
2D molybdenum disulphide nanosheets incorporated with single heteroatoms for the electrochemical hydrogen evolution reaction
Thomas H. M.
Lau
,
John S.
Foord
and
S. C. Edman
Tsang
 *
*
Department of Chemistry, University of Oxford, Oxford, OX1 3QR, UK. E-mail: edman.tsang@chem.ox.ac.uk
First published on 15th April 2020
Abstract
2D nanosheets give enhanced surface area to volume ratios in particle morphology and they can also provide defined surface sites to disperse foreign atoms. Placing atoms of catalytic interest on 2D nanosheets as Single Atom Catalysts (SAC) represents one of the novel approaches due to their unique but tunable electronic and steric characteristics. Here in this mini-review, we particularly highlight some recent and important developments on heteroatom doped MoS2 nanosheets (SAC-MoS2) as catalysts for the electrochemical hydrogen evolution reaction (HER) from water, which could lead to opening up to a flagship of important renewable technologies in future. It is shown that the nature of dopants, doping positions and the polytypes of MoS2 nanosheets are the determining factors in the overall catalytic abilities of these functionalised nanosheets. This may serve to obtain atomic models which lead to further understanding of the ‘metal–support interaction’ in catalysis.
1. Introduction
The subtle climate change due to excessive carbon emissions to the atmosphere has prompted our community to switch energy sources from oil, gas and coal to renewables such as solar and wind power, tidal waves, etc. Green H2 manufacture from electrolysis of water over an efficient electrocatalyst is the core technology to harness these renewables. The produced H2 with zero carbon emission can be directly used in combustion engines, and fuel cells, for reduction of CO2, etc. Alternatively, it can be stored as chemicals such as ammonia or methanol to facilitate transport and for use in other areas. Although noble metal catalysts give satisfactory performance in electrolysers, their high cost and scarcity have compelled chemical manufacturers to find greener and more cost-effective alternatives. Thanks to the advances in nanotechnology, two-dimensional (2D) metallic or semi-conductive materials such as graphite based nanostructures,1,2 transition metal dichalcogenides (TMDC),3–5 transition metal oxides (TMO),6,7 and transition metal nitrides (TMN)8,9 have been thoroughly studied and characterised. These atomically thick 2D nanosheets possess a much higher surface-to-volume-ratio than three-dimensional (3D) nanomaterials. Since more active sites located at the surface can be accessed for the desired reactions, they generally exhibit better catalytic performances than their 3D counterparts. However, they are still incomparable to conventional noble metal catalysts such as Pt and Ru. Thus, further modifications have been attempted to improve the material properties of these 2D catalysts. The general idea is to either increase the number of active sites available or enhance the intrinsic activity of each active site. For example, the former approach can be achieved by exposing more edge sites to terrace sites by introducing surface defects,10–14 while the latter one can involve changing the electronic structure of the responsible atoms at the active sites via strain eningeering.15–17One of the newest modification methods to functionalise 2D nanosheets is to incorporate single heteroatoms onto the large basal surface of 2D catalysts. Generally, single-atom catalysts (SAC) refers to foreign single atoms deposit on support materials, the atomic dopants act as the active sites and contribute primarily towards the catalytic activities in the reactions of interest.18–20 The dispersed atomic dopants maximise metal utilisation, which makes the catalyst excel in small molecule activation. Such a combination of SAC and 2D catalysts creates a new group of materials, which is multifunctional in catalysis due to the coexistence of two types of active sites on the surface, potentially making more complicated reaction mechanisms plausible. While this new type of catalyst generates much interest as it combines the knowledge of the two hottest research areas in catalysis, the research field has not yet been well explored. Several excellent review articles have been published to highlight some recent research work in the area of SAC.21–23 Similarly, there are some good reviews focusing on using 2-D materials such as graphene, molybdenum disulphide (MoS2), graphitic carbon nitride (g-C3N4), etc. as catalysts in electrochemical reactions including the HER when doped with heteroatoms. The challenges of synthesising and analysing SAC-2D catalysts have also been reviewed therein. The readers should refer to these articles for reference without being repeatedly discussed in this article. In an attempt to provide insights for future research in the field of SAC and 2D materials in combination, this mini-review will particularly examine some selected key publications on single atom doped MoS2 nanosheets (SAC-MoS2) in the HER as a short but sharply focused review. The synthetic approaches and the electrocatalytic activities of these materials will be thoroughly analysed with the aim to better understand the structure–activity relationship of SAC-MoS2.
2. Polytypes of 2D MoS2
MoS2 is a layered structure with each triatomic monolayer consisting of a hexagonal plane of molybdenum atoms sandwiched between two other hexagonal planes of sulphur atoms via strong in-plane covalent bonding. Adjacent layers are stacked together by weak out-of-plane van der Waals interactions.24 There are multiple polytypes of MoS2, 2H-MoS2, 3R-MoS2, and 1T-MoS2, which give distinctive electronic properties. The digit refers to the number of layers in the unit cell while the letters “H”, “R”, and “T” indicate hexagonal (D3h group), rhombohedral (C53v group), and tetragonal (D3d group) symmetry, respectively. Both 2H-MoS2 and 3R-MoS2 are semi-conducting while 1T-MoS2 displays a metallic behaviour. 1T-MoS2 is found to possess an electrical conductivity of around 107 times higher than that of 2H-MoS2.32 If 2H- and 3R-MoS2 crystals are exfoliated into monolayers, the polytype will then be referred to as 1H to denote a monolayer structure. 1T-MoS2 is metastable as its unit cell is the distorted form of 2H-MoS2. Thus, it needs to be stabilised or else it will be converted back to 2H-MoS2 upon heating above 95 °C.33 For example, computational calculations suggested that the 1T polytype can be distorted into a lower energy 1T′ structure due to the stabilisation of hydrogen adsorption in the HER.34,35The choice of the MoS2 polytype not only affects the overall electrical conductivity but also the density state of the active sites. During catalysis, the Mo edges (101![[0 with combining macron]](https://www.rsc.org/images/entities/char_0030_0304.gif) ) and the S edges (110) of MoS2 can accept and donate electrons like a metal surface.36 The contribution by the edge sites however is far less important for 2D MoS2 due to the presence of a larger basal surface. The basal plane (0001) of semiconducting MoS2 has been verified to be chemically inert both experimentally15 and computationally37 as further modification is required to activate it. In contrast, the metallic feature of 1T-MoS2 has made the basal sites highly active for electrocatalysis.38,39 Top-down and bottom-up methods are two major approaches to obtain 2D MoS2 nanosheets. The former method is to isolate monolayers from bulk MoS2 crystals by weakening the interlayer van der Waals interactions. Lithium intercalation is one example to create monolayer 1T-MoS2 with rich S-defects.40–42 The latter method involves reacting Mo precursors with S sources to grow MoS2 unit by unit. Chemical vapour deposition (CVD)43–46 and one-pot hydrothermal/solvothermal synthesis47–49 are often used to grow high-quality ultrathin 2H-MoS2 or monolayer 1H-MoS2.
) and the S edges (110) of MoS2 can accept and donate electrons like a metal surface.36 The contribution by the edge sites however is far less important for 2D MoS2 due to the presence of a larger basal surface. The basal plane (0001) of semiconducting MoS2 has been verified to be chemically inert both experimentally15 and computationally37 as further modification is required to activate it. In contrast, the metallic feature of 1T-MoS2 has made the basal sites highly active for electrocatalysis.38,39 Top-down and bottom-up methods are two major approaches to obtain 2D MoS2 nanosheets. The former method is to isolate monolayers from bulk MoS2 crystals by weakening the interlayer van der Waals interactions. Lithium intercalation is one example to create monolayer 1T-MoS2 with rich S-defects.40–42 The latter method involves reacting Mo precursors with S sources to grow MoS2 unit by unit. Chemical vapour deposition (CVD)43–46 and one-pot hydrothermal/solvothermal synthesis47–49 are often used to grow high-quality ultrathin 2H-MoS2 or monolayer 1H-MoS2.
3. Synthetic methods of SAC-MoS2
Table 1 lists the synthetic approaches of selected publications that were adopted in preparing different SAC-MoS2 for the electrochemical HER. It has been shown that the synthesis of SAC-MoS2 nanosheets is predominantly achieved by two different synthetic routes: (1) incorporating single heteroatoms onto the as-prepared 2D MoS2 nanosheets with the desired polytypes and (2) directly growing the catalysts through the hydrothermal/solvothermal reaction. The synthetic route will largely influence the doping position of the heteroatoms adopted in the SAC-MoS2 catalysts. It was revealed that there are four general doping sites for single molecular layer MoS2, namely the Mo atop site, S substitution site, Mo substitution site, and hollow site.50 For the first route, MoS2 nanosheets are first synthesised beforehand usually by lithium intercalation (Method 1) or by one-pot synthesis (Method 2). As mentioned, lithium intercalation can be applied to exfoliate bulk MoS2 into 1T-MoS2. However, a prolonged heating above the 95 °C threshold using hydrothermal techniques on non-stabilised 1T-MoS2 would reconvert the structure from metallic 1T- to semiconducting 2H-MoS2. For the one-pot hydrothermal/solvothermal synthesis, nanosheets are being built-up unit by unit through reactions between the Mo and S precursors. The heteroatoms are then incorporated onto the as-prepared nanosheets afterwards. Since the six-coordinated Mo atoms are well located inside the intact MoS2 unit cells, the incoming heteroatom dopants during the incorporation process are less likely to penetrate into the outer S layer and substitute the Mo atom. Therefore, they are placed at the Mo atop sites or S substitution sites unless extra treatment such as high temperature annealing is applied.| Entry | Catalyst | Synthetic method | Polytype | Doping concentration and preferable doping position | Ref. |
|---|---|---|---|---|---|
| 1A | MoS2 | Mechanical exfoliation under sonication | 2H | — | 25 |
| 1B | MoS2 | Lithium intercalation | 1T | — | 25 |
| 2A | Co-MoS2 | One pot solvothermal reaction at 400 °C, 4 h | 2H | 1.7% Co, Mo substitution sites at edge sites | 26 |
| 2B | Pt-MoS2 | One pot solvothermal reaction at 400 °C, 4 h | 2H | 1.7% Pt, Mo substitution sites at edge sites | 26 |
| 3 | Co-MoS2 with S defects | Lithium intercalation, hydrothermal doping at 180 °C, 12 h | 1H | 3% Co, Mo atop sites at the basal plane | 27 |
| 4 | Pd-MoS2 | One pot solvothermal reaction at 180 °C, 24 h | 1T | 1% Pd, Mo substitution sites at the basal plane | 28 |
| 5A | Pt-MoS2 | Lithium intercalation, sonochemical doping at room temperature, 24 h | 1T | 3% Pt, Mo atop sites at the basal plane | 29 |
| 5B | Pd-MoS2 | Lithium intercalation, sonochemical doping at room temperature, 24 h | 1T | 3% Pd, Mo atop sites at the basal plane | 29 |
| 6 | P-MoS2 | One pot solvothermal reaction at 200 °C, 24 h | 1T | 5% P, S substitution at the basal plane | 30 |
| 7 | N-MoS2 | One pot solvothermal reaction at 200 °C, 24 h | 1T | 5.7% N, Mo substitution sites at the basal plane | 31 |
For the second route, the one pot bottom-up synthetic approach can also be adopted to directly synthesise SAC-MoS2 through the reactions between Mo, S, and the dopant precursors. The heteroatoms can be embedded inside the MoS2 nanosheets as a result due to their presence during the unit by unit construction of MoS2 nanosheets. However, it should be noted that a precise control of the molar concentration for the three precursors is vital for obtaining monolayer nanosheets confined with single heteroatoms within the tolerance of the thermodynamic maximum limits. An excess amount of single metal atom dopants may result in the formation of clusters or nanoparticles28 while an insufficient amount of Mo or S sources can lead to the creation of surface defects.30
Several selected publications listed in Table 1 (entries 1, 3 and 5) adopted the first route to synthesise SAC-MoS2 (Fig. 1, Method 1). Lithium intercalation was applied to obtain 1T-MoS2 nanosheets. The delicate 1T polytype was then preserved when the as-prepared nanosheets underwent doping in a sonochemical reaction under ambient conditions to prevent the conversion back to 2H.29 The same hydrothermal/solvothermal techniques are also being applied in the one pot synthesis of SAC-MoS2 (Table 1, entries 2, 3, 4, 6 and 7) (Fig. 1, Method 3). While the reaction temperature in all cases is at least above 180 °C, the metal dopants embedded into the as-grown MoS2 can provide extra kinetic stability to prevent the conversion of 1T polytypes. However, this only applies to a certain extent as the MoS2 nanosheets are grown at a high temperature of 400 °C, which will still drive to the 2H polytype with a multiple-layer thickness. Therefore, the synthetic condition also plays a crucial role in determining the resulting polytypes. Overall, the doping concentration, doping position, and polytypes largely depend on the synthetic route and condition. Since there are pros and cons for each discussed synthetic method, the choice should depend on the structure of the SAC-MoS2 material desired as it ultimately determines the catalytic performance.
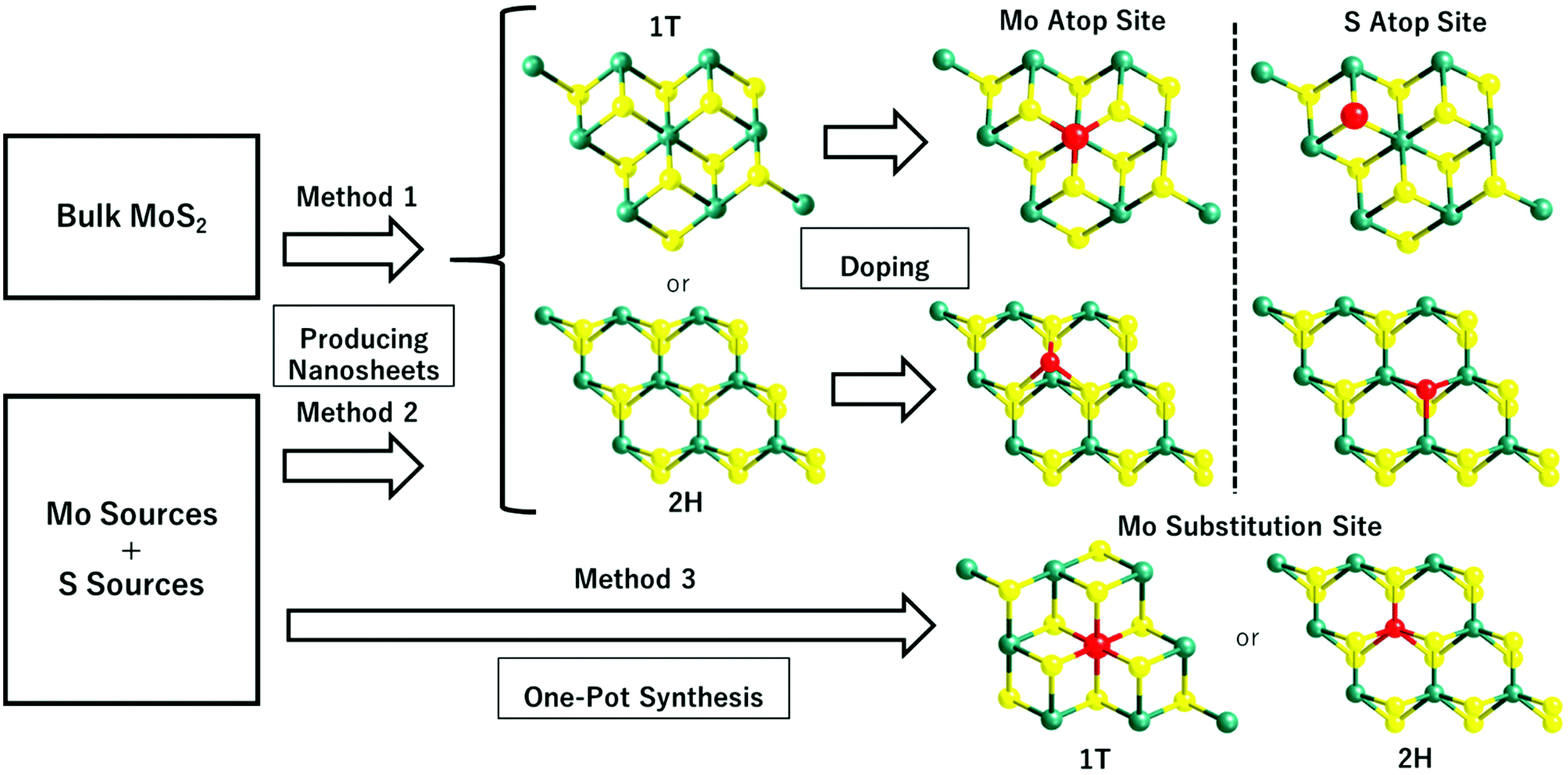 | ||
| Fig. 1 Summary of the synthetic routes of SAC-MoS2 adopted in recent highlighted publications. Method 1 uses the top-down exfoliation of bulk MoS2 to create MoS2 nanosheets, while method 2 uses the bottom-up hydrothermal/solvothermal reactions to make the nanosheets directly from Mo and S sources. The pure MoS2 created with method 1 and method 2 will then be doped with the heteroatoms to form SAC-MoS2. Method 3 uses one-pot synthesis of the SAC-MoS2via hydrothermal/solvothermal reactions. For all the MoS2 structural models shown in the above figure, the green, yellow, and red spheres represent Mo atoms, S atoms, and dopant atoms, respectively. | ||
4. Catalytic performance of SAC-MoS2
The electrocatalytic performance of catalystcs can be evaluated by several key parameters. An ideal HER catalyst should possess a low overpotential, a low Tafel slope, and high stability while the synthetic cost should remain minimal. In theory, the HER can take place at a cell voltage of 0 V but actually requires extra potential to initiate the reaction due to the resistance in the real electrochemical system. This extra potential is called overpotential (η), which has been commonly used for assessing the strength of the catalyst to reduce the activation energy of the HER. However, it should be noted that the value can also be largely affected by the pH value of the electrolyte and the molar concentration of the analytes. As a result, the research highlighted in this article mainly employed an acidic medium in their electrochemical systems. The Tafel slope is another characteristic indicator to study the activity of the electrocatalyst. It reflects how the logarithm of the current density varies with the overpotential: the derived Tafel slope is inversely proportional to the charge transfer coefficient. Thus, the small value of the Tafel slope indicates a high charge transfer ability.51 The Tafel slope can also be used to reveal the rate determining step (RDS) of the reaction. It is well known that the reaction mechanism of hydrogen evolution will be changed under different pH conditions. Acid solutions are generally used as electrolytes as they show higher ionic conductivity and no carbonate will be formed in the process, as compared with alkaline medium.52 There are three fundamental reaction steps involved in the overall HER (Fig. 2) in acidic medium. The Volmer step describes the generation of an adsorbed hydrogen atom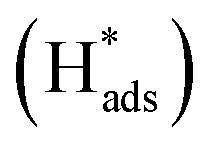 at the catalyst (H+(aq) + e− →
at the catalyst (H+(aq) + e− → 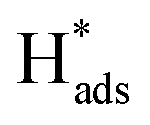 ).
).
 | ||
| Fig. 2 (a) DOS of MoS2, Pt-MoS2, and H adsorbed Pt-MoS2. The dashed line represents the Fermi level. (b) HER process through the Volmer–Heyrovsky pathway on a Pt-MoS2 catalyst. (c) Volcano plot of 2H-MoS2 doped with different transition metals at the Mo substitution site.26 | ||
The reaction then proceeds with either the Tafel step or the Heyrovsky step, which is usually dependent on the amount of 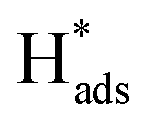 available. In the Tafel step, two absorbed hydrogen atoms recombine to yield H2 gas (
available. In the Tafel step, two absorbed hydrogen atoms recombine to yield H2 gas ( → H2(g)), whereas for the Heyrovsky step,
→ H2(g)), whereas for the Heyrovsky step, 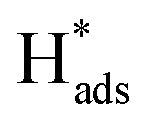 will interact with another electron and proton pair to form H2 (
will interact with another electron and proton pair to form H2 (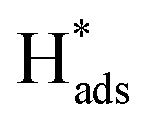 + H+(aq) + e− → H2(g)). A Tafel slope value of 120, 40, and 30 mV per decade indicates that the RDS is characterised by the Volmer step, the Heyrovsky step, and the Tafel step, respectively, for MoS2 based materials.53 Thus, through obtaining the Tafel slope values, it can be predicted that which reaction pathway, the Volmer–Heyrovsky reaction or Volmer–Tafel reaction, is playing the important role in affecting the RDS. Table 2 lists out the overpotentials and the Tafel slope values of different SAC-MoS2 from the selected publications. It can be observed that undoped MoS2 with 1T polytypes has a much smaller Tafel slope value (40 mV per decade) than semiconducting 2H counterparts (180 mV dec−1) (Table 2, entries 1A and 1B). As explained earlier, SAC-MoS2 nanosheets with 1T polytypes are metallic and surface-active.
+ H+(aq) + e− → H2(g)). A Tafel slope value of 120, 40, and 30 mV per decade indicates that the RDS is characterised by the Volmer step, the Heyrovsky step, and the Tafel step, respectively, for MoS2 based materials.53 Thus, through obtaining the Tafel slope values, it can be predicted that which reaction pathway, the Volmer–Heyrovsky reaction or Volmer–Tafel reaction, is playing the important role in affecting the RDS. Table 2 lists out the overpotentials and the Tafel slope values of different SAC-MoS2 from the selected publications. It can be observed that undoped MoS2 with 1T polytypes has a much smaller Tafel slope value (40 mV per decade) than semiconducting 2H counterparts (180 mV dec−1) (Table 2, entries 1A and 1B). As explained earlier, SAC-MoS2 nanosheets with 1T polytypes are metallic and surface-active. 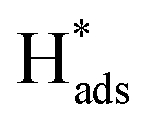 will be readily formed at the basal plane and the Volmer step will not be the RDS in this case. In contrast, it is difficult for H adsorption to take place on the basal plane of 2H/1H-MoS2. The overall reaction rate is hindered by insufficient H adsorption, which also suggested why the overpotential of 2H-MoS2 is larger than that of 1T. As a result, the metallic 1T-MoS2 based structures appear to be more useful than 2-H counterparts for the electrochemical hydrogen production due to the lower impedance encountered, giving higher energy conversion. However, the long-term stability of this structure under practical conditions may have to be ascertained for application in electrolysers.
will be readily formed at the basal plane and the Volmer step will not be the RDS in this case. In contrast, it is difficult for H adsorption to take place on the basal plane of 2H/1H-MoS2. The overall reaction rate is hindered by insufficient H adsorption, which also suggested why the overpotential of 2H-MoS2 is larger than that of 1T. As a result, the metallic 1T-MoS2 based structures appear to be more useful than 2-H counterparts for the electrochemical hydrogen production due to the lower impedance encountered, giving higher energy conversion. However, the long-term stability of this structure under practical conditions may have to be ascertained for application in electrolysers.
| Entry | Catalyst | Overpotential (η) (mV@10 mA cm−2) vs. RHE | Tafel slope (mV per decade) | Ref. |
|---|---|---|---|---|
| 1A | MoS2 | ∼−375 mV | 186 | 25 |
| 1B | MoS2 | ∼−250 mV | 40 | 25 |
| 2A | Co-MoS2 | ∼−285 mV | N/A | 26 |
| 2B | Pt-MoS2 | ∼−185 mV | 96 | 26 |
| 3 | Co-MoS2 with S defects | −300 mV | 92 | 27 |
| 4 | Pd-MoS2 | −89 mV | 62 | 28 |
| 5A | Pt-MoS2 | −223 mV | 57 | 29 |
| 5B | Pd-MoS2 | −140 mV | 50 | 29 |
| 6 | P-MoS2 | −43 mV | 34 | 30 |
| 7 | N-MoS2 | −168 mV | 41 | 31 |
To understand the effect of single metal dopants on MoS2 for the HER, Deng et al. were among the pioneer groups to conduct electrochemical scan tests of 2H-MoS2 doped with several types of single transition metal atoms (Pt, Co, and Ni) at Mo substitution sites for the HER (Table 2, entry 2).26 According to the experimental results, the Pt doped one exhibited the best catalytic performance among three with the smallest overpotential of −185 mV at 10 mA cm−2. The Density of States (DOS) simulation provided an explanation that the introduction of Pt atoms into the Mo substitution site induced more hybridized electronic states to occur near the Fermi level, which enhanced the H adsorption ability of basal S atoms and thus the catalytic performance for the HER (Fig. 2a and b).
Density functional theory (DFT) calculations are also applied to calculate the Gibbs energy for hydrogen adsorption (ΔGH*) of the catalytic or electrocatalytic system when doped with different single transition metal atoms in order to predict the overall trend. According to the Sabatier principle, the H adsorption process is optimal when ΔGH* is close to zero.54 When the value of ΔGH* is too negative, the hydrogen atom will bind too strongly onto the surface, which inhibits the subsequent desorption process for the formation of H2 gas until thermal or electrical energy is supplied. In contrast, if the value of ΔGH* is too positive, the catalyst surface will not favour the adsorption process for either the catalytic or electrocatalytic processes. It should be noted that this principle offers good predictive values to metallic phases with no impedance or diffusional limitations imposed between the active sites. Pt is known to be at the apex of the Sabatier volcano for H species. On the other hand, for composite materials such as SAC-MoS2 one should consider the composite surface for the H interactions. For unmodified single molecular 2H-MoS2 monolayers, the value of the Tafel slope (96 mV per decade) indicated that H adsorption is predominately the RDS for the overall HER. A plot of the reaction rate of 2H-MoS2 when doped with different metal atoms (in terms of exchange current density) against their ΔGH* in the HER was shown to have a volcano shape with Pt at the apex, indicating the dominant role of Pt compared to the 2H-MoS2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26 (Fig. 2c). The excellent experimental results of Co-doped MoS2 compared with other metals further supported the volcano plot. This suggests that this inexpensive non-noble metal can be effectively used without much activity compromise compared to using Pt. However, this work did not give a satisfactory explanation for the surprisingly poor Ni performance, which is electronically similar to Co atoms as they are neighbouring elements to each other. Another independent work also studied how single transition metal atoms (Co, Ni, Ag, Cu) doped at the Mo atop sites would affect the semiconducting 2H-MoS2, but with a somewhat different conclusion drawn. Co-MoS2 was confirmed to be the best HER catalyst among four transition metals with a Tafel slope value of 92 mV per decade and an overpotential of −300 mV at 10 mA cm−2 (Table 2, entry 3).27 The DOS calculations suggested that Co dopants help shifting the conduction band close to the Fermi level, which ultimately makes the surrounding S atoms at the basal planes better active sites for H adsorption. Also, no nice volcano relationship was noted when considering theoretical electronic factors of the metal dopers. The extended X-ray absorption fine structure (EXAFS) analysis was in fact conducted to study the chemical environment of the dopants. Interestingly, it was found that Co atoms anchored at the Mo atop site did not form a Co–Mo bond due to geometric constraint whereas the Ni–Mo formed negatively influenced the local S atoms in terms of the H adsorption ability. Thus, this clearly demonstrates that the metal doper can have different interactions with the single molecular layer MoS2. Therefore, basing solely on the electronic factor it is difficult to predict the trend without considering the precise position of the dopers and the steric constraints of the atoms in the composite structures. Regardless, the two pieces of research have interestingly shown that even when the same metal atoms are doped, the different doping position (Mo substitution site and Mo atop site) will largely affect the electronic properties of the resulting SAC-MoS2 with 1H/2H polytypes.
26 (Fig. 2c). The excellent experimental results of Co-doped MoS2 compared with other metals further supported the volcano plot. This suggests that this inexpensive non-noble metal can be effectively used without much activity compromise compared to using Pt. However, this work did not give a satisfactory explanation for the surprisingly poor Ni performance, which is electronically similar to Co atoms as they are neighbouring elements to each other. Another independent work also studied how single transition metal atoms (Co, Ni, Ag, Cu) doped at the Mo atop sites would affect the semiconducting 2H-MoS2, but with a somewhat different conclusion drawn. Co-MoS2 was confirmed to be the best HER catalyst among four transition metals with a Tafel slope value of 92 mV per decade and an overpotential of −300 mV at 10 mA cm−2 (Table 2, entry 3).27 The DOS calculations suggested that Co dopants help shifting the conduction band close to the Fermi level, which ultimately makes the surrounding S atoms at the basal planes better active sites for H adsorption. Also, no nice volcano relationship was noted when considering theoretical electronic factors of the metal dopers. The extended X-ray absorption fine structure (EXAFS) analysis was in fact conducted to study the chemical environment of the dopants. Interestingly, it was found that Co atoms anchored at the Mo atop site did not form a Co–Mo bond due to geometric constraint whereas the Ni–Mo formed negatively influenced the local S atoms in terms of the H adsorption ability. Thus, this clearly demonstrates that the metal doper can have different interactions with the single molecular layer MoS2. Therefore, basing solely on the electronic factor it is difficult to predict the trend without considering the precise position of the dopers and the steric constraints of the atoms in the composite structures. Regardless, the two pieces of research have interestingly shown that even when the same metal atoms are doped, the different doping position (Mo substitution site and Mo atop site) will largely affect the electronic properties of the resulting SAC-MoS2 with 1H/2H polytypes.
For SAC-MoS2 with 1T polytypes, Luo et al. suggested a significantly much stronger interaction between the transition metal doper and the 1T-MoS2 support. They showed that the inclusion of 1% Pd atoms into the 1T structure can dramatically boost the HER activity (Table 2, entry 4).28 It is confirmed by X-ray photoelectron spectroscopy (XPS), X-ray powder diffraction and EXAFS analysis that Pd atoms were successfully grown at the Mo substitution sites (Fig. 3). It is also possible to help create S defects during the synthesis. DFT calculations suggested that the ΔGH of the S adjacent to Pd (Pd–S*–Mo) exhibited an almost thermoneutral value of −0.02 eV. Similar to the aforementioned metal atoms doped on 2H-MoS2, the major function of Pd atoms helps activating the adjacent S atoms (Pd–S*–Mo) at the basal plane of 1T-MoS2. As suggested by the durability test, the highly stable Pd–S bonds also provide overall stability, which is another important criterion for a good heterocatalyst. However, it also should be noted that since the metastable 1T polytype is just a distorted form of 2H polytypes, both 1T and 2H polytypes will always coexist upon the incorporation of heteroatoms. Most key research studies in the literature have tended to neglect this important factor as the degree of coexistence (1T:2H ratios) was usually not provided nor addressing their correlation with activity. Another independent research work also studied the catalytic performance of Pd doped on 1T-MoS2. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM, Fig. 4) combined with other structural characterisation techniques and computational calculations suggested that the Pd atoms are doped at the Mo atop site rather than the Mo substitution site.29 The Tafel slope analysis and DFT modelling reconfirmed that the H adsorption was readily facilitated by the 1T surface and thus the Volmer step is not the RDS. More importantly, the incorporated Pd atoms here could act as the active sites to accelerate the rate limiting recombination step (Heyrovsky step or Tafel step) to form hydrogen gas. Overall, it was suggested that H adsorption would preferably take place on the 1T surface. The 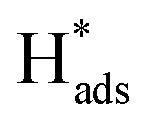 then hopped to the single Pd atoms to be recombined to H2. The difference in the roles of the Pd atoms for the HER from these two different studies probably originates from the two different doing positions due to their difference in synthetic methods used. The Pd atoms substituting Mo atoms usually formed stable Pd–S bonds which may offer steric hindrance to prevent any interactions with the
then hopped to the single Pd atoms to be recombined to H2. The difference in the roles of the Pd atoms for the HER from these two different studies probably originates from the two different doing positions due to their difference in synthetic methods used. The Pd atoms substituting Mo atoms usually formed stable Pd–S bonds which may offer steric hindrance to prevent any interactions with the 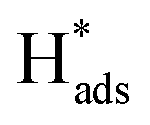 while uncoordinated Pd atoms at the Mo atop site are electronically and spatially freely to undergo redox reactions, making Pd-1T-MoS2 a bifunctional catalyst for the HER.
while uncoordinated Pd atoms at the Mo atop site are electronically and spatially freely to undergo redox reactions, making Pd-1T-MoS2 a bifunctional catalyst for the HER.
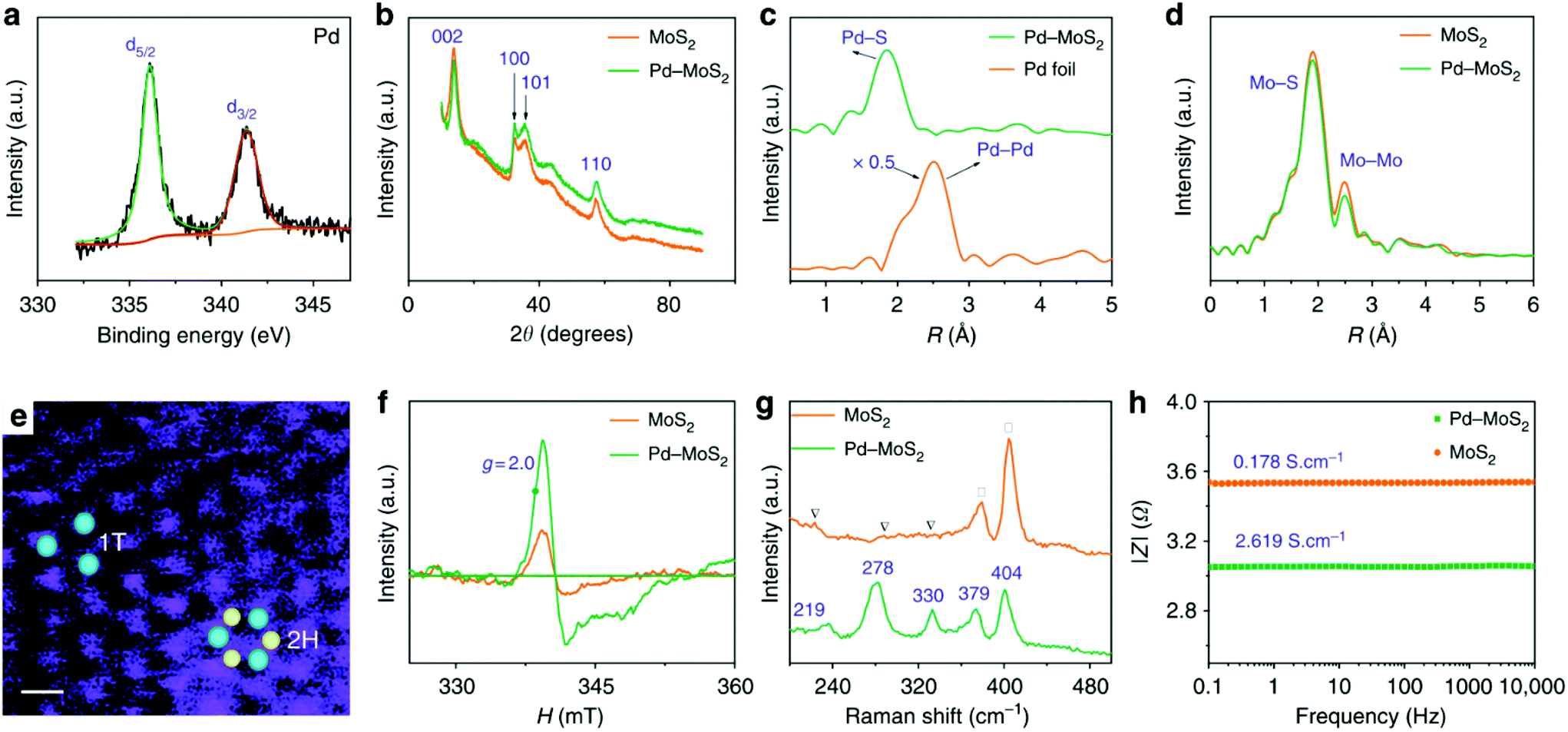 | ||
| Fig. 3 Structural analysis of 1%Pd-MoS2 and MoS2. (A) Pd-3d XPS and (B) XRD spectrum of the 1%Pd-MoS2. Fourier transform of the k3-weighted (C) Pd K-edge and (D) Mo K-edge of the EXAFS spectra. (E) Dark-field scanning transmission electron microscopy image of the 1%Pd-MoS2. Blue and yellow dots represent Mo and S atoms respectively. The white line is the scale bar of 1 nm. (f) ESR spectra and (g) Raman spectra of 1%Pd-MoS2 and MoS2. (h) Bode spectra with a frequency range from 0.1 Hz to 10 kHz and an amplitude of 5.0 mV.28 | ||
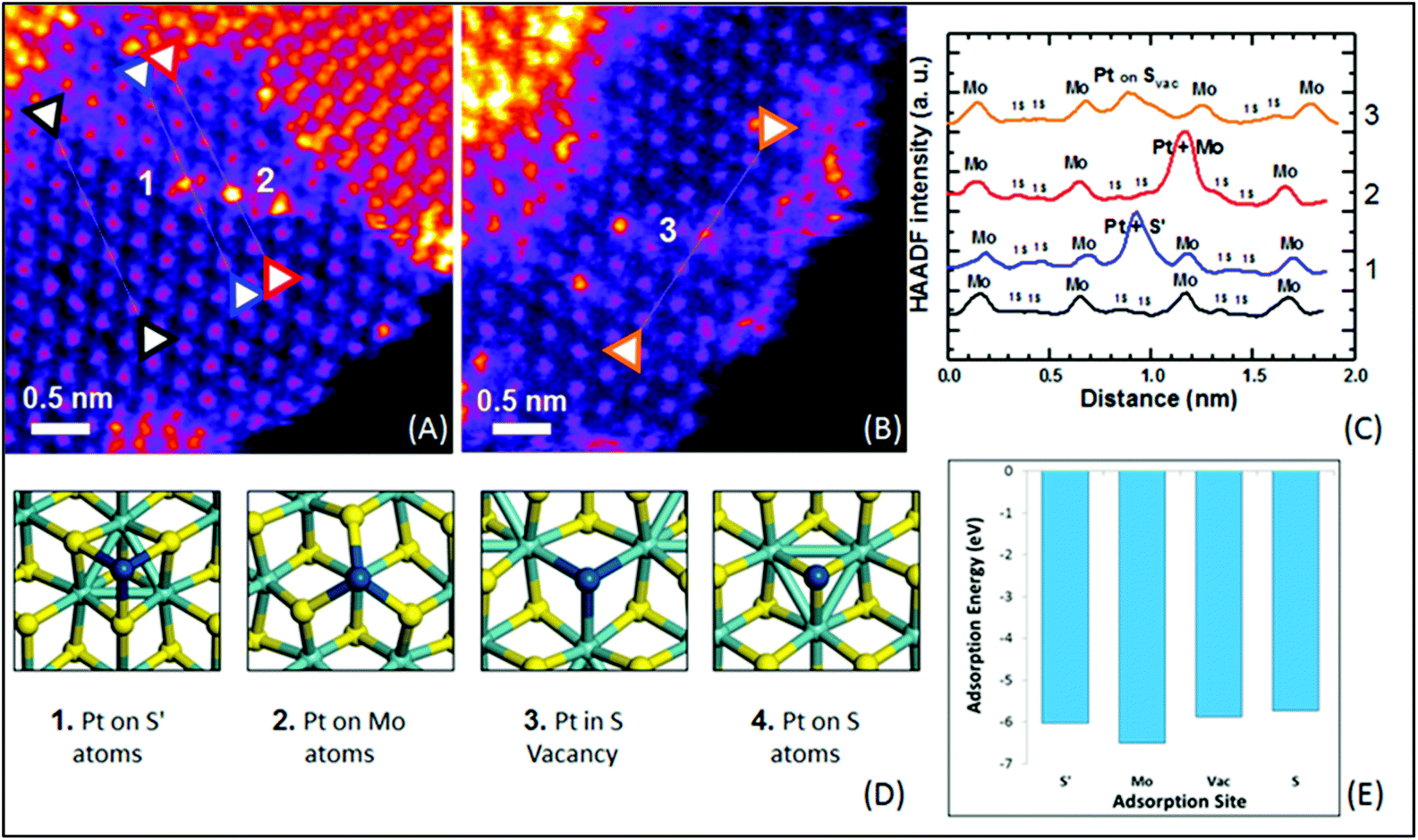 | ||
| Fig. 4 (a) and (b) HAADF-STEM of Pt-1T-SMoS2. (c) ADF intensity line profiles taken along the corresponding coloured line of black, blue, red, and orange in the 〈110〉 direction. (d) The proposed model of the Pt atom doping at 4 different adsorption sites. Numbers 1, 2 and 3 correspond to the experimentally observed Pd position shown in (a) and (b). (e) Adsorption energy for Pt atoms on each proposed adsorption site.29 | ||
It is interesting to note that non-metal P atom doped 1T MoS2 nanosheets prepared by Liu and co-workers exhibited a small Tafel slope of 34 mV per decade with an excellent overpotential of 43 mV at 10 mA cm−2 (Table 2, entry 6). Their catalytic performance is comparable to that of Pt doped graphene (with an overpotential of 45 mV and a Tafel slope of 29 mV per decade) which is considered to be one of the best SAC electrocatalysts reported.55 The DFT calculation suggested the possibility that the P atoms incorporated at S substitution sites (S vacancy sites) could both act as new main active sites for H adsorption and activate the basal S atoms of MoS2 at the same time. This offers a new opportunity to develop a non-metal based catalyst with no metal leaching issues during electrolysis of water at high potentials. The DOS and partial DOS simulations also suggested that more electronic states are shifted near the Fermi level after introducing P atoms (Fig. 5), which ultimately increased the electrical conductivity at the basal plane for an enhanced HER. In contrast, non-metal N atoms doped at the Mo substitution site once again did not directly participate in the HER but helped activate the surrounding S atoms to be more active for the H adsorption (Table 2, entry 7). Similarly, more DOS can be observed at the Fermi level when N atoms were embedded into MoS2, which also increases the electrical conductivity of basal sites. This once again reinforces the emphasis on how important the nature of the doper and the doping position can be when tailoring the SAC-MoS2 for hydrogen reduction.
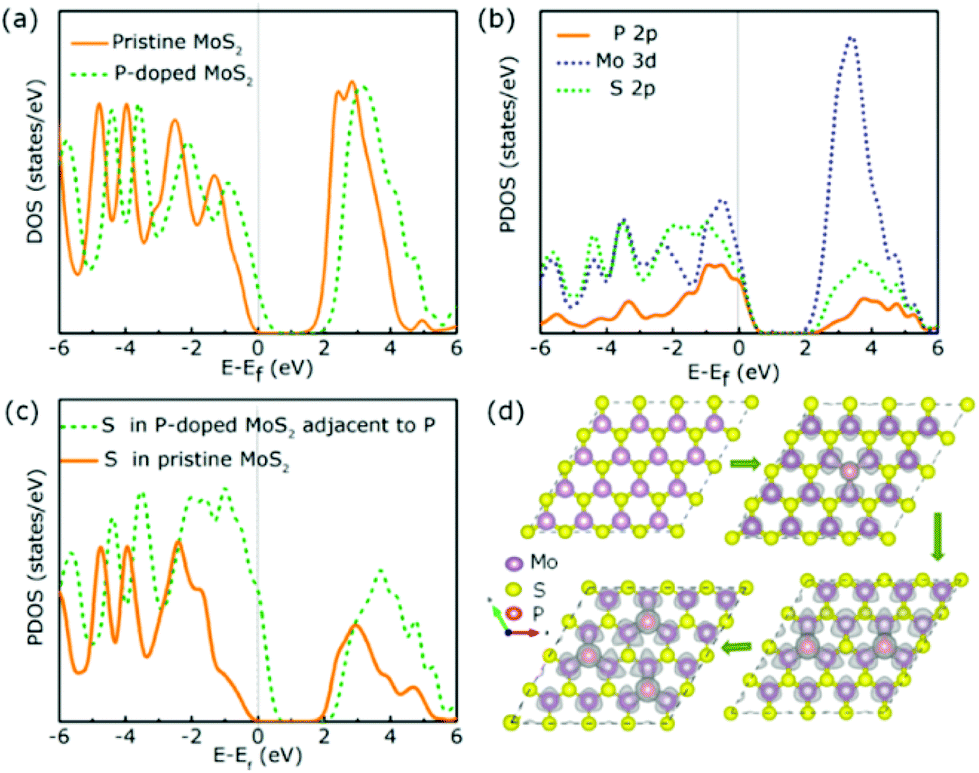 | ||
| Fig. 5 (a) Total DOS of pure MoS2 and P doped MoS2. (b) Partial DOS of P-MoS2. (c) Partial DOS for S atoms. The vertical grey lines represent the Fermi level. (d) Partial charge density of P-MoS2 when doped with 0–3 P atoms. | ||
5. Conclusions
The effective use of catalyst components to convert substrate molecules to a product is important in catalysis. As stated, the use of SACs on 2D materials is a hot topic of research due to their high characteristic surface area to volume ratios and the electronic and steric influence on catalytically active atoms can be made clearer over these defined nanostructures. However, at this early stage, there are still limited amounts of high-quality data with corresponding details of experimental analysis, and computational calculations to lead to the understanding of the catalyst structure and activity relationship at the atomic level.Here, through reviewing some selected key publications of SAC-MoS2 for the HER, we can draw some conclusions with new insights into the otherwise commonly used term ‘metal–support interactions’ in heterogeneous catalysis. Also, understanding the HER over the catalyst surface for H2 production from water is also very important in developing new catalytic technologies to harness renewable energies. It is shown that the polytypes of 2D MoS2 (the specific support material with characteristic structure and properties), the choice of single heteroatoms (i.e. both metal or non-metal atoms), and the doping position (defined site for specific electronic and steric perturbation) are the three most important factors for deciding the catalytic performance of the resulting SAC-MoS2 in the HER. Conventional electrochemical scan tests and volcano plots can be used to gauge which metal dopants on the 2H-MoS2 support could potentially help enhancing the overall HER performance. DFT modelling is important to characterise the electronic and steric properties of the atoms as the catalyst as well as the influence from the support. However, such a trend can subtly be changed if the chemical nature of dopers, relative sizes, and dwelling positions are altered. As a result, atomic positions on defined structures with defined interactions are required to obtain further understanding of structure–activity relationships. Specifically to the HER, the heteroatoms that replace Mo atoms can activate the adjacent in-plane S atoms for H adsorption which ultimately enhance the ability for H adsorption. They are anticipated to be more stable and durable in position since they are stably embedded into the MoS2 structure, whereas heteroatom doping at the Mo atop sites or S vacancy sites through interacting with the outer S layers can act as the active sites themselves and also activate the surrounding S through electronic perturbation for the HER. In the case where the basal S atoms have already been activated for H adsorption by other reasons, for example the heteroatoms are doped on a S defect rich 1T-MoS2, the heteroatoms may become the active sites for recombination of  to H2 if preferable. Overall, all these parameters could be tailored by choosing the correct synthetic methods, which makes SAC-MoS2 an interesting material for various electrochemical applications. This also provides insight into developing other 2D nanomaterials confined with different heteroatoms for various catalytic uses.
to H2 if preferable. Overall, all these parameters could be tailored by choosing the correct synthetic methods, which makes SAC-MoS2 an interesting material for various electrochemical applications. This also provides insight into developing other 2D nanomaterials confined with different heteroatoms for various catalytic uses.
Conflicts of interest
There are no conflicts to declare.Notes and references
- W. Choi, I. Lahiri, R. Seelaboyina and Y. S. Kang, Crit. Rev. Solid State Mater. Sci., 2010, 35, 52–71 CrossRef.
- D. Akinwande, C. J. Brennan, J. S. Bunch, P. Egberts, J. R. Felts, H. Gao, R. Huang, J.-S. Kim, T. Li and Y. Li, et al. , Extreme Mech. Lett., 2017, 13, 42–77 CrossRef.
- M. Chhowalla, H. S. Shin, G. Eda, L. J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- H. Pan, Sci. Rep., 2014, 4, 1–6 Search PubMed.
- M. Chhowalla, Z. Liu and H. Zhang, Chem. Soc. Rev., 2015, 44, 2584–2586 RSC.
- A. Yusuf, C. Snape, J. He, H. Xu, C. Liu, M. Zhao, G. Z. Chen, B. Tang, C. Wang, J. Wang and S. N. Behera, Catal. Rev. - Sci. Eng., 2017, 59, 189–233 CrossRef CAS.
- S. Royer and D. Duprez, ChemCatChem, 2011, 3, 24–65 CrossRef CAS.
- M. Khazaei, M. Arai, T. Sasaki, C. Y. Chung, N. S. Venkataramanan, M. Estili, Y. Sakka and Y. Kawazoe, Adv. Funct. Mater., 2013, 23, 2185–2192 CrossRef CAS.
- J. Xie and Y. Xie, Chem. – Eur. J., 2016, 22, 3588–3598 CrossRef CAS PubMed.
- D. Kong, H. Wang, J. J. Cha, M. Pasta, K. J. Koski, J. Yao and Y. Cui, Nano Lett., 2013, 13, 1341–1347 CrossRef CAS PubMed.
- Y. Zhang, Q. Ji, G. F. Han, J. Ju, J. Shi, D. Ma, J. Sun, Y. Zhang, M. Li, X. Y. Lang, Y. Zhang and Z. Liu, ACS Nano, 2014, 8, 8617–8624 CrossRef CAS PubMed.
- H. Nan, Z. Wu, J. Jiang, A. Zafar, Y. You and Z. Ni, J. Phys. D: Appl. Phys., 2017, 50, 154001 CrossRef.
- M. R. Islam, N. Kang, U. Bhanu, H. P. Paudel, M. Erementchouk, L. Tetard, M. N. Leuenberger and S. I. Khondaker, Nanoscale, 2014, 6, 10033–10039 RSC.
- L. Tao, X. Duan, C. Wang, X. Duan and S. Wang, Chem. Commun., 2015, 51, 7470–7473 RSC.
- H. Li, C. Tsai, A. L. Koh, L. Cai, A. W. Contryman, A. H. Fragapane, J. Zhao, H. S. Han, H. C. Manoharan, F. Abild-Pedersen, J. K. Nørskov and X. Zheng, Nat. Mater., 2015, 15, 48–53 CrossRef PubMed.
- H. Li, A. W. Contryman, X. Qian, S. M. Ardakani, Y. Gong, X. Wang, J. M. Weisse, C. H. Lee, J. Zhao, P. M. Ajayan, J. Li, H. C. Manoharan and X. Zheng, Nat. Commun., 2015, 6, 7381 CrossRef PubMed.
- Y. Tan, P. Liu, L. Chen, W. Cong, Y. Ito, J. Han, X. Guo, Z. Tang, T. Fujita, A. Hirata and M. W. Chen, Adv. Mater., 2014, 26, 8023–8028 CrossRef PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- G. Vilé, D. Albani, M. Nachtegaal, Z. Chen, D. Dontsova, M. Antonietti, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2015, 54, 11265–11269 CrossRef PubMed.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef PubMed.
- A. Alarawi, V. Ramalingam and J. H. He, Mater. Today Energy, 2019, 11, 1–23 CrossRef.
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Angew. Chem., Int. Ed., 2017, 56, 13944–13960 CrossRef PubMed.
- K. C. Kwon, J. M. Suh, R. S. Varma, M. Shokouhimehr and H. W. Jang, Small Methods, 2019, 3, 1–20 Search PubMed.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef.
- D. Voiry, M. Salehi, R. Silva, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nano Lett., 2013, 13, 6222–6227 CrossRef CAS PubMed.
- J. Deng, H. Li, J. Xiao, Y. Tu, D. Deng, H. Yang, H. Tian, J. Li, P. Ren and X. Bao, Energy Environ. Sci., 2015, 8, 1594–1601 RSC.
- T. H. M. Lau, X. Lu, J. Kulhavý, S. Wu, L. Lu, T. S. Wu, R. Kato, J. S. Foord, Y. L. Soo, K. Suenaga and S. C. E. Tsang, Chem. Sci., 2018, 9, 4769–4776 RSC.
- Z. Luo, Y. Ouyang, H. Zhang, M. Xiao, J. Ge, Z. Jiang, J. Wang, D. Tang, X. Cao, C. Liu and W. Xing, Nat. Commun., 2018, 9, 1–8 Search PubMed.
- T. H. M. Lau, S. Wu, R. Kato, T. S. Wu, J. Kulhavý, J. Mo, J. Zheng, J. S. Foord, Y. L. Soo, K. Suenaga, M. T. Darby and S. C. E. Tsang, ACS Catal., 2019, 9, 7527–7534 CrossRef CAS.
- P. Liu, J. Zhu, J. Zhang, P. Xi, K. Tao, D. Gao and D. Xue, ACS Energy Lett., 2017, 2, 745–752 CrossRef CAS.
- R. Li, L. Yang, T. Xiong, Y. Wu, L. Cao, D. Yuan and W. Zhou, J. Power Sources, 2017, 356, 133–139 CrossRef.
- M. Acerce, D. Voiry and M. Chhowalla, Nat. Nanotechnol., 2015, 10, 313–318 CrossRef.
- F. Wypych and R. Schollhorn, J. Chem. Soc., Chem. Commun., 1992, 1386–1388 RSC.
- D. B. Putungan, S. Lin and J. Kuo, Phys. Chem. Chem. Phys., 2015, 17, 21702–21708 RSC.
- J. C. Torres-Guzmán, G. Martínez-Mekler and M. F. Müller, J. Chem. Phys, 2016, 174702 CrossRef.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef PubMed.
- H. Tang and S. Roy Morrison, Thin Solid Films, 1993, 227, 90–94 CrossRef.
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef PubMed.
- D. Voiry, M. Salehi, R. Silva, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nano Lett., 2013, 13, 6222–6227 CrossRef PubMed.
- C. A. Papageorgopoulos and W. Jaegermann, Surf. Sci., 1995, 338, 83–93 CrossRef.
- Y. Wang, J. Z. Ou, S. Balendhran, A. F. Chrimes, M. Mortazavi, D. D. Yao, M. R. Field, K. Latham, V. Bansal, J. R. Friend, S. Zhuiykov, N. V. Medhekar, M. S. Strano and K. Kalantar-Zadeh, ACS Nano, 2013, 7, 10083–10093 CrossRef.
- X. Fan, P. Xu, D. Zhou, Y. Sun, Y. C. Li, M. A. T. Nguyen, M. Terrones and T. E. Mallouk, Nano Lett., 2015, 15, 5956–5960 CrossRef PubMed.
- X. Ma, P. Zhang, Z. Wang, R. Wu, T. Jiang, H. Wang, Y. Zhao, S. Wu, Y. Xie, Y. Zhan and T. Nan, Nanotechnology, 2016, 28, 084001 Search PubMed.
- T. Payam, W. Jieqiong, X. Hui, F. D. Joel, A. Mumtaz Murat, Z. Chuan, K. Kaifei, B. Brett, Z. Lijie, Z. Puqin, H. Shaoming, Y. Sen, V. B. Frank, C. John and Z. Hao, Mater. Res. Express, 2016, 3, 75009 CrossRef.
- Z. He and W. Que, Appl. Mater. Today, 2016, 3, 23–56 CrossRef.
- L. Samad, S. M. Bladow, Q. Ding, J. Zhuo, R. M. Jacobberger, M. S. Arnold and S. Jin, ACS Nano, 2016, 10, 7039–7046 CrossRef.
- X. Feng, Q. Tang, J. Zhou, J. Fang, P. Ding, L. Sun and L. Shi, Cryst. Res. Technol., 2013, 48, 363–368 CrossRef.
- C. Perumal Veeramalai, F. Li, H. Xu, T. W. Kim and T. Guo, RSC Adv., 2015, 5, 57666–57670 RSC.
- X. Feng, Q. Tang, J. Zhou, J. Fang, P. Ding, L. Sun and L. Shi, Cryst. Res. Technol., 2013, 48, 363–368 CrossRef.
- G. Liu, A. W. Robertson, M. M.-J. J. Li, W. C. H. H. Kuo, M. T. Darby, M. H. Muhieddine, Y.-C. C. Lin, K. Suenaga, M. Stamatakis, J. H. Warner and S. C. E. Tsang, Nat. Chem., 2017, 9, 810–816 CrossRef.
- R. G. Compton and C. E. Banks, Understanding voltammetry, World Scientific, 2011 Search PubMed.
- S. Park, Y. Shao, J. Liu and Y. Wang, Energy Environ. Sci., 2012, 5, 9331–9344 RSC.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS.
- S. Trasatti, J. Electroanal. Chem., 1972, 39, 163–184 CrossRef CAS.
- N. Cheng, S. Stambula, D. Wang, M. N. Banis, J. Liu, A. Riese, B. Xiao, R. Li, T. K. Sham, L. M. Liu, G. A. Botton and X. Sun, Nat. Commun., 2016, 7, 1–9 Search PubMed.
| This journal is © The Royal Society of Chemistry 2020 |