
Modified gaphene oxide (GO) particles in peptide hydrogels: a hybrid system enabling scheduled delivery of synergistic combinations of chemotherapeutics†
John D.
Schneible
 a,
Kaihang
Shi
a,
Ashlyn T.
Young
b,
Srivatsan
Ramesh
a,
Nanfei
He
c,
Clay E.
Dowdey
a,
Jean Marie
Dubnansky
a,
Radina L.
Lilova
a,
Wei
Gao
c,
Erik
Santiso
a,
Michael
Daniele
*bd and
Stefano
Menegatti
*a
a,
Kaihang
Shi
a,
Ashlyn T.
Young
b,
Srivatsan
Ramesh
a,
Nanfei
He
c,
Clay E.
Dowdey
a,
Jean Marie
Dubnansky
a,
Radina L.
Lilova
a,
Wei
Gao
c,
Erik
Santiso
a,
Michael
Daniele
*bd and
Stefano
Menegatti
*a
aDepartment of Chemical and Biomolecular Engineering, North Carolina State University, 911 Partners Way, Raleigh, North Carolina, USA. E-mail: smenega@ncsu.edu
bJoint Department of Biomedical Engineering, North Carolina State University – University of North Carolina Chapel Hill, North Carolina, USA
cDepartment of Textile Engineering, Chemistry, and Science, 1020 Main Campus Drive, Raleigh, North Carolina, USA
dDepartment of Electrical and Computer Engineering, North Carolina State University, 890 Oval Drive, Raleigh, North Carolina, USA. E-mail: mdaniel6@ncsu.edu
First published on 3rd March 2020
Abstract
The scheduled delivery of synergistic drug combinations is increasingly recognized as highly effective against advanced solid tumors. Of particular interest are composite systems that release a sequence of drugs with defined kinetics and molar ratios to enhance therapeutic effect, while minimizing the dose to patients. In this work, we developed a homogeneous composite comprising modified graphene oxide (GO) nanoparticles embedded in a Max8 peptide hydrogel, which provides controlled kinetics and molar ratios of release of doxorubicin (DOX) and gemcitabine (GEM). First, modified GO nanoparticles (tGO) were designed to afford high DOX loading and sustained release (18.9% over 72 h and 31.4% over 4 weeks). Molecular dynamics simulations were utilized to model the mechanism of DOX loading as a function of surface modification. In parallel, a Max8 hydrogel was developed to release GEM with faster kinetics and achieve a 10-fold molar ratio to DOX. The selected DOX/tGO nanoparticles were suspended in a GEM/Max8 hydrogel matrix, and the resulting composite was tested against a triple negative breast cancer cell line, MDA-MB-231. Notably, the composite formulation afforded a combination index of 0.093 ± 0.001, indicating a much stronger synergism compared to the DOX–GEM combination co-administered in solution (CI = 0.396 ± 0.034).
Introduction
Combination chemotherapy is currently the most widely utilized, clinically established route to fight cancer, owing to its distinct advantages over current single-drug chemotherapy. Single-drug therapies cause acquired drug resistance upon prolonged administration,1,2 which cause non-responsiveness to therapy and the need of increased dosage.3,4 This results in poorer quality of life during treatment and decreased life expectancy.4–6 Combination chemotherapy offers superior therapeutic outcomes by operating through the simultaneous disruption of multiple metabolic pathways in cancer cells; these provide three mechanistic outcomes: synergistic, additive, and potentiation.7 Among these mechanistic outcomes, synergism, whereby the efficacy of multiple chemotherapeutics administered together is greater than the sum of individual therapies administered independently, represents the most favorable therapeutic outcome.7 Historically, synergism has been attributed to the combination of the drugs utilized and their molar ratio in the chemotherapeutic cocktail.7,8 More recently, however, the administration schedule, that is the sequence and timing by which every drug is administered, has been shown to be critical – together with molar ratio – to achieve therapeutic synergism.9–11Recent research on the optimization of molar ratio and schedule of chemotherapeutic regimens has demonstrated that a more favorable therapeutic outcome can be achieved relative to single-drug therapy. This has been well-documented in pre-clinical studies, both in vitro and in animal models,10,12–18 as well as in clinical settings.19–22 Current methods to administer scheduled combined chemotherapy rely on subsequent injections, which require prolonged patient hospitalization and extended observation by trained specialists, and result in higher medical costs and discomfort for patients. It is now anticipated that next-generation treatments will rely on engineered solutions, comprising multiple biocompatible materials that (i) are seamlessly integrated into stable systems and (ii) release the various therapeutic ingredients at tunable amounts and rates to provide the desired therapeutic sequence, ratio, and kinetics.
To this end, researchers have investigated a myriad of drug delivery systems (DDSs), ranging from nano-/micro-particles and liposomes,23,24 to polymer conjugates (PDC's)10,12,13,18,25,26 and hydrogels.14,27–29 Among these systems, nanoparticles and hydrogels possess distinct advantages, mainly loading capacity and tenability of release.30–36
A nanoscale carrier that has attracted considerable attention as a DDS is graphene oxide (GO). GO features a high specific surface area (up to 500 m2 g−1),37 which is ideal for loading large quantities of poorly bioavailable drugs, such as taxanes, anthracyclines, and camptothecan analogues.38–49 Additionally, the surface of GO features an abundance of carboxylic acids, epoxides, and hydroxyl groups that can be modified to introduce stimuli-responsive behavior, enable active targeting, and prolong circulation.31,50,51 Drug adsorption and release on native GO has also been studied in silico using model drugs doxorubicin (DOX), paclitaxel, 5-fluorouracil, and thioguanine.52–55 These simulations have proven very helpful in designing GO-based drug delivery systems by guiding the choice of type and degree of surface modification, and the loading conditions that maximize therapeutic efficacy.
Hydrogels with shear-thinning and thixotropic behavior are also of interest in drug delivery, as they can be easily introduced by direct injection into surgical cavities to form “depots” that provide sustained drug release.34,35 This bypasses the pharmacokinetic limitations inherent in many chemotherapeutic agents, as well as the bio-distribution limitations of intravenously administered systemic delivery systems.34,35,56–58 Owing to their flexible molecular architecture, hydrogels enable release kinetics that can easily be tuned by adjusting the polymer/water ratio in cross-linked gels.59,60 “Designer” peptides have received strong interest to construct hydrogel-based DDSs. Designer peptides are inherently biocompatible, bio-degradable, and feature rapid gelation via hierarchal self-assembly.61 Gelation kinetics can be tuned effectively by varying the amino acid composition of the peptide and the aqueous environment (pH, ionic strength, temperature).62–66 A designer peptide widely used is the Max8 sequence, constituted by VKVKVKVKVDPPTKVEVKVKV-NH2, wherein DP is D-proline. The Max8 peptide is a stimuli-responsive peptide that is stable in aqueous solutions at low ionic strength and undergoes gelation in to physiological conditions by self-assembling into 3.2 nm diameter β-hairpin nanofibers.64 At low gel fractions (0.5–2 w/w) the Max8 hydrogel imposes little resistance to the transport of both small and large molecules, as demonstrated by Branco et al. with fluorescein–dextran conjugates.63 In the context of scheduled delivery of multiple drugs, the high permeability of the hydrogel makes it an ideal depot for the release of the first drug in a sequential system; the limited control of the relative flux of multiple diffusing drugs, however, limits the applicability of hydrogels alone for scheduled combined chemotherapy. Rather, composite systems combining drug-loaded nanocarriers suspended in a drug-loaded peptide hydrogel hold great promise for precise delivery kinetics of multiple payloads.67–70
In this work, we demonstrate scheduled and synergistic release of combined chemotherapeutics through the development of a composite hydrogel system comprising DOX-loaded modified-GO nanoparticles suspended in a gemcitabine (GEM)-loaded Max8 hydrogel (Fig. 1). The synergism of DOX and GEM has been extensively studied in terms of molar ratio and delivery kinetics,9,10,71–73 demonstrating that the combination is synergistic for molar ratios (GEM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DOX) > 1, and optimal synergism occurs at a ratio of 10:1, with GEM being administered prior to DOX.9,10 We initially investigated different types and levels of chemical modification to tune the surface charge and hydrophobicity of GO nanoparticles and evaluated the corresponding DOX loading and release at different values of solution pH and ionic strength. Notably, GO modification with tris(2-aminoethyl) amine (TREN) achieved high loading, ranging from 0.2–0.6 mg DOX per mg GO, and afforded an initially rapid release of 18.9% of the loaded DOX within 72 h followed by sustained release of 31.4% over the course of 4 weeks. To gain molecular-level insight into the phenomena governing the adsorption and release of DOX on TREN-GO, we developed a computational model that quantitatively describes the DOX:TREN-GO interaction at different modification degrees, in terms of (i) molarity of loading and release (i.e., water/GO partition coefficient of DOX), and (ii) reversible formation and adsorption/desorption of DOX aggregates. Selected DOX–TREN-GO nanoparticles were then suspended in a GEM/Max8 hydrogel, and the composite formulation was evaluated on a triple negative breast cancer cell line (MDA-MB-231) demonstrating high therapeutic efficacy. Notably, the DOX–GEM-loaded composite system afforded a combination index of 0.093 ± 0.001, considerably lower than the free drug DOX–GEM combination (CI = 0.396 ± 0.034) at the same concentration and molar ratio (DOX:GEM = 1:10). This indicates that the differential release induced from the composite system provides the desired therapeutic increment above the molar-based synergism.
:DOX) > 1, and optimal synergism occurs at a ratio of 10:1, with GEM being administered prior to DOX.9,10 We initially investigated different types and levels of chemical modification to tune the surface charge and hydrophobicity of GO nanoparticles and evaluated the corresponding DOX loading and release at different values of solution pH and ionic strength. Notably, GO modification with tris(2-aminoethyl) amine (TREN) achieved high loading, ranging from 0.2–0.6 mg DOX per mg GO, and afforded an initially rapid release of 18.9% of the loaded DOX within 72 h followed by sustained release of 31.4% over the course of 4 weeks. To gain molecular-level insight into the phenomena governing the adsorption and release of DOX on TREN-GO, we developed a computational model that quantitatively describes the DOX:TREN-GO interaction at different modification degrees, in terms of (i) molarity of loading and release (i.e., water/GO partition coefficient of DOX), and (ii) reversible formation and adsorption/desorption of DOX aggregates. Selected DOX–TREN-GO nanoparticles were then suspended in a GEM/Max8 hydrogel, and the composite formulation was evaluated on a triple negative breast cancer cell line (MDA-MB-231) demonstrating high therapeutic efficacy. Notably, the DOX–GEM-loaded composite system afforded a combination index of 0.093 ± 0.001, considerably lower than the free drug DOX–GEM combination (CI = 0.396 ± 0.034) at the same concentration and molar ratio (DOX:GEM = 1:10). This indicates that the differential release induced from the composite system provides the desired therapeutic increment above the molar-based synergism.
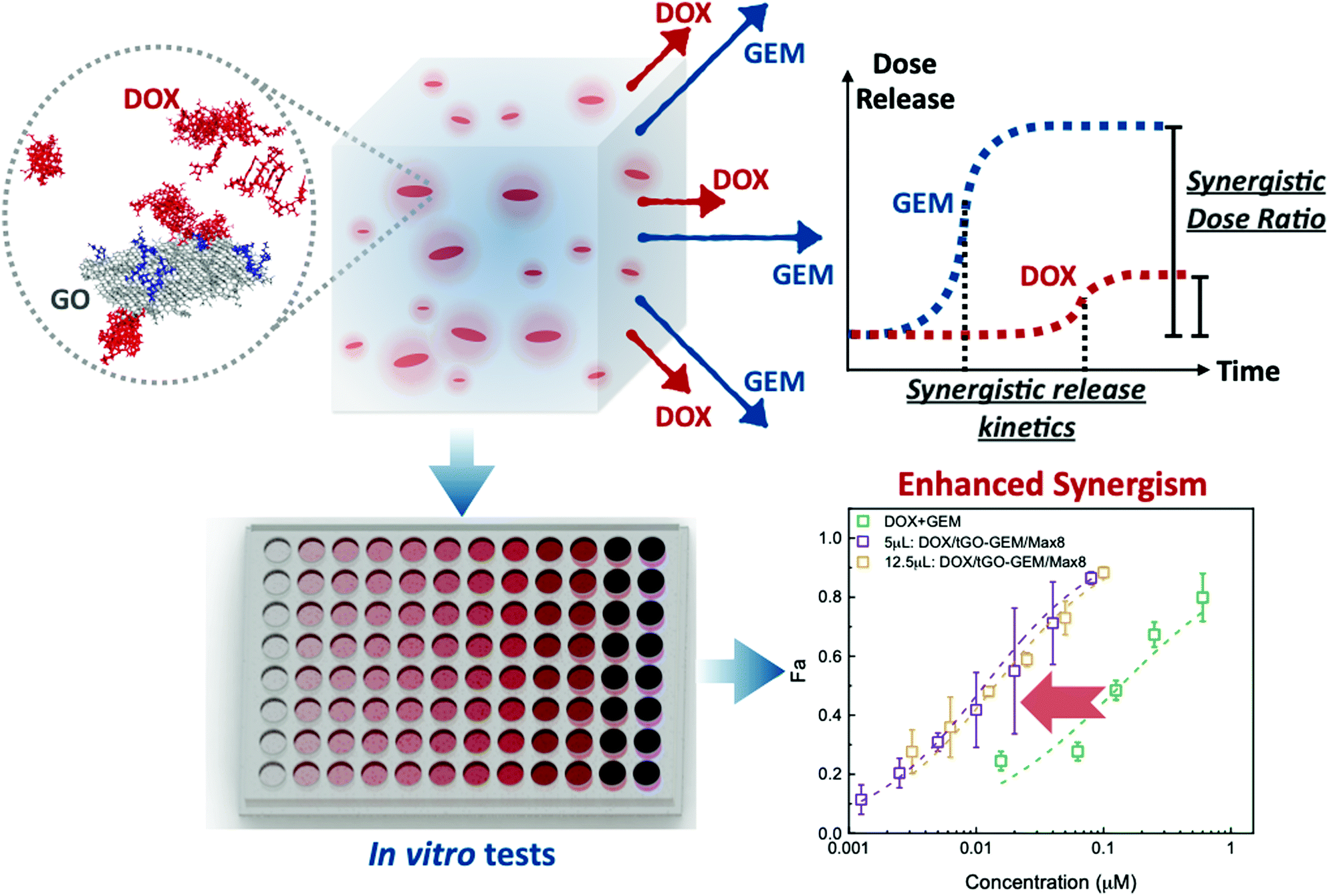 | ||
| Fig. 1 Design and proposed mechanism of dual release of DOX and GEM from the hybrid system comprising modified GO nanoparticles suspended in a Max8 hydrogel. The combination of drug release at controlled relative molar ratio and kinetics enhances the inherent synergism of DOX and GEM beyond the simple effect of codelivery. | ||
Experimental
Materials
Graphite powder (4827) was sourced from Asbury Graphite Mills (Lumberton, NC). Doxorubicin hydrochloride (DOX) and Gemcitabine hydrochloride (GEM) were obtained from LC Laboratories (Woburn, MA). The Max8 peptide (VKVKVKVKVDPPTKVEVKVKV-NH2),63,74,75 where DP represents D-Proline and –NH2 indicates an amidated C-terminus, was obtained from Genscript (Nanjing, China) at >95% purity. 1,1-Carbonyldiimidazole (CDI), dimethylaminopyridine (DMAP), ethylenediamine (EDA), tris(2-aminoethyl) amine (TREN), acetic anhydride (Ac2O), benzylamine (BA), succinic anhydride (SA), anhydrous tetrahydrofuran (THF), phosphate buffered saline (PBS), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), potassium permanganate (KMnO4), and hydrogen peroxide (H2O2) were purchased from Sigma (St. Louis, MO). Triple negative breast cancer cells MDA-MB-231 were purchased from ATCC (Manassas, VA). Dulbecco's Modified Eagle Medium (DMEM). Fetal bovine serum (FBS) was obtained from Genesee Scientific (San Diego, CA). Penicillin Streptomycin (Pen Strep) was obtained from Gibco (Gaithersburg, MD). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), was purchased from Invitrogen (Carlsbad, CA). All other chemicals were of reagent grade or higher.Synthesis and characterization of modified graphene oxide (GO)
Synthesis and characterization of TREN-GO (tGO) library
Additionally, both GO and tGO particles were analyzed by Fourier Transform Infrared (FTIR) Spectroscopy using a Nicolet 6700 FTIR Spectrophotometer (ThermoFisher Scientific, Waltham, MA). First, the particles were finely ground in potassium bromide (dried in a vacuum oven for 3 h at 60 °C) and placed in the optical bench, which was purged continuously using dry air. The samples were then analyzed using the following data acquisition parameters: accumulation of 512 interferograms with a resolution of 4 cm−1, wavenumber range of 4000–400 cm−1. The resulting spectra were analyzed using the OMNIC Spectra Software (Thermo Scientific).
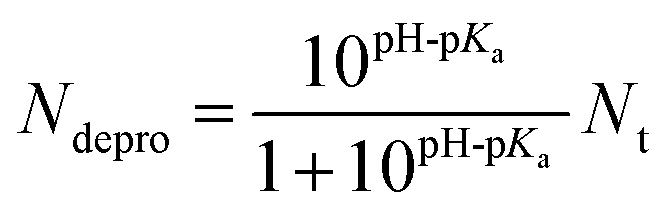 | (1) |
 | (2) |
| Functional groups | Elements | RESP charges (this work) | SESP charges92 | AM1-BCC charges (this work)a |
|---|---|---|---|---|
| a AM1-BCC charges were determined for the whole molecule, so the total charge of the functional group fragment is not necessarily equal to 0 (neutral state) or −1 (deprotonated state). | ||||
| Epoxy | C (GO) | +0.1357 | +0.18 | +0.15 |
| O | −0.2714 | −0.36 | −0.28 | |
| Hydroxyl | C (GO) | +0.1904 | +0.18 | +0.30 |
| O | −0.5689 | −0.57 | −0.58 | |
| H | +0.3785 | +0.39 | +0.41 | |
| Carboxyl (protonated) | C (GO) | +0.0642 | — | −0.11 |
| C | +0.6655 | — | +0.65 | |
| O (sp2) | −0.5649 | — | −0.55 | |
| O (sp3) | −0.6080 | — | −0.60 | |
| H | +0.4432 | — | +0.44 | |
| Carboxyl (deprotonated) | C (GO) | −0.3044 | — | −0.03 |
| C | +0.8408 | — | +0.90 | |
| O | −0.7682 | — | −0.81 | |
| O | −0.7682 | — | −0.81 | |
To model the TREN-modified GO (tGO) surface, we tethered TREN moieties on the GO through carbamate bonds. While we recognized that TREN groups can be added to the surface of the particles through other functional groups on the native GO surface (e.g., hydroxyl groups and epoxy groups), we showed that our proposed scheme of TREN modification of the surface is sufficient to investigate the tGO–DOX interaction during both adsorption and release. The TREN density on the model tGO surface (molecules per nm2) was calculated from the experimental values measured by Kaiser's test, assuming 500 m2 g−1 as specific surface area for the GO particles.37 As the surface area of our GO model is 50 nm2, the number of TREN groups on the model surface ranges between 18 and 24. The pKa values of the two primary amines and the tertiary amine on TREN are 10.6 and 10.9, respectively. Under weak neutral conditions (pH = 6), all amine groups in the TREN are protonated and the TREN molecule carries a +3 charge. The initial configuration of the system was created using the PACKMOL package95 and the simulation box was filled using the TIP3P water model.96 Sixty-four DOX molecules were initially randomly distributed at a distance from the GO surface ranging between 0 and 45 Å. Following initial geometry optimization, the system was slowly heated from 0 K to room temperature (298 K) in an NVT ensemble for 200 ps with 2 fs time steps, while restraining the solid surface and the DOX molecules in their initial position by a harmonic spring force. The DOX molecules were then released, and an NPT simulation was performed to equilibrate the density of the system at 1 bar, 298 K for 2 ns. Throughout the simulation, the pressure was maintained constant using the Berendsen barostat with pressure relaxation time of 2 ps, while the temperature was controlled using the Langevin thermostat with collision frequency γ = 1 ps−1; the Langevin thermostat has been shown to be more efficient in system equilibration than the Berendsen temperature coupling scheme, but the Berendsen thermostat is more stable in reproducing the correct dynamics of the system.97 Starting from the last equilibrated frame, a production NPT MD was performed wherein all TREN groups on the surface were allow to relax while the base GO sheet was restrained using a weak harmonic constraint of 10 kcal mol−1 Å−2. The Berendsen thermostat was used during the simulation with the temperature coupling time of 10 ps. All simulations were run over 70 ns, at which time the number of DOX molecules adsorbed on either the native GO or tGO surface reaches a plateau. At least two independent MD simulations were run for the native GO and three tGO (0.359, 0.435, and 0.491 TREN molecules per nm2 corresponding to ζ potential of −8.9, 10.1, and 20.7 mV, respectively) models to calculate the average number of DOX molecules adsorbed on different GO surfaces for subsequent comparison with experimental data.
Synthesis and characterization of tGO–Max8 composite
590 nm and λem = 618 nm.
:1 GEM:DOX molar ratio in solution upon release, based on the data of DOX release obtained. Samples including gels were prepared using a total gel volume of 200 μL. All release tests were performed by contacting every system with 1 mL of 10 mM PBS at pH 5 at 37 °C. At set time points (8, 16, 32, 48, and 72 h), 200 μL of supernatant was collected and replenished with fresh buffer. The collected samples were analyzed by liquid chromatography using a reverse-phase Aeris 3.6 μm C18 column (50 × 4.6 mm) installed on a Waters 2690 HPLC system (Waters, Milford, MA). The chromatographic method utilized a 5–100% gradient of acetonitrile (0.1% v/v formic acid) in water (0.1% v/v formic acid) over 10 minutes, while monitoring the effluent at 290 nm and 480 nm to monitor GEM and DOX, respectively. The concentrations of GEM and DOX were determined by peak-area integration of the resulting 290 nm and 480 nm chromatograms relative to the respective standard curves.
In vitro characterization
Statistical analysis
Statistical significance was determined by evaluation of the Student's t-test using Microsoft Excel. Statistical significance for samples was evaluated against the control test condition, and p < 0.05 was considered significant. For analysis *, **, and *** represent p < 0.05, 0.01, 0.001, respectively.Results and discussion
Synthesis and characterization of modified graphene oxide (GO)
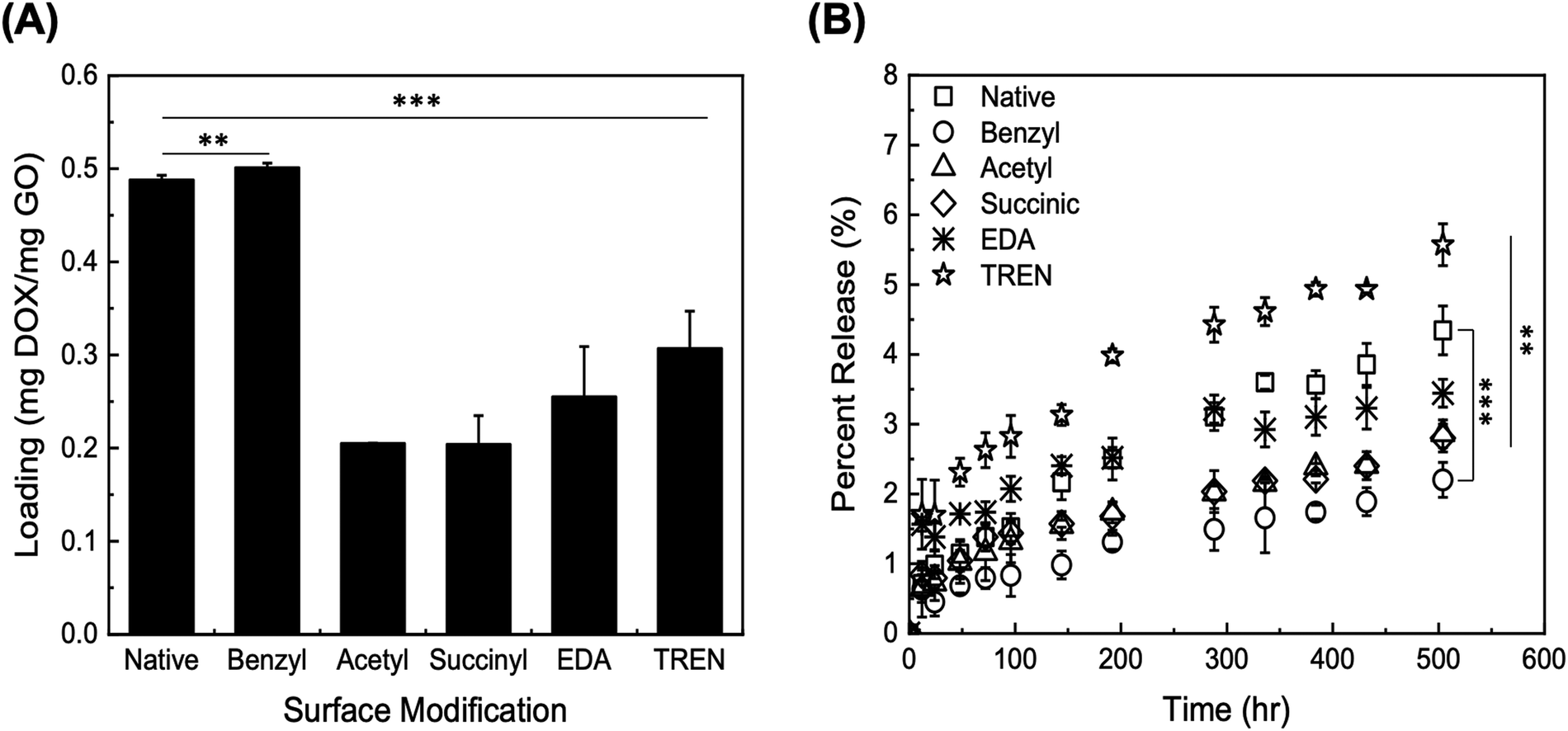 | ||
| Fig. 2 Initial screening studies of (A) DOX loading on GO particles with different chemical modifications (listed in the abscissa); (B) DOX release from modified GO particles within 500 h. Error bars represent mean ± 99% CI (n ≥ 3). ** indicates p < 0.01, and *** indicates p < 0.001, as obtained from a Student's t-test. | ||
The release of DOX from the modified GO particles was performed in PBS at pH 7.4 over a total collection time of 500 h. The values of percent release (DOX released vs. loaded) are presented in Fig. 2B; the values of percent release obtained through the initial 100 h are presented in Fig. S2 (ESI†). The GO particles modified with TREN and EDA returned the highest values of percent release, at both 72 h and 500 h. The electrostatic repulsion between DOX and TREN/EDA moieties, both of which are positively charged at pH 7.4, combined with a high concentration gradient between the GO surface and the solution phase, likely play a predominant role in release. GO particles modified with TREN (2 primary and 1 tertiary amine), in fact, released 2.63 ± 0.3% after 72 h and 5.57 ± 0.3% after 504 h, whereas GO modified with EDA (1 primary amine) released 1.74 ± 0.2% after 72 h and 3.44 ± 0.2% after 504 h. Comparatively, at the 72 h and 504 h time points, native GO released 1.38 ± 0.2% and 4.34 ± 0.4%, acetyl-GO released 1.16 ± 0.4% and 2.86 ± 0.2%, succinyl-GO released 1.38 ± 0.2% and 2.80 ± 0.2%, and benzyl-GO released 0.8 ± 0.1% and 2.20 ± 0.3%. Notably, the loading of DOX on TREN-GO was only slightly lower compared to compared to that of native GO. These results indicate that modification of GO particles with cationic moieties is essential towards ensuring both a favorable loading and high release of DOX. Accordingly, we selected TREN as the surface modification moiety in all subsequent studies.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1736 cm−1, aromatic CC stretching at 1625 cm−1, alkoxy C–O stretching at 1064 cm−1, and epoxy C–O stretching at around 854 cm−1.99 In tGO, the appearance of overlapping peaks of CO stretching and N–H bending of the amide functional group at around 1640 cm−1, and the appearance of a C–N stretching peak at 1460 cm−1,99 confirms the conjugation of TREN via formation of amide bonds. Furthermore, the presence of a carboxyl CO stretch peak at 1730 cm−1 in native GO and the absence of this peak in tGO indicates that all distal carboxylic acids are implicated in the formation of amide bonds with TREN. At the same time, the high values of TREN surface density measured on tGO particles and the exiguity of distal carboxylic acids typical of native GO also suggests that TREN conjugation also occurs through the formation of carbamate bonds, which result from the activation of the hydroxyl groups on GO with CDI; however, because carbamate and amide bonds have the same FTIR signature and the adsorption of air moisture by tGO prevents the quantification of the hydroxyl groups converted in carbamate bonds,100 it is not possible to quantitatively distinguish the two conjugation routes. Finally, the persistence of the epoxy peak at ∼850 cm−1 in the tGO sample indicates that TREN reaction to epoxide groups is unlikely.
O stretching at 1736 cm−1, aromatic CC stretching at 1625 cm−1, alkoxy C–O stretching at 1064 cm−1, and epoxy C–O stretching at around 854 cm−1.99 In tGO, the appearance of overlapping peaks of CO stretching and N–H bending of the amide functional group at around 1640 cm−1, and the appearance of a C–N stretching peak at 1460 cm−1,99 confirms the conjugation of TREN via formation of amide bonds. Furthermore, the presence of a carboxyl CO stretch peak at 1730 cm−1 in native GO and the absence of this peak in tGO indicates that all distal carboxylic acids are implicated in the formation of amide bonds with TREN. At the same time, the high values of TREN surface density measured on tGO particles and the exiguity of distal carboxylic acids typical of native GO also suggests that TREN conjugation also occurs through the formation of carbamate bonds, which result from the activation of the hydroxyl groups on GO with CDI; however, because carbamate and amide bonds have the same FTIR signature and the adsorption of air moisture by tGO prevents the quantification of the hydroxyl groups converted in carbamate bonds,100 it is not possible to quantitatively distinguish the two conjugation routes. Finally, the persistence of the epoxy peak at ∼850 cm−1 in the tGO sample indicates that TREN reaction to epoxide groups is unlikely.
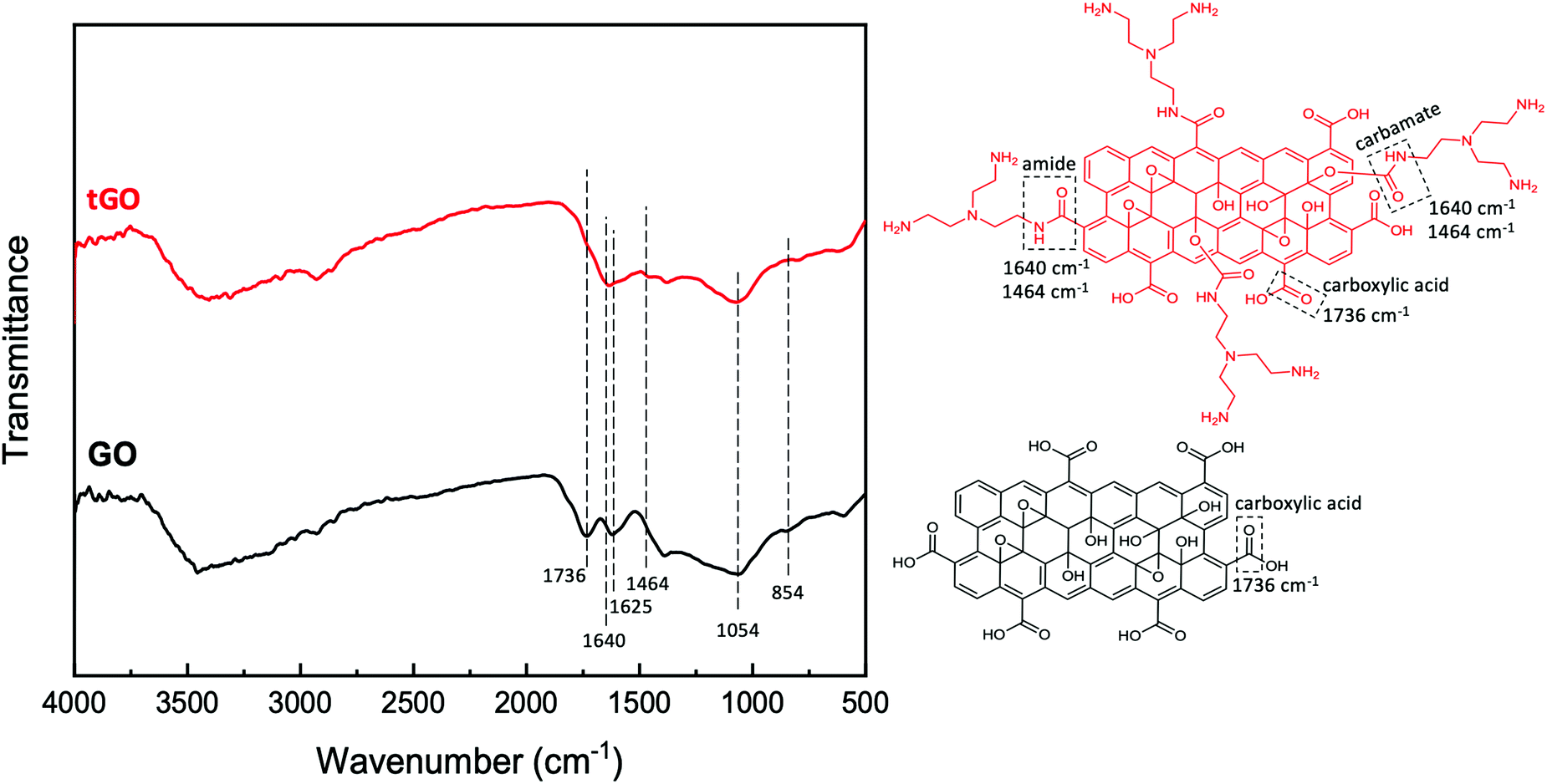 | ||
| Fig. 3 FTIR analysis of native GO and tGO particles and corresponding model structures. | ||
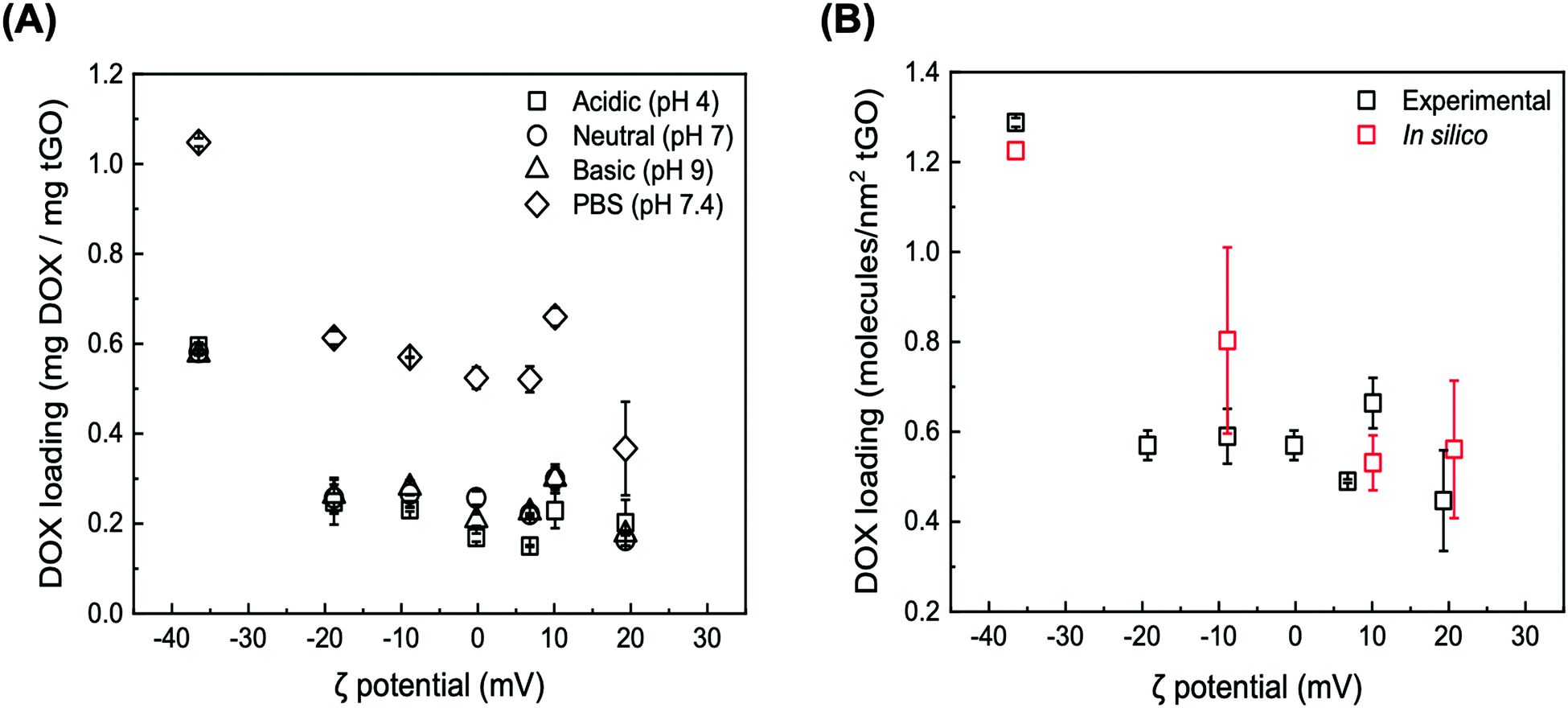 | ||
| Fig. 4 (A) Loading of DOX onto tGO particles as a function of ζ potential at different loading conditions: Milli-Q water (titrated with HCl or NaOH) at pH 4 (squares), pH 6 (circles), and pH 9 (triangles), and PBS at pH 7.4 (diamonds), where error bars represent mean ± 99% CI (n ≥ 3); (B) comparison between the values of DOX loading measured at pH 6 (black) vs. predicted by the MD simulations at pH 6.1 (red). The experimental error is presented as mean ± 99% CI (n ≥ 3), while the error from the simulations is presented as mean ± S.D. for n ≥ 4 independent simulations with distinct initial configurations. | ||
At low ionic strength, DOX loading at neutral and basic pH displays the same downward trend, featuring an initial sharp decrease from 0.581 ± 0.004 and 0.576 ± 0.011 mg mg−1 GO (ζ potential = −36.5 mV) to 0.257 ± 0.030 mg mg−1 GO and 0.262 ± 0.040 (ζ potential = −18.8 mV) followed by a shallowed decrease, reaching 0.163 ± 0.012 mg mg−1 GO and 0.176 ± 0.002 at ζ potential = 19.3 mV. At the same time, a subtle yet statistically significant increase in DOX loading at neutral and basic pH vs. acidic pH is observed on the tGO samples with ζ potential between −10 mV and +10 mV. As the pH of the aqueous phase increases, in fact, the positive charge carried by DOX83 and TREN is softened, and consequently DOX loading is improved.
DOX loading conducted at higher ionic strength (PBS, pH 7.4) exhibited the same trend observed in the low salt concentration regimes, but consistently at a higher magnitude. Specifically, the loading of DOX on native GO nearly doubles, reaching 1.048 ± 0.009 mg mg−1 GO; on tGO samples with higher ζ potential, DOX loading decreases to within a range of values between 0.613 ± 0.015 mg mg−1 GO and 0.367 ± 0.104 mg mg−1 GO.
These results collectively indicate two prominent trends. First, as the ζ potential of tGO increases with the modification level, the electrostatic repulsion inhibits the adsorption of DOX molecules onto the surface of tGO. Second, increasing the ionic strength of the loading solution partially screens DOX/tGO electrostatic repulsion and concurrently promotes DOX/tGO hydrophobic interactions, resulting in a constant upward shift in DOX loading across the entire range of TREN modification.
To visualize these phenomena at the molecular level, we performed a number of molecular dynamic (MD) simulations of DOX adsorption onto tGO model surfaces constructed by appending TREN moieties onto a flat, two-faced 4.8 nm × 5.1 nm GO surface. Coherently with the FTIR results, the TREN moieties were appended through carbamate bonds to the hydroxyl groups on the GO surface. All MD simulations were performed at nearly neutral pH and in absence of salt, since the modeling of buffered aqueous systems is computationally complex and outside the scope of this work.102–104 As expected, the values of DOX loading predicted by the MD simulations describe a downward trend with ζ potential (Fig. 4B), caused by the electrostatic repulsion between the positively charged daunosamine moiety of DOX and the TREN moieties on tGO. On the other hand, the spacing between TREN moieties on the tGO surface is sufficient to allow adsorption of DOX molecules on surface patches of native graphene oxide (Fig. S4, ESI†) across the entire range of degree of modification (0.359–0.491 TREN molecules nm−2 of tGO surface).
Notably, the values of DOX loading measured experimentally and those obtained in silico show excellent quantitative agreement (Fig. 4B), indicating that the proposed in silico model portrays accurately the mechanism of DOX adsorption onto tGO particles. The simulations also indicate the formation of aggregated stacks of DOX molecules onto the tGO surface at the binding equilibrium (Fig. S4, ESI†); the presence of aggregate aggregates will be functional to formulate a mechanism explaining the experimental data of DOX release presented subsequently.
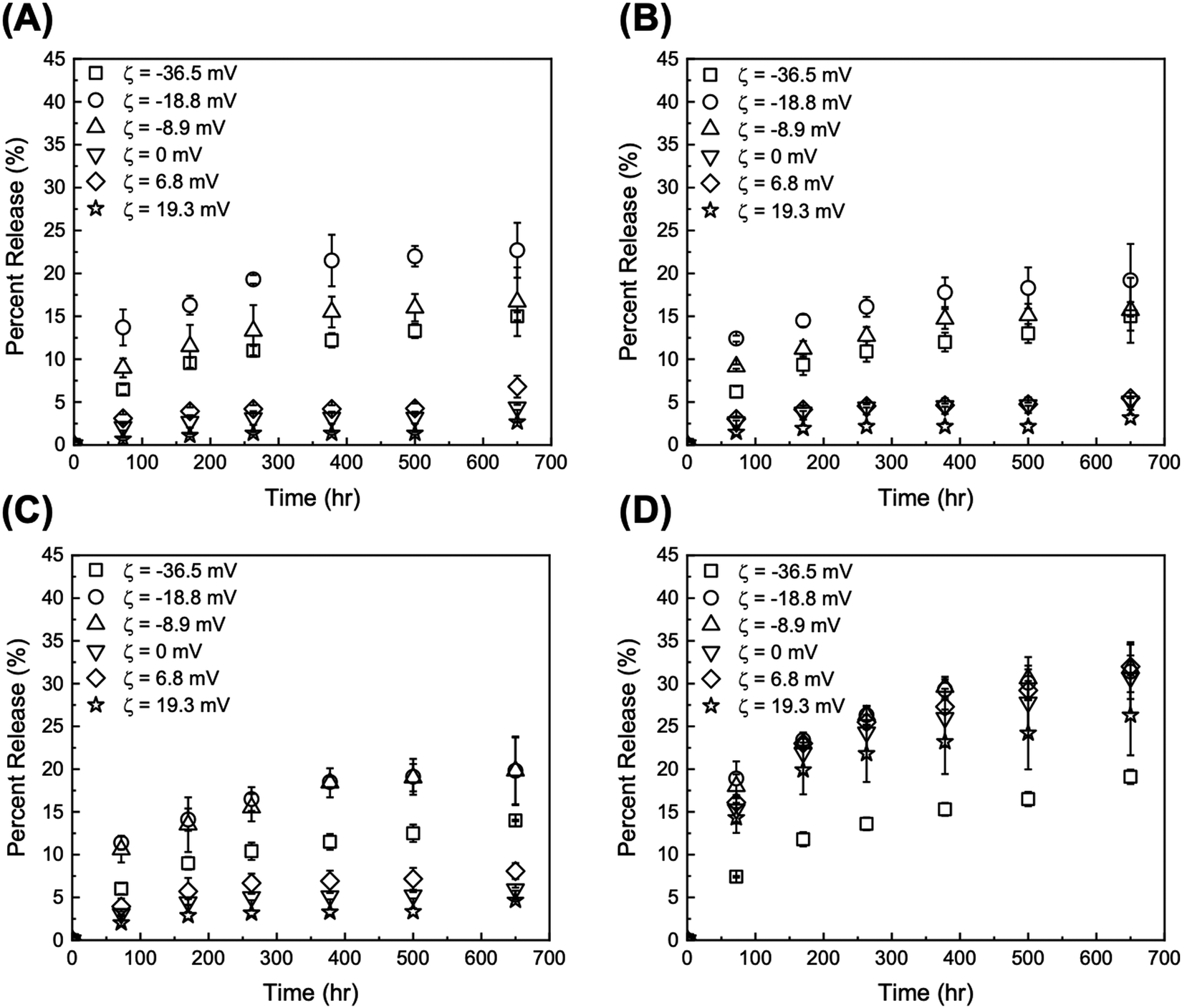 | ||
| Fig. 5 Release profiles at pH 5 from DOX/tGO particles loaded at (A) pH 4, (B) pH 6, (C) pH 9, and (D) PBS at pH 7.4. Error bars represent mean ± 99% CI (n ≥ 3). | ||
Two time scales are of relevance in this study: (i) short term (72 h), which is representative of a typical therapeutic window for in vitro DDS validation, and (ii) long term (>600 h), which fits the time scale of maintenance chemotherapy. The values of DOX percent release from GO and tGO particles at both pH 5 and 7.4 for 72 and 650 h are reported in Tables S2 and S3 (ESI†), respectively. As anticipated, the release experiments conducted at pH 5 afforded higher values of DOX percent release compared to those at pH 7.4. The lower pH promotes the positive charge on both DOX and TREN moieties, which causes DOX–DOX and DOX–TREN repulsion thereby triggering release. Because drug release in a tumor-mimetic environment is more relevant in the context of this work, our analysis focuses on the release data generated at pH 5. The comparison of DOX release from the particles loaded at low ionic strength shows that only two tGO particles outperform the native GO particles, namely tGO with ζ potential = −18.8 mV and −8.9 mV. The former afforded a 72 h-release of 13.7 ± 2.1% (when loaded pH 4), 12.4 ± 0.4% (pH 6), and 11.4 ± 0.8% (pH 9), and a 650 h-release of 22.7 ± 3.2% (pH 4), 19.2 ± 4.2% (pH 6), and 19.8 ± 4.0% (pH 9). The latter showed a 72 h-release of 9.0 ± 1.1% (pH 4), 9.2 ± 0.3% (pH 6), and 10.6 ± 1.5% (pH 9), and a 650 h-release of 16.7 ± 4.0% (pH 4), 15.7 ± 3.8% (pH 6), and 19.8 ± 3.9% (pH 9). Comparatively, the native GO released only ∼6% DOX after 72 h and ∼15% after 650 h. On the other hand, tGO carriers with higher ζ potential afforded a lower release of DOX.
Given the negligible dependence of DOX loading upon pH at low salt concentration as well as DOX release from the resulting particles, only the DOX/GO systems loaded at neutral pH (low vs. high salt concentrations) were considered in the rest of this study. The values of DOX percent release at pH 5 for 72 h and 650 h plotted against the values of ζ potential of the corresponding GO and tGO particle carriers are presented in Fig. 6A and B, respectively; the analogous plots for release at pH 7.4 are presented in Fig. S6 (ESI†).
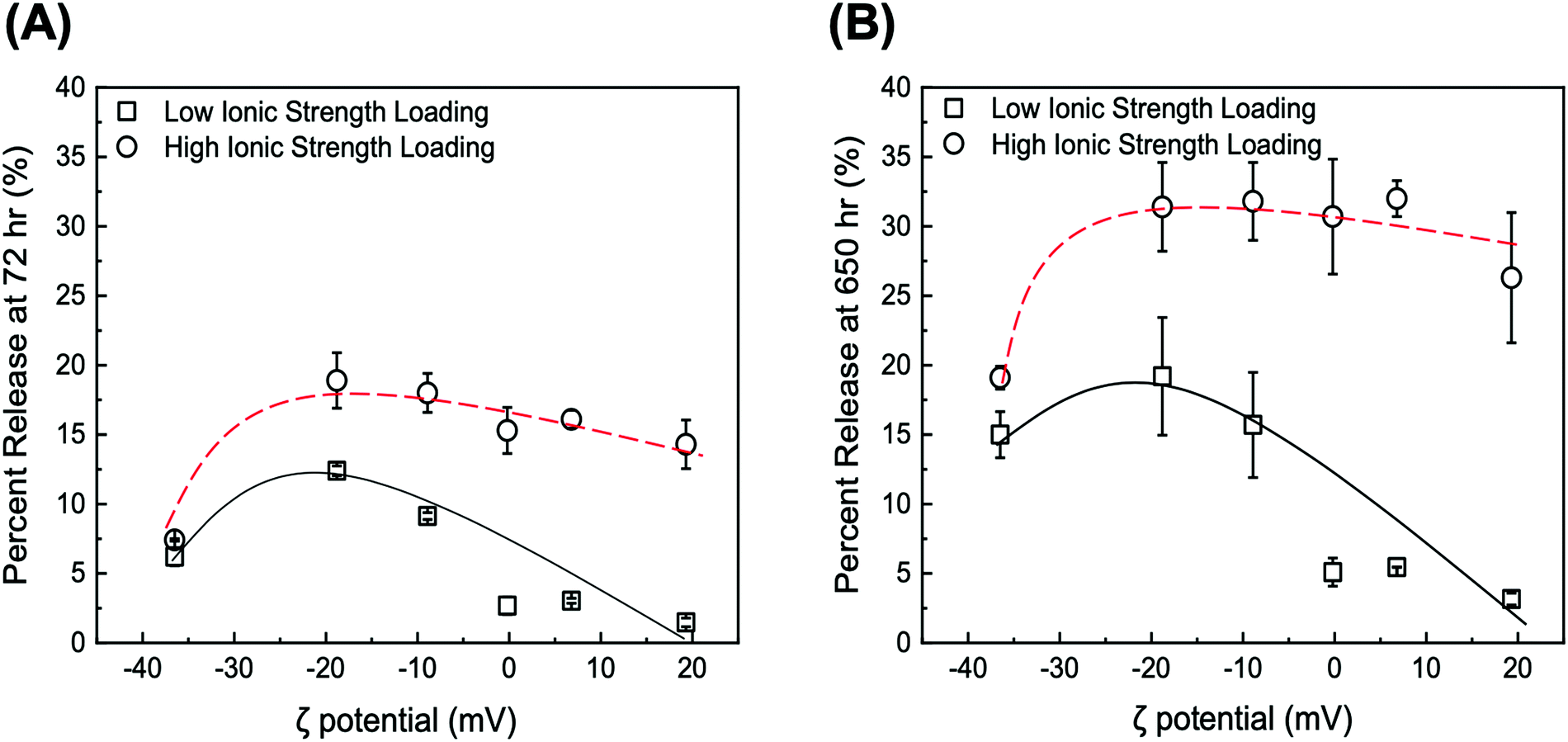 | ||
| Fig. 6 Values of DOX percent release at pH 5 after (A) 72 h and (B) 650 h from DOX/GO and DOX/tGO particles loaded at neutral pH and low ionic strength (squares) or high ionic strength (circles). Hashed are added to guide the visualization of the results. Error bars represent mean ± 99% CI (n ≥ 3). | ||
It is immediately evident that DOX release is determined by two parameters, namely (i) the ionic strength of DOX solution during loading and (ii) the ζ potential of the particles.
First, regarding the ionic strength of the loading solution, a significant increase is observed in the values of DOX percent release, which shift from a range of 1.48–12.4% (low salt loading) to 7.42–18.9% (high salt loading) for 72 h release, and from 3.16–19.2% (low salt loading) to 19.1–31.8% (high salt loading) for 650 h release. In this regard, the in silico modeling of DOX adsorption indicated the formation of DOX aggregates both in solution and onto the GO surface. The extent of aggregation is likely enhanced at high salt conditions, where electrostatic repulsion is softened and hydrophobic interactions are promoted, as observed with drug-like molecules;105 accordingly, we hypothesize that DOX adsorbs onto the tGO surface in a predominantly mono-/di-meric form when loaded in a low ionic strength environment, and in a rather multimeric form at higher ionic strength. When exposed to pH 5, DOX aggregates are considerably less stable than monomeric DOX, due to the combination of DOX–DOX and DOX–TREN electrostatic repulsion, and are released more easily, resulting in an upward shift in percent release.
The dependence of DOX percent release on ζ potential, on the other hand, is rather counterintuitive. In place of a monotonic trend reverse to that of DOX loading (Fig. 4B), in fact, concave downward curves were obtained (Fig. 6A and B). To explain this behavior, one must consider the charge environment onto the GO particles and in the layer of charges surrounding them (Fig. 7). The surface of GO is inherently negatively charged, due to the numerous oxygen-containing moieties;106 on this surface, the TREN moieties and the DOX molecules form a layer of “condensed” positive counter-ions (Stern layer), respectively covalently linked and non-covalently adsorbed. The enveloping Gouy layer, framed by the Stern plane and the slipping plane, at which the ζ potential is measured, contains a mixture of positive (e.g., H+ and Na+) and negative (e.g., OH− and Cl−) counter-ions. On virgin GO, DOX molecules adsorbed at pH 6 are strongly retained by the negatively charged moieties on the surface of GO (Fig. 7A); as the environment surrounding the DOX/GO particles is adjusted to pH 5, the softening of the inherent negative charge on the surface of GO and the DOX–DOX electrostatic repulsion molecules triggers release. On tGO particles with low TREN modification (ζ potential <0), DOX molecules are adsorbed on patches of native GO between TREN moieties (Fig. 7B); at pH 5, the softening of the negative charge on GO, and the combined DOX–DOX and DOX–TREN repulsion favors DOX desorption, resulting in an increment of percent release. On tGO particles with high TREN modification (ζ potential ≈ or ≥0), the higher density of TREN moieties in the Stern layer drives the accumulation of negative (OH− and Cl−) counter-ions within the Gouy layer, resulting in the electric stabilization of the cationic DOX molecules adsorbed onto the surface of GO (note the cyan spheres representing Cl− ions intercalated between DOX and TREN molecules in Fig. 7C and D); this results in a softening of DOX–TREN repulsions at pH 5, which translates in a decrease of percent release. These phenomena are evident when DOX loading is performed at low ionic strength. When DOX loading is operated in PBS, in fact, the TREN moieties are stabilized by counter-anions prior to DOX adsorption and DOX release at pH 5 is mostly triggered by the disassembly of drug aggregates by DOX–DOX electrostatic repulsion. This makes the values of percent release much less dependent upon ζ potential. Native GO is an exception, given its complete lack of TREN modification.
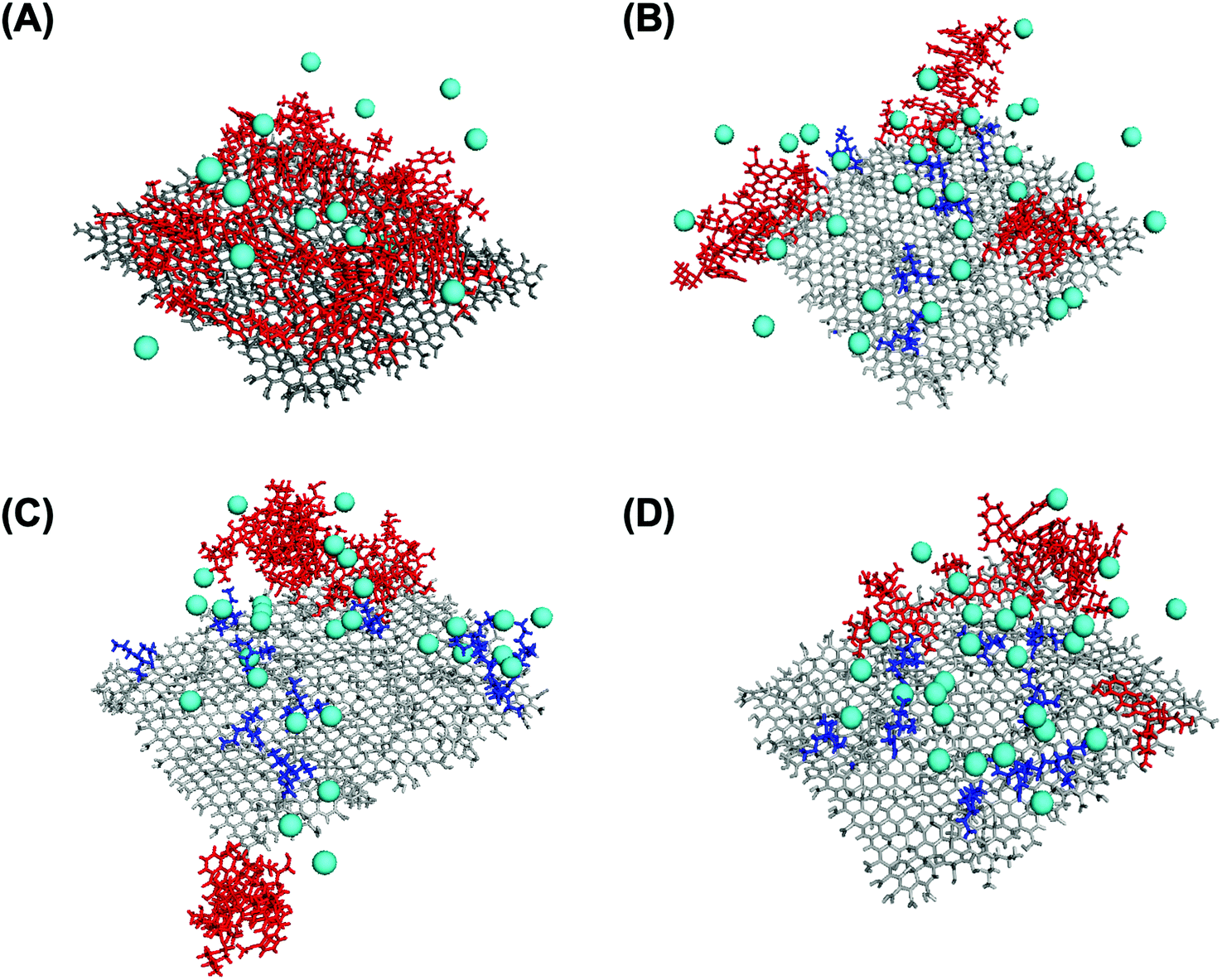 | ||
| Fig. 7 Snapshots from DOX (red frame) loading on TREN- (blue frame) modified GO (grey frame) and the dispersion of Cl− ions (cyan spheres) on (A) native GO (ζ = −36.5 mV), (B) tGO (ζ = −8.9 mV), (C) tGO (ζ = 10.1 mV), and (D) tGO (ζ = 20.7 mV). | ||
Synthesis and characterization of tGO–Max8 composite
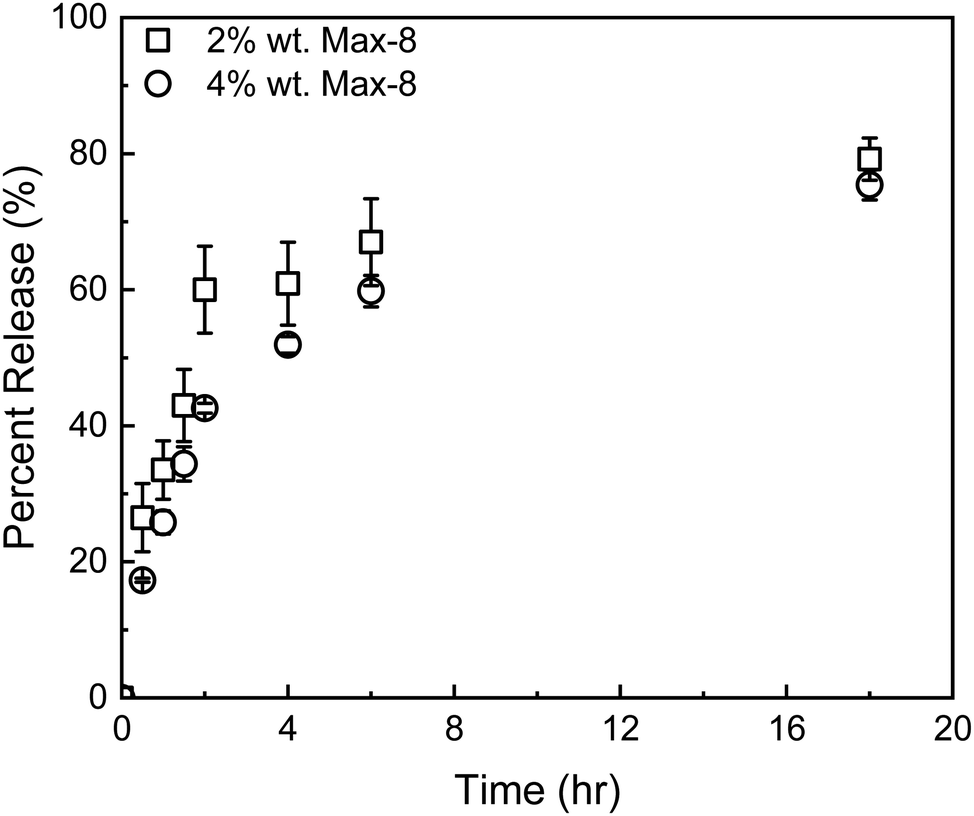 | ||
| Fig. 8 Release profiles of GEM from a Max8 hydrogel at 2 w/w (squares) and 4 w/w (circles). Error bars represent mean ± 99% CI (n ≥ 3). | ||
The composite DDS was prepared by dissolving Max8 peptide at 2 w/w in a sonicated aqueous suspension of the selected DOX/tGO particles. The instantaneous formation of the peptide hydrogel resulted in a homogeneous dispersion of the particles. Confocal fluorescence microscopy images of the control tGO–Max8 composite (Fig. S7, ESI†) and DOX/tGO–Max8 composite immediately after gel formation (Fig. S7, ESI†) confirms the homogeneous dispersion of the tGO particles; further, Fig. S7 (ESI†) indicates DOX remains bound to the tGO particles upon gel formation.
The release kinetics of the composite tGO–Max8 systems are presented in Fig. 9. It is first noted that the peptide hydrogel poses a notable resistance to the transport of DOX, resulting in a 3-fold reduction of the percent release in the aqueous phase surrounding the hydrogel, from the 19.0 ± 0.1% 72 h-release given by free DOX/tGO particles in solution to the 6.2 ± 0.2% given by DOX/tGO particles embedded in the Max8 scaffold. This is likely caused by the cationic and amphiphilic nature of the Max8 peptide (rich in positively charged lysine and hydrophobic valine residues), which limits the partitioning of DOX – a positively charged molecule – from the tGO surface to the hydrogel phase and its migration through the hydrogel matrix, as noted in prior work.107 We have observed that the display of hydrophobic moieties in polycationic hydrogels hinders the migration of DOX. Second, we observed a drastic change in GEM release after 72 h, from 95.5 ± 1.1% from the GEM/Max8 hydrogel alone compared to 49.4 ± 0.2% from the composite system. This is likely caused by the interaction between GEM and the DOX/tGO nanoparticles during its diffusion pathway. Finally, the concurrent migration of the two drugs results in a further decrease of DOX release, from the 6.2 ± 0.2% given by the GEM-free composite to the 3.3 ± 0.1% given by the GEM-loaded hydrogel after 72 h. This is attributed to DOX–GEM interactions that have been documented to occur and drastically change diffusion properties of the DOX–GEM pair through poly-cationic hydrogel matrices.107
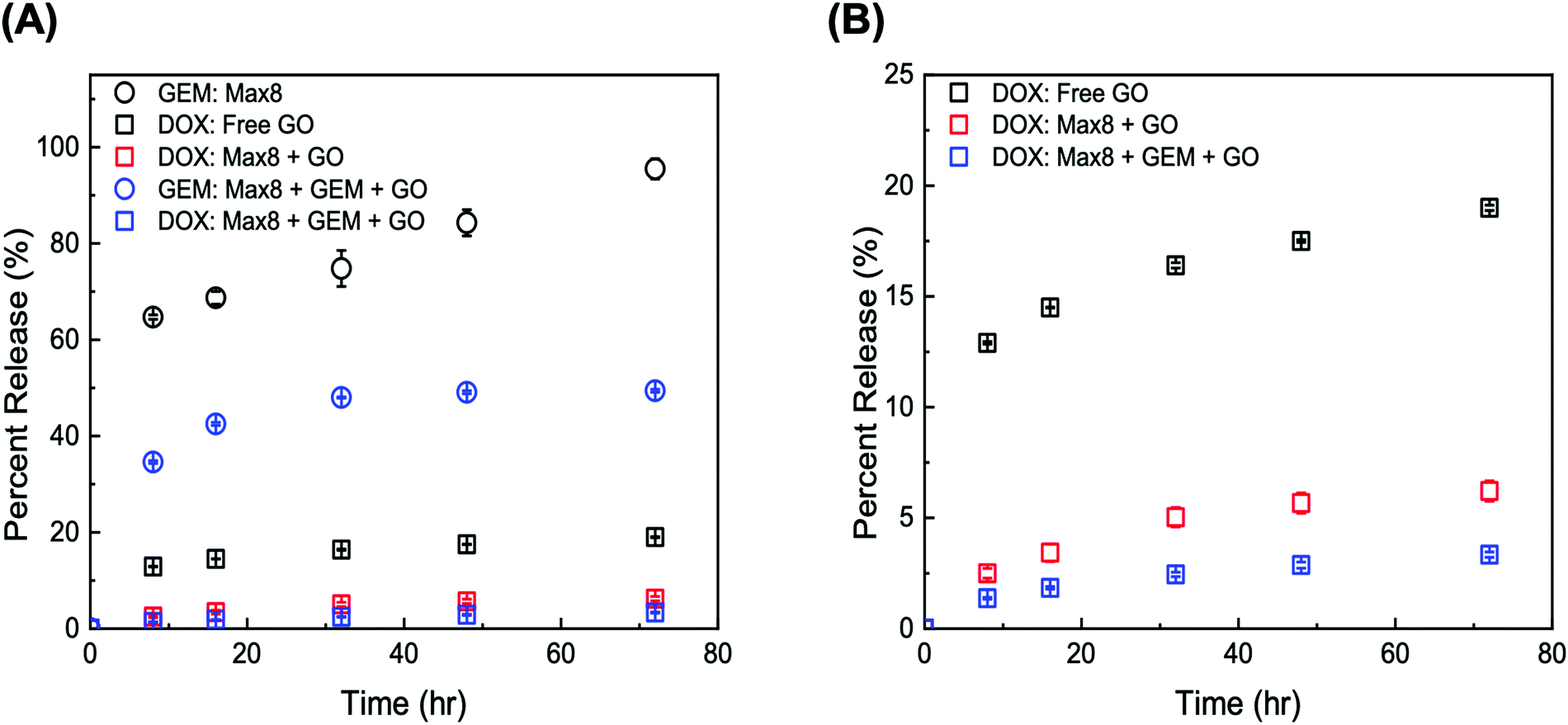 | ||
| Fig. 9 (A) Release profiles of GEM from 2 w/w GEM/Max8 (black circles), DOX from free DOX/tGO particles (black squares), DOX from DOX/tGO particles embedded in a 2 w/w Max8 hydrogel (red squares), GEM from a 2 w/w GEM/Max8 hydrogel loaded with tGO particles (blue circles), and DOX from DOX/tGO particles embedded in a 2 w/w GEM/Max8 hydrogel (blue); (B) release of DOX from the system listed in panel (A). Error bars represent mean ± 99% CI (n ≥ 3). | ||
In vitro characterization
To determine the efficacy of the composite system relative to free single drug and drug combination treatments, we conducted in vitro evaluation of drug-loaded DOX/tGO–GEM/Max8 composites using the triple negative breast cancer cell line MDA-MB-231. The results are presented as dose–response curves in Fig. 10A, fit with the median-effect equation.108 The corresponding IC50 values are presented in Fig. 10D. Free DOX and free GEM yielded IC50 values of 0.399 ± 0.04 μM and >20 μM, respectively, in line with published data.9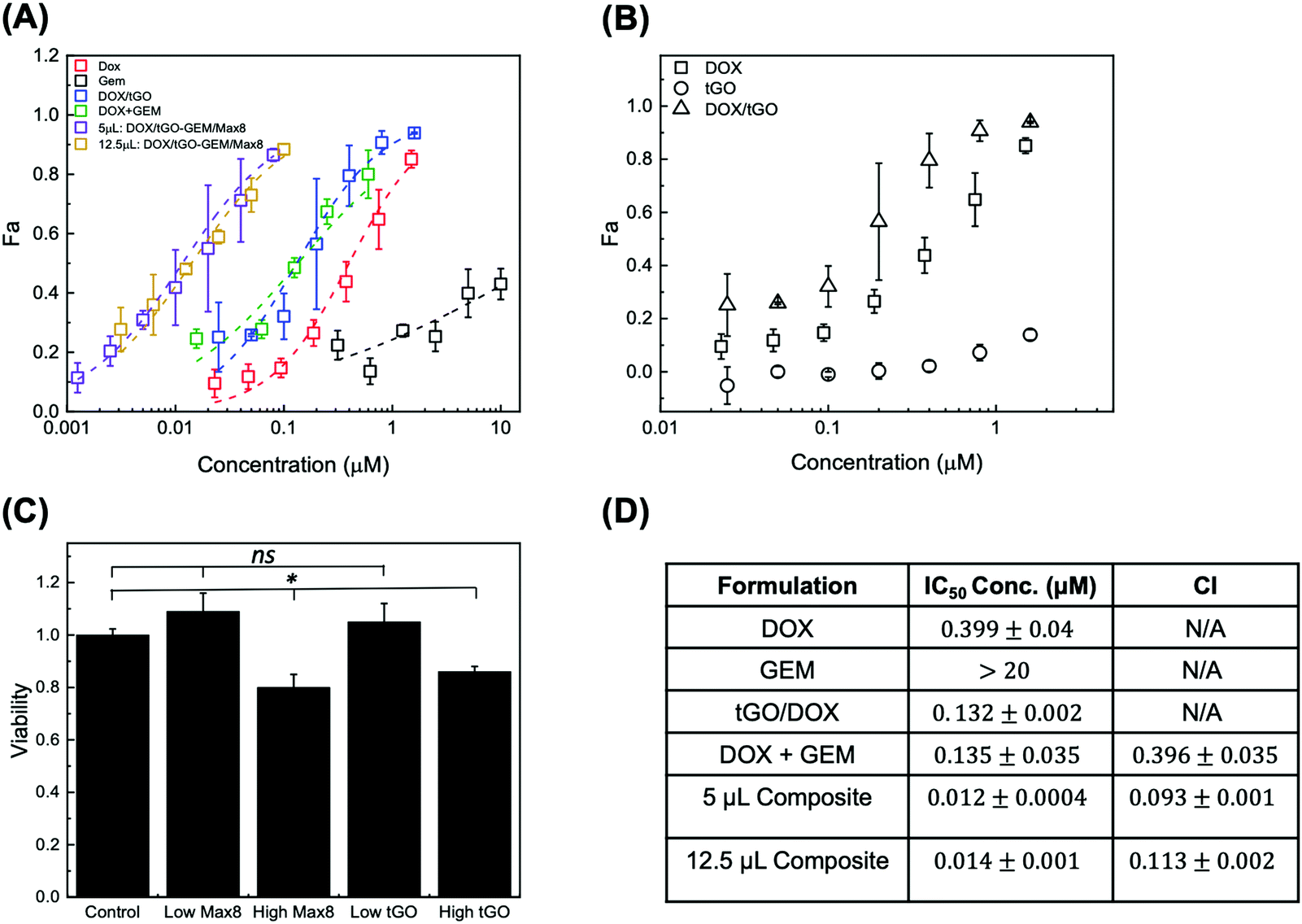 | ||
| Fig. 10 (A) Dose–response curves for free DOX (red), free GEM (black), DOX/tGO particles (blue), 10:1 molar ratio free drug combination of GEM/DOX (green), 10:1 molar ratio free drug combination of GEM/DOX from composite system with 5 μL (purple), and 12.5 μL (yellow) of DOX/tGO–GEM/Max8 composite gel; (B) dose–response curves for DOX (squares), DOX/tGO (triangles), and tGO (circles); (C) cytotoxicity of Max8 hydrogel (low – 5 μL and high – 12.5 μL) and tGO particles (low – 0.2 μg mL−1 and high – 14.6 μg mL−1) relative to untreated cells (control), and (D) calculated IC50 values for the various formulations and the corresponding combination index (CI). Error bars represent mean ± SD. (n ≥ 3). ns indicates no significance, and * indicates p < 0.05, as obtained from a Student's t-test. | ||
In testing the free DOX/tGO particles, we referred to the values of 72 h-release at pH 5 to determine the concentration of particles affording a release of DOX at a dose equivalent to that utilized in the free drug study and accounted for the possibility of uptake of the DOX/tGO particles. The range of DOX concentration utilized in this study, 0.025–1.6 μM, translated in a dose of 0.2–14.2 μg tGO mL−1 of total release volume. The same volume of drug-free particles was also tested as a control to evaluate their basal cytotoxicity (Fig. 10C). Notably, the DOX/tGO particles featured a cytotoxic activity comparable to that of free DOX, with an IC50 value of 0.131 ± 0.002 μM, corresponding to ∼1.7 μg of DOX/tGO particles mL−1 (Fig. 10B). At the same time, the tGO particles alone proved relatively biocompatible, yielding a cell survival rate above 90% when employed at concentrations below 14.2 μg mL−1. Most importantly, at the dose of 1.7 μg mL−1 needed for the DOX-loaded particles to match the IC50 of free DOX, the tGO particles alone show a cell survival rate of ∼99%.
The cytotoxicity of the DOX–GEM combination was then evaluated at the reference GEM:DOX molar ratio for of 10:1, reported as synergistic in prior work (Fig. 10D).9 For the free-drug combination, we observed an IC50 value of 0.135 ± 0.035 μM, relative to DOX concentration. This corresponded to a combination index (CI), calculated utilizing the Chou–Talalay method,109 of 0.396 ± 0.035, confirming strong synergism of the drug pair, consistent with that obtained by Vogus et al.9 using the same drug pair and concentration, and cancer cell line. Lastly, we sought to demonstrate that the dual-drug loaded tGO–Max8 composite system is more therapeutically efficacious than the free drug combination. Accordingly, the reference 10:1, GEM:DOX molar ratio was utilized in these release experiments. We observed an IC50 value of 0.0116 ± 0.0004 μM, relative to DOX concentration, corresponding to a CI of 0.093 ± 0.001, indicating a stronger synergism than the free drug pair. It is noted that the reported values of CI are comparable to those obtained by Vogus et al. of CI = ∼0.12 with sequential administration of this drug pair (GEM for 24 h, then DOX for 48 h).9 We attribute this further reduction in CI (higher synergism) to the ability of the composite tGO–Max8 system to control the release kinetics and achieve the postulated scheduled release, known to improve synergism.9,10 Notably, the IC50 value of the composite system corresponds to a dose of 16 μg mL−1 of tGO particles and 5 μL of Max8 hydrogel, both proven to be non-cytotoxic per se (Fig. 10C).
Conclusions
Scheduled delivery of synergistic combinations of chemotherapeutics is increasingly regarded as a highly effective treatment for aggressive solid tumors. The precise control of molar ratio, sequence, and rate of delivery, in fact, enhances drug synergism and enables a drastic reduction of the required therapeutic doses, while maintaining a highly efficacious outcome. To date, two main approaches to achieve scheduled delivery appear in clinical settings: portable devices that accompany the patient through therapy and inject different drugs based on a set timetable, or engineered materials featuring fine-tuned drug release kinetics. The design and optimization of the chemical functionalization and organization of composite materials as drug delivery systems (DDS) greatly benefits from the integration of in silico design and experimental evaluation. By providing a deeper insight into the complex physicochemical interactions between drugs and materials, integrated computational-experimental methods enhance the quality and accelerate the process of discovery and validation of therapeutic products. This study aims to apply this concept to the design of a homogenous hydrogel-nanoparticle composite capable of delivering a recognized synergistic drug pair (DOX and GEM) with precise molar ratio and release kinetics. In this context, we resolved to (i) adopt materials that combine biocompatibility and affordability with chemically versatility, and (ii) implement scalable functionalization strategies to ensure the translational potential of the resulting DDS. GO nanoparticles and Max8 peptide hydrogel fulfill the requirements on the materials, while TREN modification of the GO surface and the tuning of the Max8/water ratio represent a scalable approach to achieve precise control over the ratio and kinetics of drug delivery. Crucial to the design and understanding of the proposed DDS has been the development of an MD model capable of describing quantitatively the DOX loading on and release from the modified GO particles. The homogeneous distribution of DOX–tGO particles and the thixotropic nature of the Max8 hydrogel matrix make the proposed formulation an exquisitely injectable one, ideal for the treatment of solid tumors through both first-line or consolidation chemotherapy. In this regard, the MDA-MB-231 triple negative breast cancer (TNBC) cell line adopted in this work represents an ideal target for the preclinical testing of the proposed DDS. Patients with metastatic TNBC, in fact, have a poor prognosis and a median overall survival of ∼13 months upon treatment. In the attempt to contribute to the development of a therapeutic arsenal against TNBC, we have tailored our composite system to release GEM at 47-fold rate and a 10-fold molar ratio compared to DOX, which the literature indicates as highly synergistic. The results presented herein demonstrate the validity of our design: the composite DDS afforded a remarkable combination index of 0.093 ± 0.001. This value is not only lower than that provided by the DOX–GEM combination as free drugs in solution (CI = 0.396 ± 0.034) but is lower than any value reported in the literature for this and similar drug combinations delivered by either microfluidic devices or engineered materials.6,9,10,110 Together with reinforcing the value of combining delivery schedule and molar ratio towards increasing therapeutic synergism, this study presents a step forward in the development of translatable (effective, affordable, and scalable) solutions enabling successful treatment of unmet oncological diseases. To this end, future work on different cell lines (e.g., renal cell carcinoma Caki-2 cells or bladder carcinoma MB49 cells) will be undertaken to demonstrate the flexibility of the proposed DDS towards the treatment of different forms of cancer. Selected formulations will be characterized in vivo to evaluate, together with therapeutic efficacy, biodistribution and potential adverse effects; these, however, are not anticipated, given the biocompatibility of Max8 and the low amount of modified GO particles present in the formulation. With its focus on material design and understanding, this contribution lays the ground for the future optimization of novel composites targeting aggressive metastatic solid tumors that – to this day – impact the health and quality of life of millions of people worldwide.Author contributions
Conceptualization, J. D. S., K. S., E. S., M. D., and S. M.; methodology, J. D. S., K. S., A. T. Y., R. L. L., E. S., M. D., and S. M.; investigation, J. D. S., K. S., A. T. Y., S. R., N. H., C. E. D., J. M. D., and R. L. L.; formal analysis, J. D. S. and K. S.; writing – original draft, J. D. S., K. S., E. S., and S. M.; writing – review & editing, J. D. S, K. S., E. S., M. D., and S. M.; funding acquisition, W. G., E. S., M. D., and S. M.; resources, N. H., W. G., E. S., M. D., and S. M.; supervision, E. S., M. D., and S. M.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by start-up funds provided by NC State University. SM, SR, CED and JDS kindly acknowledge support from the Department of Education Graduate Assistance in Areas of National Need (GAANN) in Molecular Biotechnology. ATY acknowledges support from the NIH Integrative Vascular Biology Traineeship (NIH T32HL069768). KS thanks Dr. Nathan Duff and Dr. HoShin Kim for helpful discussions.Notes and references
- J. G. Croissant, D. Zhang, S. Alsaiari, J. Lu, L. Deng, F. Tamanoi, A. M. AlMalik, J. I. Zink and N. M. Khashab, J. Controlled Release, 2016, 229, 183–191 CrossRef CAS PubMed.
- T. Anajafi, M. D. Scott, S. You, X. Yang, Y. Choi, S. Y. Qian and S. Mallik, Bioconjugate Chem., 2016, 27, 762–771 CrossRef CAS PubMed.
- A. C. Palmer and P. K. Sorger, Cell, 2017, 171, 1678–1691 CrossRef CAS PubMed.
- K. Goto, Y. Ohe, T. Shibata, T. Seto, T. Takahashi, K. Nakagawa, H. Tanaka, K. Takeda, M. Nishio and K. Mori, et al. , Lancet Oncol., 2016, 17, 1147–1157 CrossRef CAS PubMed.
- Y.-G. Lee, J. H. Lee, S.-H. Kim, Y. J. Kim, H. Lee, S. Ahn, J.-S. Jang, J.-S. Lee and J. H. Kim, Lung Cancer, 2018, 122, 88–93 CrossRef PubMed.
- A. Pusuluri, V. Krishnan, D. Wu, C. W. Shields, L. W. Wang and S. Mitragotri, Bioeng. Transl. Med., 2019, 4, e10129 Search PubMed.
- Q. Hu, W. Sun, C. Wang and Z. Gu, Adv. Drug Delivery Rev., 2016, 98, 19–34 CrossRef CAS PubMed.
- W. Fan, B. Yung, P. Huang and X. Chen, Chem. Rev., 2017, 117, 13566–13638 CrossRef CAS PubMed.
- D. R. Vogus, A. Pusuluri, R. Chen and S. Mitragotri, Bioeng. Transl. Med., 2018, 3, 49–57 CrossRef CAS PubMed.
- D. R. Vogus, M. A. Evans, A. Pusuluri, A. Barajas, M. Zhang, V. Krishnan, M. Nowak, S. Menegatti, M. E. Helgeson and T. M. Squires, et al. , J. Controlled Release, 2017, 267, 191–202 CrossRef CAS PubMed.
- Y.-X. Bao, X.-D. Zhao, H.-B. Deng, C.-L. Lu, Y. Guo, X. Lu and L.-L. Deng, Cell. Oncol., 2016, 39, 343–352 CrossRef CAS PubMed.
- Z. Zhang, S. Liu, Y. Qi, D. Zhou, Z. Xie, X. Jing, X. Chen and Y. Huang, J. Controlled Release, 2016, 235, 125–133 CrossRef CAS PubMed.
- X. Li, Y. He, J. Hou, G. Yang and S. Zhou, Small, 2019, e1902262 Search PubMed.
- L. Wei, J. Chen, S. Zhao, J. Ding and X. Chen, Acta Biomater., 2017, 58, 44–53 CrossRef CAS PubMed.
- H. Wang, J. Wu, K. Xie, T. Fang, C. Chen, H. Xie, L. Zhou and S. Zheng, ACS Appl. Mater. Interfaces, 2017, 9, 10567–10576 CrossRef CAS PubMed.
- S. Narayanan, U. Mony, D. K. Vijaykumar, M. Koyakutty, B. Paul-Prasanth and D. Menon, Nanomedicine, 2015, 11, 1399–1406 CrossRef CAS PubMed.
- T. Wang, D. Wang, J. Liu, B. Feng, F. Zhou, H. Zhang, L. Zhou, Q. Yin, Z. Zhang and Z. Cao, et al. , Nano Lett., 2017, 17, 5429–5436 CrossRef CAS PubMed.
- L. Li, W. Sun, Z. Zhang and Y. Huang, J. Controlled Release, 2016, 232, 62–74 CrossRef CAS PubMed.
- J.-Y. Douillard, A. Sobrero, C. Carnaghi, P. Comella, E. Díaz-Rubio, A. Santoro and E. Van Cutsem, Ann. Oncol., 2003, 14, ii7–ii12 CrossRef PubMed.
- L. H. J. Simkens, H. van Tinteren, A. May, A. J. ten Tije, C.-J. M. Creemers, O. J. L. Loosveld, F. E. de Jongh, F. L. G. Erdkamp, Z. Erjavec and A. M. E. van der Torren, et al. , Lancet, 2015, 385, 1843–1852 CrossRef CAS.
- M. Ducreux, D. Malka, J. Mendiboure, P.-L. Etienne, P. Texereau, D. Auby, P. Rougier, M. Gasmi, M. Castaing and M. Abbas, et al. , Lancet Oncol., 2011, 12, 1032–1044 CrossRef CAS PubMed.
- F. Cardoso, P. L. Bedard, E. P. Winer, O. Pagani, E. Senkus-Konefka, L. J. Fallowfield, S. Kyriakides, A. Costa, T. Cufer and K. S. Albain, J. Natl. Cancer Inst., 2009, 101, 1174–1181 CrossRef CAS PubMed.
- X. Liang, C. Gao, L. Cui, S. Wang, J. Wang and Z. Dai, Adv. Mater., 2017, 29, 1703135 CrossRef PubMed.
- K. M. Camacho, S. Menegatti, D. R. Vogus, A. Pusuluri, Z. Fuchs, M. Jarvis, M. Zakrewsky, M. A. Evans, R. Chen and S. Mitragotri, J. Controlled Release, 2016, 229, 154–162 CrossRef CAS PubMed.
- K. M. Camacho, S. Menegatti and S. Mitragotri, Nanomedicine, 2016, 11, 1139–1151 CrossRef CAS PubMed.
- K. M. Camacho, S. Kumar, S. Menegatti, D. R. Vogus, A. C. Anselmo and S. Mitragotri, J. Controlled Release, 2015, 210, 198–207 CrossRef CAS PubMed.
- Y. Zheng, Y. Cheng, J. Chen, J. Ding, M. Li, C. Li, J. Wang and X. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 3487–3496 CrossRef CAS PubMed.
- P. S. Yavvari, S. Pal, S. Kumar, A. Kar, A. K. Awasthi, A. Naaz, A. Srivastava and A. Bajaj, ACS Biomater. Sci. Eng., 2017, 3, 3404–3413 CrossRef CAS.
- B. Balakrishnan, N. Joshi, K. Thorat, S. Kaur, R. Chandan and R. Banerjee, J. Mater. Chem. B, 2019, 7, 2920–2925 RSC.
- Y. Wang, Q. Zhao, N. Han, L. Bai, J. Li, J. Liu, E. Che, L. Hu, Q. Zhang and T. Jiang, et al. , Nanomedicine, 2015, 11, 313–327 CrossRef CAS PubMed.
- S. F. Kiew, L. V. Kiew, H. B. Lee, T. Imae and L. Y. Chung, J. Controlled Release, 2016, 226, 217–228 CrossRef CAS PubMed.
- S. Parveen, R. Misra and S. K. Sahoo, Nanomedicine, 2012, 8, 147–166 CrossRef CAS PubMed.
- F. Masood, Mater. Sci. Eng., C, 2016, 60, 569–578 CrossRef CAS PubMed.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 CrossRef CAS PubMed.
- R. Dimatteo, N. J. Darling and T. Segura, Adv. Drug Delivery Rev., 2018, 127, 167–184 CrossRef CAS PubMed.
- A. Fakhari and J. Anand Subramony, J. Controlled Release, 2015, 220, 465–475 CrossRef CAS PubMed.
- B. Shen, D. Lu, W. Zhai and W. Zheng, J. Mater. Chem. C, 2013, 1, 50–53 RSC.
- A. Deb and R. Vimala, J. Drug Delivery Sci. Technol., 2018, 43, 333–342 CrossRef CAS.
- A. Deb, N. G. Andrews and V. Raghavan, Int. J. Biol. Macromol., 2018, 113, 515–525 CrossRef CAS PubMed.
- T. H. Tran, H. T. Nguyen, T. T. Pham, J. Y. Choi, H.-G. Choi, C. S. Yong and J. O. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 28647–28655 CrossRef CAS PubMed.
- Y.-S. Huang, Y.-J. Lu and J.-P. Chen, J. Magn. Magn. Mater., 2017, 427, 34–40 CrossRef CAS.
- M. Hashemi, A. Yadegari, G. Yazdanpanah, S. Jabbehdari, M. Omidi and L. Tayebi, RSC Adv., 2016, 6, 74072–74084 RSC.
- Z. Xu, S. Zhu, M. Wang, Y. Li, P. Shi and X. Huang, ACS Appl. Mater. Interfaces, 2015, 7, 1355–1363 CrossRef CAS PubMed.
- F. Nasrollahi, J. Varshosaz, A. A. Khodadadi, S. Lim and A. Jahanian-Najafabadi, ACS Appl. Mater. Interfaces, 2016, 8, 13282–13293 CrossRef CAS PubMed.
- Y. Lv, L. Tao, S. W. Annie Bligh, H. Yang, Q. Pan and L. Zhu, Mater. Sci. Eng., C, 2016, 59, 652–660 CrossRef CAS PubMed.
- L. He, S. Sarkar, A. Barras, R. Boukherroub, S. Szunerits and D. Mandler, Chem. Commun., 2017, 53, 4022–4025 RSC.
- D. de Melo-Diogo, E. C. Costa, C. G. Alves, R. Lima-Sousa, P. Ferreira, R. O. Louro and I. J. Correia, Eur. J. Pharm. Biopharm., 2018, 131, 162–169 CrossRef CAS PubMed.
- Y. T. Fong, C.-H. Chen and J.-P. Chen, Nanomaterials, 2017, 7, E388 CrossRef PubMed.
- W. Zhang, Z. Guo, D. Huang, Z. Liu, X. Guo and H. Zhong, Biomaterials, 2011, 32, 8555–8561 CrossRef CAS PubMed.
- K. Yang, L. Feng and Z. Liu, Adv. Drug Delivery Rev., 2016, 105, 228–241 CrossRef CAS PubMed.
- Q. Zhang, Z. Wu, N. Li, Y. Pu, B. Wang, T. Zhang and J. Tao, Mater. Sci. Eng., C, 2017, 77, 1363–1375 CrossRef CAS PubMed.
- Z. Hasanzade and H. Raissi, Appl. Surf. Sci., 2017, 422, 1030–1041 CrossRef CAS.
- M. Mahdavi, F. Rahmani and S. Nouranian, J. Mater. Chem. B, 2016, 4, 7441–7451 RSC.
- F. Safdari, H. Raissi, M. Shahabi and M. Zaboli, J. Inorg. Organomet. Polym., 2017, 27, 805–817 CrossRef CAS.
- Z. Hasanzade and H. Raissi, J. Mol. Model., 2017, 23, 36 CrossRef PubMed.
- M. W. Tibbitt, J. E. Dahlman and R. Langer, J. Am. Chem. Soc., 2016, 138, 704–717 CrossRef CAS PubMed.
- A. P. Mathew, S. Uthaman, K.-H. Cho, C.-S. Cho and I.-K. Park, Int. J. Biol. Macromol., 2018, 110, 17–29 CrossRef CAS PubMed.
- K. Park, J. Controlled Release, 2016, 240, 2–8 CrossRef CAS PubMed.
- J. Huang, W.-J. Wang, B.-G. Li and S. Zhu, Macromol. Mater. Eng., 2013, 298, 391–399 CrossRef CAS.
- H. Hezaveh and I. I. Muhamad, J. Taiwan Inst. Chem. Eng., 2013, 44, 182–191 CrossRef CAS.
- A. F. Dexter, N. L. Fletcher, R. G. Creasey, F. Filardo, M. W. Boehm and K. S. Jack, RSC Adv., 2017, 7, 27260–27271 RSC.
- K. Rajagopal, B. Ozbas, D. J. Pochan and J. P. Schneider, Eur. Biophys. J., 2006, 35, 162–169 CrossRef CAS PubMed.
- M. C. Branco, D. J. Pochan, N. J. Wagner and J. P. Schneider, Biomaterials, 2009, 30, 1339–1347 CrossRef CAS PubMed.
- L. Haines-Butterick, K. Rajagopal, M. Branco, D. Salick, R. Rughani, M. Pilarz, M. S. Lamm, D. J. Pochan and J. P. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7791–7796 CrossRef CAS PubMed.
- Y. Chen, H. X. Gan and Y. W. Tong, Macromolecules, 2015, 48, 2647–2653 CrossRef CAS.
- P. Kumar, V. Pillay, G. Modi, Y. E. Choonara, L. C. du Toit and D. Naidoo, Recent Pat. Drug Delivery Formulation, 2011, 5, 24–51 CrossRef CAS.
- A. Vashist, A. Kaushik, A. Ghosal, J. Bala, R. Nikkhah-Moshaie, W. A. Wani, P. Manickam and M. Nair, Gels, 2018, 4, 75 CrossRef CAS PubMed.
- M. Liu, P. Huang, W. Wang, Z. Feng, J. Zhang, L. Deng and A. Dong, J. Mater. Chem. B, 2019, 7, 2667–2677 RSC.
- J. Basso, A. Miranda, S. Nunes, T. Cova, J. Sousa, C. Vitorino and A. Pais, Gels, 2018, 4, 62 CrossRef PubMed.
- M. Constantin, S.-M. Bucatariu, F. Doroftei and G. Fundueanu, Carbohydr. Polym., 2017, 157, 493–502 CrossRef CAS PubMed.
- D. Liu, Y. Chen, X. Feng, M. Deng, G. Xie, J. Wang, L. Zhang, Q. Liu and P. Yuan, Colloids Surf., B, 2014, 113, 158–168 CrossRef CAS PubMed.
- C. Wang, G. Zhang, G. Liu, J. Hu and S. Liu, J. Controlled Release, 2017, 259, 149–159 CrossRef CAS PubMed.
- T. Lammers, V. Subr, K. Ulbrich, P. Peschke, P. E. Huber, W. E. Hennink and G. Storm, Biomaterials, 2009, 30, 3466–3475 CrossRef CAS PubMed.
- S. R. Leonard, A. R. Cormier, X. Pang, M. I. Zimmerman, H.-X. Zhou and A. K. Paravastu, Biophys. J., 2013, 105, 222–230 CrossRef CAS PubMed.
- C. Yan, A. Altunbas, T. Yucel, R. P. Nagarkar, J. P. Schneider and D. J. Pochan, Soft Matter, 2010, 6, 5143–5156 RSC.
- N. He, O. Yildiz, Q. Pan, J. Zhu, X. Zhang, P. D. Bradford and W. Gao, J. Power Sources, 2017, 343, 492–501 CrossRef CAS.
- E. Kaiser, R. L. Colescott, C. D. Bossinger and P. I. Cook, Anal. Biochem., 1970, 34, 595–598 CrossRef CAS PubMed.
- A. M. Baptista, V. H. Teixeira and C. M. Soares, J. Chem. Phys., 2002, 117, 4184–4200 CrossRef CAS.
- A. Adnan, R. Lam, H. Chen, J. Lee, D. J. Schaffer, A. S. Barnard, G. C. Schatz, D. Ho and W. K. Liu, Mol. Pharmaceutics, 2011, 8, 368–374 CrossRef CAS PubMed.
- L. J. Henderson, Am. J. Physiol., 1908, 21, 173–179 CrossRef CAS.
- K. A. Hasselbalch, Die Berechnung der Wasserstoffzahl des Blutes aus der freien und gebundenen Kohlensäure desselben, und die Sauerstoffbindung des Blutes als Funktion der Wasserstoffzahl, Julius Springer, Berlin, 1916 Search PubMed.
- B. Konkena and S. Vasudevan, J. Phys. Chem. Lett., 2012, 3, 867–872 CrossRef CAS PubMed.
- G. Speelmans, R. W. H. M. Staffhorst, B. de Kruijff and F. A. de Wolf, Biochemistry, 1994, 33, 13761–13768 CrossRef CAS.
- C.-J. Shih, S. Lin, R. Sharma, M. S. Strano and D. Blankschtein, Langmuir, 2011, 28, 235 CrossRef PubMed.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- F.-Y. Dupradeau, A. Pigache, T. Zaffran, C. Savineau, R. Lelong, N. Grivel, D. Lelong, W. Rosanski and P. Cieplak, Phys. Chem. Chem. Phys., 2010, 12, 7821 RSC.
- E. Vanquelef, S. Simon, G. Marquant, E. Garcia, G. Klimerak, J. C. Delepine, P. Cieplak and F.-Y. Dupradeau, Nucleic Acids Res., 2011, 39, W511–W517 CrossRef CAS PubMed.
- A. Jakalian, B. L. Bush, D. B. Jack and C. I. Bayly, J. Comput. Chem., 2000, 21, 132–146 CrossRef CAS.
- A. Jakalian, D. B. Jack and C. I. Bayly, J. Comput. Chem., 2002, 23, 1623–1641 CrossRef CAS PubMed.
- D. Stauffer, N. Dragneva, W. B. Floriano, R. C. Mawhinney, G. Fanchini, S. French and O. Rubel, J. Chem. Phys., 2014, 141, 044705 CrossRef CAS PubMed.
- H. Lei, X. Wang and C. Wu, J. Mol. Graphics Modell., 2012, 38, 279–289 CrossRef CAS PubMed.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, Abstr. Pap., Am. Chem. Soc., 2001, 222, U403 Search PubMed.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926 CrossRef CAS.
- J. E. Basconi and M. R. Shirts, J. Chem. Theory Comput., 2013, 9, 2887–2899 CrossRef CAS PubMed.
- K. R. Karnati and Y. Wang, Phys. Chem. Chem. Phys., 2018, 20, 9389–9400 RSC.
- A. Yang, J. Li, C. Zhang, W. Zhang and N. Ma, Appl. Surf. Sci., 2015, 346, 443–450 CrossRef CAS.
- T. Szabó, O. Berkesi and I. Dékány, Carbon, 2005, 43, 3186–3189 CrossRef.
- S. Chen, Y. Itoh, T. Masuda, S. Shimizu, J. Zhao, J. Ma, S. Nakamura, K. Okuro, H. Noguchi and K. Uosaki, et al. , Science, 2015, 348, 555–559 CrossRef CAS PubMed.
- Z. Yue, C. Li, G. A. Voth and J. M. J. Swanson, J. Am. Chem. Soc., 2019, 141, 13421–13433 CrossRef CAS PubMed.
- J. Hong, R. J. Hamers, J. A. Pedersen and Q. Cui, J. Phys. Chem. C, 2017, 121, 3584–3596 CrossRef CAS.
- M. Vatanparast and Z. Shariatinia, J. Mol. Graphics Modell., 2019, 89, 50–59 CrossRef CAS PubMed.
- Y. Hu, T. Guo, X. Ye, Q. Li, M. Guo, H. Liu and Z. Wu, Chem. Eng. J., 2013, 228, 392–397 CrossRef CAS.
- M. Li, C. Liu, Y. Xie, H. Cao, H. Zhao and Y. Zhang, Carbon, 2014, 66, 302–311 CrossRef CAS.
- J. D. Schneible, A. Singhal, R. L. Lilova, C. K. Hall, A. Grafmüller and S. Menegatti, Biomacromolecules, 2019, 20, 3126–3141 CrossRef CAS PubMed.
- T.-C. Chou, Am. J. Cancer Res., 2011, 1, 925–954 Search PubMed.
- T.-C. Chou, Cancer Res., 2010, 70, 440–446 CrossRef CAS PubMed.
- D. R. Budman, A. Calabro, L. Rosen and M. Lesser, Anticancer Drugs, 2012, 23, 272–279 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Short time scale DOX release from modified GO library, zeta potential – amine surface density relationship, DOX loading for tGO at various zeta potential and loading buffer composition, simulation snapshots of DOX loading, DOX release at pH 7.4, and confocal images of composite hydrogel. See DOI: 10.1039/d0tb00064g |
| This journal is © The Royal Society of Chemistry 2020 |