
Dynamic covalent polymers for biomedical applications
Yan
Zhang
 *a,
Yunchuan
Qi
b,
Sébastien
Ulrich
*c,
Mihail
Barboiu
*d and
Olof
Ramström
*be
*a,
Yunchuan
Qi
b,
Sébastien
Ulrich
*c,
Mihail
Barboiu
*d and
Olof
Ramström
*be
aKey Laboratory of Carbohydrate Chemistry and Biotechnology, Ministry of Education, School of Pharmaceutical Sciences, Jiangnan University, Wuxi, 214122, P. R. China. E-mail: zhangyanyz@jiangnan.edu.cn
bDepartment of Chemistry, University of Massachusetts Lowell, One University Ave., Lowell, MA 01854, USA. E-mail: olof_ramstrom@uml.edu
cInstitut des Biomolécules Max Mousseron (IBMM), CNRS, Université de Montpellier, ENSCM, Montpellier, France. E-mail: sebastien.ulrich@enscm.fr
dInstitut Européen des Membranes, Adaptive Supramolecular Nanosystems Group, University of Montpellier, ENSCM, CNRS, Place Eugène Bataillon, CC 047, F-34095, Montpellier, France. E-mail: mihail-dumitru.barboiu@umontpellier.fr
eDepartment of Chemical and Biomedical Sciences, Linnaeus University, SE-39182 Kalmar, Sweden
First published on 3rd December 2019
Abstract
The rapid development of supramolecular polymer chemistry and constitutional dynamic chemistry over the last decades has made tremendous impact on the emergence of dynamic covalent polymers. These materials are formed through reversible covalent bonds, endowing them with adaptive and responsive features that have resulted in high interest throughout the community. Owing to their intriguing properties, such as self-healing, shape-memory effects, recyclability, degradability, stimuli-responsiveness, etc., the materials have found multiple uses in a wide range of areas. Of special interest is their increasing use for biomedical applications, and many examples have been demonstrated in recent years. These materials have thus been used for the recognition and sensing of biologically active compounds, for the modulation of enzyme activity, for gene delivery, and as materials for cell culture, delivery, and wound-dressing. In this review, some of these endeavors are discussed, highlighting the many advantages and unique properties of dynamic covalent polymers for use in biology and biomedicine.
 Yan Zhang | Yan Zhang received her BSc degree from Central South University, P. R. China. In 2013, she obtained her PhD degree from the Royal Institute of Technology, Sweden, under the supervision of Prof. Olof Ramström. After a postdoctoral period at the Institut Européen des Membranes (IEM), Montpellier, France, with Dr Mihail Barboiu, she is now a professor at Jiangnan University, School of Pharmaceutical Sciences, P. R. China. Her current research is focused on biomaterials based on dynamic covalent polymers. |
 Yunchuan Qi | Yunchuan Qi graduated from Shaanxi Normal University in 2014 and received his MSc in 2017 from Beijing Normal University. He started his PhD research in Prof. Olof Ramström's group at University of Massachusetts Lowell in 2017, focusing on dynamic covalent polymers. |
 Sébastien Ulrich | Sébastien Ulrich carried out his PhD with Prof. Jean-Marie Lehn (Université de Strasbourg, France), and post-docs with Prof. Harry L. Anderson (Oxford University, UK) and Prof. Eric T. Kool (Stanford University, CA, USA). In 2011 he joined the group of Prof. Pascal Dumy, first in Grenoble, then in Montpellier, France where he was recruited by the CNRS in 2012 to develop his current research interests in the field of supramolecular bioorganic chemistry. In 2017, he was awarded the CNRS Bronze Medal. |
 Mihail Barboiu | Mihail Barboiu graduated from University Politehnica of Bucharest and received his PhD in 1998 from the University of Montpellier, before being post-doctoral researcher at the University Louis Pasteur in Strasbourg. He is CNRS Research Director at the Institut Europeen des Membranes in Montpellier and Fellow of Royal Society of Chemistry. Since 2014 he is also Extraordinary Professor in Sun-Yat-sen University in Guangzhou, China. A major focus of his research is Dynamic Constitutional Chemistry toward Dynamic Interactive Systems: adaptive biomimetic membranes, delivery devices etc. author of more than 280 scientific publications, 3 books and 22 chapters and 420 conferences and lectures. Dr Barboiu has received in 2004 the EURYI Award in Chemistry and in 2015 the RSC Surfaces and Interfaces Award for the Development of Artificial Water Channels. |
 Olof Ramström | Olof Ramström received his MSc and PhD degrees from Lund Institute of Technology/Lund University, Sweden. After a period at the Louis Pasteur University, Strasbourg, France, he returned to Sweden and the Royal Institute of Technology, Stockholm. He is currently active at University of Massachusetts, Lowell, USA, and Linnaeus University, Sweden. |
1. Introduction
During the last decades, dynamic covalent chemistry has emerged as a powerful means to generate controlled molecular systems and networks, designed self-processes, and complex architectures.1–5 It expands the adaptive features of supramolecular chemistry to the molecular level,6 thereby forming a constitutional dynamic chemistry encompassing dynamically interchanging connections between different components.5,7 Compared to supramolecular interactions, component reshuffling through dynamic covalent bonds usually needs physical or chemical triggers and takes longer time, but stronger, optimal connections can generally be established, leading to responsive systems with controllable stability under different conditions or in the presence of stimuli. These features are often of advantage, and have led to applications in a range of areas, such as organic synthesis, catalysis, materials sciences, and biomedicine.8–11Dynamic covalent polymers (dynamers) are polymeric structures in which the units are linked through reversible covalent bonds, thus in principle giving rise to constitutional dynamics.12–15 Such dynamic polymers show intriguing malleability, self-healing, and shear-thinning effects, as well as tunable mechanical and optical properties.16,17 Moreover, when based on repeating units of embedded functional groups, they often display multivalent effects, resulting in higher binding affinity compared to their corresponding monomers, a feature that is especially useful in biorecognition.
A number of summarizing accounts on the topic of dynamic covalent polymers have been reported, however generally emphasizing their preparations, physicochemical nature, and applications as self-healing materials, shape-memory matrices, stimuli-responsive structures, etc.10,18–22 However, in parallel to the rich development of new dynamic materials bestowed with multiple functionalities and able to operate under a wide range of conditions, biomedical applications have increasingly become a target.
In this review, we provide a short background on dynamic polymer features, preparation, and distinctive properties, that will help the reader to focus on the biomedical applications of this new type of adaptive systems. Extensive studies have been directed in recent years with the goal and control molecular structure and to generate related biofunctions in fully adaptive synthetic systems. A range of intriguing examples will be described, involving different aspects of gene and drug delivery, enzyme activation, recognition of bioactive compounds, cell culture and delivery, as well as antibacterial and wound-dressing materials.
2. Synthesis of dynamic covalent polymers
In addition to selecting suitable main-chain (backbone) and potential side-chain structures, the key step in preparing dynamic polymers is the inclusion of elements carrying the ability to form dynamic covalent bonds. The features of the various reversible bond types have to be considered in great detail, as they are essential to ensure sufficient structural control and functionality of the final polymeric structures. Especially for applications in the biomedical area, the exchange rates and biocompatibility of the reversible reactions can be critical. With a judicious selection and design of the building blocks, their arrangement in a preferred order is in principle possible, thereby obtaining materials with desired functions.2.1. Types of reversible reactions used for the construction of dynamic polymers
Kinetically unstable bond types leading to reversibility and reaction equilibria are essentially known since the dawn of modern (organic) chemistry. However, in response to the conceptualization of constitutional dynamic chemistry (cf. also dynamic combinatorial chemistry, etc.) in the 1990s and early 2000s,23–26 the scope of reversible reaction types has been continuously enriched and expanded. Herein, we limit the description in giving a very brief introduction to each of the frequently used reversible reactions applied to dynamic polymers, especially those that are preferred for biomedical systems (Table 1), and the reader is recommended to consult with other sources for more detail.10,27,28| Imine formation |

|
| Disulfide formation |
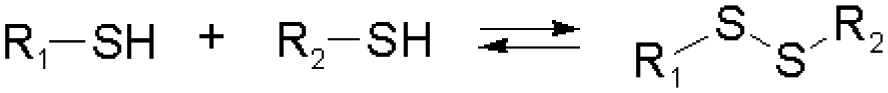
|
| Transesterification |
|
| Conjugate addition |
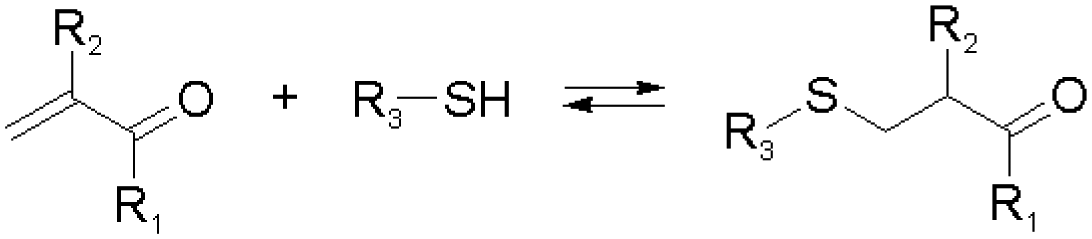
|
| Diels–Alder cycloaddition |
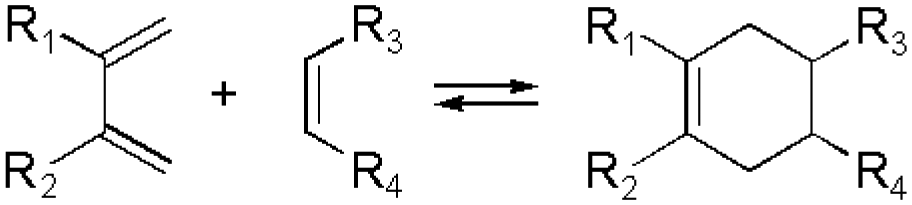
|
| Boronic ester formation |

|
| Olefin metathesis |
|
The various types of imine bonds (incl. hydrazones, acylhydrazones, etc.) constitute one of the most commonly used tools to build dynamic polymers.29,30 The condensation between aldehydes and primary amines leads to imine products, which can undergo the reversed process with or without the addition of catalysts. This facilitates further polymeric component exchange, while enabling an element of control. Depending on their structures, imines often display high formation/hydrolysis rates under physiological conditions as well as good biocompatibility. pH-control is here important, leading to modulation between the imine and the hydrolyzed states, enabling microenvironmental changes in biological systems. Moreover, aldehydes may reversibly interact with peptides in biological systems. Imines have therefore become very attractive for biomedical applications.
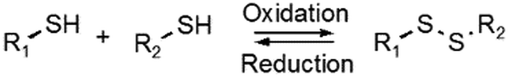
Disulfide formation and exchange reactions are also frequently to construct dynamic polymers.31 Both anionic (thiolate) and radical pathways can in this case lead to disulfide recomposition. In the former case, a redox balance between thiolate and disulfide species leads to exchange. Concerning the radical pathway, light or radical initiators can cause S–S bond cleavage, leading to disulfide scrambling following radical recombination. Similarly, Se–Se and Se–N bonds can be used in reversible, radical-based exchange processes.32,33 Such disulfide-linked polymers have, for example, been developed to target cysteine groups in biological systems, leading to degradation of the polymers or release of the encapsulated entities. The presence of redox agents or light sources can also introduce corresponding structural and functional changes, which can be compatible with in vitro and in vivo analyses.34,35
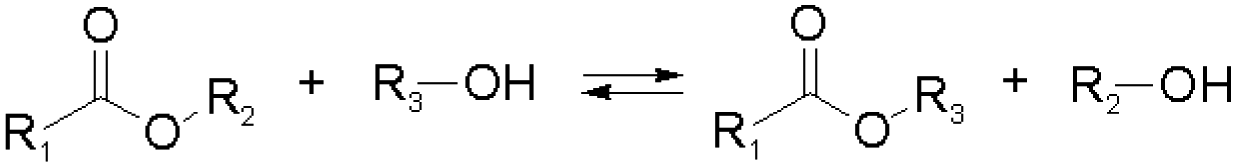
Other than imines, nucleophilic addition/substitution reactions are often sufficiently reversible, especially when performed under the catalysis of acids or bases. Examples include trans(thiol)esterification, hemiacetal/hemiaminal reactions, nitroaldol addition, and conjugate addition reactions, also susceptible to biological functional groups, such as thiols, and amines. To accelerate the exchange processes, a wide range of base catalysts have been exemplified, ranging from the weakly basic triethylamine to the stronger base 1,1,3,3-tetramethylguanidine (TMG). A variety of Lewis acids, such as zinc acetate, have furthermore been demonstrated.36,37
The Diels–Alder (DA) cycloaddition reaction remains a most powerful tool to generate C–C bonds and ring structures, typically through the reaction between an electron-deficient dienophile and a suitable electron-rich diene. The reaction is often moderately reversible, but efficient retro-DA reactions can occasionally be achieved at more elevated temperatures. Nevertheless, cycloreversion under mild, ambient conditions have been reported, for example involving cyano-functionalized dienophiles or N-phenyltriazolinediones, significantly expanding the application scope of this system,38,39 Thus, temperature control can, in principle, be used to tune the properties of biomaterials based on DA reactions.
Boronic ester formation, especially the reaction between boronic acids and cis-1,2/1,3-diols, is another widely used reaction in dynamic chemistry. The formation is compatible with aqueous conditions, and the reversibility can be controlled by pH.40,41 Considering the presence of diol motives in natural carbohydrates, this type of reaction is often related to biomedical applications. For example, boronic acids grafted to biomaterials can lead to capture and release of cells upon addition of carbohydrates.
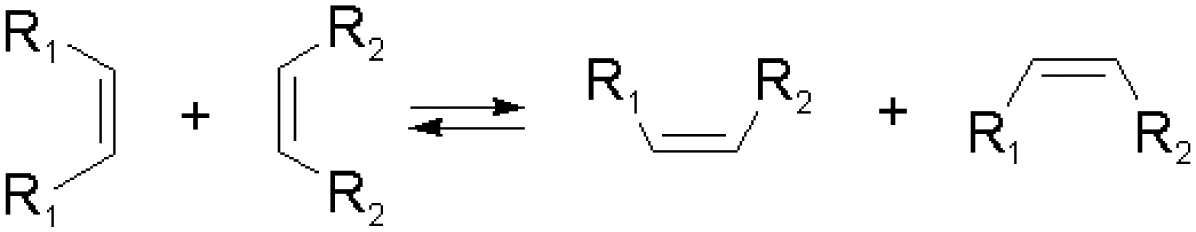
Olefin metathesis is another important approach to forming reversible C–C bonds. The reaction has, for example, been very successfully used to synthesize bioactive macrocyclic compounds and organic polymers, and has also been applied in dynamic chemistry protocols. The reaction can be accelerated by catalysts at room temperature, through a cycloaddition/cycloreversion mechanism.27 Compared to other reversible reactions, olefin metathesis provided a highly efficient C–C bond shuffling under ambient conditions.42
In addition to these commonly used reactions, a number of alternative transformations have been proposed for the fabrication of dynamic covalent polymers. These include some more recently developed reversible reaction types, such as the silyl ether formation/exchange, reversible diarylbisbenzo-furanone crosslinking, and urea/urethane exchange.43–45 This development has greatly enriched the variety of dynamic chemistry and brought new features to applications regarding biomaterials design, molecular sensing and cell interactions.
2.2. Design strategies of polymer architectures
The reversible reaction type being the most important element of the final structures, other considerations have to be made in order to achieve the desired materials. Special attention has to paid regarding interactions with biological targets, where the binding affinities may be considerably affected by the polymer structures. In analogy to traditional polymers, the dynamic covalent counterparts can thus be built up through different arrangements of the connections, leading to various types of polymer architectures.Typically, there are two approaches for reversible bonds to be inserted into polymeric structures: main-chain and side-chain incorporation (Fig. 1).19 The former strategy can lead to formation and collapse of polymer structures under specific conditions, resulting in degradable or self-healing biomaterials, whereas the latter approach can modulate the binding effects during the biorecognition process. When the polymer main-chain is composed of reversible covalent reactions, the resulting polymer structure can be either linear or cross-linked. In the linear case, homo- or hetero-ditopic complementary building blocks are adopted, whereas tritopic or even tetratopic components are required for construction of cross-linked polymers. For example, trialdehyde and tris(2-amino-ethyl)amine are often observed in three dimensional dynamic architectures, leading to more stabilized complex reaction networks, which can provide additional information regarding the interaction modes to the active sites of biological targets.46–48 In the case of main-chain dynamic covalent polymers, it is important to notice that their length can adapt by induced polymerization and depolymerization processes. On the other hand, when the side-chain is grafted into polymers through reversible covalent bonds, various types of functional groups can be attached, forming brush-like polymeric structures for more specific interactions with biological targets. One advantage with this side-chain attachment is that the functional entities can be easily switched, without interrupting the main-chain backbone which is imposed by a non-dynamic polymer scaffold.49 This can furthermore lead to functional group repositioning to further optimize biological interactions and changes of the associated biological activities.
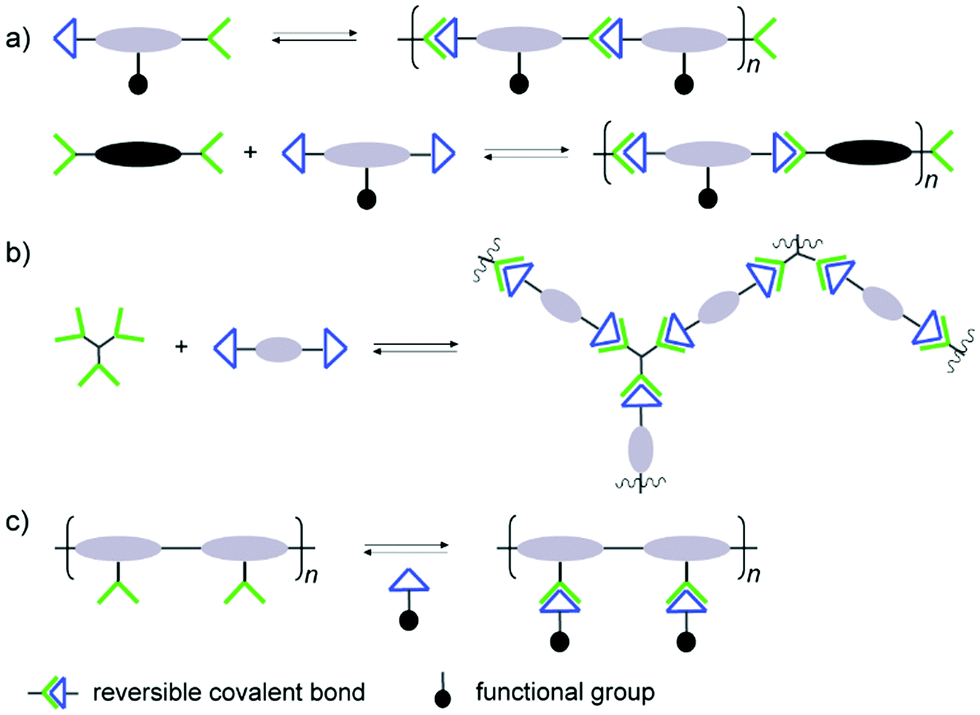 | ||
| Fig. 1 Different types of architectures in dynamic polymers with (a) linear main-chain; (b) cross-linked main-chain; (c) side-chain connections.19 Copyright 2017 American Chemical Society. | ||
Furthermore, if different reversible reactions are amended into one polymer, dynamic architectures with much higher complexity can be achieved, making it possible to endow both the backbone and sidechain with dynamic character. Such multi-step fabrication of dynamic polymers with different reaction types can contribute to smart biomaterials displaying multi-responsiveness to various stimuli inside the biological system or other external factors. For example, hydrazone and boronic ester bonds within a linear polymer were aligned with orthogonal disulfide exchanges in a complex dynamic network, where different bonds responded to different pH conditions, leading to precisely controlled functionally tunable systems.50
3. Properties of dynamic covalent polymers
The kinetic instability of dynamic covalent linkages under defined conditions, coupled with their restoration, give them unique inherent features that distinguish them from other covalent bonds.19 When introduced in polymers, such linkage types endow the materials with the same unique bond features, and can also give rise to important emerging properties not present in the individual building blocks. Dynamic covalent polymers have thus been shown to demonstrate many unique properties, such as self-healing ability, recyclability, stimuli-responsiveness, shape memory behavior, degradability, etc., which will be discussed in the following sections.51–573.1. Reversibility
The reversible bond formation is the key feature of dynamic covalent chemistry.29 In a dynamic covalent system, the forward reaction proceeds in parallel with the backward reaction, and all species will eventually settle into the most thermodynamically stable states.10 In a dynamic covalent polymer, the collection of the reversible reactions in each dynamic connection leads to multiple equilibria, which have to be properly controlled to form the desired material. The polymer-forming reaction should thus be predominating, either by selecting reaction types and/or building blocks with a high thermodynamic propensity of forming the product, or by installing secondary processes that funnel the products out of an unfavorable equilibrium. In the first scenario, the material will be sufficiently stable while maintaining its dynamic properties under normal conditions. This will lead to a situation where the equilibrium can be re-established in the system if the polymer has been damaged by external forces and the dynamic covalent bonds have been broken. In principle, the dynamic bonds would thus be reformed and restore the polymer in its original physical form and functionality.22The unique properties of dynamic covalent polymers have been explored in a multitude of studies and applications in recent years, and in the majority of cases the desired functionalities have been designed to originate from the covalent bond reversibility.32,58–60
Self-healing is one of the most widely studied topics of dynamic covalent polymers and an excellent demonstration of the reversibility of dynamic covalent chemistry.18 Conventional self-healing materials can recover automatically from mechanical damages, but require additives or encapsulated agents for the healing process.61 On the other hand, because of the reversible nature of reversible covalent bonds, dynamic polymers are in principle able to spontaneously recover from damage to the original form without external agents (Fig. 2).62 Dynamic covalent polymers are thus bestowed with an inherent, self-healable ability, which has been demonstrated for a variety of different dynamers in recent years.63–65 These polymers have been based on a range of dynamic covalent linkages, including imine bonds/acylhydrazone linkages,51,63,66–69 boronic ester linkages,59,70,71 Diels–Alder adducts,58,64,72etc. Materials used for biological applications, such as cell culture and wound-dressing,73,74 have taken advantage of the self-healing property of dynamic covalent polymers to achieve their specific functionalities.
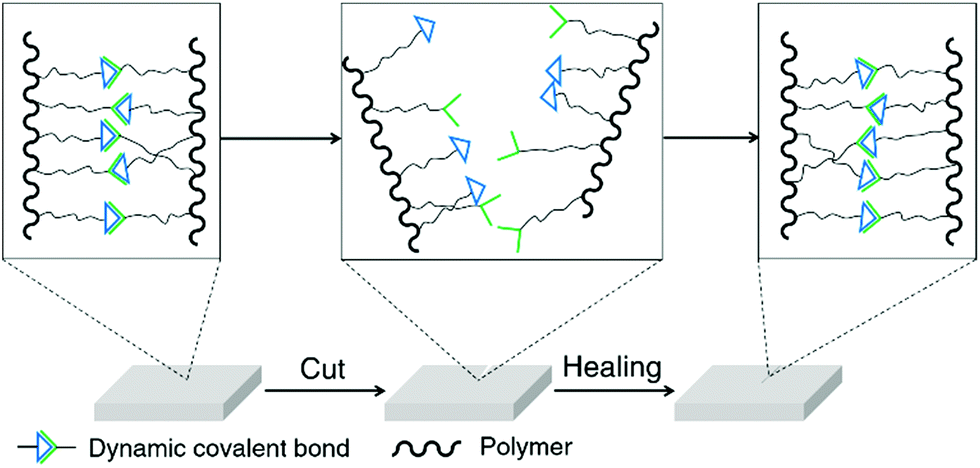 | ||
| Fig. 2 Illustration of self-healing dynamic covalent polymers. | ||
Recyclability is another important characteristic of dynamic covalent polymers, again enabled by the properties of the incorporated reversible bonds.57 Common covalent polymers cannot be fully restored to their original physical structures or mechanical performances after recycling. In contrast, many non-covalent cross-linked materials can be easily recycled, however generally forming less mechanically robust structures.56 For dynamic covalent polymers, on the other hand, both high recyclability and robust structure can be maintained due to the selected reversible covalent bonds (Fig. 3).60,65
 | ||
| Fig. 3 Illustration of the recyclability of dynamic covalent polymers. | ||
3.2. Responsiveness
Generally, the structures of the dynamic covalent polymers are controlled by thermodynamics, and can be affected by changing the surrounding environmental factors.22 This makes dynamic covalent polymers responsive to external stimuli, such as temperature, light, pH of the medium, and redox reactions, leading to systemic adaptation and associated changes of the physical or chemical properties (Table 2).67,75–77 Some of these responsive factors, especially pH changes and redox reactions, are often present in biological systems, and enable the dynamic covalent polymers with attractive properties as biofunctional materials, suitable for gene delivery, biorecognition, etc.78,79| Thermoresponsive |
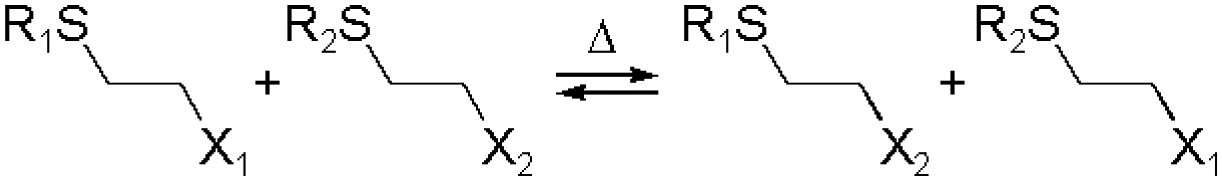
|
| pH responsive |
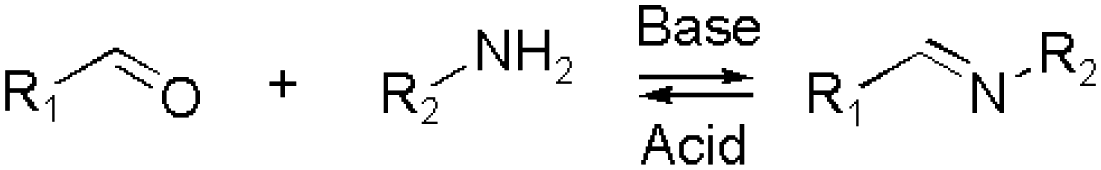
|
| Redox responsive |
|
Temperature changes have direct effects on the thermodynamics of the dynamic covalent system. For example, the self-healing process of certain dynamic covalent polymers can be triggered or accelerated by mild heating.64,80 Furthermore, dynamic covalent bonds are typically sensitive to reversible breakage with increasing temperatures, which will cause chemical and physical changes in the polymers.45
The pH of the medium is another important stimulus, and several dynamic covalent bond types are sensitive to the acidity of the medium. In addition, the remaining part of the polymer can often contain pH-sensitive groups, which can result in both conformational and constitutional dynamics of the materials. The pH-responsiveness is an important feature of some materials used for biorecognition, for which the formation and breaking of the dynamic covalent bonds under different pH conditions is essential for their performances.79
Moreover, redox-responsive dynamic polymers have been explored in multiple studies, often based on disulfide bonds. For example, redox-sensitive biomaterials for gene delivery have been developed.81
3.3. Mechanical properties
Dynamic covalent polymers combine the advanced mechanical properties of traditional polymers with the unique features of dynamic covalent bonds, and this can lead to intriguing new effects and altered mechanical performances of the polymers.22 Such mechanical features of dynamic covalent polymers can also be applied in biological systems, for example, by maintaining robust structures in different environments.82Conventional shape-memory polymers are able to recover the original (permanent) geometry after having been persuaded (programmed) into a temporary shape. However, the permanent shape cannot be altered once it is fabricated due to the rigid, covalently cross-linked polymer network.10 On the other hand, the original geometry of shape-memory polymers containing dynamic covalent bonds can be relatively easily reconfigured, taking advantage of the dynamic nature of the bonds, thus enabling alternative programming pathways (Fig. 4).83 By allowing the dynamic connections to undergo topological rearrangement, leading to solid-state plasticity effects, the permanent shape of the material can be remolded.84 This improves the overall performance and can also reduce the cost of such high-performance polymers.
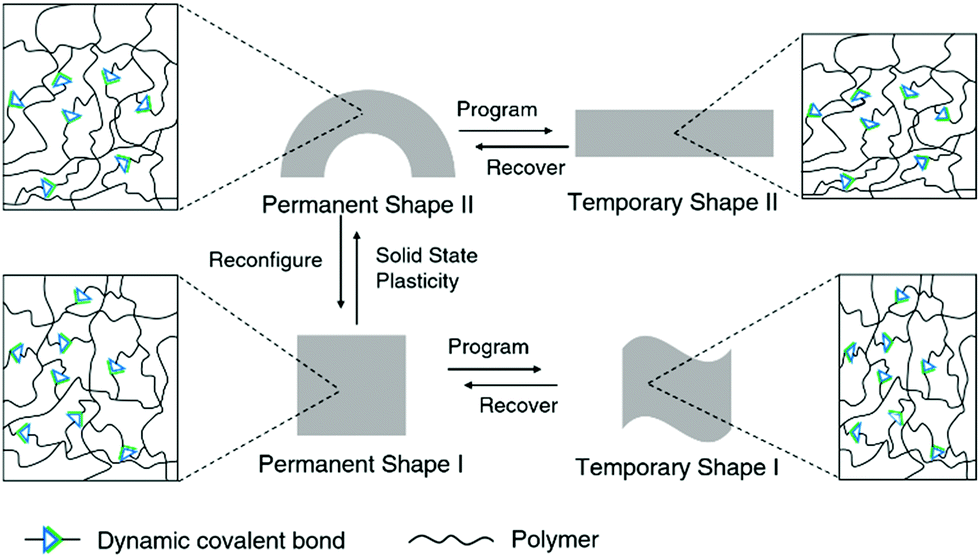 | ||
| Fig. 4 Reconfigurable shape memory dynamic covalent polymers. | ||
High stretchability is another feature of dynamic covalent polymers. In order to achieve good stretchability, a common strategy is the embodiment of sacrificial units within the materials.63 Since dynamic covalent bonds can be controlled to break more easily than their normal counterparts, owing to the weaker bond strengths, they are inherently good candidates for use in sacrificial units.20
3.4. Degradability
Degradability is one of the most important advantages of dynamic covalent polymers, which, again, is mainly due to the relatively weak strength of the reversible bonds.57,85,86 All such polymers can in principle undergo degradation to their respective reaction components under suitable conditions favoring bond breakage. For example, in analogy to traditional polyesters, dynamers based on ester linkages can typically also be degraded under relatively mild hydrolytic conditions. Thus, degradability is one of the favorable features of biological materials.4. Bioapplications of dynamic covalent polymers
4.1. Gene delivery
The non-covalent recognition of nucleic acids by synthetic vectors requires combining multiple weak electrostatic interactions in order to achieve a stable interaction in biological media. This multivalent binding is best achieved using macromolecules such as polymers87–90 and dendrimers.91 Nucleic acids condensation92 is a complex multi-step process (interaction, complexation, folding) that results in the formation of “polyplexes” nanoparticles that can penetrate cells and deliver functional nucleic acids like pDNA, siRNA or mRNA, provided they have appropriate sizes and surface charges. However, standard polymers and dendrimers, which do not degrade, tend to poorly release their cargo once inside cells and to accumulate, often leading to toxic side-effects. Dynamic covalent chemistry provides a unique opportunity in this field.93 Indeed, the use of reversible covalent bonds, operating chemo-selectively in aqueous media under mild reaction conditions should grant access to self-assembled artificial vectors that can adapt and dynamically respond to chemical effectors, thus mimicking some of the features of viral gene delivery.94,95 In this context, condensation reactions such as imine bond formation (incl. (acyl)hydrazones and oximes).96,97 have become very popular as pH-sensitive reversible linkages since cell entry through endocytosis occur through passage by late endosomes which are slightly acidic nano-compartments (pH ≈ 5.0–5.5).98 Disulfides99 have also attracted a strong interest as redox-sensitive reversible linkages due the high (mM) intracellular concentration of reducing agent glutathione.Ulrich and coworkers have reported linear main-chain dynamic covalent polymers self-assembled by polycondensation through acylhydrazone linkages (Fig. 5).78 This design was shown to endow pH-sensitivity to the system, undergoing a much faster depolymerization at acidic pH (4–5) than at neutral pH. Thanks to the multivalency expressed by the system, which combines alternatively cationic and PEG-like monomers, a strong DNA complexation was evidenced. This initial design was recently extended to biomolecular dynamic covalent polymers made of amino acid derivatives, thereby significantly expanding the scope of the approach in terms of chemical functionalities that can be introduced. siRNA delivery in live cells was successfully demonstrated.100
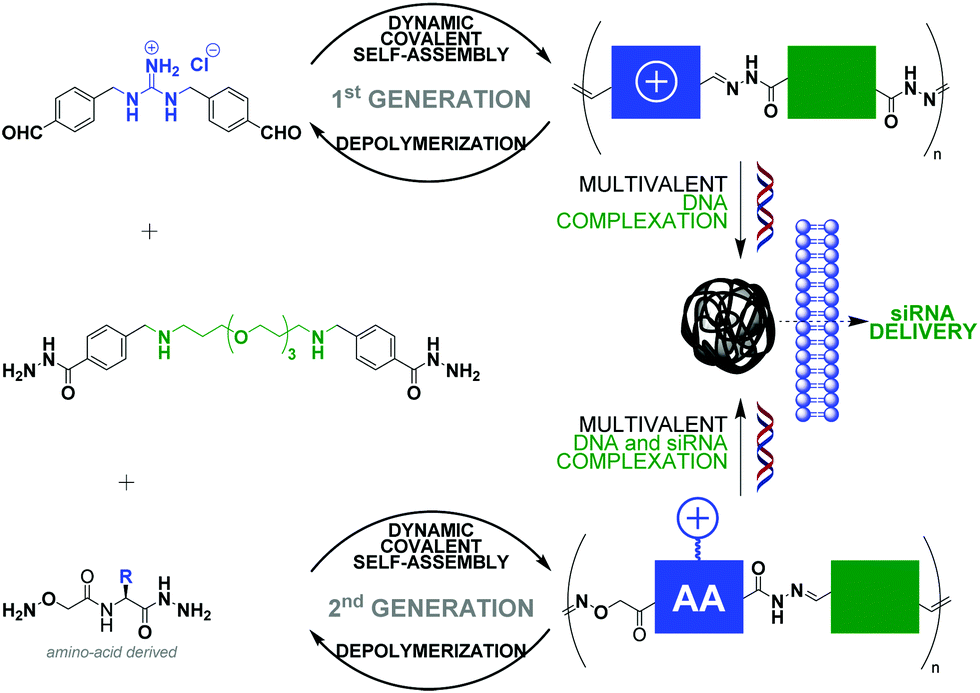 | ||
| Fig. 5 Dynamic self-assembly of linear dynamic covalent polymers, with reversible covalent linkages inserted within their main-chain, for multivalent DNA complexation and siRNA delivery. Dynamic covalent polymer formation occurs through poly-condensation between bisaldehyde and bishydrazide complementary monomers. AA: amino-acid. | ||
Peptide building blocks can also be self-assembled into multimeric dynamic covalent polymers using disulfide bond formation, simply through thiol oxidation. For instance, DMSO-promoted oxidation of Cys(Lys)10Cys yields, by step-growth polymerization, reductively cleavable linear polycations of MW up to 187![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da that can complex and deliver pDNA in cells (Fig. 6).81
000 Da that can complex and deliver pDNA in cells (Fig. 6).81
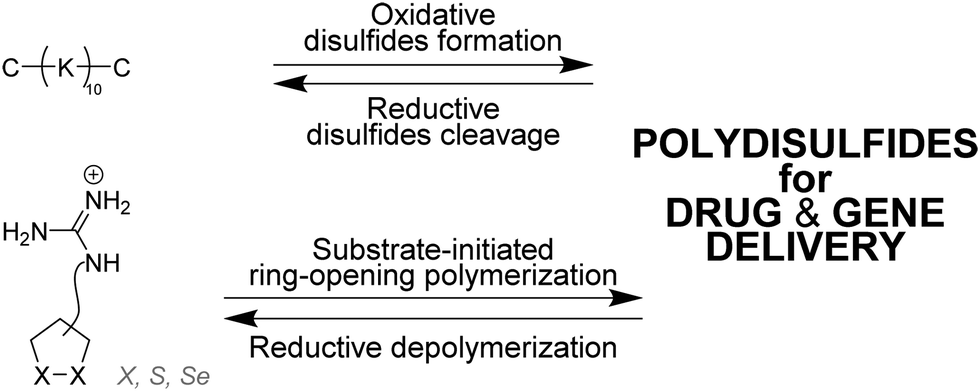 | ||
| Fig. 6 Strategies for the dynamic self-assembly of disulfide-based linear dynamic covalent polymers, based on (i) oxidative disulfide formation (top), and (ii) ring-opening polymerization (bottom). C: cysteine, K: lysine. | ||
Matile and coworkers have used ring-opening polymerization of strained cyclic disulfides101,102 and diselenides103,104 to generate redox-sensitive main-chain dynamic covalent polymers (Fig. 6).105 The process is initiated by a substrate – which can be a drug for instance106 – and yield spontaneously cell-penetrating cationic poly(disulfide)s which can depolymerize within minutes after intracellular reduction.106–108 Delivery of fluorescence dyes, drugs, proteins,109 liposomes and polymersomes110 has been achieved using this approach.111
In all these examples, it is important to note that polymer formation always competes with macrocycle formation. The way most commonly used to bias the system toward polymer formation was to carry out the self-assembly at high concentration, typically above 50 mM. However, template effects can also potentially shift the ring-chain equilibria. Hashim et al. reported that, indeed, a small water-soluble tetraguanidylated dithiol undergoes a nucleic acid-templated oxidative polymerization (Fig. 7). Using siRNA as templates, they demonstrated the formation of polydisulfide nanocaplets of small and uniform size which depolymerize in the reductive cytosolic environment and liberate the packaged siRNA.112
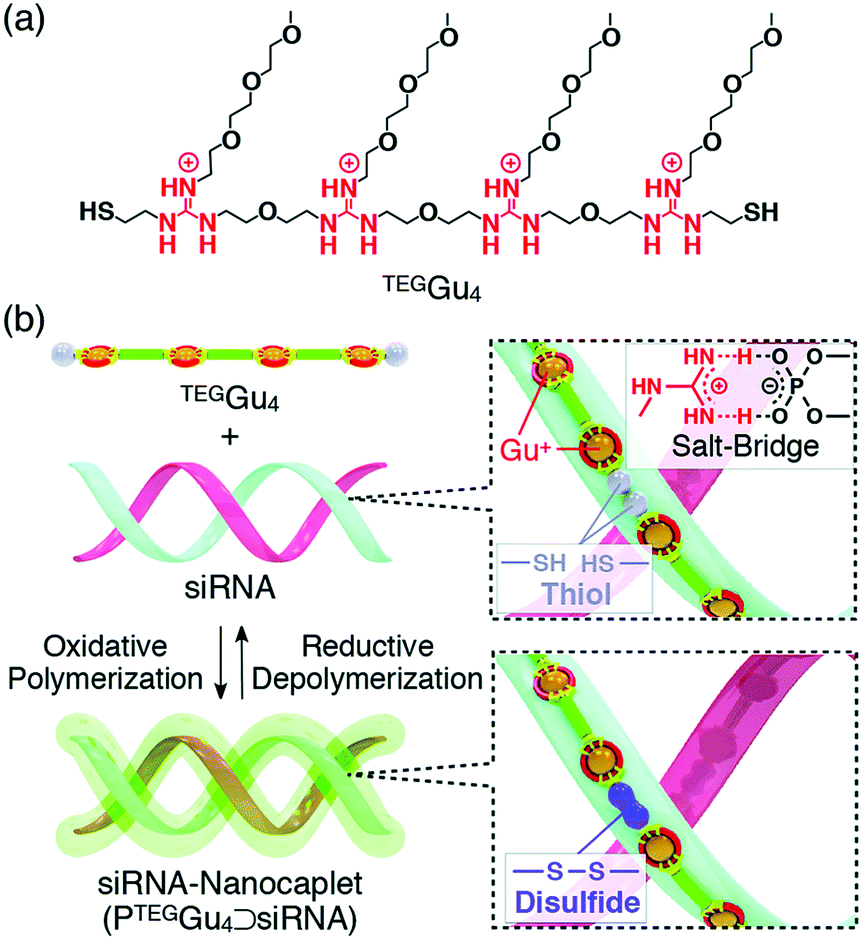 | ||
| Fig. 7 Nucleic acid-templated formation of redox-sensitive polydisulfide dynamic covalent polymers for siRNA delivery. (a) Molecular structure of water-soluble tetraguanidylated dithiol building block TEGGu4; (b) principle of siRNA-templated oxidative polymerization and reductive depolymerization. Reproduced from ref. 115 and 116 Copyright 2015, American Chemical Society. | ||
While dynamic covalent chemistry can happen between building blocks associated onto nucleic acids templates, Matile and coworkers have also found that dynamic covalent exchange can take place during the delivery process, evidencing a covalent and transient binding, through disulfide exchange, with the transferrin receptor which in turns promotes further cell penetration.102,113,114
Barboiu and coworkers used imines as reversible covalent linkage for generating cross-linked dynamic covalent polymers, coined Dynamic Constitutional Frameworks (DCFs).117–120 The design uses commercially-available 1,3,5-benzenetrialdehyde, a building block which can be connected to mono-, di- and poly-amines through imine-bond formations (Fig. 8). Varying the ratio of the latter enables tuning the density of functional positive groups brought by the monoamine (PEG-ylated squalene-PEI, Girard's reagent T, N,N-dimethylethylene amine, amino-guanidine), the length of the linear chains imposed by the diamine (poly-(ethylene-glycol)-bis(3-amino-propyl)-terminated, Mn ≈ 1500 g mol−1; Jeffamine-400, Jeffamine-2000), and the degree of cross-links given by the polyamine (branched polyethyleneimine 800 and 2000), within the dynamic frameworks, thereby allowing screening effectively, by self-assembly, subtle changes in dendrimer structures that may have a profound effect on nucleic acid complexation and delivery.127 In the end, the authors illustrated the potential of their approach by demonstrating effective dsDNA delivery on cell cultures.121
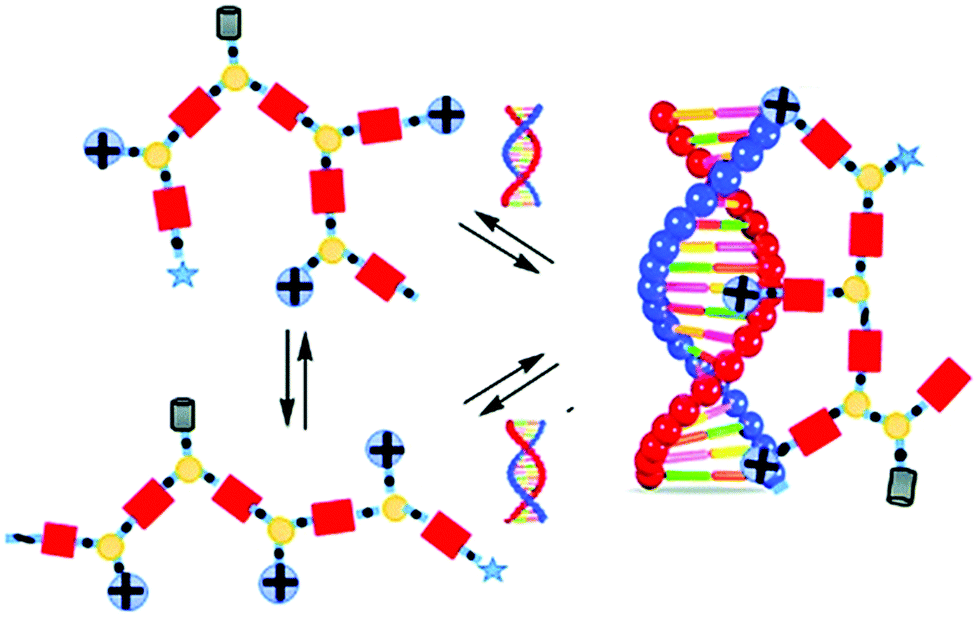 | ||
| Fig. 8 Dynamic covalent frameworks for pDNA complexation and delivery. Reproduced from ref. 117 Copyright 2015 The Royal Society of Chemistry. | ||
Finally, Fernandez-Trillo, Montenegro and coworkers explored dynamic covalent polymers where reversible linkages are installed as side-chain connections.122,123 They used polyhydrazide polymer scaffolds (poly-acryloyl hydrazide) which can be coupled, through acylhydrazone chemistry, with multiple aldehydes, bearing cationic head groups and lipophilic tails (Fig. 9). Here also, the self-assembly approach alleviates the screening of multiple combinations of different aldehydes, and led to the identification of amphiphilic transporters for pDNA/siRNA/mRNA delivery.124–126 This approach has then been extended using more structured scaffolds such as α-helical peptide scaffolds,127 with a successful implementation for the delivery of gene editing tools.128
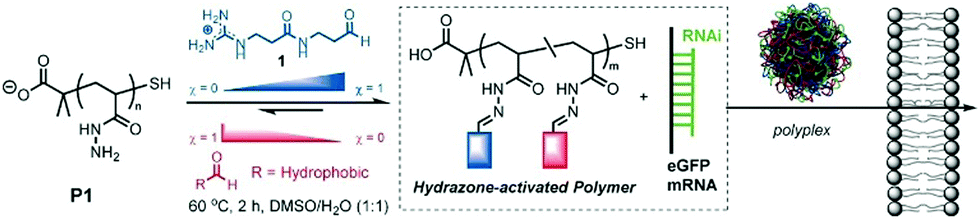 | ||
| Fig. 9 Dynamic covalent polymers featuring reversible covalent post-polymerization with different ratio (χ) of cationic and hydrophobic aldehydes, yielding amphiphilic delivery vectors for nucleic acids. Reproduced from ref. 126 Copyright 2016 Wiley-VCH. | ||
Although this approach does not allow for adaptive growth and decay as in the previous examples – length of polymer conjugate and density of functional groups being imposed by the structure of the initial polymer scaffold125 – constitutional adaptation can take place in the organization of the aldehydes fragments within the polymer structure (e.g. alternating vs. block distribution).129–132 However, such constitutional templating effects remain to be demonstrated within the context of gene delivery.
4.2. Enzyme activation
Enzymes are well-suited for integration with dynamic systems, and have been efficiently applied to asymmetric synthesis and identification protocols (cf. dynamic covalent kinetic resolution (DCKR) and dynamic systemic resolution (DSR)).8,133–135 This development has recently been reviewed,136 and will not be further discussed here.Another novel application is using dynamic polymers as additive matrices for enzyme encapsulation and activation as shown by Barboiu and coworkers. The microenvironment of enzyme is critical for its structural stability and catalytic activity. Normally, strategies such as protein engineering and chemical modification are required to improve enzyme performances, but long-term screening processes are usually required. In recent years, a straightforward method has been developed based on direct addition of functionalized dynamic polymers to the reaction media. The polymers form an adaptable matrix around the enzyme molecules, thereby leading to a significantly increased enzyme activity (Fig. 10).137–139
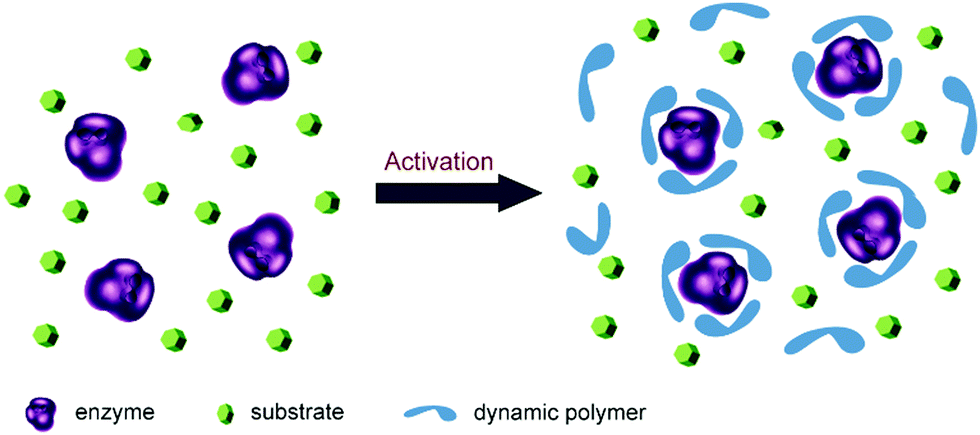 | ||
| Fig. 10 Concept of enzyme activation by direct addition of dynamers. The dynamic polymers (blue) formed an interactive protecting layer around enzyme (purple) with enzyme favored microenvironment, leading to enhanced catalytic performances.137 Copyright 2016 The Royal Society of Chemistry. | ||
For example, the dynamic covalent polymers can be synthesized by connecting a polyvalent aldehyde (e.g., 1,3,5-benzenetrialdehyde (1)), a bivalent polymeric amine (e.g., bis(3-aminopropyl)-terminated poly(ethylene glycol) (EG, 2)), and different functionalized amines (3–4) through reversible imine reactions (Fig. 11).137,138 Among these components, the trialdehyde serves as the tritopic center for three dimensional cross-linking, providing nonlinear complex network structures; EG provides the water-soluble backbone, and the amino-containing small molecules interact with the enzyme surfaces. Typical functional amines can be N-(2-aminoethyl)acetamide (3) and poly(amidoamine) dendrimer 4 (ethylene diamine core, generation 1.0), for enhanced H-bonding with the target enzyme.
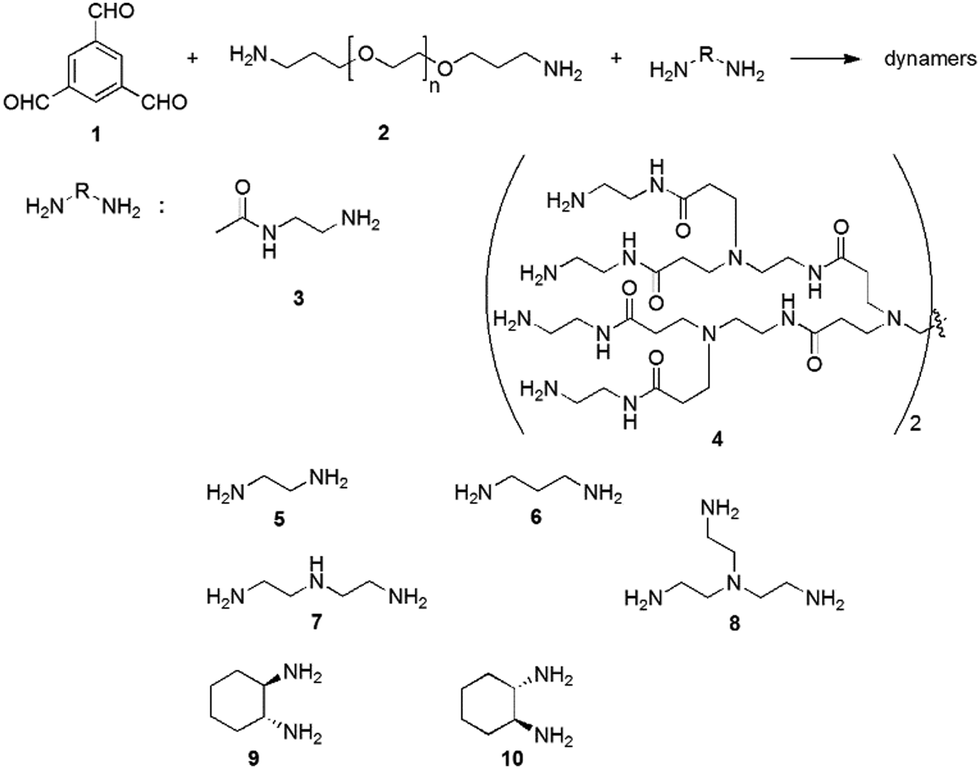 | ||
| Fig. 11 Syntheses of functional dynamic polymers for enzyme activation through imine formation reaction.21,139 Copyright 2016 The Royal Society of Chemistry. | ||
When this type of dynamic covalent network polymer was added into the enzyme reaction solutions, the catalytic activities could be increased, as exemplified for both carbonic anhydrase (CA) and lipases. Activation constants of around 1 mM could, e.g., be estimated for carbonic anhydrase. Moreover, the polymer containing multi-amide-functionalized dynamer 1·2·4 showed higher activation effect than the dynamic polymer with single-amide substituted dynamer 1·2·3, demonstrating the multivalent effect from the amide groups. As a reference, a dynamic covalent polymer with only the core aldehyde and the EG linker was also synthesized, resulting in no obvious activation of the enzyme performances.
The interactions between the dynamers and the surfaces of the enzymes were furthermore confirmed in a fluorescence titration study. With increasing amounts of dynamers added into the enzyme solution, the fluorescence intensity of the enzyme was gradually decreased, leading to association constants of around 4000 M−1 (Fig. 12). Circular dichroism spectroscopy studies furthermore revealed a high maintenance of the protein secondary structures by assistance of the dynamers.
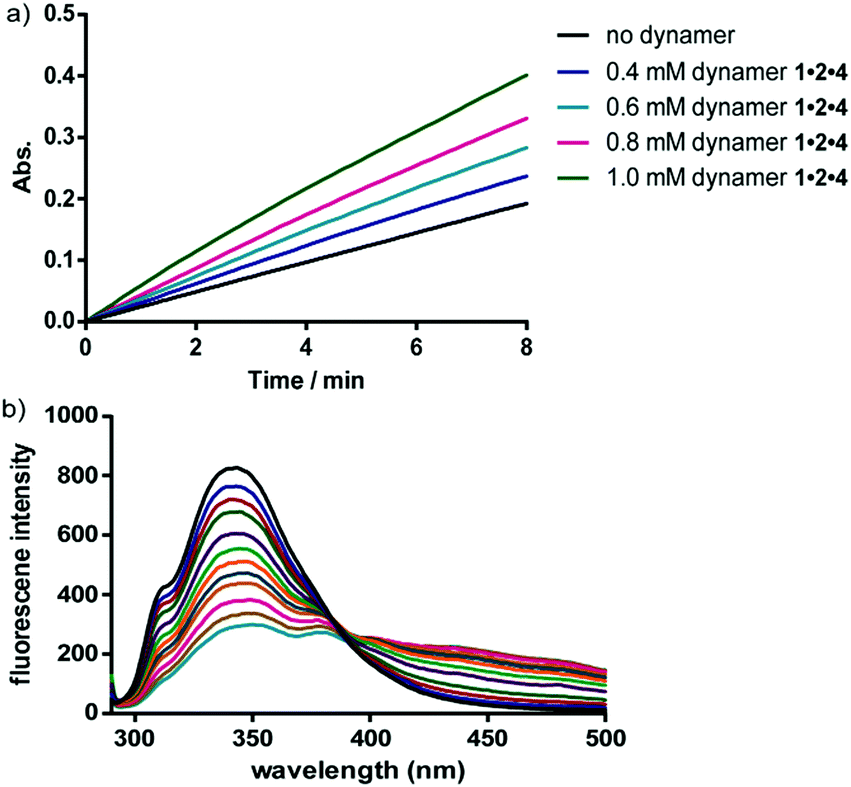 | ||
| Fig. 12 (a) Representative UV time-course study of CA activity changes upon hydrolysis reaction of p-nitrophenyl acetate, showing increased activity by adding more dynamer 1·2·4; (b) representative fluorescent titration study for CA-dynamer 1·2·4 interactions, the fluorescence intensity was gradually quenched with increasing amount of dynamer 1·2·4.137 Copyright 2016 The Royal Society of Chemistry. | ||
Other amines, including the linear 1,3-diaminopropane (5), the branched tris(2-aminoethyl)amine (7), and the chiral 1,2-diamino cyclohexane (9–10), have also been used to synthesize dynamic covalent polymers (Fig. 11), and tested for their influences on the activity of carbonic anhydrase.139 All these amines also resulted in decent activation effects, but generally with lower activation constants than amines 3–4, demonstrating the importance of amide-induced H-bonding effects. Unexpectedly, an exponential increase in the enzyme-catalyzed product formation was observed for the polymeric structures with optical activity, such as dynamers based on amines 9–10, indicating the requirement for a pre-organization period that enable the adaptive encapsulation of the enzyme by the dynameric matrices.
4.3. Recognition of biologically active compounds
Dynamic covalent polymers exhibiting selective recognition of compounds playing important roles in biological systems is another important development. In this context, the boronic acid–carbohydrate interaction has proven to be a particularly powerful tool. Various boronate-based sensors have thus been designed and applied to carbohydrate-mediated recognition studies, not only at the small-molecule level,41 but also involving polymeric and dynamic structures, where the polymer-grafted boronic acids can reversibly respond to carbohydrates through the side-chains.Since boronic ester formation is strongly regulated by pH, carbohydrate/pH dual-responsive materials have been investigated. For example, two pieces of poly(acrylamide) gels, respectively functionalized with phenylboronic acid and catechol moieties, could be joined in basic aqueous media and disconnected in solutions of a lower pH of 4. Furthermore, the gel adhesion strength could also be tuned by competitive monosaccharide molecules, such as glucose and galactose.79
The dynamic property of the boronic acid–diol reaction can be further applied to the reversible capture and release of cells. Considering the overexpressed sialic acid in many cancer cell types, this chemistry further lends itself to selective targeting of cancerous tissue. An example of a polymeric matrix presenting boronate units is highlighted in Fig. 13. A silicon nanowire array was thus produced and poly(acrylamidophenylboronic acid) brushes were grafted to the surfaces.75 At a lower pH-value of 6.8, the matrix was responsively switched to an adhesive mode, and cancer cells could be captured by the material. However, upon addition of 70 mM glucose at pH 7.8, the cell-repulsive state was turned on due to the competitive glucose binding on the surface. This setup thus, shows great potential in cell-based diagnostics, in vivo drug delivery, etc.
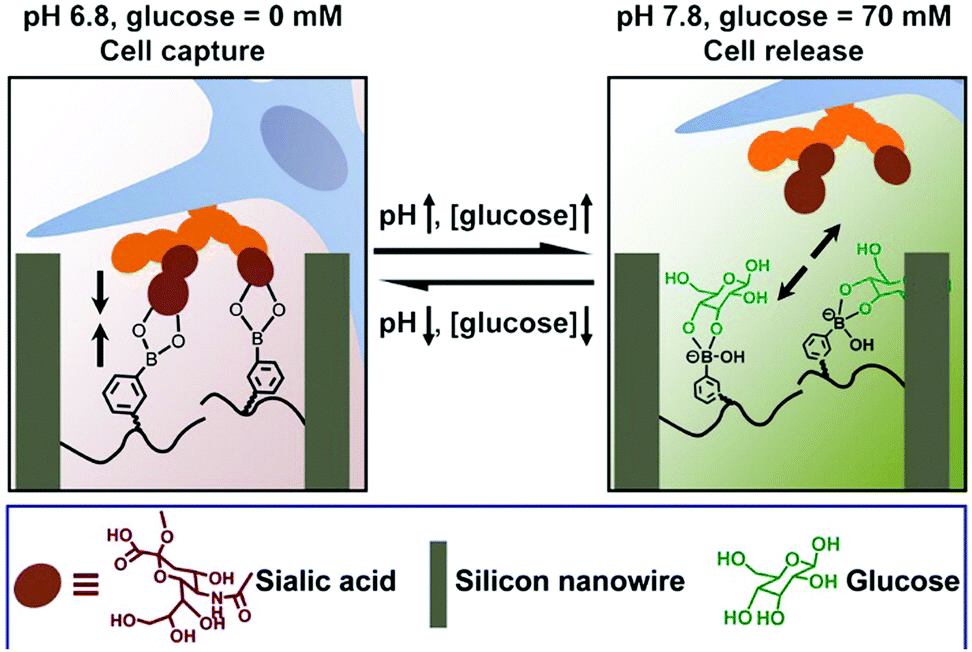 | ||
| Fig. 13 Cell capture and release induced by pH and glucose. At pH 6.8, the silicon nanowire surface (brown) can capture cells (blue) through boronic acid–sialic acid (orange) interactions; by increasing pH to 7.8 and the addition of glucose (green), the replaced boronic acid–glucose binding leads to release of cells.75 Copyright 2013 American Chemical Society. | ||
The reversible boronic ester formation reaction can also be coupled with supramolecular electrostatic interactions, leading to controlled cell membrane imaging.140 The main sensing structure was cationic conjugated polyfluorene tagged with phenyl-boronic acid groups (PFP–PBA), while the cationic part was embedded to target the negatively charged cell membrane (Fig. 14). The boronic acid groups were in this case inserted to bind to the diol units of cell-surface glycoproteins. The study revealed that PBA changed from neutral to negative state during the boronic acid–diol reaction. Therefore, in response to the dual dynamic covalent and electrostatic interactions between sensor and cells, the imaging of cell membrane was turned on. Moreover, D-glucose could be used as a competitor and block the binding sites of PFP–PBA, thereby turning off the cell membrane imaging.
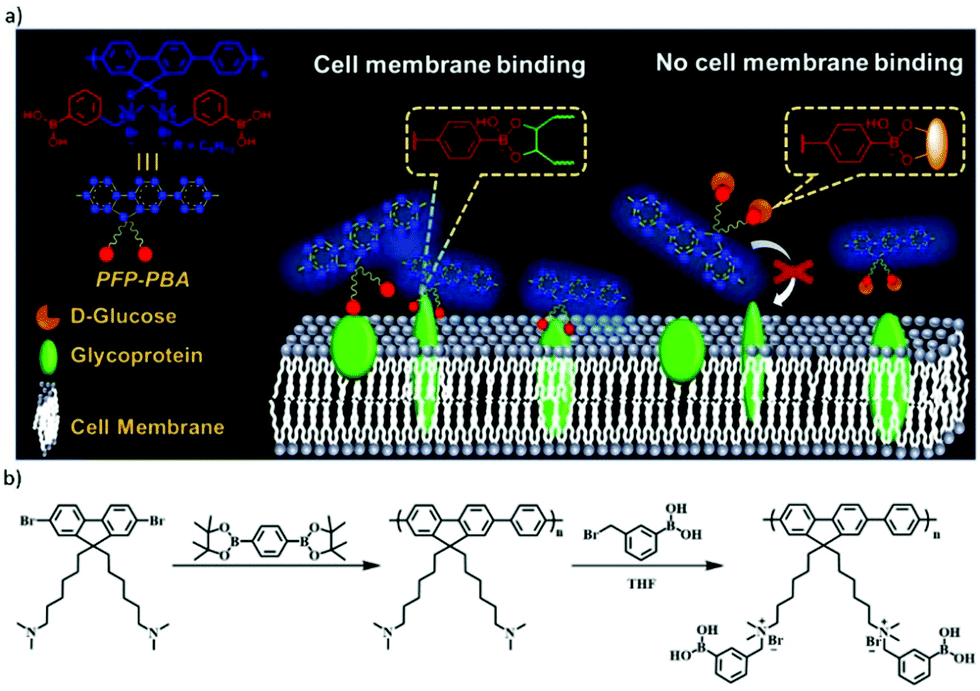 | ||
| Fig. 14 (a) Concept of PFP–PBA for controllable imaging of cell membrane, the boronic ester bond between PFP–PBA and glycoprotein can turn on the imaging of the cell membrane, meanwhile, with the presence of D-glucose, the imaging is turned off; (b) synthetic route for PFP–PBA.140 Copyright 2019 American Chemical Society. | ||
Concerning the multiple diols in the complex structures of carbohydrates, in some cases, the specific binding mode is important for further applications. However, the signals in other methods, for example, NMR spectra, are usually too weak to be detected. In a recent study, a peptide amphiphile with a benzoboroxole (BOB) group attached at the hydrophilic end was assembled into fibers, leading to aligned BOB/saccharide pairs with low molecular mobility, high density and multivalent effect.141 Thus, enhanced nuclear Overhauser effects could be observed by NMR (NOESY), confirming the binding between BOB and mannopyranoside on the site of diols at 2,3-position instead of 4,6-position.
Besides carbohydrate structures, the dynamic polymers can also be used to detect other biomolecules. For example, dynamic covalent polymers containing entities displaying aggregation-induced emission (AIE) effects have been used for selective detection of ATP.142 The typical AIE luminogen tetraphenylethene (TPE) was first derivatized to yield a tetra-aldehyde, and then linked with an amine-terminated poly(ethylene glycol) structure (EG) through imine formation. Upon further functionalization with a positively charged aminoguanidinium group, a dynameric framework (G2) was produced (Fig. 15a). In this case, both the main-chain connection and the side-chain functional group decoration were linked by dynamic imine bonds, leading to higher degrees of structural freedom for spontaneous responses. As a result, owing to the stronger electrostatic interactions, the G2-ATP aggregate displayed much higher fluorescence intensity than the corresponding aggregates with ADP or AMP (Fig. 15b). On the other hand, compared to the non-crosslinked G1 structure, prepared in the absence of the EG linker, Dynamer G2 showed higher fluorescent signals, potentially emanating from the multivalent effect of the polymerized structure. Moreover, when the imine bonds in dynamer G2 were reduced to secondary amines, lower efficiency in ATP detection was recorded (Fig. 15c), demonstrating the advantages of applying dynamers for adaptively optimized molecular recognition.
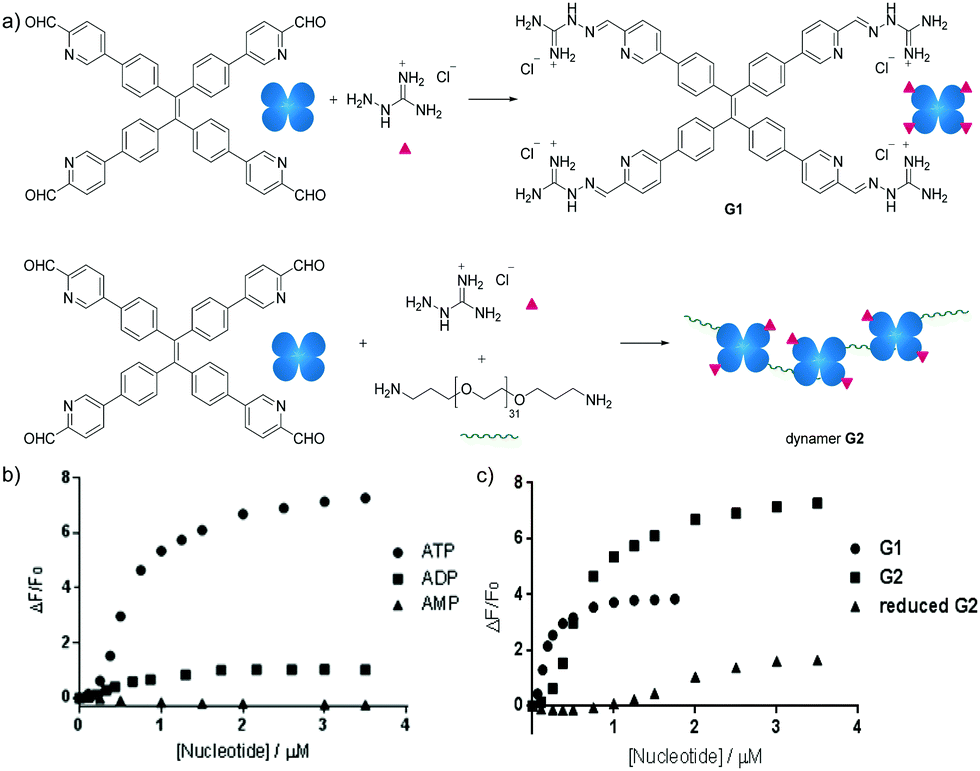 | ||
| Fig. 15 (a) Syntheses of AIE based sensor G1 and dynamer G2; ATP sensing study by fluorescence spectroscopy of (b) selective detection of ATP among other nucleotides by dynamer G2; (c) ATP detection of dynamer G2, compared to monomer G1 and imine reduced G2, demonstrating the importance of multivalent and dynameric effects.142 Copyright 2018 Wiley-VCH. | ||
4.4. Cell culture and delivery
During the last decade, hydrogels with high water content and optimal 3D structures have been playing important roles in mimicking the extracellular matrix (ECM), leading to improved performances in cell delivery and cell culture compared to the corresponding 2D environments. With embedded reversible covalent bonds, where the crosslinking can be rapidly breaking and reforming, dynamic hydrogels can be formed that not only provide adaptively changeable micro-structures along with cell growth, but also facilitate various stimuli responsive reactions, leading to a delicate platform for studying chemical and mechanical cues in ECM.143In recent years, there have been extensive studies of dynamic hydrogels in cell culture applications. Structurally, the materials are often composed of naturally derived polymers, such as collagen, alginate, hyaluronic acid, etc.; or based on certain synthetic polymers, with poly(ethylene glycol) (PEG) and poly(acrylamide) as typical representatives.144 Among all the reversible reaction types adopted for dynamic connections, imines (incl. hydrazones) and disulfides are the most frequently used, owing to their operation under mild reaction conditions and their reasonable cytocompatibility.
While the reversible reactions have been primarily intended for direct use in constructing dynamic hydrogels for cell culture, an in-depth evaluation of the related kinetic and thermodynamic properties have been performed, with further application in mimicking the viscoelastic properties of native tissues.145 Starting from hydrazine-terminated PEG and aldehyde-terminated multi-arm PEG, main-chain cross-linked dynamic hydrogels composed of reversible hydrazone bonds were rapidly formed at physiological pH and temperature (Fig. 16a). With calculated rate and equilibrium constants of model hydrazone reactions, the equilibrium modulus and stress relaxation characteristics of the resulting hydrogels could be estimated, the compatibility regarding timescales of reversible reaction, hydrogel formation and cell encapsulation could be demonstrated. Moreover, the viscoelastic properties of the materials were comparable to mouse gastrocnemius muscles, an aspect of native tissue that is difficult to capture with traditional, static hydrogels. The cytocompatibility of the dynamic hydrogels was also examined (Fig. 16b and c), following the encapsulation and culture of mouse C2C12 myoblasts along with the development of physiologically relevant morphologies.
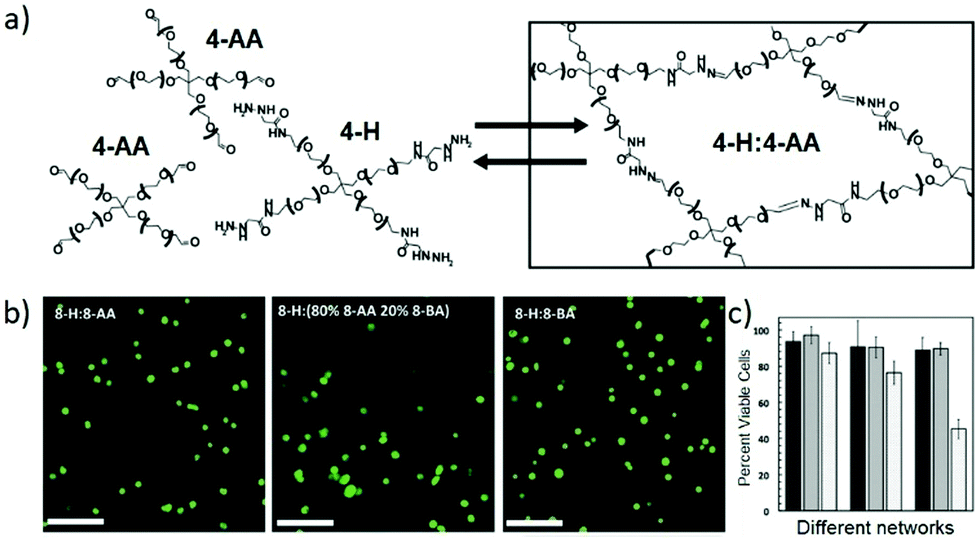 | ||
| Fig. 16 (a) Hydrogel structures synthesized from aldehyde and hydrazine terminated multi-arm PEG, for 3D cell culture; (b) images of encapsulated C2C12 myoblasts in the dynamic hydrogels, stained with calcium-AM (green, live); (c) percentages of cell viability encapsulated inside the different hydrogel networks, quantified at 24 hours (dark bars), 72 hours (medium bars) and 240 hours (light bars).145 Copyright 2014 Wiley-VCH. | ||
Among the various examples of dynamic chemistry in cell culture, of high interest are gradient hydrogels that mimic the spatiotemporal differences of multiple gradient cues in tissues.73 The basic principle is taking advantage of the self-healing feature of the materials to manually connect different modules of gradually changing components together, forming a unique type of gradient hydrogels. Initially, the dually crosslinked hydrogel network was generated through imine formation reaction between N-carboxyethyl chitosan (CEC) and oxidized acrylated hyaluronic acid (OAHA), where OAHA was first functionalized with matrix metalloproteinase (MMP)-sensitive cross-linker and RGD peptides (Fig. 17a). After transforming the resulted injectable and self-healing CEC–OAHA–MMP material into customized gradient hydrogels (Fig. 17b), the resulting gels were further applied to encapsulation of sarcoma cells. As a result, the cells showed good responsiveness to the gradient cues of the RGD peptides and the MMP-sensitive cross-linkers inside the hydrogels.
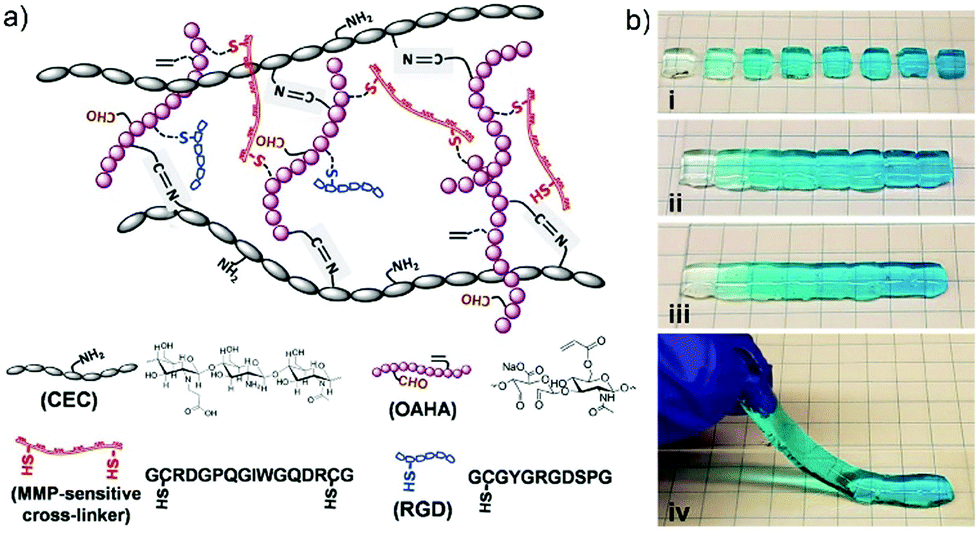 | ||
| Fig. 17 (a) The structure and different components of CEC–OAHA–MMP hydrogels connected through reversible imine bonds and dithiol MMP-sensitive cross-linker; (b) combining hydrogel modules stained with gradient blue dye (i) together, at 37 °C for 20 min (ii), the resulted a one-piece hydrogel was self-healed without boundaries between the different modules (iii and iv).73 Copyright 2017 Wiley-VCH. | ||
Similar to cell culture, cell delivery using hydrogel vehicles has begun to attract increased attention, especially for stem-cell applications. However, the interference by sensitive cells and the narrow gelation time window can be problematic. Under such circumstances, dynamic hydrogels with shear-thinning and self-healing properties turned out to be a superior option.
For example, combining reversible covalent crosslinking with thermoresponsive engineered proteins, injectable hydrogels were produced and used for stem cell delivery.146 Structurally, the designed hydrogels were composed of dual crosslinking: the first of which achieved by mixing hydrazine-modified elastin-like protein (ELP-HYD) and aldehyde-modified hyaluronic acid (HA-ALD) through reversible hydrazone bonds, while the secondary crosslinking occurred upon heating through the thermoresponsive phase segregation of ELP, reinforcing the whole network (Fig. 18). As a result, the generated hydrogels showed significant protection of the stem cells during injection and maintenance of the stem cell-differentiation potentials posterior to injection.
 | ||
| Fig. 18 Injectable ELP–HA hydrogels. (a) The structures of hydrazine-modified elastin-like protein (ELP-HYD) and aldehyde-modified hyaluronic acid (HA-ALD); (b) schematic of ELP–HA hydrogel formation, through the first network of hydrazone formation and second network of ELP thermal assembly; (c) photographs demonstrating the injectable and self-healing properties of the ELP–HA hydrogels.146 Copyright 2017 Wiley-VCH. | ||
Similar ELP-HYD hydrogels have also been used in a cartilage regeneration study.82 Different from other matrix materials, dynamic hydrogels linearly linked by hydrazone bonds provided a comparable stiffness upon varied hyaluronic acid concentrations. Thus, by encapsulation of chondrocytes within these hydrogels, the effects of the hyaluronic acid concentration on the cell responsiveness could be better evaluated without the influences of mechanical and biochemical cues.
Other examples of dynamic hydrogels for cell culture or delivery have been reported in literature, including boronic acid-based hydrogels for 3D-culture of breast cancer cells and pulmonary fibroblasts,147 polysaccharide-based hydrogels with double crosslinked imine bonds/Diels–Alder cycloadducts for 3D cell encapsulation of fibroblasts,148 hydrazone-linked poly(vinyl alcohol)-heparin hydrogels for controlled cell- and growth factor delivery,149 poly(amidoamine)-based nanocomposite hydrogels with porous structures for cell growth and proliferation,149etc. Moreover, hyaluronic acid–collagen hydrogels crosslinked by hydrazone groups proved to promote cell spread, fiber remodeling and focal adhesion formation during 3D cell culture.150 Further application of these self-healing dynamic biomaterials into cell-laden bioinks has also been developed.151
4.5. Antimicrobial materials
In the battle of fighting antimicrobial resistance with new antibiotics, contributions involving dynamic materials have also been demonstrated. In these cases, the antibacterial activities primarily emanate from the dynamic responsiveness of the materials and the polymeric multivalent effects.For the construction of antimicrobial dynamic materials, the most straightforward method is encapsulation of antibiotic structures into the polymers, leading to functional materials with desired medical efficacies. For example, the reaction between the antifungal agent vanillin with amino-containing chitosan resulted in dynamers containing reversible imine bonds, where the obtained dynamic films showed improved antifungal activity towards Candida albicans compared to vanillin alone.152 In another example, the antimicrobial agent vancomycin was loaded into dynamic hydrogels, which were cross-linked by two types of branched PEG-based polymers functionalized with amino- or aldehyde group, respectively. The resulting imine-linked hydrogels showed improved skin adhesiveness and demonstrated usage in rapid hemorrhage control and infection prevention.153
More recently, a unique type of antibacterial peptide quaternary assemblies have been designed.154 By incorporation of aryl aldehyde and acryl hydrazide groups into peptide oligomer backbones, which served as hetero-ditopic building blocks, peptide–peptide intermolecular macrocycles with tunable ring sizes could be formed, mediated by hydrazone linkages in aqueous media under neutral pH conditions (Fig. 19). Among the diverse peptide assemblies, quaternary structures of dumbbell-, zipper-like- and multi-loop shapes were selectively formed. With careful control of the component proportions, higher-molecular-weight (>14 kDa) ladder polymers could be synthesized, showing amplified antimicrobial activity against the Gram-positive Staphylococcus aureus. Later on, the development was expanded to also encompass simple dialdehyde or dihydrazide small-molecule cross-linkers, or even tetra-/hexa-functionalized oligomers.155 Together with the cationic antimicrobial peptide buforin II, the dynamic covalent peptide-based polymers showed enhanced inhibition effects towards the growth of Gram-negative and drug-resistant bacterial strains.
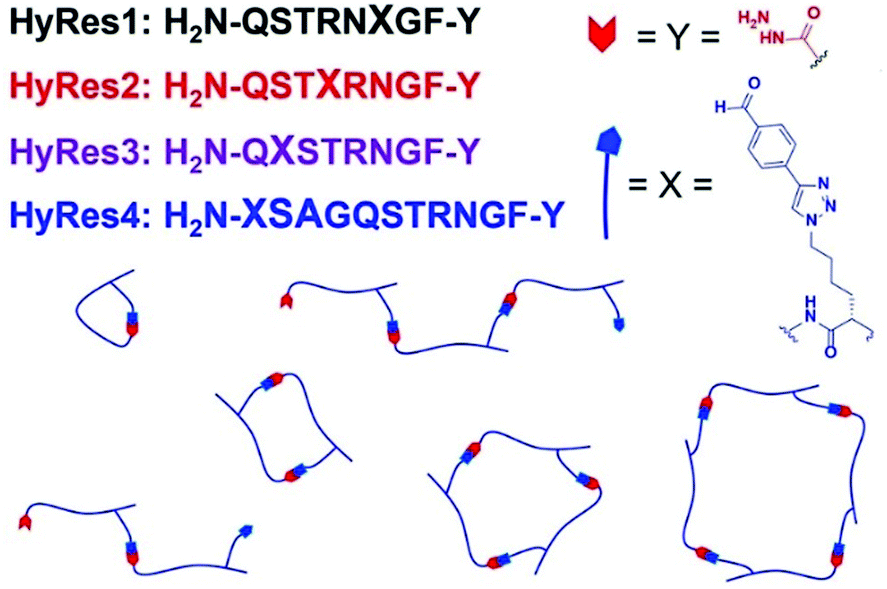 | ||
| Fig. 19 Dynamic covalent peptides synthesized on hydrazide C-terminal resins (HyRes) were assembled into various macrocyclic and oligomeric structures through hydrazine formation reactions.155 Copyright 2018 American Chemical Society. | ||
Dynamic covalent polymers can also be attached onto surfaces, where the tunable nature can be used to switch the corresponding surface bioactivities. One example is phenylboronic acid containing polymer-grafted gold surfaces (Au-PBH), which was further crosslinked with functionalized β-cyclodextrin (CD) derivatives through boronate bonds (Fig. 20).156 When the attached group was a lysine residue, the construction showed binding and release ability to protein Plg, where the responsiveness could be controlled by addition of specific carbohydrate structures. When the attached group was changed to a quaternary ammonium salt residue (QAS), known to have biocidal effects, the materials demonstrated the potential to kill bacteria and release the dead bacteria upon treatment with carbohydrates. A similar study was performed using surfaces to which acyl hydrazide structures had been anchored.49 By changing aldehyde-containing functional components, which could be reversibly linked to the brush-like surfaces via acylhydrazone bonds, the switching between different activities could be easily controlled. For example, sulfonic acid-containing groups that mimic heparin structures could be attached to turn on blood compatibility, quaternary ammonium groups could contribute to the antibacterial effects; and poly(ethylene glycol)methyl ether methacrylate groups could add antifouling properties to the materials.
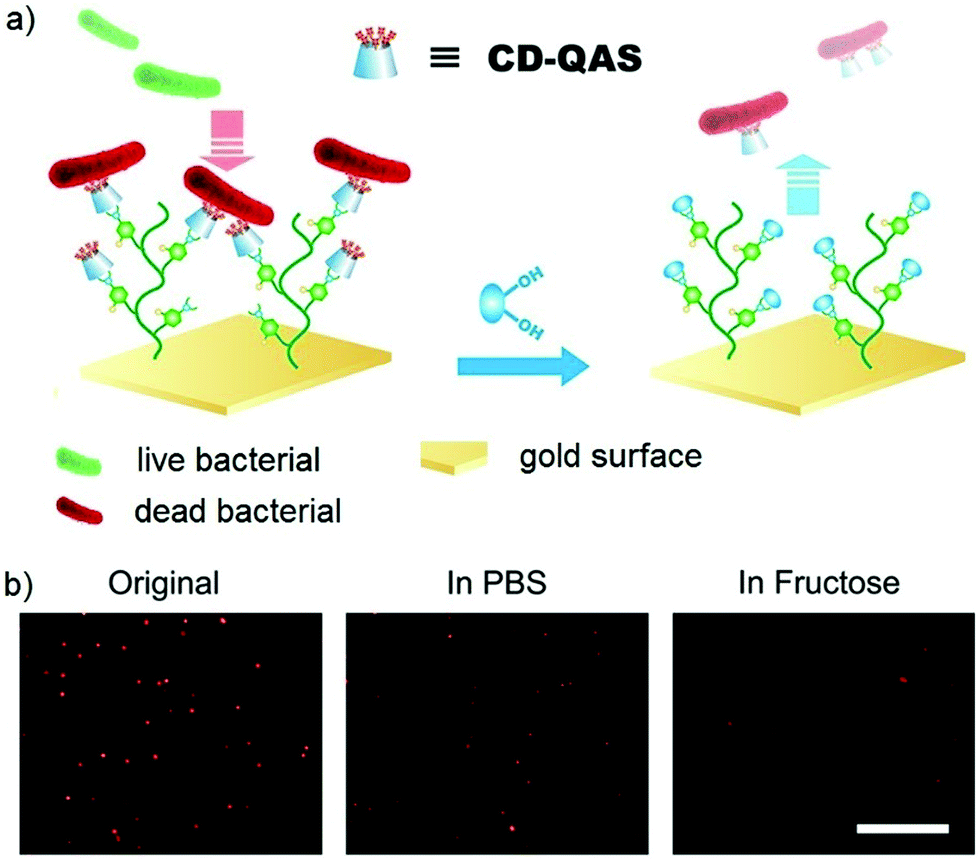 | ||
| Fig. 20 (a) Sugar-triggered bacterial release from the Au-PBH/CD-QAS surface; (b) fluorescence images of bacteria on Au-PBH/CD-QAS surfaces before and after the treatment of fructose (in PBS), stained with propidium iodide (red color), showing dramatic decrease of bacterial density in the presence of fluctose.156 Copyright 2018 American Chemical Society. | ||
Due to the ever-increasing antibiotic resistance problem, a narrow-spectrum antibiotic that specifically targets the disease-causing strain can be important for the treatment of infections. Thus, a dynamic systemic approach in search for the most suitable peptide probes for specific bacterial strains was designed, based on phage display platform.157
2-Acetylphenylboronic acid-moieties were incorporated into phage display peptides to elicit binding against live bacterial cells through reversible iminoboronate formation (Fig. 21). With the combined effects from dynamic covalent, hydrophobic and electrostatic interactions, peptide structures that bind S. aureus with submicromolar affinity were successfully identified. Furthermore, the peptide hits were converted to bactericidal agents with high antibiotic potency and specificity.
 | ||
| Fig. 21 Illustration of a phage binding to bacterial cells via combined dynamic covalent and supramolecular interactions, more specifically, iminoboronate formation, hydrophobic and electrostatic interactions.157 Copyright 2018 American Chemical Society. | ||
4.6. Wound-dressing materials
For wound dressing applications, hydrogels constitute one of the most investigated materials in recent years. Especially when dynamic covalent bonds are involved, the self-healing, injectable, moldable and degradable properties of the dynamic materials often lead to important biomedical functions.A variety of dynamic hydrogels have been designed and applied for wound dressing. For example, a hydrogel was formed by crosslinking hydrazide-modified hyaluronic acid and benzaldehyde terminated F127 triblock copolymers through acyl hydrazone bonds;74 another agarose-based hydrogel was prepared by linearly connecting an agarose–ethylenediamine conjugate and a dialdehyde-functionalized polyethylene glycol and forming imine linkages (Fig. 22).158 Both of the hydrogels exhibited rapid gelation time, good mechanical strength, self-healing and tissue adhesion properties, accompanied with excellent biocompatibility and wound repair effectiveness.
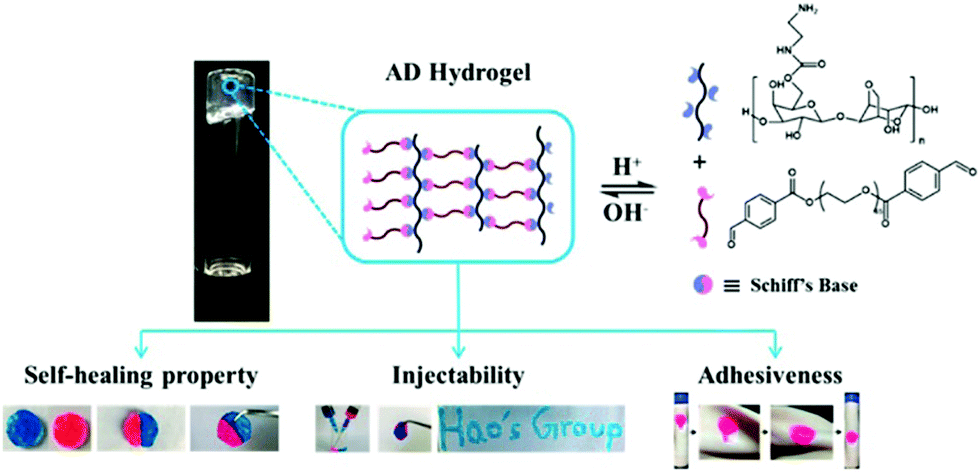 | ||
| Fig. 22 Schematic illustration of Schiff's base linked dynamic hydrogels, formed between hydrazide-modified hyaluronic acid (blue) and benzaldehyde terminated copolymers (pink), and the related properties.158 Copyright 2018 American Chemical Society. | ||
A moldable and removable hydrogel for wound dressing has also been developed.159 The material was dynamically cross-linked by mixing a thiol-functionalized PEG structure with aldehyde groups in oxidized dextran through hemithioacetal formation (Fig. 23). Besides the aforementioned advantageous characteristics, the resulting dynamic hydrogels displayed adaptivity features and shape malleability, offering a suitable time window for optimized wound coverage, which dramatically eased the operation for healing. Moreover, the labile hemithioacetal bonds could readily undergo exchange with a competitive thiol source, glutathione, leading to postremoval ability and straightforward replacement of the wound dressings, thus, avoiding secondary injuries to the dressed skin.
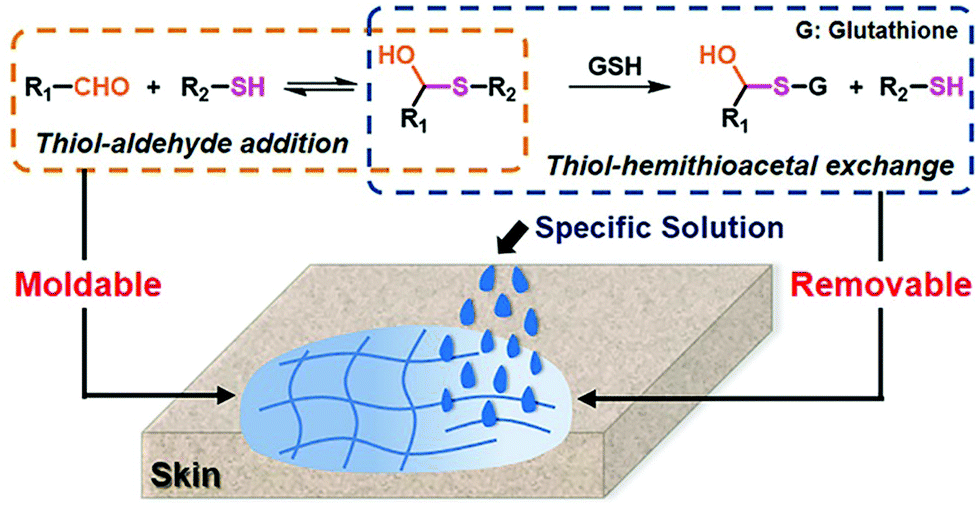 | ||
| Fig. 23 Fabrication of moldable hydrogels as wound dressing material by reversible hemithioacetal addition (orange dots framed) reaction; the removable of the hydrogels from skins can be realized by the addition of glutathione, through the hemithioacetal exchange reaction (blue dots framed).159 Copyright 2019 American Chemical Society. | ||
Recently, an injectable, self-healing hydrogel combined with the functions of antibacterial activity and sequential drug release has been reported.11 The hydrogel was constructed with aminated gelatin (NGel), adipic acid dihydrazide (ADH) and oxidized dextran (ODex) to form complex reversible imine/hydrazone networks under physiological conditions. Meanwhile, microspheres containing basic fibroblast growth factor (bFGF) and poly(lactic-co-glycolic acid) (PLGA) were encapsulated into the hydrogels together with the antiseptic drug chlorhexidine acetate (CHA) (Fig. 24). After being injected at the wound site, the sequential release of CHA from the hydrogel could effectively prevent infection at the early stages, while accelerated cell proliferation and wound healing could be achieved by the release of growth factor bFGF.
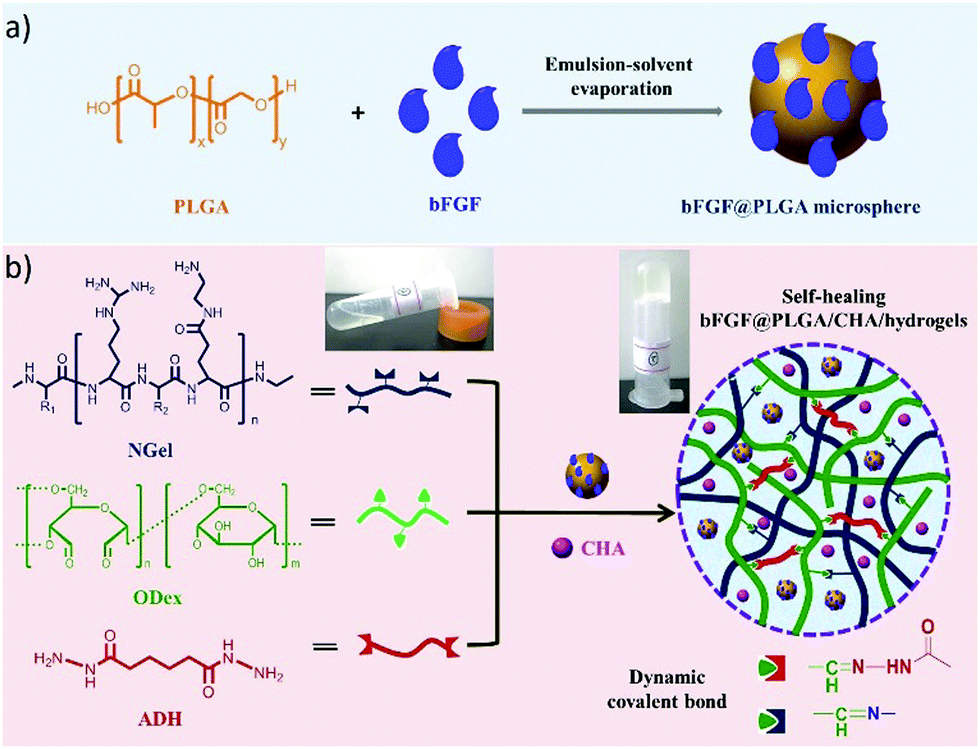 | ||
| Fig. 24 Schematic presentation of (a) fabrication of bFGF@PLGA microspheres; (b) preparation of bFGF@PLGA/CHA/hydrogels through formation of dynamic imine/hydrazone bonds between aminated gelatin (NGel), adipic acid dihydrazide (ADH) and oxidized dextran (ODex), and further encapsulation of bFGF@PLGA microspheres and drug chlorhexidine acetate (CHA), leading to sequential release of CHA during wound healing.11 Copyright 2019 Elsevier. | ||
5. Summary and outlook
Since their inception, dynamic covalent polymers have revealed unique features – adaptive self-fabrication, dynamic responsiveness to physico-chemical stimuli, etc. – that turned out to be of great benefit for a wide range of applications, from material science to biomedical applications. In reviewing the recent advances on the latter, we summarized the different features (responsiveness to pH, heat, light, or redox potential) that can be endowed, by a proper choice of reversible covalent bonds, to dynamic covalent polymers and which can be useful for bio-applications. Different polymer architectures – linear, branched, bottlebrush – can be easily explored by changing the design of the building blocks, thereby expressing different degrees of multivalency that is often key for achieving bioactivity. As discussed in this review, dynamic covalent polymers are finding increasing applications in the biomedical fields, for the recognition and sensing of biologically active compounds, for the modulation of enzyme activity, for gene delivery, and as materials for cell culture, delivery, and wound-dressing. Their self-fabrication by covalent self-assembly is a tremendous advantage, and their adaptive behavior qualifies them as smart materials that can adapt to the biological partners and environments they encounter.Dynamic covalent polymers form a class of adaptive systems that can be efficiently used for biomedical applications. The scope of such polymers needs further exploration, and a deeper understanding of the polymer architectures and the polymerization/aggregation formats are warranted. In this context, comparisons with supramolecular materials can be constructive. These systems share some of the features of dynamic covalent polymers and have been extensively used to construct complementary multifunctional platforms for applications in various biomedical systems.160–168 The well-defined size, shape and surface functionality of such dynamic materials, well matching nano/micro-sized biological targets, can result in not only multivalent behavior, but also new properties in applications, such as biorecognition, drug delivery, and bio-imaging. The common features of all these systems: controlled self-generation via reversible interconversions through component exchange, dynamic diversity, and potential addressability, make species and processes such as those presented here of interest for the development of a constitutional dynamic “self-fabrication” approach for biological functions, toward systems of increasing dimensional complexity. There are still challenges to address in the field, for example, accurately designed structural complexes, real clinical applications of the current dynamic materials, delineation of mechanisms/modes-of-action regarding the biological effects, etc. However, cooperative, dynamic contributions from all aspects of the area, including more variations of the structural architectures, broadened biological applications and deepened insights into the bio-interaction mechanisms, will arguably lead to new breakthroughs in the field.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The study was in part supported by the Natural Science Foundation of Jiangsu Province (BK20180625, to YZ), the National First-class Discipline Program of Light Industry Technology and Engineering (LITE2018-20, to YZ), Agence National de la Recherche (ANR-17-CE07-0042-01, to SU; ANR DYNAFUN, ANR-15-CE29-0009-02, to MB), and the National Institutes of Health (1R21AI140418, to OR). YQ thanks the China Scholarship Council for a special scholarship award.References
- W. Zhang and Y. Jin, Dynamic Covalent Chemistry: Principles, Reactions, and Applications, Wiley-VCH, Weinheim, 2017 Search PubMed.
- Y. H. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- J.-M. Lehn, Chem. – Eur. J., 1999, 5, 2455–2463 CrossRef CAS.
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- J. M. Lehn, Angew. Chem., Int. Ed., 2015, 54, 3276–3289 CrossRef CAS PubMed.
- Topics in Current Chemistry: Constitutional Dynamic Chemistry, ed. M. Barboiu, Springer, 2012 Search PubMed.
- Y. Zhang, F. Schaufelberger, M. Sakulsombat, C. Liu and O. Ramström, Tetrahedron, 2014, 70, 3826–3831 CrossRef CAS.
- H. Liu, J. Feng, J. Zhang, P. W. Miller, L. Chen and C. Y. Su, Chem. Sci., 2015, 6, 2292–2296 RSC.
- P. Chakma and D. Konkolewicz, Angew. Chem., Int. Ed., 2019, 58, 9682–9695 CrossRef CAS PubMed.
- M. Chen, J. Tian, Y. Liu, H. Cao, R. Li, J. Wang, J. Wu and Q. Zhang, Chem. Eng. J., 2019, 373, 413–424 CrossRef CAS.
- Y. Liu, J. M. Lehn and A. K. H. Hirsch, Acc. Chem. Res., 2017, 50, 376–386 CrossRef CAS PubMed.
- N. Roy, B. Bruchmann and J. M. Lehn, Chem. Soc. Rev., 2015, 44, 3786–3807 RSC.
- A. K. H. Hirsch, E. Buhler and J.-M. Lehn, J. Am. Chem. Soc., 2012, 134, 4177–4183 CrossRef CAS PubMed.
- J. M. Lehn, Aust. J. Chem., 2010, 63, 611–623 CrossRef CAS.
- F. Garcia and M. M. J. Smulders, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3551–3577 CrossRef CAS PubMed.
- T. Maeda, H. Otsuka and A. Takahara, Prog. Polym. Sci., 2009, 34, 581–604 CrossRef CAS.
- N. Roy, B. Bruchmann and J.-M. Lehn, Chem. Soc. Rev., 2015, 44, 3786–3807 RSC.
- Y. Liu, J.-M. Lehn and A. K. H. Hirsch, Acc. Chem. Res., 2017, 50, 376–386 CrossRef CAS PubMed.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Adv. Mater., 2017, 29, 1606100 CrossRef PubMed.
- Y. Jin, Q. Wang, P. Taynton and W. Zhang, Acc. Chem. Res., 2014, 47, 1575–1586 CrossRef CAS PubMed.
- F. García and M. M. J. Smulders, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3551–3577 CrossRef PubMed.
- I. Huc and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2106–2110 CrossRef CAS PubMed.
- J.-M. Lehn, Chem. Eng. J., 1999, 5, 2455–2463 CAS.
- J.-M. Lehn and A. V. Eliseev, Science, 2001, 291, 2331–2332 CrossRef CAS PubMed.
- O. Ramström and J.-M. Lehn, Nat. Rev. Drug Discovery, 2002, 1, 26–36 CrossRef PubMed.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- H. Vardhan, A. Mehta, I. Nath and F. Verpoort, RSC Adv., 2015, 5, 67011–67030 RSC.
- S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Chem. Soc. Rev., 2014, 43, 1861–1872 RSC.
- M. Irigoyen, A. Fernández, A. Ruiz, F. Ruipérez and J. M. Matxain, J. Org. Chem., 2019, 84, 4200–4210 CrossRef CAS PubMed.
- Y. Yi, H. Xu, L. Wang, W. Cao and X. Zhang, Chem. – Eur. J., 2013, 19, 9506–9510 CrossRef CAS PubMed.
- M. Hebel, A. Riegger, M. M. Zegota, G. Kizilsavas, J. Gacanin, M. Pieszka, T. Luckerath, J. A. S. Coelho, M. Wagner, P. M. P. Gois, D. Y. W. Ng and T. Weil, J. Am. Chem. Soc., 2019, 141, 14026–14031 CrossRef CAS PubMed.
- H. Faustino, M. Silva, L. F. Veiros, G. J. L. Bernardes and P. M. P. Gois, Chem. Sci., 2016, 7, 5052–5058 RSC.
- Y. Zhang, P. Vongvilai, M. Sakulsombat, A. Fischer and O. Ramström, Adv. Synth. Catal., 2014, 356, 987–992 CrossRef CAS PubMed.
- P. Vongvilai, M. Angelin, R. Larsson and O. Ramström, Angew. Chem., Int. Ed., 2007, 46, 948–950 CrossRef CAS PubMed.
- N. Roy and J.-M. Lehn, Chem. – Asian J., 2011, 6, 2419–2425 CrossRef CAS PubMed.
- P. J. Boul, P. Reutenauer and J.-M. Lehn, Org. Lett., 2005, 7, 15–18 CrossRef CAS PubMed.
- W. L. A. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375–1397 CrossRef CAS PubMed.
- Y. Kubo, R. Nishiyabu and T. D. James, Chem. Commun., 2015, 51, 2005–2020 RSC.
- Y.-X. Lu and Z. Guan, J. Am. Chem. Soc., 2012, 134, 14226–14231 CrossRef CAS PubMed.
- K. Imato, J. C. Natterodt, J. Sapkota, R. Goseki, C. Weder, A. Takahara and H. Otsuka, Polym. Chem., 2017, 8, 2115–2122 RSC.
- S. Debnath, R. R. Ujjwal and U. Ojha, Macromolecules, 2018, 51, 9961–9973 CrossRef CAS.
- S. Mukherjee, M. R. Hill and B. S. Sumerlin, Soft Matter, 2015, 11, 6152–6161 RSC.
- Y. Zhang and M. Barboiu, Chem. Commun., 2015, 51, 15925–15927 RSC.
- P. R. Christensen, A. M. Scheuermann, K. E. Loeffler and B. A. Helms, Nat. Chem., 2019, 11, 442–448 CrossRef CAS.
- Y. Zhang and M. Barboiu, ACS Omega, 2018, 3, 329–333 CrossRef CAS PubMed.
- J. Deng, X. Liu, L. Ma, C. Cheng, S. Sun and C. Zhao, J. Mater. Chem. B, 2016, 4, 694–703 RSC.
- K.-D. Zhang and S. Matile, Angew. Chem., Int. Ed., 2015, 54, 8980–8983 CrossRef CAS PubMed.
- M. M. Iftime, S. Morariu and L. Marin, Carbohydr. Polym., 2017, 165, 39–50 CrossRef CAS PubMed.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS PubMed.
- Y. Wang, P. Xing, W. An, M. Ma, M. Yang, T. Luan, R. Tang, B. Wang and A. Hao, Langmuir, 2018, 34, 13725–13734 CrossRef CAS PubMed.
- Z. Wen, M. K. McBride, X. Zhang, X. Han, A. M. Martinez, R. Shao, C. Zhu, R. Visvanathan, N. A. Clark, Y. Wang, K. Yang and C. N. Bowman, Macromolecules, 2018, 51, 5812–5819 CrossRef CAS.
- H. Lei, S. Wang, D. J. Liaw, Y. Cheng, X. Yang, J. Tan, X. Chen, J. Gu and Y. Zhang, ACS Macro Lett., 2019, 582–587 CrossRef CAS.
- C. Zhu, Z. Zou, Y. Li, X. Lei, W. Zhang and J. Xiao, Sci. Adv., 2018, 4, eaaq0508 CrossRef.
- L. Lu, J. Pan and G. Li, J. Mater. Chem. A, 2017, 5, 21505–21513 RSC.
- J. Xu, S. Ye and J. Fu, J. Mater. Chem. A, 2018, 6, 24291–24297 RSC.
- J. Tang, J. Yang, H. Yang, R. Miao, R. Wen, K. Liu, J. Peng and Y. Fang, J. Mater. Chem. C, 2018, 6, 12493–12497 RSC.
- Q. An, I. D. Wessely, Y. Matt, Z. Hassan, S. Bräse and M. Tsotsalas, Polym. Chem., 2019, 10, 672–678 RSC.
- B. S. Sumerlin, Science, 2018, 362, 150–151 CrossRef CAS PubMed.
- H. Liu, X. Sui, H. Xu, L. Zhang, Y. Zhong and Z. Mao, Macromol. Mater. Eng., 2016, 301, 725–732 CrossRef CAS.
- R. Mo, J. Hu, H. Huang, X. Sheng and X. Zhang, J. Mater. Chem. A, 2019, 7, 3031–3038 RSC.
- Y. Peng, Y. Yang, Q. Wu, S. Wang, G. Huang and J. Wu, Polymer, 2018, 157, 172–179 CrossRef CAS.
- L. Cao, J. Fan, J. Huang and Y. Chen, J. Mater. Chem. A, 2019, 7, 4922–4933 RSC.
- F. Ding, S. Wu, S. Wang, Y. Xiong, Y. Li, B. Li, H. Deng, Y. Du, L. Xiao and X. Shi, Soft Matter, 2015, 11, 3971–3976 RSC.
- Z. Guo, W. Ma, H. Gu, Y. Feng, Z. He, Q. Chen, X. Mao, J. Zhang and L. Zheng, Soft Matter, 2017, 13, 7371–7380 RSC.
- H. Yang, J. Tang, C. Shang, R. Miao, S. Zhang, K. Liu and Y. Fang, Macromol. Rapid Commun., 2018, 39, 1700679 CrossRef.
- D. E. Apostolides, C. S. Patrickios, E. Leontidis, M. Kushnir and C. Wesdemiotis, Polym. Int., 2014, 63, 1558–1565 CrossRef CAS.
- M. Nakahata, S. Mori, Y. Takashima, H. Yamaguchi and A. Harada, Chem, 2016, 1, 766–775 CAS.
- X. Zhi, C. Zheng, J. Xiong, J. Li, C. Zhao, L. Shi and Z. Zhang, Langmuir, 2018, 34, 12914–12923 CrossRef CAS PubMed.
- K. K. Oehlenschlaeger, J. O. Mueller, J. Brandt, S. Hilf, A. Lederer, M. Wilhelm, R. Graf, M. L. Coote, F. G. Schmidt and C. Barner-Kowollik, Adv. Mater., 2014, 26, 3561–3566 CrossRef CAS.
- Z. Wei, D. M. Lewis, Y. Xu and S. Gerecht, Adv. Healthcare Mater., 2017, 6, 1700523 CrossRef.
- Z. Li, F. Zhou, Z. Li, S. Lin, L. Chen, L. Liu and Y. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 25194–25202 CrossRef CAS PubMed.
- H. Liu, Y. Li, K. Sun, J. Fan, P. Zhang, J. Meng, S. Wang and L. Jiang, J. Am. Chem. Soc., 2013, 135, 7603–7609 CrossRef CAS.
- Y. Li, O. Rios, J. K. Keum, J. Chen and M. R. Kessler, ACS Appl. Mater. Interfaces, 2016, 8, 15750–15757 CrossRef CAS PubMed.
- G. Das, S. Nagaraja, V. Sridurai, D. B. Shinde, M. Addicoat, T. Prakasam, F. Gándara, F. Ravaux, S. K. Sharma, G. G. Nair, Z. Lai, R. Jagannathan, M. A. Olson and A. Trabolsi, Chem. Mater., 2019, 31, 4148–4155 CrossRef CAS.
- C. Bouillon, D. Paolantoni, J. C. Rote, Y. Bessin, L. W. Peterson, P. Dumy and S. Ulrich, Chem. – Eur. J., 2014, 20, 14705–14714 CrossRef CAS PubMed.
- M. Nakahata, S. Mori, Y. Takashima, A. Hashidzume, H. Yamaguchi and A. Harada, ACS Macro Lett., 2014, 3, 337–340 CrossRef CAS.
- B. Zhang, Z. A. Digby, J. A. Flum, P. Chakma, J. M. Saul, J. L. Sparks and D. Konkolewicz, Macromolecules, 2016, 49, 6871–6878 CrossRef CAS.
- D. Oupicky, A. L. Parker and L. W. Seymour, J. Am. Chem. Soc., 2002, 124, 8–9 CrossRef CAS PubMed.
- S.-H. Kim, S.-H. Lee, J.-E. Lee, S. J. Park, K. Kim, I. S. Kim, Y.-S. Lee, N. S. Hwang and B.-G. Kim, Biomaterials, 2018, 178, 401–412 CrossRef CAS PubMed.
- Z. Fang, N. Zheng, Q. Zhao and T. Xie, ACS Appl. Mater. Interfaces, 2017, 9, 22077–22082 CrossRef CAS PubMed.
- G. Zhang, Q. Zhao, L. Yang, W. Zou, X. Xi and T. Xie, ACS Macro Lett., 2016, 5, 805–808 CrossRef CAS.
- M. B. Sims, K. Y. Patel, M. Bhatta, S. Mukherjee and B. S. Sumerlin, Macromolecules, 2018, 51, 356–363 CrossRef CAS.
- C. Ninh and C. J. Bettinger, Biomacromolecules, 2013, 14, 2162–2170 CrossRef CAS PubMed.
- S. K. Samal, M. Dash, S. V. Vlierberghe, D. L. Kaplan, E. Chiellini, C. V. Blitterswijk, L. Moroni and P. Dubruel, Chem. Soc. Rev., 2012, 41, 7147–7194 RSC.
- D. N. Nguyen, J. J. Green, J. M. Chan, R. Longer and D. G. Anderson, Adv. Mater., 2009, 21, 847–867 CrossRef CAS PubMed.
- S. Y. Wong, J. M. Pelet and D. Putnam, Prog. Polym. Sci., 2007, 32, 799–837 CrossRef CAS.
- T. Merdan, J. Kopecek and T. Kissel, Adv. Drug Delivery Rev., 2002, 54, 715–758 CrossRef CAS.
- X. X. Liu, P. Rocchi and L. Peng, New J. Chem., 2012, 36, 256–263 RSC.
- A. Estévez-Torres and D. Baigl, Soft Matter, 2011, 7, 6746–6756 RSC.
- S. Ulrich, Acc. Chem. Res., 2019, 52, 510–519 CrossRef CAS PubMed.
- K. Miyata, N. Nishiyama and K. Kataoka, Chem. Soc. Rev., 2012, 41, 2562–2574 RSC.
- E. Wagner, Acc. Chem. Res., 2012, 45, 1005–1013 CrossRef CAS PubMed.
- D. K. Kölmel and E. T. Kool, Chem. Rev., 2017, 117, 10358–10376 CrossRef PubMed.
- S. Ulrich, D. Boturyn, A. Marra, O. Renaudet and P. Dumy, Chem. – Eur. J., 2014, 20, 34–41 CrossRef CAS.
- S. Mukherjee, R. N. Ghosh and F. R. Maxfield, Physiol. Rev., 1997, 77, 759–803 CrossRef CAS.
- S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Chem. Soc. Rev., 2014, 43, 1861–1872 RSC.
- C. Bouillon, Y. Bessin, F. Poncet, M. Gary-Bobo, P. Dumy, M. Barboiu, N. Bettache and S. Ulrich, J. Mater. Chem. B, 2018, 6, 7239–7246 RSC.
- Y. Y. Cheng, L. L. Zong, J. Lopez-Andarias, E. Bartolami, Y. Okamoto, T. R. Ward, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2019, 58, 9522–9526 CrossRef CAS.
- L. L. Zong, E. Bartolami, D. Abegg, A. Adibekian, N. Sakai and S. Matile, ACS Cent. Sci., 2017, 3, 449–453 CrossRef CAS PubMed.
- E. Bartolami, D. Basagiannis, L. L. Zong, R. Martinent, Y. Okamoto, Q. Laurent, T. R. Ward, M. Gonzalez-Gaitan, N. Sakai and S. Matile, Chem. – Eur. J., 2019, 25, 4047–4051 CrossRef CAS PubMed.
- N. Chuard, A. I. Poblador-Bahamonde, L. L. Zong, E. Bartolami, J. Hildebrandt, W. Weigand, N. Sakai and S. Matile, Chem. Sci., 2018, 9, 1860–1866 RSC.
- G. Gasparini, E.-K. Bang, J. Montenegro and S. Matile, Chem. Commun., 2015, 51, 10389–10402 RSC.
- E. K. Bang, G. Gasparini, G. Molinard, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2013, 135, 2088–2091 CrossRef CAS PubMed.
- N. Chuard, G. Gasparini, A. Roux, N. Sakai and S. Matile, Org. Biomol. Chem., 2015, 13, 64–67 RSC.
- G. Gasparini, E. K. Bang, G. Molinard, D. V. Tulumello, S. Ward, S. O. Kelley, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 6069–6074 CrossRef CAS PubMed.
- G. Gasparini and S. Matile, Chem. Commun., 2015, 51, 17160–17162 RSC.
- N. Chuard, G. Gasparini, D. Moreau, S. Lorcher, C. Palivan, W. Meier, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2017, 56, 2947–2950 CrossRef CAS PubMed.
- E. K. Bang, M. Lista, G. Sforazzini, N. Sakai and S. Matile, Chem. Sci., 2012, 3, 1752–1763 RSC.
- P. K. Hashim, K. Okuro, S. Sasaki, Y. Hoashi and T. Aida, J. Am. Chem. Soc., 2015, 137, 15608–15611 CrossRef CAS PubMed.
- D. Abegg, G. Gasparini, D. G. Hoch, A. Shuster, E. Bartolami, S. Matile and A. Adibekian, J. Am. Chem. Soc., 2017, 139, 231–238 CrossRef CAS PubMed.
- G. Gasparini, G. Sargsyan, E. K. Bang, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2015, 54, 7328–7331 CrossRef CAS PubMed.
- R. Mogaki, P. K. Hashim, K. Okuro and T. Aida, Chem. Soc. Rev., 2017, 46, 6480–6491 RSC.
- P. K. Hashim, K. Okuro, S. Sasaki, Y. Hoashi and T. Aida, J. Am. Chem. Soc., 2015, 137, 15608–15611 CrossRef CAS PubMed.
- R. Catana, M. Barboiu, I. Moleavin, L. Clima, A. Rotaru, E.-L. Ursu and M. Pinteala, Chem. Commun., 2015, 51, 2021–2024 RSC.
- L. Marin, D. Ailincai, M. Cahn, D. Stan, C. A. Constantinescu, L. Ursu, F. Doroftei, M. Pinteala, B. C. Simionescu and M. Barboiu, ACS Biomater. Sci. Eng., 2016, 2, 104–111 CrossRef CAS.
- I. A. Turin-Moleavin, F. Doroftei, A. Coroaba, D. Peptanariu, M. Pinteala, A. Salic and M. Barboiu, Org. Biomol. Chem., 2015, 13, 9005–9011 RSC.
- L. Clima, D. Peptanariu, M. Pinteala, A. Salic and M. Barboiu, Chem. Commun., 2015, 51, 17529–17531 RSC.
- M. A. Deriu, N. Tsapis, M. Noiray, G. Grasso, N. El Brahmi, S. Mignani, J. P. Majoral, E. Fattal and A. Danani, Nanoscale, 2018, 10, 10952–10962 RSC.
- I. Lostalé-Seijo and J. Montenegro, Nat. Rev. Chem., 2018, 2, 258–277 CrossRef.
- A. Fuertes, M. Juanes, J. R. Granja and J. Montenegro, Chem. Commun., 2017, 53, 7861–7871 RSC.
- M. Juanes, O. Creese, P. Fernandez-Trillo and J. Montenegro, MedChemComm, 2019, 10, 1138–1144 RSC.
- J. M. Priegue, I. Lostale-Seijo, D. Crisan, J. R. Granja, F. Fernandez-Trillo and J. Montenegro, Biomacromolecules, 2018, 19, 2638–2649 CrossRef CAS PubMed.
- J. M. Priegue, D. N. Crisan, J. Martinez-Costas, J. R. Granja, F. Fernandez-Trillo and J. Montenegro, Angew. Chem., Int. Ed., 2016, 55, 7492–7495 CrossRef CAS PubMed.
- I. Louzao, R. Garcia-Fandino and J. Montenegro, J. Mater. Chem. B, 2017, 5, 4426–4434 RSC.
- I. Lostale-Seijo, I. Louzao, M. Juanes and J. Montenegro, Chem. Sci., 2017, 8, 7923–7931 RSC.
- C. S. Mahon, C. Sakonsinsiri, M. A. Fascione, T. McAllister, W. B. Turnbull and D. A. Fulton, Org. Biomol. Chem., 2015, 13, 2756–2761 RSC.
- C. S. Mahon and D. A. Fulton, Nat. Chem., 2014, 6, 665–672 CrossRef CAS PubMed.
- C. S. Mahon and D. A. Fulton, Chem. Sci., 2013, 4, 3661–3666 RSC.
- C. S. Mahon, A. W. Jackson, B. S. Murray and D. A. Fulton, Chem. Commun., 2011, 47, 7209–7211 RSC.
- Y. Zhang, L. Hu and O. Ramström, Chem. Commun., 2013, 49, 1805–1807 RSC.
- Y. Zhang and O. Ramström, Chem. – Eur. J., 2014, 20, 3288–3291 CrossRef CAS PubMed.
- Y. Zhang, H. S. N. Jayawardena, M. Yan and O. Ramström, Chem. Commun., 2016, 52, 5053–5056 RSC.
- Y. Zhang, Y. Zhang and O. Ramström, Catal. Rev., 2019, 1–30 Search PubMed.
- Y. Zhang, Y.-M. Legrand, E. Petit, C. T. Supuran and M. Barboiu, Chem. Commun., 2016, 52, 4053–4055 RSC.
- Y. Zhang, W.-X. Feng, Y.-M. Legrand, C. T. Supuran, C.-Y. Su and M. Barboiu, Chem. Commun., 2016, 52, 13768–13770 RSC.
- Y. Zhang, C. T. Supuran and M. Barboiu, Chem. – Eur. J., 2018, 24, 715–720 CrossRef CAS PubMed.
- H. Zhao, K. Peng, F. Lv, L. Liu and S. Wang, ACS Appl. Bio Mater., 2019, 2, 1787–1791 CrossRef CAS.
- J. Wang, Z. Gao, W. Qi, Y. Zhao, P. Zhang, M. Lin, Z. Li, G. Chen and M. Jiang, ACS Biomater. Sci. Eng., 2018, 4, 2061–2066 CrossRef CAS.
- Y. Zhang, Y. Li, C. Su and M. Barboiu, ChemPlusChem, 2018, 83, 506–513 CrossRef CAS.
- A. M. Rosales and K. S. Anseth, Nat. Rev. Mater., 2016, 1, 15012 CrossRef CAS PubMed.
- S. R. Caliari and J. A. Burdick, Nat. Methods, 2016, 13, 405 CrossRef CAS PubMed.
- D. D. McKinnon, D. W. Domaille, J. N. Cha and K. S. Anseth, Adv. Mater., 2014, 26, 865–872 CrossRef CAS PubMed.
- H. Wang, D. Zhu, A. Paul, L. Cai, A. Enejder, F. Yang and S. C. Heilshorn, Adv. Funct. Mater., 2017, 27, 1605609 CrossRef.
- M. E. Smithmyer, C. C. Deng, S. E. Cassel, P. J. LeValley, B. S. Sumerlin and A. M. Kloxin, ACS Macro Lett., 2018, 7, 1105–1110 CrossRef CAS.
- S. Li, J. Yi, X. Yu, H. Shi, J. Zhu and L. Wang, ACS Biomater. Sci. Eng., 2018, 4, 872–883 CrossRef CAS.
- J. J. Roberts, P. Naudiyal, L. Jugé, L. E. Bilston, A. M. Granville and P. J. Martens, ACS Biomater. Sci. Eng., 2015, 1, 1267–1277 CrossRef CAS.
- J. Lou, R. Stowers, S. Nam, Y. Xia and O. Chaudhuri, Biomaterials, 2018, 154, 213–222 CrossRef CAS PubMed.
- S. W. Kim, D. Y. Kim, H. H. Roh, H. S. Kim, J. W. Lee and K. Y. Lee, Biomacromolecules, 2019, 20, 1860–1866 CrossRef CAS PubMed.
- L. Marin, I. Stoica, M. Mares, V. Dinu, B. C. Simionescu and M. Barboiu, J. Mater. Chem. B, 2013, 1, 3353–3358 RSC.
- Y. Bu, L. Zhang, J. Liu, L. Zhang, T. Li, H. Shen, X. Wang, F. Yang, P. Tang and D. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 12674–12683 CrossRef CAS PubMed.
- J. F. Reuther, J. L. Dees, I. V. Kolesnichenko, E. T. Hernandez, D. V. Ukraintsev, R. Guduru, M. Whiteley and E. V. Anslyn, Nat. Chem., 2017, 10, 45 CrossRef PubMed.
- J. F. Reuther, A. C. Goodrich, P. R. Escamilla, T. A. Lu, V. Del Rio, B. W. Davies and E. V. Anslyn, J. Am. Chem. Soc., 2018, 140, 3768–3774 CrossRef CAS PubMed.
- W. Zhan, Y. Qu, T. Wei, C. Hu, Y. Pan, Q. Yu and H. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 10647–10655 CrossRef CAS PubMed.
- K. A. McCarthy, M. A. Kelly, K. Li, S. Cambray, A. S. Hosseini, T. van Opijnen and J. Gao, J. Am. Chem. Soc., 2018, 140, 6137–6145 CrossRef CAS PubMed.
- Z. Zhang, X. Wang, Y. Wang and J. Hao, Biomacromolecules, 2018, 19, 980–988 CrossRef CAS PubMed.
- Y. Hua, Y. Gan, P. Li, L. Song, C. Shi, C. Bao, Y. Yang, Q. Zhou, Q. Lin and L. Zhu, ACS Biomater. Sci. Eng., 2019, 5, 4048–4053 CrossRef CAS.
- R. Dong, Y. Zhou, X. Huang, X. Zhu, Y. Lu and J. Shen, Adv. Mater., 2015, 27, 498–526 CrossRef CAS PubMed.
- A. Harada, Y. Takashima and H. Yamaguchi, Chem. Soc. Rev., 2009, 38, 875–882 RSC.
- G. Yu, K. Jie and F. Huang, Chem. Rev., 2015, 115, 7240–7303 CrossRef CAS PubMed.
- E. Moulin, J. J. T. Armao and N. Giuseppone, Acc. Chem. Res., 2019, 52, 975–983 CrossRef CAS PubMed.
- T. Xiao, W. Zhong, L. Xu, X. Q. Sun, X. Y. Hu and L. Wang, Org. Biomol. Chem., 2019, 17, 1336–1350 RSC.
- L. Zhao, Y. Liu, R. Chang, R. Xing and X. Yan, Adv. Funct. Mater., 2019, 29, 1806877 CrossRef.
- D. P. Goronzy, M. Ebrahimi, F. Rosei, Arramel, Y. Fang, S. De Feyter, S. L. Tait, C. Wang, P. H. Beton, A. T. S. Wee, P. S. Weiss and D. F. Perepichka, ACS Nano, 2018, 12, 7445–7481 CrossRef CAS PubMed.
- A. Martinez, C. Ortiz Mellet and J. M. Garcia Fernandez, Chem. Soc. Rev., 2013, 42, 4746–4773 RSC.
- J. Zhou, G. Yu and F. Huang, Chem. Soc. Rev., 2017, 46, 7021–7053 RSC.
| This journal is © the Partner Organisations 2020 |