
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTetrazine- and trans-cyclooctene-functionalised polypept(o)ides for fast bioorthogonal tetrazine ligation†
Kerstin
Johann
a,
Dennis
Svatunek
 b,
Christine
Seidl
a,
Silvia
Rizzelli
a,
Tobias A.
Bauer
a,
Lydia
Braun
a,
Kaloian
Koynov
c,
Hannes
Mikula
b and
Matthias
Barz
*a
b,
Christine
Seidl
a,
Silvia
Rizzelli
a,
Tobias A.
Bauer
a,
Lydia
Braun
a,
Kaloian
Koynov
c,
Hannes
Mikula
b and
Matthias
Barz
*a
aDepartment of Chemistry, Johannes Gutenberg University Mainz, Duesbergweg 10-14, 55128 Mainz, Germany. E-mail: barz@uni-mainz.de
bInstitute of Applied Synthetic Chemistry, Technische Universität Wien, Getreidemarkt 9, 1060 Vienna, Austria
cMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany
First published on 29th June 2020
Abstract
The inverse electron demand Diels–Alder (IEDDA) reaction-initiated ligation between 1,2,4,5-tetrazines (Tz) and trans-cyclooctenes (TCO) is one of the fastest bioorthogonal reactions known today and is therefore increasingly used for in vivo click chemistry. Described herein is the synthesis of Tz- and TCO-functionalised polypeptides, polypeptoids and polypeptide-block-polypeptoids (polypept(o)ides) by ring-opening polymerisation of the corresponding N-carboxyanhydrides using Tz- or TCO-functional amine initiators. Despite the reactivity of tetrazines, polymers with low dispersity and high end group integrity can be obtained as observed by gel permeation chromatography (GPC), nuclear magnetic resonance (NMR) spectroscopy and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry. Amphiphilic Tz-functionalised block copolypept(o)ides were used to prepare polymeric micelles and organic colloids by miniemulsion techniques, which may find an application as clearing agents in pretargeted nuclear imaging and therapy using efficient in vivo click chemistry. The reaction kinetics of the tetrazine ligation using the synthesised polymers and the accessibility of the Tz groups on the polymeric nanoparticles were evaluated using UV–Vis and fluorescence correlation spectroscopy (FCS), and second-order rate constants were determined by stopped-flow spectrophotometry ensuring quantitative conversions in seconds at sub-millimolar concentrations (10–30 s).
Introduction
In the last decade, the development of fast chemoselective reactions for efficient labelling of molecules in vitro and also in vivo, so-called bioorthogonal reactions, has seen significant advances.1 Bertozzi et al. were the first to show that the bioorthogonal reaction between azides and alkynes can be accelerated by using strained reactants.2 In 2008, the Fox and Weissleder lab independently reported on the inverse electron demand Diels–Alder (IEDDA) reaction of 1,2,4,5-tetrazines (Tz) with strained alkenes as fast bioorthogonal reactions for in vivo click chemistry.3 This type of reaction has since been intensively investigated and the reactants have been further optimized in order to increase the rates of the reaction while retaining stability under physiological conditions.4,5 Today, the fastest known click reactions occur between Tz and trans-cyclooctenes (TCO) with second-order rate constants of up to 3.3 × 106 M−1 s−1 and between Tz and trans-1-sila-4-cycloheptene with k2 = 1.14 × 107 M−1 s−1.4,6 It is therefore not surprising that the tetrazine ligation has been increasingly used in biomedical research, for example in pretargeting strategies,7,8 in peptide chemistry,9 for site-specific labelling of proteins,10 as bioorthogonal cell glue11 or for triggered drug release via “click-to-release” chemistry.12With regard to the use of IEDDA reactions in polymer chemistry, the Tz-norbornene reaction was for example utilised for the post-polymerisation functionalisation of norbornene-functionalised polylactide13 and poly(carbonate)s14 or polymer–polymer coupling of poly(ethylene glycol) (PEG)-Tz and polystyrene-norbornene.15 The Tz-TCO ligation, however, has not been widely used in the field of polymer science yet. This reaction has for instance been employed for site-selective protein modification,16 to synthesise homodimeric protein-polymer conjugates17 or for surface functionalisation.18 Furthermore, Tz-modified dextran polymers have been applied in pretargeted nuclear imaging approaches using the tetrazine ligation, both as a secondary imaging agent and as masking agent.19,20 Clearing agents for pretargeting strategies have also been prepared using Tz-functionalised albumin and polystyrene beads with albumin-Tz coating.21 Kara et al. developed a synthetic approach for direct tetrazine synthesis on the backbone of poly(N-isopropyl acrylamide) (PNIPAAm) and PEG-based polymers.22 Recently, Kramer et al. synthesised N-(2-hydroxypropyl)methacrylamide (HPMA)-based block copolymers functionalised with Tz groups either at the chain end by using a Tz-functional chain transfer agent or randomly distributed by post-polymerisation modification.23 In a recent work, we developed TCO-functionalised molecular polymer brushes (PeptoBrushes), polypeptide-graft-polypeptoid polymers functionalised with multiple TCO groups, as tumour targeting agents for pretargeted nuclear imaging.8
While polypeptides are an established class of polymer materials,24,25 polypeptoids26,27 and polypept(o)ides28 are just recently investigated in greater detail. Polypept(o)ides, hybrid materials combining polypeptides with the polypeptoid polysarcosine (pSar; poly(N-methylated glycine)), combine chemical versatility with high biocompatibility, which seems to be particularly interesting for applications in nanomedicine.29,30 Both polypeptides and polypeptoids can be conveniently synthesised by ring-opening N-carboxyanhydride (NCA) polymerisation yielding polymers with narrow molecular weight distributions and with the possibility to introduce functional end groups by the use of functional initiators.31–33 The non-ionic, hydrophilic pSar has shown stealth-like properties comparable to those of PEG, i.e. high solubility in water and low cellular toxicity and immunogenicity in various preclinical models.8,27,30,34,35 It represents exclusively an H-bond acceptor without donor properties and thus follows the Whitesides’ rules for protein resistant materials.36 In contrast, polypeptides can also be of anionic, cationic or hydrophobic nature. They can be stimuli-responsive, biodegradable and undergo secondary structure-directed self-assembly in solution.25,37 We previously reported on the synthesis of end group functionalised polysarcosine and polypeptides for the application in copper(I)-catalysed azide–alkyne coupling (CuAAC), strain-promoted azide–alkyne coupling (SPAAC) and native chemical ligation (NCL).33
In this work, we describe the straightforward synthesis of Tz- and TCO-functionalised pSar, poly(γ-benzyl-L-glutamate) (pGlu(OBn)) and the amphiphilic block copolypept(o)ide pSar-block-pGlu(OBn), by using the respective functional initiators, as well as their use in the tetrazine ligation. Polymeric micelles and organic colloids based on poly(D,L-lactide) were also prepared from Tz-pSar-block-pGlu(OBn). It will be demonstrated that the Tz groups preserve their reactivity and can be used for efficient functionalisation of the synthesised polymers, block copolymer synthesis and the functionalisation of polymeric nanoparticles.
Experimentals
Materials and methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–28000 g mol−1) was purchased from Sigma Aldrich. Deuterated solvents were obtained from Deutero GmbH (Kastellaun, Germany). Milli-Q (MP) water (Millipore) with a resistance of 18.2 MΩ and TOC <3 ppm was used throughout the experiments. Dialysis was performed with Spectra/Por membranes (Roth) with a nominal molecular weight cut-off of 3500 g mol−1. Cy5-TCO was purchased from Click Chemistry Tools (Scottsdale, Arizona, US).
000–28000 g mol−1) was purchased from Sigma Aldrich. Deuterated solvents were obtained from Deutero GmbH (Kastellaun, Germany). Milli-Q (MP) water (Millipore) with a resistance of 18.2 MΩ and TOC <3 ppm was used throughout the experiments. Dialysis was performed with Spectra/Por membranes (Roth) with a nominal molecular weight cut-off of 3500 g mol−1. Cy5-TCO was purchased from Click Chemistry Tools (Scottsdale, Arizona, US).
Analytical hexafluoroisopropanol (HFIP) gel permeation chromatography (GPC) was carried out at a flow rate of 0.8 mL min−1 at 40 °C with 3 g L−1 potassium trifluoroacetate added to the eluent. The GPC system was equipped with a UV detector (Jasco UV-2075 Plus) set at a wavelength of 230 nm and an RI detector (Jasco RI-2031). Modified silica gel columns (PFG columns, particle size: 7 μm, porosity: 100 Å and 4000 Å) were used. Molecular weights were determined by using a calibration with poly(methyl methacrylate) (PMMA) standards (Polymer Standards Service GmbH Mainz) and toluene as an internal standard. The degree of polymerisation (DP) of polysarcosine (pSar) was determined by calibration of apparent Mn against a series of pSar standards characterised by static light scattering to obtain absolute molecular weights.34 Prior to measurement, the samples were filtered through polytetrafluoroethylene (PTFE) syringe filters with a pore size of 0.2 μm. The elution diagram was analysed with WinGPC software (Polymer Standards Service GmbH Mainz).
Attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy was performed on an FT/IR-4100 (JASCO Corporation) with an ATR sampling accessory (MIRacleTM, Pike Technologies). The IR spectra were analysed with the software Spectra Manager version 2.02.05 (JASCO Corporation). 16 scans were performed per measurement.
Single-angle dynamic light scattering (DLS) measurements were performed on a Zetasizer Nano ZS (Malvern Instruments Ltd, Worcestershire, UK) at an angle of 173° and a wavelength of 633 nm at 25 °C. Three measurements were performed per sample at a concentration of 0.3 mg mL−1 in 10 mM NaCl solution, and size distribution (intensity-weighted) histograms were calculated based on the autocorrelation function of samples, with automated attenuator adjustment and multiple scans (typically 10–15 scans). For the analysis of the data, Malvern Zetasizer Software version 7.12 was used.
Ultraviolet–visible (UV–Vis) spectroscopy was carried out on a Jasco V-630 spectrophotometer. The UV–Vis spectra were analysed with the software Spectra Manager version 2.07.02 (JASCO Corporation).
Melting points were measured using a Mettler FP62 melting point apparatus at a heating rate of 1 °C min−1.
Reaction kinetics of Tz-polymers with PEG4-TCO were determined using pseudo-first-order measurements with an excess of PEG4-TCO in PBS (pH = 7.4) at 37.0 ± 0.1 °C following the decrease of tetrazine absorbance at 535 nm. Measurements were performed in triplicates using an SX20 stopped-flow photometer (Applied Photophysics, UK) equipped with a 535 nm LED light source. Data analysis was performed using Prism 6 (Graphpad) to determine the observed rate constants which were converted into second-order rate constants through dividing by the TCO concentration. Reaction kinetics of the reference compounds HELIOS 347Me and HELIOS 388Me with PEG4-TCO or TCO-polymers were determined using pseudo-first-order measurements with an excess of the TCO compound in PBS (pH = 7.4) at 37.0 ± 0.1 °C following the increase of fluorescence at >400 nm. Measurements were performed in triplicates using an SX20 stopped-flow photometer (Applied Photophysics, UK) equipped with a 360 nm LED light source and a photomultiplier type R374 in combination with a 400 nm longpass filter as detector. Data analysis was performed using Prism 6 (Graphpad) to determine the observed rate constants which were converted into second-order rate constants through dividing by the TCO concentration. The used concentrations, observed rate constants and calculated second-order rate constants are shown in Table 2.
MALDI-TOF mass spectra were recorded on a Shimadzu Axima CFR MALDI-TOF mass spectrometer equipped with a nitrogen laser delivering 3 ns pulses at a wavelength of 337 nm or on a Bruker rapifleX MALDI-TOF/TOF mass spectrometer equipped with a 10 kHz scanning smartbeam 3D laser. HABA (2-(4′-hydroxybenzeneazo)benzoic acid) or dithranol were used as matrices for pSar or pGlu(OBn) respectively. Samples were prepared by dissolving the polymer in methanol or dichloromethane at a concentration of 1 mg mL−1. 10 μL of the sample solution and 10 μL of matrix solution (10 mg mL−1) in methanol or dichloromethane were added to a multistage target and the solvents were evaporated to create a thin matrix/analyte film. The samples were measured in linear or reflectron mode and analysed using mMass software version 5.5.0. Additionally, spectra were analysed using MATLAB. After baseline correction and peak assignment, dispersities of the molecular weight distribution were calculated for each polymer.
Fluorescence correlation spectroscopy (FCS) experiments were performed using a commercial FCS setup (Zeiss, Germany) consisting of the module ConfoCor 2 and an inverted microscope model Axiovert 200 with a Zeiss C-Apochromat 40×/1.2 W water immersion objective. The excitation was done by the 633 nm spectral line of a He/Ne laser and the collected fluorescence was filtered through an LP650 long pass emission filter before reaching the detector, an avalanche photodiode that enables single-photon counting. Eight-well polystyrene-chambered coverglass (Laboratory-Tek, Nalge Nunc International) was used as a sample cell. For each solution, a series of 10 measurements with a total duration of 5 min were performed. The obtained experimental autocorrelation curves were fitted with the theoretical model function for an ensemble of m different types of freely diffusing fluorescence species. The fits yielded the diffusion times of the fluorescent species from which the respective diffusion coefficients were evaluated.38 Finally, the hydrodynamic radii (Rh) were calculated (assuming spherical particles) by using the Stokes–Einstein relation:
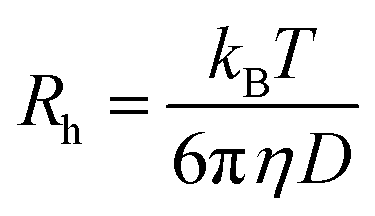
In this equation, kB is the Boltzmann constant, T is the temperature, D the diffusion coefficient and η is the viscosity of water. A reference standard with known diffusion coefficient (Alexa Fluor 647) was used for calibration.
Scanning electron microscopy (SEM) images were recorded on a field emission microscope (LEO 1530 Gemini) working at an acceleration voltage of 0.7 kV. Samples were dried on a silica wafer at a concentration of approximately 0.1 mg mL−1. The images were analysed using ImageJ.
Atomic force microscopy (AFM) images were obtained on a Cypher AFM (Asylum Research) at a scan rate of 1.0 Hz. Samples were prepared by placing a drop of the particle solution with a concentration of approximately 0.1 mg mL−1 onto freshly cleaned mica. The recorded images were analysed using Gwyddion software, version 2.49.
Synthetic procedures
1H NMR (400 MHz, DMSO-d6): δ [ppm] = 8.47–8.37 (m, 2H, CHarom,mTz), 7.60–7.40 (m, 2H, CHarom,mTz), 4.55–3.75 (m, 198H (2n), br, –NCH3–CH2–CO–), 3.10–2.60 (m, 303H (3n), br, –NCH3).
GPC in HFIP (vs. PMMA standards): Mn = 23.8 kg mol−1, Đ = 1.14. Degree of polymerisation (DP) was determined to be 101 by calibration of apparent Mn against a series of pSar standards characterised by static light scattering to obtain absolute molecular weights.34
1H NMR (400 MHz, DMSO-d6): δ [ppm] = 7.80 (s, 1H, NH), 6.94 (s, 1H, NH), 5.64–5.36 (m, 2H, –CH–CH–), 4.72–3.78 (m, 120H (2n), br, –NCH3–CH2–CO–), 3.17–2.60 (m, 184H (3n), br, –NCH3), 2.37–2.15 (m, 4H, –CH2–), 1.97–1.73 (m, 4H, –CH2–), 1.70–1.38 (m, 6H, –CH2–).
GPC in HFIP (vs. PMMA standards): Mn = 17.5 kg mol−1, Đ = 1.10. Degree of polymerisation (DP) was determined to be 66 by calibration of apparent Mn against a series of pSar standards characterised by static light scattering to obtain absolute molecular weights.34
General procedure for the synthesis of mTz-poly(γ-benzyl-L-glutamate) (mTz-pGlu(OBn)) using the example of mTz-pGlu(OBn)62.
Polymerisation of Glu(OBn) NCA using mTz-NH2 as initiator was carried out at 0 °C analogously to the synthesis of mTz-pSar. Polymer end groups were acetylated by addition of 20 eq. triethylamine and 10 eq. acetic anhydride when complete conversion of the reaction was indicated by IR spectroscopy. The solution was stirred overnight at 0 °C and afterwards precipitated in diethyl ether as described for pSar. mTz-pGlu(OBn)62 was obtained as a pink solid in a yield of 99%.
1H NMR (400 MHz, DMSO-d6): δ [ppm] = 8.81–7.82 (55H (1n), br, NH), 7.62–7.52 (m, 2H, CHarom,mTz), 7.50–6.94 (m, 305H (5n), –CHarom.), 5.30–4.72 (m, 124H (2n), –CH2–C6H5), 4.33–3.70 (m, 62H (1n), NH–CH–CO–), 2.98 (s, 3H, –CH3), 2.42–1.62 (m, 190H (4n), –CH2–CH2–).
GPC in HFIP (vs. PMMA standards): Mn = 16.2 kg mol−1, Đ = 1.12.
1H NMR (400 MHz, DMSO-d6): δ [ppm] = 8.55–7.68 (m, 19H (1n), br, NH), 7.50–6.94 (m, 112H (5n), –CHarom.), 5.64–5.30 (m, 2H, –CH–CH–), 5.29–4.67 (m, 46H (2n), –CH2–C6H5), 4.22–3.72 (m, 23H (1n), NH–CH–CO–), 3.16–2.89 (m, 4H, –CH2–), 2.45–1.75 (m, 78H (4n), –CH2–CH2–), 1.69–1.42 (m, 6H, –CH2–).
GPC in HFIP (vs. PMMA standards): Mn = 9.7 kg mol−1, Đ = 1.15.
1H NMR (400 MHz, DMSO-d6): δ [ppm] = 8.47–8.38 (m, 2H, CHarom,mTz), 8.16–7.90 (5H (1n), NH), 7.60–7.47 (m, 2H, CHarom,mTz), 7.40–7.10 (m, 104H (5n), –CHarom.), 5.15–4.85 (m, 40H (2n), –CH2–C6H5), 4.63–3.57 (m, 454H (1n + 2m), NH–CH–CO–, –NCH3–CH2–CO–), 3.03–2.64 (m, 682H (3m), –NCH3), 2.42–1.62 (m, 72H (4n), –CH2–CH2–).
GPC in HFIP (vs. PMMA standards): Mn = 40.2 kg mol−1, Đ = 1.21.
To measure the efficiency and kinetics of the tetrazine ligation by UV–Vis spectroscopy (λ = 540 nm), 70 μL of stock solution of a Tz-functionalised polymer were directly added to a cuvette and measured once. Then, 70 μL of stock solution of a TCO-functionalised polymer at the same concentration were added, mixed with a pipette and measured at the specified time points.
For FCS measurements, Tz-functionalised polymers or nanoparticles were fluorescently labelled using Cy5-TCO. Briefly, Cy5-TCO was added to a hundredfold excess of Tz-functionalised polymer or nanoparticle solution in water from a stock solution, shaken at room temperature for 2 h and stored at 4 °C until measurement. Samples were diluted to a final concentration of 10–100 nM Cy5 prior to measurement.
Nanoparticle preparation
000–28000 g mol−1) were dissolved in 1.25 g chloroform and 10 mg mTz-functionalised block copolymer were dissolved in 3 mL MP-water. The organic phase was added to the aqueous phase and subjected to ultrasonication in a glass vial under ice-cooling for 180 s at 70% amplitude in a pulse regime (10 s sonication, 10 s pause). A Branson Digital Sonifier W-250 with a 1/4′′ tip was used for ultrasonication. The obtained miniemulsion was slowly stirred overnight at room temperature until the evaporation of the organic solvent was completed. To separate the nanoparticles from bigger aggregates, the colloidal solution was filtered through syringe filters with a pore size of 1–2 μm (Rotilabo®-fibre glass syringe filters) and afterwards stored at +4 °C or lyophilised. The lyophilised colloids can be re-dissolved before further application.
Results and discussion
In this work, we present a direct synthetic pathway to obtain Tz- and TCO-functionalised polypeptides, polypeptoids and polypept(o)ides by nucleophilic ring-opening polymerisation (ROP), thereby developing new biocompatible polymer systems which can be utilised in tetrazine ligations. The presence of a terminal Tz or TCO group on a polymer allows the introduction of additional functionalities or to synthesise block copolymers. Tz-functionalised amphiphilic block copolypept(o)ides are of particular interest as they can be used to prepare Tz-modified nanoparticles such as polymeric micelles or organic colloids, in which the Tz groups should be easily accessible due to their localization at the end of the corona-forming hydrophilic block. Due to the high reaction rates of tetrazine ligations, nanoparticles bearing the respective functional groups can be quickly functionalised in a bioorthogonal manner, even in vivo, which offers interesting possibilities for several diagnostic or therapeutic applications.39 For example, they could be used as imaging probes in pretargeted imaging approaches or as masking or clearing agents to inactivate or remove unreacted tracer from the circulation.20,21,40 For these in vivo applications, biocompatible and preferably biodegradable polymeric nanoparticles are needed, which is why we chose to use polypept(o)ide-based systems. Scheme 1 provides an overview of the performed syntheses and further application of the synthesised polymers in nanoparticle preparation and ligation experiments. Tz and TCO groups are not only stable during nucleophilic ring-opening polymerisation but moreover easily accessible for post-polymerisation modification.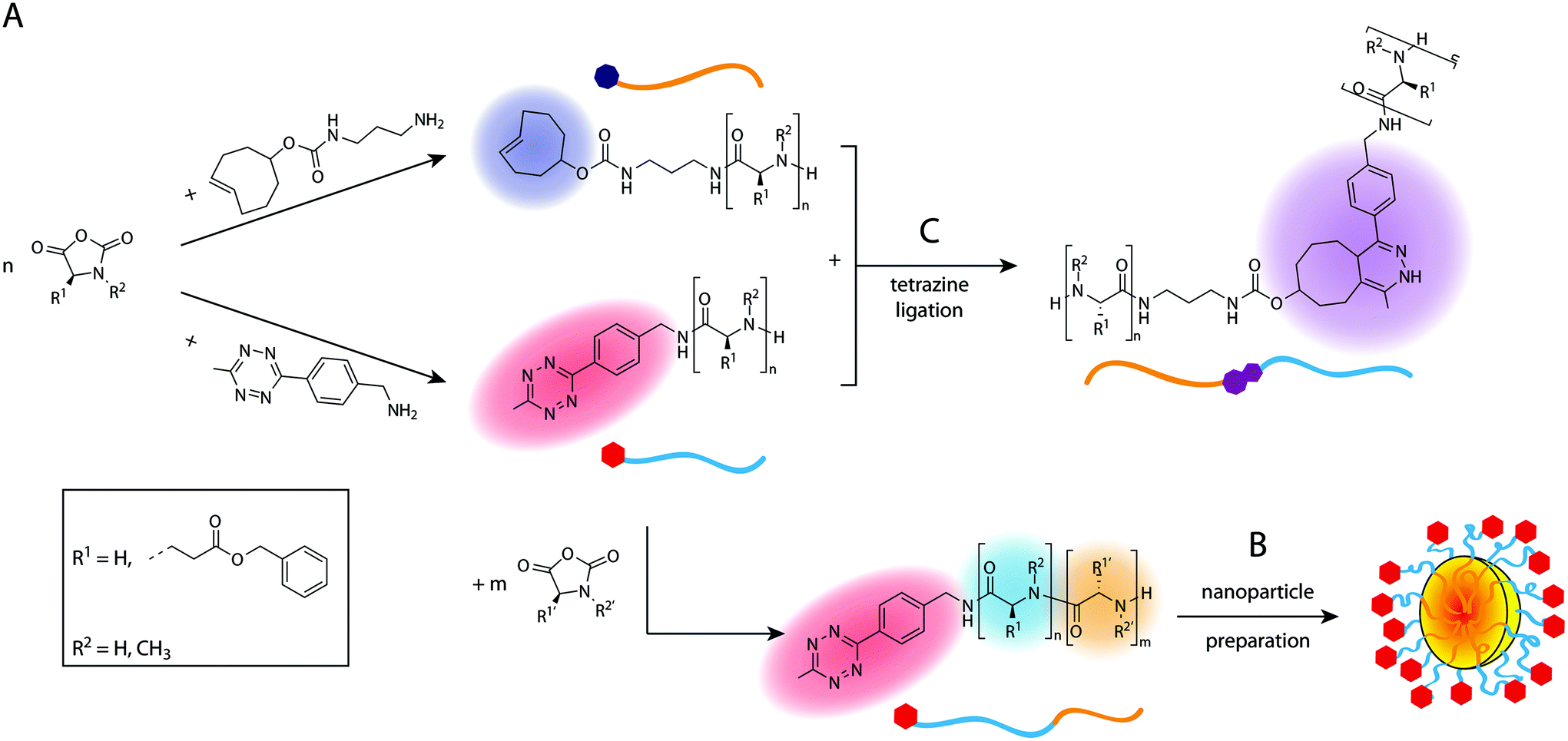 | ||
| Scheme 1 Overview of polymer synthesis (A) and nanoparticle preparation (B) with subsequent tetrazine ligation experiments (C). | ||
Polymer synthesis
Polypeptides and polypeptoids can both be synthesised by ROP of N-carboxyanhydrides (NCAs) using primary amines as polymerisation initiators. The use of a functional initiator allows the introduction of the respective functional group into the polymer in α-position as long as the respective initiator does not interfere with the polymerisation itself. An alternative route for the incorporation of terminal functionalities is to convert the terminal amino group present in both polymer types into the desired reactive groups by using peptide coupling chemistry. Directly using the functional initiator is usually preferable to any post-polymerisation functionalisation in terms of quantitative end group conversion, as long as the desired group is stable under the polymerisation conditions or suitable protecting groups are available. Amine-functional TCO and commercially available 6-methyl-tetrazine (mTz) were used as initiators in this work. We chose this particular Tz-initiator because methyl-substituted Tz possess high stability in blood serum as compared to more reactive Tz, which are prone to degradation (e.g. >90% mTz vs. 40% HTz remaining in fetal bovine serum at 37 °C after 10 h).41 Stability under physiological conditions is an important property with regard to potential in vivo applications. Both initiators were dried in high vacuum with azeotropic removal of water with benzene before the polymerisation. Polysarcosine (pSar) as a hydrophilic polypeptoid and poly(γ-benzyl-L-glutamate) (pGlu(OBn)) as hydrophobic polypeptide were synthesised using both initiators respectively. Additionally, the block copolypept(o)ide pSar-b-pGlu(OBn) was synthesised bearing an mTz group at the hydrophilic pSar block. This amphiphilic polymer was further utilised for the preparation of nanoparticles with Tz groups attached to the hydrophilic corona, which could potentially be used as clearing agents in pretargeted imaging. Table 1 gives an overview of all synthesised mTz- and TCO-functionalised polymers.| Polymer | Initiator | X n(target) | X n(MALDI) | X n(NMR) | X n(GPC) | M n [g mol−1] | M n(GPC) [g mol−1] | Đ (GPC) | |
|---|---|---|---|---|---|---|---|---|---|
| a Highest peak observed in the respective MALDI-TOF spectrum. b Determined by 1H NMR spectroscopy in DMSO-d6. c Determined by GPC in HFIP relative to pSar standards. d The number average molecular weight calculated from the determined chain lengths. e Determined by GPC in HFIP relative to PMMA standards. f Determined by 1H NMR spectroscopy in DMSO-d6 relative to pSar chain length determined by GPC. | |||||||||
| P1 | pSar | mTz-NH2 | 25 | 23 | 28 | 19 | 1600 | 8200 | 1.16 |
| P2 | pSar | mTz-NH2 | 50 | 46 | 53 | 51 | 3800 | 15300 |
1.15 |
| P3 | pSar | mTz-NH2 | 100 | 82 | 99 | 99 | 7200 | 23800 |
1.14 |
| P4 | pSar | mTz-NH2 | 200 | 128 | 255 | 179 | 12900 |
27900 |
1.26 |
| P5 | pSar | TCO-NH2 | 50 | 52 | 60 | 66 | 4900 | 17500 |
1.10 |
| P6 | pSar | TCO-NH2 | 100 | 92 | 113 | 118 | 8600 | 24000 |
1.19 |
| P7 | pSar | TCO-NH2 | 200 | 130 | 266 | 179 | 12900 |
28100 |
1.23 |
| P8 | pGlu(OBn) | mTz-NH2 | 30 | 30 | 38 | — | 8600 | 10200 |
1.12 |
| P9 | pGlu(OBn) | mTz-NH2 | 60 | 49 | 62 | — | 13800 |
16200 |
1.12 |
| P10 | pGlu(OBn) | TCO-NH2 | 25 | 28 | 23 | — | 5300 | 9700 | 1.15 |
| P11 | pGlu(OBn) | TCO-NH2 | 50 | 58 | 39 | — | 8800 | 21250 |
1.16 |
| P12 | pSar-b-pGlu(OBn) | mTz-NH2 | 200/25 | — | —/20f | 217/— | 20200 |
40200 |
1.21 |
All polymers were characterised by NMR spectroscopy, GPC and MALDI-TOF mass spectrometry. GPC shows narrow molecular weight distributions for all polymers (Fig. 1), but especially for lower degrees of polymerisation (Xn), the obtained curves have a high molecular weight tailing, which vanishes with increasing polymer length. Interestingly, the tailing is absent in case of amphiphilic block copolymers with a molecular weight of approximately 20000 g mol−1. It is thus likely that the observed bimodal distributions are due to aggregation behaviour of end group modified polymers in HFIP and not due to side reactions during the polymerisation. Competing initiation by water or other impurities present in the reaction mixture should have an increased impact for polymers with a higher targeted Xn due to the lower amounts of the desired initiator applied. 1H and DOSY NMR spectra confirmed the incorporation of mTz or TCO groups into the polymers (Fig. S1–S7†). Especially for higher Xn, it has to be taken into account, that the determination of the chain length by 1H NMR spectroscopy can be inaccurate due to low intensities of the initiator signals. Thus, the largest difference between Xn observed by GPC and NMR is found for the highest targeted degree of polymerisation of 200. While DOSY confirms that all visible signals of the functional groups possess the same diffusion coefficient and are hence indeed attached to a single polymer species, it does not prove that all polymer chains bear the desired functionalities. MALDI-TOF mass spectrometry was performed to further analyse the composition of the synthesised polymers, especially with regard to end group integrity, and to analyse the presence of possible side products, e.g. polymers initiated by water and thus lacking the functional end group (Fig. 2 and Fig. S12–S15†). All MALDI-TOF spectra show narrow molecular weight distributions with dispersities below 1.03. These values are lower than the dispersities observed with GPC analysis and some are even below the theoretical limit, which is a behaviour sometimes found in MALDI-TOF analysis of polypeptides, polypeptoids and polypept(o)ides caused presumably by mass discrimination effects.42 More importantly, all peaks could be assigned to the TCO- or mTz-functionalised polymer species that were targeted. The sub-distributions correspond to the same polymer species with different counterions (Fig. S12–S15†). In summary, the combined analytical data underline the successful synthesis of mTz- and TCO-functionalised pSar and pGlu(OBn) and mTz-pSar-b-pGlu(OBn) polymers by controlled living nucleophilic ring-opening polymerisation.
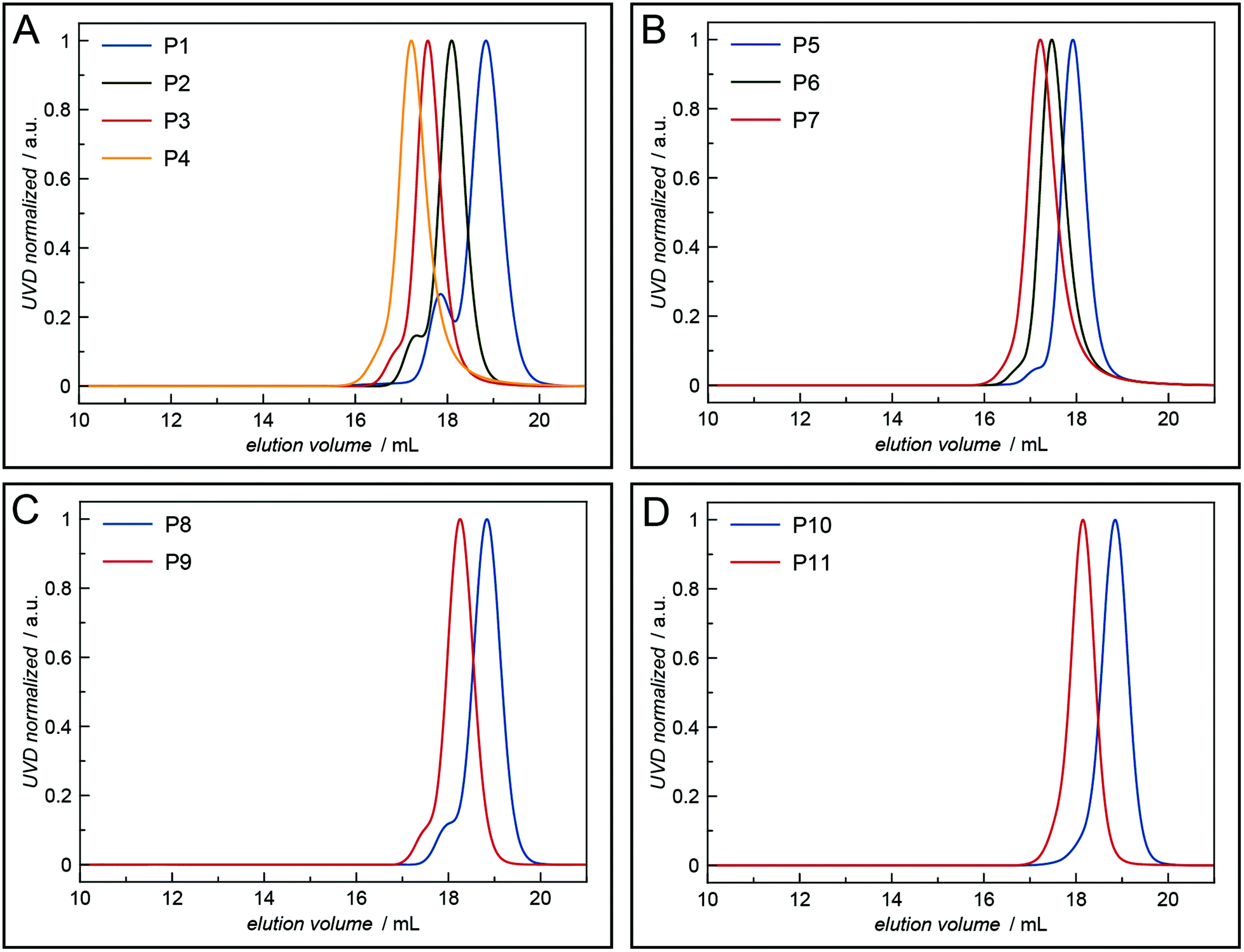 | ||
| Fig. 1 HFIP GPC elugrams of the synthesised mTz- and TCO-functionalised polymers: (A) mTz-pSar, (B) TCO-pSar, (C) mTz-pGlu(OBn), (D) TCO-pGlu(OBn). | ||
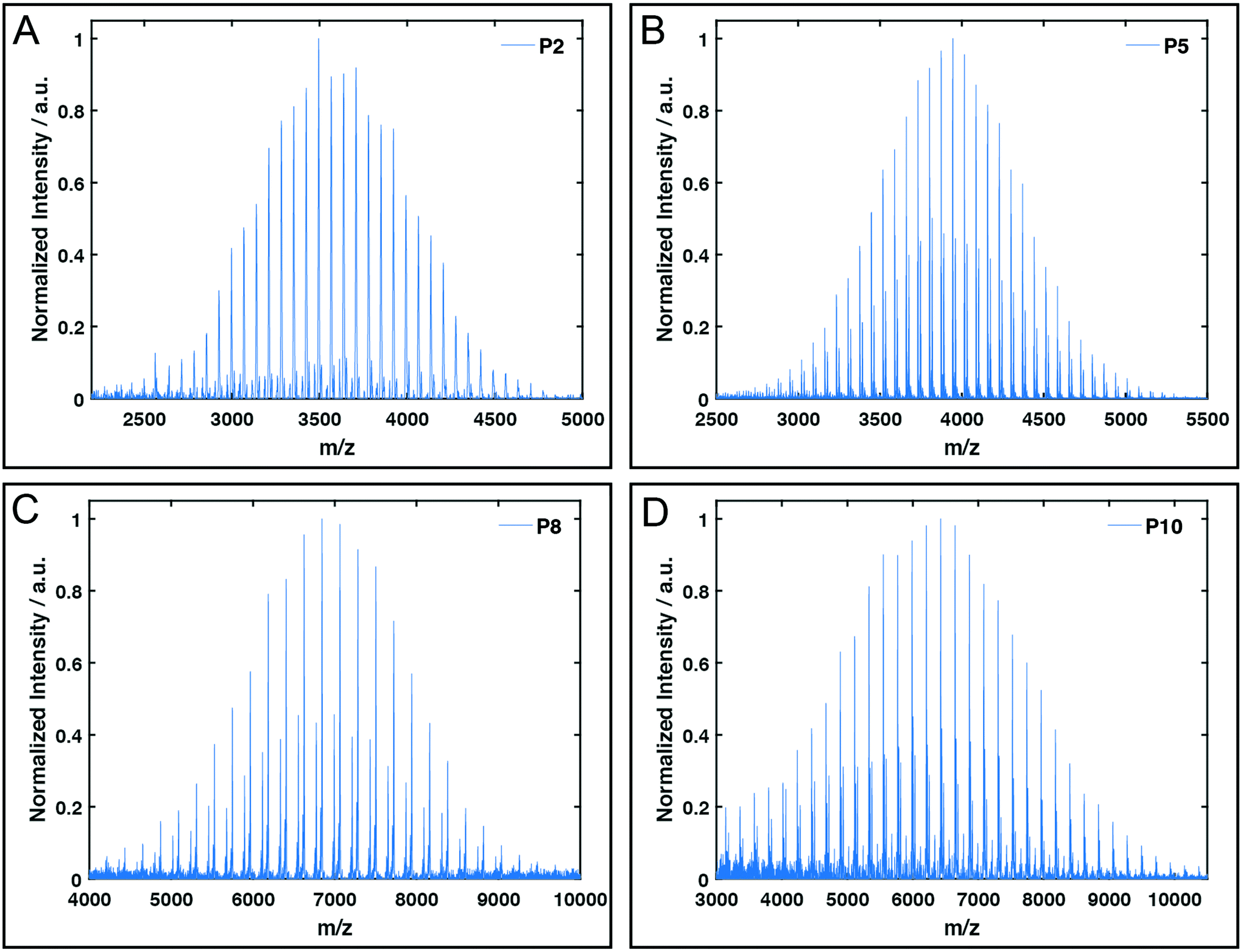 | ||
| Fig. 2 Exemplary MALDI-TOF plots of mTz-pSar51 (P2, A), TCO-pSar66 (P5, B), mTZ-pGlu(OBn)38 (P8, C) and TCO-pGlu(OBn)23 (P10, D). | ||
Tetrazine ligation experiments
After the synthesis of polymers containing reactive groups, which can be applied to tetrazine ligations with their respective counterparts, it was important to analyse whether these polymers would indeed react both with low molecular weight compounds and also with other polymers to form block copolymers and to which extent reaction kinetics are altered by steric constraints. The synthesis of block copolymers was performed by mixing equimolar solutions of the polymers in DMF, or also in water in the case of the reaction between two pSar polymers, and stirring overnight. GPC analysis demonstrates that for all combinations of pSar and pGlu(OBn) the respective block copolymers can be obtained (Fig. S8–S11†). Minor fractions of remaining homopolymer of one of the reaction partners originate from the fact that for the small amounts of polymer used here, the ratio of the reaction partners cannot be perfectly adjusted. Moreover, achieving equimolar ratios for both polymers is intrinsically difficult, because molecular weights cannot be accurately determined.In first experiments, the reaction kinetics of the tetrazine ligation were monitored by UV–Vis spectroscopy using the specific absorption band of the tetrazine group around 530 nm. Briefly, a solution in DMF or water of the Tz-functionalised polymer, to which a solution of TCO-functionalised polymer was added, was analysed at different time points (Fig. S16†).
However, this method with manual addition of the second reactant solution failed to determine second-order rate constants, because the reaction was almost completed already at the first time point at 10 s for reactions in water. Therefore, stopped-flow spectrophotometry was applied to analyse the reaction kinetics of tetrazine ligations between different polymers and low-molecular-weight compounds more accurately. Polymers were reacted among themselves and with TCO-PEG4 or two fluorogenic “turn-on” Tz derivatives (HELIOS 347Me and 388Me;43 Fig. S17†), which show significantly increased fluorescence upon reaction with a dienophile. This increase in fluorescence allows for real-time monitoring of the reaction progress. Measurements in PBS (pH = 7.4) revealed rates ranging from 463 M−1 s−1 for the reaction between mTz-pSar20 and TCO-PEG4 to 1806 M−1 s−1 for HELIOS 347Me and TCO-pSar66 (Fig. 3A, C and Table 2).
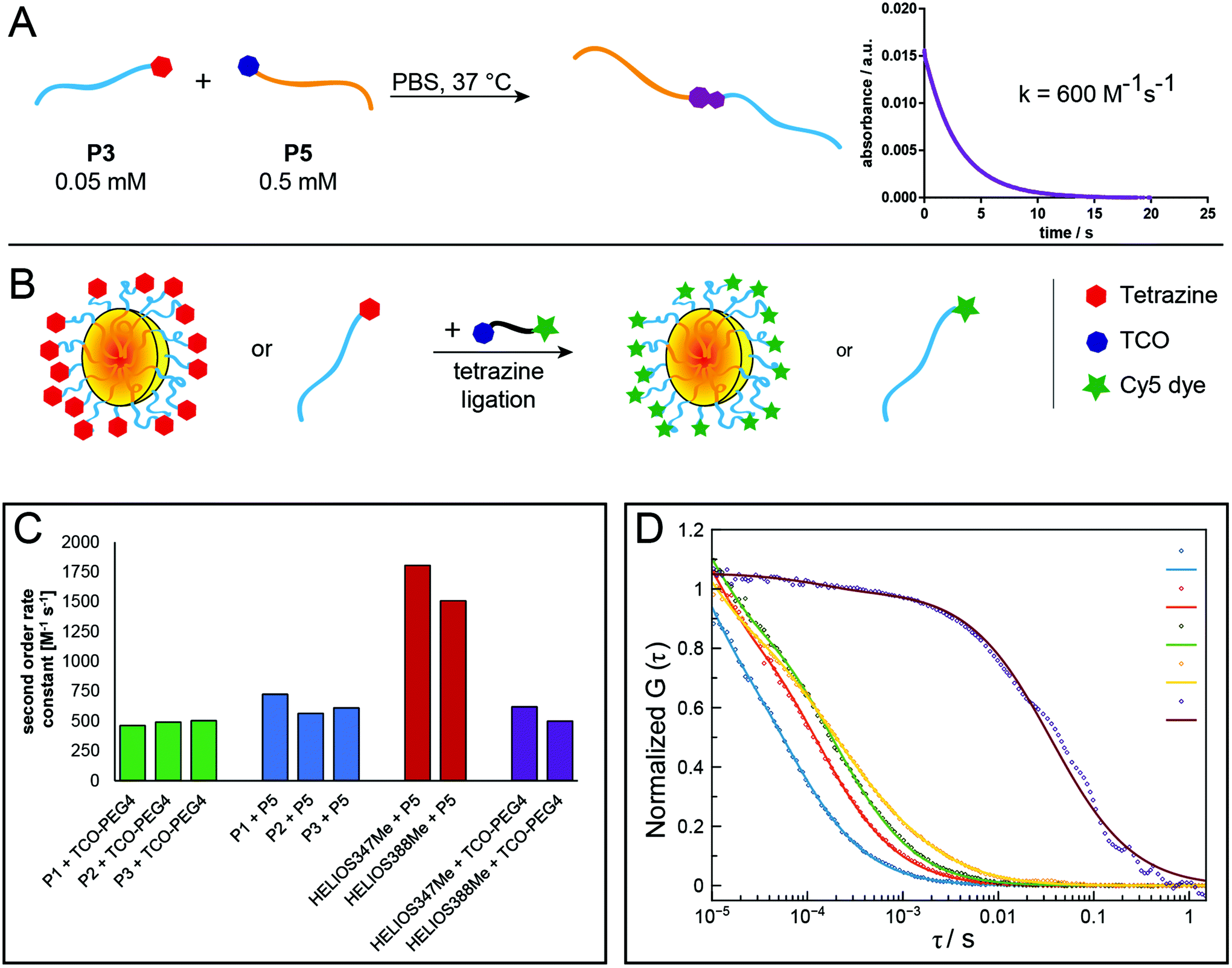 | ||
| Fig. 3 Schematic representation of the ligation procedure for stopped-flow kinetics measurements with exemplary graph of the reaction rate measurement between P3 and P5 (<9 s to 95% conversion, A). Schematic representation of polymer and NP labelling with the fluorescent dye Cy5 prior to FCS measurements (B). Second-order rate constants determined by stopped-flow kinetics measurements (C) and FCS correlation curves after labelling of P2, P4, polymeric micelles and organic colloids made from P12 with Cy5-TCO. (D) Squares: measured values, lines: corresponding fits. Blue: Cy5-TCO; Red: P2; Green: P4; Yellow: polymeric micelles; Purple: colloids. | ||
| Tz component | TCO component | TCO concentration [mM] | Observed rate [s−1] | k 2 [M−1 s−1] | Standard deviationa [M−1 s−1] |
|---|---|---|---|---|---|
| a Values represent the deviation of n = 3. | |||||
| P1 | TCO-PEG4 | 2.50 | 1.158 | 460 | 0.2 |
| P2 | TCO-PEG4 | 2.50 | 1.230 | 490 | 0.2 |
| P3 | TCO-PEG4 | 2.50 | 1.260 | 500 | 0.2 |
| P1 | P5 | 0.55 | 0.402 | 730 | 0.3 |
| P2 | P5 | 0.55 | 0.313 | 560 | 0.6 |
| P3 | P5 | 0.55 | 0.338 | 610 | 0.3 |
| HELIOS 347Me | P5 | 0.06 | 0.100 | 1800 | 1.2 |
| HELIOS 388Me | P5 | 0.06 | 0.084 | 1500 | 2.5 |
| HELIOS 347Me | TCO-PEG4 | 0.05 | 0.031 | 620 | 0.5 |
| HELIOS 388Me | TCO-PEG4 | 0.05 | 0.025 | 500 | 2.3 |
While most reaction rates were in the same range as those of the model reactions between TCO-PEG4 and the two HELIOS Tzs (500–620 M−1 s−1), TCO-pSar66 reacted significantly faster with both HELIOS Tzs (1500–1800 M−1 s−1). We could recently show that the formation of hydrophobic TCO patches on polymer brushes leads to a significant enhancement of reaction rates of the tetrazine ligation.8 A comparable effect, i.e. the formation of micellar assemblies of TCO-pSar, could presumably be the reason for the high reaction rates observed here. Furthermore, conjugation of the hydrophobic TCO to highly hydrophilic pSar could increase the solubility of the TCO compared to TCO-PEG4 and thus allow for faster tetrazine ligation reactions in aqueous media. These results show that Tz and TCO attached to pSar are not only accessible for tetrazine ligation reactions but this combination can even increase the rates of the reaction.
Nanoparticle preparation and conjugation studies
The amphiphilic block copolypept(o)ide mTz-pSar217-b-pGlu(OBn)20 was synthesised to be used in the preparation of two different types of polymeric nanoparticles (NPs), namely polymeric micelles and block copolymer-coated organic colloids (Fig. 4 and Scheme S7†), analogously to the respective non-functional NPs.30,31 Having the mTz group attached to the hydrophilic pSar block allows for the preparation of NPs with multiple mTz groups attached to the hydrophilic particle corona, which could be useful tools for the attachment of functional moieties ex or even in vivo. For this purpose, it is important to show whether the Tz groups on the particle surface are still reactive and can be addressed by their TCO counterparts. Therefore, we intended to establish a generally applicable synthesis of polypept(o)ide-based NPs and subsequently investigate their reactivity in tetrazine ligations. Polymeric micelles were prepared by dual asymmetric centrifugation44 yielding spherical micelles with a z-average hydrodynamic diameter of 112 nm (PDI = 0.15; measured by single-angle DLS) which are on average slightly larger than their non-functionalised analogues (Dh = 102 nm (single-angle DLS)).30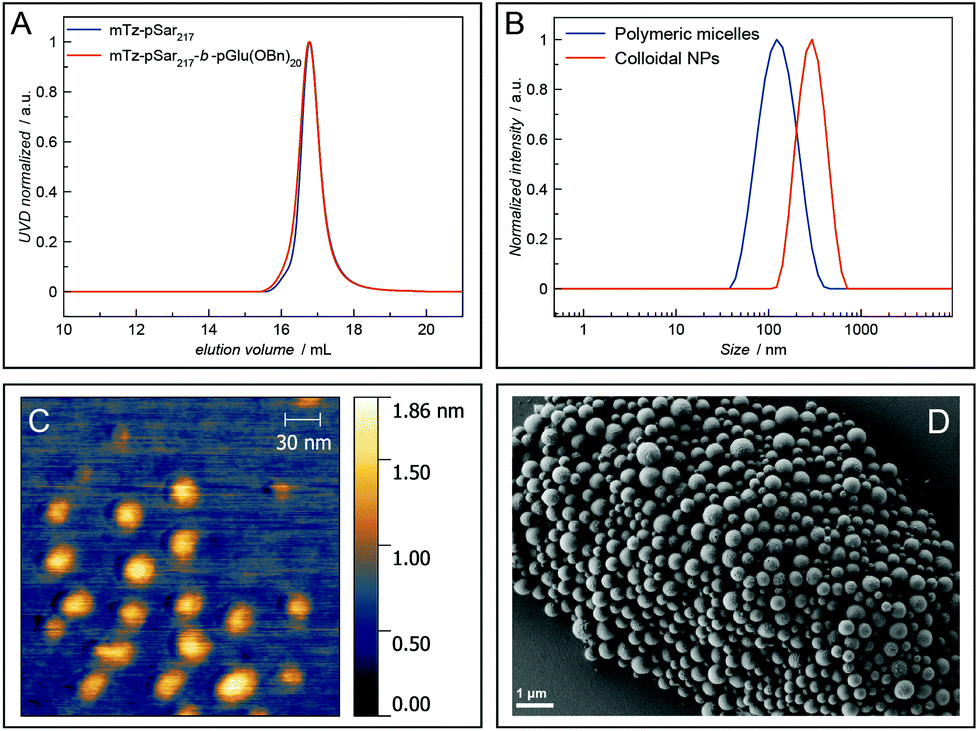 | ||
| Fig. 4 HFIP GPC elugram of the block copolymer P12 (A), single-angle DLS plots of polymeric micelles and organic colloids prepared from this polymer (B), AFM image of polymeric micelles (C, scale bar 30 nm) and SEM image of organic colloids (D, scale bar 1 μm). | ||
mTz-functionalised organic colloids with a core of poly(D,L-lactide) (PDLLA) and mTz-pSar217-b-pGlu(OBn)20 as polymeric surfactant were also obtained in a similar size regime (Dh = 272 nm, PDI = 0.20) as their non-functionalised analogues by miniemulsion technique in combination with solvent evaporation.31
Polymeric micelles were additionally analysed by atomic force microscopy (AFM), while organic colloids were visualised by scanning electron microscopy (SEM). Spherical particles could be detected in both cases. The diameter of the polymeric micelles determined from the AFM measurements was in the range of 25 nm to 35 nm and thus considerably smaller than the hydrodynamic diameter determined by single-angle DLS measurements (Fig. 4C). This observation can be attributed to two effects. First, drying effects may occur during sample preparation and a collapse of the corona results in the particles appearing smaller. Secondly, there are some larger particles, possibly compound micelles, which are few in number but contribute disproportionally to the scattering intensities measured by DLS which are proportional to the power of six of the particle radius. SEM images of the organic colloids (Fig. 4D) show a broader particle size distribution in the range of 200–500 nm, also with some larger particles.
Fluorescence correlation spectroscopy (FCS) was used to further evaluate the ability of the prepared polymers and NPs to undergo tetrazine ligations. For this purpose, Cy5-TCO was added to polymer or NP samples in aqueous solution and then analysed by FCS. Here, the presence of a fluorescent polymer or nanoparticle species indicates the successful attachment of the fluorescent dye Cy5 to the respective species via the tetrazine ligation (Fig. 3B and D). The hydrophilic polymers P2 and P4 and the mTz-functional polymeric micelles and organic colloids were reacted with Cy5-TCO in this manner and the respective hydrodynamic radii determined by FCS were 1.4 nm and 2.2 nm for the polymers, 31.2 nm for the polymeric micelles and 134.2 nm for the organic colloids. Thus, the hydrodynamic diameter of the polymeric micelles determined by FCS (62.4 nm) is in between the sizes determined by AFM and single-angle DLS. This value presumably best approximates reality, since with this method the average is determined independent of the particle size and the particle sizes are not altered by drying effects.
The combined results demonstrate that via the synthetic pathway established in this work Tz- and TCO-functionalised polypeptides, polypeptoids, polypept(o)ides and Tz-functionalised nanoparticles can be obtained in which the Tz groups are accessible for bioorthogonal tetrazine ligation reactions. Consequently, this approach provides a synthetic route to potential clearing or masking agents for pretargeted nuclear imaging, which are based on the highly biocompatible material polypept(o)ides.
Conclusions
In summary, we present the first synthetic pathway to obtain Tz- or TCO-functionalised polypeptides, polypeptoids and polypept(o)ides by nucleophilic ring-opening polymerisation using the respective functional initiators. Incorporation of the functional groups into well-defined homo- and block copolymers was demonstrated by NMR spectroscopy, GPC and MALDI-TOF mass spectrometry. As the integrity of the reactive groups after the polymerisation process is of great importance for further use of these polymers in bioorthogonal tetrazine ligations, ligation experiments were carried out using the synthesised polymers. The functionalised polymers were shown to react among themselves as well as with small molecules bearing the complementary functionality. Second-order rate constants are within the same order of magnitude or higher as those of low molecular weight model compounds used in a reference reaction (500–1800 M−1 s−1). Additionally, Tz-functionalised polymeric micelles and organic colloids were prepared. These nanoparticles readily reacted with TCO-Cy5 in a model reaction as verified by FCS, indicating stability of the Tz group during the nanoparticle preparation, which enables the fast and efficient modification of nanoparticles by the tetrazine ligation. Future work will be dedicated to determining whether these Tz-functionalised polymers and nanoparticles maintain their reactivity in vivo and may be employed as clearing or masking agents in pretargeted nuclear imaging strategies for diagnosis and therapy.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This project has received funding from the European Union's EU Framework Programme for Research and Innovation Horizon 2020, under grant agreement no. 668532. M. B. acknowledges funding from the CRC 1066-2. This work was supported by the Max Planck Graduate Center with the Johannes Gutenberg-Universität Mainz (MPGC). We thank J. Schnee and S. Türk for assistance with the MALDI-TOF measurements and G. Glaßer for assistance with SEM measurements.References
-
(a) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed
; (b) S. Mayer and K. Lang, Synthesis, 2017, 49, 830–848 CAS
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed
-
(a) M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed
- A. Darko, S. Wallace, O. Dmitrenko, M. M. Machovina, R. A. Mehl, J. W. Chin and J. M. Fox, Chem. Sci., 2014, 5, 3770–3776 RSC
-
(a) C. Denk, D. Svatunek, T. Filip, T. Wanek, D. Lumpi, J. Fröhlich, C. Kuntner and H. Mikula, Angew. Chem., Int. Ed., 2014, 53, 9655–9659 CrossRef CAS PubMed
- Y. Fang, H. Zhang, Z. Huang, S. L. Scinto, J. C. Yang, C. W. am Ende, O. Dmitrenko, D. S. Johnson and J. M. Fox, Chem. Sci., 2018, 9, 1953–1963 RSC
-
(a) R. Rossin, P. R. Verkerk, S. M. van den Bosch, R. C. M. Vulders, I. Verel, J. Lub and M. S. Robillard, Angew. Chem., Int. Ed.Clin. Cancer Res., 2010, 49, 3375–3378 CrossRef CAS PubMed
- E. J. L. Stéen, J. T. Jørgensen, K. Johann, K. Norregaard, B. Sohr, D. Svatunek, A. Birke, V. Shalgunov, P. E. Edem, R. Rossin, C. Seidl, F. Schmid, M. S. Robillard, J. L. Kristensen, H. Mikula, M. Barz, A. Kjaer, M. M. Herth, K. Nørregaard and A. Kjær, ACS Nano, 2020, 14, 568–584 CrossRef PubMed
- M. Pagel, J. Pept. Sci., 2019, 25, e3141 CrossRef PubMed
- J. W. Wollack, B. J. Monson, J. K. Dozier, J. J. Dalluge, K. Poss, S. A. Hilderbrand and M. D. Distefano, Chem. Biol. Drug Des., 2014, 84, 140–147 CrossRef CAS PubMed
- H. Koo, M. Choi, E. Kim, S. K. Hahn, R. Weissleder and S. H. Yun, Small, 2015, 11, 6458–6466 CrossRef CAS PubMed
-
(a) R. Rossin, S. M. J. van Duijnhoven, W. ten Hoeve, H. M. Janssen, L. H. J. Kleijn, F. J. M. Hoeben, R. M. Versteegen and M. S. Robillard, Bioconjugate Chem., 2016, 27, 1697–1706 CrossRef CAS PubMed
- I. A. Barker, D. J. Hall, C. F. Hansell, F. E. Du Prez, R. K. O'Reilly and A. P. Dove, Macromol. Rapid Commun., 2011, 32, 1362–1366 CrossRef CAS PubMed
- R. J. Williams, I. A. Barker, R. K. O'Reilly and A. P. Dove, ACS Macro Lett., 2012, 1, 1285–1290 CrossRef CAS
- C. F. Hansell, P. Espeel, M. M. Stamenović, I. A. Barker, A. P. Dove, F. E. Du Prez and R. K. O'Reilly, J. Am. Chem. Soc., 2011, 133, 13828–13831 CrossRef CAS PubMed
- L. Xu, M. Raabe, M. M. Zegota, J. Nogueira, V. Chudasama, S. L. Kuan, T. Weil, M. M. Zegota and J. C. F. Nogueira, Org. Biomol. Chem., 2020, 18, 1140–1147 RSC
- M. M. Lorenzo, C. G. Decker, M. U. Kahveci, S. J. Paluck and H. D. Maynard, Macromolecules, 2016, 49, 30–37 CrossRef CAS PubMed
- O. Roling, A. Mardyukov, S. Lamping, B. Vonhören, S. Rinnen, H. F. Arlinghaus, A. Studer and B. J. Ravoo, Org. Biomol. Chem., 2014, 12, 7828–7835 RSC
-
(a) N. K. Devaraj, G. M. Thurber, E. J. Keliher, B. Marinelli and R. Weissleder, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4762–4767 CrossRef CAS PubMed
- J.-P. Meyer, K. M. Tully, J. Jackson, T. R. Dilling, T. Reiner and J. S. Lewis, Bioconjugate Chem., 2018, 29, 538–545 CrossRef CAS PubMed
- R. Rossin, T. Läppchen, S. M. van den Bosch, R. Laforest and M. S. Robillard, J. Nucl. Med., 2013, 54, 1989–1995 CrossRef CAS PubMed
- S. S. Kara, M. Y. Ateş, G. Deveci, A. Cetinkaya and M. U. Kahveci, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 673–680 CrossRef CAS
- S. Kramer, D. Svatunek, I. Alberg, B. Gräfen, S. Schmitt, L. Braun, A. H. A. M. van Onzen, R. Rossin, K. Koynov, H. Mikula and R. Zentel, Biomacromolecules, 2019, 20, 3786–3797 CrossRef CAS PubMed
-
(a) T. Aliferis, H. Iatrou and N. Hadjichristidis, Biomacromolecules, 2004, 5, 1653–1656 CrossRef CAS PubMed
- C. Bonduelle, Polym. Chem., 2018, 9, 1517–1529 RSC
-
(a) A. Birke, J. Ling and M. Barz, Prog. Polym. Sci., 2018, 81, 163–208 CrossRef CAS
- J. Sun and R. N. Zuckermann, ACS Nano, 2013, 7, 4715–4732 CrossRef CAS PubMed
- K. Klinker and M. Barz, Macromol. Rapid Commun., 2015, 36, 1943–1957 CrossRef CAS PubMed
- P. Heller, A. Birke, D. Huesmann, B. Weber, K. Fischer, A. Reske-Kunz, M. Bros and M. Barz, Macromol. Biosci., 2014, 14, 1380–1395 CrossRef CAS PubMed
- F. Fenaroli, U. Repnik, Y. Xu, K. Johann, S. van Herck, P. Dey, F. M. Skjeldal, D. M. Frei, S. Bagherifam, A. Kocere, R. Haag, B. G. de Geest, M. Barz, D. G. Russell and G. Griffiths, ACS Nano, 2018, 12, 8646–8661 CrossRef CAS PubMed
- A. Birke, D. Huesmann, A. Kelsch, M. Weilbächer, J. Xie, M. Bros, T. Bopp, C. Becker, K. Landfester and M. Barz, Biomacromolecules, 2014, 15, 548–557 CrossRef CAS PubMed
-
(a) R. Holm, K. Klinker, B. Weber and M. Barz, Macromol. Rapid Commun., 2015, 36, 2083–2091 CrossRef CAS PubMed
- K. Klinker, R. Holm, P. Heller and M. Barz, Polym. Chem., 2015, 6, 4612–4623 RSC
- B. Weber, A. Birke, K. Fischer, M. Schmidt and M. Barz, Macromolecules, 2018, 51, 2653–2661 CrossRef CAS
-
(a) P. H. Maurer, D. Subrahmanyam, E. Katchalski and E. R. Blout, J. Immunol., 1959, 83, 193–197 CAS
- E. Ostuni, R. G. Chapman, R. E. Holmlin, S. Takayama and G. M. Whitesides, Langmuir, 2001, 17, 5605–5620 CrossRef CAS
-
(a) D. Huesmann, K. Klinker and M. Barz, Polym. Chem., 2017, 8, 957–971 RSC
-
R. Rigler and E. S. Elson, Fluorescence Correlation Spectroscopy. Theory and Applications, Springer, Berlin, Heidelberg, 2001, vol. 65 Search PubMed
-
(a) H. J. Chung, T. Reiner, G. Budin, C. Min, M. Liong, D. Issadore, H. Lee and R. Weissleder, ACS Nano, 2011, 5, 8834–8841 CrossRef CAS PubMed
-
(a) S. M. J. van Duijnhoven, R. Rossin, S. M. van den Bosch, M. P. Wheatcroft, P. J. Hudson and M. S. Robillard, J. Nucl. Med., 2015, 56, 1422–1428 CrossRef CAS PubMed
- M. R. Karver, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2011, 22, 2263–2270 CrossRef CAS PubMed
-
(a) C. Muhl, O. Schäfer, T. Bauer, H.-J. Räder and M. Barz, Macromolecules, 2018, 51, 8188–8196 CrossRef CAS
- L. G. Meimetis, J. C. T. Carlson, R. J. Giedt, R. H. Kohler and R. Weissleder, Angew. Chem., Int. Ed., 2014, 53, 7531–7534 CrossRef CAS PubMed
-
(a) B. Weber, C. Kappel, M. Scherer, M. Helm, M. Bros, S. Grabbe and M. Barz, Macromol. Biosci., 2017, 17, 1700061 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Procedures of the monomer synthesis, additional reaction schemes and structures, supplementary NMR, MALDI-TOF and UV–Vis spectra and GPC elugrams. See DOI: 10.1039/d0py00375a |
| This journal is © The Royal Society of Chemistry 2020 |