
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in the mechanisms of the hydrogen evolution reaction by non-innocent sulfur-coordinating metal complexes
Maria
Drosou
 ,
Fotios
Kamatsos
and
Christiana A.
Mitsopoulou
*
,
Fotios
Kamatsos
and
Christiana A.
Mitsopoulou
*
Inorganic Chemistry Laboratory, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis, Zografou 15771, Greece. E-mail: cmitsop@chem.uoa.gr
First published on 1st November 2019
Abstract
Efficient hydrogen generation from aqueous protons through direct conversion of solar energy is an essential prerequisite of a future hydrogen economy and easily accessible renewable energy. For achieving this goal, the effective synthesis of a catalyst that can promote the hydrogen evolution reaction (HER) is in demand. Several mononuclear non-noble metal complexes carrying non-innocent S donating ligands have been reported as efficient photocatalysts and electrocatalysts for proton reduction. A thorough understanding of the elementary steps of hydrogen formation allows one to derive structure–property relationships that can guide future catalyst design. In this review, we highlight the mechanisms of the homogeneous HER for these catalysts in the light of photocatalytic, electrocatalytic and computational data with the main concern of elucidating the interplay between the localization of the electron density and the hydrogen evolution reaction pathway. In addition, the effects of π-conjugation, heteroatoms and electron accepting/donating moieties on the basicity of the metal center and catalyst overpotential are discussed to rationalize the formation of a metal or ligand hydrid intermediate. Moreover, some suggestions are provided for future use in the design of effective HER catalysts.
 Maria Drosou | Maria Drosou obtained her Master of Science degree in Inorganic Chemistry from the University of Athens in 2018. She is currently pursuing her Ph.D. in Inorganic Chemistry under the guidance of Prof. Christiana Mitsopoulou. Her current research interests are focused on computational investigation of mechanistic processes in organometallic chemistry. |
 Fotios Kamatsos | Fotios Kamatsos earned his Bachelor's degree in Chemistry at the National and Kapodistrian University of Athens in 2016. During his Master's studies, he worked for Prof. Christiana Mitsopoulou at the Chemistry Department where he developed his passion for the synthesis and photocatalysis research field. Fotis is currently carrying out doctoral research under the supervision of Prof. Christiana Mitsopoulou and focuses on improving photocatalytic systems for hydrogen production, through the design and synthesis of new catalysts. |
 Christiana A. Mitsopoulou | Prof. Christiana A. Mitsopoulou received her Ph.D. (1989) from the Chemistry Department of the National and Kapodistrian University of Athens, following academic posts at Queen Mary and UCL in liquid and solid NMR. She joined the Inorganic Lab of N.K.U.A. in 1981 and was promoted to Professor in 2010. She is Director of the Inorganic Laboratory and the MSc Program “Inorganic Chemistry and its Industrial Applications”. She contributed towards providing insight into dithiolene chemistry and photosplitting of water using both experiments and computational data. Her current research interests are focused on ligand and metal complex design for photodynamic therapies and catalysts for photocatalysis and electrocatalysis. |
1. Introduction
The globally increasing energy demand and limited fossil fuel resources intensify the necessity to pursue alternative energy sources that are both clean and renewable.1 Hydrogen, as a key energy carrier, is considered a promising alternative in our transition away from the present-day hydrocarbon economy. H2 production via electrochemical water splitting is a sustainable method of electrical energy conversion to clean fuel. However, energy acquired via fossil-fuel cracking is environmentally unsustainable due to the increase in atmospheric CO levels and perpetual lowering of global carbon reserves. Thus, a major challenge is the direct conversion of solar energy to H2 fuel via the light-driven generation of hydrogen from aqueous protons.2–5Nature achieves the conversion of protons and electrons into molecular H2 (2H+ + 2e− → H2) using the Earth abundant metals Ni and/or Fe in their active sites under ambient pressure and temperature.6,7 Over the years, several groups have developed and studied synthetic metal complexes with excellent performances for electrocatalytic hydrogen production and some of them also provide highly efficient photocatalytic systems.8–10 The development of inexpensive proton reduction catalysts based on Earth-abundant elements11,12 such as Fe,13 Ni,14–16 Cu,17–19 and Co20,21 to replace the less abundant and high cost platinum-based materials for catalytic proton reduction is a great scientific challenge and a significant step towards sustainable solar energy conversion. Some of the most efficient H2 evolution homogeneous catalysts designed are the distorted octahedral Ni(II) complex [Ni(bztpen)]2+ (bztpen = N-benzyl-N,N′,N′-tris(pyridine-2-ylmethyl)ethylenediamine), which shows a very high TON of 308![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 over 60 h electrolysis with an applied potential of −1.25 V vs. SHE22 and the distorted octahedral Co(II) complex of 2-bis(2-pyridyl)(methoxy)methyl-6-pyridylpyridine, which produces >55000 moles of H2 per mole of catalyst over 60 h under −1.30 V vs. SHE applied potential.23
000 over 60 h electrolysis with an applied potential of −1.25 V vs. SHE22 and the distorted octahedral Co(II) complex of 2-bis(2-pyridyl)(methoxy)methyl-6-pyridylpyridine, which produces >55000 moles of H2 per mole of catalyst over 60 h under −1.30 V vs. SHE applied potential.23
Based on the kinetic and thermodynamic information obtained by spectroscopic and electrochemical methods, a possible catalytic mechanism of a homogeneous proton reduction system can be proposed. In parallel to the development of experimental strategies, computational methods based on quantum chemistry provide significant insight into the elementary steps leading to H2 evolution from protons in a solution.24–26 Accurate identification of the H2 evolution reaction (HER) active sites on catalytically active complexes could promote the development of even more robust and efficient catalysts by rational design. The origin of the catalytic reactivity of molecular catalysts can be uncovered by correlating certain activity descriptors derived on the basis of DFT calculations on the minima and transition state structures of catalytically relevant intermediates, leading to the design of more efficient candidates for the HER.27
Recently, it has been shown that synergy between metal- and ligand-based redox activities can improve the performance of metal complex catalysts.28–30 The combination of the redox activities of a dithiolato ligand31 and a metal enables complex redox behaviour involving multi-step electron transfer processes, between electrons delocalized over hybrid ligand π orbitals and metal d orbitals. The electron-rich and π-back-donating sulfur donor character is favorable for the stabilization of otherwise labile low-oxidation-state metals, facilitating metal hydride intermediates for the HER. Dithiolene complexes are found as biological cofactors32 and are involved in bioinorganic processes at the catalytic centers of Mo and W oxotransferases.33 The chemistry and potential applications of these compounds have been extensively studied in the last 60 years34–36 and they have been used successfully as photosensitizers and catalysts for proton reduction.37,38
The main aim of this work is to provide a comprehensive account on dithiolene, thiolate and thiosemicarbazone based HER mononuclear metal catalysts, investigating both experimental and computational aspects of the respective HER mechanisms, toward a better/deeper understanding of their fascinating chemistry. The catalytic reactivity of dithiolene and thiolate metal complexes is compared under electrocatalytic and photocatalytic conditions in terms of the turnover number (TON), turnover frequency (TOF) and overpotentials. The role of the nitrogen heteroatoms in thiolate and thiosemicarbazone ligands is revealed in the light of the multiple electron and protonation steps in the mechanism of the HER which minimize the energy of the transients, in accordance with photosynthesis.
2. Electronic structures of metal complexes carrying non-innocent S-donating ligands
The term non-innocent ligand refers to redox-active molecules coordinated with redox-active transition metals, to form metal complexes with ambiguous metal and ligand oxidation-states.39 Thiolate ligands possess a σ-donor orbital and a filled π orbital that are mostly sulphur 3p in character and their symmetry allows such ligands to act as σ- and π-donors to the metal center.40 Early studies on the electronic structures of transition metal complexes bearing thiolate ligands, [LnM(SR)], revealed that the frontier orbitals have greater sulphur than metal character.40–42 One-electron oxidation of [LnM(SR)] yields the monocation [LnM(˙SR)]+, which can be formulated as a thiyl radical with the unpaired electron localized mostly on the sulphur p non-bonding orbital. Such radicals are very reactive, as the M–˙SR bond weakens and dissociates in solution to generate polynuclear disulfide-bridged [R–S–S–R]2+ species.41,43,44 The thiyl radical cations [CpFe(CO)2SR]+ are also reactive towards alkyl halides achieving nucleophilic displacement of the halide by the thiolate ligand.40 The nucleophilicity of thiolate complexes, mainly attributed to the metal–sulphur π interaction, plays a major role in the catalytic hydrogen evolution pathways.Among the various redox-active ligands, dithiolene and ene-1,2-dithiolate ligands are considered to be prototypical examples of redox non-innocent ligands. The delocalized π-systems of the dithiolene ligands in conjunction with the π-back donation from the metal center results in the formation of extended networks of π–π interactions involving metal and ligand orbitals. Thus, these systems display interesting chemical properties such as ferromagnetism, spin-ladder behavior, superconducting properties and catalytic activity.45–47 Since non-innocent ligands store reducing equivalents in the catalytic complex, they can lower the required overpotential to form hydrogen from solution protons. Elucidation of the nature of the metal–sulfur bond is important in gaining insight into the structure/function relationships of thiolate catalysts.
Dithiolene ligands are strong electron donors and upon coordination to a metal they form a square planar complex (i.e. Ni, Co, Fe, Pd or Pt) and the filled S px orbitals interact with the metal dx2−y2 to form σ bonds, while the metal donates electron density back to the ligand through the dxz orbital that interacts with the pz S orbitals. This π-back-donation leads to increased mixing of metal and ligand orbitals. Thus, the M–S bond is highly covalent and high accuracy methods are needed to determine the exact localization of the electron density. The electronic structure of transition metal dithiolene complexes has long been debated48–50 because the metal and ligand oxidation states were difficult to determine using traditional characterization techniques. An array of experimental and theoretical methods such as X-ray diffraction, IR/Raman, UV-Vis-NIR, EPR, and XAS techniques and DFT and ab initio electronic structure calculations is typically advised to establish the nature of the M–S bonds of dithiolates.
Metal and ligand oxidation-states and bond orders are reflected in the bond lengths and the geometry of the complex, which can be measured by X-ray crystallography. Furthermore, associated infrared (IR) stretching frequencies increase and decrease upon strengthening and weakening of the corresponding bond, respectively.51 However, delocalized systems show more complicated behavior and the structural differences are often small and may not be detectable by those techniques.52 The EPR information is helpful in measuring the participation of the metal in the singly occupied molecular orbital (SOMO). Ligand K-edge XAS53 probes the 1s → valence np transitions and the intensity of the corresponding peak quantifies54 the amount of the ligand character on a specific metal–ligand bond. In contrast to the metal K-edge XAS, which probes the weak (quadrupole allowed) metal's 1s → 3d transitions, ligand 1s → np transitions are electric dipole allowed, and thus more intense.55–57 An increase in the ligand character of the ground state wave function corresponds to more covalent metal–ligand bonding. Based on these studies, Solomon and co-workers described the dithiolene-metal electronic structure as “inverted” from the usual type of bonding in metal complexes, because the LUMO is mostly localized on the ligand and the HOMO has an increased metal character.58 Thus the same ligand could behave as innocent or noninnocent depending on the metal center of the complex under study.
Complexes bearing dithiolato ligands can exhibit two-step redox behavior. Based on experimental and computational data on square planar dithiolate complexes of M2+ = Ni2+, Pd2+, Pt2+, Co2+, Cu2+, Zn2+ and Au2+, it was suggested that the neutral complex is best described by three resonance structures, where the true electronic structure lies somewhere between these extremes (Scheme 1). The first one describes a delocalized resonance pair between enedithiolate and dithioketone ligand forms, while the second one assumes a ligand diradical character,59 where each ligand has an unpaired electron and these are antiferromagnetically coupled. It is important to differentiate between separated valence tautomer equilibrium situations,60 which involve two (or more) different species in equilibrium separated by an activation barrier, and barrierless delocalized “resonance” cases, involving two (or more) resonance forms, which describe only one species with one energy potential minimum.61 In the first case, the degree of covalency in the metal–ligand bond is low, while their frontier orbitals are nearly degenerate. In the second case, the complexes exhibit delocalized electronic structures and the frontier molecular orbitals of the complex are a result of metal and ligand orbital mixing. The frontier molecular orbitals of the dithiolene ligand framework were obtained computationally employing the BP86 DFT functional.62 Upon reduction of the neutral complex, the singly occupied molecular orbital (SOMO) was identified as the antibonding combination of the metal dxz orbital and the b2g orbital of the dithiolene ligand framework (Scheme 1). The non-innocent character of the dithiolene ligand in a square planar bis(dithiolato) complex is mainly attributed to the interaction of the b2g orbital of the ligands with the metal dxz orbital. The amount of mixing is controlled by the relative energies of the metal and ligand fragment orbitals. The b2g molecular orbital is filled in dianionic bis(dithiolene) complexes.
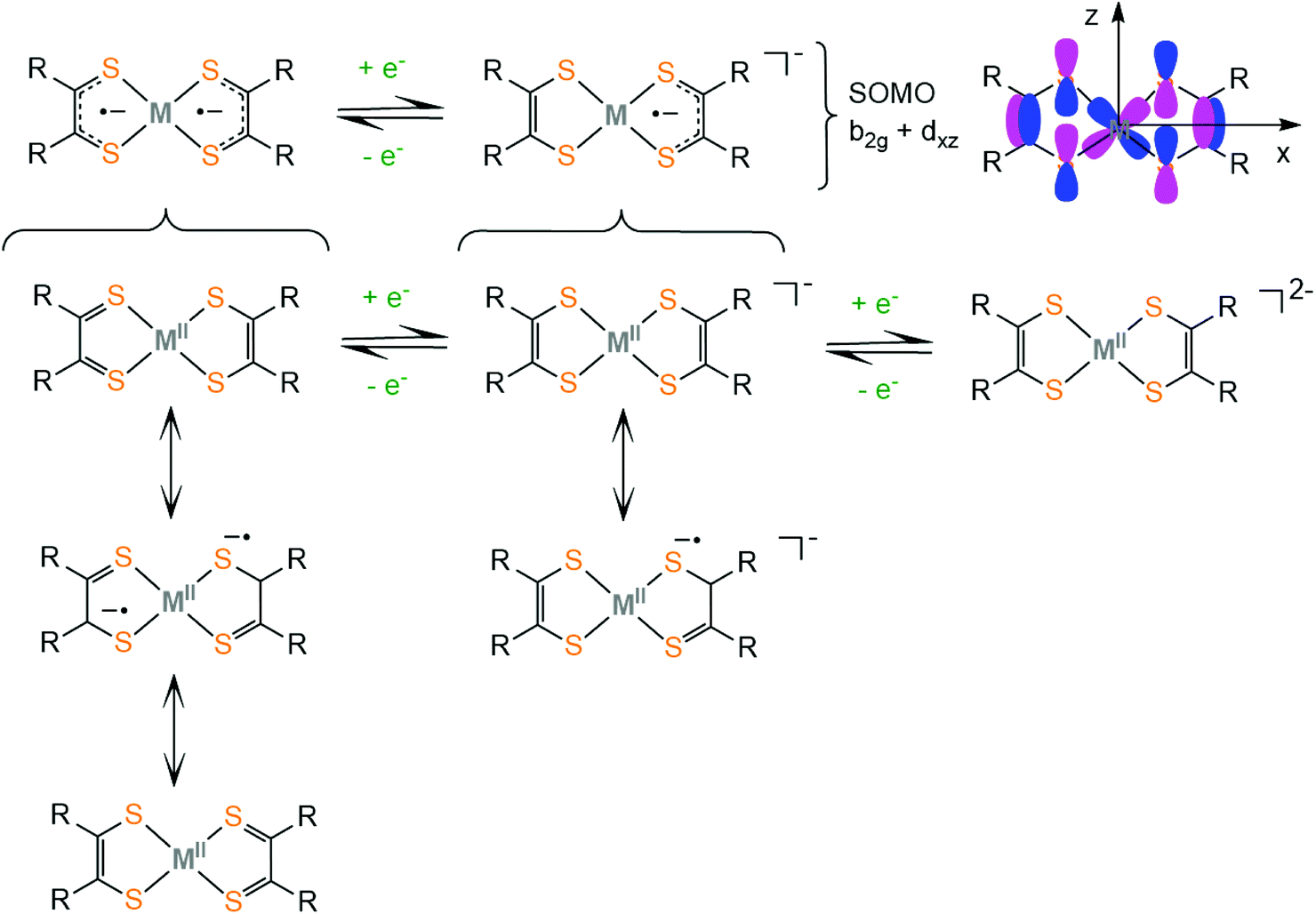 | ||
| Scheme 1 Redox structures of bis-dithiolato metal complexes. | ||
In neutral dithiolate complexes with two equivalent redox-active ligands an intense, low energy (near IR) electronic transition is attributed to the HOMO–LUMO inner-π-transition b1u → b2g. This characteristic absorption band usually undergoes a solvatochromic shift and has an interligand character, between differently charged ligands in the excited state (ligand to ligand charge transfer, LLCT), with some metal contribution.51,63–67
Similar to dithiolenes, o-quinone ligands (O instead of S) may exist as neutral quinones, radical semiquinones or dianionic catecholates and o-quinonediimine ligands (N–R instead of S) may exist as neutral quinonediimines, radical semiquinonediimines or dianionic diamides. Theoretical studies based on first principles showed that substitution of a sulphur atom of the dithiolene ligand with N or O leads to increased participation of the ligands in the redox active orbitals. Neese and Wieghardt employed DFT and multireference ab initio calculations (CASSCF) to investigate the electronic structure of ortho-semiquinonato type Ni complexes with O-, NH-, and S-ligands. Using the broken symmetry formalism, they found that the singlet diradical character of square planar Ni2+ complexes of o-semiquinonato ligands increases as the ligating atoms are S < N < O.68 Notably, Wieghardt and his group experimentally and computationally showed that Ni2+, Pt2+ and Pd2+ neutral complexes with o-diiminobenzosemiquinonato ligands also exist as π radicals in the ground state.69
The electronic structure of the octahedral ruthenium and rhenium complexes, [Ru(DPPBT)3]n and [Re(DPPBT)3]n, bearing the non-innocent ligand 2-diphenylphosphino-benzenethiolate (DPPBT), which contains S and P ligating donor atoms, is similar to the non-innocent dithiolene framework. The cationic [Ru(DPPBT)3]+ possesses a singlet diradical character with unpaired electrons delocalized over the metal d orbitals with appropriate symmetry and the sulphur p orbitals.70 The orientation and energy of the S p orbitals allow the reversible ethylene addition to [Ru(DPPBT)3]+, while [Re(DPPBT)3]n does not show the same activity.71 Computational analysis of their electronic structure based on DFT and ab initio calculations predicted that [Re(DPPBT)3]0 has less thiyl-diradical character than [Ru(DPPBT)3]+, which explains the differences in their activity.72
The electronic structure of first row transition metal complexes bearing the non-innocent glyoxal-bis(2-mercaptoanil), gma (Scheme 2), ligand has also been investigated. The (gma)2− ligand has an exceptionally low lying LUMO π orbital which is located on the α-diimine unit (b2 orbital) and can be easily reduced to generate the (gma˙)3− radical. The unpaired electron is delocalized over the π bond between the carbons and over the pz orbitals of the nitrogen atoms, inducing strengthening of the C–C bond and weakening of the two C–N bonds, as indicated by crystal structure analysis, spectroscopic measurements and DFT calculations. When the effective nuclear charge on the metal center is low (Fe and Ni) and thus the energy of the metal d orbitals is relatively high, the dyz orbital transfers the electron density to the ligand via a π-backbonding interaction, while metals with higher effective nuclear charge (Zn) participate less in the SOMO.73 Similar electronic structures have been reported for nickel complexes bearing thiosemicarbazone ligands, where the SOMO of the reduced complexes is 90% localized on the ligand and 10% on the metal.74 As will be shown in section 4, complexes based on the strongly non-innocent thiosemicarbazone macrocyclic ligands have been successfully used as hydrogen evolution catalysts. Knowledge of the factors which govern the π-electron distribution in thiolate complexes will allow chemists to tailor catalysts with predictable properties, such as protonation sites, reduction potentials and metal hydricity values.
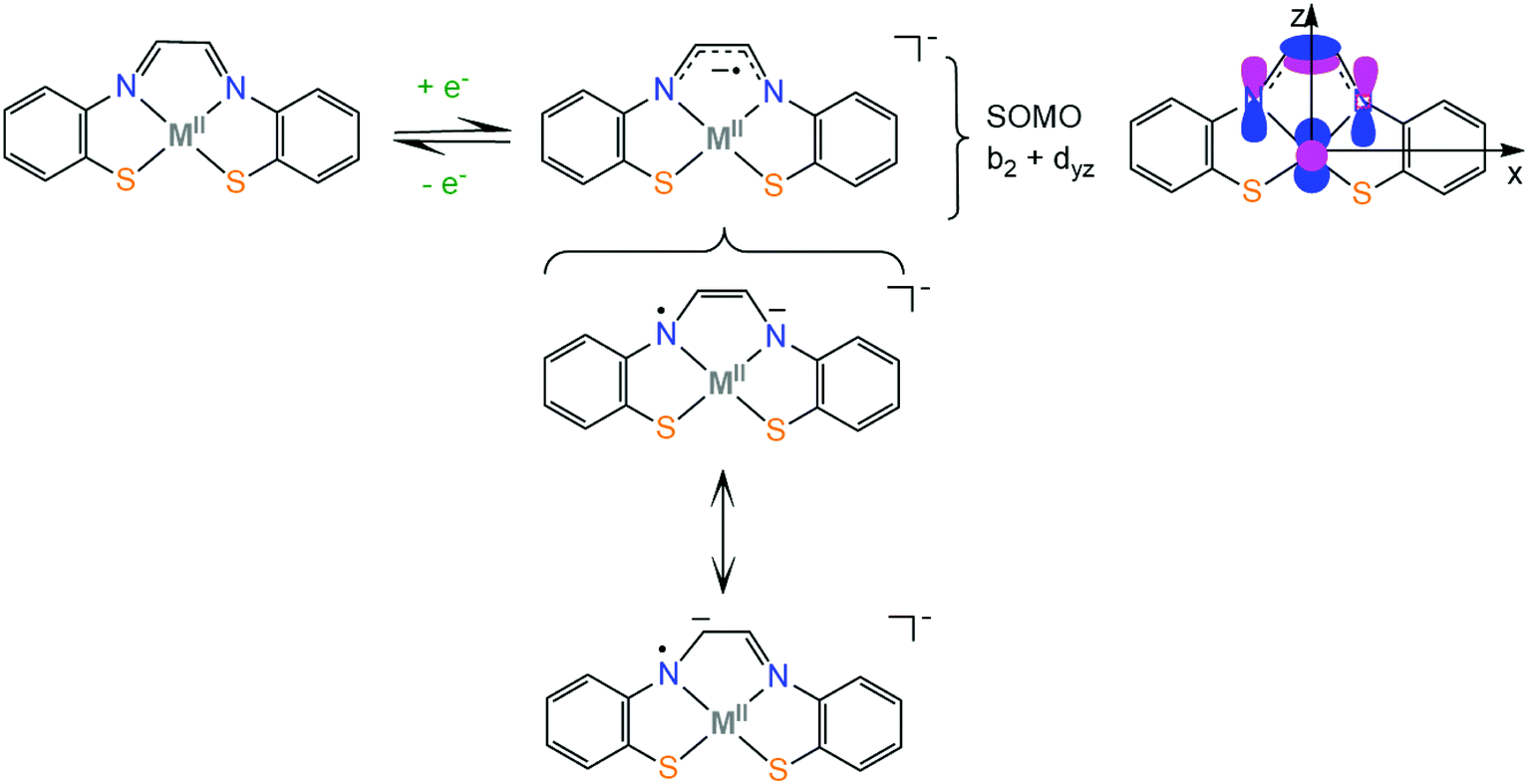 | ||
| Scheme 2 Resonance structures of glyoxal-bis(2-mercaptoanil) metal complexes. | ||
3. Hydrogen evolution by molecular systems
3.1 HER mechanisms
The general mechanism of the photocatalytic hydrogen production includes excitation of a photosensitizer75 using visible light and subsequently electron transfer from the excited photosensitizer to the proton reduction catalyst.76,77 The photosensitizer is regenerated by a sacrificial electron donor, such as triethylamine (TEA), triethanolamine (TEOA) and ascorbic acid. In an electrocatalytic hydrogen evolution system, the cathodic potential is applied to a solution of the catalyst in the presence of a proton source, to provide the driving force for the proton reduction. The role of the catalyst is to lower the activation barrier for the reduction of two protons to H2. Most of the reported H2 evolving homogeneous electrocatalytic systems operate in organic solvents, such as acetonitrile (CH3CN), dimethylformamide (DMF), dichloromethane (CH2Cl2) and tetrahydrofuran (THF), in the presence of various acids, such as ascorbic acid, acetic acid, trifluoroacetic acid (TFA) and toluenesulfonic acid (TsOH), acting as proton donors. However, pure aqueous systems are preferred, since they are environmentally friendly and low cost. Although photocatalysis is different in nature from electrocatalysis, the latter provides us with necessary details in order to elucidate the photocatalytic mechanism.Based on the extensive experimental and theoretical studies, several mechanistic processes which lead to hydrogen evolution mediated by transition metal-containing molecular catalysts have been proposed.78–81 In most cases, the H2 evolving species is believed to be a metal hydride intermediate, generated by either consecutive or coupled proton and electron transfer (PCET).82 The possible mechanisms for the formation of the metal-hydride mediated H–H bond are depicted in Scheme 3. Usually, the HER catalytic cycle starts with a 1e− reduction of the catalyst followed by either protonation to form H–M(n−1)+ or a second reduction.83 In the first case, a possible bimetallic route involves the reaction between two metal hydrides H–Mn+ to release H2via reductive elimination. Alternatively, the H–Mn+ is further reduced to H–M(n−1)+, which can evolve H2 either by a homolytic bimetallic route or by a heterolytic pathway.76 The tendency of the metal hydride H–M(n−1)+ to react with a proton and release H2 depends on the hydricity of the metal hydride, the acidity of the proton source, the hydricity of H2 and the activation energy of the reaction.84,85 Hydride donor ability, or hydricity 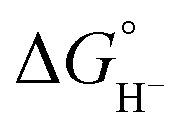 , of a metal hydride [M − H]+ is formally defined as the heterolytic bond dissociation free energy of the metal hydride bond to yield the parent metal complex [M]2+ and the hydride anion.86 Notably, homolytic and heterolytic pathways may occur simultaneously and the dominant route depends on experimental conditions. While the metal-hydride mechanisms are a successful approach, catalyst candidates of this type are often prone to decomposition and nanoparticle formation, decreasing the system's longevity.87
, of a metal hydride [M − H]+ is formally defined as the heterolytic bond dissociation free energy of the metal hydride bond to yield the parent metal complex [M]2+ and the hydride anion.86 Notably, homolytic and heterolytic pathways may occur simultaneously and the dominant route depends on experimental conditions. While the metal-hydride mechanisms are a successful approach, catalyst candidates of this type are often prone to decomposition and nanoparticle formation, decreasing the system's longevity.87
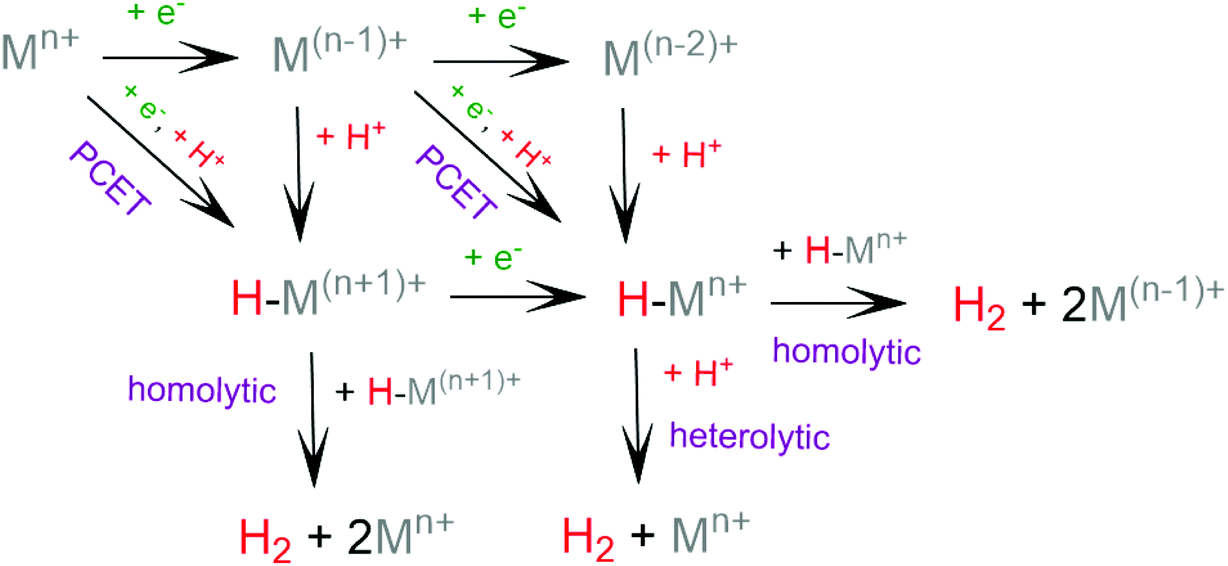 | ||
| Scheme 3 Possible reaction pathways for the homolytic and heterolytic H2 formation via a metal-hydride intermediate. | ||
Recently, scientific efforts have turned to the development of redox-active ligand frameworks that participate in metal-assisted ligand-centered or ligand-assisted metal-centered H2 formation.88 Non-innocent ligands participate in the catalytic activities of several metalloenzymes, such as hydrogenase89 and galactose oxidase.90 The possible mechanisms for the formation of H–H bonds facilitated by non-innocent ligands are depicted in Scheme 4. A ligand centered reduction can facilitate the formation of a metal hydride by increasing the electron density on the metal and by acting as a proton relay, leading to ligand-assisted metal-centered reactivity. Alternatively, a low-valence metal center favours ligand protonation, possibly leading to metal-assisted ligand-centered reactivity. Ligand-centered reactivity can occur on redox active ligands.91,92 Some of these ligands can also catalyse proton reduction even in metal free systems. Ligand centered HER catalytic mechanisms have been recently reviewed.93
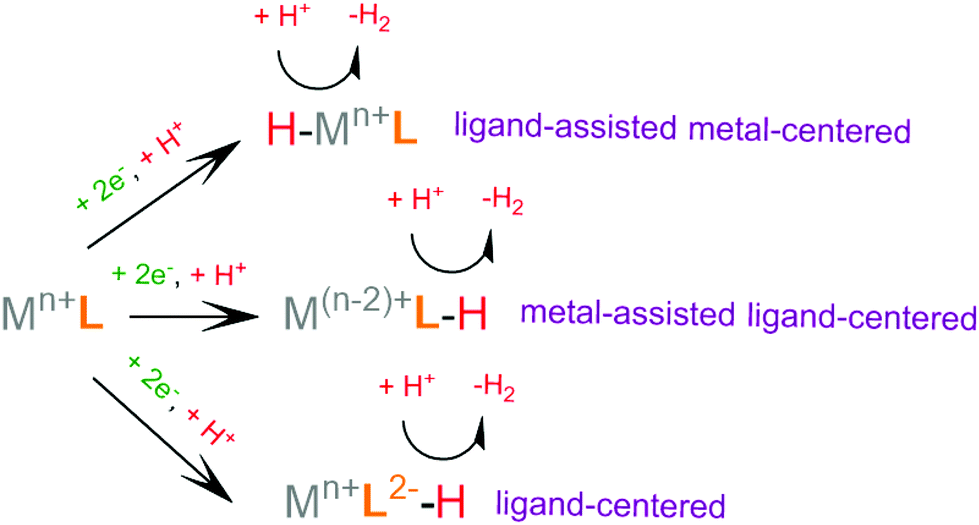 | ||
| Scheme 4 Possible catalytic pathways of non-innocent ligand mediated H2 formation. | ||
For H2 formation to take place, two electron transfer (electrochemical step, E) processes and two proton transfer (chemical step, C) processes are required. The sequence of these steps may be alternating, ECEC or CECE, resulting in two sequential net hydrogen atom transfers, avoiding the build-up of charge. Alternatively, a mechanism consisting of double protonation followed by reduction, CCEE, might be preferable, since reduction of protonated species tends to require more anodic potential. Electrophilic moieties on the ligands may facilitate the reduction of the catalyst, leading to a doubly reduced intermediate, which tends to be basic; thus an EECC pathway may take place. Electron and proton transfer may be coupled in a single PCET step, a process that usually avoids high energy intermediates.82 From the various possible reaction pathways, the system follows the one that includes intermediate species with low relative free energies and with low free energy barriers separating the intermediates. In an efficient H2 evolving cycle, the energies of the catalytic intermediates would gradually decrease from the energy of the reacting protons, electrons and catalyst to the energy of the products. An ideal catalyst does not produce high and low energy intermediates, which typically involve high activation energies that limit the reaction rate.94 The mechanism of a given catalyst depends on its metal center and ligand framework, as well as the experimental conditions, most importantly the pKa of the acid used as a source of protons, the solvent, the applied potential during electrocatalysis or the photosensitizer of the photocatalytic system.
The electronic structure computational methods can provide valuable information about the thermodynamic and kinetic aspects of the catalytic cycle, by calculating the relative free energies of possible stable intermediates and transition states.82 The proton transfer rate constants can be calculated using transition state theory or other theoretical models that include proton tunnelling.95,96 The calculation of electron transfer rate constants is based on the Marcus theory and includes the calculation of reorganization energies.97 The accuracy of a theoretical study is verified by the agreement of the calculated data with the corresponding observable values.
Electrochemical and spectroscopic methods provide the most important mechanistic insight into both photochemical and electrochemical proton reduction reactions.98 The onset potential of the catalytic peak reflects the driving force needed for proton reduction. The shape and behaviour of the catalytic peak under several experimental conditions, such as the acid concentration, scan rate and catalyst concentration, can provide valuable mechanistic insight. In the cases where a new reduction peak appears upon addition of an acid, electron transfer is associated with proton transfer, either via a PCET process or by a chemical step (C) followed by an electrochemical step (E). Furthermore, single-crystal X-ray structure determination of stable intermediates isolated from catalytic systems also provides invaluable insight into the structures of intermediates. Spectroscopic techniques are usually employed for in situ identification of catalytic intermediates or by-products under experimental conditions. Isotopic labelling studies are crucial for probing the catalytically active sites and the rate determining step of the catalytic cycle. In particular, the deuterium kinetic isotope effect (KIE = kH/kD), where kH is the rate of proton reduction and kD the rate of catalytic reduction of a deuterated acid, is considered an essential tool for the study of H2 evolution reaction mechanisms. A large KIE indicates a ligand centred process,99–101 while an inverse value KIE < 1 is consistent with a metal hydride intermediate.96,102
A multiproton and multielectron electrocatalytic reaction may involve several protonation and oxidation states with different solubility and stability. Since the system cannot be considered homogeneous if an intermediate species forms a material103 which deposits on the surface of the electrode and is responsible for catalytic performance, an understanding of catalyst–electrode interactions under electrocatalytic conditions is vital.87 A number of reports show that Ni–S and Co–S bonds are susceptible to decomposition. Cleavage of the C–S bonds of those complexes is also favored under the applied cathodic potential and/or acidic conditions.104,105 Usually, the diagnostic criteria employed to establish whether proton reduction is truly catalytic, or attributed to the deposited material are the rinse tests and the construction of Cottrell plots. During the rinse test, the working electrode is transferred from the catalyst solution, after a potential is applied for a period of time, to a fresh solution containing only the acid used as the proton source. When the ratio of the catalytic peak current under acid saturated conditions to the current in the absence of an acid shifts linearly with the square of the scan rate, the reaction rate is diffusion controlled and thus the reaction is thought to be homogeneous.
3.2 Basic concepts on evaluating photocatalytic and electrocatalytic systems
Comparative evaluation of the performances of different catalytic systems is possible via the use of certain key parameters. The turnover number (TON) represents the total number of moles of H2 formed per mole of the catalyst (eqn (1)), either electrocatalytically using bulk controlled potential electrolysis (CPE) or upon irradiation of a photocatalytic system. The turnover frequency (TOF) is defined as the TON per unit of time (eqn (2)):| TON = nH2/ncat | (1) |
| TOF = TON/t | (2) |
Faradaic efficiency (FE) is given by eqn (3) and can be used to estimate the amount of electric potential energy consumed in side reactions:
| FE = 200 F nH2/Q | (3) |
485 C mol−1), and Q is the total charge passed.76,79,106,107
The relatively low TON, with respect to the TOF, usually observed with controlled potential electrolysis (CPE) experiments, is attributed to the different methods used to estimate the two parameters. For the TOF estimation only the catalyst at the electrode surface is taken into account, but the bulk concentration of the catalyst is used to calculate the TON values.
The overpotential (η)106,108,109 of a proton reduction electrocatalyst is obtained by subtracting the standard reduction potential, EH+/H2, of the H+/H2 couple under the working conditions from the onset potential, E (eqn (4)), and it represents the driving force required for the H2 formation beyond the thermodynamic potential:
| η = E − EH+/H2 | (4) |
For a HER catalyst to be considered efficient it should exhibit a large catalytic current at a small overpotential. The overpotential at which a catalyst operates is the most commonly used measure of its activity.110
Quantum yield (Φ) is defined as the ratio of the number of electrons used for proton reduction to the number of photons absorbed by the system per unit of time (eqn (4)) and, as with faradaic efficiency, it is a measure of the energy loss pathways of the photocatalytic process.
| Φ = 2H2 (moles per s)/(absorbed light intensity/ Einstein per s) |
In summary, research is directed towards the development of noble metal-free, robust molecular catalysts that work efficiently in aqueous solutions, with small overpotentials and high faradaic yields and turnover frequencies.
3.3 Hydrogenases
Hydrogenases are a group of metal-containing enzymes that catalyse the H2 oxidation or production reaction. Their structures serve as an inspiration for the design of bioinspired synthetic catalysts. Several hydrogenases mimicking metal complexes based on Earth-abundant metals have been reported since the unraveling of the structure of hydrogenases from bacteria such as Desulfovibrio gigas and Clostridium pasteurianum.111–117 Three main classes of hydrogenases are known; these are [NiFe]-hydrogenases, [FeFe]-hydrogenases and [Fe]-hydrogenases. Only bimetallic [NiFe] and [FeFe] hydrogenases can catalyze the reversible H2 oxidation into protons and electrons, while [NiFe] hydrogenases are more proficient in H2 oxidation and [FeFe] hydrogenases are more efficient H2 producers. However, a major disadvantage in using hydrogenases for H2 production is their inactivation in the presence of O2.6 Even though the detailed reaction pathway and intermediate structures are still under debate, in depth studies of the mechanistic aspects of these natural systems have revealed repeating patterns of metal–ligand cooperative catalytic activity that guide the design of thiolate ligands for hydrogen evolution catalysts.The active sites of [FeFe]-hydrogenases consist of a binuclear Fe–Fe active site, in which each Fe ion is coordinated by two inorganic ligands composing a [Fe(CN)(CO)] moiety. Two thiolates of cysteine residues and an additional CO ligand bridge the two metals. Furthermore, one Fe center is coordinated by a thiolate of the cysteine residue in a terminal fashion. The two bridging sulphur atoms are connected by a pendant amine that interacts with the surrounding protein environment through hydrogen-bonding interactions. The pendant amine acts as an internal base and relays protons to the metal center to facilitate the heterolytic formation of the H–H bond between the metal-hydride and the amine proton (Scheme 5). Initial proton and electron transfer, followed by a proton migration to a Fe atom, leads to the Fe(II)-hydride118 intermediate, which was spectroscopically characterized.119,120 Subsequently, the amine is reduced121 and accepts another proton. The H+ component of the pendant amine and the H− of the Fe(II)-hydride are positioned within a favourable distance for heterolytic coupling to form the H–H bond.122
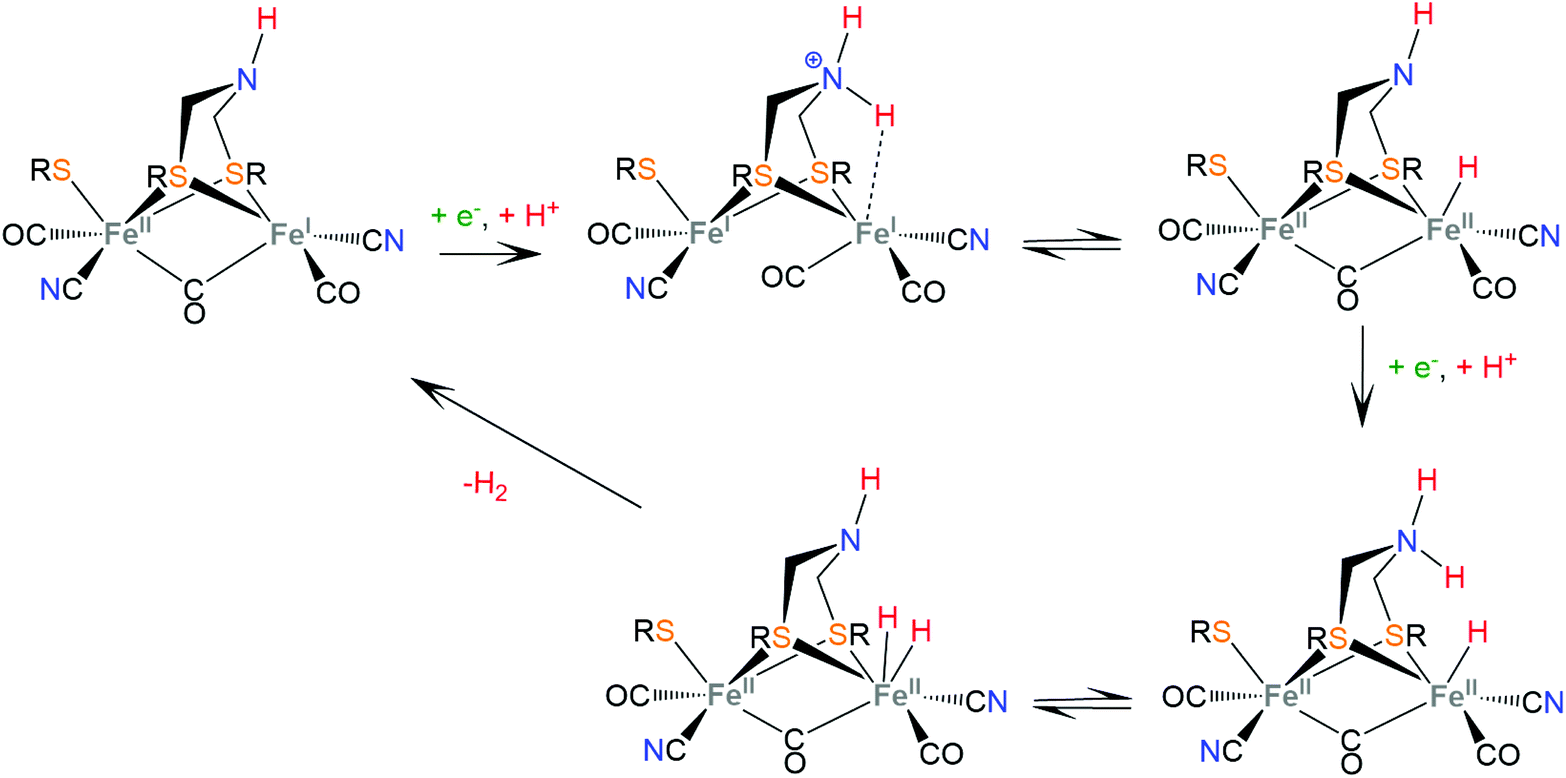 | ||
| Scheme 5 Proposed mechanism for H2 evolution from [FeFe] hydrogenases. | ||
The active sites of [NiFe]-hydrogenases are composed of a bimetallic four-member ring that connects the Ni and Fe metal centers via two thiolate cysteine residues. Two exo-cyclic thiolate cysteine residues are bound to Ni and the Fe ion is coordinated by three inorganic ligands composing a [Fe(CN)2(CO)] moiety. The low-spin Fe(II) ion is redox inactive throughout catalysis, whereas Ni changes its oxidation state ranging from Ni(III) to Ni(I). In the proposed proton reduction mechanism, a NiII/NiI reduction is accompanied by a terminal thiolate protonation (Scheme 6). The sulphur donor acts as a proton relay and transfers the proton to the Ni center, to form a Ni(III)-hydride, where the hydride takes a bridging position between the two metal ions, attributed to spontaneous rearrangements of the electron density.123 A second protonation of the sulphur donor and a nickel centered NiIII/NiII reduction lead to heterolytic H–H bond formation in the Ni center. Notably, synthetic [NiFe]-hydrogenase mimics operate via a Fe(II)-hydride formation, even though in natural systems the Ni-hydride pathway seems to be favorable.124,125
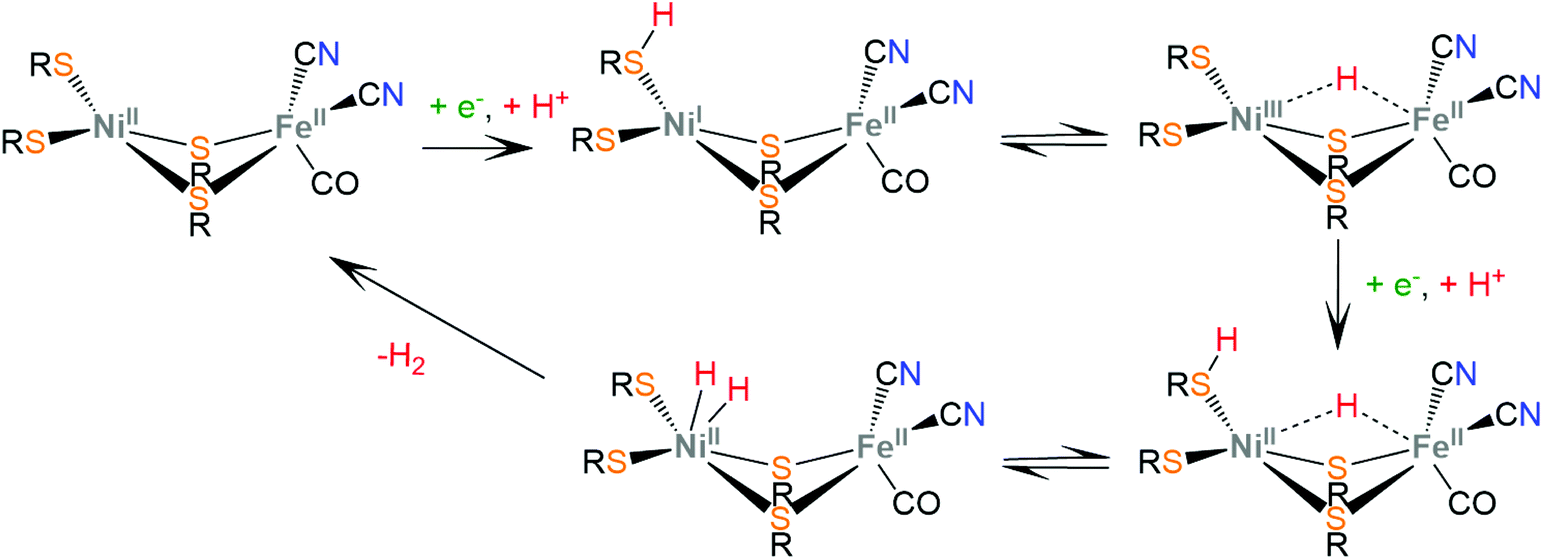 | ||
| Scheme 6 Proposed mechanism for H2 evolution from [NiFe] hydrogenases. | ||
Synthetic hydrogenase mimics have been recently reviewed.2,9 To sum up, nature achieves the reversible proton reduction reaction employing a system that avoids the formation of Fe(0) or Ni(0) oxidation states, contains several groups available to act as proton relays for hydride formation and avoids high overpotential values by combining proton and electron transfer steps into a single PCET step,126 resulting in the net transfer of two hydrogen atoms.
4. Mononuclear complexes
4.1 Homoleptic complexes with dithiolene ligands
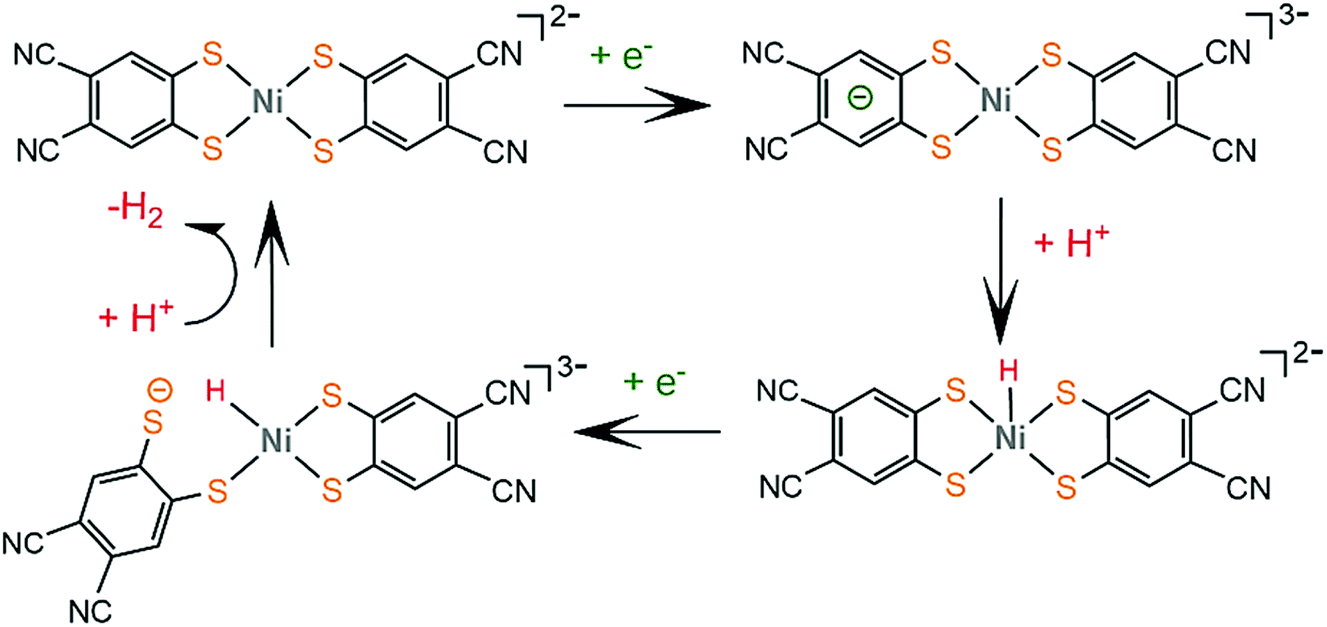
Bis-dithiolene complexes exhibit exceptionally high rates of hydrogen generation and total turnover numbers (TONs). Among the earliest reported dithiolene catalysts for hydrogen generation in aqueous media were the monoanions of the aryl-dithiolene complexes of W(VI).77 In 2012, Holland and Eisenberg reported that the Co(II) 1,2-benzenedithiolate monoanion (1) catalyzes the production of H2 from protons achieving >2700 TONs and an initial TOF of 880 h−1 in 1:1 water–acetonitrile mixtures with [Ru(bpy)3]2+ as the photosensitizer and ascorbic acid as the sacrificial electron donor, under photocatalytic conditions.127 The complex is also an active electrocatalyst in the presence of trifluoroacetic acid (TFA) or toluenesulfonic acid (TsOH) proton donors at −1.01 V vs. Fc+/Fc, respectively, glassy carbon working electrode (GCE). Introduction of various groups on the bdt aromatic ring led to the cobalt complexes 4, 7 and 11, which also exhibit catalytic activity for proton reduction on similar systems.128 Complex 11 is the most active photocatalyst, achieving almost 9000 TON, with an initial TOF of 3400 h−1, while 7 reaches 6000 TON and 4 2300 TON, under optimal photocatalytic conditions. Complex 11 exists as a dimer in the solid form, but upon reduction at −0.04 V vs. SCE it forms a dianionic monomer. A particularly interesting result of this study is that 11 shows the highest overpotential, even though it functions best as a photocatalyst and it has electron withdrawing ligands that anodically shift its reduction potential, suggesting that it follows a different reaction pathway. This was explained by calculating the pKa values of the four catalysts in their dianionic forms using density functional theory methods.129 The authors suggest that catalysts 1, 4 and 7 follow an ECCE pathway, where a metal centred CoIII/II reduction is followed by two protonations of adjacent sulphur ligating atoms, which leads to easily reduced doubly protonated species (Scheme 7).
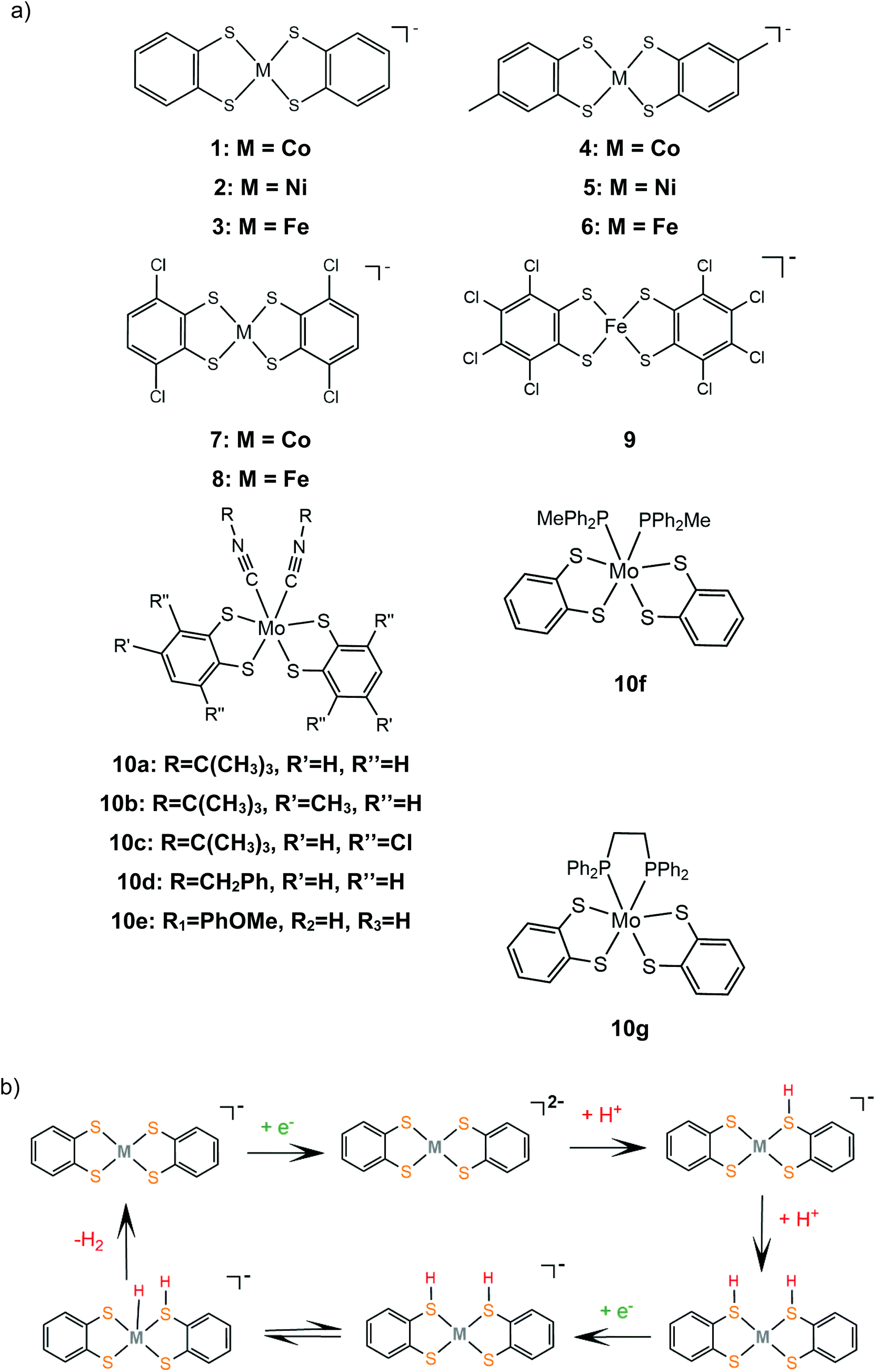 | ||
| Scheme 7 (a) Metal complexes [M(bdt)2] acting as HER catalysts. (b) Proposed ECCE mechanism catalysed by complexes 1, 2 and 3. | ||
In contrast, catalyst 11 follows an ECEC pathway, because the S atoms of the electron withdrawing maleonitrile-2,3-dithiolate ligand are less nucleophilic and only one of them is likely to be protonated after one electron reduction of the complex (Scheme 8). The monoprotonated complex shows a more cathodic reduction potential than its doubly protonated analogues. The last step of both proposed catalytic cycles is intramolecular proton transfer from a sulphur atom to CoI to form a hydrogen evolving Co(III)-hydride. Notably, a study in 2017 proved that upon addition of an acid, complex 1 gets adsorbed onto the glassy carbon electrode, though on reducing the applied potential the molecular protonated species regenerate. This indicates that electrode-adsorbed species are a catalytic intermediate in the proton reduction catalytic cycle of 1.130
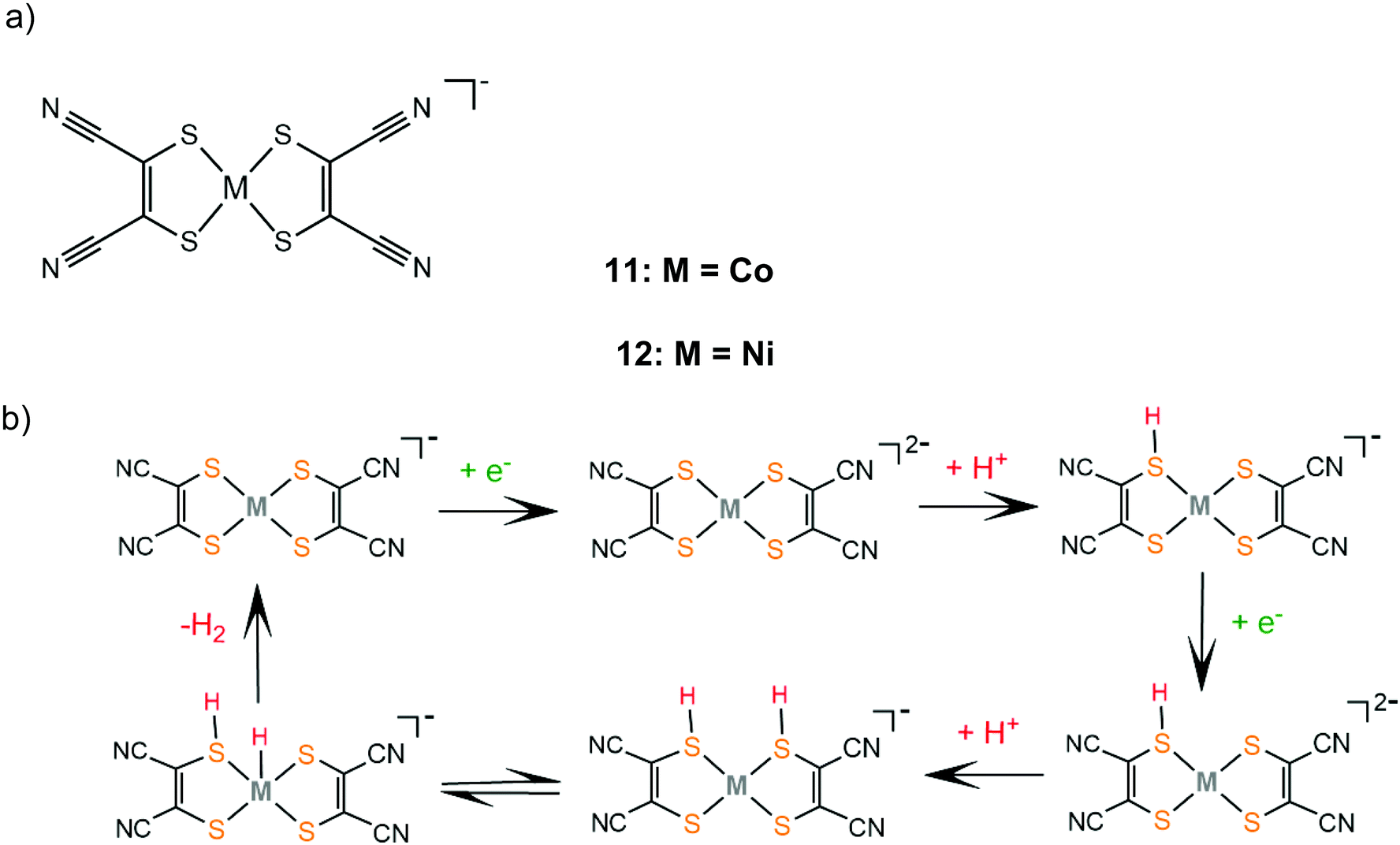 | ||
| Scheme 8 (a) Metal complexes [M(mnt)2] acting as HER catalysts. (b) Proposed ECEC mechanism catalysed by complexes 11 and 12. | ||
Similar reaction pathways have been reported for other metal benzenethiolates 2, 3, 5, 6, 8 and 9 and maleonitrile-2,3-dithiolates 12 and 13. The Ni(II) 1,2-benzenedithiolate 2 has been reported by Mitsopoulou and Artero to reduce protons in acetonitrile at −1.42 and −1.68 V vs. Fc+/Fc with a faradaic efficiency of 71% (TON: 113), in the presence of TFA (pKa = 12.7), after 3 h CPE with an applied potential of −1.2 V vs. Fc+/Fc.131 In the presence of the weaker acid triethylammonium tetrafluoroborate (pKa = 18.6) no activity was observed. Furthermore, catalysts 5 and 12 were also evaluated as possible proton reduction electrocatalysts under the same conditions. Catalyst 5 achieves a TON of 158 and an overpotential of 0.467 V with a faradaic yield of 88%. Theoretical calculations reveal that for catalysts 2 and 5 ECEC and ECCE mechanisms take place simultaneously. However, the ECCE pathway is strongly unfavourable thermodynamically for complex 12 and it is the least active electrocatalyst, with an overpotential of 0.567 V and a TON of 6 (Scheme 8). They also investigated the acid induced decomposition of the catalysts to exclude the possibility of heterogeneous catalysis. The linear dependence of the catalytic current on the square root of the scan rate and rinse test results indicate that complexes 2 and 5 are molecular electrocatalysts in the presence of TFA, even though the system becomes heterogeneous in the presence of the stronger acid 4-bromo-anilinium tetrafluoroborate (pKMeCNa = 9.43) because of the electrodeposition of a Ni–S film on the electrode surface.131,132
In 2016, Eisenberg and coworkers described the Fe(II) 1,2-benzenedithiolate (3)133 and 6, 8 and 9 derivatives, which have been investigated as homogeneous photocatalysts for water reduction using water-soluble CdSe QDs as the photosensitizer and ascorbic acid as the sacrificial electron donor in a 1:1 ethanol–water solution, with maximum efficiencies of 29400, 20600, 15200 and 8000 TON, for 6, 3, 8 and 9, respectively.134 The complexes were isolated as dianionic dimers, as shown by single-crystal X-ray diffraction structure determination. In the absence of an acid, the CV of 3 exhibited a reversible wave with E1/2 = −0.723 V vs. SCE, attributed to the reduction of the dimer to generate the corresponding catalytically active dianionic monomer.135 When titrated with TFA in acetonitrile solution containing 1 M H2O the reversible reduction wave became catalytic, indicating that the catalytic cycle starts with an electrochemical step. In an earlier study, Sellmann and coworkers,133 using 1H NMR and electrochemical experiments, demonstrated that when the dianionic monomer is protonated on a ligating sulphur atom, a subsequent protonation on another ligating sulphur or on the metal center takes place. This suggests that 3 also follows the proton reduction mechanisms depicted in Scheme 7.
Eckenhoff and Eisenberg reported seven hexacoordinated Mo(IV) complexes, 10a–g, bearing two bdt chelates and two additional isocyanide or phosphine ligands to complete the coordination sphere.136 In the cyclic voltammograms of the complexes in aqueous acetonitrile the MoIV/MoIII and MoIII/MoII couples are observed and in the presence of TFA the second peak becomes catalytic. During the metal centered reductions, the additional isocyanide ligands dissociate, as observed from 1H NMR data, and [Mo(bdt)2]2− is formed, which evolves H2 after two sequential protonations. Thus, complex [Mo(bdt)2] is the active catalyst in the H2 evolving catalytic cycle. The complexes are active photocatalysts in the presence of [Ru(bpy)3]2+ as the photosensitizer and ascorbic acid as the sacrificial electron donor. Since complexes 10a–g act as precatalysts and the active dithiolene catalysts are generated in situ, the relative efficiencies are determined from their reduction potentials and their stability. Isocyanide substituted catalysts 10a, b and d show similar catalytic efficiencies, reaching 520, 475 and 455 TON, respectively, after 24 h of irradiation, while 10c and e, whose reduction potential is anodically shifted by ∼250 mV, reach 100 and 260 TON, respectively. Complexes 10f and g, bearing phosphine ligands, reach 135 and 402 TON, respectively, even though they show the highest initial rates of hydrogen production, which indicates fast decomposition.
As is evident from the above references, incorporation of electron-accepting moieties on dithiolene ligands leads to reduced electron density on the ligating sulphur atoms; thus it anodically shifts the reduction potentials of the respective complexes. However, both complexes 11 and 12 display higher overpotentials than their corresponding cobalt and nickel benzenedithiolate analogues, because they follow an ECEC reaction pathway, in contrast to the ECCE pathway followed by 1, 2 and 3 (Schemes 7 and 8).
In an early study by Sakai and coworkers, the dinuclear iron-maleonitriledithiolate complex 13a was reported to exhibit catalytic activity for the electrocatalytic reduction of protons in aqueous acetate buffer solution with an overpotential of 0.560 V.137 Electrochemical and X-ray diffraction experiments showed that the dimer 13a gets adsorbed over the glassy carbon electrode surface and the adsorption efficiency depends on the counterion. After 5 h of CPE at an applied potential of −1.2 V vs. SCE, the adsorbed catalyst achieved a TON of 3900 and a faradaic yield of 99%. Later, the dianionic iron maleonitrile dithiolate complex 13b was presented and its structure was crystallographically determined.138 The thiophenol moiety completes the coordination sphere of the Fe(III) center and the complex takes a distorted square pyramidal geometry, where a thiophenol benzene hydrogen atom interacts with two ligating sulphur atoms. Complex 13b catalyses proton reduction at a relatively low reduction potential of −0.309 V vs. Ag/AgCl (−0.67 V vs. Fc+/Fc) in acetonitrile and at −0.53 V vs. Ag/AgCl in water with TsOH as a source of protons. Based on UV/Vis and EPR experiments the authors propose that in the presence of TsOH two sulphur ligating atoms of the maleonitrile thiolate and thiophenol ligands are protonated and an H2 molecule can be evolved (Scheme 9).
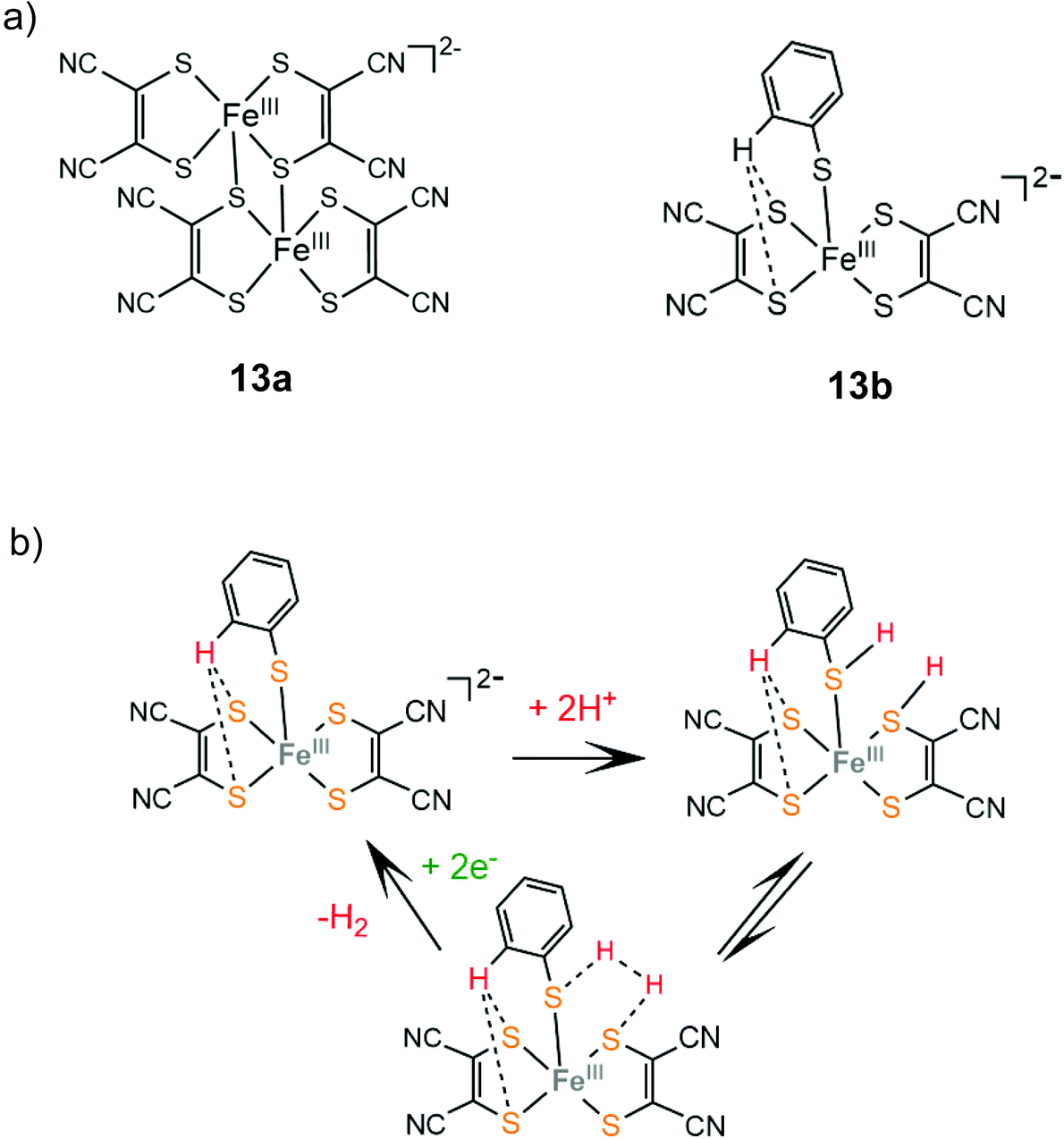 | ||
| Scheme 9 (a) Molecular structures of iron dithiolate complexes 13a and 13b. (b) Proposed mechanism catalysed by complex 13b. | ||
The radical nature of certain dithiolene ligands plays an important role in lowering the reduction potential of the respective complexes. In an early study by Sarkar and coworkers, complex 14 bearing 1,2-dicarbomethoxyethylene-dithiolate ligands was synthesized and spectroscopically characterized and its electrocatalytic activity in proton reduction was evaluated.139 In the presence of TsOH in acetonitrile, a ligand centered reduction at −0.341 V vs. Ag/AgCl (−0.71 V vs. Fc+/Fc) is followed by a catalytic peak at −0.69 V vs. Ag/AgCl, attributed to the protonated catalyst. Crystallographic characterization of complex 14 revealed that one of the ligands exists as a radical, which may facilitate reduction. Experimental characterization of catalytic intermediates using various spectroscopic techniques revealed that the ligating sulphur atoms are the protonation sites of the catalyst.
The group of Fontecave incorporated the dimercaptomaleonitrile and 1,2-dicarbomethoxyethylene-dithiolate ligands to tungsten oxo complexes.140 Catalysts 15 and 16 can evolve hydrogen at −1.83 V vs. Ag/AgCl reaching TOF values of 63 and 143 s−1, respectively, in the presence of acetic acid (pKMeCNa = 22.3) as a proton donor in acetonitrile. The complexes are also active photocatalysts achieving TONs of 12 and 35, for 15 and 16, respectively, when coupled with [Ru(bpy)3]2+ as a photosensitizer and ascorbic acid as the sacrificial electron donor in a 1:1 acetonitrile–water solution. Electrochemical experiments showed that under acid titration a new reduction peak appears at −1.2 V, attributed to PCET on one of the oxo ligands. The catalytic system was also investigated computationally in order to elucidate the detailed reaction pathway (Scheme 10). DFT results suggest that hydrogen formation takes place exclusively in the W-oxo unit, after four consecutive PCET events, which lead to abstraction of one oxo ligand and formation of a metal hydride.
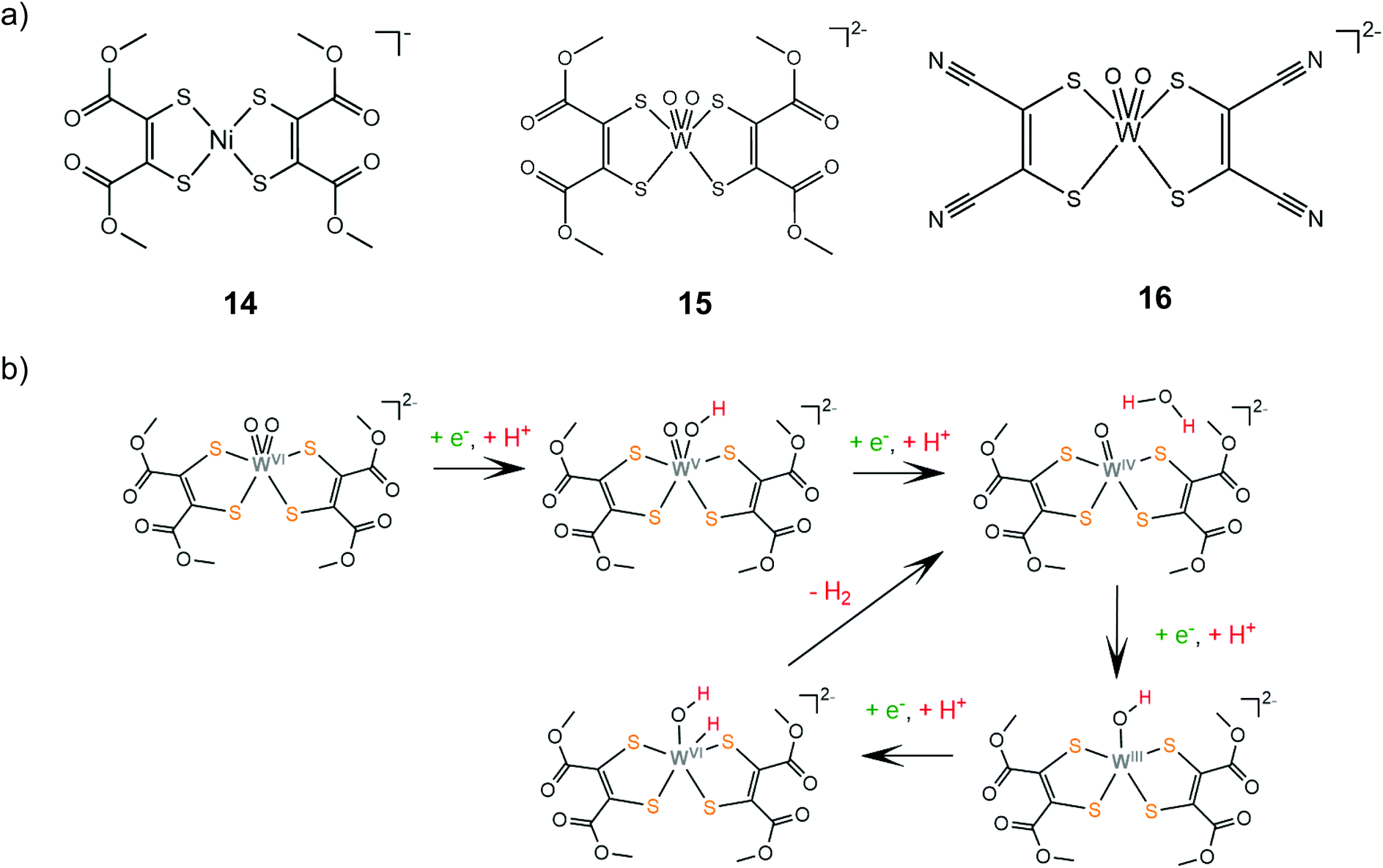 | ||
| Scheme 10 (a) HER catalysts carrying 1,2-dicarbomethoxyethylene-dithiolate ligands. (b) Proposed mechanism catalysed by complex 15. | ||
The proton reduction mechanistic steps of cobalt and nickel bis-aryldithiolene complexes, 17 and 18, were thoroughly examined experimentally and theoretically, elucidating the role of the metal center in the thermodynamically preferred reaction pathway. A series of cobalt bis-aryldithiolene complexes, 17a–c, have been reported to reduce protons in DMF when anilinium tetrafluoroborate (AnBF4, pKDMFa = 4.3) is added under applied potentials of −1.46 V, −1.37 V and −1.45 V, for 17a, b and c respectively.141 The catalytic cycle begins with a metal centred reduction to afford the dianionic complex and a subsequent Co-hydride formation (Scheme 11). A ligating sulphur atom acts as a proton relay and transfers a second proton to the metal, leading to a Co-dihydrogen transition state.
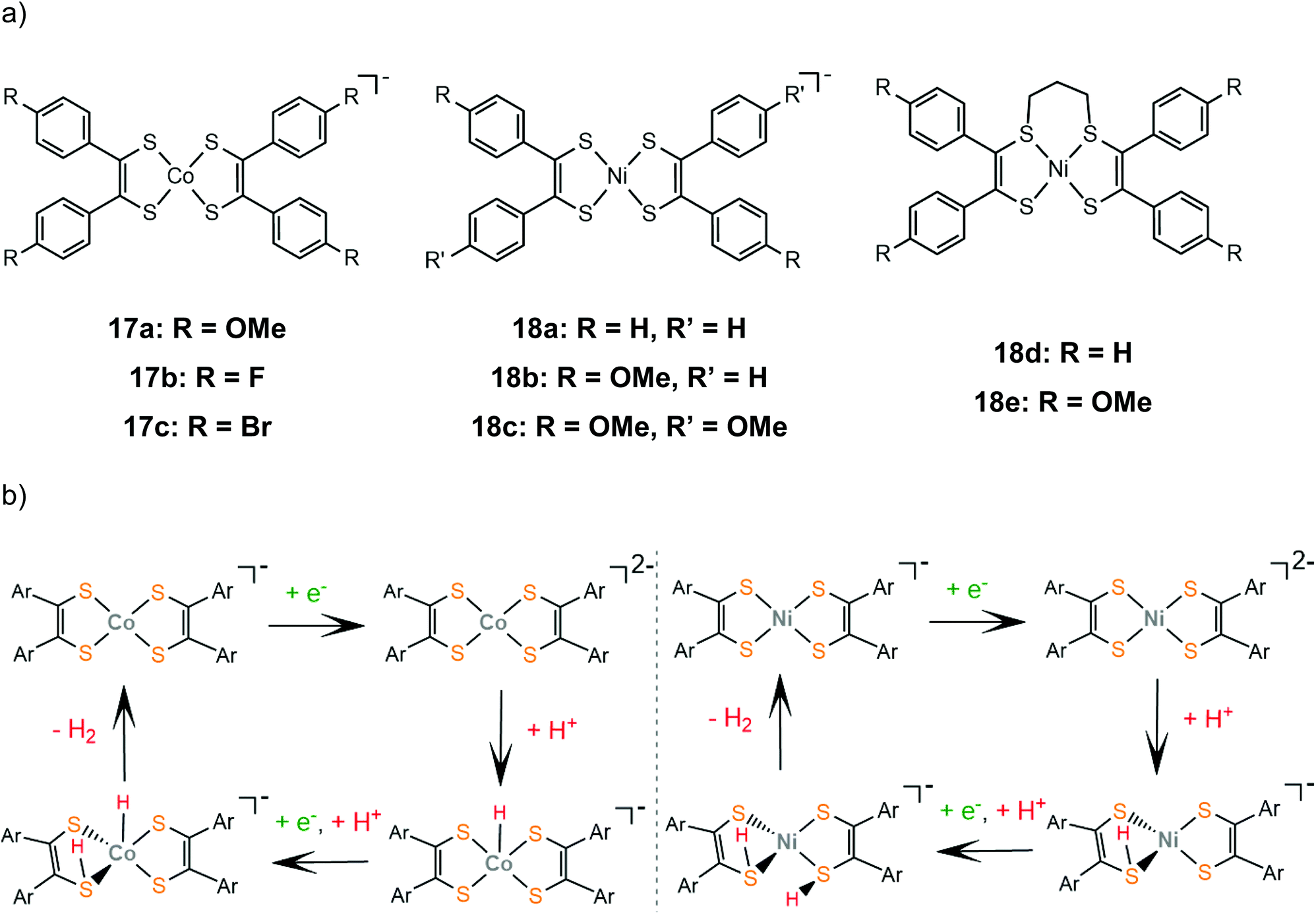 | ||
| Scheme 11 (a) Structures of aryl-benzene complexes. (b) Proposed mechanisms catalysed by complexes 17a (left) and 18a (right). | ||
Later, the electrocatalytic activity of a series of nickel complexes, 18a–c, was studied by Mitsopoulou and Artero.142 The molecular catalysts reach 15, 25 and 12 TON, for 18a, b and c, respectively, in a 3 h period of CPE in DMF and in the presence of TFA as a source of protons, while faradaic yields range between 66 and 83%. In contrast to the cobalt analogues, DFT calculations show that the dianionic species is preferably protonated on a sulphur atom (Scheme 11). The second protonation also takes place on a sulphur donor, to generate the hydrogen evolving species, without the formation of a stable nickel hydride. In a later study by the same group, the sulphur-alkylated nickel bis-aryldithiolene complexes, 18d and 18e, were also investigated to elucidate the effect of the propyl group on the electronic structure and catalytic properties of nickel dithiolenes.143 The optimized photocatalytic system containing 18d and 18e as proton reduction photocatalysts in an acetonitrile–water mixture in the presence of [Ru(bpy)3]2+ as a photosensitizer and ascorbate as a sacrificial electron donor, reached TON of 25 for 18d and 73 for 18e, respectively. Spectroscopic and electrochemical observations suggest that alkylation at two adjacent sulphur sites stabilize the reduced forms of the dithiolene ligands, increasing the electron density on the NiS4 core.
As has already been observed, it is possible to tailor the redox properties of a dithiolene catalyst by choosing appropriate substituents on the bidentate dithiolene ligands. It has been shown that increasing the conjugation of the aromatic systems can stabilize the reduced state of a metal complex, because it induces greater charge delocalization away from the metal. The extension of the dithiolene ligand π-system was investigated using the Co and Ni complexes of (R)-1,1′-binaphthalene-2,2′-dithiol, 19 and 20.144 Both complexes catalyze proton reduction when a acetonitrile/water solution of complex 19/20, eosin photosensitizer and TEA as a sacrificial electron donor is irradiated under visible light. The authors optimized the photocatalytic system reaching maximum hydrogen TONs of 495 for complex 19 and 676 for 20. Electrochemical investigation of the catalysts in the presence of TFA shows that initial reduction at −0.72 and −0.68 V vs. SCE, for complexes 19 and 20, respectively (−1.04 and −1.00 V vs. Fc+/Fc), is followed by a chemical protonation step, suggesting an ECEC pathway. Indeed, extension of the π-system lowers the 19/20− → 19/202− reduction potentials, when compared to those of complexes 17a and 18a.
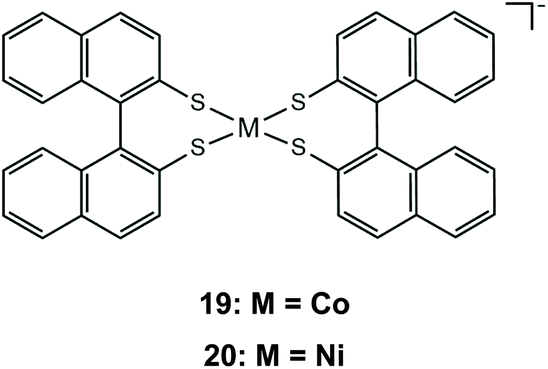
Complexes 21 and 22 are active proton reduction photocatalysts, achieving turnover numbers of 50 and 500, respectively, when combined with the [Ru(bpy)3]2+ photosensitizer and ascorbic acid as a sacrificial electron donor in an acetonitrile–water 1:1 solvent mixture. Complex 21 is more effective when combined with TEOA as both a proton donor and a sacrificial electron donor in acetonitrile, reaching a TON of 190 in 5 h with an initial TOF of 163 h−1. Bulk electrolysis of catalyst 21 in acetonitrile and in the presence of acetic acid at an applied potential of −1.60 V vs. Ag/AgCl (−1.96 V vs. Fc+/Fc) for 22 h resulted in 700 TON of H2 with a faradaic efficiency of 90% and a TOF of 5570 s−1.146 Complex 22 is also an active electrocatalyst for proton reduction under the same conditions with the catalytic peak at −1.30 V vs. Ag/AgCl, achieving a TOF value of 1030 s−1 with a faradaic yield of 92% after 1.5 h.147 DFT calculations provide insight into the mechanism for proton reduction by catalyst 21 (Scheme 12). Firstly, two metal centered reduction events, concomitant with the protonation of a quinoxaline nitrogen atom, take place, to generate the active catalyst. Next, the complex is further reduced and protonated to afford a Co(II)-hydride. A ligating sulfur atom acts, once again, as a proton relay and transfers a solution proton to Co, which results in a Co-dihydrogen low energy (+5.8 kcal mol−1) transition state.146
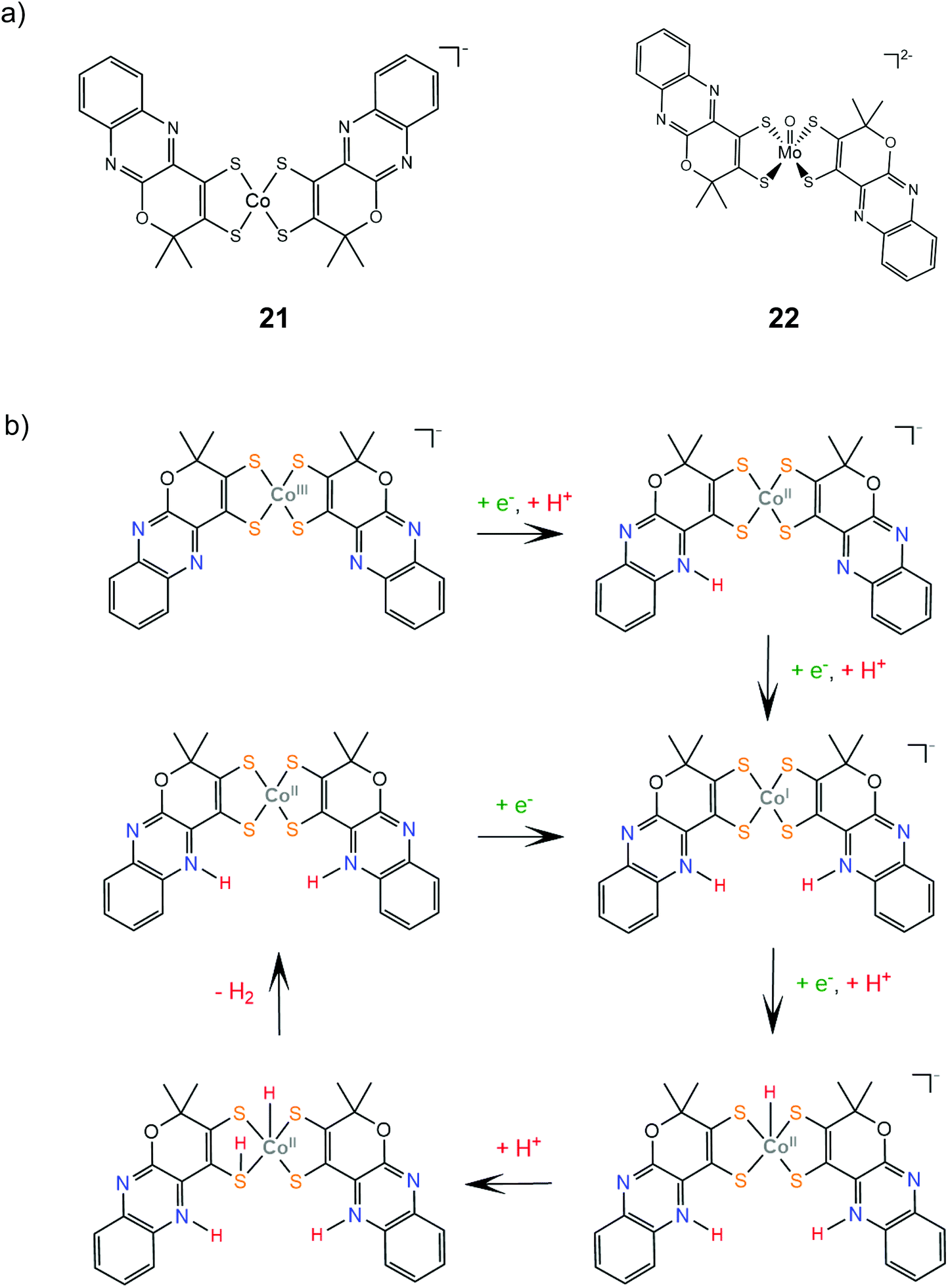 | ||
| Scheme 12 (a) Molecular structures of complexes carrying quinoxaline–pyran-fused dithiolene ligands. (b) Proposed mechanism catalysed by complex 21. | ||
The respective molybdenum complex, 22, follows a different catalytic pathway, since catalysis takes place mainly on the Mo-oxo site. The electrochemical data suggest that protonation takes place before complex reduction. Computational data reveal that the site of first protonation is a quinoxaline nitrogen atom, similar to complex 21, while the next steps involve two consecutive reduction and protonation steps, similar to W-oxo complex 15147 (Scheme 13).
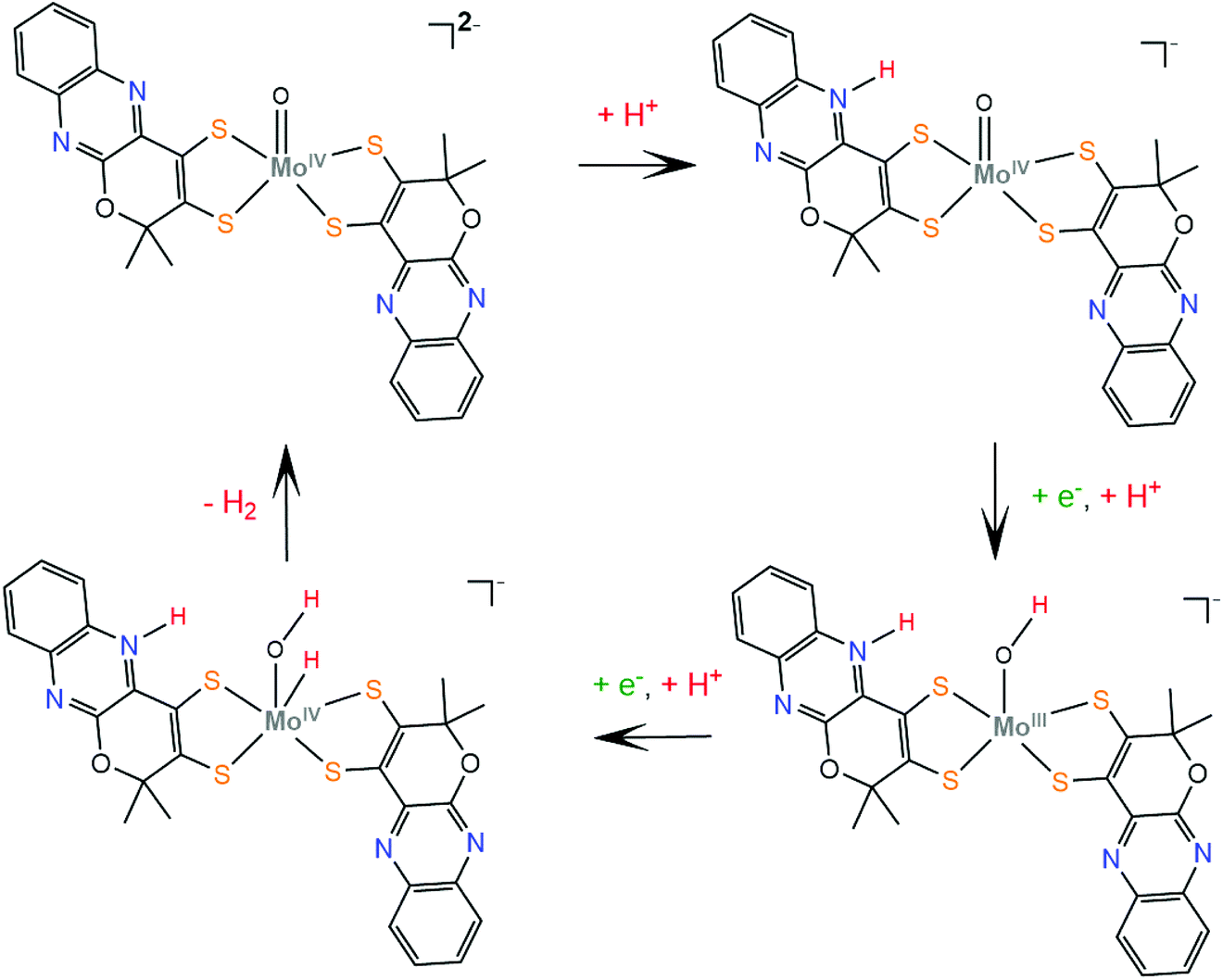 | ||
| Scheme 13 Proposed mechanism catalysed by complex 22. | ||
Sakai and coworkers studied extensively the ligand centered proton coupled electron transfer (PCET) processes in nickel dithiolene proton reduction electrocatalysts bearing pyrazine donors. Complex 23 is an efficient molecular electrocatalyst in aqueous solution achieving a TON of 20000 over 24 h CPE with the faradaic efficiency reaching 100% when the applied potential is −0.95 V vs. SCE (−1.27 V vs. Fc+/Fc).148 DFT calculations and electrochemical experiments showed that a protonation and two consecutive PCET events lead to protonation of three pyrazine nitrogen atoms, which facilitates a NiIII-hydride formation via an endergonic intramolecular proton transfer step149 (Scheme 14). In this way, the system avoids the formation of low valent NiI and Ni0 species achieving overpotential values smaller than 0.4 V at pH 5. The electrocatalytic hydrogen evolution mechanism of complex 24 was also investigated by the same group.150 The catalyst evolves hydrogen in a DMF and acetic acid solution at an applied potential of −2.5 V. The reaction is proposed to follow an ECEC pathway, involving an initial ligand centered reduction and subsequent Ni(III)-hydride formation (Scheme 15).
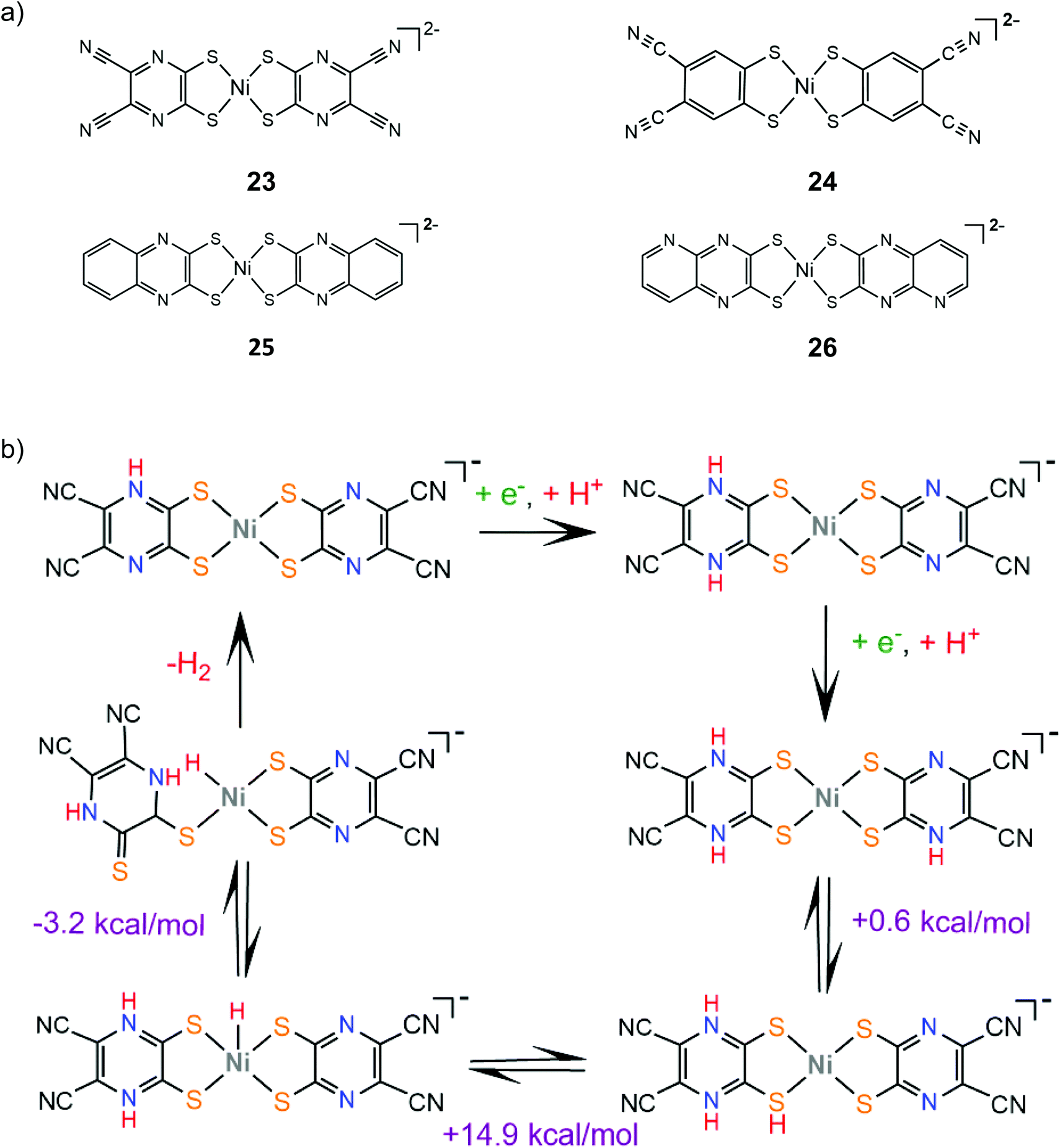 | ||
| Scheme 14 (a) HER catalysts carrying substituent qdt ligands. (b) Proposed mechanism catalysed by complex 23. | ||
 | ||
| Scheme 15 Proposed ECEC mechanism catalysed by complex 24. | ||
The large overpotential needed for catalyst 24 underlines the importance of pyrazine nitrogen atoms of catalyst 23, which promote the lower energy PCET process instead of the stepwise electrochemical and chemical steps. Although PCET processes are usually thermodynamically favorable, because they avoid high energy intermediates, leading to lower overpotentials, their activation energies depend on various factors including reactant and product geometry differences and inner/outer sphere reorganization energies.82,151 Thus, designing non-innocent ligands able to undergo efficient PCET events is important for the design of active catalysts.
The stability of the PCET state was computationally found to increase with the Hammett constant of the pyrazinedithiolate substituent group on a series of pyrazinedithiolate nickel complexes, attributed to increased charge delocalization.152 Complexes 25 and 26, where the π-system of the pyrazine ring is extended, were also synthesized and their catalytic performances in aqueous solutions were evaluated and compared.153 Indeed, complexes 25 and 26 achieve overpotential values of 0.17 V and 0.23 V, respectively, at pH 9.0, which indicates a large stabilization of the PCET state when compared to complex 23. The stabilization can be attributed to the formation of thioamide resonance structures, where the electron density is delocalized among the metal, the ligating sulfur atoms and the protonated nitrogen atoms.154 Notably, under higher overpotential conditions, the catalytic efficiencies of 25 and 26 increase considerably, possibly because different reaction pathways are thermodynamically favored under more negative potentials.153
4.2 Heteroleptic complexes with dithiolene ligands
Mechanistic investigation of homoleptic dithiolene HER catalysts shows that the strongly donating nature of the sulfur ligating atoms can facilitate metal hydride formation. Two heteroleptic complexes bearing the bdt ligand and a sterically demanding diphosphine were synthesized and studied as potential proton reduction electrocatalysts.155 Electrochemical characterization revealed that the NiII/NiI reduction potential of complex 27 is 0.75 V more cathodic than that of complex 28, which suggests that the electron donating ferrocene group destabilizes the reduced form of the catalyst. Complex 27 catalyzes proton reduction at an applied potential of −1.81 V in THF in the presence of acetic acid (pKTHFa = 22.48) as a proton source, reaching a TOF of 1240 and an overpotential of 0.27 V. DFT calculations suggest an ECEC pathway, involving a transition state where two hydrogen atoms are bound to the metal center (Scheme 16).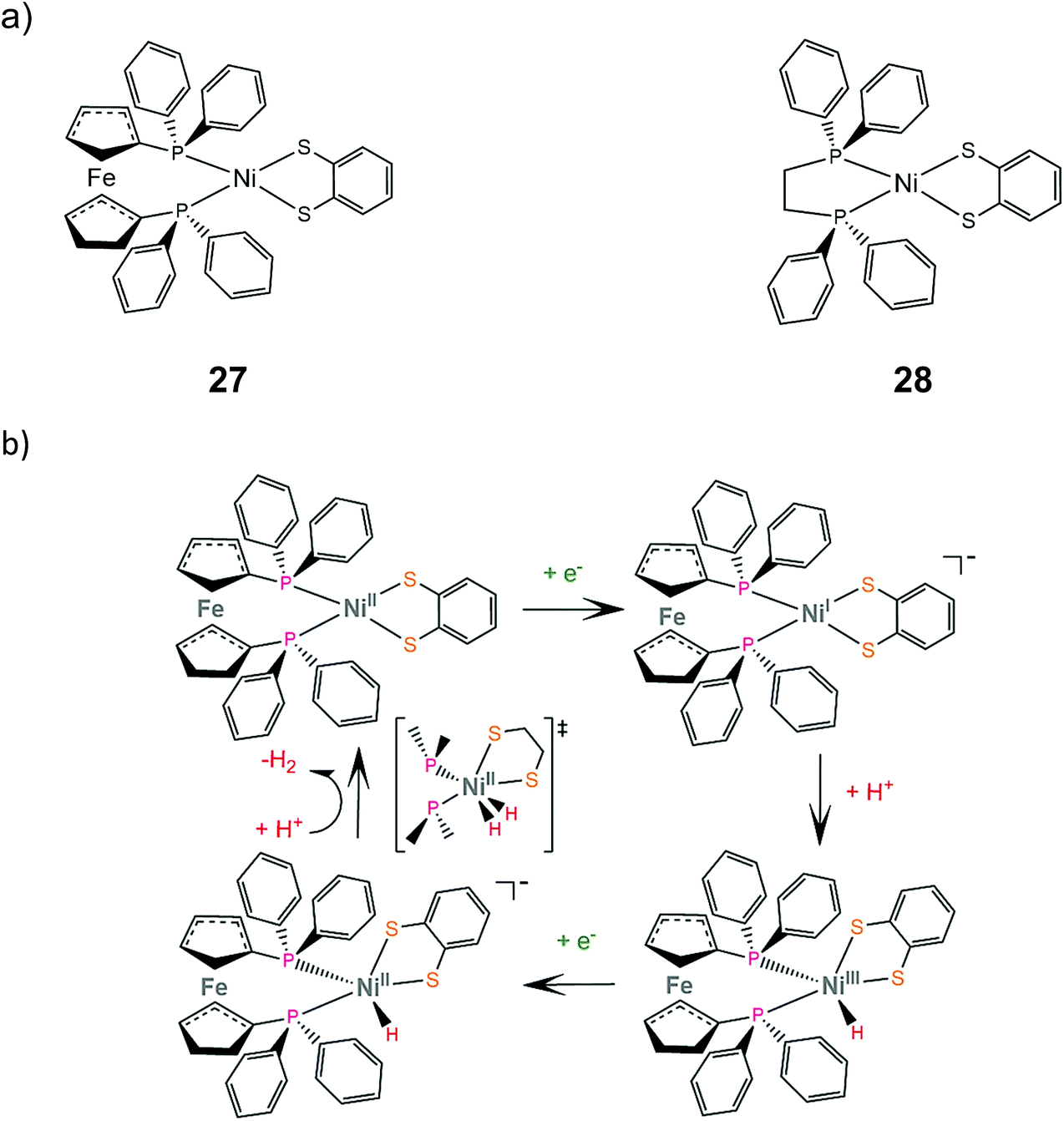 | ||
| Scheme 16 (a) Molecular structures of heteroleptic dithiolene–diphosphine complexes. (b) Proposed ECEC mechanism catalysed by complex 27. | ||
The electrocatalytic performance of the first heteroleptic diimine–dithiolene complex (29) was evaluated and the hydrogen evolution reaction pathway was compared to that of the corresponding homoleptic dithiolene complex 24.150 Complex 29 catalyzes proton reduction in DMF at potential values that depend on the strength of the acid used as a proton donor, in a range of −2.11 V for triethylammonium chloride (pKDMFa = 9.2) to −2.24 V for acetic acid (pKDMFa = 13.5). The dependence of the catalyst's overpotential on the proton donor pKa led the authors to propose an EECC pathway, where the initial metal centered reduction at −1.5 V is followed by a PCET process which results in the formation of a square planar Ni(II)-hydride (Scheme 17). On the basis of DFT calculations, this species can react with a proton in solution to achieve heterolytic hydrogen formation or it can be further reduced and follow an ECEC mechanism.
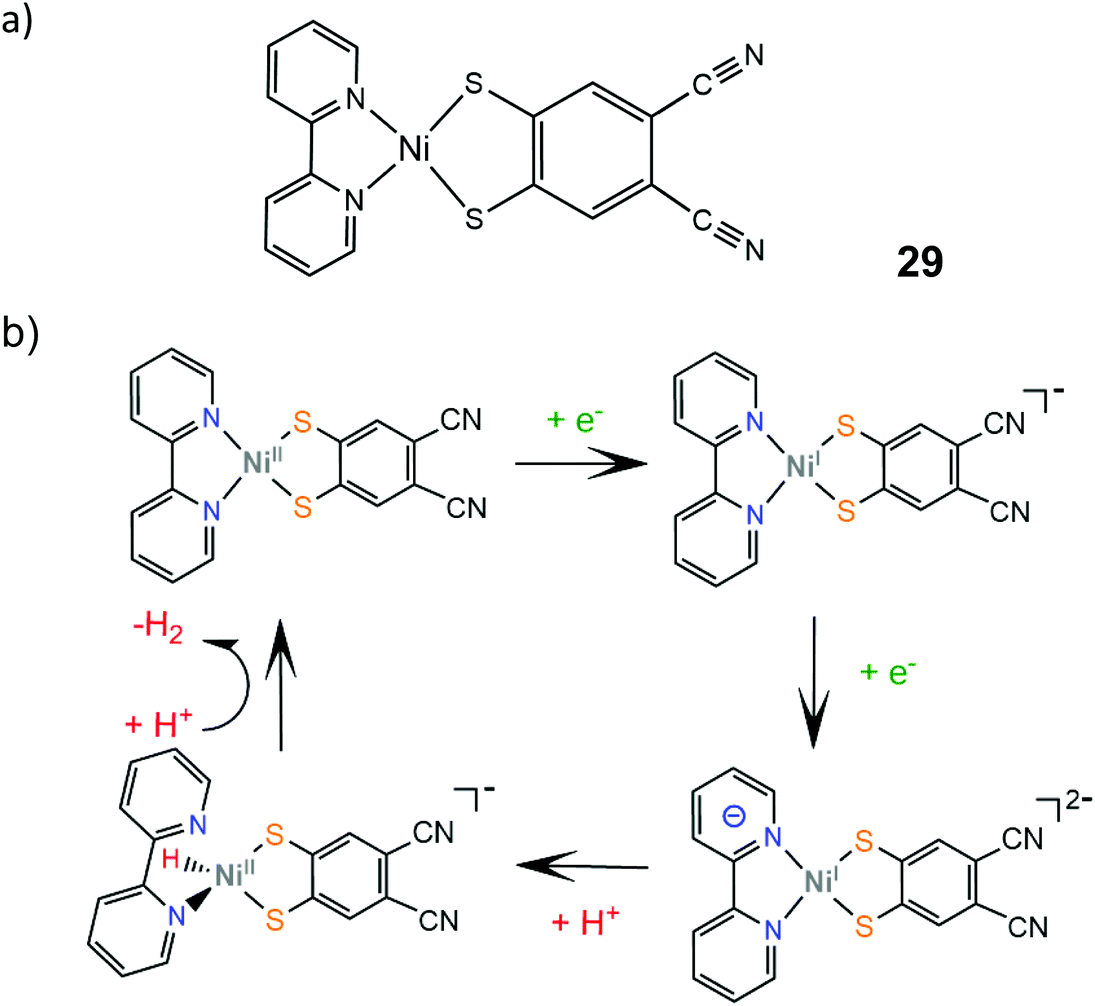 | ||
| Scheme 17 (a) Molecular structure of Ni(bpy)(dcbdt). (b) Proposed EECC mechanism catalysed by complex 29. | ||
4.3 SN- and SO-type bidentate ligands
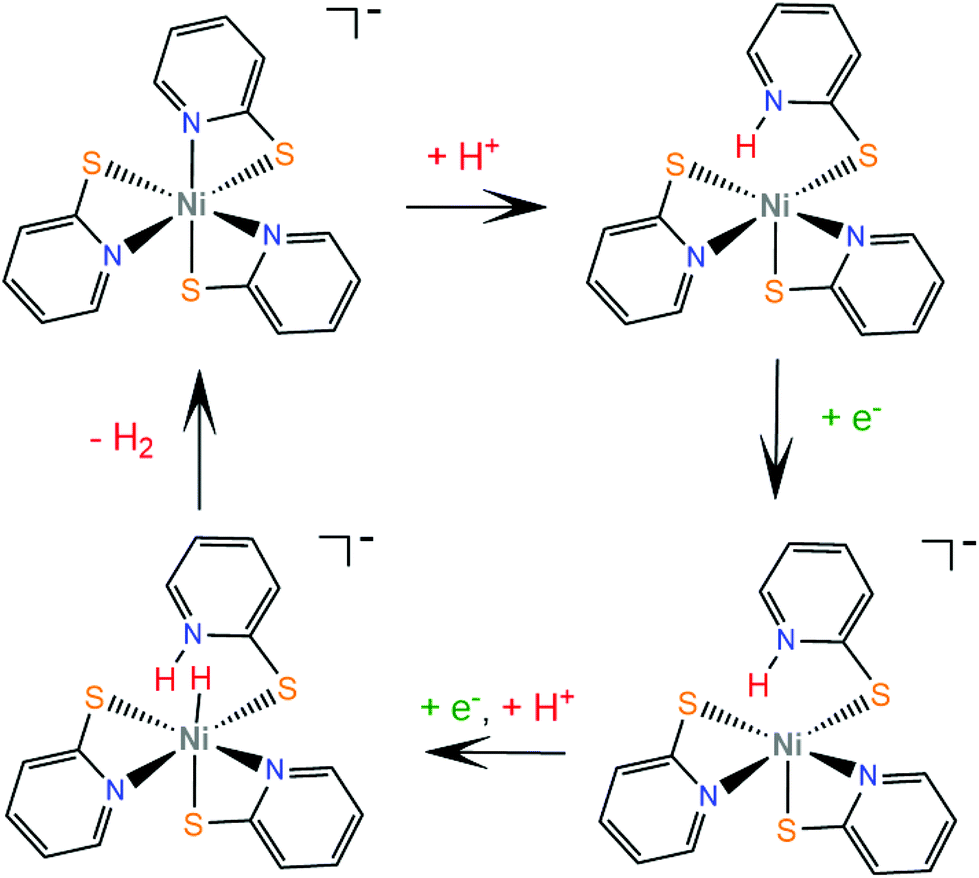
Numerous metal pyridine–thiolate complexes have been studied as HER catalysts (Scheme 18). Eisenberg and his group reported for the first time the use of pyridine–thiolate nickel(II) complexes as proton reduction catalysts.156 Irradiation of a solution of fluorescein, TEA and complex 30 in 1:1 ethanol–water at 520 nm for 40 h results in the production of 5500 TONs of H2 and an initial TOF of 250 h−1 under optimized conditions. At a high concentration of the sacrificial electron donor, a reductive quenching pathway is favoured, while at a low concentration, oxidative quenching dominates, increasing the system's lifetime. Cyclic voltammetry experiments and UV/Vis acid titration experiments showed that protonation of the catalyst initiates the catalytic cycle. Computational studies157 reveal that protonation of 30 preferably takes place at the pyridyl nitrogen, accompanied by dechelation of the protonated ligand (Scheme 19). The protonated complex undergoes a metal centred reduction and a subsequent PCET process results in the formation of a Ni-hydride, which evolves H2via a low energy transition state.
 | ||
| Scheme 18 Molecular structures of metal pyridine–thiolate HER catalysts. | ||
 | ||
| Scheme 19 Proposed mechanism catalysed by complex 30. | ||
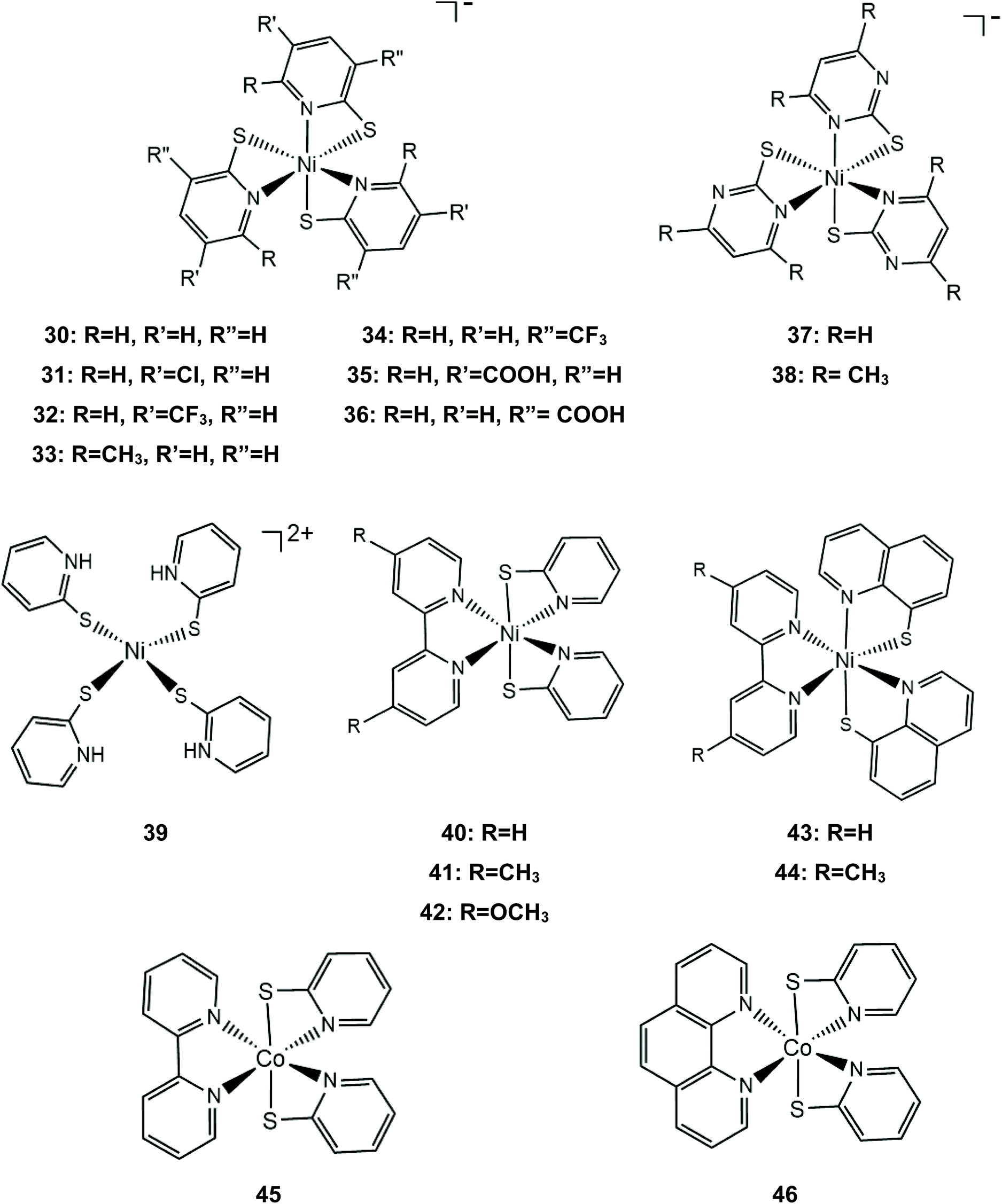
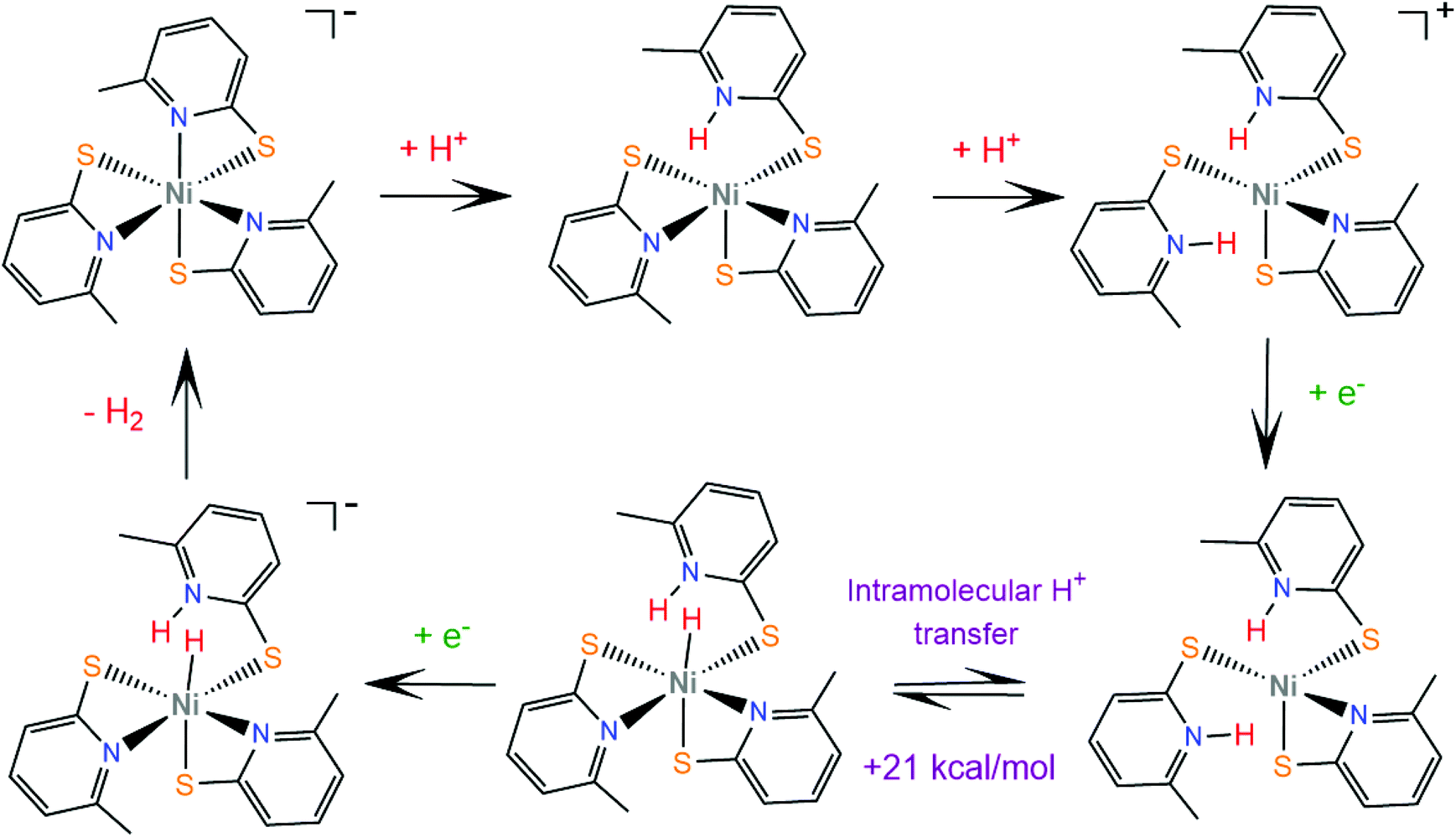
Later, the same group extended the study of pyridine–thiolate nickel complexes and presented the complexes 31–33 and 37–42.158 The catalytic ability for the photogeneration of hydrogen from water was examined on their optimized fluorescein-TEA system. The relative activities of the catalysts and their electrochemical behaviours provide insight into the mechanisms of the catalytic processes. Complex 41 achieves the highest TON of 7335 after 30 h of irradiation with a TOF of 312 h−1, complex 33 follows, reaching a TON of 5020 and a TOF of 317 h−1 and, notably, complex 37 is the least active with a TON of 1660 and a TOF of 103 h−1. It is evident that electron-donating moieties can enhance the catalytic activity of the nickel thiolate complexes. The cyclic voltammograms of the catalysts in 1:1 ethanol–water solutions show that electron donating moieties shift cathodically the reduction potential of the corresponding complexes. Computational studies by McCormick and her group suggest that the incorporation of electron donating substituents facilitates protonation of the pyridine nitrogen.159 Using DFT calculations, they showed that the highly efficient catalyst 33 does not follow the CECE mechanism of 30, but in the presence of acetic acid it undergoes a second protonation at another pyridine N atom (Scheme 20). Subsequently, the complex is reduced and an endergonic intramolecular proton transfer from the ligand to Ni, coupled with a second reduction, leads to a hydrogen evolving Ni hydride. Furthermore, they studied three pyridine thiolate nickel catalysts, 34–36, to investigate the interplay among pKa, reduction potential and catalytic efficiency with the σ-Hammett constant and the position of the moiety on the pyridine ring. Interestingly, the electrocatalytic activities of the catalysts follow the order 34 < 35 = 36, with 35 showing a smaller overpotential than catalyst 36. The pKa of the second protonation increases with the electron donating character of the substituent. Also, the position of the substituents on the pyridine ring is important only in the case of the –COOH moiety, as the ortho-COOH substituted pyridine–thiolate complex 36 shows a much larger calculated pKa value than the corresponding para-COOH complex 35, because the carboxyl oxygen might act as a proton shuttle in this specific geometric arrangement.
 | ||
| Scheme 20 Proposed CCEE mechanism catalysed by complex 33. | ||
Nickel complexes 43 and 44 were also found to be active photocatalysts and electrocatalysts for proton reduction.160 In a photocatalytic system similar to that used by Eisenberg,158 the catalysts achieve 5923 and 7634 TON values, respectively, consistent with the electron donating nature of the methyl substituted bipyridine. When eosin was used instead of fluorescein, half of the amount of hydrogen was produced, due to the decomposition of the photosensitizer. The authors suggest that the reduced fluorescein transfers an electron to the protonated catalyst and hydrogen is evolved via a pathway similar to that of catalyst 30.160 The cobalt analogue of complex 40, cobalt bipyridine pyridine–thiolate (45) and cobalt 1,10-phenanthroline pyridine–thiolate (46) were used as photocatalysts in the above system, reaching 210.3 and 163.2 μmol h−1 of hydrogen evolution.161
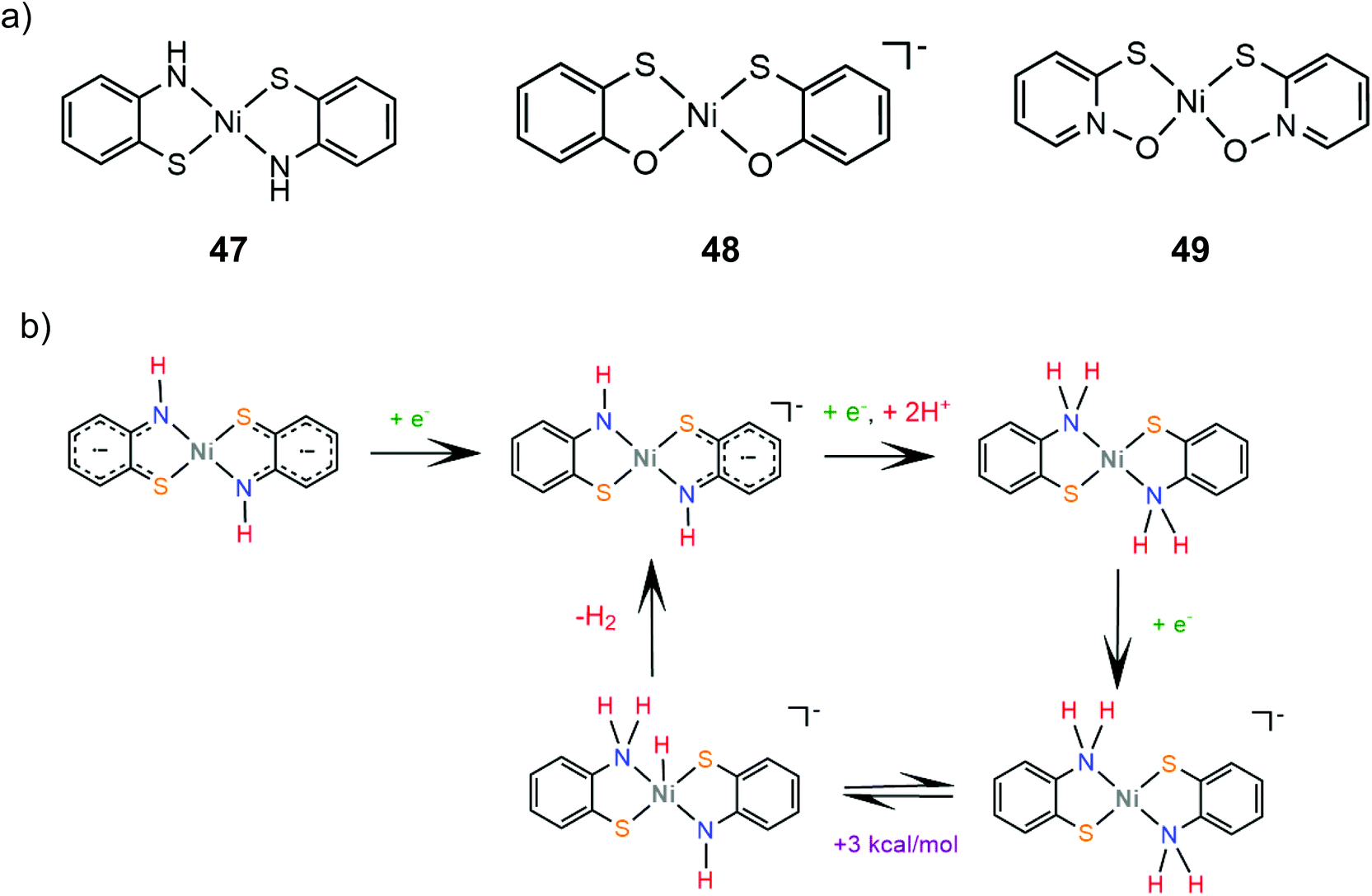
The group of Eisenberg also studied the catalytic activity of the nickel thiolate complexes 47–49 (Scheme 21a).162 Irradiation (λ = 520 nm) of an aqueous basic solution of fluorescein as a photosensitizer, triethanolamine as the sacrificial electron donor and a catalyst results in 6190, 5600 and 5900 moles of H2 per mole of catalysts 47, 48 and 49, respectively. Furthermore, the complexes also show remarkable catalytic activity for the photogeneration of hydrogen from water in a system containing CdSe quantum dots as a photosensitizer and ascorbic acid as a sacrificial electron donor in water under pH 4.5, achieving TON values of 293400, 281800 and 308000 for 47, 48 and 49, respectively. Importantly, it should be noted that complex 2 (nickel benzenedithiolate) was also found to be efficient in the same system, showing a TON of 105300 and a TOF of 730 h−1, even though when the system based on fluorescein was employed, it did not show any catalytic activity. This observation underlines the importance of the nature of the photosensitizer not only concerning the value of its standard potential – which is crucial for a photocatalytic process –but also for its reactivity towards the molecular catalyst. In our opinion an electrocatalyst can also act as a photocatalyst in HER catalysis if the right photosensitizer is used.
 | ||
| Scheme 21 (a) Molecular structures of Ni–thiolate complexes. (b) Proposed ECEC mechanism catalysed by complex 47. | ||
Electrochemical experiments in water show that complexes 47, 48 and 49 are active electrocatalysts with overpotentials of 0.8 V for 47 and 48 and 0.7 V for 49 in an aqueous acetic acid solution (Scheme 21a). Notably, nickel benzenedithiolate (complex 2) shows an overpotential of 1.4 V in a similar system, which suggests that nitrogen and oxygen ligating atoms reduce the catalyst overpotential. A possible electrocatalytic mechanism was proposed for catalyst 47 based on spectroscopic and electrochemical data. In the presence of an acid and a reducing agent, complex 47 is protonated on the nitrogen ligating atoms, concomitant with two electron reduction, to form a neutral diamagnetic intermediate, whose structure was crystallographically determined (Scheme 21b). Chemically induced reduction of this intermediate leads to H2 generation, which indicates that an intramolecular proton transfer is taking place to form an H2 evolving Ni-hydride.162 The nickel-hydride was calculated to be 3 kcal mol−1 higher energetically that the ligand protonated intermediate.163
4.4 Diphenylphosphinobenzenethiolate ligand
Grapperhaus and coworkers examined the catalytic activity for proton reduction and hydrogen oxidation of the rhenium diphenylphosphinobenzenethiolate complex, 50,100 which has been found to show reversible binding of ethylene on the ligating sulphur donors as a function of the charge state.71,164 Complex 50 catalytically reduces protons to H2 with an overpotential of 380 mV, a TOF of 9 h−1 and a TON of 54 after 6 h CPE in dichloromethane with acetic acid as a proton source. The first step of the catalytic cycle is ligand sulphur protonation, which is accompanied by reduction (Scheme 22). The monoprotonated complex cation was isolated and its structure was determined crystallographically. Subsequent sulphur protonation and reduction lead to the hydrogen evolving complex. The mer-arrangement of the thiolate chelates of the octahedral complex orients two of the sulphur lone pairs in a way that favours H2 evolution or abstraction. The rate determining step is found by kinetic experiments to be the H2 evolving process.100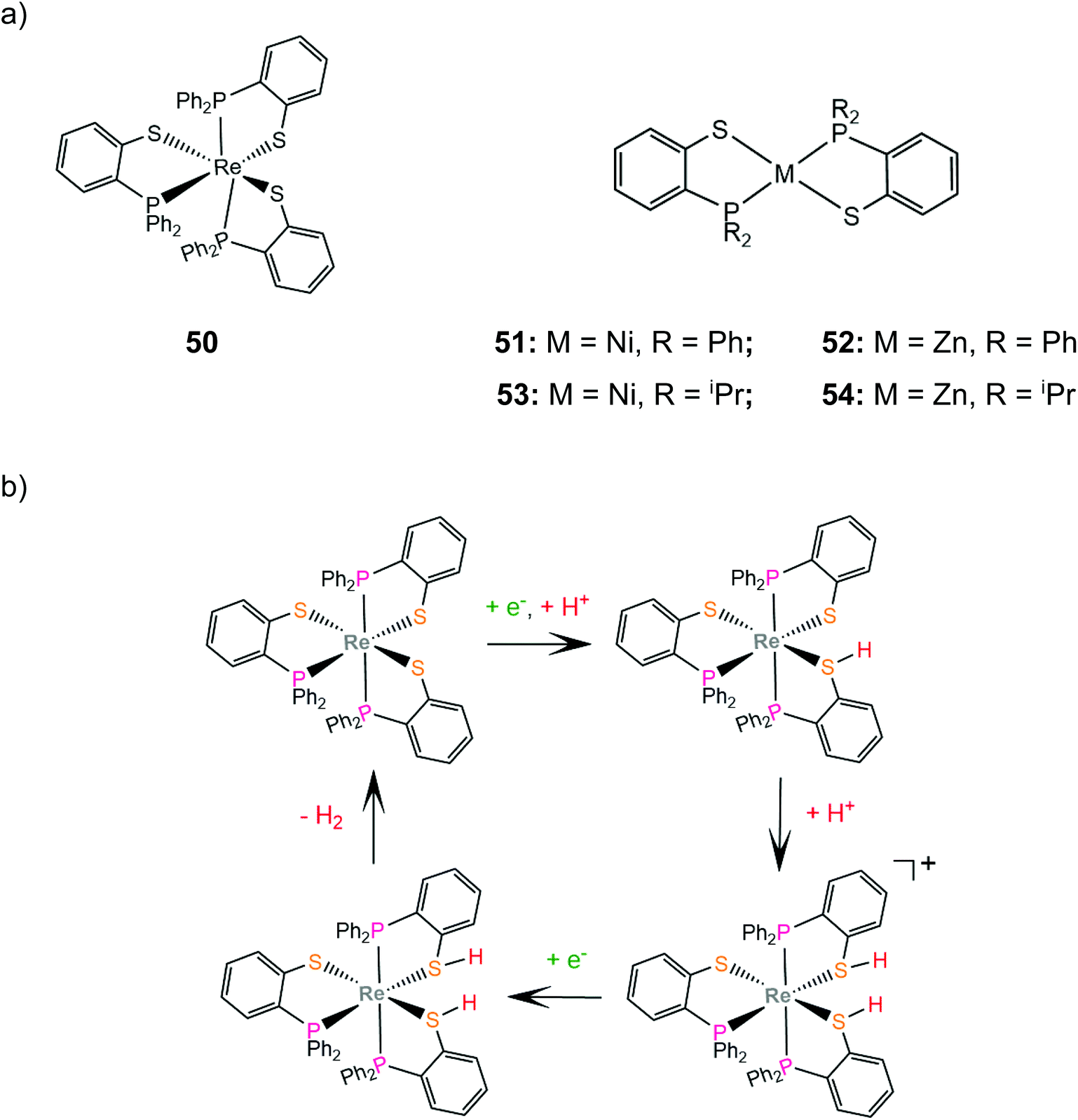 | ||
| Scheme 22 (a) Molecular structures of complexes carrying diphenylphosphinobenzenethiolate ligands. (b) Proposed ligand centred mechanism catalysed by complex 50. | ||
In another paper by the group of Grapperhaus, the homoleptic nickel and zinc complexes of diphenylphosphinobenzenethiolates 51 and 52 and dipropylphosphinobenzenethiolates 53 and 54 were synthesized.165 Complex 51 reaches a TON of 3.4 over 24 h of CPE at an applied potential of −1.9 V vs. Fc+/Fc with an overpotential of 1.1 V in dichloromethane with acetic acid as a proton source, while 51–54 did not show electrocatalytic activity. The catalyst undergoes a metal centered reduction at −2.15 V vs. Fc+/Fc and kinetic studies with deuterated acid indicate the formation of a metal hydride intermediate (Scheme 23). Notably, complex 51 also serves as an electrocatalyst for the hydrogen evolution similar to rhenium diphenylphosphinobenzenedithiolate but the latter acts without the formation of a metal hydride because of the largest basicity of S atoms caused by the transfer of electron density from the formal Re(I) to the thiol sulphurs.
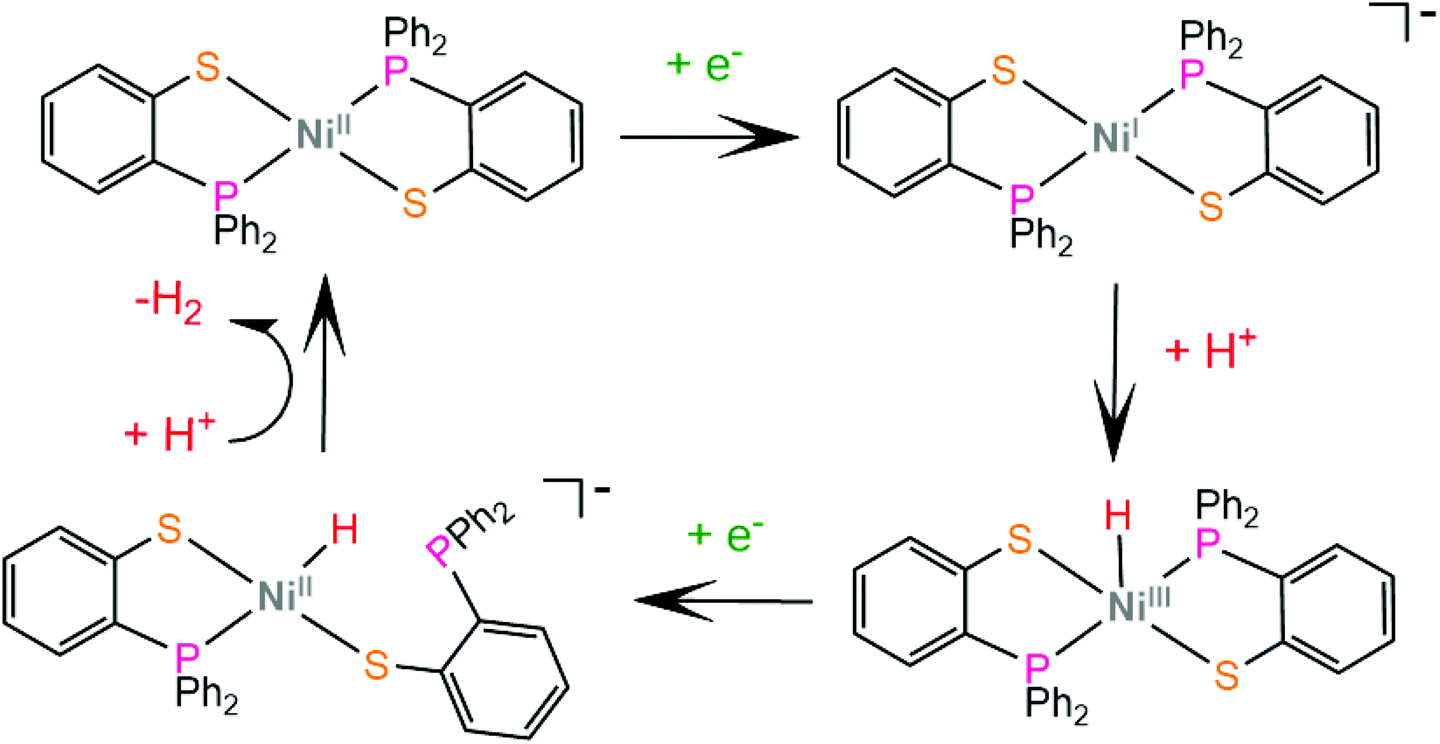 | ||
| Scheme 23 Proposed ECEC mechanism catalysed by complex 51. | ||
4.5 Macrocyclic thiolates
Transition metal thiolate complexes with N2S2 macrocyclic saturated ligands have been used as catalysts for oxygen capture.166 An N,N′-dimethyl-N,N′-bis(2-sulfanylethyl)ethylenediamine ligand was combined with nickel to provide a square planar macrocyclic complex, 55, which acts as a highly efficient catalyst for photocatalytic and electrocatalytic proton reduction.167 An aqueous solution of fluorescein as a photosensitizer, TEA as a sacrificial electron donor and complex 55 achieves a TON of 1510 in 24 h of irradiation in pH 11.6, with an initial TOF of 100 h−1. The electrocatalytic activity of 55 was evaluated in MeCN with acetic acid (pKMeCNa = 23.51) as a source of protons. Two catalytic peaks at −1.61 and −1.92 V vs. SCE are observed with TOF values of 1441 s−1 and 5575 s−1, and overpotentials of 0.560 and 0.670 V, respectively. The HER mechanism was further examined computationally to reveal an ECCE pathway involving a metal centered reduction, followed by two consecutive sulfur protonation steps (Scheme 24). The final step is a reduction concomitant with an intramolecular proton transfer, to form a square planar Ni(II)-hydride.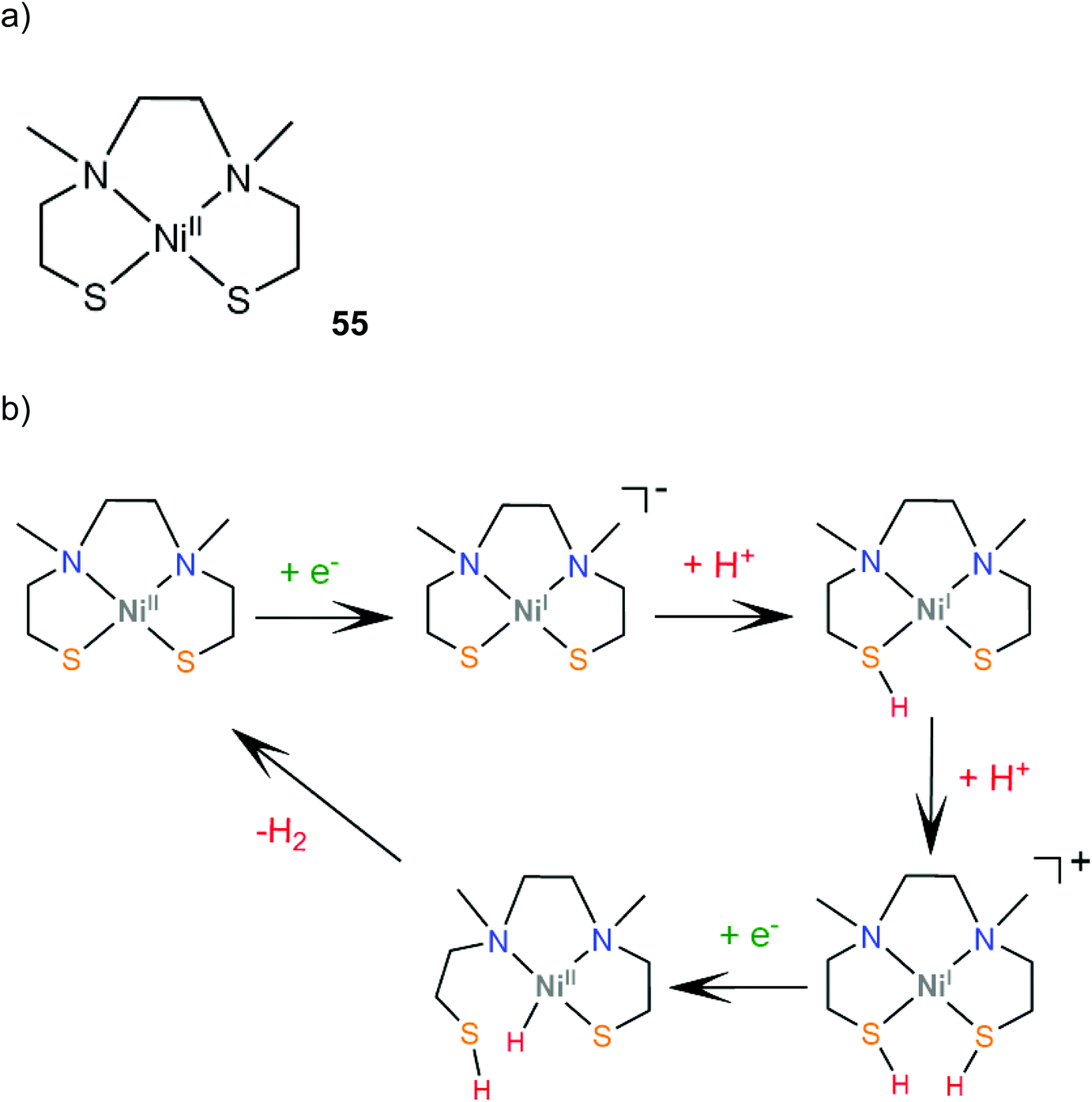 | ||
| Scheme 24 (a) Structure of the Ni–thiolate complex. (b) Proposed ECCE mechanism catalysed by complex 55. | ||
Six nickel complexes with an S2N2-ligand framework similar to complex 47 were synthesized and used as photocatalysts and electrocatalysts for hydrogen production.168 Complexes 57a–c possess Schiff-base type macrocyclic ligands and are found to be in an equilibrium state with their non-innocent type 56a–c isomers in THF solution169 (Scheme 25a). Irradiation (λ > 400 nm) together with [Ir(ppy)2(bpy)]+ (ref. 170) as a photosensitizer in H2O:THF (1:3) containing TEOA gave 414 and 416 moles of H2 per mole of catalyst for 56b and 57c, though a TON of 234 was also produced with equimolar Ni(OAc)2. The presence of metallic mercury limited the catalytic activity, implicating a heterogeneous active catalyst. The catalysts are suggested to follow essentially the same reaction pathway. Cyclic voltammograms in THF solutions in the presence of acetic acid as a source of protons show two cathodic peaks, attributed to consecutive one electron reduction of each complex, and a catalytic peak near −2.4 V vs. SCE. The proposed electrocatalytic mechanism of 56a shown in Scheme 25b was based on electrochemical data and DFT calculations. The doubly reduced complex is protonated on a ligating sulphur atom and reduction of the protonated species induces a thermodynamically favoured intramolecular proton transfer, to generate a Ni(III)-hydride. The Ni(II)-hydride formed upon reduction of these species can evolve H2 using a sulphur atom as a proton relay. Notably, these macrocyclic complexes have much larger overpotentials than both their amine-thiolate analogue, 47, probably because nitrogen protonation is coupled with reduction in the latter case.168
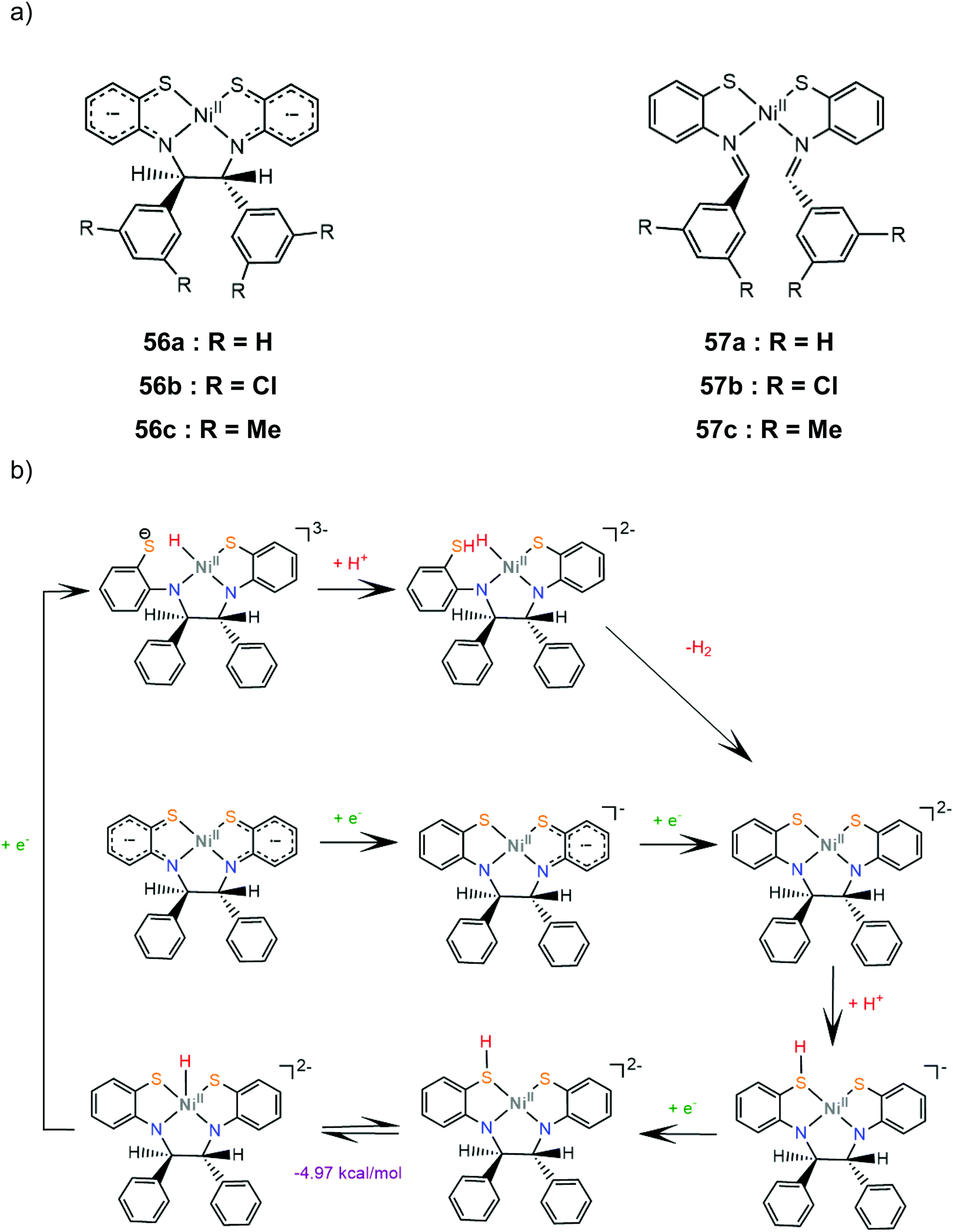 | ||
| Scheme 25 (a) Molecular structures of complexes carrying Schiff based ligands. (b) Proposed mechanism catalysed by complex 56a. | ||
In another study, two S2N2-type Ni(II) complexes bearing different chelating-ring sizes, 58 and 59, were synthesized and examined as photocatalysts and electrocatalysts for proton reduction. In the photocatalytic system [Ru(bpy)3]2+ was used as a photosensitizer and sodium ascorbate as a sacrificial electron donor in aqueous solution at pH 4.0.171 After 4 h, catalyst 58 produced a TON of 36 with a quantum yield of 0.78% and 59 a TON of 3.8. During 5 h CPE of aqueous solutions of the complexes in the presence of ascorbate buffer, complex 58 achieved 1100 TON of H2 with a TOF value of 230 h−1, while complex 59 showed remarkably lower activity, reaching a TON of 19 and a TOF of 3.6 h−1. Kinetic analysis and DFT calculations were performed to elucidate the hydrogen evolution step. The authors suggest that an initial metal centered reduction followed by a PCET process leads to an octahedral Ni(II)-hydride that reacts with H3O+ to release H2 (Scheme 26). The difference in the efficiencies of the two catalysts is in the activation energy of the H–H bond formation, calculated to be 6.7 kcal mol−1 for 58 and 9.3 for 59.171 A similar catalyst to complex 58, with Cl atoms instead of acetonitrile groups to fill the coordination sphere, has also been reported to photocatalyze hydrogen generation in an aqueous solution with CdS nanorods as a photosensitizer and ascorbic acid as a sacrificial electron donor and produces 24900 mol of H2 per mole of catalyst during 83 h irradiation, with a TOF of 300 h−1 and the quantum yield reaching 24% at 420 nm.172
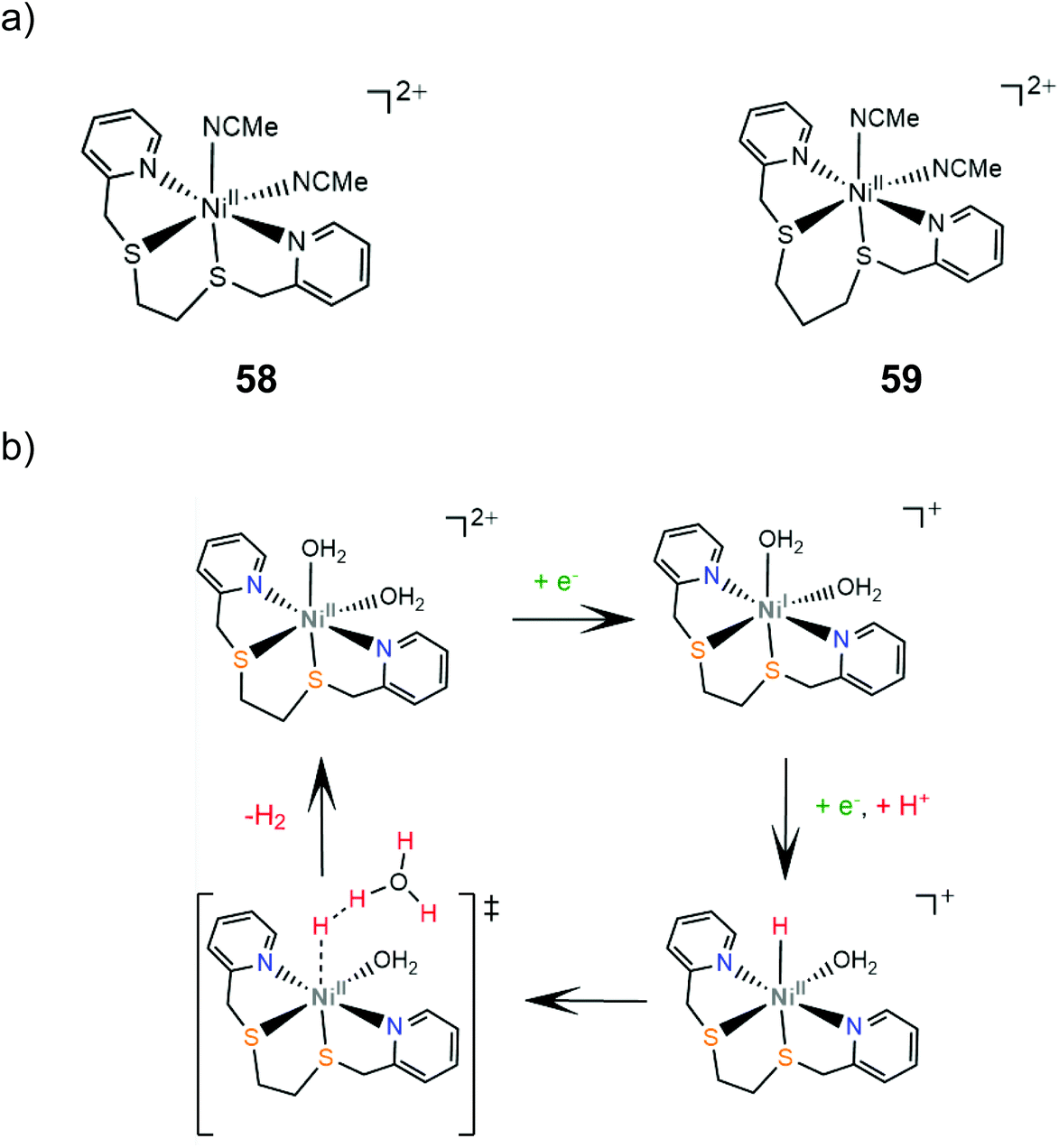 | ||
| Scheme 26 (a) Molecular structures of six-coordinated Ni(II) complexes as HER catalysts. (b) Proposed mechanism catalysed by complex 58. | ||
4.6 Thiosemicarbazone ligands
A particularly interesting class of redox active ligands involves thiosemicarbazone derivatives, a class of Schiff base polydentate ligands. Since they possess unsaturated and delocalized electronic structures, a clear definition of the metal oxidation state is not straightforward. These non-innocent sulphur donating ligands have recently attracted attention in the design of HER catalysts due to their strong chelating ability, redox activity, and bio-relevance.173McNamara and coworkers reported that a distorted square planar nickel complex containing a bis-dithiocarbazate ligand (60) exhibited a faradaic yield of 98% at an applied potential of −1.8 V vs. Fc+/Fc in the presence of trifluoroacetic acid in acetonitrile and a rinse test showed no catalytically active films on the electrode surface.174 The HER mechanism is proposed to involve initially an electrochemical reduction of the metal Ni(II/I) at −1.25 V vs. Fc+/Fc and subsequent protonation of either the metal or the coordinated sulfur atom. Photolysis of a solution of fluorescein, triethylamine and 60 in 1:1 EtOH:H2O resulted in H2 generation corresponding to a turnover number (TON) of 3300 in 70 h. Notably, spectroscopic studies indicated an interaction between complex 60 and fluorescein, tentatively attributed to coordination of the latter's negatively charged oxygen atom on Ni, which may be responsible for the high stability of the system.
A square planar diamagnetic Ni(II) complex (61) bearing the ligand 2-(2-(diphenylphosphino)benzylidene)hydrazinecarbothioamide (HthioP) was prepared and X-ray diffraction analysis revealed a tridentate NSP chelator and a second HthioPO oxidized ligand bound to Ni via the monosulphur donor.175 Complex 61 acts as an effective electrocatalyst in the presence of Et3NHCl in acetonitrile and shows remarkable results in photocatalytic hydrogen evolution using fluorescein as a photosensitizer and triethylamine as a sacrificial electron source in a 1:1 ethanol: water mixture. Under optimal photocatalytic conditions, the TON with respect to 61 after 24 h was 8000 with an initial TOF of 500 h−1. DFT methods were employed to find a possible H2 formation mechanism, which is shown in Scheme 27. A long-distance Ni⋯H interaction between the metal centre and the amide group proton is supported by crystal X-ray structural analyses and geometry optimization at the 6-31+g(d,p) level of theory. Thus, upon reduction of Ni(II/I) the amide proton transfer on Ni(I) is thermodynamically favoured by 45.18 kcal mol−1. The Ni-hydride formed reacts with a proton in solution to evolve H2 and complex 61 is regenerated after imide group protonation to restart the catalytic cycle. The reaction of the HthioP ligand with the Co precursor Co(BF4)2·6H2O resulted in the formation of the octahedral Co(II) complex 62 with two NSP tris-chelate ThioP ligands. Complex 62 undergoes also a metal centre pseudoreversible CoII/CoI redox process at −0.46 V (vs. Ag/AgCl) and generates H2 at −0.85 V (−1.1 V vs. Fc+/Fc), similar to 61. In contrast, the first reduction of complex 61 occurs at −0.78 V. Complex 62 produces H2 under almost identical photocatalytic conditions, with the initial turnover frequency (TOF) of about 200 h−1, while the turnover number (TON) is 2000 after 24 h, which highlights the importance of the Ni⋯H interaction and the open Ni coordination site in the catalytic efficiency.
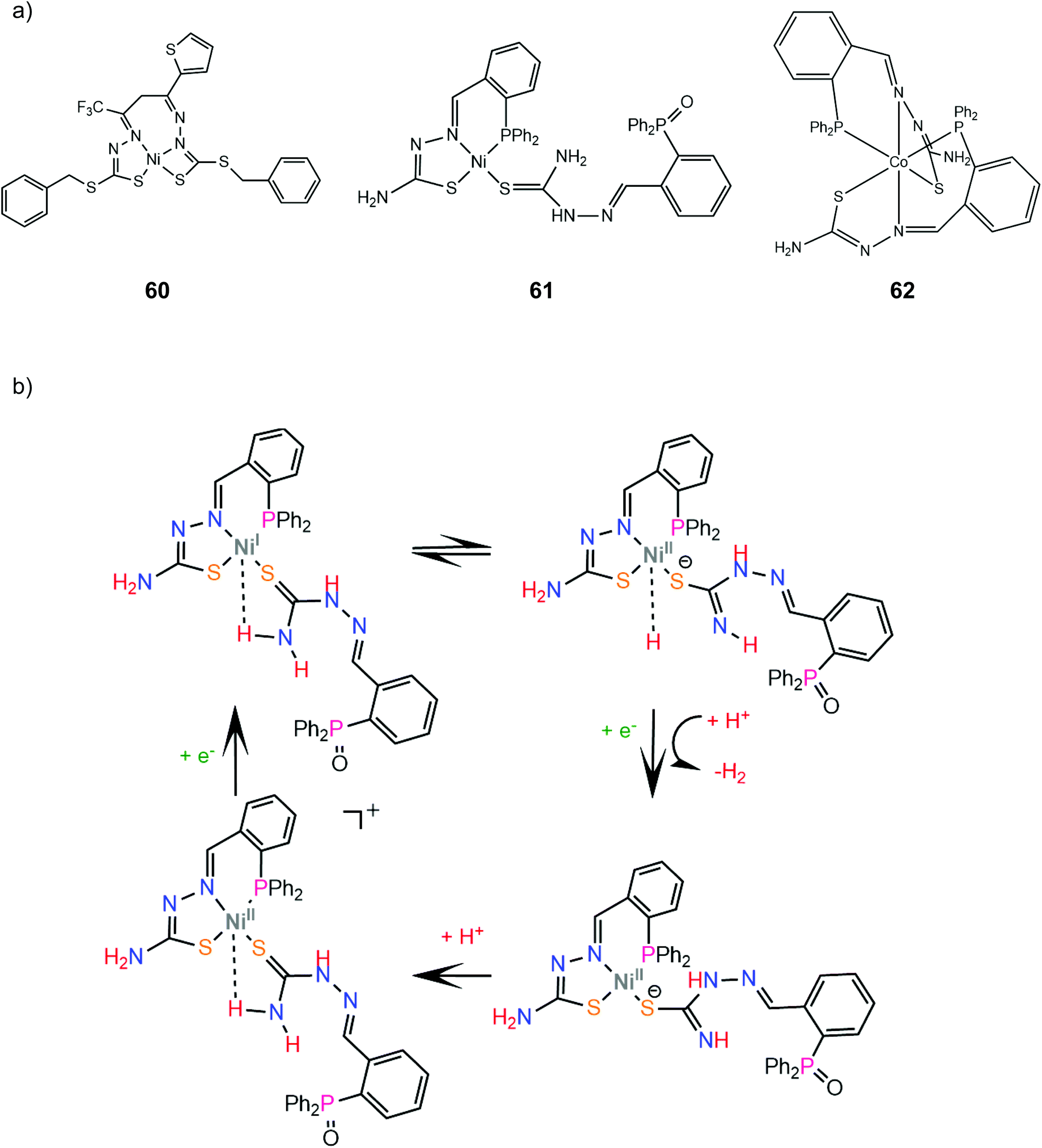 | ||
| Scheme 27 (a) Molecular structures of metal complexes carrying thiosemicarbazone ligands. (b) Proposed mechanism catalysed by complex 61. | ||
Two octahedral tris(thiosemicarbazide) cobalt(III) geometric fac- and mer-isomers (63 and 64) were synthesized and used as HER catalysts176 (Scheme 28). They both act as effective homogeneous catalysts for H2 evolution electrolysis of a MeOH solution in the presence of acetic acid (pKa = 9.7) at −1.28 V vs. SCE (−1.6 V vs. Fc+/Fc). Based on the icat/ip value of 23 in the acid-independent region the TOF of 63 was calculated to be 210 s−1 and the overpotential η was 560 mV at pH 7. Aqueous solutions of 63 and 64 containing fluorescein and triethylamine were photoactive for 16 h with final TON values of 900 and 890, respectively. Interestingly, the two geometric isomers (63 and 64) show the same electrochemical behaviour and photocatalytic efficiency, which suggests a common HER mechanism. The authors proposed an EECC mechanism involving two consecutive metal centre reductions leading to a Co(I) intermediate, which can be doubly protonated on the metal to form a Co(III)-hydride. The amine chelating groups are proposed to act as a proton relay on the metal. The H2 evolving species is the Co(III)-hydride intermediate bearing a pendant protonated amine.
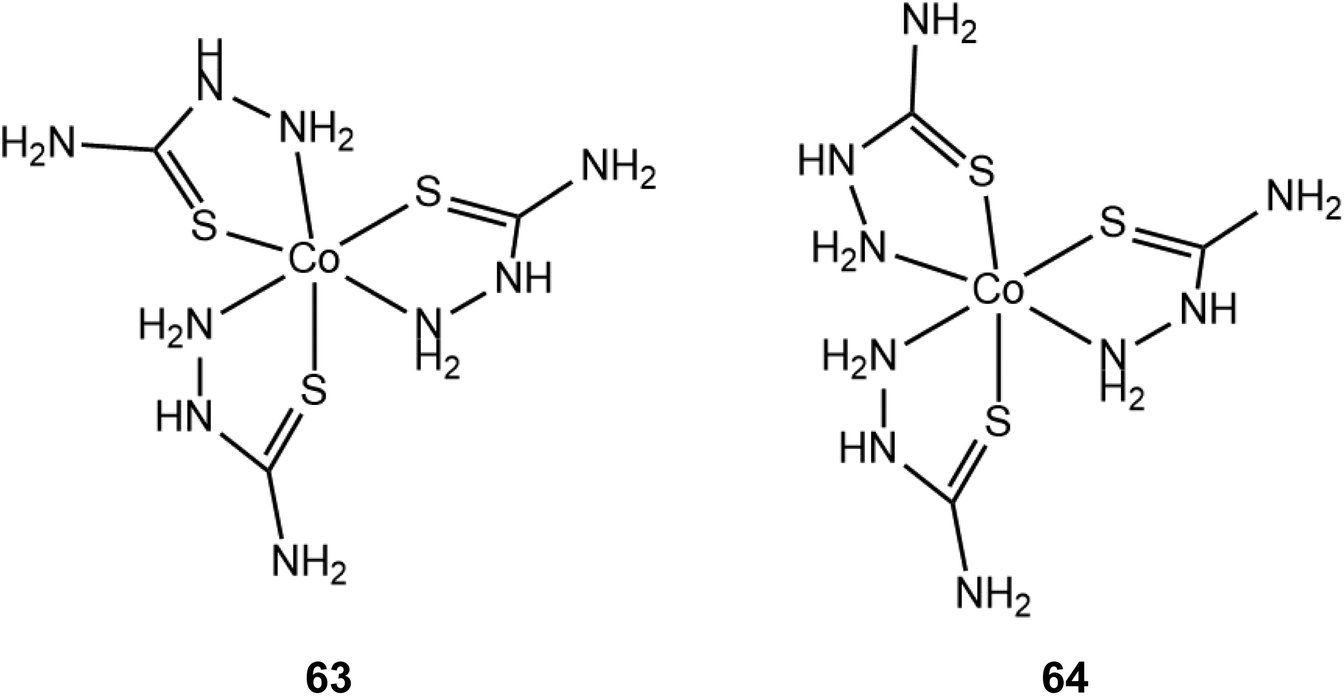 | ||
| Scheme 28 Molecular structures of tris(thiosemicarbazide) cobalt(III) complexes. | ||
Diacetyl-bis(N-4-methyl-3-thiosemicarbazone) was the first example of a metal free ligand acting as a homogeneous electrocatalyst for the HER, as was reported in 2016 by Grapperhaus and co-workers.101 The ligand is an efficient solution electrocatalyst for H2 production, with a TOF of 1320 s−1 and an overpotential of 1.430 V using acetic acid as the proton source in methanol solution. The non-innocent bis-thiosemicarbazone ligand framework was combined with various metal centers and the corresponding complexes were also reported to act as electrocatalysts for H2 evolution with a maximum TOF of 1170 s−1 at an overpotential of 756 mV; this overpotential implies the necessity of a metal ion for reducing the catalytic potential of a free organic ligand. Compound 65 exhibits a catalytic peak at −1.7 V vs. Fc+/Fc in MeOH with acetic acid (pKa = 9.7) as the source of protons, while in acetonitrile (pKa of acetic acid = 22.3) the catalytic peak arises at −2.3 V, very close to the reduction potential of the free ligand, indicating that the complex is protonated in the MeOH acidic solution. Notably, both the complex and the free ligand also catalyse the hydrogen oxidation reaction (HOR). Compound 65 was proposed to produce H2 catalytically via the CECE reaction mechanism through a ligand-centred HER, involving either the homocoupling of reduced ligand-protolated radicals or the heterocoupling of one reduced protonated radical with a reduced doubly protonated cationic radical (Scheme 29).
 | ||
| Scheme 29 (a) Molecular structures of metal diacetyl-bis(N-4-methyl-3-thiosemicarbazone) complexes. (b) Proposed mechanism catalysed by complex 65. | ||
In the same year, the square planar Ni(II) complex 70 bearing the thiosemicarbazone derivative resulting from the incorporation of a p-methoxyphenyl moiety on the amine group, {bis[4-(p-methoxyphenyl)thiosemicarbazone]}-2,3-butane, was synthesized, characterized and studied as a HER electrocatalyst in DMF solution using TFA as a source of protons.177 During a 16 h CPE experiment under a potential of −0.8 V vs. Ag/AgCl (−1.2 V vs. Fc+/Fc) the faradaic efficiency observed was 80% and 21 TON of H2 was produced. Notably, this modified thiosemicarbazone did not show HER catalytic activity, as opposed to the diacetyl-bis(N-4-methyl-3-thiosemicarbazone) ligand previously discussed. A proposed mechanism for the H2 formation is an ECEC pathway, where the first protonation takes place on the coordinating nitrogen atom and the second protonation results in the formation of a Ni(III) hydride species.
In 2017, the copper(I) thiosemicarbazone complex was reported, which is also found to promote H2 evolution from protons. In the absence of an acid, the CV of 66 in acetonitrile and dimethylformamide exhibited a reversible Cu(II/I) wave with E1/2 = −1.20 V vs. Fc+/Fc.88 When titrated with acetic acid (pKACNa = 22.3), the Cu(II/I) peak became 250 mV more positive, indicating that the complex is protonated in the weak acid solution and a catalytic peak appeared at −2.1 V. The catalyst is active for the HER with a stable performance over 23 h during CPE at an 800 mV overpotential and 73 TON and with a hydrogen generation rate of 10000 s−1, resulting in 81% faradaic efficiency. On the basis of DFT calculations they suggested that the ligand could play a primal role in the metal centre, with electrons and protons stored in the extended thiosemicarbazone π-system and nitrogen atoms of the ligand. The authors assumed that the key intermediate in H2 evolution for 66 was not a traditional copper hydride, but the doubly reduced Cu(I) complex protonated on the coordinating nitrogen atom and the adjacent nitrogen atom. Thus, a CECE pathway was proposed for the catalytic cycle (Scheme 30).
 | ||
| Scheme 30 Proposed CECE mechanism catalysed by complex 66. | ||
Analogous ligand assisted catalytic activity for the HER was found for the cobalt(III) complex 68. Crystal X-ray diffraction analysis revealed that 68 exists as a dimer, which dissociates upon two-electron reduction.178 Complex 68 is able to catalyse proton reduction when titrated with Et3NHBF4 (pKa = 9.2) in DMF, as it exhibits a TON of 9 with a faradaic yield of 65% under CPE with an applied potential of −1.60 vs. Fc+/Fc for 4 h. Combined electrochemical, theoretical and kinetic analyses suggested a possible catalytic mechanism involving a three-electron-reduced Co(I) species protonated on the coordinating nitrogen of the thiosemicarbazone ligand. The last step of the HER mechanism is the second protonation, proposed by the authors to take place on the Co(I) centre. Notably, the corresponding Zn complex is not reported to be an active HER catalyst, underlying the impact of the thiosemicarbazone amine substituents.
Recently, the Ni(II) complex of thiosemicarbazone was also studied as a potential electrocatalyst for H2 formation.179 Complex 67 undergoes ligand centred reduction at −1.83 V vs. Fc+/Fc and the Ni(II/I) reduction takes place at −2.45 V. The electrocatalytic activity of 67 was examined in acetonitrile and dimethylformamide with acetic acid and TFA as the proton sources. In acetonitrile, 67 was reported to catalyse H2 evolution at −2.35 V vs. Fc+/Fc when acetic acid (pKa = 23.5) was introduced, with a TON of 48 over 4 h of CPE and an initial TOF of 4200 s−1. When the stronger acid TFA (pKa = 12.7) was used instead, a lower overpotential but also lower efficiency (TON of 24 and TOF of 1300 s−1) were observed. Electrocatalytic experiments performed in DMF solution also indicate better catalytic activity in the presence of acetic acid, but the efficiency is considerably lower. The molecular catalyst 67 was non-covalently incorporated onto a p-Si electrode to serve as a proton reduction co-catalyst in a photocathodic hydrogen evolution device, achieving a 100 mV lower overpotential relative to the Ni metal deposits.180 Mechanistic investigations of the electrocatalytic HER showed a ECEC pathway (Scheme 31). In contrast to the observations for the corresponding Co complex 68, theoretical studies indicated that the formation of the nickel–hydride intermediate is thermodynamically favourable after the second reduction. The hydride subsequently reacts with a proton in solution to form H2via a low energy (10.5 kcal mol−1) transition state and re-enter the catalytic cycle.179
 | ||
| Scheme 31 Proposed ECEC mechanism catalysed by complex 67. | ||
The {bis[4-(p-methoxyphenyl)thiosemicarbazone]}-2,3-butane ligand was further investigated with Pd(II) as the metal centre, forming the square planar diamagnetic complex 69.179 The coordinated ligand is reduced at −1.02 V vs. Ag/AgCl (−1.4 V vs. Fc+/Fc) and Pd(II/I) reduction is possible at −1.64 V. Note that the free ligand is reduced at −1.6 V vs. Ag/AgCl. Complex 69 achieved proton reduction to form H2 in DMF using TFA (pKa = 6) as the proton source. A bulk electrolysis experiment showed a TON of H2 evolution of 2 in a 4 h experiment with a faradaic yield of 35% at a controlled potential of −1.2 V vs. Ag/AgCl (−1.55 V vs. Fc+/Fc). The low catalytic efficiency is attributed to the decomposition of the catalyst after 1 h of electrolysis. A mercury pool electrode was used during this experiment, in order to exclude the possibility of active Pd nanoparticle formation. The HER of molecular complex 69 is proposed to follow an ECEC pathway (Scheme 32), similar to its Ni and Co analogues 67 and 68, but the proton reduction is a ligand centred process, as the H2 evolving intermediate is a Pd(I) triplet species with the coordinating and the hydrazino nitrogen atoms protonated, similar to the Cu thiosemicarbazone complex 66.
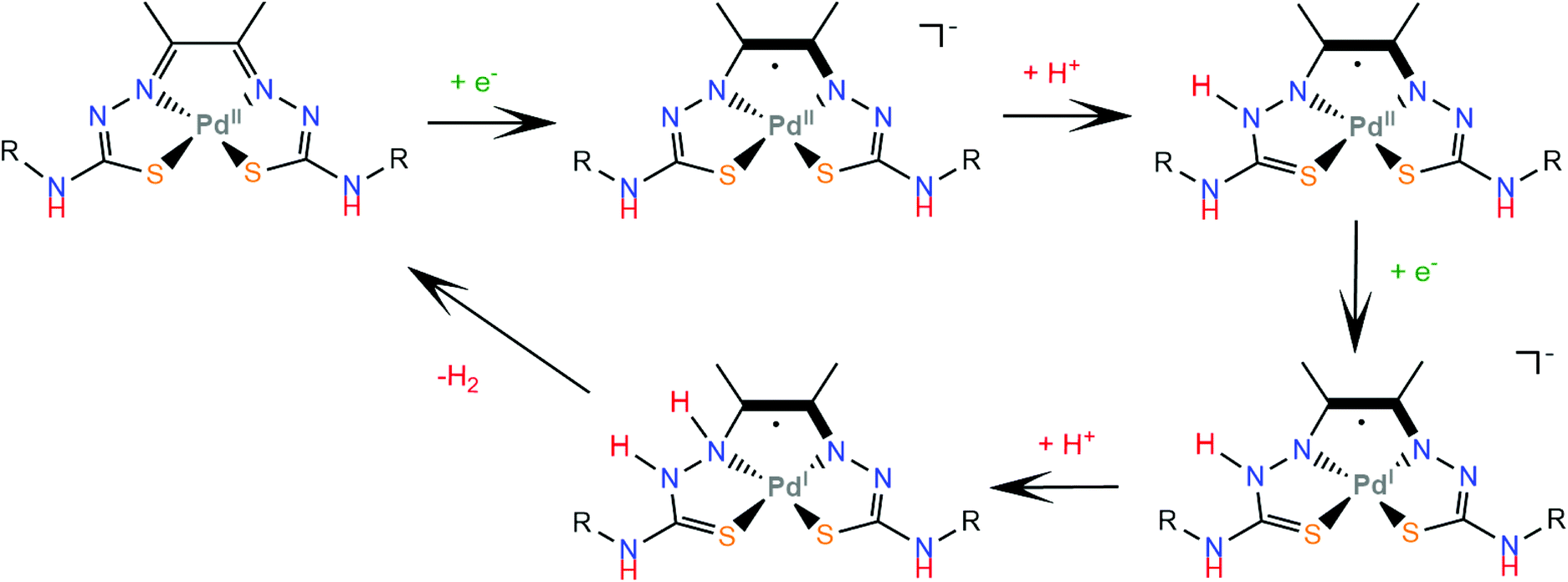 | ||
| Scheme 32 Proposed ECEC mechanism catalysed by complex 69. | ||
Recently, the group of Grapperhaus presented the nickel complexes 71–74 bearing pendant amines on the thiosemicarbazone ligand framework181 (Scheme 33a). Electrochemical characterization of the synthesized complexes shows that alkylation of the amine group anodically shifts the ligand centered reduction potential by 0.08 V for 71 and 72 and by 0.14 V for 73 and 74, attributed to the presence of a positively charged, electrophilic moiety. Alkylation also facilitates metal centered reduction by 0.09 V and 0.16 V, respectively, with the symmetric complex again showing the largest anodic shift. Catalyst 73 shows the highest catalytic efficiency for proton reduction in acetonitrile with acetic acid as a proton source, achieving an overpotential of 0.560 V, a TOF of 6300 s−1 and a TON of 18 after 4 h CPE. Under the same homogeneous conditions, catalysts 71, 72 and 74 show TOF values of 2900, 1500 and 2400 s−1 and overpotentials of 0.590, 0.660 and 0.670 V, respectively. Ligand centered reduction of complex 73 takes place at an applied potential of −1.72 V vs. Fc+/Fc and metal centered reduction at −2.31 V. In the presence of an acid, metal centered reduction shifts anodically, indicating an ECEC reaction pathway. Based on the relative catalytic efficiencies of complexes 71–74 and the observation that the overpotentials of the catalysts increase, when the scan rate is increased, the authors proposed that metal center reduction is coupled with an intramolecular proton transfer from the pendant amine to the metal center, to generate a Ni-hydride that can react with a proton in solution to evolve hydrogen (Scheme 33b).
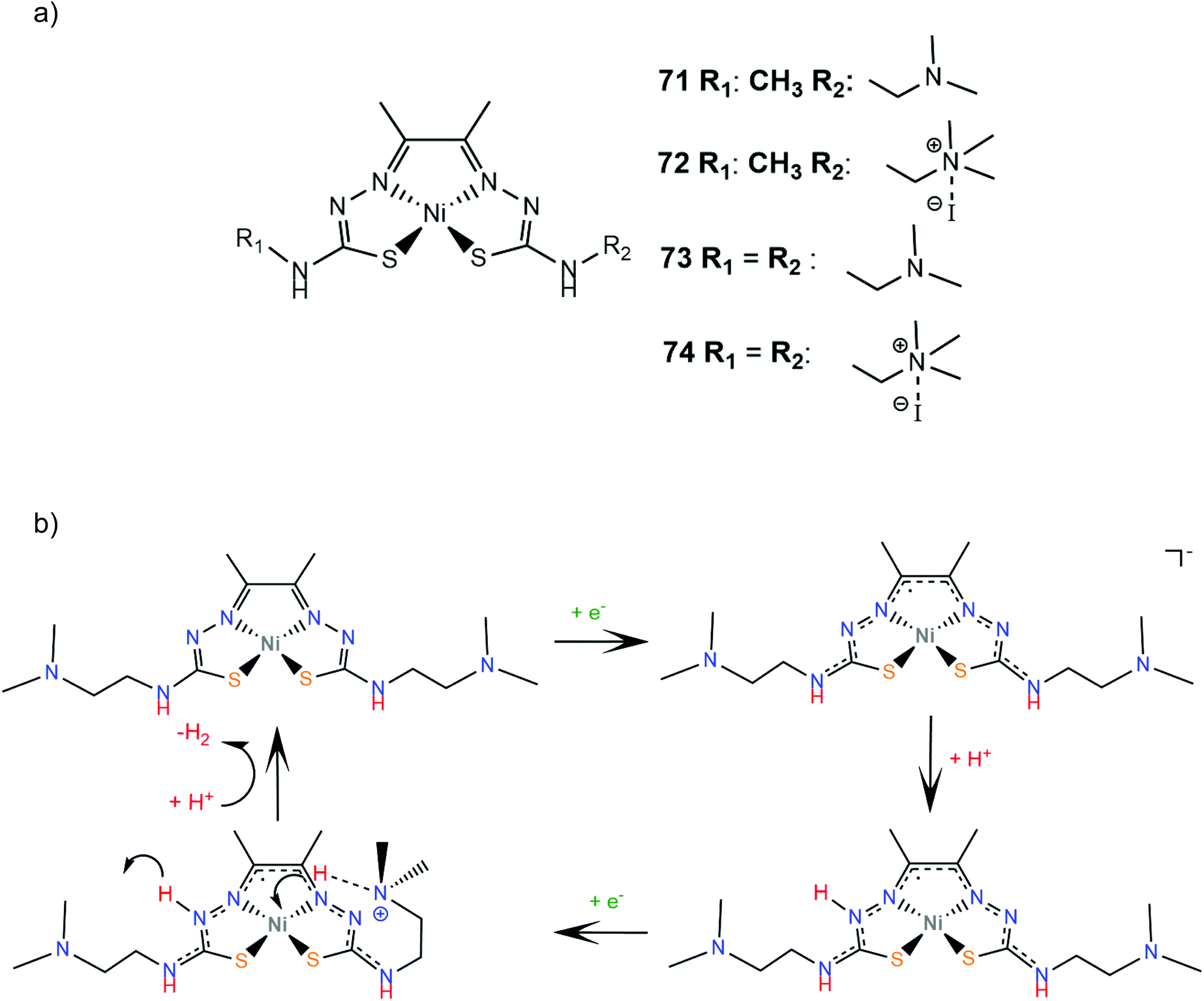 | ||
| Scheme 33 (a) Her catalysts carrying pyrazine substituents on their thiosemicarbazone ligands. (b) Proposed ECEC mechanism catalysed by complex 73. | ||
5. Summary and perspectives
In conclusion, we have summarized the design, catalytic properties and hydrogen evolution reaction pathways of dithiolene and thiolate complexes as well as complexes that carry non-innocent ligands with sulphur chelating atoms. All these complexes serve as molecular electrocatalysts and photocatalysts for proton reduction. Experimental observations, including electrochemical studies and spectroscopic characterization of catalytic intermediates, in conjunction with computational studies of possible intermediate structures, elucidate several mechanistic details and reveal the role of the redox-active, non-innocent thiolate ligands. The electronic structures of frontier orbitals determine the alternative sequences of the various proton and electron transfer steps and the possible sites for electron and proton uptake. Electron density localization is determined by the energy levels of metal d orbitals relative to those of ligand-based p orbitals. DFT studies together with experimental data reveal that there is a clear synergistic effect of different components, which function together to activate dithiolene/thiolate complexes and H+/H2O and to promote the electron transfer in hydrogen evolution.For effective future design of proton reduction catalysts the following comments can be taken into account. The combination of multi-step metal–ligand cooperative redox activity and extended π-delocalization over metal–ligand bonds play central roles in achieving lower overpotentials. For example, the redox active thiosemicarbazone analogues upon reduction stabilize the unpaired electron via the alternating π-bonds of the hydrazone bridge. Furthermore, the presented results shed light on the ability of ligand donor atom pKa values to tune the HER mechanism toward ligand- or hydride-based H2 formation. Thiolate ligands bearing electron donating moieties tend to enhance hydrogen evolution efficiencies, because they favor metal or ligand protonation, which is either coupled with or followed by complex reduction. Thus, increasing the nucleophilicity of possible protonation sites can contribute to a lower overpotential by changing the catalytic pathway. Moreover, the ligand-based proton coupled electron transfer pathway leads to H2 formation without applying the highly negative potential required to generate low-valent metal intermediates, since the electron accepting abilities of the protonated ligands allow the metal centre to have higher formal charge. The electron donating ability, and consequently the proton affinity, of the ligand is strongly influenced by the metal electron density, as can be seen from the very large HER overpotential of the free thiosemicarbazone ligand. Thus, the role of the metal ion is important not only for the modulation of the structure of the catalysts but also for the reactivity of the coordinated atoms towards H+.
On the other side, non-innocent ligands that contain both N and S ligating atoms tend to perform better than dithiolene ligands, because N donors are more easily protonated and thus they efficiently relay protons to the metal center. However, one can modulate the electronic structure and density around the catalytic center by selecting more electron donating ligating atoms, such as sulphur or phosphorus atoms, which facilitate initial hydride formation. Furthermore, inspired by nature, functionalization of the ligands to expose more proton relay sites can lower the activation energy of metal hydride formation. Care has to be taken as many thiolate complexes tend to decompose under the applied anodic potential to form M0 or other inorganic nanoparticles that lead to a heterogeneous catalytic system. Since the understanding of the elementary mechanistic steps of hydrogen formation is essential for novel catalyst design, it is important to evaluate the stability of the catalysts under experimental conditions. Decomposition of bis-dithiolene complexes depends on the strength of the acid employed and the potential applied and most probably takes place via cleavage of M–S or C–S bonds when the formation of intermediates with highly distorted geometries is reached.
Although reasonable progress has been made in the field of catalytic hydrogen evolution by dithiolene/thiolate complexes, further advancements are needed to use this class of catalysts for practical applications. A deeper understanding of the mechanisms of H2 formation can give better insight into some important structure–function relationships of dithiolene and thiolate complexes that will inspire future design of highly efficient proton reduction catalysts. Since on one hand, the influence of the metal ions and the electron accepting or donating substituents on the dithiolene or thiolate chelating ligands affects not only the structures of complexes but also the photocatalytic or electrocatalytic mechanisms with multi-electron and proton steps necessary for water reduction – a two electron process – and on the other hand, more properties (such as the hydricity of the formed hydride, the acidity of the proton source, the hydricity of H2 and the activation energy of the reaction) have to be taken into account for the design of an effective low cost catalyst, it may be the time for a systematic, data-driven approach on HER catalyst design, where experimental and theoretical data could be used to construct machine learning models for structure/catalytic property relationships.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank the Special Research Account of NKUA for financial support. M. D. is also grateful for the support from the Onassis Foundation – Scholarship (ID: G ZO 005-1/2018-2019). F. K. is also grateful for the co-funding by Greece and the European Union (European Social Fund – ESF) through the Operational Programme “Human Resources Development, Education and Lifelong Learning” in the context of the project “Strengthening Human Resources Research Potential via Doctorate Research” (MIS-5000432), implemented by the State Scholarships Foundation (IKY).References
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2003, 103, 15729–15735 CrossRef.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119(4), 2752–2875 CrossRef CAS.
- V. R. Bakuru, M. E. DMello and S. B. Kalidindi, ChemPhysChem, 2019, 20(10), 1177–1215 CrossRef CAS.
- T. Wang, H. Xie, M. Chen, A. D'Aloia, J. Cho, G. Wu and Q. Li, Nano Energy, 2017, 42, 69–89 CrossRef CAS.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1(1), 0003 CrossRef CAS.
- L. Yuan and J. Koo, Biotechnol. Bioeng., 2019, 116(11), 3124–3135 CrossRef.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114(8), 4081–4148 CrossRef CAS.
- Y. Xu and B. Zhang, Chem. Soc. Rev., 2014, 43(8), 2439–2450 RSC.
- W. T. Eckenhoff, Coord. Chem. Rev., 2018, 373, 295–316 CrossRef CAS.
- S. Fukuzumi, Y. M. Lee and W. Nam, Coord. Chem. Rev., 2018, 355, 54–73 CrossRef CAS.
- J. R. McKone, S. C. Marinescu, B. J. R. Brunschwig, B. S. Winkler and H. B. Gray, Chem. Sci., 2014, 5(3), 865–878 RSC.
- P. Sutra and A. Igau, Curr. Opin. Green Sustainable Chem., 2018, 10, 60–67 CrossRef.
- I. Bhugun, D. Lexa and J. M. Savéant, J. Am. Chem. Soc., 1996, 118(16), 3982–3983 CrossRef CAS.
- Y. Xu and R. Xu, Appl. Surf. Sci., 2015, 351, 779–793 CrossRef CAS.
- R. M. Bullock, A. M. Appel and M. L. Helm, Chem. Commun., 2014, 50(24), 3125–3143 RSC.
- Y. Han, H. Fang, H. Jing, H. Sun, H. Lei, W. Lai and P. Cao, Angew. Chem., Int. Ed., 2016, 55(18), 5457–5462 CrossRef CAS.
- H. Lei, H. Fang, Y. Han, W. Lai, X. Fu and R. Cao, ACS Catal., 2015, 5(9), 5145–5153 CrossRef CAS.
- P. Zhang, M. Wang, Y. Yang, T. Yao and L. Sun, Angew. Chem., Int. Ed., 2014, 53(50), 13803–13807 CrossRef CAS.
- J. Wang, C. Li, Q. Zhou, W. Wang, Y. Hou, B. Zhang and X. Wang, Dalton Trans., 2016, 45(13), 5439–5443 RSC.
- B. D. Stubbert, J. C. Peters and H. B. Gray, J. Am. Chem. Soc., 2011, 133(45), 18070–18073 CrossRef CAS.
- S. Aroua, T. K. Todorova, V. Mougel, P. Hommes, H. U. Reissig and M. Fontecave, ChemCatChem, 2017, 9(12), 2099–2105 CrossRef CAS.
- P. Zhang, M. Wang, Y. Yang, D. Zheng, K. Han and L. Sun, Chem. Commun., 2014, 50(91), 14153–14156 RSC.
- Y. Sun, J. P. Bigi, N. A. Piro, M. L. Tang, J. R. Long and C. J. Chang, J. Am. Chem. Soc., 2011, 133(24), 9212–9215 CrossRef CAS PubMed.
- R. Z. Liao, M. Wang, L. Sun and P. E. Siegbahn, Dalton Trans., 2015, 44(21), 9736–9739 RSC.
- K. Ye, Y. Y. Li and R. Z. Li, RSC Adv., 2016, 6(93), 90035–90045 RSC.
- B. H. Solis and S. Hammes-Schiffer, Inorg. Chem., 2011, 50, 11252–11262 CrossRef CAS.
- J. T. Muckerman and E. Fujita, Chem. Commun., 2011, 47, 12456–12458 RSC.
- J. W. Jurss, R. S. Khnayzer, J. A. Panetier, K. A. E. Roz, E. M. Nichols, M. Head-Gordon, J. R. Long, F. N. Castellano and C. J. Chang, Chem. Sci., 2015, 6, 4954–4972 RSC.
- V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2(2), 270–279 CrossRef CAS.
- O. R. Lucaa and R. H. Crabtree, Chem. Soc. Rev., 2013, 42(4), 1440–1459 RSC.
- J. A. Denny and M. Y. Darensbourg, Chem. Rev., 2015, 115, 5248–5273 CrossRef CAS.
- H. Dobbek, L. Gremer, R. Kiefersauer, R. Huber and O. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(25), 15971–15976 CrossRef CAS PubMed.
- J. M. Tunney, J. McMaster and C. D. Garner, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier–Pergamon, Oxford, 2nd edn, 2004, vol. 8, p. 459 Search PubMed.
- M. J. Baker-Hawkes, E. Billig and H. B. Gray, Am. Chem. Soc., 1966, 88(21), 4870–4875 CrossRef CAS.
- D. J. Harrison, N. Nguyen, A. J. Lough and U. Fekl, J. Am. Chem. Soc., 2006, 128(34), 11026–11027 CrossRef CAS.
- R. Sarangi, S. DeBeer, G. Deanne, J. Rudd, R. K. Szilagyi, X. Ribas, C. Rovira, M. Almeida, K. O. Hodgson, B. Hedman and E. I. Solomon, J. Am. Chem. Soc., 2007, 129(8), 2316–2326 CrossRef CAS.
- A. Zarkadoulas, E. Koutsouri and C. A. Mitsopoulou, Coord. Chem. Rev., 2012, 256, 2424–2434 CrossRef CAS.
- E. Lyris, D. Argyropoulos, C. Mitsopoulou, D. Katakis and E. Vrachnou, J. Photochem. Photobiol., A, 1997, 108, 51–54 CrossRef CAS.
- C. K. Jørgensen, Coord. Chem. Rev., 1966, 1(1–2), 164–178 CrossRef.
- M. T. Ashby, J. H. Enemark and D. L. Lichtenberger, Inorg. Chem., 1988, 27(1), 191–197 CrossRef CAS.
- J. Springs, C. Janzen, M. Y. Darensbourg, J. C. Calabrese, P. J. Krusic, J. N. Verpeaux and C. Amatore, J. Am. Chem. Soc., 1990, 112(15), 5789–5797 CrossRef CAS.
- P. L. Braunwarth, G. Huttner, D. Guenauer, K. Evertz, W. Imhof, C. Emmerich and L. Zsolnai, Organometallics, 1991, 10(11), 3861–3873 CrossRef.
- B. Albela, E. Bothe, O. Brosch, K. Mochizuki, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 1999, 38(22), 5131–5138 CrossRef CAS.
- S. Kimura, E. Bill, E. Bothe, T. Weyhermuller and K. Wieghardt, J. Am. Chem. Soc., 2001, 123, 6025–6039 CrossRef CAS.
- T. Kusamoto and H. Nishihara, Coord. Chem. Rev., 2019, 380, 419–439 CrossRef CAS.
- R. A. L. Silva, I. C. Santos, S. Rabaça, E. B. Lopes and V. Gama, Crystals, 2018, 8, 141 CrossRef.
- N. Robertson and L. Cronin, Coord. Chem. Rev., 2002, 227, 93–127 CrossRef CAS.
- R. Eisenberg and H. B. Gray, Inorg. Chem., 2011, 50, 9741–9751 CrossRef CAS.
- S. Sproules and K. Wieghardt, Coord. Chem. Rev., 2011, 255, 837–860 CrossRef CAS.
- (a) D. Argyropoulos, E. Lyris, C. A. Mitsopoulou and D. Katakis, J. Chem. Soc., Dalton Trans., 1997, 4, 615–621 RSC; (b) D. Argyropoulos, C. A. Mitsopoulou and D. Katakis, Inorg. Chem., 1996, 35, 5549–5554 CrossRef CAS PubMed.
- C. Lauterbach and J. Fabian, Eur. J. Inorg. Chem., 1999, 11, 1995–2004 CrossRef.
- E. Bill, E. Bothe, P. Chaudhuri, K. Chlopek, D. Herebian, S. Kokatam, K. Ray, T. Weyhermüller, F. Neese and K. Wieghardt, Chem. – Eur. J., 2004, 11(1), 204–224 CrossRef CAS.
- T. Glaser, B. Hedman, K. O. Hodgson and E. I. Solomon, Acc. Chem. Res., 2000, 33(12), 859–868 CrossRef CAS.
- F. Neese, B. Hedman, K. O. Hodgson and E. I. Solomon, Inorg. Chem., 1999, 38, 4854–4860 CrossRef CAS.
- E. I. Solomon, B. Hedman, K. O. Hodgson, A. Dey and R. K. Szilagyi, Coord. Chem. Rev., 2005, 249, 97–129 CrossRef CAS.
- B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1990, 112(4), 1643–1645 CrossRef CAS.
- T. Glaser, B. Hedman, K. O. Hodgson and E. I. Solomon, Acc. Chem. Res., 2000, 33(12), 859–868 CrossRef CAS.
- R. K. Szilagyi, B. S. Lim, T. Glaser, R. H. Holm, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 2003, 125, 9158–9169 CrossRef CAS.
- L. Salem and C. Ronland, Angew. Chem., Int. Ed. Engl., 1972, 11(2), 92–111 CrossRef CAS.
- E. Evangelio and D. Ruiz-Molina, C. R. Chim., 2008, 11, 1137–1154 CrossRef CAS.
- W. Kaim, Inorg. Chem., 2011, 50, 9752–9765 CrossRef CAS.
- K. Ray, T. Weyhermuller, F. Neese and K. Wieghardt, Inorg. Chem., 2005, 44, 5345 CrossRef CAS.
- L. Pilia, D. Espa, A. Barsella, A. Fort, C. Makedonas, L. Marchiò, M. L. Mercuri, A. Serpe, C. A. Mitsopoulou and P. Deplano, Inorg. Chem., 2011, 50(20), 10015–10027 CrossRef CAS.
- D. Espa, L. Pilia, C. Makedonas, L. Marchiò, M. L. Mercuri, A. Serpe, A. Barsella, A. Fort, C. A. Mitsopoulou and P. Deplano, Inorg. Chem., 2014, 53(2), 1170–1183 CrossRef CAS.
- C. Makedonas and C. A. Mitsopoulou, Inorg. Chim. Acta, 2007, 360, 3997–4009 CrossRef CAS.
- P. Deplano, L. Pilia, D. Espa, M. Laura Mercuri and A. Serpe, Coord. Chem. Rev., 2010, 254, 1434–1447 CrossRef CAS.
- C. A. Mitsopoulou, Coord. Chem. Rev., 2010, 254(13), 1448–1456 CrossRef CAS.
- V. Bachler, G. Olbrich, F. Neese and K. Wieghardt, Inorg. Chem., 2002, 41(16), 4179–4193 CrossRef CAS PubMed.
- D. Herebian, E. Bothe, F. Neese, T. Weyhermuller and K. Wieghardt, J. Am. Chem. Soc., 2003, 125(30), 9116–9128 CrossRef CAS PubMed.
- C. A. Grapperhaus, P. M. Kozlowski, D. Kumar, H. N. Frye, K. B. Venna and S. Poturovic, Angew. Chem., Int. Ed., 2007, 46, 4085–4088 CrossRef CAS PubMed.
- K. Ouch, M. S. Mashuta and C. A. Grapperhaus, Inorg. Chem., 2011, 50(20), 9904–9914 CrossRef CAS PubMed.
- H. Tang, E. N. Brothers and M. B. Hall, Inorg. Chem., 2017, 56(1), 583–593 CrossRef CAS.
- P. Ghosh, E. Bill, T. Weyhermuller, F. Neese and K. Wieghardt, J. Am. Chem. Soc., 2003, 125, 1293–1308 CrossRef CAS PubMed.
- A. Kochem, G. Gellon, O. Jarjayes, C. Philouze, A. M. Hardemare, M. van Gastel and F. Thomas, Dalton Trans., 2015, 44(28), 12743–12756 RSC.
- Y. J. Yuan, Z. T. Yu, D. Q. Chen and Z. G. Zou, Chem. Soc. Rev., 2016, 46(3), 603–631 RSC.
- W. T. Eckenhoff and R. Eisenberg, Dalton Trans., 2012, 41(42), 13004–13021 RSC.
- C. Mitsopoulou, J. Konstantatos, D. Katakis and E. Vrachnou, J. Mol. Catal., 1991, 67, 137–146 CrossRef CAS.
- M. Wang, L. Chen and L. Sun, Energy Environ. Sci., 2012, 5(5), 6763–6778 RSC.
- V. S. Thoi, Y. Sun, J. R. Long and C. J. Chang, Chem. Soc. Rev., 2013, 42(6), 2388–2400 RSC.
- S. Berardi, S. Drouet, L. Francas, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, C. Richmond, T. Stolla and A. Llobet, Chem. Soc. Rev., 2014, 43(22), 7501–7519 RSC.
- S. Fukuzumi, T. Suenobu and Y. Yamada, Organometallics and Related Molecules for Energy Conversion, Springer, Berlin, Heidelberg, 2015, pp. 313–345 Search PubMed.
- B. H. Solis and S. Hammes-Schiffer, Inorg. Chem., 2014, 53(13), 6427–6443 CrossRef CAS.
- R. H. Morris, Chem. Rev., 2016, 116(15), 8588–8654 CrossRef CAS.
- N. V. Belkova, O. A. Filippov and E. S. Shubina, Chem. – Eur. J., 2018, 24, 1464–1470 CrossRef CAS.
- E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. M. Bullock, A. J. M. Miller and A. M. Appel, Chem. Rev., 2016, 116, 8655–8692 CrossRef CAS.
- Y. Matsubara, E. Fujita, M. D. Doherty, J. T. Muckerman and C. Creutz, J. Am. Chem. Soc., 2012, 134, 15743–15757 CrossRef CAS.
- V. Artero and M. Fontecave, Chem. Soc. Rev., 2013, 42(6), 2338–2356 RSC.
- A. Z. Haddad, S. P. Cronin, M. S. Mashuta, R. M. Buchanan and C. A. Grapperhaus, Inorg. Chem., 2017, 56, 11254–11265 CrossRef CAS.
- A. Volbeda, E. Garcin, C. Piras, A. L. de Lacey, V. M. Fernandez, E. C. Hatchikian, M. Frey and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 1996, 118(51), 12989–12996 CrossRef CAS.
- F. Himo, L. A. Eriksson, F. Maseras and P. E. M. Siegbahn, J. Am. Chem. Soc., 2000, 122(33), 8031–8036 CrossRef CAS.
- B. H. Solis, A. G. Maher, T. Honda, D. C. Powers, D. G. Nocera and S. Hammes-Schiffer, ACS Catal., 2014, 4(12), 4516–4526 CrossRef CAS.
- E. J. Thompson and L. A. Berben, Angew. Chem., Int. Ed., 2015, 54, 11642–11646 CrossRef CAS.
- G. G. Luo, H. L. Zhang, Y. W. Tao, Q. Y. Wu, D. Tian and Q. Zhang, Inorg. Chem. Front., 2019, 6(2), 343–354 RSC.
- M. R. DuBois and D. L. DuBois, Chem. Soc. Rev., 2009, 38, 62–72 RSC.
- D. Borgis and J. T. Hynes, Chem. Phys., 1993, 170(3), 315–346 CrossRef CAS.
- S. Fukuzumi, T. Kobayashi and T. Suenobu, J. Am. Chem. Soc., 2010, 132(5), 1496–1497 CrossRef CAS.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65(3), 599 CrossRef CAS.
- K. J. Lee, N. Elgrishi, B. Kandemir and J. L. Dempsey, Nat. Rev. Chem., 2017, 1(5), 0039 CrossRef CAS.
- Y. Wu, N. Rodríguez-López and D. Villagrán, Chem. Sci., 2018, 9, 4689–4695 RSC.
- A. Z. Haddad, D. Kumar, K. O. Sampson, A. M. Matzner, M. S. Mashuta and C. A. Grapperhaus, J. Am. Chem. Soc., 2015, 137(29), 9238–9241 CrossRef CAS.
- A. Z. Haddad, B. D. Garabato, P. M. Kozlowski, R. M. Buchanan and C. A. Grapperhaus, J. Am. Chem. Soc., 2016, 138(25), 7844 CrossRef CAS.
- C. P. Casey and J. B. Johnson, Can. J. Chem., 2005, 83(9), 1339–1346 CrossRef CAS.
- B. Chaudret and K. Philippot, Oil Gas Sci. Technol., 2007, 62, 799–817 CrossRef CAS.
- N. Elgrishi, B. D. McCarthy, E. S. Rountree and J. L. Dempsey, ACS Catal., 2016, 6, 3644–3659 CrossRef CAS.
- K. J. Lee, B. D. McCarthy and J. L. Dempsey, Chem. Soc. Rev., 2019, 48, 2927 RSC.
- V. Artero and J. M. Savéant, Energy Environ. Sci., 2014, 7, 3808–3814 RSC.
- C. Costentin, S. Drouet, M. Robert and J. M. Saveant, J. Am. Chem. Soc., 2012, 134, 11235–11242 CrossRef CAS PubMed.
- V. Fourmond, P. A. Jacques, M. Fontecave and V. Artero, Inorg. Chem., 2010, 49, 10338–10347 CrossRef CAS.
- A. M. Appel and M. L. Helm, ACS Catal., 2014, 4, 630–633 CrossRef CAS.
- P. A. Jacques, V. Artero, J. Pecaut and M. Fontecave, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20627–20632 CrossRef CAS.
- M. D. Wodrich and X. Hu, Nat. Rev. Chem., 2018, 2(1), 0099 CrossRef CAS.
- T. B. Rauchfuss, Acc. Chem. Res., 2015, 48(7), 2107–2116 CrossRef CAS.
- K. Weber, T. Krämer, H. S. Shafaat, T. Weyhermüller, E. Bill, M. van Gastel, F. Neese and W. Lubitz, J. Am. Chem. Soc., 2012, 134(51), 20745–20755 CrossRef CAS.
- T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Coord. Chem. Rev., 2014, 270, 127–150 CrossRef.
- C. Wombwell, C. A. Caputo and E. Reisner, Acc. Chem. Res., 2015, 48(11), 2858–2865 CrossRef CAS.
- T. B. Rauchfuss, Inorg. Chem., 2004, 43(1), 14–26 CrossRef CAS.
- J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107(11), 5411 CrossRef CAS.
- E. S. Wiedner, J. Am. Chem. Soc., 2019, 141(18), 7212–7222 CrossRef CAS PubMed.
- D. W. Mulder, Y. Guo, M. W. Ratzloff and P. W. King, J. Am. Chem. Soc., 2016, 139(1), 83–86 CrossRef.
- E. J. Reijerse, C. C. Pham, V. Pelmenschikov, R. G. Wilson, A. A. Venkatesh, J. F. Siebel, L. Gee, Y. Yoda, K. Tamasaku, W. Lubitz, T. B. Rauchfuss and S. P. Cramer, J. Am. Chem. Soc., 2017, 139(12), 4306–4309 CrossRef CAS.
- A. A. Silakov, C. Lambertz, O. Rüdiger, T. Happe, E. Reijerse and W. Lubitz, Angew. Chem., Int. Ed., 2013, 51(46), 11458–11462 Search PubMed.
- V. Pelmenschikov, J. A. Birrell, C. C. Pham, N. Mishra, H. Wang, C. Sommer, E. Reijerse, C. P. Richers, K. Tamasaku, Y. Yoda, T. B. Rauchfuss, W. Lubitz and S. P. Cramer, J. Am. Chem. Soc., 2017, 139(46), 16894–16902 CrossRef CAS.
- S. Qiu, L. M. Azofra, D. R. MacFarlane and C. Sun, Phys. Chem. Chem. Phys., 2016, 18, 15369–15374 RSC.
- H. Tang and M. B. Hall, J. Am. Chem. Soc., 2017, 139(49), 18065–18070 CrossRef CAS.
- S. Qiu, L. M. Azofra, D. R. MacFarlane and C. Suna, ACS Catal., 2016, 6(8), 5541–5548 CrossRef CAS.
- B. L. Greene, C. H. Wu, P. M. McTernan, M. W. W. Adams and R. B. Dyer, J. Am. Chem. Soc., 2015, 137, 4558–4566 CrossRef CAS.
- W. R. McNamara, Z. Han, P. J. Alperin, W. W. Brennessel, P. L. Holland and R. Eisenberg, J. Am. Chem. Soc., 2011, 133, 15368–15371 CrossRef CAS.
- W. R. McNamara, Z. Han, C. J. M. Yin, W. W. Brennessel, P. L. Holland and R. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(39), 15594–15599 CrossRef CAS.
- B. H. Solis and S. Hammes-Schiffer, J. Am. Chem. Soc., 2012, 134(37), 15253–15256 CrossRef CAS.
- K. J. Lee, B. D. McCarthy, E. S. Rountree and J. L. Dempsey, Inorg. Chem., 2017, 56(4), 1988–1998 CrossRef CAS.
- A. Zarkadoulas, M. J. Field, V. Artero and C. Mitsopoulou, ChemCatChem, 2017, 9(12), 2308–2317 CrossRef CAS.
- M. Fang, M. H. Engelhard, Z. Zhu, M. L. Helm and J. A. S. Roberts, ACS Catal., 2014, 4, 90–98 CrossRef CAS.
- D. Sellmann, M. Geck and M. Moll, J. Am. Chem. Soc., 1991, 113(14), 5259–5264 CrossRef CAS.
- H. Lv, T. P. A. Ruberu, V. E. Fleischauer, W. W. Brennessel, M. L. Neidig and R. Eisenberg, J. Am. Chem. Soc., 2016, 138, 11654–11663 CrossRef CAS.
- B. S. Kang, L. H. Weng, D. X. Wu, F. Wang, R. Huang, Z. Y. Huang and H. Q. Liu, Inorg. Chem., 1988, 27(7), 1128–1130 CrossRef CAS.
- W. T. Eckenhoff, W. W. Brennessel and R. Eisenberg, Inorg. Chem., 2014, 53(18), 9860–9869 CrossRef CAS.
- T. Yamaguchi, S. Masaoka and K. Sakai, Chem. Lett., 2009, 38(5), 434–435 CrossRef CAS.
- A. Begum and S. Sarkar, Eur. J. Inorg. Chem., 2012, 40–43 CrossRef CAS.
- A. Begum, G. Moula and S. Sarkar, Chem. – Eur. J., 2010, 16, 12324–12327 CrossRef CAS.
- M. Gomez-Mingot, J. P. Porcher, T. K. Todorova, T. Fogeron, C. Mellot-Draznieks, Y. Li and M. Fontecave, J. Phys. Chem., 2015, 119, 13524–13533 CrossRef CAS PubMed.
- C. S. Letko, J. A. Panetier, M. Head-Gordon and T. D. Tilley, J. Am. Chem. Soc., 2014, 136(26), 9364–9376 CrossRef CAS.
- A. Zarkadoulas, M. J. Field, C. Papatriantafyllopoulou, J. Fize, V. Artero and C. A. Mitsopoulou, Inorg. Chem., 2015, 55(2), 432–444 CrossRef.
- A. Zarkadoulas, E. Koutsouri, E. Semidalas, V. Psycharis, C. P. Raptopoulou and C. A. Mitsopoulou, Polyhedron, 2018, 152, 138–146 CrossRef CAS.
- H. Rao, Z. Y. Wang, H. Q. Zheng, X. B. Wang, C. M. Pan, Y. T. Fan and H. W. Hou, Catal. Sci. Technol., 2015, 5(4), 2332–2339 RSC.
- F. J. Hine, A. J. Taylor and C. D. Garner, Coord. Chem. Rev., 2010, 254(13–14), 1570–1579 CrossRef CAS.
- T. Fogeron, J. P. Porcher, M. Gomez-Mingot, T. K. Todorova, L. M. Chamoreau, C. Mellot-Draznieks, Y. Li and M. Fontecave, Dalton Trans., 2016, 45(37), 14754–14763 RSC.
- J. P. Porcher, T. Fogeron, M. Gomez-Mingot, E. Derat, L. M. Chamoreau, Y. Li and M. Fontecave, Angew. Chem., Int. Ed., 2015, 54, 14090–14093 CrossRef CAS.
- K. Koshiba, K. Yamauchi and K. Sakai, Angew. Chem., Int. Ed., 2017, 56, 1–6 CrossRef PubMed.
- K. Koshiba, K. Yamauchi and K. Sakai, Dalton Trans., 2019, 48, 635 RSC.
- K. Koshiba, K. Yamauchi and K. Sakai, ChemElectroChem, 2019, 6(8), 2273–2281 CrossRef CAS.
- M. Hang, V. Huynh and T. J. Meyer, Chem. Rev., 2007, 107, 5004–5064 CrossRef.
- Y. Kimura, M. Hayashi, Y. Yoshida and H. Kitagawa, Inorg. Chem., 2019, 58(6), 3875–3880 CrossRef CAS.
- Y. Aimoto, K. Koshiba, K. Yamauchi and K. Sakai, Chem. Commun., 2018, 54, 12820–12823 RSC.
- S. H. Schlindwein, M. R. Ringenberg, M. Nieger, D. Gudat and Z. Anorg, Allg. Chem., 2017, 643, 1628–1634 CrossRef CAS.
- L. Gan, T. L. Groy, P. Tarakeshwar, S. K. S. Mazinani, J. Shearer, V. Mujica and A. K. Jones, J. Am. Chem. Soc., 2015, 137(3), 1109–1115 CrossRef CAS.
- Z. Han, W. R. McNamara, M. S. Eum, P. L. Holland and R. Eisenberg, Angew. Chem., Int. Ed., 2012, 51, 1667–1670 CrossRef CAS.
- C. N. Virca and M. McCormick, Dalton Trans., 2015, 44, 14333 RSC.
- Z. Han, L. Shen, W. W. Brennessel, P. L. Holland and R. Eisenberg, J. Am. Chem. Soc., 2013, 135(39), 14659–14669 CrossRef CAS.
- C. N. Virca, J. R. Lohmolder, J. B. Tsang, M. M. Davis and T. M. McCormick, J. Phys. Chem. A, 2018, 122(11), 3057–3065 CrossRef CAS.
- H. Rao, W. Q. Yu, H. Q. Zheng, J. Bonin, Y. T. Fan and H. W. Hou, J. Power Sources, 2016, 324, 253–260 CrossRef CAS.
- C. Zhang, G. Li and X. Cai, Int. J. Energy Res., 2018, 42, 977–984 CrossRef CAS.
- A. Das, Z. Han, W. W. Brennessel, P. L. Holland and R. Eisenberg, ACS Catal., 2015, 5(3), 1397–1406 CrossRef CAS.
- S. Koroidov, K. Hong, K. S. Kjaer, L. Li, K. Kunnus, M. Reinhard, R. W. Hartsock, D. Amit, R. Eisenberg, C. D. Pemmaraju, K. J. Gaffney and A. A. Cordones, Inorg. Chem., 2018, 57(21), 13167–13175 CrossRef CAS.
- C. A. Grapperhaus, K. Ouch and M. S. Mashuta, Redox-Regulated Ethylene Binding to a Rhenium-Thiolate Complex, J. Am. Chem. Soc., 2009, 131(1), 64–65 CrossRef CAS.
- R. Jain, M. S. Mashuta, R. M. Buchanan and C. A. Grapperhaus, Eur. J. Inorg. Chem., 2017, 3714–3719 CrossRef CAS.
- C. A. Grapperhaus and M. Y. Darensbourg, Acc. Chem. Res., 1998, 31(8), 451–459 CrossRef CAS.
- G. G. Luo, Y. H. Wang, J. H. Wang, J. H. Wua and R. B. Wu, Chem. Commun., 2017, 53(52), 7007–7010 RSC.
- S. Inoue, M. Mitsuhashi, T. Ono, Y. N. Yan, Y. Kataoka, M. Handa and T. Kawamoto, Inorg. Chem., 2017, 56, 12129–12138 CrossRef CAS.
- T. Kawamoto, N. Suzuki, T. Ono, D. Gong and T. Konno, Chem. Commun., 2013, 49(7), 668–670 RSC.
- L. L. Tinker, N. D. McDaniel, P. N. Curtin, C. K. Smith, M. J. Ireland and S. Bernhard, Chem. – Eur. J., 2007, 13, 8726–8732 CrossRef CAS.
- D. Hong, Y. Tsukakoshi, H. Kotani, T. Ishizuka, K. Ohkubo, Y. Shiota, K. Yoshizawa, S. Fukuzumi and T. Kojima, Inorg. Chem., 2018, 57(12), 7180–7190 CrossRef CAS.
- J. M. Lei, S. P. Luo and S. Z. Zhan, Int. J. Hydrogen Energy, 2018, 43(41), 19047–19056 CrossRef CAS.
- B. M. Paterson and P. S. Donnelly, Chem. Soc. Rev., 2011, 40(5), 3005–3018 RSC.
- C. F. Wise, D. Liu, K. J. Mayer, P. M. Crossland, C. L. Harley and W. R. McNamara, Dalton Trans., 2015, 44(32), 14265–14271 RSC.
- X. Jing, P. Wu, X. Liu, L. Yang, C. He and C. Duan, New J. Chem., 2015, 39, 1051 RSC.
- Y. Zhao, Y. Wang, Q. Wu, J. Lin, S. Wu, W. Hou, R. Wu and G. Luo, Chin. J. Catal., 2018, 39, 517–526 CrossRef CAS.
- T. Straistari, J. Fize, S. Shova, M. Rglier, V. Artero and M. Orio, ChemCatChem, 2016, 8, 1–8 CrossRef.
- T. Straistari, R. Hardre, J. Fize, S. Shova, M. Giorgi, M. Reglier, V. Artero and M. Orio, Chem. – Eur. J., 2018, 24, 8779–8786 CrossRef CAS.
- T. Straistari, R. Hardré, J. Massin, M. Attolini, B. Faure, M. Giorgi, M. Réglier and M. Orio, Eur. J. Inorg. Chem., 2018,(20–21), 2259–2266 CrossRef CAS.
- S. Gulati, O. Hietsoi, C. A. Calvary, J. M. Strain, S. Pishgar, H. C. Brun, C. A. Grapperhaus, R. M. Buchanan and J. Spurgeon, Chem. Commun., 2019, 55(64), 9440–9443 RSC.
- C. A. Calvary, O. Hietsoi, J. M. Strain, M. S. Mashuta, J. M. Spurgeon, R. M. Buchanan and C. A. Grapperhaus, Eur. J. Inorg. Chem., 2019, 33, 3782–3790 CrossRef.
| This journal is © the Partner Organisations 2020 |