
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRoadmap towards solar fuel synthesis at the water interface of liposome membranes†
Andrea
Pannwitz‡
 *ab,
David M.
Klein‡
a,
Santiago
Rodríguez-Jiménez
c,
Carla
Casadevall
c,
Hongwei
Song
d,
Erwin
Reisner
*c,
Leif
Hammarström
*d and
Sylvestre
Bonnet
*a
*ab,
David M.
Klein‡
a,
Santiago
Rodríguez-Jiménez
c,
Carla
Casadevall
c,
Hongwei
Song
d,
Erwin
Reisner
*c,
Leif
Hammarström
*d and
Sylvestre
Bonnet
*a
aLeiden Institute of Chemistry, Leiden University, Einsteinweg 55, Leiden, 2333 CC, The Netherlands. E-mail: andrea.pannwitz@uni-ulm.de; bonnet@chem.leidenuniv.nl
bInstitute of Inorganic Chemistry I, Ulm University, Albert-Einstein-Allee 11, 89081 Ulm, Germany
cYusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: er376@cam.ac.uk
dDepartment of Chemistry – Angstrom Laboratory, Uppsala University, Box 523, 751 20 Uppsala, Sweden. E-mail: leif.hammarstrom@kemi.uu.se
First published on 4th March 2021
Abstract
Artificial photosynthesis has experienced rapid developments aimed at producing photocatalytic systems for the synthesis of chemical energy carriers. Conceptual advances of solar fuel systems have been inspired by improved understanding of natural photosynthesis and its key operational principles: (a) light harvesting, (b) charge separation, (c) directional proton and electron transport between reaction centres and across membranes, (d) water oxidation and (e) proton or CO2 reduction catalysis. Recently, there has been a surge of bio-inspired photosynthetic assemblies that use liposomes as nanocompartments to confine reaction spaces and enable vectorial charge transport across membranes. This approach, already investigated in the 1980s, offers in principle a promising platform for solar fuel synthesis. However, the fundamental principles governing the supramolecular assemblies of lipids and photoactive surfactant-like molecules in membranes, are intricate, and mastering membrane-supported photochemistry requires thorough understanding of the science behind liposomes. In this review, we provide an overview of approaches and considerations to construct a (semi)artificial liposome for solar fuel production. Key features to consider for the use of liposomes in solar fuel synthesis are highlighted, including the understanding of the orientation and binding of different components along the membrane, the controlled electron transport between the reaction centres, and the generation of proton gradients as driving force. Together with a list of experimental techniques for the characterisation of photoactive liposomes, this article provides the reader with a roadmap towards photocatalytic fuel production at the interface of lipid membranes and aqueous media.
 Andrea Pannwitz | Andrea Pannwitz studied chemistry at the Georg-August-University Göttingen in Germany and obtained her PhD in 2017 at the University of Basel in Switzerland. Her PhD studies with Oliver Wenger focused on light-induced proton-coupled electron transfer. She pursued her postdoctoral studies together with Sylvestre Bonnet at Leiden University in the Netherlands, partly supported by an Early Postdoc. Mobility fellowship by the SNSF. In 2020 she started her independent career as assistant professor for Inorganic Chemistry and Energy Conversion at Ulm University with a major interest in photochemical conversions at membrane interfaces. |
 David M. Klein (left) and Sylvestre Bonnet (right) | David M. Klein received his BSc's degree in Molecular Science & Technology from the University of Leiden and Delft in 2015 and his MSc's degree in Chemistry in 2017 from the University of Leiden. He is currently working as a PhD in the laboratory of Sylvestre Bonnet at Leiden University supported by an HRSMC fellowship. His work focusses on the design of liposomes functionalised for artificial photosynthetic purposes, such as transmembrane electron transfer and photocatalytic water splitting. Sylvestre Bonnet is Professor for bioinorganic chemistry at Leiden University. His research focuses on photoactive metal complexes in biological and biomimetic environments, with particular interest in photocatalysis at lipid bilayers, upconversion, and light-activated metallodrugs. |
 From left to right: Santiago Rodríguez-Jiménez, Carla Casadevall and Erwin Reisner | Santiago Rodríguez-Jiménez obtained his PhD in chemistry at the University of Otago under the supervision of Prof. Sally Brooker, where he worked on the design and synthesis of spin-switching iron complexes for chemo-sensing applications. After his PhD, he worked in the same group as a postdoctoral research fellow focusing on the synthesis and electrode immobilisation of molecular hydrogen evolution electrocatalysts. Subsequently, he joined the group of Erwin Reisner as a postdoctoral researcher working on artificial photosynthetic self-assembled molecular systems. Carla Casadevall obtained her PhD degree in chemistry in 2019 at the Institute of Chemical Research of Catalonia (ICIQ) under the guidance of Prof. Julio Lloret-Fillol. Her PhD sought a fundamental understanding of the mechanisms involved in artificial photosynthesis, as well as the development of new sustainable methodologies to produce solar fuels and fine chemicals. Then, she joined the group of Erwin Reisner as a BBSRC postdoctoral researcher and now she has started in the same group as a Marie Curie fellow, working on hybrid-materials for solar fuel production. Erwin Reisner is Professor of Energy and Sustainability at the University of Cambridge and his research focusses on the development of solar-driven processes for the sustainable synthesis of fuels and chemicals. |
 Hongwei Song (left) and Leif Hammarström (right) | Hongwei Song obtained his PhD degree in Physical Chemistry in 2019 at Institute of Chemistry, Chinese Academy of Sciences (ICCAS) under the supervision of Prof. Andong Xia, where his work focused on the understanding of ultrafast elementary photochemical processes of organic molecules in liquid solution, as well as the development of new optical spectroscopic techniques. Then, he joined the group of Prof. Leif Hammarström as a postdoctoral researcher, working on mechanisms of artificial photosynthesis. Leif Hammarström obtained his PhD degree in Physical Chemistry at Uppsala University in 1995, for his thesis “Electron Transfer in Lipid Vesicles”. After postdoctoral work on inorganic photochemistry at University of Bologna, he began his independent career in Uppsala, where he since 2004 is Full Professor. His main research activities concern mechanisms of artificial photosynthesis: photoinduced electron transfer, proton-coupled electron transfer, mechanisms of solar fuel catalysts and dye-semiconductor systems. He is Chairperson of the Swedish Consortium for Artificial Photosynthesis. |
Key learning points(1) Photocatalysis on lipid membranes allows to mimic and understand natural photosynthesis and the role of membranes in it.(2) The physical–chemical properties of the membrane, as well as the strategy used to anchor catalysts and photosensitizers to it, both influence the catalytic outcomes of membrane-embedded photocatalytic systems. (3) Membranes are thick with respect to electron and proton transfer, and transferring them through membranes requires molecular charge carriers or transmembrane assemblies with appropriate hydro/lipophilicity. (4) When working with dissymmetric photoactive liposomes, ion leakage and osmotic stress on the membrane should be controlled. (5) Two-dimensional diffusion of membrane-embedded species is inherently different from 3D diffusion in bulk aqueous phases, which influences the kinetics of energy and electron transfer within lipid membranes. |
1. Introduction to photocatalysis on lipid bilayers
1.1 Motivation
Natural photosynthesis provides a biological blueprint for a scalable process to store solar energy in the chemical bonds of complex organic molecules. In the light-dependent reactions of oxygenic photosynthesis, photon absorption in the thylakoid membrane drives water oxidation (eqn (1)), which provides electrons and protons for the generation of the biological reductant dihydronicotinamide adenine dinucleotide (phosphate) (NAD(P)H) together with energy carrier molecules of adenosine triphosphate (ATP). The latter are subsequently used in the dark reaction of the Calvin cycle to fix CO2 as carbohydrates (eqn (2)), or to reduce protons (eqn (3)) in photobiological H2 production by redirecting reduced NAD(P)H towards hydrogenases in some unicellular algae and cyanobacteria under special conditions.1,2Water oxidation half reaction E°′ (V) vs. SHE at pH 7
| 2H2O → O2 + 4H+ + 4e− 0.81 | (1) |
Proton and selected CO2 reduction half reactions E°′ (V) vs. SHE at pH 7
| 6CO2 + 24H+ + 24e− → C6H12O6 + 6H2O −0.43 | (2) |
| 2H+ + 2e− → H2 −0.41 | (3) |
| CO2 + 2H+ + 2e− → CO + H2O −0.53 | (4) |
| CO2 + H+ + 2e− → HCO2− −0.39 | (5) |
| CO2 + 6H+ + 6e− → CH3OH + H2O −0.38 | (6) |
The thylakoid membrane consists of the supramolecular assembly of lipids and additives into a flexible, fluid two-dimensional membrane that hosts an array of protein units that organise the light reactions and separate them from the dark reactions. The absorption of solar energy occurs mainly via chlorophylls and accessory pigments in the light harvesting complexes (LHCs), which act as antenna for light collection (Fig. 1, left). The chlorophylls pass energy through a series of chromophores on to the reaction centres of Photosystems II (PSII) and I (PSI), where the central chlorophyll pigments P680 and P700, respectively, induce charge separation and subsequent unidirectional electron transfer. Such electron transfer occurs over a long distance, and follows an electron transport chain of small-distance electron transfer steps within the protein scaffold and redox mediators of the thylakoid membrane.3 Overall, long-lived charge separation across the thylakoid membrane is achieved with high quantum efficiency via a Z-scheme energy-storage architecture. Electrons are first transferred from the photoexcited P680 in PSII to the plastoquinones (PQs) embedded in the thylakoid membrane, then to the Cytochrome b6f complex (Cytb6f), where plastocyanin collects the electrons to reduce P700+ in PSI.3 The latter is an oxidised species generated from photoexcitation of PSI and electron transfer to ferredoxin (Fd, an iron–sulfur protein) and ferredoxin NAD(P)+ reductase (FNR) in the stroma of the chloroplasts. Ultimately, P680 is regenerated in PSII by electrons liberated from water oxidation in the lumen. The unidirectional flow of electrons and protons across the membrane generate a proton gradient that powers the conversion of ADP to ATP by ATP synthase (ATPase, Fig. 1, left).
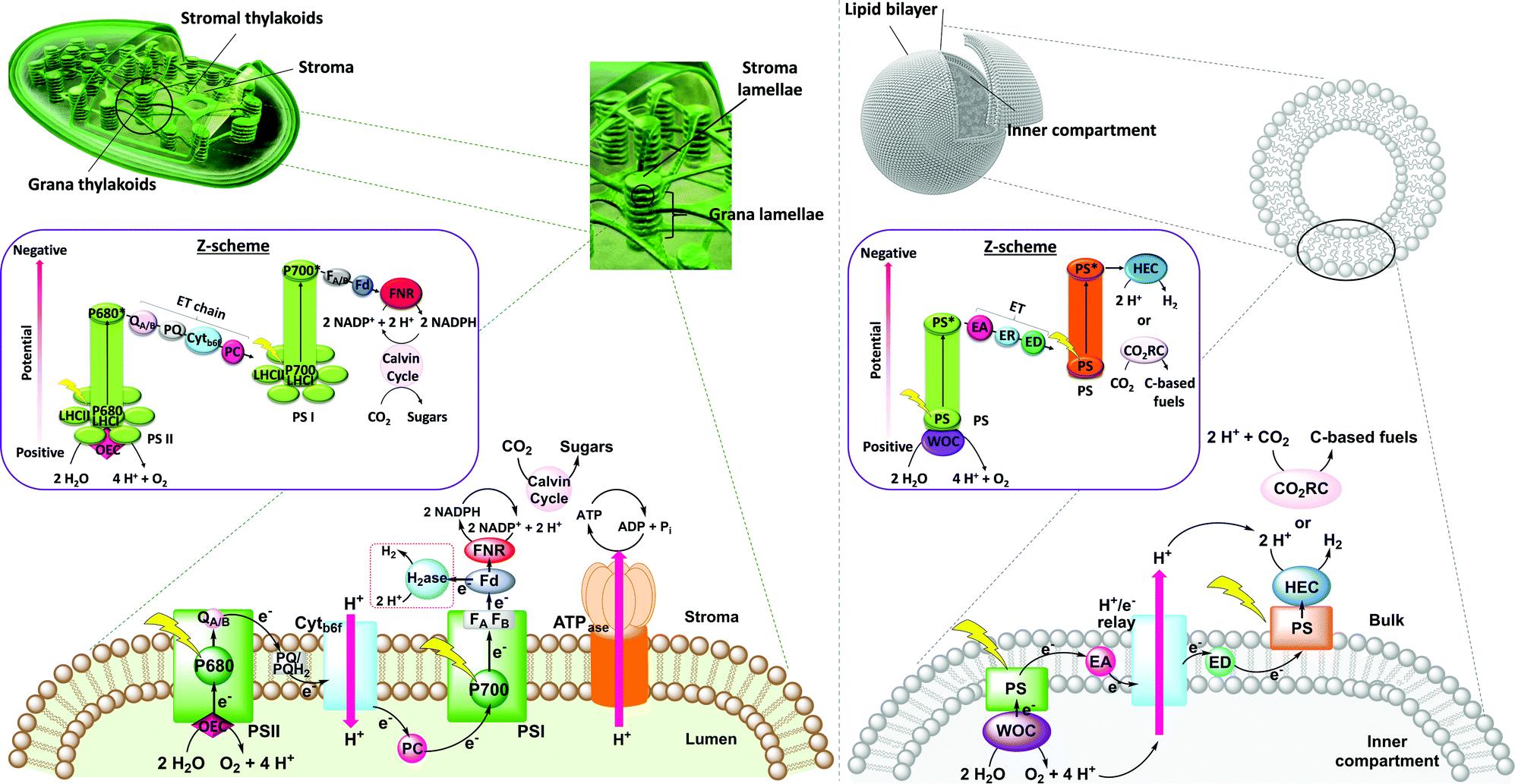 | ||
| Fig. 1 Left: Scheme of a natural chloroplast showing light-induced charge transfer across the thylakoid membrane, connecting PSII, PSI and the reaction centres for CO2 reduction and ATP generation. The left inset shows the respective energy-storing Z-scheme pathway. Right: Scheme of a synthetic vesicle as a platform for an artificial photosynthetic assembly. The right inset shows the respective Z-scheme. PSI = photosystem I, PSII = photosystem II, Ph = pheophytin, PQ = plastoquinone, PC = plastocyanin, b6f = cytochrome b6f, Fd = ferredoxin, FNR = ferredoxin-NADP+ reductase, LHCI = light-harvesting complex I, LHCII = light-harvesting complexes II, OEC = oxygen evolving complex, P680 and P700 = reaction centres in PSII and PSI respectively, WOC = water oxidation catalyst, PS = photosensitiser, EA = electron acceptor, ED = electron donor, HEC = hydrogen evolution catalyst, CRC = CO2 reduction catalyst, and ER = electron relay. | ||
A crucial feature of the thylakoid membrane is that it separates the oxidative from the reductive catalytic centres by compartmentalisation of the different redox half-reactions within the chloroplast lumen and stroma (Fig. 1, left). Such organisation minimises charge recombination and chemical back reactions, which by recombining holes and electrons, or oxidised and reduced molecular species, are detrimental for the reaction. PSI and ATPase do not fit into the space between the stacked membranes and are therefore mainly located in the stromal lamellae. PSII is located in the grana lamellae and thereby spatially separated from PSI and ATPase, preventing the uncontrolled spill-over of excitons from PSII to PSI.4 This heterogeneity of the thylakoid membrane maximises the packing density of photosynthetic complexes and plays an important role in optimising light harvesting and energy transfer under changing light conditions. In contrast, established approaches in artificial photosynthesis rarely take compartmentalisation into account.5–7
Mimicry of natural photosynthesis by synthetic design is an appealing strategy for the development of sustainable methodologies to produce solar fuels. Apart from the electrocatalytic oxidation of water into oxygen with the concomitant release of protons, and the reduction of CO2 (eqn (4–6)) and water (eqn (3)) into carbon-based fuels and H2, respectively, light harvesting with various photosensitisers and charge separation have been investigated. The concept of using self-assemblies in biomimetic lipid bilayer membranes for artificial photosynthesis was already approached in the 70's and 80's,7 but is receiving increased attention today due to the dramatically improved choice of catalysts and photosensitisers that can be spatially arranged to generate transmembrane potentials and compartmentalised artificial photosynthetic systems (Fig. 1, right).7,8 The use of the spherical membranes of liposomes as biologic mimics of the thylakoid membrane is a promising approach to confine redox half reactions, facilitate charge separation, and avoid cross-reactivity. While synthetic catalysts are continuously improving in terms of stability and activity in solar-to-chemical conversion, nature also provides evolutionarily optimised biological catalysts (enzymes) that are also appealing for building semiartificial photosynthetic systems due to their excellent catalytic activity.9 Amphiphilic polymers have also been proposed as alternative for lipids, as they can be tuned to self-assemble into thicker and often more solid membranes, though sometimes at the cost of biocompatibility (e.g., to accommodate transmembrane proteins). Photocatalytic polymersomes are also part of a larger effort to develop compartmentalised vesicle-based catalytic systems10–13 for tandem catalysis, which has been reviewed elsewhere and remains out of the scope of this article. This roadmap focuses on lipid-based photocatalytic vesicles for biomimetic solar fuel production, provides guidelines on how to construct such liposomes, and helps understanding the basic physical–chemical concepts, opportunities, and challenges, of using self-assembled lipid bilayer membranes as a supramolecular platform for (semi)artificial photosynthesis.
1.2 Lipid bilayers
Lipid bilayers are supramolecular self-assemblies formed from amphipathic lipids in an aqueous solution. Generally speaking, amphipathic (or amphipolar) molecules consist of a polar moiety (i.e., a hydrophilic head group), covalently connected to a non-polar hydrophobic part (e.g., an alkyl chain). Usually, lipids are also amphiphilic in the sense that they possess affinity for both aqueous and oil-like phases, with their preference expressed by their log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P value (see below). Most amphipathic molecules are also amphiphilic, and consequently both terms “amphiphilic” and “amphipathic” are usually used as synonyms, although they are conceptually different. One should also note that some lipids can be plain hydrophobic, such as cholesterol or fat (triglycerids), in which case they are not amphipathic enough to generate bilayers in water. In all cases, for amphiphilic lipid molecules the polar head groups interact with water via ion–dipole or dipole–dipole interactions when exposed to an aqueous environment, whereas the interaction of the hydrophobic alkyl chain is unfavourable with water, leading to the hydrophobic effect that causes the hydrophobic groups to interact. In the case of lipids, the resulting self-assembled structure is a bilayer membrane, which generates an interface between the water phase and the more hydrophobic environments of the interior of the membrane. This hydrophobic environment can also be observed in oil–water emulsions, in foams, or in micelles-containing aqueous solutions.
P value (see below). Most amphipathic molecules are also amphiphilic, and consequently both terms “amphiphilic” and “amphipathic” are usually used as synonyms, although they are conceptually different. One should also note that some lipids can be plain hydrophobic, such as cholesterol or fat (triglycerids), in which case they are not amphipathic enough to generate bilayers in water. In all cases, for amphiphilic lipid molecules the polar head groups interact with water via ion–dipole or dipole–dipole interactions when exposed to an aqueous environment, whereas the interaction of the hydrophobic alkyl chain is unfavourable with water, leading to the hydrophobic effect that causes the hydrophobic groups to interact. In the case of lipids, the resulting self-assembled structure is a bilayer membrane, which generates an interface between the water phase and the more hydrophobic environments of the interior of the membrane. This hydrophobic environment can also be observed in oil–water emulsions, in foams, or in micelles-containing aqueous solutions.
Due to their amphiphilicity, lipids are also surface-active and hence surfactants. Surface activity and interfacial behaviour can be measured via tensiometry (surface pressure measurements). At a low concentration in water, surfactant amphiphiles concentrate at the interface to air, and with increasing surfactant bulk concentration, the surface tension is increasingly lowered with no aggregation being observed in the bulk solution. Increasing the surfactant concentration further will reach a point known as the critical micelle concentration (CMC), where aggregates start forming in the bulk aqueous phase. At concentrations above the CMC, only a very small fraction of the surfactant is dissolved as monomers in water (cmax = CMC), while all other molecules are dynamically forming self-assembled aggregates. Depending on the surfactant's molecular geometry, different self-assemblies may form, such as micelles, reverse micelles, bilayers, and vesicles. Lipids typically form bilayer membranes and vesicles. Depending on the lipids, addition of salts and differences in temperature may induce changes in aggregation dynamics and hence in the CMC.
Lipids can generally be classified according to their head groups: zwitterionic, anionic, cationic, and non-ionic.14 Examples for each class of bilayer-forming lipids are depicted in Fig. 2: the zwitterionic 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), the anionic 1,2-dimistroyl-sn-glycero-3-phosphonate(1′-rac-glycerol) (sodium salt) (DMPG), the cationic 1,2-dioleoyl-sn-glycero-3-[phospho-rac-(3-lysyl(1-glycerol))] (chloride salt) (O-Lysyl PG), and the non-ionic 1-palmitoyl-2-oleoyl-3-bis(β-D-glucosyl)-sn-glycerol (16:0–18:1 GlcGlcDAG). Further distinction can be made according to the counterion (Cl−, Br−, I−, Na+, K+etc.) or the chemical character of the alkyl chain (e.g., hydrocarbon or fluorocarbon, saturated or unsaturated).
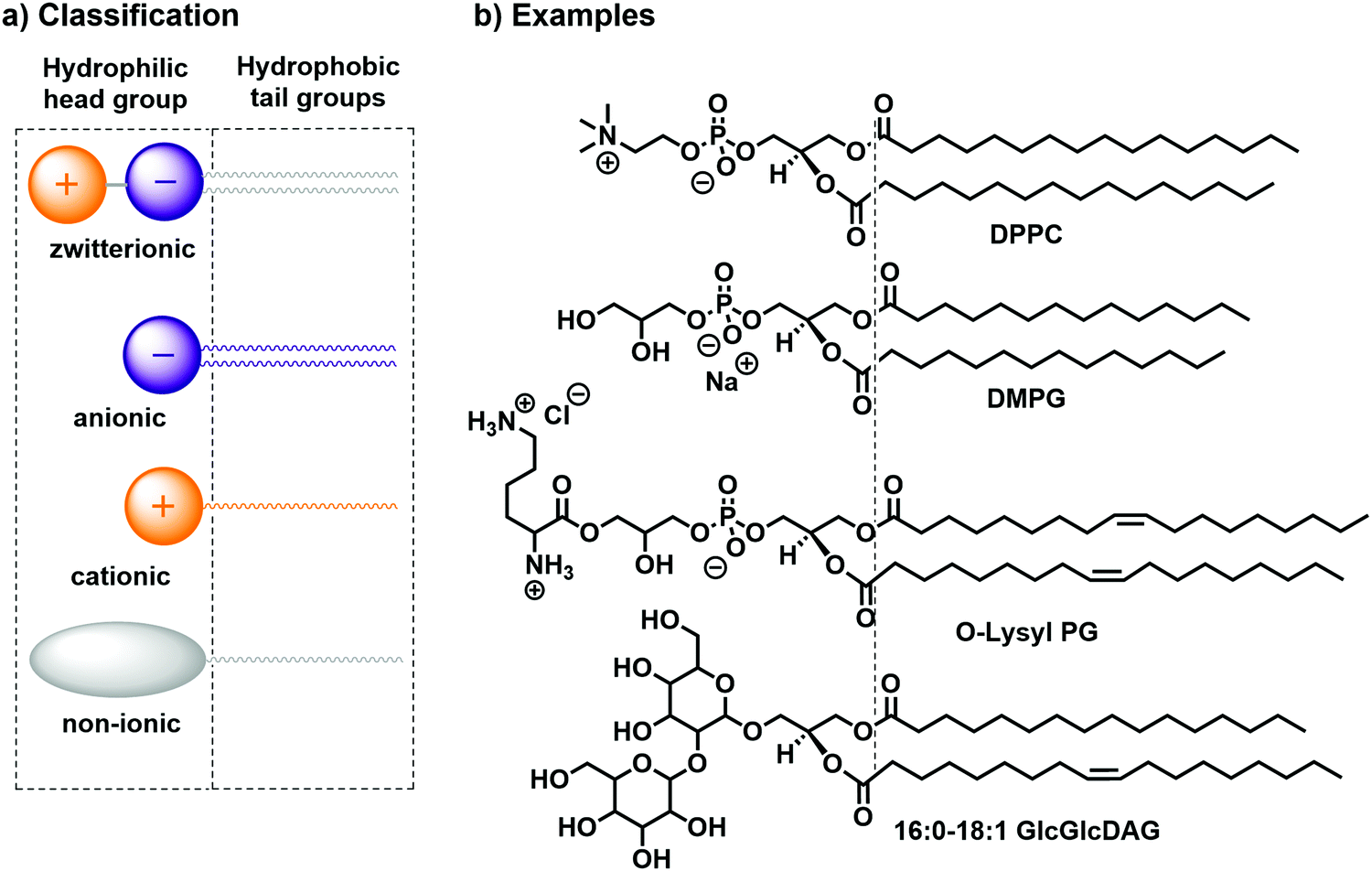 | ||
| Fig. 2 (a) Classification14,15 and (b) practical examples of bilayer-forming amphiphiles: DPPC, DMPG, O-Lysyl PG, and 16:0–18:1 GlcGlcDAG. | ||
A common way to characterise the hydrophilicity and hydrophobicity of a molecule is to determine its water–octanol partition coefficient (logP). This dimensionless number is obtained by measuring experimentally the concentrations coct and cwater of the molecule in each phase of a biphasic n-octanol/water mixture at the thermodynamic equilibrium. Then, the decadic logarithm, logP, is calculated as follows:
| logP = log(coct/cwater) | (7) |
Typically, hydrophilic compounds like tetraethylammonium iodide (Et4N+I−) have a low negative logP value (−2.82)16 and more hydrophobic compounds like triethylamine (Et3N) have a high, positive logP value (+1.44).16 Amphiphilic molecules have a logP value close to zero, i.e. at the equilibrium, they distribute in similar quantities in the octanol and aqueous phases. For example, nitromethane has a logP = 0.08,16 even if this molecule is not expected to have surface-active properties. On the other hand, decyltrimethylammonium iodide (C13, logP = −0.16)16 has one carbon less than the water-soluble tributylethylammonium iodide (C14, logP = −1.30),16 but due to its single long alkyl tail it is more hydrophobic, also reaching a zone of less negative logP values typical for amphiphilic molecules. In addition, due to its dissymmetric “head–tail” geometry it has surface-active properties, which is also typical for lipids. One should note that other concepts to characterise surfactant-like molecules have been proposed, such as the “hydrophilic lipophilic balance” (HLB). This numerical approximation characterises the interfacial behaviour of an amphiphile in the presence of a mixture of an organic solvent and water, as described in details elsewhere.14
In absence of organic co-solvents, amphiphiles dissolved in water usually form either membrane bilayers or micelles, depending on their geometry and steric properties.17 For bilayer formation the most suitable geometry of an amphiphile is close to a cylindrical shape (Fig. 3a). On the other hand, to obtain spherical and rod-shaped micelles a conical shape of the amphiphile is required (Fig. 3b). To assess a priori if an amphiphile forms bilayers or micelles, the packing parameter can be used:
| x = v/(a0·lc) | (8) |
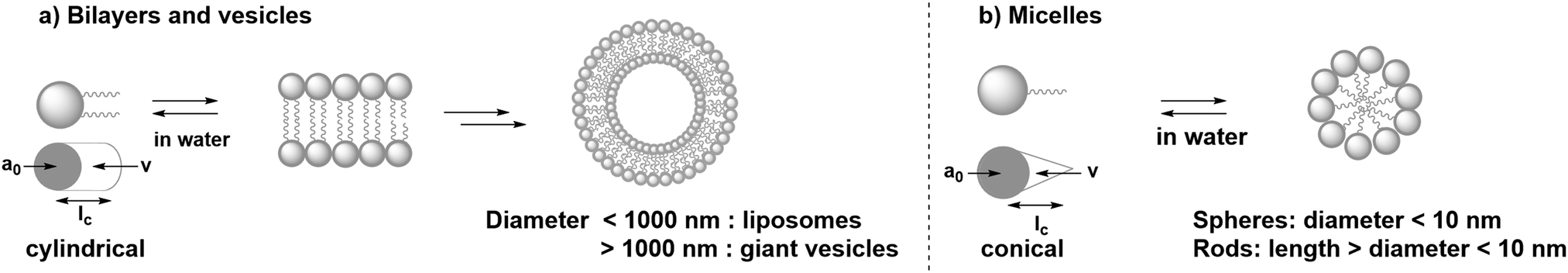 | ||
| Fig. 3 (a) Self-assembly of cylindrical amphiphiles in water into bilayers and vesicles. (b) Self-assembly of conical amphiphiles in water into spherical and rod-shaped micelles. | ||
1.3 Individual components for artificial photosynthesis at lipid bilayers
Several synthetic or biological functional components are necessary to build a compartmentalised system for artificial photosynthesis.6 Basically, these functional molecules can be divided into four main categories: photosensitisers, catalysts, sacrificial reagents, and charge carriers. Important considerations have been established over the years to target these components towards different regions of the lipid bilayer and construct photocatalytic membranes. These consideration can be sorted into the four following general strategies (Fig. 4): (a) covalent attachment of the functional group to a lipid that inserts in the membrane, (b) electrostatic attraction of charged functional molecules to the charged head groups of the lipids, (c) integration of the functional group into an amphiphilic molecule, which targets the interface between the bulk water phase and the hydrophobic core of the lipid membrane, and (d) building enough hydrophobicity in the functional molecule to embed it within the hydrophobic core of the membrane.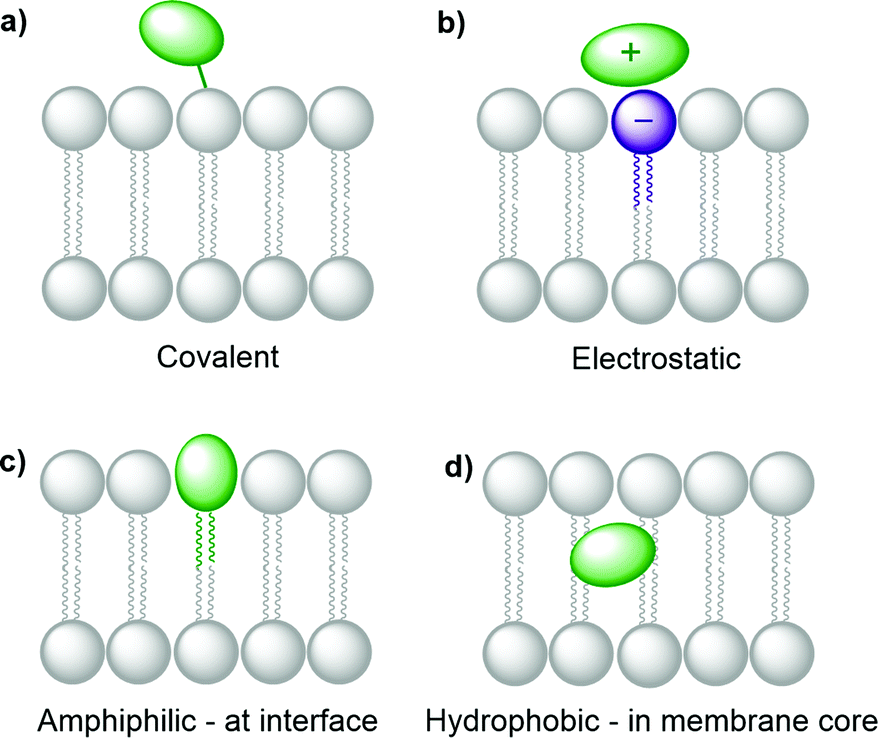 | ||
| Fig. 4 Strategies to integrate functional molecules in lipid bilayer membranes. | ||
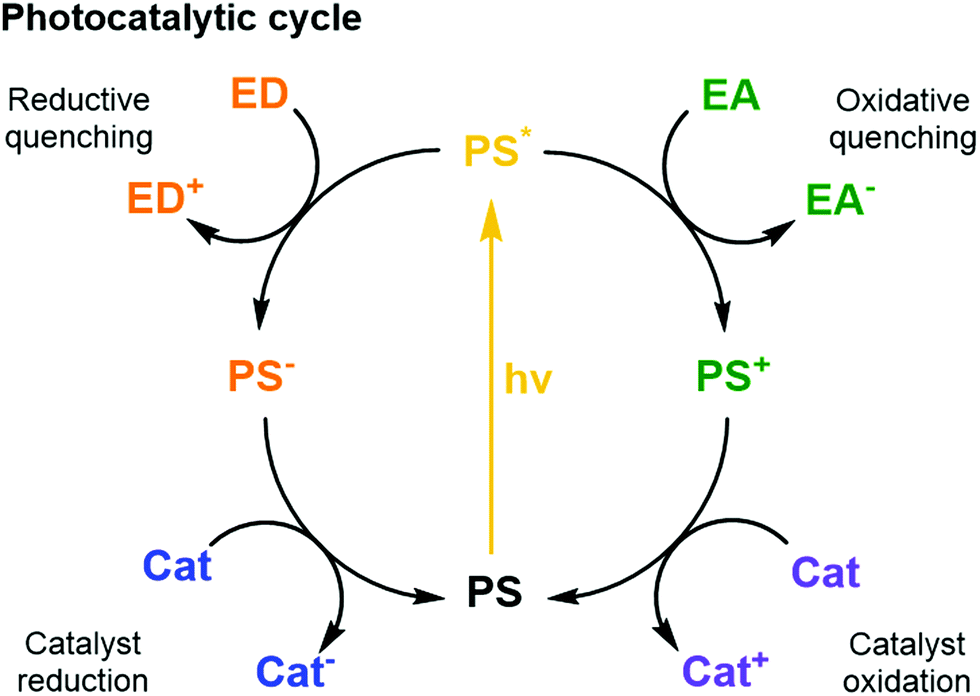 | ||
| Fig. 5 Schematic photocatalytic cycles of a PS including quenching reactions with an ED or EA and the reductive and oxidative reactions with a catalyst. | ||
Thermodynamics (i.e., redox potentials in ground and excited states), and kinetics (i.e., excited state lifetime and charge transfer dynamics) must be well orchestrated in different PSs to couple efficient light collection with electron transfer and catalysis within the assembled components.6 Furthermore, the spectral properties of the PS influence its ability to absorb photons. The most relevant properties here are how the absorption spectrum (extinction coefficient) of the PS overlaps with the irradiance spectrum of the light source (e.g., the sun); what are the relative quantum yields for non-radiative deactivation, electron transfer, and photon emission; and last but not least, what is the photostability of the PS under photocatalytic conditions. Ideally, the other components of the system should show little absorption where the PS absorbs most, to avoid competition for light (filter effect) and photon loss.
One of the most widely used type of PS molecules are polypyridine ruthenium(II) complexes such as [Ru(bpy)3]Cl2 (PS1, bpy = 2,2′-bipyridine, Fig. 6a).6,18 The positive charge of this complex and the limited hydrophobicity of the bipyridine ligands makes it quite hydrophilic (logP = −2.50),21 which prevents it from interacting strongly with positively charged or neutral lipid bilayers. A negatively charged membrane is therefore required to attract PS1 electrostatically (Fig. 4b). An alternative strategy consists in functionalising the bpy ligand in PS1 with hydrophobic groups such as alkyl chains, to yield the amphiphilic analogues PS2 and PS3 (Fig. 6a). These molecules can be readily immobilised within lipid bilayers (Fig. 4c). For instance, PS2 was employed in lipid vesicles for photocatalytic hydrogen evolution19 and water oxidation reactions,22,23 whereas PS3 was used in unilamellar vesicles for photocatalytic carbon dioxide reduction.24 In the presence of a lipid bilayer these amphiphilic molecules may either assemble at the interface (Fig. 4c), if the alkyl chains in PS3 are not too long and the counter anion (e.g. Cl−) solubilises the dicationic head in the water phase; or, they may integrate deeper within the membrane (Fig. 4d) with longer, more hydrophobic alkyl chains and more hydrophobic counter anions (e.g., PF6−). Another classical family of PSs consists in metal porphyrins and includes for example PS4, PS5 and Chla (Fig. 6a). PS4 and PS5 were immobilised in charged vesicles to study charge recombination25 and transmembrane charge transport,26 respectively. The biological PS Chla (Fig. 6a) acts as a visible-light harvesting unit in PSI and PSII, and was utilised in charged micelles for photocatalytic hydrogen evolution.27
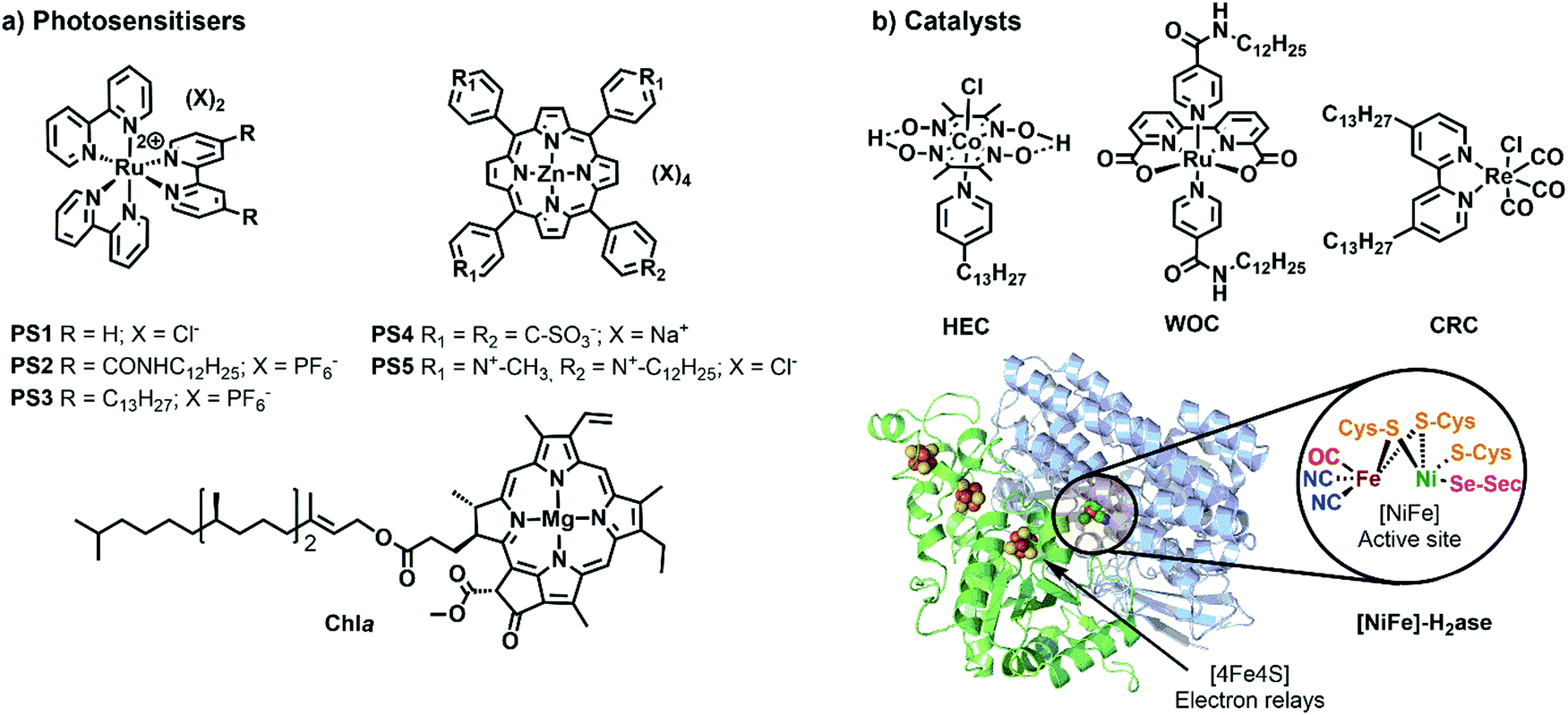 | ||
| Fig. 6 Synthetic and biological (a) PSs and (b) catalysts used to build photocatalytic vesicular and micellar systems. X = counterion, HEC = hydrogen evolution catalyst, WOC = water oxidation catalyst, CRC = CO2 reduction catalyst, H2ase = crystal structure of oxygen-tolerant [Ni–Fe]-hydrogenase enzyme, obtained from bacterium Desulfomicrobium baculatum (pdb: 1CC1). | ||
It should be noted that the integration of the PS into the membrane has often a significant effect on its electronic and photochemical properties. Despite the currently unpredictable effects of the membrane environments on the properties of the integrated components, ligand modifications (e.g. covalent attachment of alkyl groups) can tune the photophysical and redox behaviour of the PSs as well,18 or that of other active components (see below), making the spectroscopic and electrochemical characterisation of newly synthesised components fundamental to reach an efficient self-assembled system. Last but not least, the primary input in a photocatalytic system is the rate of formation of excited states, or rate of photon absorption. This photochemical input depends not only on the PS concentration, but also on the molar absorption coefficient of the PS at the irradiation wavelength, and on light intensity. The quantum yield of product formation can be calculated by dividing the measured rate of product formation by the rate of photon absorption, and multiplying this number by the number of electrons transferred photocatalytically (e.g., two for the reduction of CO2 into CO, or four for water oxidation into O2).
Catalysts for solar fuel production are commonly redox active metal complexes that can accumulate charges (either electrons and/or protons) to facilitate bond-making and bond-breaking reactions such as hydrogen evolution, CO2 reduction, or water oxidation. Examples for catalysts that have been supported on lipid bilayers and micelles are depicted in Fig. 6b. Hydrogen evolution was performed with an alkylated cobaloxime-based hydrogen evolution catalyst (HEC) in zwitterionic vesicles and with a hydrogenase in charged micelles.19,28 An alkylated rhenium CO2 reduction catalyst (CRC) was employed to study photocatalytic CO2 reduction in zwitterionic vesicles.24 Photocatalytic water oxidation in vesicles was achieved with an alkylated version of a Ru-based water oxidation catalyst (WOC).23 More examples of catalysts, which could be potentially employed upon ligand modification in vesicles, are reported elsewhere.6,29
To select a suitable catalyst, several performance metrics such as catalytic activity, product selectivity, and stability, need to be considered. The catalytic activity is characterised by the turnover number (TONCat) and turnover frequency (TOFCat). TOFCat is a measure of the product generation rate, given as the number of moles of products produced per mole of catalyst in the system and per unit time. The TONCat at a certain time t, is the total number of product molecules generated at that moment per molecule of catalyst initially introduced in the system. While in catalysis under dark conditions, catalyst decomposition often explains why the reaction stops, in photocatalysis the end of a photocatalytic reaction may also be due to PS degradation. For photocatalytic systems it is thus equally important to assess the PS stability. Here, we refer to TONPS and TOFPS when referencing the TON or TOF of the system to the amount of PS initially introduced. It should also be noted that sometimes, pH changes or consumption of the sacrificial reagent may also be involved in the end of the photocatalytic reaction.
A catalyst requires a certain driving force (i.e., redox potential) to generate the catalytically active species. The difference between this catalytic onset potential and the thermodynamic potential (see eqn (1)–(6)) is known as the overpotential. In an ideal scenario, a high reaction rate is reached at a small overpotential. In a photochemical system, the “applied” potential often corresponds to the redox potential of the reduced or oxidised ground state of the PS, and it should match the overpotential requirement of the catalyst. Particularly important in CO2 reduction is product selectivity, because co-generation of H2 and other carbon-based reduction products readily occurs in CO2-saturated aqueous solutions. Stability is especially challenging in water oxidation catalysis, where organic ligands often degrade under the highly oxidising conditions and evolved O2.30 The performance of catalysts is commonly assessed in homogeneous solution, but the hydrophobic anchoring groups and the membrane environment will affect the catalytic properties.29
Biological catalysts (enzymes) operate commonly with intrinsically low overpotentials and are therefore efficient model catalysts in semiartificial photocatalytic systems,2,10 including semiartificial photocatalytic micellar systems and electrocatalytic bilayer lipid membranes.29,31 These excellent catalytic capabilities are mainly due to their evolutionary-optimised architecture in which the protein environment facilitates the catalytic transformation at the active site by controlling substrate access and orientation as well as stabilising transition states of catalytic intermediates. Some examples are hydrogenase (H+↔H2; Fig. 6b), formate dehydrogenase (CO2↔formate), carbon monoxide dehydrogenase (CO2↔CO) enzymes, and PSII for light-driven water oxidation.1,2,9 Despite their excellent performance, these enzymes are costly to isolate, fragile, and often display O2-sensitivity, which can make them difficult to handle. Their suitability for the integration in photocatalytic systems also requires consideration of cofactor dependence (e.g. NADH) and substrate affinity. The latter is expressed by the Michaelis–Menten constant (KM), which is low for high substrate affinity. The use of compartmentalised vesicles allows the encapsulation of enzymes within the vesicles with low substrate affinity and provide them with the required high local concentration of substrate as in living cells. The reader is referred to references1,9 for a more detailed discussion on selection criteria for enzymes.
Sacrificial ED and EA reagents are widely used in artificial photosynthesis when studying half-reactions, although the ultimate goal is to use a sustainable source of charge to drive a closed redox cycle.32 They serve as reductive and oxidative equivalents used to quench a photoexcited PS (PS*, Fig. 5), or regenerate the PS after reductive or oxidative quenching by a primary quencher. This reduces the possibility for charge recombination as they commonly decompose quickly and irreversibly. The use of sacrificial reagents allows investigating and optimising individual half-reactions such as proton/CO2 reduction or water oxidation without the demanding requirement to couple both half reactions with a single charge carrier. The choice of ED and EA is generally based on their redox potentials, rapid photooxidation and reduction in the presence of PS* and, ideally, producing harmless intermediates and products, although non-innocent decomposition by-products are sometimes generated. Some of the most commonly used ED in aqueous media for light-driven proton and CO2 reduction are mild reducing agents such as triethanolamine, ethylenediaminetetraacetic acid (EDTA), and ascorbic acid.32 On the other hand, one of the most common EA used in light-driven water oxidation reactions is the strong oxidising agent sodium peroxodisulphate (Na2S2O8);6 [Co(NH3)5Cl]Cl2 has been proposed as well, but its propensity to form catalytically active cobalt oxides is problematic, in particular in the context of the water oxidation reaction. One should note that the conditions used for many photocatalytic water oxidation experiments, i.e. near-neutral pH and Na2S2O8 as EA, are drastically different from the conditions in which chemically driven water oxidation catalysts are usually tested, which involves strong concentrations of cerium ammonium nitrate (CAN) and highly acidic solution (pH = 1).
Charge carriers are redox active species capable of transporting reducing or oxidising equivalents between different components of a photocatalytic system (Fig. 7a). A typical example is the water-soluble, dicationic methyl viologen (MV2+), which can accept one electron to form the blue-coloured cation radical MV˙+. In some cases, charge carriers can undergo proton-coupled electron transfer (PCET) and carry electrons and protons across lipid membranes; good examples are the membrane-soluble 1-methoxy-N-methylphenazinium cation (MMP+), or the quinones 2,5-diphenyl-1,4-benzoquinone (Qs) and decylubiquinone (dQ).26,33 The thermodynamic and kinetic features of charge carriers must also be carefully considered to ensure an efficient unidirectional electron transfer. Furthermore, the chemical stability and solubility of the charge carrier must be high in the relevant redox and protonation states (Fig. 7a). For example, in presence of O2 the one-electron reduced species MV˙+ is re-oxidised into MV2+, which can be problematic in full water-splitting schemes also producing O2.
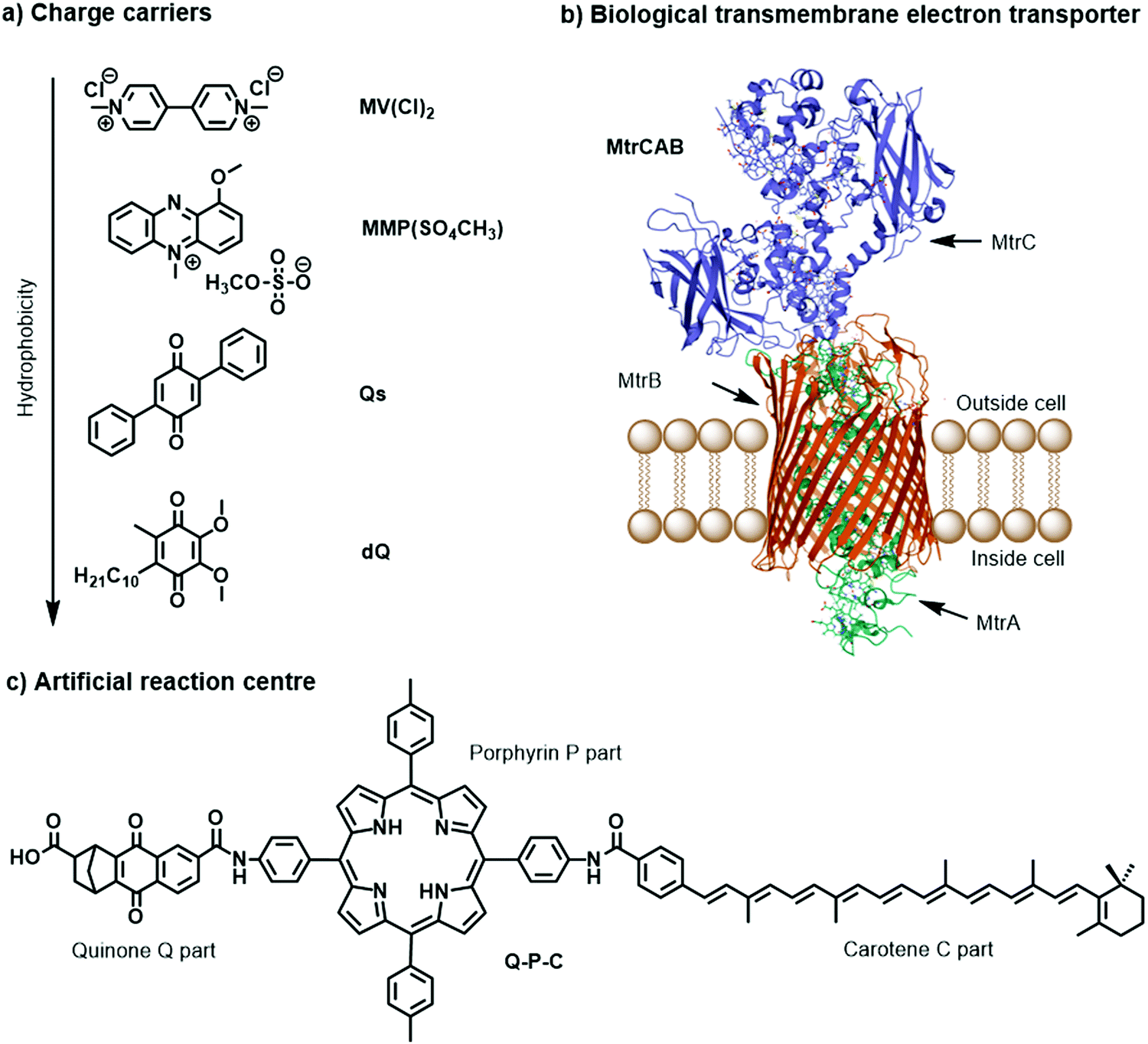 | ||
| Fig. 7 Molecular structures of (a) metal-free charge carriers, (b) crystal structure of biological electron transmembrane transporter icosa-heme cytochrome protein MtrCAB from bacterium Shewanella baltica (pdb: 6R2Q), with cellular lipid bilayers and its three subunits, and (c) artificial reaction centre Q–P–C used to light-induce proton transport across vesicle membranes.33,35 | ||
The electrostatic charge of a carrier can also be exploited to enhance ET kinetics or minimise charge recombination due to electrostatic repulsion or attraction.25 Other features to consider are whether the reduced or oxidised charge charrier acts innocently, i.e. it exclusively engages in an outer-sphere ET reaction, or non-innocently, i.e. it can undergo side-reactions such as a disproportionation reaction (e.g. 2 MV+ → MV2+ + MV0) or reacts with O2 present in solution.34 The affinity of the charge carrier for the catalyst should also be considered, as it could disrupt catalysis by coordinatively binding to the catalytically active metal centre. Another aspect to consider are changes in the pKa value due to reduction or oxidation of the charge carrier, which could influence catalysis. Moreover, the effect of membrane environments can tune features of the catalyst such as its redox potentials, which are typically measured electrochemically in bulk organic solutions. In addition to the presented metal-free charge carriers (Fig. 7a), water-soluble metal-based complexes, such as cationic cobalt tris-bipyridine complex, [Co(bpy)3]2+/3+, could be conceptually appropriate reductive and oxidative equivalent carriers, however because of the similarity with other synthetic components (e.g. the water oxidation or proton reduction catalyst) they may potentially act undesirably. Other hydrophilic electron relays such as I−/IO3− and Fe3+/Fe2+ have also been used. Compartmentalised vesicles have also been proposed that use lipophilic charge carriers to transport electrons and protons across vesicle membranes between spatially separated reaction centres. For example, membrane-bound cytochrome MtrCAB proteins (Fig. 7b) have been proposed as biological molecular wires and membrane-embedded artificial reaction centres, which artificial quinone–porphyrin–carotenoid triads (Q-P-C, Fig. 7c)33,35 try to mimic. Overall, transporting the reductive equivalent from the site of water oxidation to the site of proton or CO2 reduction requires charge carriers that are metastable in two different oxidation states, and do not react too quickly with the photogenerated products O2, H2, and/or reduced form of carbon dioxide.
To conclude, not only the position in the membrane, but also the number of electrons exchanged by the different components of the membrane during photocatalysis, should be considered carefully. Overall, natural photosynthesis requires at least eight photons in both photosystems to extract four electrons and protons from two water molecules in PSII, and to deliver these electrons one by one for the two-electron reduction of two NAD(P)+ molecules via Cytochrome b6f (Cyt b6f) to PSI, while protons are passed one by one through the membrane by stepwise rotation of ATPase, thereby generating ATP (Fig. 1, left). Likewise, there is often a mismatch in the number of electrons (or charges) exchanged by the different components of an artificial photosynthesis system (Fig. 1, right). Typically, one electron is exchanged between an excited photosensitizer molecule and a charge carrier such as ascorbate or MV2+, while two electrons are needed for long-distance charge transporters such as quinones or MMP+, two to eight electrons are needed for catalytic fuel production, and four electrons are provided by water oxidation. This discrepancy in principle requires careful analysis of the kinetics of each electron transfer step, to understand how they may integrate in a solar fuel production system.
2. The hydrophobic membrane
2.1 Properties of the membrane
Natural cell membranes are two-dimensional (2D) flexible liquid crystals that can adjust easily to environmental mechanic stress and provide a fluid “solvent” for membrane proteins as well as high mobility for mass transport within the membrane. Depending on the application, more rigid membranes can also be prepared.6,24,26 Most membranes indeed can exist in different phases, typically a liquid-crystalline phase with high lateral diffusion of the lipids, and a gel-like or even “solid” phase, characterised by much lower lateral diffusion properties (Fig. 8). A membrane with the desired phase, flexibility, and mobility, can be constructed from either pure lipids or from lipid mixtures. In the former case, transition temperatures (Tm) from the gel phase to the liquid crystalline phase are tabulated for most lipids (see Table 1).15 Additives such as cholesterol and lipid mixtures can fine-tune the properties of the membrane. Experimentally, Tm can for example be obtained from fluorescence anisotropy measurements with a membrane-solubilised probe,34 or differential scanning calorimetry.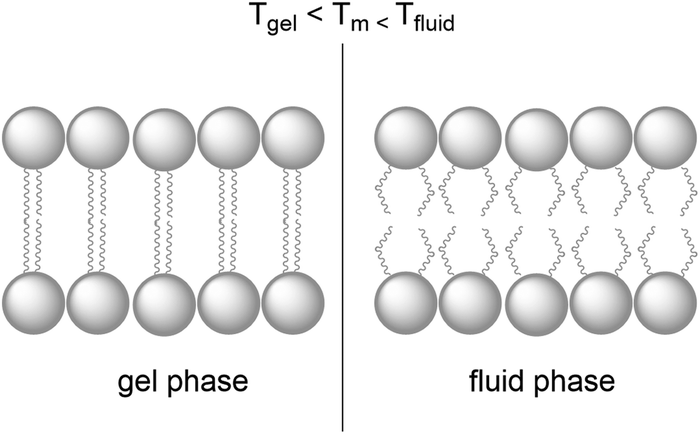 | ||
| Fig. 8 A lipid bilayer changes from the rigid gel phase (left) to the mobile fluid phase (right) above the transition temperature (Tm) of the lipid. | ||
| Lipids | T m (°C) |
|---|---|
| DSPC | 55 |
| DPPC | 41 |
| SMPC | 31 |
| DMPG | 23 |
| DOPC | −17 |
| Egg lecithin | −10 |
Molecularly, the phase of the lipid bilayer is influenced by the number and length of the alkyl chains, their degree of unsaturation, the headgroup structure (i.e. its size, electrical charge, and polarity), and the presence of additives such as other amphiphiles or cholesterol in the membrane. Saturated chains (such as DPPC) allow for closer packing than the less flexible and bulkier unsaturated chains (such as 1,2-dioleoyl-sn-glycero-3-phosphocholine, DOPC), and longer chains maximise hydrophobic interactions. These features explain the large variation in gel-to-liquid crystalline transition temperatures Tm between lipids such as DPPC and DOPC. The lateral and transverse diffusion of lipids and active components in the lipid bilayer are influenced by the mobility of the medium and hence the phase of the lipid bilayer. Diffusion coefficients for 2D lateral mobility in liquid crystalline bilayers are on the order of μm2 s−1, while the values in the gel phase are several orders of magnitude lower. The lateral mobility describes the mobility of a lipid molecule (or photoactive component in the membrane) parallel to the water–membrane interface and within one of the monolayers forming the membrane. Transversal diffusion from one monolayer to the other, also called “flip-flop”, requires intermediates states where the hydrophilic head of the molecule is located in the hydrophobic core of the membrane; it is hence a slow process, that takes place within several hours to weeks, depending on the lipid composition and temperature.15
Unsaturated lipids in a lipid mixture usually decrease its phase transition temperature Tm and increase the lateral fluidity of a membrane, which is in principle good for electron transfer processes within the membrane. However, they also increase the photochemical membrane instability, as double bonds are prone to oxidation in the presence of light and O2, in particular with transition-metal based PSs that can quickly generate long-lived triplet excited states. In the presence of O2, these triplet excited states are indeed prone to generate singlet oxygen (1O2) by energy transfer, or radical oxygen-based species (such as the superoxide ion O2˙−) by electron transfer, which in turn can oxidise the unsaturated moieties in the lipid tails into peroxides, alcohols, or conjugated ketones.36 Lipid photooxidation can culminate into C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond breaking and generation of aldehydes that destabilise the membrane and make it leaky, which is counter-productive for compartmentalised photocatalysis. The introduction of unsaturated parts in photocatalytic membranes needs hence to be carefully considered, avoided if possible, and when utilised, the integrity of the membranes should be carefully verified.
C double bond breaking and generation of aldehydes that destabilise the membrane and make it leaky, which is counter-productive for compartmentalised photocatalysis. The introduction of unsaturated parts in photocatalytic membranes needs hence to be carefully considered, avoided if possible, and when utilised, the integrity of the membranes should be carefully verified.
The role of membrane fluidity in semi-artificial vesicular systems was investigated in a few examples. Photocatalytic hydrogen evolution in vesicles, containing membrane-bound PS2 and a homogeneous [FeFe]-H2ase subunit mimic catalyst, performed with higher TON of H2 with low Tm lipids, such as DOPC and DMPC, than with DPPC or 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC).37 On the other hand, photocatalytic water oxidation in vesicles,22 containing membrane-bound PS2 and [Ru(ddp)(4-picoline)3] (ddp = 4-dodecyl-2,6-dipicolinate) catalyst, showed the opposite trend, i.e. lower TON of O2 were obtained with the low Tm lipid DOPC than with lipids with higher Tm such as DMPC and 1-stearoyl-2-myristoyl-sn-glycero-3-phosphocholine (SMPC). Thus, although basic understanding of the physical–chemical principles underlying photochemistry in unsaturated membrane is available, there has been no thorough investigation of the detailed role of unsaturations in lipids and of Tm on the performances of photocatalytic membranes.
2.2 Structural integrity of liposomes
Although liposomes are well-defined supramolecular systems with excellent kinetic stability in solution, photoreactive liposomes sometimes become unstable in the dark or during photocatalysis. For example, they may coalescence into very large assemblies by aggregation, in particular with uncharged lipids; alternatively, the membrane may rupture due to the formation of holes or due to photochemical oxidation of its lipids components; or the membrane may become destabilised by external agents, for example, a detergent, or a photoproduct. These phenomena may be prevented by the following “tricks”:(a) The bilayer may be strengthened by introducing additives, e.g. 20 percent cholesterol;
(b) Electrostatic repulsion may be embedded by design, e.g. by doping the liposomes with positively or negatively charged lipids to generate charged membrane interfaces. For example, 20% charged lipids were mixed in neutral or zwitterionic lipids,38 or amphiphilic active components with charged head groups were added to the membrane.25
(c) Steric hindrance may be added by introducing polyethylene glycol-containing lipids in the lipid mixture forming the membrane. Here as well, the PEG group prevents the liposomes to aggregate. A typical example is the anionic lipid 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (NaDSPE-PEG2K), which is added (∼1 mol%) to for example zwitterionic lipid membranes based on e.g. 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC).39
2.3 Osmotic pressure
Controlling the bulk concentrations of membrane-impermeable solutes, and hence the osmotic pressure of the aqueous solutions on both sides of the liposome membrane, is a crucial consideration when preparing photocatalytic liposomes, especially where the inner aqueous compartment and outer bulk solution do not have the same composition (see Section 3.1). The osmotic pressure of a solution onto a bilayer membrane expresses the pressure which is required to stop water from diffusing through the lipid bilayer by osmosis. The two solutions on both sides of the membrane may have different or equal osmotic pressures, which will determine the electrochemical and mechanical balance of the membrane (see below). For each solution, the contribution πi (in Pa) of each solute i to the total osmotic pressure π can in principle be calculated by the following equation:| πi = ji·c·R·T | (9) |
| ji = 1 + αi(ni − 1) | (10) |
Eqn (10) gives, for a simple solution containing a non-dissociating solute such as glucose (ni = 1), αi = 0 and ji = 1. A fully dissociating solute has αi = 1, thus NaCl (ni = 2) yields ji = 2, while MgCl2 (ni = 3) yields ji = 3. However, for any solute that is dissociating incompletely (0 < αi < 1), such as carboxylic acids, or for the phosphates of a phosphate buffer, it may become difficult to calculate ji and hence πi. In addition, eqn (9) and (10) account for perfect solutions only, and might deviate from reality when ions have multiple charges, or when concentrations are not low (>10−2 mol L−1). Last but not least, buffer solutions might be too complex, or have unknown components, which makes the calculation of its osmotic pressure impossible. In such cases, the total osmotic pressure of a solution can simply be measured experimentally, in osmole per litre (abbreviated as Osm L−1, a non-SI unit), using an osmometer. For example, a 0.125 M ethylenediaminetetraacetic acid buffer at pH = 8 has an osmotic pressure of 374 mOsm.23 To obtain mechanically stable liposomes, the experimental osmotic pressures of the two solutions on both sides of the liposome membrane, whatever they contain, should be equal.
In a liposome, three scenarios can indeed be distinguished: a hypotonic, a hypertonic, and an isotonic situation (Fig. 9). In a hypotonic scenario, the solution on the outside of the lipid bilayer has a lower concentration of membrane-impermeable solutes. This scenario results in a net flow of water from the bulk towards the inner aqueous compartment, in turn leading to the swelling of the liposome (Fig. 9a). In extreme cases, the liposomes may burst, releasing their interior towards the outside. A hypertonic situation describes the opposite scenario, which causes a net flow of water from the inner compartment towards the bulk resulting in liposome shrinkage (Fig. 9b). For instance, liposomes based on egg yolk phosphatidylcholine with a typical hydrodynamic diameter of 140–150 nm as determined by DLS, decreased by 20 nm upon addition of NaOH (to pH > 11) to the bulk (outside) solution.40 This physical behaviour was assigned to the impermeability of the lipid bilayer towards Na+. Finally, in the isotonic situation (Fig. 9c) the total contribution to the osmotic pressure of all membrane-impermeable solutes in the inner aqueous compartment and in the bulk, are balanced, resulting in a net-zero movement of water, and a time-independent size of the liposomes. A hypothetical dilution of such a liposome sample with pure water would result in a decrease of the bulk concentration of NaCl and hence yield a hypotonic situation and swelling of the liposomes. To conclude, liposomes are very dynamic supramolecular systems and simple dilutions with MilliQ water for example can lead to drastic changes in the size and integrity of the liposomes. In principle, addition of redox or photoactive components on one side of a liposome, should always be done with isotonic solutions, prepared with an osmometer.
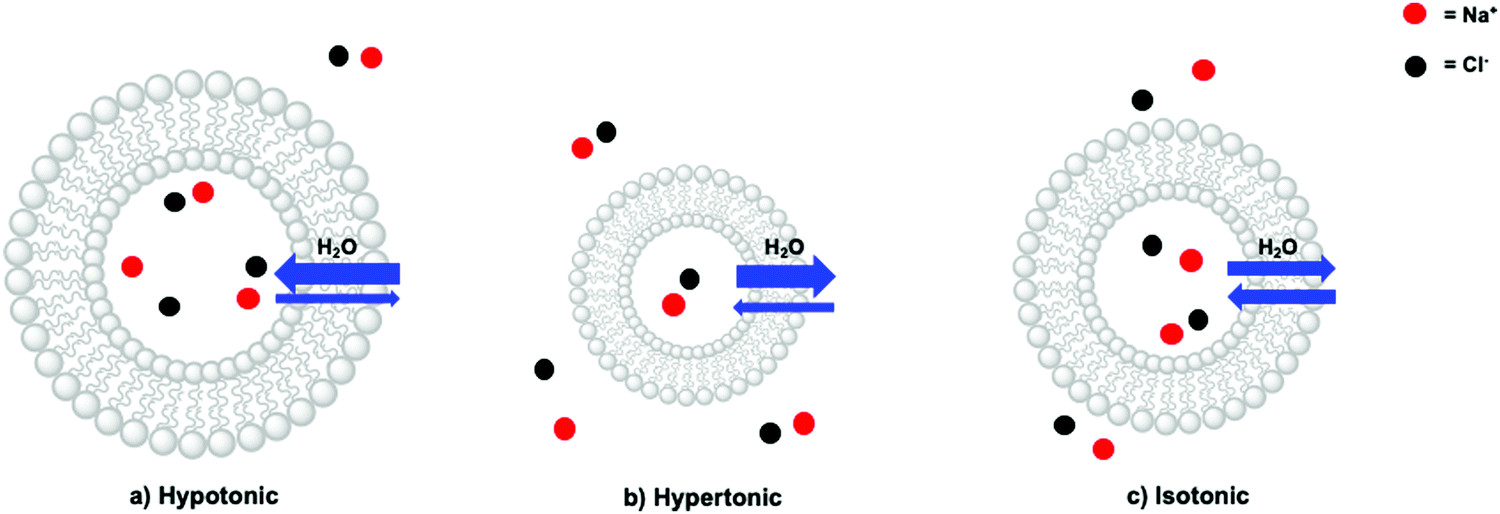 | ||
| Fig. 9 Different ions and ion concentrations lead to osmotic pressure, which results in (a) a hypotonic, (b) hypertonic, or (c) isotonic situation. Amount of red and black dots represent relative concentrations. | ||
2.4 Membrane permeability and leakage
| Ji = Ki·A·(Ci,outside − Ci,inside) | (11) |
| Ki = (P·Di)/d | (12) |
P measured at n-octanol/water biphasic mixtures, Di is the 2D diffusion constant of solute i (in cm2 s−1), and d is membrane thickness (in cm). As can be observed from eqn (11), a high concentration gradient and a high surface area of the membrane result in a higher rate of diffusion, whereas a thick membrane results in a lower rate of diffusion. The thickness of the membrane depends on the character of the head group, the length of the alkyl tails, and the packing of the lipids.15 For example, it was demonstrated that the permeability of phospholipid bilayers in their fluidic phase (T > Tm) to protons, potassium ions, water, urea, and glycerol, decreases with chain length (from 14 to 24 carbon atoms) for unsaturated lipids; for instance, proton permeation was reduced from Ki = 1.3 × 10−2 cm s−1 to 4.9 × 10−5 cm s−1, when increasing membrane thickness from 2.0 nm to 3.8 nm.41
The permeability coefficient (P) varies per molecule and is dependent on the charge, polarity, size and molecular mass of the molecule in motion. If ΔG is the change in free Gibbs enthalpy (kJ mol−1) involved when moving a solute from the bulk aqueous phase to the hydrophobic core of the membrane, one can estimate whether a molecule can cross the lipid bilayer (ΔG ≤ 0) by using logP as a guideline (Fig. 10). In general, very hydrophobic molecules dissolve well inside the hydrophobic lipid bilayers and accumulate there, such as DPPC and cholesterol (logP ≫ 0, Fig. 10c). Amphiphilic small molecules characterised by logP ≈ 0, which includes gas molecules relevant to artificial photosynthesis (O2, CO2, N2, or H2), can either accumulate or diffuse across the lipid bilayer (Fig. 10b). Neutral polar molecules, such as H2O, can also diffuse through a lipid bilayer albeit much slower than small hydrophobic molecules, because they prefer to be solubilised by water.40 On the other hand, hydrophilic, charged molecules such as PS1 or calcein characterised by a logP ≪ 0, are poorly permeable to the lipid membrane because it is too costly to dissolve them in the hydrophobic core of the membrane (Fig. 10a).21,42 Similarly, ions, protons, and electrons, are poorly permeable to the membrane and therefore their transfer across a lipid bilayer has to be facilitated by other means such as membrane channels or ion pumps (see Section 2.3 for more details). To illustrate these points with some numbers, the permeability coefficients through lecithin-based lipid bilayers are generally in the order of 10−3 to 10−4 cm s−1 for H2O, 10−11 to 10−12 cm s−1 for anions, and 10−12 to 10−14 cm s−1 for cations that are commonly utilised such as Na+.40 In some other cases certain charged molecules, such as the charge carrier cetyl methyl viologen (CMV), have a different mechanism to cross the lipid bilayer. The stable and twofold positively charged form, CMV2+, is water soluble and barely penetrates the lipid bilayer. Likewise, the singly reduced species CMV+ is still positively charged and anticipated to rest in the aqueous phase. However, upon a disproportionation reaction two singly-charged CMV+ molecules can generate a CMV2+ and a neutral CMV0 molecule, which can freely diffuse across a lipid bilayer.43
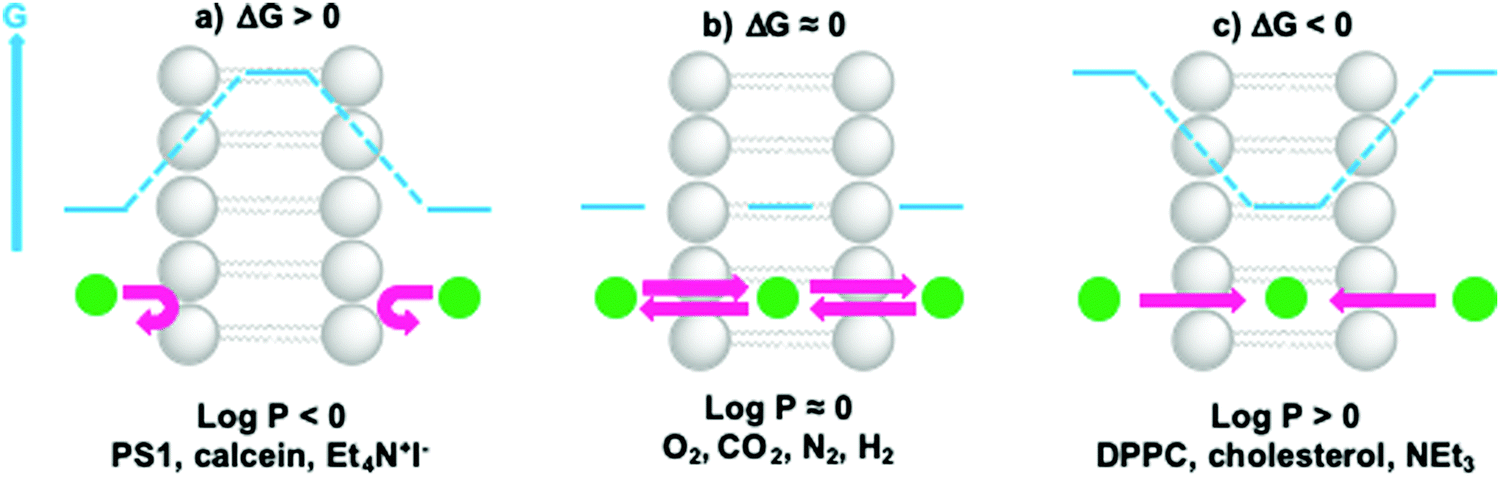 | ||
| Fig. 10 Membrane permeability and residence time of ions and molecules depends on their hydrophilicity. When (a) logP ≪ 0, ΔG > 0 and molecules cannot cross or enter the lipid bilayer; when (b) logP ≈ 0, ΔG ≈ 0, molecules can redistribute between the aqueous phase and the hydrophobic core of the lipid bilayer, and cross the membrane; when (c) logP ≫ 0, ΔG < 0 and molecules prefer to reside within the lipid bilayer. Actual logP values can be found in references16,21,42 or can be obtained by using logP predictors such as miLogP2.2 (http://www.molinspiration.com). For example, logP = +7.62 for cholesterol, logP = +0.09 for O2, logP = +0.02 for CO2, logP = +0.02 for N2, and logP = +0.16 for H2. | ||
Although membrane leakage has mainly been studied for liposome-based drug delivery, it is also an important aspect in artificial photosynthesis applications, especially when preparing dissymmetrical liposomes for transmembrane electron, proton, or ion transfer (see Section 2.6). For such systems, the interior of the liposome contains a chemical that is absent from the outside bulk solution, or vice-versa. To demonstrate transmembrane electron- or proton-transfer, both sides of the membrane should not exchange molecules by membrane leakage. For example, transmembrane electron transfer was initially reported for a system containing the charge carrier MV2+ (see Fig. 7a) in the inner aqueous compartment of sodium dihexadecyl phosphate (DHP) vesicles, whereas the PS1 and the sacrificial ED (EDTA) were located in the bulk.7 However, several studies pointed to the photoredox reaction between photoexcited PS1 and MV2+ actually occurring in the bulk, due to leakage of both MV˙+ and PS1 across the DHP bilayer.7
As a side note, the membrane permeability of molecules does not depend only on their logP, as water-soluble compounds may also permeate membranes via holes or ruptures in the membrane. The lipid composition, for example, was shown to play an important role in membrane leakage. Liposomes prepared from the unsaturated lipid DOPC (Tm = −17 °C) have a larger release rate at room temperature for water-soluble molecules than liposomes prepared from the saturated lipid DPPC (Tm = +41 °C).42 Generally, vesicles prepared from saturated lipids with longer alkyl chain lengths pack more tightly, possess a high gel-to-liquid crystalline transition temperature (Tm), and retain entrapped solutes better than unsaturated lipids and shorter alkyl tail lengths who have a less rigid lipid bilayer assembly. Furthermore, addition of cholesterol to the lipid composition leads to increased lipid bilayer stability and less membrane leakage. Last but not least, liposomes are leakier near their phase transition temperature Tm due to the co-existence of gel-phase and liquid-phase domains within the membrane, which generate grain junction through which polar molecules may diffuse. As a practical consequence, vesicles constructed from DMPC, which has a transition temperature Tm = +23 °C that is near room temperature, are typically leakier in room-temperature experiments, compared to DPPC-based liposomes which have Tm significantly higher than 20–25 °C (Fig. 11).44 Photocatalytic membranes based on DMPC should hence be studied at lower or higher temperature than room temperature.
 | ||
| Fig. 11 Water-soluble substrate (pink diamonds) crosses membranes more easily near the transition temperature Tm due to permeable grain junction between fluid phase and gel phase domains. | ||
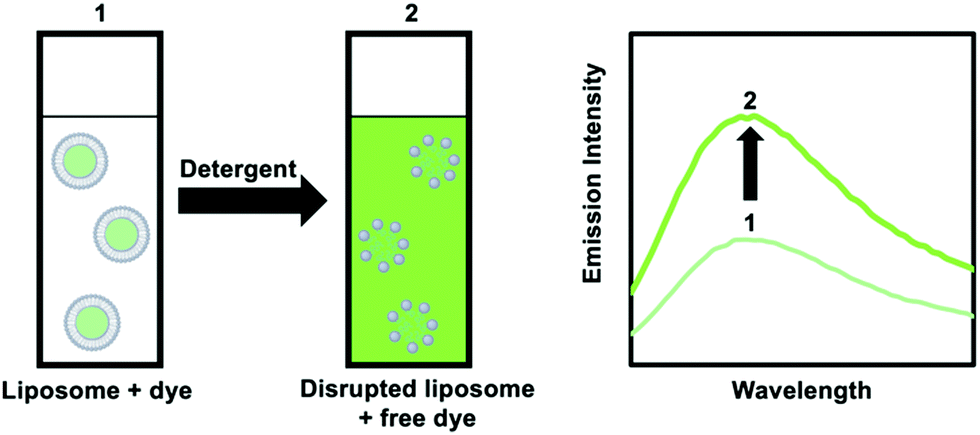 | ||
| Fig. 12 Addition of a detergent to a liposome solution with an encapsulated and weakly luminescent dye results in the release of the dye and enhanced emission intensity. | ||
An alternative method is to use dye-quencher pairs, such as 8-aminonapthalene-1,3,6-trisulfonic acid with p-xylene-bis-pyridinium bromide (ANTS/DPX), and fluorescent enhancement pairs, like Tb3+ and dipicolinic acid (Tb3+/DPA).45 For ANTS/DPX, both molecules are co-encapsulated in the vesicle and quench each other; upon disruption of the lipid bilayer by a detergent luminescence increases because quenching becomes smaller. The latter case involves one of the only positively charged probes used for leakage tests. Here, the weakly luminescent Tb3+ ions leaks from the interior of the liposome and coordinates to DPA present in the bulk outside solution, leading to the formation of a highly luminescent [Tb(DPA)3] complex.45
2.5 Shape of vesicles
Vesicles exist in different sizes and shapes and their preparation is described in Section 3.1. Vesicles that are visible with a microscope should have a micrometre diameter and are called “giant” vesicles (Fig. 13a). Vesicles that are smaller than 1 μm are either called “large” or “small” vesicles (Fig. 13b). Both types can be unilamellar (only one bilayer) or multilamellar (several bilayers) with multiple membranes forming an onion-like “vesicle within a vesicle”. Liposomes are spherical vesicles made of at least one unilamellar membrane. In principle, the larger the vesicle becomes, the more shapes are possible, which can be seen in the confocal image of membrane-stained giant vesicles in Fig. 13a. Apart from spheres, ellipsoids, tubes and other shapes have also been described. | ||
| Fig. 13 (a) Confocal microscopy image of a luminescent dye embedded in the lipid bilayer of giant DMPC vesicles. Figure adapted from ref. 39. CryoTEM images of (b) extruded DMPC (Tm = 23 °C) vesicles vitrified from 40 °C, (c) extruded DPPC (Tm = 41 °C) vesicles vitrified from 25 °C, and (d) sonicated DLPA (Tm = 31 °C) vesicles vitrified from 18 °C. Adapted with permission from M. Andersson, L. Hammarström and K. Edwards, J. Phys. Chem., 1995, 99, 14531–14538. Copyright (1995) American Chemical Society.34 | ||
The morphology of the lipid bilayer of liposomes depends on the lipid head group, the length of the hydrocarbon tails, the size of the liposomes, the transition temperature of the lipid, and the preparation methodology. Decreasing the temperature of a liposome solution to values below Tm, may result in faceted vesicles (Fig. 13c) or the creation of open bilayer fragments (Fig. 13d) instead of smooth round vesicles, as can be experimentally visualised by cryogenic transmission electron microscopy (CryoTEM, see Fig. 13b–d).34 These morphological changes have an effect on photoredox reactions, such as the reduction of CMV by dithionite (S2O42−).34 In the case of smooth vesicles, monoexponential reduction kinetics can be observed, whereas the presence of bilayer irregularities and non-vesicular structures can lead to double-exponential reduction kinetics.34 Therefore, in order to ease the study of complex photocatalytic reactions occurring at the membrane–water interface, such as electron, proton, or ion transport across a lipid bilayer,34 one should aim to use regular unilamellar vesicles either in the fluid or the gel phase.
2.6 Transport of electrons, protons, and ions across a lipid bilayer
Biological transmembrane electron transfer takes place in cell compartments of photosynthetic and respiratory organelles to allow for redox reactions to occur and to generate an imbalance in electrochemical potential across the membrane. This imbalance is translated into a proton motive force that is ultimately used as an energy source to drive endergonic transformations such as the generation of ATP by the membrane-embedded ATPase. There are also examples for ion transfer that act without proton motive forces, such as simple passive transmembrane protein channels such as gramicidin A, aquaporin, the prokaryotic potassium channels of streptomyces A, or even artificial ion channels or transporters. In this review, we only consider the light-induced transport of electrons, protons, and ions across lipid bilayers for artificial photosynthesis.Electron transfer through a lipid bilayer cannot generally occur by direct tunnelling between reactants on either side of the membrane as the thickness of the hydrophobic core of typically ca. 30–35 Å is too high to allow direct charge transfer at reasonable rate. The electron transfer rate, k, is given by eqn (13):
| k = A·e−βr | (13) |
To mimic natural transmembrane redox processes the following strategies are possible:
(a) To incorporate biologic components into the lipid bilayer such as natural reaction centres and membrane-bound electron transfer proteins (Fig. 14a–e).
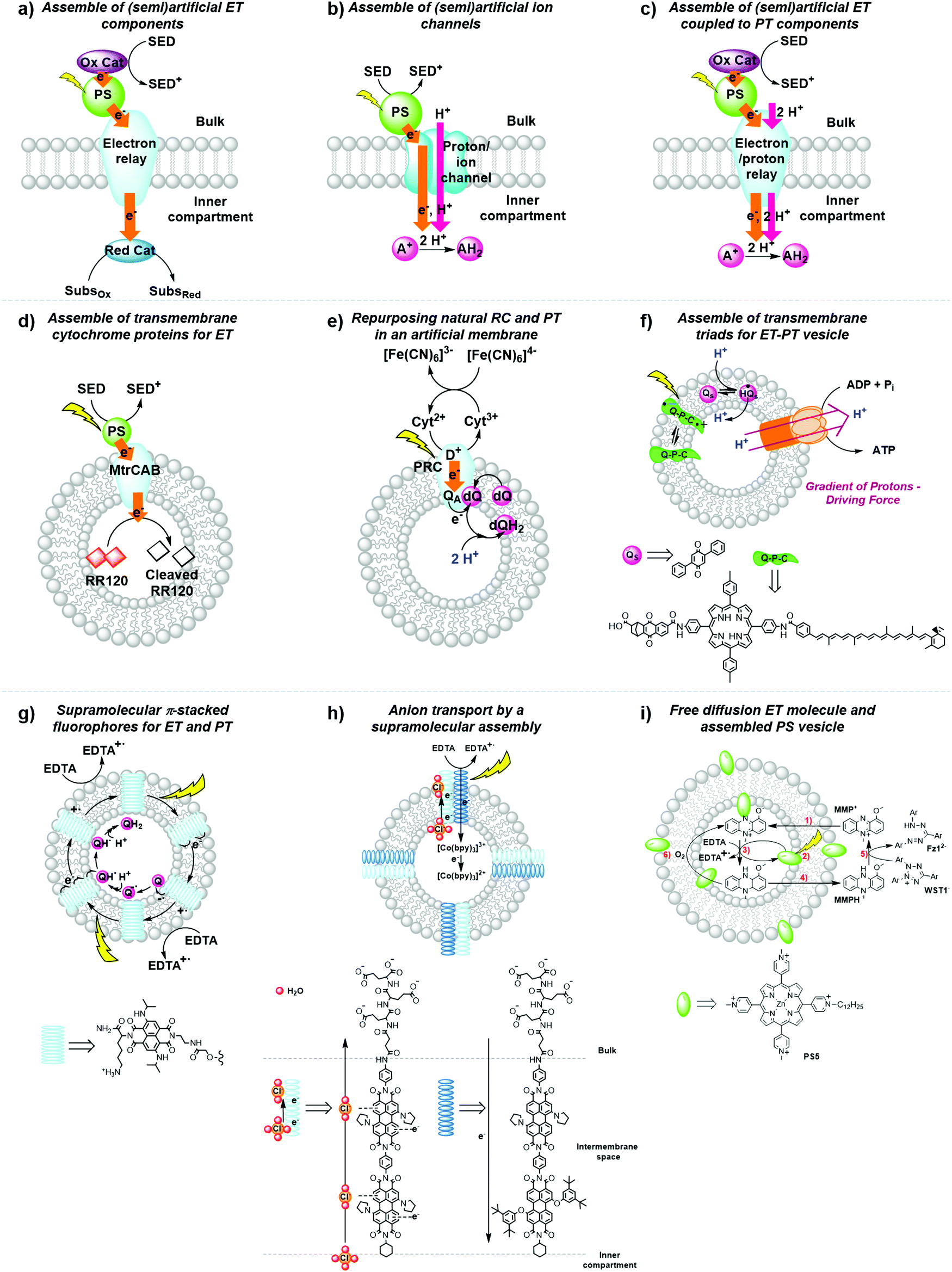 | ||
| Fig. 14 General strategies for the development of (semi)artificial liposome systems (a–c) and selected examples (d–i) for proton and electron transfer across lipid bilayers. Assemblies of (a) (semi)artificial electron transfer (ET) components, (b) (semi)artificial ET coupled to proton transfer (PT) components across the membrane, (c) (semi)artificial ion channels, (d) transmembrane cytochrome (MtrCAB) proteins for ET,5 (e) repurposed natural photosynthetic reaction centres (PRC) and proton transfer components in an artificial membrane,47 (f) transmembrane quinone–porphyrin–carotenoid triad (Q–P–C) for coupled ET-PT across the membrane,33,35 (g) supramolecular π-stacked fluorophores for coupled ET and PT,48 (h) supramolecular transmembrane rods for electron and anion transport,49 and (i) membrane-embedded PSs combined with a diffusion-based ET molecule.26 Ox. Cat. = oxidation catalyst; Red. Cat. = reduction catalyst; SED = sacrificial electron donor; SubsOx = oxidised substrate; SubsRed = reduced substrate; A+ = electron acceptor; AH2 = reduced and protonated electron acceptor; Qs and dQ = membrane-soluble quinone (Fig. 7), D+ = donor; QA = quinone A from the isolated reaction centre. | ||
(b) To synthesise transmembrane molecules spanning the lipid bilayer, such as rigid molecular triads or supramolecular assemblies of electron conduits (Fig. 14f–h).
(c) To organise the diffusion of molecular charge carriers embedded within the lipid bilayer of artificial membranes (Fig. 14e, f and i).
Although many examples have been reported,7 we only present here a selection of examples to illustrate the three strategies discussed above.
Strategy a is best represented by a semiartificial compartmentalised liposome system (Fig. 14d) that used the proton-coupled icosa-haem transmembrane electron transfer protein complex, MtrCAB (analogous to S. Baltica OS185's structure shown in Fig. 7c).5 MtrCAB is a cytochrome-type protein complex found in Shewanella oneidensis MR-1 bacteria, where it acts as a natural transporter of electrons to external minerals. In the reported system, phosphonated ruthenium(II) tris-bipyridine ([Ru(bpy)2(4,4′-(PO3H2)2bpy)]2+, RuP) photosensitised titanium oxide nanoparticles (RuP-TiO2), and carbon nanodots were employed as synthetic light harvesters placed outside the liposome. To demonstrate spectroscopically transmembrane electron transfer with MtrCAB embedded in a synthetic liposome, the redox active dye, reactive red 120 (RR120), was encapsulated inside the liposome and showed the expected bleaching of the band at 539 nm upon photo-reduction. The sacrificial electron donor EDTA regenerated the hole left in the light-harvesting nanoparticles. The rate limiting step was identified as the electron transfer from the light-harvesting nanoparticles to the MtrCAB electron relay.5 In a similar example, a re-purposed photosynthetic reaction centre, isolated from R. sphaeroides bacteria, was embedded in the membrane of a giant unilamellar vesicle with the desired physiological orientation by following a droplet transfer synthetic method (Fig. 14e).47 With this protocol roughly 90% of the photosynthetic reaction centre was facing the aqueous bulk solution and, thus, 90% of the quinone sites (QB) were facing the inner space of the lipid bilayer, thus creating an artificial lumen that enabled the generation of a light-driven proton gradient across the membrane. This semiartificial vesicle was used in combination with the water-soluble ED cytochrome c2, the natural electron donor to this reaction centre, as well as ferrocyanide ([Fe(CN)6]4−) in the bulk; on the other hand, the quinone on the inner monolayer of the vesicle was used as final EA. Under constant red-light irradiation (λ = 865 nm) the repurposed reaction centres converted light energy to the energy of a proton gradient across the lipid bilayer at about 0.061 pH min−1, which is equivalent to a proton motive force of 3.6 mV min−1. Hence, the reduction and subsequent protonation of the quinone within the membrane induced a pH gradient in addition to the electrochemical potential.47
In strategy b, fully synthetic examples were developed to couple transmembrane electron transfer to transmembrane proton transfer. A prominent example is the synthetic molecular triad (Q-P-C, Fig. 14f) consisting of a naphthoquinone moiety as electron acceptor (Q), the PS tetraarylporphyrin (P), and a carotenoid as electron donor (C). This triad was embedded into a lipid bilayer and transported electrons across a membrane upon light irradiation.33 Control titration studies showed that the majority of triads were arranged with their Q ends facing the aqueous bulk, while the C moiety spanned the lipid bilayer towards the inner aqueous compartment. Irradiation of the triad with laser pulses at 430 nm excited the porphyrin group, which results in the formation of a charge separated species Q−–P–C+ that could be detected by the transient absorbance of the carotenoid radical cation at 930 nm. This biradical was generated with a quantum yield of 0.1 and had a lifetime of 60 ns within the lipid bilayer in the presence of freely diffusing quinones (Qs), which established a reduction potential near the outer surface of the bilayer and an oxidation potential near its inner surface. In response to that, Qs accepted an electron from the Q− part of the triad generating a reduced and anionic quinone that got protonated and hence transferred protons across the bilayer by electron transfer to the oxidised C− part of the triad, with an overall quantum yield of 0.004.33 This process turned over during light irradiation, generating a pH gradient between the inner compartment and the bulk.33 Ultimately, in a follow-up study, the proton motive force generated by this artificial liposome was coupled to a membrane-bound ATP synthase complex in the same lipid bilayer, which upon irradiation resulted in the synthesis of ATP with an initial quantum yield at low-light irradiation of more than 0.07 for the whole process within the new assembly.35 In another example, p-octiphenyl rods were used to create a helical tetrameric supramolecular assembly with π-stacked blue and red-fluorescent naphthalene diimides spanning the lipid bilayer of a liposome, which also contained freely diffusing quinones as EA (Fig. 14g). The functionalised liposomes were immersed in an aqueous solution containing EDTA as ED in the bulk. The irradiation of the π-stacked dyes with visible light generated a fast electron transport across the interacting chromophores until the electron was finally accepted by the quinone in the inner aqueous compartment, which was accompanied by proton transfer to the quinone. The long-lived charge separated state across the membrane was hence translated into an oxidation potential near the outer surface of the membrane and a reduction potential near the inner surface, as well as a proton gradient across the membrane.48 In a third and last example of coupled electron and ion transmembrane transfer, rigid oligo(p-phenyl-ene)-N,N-perylenediimide rods were embedded in egg yolk phosphatidylcholine based vesicles (Fig. 14h).49 These rods were able to perform photoinduced transmembrane electron transfer from EDTA in the bulk to the electron-accepting charge carrier [Co(bpy)3]2+ in the inner aqueous phase, while simultaneously transporting Cl−, OH− or SO42− anions, in the reverse direction. Interestingly, the rods failed to transport cations at all and its selectivity for anion transport was thus proposed to result from anion-π interactions along the π-acidic environments of the oligo-aromatic rods.49
Finally, in strategy c the membrane-soluble molecular charge carrier MMP+ was employed for unidirectional transmembrane electron transfer in liposomes. In this system, PS5 was embedded in the membrane, EDTA was placed in the inner space of the liposome as ED, and 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium anion (WST1−) was finally added in the exterior aqueous bulk solution as EA (Fig. 14i).26 In an anaerobic atmosphere, MMP+ catalytically transported photoelectrons across the membrane without limiting the overall reaction rate. In such conditions late charge recombination of reduced WST1− with ED+, as well as quenching of PS5* excited states by the reduced WST1− were avoided by their physical separation with the liposomal membrane. However, as soon as oxygen was introduced in the system transmembrane photoelectron transfer stopped occurring, because MMPH, the reduced form of MMP+, reacted with O2 faster than with WST1−. This limitation shows how tricky it is to avoid charge recombination even at a late stage of the photocatalytic cycle.
3. The water membrane interface
3.1 Preparation of liposomes and generation of confined aqueous reaction spaces
Vesicles provide three different reaction spaces: the inner aqueous compartment, the hydrophobic core of the membrane, and the outer aqueous bulk solution. These three spaces can be addressed separately, loaded with different photocatalytic components, and characterised via different experimental techniques. Table S1 (ESI†) lists these techniques and which information one can obtain from them.By self-assembly of lipid bilayers into vesicles a high local concentration of reactants can be achieved in the bilayer or by the encapsulation of the substrates in the inner aqueous compartment, even upon dilution of the vesicle solution. As an example, a vesicle with a diameter of 100 nm has an internal volume of approximately 10−13 μL. Confinement within the vesicle can be important because the local concentration of water soluble substrates and products influences reaction dynamics in water and at the interface of the membrane.1,9 Furthermore, the hydrophilic products from water oxidation and CO2 reduction, such as protons and formate, respectively, might affect membrane stability due to changes of the osmotic pressure. For example, when oxidising two water molecules to one molecule of O2 that diffuses away, four protons are generated as well, which changes the pH and osmotic pressure.
For unidirectional electron transfer through membranes, it can be particularly appealing to differentiate the inner and outer reaction spaces in vesicles, for example by loading the interior space of the vesicles with functional molecules or substrate, or by adding after liposome preparation, compounds that will only be present on the outside. Among all methods to prepare liposomes, a standard one is depicted in Fig. 15. Firstly, a solution of lipids and membrane-soluble components, such as PSs and catalysts (see Section 1.3) dissolved in organic solvents, is transferred to a glass tube or flask. Subsequently, the organic solvents are removed under reduced pressure, resulting in the formation of a thin lipid film on the glass wall of the tube or flask. In the next step, the vacuum dried thin lipid film is hydrated using an aqueous buffer containing water-soluble components, such as sacrificial reagents or charge carriers (Buffer 1). Several freeze–thaw cycles (in some cases also sonication) induce the formation of giant multilamellar vesicles (diameter > 1 μm). To obtain liposomes (diameter < 1 μm), extrusion with a suitable pore size filter, sonication, or dialysis, can be performed. Extrusion typically provides relatively narrow hydrodynamic diameter distributions characterised by low polydispersity index (PDI) in DLS or cryo-TEM analysis (PDI < 0.1). At this stage both sides of the membrane are identical; however, the bulk solution can be replaced by a second and different aqueous buffer solution (Buffer 2) containing water-soluble components that are absent from the interior; using size exclusion chromatography (SEC), it is then possible to isolate the liposomes filled with Buffer 1 from excess Buffer 1. Dissymmetrical liposomes are then obtained, filled with Buffer 1 and surrounded with Buffer 2. As mentioned above, with such liposomes it is critical to use two Buffers 1 and 2 with identical osmotic pressure, as measured by an osmometer. Otherwise, differences in osmotic pressure on both sides of the membrane lead to the uptake or release of water from the inner aqueous compartment, which may make the membrane leaky (see Section 2.3).
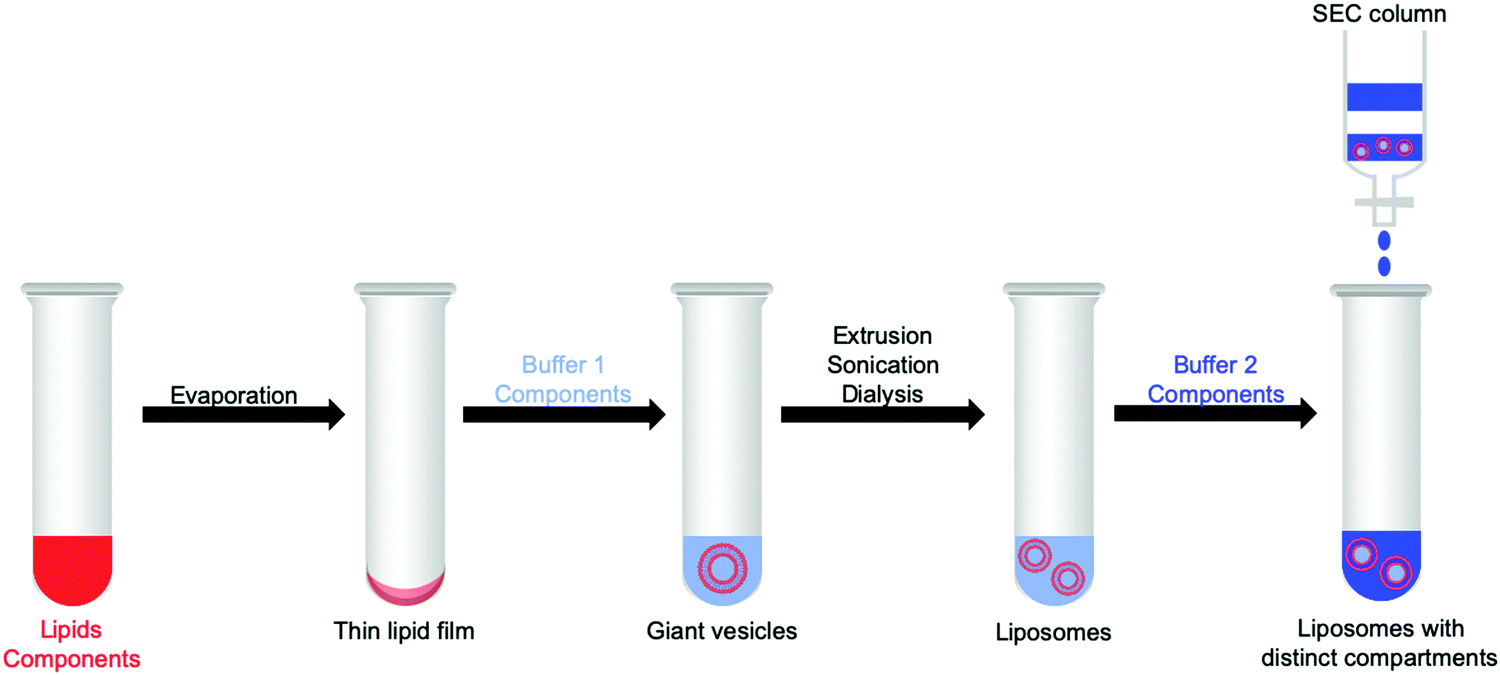 | ||
| Fig. 15 Method for the preparation of liposomes with distinct compartments that are functionalised with active components for artificial photosynthesis. SEC = size exclusion chromatography. Membrane soluble components are shown in red, whereas water-soluble components are shown in light or dark blue. Light blue shows Buffer 1, dark blue Buffer 2 (see text). | ||
3.2 Properties of the interface
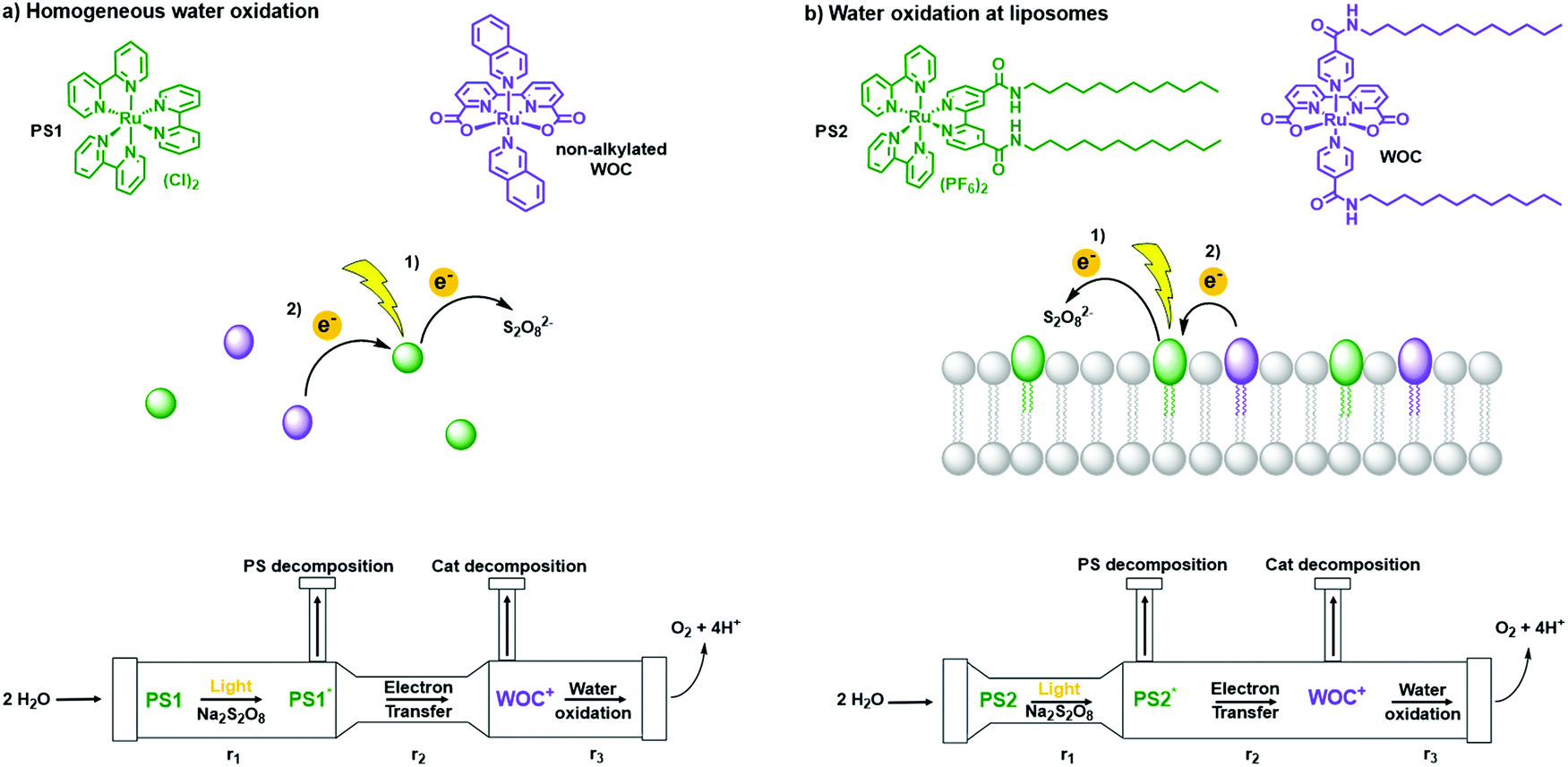 | ||
| Fig. 16 Water oxidation in (a) homogeneous solution and at (b) liposomes. (top) Photosensitisers (green) PS1 and PS2 and catalysts (purple) non-alkylated WOC and WOC. (middle) Schematic step-by-step electron transfer in homogeneous conditions and at liposomes. (bottom) Scheme of three rate determining steps (RDS) in photocatalytic water oxidation under homogeneous and liposome conditions. Figure adapted with permission from ref. 50. | ||
Another surface concentration effect that should be noted is related to protons; proton management is indeed important, as most redox reactions in artificial photosynthesis are coupled to proton transfer (PCET, see for example eqn (1)–(6)). At lipid bilayer interfaces the protonation–deprotonation dynamics are different than in homogeneous solution. This protonation–deprotonation equilibrium is described by the acidity constant, pKa. The pKa of a proton-donating or accepting group at the bilayer-water interface differs from the value in bulk water solutions, essentially due to different surface polarities (dielectric constant), and the electrostatic enhancement or reduction of proton concentration at the bilayer interface due to surface charge changes. For example, while the acidity constants of most lipids are tabulated,15 it should be noted that pKa values at a membrane can shift by up to ΔpKa = 2.5.15
:1 POPC:POPS (i.e. 20% content of anionic lipid) had higher adsorption levels of monovalent cations, in comparison with zwitterionic neutral vesicles. Furthermore, Na+ was found to be the strongest cation adsorbed, followed by K+ and then Cs+. The rigidification of the membranes was explained by the formation of lipid-cation complexes via the coordinating oxygen atoms in carbonyl groups of hydrophilic lipid head groups to the respective cations.
3.3 Adsorption of active molecules at the water–membrane interface
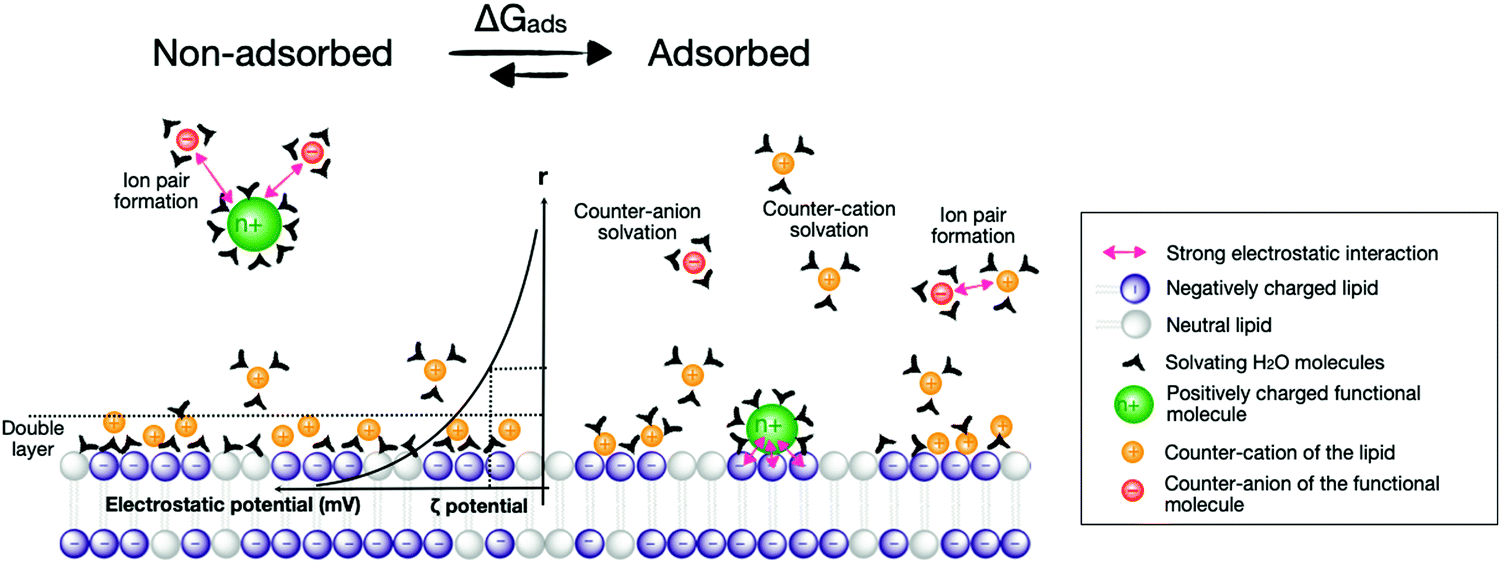
Considering the adsorption of active components to membrane surfaces is important both during the construction of photoactive liposomes and during the light-induced chemical reactions itself, electrostatic and hydrophobic interactions play a major role in the overall photocatalytic scenario. In terms of energy, the supramolecular adsorption of molecules onto lipid bilayers can be described in terms of a solvation–desolvation process, where all supramolecular forces describing the interaction between the functional molecule and the membrane have to be considered, i.e., charges and hydrophobicity indeed, but also π–π stacking, dipolar interactions, van der Waals interactions, hydrogen bonding, and ligand coordination to metals. Altogether, these interactions combine into a free Gibbs energy of adsorption of the photoactive molecule to the membrane (ΔGads) that is composed of an enthalpy (ΔHads) and an entropy (ΔSads) term (eqn (14), Fig. 17).| ΔGads = ΔHads − T·ΔSads | (14) |
 | ||
| Fig. 17 Adsorption of charged photoactive molecules (in green) onto a negatively charged membrane (negatively charged lipids in purple). The left side represents the non-adsorbed state, the right side the adsorbed state. Counter anions of the functional molecules (in red) and counter cations of the lipids (in orange) are also exchanged and re-solvated upon adsorption, which plays a major contribution in the free Gibbs energy of adsorption (ΔGads) of the functional molecule onto the membrane. Black symbols represent (a selection of) the water molecules solvating the charged species, either in the bulk, or at the membrane. Pink double arrows represent electrostatic attraction between charged species in the system. The double layer and evolution of the electrostatic potential and the zeta (ζ) potential are shown as a function of the distance (r) from the membrane. | ||
Both ΔHads and ΔSads have the following contributions:
(a) Supramolecular interaction between the functional molecule and lipid membrane.
(b) Solvation of the functional molecule and its counter anions/cations in the aqueous phase.
(c) Solvation of the lipid membrane and its counter anions/cations in the aqueous phase.
(d) Ion pair formation between the counter anions and counter cations, which are released from the membrane upon adsorption.
Negatively charged lipid bilayers are normally surrounded by NH4+, Na+ or K+ counter-cations, and positively charged metal complexes bring along anions such as Cl− or PF6−. Upon adsorption of a positively charged functional molecule to the surface of negatively charged lipid bilayers the counter-ions can be exchanged, a process that has thermodynamic consequences. In the exemplary case of the adsorption of one dicationic functional molecule such as [Ru(tpy)(bpy)(OH2)]2+ (tpy = 2,2′:6′,2′′-terpyridine) onto negatively charged DMPG based lipid bilayers involves the desorption of two Na+ from the bilayer surface.51 Depending on the respective ion pairing and solvation energies and entropies, this process can either be endothermic or exergonic due to a significant energetic contribution from the entropy term of ion release (two equivalents of Na+ cations) to the bulk.51
3.4 Reaction dynamics at water–membrane interfaces
Reaction dynamics differ between vesicles-supported reactions and reactions in homogeneous solutions. In a homogeneous solution, components interact in a three-dimensional manner, whereas in vesicles components are normally immobilised on a two-dimensional lipid bilayer, which forms the platform of the photoreactions. One method to study the effects of reduced dimensionality on reaction kinetics is bimolecular fluorescence quenching with PSs and quenchers attached to the membrane. For three-dimensional bimolecular reactions, fluorescence quenching with excess of quencher molecules will show a normal exponential decay of fluorescence vs. time, with a rate law that is pseudo-first order in quencher concentration [Q]. For reactants diffusing in 2D within the vesicle interface, the fluorescence decay will instead be strongly non-exponential. This can be qualitatively understood by considering the concentration profile of quenchers as a function of distance from excited molecules, [Q] = f(r) (Fig. 18). At the time of excitation (t = 0) the quenchers are randomly distributed in solution and [Q] will be independent on r. The excited PSs that are next to a quencher will react rapidly, and the remaining excited PSs will be the ones with larger distance to the nearest Q. With time, the reaction will consume all quenchers at a short distance, leaving a “concentration hole” around the remaining excited states. At the same time, diffusion of quenchers from the bulk to the vesicle interface will occur to balance the concentration. In a three-dimensional, homogeneous solution, reaction and diffusion rates rapidly equilibrate to set up a steady-state concentration profile of quenchers that then remains for the rest of the reaction, resulting in single-exponential kinetics. When the concentration profile still changes, however, quenching is faster than given by the long-time rate constant. This transient effect in fluorescence quenching is well established52 and is important at times after excitation t ≪ R2/DPSQ, where R is the intermolecular reaction distance and DPSQ is the sum of the diffusion coefficients of PS and quencher. For small molecules in a low-viscosity solvent, this is typically at t < 100 ps.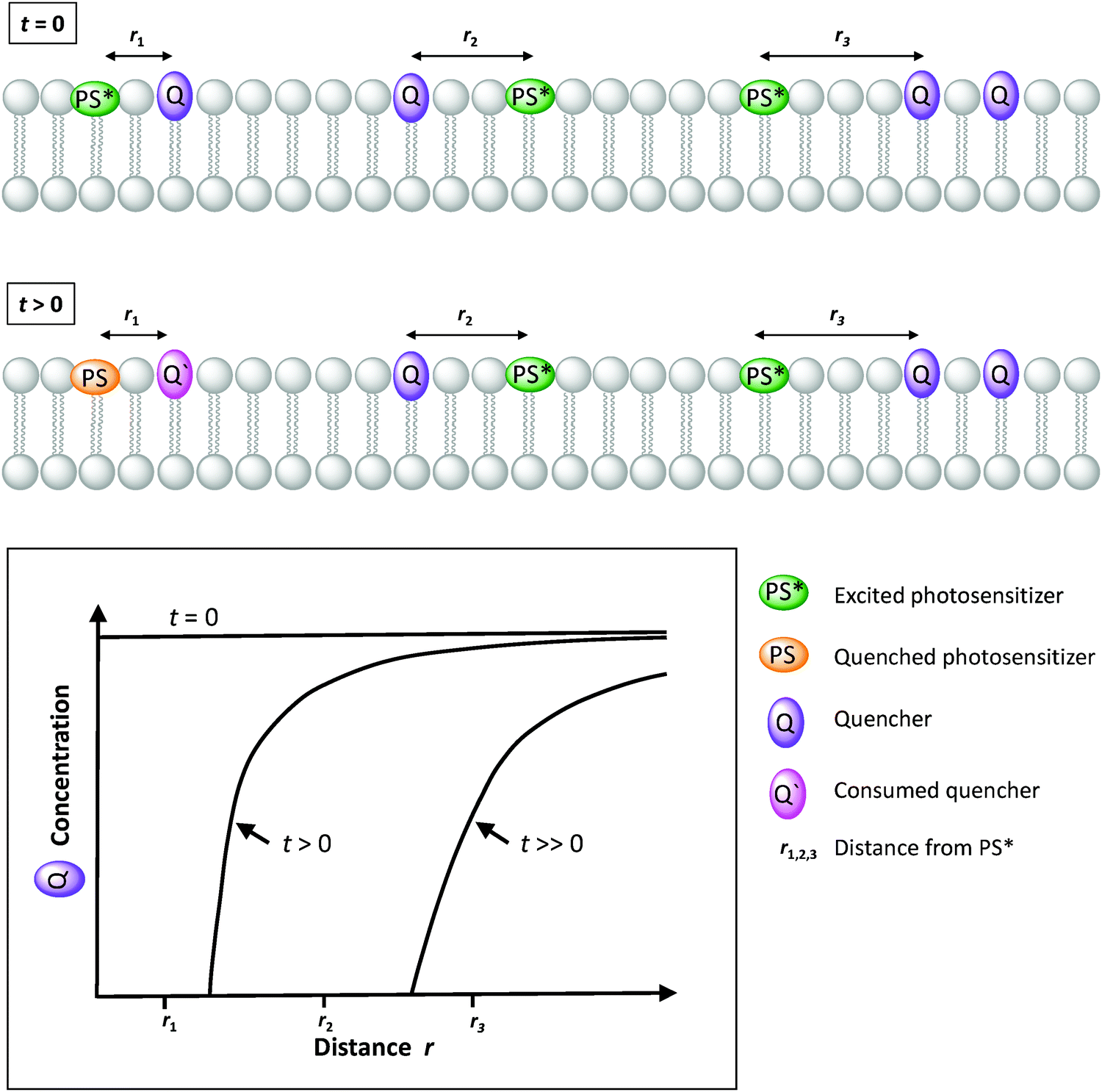 | ||
| Fig. 18 Qualitative diffusion-reaction dynamics at lipid bilayer surfaces containing PS and quenchers (Q) at various distances (r1,2,3) leads to a time-dependent concentration profile of Q as a function of distance from PS*. For three-dimensional (homogeneous) solution, the concentration profile rapidly becomes constant (qualitatively similar to the profile labelled “t > 0”), and the usual pseudo-first order rate constant is obtained. With two-dimensional diffusion instead, the concentration profile keeps evolving and the kinetics is non-exponential. The concentration profiles shown are qualitative and not mathematically exact. | ||
For diffusion in lower dimensions, such as within a two-dimensional lipid bilayer, diffusion can never keep up with reaction, and the “concentration hole” will keep growing as the reaction proceeds. The fluorescence decay is then much more complicated, so that numerical fitting of experimental data becomes unfeasible. An approximate solution for the time dependence of the fluorescence intensity in the case of diffusion-controlled, two-dimensional quenching is given in eqn (15),53 where τ0 is the lifetime of the PS in the absence of quencher and [Q] is the concentration of quencher. The solution for the case of reaction-control (slower than the diffusion limit) is somewhat more complex.54 For very slow quenching, i.e. low reactivity even at contact distance, [Q] near the PSs will be approximately equal to the bulk value, and the quenching kinetics should again be simple and follow a single exponential.
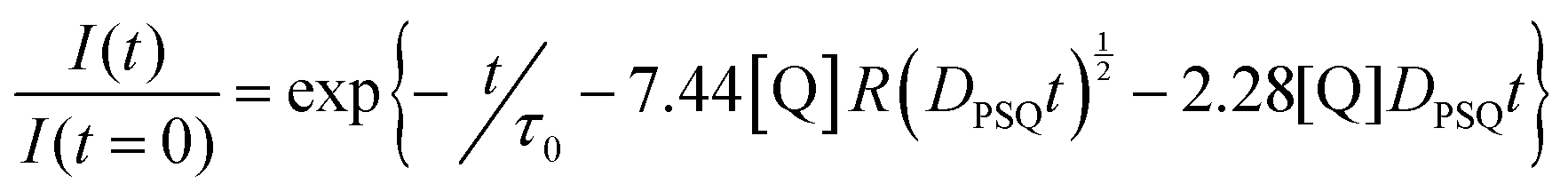 | (15) |
An example where eqn (15) was used to study electron transfer at the interface of vesicles is shown in Fig. 19. The ruthenium PS [Ru(dcb)3]4− (dcb = 4,4′-dicarboxylate-2,2′-bipyridine) was electrostatically associated to egg lecithin vesicles doped with cationic surfactants, and amphiphilic CMV2+ as quenchers.55 The good agreement with eqn (15) suggests that [Ru(dcb)3]4− was localised very close to the surface, with negligible diffusion orthogonal to the membrane surface. The diffusion coefficient obtained from a fit to the data was comparable to that of amphiphiles in bilayers, (6 ± 2) × 10−11 m2 s−1.55 For comparison: diffusion coefficients in homogeneous solution are on the order of 1 × 10−9 m2 s−1.56 As a last remark, these kinetic effects are not only important for energy transfer; eqn (15) is also valid for electron transfer, e.g. the non-fluorescent reactions of for instance an oxidised or reduced PS, in its ground state, with membrane-bound catalysts.
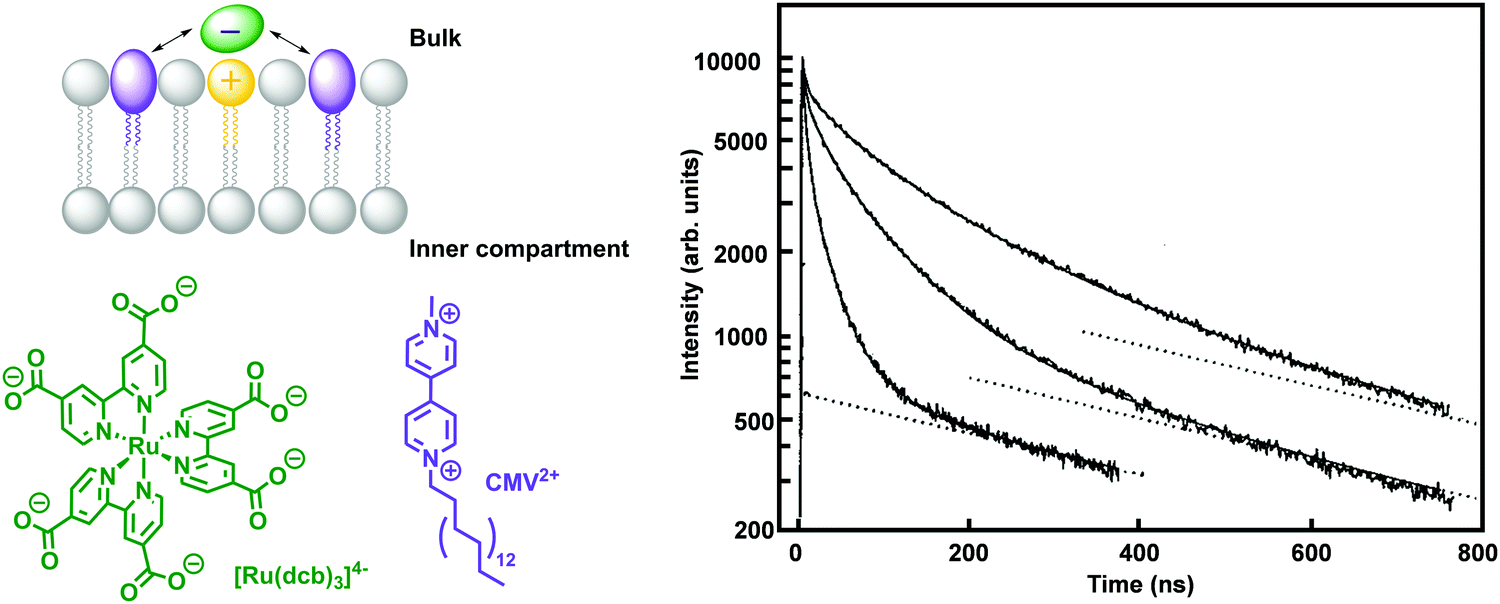 | ||
| Fig. 19 Left: Example of homogeneous PS [Ru(dcb)3]4− (green) electrostatically associated to egg lecithin vesicles doped with cationic surfactants (yellow), and amphiphilic CMV2+ (purple) as membrane-immobilised quencher. Right: Representative time-resolved luminescence decay of [Ru(dcb)3]4− in the presence of CMV2+, showing strongly non-exponential behaviour (the dotted line indicate luminescence from a fraction of [Ru(dcb)3]4− not bound to the vesicles). Adapted with permission from L. Hammarström, T. Norrby, G. Stenhagen et al., J. Phys. Chem. B, 1997, 101, 7494–7504. Copyright (1997) American Chemical Society.55 | ||
5. Conclusion and future prospects
Although it is difficult to predict whether liposomes and membrane-based artificial photosynthetic systems will play any role in solving the energy crisis, they represent an attractive and under-explored concept that complements traditional semiconductor-based developments to drive photocatalysis. In particular, they allow interrogating the coupling of two half-reactions in a single photocatalytic system, and provide a framework for developing innovative solution for avoiding charge recombination and back reactions – the main challenge in photocatalytic artificial photosynthesis. Liposome- and lipid bilayer-based photocatalytic systems also represent a great opportunity to improve our fundamental understanding of supramolecular chemistry, not only in static terms, but also dynamically. In static terms, because one needs to understand how electrostatic forces, hydrophobicity, coordination chemistry, van der Waals interactions, and π–π stacking, can be combined to organise molecules in space and achieve dissymmetric assemblies where electrons get a chance to flow faster in one direction than in the other. Photocatalytic liposomes will also improve our understanding of dynamic self-assembly, because the time-dependent nature of photochemical processes allows for probing how supramolecular chemistry evolves in time under the action of light absorption, energy transfer, and electron transfer, while covalent chemistry often offers decomposition as the final outcome of photoelectron transfer. Photocatalytic lipid bilayers also pave the way towards new strategies to regenerate photochemically or mechanically damaged components, possibly by self-healing processes. These are aspects that are very difficult to implement in covalent systems, but also in most solid-state-materials.Photochemistry on lipid bilayers, however, also represent a formidable challenge. First, photochemically active liposomes are nowadays beyond the limit of what can be modelled by computational methods. It is hence usually difficult to build an accurate theoretical model that fits experimental data. Second, they make time-resolved spectroscopy methods particularly challenging because of the high light scattering generated by liposomes, the size of which precisely fits with the wavelength of visible light. Hopefully, these challenges will be overcome by combining experimental work on photocatalytic liposomes with the knowledge generated by alternative self-assembled electro- or photo-catalytic systems. For example, hybrid photocatalytic electrodes combine small artificial molecules as electron relays and enzymes as catalysts, to transform light energy into current. Photocatalytic soap films have also been proposed, for example in the SOFIA project (GAN 828838), to convert sun energy into a chemical fuel. Vesicle-based compartmentalisation catalysis will also be very useful, as it aims at combining several molecular or enzyme-based catalyst using a vesicle-in-a-vesicle strategy. In such assemblies, small molecules do move across membranes to perform a series of chemical reactions, while the catalysts stay isolated from each other by the different membranes of the compartmentalised system. Ultimately, we are convinced that full understanding of a process as complicated as natural photosynthesis, requires scientists to be able of making, with their own hands, an artificial (or semi-artificial) photocatalytic system that can perform the same function as the natural one: transforming the fleeting, transient energy of photons in a sunlight beam, into the stable chemical energy of a chemical bond.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union*s Horizon 2020 research and innovation program FETOPEN 2018–2020 under grant agreement # 828838 – SoFiA. A. Pannwitz gratefully acknowledges the Vector Foundation. D. M. Klein thanks the Netherlands Organization for Scientific Research (NWO) for an HRSMC PhD fellowship. C. Casadevall acknowledges the European Commission for an Horizon 2020 Marie Skłodowska-Curie Individual Fellowship (890745-SmArtC). C. Casadevall and E. Reisner acknowledge the Biotechnology and Biological Sciences Research Council (BBSRC) for financial support (BB/S00159X/1).References
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- X. Fang, S. Kalathil and E. Reisner, Chem. Soc. Rev., 2020, 49, 4926–4952 RSC.
- I. Rojdestvenski, A. G. Ivanov, M. G. Cottam, A. Borodich, N. P. A. Huner and G. Oquist, Biophys. J., 2002, 82, 1719–1730 CrossRef CAS PubMed.
- B. Daum, D. Nicastro, J. Austin, J. R. McIntosh and W. Kühlbrandt, Plant Cell, 2010, 22, 1299–1312 CrossRef CAS PubMed.
- A. Stikane, E. T. Hwang, E. V. Ainsworth, S. E. H. Piper, K. Critchley, J. N. Butt, E. Reisner and L. J. C. Jeuken, Faraday Discuss., 2019, 215, 26–38 RSC.
- M. Hansen, S. Troppmann and B. König, Chem. – Eur. J., 2016, 22, 58–72 CrossRef CAS PubMed.
- J. N. Robinson and D. J. Cole-Hamilton, Chem. Soc. Rev., 1991, 20, 49–94 RSC.
- J. Flores, B. M. White, R. J. Brea, J. M. Baskin and N. K. Devaraj, Chem. Soc. Rev., 2020, 49, 4602–4614 RSC.
- F. A. Armstrong and J. Hirst, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14049–14054 CrossRef CAS PubMed.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 8572–8610 RSC.
- H.-J. Choi and C. D. Montemagno, Nano Lett., 2005, 5, 2538–2542 CrossRef CAS PubMed.
- M. C. M. van Oers, F. Rutjes and J. C. M. van Hest, Curr. Opin. Biotechnol, 2014, 28, 10–16 CrossRef CAS PubMed.
- D. Hvasanov, J. R. Peterson and P. Thordarson, Chem. Sci., 2013, 4, 3833–3838 RSC.
- T. F. Tadros, An Introduction to Surfactants, De Gruyter, 2014 Search PubMed.
- D. Marsh, Handbook of Lipid Bilayers, CRC Press, 2013 Search PubMed.
- A. Leo, C. Hansch and D. Elkins, Chem. Rev., 1971, 71, 525–616 CrossRef CAS.
- J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press, 2011 Search PubMed.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
- S. Troppmann and B. König, Chem. – Eur. J., 2014, 20, 14570–14574 CrossRef CAS PubMed.
- G. D. Scholes, G. R. Fleming, A. Olaya-Castro and R. van Grondelle, Nat. Chem., 2011, 3, 763–774 CrossRef CAS PubMed.
- C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2007, 129, 46–47 CrossRef CAS PubMed.
- M. Hansen, F. Li, L. Sun and B. König, Chem. Sci., 2014, 5, 2683–2687 RSC.
- B. Limburg, J. Wermink, S. S. Van Nielen, R. Kortlever, M. T. M. Koper, E. Bouwman and S. Bonnet, ACS Catal., 2016, 6, 5968–5977 CrossRef CAS.
- N. Ikuta, S. Y. Takizawa and S. Murata, Photochem. Photobiol. Sci., 2014, 13, 691–702 CrossRef CAS PubMed.
- B. Limburg, E. Bouwman and S. Bonnet, J. Phys. Chem. B, 2016, 120, 6969–6975 CrossRef CAS PubMed.
- B. Limburg, E. Bouwman and S. Bonnet, Chem. Commun., 2015, 51, 17128–17131 RSC.
- N. Sugiyama, M. Toyoda and Y. Amao, Colloids Surf., A, 2006, 284–285, 384–387 CrossRef.
- Y. Amao and I. Okura, J. Mol. Catal. A: Chem., 1996, 105, 125–130 CrossRef CAS.
- F. Franco, S. Fernández and J. Lloret-Fillol, Curr. Opin. Electrochem., 2019, 15, 109–117 CrossRef CAS.
- P. Garrido-Barros, C. Gimbert-Suriñach, R. Matheu, X. Sala and A. Llobet, Chem. Soc. Rev., 2017, 46, 6088–6098 RSC.
- V. Radu, S. Frielingsdorf, S. D. Evans, O. Lenz and L. J. C. Jeuken, J. Am. Chem. Soc., 2014, 136, 8512–8515 CrossRef CAS PubMed.
- Y. Pellegrin and F. Odobel, C. R. Chim, 2017, 20, 283–295 CrossRef CAS.
- G. Steinberg-Yfrach, P. A. Liddell, S.-C. Hung, A. L. Moore, D. Gust and T. A. Moore, Nature, 1997, 385, 239–241 CrossRef CAS.
- M. Andersson, L. Hammarström and K. Edwards, J. Phys. Chem., 1995, 99, 14531–14538 CrossRef CAS.
- G. Steinberg-Yfrach, J.-L. Rigaud, E. N. Durantini, A. L. Moore, D. Gust and T. A. Moore, Nature, 1998, 392, 479–482 CrossRef CAS PubMed.
- I. O. L. Bacellar, M. C. Oliveira, L. S. Dantas, E. B. Costa, H. C. Junqueira, W. K. Martins, A. M. Durantini, G. Cosa, P. Di Mascio, M. Wainwright, R. Miotto, R. M. Cordeiro, S. Miyamoto and M. S. Baptista, J. Am. Chem. Soc., 2018, 140, 9606–9615 CrossRef CAS PubMed.
- S. Troppmann, E. Brandes, H. Motschmann, F. Li, M. Wang, L. Sun and B. König, Eur. J. Inorg. Chem., 2016, 554–560 CrossRef CAS.
- P. Jurkiewicz, L. Cwiklik, A. Vojtíšková, P. Jungwirth and M. Hof, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 609–616 CrossRef CAS PubMed.
- A. Pannwitz, H. Saaring, N. Beztsinna, X. Li, M. A. Siegler and S. Bonnet, Chem. – Eur. J., 2021, 27, 3013–3018 CrossRef CAS PubMed.
- J. Sabın, G. Prieto, J. M. Ruso, R. Hidalgo-Álvarez and F. Sarmiento, Eur. Phys. J. E: Soft Matter Biol. Phys., 2006, 20, 401–408 CrossRef PubMed.
- S. Paula, A. G. Volkov, A. N. Van Hoek, T. H. Haines and D. W. Deamer, Biophys. J., 1996, 70, 339–348 CrossRef CAS PubMed.
- B. Maherani, E. Arab-Tehrany, A. Kheirolomoom, D. Geny and M. Linder, Biochimie, 2013, 95, 2018–2033 CrossRef CAS PubMed.
- L. Hammarström, M. Almgren and T. Norrby, J. Phys. Chem., 1992, 96, 5017–5024 CrossRef.
- L. M. Hays, J. H. Crowe, W. Wolkers and S. Rudenko, Cryobiology, 2001, 42, 88–102 CrossRef CAS PubMed.
- I. D. Johnson, Molecular Probes Handbook: A Guide to Fluorescent Probes and Labeling Technologies, Life Technologies Corporation, 2010 Search PubMed.
- C. C. Page, C. C. Moser, X. Chen and P. L. Dutton, Nature, 1999, 402, 47–52 CrossRef CAS PubMed.
- E. Altamura, F. Milano, R. R. Tangorra, M. Trotta, O. H. Omar, P. Stano and F. Mavelli, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3837–3842 CrossRef CAS PubMed.
- S. Bhosale, A. L. Sisson, P. Talukdar, A. Fürstanberg, N. Banetji, E. Vauthey, G. Bollot, J. Mareda, C. Röger, F. Würthner, N. Sakai and S. Matile, Science, 2006, 313, 84–86 CrossRef CAS PubMed.
- A. Perez-Velasco, V. Gorteau and S. Matile, Angew. Chem., Int. Ed., 2008, 47, 921–923 CrossRef CAS PubMed.
- B. Limburg, E. Bouwman and S. Bonnet, ACS Catal., 2016, 6, 5273–5284 CrossRef CAS.
- A. Bahreman, M. Rabe, A. Kros, G. Bruylants and S. Bonnet, Chem. – Eur. J., 2014, 20, 7429–7438 CrossRef CAS PubMed.
- T. L. Nemzek and W. R. Ware, J. Chem. Phys., 1975, 62, 477–489 CrossRef CAS.
- C. S. Owen, J. Chem. Phys., 1975, 62, 3204–3207 CrossRef CAS.
- B. Medhage and M. Almgren, J. Fluoresc., 1992, 2, 7–21 CrossRef CAS PubMed.
- L. Hammarström, T. Norrby, G. Stenhagen, J. Mårtensson, B. Åkermark and M. Almgren, J. Phys. Chem. B, 1997, 101, 7494–7504 CrossRef.
- J. Moldenhauer, M. Meier and D. W. Paul, J. Electrochem. Soc., 2016, 163, H672–H678 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cs00737d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |