolecular O–O bond formation to regenerate the initial state. This study is the first example of light-driven water oxidation by a molecular dinuclear catalyst.
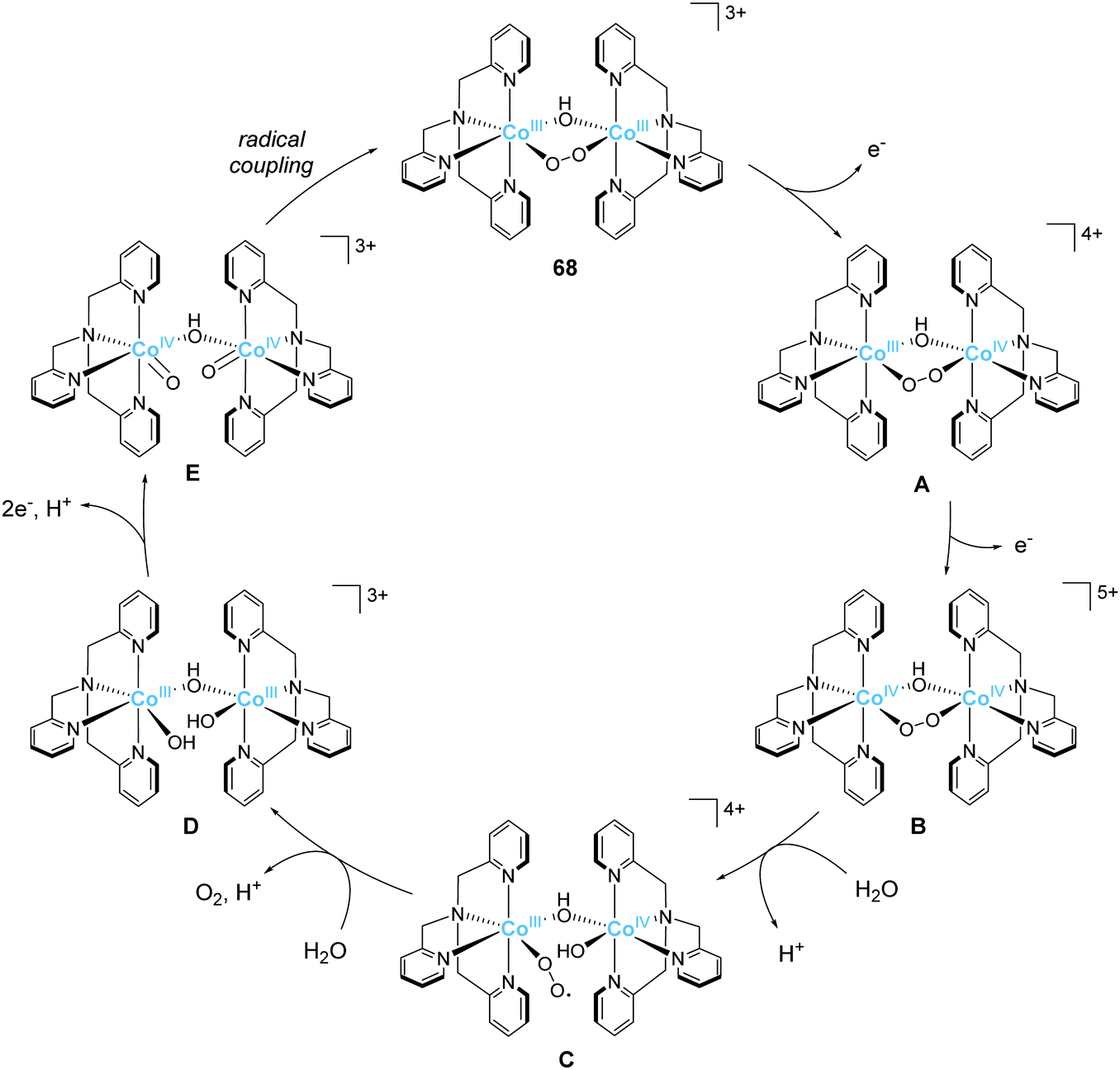 | ||
| Fig. 32 Proposed mechanism for water oxidation promoted by the μ-O2 bridged cobalt dimer complex 68. | ||
In 2015, Thummel et al. reported the water oxidation reactions catalysed by 69 and 70, which were two different types of iron-based complexes.14369 is a μ-oxo-bridged dimeric complex bearing 2-(pyrid-2′-yl)-8-(1′′,10′′-phenanthrolin-2′′-yl)quinoline (ppq) ligands, and 70 is a monomer complex comprising the bis-phenanthroline amine (dpa) ligand. In the electrochemical measurements of 69 in MeCN, the complex exhibited a single two-electron wave which was attributed to the formation of the FeIV–O–FeIV species. The species then undergoes disproportionation with the loss of two protons to afford the FeIII–O–FeVO state. In contrast, 70 displayed two reversible waves, which were assigned to FeIV/III and FeV/IV redox couples. In the cyclic voltammograms of the complexes in pH 1 aqueous buffer, large irreversible currents were observed, which indicated the catalytic activity of the complexes. Encouraged by the results, the activity of the complexes was examined by using cerium ammonium nitrate (CAN) as the chemical oxidant at pH 1. The dimeric complex exhibited a higher initial reaction rate (7920 h−1) than that of the monomeric complex (842 h−1). The group further confirmed that the dimeric catalyst remains intact during the oxidation process. While the mechanistic aspects were not discussed in this report, the results indicated that FeIII–O–FeVO species, which form via a two-electron oxidation process of the initial state, is the key intermediate which triggers the catalysis.
Das et al. reported another kind of dinuclear oxo-bridged iron complex 71, [(FeLCl)2O](FeCl4)2 (L = (2-(pyrridin-2-yl)oxazolidine-4,4-diyl)) as an electrocatalyst for water oxidation.144 They designed the catalyst with a stable metal–oxo–metal scaffold, using a redox-inactive oxazolidine ligand. The bulk electrolysis experiment at 1.0 V vs. Ag/AgCl was performed to evaluate the catalytic activity of complex 71, the TON, TOF and a faradaic efficiency were determined to be 408, 0.11 s−1, and 78.6%, respectively. They also investigated the catalytic mechanism of complex 71 employing theoretical study as well as ESI-MS analysis of the crucial steps (Fig. 33). The computational results revealed that the complex 71 undergoes dechlorination by hydroxide anions under basic medium conditions to produce intermediate A. The intermediate A can undergo dehydration by releasing 18.3 kcal mol−1 of free energy to produce intermediate B which is a bis-μ-oxo bridged iron complex. In the next step, a peroxo-bridged iron complex C forms upon complexing with two hydroxide anions as the process is thermodynamically favourable by 16.2 kcal mol−1. At this stage, two electron oxidation of C followed by dehydration can occur resulting in intermediate E from which the release of dioxygen is very facile (ΔG = −11.9 kcal mol−1) upon complexing with two hydroxide anions. In a follow-up step, electrochemical two electron oxidation of F can produce intermediate A to complete the catalytic cycle.
 | ||
| Fig. 33 Plausible catalytic mechanism for water oxidation mediated by 71. | ||
In 2016, Fukuzumi and Kojima et al. reported the light-driven water oxidation reaction catalysed by the dinuclear cobalt complex 72. The structure of the complex 72 is similar to that of 68, although the bridging ligand in 72 (doubly-bridged μ-OH/μ-O motif) is distinct from that of 68 (doubly-bridged μ-OH/μ-O2 motif). This change in the bridging moieties enables π–π interactions between the two equatorial pyridine rings, which is a unique feature of the complex. Importantly, this hydrophobic interaction is expected to stabilize the dimeric structure in aqueous media. For the catalysis experiments, the group used conditions similar to those reported by Thapper et al.; [Ru(bpy)3]2+ was used as the photosensitizer, and Na2S2O8 was the sacrificial electron acceptor at pH 9.7. Further, the TON was calculated to be 742, and the quantum yield was 44%.145 The authors were successful in developing the water oxidation reaction catalysed by 72 with the use of [Ru(bpy)3]3+ as the chemical oxidant. At pH 7.8, the dioxygen evolution was confirmed, and the TON was determined to be 4.3. The O2 yield, relative to the added oxidant, was 22%. The detailed mechanism of this chemically-driven water oxidation reaction was proposed based on DFT calculations, isotope-labelling experiments, and UV-vis absorption spectroscopy (Fig. 34). Under these conditions, one out of the two hydroxy ligands is deprotonated, and thus the resting state is Co(III)–(μ-O,μ-OH)–Co(III) (72). From this state, the two-electron oxidation involving the transfer of one proton proceeds to afford the bis-(μ-oxyl)Co(III)2 state (A). The intramolecular radical coupling between the two oxyl ligand affords μ:η2,η2-peroxo dinuclear Co(III) intermediate (B). It should be noted that this mechanism of O–O bond formation process of 72 is totally different from that of 68 (vide supra). The intermediate rapidly undergoes ligand substitution to afford a μ-peroxo–μ-hydroxo dinuclear Co(III) complex (C). Finally, the oxidation of the complex, the evolution of dioxygen, and the deprotonation reaction occur and thereby regenerate the initial state. Notably, the rate-determining step of this system is not the O–O bond formation step. This is because the two water molecules that form a dioxygen molecule are preliminary fixed between the two CoIII centres as bridging ligands. In other words, the study indicated the importance of the dinuclear structure in facilitating the smooth formation of the O–O bond.
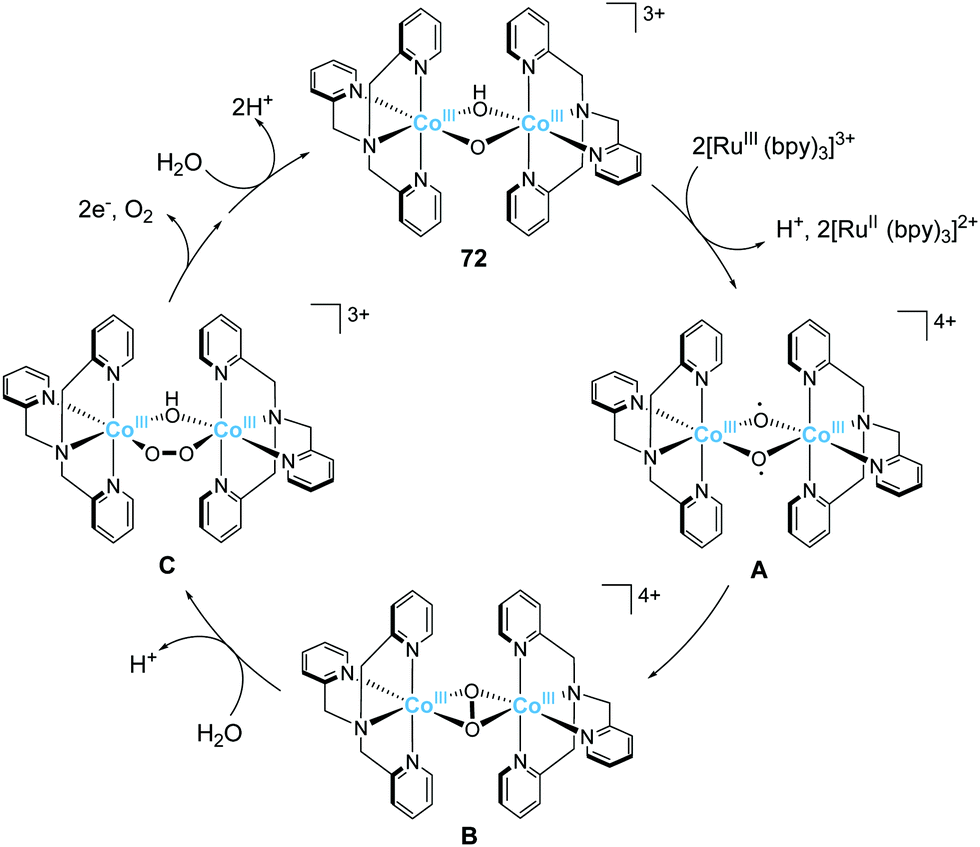 | ||
| Fig. 34 Possible catalytic pathway for water oxidation mediated by the dinuclear cobalt complex 72. | ||
Kieber-Emmons et al. developed a novel copper-based dimeric complex (73) in 2017.146 They hypothesized that it is necessary to mitigate the formation of the terminal Cu(IV)O/Cu(III)–O· intermediate for achieving efficient water oxidation. This is because late transition metal oxo species have been disfavoured due to their filled antibonding orbitals in metals with high d electron counts.79 Therefore, the terminal Cu(IV)O/Cu(III)–O˙ becomes unstable and/or too reactive, which is not suitable for effective water oxidation.
Based on this assumption, they screened several copper complexes with scaffolds derived from tmpa (tmpa = tris(2-pyridylmethyl)amine), and found that 73 functions as a catalyst for water oxidation. In the cyclic voltammogram of the complex with a rotating disc electrode, a large irreversible current was observed with an onset potential of ∼1.2 V (vs. NHE) at pH 12.5, suggesting the catalytic activity of the complex for water oxidation. The CPE experiment was also conducted at 1.62 V (vs. NHE), and the formation of dioxygen was confirmed. The faradaic efficiency of the reaction was determined to be 112 ± 23% by using rotating ring disc electrode voltammetry. The rate of the catalytic reaction was estimated to be 33 s−1 based on the results of the electrochemical measurements (eqn (4)) with an overpotential of 1.0 V. They also clarified that the complex functions as a homogeneous catalyst by employing several analytical techniques. The results of the electrochemical and spectroscopic studies revealed that the complex 73 undergoes partial dissociation in aqueous solutions (pH 12.5), which leads to the formation of the monomeric species. The DFT calculations indicated that the oxidation of the monomer and dimer both result in the formation of the same bis-(μ-O)Cu(III)2 intermediate, and thus the formation of terminal Cu(IV)O/Cu(III)–O˙ species is avoided (Fig. 35). Calculations further revealed that both intermolecular water nucleophilic attack and redox isomerization of bis-(μ-O)Cu(III)2 species (E) are energetically accessible pathways for O–O bond formation. Finally, a hydroxyl anion attacks the common intermediate of these two pathways, Cu(II)–OO˙ species (G), which leads to the formation of dioxygen with the concomitant regeneration of the monomeric species. This study indicates that the use of the dimeric structure is an important strategy for obtaining copper-based catalysts for water oxidation because the formation of the energetically unfavoured copper–oxo/oxyl radical species can be avoided by employing such structures.
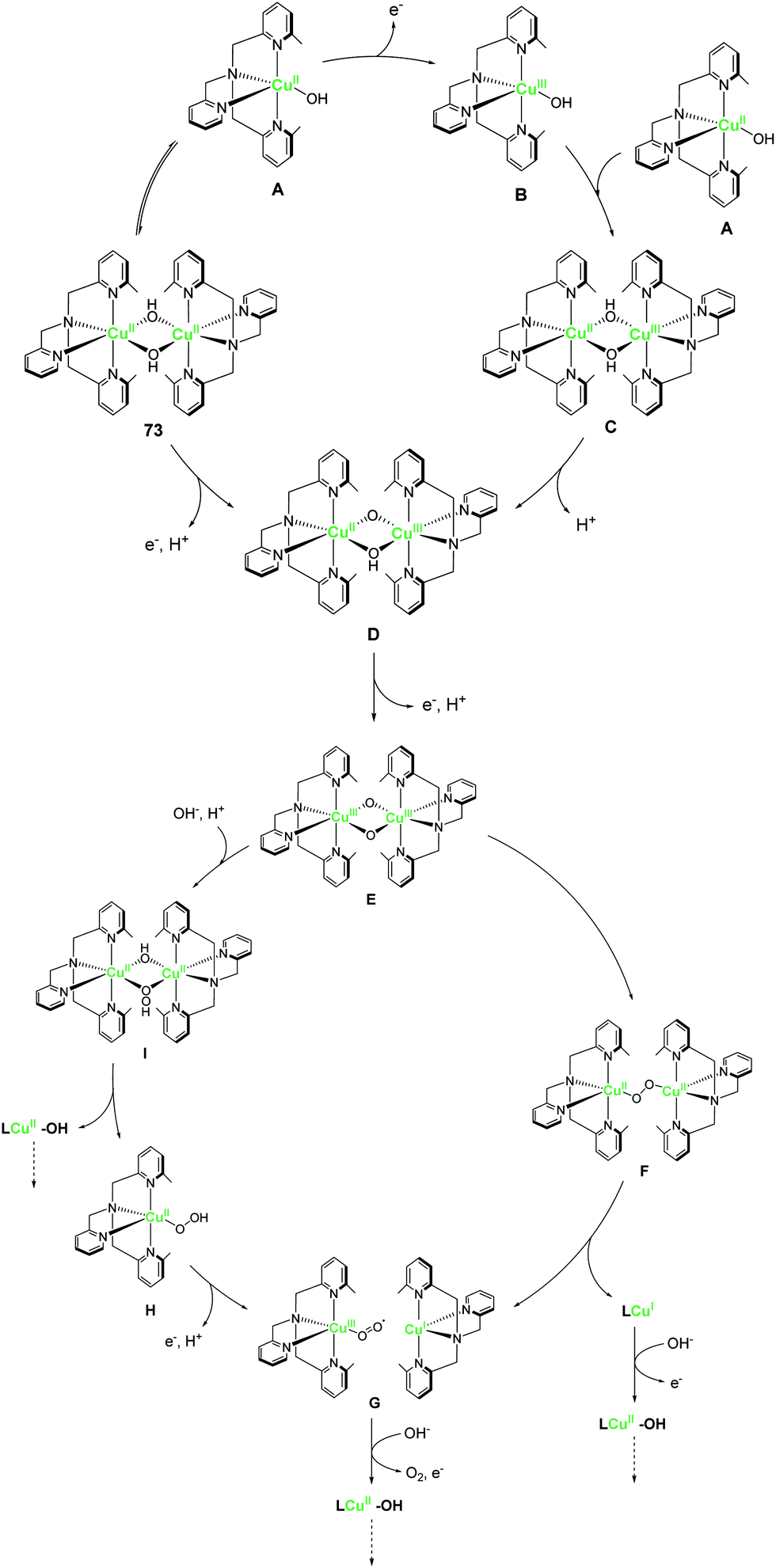 | ||
| Fig. 35 Plausible catalytic mechanism for water oxidation by the copper-based dimeric complex 73. | ||
In 2015, Zhang et al. reported a dimeric copper complex for water oxidation. Before the discovery of this complex, the copper-complex-catalysed water oxidation reaction was known to require basic conditions. This is the first example of a copper complex-based water oxidation catalyst (74) which operates in neutral aqueous solutions.147 The complex, 74, exhibited an irreversible catalytic current with an onset potential at 1.6 V (vs. NHE) in a phosphate buffer solution (pH 7), indicating its activity for water oxidation. The onset potential of the catalytic current shift depended on the pH of the solutions, with the slope of −59 mV per pH, which is consistent with PCET oxidation. The overpotential was approximately 0.8 V at pH 7. The CPE experiment at 1.87 V for 0.5 h confirmed the oxygen evolution with a faradaic efficiency of 98%. The homogeneous nature and the stability of the complex during the catalysis were confirmed by UV-vis absorption spectroscopy and electrochemical measurements. The TOF value of the reaction was estimated to be 0.6 s−1 based on electrochemical measurements (eqn (4)). The mechanism of water oxidation mediated by the complex was constructed based on the DFT calculations and electrochemical measurements and is shown in Fig. 36. When the complex is dissolved in water, one of the pyridyl groups dissociate from the copper centre, and a water molecule coordinates at the formed vacant site. In this state, hydrogen bonding exists between the dissociated pyridyl moiety and the coordinated aqua ligand. This initial state undergoes two sequential PCET reactions to form the Cu(III)–O–Cu(III) species (B). Subsequently, the intramolecular hydrogen atom transfer reaction from the hydroxy ligand to the pyridyl moiety occurs leading to the formation of the O–O bond in an intramolecular fashion (B → D). Further PCET reaction (D → E) and oxidation of the complex induce the liberation of O2 from the complex (E → F), and the coordination of a water molecule regenerates the initial state of the complex (F → 74). Similar to the catalytic cycle of 73, the formation of Cu(IV)O species is prevented in this catalytic cycle. This study highlights the importance of a cooperative interaction between the two copper centres in facilitating O–O bond formation without generating the unstable Cu(IV)O state. It should be noted that the oxidation resistance of the ligand is vital for driving the reaction.
 | ||
| Fig. 36 Proposed mechanism of water oxidation catalysed by the dimeric copper complex 74, which operates in neutral aqueous solutions. | ||
In 2018, Zhang et al. also reported the catalytic activity of an unsymmetrical dimeric copper complex (75) that has a structure similar to that of 74.148 This complex served as an electrocatalyst for water oxidation in phosphate buffer (pH = 7.0). The electrochemical measurements of the complex revealed an irreversible current at 1.6 V (vs. NHE), indicating its catalytic activity. The CPE experiment conducted at 1.87 V (vs. NHE) confirmed the evolution of dioxygen. The molecular integrity of the complex was confirmed by electrochemical measurements, SEM, XPS, and UV-vis absorption spectroscopy. The overpotential and TOF value based on (eqn (4)) were estimated to be 780 mV and 0.78 s−1, respectively. The unsymmetrical structure of 75 is its unique attribute, wherein one of the copper centres has an aqua ligand in the initial state. Further, this unsymmetric structure is the origin of the unique electrochemical behaviour of the complex. The DPV experiment displayed two oxidation peaks. Although the first peak at 1.75 V (vs. NHE) did not show any pH dependence, the second peak shifted with variations in solution pH with a slope of −64 mV per pH, which indicates that the second oxidation process is a PCET reaction. The kinetic isotope effect for the reaction was 2.04, suggesting that the transfer of a proton is involved in the rate-determining step. Note that the concentration of the phosphate anion largely affects the catalytic activity, indicating the role of phosphate anion as a proton acceptor. The catalytic cycle of 75 proposed by considering the results of aforementioned experiments (Fig. 37) is distinct from the that of 74. One-electron oxidation and the subsequent PCET reaction of the initial complex generates the OH–Cu(III)–(μ-O)–Cu(III) intermediate (B). The proton on the hydroxy ligand of this intermediate interacts with the phosphate anion via hydrogen bonding and promotes the intramolecular O–O bond formation. The formed intermediate (C) is then oxidised to the superoxide species (D), and the additional oxidation of the intermediate generates dioxygen to close the catalytic cycle. The results described in the study demonstrated that the introduction of an open site on the metal centre drastically changes the catalysis mechanism.
 | ||
| Fig. 37 Proposed mechanism for water oxidation by the unsymmetrical dimeric copper complex 75. | ||
As described above, there have been several reports on water oxidation catalysts based on dinuclear complexes. Importantly, the O–O bond formation process proceeds mainly in an intramolecular fashion, whereas the mononuclear metal complexes mainly form O–O bonds via WNA to form high-valent metal–oxo species. Such features of the dimeric complex should be of importance because the O–O bond formation process is often the rate-determining step in water oxidation. The formation of the bond via an intramolecular pathway would lead to the acceleration of the rate-determining step in the entire reaction. Overall, these results indicate that the use of dinuclear structures is a significant strategy for designing molecular water oxidation catalysts (Chart 13). It should be noted that appropriate design of the ligand for stabilizing high valent species, such as by the introduction of an anionic moiety, and improvement of their oxidation resistance is essential for achieving water-oxidation with high efficiency.
 | ||
| Chart 13 Summary of features of water oxidation catalysts with dimeric structures. | ||
3.7 Multinuclear water oxidation catalysts
The natural enzyme that catalyses the water oxidation reaction (OEC) contains the multinuclear Mn4CaO5 complex as the catalytic centre. Therefore, artificial multinuclear metal complexes are expected to be excellent candidates for the development of water oxidation catalysts. Moreover, the OEC contains the earth-abundant first-row transition metal manganese ions, and thus the use of multinuclear complexes containing such metal ions is an important approach.The detailed structure of the Mn4CaO5 cluster was elucidated using synchrotron X-ray crystallography.50,51 The cluster is stabilised by six carboxylates, one histidyl group of the surrounding amino acid residues, and water molecules coordinated to Mn and Ca ions (Fig. 38).
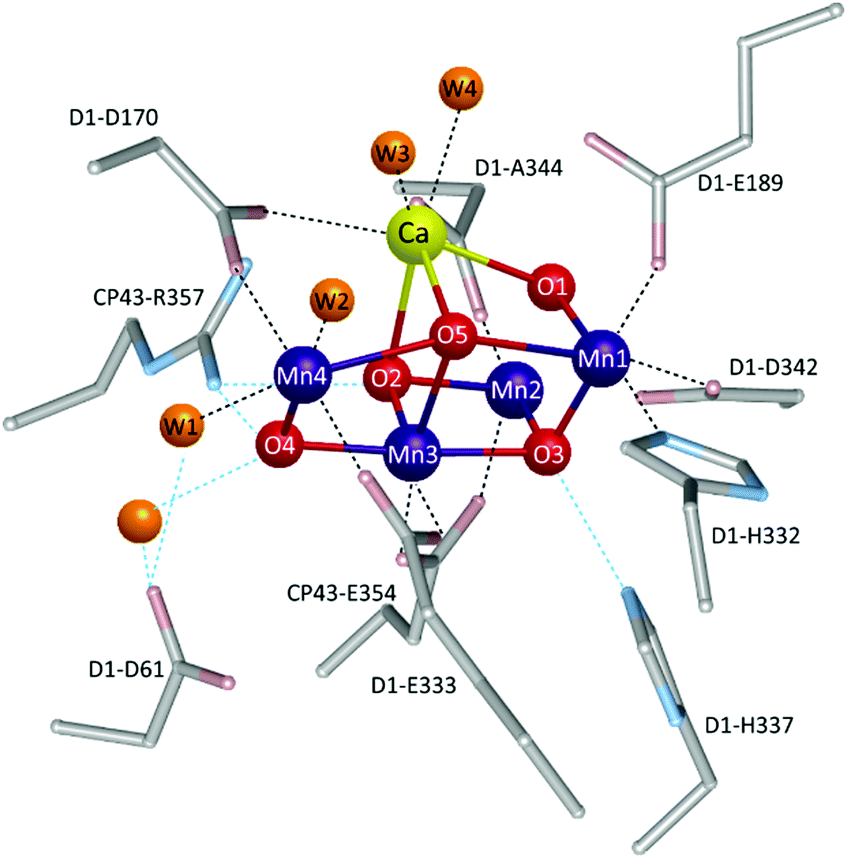 | ||
| Fig. 38 The structure of the Mn4CaO5 cluster and its ligand environment in the OEC. Manganese, purple; calcium, yellow; oxygen, red; oxygen atoms in water molecules, orange. | ||
The mechanism of water oxidation catalysed by the OEC has been extensively investigated. The mechanism includes two important steps: accumulation of multiple charges and the O–O bond formation. During the accumulation of charges, four manganese ions are oxidised in a stepwise fashion with the involvement of the PCET processes. The existence of multiple redox centres is regarded essential for promoting the rapid electron transfer reaction. While the details of the O–O bond formation process has not been clarified yet, recent studies indicate that the presence of the neighbouring water coordination sites is important for accelerating the process.149,150 Thanks to these two features, the OCE can efficiently catalyse water oxidation.47
Based on the aforementioned background, several studies on the structural mimicry of the OEC have been reported. Particularly, the cubane-type manganese complexes are the most intensively investigated. Development of such complexes is an interesting research target because such studies can contribute both to the understanding of the features of the OEC, and the construction of artificial water oxidation catalysts. However, most of the structural models of the OEC are inactive for water oxidation, although excellent structural mimics were obtained.46–49
Dismukes et al. disclosed the only example of a manganese-cubane complex that is active for water oxidation. In 1997, they reported the synthesis of complex comprising the Mn4O4 scaffold stabilised by the phenylphosphonic acid ligands (76 in Chart 14).151 In a subsequent study, the ligand photodissociation reaction of the complex in the gas phase was investigated.152,153 In this study, irradiation with a laser (337 nm) resulted in the formation of the Mn4O2 core structure, which indicates the dissociation of dioxygen from the complex by photoirradiation, although the process is not catalytic. However, in 2008, Spiccia et al. found that the Mn4O4 complex stabilized by bis(4-methoxyphenyl)phosphinate can serve as a catalyst for water oxidation.154 In their study, the complex was deposited into a thin Nafion membrane deposited on a conducting electrode. The irradiation of white light (270–750 nm) on the electrode with polarization at 1.00 V resulted in the formation of dioxygen, which was confirmed by a Clarke electrode. They also performed an isotope labelling experiment and confirmed that the source of dioxygen is water.
 | ||
| Chart 14 Structures of multinuclear water oxidation catalysts. | ||
The mechanism of catalysis was studied by quantum chemical calculations.155 The mechanism proposed in the study involves the dissociation of a phosphate group from the ligand upon photoirradiation, which renders the Mn4O4 core more compact. In this state, the distance between two oxygen atoms becomes significantly less, and the O–O bond formation proceeds. Subsequently, dioxygen is dissociated from the complex, and the coordination of the phosphate ligand affords the Mn4O2 (‘butterfly’) structure. This complex is also utilized as the water oxidation catalyst in the water-splitting cell.45,156,157 Photoirradiation of this water-splitting cell with the RuII dye injects and transfers the electrons into the TiO2 conduction band, causing the formation of the RuIII species. Then, the species oxidises the cubane complex to drive the water oxidation reaction. In other words, the reaction proceeds only upon irradiation of visible light, in the absence of external bias. The performance of the cell was investigated using various techniques, and the formation of dioxygen was confirmed. The TOF of the reaction was estimated to be 47 ± 10 h−1. The authors also studied the stability of the cubane-structure during the catalysis. Initially, it was suggested that the Mn4O4 structure is retained during the reaction based on the results of EPR and electrochemical measurements.158,159 However, a close inspection revealed that the true catalytic active species is the manganese-based nanostructure that is similar to manganese oxide lattice.160 From these results, it is concluded that the complex 76 serves as a precursor alone and forms the active nanostructure. The heterogeneous species formed by the dissociation of the complex serves as an active and robust self-healing catalyst for water oxidation. Although the complex cannot function as a homogeneous catalyst, the study demonstrated that a high-surface-area material with high catalytic activity can be obtained using the complex as a pre-catalyst.
Inspired by the studies on the structural models of the OEC, the cubane-complexes containing cobalt ions were also studied as water oxidation catalysts.161–164 In these early studies, synthesis, structural determination, and electrochemical properties of the cobalt–cubane complexes were mainly reported. In 2011, Dismukes et al. reported the water oxidation reaction catalysed by the complex 77.165 This is the first example of a cobalt–cubane complex that showed catalytic activity for water oxidation. The complex can be synthesized from Co(NO3)2, NaOAc, and pyridine in a straightforward manner, and characterized by comparing the results of 1H NMR, cyclic voltammetry, and UV-vis absorption and mass spectroscopy with those reported in the previous studies. In their study, the catalytic activity of the complex under photoirradiation was investigated using [Ru(bpy)3]2+ as a photosensitizer, and Na2S2O8 as a sacrificial electron acceptor. The TOF and TON (1 h) values of the reaction were calculated to be 40 ± 2 and 0.02 s−1, respectively. In this catalytic system, the evolution of dioxygen was terminated after 1 h probably due to the photodecomposition of [Ru(bpy)3]2+.166 The robustness of the catalyst was confirmed by 1H NMR spectroscopy. The control experiment using Co(II) salt did not afford dioxygen. These results clearly demonstrate that the complex 75 remains intact during the catalysis and is responsible for water oxidation. This is a remarkable example which shows that the artificial M4O4–cubane structure can function as a molecular catalyst for water oxidation.
Later, Bonchio et al. prepared several cobalt cubane complexes (78–83) with structures similar to that of 77.167 The aim of their study was to improve the stability and efficiency of the catalytic system by introducing para-substituted pyridines to the Co4O4 cubane structure as terminal ligands. In the cyclic voltammograms of the complexes, the overpotentials which varied in a narrow range (0.50–0.57 V) were detected; however, no apparent ordering effect as a function of the pyridine substituent was observed. In contrast, the rate constant of photoinduced electron transfer reaction largely depended on the substituents. Hammett linear free energy plots revealed a correlation between the photoinduced electron transfer constants and the electron-donating ability of the ligand. This result indicates that the introduction of an electron-donating group can enhance the rate of photoinduced electron transfer. They also optimized the reaction conditions to prevent the decomposition of [Ru(bpy)3]2+ during the photolysis, which was a major problem in the catalytic system developed by Dismukes et al.; therefore, oxygen evolution occurred until quantitative consumption of the sacrificial electron acceptors for all complexes and the TON reached 140. However, the effect of substituents on the quantum yield was not straightforward. This is probably related to an overall balance of competing factors governing the diverse steps before oxygen evolution. This rigorous study successfully provides an effective photocatalytic system for water oxidation based on the cobalt–cubane complex.
In 2015, Tilley et al. reported the reaction mechanism of water oxidation mediated by the cobalt cubane complex 77. Their study indicated that the stoichiometric reaction of the one-electron oxidised species of 77 (the Co3(III)Co(IV) state) with hydroxy anion affords the formation of O2 with quantitative regeneration of the initial state. They also indicated that Co3(III)Co(IV) species undergoes disproportionation to form the species with higher valent, Co3(III)Co(V) or Co2(III)Co2(IV). The proposed mechanism of water oxidation reaction mediated by 77 is shown in Fig. 39.168,169 The mechanism was formulated based on the results of the aforementioned experiments and DFT calculations.170 Several reaction pathways were considered for the reaction. Among them, the lowest energy pathway involved the formation of the Co(V)O (C) species, which can also be regarded as the Co(IV)–oxyl radical species. The nucleophilic attack of the hydroxy anion on the Co(V)O intermediate forms the O–O bond (D). Further oxidation gives the peroxo radical species (E), and the ligand exchange reaction with hydroxy anion generates dioxygen. This proposed catalytic cycle indicates that the +2 oxidation state of the cobalt centre is not necessary for the reaction. Therefore, structures that can stabilise the higher oxidation states might be essential for obtaining efficient catalysts. These studies provide important insights into the water oxidation reactions catalysed by multinuclear metal complexes.
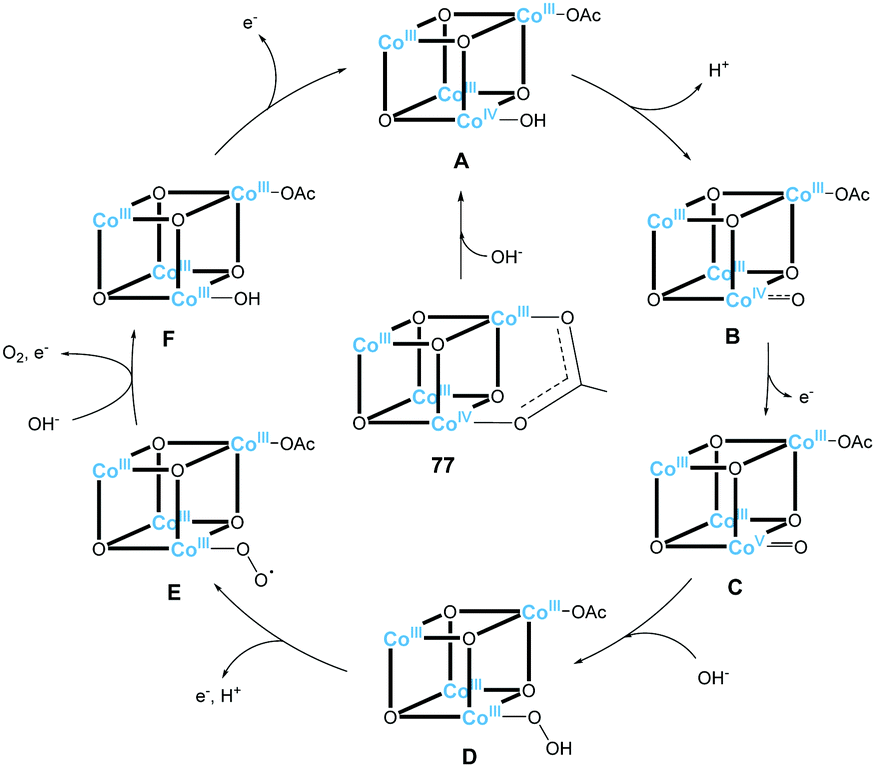 | ||
| Fig. 39 Plausible catalytic cycle of water oxidation facilitated by the cobalt cubane complex 77. | ||
Given that the multinuclear structure is essential for driving the water oxidation reaction in the OEC, the use of manganese-based multinuclear complexes can be an effective strategy. In 2018, Maayan and Christou et al. reported the catalytic activity of a dodecanuclear manganese complex (84).171 The previous studies demonstrated that the MnIV–O–MnIII–H2O motif in the Mn4O5Ca cluster of the OEC plays an important role in the catalysis. Interestingly, this motif is a multiple structural element in the complex 82, and thus it is expected that the complex can exhibit activity for water oxidation. However, the previously reported relevant complexes that have the Mn12 core are insoluble or unstable in the presence of water. Based on this background, the authors designed a new Mn12 complex bearing the bulky benzoate ligands to protect the core, but with at least two hydrophilic groups on each ligand to enhance the stability of the complex in water. The structure of the complex was confirmed by FT-IR, XPS, and UV-vis absorption spectroscopies. The electrochemical measurements of the complex in dry MeCN exhibited the four reversible one-electron processes, which are assignable to sequential electron transfer of the manganese ions. The results clearly demonstrated that the complex possesses the ability to transfer multiple electrons. In the cyclic voltammograms of the complex in acetate buffer (pH 6.0), the reduction peak of O2 to superoxide was observed at −0.45 V (vs. NHE), which indicated dioxygen evolution. To further verify the catalytic activity, the CPE experiment was conducted at 1.21 V (vs. NHE) at pH 6, and the formation of dioxygen was confirmed. Faradaic efficiency in the first 20 min of the CPE was 77.9%. TOF and TON values were estimated from a foot-of-the-wave analysis to be 0.035 s−1 and 629 (5 h), respectively. It should also be noted that the catalyst can promote the reaction with substantially low overpotential (334 mV). The CV curve of the complex after multiple scans remained unchanged, indicating the high stability of the complex. The results of UV-vis absorption spectroscopy also support the robustness of the complex. The catalytic activity of the complex is attributed to the multinuclear structure and hydroxy substituents. First, the existence of multiple manganese centres provides surface sites with a core that serves as a charge reservoir. Second, the hydroxy groups on the ligands provide multiple shuttle pathways through hydrogen bonding, and transfer H2O to the vicinity of the Mn ions and remove generated H+ ions from the catalytic centres. This excellent study highlights the importance of a multinuclear metal complex for achieving water oxidation under mild conditions.
In 2015, Luber and Patzke et al. reported a family of cobalt-lanthanoid cubane complexes (85–88) for water oxidation.172 This is the first example of artificial catalysts that contain both 3d and 4f transition metal ions. Redox inactive Ln3+ (Ln = Ho–Yb) cations are introduced into the cubane framework to mimic a Ca2+ ion in the OEC. The catalytic activity of these complexes was investigated in the presence of [Ru(bpy)3]2+ as a photosensitizer and S2O82− as a sacrificial electron acceptor. The production of dioxygen was confirmed, and the control experiments revealed that all of the three components, catalyst, photosensitizer, and sacrificial electron acceptor, are essential for promoting the reaction. Under optimised condition, the maximum TON and TOF were 211 and 9.6 s−1, respectively. Among the complexes, the structures with Er3+ and Ho3+ exhibited the highest activity. The electrochemical studies of the complexes and the relevant cobalt–cubane complex which did not contain the Ln3+ ions were conducted. All of the complexes exhibited pH-dependent redox processes which were attributed to the CoIII/CoII redox couple. The redox potentials of the cubane complexes with Ln3+ ions are located in the more positive potential region compared to the relevant cobalt–cubane complex, indicating the significant effect of the lanthanide ions on the redox potentials. Note that the catalytic activity of the cobalt–lanthanide complexes was higher than the cobalt–cubane complex, and thus the redox-tuning of the Lewis acidity of Ln3+ is essential for enhancing the activity. They also tested the stability of the complexes using various experiments including DLS, recycling test, ICP-MS, UV-vis absorption spectroscopy, XANES/EXAFS, and HR-ESI-MS analysis, and confirmed the molecular integrity of the complexes during the catalysis. Isotope labelling experiments using 10% H218O confirmed that the source of oxygen atoms is water. Mechanistic studies based on the results of the photocatalytic experiment and quantum chemical calculations revealed another role of the Ln3+ ion during the catalysis. As Ln3+ ions have flexible ligand binding modes, which is in close analogy with the mechanism of the OEC, smooth ligand exchange reactions were possible during the catalysis which contributed to the enhancement of the catalytic activity. Collectively, this study demonstrates the importance of the incorporation of Lewis acidic and redox inactive metal ions into the cubane structure to improve the performance. This finding is also impressive for providing insights into the catalytic mechanism of the OEC.
As a multinuclear metal complex, polyoxometalates (POMs) is also important materials for the development of efficient water oxidation catalysts. POMs, which have structurally rigid and stable molecular metal–oxo cores, are known as efficient water oxidation catalysts due to its ability to drive fast and reversible electron transfer reactions maintaining its structure.173–175 Recently, the development of POM-based water oxidation catalysts containing earth-abundant first-row transition metal ions such as Mn,176–178 Fe,179,180 Co,181 Ni182–184 and Cu185 as catalytic centers is also reported In this review, we will not discuss the details of these systems because the catalytic centers in the system are supported by POMs containing 2nd or 3rd-row transition metal ions. The recent representative reviews186–188 will be helpful for further understanding the nature of these catalysts.
In 2016, we reported the catalytic activity of the pentanuclear iron complex (89).189 The complex is not a structural mimic of the OEC but possesses the essential features of the OEC; multinuclear redox centres, and neighbouring water activation sites. The complex consists of the [Fe3(μ3-O)] core wrapped by two [Fe(μ-bpp)3] units (Hbpp = bis(pyridyl)pyrazole). Although the metal ions at the apical positions are coordinatively saturated in the hexacoordinated structure, the iron ions in the [Fe3(μ3-O)] core are in a pentacoordinated (i.e. coordinatively unsaturated) structure. Therefore, the iron ions at the core are expected to serve as neighbouring water activation sites. The CV of the complex in the absence of the substrate displayed five redox waves attributed to the oxidation of the iron centres, which indicates that the complex has multi-electron-transfer ability. Upon addition of the water as a substrate to the solution, a large irreversible current was observed coupled with the fourth oxidation wave. The CPE experiment of the complex at 1.42 V (vs. Fc/Fc+) in MeCN–H2O (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) confirmed the evolution of dioxygen with a faradaic efficiency of 96%. The TOF value of the reaction was calculated using eqn (4) and determined to be 1900 s−1. This value is about 1000 times higher than those of other reported iron-based molecular catalysts and higher than that of the OEC (400 s−1). The TON estimated from the CPE results was approximately 1000000 to 10000000 for 120 min based on the assumption that the thickness of the reaction–diffusion layer adjacent to the electrode surface (μ) is of the order of
:1 v/v) confirmed the evolution of dioxygen with a faradaic efficiency of 96%. The TOF value of the reaction was calculated using eqn (4) and determined to be 1900 s−1. This value is about 1000 times higher than those of other reported iron-based molecular catalysts and higher than that of the OEC (400 s−1). The TON estimated from the CPE results was approximately 1000000 to 10000000 for 120 min based on the assumption that the thickness of the reaction–diffusion layer adjacent to the electrode surface (μ) is of the order of  ,190 where D (cm2 s−1) is the diffusion coefficient of the catalyst. Similar to other molecular catalysts based on earth-abundant first-row transition metal ions, the decomposition of the pentairon catalyst during the catalysis is also discussed in this system,191,192 although the results of several measurements including electrochemical study, UV-vis absorption spectroscopy and XPS fully evidenced the stability of the complex and no formation of heterogeneous catalytic active species under our experimental condition.193
,190 where D (cm2 s−1) is the diffusion coefficient of the catalyst. Similar to other molecular catalysts based on earth-abundant first-row transition metal ions, the decomposition of the pentairon catalyst during the catalysis is also discussed in this system,191,192 although the results of several measurements including electrochemical study, UV-vis absorption spectroscopy and XPS fully evidenced the stability of the complex and no formation of heterogeneous catalytic active species under our experimental condition.193
The catalytic cycle for 89 was proposed based on the results of electrochemical and spectroelectrochemical measurements, 57Fe Mössbauer spectroscopy, and quantum chemical calculations (Fig. 40). Initially, the four sequential oxidations of iron ions afforded the four-electron oxidised species (S4). The S4 state then reacts with water to afford the Fe(III)5(OH2) state. Note that the reaction barrier for this step was estimated to be ca. 15 kcal mol−1. Coordination with another water molecule, together with sequential or simultaneous deprotonation, produces the bis-oxo species as a key reaction intermediate. Quantum calculations indicated that the species is in the mixed- valence state, Fe(II)2Fe(III){Fe(IV)O}2. Subsequently, the O–O bond formation process proceeds between the Fe(IV)O moieties in an intramolecular fashion with an estimated reaction barrier of less than 10 kcal mol−1. Then, O2 is evolved from the peroxo intermediate to complete the catalytic cycle. Notably, this proposed catalytic cycle has several similarities with the reaction mechanism of the OEC; the accumulation of multiple charges and the subsequent intramolecular formation of O–O bond. These results indicate that the functional mimicry of the OEC can be an innovative strategy for the development of highly active catalysts for water oxidation.
 | ||
| Fig. 40 Plausible catalytic cycle water oxidation promoted by the pentanuclear iron complex 89. | ||
As shown above, the arrangement of multiple metal ions in close proximity, such as in the OEC, can be considered an important strategy for the construction of water oxidation catalysts. Multinuclear metal complexes are an intriguing class of compounds (Chart 15). It was also revealed that the use of a scaffold with appropriate functions is important for this class of materials, rather than the simple imitation of the OEC structure. As evidenced by our study,189,194,195 such a functional mimic of the OEC can afford the catalyst with remarkable efficiency. Although the number of examples is currently limited, further exploration of this field should pave the way for the development of efficient water oxidation catalysts.
 | ||
| Chart 15 Summary of features of water oxidation catalysts with multinuclear structures. | ||
4. Conclusions
This review summarized the recent progress in molecular water oxidation catalysts of earth-abundant first-row transition metal ions. Table 1 summarises the TON, TOF, and overpotential of water oxidation catalysts introduced in this review. As the natural enzyme for water oxidation contains a Mn4CaO5 cluster as the catalytic centre, the complexes with earth-abundant first-row transition metal ions are expected to be excellent candidates for artificial catalysts. Moreover, numerous examples of synthetic catalysts based on ruthenium complexes provided an overview of the following key factors that guide successful artificial water oxidation: (1) accumulation of four oxidation equivalents; (2) formation of a high-valent key intermediate wherein an oxygen atom is bound to the metal centre; and (3) O–O bond formation, which is typically the rate-determining step in the catalysis. However, a concrete strategy for achieving these key requirements in metal complexes with earth-abundant first-row transition metal ions has not been well-developed, mainly due to high lability of the complexes, which behave in complete contrast to the ruthenium-complex-based catalysts for water oxidation.| Cat. | Oxidant | η/mV | TON | TOF/s | Ref. |
|---|---|---|---|---|---|
η: onset overpotential, turnover numbers (TONs) are defined as moles of produced product per mole of catalyst, nO2/ncat., turnover frequencies (TOFs) are defined as moles of produced product per mole of catalyst per s.a Electrolysis at a potential of 1.35 V vs. NHE (pH 12. 5).b Electrolysis at a potential of 1.13 V vs. NHE (pH 12.4).c Electrolysis at a potential of 1.1 V vs. NHE (pH > 10.4).d Photochemical oxidation using [Ru(bpy)3]Cl2 as photosensitizer and Na2S2O8 as the sacrificial electron acceptor.e Electrolysis at a potential of 1.59 V vs. NHE (pH 9.2).f Electrolysis at a potential of 1.3 V vs. NHE (pH 8, 9).g Photochemical oxidation using [Ru(bpy)3](ClO4)2 as photosensitizer and Na2S2O8 as the sacrificial electron acceptor.h Electrolysis at a potential of 1.5 V vs. NHE (pH 10.8).i Electrolysis at a potential of 1.23 V vs. SHE (pH 12.2).j Electrolysis at a potential of 1.60 V vs. NHE (pH 6.5).k Electrolysis at a potential of 1.4 V vs. NHE (pH 11).l Electrolysis at a potential of 1.58 V vs. NHE.m Electrolysis at a potential of 1.4 V vs. NHE (pH 8).n Electrolysis at a potential of 1.30 V vs. NHE (pH 11.5).o Electrolysis at a potential of 1.55 V vs. NHE.p Electrolysis at a potential of 1.64 V vs. NHE.q Electrolysis at a potential of 1.2–2.0 V vs. Ag/Ag+.r Electrolysis at a potential of 1.60 V vs. NHE (pH 7).s Photochemical oxidation using [Ru(bpy)3](NO3)2 as photosensitizer and Na2S2O8 as the sacrificial electron acceptor.t The catalyst was coated onto an ITO electrode and subjected to electrolysis at a potential of 1.60 V vs. NHE (pH 7).u Photochemical oxidation using [Ru(bpy3)](PF6)2 as photosensitizer and Na2S2O8 as the sacrificial electron acceptor.v Photochemical oxidation using [Ru(bpy2)(deeb)](PF6)2 as photosensitizer and Na2S2O8 as the sacrificial electron acceptor.w Electrolysis at a potential of 1.62 V vs. NHE (pH 12.5).x Electrolysis at a potential of 1.87 V vs. NHE (pH 7).y Photochemical oxidation using [Ru(bpy)3]3+ as photosensitizer and Na2S2O8 as the sacrificial electron acceptor.z Electrolysis at a potential of 1.21 V vs. NHE (pH 6).aa Electrolysis at a potential of 1.42 V vs. Fc/Fc+.ab Estimated based on the assumption that the thickness of the reaction–diffusion layer adjacent to the electrode surface (μ) is on the order of 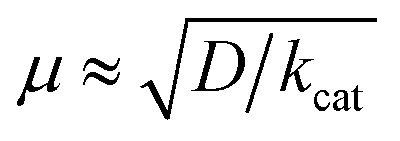 .ac Determined based on the profile of cyclic voltammograms (eqn (4)).ad Based on the results of foot-of-the-wave analysis (FOWA).ae Obtained as a roughly estimated value by considering the thickness of the reaction–diffusion layer during the controlled potential electrolysis. .ac Determined based on the profile of cyclic voltammograms (eqn (4)).ad Based on the results of foot-of-the-wave analysis (FOWA).ae Obtained as a roughly estimated value by considering the thickness of the reaction–diffusion layer during the controlled potential electrolysis. |
|||||
| 1 | Electrochemical | ∼750 | >30a | ∼100ac | 62 |
| 2 | Electrochemical | 510–560 | ∼1b | 0.40ac | 66 and 67 |
| 3 | Electrochemical | 560–580 | >30c | 0.7ac | 68 |
| 4 | [Ru(bpy)3](ClO4)3 | — | 180 | 4.0 | 71 |
| Photochemical | — | 335d | — | ||
| 5 | Electrochemical | 500 | — | 79e | 72 and 73 |
| 6 | Electrochemical | 540, 510f | — | — | 74 |
| Photochemical | — | 51g | 1.3g | ||
| [Ru(bpy)3](ClO4)3 | — | 15 | — | ||
| m-CPBA | — | 5 | — | ||
| 7 | CAN | — | 16 | 0.75 | 75 |
| [Ru(bpy)3](ClO4)3 | — | 7 | 0.9 | ||
| Photochemical | — | 20g | — | ||
| 8 | CAN | — | 5 | 0.53 | 75 |
| [Ru(bpy)3](ClO4)3 | — | 26.5 | 2.2 | ||
| Photochemical | — | 43.5g | 0.6g | ||
| 9 | Electrochemical | 800 | 19h | 145ac | 76 and 77 |
| 10 | CAN | — | 82 | 0.063 | 84 |
| 11 | CAN | — | 360 | 0.23 | 84 |
| 12 | CAN | — | 63 | 0.046 | 84 |
| 13 | CAN | — | 145 | 0.14 | 84 |
| 14 | CAN | — | 40 | 0.015 | 84 |
| 15 | Inactive | — | — | — | 84 |
| 16 | Inactive | — | — | — | 84 |
| 17 | CAN | — | 3800 | 0.41 | 85 |
| 18 | Inactive | — | — | — | 86 |
| 19 | Inactive | — | — | — | 86 |
| 20 | Electrochemical | 800 | 16–24i | — | 86–88 |
| 21 | Electrochemical | 750 | 3.4j | — | 89 |
| 22 | Oxone | — | ∼3–4 | 0.010 | 91 |
| H2O2 | 0.014 | ||||
| 23 | H2O2 | — | ∼3–4 | 0.0034 | 91 |
| 24 | Electrochemical | 520 | 13k | 33ac | 92 |
| 25 | Electrochemical | 640 | >29l | 0.15ac | 94 |
| 26 | Electrochemical | 542 | — | 3 × 10−6m |
96 |
| 27 | Inactive | — | — | — | 97 |
| 28 | CAN | — | 2.5 | — | 97 |
| 29 | CAN | — | 5.8 | — | 97 |
| 30 | CAN | — | 7.5 | — | 97 |
| 31 | CAN | — | 17 | >1.3 | 97 and 98 |
| 32 | CAN | — | 10 | — | 99 |
| Photochemical | — | 220d | 0.76d | ||
| 33 | CAN | — | 17 | — | 99 |
| Photochemical | — | 60d | 0.21d | ||
| 34 | Electrochemical | — | — | 5.68ac | 100 |
| 35 | Electrochemical | 380 | — | 7.53ac | 101 |
| 36 | Electrochemical | — | — | 7.5ac | 101 |
| 37 | Electrochemical | — | — | 8.81ac | 101 |
| 38 | Inactive | — | — | — | 101 |
| 39 | Electrochemical | 700n | — | 3.56ad | 102 |
| 40 | Electrochemical | 400n | — | 3.58ad | 102 |
| 41 | Electrochemical | 270n | — | 0.43ad | 102 |
| 42 | Electrochemical | 170n | — | 0.16ad | 102 |
| 44 | Electrochemical | 170 | 15o | — | 90 and 106–108 |
| 45 | Electrochemical | 610 | 362p | 30ac | 104 |
| 46 | Electrochemical | — | — | 0.0005q | 119 and 120 |
| 47 | Electrochemical | — | — | 0.0007q | 119 and 120 |
| 48 | Electrochemical | — | 9.2q | 0.0018q | 119 and 120 |
| 49 | Electrochemical | — | — | — | 121 |
| 50 | Electrochemical | — | — | — | 121 |
| 51 | TBHP | — | ∼0.02 | — | 122 |
| 52 | Electrochemical | — | — | — | 124 |
| 53 | Electrochemical | — | — | 0.81r | 124 |
| 54 | Photochemical | — | 89s | 0.12s | 127 |
| 55 | Photochemical | — | 10s | 0.14s | 127 |
| 56 | Photochemical | — | 122s | 0.17s | 127 |
| 57 | Electrochemical | — | — | 1400ac | 128 |
| 58 | Electrochemical | — | — | — | 128 |
| 59 | Electrochemical | — | — | — | 128 |
| 60 | Electrochemical | — | — | 0.20t | 129 |
| 61 | Electrochemical | — | 0.67ac | 130 | |
| 62 | Electrochemical | — | 30ac | 131 | |
| 63 | NaClO | — | 4 | 0.0033 | 132 |
| 64 | TBHP | — | 20 | — | 137–139 |
| Oxone | — | 0.0006 | |||
| 65 | [Ru(bpy)3](PF6)3 | — | 25 | 0.027 | 140 |
| Photochemical | — | 1u | — | ||
| Photochemical | — | 4v | — | ||
| 66 | Electrochemical | — | — | — | 141 |
| 67 | Electrochemical | — | — | — | 141 |
| 68 | Photochemical | — | 58g | 1.4g | 142 |
| Electrochemical | 540 | — | — | ||
| 69 | CAN | — | — | 2.2 | 143 |
| 70 | CAN | — | — | 0.23 | 143 |
| 72 | Photochemical | — | 742g | — | 145 |
| [Ru(bpy)3](ClO4)3 | — | 4.3 | — | ||
| 73 | Electrochemical | 1000w | — | 33ac | 146 |
| 74 | Electrochemical | 800x | — | 0.6ac | 147 |
| 75 | Electrochemical | 780x | — | 0.78ac | 148 |
| 76 | Inactive | — | — | — | 151 |
| 77 | Photochemical | 550 | 40y | 0.02y | 165,168 and 169 |
| 78 | Electrochemical | 520ac | — | — | 167 |
| 79 | Electrochemical | 500ac | — | — | 167 |
| 80 | Electrochemical | 570ac | — | — | 167 |
| 81 | Electrochemical | 530ac | — | — | 167 |
| 82 | Electrochemical | 510ac | — | — | 167 |
| 83 | Electrochemical | 510ac | — | — | 167 |
| 84 | Electrochemical | 334z | 629ad | 0.035ad | 171 |
| 85 | Photochemical | — | 163y | 9.6y | 172 |
| 86 | Photochemical | — | — | — | 172 |
| 87 | Photochemical | — | 211y | 8.9y | 172 |
| 88 | Photochemical | — | — | — | 172 |
| 89 | Electrochemical | 520 | >78aa | 1900ac | 189 |
| 1 × 106aa,ab |
140–1400ae | ||||
In this study, the design, activity, and mechanism of molecular catalysts of earth-abundant metal ions were reviewed with a key focus on the structures of the ligands, and the essential factors required for the development of effective catalysts were established. First, the catalysts require robust structures that are resistant to oxidation. As mentioned above, high lability is one of the inherent features of earth-abundant first-row transition metal ions. Therefore, strategies to mitigate the demetallation of the ligand should be applied while constructing the catalysts. In this context, the use of multidentate ligands, ideally tetra- or pentadentate ligands, is critical. Several kinds of multidentate ligands such as the PY5,72–77 alkylamine-pyridine,84–89,91,92,94,96 TAMLs,90,97,99–105 and porphyrins119–122,124,127–131 have been widely employed. Importantly, the ligands themselves need to be oxidation resistant. Therefore, ligands with oxidation-prone moieties, such as alkyl groups, should be avoided. In another approach, the introduction of the appropriate substituents72–77 or deuteration85 of the oxidation-prone ligands may be pursued to mitigate ligand-oxidation.
Second, to enhance the multi-electron transfer reaction for accumulating the charge, the utilization of redox non-innocent ligands is promising. Alternatively, an increase in the number of metal centres by using di- or multinuclear complexes can promote multi-electron transfer. Another important strategy is to adopt the PCET approach. As the PCET reaction releases electrons and protons at the same time, the total charge of the complex upon oxidation remains unchanged. Therefore, the oxidation reaction occurs under milder conditions and facilitates the formation of the highly oxidised species. Furthermore, the stabilisation of the oxidised species is also an important factor. The charge delocalisation by redox non-innocent ligands or multiple metal centres and the suppression of the total charge increases by PCET will be beneficial for this purpose.
Third, high-valent key reaction intermediates should be stabilised to avoid decomposition or undesired side reactions, while retaining substantial reactivity for water oxidation. In this context, the negatively-charged anionic ligands, and ligands that can provide strong ligand field to the resultant complexes are useful. Ligands with PCET abilities are also promising because these ligands become negatively charged via proton dissociation upon oxidation of metal centres and thus function as anionic ligands. To improve the reactivity of high-valent key intermediates, the introduction of electron-withdrawing groups into ligands is another valuable strategy. Electron-withdrawing groups can enhance the reactivity of the metal centres by increasing the positive charge on the metal centres.
Finally, the O–O bond formation process should be accelerated. As mentioned earlier, this process is usually the rate-determining step, and thus determines the reaction rate. This is mainly because the O–O bond formation proceeds in an intermolecular fashion. The use of conditions that favour the intramolecular O–O bond formation process is a potential solution to this problem. Intramolecular O–O formation processes do not require the diffusion of molecules and benefits from enhanced kinetics. To achieve such a process, the use of complexes with two substitution-labile sites which are oriented in a cis configuration is a promising strategy.84 The design and incorporation of neighbouring coordination sites and the use of di- or multinuclear metal complexes is also beneficial.145,189
The compatibility between the ligand structures and metal ions is also vital for catalyst design. Manganese complexes are expected to be good candidates for water oxidation catalysis because the catalytic centre of the natural enzyme, the OEC, contains manganese ions. However, most of the reported manganese-complex-based catalysts require oxygen transfer reagents such as H2O2 and Oxone for O2 evolution, and true H2O oxidation, wherein the H2O contributes both the oxygen atoms for generating O2, is hard to achieve. This is maybe because the formation of reactive high-valent key intermediates is difficult and/or the stability of the intermediates is not sufficient. To overcome the drawback, the use of the complexes with anionic ligands and multinuclear complexes is a valuable approach for obtaining manganese-based catalysts.140 This is because negatively charged ligands can stabilise high-valent key intermediates, and multinuclear structures promote multi-electron transfer reactions and stabilise the formed high-valent species. Iron complexes are potential candidates for water oxidation catalysts because these complexes can access the reactive FeVO intermediate owing to the high redox flexibility of the metal ion, which is similar to that of the ruthenium ion. Moreover, the development of water oxidation catalysts based on iron complexes using various kinds of ligand such as PY5,75 alkylamine-pyridyl ligands,84,85,94 and TAMLs97,99 has been reported. Furthermore, successful water oxidation has been achieved using chemical oxidants, electrochemical conditions, and photochemical conditions. It is noteworthy that a multinuclear metal complex that exhibits a remarkable reaction rate and stability was developed using iron ions.189 In the catalysis mediated by iron-based complexes, the existence of substitution labile sites in cis configuration84 or the presence of neighbouring water activation sites189 can accelerate the O–O bond formation process in the intramolecular reaction pathway. The complexes containing cobalt anions can catalyse photochemical and electrochemical water oxidation reactions. In the case of cobalt complexes, the reactivity of the high-valent intermediates is low. Therefore, molecular design to improve the reactivity of these intermediates is essential. Notably, the introduction of strong electron-withdrawing groups into the ligands has enhanced the catalytic activity of the cobalt-based catalysts.123,124,128 Also, the employment of multinuclear structure is a niche approach for forming the reactive intermediate by accumulating multiple charges among the metal ions.165,167–169 Complexes containing the late 3d transition metal ions, such as Ni and Cu, can also catalyse electrochemical water oxidation. Accessing the high-valent state may be difficult with these classes of compounds. Therefore, the active species for water oxidation is mainly the MIII–O· intermediate of these complexes.66–68,92,102,104,108,130,146–148 To accumulate enough charge to drive the reaction, the utilization of redox-active ligands should be advantageous for such scenarios.66–68,97,99,102,128–131 Dinuclear complexes are another set of attractive targets for storing multiple oxidation equivalents for enabling water oxidation.146–148
The development of molecular catalysts containing earth-abundant first-row transition metal ions is cutting-edge research in the field of water oxidation. While the number of catalysts that exhibit high stability and reaction rate comparable to that of ruthenium-based catalysts is still limited, the recent progress unveils their potential for the development of the next-generation water oxidation catalysts. In summary, the strategies for designing active water oxidation catalysts are becoming more evident. Further studies and efforts in this area are expected to induce a paradigm shift in this research toward the ideal water oxidation catalyst for realizing sustainable development.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by KAKENHI (17H06444 and 19H00903 (S. M.); and 17K19185, 17H05391, 19H04602, and 20H02754 (M. K.)) from the Japan Society for the Promotion of Science. This work was also supported by JST ACT-C, Grant Number JPMJCR12YH, Japan, JST PRESTO (Grant Number JPMJPR20A4 (M. K.)), and JST CREST (Grant Number JPMJCR20B6 (S. M.)), Japan. Research Grants in the Natural Sciences from the Mitsubishi Foundation and the Tokuyama Science Foundation.Notes and references
- T. J. Meyer, Acc. Chem. Res., 1989, 22, 163–170 CrossRef CAS.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- D. G. Nocera, Inorg. Chem., 2009, 48, 10001–10017 CrossRef CAS PubMed.
- D. Gust, T. Moore and A. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed.
- A. Magnuson, M. Anderlund, O. Johansson, P. Lindblad, R. Lomoth, T. Polivka, S. Ott, K. Stensjö, S. Styring, V. Sundström and L. Hammarström, Acc. Chem. Res., 2009, 42, 1899–1909 CrossRef CAS PubMed.
- J. J. Concepcion, R. L. House, J. M. Papanikolas and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15560–15564 CrossRef CAS PubMed.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116, 14120–14136 CrossRef CAS PubMed.
- S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029–4030 CrossRef CAS.
- J. J. Concepcion, J. W. Jurss, M. K. Brennaman, P. G. Hoertz, A. O. T. Patrocinio, N. Y. M. Iha, J. L. Templeton and T. J. Meyer, Acc. Chem. Res., 2009, 42, 1954–1965 CrossRef CAS PubMed.
- M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed.
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS PubMed.
- L. Duan, L. Wang, F. Li, F. Li and L. Sun, Acc. Chem. Res., 2015, 48, 2084–2096 CrossRef CAS PubMed.
- M. Okamura and S. Masaoka, Chem. – Asian J., 2015, 10, 306–315 CrossRef CAS PubMed.
- H. Tseng, R. Zong, J. T. Muckerman and R. Thummel, Inorg. Chem., 2008, 47, 11763–11773 CrossRef CAS PubMed.
- J. J. Concepcion, J. W. Jurss, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2008, 130, 16462–16463 CrossRef CAS PubMed.
- S. Masaoka and K. Sakai, Chem. Lett., 2009, 38, 182–183 CrossRef CAS.
- M. Yoshida, S. Masaoka and K. Sakai, Chem. Lett., 2009, 38, 702–703 CrossRef CAS.
- M. Yoshida, S. Masaoka, J. Abe and K. Sakai, Chem. – Asian J., 2010, 5, 2369–2378 CrossRef CAS.
- H. Yamazaki, T. Hakamata, M. Komi and M. Yagi, J. Am. Chem. Soc., 2011, 133, 8846–8849 CrossRef CAS PubMed.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- A. Kimoto, K. Yamauchi, M. Yoshida, S. Masaoka and K. Sakai, Chem. Commun., 2012, 48, 239–241 RSC.
- M. Okamura, M. Yoshida, R. Kuga, K. Sakai, M. Kondo and S. Masaoka, Dalton Trans., 2012, 41, 13081–13089 RSC.
- R. Matheu, M. Z. Ertem, J. Benet-Buchholz, E. Coronado, V. S. Batista, X. Sala and A. Llobet, J. Am. Chem. Soc., 2015, 137, 10786–10795 CrossRef CAS PubMed.
- M. Yoshida, M. Kondo, S. Torii, K. Sakai and S. Masaoka, Angew. Chem., Int. Ed., 2015, 54, 7981–7984 CrossRef CAS PubMed.
- Y. Xie, D. W. Shaffer, A. Lewandowska-Andralojc, D. J. Szalda and J. J. Concepcion, Angew. Chem., Int. Ed., 2016, 55, 8067–8071 CrossRef CAS.
- M. Yoshida, M. Kondo, M. Okamura, M. Kanaike, S. Haesuwannakij, H. Sakurai and S. Masaoka, Faraday Discuss., 2017, 198, 181–196 RSC.
- R. Matheu, M. Z. Ertem, C. Gimbert-Suriñach, J. Benet-Buchholz, X. Sala and A. Llobet, ACS Catal., 2017, 7, 6525–6532 CrossRef CAS.
- R. Matheu, M. Z. Ertem, M. Pipelier, J. Lebreton, D. Dubreuil, J. Benet-Buchholz, X. Sala, A. Tessier and A. Llobet, ACS Catal., 2018, 8, 2039–2048 CrossRef CAS.
- Q. Daniel, L. Duan, B. J. J. Timmer, H. Chen, X. Luo, R. Ambre, Y. Wang, B. Zhang, P. Zhang, L. Wang, F. Li, J. Sun, M. Ahlquist and L. Sun, ACS Catal., 2018, 8, 4375–4382 CrossRef CAS.
- M. Kondo and S. Masaoka, Chem. Lett., 2016, 45, 1220–1231 CrossRef CAS.
- P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012–6021 RSC.
- A. Singh and L. Spiccia, Coord. Chem. Rev., 2013, 257, 2607–2622 CrossRef CAS.
- A. R. Parent and K. Sakai, ChemSusChem, 2014, 7, 2070–2080 CrossRef CAS PubMed.
- M. D. Kärkäs and B. Åkermark, Dalton Trans., 2016, 45, 14421–14461 RSC.
- N. Wang, H. Zheng, W. Zhang and R. Cao, Chin. J. Catal., 2018, 39, 228–244 CrossRef CAS.
- D. Lukács, Ł. Szyrwiel and J. S. Pap, Catalysts, 2019, 9, 83–128 CrossRef.
- T. Liu, B. Zhang and L. Sun, Chem. – Asian J., 2019, 14, 31–43 CrossRef CAS PubMed.
- Non-Noble Metal Catalysis: Molecular Approaches and Reactions, ed. R. J. Klein Gebbink and M. C. Moret, Wiley-VCH, Weinheim, 2019 Search PubMed.
- H. Lee, X. Wu and L. Sun, Nanoscale, 2020, 12, 4187–4218 RSC.
- G. C. Dismukes, R. Brimblecombe, G. A. N. Felton, R. S. Pryadun, J. E. Sheats, L. Spiccia and G. F. Swiegers, Acc. Chem. Res., 2009, 42, 1935–1943 CrossRef CAS PubMed.
- H. Dau and I. Zaharieva, Acc. Chem. Res., 2009, 42, 1861–1870 CrossRef CAS PubMed.
- J. S. Kanady, E. Y. Tsui, M. W. Day and T. Agapie, Science, 2011, 333, 733–736 CrossRef CAS PubMed.
- E. Y. Tsui, R. Tran, J. Yano and T. Agapie, Nat. Chem., 2013, 5, 293–299 CrossRef CAS PubMed.
- C. Zhang, C. Chen, H. Dong, J. R. Shen, H. Dau and J. Zhao, Science, 2015, 348, 690–693 CrossRef CAS PubMed.
- B. Gerey, E. Couré, J. Fortage, J. Pécaut and M.-N. Collomb, Coord. Chem. Rev., 2016, 319, 1–24 CrossRef CAS.
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago and J. R. Shen, Nature, 2015, 517, 99–103 CrossRef CAS PubMed.
- E. I. Solomon, T. C. Brunold, M. I. Davis, J. N. Kemsley, S.-K. Lee, N. Lehnert, F. Neese, A. J. Skulan, Y. S. Yang and J. Zhou, Chem. Rev., 2000, 100, 235–349 CrossRef CAS PubMed.
- J. P. Collman, R. Boulatov, C. J. Sunderland and L. Fu, Chem. Rev., 2004, 104, 561–588 CrossRef CAS PubMed.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que Jr, Chem. Rev., 2004, 104, 939 CrossRef CAS PubMed; M. M. Abu-Omar, A. Loaiza and N. Hontzeas, Chem. Rev., 2005, 105, 2227–2252 CrossRef PubMed.
- I. G. Denisov, T. M. Makris, S. G. Sligar and I. Schlichting, Chem. Rev., 2005, 105, 2253–2278 CrossRef CAS PubMed.
- W. Nam, Acc. Chem. Res., 2007, 40(7), 465 CrossRef CAS.
- C. E. Tinberg and S. J. Lippard, Acc. Chem. Res., 2011, 44, 280–288 CrossRef CAS PubMed.
- W. Nam, Y.-M. Lee and S. Fukuzumi, Acc. Chem. Res., 2014, 47(4), 1146–1154 CrossRef CAS PubMed.
- W. Nam, Acc. Chem. Res., 2015, 48(8), 2415–2423 CrossRef CAS PubMed.
- L. M. Mirica, X. Ottenwaelder and T. D. P. Stack, Chem. Rev., 2004, 104, 1013–1046 CrossRef CAS PubMed.
- E. A. Lewis and W. B. Tolman, Chem. Rev., 2004, 104, 1047–1076 CrossRef CAS PubMed.
- S. M. Barnett, K. I. Goldberg and J. M. Mayer, Nat. Chem., 2012, 4, 498–502 CrossRef CAS PubMed.
- S. Fukuzumi and D. Hong, Eur. J. Inorg. Chem., 2014, 645–659 CrossRef CAS.
- M. M. Najafpour and S. I. Allakhverdiev, J. Photochem. Photobiol., B, 2015, 152, 127–132 CrossRef CAS.
- M. M. Najafpour, G. Renger, M. Hołyńska, A. N. Moghaddam, E.-M. Aro, R. Carpentier, H. Nishihara, J. J. Eaton-Rye, J.-R. Shen and S. I. Allakhverdiev, Chem. Rev., 2016, 116, 2886–2936 CrossRef CAS.
- T. Zhang, C. Wang, S. Liu, J. L. Wang and W. Lin, J. Am. Chem. Soc., 2014, 136, 273–281 CrossRef CAS PubMed.
- D. L. Gerlach, S. Bhagan, A. A. Cruce, D. B. Burks, I. Nieto, H. T. Truong, S. P. Kelley, C. J. Herbst-Gervasoni, K. L. Jernigan, M. K. Bowman, S. Pan, M. Zeller and E. T. Papish, Inorg. Chem., 2014, 53, 12689–12698 CrossRef CAS PubMed.
- K. J. Fisher, K. L. Materna, B. Q. Mercado, R. H. Crabtree and G. W. Brudvig, ACS Catal., 2017, 7, 3384–3387 CrossRef CAS.
- J. M. Thomsen, D. L. Huang, R. H. Crabtree and G. W. Brudvig, Dalton Trans., 2015, 44, 12452–12472 RSC.
- H. A. Younus, Y. Zhang, M. Vandichel, N. Ahmad, K. Laasonen, F. Verpoort, C. Zhang and S. Zhang, ChemSusChem, 2020, 13, 5088–5099 CrossRef CAS PubMed.
- C.-F. Leung, S.-M. Ng, C.-C. Ko, W.-L. Man, J. Wu, L. Chen and T.-C. Lau, Energy Environ. Sci., 2012, 5, 7903–7907 RSC.
- D. J. Wasylenko, R. D. Palmer, E. Schott and C. P. Berlinguette, Chem. Commun., 2012, 48, 2107–2109 RSC.
- D. W. Crandell, S. Ghosh, C. P. Berlinguette and M.-H. Baik, ChemSusChem, 2015, 8, 844–852 CrossRef CAS PubMed.
- B. Das, A. Orthaber, S. Ott and A. Thapper, Chem. Commun., 2015, 51, 13074–13077 RSC.
- B. Das, A. Orthaber, S. Ott and A. Thapper, ChemSusChem, 2016, 9, 1178–1186 CrossRef CAS PubMed.
- L. Wang, L. Duan, R. B. Ambre, Q. Daniel, H. Chen, J. Sun, B. Das, A. Thapper, J. Uhlig, P. Diner and L. Sun, J. Catal., 2016, 335, 72–78 CrossRef CAS.
- H. Pan, L. Duan and R.-Z. Liao, ChemCatChem, 2020, 12, 219–226 CrossRef CAS.
- Note that the Ni(IV)O species can be unstable in some cases. For details, see also ref. 79.
- In general, the interaction of an oxygen atom with a metal center lower the t2g orbital symmetry to b2(dxy) and e(dxz,dyz) and the eg orbital symmetry to b1(dx2−y2) and a1(dz2) levels. The stability (and reactivity) of the oxo is largely determined by the d electron count. For late transition metals of reasonable oxidation states (less than M4+) such as Co, Cu, and Ni, the e(dxz,dyz) orbital sets are filled. The oxo thus cannot donate its pπ electron pairs to the metal and a multiple metal–oxo bond cannot be supported. For details, see also, T. A. Betley, Q. Wu, T. V. Voorhis and D. G. Nocera, Inorg. Chem., 2008, 47, 1849–1861 CrossRef CAS.
- G. Chen, L. Chen, S. M. Ng, W. L. Man and T. C. Lau, Angew. Chem., Int. Ed., 2013, 52, 1789–1791 CrossRef CAS.
- A. Company, L. Gómez, M. Güell, X. Ribas, J. M. Luis, L. Que Jr and M. Costas, J. Am. Chem. Soc., 2007, 129, 15766–15767 CrossRef CAS PubMed.
- A. Company, I. Prat, J. R. Frisch, D. R. Mas-Ballesté, M. Güell, G. Juhasz, X. Ribas, D. E. Münck, J. M. Luis, L. Que Jr and M. Costas, Chem. – Eur. J., 2011, 17, 1622–1634 CrossRef CAS PubMed.
- I. Garcia-Bosch, A. Company, C. W. Cady, S. Styring, W. R. Browne, X. Ribas and M. Costas, Angew. Chem., Int. Ed., 2011, 50, 5648–5653 CrossRef CAS PubMed.
- J. L. Fillol, Z. Codola, I. Garcia-Bosch, L. Gomez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3, 807–813 CrossRef CAS.
- Z. Codolá, I. Gamba, F. Acuña-Pares, C. Casadevall, M. Clemancey, J.-M. Latour, J. M. Luis, J. Lloret-Fillol and M. Costas, J. Am. Chem. Soc., 2019, 141, 323–333 CrossRef PubMed.
- W.-T. Lee, S. B. MuÇoz, D. A. Dickie and J. M. Smith, Angew. Chem., Int. Ed., 2014, 53, 9856–9859 CrossRef CAS.
- D. W. Crandell, S. Xu, J. M. Smith and M. H. Baik, Inorg. Chem., 2017, 56, 4435–4445 CrossRef CAS PubMed.
- Y.-Y. Li, K. Y. Per, E. M. Siegbahn and R.-Z. Liao, ChemSusChem, 2017, 10, 903–911 CrossRef CAS PubMed.
- G. Y. Luo, H. H. Huang, J. W. Wang and T. B. Lu, ChemSusChem, 2016, 9, 485–491 CrossRef CAS PubMed.
- M. Zhang, M. T. Zhang, C. Hou, Z. F. Ke and T. B. Lu, Angew. Chem., Int. Ed., 2014, 53, 13042–13048 CrossRef CAS PubMed.
- K. J. Young, M. K. Takase and G. W. Brudvig, Inorg. Chem., 2013, 52, 7615–7622 CrossRef CAS PubMed.
- M. T. Zhang, Z. Chen, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 2048–2051 CrossRef CAS PubMed.
- The stability of CuIVO can be much lower compared to RuV–oxo intermediate. For details, see also ref. 79.
- M. K. Coggins, M.-T. Zhang, A. K. Vannucci, C. J. Dares and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 5531–5534 CrossRef CAS PubMed.
- D. Hong, S. Mandal, Y. Yamada, Y.-M. Lee, W. Nam, A. Llobet and S. Fukuzum, Inorg. Chem., 2013, 52, 9522–9531 CrossRef CAS PubMed.
- M. K. Coggins, M.-T. Zhang, Z. Chen, N. Song and T. J. Meyer, Angew. Chem., Int. Ed., 2014, 53, 12226–12230 CrossRef CAS PubMed.
- W. C. Ellis, N. D. McDaniel, S. Bernhard and T. J. Collins, J. Am. Chem. Soc., 2010, 132, 10990–10991 CrossRef CAS.
- R.-Z. Liao, X.-C. Li and P. E. M. Siegbahn, Eur. J. Inorg. Chem., 2014, 728–741 CrossRef CAS.
- C. Panda, J. Debgupta, D. D. Díaz, K. K. Singh, S. S. Gupta and B. B. Dhar, J. Am. Chem. Soc., 2014, 136, 12273–12282 CrossRef CAS PubMed.
- D. Das, S. Pattanayak, K. K. Singh, B. Garaib and S. S. Gupta, Chem. Commun., 2016, 52, 11787–11790 RSC.
- H. Y. Du, S. C. Chen, X. J. Su, L. Jiao and M. T. Zhang, J. Am. Chem. Soc., 2018, 140, 1557–1565 CrossRef CAS PubMed.
- P. Garrido-Barros, I. Funes-Ardoiz, S. Drouet, J. Benet-Buchholz, F. Maseras and A. Llobet, J. Am. Chem. Soc., 2015, 137, 6758–6761 CrossRef CAS PubMed.
- P. Garrido-Barros, D. Moonshiram, M. Gil-Sepulcre, P. Pelosin, X. Gimbert-Suriñach, J. Benet-Buchholz and A. Llobet, J. Am. Chem. Soc., 2020, 142, 17434–17446 CrossRef CAS PubMed.
- F. Yu, F. Li, J. Hu, L. Bai, Y. Zhua and L. Sun, Chem. Commun., 2016, 52, 10377–10380 RSC.
- B. J. Fisher and R. Eisenberg, J. Am. Chem. Soc., 1980, 102, 7361–7363 CrossRef CAS.
- E. Zeigerson, G. Ginzburg, N. Schwartz, Z. Luz and D. Meyerstein, J. Chem. Soc., Chem. Commun., 1979, 241–243 RSC.
- J. Schneider, H. Jia, K. Kobiro, D. E. Cabelli, J. T. Muckerman and E. Fujita, Energy Environ. Sci., 2012, 5, 9502–9510 RSC.
- E. Colacio, I. B. Maimoun, R. Kivekäs, R. Sillanpää and J. Suárez-Varela, Inorg. Chim. Acta, 2004, 357, 1465–1470 CrossRef CAS.
- D. Kuciauskas, P. A. Liddell, S. Lin, T. E. Johnson, S. J. Weghorn, J. S. Lindsey, A. L. Moore, T. A. Moore and D. Gust, J. Am. Chem. Soc., 1999, 121, 8604–8614 CrossRef CAS.
- T. Dhanasekaran, J. Grodkowski, P. Neta, P. Hambright and E. Fujita, J. Phys. Chem. A, 1999, 103, 7742–7748 CrossRef CAS.
- G. Kodis, P. A. Liddell, L. de la Garza, P. C. Clausen, J. S. Lindsey, A. L. Moore, T. A. Moore and D. Gust, J. Phys. Chem. A, 2002, 106, 2036–2048 CrossRef CAS.
- D. Kim and A. Osuka, J. Phys. Chem. A, 2003, 107, 8791–8816 CrossRef CAS.
- H. Imahori, Y. Sekiguchi, Y. Kashiwagi, T. Sato, Y. Araki, O. Ito, H. Yamada and S. Fukuzumi, Chem. – Eur. J., 2004, 10, 3184–3196 CrossRef CAS PubMed.
- Y. Nakamura, N. Aratani and A. Osuka, Chem. Soc. Rev., 2007, 36, 831–845 RSC.
- H. Imahori, T. Umeyama and S. Ito, Acc. Chem. Res., 2009, 42, 1809–1818 CrossRef CAS PubMed.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS.
- J. P. Collman, X. Zhang, V. J. Lee, E. S. Uffelman and J. I. Brauman, Science, 1993, 261, 1404–1411 CrossRef CAS PubMed.
- C.-M. Che, V. K.-Y. Lo, C.-Y. Zhou and J.-S. Huang, Chem. Soc. Rev., 2011, 40, 1950–1975 RSC.
- Y. Naruta, M. Sasayama and T. Sasaki, Angew. Chem., Int. Ed. Engl., 1994, 33, 1839–1841 CrossRef.
- Y. Shimazaki, T. Nagano, H. Takesue, B. H. Ye, F. Tani and Y. Naruta, Angew. Chem., Int. Ed., 2004, 43, 98–100 CrossRef PubMed.
- Y. Gao, J. Liu, M. Wang, Y. Na, B. Åkermarkb and L. Sun, Tetrahedron, 2007, 63, 1987–1994 CrossRef CAS.
- Y. Gao, T. Åkermark, J. Liu, L. Sun and B. Åkermark, J. Am. Chem. Soc., 2009, 131, 8726–8727 CrossRef CAS PubMed.
- R. McGuire Jr, D. K. Dogutan, T. S. Teets, J. Suntivich, Y. Shao-Horn and D. G. Nocera, Chem. Sci., 2010, 1, 411–414 RSC.
- D. K. Dogutan, R. McGuire and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 9178–9180 CrossRef CAS PubMed.
- M. Z. Ertem and C. J. Cramer, Dalton Trans., 2012, 41, 12213–12219 RSC.
- W. Lai, R. Cao, G. Dong, S. Shaik, J. Yao and H. Chen, J. Phys. Chem. Lett., 2012, 3, 2315–2319 CrossRef CAS PubMed.
- T. Nakazono, A. R. Parentab and K. Sakai, Chem. Commun., 2013, 49, 6325–6327 RSC.
- D. Wang and J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15579–15584 CrossRef CAS.
- H. Lei, A. Han, F. Li, M. Zhang, Y. Han, P. Du, W. Lai and R. Cao, Phys. Chem. Chem. Phys., 2014, 16, 1883–1893 RSC.
- Y. Han, Y. Wu, W. Lai and R. Cao, Inorg. Chem., 2015, 54, 5604–5613 CrossRef CAS PubMed.
- Y. Liu, Y. Han, Z. Zhang, W. Zhang, W. Lai, Y. Wang and R. Cao, Chem. Sci., 2019, 10, 2613–2622 RSC.
- J. Limburg, J. S. Vrettos, L. M. Liable-Sands, A. L. Rheingold, R. H. Crabtree and G. W. Brudvig, Science, 1999, 283, 1524–1527 CrossRef CAS PubMed.
- R. Tagore, H. Chen, H. Zhang, R. H. Crabtree and G. W. Brudvig, Inorg. Chim. Acta, 2007, 360, 2983–2989 CrossRef CAS PubMed.
- C. Baffert, S. Romain, A. Richardot, J.-C. Leprêtre, B. Lefebvre, A. Deronzier and M.-N. Collomb, J. Am. Chem. Soc., 2005, 127, 13694–13704 CrossRef CAS PubMed.
- M. M. Najafpour, A. N. Moghaddam, H. Dau and I. Zaharieva, J. Am. Chem. Soc., 2014, 136, 7245–7248 CrossRef CAS PubMed.
- M. M. Najafpour and A. N. Moghaddam, Dalton Trans., 2012, 41, 10292–10297 RSC.
- C. Baffert, M. N. Collomb, A. Deronzier, S. K. Knudsen, J. M. Latour, K. H. Lund, C. J. McKenzie, M. Mortensen, L. P. Nielsenb and N. Thorup, Dalton Trans., 2003, 1765–1772 RSC.
- A. K. Poulsen, A. Rompel and C. J. McKenzie, Angew. Chem., Int. Ed., 2005, 44, 6916–6920 CrossRef CAS PubMed.
- W. M. C. Sameera, C. J. McKenzie and J. E. McGrady, Dalton Trans., 2011, 40, 3859–3870 RSC.
- E. A. Karlsson, B.-L. Lee, T. Åkermark, E. V. Johnston, M. D. Kärkäs, J. Sun, Ö. Hansson, J. E. Bäckvall and B. Åkermark, Angew. Chem., Int. Ed., 2011, 50, 11715–11718 CrossRef CAS PubMed.
- M. L. Rigsby, S. Mandal, W. Nam, L. C. Spencer, A. Llobet and S. S. Stahl, Chem. Sci., 2012, 3, 3058–3062 RSC.
- H.-Y. Wang, E. Mijangos, S. Ott and A. Thapper, Angew. Chem., Int. Ed., 2014, 53, 14499–14502 CrossRef CAS PubMed.
- L. D. Wickramasinghe, R. Zhou, R. Zong, P. Vo, K. J. Gagnon and R. P. Thummel, J. Am. Chem. Soc., 2015, 137, 13260–13263 CrossRef CAS PubMed.
- S. Karim, A. Chakraborty, D. Samanta, E. Zangrando, T. Ghosh and D. Das, Catal. Sci. Technol., 2020, 10, 2830–2837 RSC.
- T. Ishizuka, A. Watanabe, H. Kotani, D. Hong, K. Satonaka, T. Wada, Y. Shiota, K. Yoshizawa, K. Ohara, K. Yamaguchi, S. Kato, S. Fukuzumi and T. Kojima, Inorg. Chem., 2016, 55, 1154–1164 CrossRef CAS PubMed.
- S. J. Koepke, K. M. Light, P. E. VanNatta, K. M. Wiley and M. T. Kieber-Emmons, J. Am. Chem. Soc., 2017, 139, 8586–8600 CrossRef CAS PubMed.
- X.-J. Su, M. Gao, L. Jiao, R.-Z. Liao, P. E. M. Siegbahn, J.-P. Cheng and M.-T. Zhang, Angew. Chem., Int. Ed., 2015, 54, 4909–4914 CrossRef CAS PubMed.
- Q.-Q. Hu, X.-J. Su and M.-T. Zhang, Inorg. Chem., 2018, 57, 10481–10484 CrossRef CAS.
- M. Suga, F. Akita, M. Sugahara, M. Kubo, Y. Nakajima, T. Nakane, K. Yamashita, Y. Umena, M. Nakabayashi, T. Yamane, T. Nakano, M. Suzuki, T. Masuda, S. Inoue, T. Kimura, T. Nomura, S. Yonekura, L.-J. Yu, T. Sakamoto, T. Motomura, J.-H. Chen, Y. Kato, T. Noguchi, K. Tono, Y. Joti, T. Kameshima, T. Hatsui, E. Nango, R. Tanaka, H. Naitow, Y. Matsuura, A. Yamashita, M. Yamamoto, O. Nureki, M. Yabashi, T. Ishikawa, S. Iwata and J.-R. Shen, Nature, 2017, 543, 131–135 CrossRef CAS PubMed.
- M. Suga, F. Akita, K. Yamashita, Y. Nakajima, G. Ueno, H. Li, T. Yamane, K. Hirata, Y. Umena, S. Yonekura, L.-J. Yu, H. Murakami, T. Nomura, T. Kimura, M. Kubo, S. Baba, T. Kumasaka, K. Tono, M. Yabashi, H. Isobe, K. Yamaguchi, M. Yamamoto, H. Ago and J.-R. Shen, Science, 2019, 366, 334–338 CrossRef CAS PubMed.
- W. F. Ruettinger, C. Campana and G. C. Dismukes, J. Am. Chem. Soc., 1997, 119, 6670–6671 CrossRef CAS.
- J.-Z. Wu, F. De Angelis, T. G. Carrell, G. P. A. Yap, J. Sheats, R. Car and G. C. Dismukes, Inorg. Chem., 2006, 45, 189–195 CrossRef CAS PubMed.
- C. W. Cady, R. H. Crabtree and G. W. Brudvig, Coord. Chem. Rev., 2008, 252, 444–455 CrossRef CAS.
- R. Brimblecombe, G. F. Swiegers, G. C. Dismukes and L. Spiccia, Angew. Chem., Int. Ed., 2008, 47, 7335–7338 CrossRef CAS PubMed.
- A. E. Kuznetsov, Y. V. Geletii, C. L. Hill and D. G. Musaev, J. Phys. Chem. A, 2010, 114, 11417–11424 CrossRef CAS PubMed.
- R. Brimblecombe, A. Koo, G. C. Dismukes, G. F. Swiegers and L. Spiccia, J. Am. Chem. Soc., 2010, 132, 2892–2894 CrossRef CAS.
- R. Brimblecombe, A. Koo, G. C. Dismukes, G. F. Swiegers and L. Spiccia, ChemSusChem, 2010, 3, 1146–1150 CrossRef CAS PubMed.
- R. Brimblecombe, D. R. J. Kolling, A. M. Bond, G. C. Dismukes, G. F. Swiegers and L. Spiccia, Inorg. Chem., 2009, 48, 7269–7279 CrossRef CAS.
- R. Brimblecombe, A. M. Bond, G. C. Dismukes, G. F. Swiegers and L. Spiccia, Phys. Chem. Chem. Phys., 2009, 11, 6441–6449 RSC.
- R. K. Hocking, R. Brimblecombe, L.-Y. Chang, A. Singh, M. H. Cheah, C. Glover, W. H. Casey and L. Spiccia, Nat. Chem., 2011, 3, 461–466 CrossRef CAS.
- K. Dimitrou, K. Folting, W. E. Streib and G. Christou, J. Am. Chem. Soc., 1993, 115, 6432–6433 CrossRef CAS.
- J. K. Beattie, T. W. Hambley, J. A. Klepetko, A. F. Masters and P. Turner, Polyhedron, 1998, 17, 1343–1354 CrossRef CAS.
- K. Dimitrou, A. D. Brown, T. E. Concolino, A. L. Rheingold and G. Christou, Chem. Commun., 2001, 1284–1285 RSC.
- R. Chakrabarty, S. J. Bora and B. K. Das, Inorg. Chem., 2007, 46, 9450–9462 CrossRef CAS.
- N. S. McCool, D. M. Robinson, J. E. Sheats and G. C. Dismukes, J. Am. Chem. Soc., 2011, 133, 11446–11449 CrossRef CAS PubMed.
- G. La Ganga, F. Puntoriero, S. Campagna, I. Bazzan, S. Berardi, M. Bonchio, A. Sartorel, M. Natali and F. Scandola, Faraday Discuss., 2012, 155, 177–190 RSC.
- S. Berardi, G. La Ganga, M. Natali, I. Bazzan, F. Puntoriero, A. Sartorel, F. Scandola, S. Campagna and M. Bonchio, J. Am. Chem. Soc., 2012, 134, 11104–11107 CrossRef CAS.
- A. I. Nguyen, M. S. Ziegler, P. Oña-Burgos, M. SturzbecherHohne, W. Kim, D. E. Bellone and T. D. Tilley, J. Am. Chem. Soc., 2015, 137, 12865–12872 CrossRef CAS.
- X. Li and P. E. M. Siegbahn, J. Am. Chem. Soc., 2013, 135, 13804–13813 CrossRef CAS PubMed; A. Fernando and C. M. Aikens, J. Phys. Chem. C, 2015, 119, 11072–11085 CrossRef.
- A. I. Nguyen, M. S. Ziegler, P. O. Burgos, M. S. Hohne, W. Kim, D. E. Bellone and T. D. Tilley, J. Am. Chem. Soc., 2015, 137, 12865–12872 CrossRef CAS PubMed.
- G. Maayan, N. Gluz and G. Christou, Nat. Catal., 2018, 1, 48–54 CrossRef CAS.
- F. Evangelisti, R. Moré, F. Hodel, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2015, 137, 11076–11084 CrossRef CAS PubMed.
- H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaevb and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572–7589 RSC.
- J. M. Sumliner, H. Lv, J. Fielden, Y. V. Geletii and C. L. Hill, Eur. J. Inorg. Chem., 2014, 635–644 CrossRef CAS.
- S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS.
- R. Al-Oweini, A. Sartorel, B. S. Bassil, M. Natali, S. Berardi, F. Scandola, U. Kortz and M. Bonchio, Angew. Chem., Int. Ed., 2014, 53, 11182–11185 CrossRef CAS PubMed.
- B. Schwarz, J. Forster, M. K. Goetz, D. Yücel, C. Berger, T. Jacob and C. Streb, Angew. Chem., Int. Ed., 2016, 55, 6329–6333 CrossRef CAS.
- L. Yu, Y. Ding and M. Zheng, Appl. Catal., B, 2017, 209, 45–52 CrossRef CAS.
- J.-W. Zhao, J. Zhang, S.-T. Zheng and G.-Y. Yang, Inorg. Chem., 2007, 46, 10944–10946 CrossRef CAS PubMed.
- X. Du, Y. Ding, F. Song, B. Ma, J. Zhao and J. Song, Chem. Commun., 2015, 51, 13925–13928 RSC.
- For representative studies, see the following references Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS PubMed; Z. Huang, Z. Luo, Y. V. Geletii, J. W. Vickers, Q. Yin, D. Wu, Y. Hou, Y. Ding, J. Song, D. G. Musaev, C. L. Hill and T. Lian, J. Am. Chem. Soc., 2011, 133, 2068–2071 CrossRef; H. Lv, J. Song, Y. V. Geletii, J. W. Vickers, J. M. Sumliner, D. G. Musaev, P. Kögerler, P. F. Zhuk, J. Bacsa, G. Zhu and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 9268–9271 CrossRef PubMed.
- G. Zhu, E. N. Glass, C. Zhao, H. Lv, J. W. Vickers, Y. V. Geletii, D. G. Musaev, J. Song and C. L. Hill, Dalton Trans., 2012, 41, 13043–13049 RSC.
- X.-B. Han, Y.-G. Li, Z.-M. Zhang, H.-Q. Tan, Y. Lu and E.-B. Wang, J. Am. Chem. Soc., 2015, 137, 5486–5493 CrossRef CAS.
- L. Yu, Y. Ding, M. Zheng, H. Chen and J. Zhao, Chem. Commun., 2016, 52, 14494–14497 RSC.
- L. Yu, X. Du, Y. Ding, H. Chen and P. Zhou, Chem. Commun., 2015, 51, 17443–17446 RSC.
- Z.-J. Liu, X.-L. Wang, C. Qin, Z.-M. Zhang, Y.-G. Li, W.-L. Chen and E.-B. Wang, Coord. Chem. Rev., 2016, 33, 94–110 CrossRef.
- Q. Han and Y. Ding, Dalton Trans., 2018, 47, 8180–8188 RSC.
- D. Gao, I. Trentin, L. Schwiedrzik, L. Gonźalez and C. Streb, Molecules, 2020, 25, 157–176 CrossRef CAS.
- M. Okamura, M. Kondo, R. Kuga, Y. Kurashige, T. Yanai, S. Hayami, V. K. K. Praneeth, M. Yoshida, K. Yoneda, S. Kawata and S. Masaoka, Nature, 2016, 530, 465–468 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, M. Robert and J. M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- P. Pelosin, M. Gil-Sepulcre, P. Garrido-Barros, D. Moonshiram, J. Benet-Buchholz, C. Gimbert-Suriñach and A. Llobet, iScience, 2020, 23(8), 101378 CrossRef CAS PubMed.
- S. Mehrabani, R. Bikas, Z. Zand, Y. Mousazade, S. I. Allakhverdiev and M. M. Najafpour, Int. J. Hydrogen Energy, 2020, 45(35), 17434–17443 CrossRef CAS.
- See the ESI of ref. 189 for details.
- V. K. K. Praneeth, M. Kondo, M. Okamura, T. Akai, H. Izu and S. Masaoka, Chem. Sci., 2019, 10, 4628–4639 RSC.
- M. Kondo and S. Masaoka, Acc. Chem. Res., 2020, 53, 2140–2151 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |