
Covalent organic framework photocatalysts: structures and applications
Han
Wang†
 a,
Hui
Wang†
b,
Ziwei
Wang†
a,
Lin
Tang†
a,
Guangming
Zeng
*a,
Piao
Xu
*a,
Ming
Chen
a,
Ting
Xiong
a,
Chengyun
Zhou
a,
Xiyi
Li
b,
Danlian
Huang
a,
Yuan
Zhu
a,
Zixuan
Wang
a and
Junwang
Tang
*b
a,
Hui
Wang†
b,
Ziwei
Wang†
a,
Lin
Tang†
a,
Guangming
Zeng
*a,
Piao
Xu
*a,
Ming
Chen
a,
Ting
Xiong
a,
Chengyun
Zhou
a,
Xiyi
Li
b,
Danlian
Huang
a,
Yuan
Zhu
a,
Zixuan
Wang
a and
Junwang
Tang
*b
aCollege of Environmental Science and Engineering, Hunan University and Key Laboratory of Environmental Biology and Pollution Control (Hunan University), Ministry of Education, Changsha 410082, P. R. China. E-mail: zgming@hnu.edu.cn; piaoxu@hnu.edu.cn
bDepartment of Chemical Engineering, University College London, Torrington Place, London, WC1E7JE, UK. E-mail: junwang.tang@ucl.ac.uk
First published on 18th May 2020
Abstract
In the light of increasing energy demand and environmental pollution, it is urgently required to find a clean and renewable energy source. In these years, photocatalysis that uses solar energy for either fuel production, such as hydrogen evolution and hydrocarbon production, or environmental pollutant degradation, has shown great potential to achieve this goal. Among the various photocatalysts, covalent organic frameworks (COFs) are very attractive due to their excellent structural regularity, robust framework, inherent porosity and good activity. Thus, many studies have been carried out to investigate the photocatalytic performance of COFs and COF-based photocatalysts. In this critical review, the recent progress and advances of COF photocatalysts are thoroughly presented. Furthermore, diverse linkers between COF building blocks such as boron-containing connections and nitrogen-containing connections are summarised and compared. The morphologies of COFs and several commonly used strategies pertaining to photocatalytic activity are also discussed. Following this, the applications of COF-based photocatalysts are detailed including photocatalytic hydrogen evolution, CO2 conversion and degradation of environmental contaminants. Finally, a summary and perspective on the opportunities and challenges for the future development of COF and COF-based photocatalysts are given.
 Han Wang | Han Wang received her ME from Chinese Academy of Science in 2016. Currently, she is a PhD candidate under the supervision of Prof. Guangming Zeng in College of Environmental Science and Engineering, Hunan University, China. Her current research interests focus on the synthesis and application of functional covalent organic materials in photocatalysis and biosensing. |
 Hui Wang | Hui Wang received BSc and MSc in Environmental Engineering from Hunan University, focusing on photocatalyst fabrication for environmental purification. At present, she is pursuing her PhD study in Prof. Junwang Tang's group at the UCL Department of Chemical Engineering. Her current research focuses on photocatalytic nitrogen fixation. |
 Guangming Zeng | Prof. Guangming Zeng has been teaching courses and performing research on Environmental Science and Health at Hunan University since 1988. He has been the head of the School of Environmental Science and Engineering since 1994 at the same University. He is one of 2018 Highly Cited Researchers in the world issued by Clarivate Analytics. His current research interests focus on the synthesis and application of functional nanomaterials in the field of environment and energy. |
 Piao Xu | Piao Xu received her PhD degree from Hunan University in 2016. She is currently an assistant professor in the College of Environmental Science and Engineering, Hunan University. She has published more than 100 academic papers with more than 6705 citations and with a H factor of 41 (according to the ES database updated as of 10 April 2020). Her major research focus is on the development of nanomaterials and application in environmental remediation. |
 Junwang Tang | Dr Junwang Tang is the Director of UCL Materials Hub and Professor of Materials Chemistry in the Department of Chemical Engineering at UCL. He obtained his PhD in Physical Chemistry in 2001. After that, he was appointed as a JSPS Fellow at NIMS, Japan, and as a senior researcher in the Department of Chemistry at Imperial College London, working on solar fuel synthesis and mechanistic studies by time-resolved spectroscopy. After that, he took a faculty position at UCL. His current research interests lie in photocatalytic CH4 transformation, ammonia synthesis, solar water splitting, CO2 conversion by artificial photosynthesis and mechanistic aspects of solar energy conversion, and water treatment, as well as microwave-assisted intensified flow chemistry. |
1. Introduction
With the rapid industrialization and urbanization, there has been a greater concern about the sustainable supply of fossil fuels (e.g. oil, coal and gas) and the severe environmental issues caused by utilization of these fossil fuels.1–3 Thus, it is urgent to secure an alternative, sustainable, clean energy source by an operative and scalable technology to address the environment and energy issues. Solar driven chemical processes including water splitting, CO2 conversion, photocatalytic degradation, etc., have attracted substantial interest since solar energy is an abundant and inexhaustible source. One method for capturing solar energy is photocatalysis in which the electrons from the valence band (VB) can be excited to the conduction band (CB) when the energy of photons is greater than the semiconductor band gap, creating electron/hole pairs in the photocatalysts, and the electrons and holes migrate to the surface to participate in chemical reactions.In the 1970s, Fujishima and Honda realized water splitting under ultraviolet (UV) radiation by using a titanium dioxide (TiO2) electrode for the first time.4 And Carey et al. carried out the photocatalytic degradation of organic pollutants with TiO2 in aqueous suspensions four years later.5 These have sparked intense interest among researchers in artificial photosynthesis. Traditional inorganic semiconductor photocatalysts such as TiO2,6–8 cadmium sulphide (CdS),9–11 zinc oxide (ZnO)12,13 and silver phosphate (Ag3PO4)14 have occupied a leading position over the past several decades. Among them, TiO2 is the most important and well-known photocatalyst due to its low cost, relatively high availability and durability. However, its wide band gap of 3.2 eV that only allows for ultraviolet light absorption limits its utilization of the solar spectrum, leading to low photocatalytic efficiency and photocurrent quantum yield.15 Besides, Ag3PO4, CdS and other transition metal sulfides and oxides with a suitable band gap to absorb visible light and with good carrier transportation capacity have stimulated the attention on photocatalytic studies, whereas the heavy metal toxicity and photo-corrosion effect block their practical applications.9 As the research progressed, organic semiconductors like graphitic carbon nitride (g-C3N4),16,17 metal–organic frameworks (MOFs),18,19 and covalent organic frameworks (COFs)20–24 have been used as the photocatalyst and show promising performance towards solar energy conversion. g-C3N4 as a metal-free polymer possesses many fascinating features including “earth-abundant” nature, high physicochemical stability and favorable band gap structure. However, there are drawbacks: its synthesis is often conducted at high temperature (>500 °C) and its molecular backbone consists of either triazine or heptazine units, leading to limited structural diversity.25 As a type of porous crystalline materials, MOFs constructed from organic linkers and transition-metal nodes are attractive due to their large surface area, structural tailorability and easy pore functionalization. Unfortunately, most MOFs are unstable and can easily deteriorate under humid conditions which limits their repeated use.19
Covalent organic frameworks (COFs), as newly developed organic polymers, have caused ripples of excitement among researchers striving to exploit their promising photocatalytic potential. COFs with low density are crystalline porous materials composed of organic molecules linked by covalent bonds through reticular chemistry, and they have been widely used in areas such as heterogeneous catalysis,26–29 gas storage and separation,30,31 energy storage and optoelectronic devices.32–34 Compared with traditional semiconductors, COFs possess not only some common features but also many special advantages pertaining to photocatalysis: (i) the structural designability of COFs enables them to realize the design of targeted structures and special properties related to photocatalytic reactions such as excellent visible-light absorption, and fast electron–hole separation and transfer; (ii) the large surface area of COFs enriches accessible catalytic sites, and the highly crystalline and porous structure endows COFs with accelerated charge transport to the surface and decreases the possibility of charge trapping caused by defects, thus contributing to suppressed electron–hole recombination; (iii) COFs with strong covalent bonds show high chemical and thermal stability, and photoactive units fixed in the robust framework can avoid photo-corrosion and enhance the lifetime of the excited states; and (iv) the extended π-conjugated structure both in-plane and in the stacking direction enables high charge carrier mobility. These fascinating inherent features endow COFs with great potential in photocatalytic energy conversion and environmental remediation, and they are deemed to match or even exceed MOFs and conventional photocatalytic semiconductors in performance. Lotsch and co-workers first reported the discovery of a COF-based photocatalyst.35 A high visible-light-induced hydrogen production efficiency has been achieved based on hydrazine-based TFPT-COF (evolution rate: 1970 μmol h−1 g−1, triethanolamine (TEOA) as a sacrificial donor), which was competitive with other representative photocatalysts including Pt-modified amorphous melon (720 μmol h−1 g−1), g-C3N4 synthesized at 600 °C (840 μmol h−1 g−1),36 and crystalline poly(triazine imide) (864 μmol h−1 g−1).37 This success has initiated the exploration of COF-based photocatalysts in the whole community (Fig. 1).
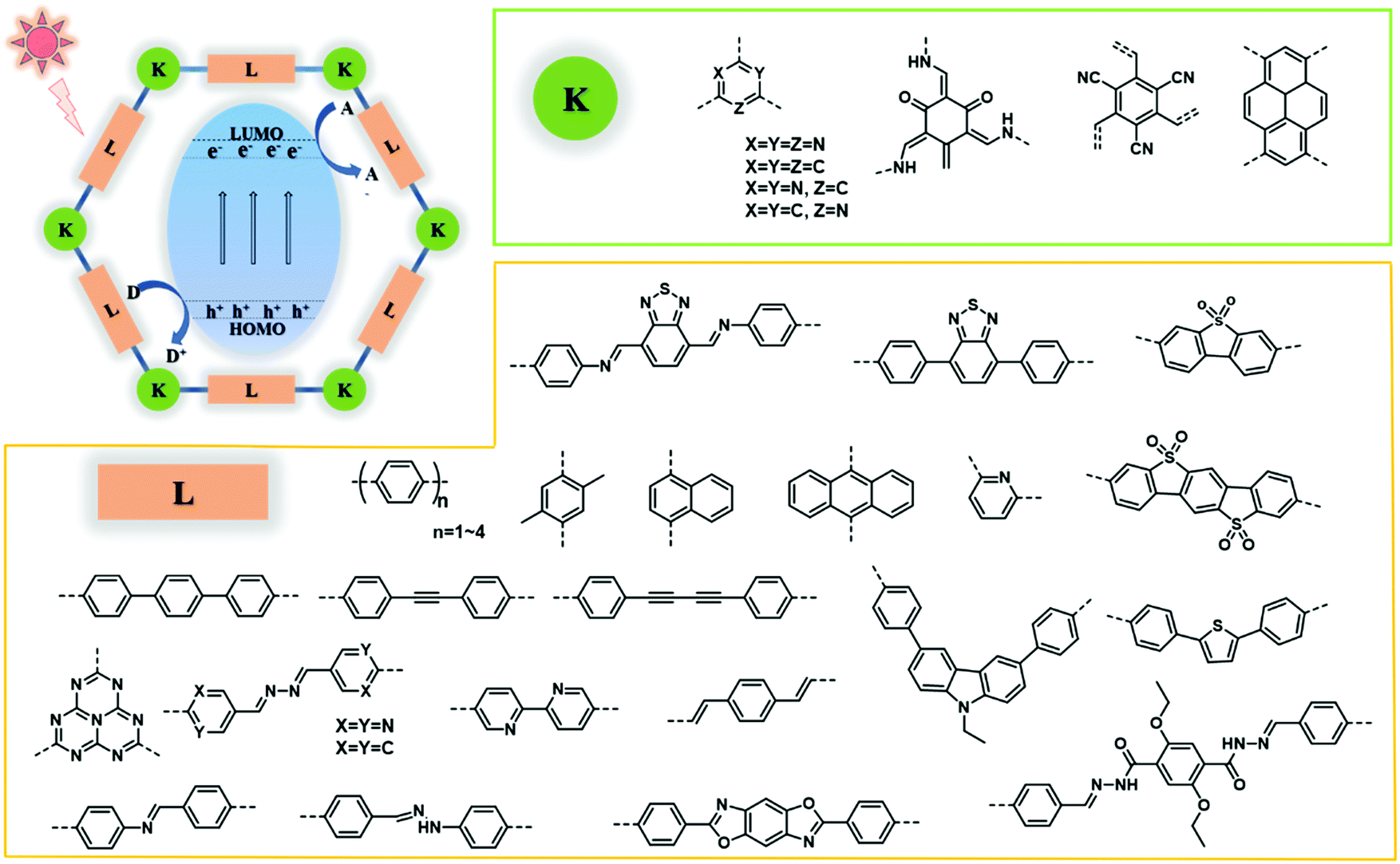 | ||
| Fig. 1 Structures of COF photocatalysts (K represents knots; L represents linkers). | ||
The number of publications in the area of COF-based photocatalysts has increased sharply, and a comprehensive review of COF photocatalysts is needed. In this review, we begin by summarizing different connections of COF building blocks including boron-containing connections, nitrogen-containing connections and double-stage connections combining imine linkages and boronate ester linkages. Subsequently, we compare the performance of COFs with different morphologies, such as 0-dimensional (0D) structures, 1-dimensional (1D) structures, 2-dimensional (2D) structures, and 3-dimensional (3D) structures. Strategies related to the enhanced photocatalytic performance of COF materials are then presented. Afterwards, the solar-driven application of COFs is discussed, including water splitting, CO2 conversion as well as photocatalytic degradation of pollutants in wastewater. Finally, a perspective on the challenges and opportunities in this area, including synthesis, functions and application, is discussed. Complementary to this review, the readers are also suggested to read another review about the design of COF if they are interested in the materials design.38–44
2. Linking chemistry of COFs
COFs are a kind of crystalline porous materials with pure organic groups connected by robust covalent bonds. Diverse covalent bonds formed from various synthetic organic reactions between theoretically unlimited building blocks endow COFs with designable crystalline structures and targeted functions. In this section, different linkages of COF building blocks are summarized, including boron-containing linkage, triazine linkage, imine linkage, β-ketoenamine linkage, hydrazone and azine linkages, and other linkages (Fig. 2).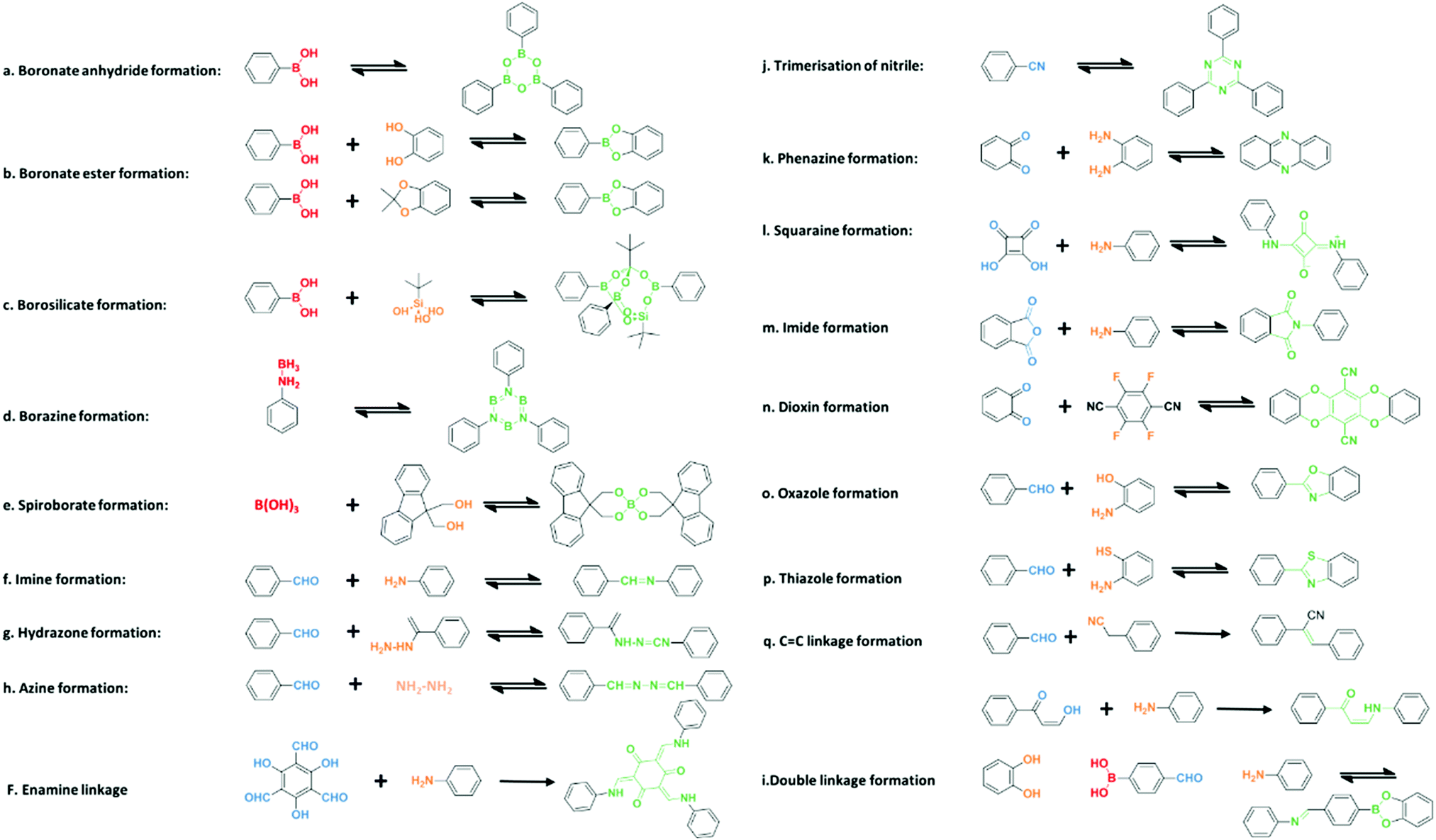 | ||
| Fig. 2 Various linkages of COF formation. | ||
2.1. Boron-containing linkage
Since the pioneering research of Yaghi and co-workers that constructed the first two COFs, namely COF-1 and COF-5, diverse syntheses of COFs linked by boron-containing linkages via the formation of boronate ester, boroxine or borazine have generated considerable interest.45 Most of the synthesized boron-based COFs could be classified into two categories: those constructed by self-condensation of single building blocks and those by co-condensation of two or more building units.As a representative example, COF-1 was designed and fabricated through the self-condensation of 1,4-benzenediboronic acid (BDBA), which was based on molecular dehydration to form six-membered boroxine connections.45 The as-prepared COF-1 possessed a layered graphitic structure with a hexagonal pore diameter of 15 Å and a Brunauer–Emmett–Teller (BET) surface area of 711 m2 g−1. In this method, it is essential to keep the reaction under a closed condition for water equilibrium to guarantee reversibility of COF formation. Similarly, the same group further successfully constructed the first 3D COFs (COF-102 and COF-103) with the self-condensation of the tetrahedral molecular building block tetra(4-dihydroxyborylphenyl)methane (TBPM) or its silane analog (TBPS).46 The crystalline COF-102 and COF-103 exhibited a higher BET surface area of 3472 m2 g−1 and 4210 m2 g−1, respectively. Since then, this self-condensation strategy has been widely used to fabricate boron-containing COFs based on various monomers, such as biphenyldiboronic acid,47 pyrene-2,7-diboronic acid (PDA),48 and 4,4′-phenylazobenzoyl diboronic acid.49
Besides self-condensation, co-condensation of two or more building blocks such as boronic acids with catechols has also been reported. The dehydration condensation of 2,3,6,7,10,11-hexahydroxytriphenylene (HHTP) and BDBA resulted in the formation of layered COF-5 with five-membered BO2C2 rings, which exhibited an eclipsed boron nitride arrangement.45 It is worth mentioning that COF-5 has been widely regarded as a representative to examine various new synthesis strategies.50–52 Likewise, the first crystalline boronate-linked 3D COFs (COF-105 and COF-108) were obtained by replacing BDBA with tetrahedral molecules TBPM and TBPS, respectively.46 COFs with different properties and functions could be designed and synthesized by a diverse combination of building units. For instance, a novel photoactive donor–acceptor TP-Por COF was prepared based on triphenylene and porphyrin units.53 The resulting TP-Por COF film with enhanced charge separation showed broad optical absorption covering the entire visible range up to 680 nm. In a conventional condensation, donor–acceptor DTP–ANDI–COF with a large pore size of 5.3 nm was obtained from N,N′-di-(4-boronophenyl)naphthalene-1,4,5,8-tetracarboxylic acid diimide and HTTP.54 The charge-separation state lifetime of 2.5 μs was determined by time-resolved electron spin resonance spectroscopy, indicating the presence of long-lived radicals produced through effective charge transfer from the donor triphenylene to the acceptor naphthalene diimide. Notably, polyfunctional catechols are easily oxidized and are difficult to dissolve in most organic solvents, leading to difficulty in the fabrication of functional building blocks and related COFs. Thus, a new Lewis acid-catalyzed strategy protecting catechols from oxidation was put forward.55 A boronate ester-linked Pc–PBBA COF with a pore size of 2.3 nm was constructed from 1,4-phenylenebis(boronic acid) (PBBA) and phthalocyanine tetra(acetonide) (Pc) in the presence of Lewis-acid catalyst BF3·OEt2. The as-prepared eclipsed COF with broad absorbance showed great potential for effective charge transfer through stacked phthalocyanines. In contrast to the conventional condensation of two components, a multiple-component (MC) strategy was also studied.56 For example, a three-component [1+2] co-condensation was attempted by using the shortest unit BDBA and a longer molecule PDA as the linkers to react with HHTP as the knots. Two MC-COFs (termed MC-COF-TP-E11E27 and MC-COF-TP-E21E17) with slipped AA stacking were generated to possess a BET surface area of 1892 and 1534 m2 g−1 and a pore size of 3.2 and 2.9 nm, respectively. This co-condensation strategy could also be used to tailor the functionality of COFs. A highly emissive 2D COF TPE-Ph COF was designed by introducing an aggregation-induced emission active tetraphenylethene (TPE) unit to condense with TPE-cored boronic acids and 1,2,4,5-tetrahydroxybenzene.57 Considering that the boronate linkages in the TPE-Ph COF formed a Lewis acid–base pair when interacted with ammonia, the TPE-Ph COF could be used as a fluorescence sensor for ammonia.
Generally, COFs with boron-containing linkages possess low density and high surface area, leading to various applications.58,59 However, boroxines and boronate esters are easily hydrolysed and oxidized, which limits their application as catalysts or their long-term usage. Still, it is undeniable that boron-containing COFs are of particular importance for mechanistic studies.50,60,61
2.2. Triazine-based linkage
In 2008, Thomas and co-workers reported the first example of triazine-based COFs (denoted as CTFs), which was achieved by cyclotrimerization of aromatic nitriles at 400 °C with the catalysis of ZnCl2.62 However, harsh synthesis conditions, such as high reaction temperature and acid solution purification, led to the destruction of long-range order. Only a few crystalline CTFs have been prepared by this method constructed from 1,3,5-tricyanobenzene, 1,4-dicyanobenzene and 2,6-dicyanonaphthalene monomers, namely CTF-0, CTF-1 and CTF-2, respectively.63,64 Moreover, limited building blocks are able to withstand the high reaction temperature, thus lowering the diversity of CTFs. Thus, other strategies based on milder conditions have been developed.65–67 A low-temperature polycondensation approach was utilized to synthesize CTFs based on a broader range of building blocks under mild conditions.68 For example, CTF-HUST-1 prepared from 1,4-benzene-dialdehyde reacted at ambient pressure and at a temperature lower than or equal to 120 °C, which also enabled the large scale synthesis. Soon afterward, a new concept was put forward to fabricate highly crystalline CTFs by in situ oxidizing alcohol to form aldehyde with controlled reaction rates. The principle behind this reaction was that decreasing the nucleation rates and lowering the concentration of nuclei could lead to relatively high crystallization.69 The as-prepared CTFs possessed much-improved crystallinity and higher photocatalytic performance compared to low crystalline or amorphous CTFs. And a controlling feeding rate method was also used to achieve highly crystalline CTFs.70Despite the crystalline problems, high BET surface area, remarkable thermal and chemical stability, and controllable C/N/H composition endow CTFs with potential for catalysis. 2D CTFs with triazine subunits can be regarded as analogs of g-C3N4, which has been studied extensively as a photocatalyst.71,72 On the one hand, the incorporated nitrogen in the backbone benefits metal nanoparticle loading, which provides a platform for the introduction of active sites for the catalytic reaction. On the other hand, the tunable structures with unlimited organic subunits allow for the controllable band alignment and optimal light absorption.73 Studies demonstrated that the photocatalytic hydrogen production of CTF-1 can be varied with different reaction conditions. For example, a well-ordered CTF-1 was synthesized via a mild microwave-assisted condensation.74 An apparent quantum efficiency (AQE) of 3.8% and 6% at 420 nm for oxygen and hydrogen evolution under visible light irradiation was determined, respectively. In particular, the oxygen evolution rate and hydrogen evolution rate of CTF-1-100 W were 140 μmol g−1 h−1 and 5500 μmol g−1 h−1, respectively, both of which are higher than those of g-C3N4.71,75 The examples verified the promising properties and applications of triazine-linked COFs. The successful synthesis of crystalline CTFs on a large scale will be the focus of future research.
2.3. Imine linkage
The polymerization of amines and aldehydes leads to the formation of imine bonds. While the resulting layered COFs with imine linkages were similar to boronic ester COFs, the imine-linked COFs showed higher stability to water, which was significantly different from boron-containing COFs. Even though their stability may be also influenced by the incorporated linkers, imine bonds have been one of the most attractive linkage motifs in COFs owing to the plenty of obtainable amine and aldehyde linkers as well as the great potential for constructing conjugated π-systems through the COF sheets. To date, imine-formation is clearly the most common synthesis strategy employed to build COFs. Early in 2009, the first imine-linked 3D COF (named as COF-300) was reported via the copolymerization of terephthaldehyde (TA) and tetra-(4-anilyl)methane (TAM) by using 1,4-dioxane as the solvent and aqueous acetic acid as the catalyst.76 The Fourier transform infrared (FT-IR) spectrum of COF-300 exhibited the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretch at 1620 and 1202 cm−1, which confirmed the formation of imine bonds. The as-prepared crystalline COF-300 with 5-fold interpenetration was stable up to 490 °C. Furthermore, TAM has been widely used in the construction of 3D COFs.23,77,78 Similarly, the first imine-linked 2D COF COF-LZU1 was synthesized through the condensation of 1,4-diaminobenzene with 1,3,5-triformylbenzene (Fig. 3).79 The as-prepared COF-LZU1 showed high stability against water and common organic solvents including acetone, dimethyl sulfoxide, tetrahydrofuran, trichloromethane, and N,N-dimethylformamide. The FT-IR spectrum of COF-LZU1 displayed a strong CN stretching mode of imines at 1618 cm−1. COF-LZU1 was demonstrated to be an ideal platform for metal ion incorporation due to the eclipsed layered-sheet arrangement. Indeed, the Pd/COF-LZU1 catalyst was successfully achieved by post-modification of COF-LZU1 with palladium acetate.
N stretch at 1620 and 1202 cm−1, which confirmed the formation of imine bonds. The as-prepared crystalline COF-300 with 5-fold interpenetration was stable up to 490 °C. Furthermore, TAM has been widely used in the construction of 3D COFs.23,77,78 Similarly, the first imine-linked 2D COF COF-LZU1 was synthesized through the condensation of 1,4-diaminobenzene with 1,3,5-triformylbenzene (Fig. 3).79 The as-prepared COF-LZU1 showed high stability against water and common organic solvents including acetone, dimethyl sulfoxide, tetrahydrofuran, trichloromethane, and N,N-dimethylformamide. The FT-IR spectrum of COF-LZU1 displayed a strong CN stretching mode of imines at 1618 cm−1. COF-LZU1 was demonstrated to be an ideal platform for metal ion incorporation due to the eclipsed layered-sheet arrangement. Indeed, the Pd/COF-LZU1 catalyst was successfully achieved by post-modification of COF-LZU1 with palladium acetate.
 | ||
| Fig. 3 Schematics for the synthesis of COF-LZU1. | ||
Various building blocks have been involved in imine-based COF formation.80–85 For instance, a highly conjugated π-electron porphyrin unit and its metal derivatives have been largely employed in the construction of functional imine-linked COFs. One study introduced two porphyrin-based COFs, termed COF-66 and COF-366, with the feature of extended planar π-conjugation.86 COF-66 and COF-366 were obtained from the solvothermal reaction of porphyrin and TA and tetrahydroxy anthracene, respectively, and the formed imine bond was characterized by FT-IR and 13C cross-polarization magic-angle spinning (CP-MAS) NMR spectroscopic techniques. Both COFs exhibited high charge carrier mobility owing to the close intermolecular π–π distances. A series of porphyrin COFs MP-DHPh COFs with varied H-bonding sites were synthesized via a three-component condensation strategy. Specifically, porphyrin derivatives (MP; M = H2, Cu, and Ni) were used to react with a mixture of TA and dihydroxyterephthalaldehyde (DHTA, H-bonding edges) at different molar ratios. As determined by ultraviolet-visible diffuse reflectance spectroscopy (UV-vis DRS), H2P-DHPh COF, CuP-DHPh COF, and NiP-DHPh COF possessed narrower band gaps of 1.31, 1.36, and 1.54 eV compared to that of 1.36, 1.40, and 1.58 eV for the corresponding amorphous MP-Ph polymers, respectively. The H2P-DHPh COF displayed higher photocatalytic singlet oxygen evolution than the CuP-DHPh COF and NiP-DHPh COF, and the photocatalytic performance of COFs increased with the increasing content of the H-bonding site. More recently, a conjugated imine-linked metalloporphyrin COF was prepared through the Schiff-base reaction of Zn-5,10,15,20-tetrakis(4-aminophenyl)-21H,23H-porphyrin (Zn-TAPP) and Cu-5,10,15,20-tetrakis(4-formylphenyl)-21H,23H-porphyrin (Cu-TFPP) in the presence of n-butanol, o-dichlorobenzene and aqueous acetic acid. The resulting ZnCu-Por-COF possessed effective π-conjugation and high charge-transfer transition.
Interestingly, COFs with two types of covalent linkage were realized by the orthogonal (interference-free) reaction strategy. Binary NTU-COF-1 with both boroxine ring and imine group was constructed from the copolymerization of 1,3,5-tris(4-aminophenyl)-benzene (TAPB) and 4-formylphenylboronic acid (FPBA), which possessed ditopic units of aldehyde and boronate. As indicated by FT-IR spectra, the appearance of B–O stretching bands (1336 cm−1 and 1305 cm−1), B–C band (1221 cm−1), B3O3 band (711 cm−1) and a strong CN band (1627 cm−1) verified the existence of B3O3 rings and imine linkage. Likewise, ternary NTU-COF-2 was successfully synthesized based on TAPB, FPBA, and HHTP with the formation of the C2O2B boronate ring and imine group. Accordingly, there are two paths for the design of bifunctional linkages. First, one of the building units possesses at least two functional moieties, which enables the simultaneous reactions of co-condensation and self-condensation with other functional building blocks, such as TATTA-FPBA COF (TATTA: 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline) and NTU-COF-1.83 Second, three functional building blocks were employed, and at least one of them has two different functional moieties to enable two non-interfering co-condensation reactions, like NTU-COF-2 and HHTP-FPBA-TATTA COF.83
2.4. β-Ketoenamine linkage
Improving the stability of COFs is of vital importance for their applications. Banerjee et al. put forward a two-step strategy to fabricate COFs with high stability when subjected to boiling water, acids and strong bases.87 Specifically, TpPa-1 and TpPa-2 with ketoenamine linkage were realized by the condensation of 1,3,5-triformylphloroglucinol (Tp) with p-phenylenediamine (Pa-1) or 2,5-dimethyl-p-phenylenediamine (Pa-2), in which Tp possesses hydroxyl groups adjacent to the formyl groups (Fig. 4). The COF formation involves two steps, that is the crystalline framework formed based on the reversible Schiff base reaction, and enhanced stability originated from irreversible enol-to-keto tautomerization. The structure of as-prepared TpPa-1 and TpPa-2 maintained integrity in boiling water and acid, and TpPa-2 was also stable in a basic medium. Notably, β-ketoenamine linked COFs usually feature low crystallinity compared to their imine counterparts as a result of an irreversible procedure in which error correction might be hindered in the COF lattice.88,89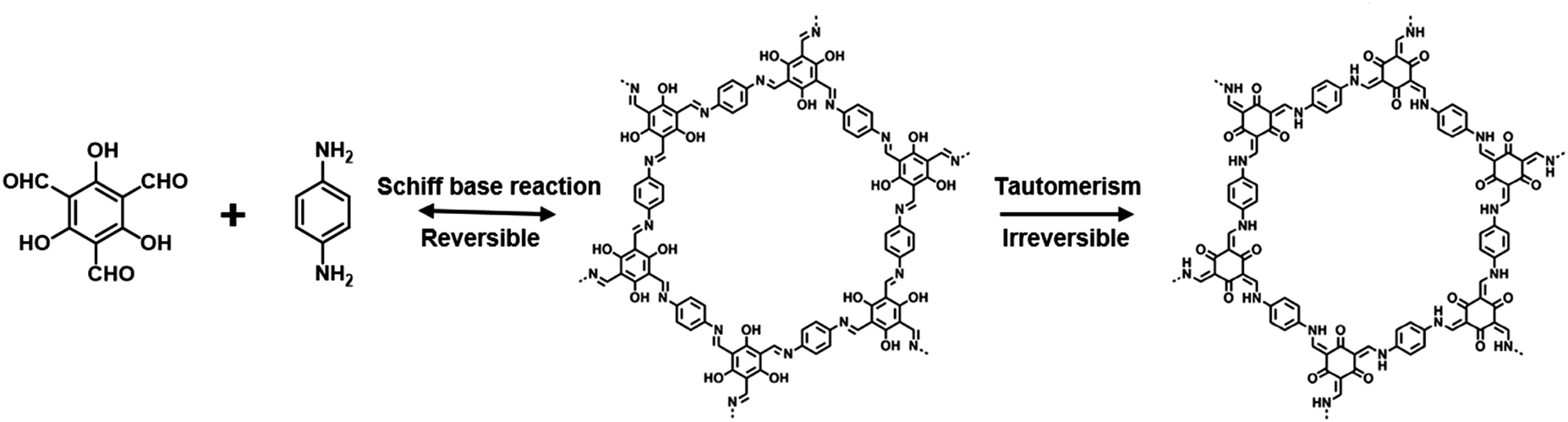 | ||
| Fig. 4 Schematic illustration of the formation of TpPa which involved the steps of reversible Schiff-base reaction and irreversible enol-to-keto tautomerism. | ||
Understandably then, the remarkable chemical stability endows β-ketoenamine linked COFs with exceptional potential for diverse applications such as photocatalytic reactions. Moreover, keto functionalities present in the β-ketoenamine core could help to enhance the lifetime of the excited triplet state.90 For example, two chemically stable β-ketoenamine COFs were prepared for photocatalytic hydrogen production.91 The designed TP-EDDA COF bearing acetylene functional groups was constructed from Tp and 4,4′-(ethyne-1,2-diyl)dianiline (EDDA), while the Tp-BDDA COF with diacetylene moieties was based on the reaction of Tp and 4,4′-(buta-1,3-diyne-1,4-diyl)dianiline (BDDA). The appearance of characteristic signals corresponding to CC and C–N bonds at ∼1451 and ∼1251 cm−1 confirmed the formation of β-ketoenamine functionalities. A much higher photocatalytic hydrogen evolution rate of TP-BDDA (324 ± 10 μmol h−1 g−1) was observed compared to that of TP-EDDA (30 ± 5 μmol h−1 g−1). Similarly, thioether-functionalized Thio-COF was fabricated via the acid-catalyzed reaction of Tp with thioether substituted diamine, which was highly stable toward water and common organic solvents (acetone, dichloromethane, ethanol, tetrahydrofuran, etc.).92 The introduction of the thioether group was beneficial for metal deposition and nanoparticle growth, paving the way for various applications, including optical and electronic devices.
In addition, Michael's addition–elimination strategy can also be used to construct β-ketoenamine linked COFs.93 A series of COFs were fabricated in a one-step process via the reaction of aromatic amines with di- and tritopic ketoenols. The disappearance of the N–H and C–N stretching at 3470, 3420, and 1206 cm−1 together with the appearance of a new C–N band at 1200 cm−1 in FT-IR spectra confirmed the formation of β-ketoenamine linkage. The obtained β-ketoenamine linked COFs exhibited improved hydrolytic stability owing to the intramolecular hydrogen bonding. The electron delocalization in these COFs generated a narrower band gap and reversible electrochemical doping. Moreover, a wide range of nucleophilic and electrophilic building units can be employed to form this kind of COFs.
2.5. Hydrazone linkage
Reversible condensation of hydrazides with the aldehyde building unit yields a crystalline hydrazone-linked structure, which offers the possibility of designing new linkages for COF synthesis. The first two hydrazone-linked COFs, COF-42 and COF-43, were reported in 2011, which were assembled via reversible dehydration of 2,5-diethoxyterephthalohydrazide and 1,3,5-triformylbenzene (TFB) or 1,3,5-tris(4-formylphenyl)benzene under solvothermal condition.94 2D trigonal layers were formed originating from the coplanar feature of the hydrazone moiety and aromatic rings. The hydrazones maintained integrity even when COF-43 was submerged in solvents such as H2O, dioxane, and dimethyl formamide.95 Moreover, the hydrazone-linked COFs have relatively weak interlayer interactions, such that they can be exfoliated into few-layer 2D polymers under mild conditions.The high robustness and easy processible nature of hydrazone COFs make them popular in various applications.96,97 The first visible-light-active COF was designed and prepared based on hydrazone linkage with the copolymerization of 2,5-diethoxy-terephthalohydrazide and 1,3,5-tris-(4-formyl-phenyl)triazine (TFPT).35 In the presence of Pt, the system produced 230–1970 μmol h−1 g−1 of hydrogen. Later, a hydrazone-linked TFB-COF was constructed from TFB and 2,5-dimethoxyterephthalohydrazide with a BET surface area of 1501 m2 g−1, which can be used as a photocatalyst for cross-dehydrogenative coupling reactions.98 Another two hydrazone COFs with rich hydroxy units were synthesized using water and then incorporated with CoII to investigate their Lewis acid catalytic activity.94 As a result, the metallated COFs were effective in catalyzing the cyanosilylation reactions of various aldehydes.
2.6. Azine linkage
The first azine-linked COF was synthesized by the condensation of 1,3,5,8-tetrakis(4-formylphenyl)pyrene with hydrazine.99 For azine linkage, hydrazine acted as a common building block to react with diverse aldehydes, which enables the formation of various functional COFs.100,101 For example, the visible-light-involved hydrogen generation from water can be achieved by the adjustment of the COF structure. Three water- and photo-stable azine-based Nx-COFs (x = 0, 1, 2, 3, represents the number of nitrogen in the central aryl ring) were synthesized by the polymerization of hydrazine and triphenylarene aldehydes.102 Raman and FT-IR spectra were employed to confirm the presence of azine CN linkage. As demonstrated, the photocatalytic hydrogen evolution was enhanced with the increased nitrogen content. Similarly, another series of azine-linked COFs with varied nitrogen atoms in the peripheral aryl ring was prepared for the investigation of photocatalytic hydrogen evolution.103 The results suggested that even very slight changes at the molecular level had a huge influence on the nanoscale morphology, atomic-scale structure, and optoelectronic properties, thereby causing significant differences in the capability of photocatalytic hydrogen production.
2.7. Imide-based linkage
In addition, a series of crystalline polyimide (PI) COFs, denoted as PI-COFs, were fabricated via reversible imidization reaction.104 Simply by extending the building molecules, the large pore size of as-prepared PI-COFs could be tuned. PI-COF-3 with a pore size of 5.3 nm and a BET surface area of 2346 m2 g−1 was designed and prepared by imidization condensation of 1,3,5-tris[4-amino(1,1-biphenyl-4-yl)]benzene (TABPB) with pyromellitic dianhydride (PMDA) in a mixed solvent of mesitylene, N-methyl-2-pyrrolidone (NMP), and isoquinoline (Fig. 5). The presence of CO stretches at 1779 and 1718 cm−1 and C–N–C stretching vibration at 1382 cm−1 in FT-IR spectra revealed the formation of imide linkage in PI-COF-3. By linking the linear building unit PMDA and the triangular building unit TABPB, PI-COF-3 was formed like a boron nitride net.105 The material remained stable in water and common organic solutions, such as acetone, ethanol, m-cresol, N,N-dimethylformamide, tetrahydrofuran, and hexanes. Notably, the large dye molecules can also be incorporated into PI-COF-3 with distinctive applications. Other functional building blocks have been utilized in the formation of imide-based COFs, such as PMDA and 1,3,5,7-tetraaminoadamantane or tetra(4-aminophenyl)methane,106 PMDA and tetramino-benzoquinone,107 and perylenetetracarboxylic dianhydride and tetra(4-aminophenyl)porphyrin (TAPP).108
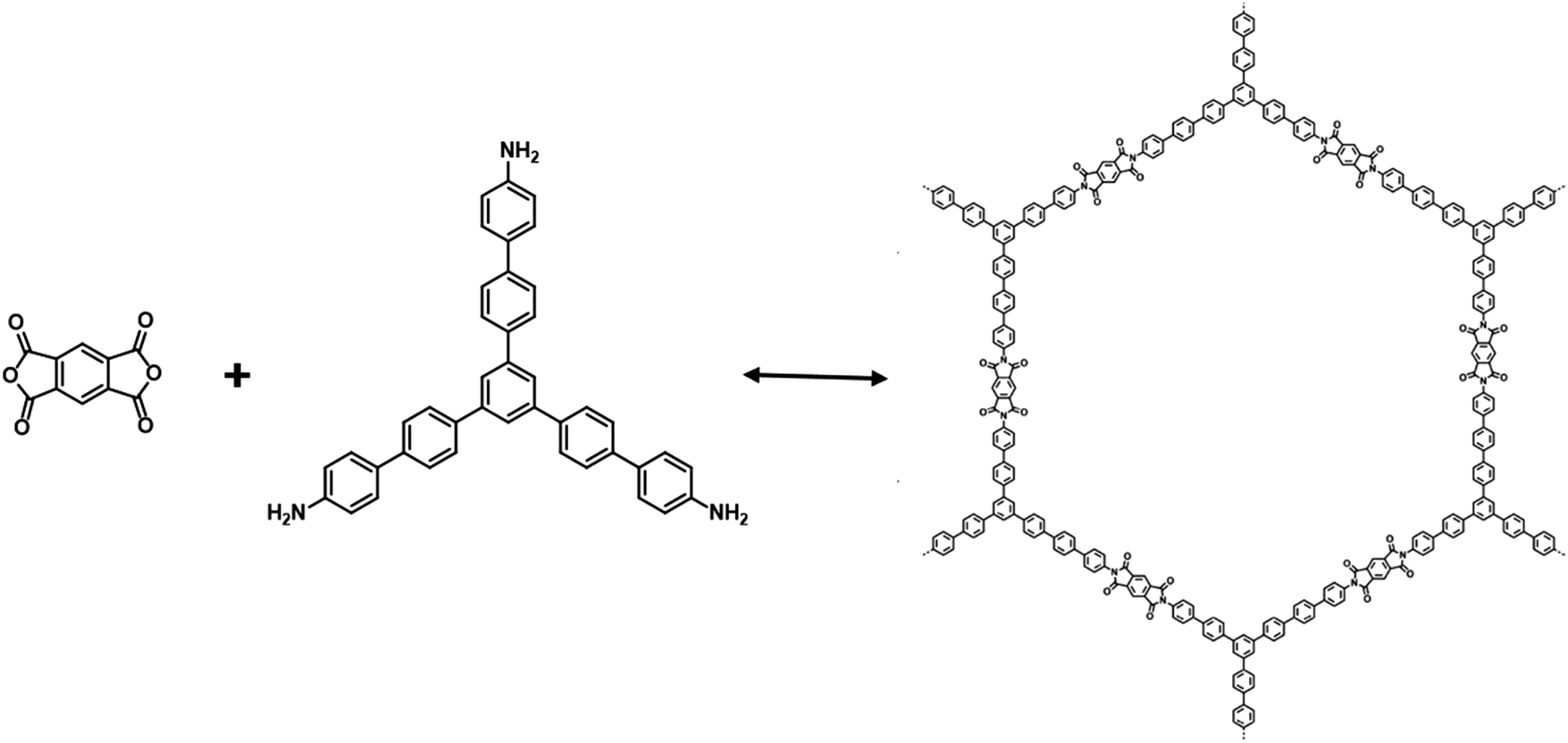 | ||
| Fig. 5 The formation of PI-COF-3. | ||
2.8. Other linkages
Besides the above-mentioned linkages, other linkages have also been used in COF fabrication such as carbamate linkage,109 borosilicate linkage,110 phenazine linkage,111 and squaraine linkage.112 For example, a 3D borosilicate-linked COF was first synthesized by condensation of tetra(4-dihydroxyboryl-phenyl)methane, tert-butylsilane triol, and tBuSi(OH)3.110 The as-prepared COF (named COF-202) possessed a BET surface area of 2690 m2 g−1 and high stability. Later, a crystalline borazine-linked COF named BLP-2(H) was prepared by thermal decomposition of 1,3,5-(p-aminophenyl)-benzene-borane.113 This as-prepared COF showed a BET surface area of 1178 m2 g−1. A squaraine-linked COF with zigzag conformation was achieved by the copolymerization of copper(II) 5,10,15,20-tetrakis(4-aminophenyl)porphyrin (TAP-CuP) and squaric acid (SQ).112 As indicated by FT-IR spectra, a CO bond (1595 cm−1) was formed with a blue-shift compared with SQ (1579 cm−1) due to the extended π-conjugation of the COF. This CuP-SQ COF with visible light absorption could act as an effective photocatalyst for singlet oxygen generation.
In addition, two crystalline benzobisoxazole-linked (BBO) COFs were prepared by the condensation of 2,5-diamino-1,4-benzenediol dihydrochloride with TFB or 1,3,5-tris(4-formylphenyl)benzene (TFPB) under the catalysis of cyanide.114 A three-step mechanism was proposed to explain the BBO linkage formation: (1) a phenolic imine linked intermediate was first formed, (2) then ring closure took place with the addition of cyanide to the imine and a benzoxazoline intermediate appeared, and finally (3) the benzoxazoline intermediate was oxidized under air, thereby promoting the BBO linkage formation.115 The resulting BBO-COF 1 and BBQ-COF 2 displayed excellent water stability and a high surface area of 891 m2 g−1 and 1106 m2 g−1, respectively. In another study, a room-temperature solution-phase reaction was employed to synthesize an azodioxy-linked COF (POR-COF) with I2-doping-enhanced photo-current generation.116 A series of spiroborate-linked ionic COFs (ICOFs) were synthesized with a high BET surface area up to 1259 m2 g−1, constructed from the transesterification of diol and trimethyl borate.117 Recently, an unsubstituted olefin-linked COF (COF-107) was first synthesized by Aldol condensation of 4,4′-biphenyldicarbaldehyde and 2,4,6-trimethyl-1,3,5-triazine (TMT).26 FT-IR and 13C CP-MAS spectroscopic techniques were utilized to verify the formation of –CHCH– linkage. The as-synthesized COF-701 possessed a BET surface area of 1715 m2 g−1 and high chemical robustness owing to the existence of unsubstituted olefin linkage.
As discussed above, various linkage motifs have been designed relating to the COF formation. Different linkages lead to different structures and properties, which usually correlated with the stability. It is easy to understand that the stability of COFs, especially in water and under light irradiation, is of crucial importance for their photocatalytic application. COFs based on boroxine and boronate ester linkages are susceptible to hydrolysis under humid conditions.118 Though enhanced stability has been achieved by protecting electron-deficient boron centers from degradation, such as in the case of the ionic spiroborate-linked COF117 and the alkylated COF-14Å,119 their applications to photocatalysis have still been hindered and limited studies have been done. Different from boron-based COFs, imine-linked and other nitrogen-containing COFs are more stable. Interlayer complementary π-interactions and intralayer hydrogen-bonding interactions have been developed to improve the stability of imine-linked COFs.120,121 Similarly, β-ketoenamine-linked COFs originating from the enol–keto tautomerization of their imine counterparts show much higher stability, and have been used in photocatalysis.87,122 The TzDTz COF (TpDTz: Tp and 4,4′-(thiazolo[5,4-d]thiazole-2,5-diyl)dianiline) was stable in boiling water and strong acids for up to 7 days, and the morphology, structure, and crystallinity were retained after a 72 h long photocatalysis experiment.123 COFs with hydrazone and azine linkages are also active in the photocatalytic process.124–126 The studies revealed that COFs obtained after photocatalysis retained connectivity and photoactivity, but lost a part of long-range order which could be ascribed to exfoliation in water and can be recovered in the original reaction conditions. Compared to the imine, hydrazine and azine COFs, triazine and phenazine-linked COFs show exceptional chemical stability, and the triazine unit as a photoactive group has been widely explored in photocatalysis.127–129 As for the newly developed CC-linked sp2 COF, extremely high stability has been found in the photocatalytic experiment. Under the light irradiation of 16 h, while imine-linked COF-LZU1 nearly lost its crystallinity in 4 h, g-C18N3-COF with CC linkages exhibited retained structure and activity despite a slight decay of crystallinity.130 Remarkably, the excellent photostability of g-C40N3 was proved by the nearly constant photocurrent density within the measurement period of 2600 s.131 Unlike MOFs, most COFs show enhanced stability because of the covalent bond, but it is still the key point to improve the water- and photostability of COF photocatalysts for practical application.
3. Morphology of COFs
One of the most common design strategies for optimizing photocatalytic performance is morphology control. Abundant building blocks and functional covalent linkages endow COFs with a designable structure. Indeed, many studies have been done to investigate the features of COFs with special structures, including 0D structures,117,132 1D structures,133,134 2D structures,135,136 and 3D structures.137,138 The morphological and structural investigations of COFs are of great importance for their photocatalytic performance. In the following section, the synthesis and photocatalytic properties of COFs on the morphologies are discussed.3.1. 0-Dimensional structures
0D-structural materials are considered as promising photocatalysts due to the large surface area. However, their photocatalytic performance still suffers from low efficiency due to the large agglomeration. In most cases, the monomers of COFs are partially soluble in reaction solvents, leading to a heterogeneous growth condition, thereby making it hard to understand the crystallization process. Currently, most of the reported COFs are synthesized with poorly controlled morphology and form insoluble and unprocessable aggregates. Recently, a strategy of homogeneous polymerization was put forward to avoid the irreversible aggregation and precipitation of crystallites, providing stable colloidal suspensions of COF nanoparticles.139 By adding a certain amount of CH3CN in a conventional solvothermal mixture of COF-5, a translucent solution with nanoparticles was obtained. CH3CN was demonstrated to stabilize the discrete crystallites and inhibit their aggregation in solution (Fig. 6a and b). Further investigation demonstrated that the interaction of the COF and the nitrile functional group was responsible for nanoparticle formation. Interestingly, the real-time growth of individual nanoparticles was observed using variable-temperature liquid cell transmission electron microscopy (VT-LCTEM) imaging (Fig. 6c). These stable porous nanoparticles with a functional internal surface were capable of site-isolated catalysis. Besides, a two-step approach was utilized to further control the formation of 2D COFs, which provided single-crystalline, micrometer-sized particles.140 When heating the COF-5 colloidal suspension, separate solutions of HHTP and PBBA were simultaneously injected, generating COF-5 nanoparticles with the sizes of 30–400 nm. To verify the generality of this strategy, the other two boronates ester-linked COF-10 and TP-COF were also studied. And later, the research was further expanded to the imine-linked COF. Colloidal TAPB-PDA COF nanoparticles were obtained by adding MeCN in the reaction system, which possessed a high BET surface area of 2070 m2 g−1.141 Similarly, considering the narrow range of nanoparticle size, separate solutions of TAPB and PDA were injected simultaneously to the TAPB–PDA COF colloid solution. The morphologies of the particles were found to vary with the different monomer addition rates. Besides, hollow spheres as an attractive modification have been studied. A crystalline hollow spherical COF, namely DhaTab, was synthesized based on 1,3,5-tris(4-aminophenyl)benzene and 2,5-dihydroxyterephthalaldehyde by self-template synthesis.142 Two steps were involved in constructing the hollow spherical structure: first, COF-DhaTab with rod-like morphology was formed within 12 h, and then it randomly self-assembled into curly or dense spheres. An inside-out Ostwald ripening mechanism was invoked in the formation of hollow spherical morphology for the next 24 h as the crystallites in the inner sphere got higher surface energy than those on the outer surface, and crystallites on the sphere wall fused to produce a smooth surface with the increase of time. Similarly, a two-step spray drying strategy was also utilized to structure imine-based COFs into spherical hollow superstructures.143 In the first stage, hollow spherical amorphous polyimine nanocrystals as precursors were prepared by the fast spray-drying process, and then, crystalline COFs with retained morphology were further obtained by submerging precursors under traditional COF synthesis conditions. | ||
| Fig. 6 (a and b) AFM of COF colloids prepared at 75% solvent concentration of CH3CN. (c) Representative VT-LCTEM image of COF-5 nanoparticles (55% growth solution). Reproduced with permission from ref. 139. Copyright 2017, American Chemical Society. (d) SEM images of g-C18N3-COF. (e) Top view SEM micrograph of g-C18N3-COF film. Reproduced with permission from ref. 139. Copyright 2019, American Chemical Society. | ||
0D structures with favorable surface speciation are deemed to display excellent photocatalytic activity compared with their bulk-phase counterparts. In addition, by reducing the particle size, the discrete energy levels arise at the band-edges of both the CB and VB considering the quantum confinement effect, thus improving the redox potential of photogenerated electrons and holes.12,144 However, the study of COF photocatalysts with 0D structure remains challenging.
3.2. 1-Dimensional structures
The research of 1D structures such as nanofibers, nanoribbons, and nanowires has increased over the years, attributable to their high surface-to-volume ratio.145 The study of COF morphology related to 1D structures is of great value. To date, solvothermal synthesis,146 vapor-assisted solid-state synthesis,147 and bottom-up microfluidic synthesis148 have been used to fabricate crystalline COF fibers. For example, novel crystalline COF nanofibers were fabricated by the solvothermal method based on the co-polymerization of 2,4,6-tris(4-aminophenyl)-pyridine (TAPP) with 2,6-dihydroxynaphthalene-1,5-dicarbaldehyde (DHNDA) at 180 °C.146 The as-prepared COF was formed as uniform nanofibers with a length of up to tens of micrometers. Interestingly, it was suggested that the morphology transformed from irregular nanoparticles to uniform nanofibers with increased crystallinity, which may be ascribed to the dissolution–recrystallization process. This transformation enabled the fabrication of COF nanohybrids with excellent optical and electrical properties. Similarly, nanofibers could also be obtained via vapor-assisted solid-state synthesis.147 Different from the solvothermal synthesis, the polycondensation of TAPP and DHNDA was carried out by exposing the mixture of monomers to solvent vapor at 120 °C for 48 h. In this method, only a small quantity of solvent vapor was needed, and the nanofibrous morphology was found to vary with the reaction time and the solvent vapor composition.Likewise, g-C18N3-COF with fibrillar morphology was prepared by Knoevenagel condensation of 1,4-diformylbenzene (DFB) with 2,4,6-trimethyl-1,3,5-triazine (TMTA) (Fig. 6d and e).130 Ultraviolet-visible diffuse reflectance spectroscopy (UV-vis DRS) displayed that the absorption band edge of g-C18N3-COF was at 450 nm, indicating a strong visible-light harvesting. And π-conjugated g-C18N3-COF with an average lifetime of 7.25 ns revealed the suppressed photogenerated electron–hole recombination. With ascorbic acid as a sacrificial agent and Pt as a co-catalyst, an average H2 production rate of 292 μmol g−1 h−1 was achieved over g-C18N3-COF. In addition, COFs bearing Tp and melamine (MA) building units with visible-light-response features were synthesized as exfoliated thin ribbon-like and interwoven thread-shaped structures under different conditions (catalyst-assisted, solvent-assisted, and liquid-free) by ball milling.134 Compared to the thread-shaped COF, the optical absorption edge of the ribbon-like COF displayed a red-shift, enhancing solar utilization efficiency, and therefore leading to a higher photocatalytic degradation rate of phenol. These findings suggested that the morphology affected the photocatalytic activity of COF-based materials, which may be ascribed to the aggregation behavior, dispersity, and incident light-harvesting capability in water.
3.3. 2-Dimensional structures
The optical, photochemical and photoelectrical features of materials could be affected directly or indirectly if their morphology and structure are changed. In addition to 0D and 1D structures, 2D structures like thin films and few-layered nanosheets have also been widely studied in the photocatalytic process.149,150 Indeed, the high smoothness and aspect ratio along with the short travel distance of photoexcited carriers render the thin films with high photocatalytic performance.151,152 In recent years, various methods have been utilized to synthesize COF thin films as free-standing forms or deposited on specific substrates, such as mechanical delamination,88,153 solvent-assisted exfoliation,154,155 solvothermal synthesis156,157 and interfacial synthesis.158,159 Among them, solvothermal synthesis is widely used because it is simple and straightforward. For example, TT-COF thin films with 200 nm thickness were prepared on a cleaned glass substrate by simply immersing the substrate in the solution of bulk TT-COF.160 As demonstrated by grazing incidence X-ray diffraction (GIXRD), the growth of 2D TT-COF thin films was parallel to the surface of the glass substrate, which indicated an ordered charge transfer pathway. A greatly enhanced photoresponse speed was observed in the well-ordered COF thin film.In another study, the BDT-ETTA COF based on amine-functionalized 1,1′,2,2′-tetra-p-aminophenylethylene (ETTA) and donor-type benzo[1,2-b:4,5-b′]-dithiophene-2,6-dicarboxaldehyde (BDT) was grown on an indium tin oxide substrate to yield BDT-ETTA COF thin films.161 The obtained COF thin films displayed strong visible light absorption with a threshold of ca. 550 nm and a band gap of 2.47 eV, indicating the photoactive potential. The results suggested that the BDT component could be the reason for the photoactivity, and the oriented COF thin films were beneficial to the photoresponse and stability. A new synthetic method was employed by directly condensing 3,4,9,10-perylenetetracarboxylic diimide (PDI) and cyanuric chloride (CC) to yield a CTF film photocatalyst.162 The CTF film with excellent photocatalytic activity showed an enhanced NADH regeneration of 75.88% and HCOOH production of 204.14 μM. Also, ultrathin 2D porphyrin nanodisks with enhanced photocatalytic activity were prepared by COF exfoliation via axial ligand incorporation. Porphyrin-containing COF DhaTph (Dha: 2,5-dihydroxyterephthalaldehyde, Tph: 5,10,15,20-tetrakis(4-aminophenyl)-21H,23H-porphyrin) was exfoliated by simultaneously incorporating 4-ethylpyridine and copper (Cu) ion ligands into the porphyrin center to yield e-CON(Cu, epy) (Fig. 7a and b).163 The resulting e-CON was further incorporated with Pt nanoparticles and reduced-graphene oxide (RGO) to obtain the composite material e-CON(Cu, epy)/Pt/RGO for photocatalytic reaction (Fig. 7c). Compared with DhaTph/Pt/RGO, an enhanced visible/NIR-light-induced hydrogen evolution of the e-CON(Cu, epy)/Pt/RGO system was observed owing to the higher surface area of e-CON and Pt/RGO. The abovementioned results demonstrated that 2-dimensional COF thin films and nanosheets with broad light absorption, optical band gap, and efficient charge separation and transfer have great potential for photocatalytic activity improvement.
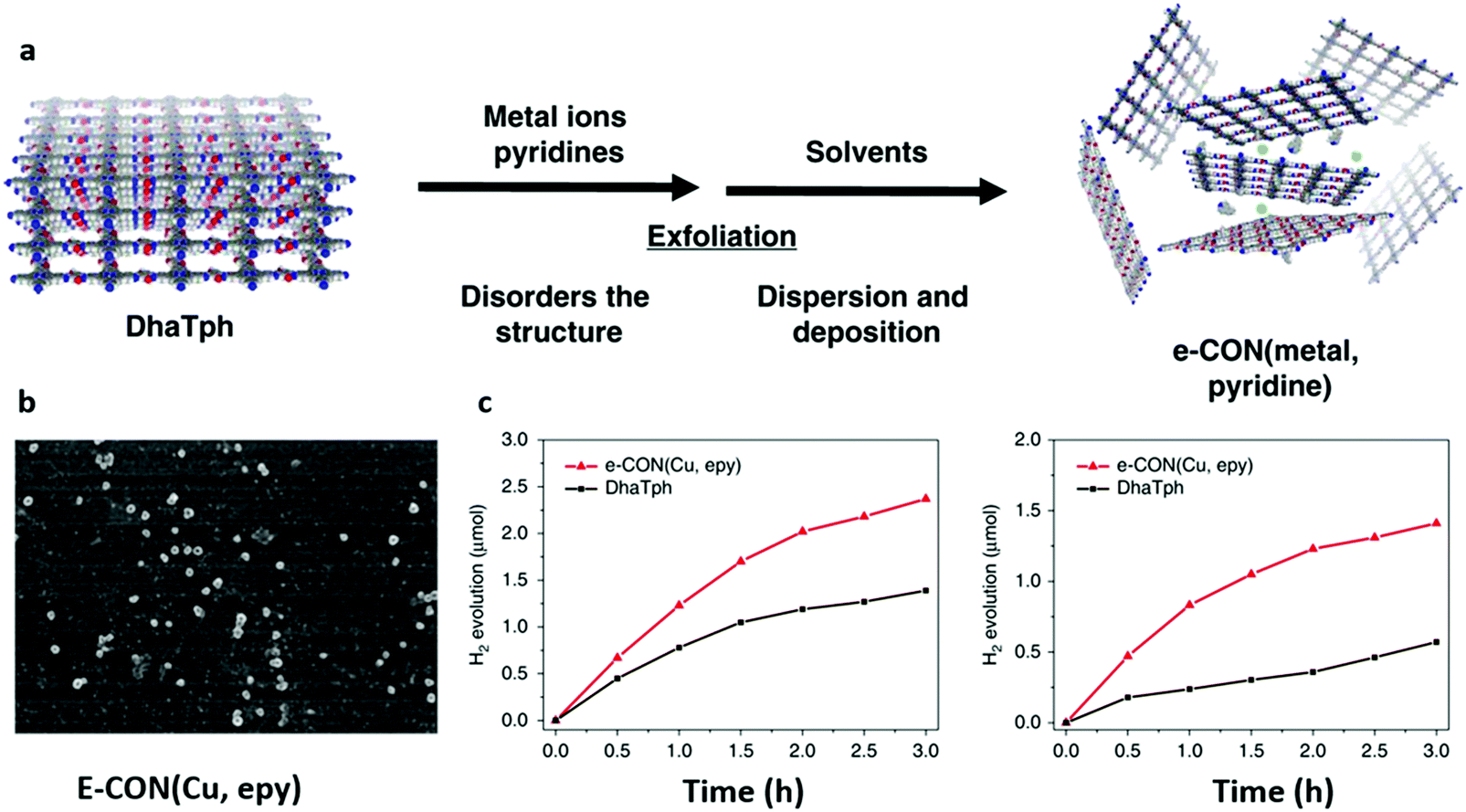 | ||
| Fig. 7 (a) Scheme for the preparation of e-CON. (b) SEM image of e-CON (Cu, epy) deposited on the silicon wafer. (c) H2 evolution upon irradiation with visible (>420 nm) and NIR (>780 nm) light using e-CON(Cu, epy)/Pt/RGO and DhaTph/Pt/RGO. Reproduced with permission from ref. 163. Copyright 2019 Springer Nature Limited. | ||
3.4. 3-Dimensional structures
COFs with 3D structures are synthesized mainly by heterogeneous nucleation and growth,164,165 template-directed approach,137,166,167 self-assembly strategy,142,165,168 and multiple-linking-site strategies.169 For example, the hollow TpPa COF was designed and synthesized with the assistance of the ZnO-nanorod template (Fig. 8).137 First, p-phenylenediamine (Pa) and Tp were dehydrated in the presence of ZnO nanorods, and then the ZnO nanorods were removed by treating the produced hybrid materials with acid (1 N HCl) for 24 h, leading to the formation of hollow TpPa nanostructures with inner and outer diameters of d = (70–130) nm and d = (60–100) nm, respectively. Another imine-linked TpBD COF containing Tp and benzidine (BD) building blocks was directly grown on Fe3O4 by a solvothermal method to form core–shell structured TpBD@Fe3O4.165 The hollow TpBD was further obtained by etching the Fe3O4 core in HCl solution, resulting in a shell thickness of ca. 50 nm. These references could offer an important process for the construction of COF-based photocatalysts. The hollow structures obtained from the template-assisted method possess ordered and uniform cavities simply by controlling the template diameter. A hollow structure with controlled porosity reduces the diffusion length and improves the contact of active sites with reactants.170,171 Moreover, the multiple reflections within the hollow cavity are beneficial for efficient light utilization, producing more photogenerated charge carriers.172,173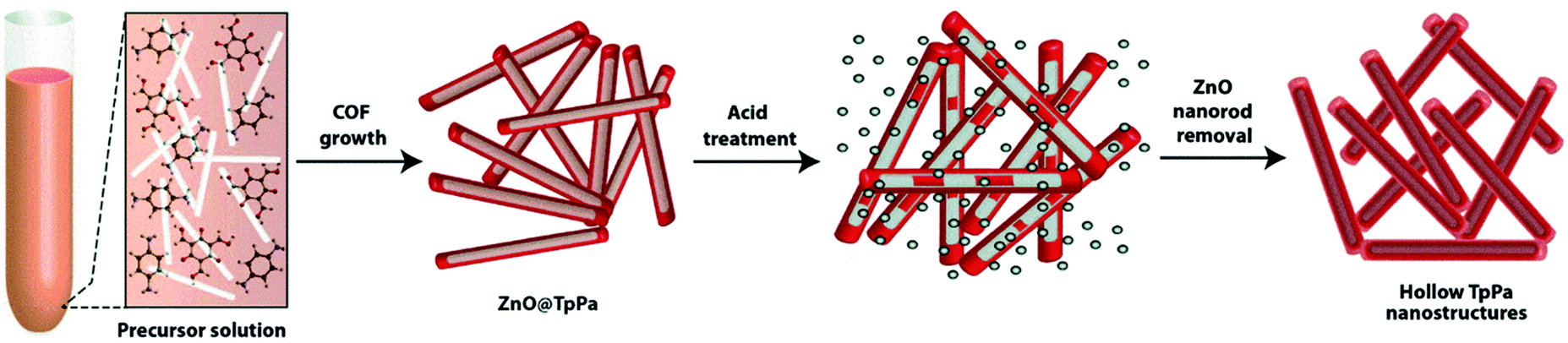 | ||
| Fig. 8 Scheme of the formation of hollow COFs via a templating strategy. Reproduced with permission from ref. 137. Copyright 2015, Royal Society of Chemistry. | ||
Apart from hollow morphology, flower-shaped morphology was also obtained by controlling the synthetic conditions of TpPa-1 and TpPa-2 COFs.87 Each flower was assigned to the aggregation of sheet-like petals with 1–3 μm in length as a result of the π–π stacking of COF layers. Specifically, the petals of TpPa-1 with spike-like tips were grown out from a core, while TpPa-2 with longer and broader petals showed a plate-like structure. Similar morphology has also been found in the Tp-Azo COF, in which petals with an average length of 40–50 nm aggregated to form a flower-like structure.174 Later, a bouquet-shaped magnetic TpPa-1 COF was successfully fabricated through a room-temperature solution-phase approach.138 Clustered Fe3O4 NPs were used as the template for growing TpPa-1 with a thread-like structure. With the increase of reaction time to 30 min, the branched TpPa-1 interconnected with each other to form bouquet-like morphology. Magnetic TpPa-1 with enhanced reactant and active site accessibility exhibited potential for photocatalysis as a result of its high porosity, large surface area and supermagnetism.
4. Strategies for enhancing the photocatalytic activity of COFs
Based on the typical photocatalytic process, the design and modification of photocatalysts with enhanced performance would involve considerations such as extended visible-light-absorbing capacity, facilitated electron–hole separation and suppressed photo-corrosion for prolonged duration. COFs as a flexible platform show promising applications in photocatalysis, and several strategies have been developed for enhancing their activity based on pristine COFs and modified COFs. To increase the visible-light absorption and decrease the recombination of photogenerated electrons and holes, the most direct way lies in the elaborate incorporation of functional building blocks to modulate the optical and electrical features of COFs. The physical and chemical properties of COFs can be changed by the selection of building blocks, which enables the control of their band gap structure at the molecular level. In addition, elemental doping including non-metal doping and metal doping can also be a facile and feasible strategy to tune the physicochemical properties of COFs at the atomic level. Band gap engineering can be realized by introducing anions and cations into the framework for improving the light-harvesting capability and tuning redox band potentials. Effectively utilizing solar energy in a large span of spectrum is critical for photocatalytic processes. To further broaden the light absorption to a higher wavelength range, the incorporation of sensitizers is another fascinating strategy to enhance the photocatalytic activity. Photosensitizers with chromophores are expected to extend the light absorption to the NIR region, and the well-matched band gap structures of COFs and sensitizers also could accelerate charge separation at their interfaces. Besides, COFs with diverse functional groups and π-stacking nanosheets act as an ideal platform for the fabrication of hybrid materials with various semiconductors. The formation of COF-based hybrid photocatalysts is deemed to be a feasible and compelling strategy for photocatalytic activity improvement, which had advantages of enlarging visible-light absorption, facilitating the electron transfer between composites, and enhancing the separation efficiency of photogenerated electron–hole pairs.4.1. Functional building block incorporation
One of the most attractive characteristics of COFs related to photocatalysis is their regular structures with unlimited building blocks, which can be applied to various reaction conditions. Based on reticular chemistry, COFs allow a predesigned pathway for precise control over their structures and properties by choosing different linkers as well as different building units containing functional groups or side chains.108,175–179 To date, functional building block incorporation is the most widely used strategy for modulating the photocatalytic performance of COFs. For example, a series of azine-linked Nx-COF photocatalysts were synthesized by selecting hydrazine as the linker and triphenylarene aldehydes as the nodes, in which the nitrogen atoms of the central aryl ring in aldehyde units varied from 0 to 3 (Fig. 9a and b).102 Replacing the carbon atoms with nitrogen atoms led to the formation of different central rings namely phenyl (N = 0), pyridyl (N = 1), pyrimidyl (N = 2), and triazine (N = 3), showing increased planarity due to the decreased dihedral angle between peripheral phenyl rings and the central aryl ring. Consequently, increased crystallinity was observed as a result of increasing nitrogen content (Fig. 9c). It was found that the increased crystallinity and improved structural definition and layer registry endowed N3-COF with enhanced exciton migration in-plane as well as along the stacking direction, thus leading to improved photocatalytic activity (Fig. 9d). In addition, N3-COF as the most nitrogen-rich COF in the system showed increased stabilization of radical anions which was shown to enhance charge separation and electron migration. Besides, π-conjugated trans-disubstituted CC linked COFs (termed g-CxNy-COFs) with different properties were designed and synthesized based on the Knoevenagel condensation of 3,5-dicyano-2,4,6-trimethylpyridine (DCTMP) with linear 4,4′′-diformyl-p-terphenyl (DFPTP), 4,4′-diformyl-1,1′-biphenyl (DFBP), or TFPB, which yielded g-C40N3-COF, g-C31N3-COF, and g-C37N3-COF, respectively.180 As demonstrated by UV-vis DRS, g-C40N3-COF showed a significant red-shift of the absorption edge compared with g-C31N3-COF and g-C37N3-COF, indicating a stronger ability of light-harvesting in the visible region, and g-C40N3-COF was found to possess a smaller optical band gap (2.36 eV) compared to g-C31N3-COF (2.40 eV) and g-C37N3-COF (2.52 eV). Time-resolved fluorescence decay spectroscopy was also used to characterize the excitation recombination with the information of the average lifetime of photo-excited electrons. g-C40N3-COF exhibited the most extended fluorescence lifetime (3.31 ns) due to the charge separation in the extended π-conjugated structure. These findings along with other optical and electronic characterization (Mott–Schottky measurement, photocurrent tests, etc.) suggested that g-C40N3-COF permitted the effective photogenerated electron–hole transfer, and thereby exhibited enhanced photocatalytic ability. These examples demonstrated that the photocatalytic performance of COFs can be enhanced by precisely selecting the building units.
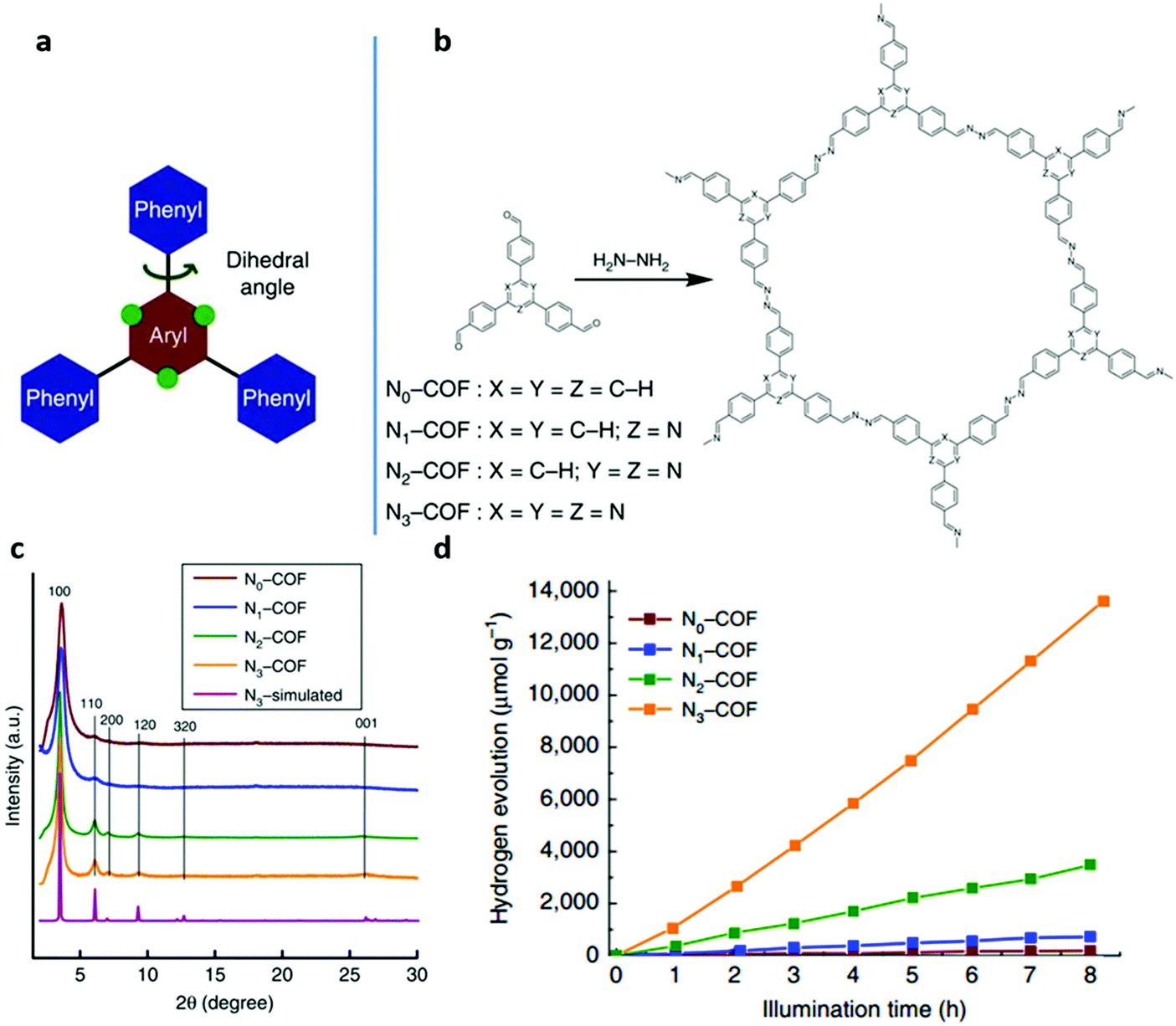 | ||
| Fig. 9 (a) A tunable triphenylarene structure. (b) Formation of Nx-COFs based on hydrazine and Nx-aldehydes. (c) PXRD patterns of Nx-COFs compared with the simulated pattern calculated for the representative N3-COF. (d) H2 production monitored over 8 h using Nx-COFs as a photocatalyst in the presence of triethanolamine as a sacrificial electron donor. Reproduced with permission from ref. 102. Copyright 2015 Macmillan Publishers Limited. | ||
Various functional building blocks such as triazine,181 sulfone,124 pyrene,182 benzothiadiazole,183 and thiophene184 have been utilized for constructing COF photocatalysts with high performance. For example, diacetylene-bridged COFs were of great interest owing to the highly conjugated structures, accessible active sites, and accelerated charge transfer.185 Porous and stable acetylene (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–) and diacetylene (–CC–CC–) functionalized β-ketoenamine COFs, TP-EDDA and TP-BDDA, were prepared and their photocatalytic properties were studied (Fig. 7).91 Ketoenamine linkage was introduced to ensure the chemical stability of the COFs. To well-determine the influence of acetylene and diacetylene functional groups, an isoreticular COF, namely, TP-DTP COF (DTP: 4,4′′-diamino-p-terphenyl), with similar pore apertures based on terphenylene edges was designed and prepared. As determined by UV-vis spectra, TP-BDDA showed an absorbance edge of 525 nm and the tail extended up to 675 nm, while the absorbance edge of TP-EDDA and TP-DTP was 520 nm and 500 nm, respectively. Similarly, the optical band gaps followed the order of TP-BDDA (2.31 eV) < TP-EDDA (2.34 eV) < TP-DTP (2.42 eV). Photocatalytic experiments indicated that the conjugated diacetylene group played a vital role in enhancing the photoactivity. Apart from narrowing the band gap, diacetylene-moieties were also considered to possess higher charge carrier mobility and enable the accelerated migration of photogenerated excitons to the surface of the photocatalyst. In addition, electron acceptors such as benzothiadiazole (BT) and electron donors such as tris-(4-aminophenyl)triazine (TAPT) and tris(4-aminophenyl)benzene (TPB) were employed to construct COFs with tailored band gaps and improved charge separation and transfer.183 The resultant BT-COFs showed extended absorption bands ranging from 400 nm to 800 nm. Compared to TAPT-BT-COF, TPB-BT-COF with a narrower band gap and a more negative conduction band was found to exhibit promoted visible-light harvesting efficiency and produce more charge carriers. And the photocurrent intensity and electrochemical impedance spectra further confirmed that the structure of TPB-BT-COF was beneficial for enhanced charge carrier separation and reduced charge transfer impedance.
C–) and diacetylene (–CC–CC–) functionalized β-ketoenamine COFs, TP-EDDA and TP-BDDA, were prepared and their photocatalytic properties were studied (Fig. 7).91 Ketoenamine linkage was introduced to ensure the chemical stability of the COFs. To well-determine the influence of acetylene and diacetylene functional groups, an isoreticular COF, namely, TP-DTP COF (DTP: 4,4′′-diamino-p-terphenyl), with similar pore apertures based on terphenylene edges was designed and prepared. As determined by UV-vis spectra, TP-BDDA showed an absorbance edge of 525 nm and the tail extended up to 675 nm, while the absorbance edge of TP-EDDA and TP-DTP was 520 nm and 500 nm, respectively. Similarly, the optical band gaps followed the order of TP-BDDA (2.31 eV) < TP-EDDA (2.34 eV) < TP-DTP (2.42 eV). Photocatalytic experiments indicated that the conjugated diacetylene group played a vital role in enhancing the photoactivity. Apart from narrowing the band gap, diacetylene-moieties were also considered to possess higher charge carrier mobility and enable the accelerated migration of photogenerated excitons to the surface of the photocatalyst. In addition, electron acceptors such as benzothiadiazole (BT) and electron donors such as tris-(4-aminophenyl)triazine (TAPT) and tris(4-aminophenyl)benzene (TPB) were employed to construct COFs with tailored band gaps and improved charge separation and transfer.183 The resultant BT-COFs showed extended absorption bands ranging from 400 nm to 800 nm. Compared to TAPT-BT-COF, TPB-BT-COF with a narrower band gap and a more negative conduction band was found to exhibit promoted visible-light harvesting efficiency and produce more charge carriers. And the photocurrent intensity and electrochemical impedance spectra further confirmed that the structure of TPB-BT-COF was beneficial for enhanced charge carrier separation and reduced charge transfer impedance.
4.2. Elemental doping
Elemental doping is another efficient strategy to regulate the surface property and electronic structure of semiconductors, thereby improving the photocatalytic activity. The element sulfur (S), as one of the most common dopants, is known to modulate the electronic structure as well as the optical absorption features of organic semiconductor photocatalysts.186,187 A series of S-doped CTFs were prepared by the annealing treatment of covalent triazine-based framework CTF-T1 with S, which were named as CTFSχ (χ = 5, 10, 20, 30).129 In this case, compared with the g-C3N4 photocatalyst, CTFSχ exhibited much better photocatalytic activity, and the highest photoactivity was obtained with CTFS10 which was about 5 times higher than that of CTF-T1. Similarly, other typical non-metal dopants such as halogens have also been utilized for photocatalysis improvement. A series of halogen (F, Cl and Br)-doped CTFs were synthesized via the thermal treatment of CTF-1 with excessive ammonium halide.188 Halogen-doped CTF-1 with decreased Nyquist plot diameter and higher photocurrent density revealed the improved efficiency of charge separation and transfer as compared with pristine CTF-1 (Fig. 10a and b). The optical band gap of CTF-1, CTFF, CTFCl and CTFBr was determined to be 2.94, 2.82, 2.48 and 2.63 eV (Fig. 10d). The narrower band gaps and facilitated electron transfer in the modified π-conjugated CTF greatly enhanced the photocatalytic performance evidenced by 7.1 times higher photocatalytic ability of CTFCl compared to pristine CTF-1 (Fig. 10c).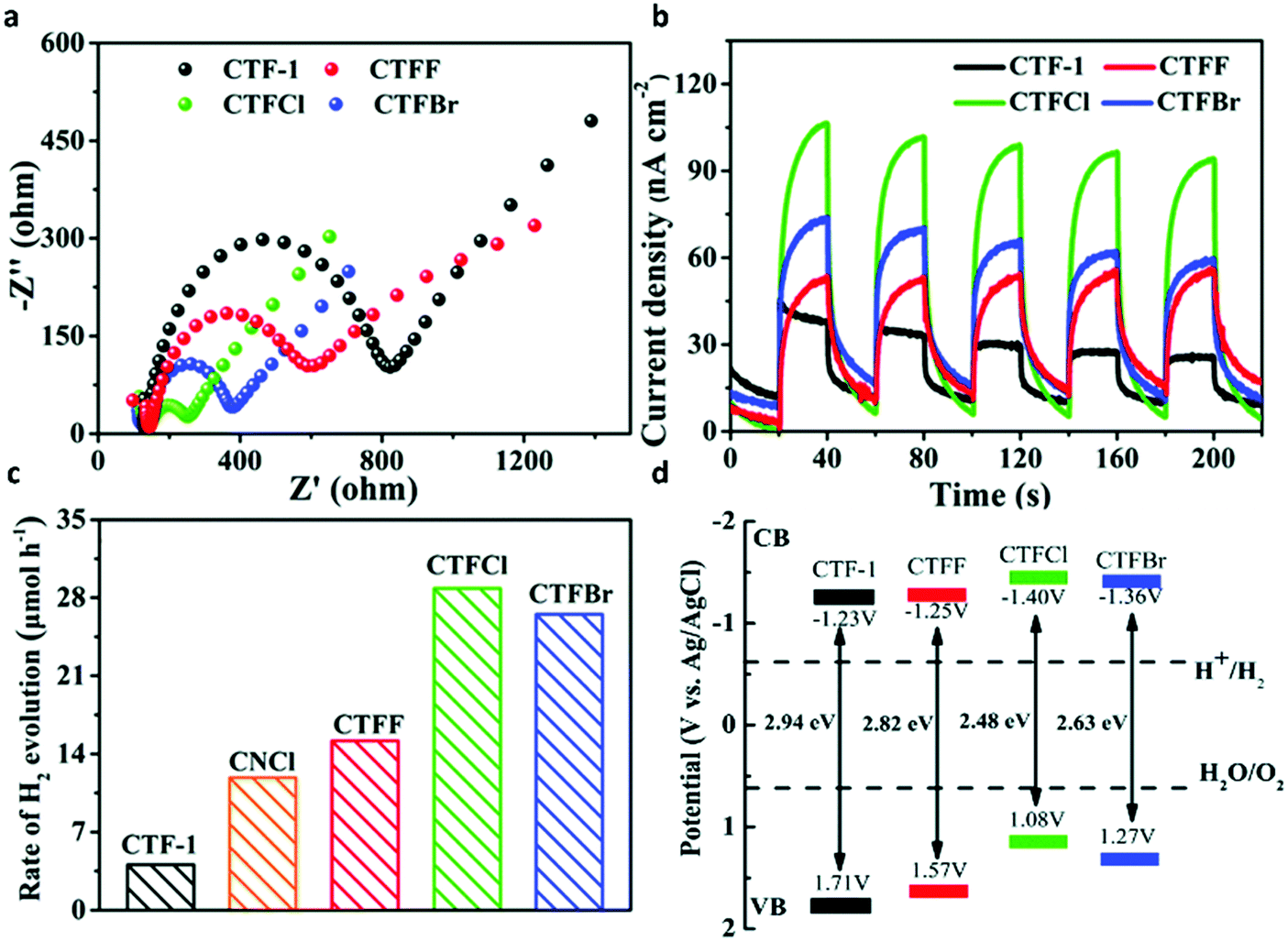 | ||
| Fig. 10 (a) Electrochemical impedance spectroscopy plots of CTF-1 and CTFX samples. (b) Photocurrent responses under visible-light irradiation of CTF-1 and CTFX samples. (c) H2 evolution rates of CTF-1, CNCl and CTFX samples. (d) The band gap structures of CTF-1 and CTFX samples. Reproduced with permission from ref. 188. Copyright 2016, Royal Society of Chemistry. | ||
In addition to the abovementioned non-metal doping, metals such as Fe, Zn, and Re have also been doped into COFs for the modulation of their optical and electrical properties by narrowing the band gap, extending visible-light absorption, facilitating electron charge transfer, and increasing the lifetime of charge carriers.189–191 The nitrogen pots in the COFs provide rich binding sites for the incorporation of metal ions via ion coordination. The inclusion of Re in CTF-py (based on 2,6-dicyanopyridine) was developed by Cao et al. for the first time.189 Compared with CTF-py, Re-modified Re-CTF-py showed a lower charge transfer resistance and a higher charge carrier separation efficiency. By incorporating Re into CTF-py, photogenerated electrons could transfer from CTF-py to Re and the recombination of electron–hole pairs was retarded, thus leading to enhanced photocatalytic activity. Furthermore, BpZn-COP was synthesized by the coordination of Zn2+ with N atoms of pyridine units in Bp-COF.191 It was found that BpZn-COP showed broader light absorption (from 550 nm to more than 600 nm) and a narrower band gap (from 2.35 eV to 2.18 eV) compared to that of Bp-COP. The presence of Zn2+ played an important role in promoting the electron transfer inside the bulk and across the interface of semiconductor and electrolyte, suppressing electron–hole recombination and improving the utilization efficiency of charge carriers. By this way, BpZn-COP displayed a much higher photocatalytic activity.
4.3. Sensitizer
Light-harvesting is one of the most important prerequisites for electron–hole generation, which greatly affects the photocatalytic performance. Photosensitizers with intense visible light absorption can be used as a co-catalyst to enhance the light absorption and the lifetime of photoinduced electron–hole pairs, thus improving the photocatalytic performance.192,193 The photocatalytic activity of COFs modified with palladium acetate was investigated by using Eosin Y (EY) as a sensitizer.194 Isoreticular COF-LZU1 and TpPa-1 were employed to facilitate energy transfer. As demonstrated, while Pd0/COF-LZU1 and Pd0/TpPa-1 were not photoactive without EY, they exhibited enhanced photocatalytic activity with the help of EY. When EY adsorbed visible light, electrons were generated and were then transferred from COFs to Pd active sites for photocatalytic reaction. Furthermore, 2D COFs with π-conjugated structures could effectively facilitate photogenerated electron transfer, leading to the improved performance.4.4. Hybrid construction
In addition, hybrid materials with synergistic effects are believed to provide versatile characters for photocatalysis. By a careful design, multicomponent heterojunction materials with improved photocatalytic efficiency could be achieved in terms of promoted charge separation and enhanced charge carrier transfer. COF-based composites have also been reported to show enhanced photocatalytic activity.163,181 A crystalline COF can serve as an attractive support matrix for nanoparticle loading due to its remarkable stability, high porosity and surface area.195 The highly stable TpPa-2 COF was employed to anchor CdS nanoparticles.196 The π-conjugated COF support was believed to enhance the photostability of the loaded CdS nanoparticles and suppress photogenerated electron–hole recombination, thus enhancing the photocatalytic performance. After combination, a yellow to reddish brown shift was observed in the absorption spectra, which indicated enhanced visible-light absorption of the CdS–COF composite. And charge carrier transfer existed between CdS and COF, leading to the decreased photogenerated electron–hole recombination. As a result, an improved photocatalytic activity was achieved as compared to bulk CdS. Likewise, CdS nanoparticle-decorated CTF-1 (CdS NPs/CTF-1) was synthesized by an one-pot solvothermal reaction.197 Size-controlled CdS NPs were uniformly dispersed on the CTF-1 layer surface with the interaction of Lewis basic nitrogen atoms in triazine groups of CTF-1. This interaction between CdS and CTF-1 endowed CdS with high stability and a nanosized structure, and simultaneously promoted the photoinduced charge separation. A higher photocatalytic capability was realized by this CdS NPs/CTF-1 hybrid when compared to pure CTF-1 and CdS under visible-light illumination. In addition, 2D layered BiOBr is frequently used in photocatalytic environment remediation and energy conversion because of its excellent electrical, optical, and catalytic features.198,199 However, its small surface area, poor light absorption, and high photoinduced electron–hole recombination limit the development. Hence, heterojunctions based on CTFs and BiOBr could be developed to enhance photocatalytic activity.200 It was revealed that BiOBr/CTF-3D-2% possessed much higher photocatalytic performance for degradation of tetracycline hydrochloride (TC-H) and ciprofloxacin (CIP) compared to pure BiOBr and CTF-3D.Similarly, a novel MOF@COF core–shell hybrid material was constructed to possess high photocatalytic performance.201 By virtue of its available amino functional groups and high stability under harsh experimental conditions, NH2-MIL-68 with 2-aminoterephthalic acid ligand and infinite chains of InO4(OH)2 was selected. As depicted in the scheme, NH2-MIL-68 was first synthesized through solvothermal reaction, and then functionalized with the tris(4-formylphenyl)amine (TFPA) molecule to obtain aldehyde-functionalized NH2-MIL-68, denoted as NH2-MIL-68(CHO). And TPA-COF was grown on the NH2-MIL-68(CHO) surface by covalently linking tris(4-aminophenyl)amine with TFPA via conventional solvothermal condensation, generating core–shell structured hybrid NH2-MIL-68@TPA-COF (Fig. 11). NH2-MIL-68@TPA-COF displayed higher photocatalytic activity, which was about 1.4 times higher than that of NH2-MIL-68, due to its large BET surface area as well as smaller band gap.
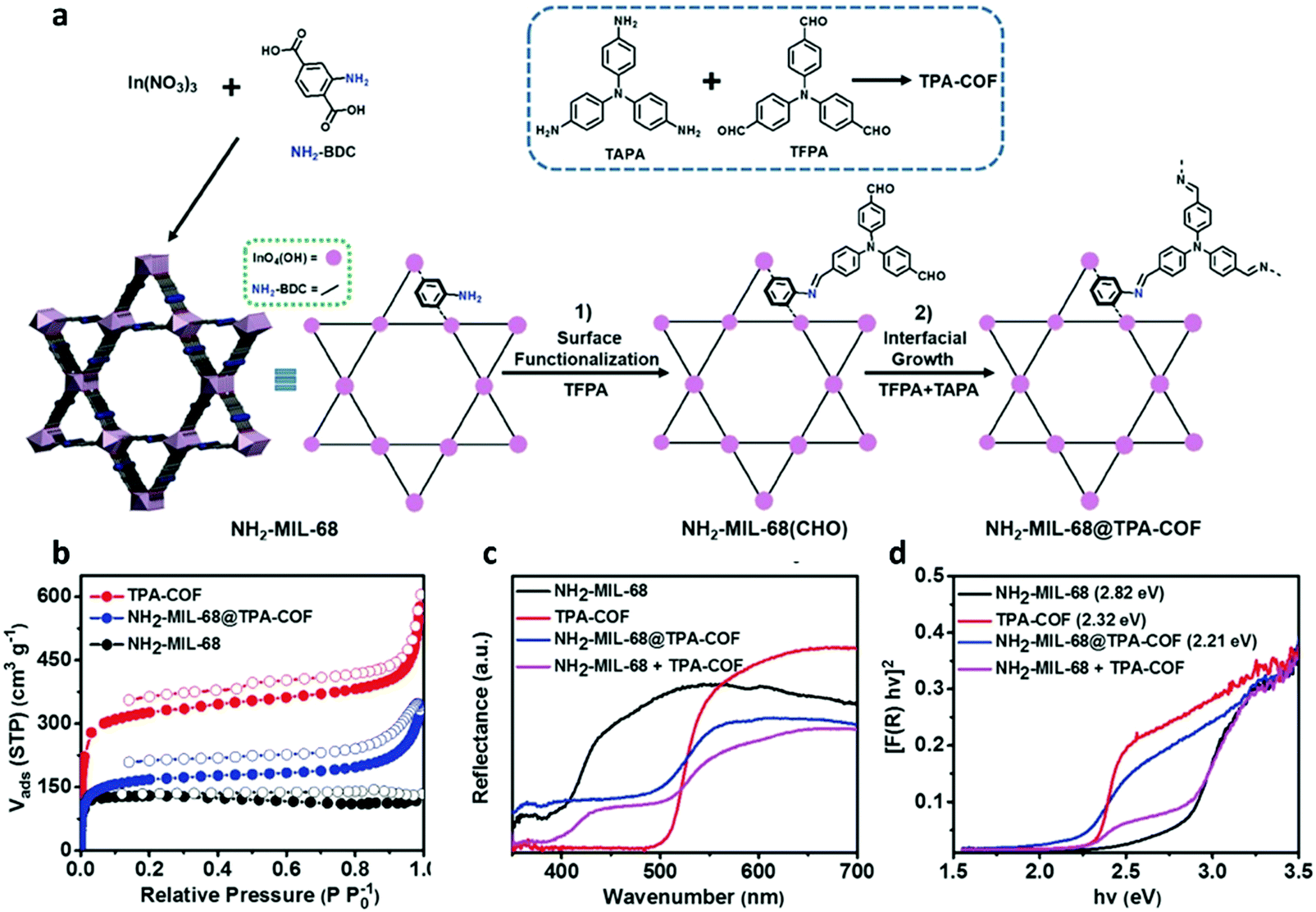 | ||
| Fig. 11 (a) General schematic of the synthesis of NH2-MIL-68@TPA-COF hybrid material. (b) N2 sorption isotherms for NH2-MIL-68, TPA-COF, and NH2-MIL-68@TPA-COF measured at 77 K. (c) UV-vis DRS spectra, and (d) the plots of the Kubelka–Munk function of NH2-MIL-68, TPA-COF, NH2-MIL-68@TPA-COF, and the mixture of NH2-MIL-68 and TPA-COF (NH2-MIL-68 + TPA-COF). Reproduced with permission from ref. 201. Copyright 2018, Wiley-VCH. | ||
As discussed above, strategies including functional building block incorporation, elemental doping, incorporation of sensitizers, and hybrid construction have been utilized in enhancing the photocatalytic performance of COFs. Among them, while functional building block incorporation as a distinctive feature of COFs has been widely used, the exploration is far from enough in view of the unlimited building molecules, and much work still needs to be done regarding the synthesis of new functional COFs. And post-synthetic modification will also be a promising strategy to utilize photoactive groups which are difficult for ab initio construction. Besides, heterojunction construction attracts considerable interest. By building suitable band positions, it is able to transfer photogenerated electron–hole pairs from the interface to the surface of two components, which leads to redox and reduction reactions. Certainly, new strategies with high performance are highly desired.
5. Photocatalytic applications
5.1. Application in photocatalytic hydrogen evolution
Nowadays, energy shortage is one of the most challenging issues, particularly in a clean and sustainable way. Hydrogen, as one of the most promising renewable energy sources, can be generated from water splitting under visible-light irradiation.202,203 COFs with diverse structural regularity, crystallinity and porosity are considered as promising photocatalytic hydrogen production platforms.130,204–206 Up to now, the highest photocatalytic hydrogen evolution rate of 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 120 μmol h−1 g−1 was reported by Tan and co-workers based on ter-CTF-0.7, which was synthesized from 4,7-bis(4-formylphenyl)-2,1,3-benzothiadiazole (M-BT), 3,6-dicarbaldehyde-N-ethylcarbazole (M-CBZ), and terephthalimidamide dihydrochloride.207
120 μmol h−1 g−1 was reported by Tan and co-workers based on ter-CTF-0.7, which was synthesized from 4,7-bis(4-formylphenyl)-2,1,3-benzothiadiazole (M-BT), 3,6-dicarbaldehyde-N-ethylcarbazole (M-CBZ), and terephthalimidamide dihydrochloride.207
The first use of a COF for photocatalytic hydrogen production was reported in 2014.35 The triazine-based building block was selected because of its high electron mobility and electron-withdrawing characteristic.208 Specifically, the crystalline hydrazone-linked COF (TFPT-COF) was prepared by condensation of 2,5-diethoxy-terephthalohydrazide with TFPT. Then, Pt as a proton reduction catalyst and TFPT-COF as the photosensitizer were integrated to form the TFPT-COF/Pt photocatalyst for visible-light-induced hydrogen evolution with sodium ascorbate or TEOA as an electron donor. A hydrogen evolution rate of 1970 μmol h−1 g−1 was achieved with 10 vol% TEOA, which was nearly 3 times higher than that achieved with other outstanding photocatalytic systems including crystalline poly(triazine imide) and Pt-modified amorphous melon.36 Moreover, the quantum efficiency was determined to be 2.2% at 500 nm. Interestingly, on the one hand, TFPT-COF with retained photoactivity lost its crystallinity after 92 h photocatalytic reaction, probably due to its exfoliation in the process; on the other hand, this filtered amorphous product could be easily reconverted to the crystalline TFPT-COF just by putting it under the original experimental conditions without additional new building units, which suggested that the connectivity and photoactivity of TFPT-COF were retained.
As discussed before, one of the most intriguing characters of COFs is structural tunability, which allows for structure-to-function design at an atomic level. Indeed, many kinds of research studies on COF-based photocatalysts for water splitting have been done by tailoring the building blocks and linkages. For instance, a series of planar pyrene-based A-TEXPY-COFs were designed and synthesized by extending alkynes with the variation of peripheral heteroaromatic building units.182 The visible-light-driven hydrogen production by COF photocatalysts was studied by using Pt as the co-catalyst and 10 vol% TEOA as a sacrificial electron donor. A-TEBPY-COF constructed from 1,3,6,8-tetrakis(4-ethynylbenzaldehyde)-pyrene (TEBPY) and hydrazine with the lowest nitrogen content and thereby the most advanced donor features exhibited the highest hydrogen production rate of 98 μmol h−1 g−1 in this system. The results were in accordance with an increasing thermodynamic driving force for hydrogen reduction with decreasing nitrogen content.
Previous studies revealed that the rigid, planar dibenzo[b,d]thiophene sulfone (DBTS) unit was conducive to visible-light-induced photocatalytic evolution.209 The DBTS unit was incorporated into ordered COFs to investigate their photocatalytic activity.124 The as-prepared FS-COF exhibited a high hydrogen generation rate, up to 16300 μmol h−1 g−1, which is almost ten times higher than that of N3-COF. Later, three ketoenamine-based COFs were prepared to investigate the effect of different groups on photocatalytic performance.210 Specifically, TpPa-COF-X (X = –H, –(CH3)2, and –NO2) were constructed from the same host backbone with different functional groups anchored on the framework. In the photocatalytic experiment, H2 evolution efficiency decreased in the order of TpPa-COF-(CH3)2 > TpPa-COF > TpPa-COF-NO2. The order was attributed to the electron-donating ability of the three groups, –CH3 > –H > –NO2, which resulted in more efficient charge transfer within the COF framework. Besides, benzothiadiazole as the electron-withdrawing unit and thiophene as the electron-donating moiety were selectively introduced into CTFs.184 The as-prepared CTF-BT/Th was dispersed in water containing 3 wt% Pt as a co-catalyst and 10 vol% TEOA as a sacrificial agent under visible-light irradiation, and it exhibited a maximum hydrogen evolution rate of 6600 μmol h−1 g−1 and an AQE of 7.3% at 420 nm. Notably, the AQE was the highest value compared to the triazine-based polymer photocatalysts that existed at that time. To further enhance the activity, an attractive COF-based hybrid material was prepared based on benzoic acid-modified CTF-1 (B-CTF-1) and NH2-MIL-125(Ti) or NH2-UiO-66(Zr).211 The results showed that the hydrogen evolution rate over 15 wt% NH2-MIL-125(Ti)/B-CTF-1 (15TBC) was 360 μmol h−1 g−1 under visible light irradiation, which was twice higher than that of B-CTF-1. This enhanced photocatalytic activity of 15TBC could be ascribed to the appearance of amide bonds between MOFs and B-CTF-1, which facilitated charge separation and improved the photocatalyst stability.
Notably, considering the charge recombination and the kinetic overpotential for hydrogen production, there is no evidence for current COFs to produce H2 without a co-catalyst. Metallic Pt with a large work function has been widely used for electron trapping among photocatalysts, which also provides efficient proton reduction sites, making the facile H2 formation.212 Thus, the COF backbone with Pt coordination sites enables the specific interaction of COF and Pt, leading to the enhanced charge transfer. However, the stability of Pt in this environment limits its development.213,214 Developing earth-abundant, scalable, low-cost co-catalysts, which are water-soluble and can also interact with a heterogeneous photoabsorber, is urgent. Apart from Pt, MoS2 quantum dots (QDs) with high quantum confinement and small-size effect also represent a prominent candidate as the hydrogen generation co-catalyst.215,216 MoS2 QD modified CTF (MoS2/CTF) composites were reported to yield higher photocatalytic hydrogen production from water under visible-light illumination. MoS2 QDs were easily distributed on the surface of CTFs uniformly via an in situ photo-deposition method.217 The obtained MoS2/CTF composites showed obviously enhanced photocatalytic hydrogen evolution compared to the original CTFs and MoS2/g-C3N4 composite. This high activity was ascribed to the interactions between CTFs and MoS2, which enabled the efficient electron–hole transfer and separation. Cobaloximes, as the most efficient transition metal-based co-catalysts, feature easy synthesis and low overpotentials for hydrogen evolution, and can be easily introduced into the photocatalytic system.218 Lotsch and co-workers firstly selected noble-metal-free cobaloximes as a co-catalyst in the N2-COF-based photocatalytic proton reduction.219 Several factors influenced the H2 evolution rate including the solvent, sacrificial donor, reaction pH, and the fundamental properties of COFs such as crystallinity and porosity. By selecting azine-linked N2-COF as the photosensitizer, chloro(pyridine)cobaloxime as the co-catalyst, and TEOA as a sacrificial donor, a H2 evolution rate of 782 μmol h−1 g−1 and a TON of 54.4 were obtained in a mixture of water and acetonitrile. Herein electrons were transferred from the LUMO of the COF to the co-catalyst, following a monometallic pathway of H2 evolution from the CoIII-hydride and/or CoII-hydride species. As the cobaloxime tends to be inactive within few hours owing to decomposition or hydrogenation, an earth-abundant, noble-metal-free nickelthiolate hexameric cluster was further employed.123 A visible-light-induced hydrogen evolution system was constructed with TzDTz COF (TpDTz: Tp and 4,4′-(thiazolo[5,4-d]thiazole-2,5-diyl)dianiline) as a photosensitizer, Ni-thiolate cluster (NiME) as a co-catalyst, and TEOA as a sacrificial agent (Fig. 12). As a result, a sustained high H2 evolution rate of 941 μmol h−1 g−1 and a TONNi > 103 were observed over 70 h visible-light illumination.
 | ||
| Fig. 12 (a) Schematic illustration of photocatalytic H2 evolution. (b) The proposed key steps of the photocatalytic H2 evolution reaction with TpDTz COF and NiME cluster cocatalyst. [Ni-L] denotes a ligand-coordinated co-catalyst state which is attained fast compared to the [R] state, [R] denotes the catalyst resting state, which is catalytically active nickel cluster species, [D] denotes the deactivated species, and [I] denotes an intermediate reduced catalyst species able to run the HER step. Reproduced with permission from ref. 123. Copyright 2019, American Chemical Society. | ||
5.2. Application in photocatalytic oxygen evolution
As mentioned above, great efforts have been made to realize the water photoreduction half-reaction by using COFs as a photocatalyst. However, water oxidation for oxygen evolution with a more complicated four-electron redox process is the rate-determining step in overall water splitting, which involves the cleavage of the O–H bond, the formation of the O–O band, and large overpotential with sluggish O–O formation kinetics.220,221 Thus, the research of photocatalytic water oxidation with COF photocatalysts is far less than that of photocatalytic hydrogen evolution. Emerging examples for photocatalytic oxygen evolution are CTFs reported by Tang et al.,128,222 sp2 carbon-conjugated COFs developed by Jiang et al. and Zhang et al.,130,131,223 and imine-linked bipyridine COFs prepared by Yang and co-workers.224 For instance, CTF-1 was synthesized via microwave-assisted condensation at different powers, and then was applied in water splitting.74 The oxygen evolution from the water was performed by using AgNO3 as a sacrificial electron acceptor and RuOχ as a co-catalyst. With visible-light illumination, the highest oxygen evolution rate of ca. 140 μmol h−1 g−1 was obtained with 3 wt% RuOx/CTF-1-100 W. Notably, the photocatalytic activity of CTF-1-100 W without a co-catalyst was even 3 times higher than that of g-C3N4 with RuOχ co-catalyst and 20 times better when CTF-1-100 W was decorated with the RuOχ co-catalyst. The AQE for oxygen production was further determined to be ca. 3.8% at 420 nm. These results suggested the considerable potential of CTFs for photocatalytic oxygen evolution under visible-light irradiation. Meanwhile, 2.01 wt% Pt/CTF-1-100 W showed a high hydrogen production of 5500 μmol h−1 g−1 with Pt as a co-catalyst and TEOA as a sacrificial electron donor under visible-light irradiation (λ ≥ 420 nm). CTF-0 based on 1,3,5-tricyanobenzene possessed the highest nitrogen to carbon ratio with alternative benzene and triazine units, which provided more active sites for oxidation reactions. Microwave-assisted synthesis and ionothermal synthesis were also used to produce CTF-0-M and CTF-0-I, respectively.222 The results suggested that the sample synthesized via ionothermal synthesis (e.g. CTF-0-I) showed a higher oxygen production. Under full arc and visible-light irradiation, CTF-0-I with Ag+ as the electron scavenger exhibited an oxygen generation of 226 and 59 μmol g−1 in the first hour, respectively. Recently, a bipyridine COF (Bp-COF) has been investigated as the first imine COF for visible-light-induced water oxidation (Fig. 13).224 The Bp-COF with visible light absorption and appropriate band gap position achieved continuous oxygen generation at a rate of 152 μmol h−1 g−1, corresponding to the AQE of 0.46% at 420 nm, in the presence of Co2+ as a co-catalyst and AgNO3 as an electron acceptor. Although the oxygen production of reported COFs was lower than that of some inorganic semiconductors and MOF-based catalysts, these results show the potential of COFs for oxygen evolution, and the photocatalytic activity of COFs for oxygen evolution could be further enhanced by precisely controlling the structural configuration to achieve suitable photoelectric properties. | ||
| Fig. 13 Photocatalytic O2 evolution of BpCo-COF under visible light irradiation (λ ≥ 420 nm). Reproduced with permission from ref. 224. Copyright 2020, Elsevier B. V. | ||
5.3. Application in the reduction of carbon dioxide
With the fast-growing population and global economy, the increasing fossil fuel consumption has led to excess emission of carbon dioxide (CO2), which causes serious environmental problems like the greenhouse effect.225–227 Many solutions such as amine and ionic liquid absorption, oxy-fuel combustion, and carbonate looping have been put forward pertaining to this dilemma.228–230 In recent years, photocatalytic reduction of CO2 to clean hydrocarbon fuels as an attractive strategy to address the environment and energy issues at the same time has aroused great interest. COFs, the promising photocatalytic candidates, with their high CO2 absorption capacity and selectivity, are recognised as a dramatic platform for photocatalytic reduction of CO2.Azine-based COFs with the existence of π-stacking aromatic units have been regarded as one of the most attractive candidates for photocatalysis. A large conjugated structure could facilitate the separation and transfer of photo-induced electrons/holes. Recently, two azine-linked crystalline COFs ACOF-1 (hydrazine, TFB) and N3-COF were utilized as photocatalysts for visible-light-induced reduction of CO2 with H2O as a hole scavenger.126 Understandably, in the reaction of CO2 photoreduction, the CO2 absorption capability of the catalyst is the key point. In this study, the high surface area of ACOF-1 (1053 m2 g−1) and N3-COF (1412 m2 g−1) with abundant accessible nitrogen sites rendered them with high CO2 absorption, leading to the facilitated photocatalytic reduction of CO2 to CH3OH. Upon 24 h visible light irradiation, the total amount of CH3OH generated over N3-COF was 13.7 μmol g−1, which was much higher than that of ACOF-1 (8.6 μmol g−1). Compared with ACOF-1, N3-COF with electron-poor triazine moieties was able to stabilize the negative charge generated on the COF which was important for the enhanced photocatalytic activity. It should be noted that the activity of these COFs outperformed that of other materials such as g-C3N4 (4.8 μmol g−1) under similar reaction conditions.231,232 Furthermore, the electronic properties and configuration of N3-COF and ACOF-1 were calculated with density functional theory (DFT). The results suggested that the potential of their LUMO was enough to drive CO2 reduction although the band gap was not suitable for the visible light response. Under visible light irradiation, the excited electrons at the LUMO energy level could reduce the adsorbed CO2 on the catalyst surface to produce methanol.
Apart from using COF itself as a photocatalyst for the reduction of CO2, crystalline COFs have also been considered as a photosensitive supporter to stabilize metallic active moieties for CO2 conversion.233 Rhenium(I) bipyridine (bpy) complexes are widely used in constructing photocatalysts to selectively reduce CO2 into CO under visible light irradiation.234,235 A pyridine-based CTF, namely CTF-py, constructed from 2,6-dicyanopyridine (DCP) with abundant N,N-chelating sites allowed for coordination of rhenium complexes targeting CO2 photoreduction. CTF-py was firstly synthesized via traditional trimerization reaction, and then the rhenium complex Re(CO)5Cl was introduced into the nitrogen sites of CTF-py to obtain Re-CTF-py through post-synthetic modification.189 The photocatalytic CO2 conversion was investigated in a solid–gas system under the irradiation of UV-Vis light, which could avoid dimerization and leaching of reactive species. The production of CO linearly increased with the irradiation time. The highest CO production rate of 353.05 μmol g−1 h−1 was observed on Re-CTF-py after 10 h continuous irradiation, while in the case of pristine CTF-py and the physical mixture it was only 13.4 μmol g−1 h−1 and 156.2 μmol g−1 h−1, respectively. The photogenerated electrons could easily transfer from CTF-py to Re via the coordination bond, indicating the efficient separation of photo-induced carriers. Using a similar strategy, a triazine COF derived from the condensation of 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline (TTA) and 2,2-bipyridyl-5,5-dialdehyde was selected as a photosensitizer to incorporate with a Re complex (Re(bpy)(CO)3Cl) for photocatalytic conversion of CO2 to CO.127 When using a Xe lamp as a light source (λ ≥ 420 nm) and TEOA as an electron donor, the resulting Re-COF showed a steady CO generation of 15 mmol g−1 for more than 20 h after the 15 min induction period with a TON of 48, which was 22 times better than that of its homogeneous Re(bpy)(CO)3Cl.
Very recently, COFs were also developed as functional supporters, like TpBpy COF, to anchor active sites for photocatalytic CO2 conversion.236–238 Compared with TpBpy, the introduction of Ni resulted in a red-shifted absorption edge and a narrower band gap due to the increased delocalization. Moreover, Ni-TpBpy helped to enhance the CO2 absorption capacity and isosteric heats, which could be ascribed to the Lewis acid–base interaction between adsorbed CO2 molecules and loaded Ni ions.217 In the experiment of Ni-TpBpy photocatalytic CO2 reduction, [Ru(bpy)]3Cl acted as a photosensitizer and TEOA served as an electron donor. Upon illumination, Ru(bpy)32+ was excited and transferred electrons to reduce the coordinated CO2 molecules on Ni-TpBpy (Fig. 14). The affinity of CO2 on Ni sites over H+ was crucial for the inhibition of H2 formation. As a result, the generated amount of H2 and CO from the Ni-TpBpy catalytic system was 170 and 4057 μmol g−1 within 5 hours, respectively, indicating a higher selectivity to CO. This CO production was comparable to other previous reported MOFs and COFs. Control experiments revealed that single Ni sites in the TpBpy framework acted as catalytic sites while TpBpy facilitated the activity as well as selectivity as a functional support.
 | ||
| Fig. 14 Schematic illustration of CO2 photoreduction over Ni-TpBpy. Reproduced with permission from ref. 217. Copyright 2019, American Chemical Society. | ||
5.4. Application in the degradation of pollutants
The unscrupulous discharge of raw sewage into the environment has led to a huge threat to ecological systems and human health. Organic pollutants, such as dyes, antibiotics and fertilizers, are one of the most persistent components to be degraded. Among various technologies,205,236 photocatalysis utilizing the most abundant solar energy is recognized to be an environmentally sustainable and effective technology for the decomposition of organic contaminants to non-hazardous products.2,239,240 Various kinds of photocatalysts such as TiO2,241,242 CdS,9,243 BiOCl,244,245 and g-C3N416,246 have been extensively studied. However, the limited structural and functional tunability hinders their development. For example, g-C3N4 based on triazine or heptazine units offers limited chemical variety and is hard for systematic post-modification. In this regard, COFs with remarkable structural regularity were supposed to be an intriguing platform for photocatalytic degradation of pollutants such as RhB, methyl blue (MB), methyl orange (MO), and tetracycline (TC).Considering the similar features of nitrogen-rich rings and π-conjugated structure to g-C3N4, COFs with visible-light catalytic active moiety C3N4 exhibited great potential to become a qualified photocatalyst. Over the years, triazine-based COFs have been explored due to their superior photodegradation efficiency compared with g-C3N4.247–249 Likewise, ultrastable TpMA with the C3N4 active center was synthesized by the co-condensation of Tp and MA under solvothermal conditions, which involved a two-step path of reversible Schiff-base reaction and irreversible enol–keto tautomerization.249 This subtly designed structure endowed TpMA with enhanced light-harvesting capability and photooxidation property as a result of the reduced band gap and positive-shifted VB position. MO was selected as a model pollutant to assess the photocatalytic performance of TpMA under visible-light illumination. MO molecules could be degraded by the TpMA photocatalyst within 40 min, whereas the bulk g-C3N4 photocatalytic system showed almost no degradation under the same conditions. In order to exclude the photosensitive effect, colorless organic contaminant phenol was also chosen to evaluate the photocatalytic performance of TpMA. Notably, 90% of phenol was decomposed by TpMA in comparison with 8% decomposition by g-C3N4 after 40 min irradiation. Upon visible light irradiation, TpMA could be excited when the energy was greater than or equal to its band gap (2.30 eV). Then the dissolved O2 quickly captured the electrons from the CB to form O2− (E0, O2/O2− = −0.33 eV vs. NHE),250–252 and the O2− radicals thus formed reacted with H2O to further produce active OH˙. Consequently, MO molecules could be effectively oxidized and mineralized by reactive oxygen species O2− and OH−. According to the total organic carbon (TOC) measurements, TpMA achieved 36.7% of MO mineralization after 40 min-irradiation.
Recently, three imine-linked COFs with a visible-light catalytic active triazine ring were prepared by condensation of three different nitrogen-containing building blocks with the same aldehyde 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)tribenzaldehyde (A), which yielded COFA+B, COFA+C, and COFA+D, respectively. Specifically, the three different monomers were 1,3,5-tris(4-aminophenyl)benzene (B), 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline (C), and 2,4,6-tris(4-hydrazinylphenyl)-1,3,5-triazine (D) (Fig. 15a).253 The MO dye and colorless phenol as model pollutants were selected to assess the photocatalytic performance of the as-prepared COFs. The BET surface area and the corresponding pore volume followed a similar trend, as COFA+D (458 m2 g−1, 0.434 cm3 g−1) < COFA+B (907 m2 g−1, 0.436 cm3 g−1) < COFA+C (1903 m2 g−1, 0.455 cm3 g−1). Although the large surface area and accessible porous nature are beneficial for mass transfer,112,254 the interaction between adsorbent and adsorbate should be given high priority, especially in the liquid phase. In the case of COFA+D, it exhibited higher absorption activity than the other two COFs due to the existence of H-bonding between azo groups of MO and hydrazine groups of COFA+D. A similar phenomenon was also observed with phenol since the N-containing group could interact with the hydroxyl groups of phenol. Therefore, the absorption of MO and phenol followed a reverse trend compared to the BET surface area, as COFA+B > COFA+D > COFA+C. MO molecules could be completely degraded by COFA+C under 30 min visible light irradiation, while only 29.6% MO could be removed by COFA+B, and COFA+D showed almost no degradation. Similarly, the photocatalytic degradation of phenol followed the order of COFA+C > COFA+B > COFA+D (Fig. 15b). The hydrazine groups on COFA+D broke the π-delocalized electron system, leading to the reduction of electron-transfer conductivity and decreasing the interfacial charge transfer; and the CB edge potential of COFA+D was too positive to reduce the molecular oxygen to O2− species, resulting in poor photocatalytic performance. On the other hand, different from COFA+B, the interdigitated triazine-benzene heterojunctions in COFA+C enabled decreased electron–hole recombination. As a result, COFA+C with a higher density of active centres and conjugation degrees showed the highest photocatalytic performance. COFA+C was excited to generate electrons and holes under visible-light illumination. The dissolved O2 captured the accumulated electrons to yield abundant O2−, and then the obtained O2− further reacted with H2O to produce OH˙. On the other hand, the holes could easily transfer to water or oxidized pollutants, which enabled effective charge separation. Thus, the finally generated reactive radicals including O2− and OH˙ could degrade pollutants effectively (Fig. 15c). In addition, COFA+C did not show any major loss of activity after four photocatalytic cycles, indicating its high stability and renewability.
 | ||
| Fig. 15 (a) Structure and band alignment of COFA+B, COFA+C and COFA+D. (b) Photocatalytic performance of COFA+B, COFA+C and COFA+D under visible light irradiation. (c) Schematic illustration of pollutant photodegradation over COFA+C under visible light irradiation. Reproduced with permission from ref. 253. Copyright 2017, Elsevier B. V. | ||
Besides, other functional building units have also been utilized to construct COFs with high photocatalytic performance. For example, a heptazine unit was embedded into the framework of CTF (forming PCN-1 and PCN-2), which was demonstrated to possess high photocatalytic performance toward degradation of RhB.255 In detail, PCN-1 was prepared by the polymerization of melem and 2,4,6-triformylphloroglucinol using a solvent of dimethyl sulfoxide, whereas PCN-2 with crystalline structure was obtained by incorporating melem moieties into CTF. Compared with the traditional polymer semiconductor g-C3N4, the PCN polymers showed broader absorption wavelength, even extended to the entire visible region. Moreover, the enhanced surface area of PCN-2 ensured more active surface sites, thereby giving more chance for reactants to access photoredox reactions. As for the photocatalytic performance, PCN-1 and CTF could degrade RhB within 120 min and 60 min, respectively, while PCN-2 could degrade RhB within 25 min under visible light irradiation. Triptycene with 3D spatial orientation containing three benzene rings is another attractive conjugated building unit for microporous materials synthesis. A triptycene-based imine-linked covalent organic polymer (TP-COP) was prepared for organic dye degradation.256 Graphene-like layered TP-COP was achieved by manual grinding of terepthaldehyde and triaminotriptycene at room temperature. The DRS analyst indicated that TP-COF responded to visible light, and a narrow band gap of ∼2.49 eV was determined by the Tauc plot. 95% of RhB degradation efficiency could be achieved within 160 min under sunlight irradiation. Meanwhile, TP-COP possessed remarkable reusability in RhB degradation without any visible performance decay.
In addition to the building block design, morphology control has also been regarded as an essential method to optimize the catalytic efficiency of photocatalysts.53,257,258 Hollow architectures have been investigated to not only promote the interaction between catalysts and substrates by decreasing the thickness of the structure but also enhance light absorption by multiple light reflections.171 As for other morphologies, TpMA with thread-like morphology could be synthesized by ball milling by varying the amount of liquid added during the process.134 With the addition of p-toluenesulfonic acid and 1 mL solvents, crystalline TpMAC(1mL) was achieved with well-defined morphology of an interwoven thread shape. When the solvent volume was increased to 3 mL, crystalline TpMAC(3mL) with thin ribbon-like morphology was presented. Both TpMAC(3mL) and TpMAC(1mL) were able to respond to visible light and the optical band gap of TpMAC(3mL) and TpMAC(1mL) was 2.29 eV and 2.56 eV, respectively. 10 mg L−1 phenol as a model environmental contaminant was selected to evaluate the photocatalytic performance of TpMAC(3mL) and TpMAC(1mL). Consequently, phenol was completely decomposed after 60 min over TpMAC(3mL) under visible light irradiation, while only 83.5% phenol was degraded by TpMAC(1mL).190,200,201,259
In addition to the morphology control, heterojunction construction has also been used to improve the photocatalytic degradation performance of COFs. For example, a Z-scheme MOF/COF heterojunction was firstly reported by the incorporation of NH2-MIL-125(Ti) with TTB-TTA (TTB: 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)tribenzaldehyde).259 The NH2-MIL-125(Ti)/TTB-TTA composite exhibited enhanced photocatalytic performance for MO degradation because of efficient charge separation through the covalent heterojunction interface. In addition, a BiOBr/CTF-3D composite was designed and prepared, showing enhanced photocatalytic activity toward antibiotic removal.200
6. Conclusions and outlook
COFs, as a kind of newly developed crystalline porous materials, possess great potential as photocatalysts. Versatile organic building blocks and various covalent bonds for COF synthesis render them with fascinating tailored functionalities. The light-harvesting antennae and photoactive chromophores can be integrated into the COF backbone which provides a platform to tailor the band gap structure for visible-light absorption and specific photocatalytic reactions. In addition, the extended in-plane conjugation along with the well-defined interlayer π-stacking structures makes COFs possess enhanced light-absorbing capacity and accelerated charge carrier mobility. In this review, the recent progress and advances related to the COF design and their photocatalytic application were presented. A growing number of covalent linkages amenable to structure and property design were briefly summarized, and controllable morphologies including 0D structures, 1D structures, 2D structures as well as 3D structures were described. Moreover, strategies for enhancing the photocatalytic activity of COF materials were discussed. In addition, the main applications of COFs as photocatalysts regarding photocatalytic H2 evolution, O2 evolution, CO2 photoreduction and photo-degradation of pollutants were presented (Table 1). While some intriguing progress and achievement have been made, the study of COFs and COF-based photocatalysts is still at its infancy stage and several issues should be solved for future development.| Photocatalysts | Building blocks of COFs | Conditions | Applications | Ref. |
|---|---|---|---|---|
| CN-COF | Tp, 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline (TTA), g-C3N4 | 100 mg of photocatalyst dispersed in 200 mL of aqueous solution containing 10 vol% TEoA as a sacrificial agent, Pt as a co-catalyst, 300 W Xe lamp with a cut-off filter (λ ≥ 420 nm) | Photocatalytic H2 evolution, 10100 μmol h−1 g−1 |
181 |
| FS-COF | 3,9-Diamino-benzo[1,2-b:4,5-b′]bis[1]benzothiophene-5,5,11,11-tetraoxide,2,4,6-triformylphloroglucinol | 5 mg of photocatalyst dispersed in 25 mL of 0.1 M ascorbic acid water solution, Pt as a co-catalyst, lactic acid (LA) as the sacrificial agent, 300 W Xe lamp with a cut-off filter | Photocatalytic H2 evolution, 16300 μmol h−1 g−1 |
124 |
| g-C40N3-COF | 4,4′′-Diformyl-p-terphenyl (DFPTP), 3,5-dicyano-2,4,6-trimethylpyridine (DCTMP) | 50 mg of photocatalyst dispersed in 100 mL of deionized water containing 10 vol% TEoA as a sacrificial agent, 3% Pt as a co-catalyst, 300 W Xe lamp with a 420 nm cut-off filter | Photocatalytic H2 evolution, 4120 μmol h−1 g−1 | 180 |
| CdS-COF (90:10) |
1,3,5-Triformylphloroglucinol (Tp), 2,5-dimethyl-p-phenylenediamine (Pa-2) | 30 mg of photocatalyst dispersed in 10 mL of deionized water, 0.5 wt% Pt as a co-catalyst, lactic acid (LA) as the sacrificial agent, 400 W xenon arc lamp with a UV-cut-off filter (λ ≥ 420 nm) | Photocatalytic H2 evolution, 3678 μmol h−1 g−1 | 196 |
| sp2c-COFERDN | 3-Ethylrhodanine, 1,3,6,8-tetrakis(4-formylphenyl)pyrene (TFPPy), 1,4-phenylenediacetonitrile (PDAN) | 50 mg of photocatalyst powder dispersed in 100 mL of aqueous solution containing 10 vol% TEoA as a sacrificial electron donor, 3 wt% Pt as a co-catalyst, 300 W Xe lamp with a water-cooling filter | Photocatalytic H2 evolution, 2120 μmol h−1 g−1 (λ ≥ 420 nm) | 223 |
| TFPT-COF | 1,3,5-Tris-(4-formyl-phenyl)triazine (TFPT), 2,5-diethoxy-terephthalohydrazide (DETH) | 4 mg of photocatalyst dispersed in 9 mL of water containing 1 mL of TEoA as a sacrificial agent, Pt as a co-catalyst, 300 W Xe lamp | Photocatalytic H2 evolution, 1970 μmol h−1 g−1 | 35 |
| N3-COF | Hydrazine hydrate, 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)tribenzaldehyde | 5 mg of photocatalyst dispersed in 10 mL of PBS containing 10 vol% TEoA as a sacrificial agent, Pt as a co-catalyst, 300 W Xe lamp with a cut-off filter (900 nm > λ > 420 nm) | Photocatalytic H2 evolution, 1703 μmol h−1 g−1 | 102 |
| N2-COF | 4,4′,4′′-(Pyrimidine-2,4,6-triyl)tribenzaldehyde, hydrazine | 5 mg of photocatalyst dispersed in 10 mL of water containing 100 μL of TEoA as a sacrificial agent, chloro(pyridine)cobaloxime(III) (Co-1) as a co-catalyst, 300 W Xe lamp | Photocatalytic H2 evolution, 782 μmol h−1 g−1 | 219 |
| g-C18N3-COF | 1,4-Diformylbenzene (DFB), 2,4,6-trimethyl-1,3,5-triazine (TMTA) | 50 mg of photocatalyst dispersed in 100 mL of 1 M aqueous ascorbic acid solution, 3% Pt as a co-catalyst, 300 W Xe lamp with a 420 nm long pass cut-off filter | Photocatalytic H2 evolution, 292 μmol h−1 g−1 | 130 |
| A-TEBPY-COF | 1,3,6,8-Tetrakis(4-ethynylbenzaldehyde)-pyrene (TEBPY), hydrazine | 10 mg of photocatalyst dispersed in 9 mL of water containing 10 vol% TEoA as a sacrificial agent, Pt as a co-catalyst, 300 W Xe lamp | Photocatalytic H2 evolution, 98 μmol h−1 g−1 | 182 |
| ter-CTF-0.7 | 4,7-Bis(4-formylphenyl)-2,1,3-benzothiadiazole (M-BT), 3,6-dicarbaldehyde-N-ethylcarbazole (M-CBZ), terephthalimidamide dihydrochloride | 50 mg of photocatalyst dispersed in 100 mL of deionized water containing 10 vol% TEoA as a sacrificial agent, Pt as a co-catalyst, 300 W Xe lamp | Photocatalytic H2 evolution, 19120 μmol h−1 g−1 |
207 |
| CdS-NP/5%CTF-1 | 2,6-Dicyanopyridine | 20 mg of photocatalyst dispersed in 80 mL of ultrapure water, 1 wt% Pt as a co-catalyst, lactic acid (LA) as the sacrificial agent, 300 W Xe lamp with a UV-cut-off filter (λ ≥ 420 nm) | Photocatalytic H2 evolution, 7500 μmol h−1 g−1 | 197 |
| CTF-BT/Th | 4,4′-(Benzothiadiazole-4,7-diyl) dibenzonitrile, 4,4′-(thiophene-2,5-diyl) dibenzonitrile | 50 mg of photocatalyst dispersed in water containing 10 vol% TEoA as a sacrificial agent, Pt as a co-catalyst, 300 W Xe lamp with a cut-off filter (λ > 420 nm) | Photocatalytic H2 evolution, 6600 μmol h−1 g−1 | 184 |
| CTF-1-100 W | 1,4-Terephthalonitrile (microwave-assisted synthesis) | 50 mg of photocatalyst dispersed in 200 mL of deionized water containing 23 mL of TEoA and 7 mL of methanol, 2.01 wt% Pt as a co-catalyst, 300 W Xe lamp with a 420 nm long pass filter | Photocatalytic H2 evolution, 5500 μmol h−1 g−1 | 74 |
| CTF-0-M2 | 1,3,5-Tricyanobenzene (microwave-assisted synthesis) | 100 mg of photocatalyst dispersed in 230 mL of water containing 10 vol% TEoA as a sacrificial agent, 3 wt% Pt as a co-catalyst, 300 W Xe lamp with a 420 nm long pass filter | Photocatalytic H2 evolution, 4900 μmol in total during seven day-long runs | 222 |
| CTF-0-I | 3-Ethylrhodanine, 1,3,6,8-tetrakis(4-formylphenyl)pyrene (TFPPy), 1,4-phenylenediacetonitrile (PDAN) | 50 mg of photocatalyst powder dispersed in 100 mL of aqueous solution containing 10 vol% TEoA as a sacrificial electron donor, 3 wt% Pt as a co-catalyst, 300 W Xe lamp with a water-cooling filter | Photocatalytic H2 evolution, 2120 μmol h−1 g−1 (λ ≥ 420 nm) | 222 |
| CTFS10 | 1,4-Dicyanobenzene (trifluoromethanesulfonic acid catalysed synthesis) | 20 mg of photocatalyst dispersed in 50 mL of aqueous solution containing 10 vol% TEoA as a sacrificial agent, Pt as a co-catalyst, 300 W Xe lamp | Photocatalytic H2 evolution, 2000 μmol h−1 g−1 | 129 |
| 15 wt% NH2-MIL-125(Ti)/B-CTF-1 (15TBC) | 1,4-Dicyanobenzene | 20 mg of photocatalyst dispersed in 80 mL of aqueous solution containing TEoA as a sacrificial agent, 3% Pt as a co-catalyst, 300 W Xe lamp with a cut-off filter (780 nm ≥ λ ≥ 420 nm) | Photocatalytic H2 evolution, 360 μmol h−1 g−1 | 211 |
| CTF-T1/CTF-T2 | 1,4-Terephthalonitrile (trifluoromethanesulfonic acid catalysed synthesis) | 80 mg of photocatalyst dispersed in 70 mL of ultrapure water containing 10 mL of TeoA as a sacrificial agent, Pt as a co-catalyst, 300 W Xe lamp with a 420 nm cut-off filter | Photocatalytic H2 evolution | 271 |
| CTFCl | 1,4-dicyanobenzene | 20 mg of photocatalyst dispersed in 50 mL of ultrapure water containing 10 vol% TEoA as a sacrificial agent, 5 wt% Pt as a co-catalyst, 300 W Xe lamp with a 420 nm band-pass filter | Photocatalytic H2 evolution | 188 |
| Pd0/TpPa-1 | Tp, p-phenylenediamine (Pa-1) | 10 mg of photocatalyst dispersed in 100 mL of water containing 10 vol% TEoA as a sacrificial agent, Eosin Y as the sensitizer, 300 W Xe lamp with a cut-off filter (λ ≥ 420 nm) | Photocatalytic H2 evolution, 10400 μmol h−1 g−1 |
194 |
| TpDTz | Tp, 4,4′-(thiazolo[5,4-d]thiazole-2,5-diyl)dianiline (DTz) | 5 mg of photocatalyst dispersed in 10 mL of water containing 10 vol% TEoA as a sacrificial agent, Ni-thiolate hexameric cluster (NiME) as a co-catalyst, 300 W Xe lamp | Photocatalytic H2 evolution, 941 μmol h−1 g−1 | 123 |
| TP-BDDA | Tp, 4,4′-(buta-1,3-diyne-1,4-diyl)dianiline (BDDA) | 10 mg of photocatalyst dispersed in 34 mL of water containing 4 mL of TEoA as a sacrificial agent, 3 wt% Pt as a co-catalyst, 300 W Xe lamp with a cut-off filter of 395 nm | Photocatalytic H2 evolution, 324 ± 10 μmol h−1 g−1 | 91 |
| e-CON(Cu, epy) | 2,5-Dihydroxyterephthalaldehyde (Dha), 5,10,15,20-tetrakis(4-aminophenyl)-21H,23H-porphine (Tph) | 0.25 mg of e-CON(Cu, epy) and 0.25 mL of Pt/RGO dispersed in 4.05 mL of water and 0.2 mL of EDTA (0.5 M), 1.0 sunlight, long-pass 420 nm filter for visible light or 780 nm filter for NIR light | Photocatalytic H2 evolution | 163 |
| g-C40N3-COF | 4,4′′-Diformyl-p-terphenyl (DFPTP), 3,5-dicyano-2,4,6-trimethylpyridine (DCTMP) | 50 mg of photocatalyst dispersed in 100 mL of deionized water containing 0.01 mol L−1 AgNO3 as an electron acceptor, 3 wt% Co2+ as a co-catalyst, 300 W Xe lamp | Photocatalytic O2 evolution, 50 μmol h−1 g−1 | 180 |
| sp2c-COF | TFPPy, PDAN | 50 mg of photocatalyst dispersed in 100 mL of aqueous solution containing AgNO3 as an electron acceptor and Co(NO3)2 as a co-catalyst, 300 W Xe lamp with a 420 nm long-pass cut-off filter | Photocatalytic O2 evolution, 22 μmol h−1 g−1 | 223 |
| CTF-1-100 W | 1,4-Terephthalonitrile (microwave-assisted synthesis) | 50 mg of photocatalyst dispersed in 200 mL of 0.02 M AgNO3 aqueous solution, 3 wt% RuOx as a co-catalyst, 300 W Xe lamp with a 420 nm long pass filter | Photocatalytic O2 evolution, 140 μmol h−1 g−1 | 74 |
| CTF-T1 | 1,4-Terephthalonitrile (trifluoromethanesulfonic acid catalysed synthesis) | 50 mg of photocatalyst dispersed in 50 mL of 0.01 M AgNO3 aqueous solution, RuO2 as a co-catalyst, 300 W Xe lamp | Photocatalytic O2 evolution | 271 |
| N3-COF | Hydrazine hydrate, 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)tribenzaldehyde | 10 mg of photocatalyst dispersed in 5 mL of deionized water, pre-injected CO2, 500 W Xe lamp with a UV and IR cut-off filter (800 nm ≥ λ ≥ 420 nm) | Photoreduction of CO2, CH3OH (13.7 μmol g−1) was generated in 24 h | 126 |
| Ni-TpBpy | 1,3,5-Triformylphloroglucinol, 5,5′-diamino-2,2′-bipyridine | 10 mg of photocatalyst dispersed in 5 mL of mixed solution of acetonitrile, H2O, and TEOA, pre-injected CO2, 300 W Xe lamp with a UV cut-off filter (λ ≥ 420 nm) | Photoreduction of CO2, CO was generated at a rate of 4057 μmol h−1 g−1 for 5 h | 218 |
| Re-CTF-py | 2,6-Dicyanopyridine | 2 mg of photocatalyst dispersed on a porous quartz film in the reaction cell, TEoA as a sacrificial agent, 300 W Xe lamp | Photoreduction of CO2, 353.05 μmol h−1 g−1 for 10 h | 189 |
| Re-TpBpy | Tp, 2,2′-bipyridine-5,5′-diamine | 15 mg of photocatalyst dispersed in the AcN/H2O mixture (10/1.8 mL) containing 0.1 M TEoA as an electron donor, 200 W Xe lamp with a high-pass filter at 390 nm | Photoreduction of CO2 to CO | 237 |
| CTF-BT | 4,4′-(Benzothiadiazole-4,7-diyl)dibenzonitrile | 5 g of photocatalyst dispersed in 4-nitrophenol solution, white LED light | Complete reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) after 50 min | 167 |
| NH2-MIL-68@TPA-COF | Tris(4-formylphenyl)amine (TFPA), tris(4-aminophenyl)amine (TAPA) | 5 mg of photocatalyst dispersed in 5 mL of Rh B aqueous solution, 300 W Xe lamp with a UV cut-off filter (λ ≥ 420 nm) | Degradation of Rh B | 201 |
| PCN-2 | Melem, 1,3,5-triformyl phloroglucinol, 2,4,6-tris(4-aminophenyl)-1,3,5-triazine | 4 mg of photocatalyst dispersed in 80 mL of Rh B, 300 W Xe lamp with a cut-off light filter (λ > 420 nm) | Complete degradation of Rh B within 25 min | 255 |
| TP-COP | Triaminotriptycene, terephthaldehyde | 100 mg of photocatalyst dispersed in 100 mL of Rh B solution, sunlight | 95% degradation of Rh B within 160 min irradiation | 256 |
| Fe–TiO2@COF | Tp, 1,1′:4′,1′′-[terphenyl]-4,4''-diamine (Ta) | 0.4 mg of photocatalyst dispersed in 4 mL of MB solution (40 mg L−1), UV light, visible light, ambient light | 96% degradation of MB after 240 min irradiation | 190 |
| COP-NT | 20 mg of photocatalyst dispersed in 20 mL of aqueous solutions of MO, RhB or MB respectively in the presence of 30% H2O2, 10 W LED | Degradation of MO, RhB and MB, 67% MO, 78% RhB and 57% MB degraded within 10 h, 4 h, and 100 min, respectively | 243 | |
| CTF-A | 1,4-Dicyanobenzene | 5 mg of photocatalyst dispersed in 50 mL of MB solution (10 mg L−1), 300 W Xe lamp with a UV cut-off filter (λ ≥ 420 nm) | Degradation of MB, totally degraded within 60 min | 248 |
| COFA+C | 4,4′,4′′-(1,3,5-Triazine-2,4,6-triyl)tribenzaldehyde (A), 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline (C) | 15 mg of photocatalyst dispersed in 50 mL of organic pollutant solution (10 mg L−1), 300 W Xe lamp with an optical cut-off filter (λ ≥ 420 nm) | Complete degradation of MO and 79% degradation of phenol after 30 min irradiation | 253 |
| TpMA | Tp, melamine (MA) | 30 mg of photocatalyst dispersed in 50 mL of MB solution (10 mg L−1), 300 W Xe lamp with a UV cut-off filter (λ ≥ 420 nm) | Complete degradation of MO within 40 min | 249 |
| BiOBr/CTF-3D | 1,4-Dicyanobenzene | 40 mg of photocatalyst dispersed in 200 mL of TC (10 mg L−1) or CIP (10 mg L−1) solution, 500 W Xe lamp | 90.9% degradation of TC within 50 min irradiation, higher degradation of CIP compared to BiOBr | 200 |
(1) The structures, morphologies and properties of COFs are most likely to be changed with different synthesis methods and reaction conditions, thereby leading to the different photocatalytic performance of COFs. Synthetic strategies such as solvothermal synthesis,260,261 ionothermal synthesis,262,263 microwave synthesis74,222,264,265 and room temperature synthesis266,267 have been developed for COF synthesis. While solvothermal synthesis is the most widely used method, the harsh synthesis conditions such as long reaction time and high temperature and pressure make it difficult for large-scale production. Ionothermal synthesis here is utilized for the synthesis of the photoactive triazine core, but the high reaction temperature and low crystalline products hamper the development. The microwave-assisted method and room temperature reaction seem to be better choices. However, only a few examples were reported,74,222,264–266 and thus further improvement is needed. Operative, low cost but effective synthetic methods with mild reaction conditions are eager to be introduced for the development of COFs with enhanced photocatalytic activity.
(2) New stable COFs with high efficiency are necessary. How to facilely control the band gap structure of COFs should be taken seriously. Efficient utilization of the solar spectrum is a significant prerequisite for photocatalysis, and efforts must be made to broaden the light absorption. Besides, the molar absorption coefficient, as a representative factor of light absorption at a specific wavelength, is highly connected to the photocatalytic activity that photocatalysts with a high molar absorption coefficient are able to utilize sunlight more effectively and generate more electron–hole pairs. Thus, constructing COF photocatalysts with enlarged light absorption as well as high molar absorption coefficient is encouraged. For example, as learnt from other traditional photocatalysts, long-wavelength-light-responsive building blocks such as lanthanide-based molecules and phthalocyanine units could be incorporated into COFs to extend the light absorption from visible light to NIR light.268–270 On the other hand, problem still exists in the high recombination rate of photogenerated charge carriers, which retards the effective transfer of electrons and holes. A two-photocatalyst system is found to replace single photocatalysts in nature to avoid inevitable back reaction. Similarly, Z-scheme systems are preferred considering that the photogenerated electrons and holes tend to be separated on divided subsystems, which minimizes the possibility of electron–hole recombination and enables longer-lived charge carriers.
(3) The fundamental mechanism of the COF-based photocatalytic system still remains unclear. Theoretical calculation as a very useful tool is capable of predicting the structures and properties as well as simulating the photocatalytic process. Physicochemical properties of COFs pertaining to the high photocatalytic activity, including surface area, crystallinity, conjugated structure, band gap configuration, visible-light absorption, charge separation and transfer, should be fully investigated. For example, by the utilization of first-principles calculations, three 2D-CTF models CTF-0,63 CTF-1,62 and CTF-264 were investigated including electronic band structures, conduction band minimum (CBM)/valence band maximum (VBM) position, work functions, and optical absorption spectra.271 As a result, 2D-CTFs with controllable construction are better candidates for visible-light-induced water splitting, which stimulated the experimental research on their photocatalytic properties. Besides, advance characterization, especially in situ and even operando technologies, should be taken into consideration to reveal the mechanism behind all the photocatalytic processes, which would provide the insight for further development of efficient COF-based photocatalysts. Technologies such as in situ FT-IR, in situ X-ray absorption spectroscopy (XPS) and in situ extended X-ray absorption fine structure (EXAFS) are highly recommended to monitor the reaction process, distinguishing reactive intermediates and investigating the active sites. More specifically, spectroscopy technologies, such as photoluminescence (PL) spectroscopy, transient absorption (TA) spectroscopy and Kelvin probe force microscopy-based spatially resolved surface photovoltage technique, are also needed for optical and electronic property analysis, corresponding to charge carrier transfer and recombination.
(4) Studies of O2 evolution and CO2 photoreduction using COF-based photocatalysts should also be strengthened in the near future, which are far less than the research on H2 evolution and pollutant degradation. It is a long-term goal to find high performance photocatalysts for visible-light-induced overall water splitting. On the other hand, as for CO2 photoreduction, increasing the product selectivity demands prompt solutions. The design of COFs with highly selective photocatalysis by elaborately choosing functional building blocks and components is highly desired. Additionally, in the current photocatalytic systems, uneconomical sacrificial electron donors and cocatalysts, such as TEOA and noble metal Pt, respectively, have often been used. Strategies like reducing the usage or using highly active but economical alternatives are to be achieved for the development of this area. Besides, the photocatalytic activity is known to be affected by varied conditions such as the amount of photocatalyst, the solvent volume, the kind of cocatalysts, the light source and intensity, and the temperature. It is difficult to compare the activity of photocatalysts reported by different groups. The standardization of photocatalytic activity evaluation has become an urgent necessity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51521006, 51709101, 51508177, 51579098, 51579096, 51779090), the National Program for Support of Top-Notch Young Professionals of China (2012, 2014), Hunan Provincial Science and Technology Plan Project (No. 2016RS3026, 2017SK2243, 2017SK2241), the Program for Changjiang Scholars and Innovative Research Team in University (IRT-13R17) and the Three Gorges Follow-up Research Project (2017HXXY-05). W. H., L. X. Y. and T. J. W. are thankful for the financial support from UK EPSRC (EP/N009533/1), Royal Society-Newton Advanced Fellowship grant (NA170422) and the Leverhulme Trust (RPG-2017-122).Notes and references
- R. Fouquet, Nat. Energy, 2016, 1, 16098 CrossRef
.
- C. Y. Zhou, C. Lai, C. Zhang, G. M. Zeng, D. L. Huang, M. Cheng, L. Hu, W. P. Xiong, M. Chen, J. J. Wang, Y. Yang and L. B. Jiang, Appl. Catal., B, 2018, 238, 6–18 CrossRef CAS
- K. He, G. Q. Chen, G. M. Zeng, A. W. Chen, Z. Z. Huang, J. B. Shi, T. T. Huang, M. Peng and L. Hu, Appl. Catal., B, 2018, 228, 19–28 CrossRef CAS
- F. A. K. Honda, Nature, 1972, 238, 37–38 CrossRef PubMed
- J. H. Carey, J. Lawrence and H. M. Tosine, Bull. Environ. Contam. Toxicol., 1976, 16, 697–701 CrossRef CAS PubMed
- W. Li, A. Elzatahry, D. Aldhayan and D. Y. Zhao, Chem. Soc. Rev., 2018, 47, 8203–8237 RSC
- J. Schneider, M. Matsuoka, M. Takeuchi, J. L. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed
- O. Ola and M. M. Maroto-Valer, J. Photochem. Photobiol., C, 2015, 24, 16–42 CrossRef CAS
- Q. Li, X. Li, S. Wageh, A. A. Al-Ghamdi and J. G. Yu, Adv. Energy Mater., 2015, 5, 1500010 CrossRef
- J. X. Low, B. Z. Dai, T. Tong, C. J. Jiang and J. G. Yu, Adv. Mater., 2019, 31, 1802981 CrossRef PubMed
- L. Cheng, Q. J. Xiang, Y. L. Liao and H. W. Zhang, Energy Environ. Sci., 2018, 11, 1362–1391 RSC
- C. B. Ong, L. Y. Ng and A. W. Mohammad, Renewable Sustainable Energy Rev., 2018, 81, 536–551 CrossRef CAS
- K. M. Lee, C. W. Lai, K. S. Ngai and J. C. Juan, Water Res., 2016, 88, 428–448 CrossRef CAS PubMed
- D. J. Martin, G. Liu, S. J. A. Moniz, Y. Bi, A. M. Beale, J. Ye and J. Tang, Chem. Soc. Rev., 2015, 44, 7808–7828 RSC
- M. D. Hernandez-Alonso, F. Fresno, S. Suarez and J. M. Coronado, Energy Environ. Sci., 2009, 2, 1231–1257 RSC
- J. W. Fu, J. G. Yu, C. J. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8, 1701503 CrossRef
- S. W. Cao and J. G. Yu, J. Phys. Chem. Lett., 2014, 5, 2101–2107 CrossRef CAS PubMed
- D. N. Jiang, P. Xu, H. Wang, G. M. Zeng, D. L. Huang, M. Chen, C. Lai, C. Zhang, J. Wan and W. J. Xue, Coord. Chem. Rev., 2018, 376, 449–466 CrossRef CAS
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, Angew. Chem., Int. Ed., 2016, 55, 5414–5445 CrossRef CAS PubMed
- Y. O. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. W. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS
- T. Banerjee, K. Gottschling, G. Savasci, C. Ochsenfeld and B. V. Lotsch, ACS Energy Lett., 2018, 3, 400–409 CrossRef CAS PubMed
- X. Chen, M. Addicoat, E. Q. Jin, L. P. Zhai, H. Xu, N. Huang, Z. Q. Guo, L. L. Liu, S. Irle and D. L. Jiang, J. Am. Chem. Soc., 2015, 137, 3241–3247 CrossRef CAS PubMed
- G. Q. Lin, H. M. Ding, R. F. Chen, Z. K. Peng, B. S. Wang and C. Wang, J. Am. Chem. Soc., 2017, 139, 8705–8709 CrossRef CAS PubMed
- X. S. Ding, J. Guo, X. A. Feng, Y. Honsho, J. D. Guo, S. Seki, P. Maitarad, A. Saeki, S. Nagase and D. L. Jiang, Angew. Chem., Int. Ed., 2011, 50, 1289–1293 CrossRef CAS PubMed
- G. P. Dong, Y. H. Zhang, Q. W. Pan and J. R. Qiu, J. Photochem. Photobiol., C, 2014, 20, 33–50 CrossRef CAS
- H. Lyu, C. S. Diercks, C. H. Zhu and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 6848–6852 CrossRef CAS PubMed
- S. C. Yan, X. Y. Guan, H. Li, D. H. Li, M. Xue, Y. S. Yan, V. Valtchev, S. L. Qiu and Q. R. Fang, J. Am. Chem. Soc., 2019, 141, 2920–2924 CrossRef CAS PubMed
- R. F. Chen, J. L. Shi, Y. Ma, G. Q. Lin, X. J. Lang and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 6430–6434 CrossRef CAS PubMed
- S. Lin, C. S. Diercks, Y. B. Zhang, N. Kornienko, E. M. Nichols, Y. B. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed
- P. Das and S. K. Mandal, Chem. Mater., 2019, 31, 1584–1596 CrossRef CAS
- S. S. Han, H. Furukawa, O. M. Yaghi and W. A. Goddard, J. Am. Chem. Soc., 2008, 130, 11580–11581 CrossRef CAS PubMed
- L. L. Wang, C. Zeng, H. Xu, P. C. Yin, D. C. Chen, J. Deng, M. Li, N. Zheng, C. Gu and Y. G. Ma, Chem. Sci., 2019, 10, 1023–1028 RSC
- Z. Meng, R. M. Stolz and K. A. Mirica, J. Am. Chem. Soc., 2019, 141, 11929–11937 CrossRef CAS PubMed
- S. Wan, J. Guo, J. Kim, H. Ihee and D. L. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8826–8830 CrossRef CAS PubMed
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC
- K. Schwinghammer, B. Tuffy, M. B. Mesch, E. Wirnhier, C. Martineau, F. Taulelle, W. Schnick, J. Senker and B. V. Lotsch, Angew. Chem., Int. Ed., 2013, 52, 2435–2439 CrossRef CAS PubMed
- J. S. Zhang, X. F. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Z. Fu, M. Antonietti and X. C. Wang, Angew. Chem., Int. Ed., 2010, 49, 441–444 CrossRef CAS PubMed
- R. P. Bisbey and W. R. Dichtel, ACS Cent. Sci., 2017, 3, 533–543 CrossRef CAS PubMed
- P. J. Waller, F. Gandara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef CAS PubMed
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC
- Y. F. Zeng, R. Q. Zou and Y. L. Zhao, Adv. Mater., 2016, 28, 2855–2873 CrossRef CAS PubMed
- S. S. Yuan, X. Li, J. Y. Zhu, G. Zhang, P. Van Puyvelde and B. Van der Bruggen, Chem. Soc. Rev., 2019, 48, 2665–2681 RSC
- H. Wang, Z. T. Zeng, P. Xu, L. S. Li, G. M. Zeng, R. Xiao, Z. Y. Tang, D. L. Huang, L. Tang, C. Lai, D. N. Jiang, Y. Liu, H. Yi, L. Qin, S. J. Ye, X. Y. Ren and W. W. Tang, Chem. Soc. Rev., 2019, 48, 488–516 RSC
- H. Wang, D. Jiang, D. Huang, G. Zeng, P. Xu, C. Lai, M. Chen, M. Cheng, C. Zhang and Z. Wang, J. Mater. Chem. A, 2019, 7, 22848–22870 RSC
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortes, A. P. Cote, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS PubMed
- J. F. Dienstmaier, D. D. Medina, M. Dogru, P. Knochel, T. Bein, W. M. Heckl and M. Lackinger, ACS Nano, 2012, 6, 7234–7242 CrossRef CAS PubMed
- S. Wan, J. Guo, J. Kim, H. Ihee and D. L. Jiang, Angew. Chem., Int. Ed., 2009, 48, 5439–5442 CrossRef CAS PubMed
- C. H. Liu, W. Zhang, Q. D. Zeng and S. B. Lei, Chem. – Eur. J., 2016, 22, 6768–6773 CrossRef CAS PubMed
- B. J. Smith and W. R. Dichtel, J. Am. Chem. Soc., 2014, 136, 8783–8789 CrossRef CAS PubMed
- B. T. Koo, R. F. Heden and P. Clancy, Phys. Chem. Chem. Phys., 2017, 19, 9745–9754 RSC
- N. L. Campbell, R. Clowes, L. K. Ritchie and A. I. Cooper, Chem. Mater., 2009, 21, 204–206 CrossRef CAS
- M. Calik, F. Auras, L. M. Salonen, K. Bader, I. Grill, M. Handloser, D. D. Medina, M. Dogru, F. Lobermann, D. Trauner, A. Hartschuh and T. Bein, J. Am. Chem. Soc., 2014, 136, 17802–17807 CrossRef CAS PubMed
- S. B. Jin, K. Furukawa, M. Addicoat, L. Chen, S. Takahashi, S. Irle, T. Nakamura and D. L. Jiang, Chem. Sci., 2013, 4, 4505–4511 RSC
- E. L. Spitler and W. R. Dichtel, Nat. Chem., 2010, 2, 672–677 CrossRef CAS PubMed
- N. Huang, L. P. Zhai, D. E. Coupry, M. A. Addicoat, K. Okushita, K. Nishimura, T. Heine and D. L. Jiang, Nat. Commun., 2016, 7, 12325 CrossRef PubMed
- S. Dalapati, E. Q. Jin, M. Addicoat, T. Heine and D. L. Jiang, J. Am. Chem. Soc., 2016, 138, 5797–5800 CrossRef CAS PubMed
- C. Jiang, M. Tang, S. L. Zhu, J. D. Zhang, Y. C. Wu, Y. Chen, C. Xia, C. L. Wang and W. P. Hu, Angew. Chem., Int. Ed., 2018, 57, 16072–16076 CrossRef CAS PubMed
- S. Wang, L. Ma, Q. Y. Wang, P. P. Shao, D. Ma, S. Yuan, P. Lei, P. F. Li, X. Feng and B. Wang, J. Mater. Chem. C, 2018, 6, 5369–5374 RSC
- V. Nguyen and M. Grunwald, J. Am. Chem. Soc., 2018, 140, 3306–3311 CrossRef CAS PubMed
- H. Y. Li, A. D. Chavez, H. F. Li, H. Li, W. R. Dichtel and J. L. Bredas, J. Am. Chem. Soc., 2017, 139, 16310–16318 CrossRef CAS PubMed
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed
- P. Katekomol, J. Roeser, M. Bojdys, J. Weber and A. Thomas, Chem. Mater., 2013, 25, 1542–1548 CrossRef CAS
- M. J. Bojdys, J. Jeromenok, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 2202–2205 CrossRef CAS PubMed
- S. Dey, A. Bhunia, D. Esquivel and C. Janiak, J. Mater. Chem. A, 2016, 4, 6259–6263 RSC
- P. Puthiaraj, S. M. Cho, Y. R. Lee and W. S. Ahn, J. Mater. Chem. A, 2015, 3, 6792–6797 RSC
- S. Y. Yu, J. Mahmood, H. J. Noh, J. M. Seo, S. M. Jung, S. H. Shin, Y. K. Im, I. Y. Jeon and J. B. Baek, Angew. Chem., Int. Ed., 2018, 57, 8438–8442 CrossRef CAS PubMed
- K. W. Wang, L. M. Yang, X. Wang, L. P. Guo, G. Cheng, C. Zhang, S. B. Jin, B. Tan and A. Cooper, Angew. Chem., Int. Ed., 2017, 56, 14149–14153 CrossRef CAS PubMed
- M. Liu, Q. Huang, S. Wang, Z. Li, B. Li, S. Jin and B. Tan, Angew. Chem., 2018, 57, 11968–11972 CrossRef CAS PubMed
- M. Y. Liu, L. P. Guo, S. B. Jin and B. E. Tan, J. Mater. Chem. A, 2019, 7, 5153–5172 RSC
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed
- A. Mishra, A. Mehta, S. Basu, N. P. Shetti, K. R. Reddy and T. M. Aminabhavi, Carbon, 2019, 149, 693–721 CrossRef CAS
- X. Jiang, P. Wang and J. J. Zhao, J. Mater. Chem. A, 2015, 3, 7750–7758 RSC
- J. J. Xie, S. A. Shevlin, Q. S. Ruan, S. J. A. Moniz, Y. R. Liu, X. Liu, Y. M. Li, C. C. Lau, Z. X. Guo and J. W. Tang, Energy Environ. Sci., 2018, 11, 1617–1624 RSC
- D. J. Martin, K. P. Qiu, S. A. Shevlin, A. D. Handoko, X. W. Chen, Z. X. Guo and J. W. Tang, Angew. Chem., Int. Ed., 2014, 53, 9240–9245 CrossRef CAS PubMed
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klock, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570–4571 CrossRef CAS PubMed
- Y. X. Ma, Z. J. Li, L. Wei, S. Y. Ding, Y. B. Zhang and W. Wang, J. Am. Chem. Soc., 2017, 139, 4995–4998 CrossRef CAS PubMed
- G. Q. Lin, H. M. Ding, D. Q. Yuan, B. S. Wang and C. Wang, J. Am. Chem. Soc., 2016, 138, 3302–3305 CrossRef CAS PubMed
- S. Y. Ding, J. Gao, Q. Wang, Y. Zhang, W. G. Song, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS PubMed
- S. B. Jin, T. Sakurai, T. Kowalczyk, S. Dalapati, F. Xu, H. Wei, X. Chen, J. Gao, S. Seki, S. Irle and D. L. Jiang, Chem. – Eur. J., 2014, 20, 14608–14613 CrossRef CAS PubMed
- T. Y. Zhou, S. Q. Xu, Q. Wen, Z. F. Pang and X. Zhao, J. Am. Chem. Soc., 2014, 136, 15885–15888 CrossRef CAS PubMed
- Z. F. Pang, S. Q. Xu, T. Y. Zhou, R. R. Liang, T. G. Zhan and X. Zhao, J. Am. Chem. Soc., 2016, 138, 4710–4713 CrossRef CAS PubMed
- X. Chen, M. Addicoat, E. Q. Jin, H. Xu, T. Hayashi, F. Xu, N. Huang, S. Irle and D. L. Jiang, Sci. Rep., 2015, 5, 14650 CrossRef CAS PubMed
- J. Y. Yue, Y. P. Mo, S. Y. Li, W. L. Dong, T. Chen and D. Wang, Chem. Sci., 2017, 8, 2169–2174 RSC
- Y. Z. Liu, Y. H. Ma, Y. B. Zhao, X. X. Sun, F. Gandara, H. Furukawa, Z. Liu, H. Y. Zhu, C. H. Zhu, K. Suenaga, P. Oleynikov, A. S. Alshammari, X. Zhang, O. Terasaki and O. M. Yaghi, Science, 2016, 351, 365–369 CrossRef CAS PubMed
- S. Wan, F. Gandara, A. Asano, H. Furukawa, A. Saeki, S. K. Dey, L. Liao, M. W. Ambrogio, Y. Y. Botros, X. F. Duan, S. Seki, J. F. Stoddart and O. M. Yaghi, Chem. Mater., 2011, 23, 4094–4097 CrossRef CAS
- S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2012, 134, 19524–19527 CrossRef CAS PubMed
- S. Chandra, S. Kandambeth, B. P. Biswal, B. Lukose, S. M. Kunjir, M. Chaudhary, R. Babarao, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 17853–17861 CrossRef CAS PubMed
- B. P. Biswal, S. Chandra, S. Kandambeth, B. Lukose, T. Heine and R. Banerjeet, J. Am. Chem. Soc., 2013, 135, 5328–5331 CrossRef CAS PubMed
- M. Bhadra, S. Kandambeth, M. K. Sahoo, M. Addicoat, E. Balaraman and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 6152–6156 CrossRef CAS PubMed
- P. Pachfule, A. Acharjya, J. Roeser, T. Langenhahn, M. Schwarze, R. Schomacker, A. Thomas and J. Schmidt, J. Am. Chem. Soc., 2018, 140, 1423–1427 CrossRef CAS PubMed
- S. L. Lu, Y. M. Hu, S. Wan, R. McCaffrey, Y. H. Jin, H. W. Gu and W. Zhang, J. Am. Chem. Soc., 2017, 139, 17082–17088 CrossRef CAS PubMed
- M. R. Rao, Y. Fang, S. De Feyter and D. F. Perepichka, J. Am. Chem. Soc., 2017, 139, 2421–2427 CrossRef CAS PubMed
- F. J. Uribe-Romo, C. J. Doonan, H. Furukawa, K. Oisaki and O. M. Yaghi, J. Am. Chem. Soc., 2011, 133, 11478–11481 CrossRef CAS PubMed
- D. N. Bunck and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 14952–14955 CrossRef CAS PubMed
- K. Gottschling, L. Stegbauer, G. Sayasci, N. A. Prisco, Z. J. Berkson, C. Ochsenfeld, B. F. Chmelka and B. V. Lotsch, Chem. Mater., 2019, 31, 1946–1955 CrossRef CAS PubMed
- X. J. Zhao, P. Pachfule, S. Li, T. Langenhahn, M. Y. Ye, C. Schlesiger, S. Praetz, J. Schmidt and A. Thomas, J. Am. Chem. Soc., 2019, 141, 6623–6630 CrossRef CAS PubMed
- W. T. Liu, Q. Su, P. Y. Ju, B. X. Guo, H. Zhou, G. H. Li and Q. L. Wu, ChemSusChem, 2017, 10, 664–669 CrossRef CAS PubMed
- S. Dalapati, S. B. Jin, J. Gao, Y. H. Xu, A. Nagai and D. L. Jiang, J. Am. Chem. Soc., 2013, 135, 17310–17313 CrossRef CAS PubMed
- Y. L. Zhu, S. Wan, Y. H. Jin and W. Zhang, J. Am. Chem. Soc., 2015, 137, 13772–13775 CrossRef CAS PubMed
- S. B. Alahakoon, C. M. Thompson, A. X. Nguyen, G. Occhialini, G. T. McCandless and R. A. Smaldone, Chem. Commun., 2016, 52, 2843–2845 RSC
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS PubMed
- F. Haase, T. Banerjee, G. Savasci, C. Ochsenfeld and B. V. Lotsch, Faraday Discuss., 2017, 201, 247–264 RSC
- Q. R. Fang, Z. B. Zhuang, S. Gu, R. B. Kaspar, J. Zheng, J. H. Wang, S. L. Qiu and Y. S. Yan, Nat. Commun., 2014, 5, 4503 CrossRef PubMed
- A. P. Cote, H. M. El-Kaderi, H. Furukawa, J. R. Hunt and O. M. Yaghi, J. Am. Chem. Soc., 2007, 129, 12914–12915 CrossRef CAS PubMed
- Q. R. Fang, J. H. Wang, S. Gu, R. B. Kaspar, Z. B. Zhuang, J. Zheng, H. X. Guo, S. L. Qiu and Y. S. Yan, J. Am. Chem. Soc., 2015, 137, 8352–8355 CrossRef CAS PubMed
- Z. Luo, L. Liu, J. Ning, K. Lei, Y. Lu, F. Li and J. Chen, Angew. Chem., 2018, 57, 9443–9446 CrossRef CAS PubMed
- C. L. Zhang, S. M. Zhang, Y. H. Yan, F. Xia, A. N. Huang and Y. Z. Xian, ACS Appl. Mater. Interfaces, 2017, 9, 13415–13421 CrossRef CAS PubMed
- S. J. Lyle, T. M. O. Popp, P. J. Waller, X. K. Pei, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 11253–11258 CrossRef CAS PubMed
- J. R. Hunt, C. J. Doonan, J. D. LeVangie, A. P. Cote and O. M. Yaghi, J. Am. Chem. Soc., 2008, 130, 11872–11873 CrossRef CAS PubMed
- J. Guo, Y. H. Xu, S. B. Jin, L. Chen, T. Kaji, Y. Honsho, M. A. Addicoat, J. Kim, A. Saeki, H. Ihee, S. Seki, S. Irle, M. Hiramoto, J. Gao and D. L. Jiang, Nat. Commun., 2013, 4, 2736 CrossRef PubMed
- A. Nagai, X. Chen, X. Feng, X. S. Ding, Z. Q. Guo and D. L. Jiang, Angew. Chem., Int. Ed., 2013, 52, 3770–3774 CrossRef CAS PubMed
- K. T. Jackson, T. E. Reich and H. M. El-Kaderi, Chem. Commun., 2012, 48, 8823–8825 RSC
- D. A. Pyles, J. W. Crowe, L. A. Baldwin and P. L. McGrier, ACS Macro Lett., 2016, 5, 1055–1058 CrossRef CAS
- Y. H. Cho, C. Y. Lee, D. C. Ha and C. H. Cheon, Adv. Synth. Catal., 2012, 354, 2992–2996 CrossRef CAS
- B. Nath, W. H. Li, J. H. Huang, G. E. Wang, Z. H. Fu, M. S. Yao and G. Xu, CrystEngComm, 2016, 18, 4259–4263 RSC
- Y. Du, H. S. Yang, J. M. Whiteley, S. Wan, Y. H. Jin, S. H. Lee and W. Zhang, Angew. Chem., Int. Ed., 2016, 55, 1737–1741 CrossRef CAS PubMed
- H. F. Li, H. Y. Li, Q. Q. Dai, H. Li and J. L. Bredas, Adv. Theory Simul., 2018, 1, 1700015 CrossRef
- L. M. Lanni, R. W. Tilford, M. Bharathy and J. J. Lavigne, J. Am. Chem. Soc., 2011, 133, 13975–13983 CrossRef CAS PubMed
- S. Kandambeth, D. B. Shinde, M. K. Panda, B. Lukose, T. Heine and R. Banerjee, Angew. Chem., Int. Ed., 2013, 52, 13052–13056 CrossRef CAS PubMed
- H. Xu, J. Gao and D. L. Jiang, Nat. Chem., 2015, 7, 905–912 CrossRef CAS PubMed
- Y.-P. Zhang, H.-L. Tang, H. Dong, M.-Y. Gao, C.-C. Li, X.-J. Sun, J.-Z. Wei, Y. Qu, Z.-J. Li and F.-M. Zhang, J. Mater. Chem. A, 2020, 8, 4334–4340 RSC
- B. P. Biswal, H. A. Vignolo-Gonzalez, T. Banerjee, L. Grunenberg, G. Savasci, K. Gottschling, J. Nuss, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2019, 141, 11082–11092 CrossRef CAS PubMed
- X. Y. Wang, L. J. Chen, S. Y. Chong, M. A. Little, Y. Z. Wu, W. H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed
- L. Stegbauer, S. Zech, G. Savasci, T. Banerjee, F. Podjaski, K. Schwinghammer, C. Ochsenfeld and B. V. Lotsch, Adv. Energy Mater., 2018, 8, 1180–1189 Search PubMed
- Y. H. Fu, X. L. Zhu, L. Huang, X. C. Zhang, F. M. Zhang and W. D. Zhu, Appl. Catal., B, 2018, 239, 46–51 CrossRef CAS
- S. Z. Yang, W. H. Hu, X. Zhang, P. L. He, B. Pattengale, C. M. Liu, M. Cendejas, I. Hermans, X. Y. Zhang, J. Zhang and J. E. Huang, J. Am. Chem. Soc., 2018, 140, 14614–14618 CrossRef CAS PubMed
- J. Xie, S. A. Shevlin, Q. Ruan, S. J. A. Moniz, Y. Liu, X. Liu, Y. Li, C. C. Lau, Z. X. Guo and J. Tang, Energy Environ. Sci., 2018, 11, 1617–1624 RSC
- L. Y. Li, W. Fang, P. Zhang, J. H. Bi, Y. H. He, J. Y. Wang and W. Y. Su, J. Mater. Chem. A, 2016, 4, 12402–12406 RSC
- S. C. Wei, F. Zhang, W. B. Zhang, P. R. Qiang, K. J. Yu, X. B. Fu, D. Q. Wu, S. Bi and F. Zhang, J. Am. Chem. Soc., 2019, 141, 14272–14279 CrossRef CAS PubMed
- S. Bi, C. Yang, W. B. Zhang, J. S. Xu, L. M. Liu, D. Q. Wu, X. C. Wang, Y. Han, Q. F. Liang and F. Zhang, Nat. Commun., 2019, 10, 2467 CrossRef PubMed
- X. D. Zhuang, W. X. Zhao, F. Zhang, Y. Cao, F. Liu, S. Bia and X. L. Feng, Polym. Chem., 2016, 7, 4176–4181 RSC
- X. L. Li, C. L. Zhang, S. L. Cai, X. H. Lei, V. Altoe, F. Hong, J. J. Urban, J. Ciston, E. M. Chan and Y. Liu, Nat. Commun., 2018, 9, 2998 CrossRef PubMed
- H. Z. Lv, X. L. Zhao, H. Y. Niu, S. J. He, Z. Tang, F. C. Wu and J. P. Giesy, J. Hazard. Mater., 2019, 369, 494–502 CrossRef CAS PubMed
- H. Y. Liu, J. Chu, Z. L. Yin, X. Cai, L. Zhuang and H. X. Deng, Chem, 2018, 4, 1696–1709 CAS
- G. Das, B. P. Biswal, S. Kandambeth, V. Venkatesh, G. Kaur, M. Addicoat, T. Heine, S. Verma and R. Banerjee, Chem. Sci., 2015, 6, 3931–3939 RSC
- P. Pachfule, S. Kandmabeth, A. Mallick and R. Banerjee, Chem. Commun., 2015, 51, 11717–11720 RSC
- S. J. He, T. Zeng, S. H. Wang, H. Y. Niu and Y. Q. Cai, ACS Appl. Mater. Interfaces, 2017, 9, 2959–2965 CrossRef CAS PubMed
- B. J. Smith, L. R. Parent, A. C. Overholts, P. A. Beaucage, R. P. Bisbey, A. D. Chavez, N. Hwang, C. Park, A. M. Evans, N. C. Gianneschi and W. R. Dichtel, ACS Cent. Sci., 2017, 3, 58–65 CrossRef CAS PubMed
- A. M. Evans, L. R. Parent, N. C. Flanders, R. P. Bisbey, E. Vitaku, M. S. Kirschner, R. D. Schaller, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Science, 2018, 361, 53–57 Search PubMed
- R. L. Li, N. C. Flanders, A. M. Evans, W. Ji, I. Castano, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Chem. Sci., 2019, 10, 3796–3801 RSC
- B. Gole, V. Stepanenko, S. Rager, M. Grune, D. D. Medina, T. Bein, F. Wurthner and F. Beuerle, Angew. Chem., Int. Ed., 2018, 57, 846–850 CrossRef CAS PubMed
- L. Garzon-Tovar, C. Avci-Camur, D. Rodriguez-San-Miguel, I. Imaz, F. Zamora and D. Maspoch, Chem. Commun., 2017, 53, 11372–11375 RSC
- D. Beydoun, R. Amal, G. Low and S. McEvoy, J. Nanopart. Res., 1999, 1, 439–458 CrossRef CAS
- X. F. Duan and C. M. Lieber, Adv. Mater., 2000, 12, 298–302 CrossRef CAS
- W. Huang, Y. Jiang, X. Li, X. J. Li, J. Y. Wang, Q. Wu and X. K. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 8845–8849 CrossRef CAS PubMed
- Y. Jiang, W. Huang, J. Y. Wang, Q. Wu, H. J. Wang, L. L. Pan and X. K. Liu, J. Mater. Chem. A, 2014, 2, 8201–8204 RSC
- D. Rodriguez-San-Miguel, A. Abrishamkar, J. A. R. Navarro, R. Rodriguez-Trujillo, D. B. Amabilino, R. Mas-Balleste, F. Zamora and J. Puigmarti-Luis, Chem. Commun., 2016, 52, 9212–9215 RSC
- B. Luo, G. Liu and L. Z. Wang, Nanoscale, 2016, 8, 6904–6920 RSC
- J. Di, J. Xiong, H. M. Li and Z. Liu, Adv. Mater., 2018, 30, 1704548 CrossRef PubMed
- K. Nakata and A. Fujishima, J. Photochem. Photobiol., C, 2012, 13, 169–189 CrossRef CAS
- S. Ida and T. Ishihara, J. Phys. Chem. Lett., 2014, 5, 2533–2542 CrossRef CAS PubMed
- K. X. Yao, Y. L. Chen, Y. Lu, Y. F. Zhao and Y. Ding, Carbon, 2017, 122, 258–265 CrossRef CAS
- I. Berlanga, M. L. Ruiz-Gonzalez, J. M. Gonzalez-Calbet, J. L. G. Fierro, R. Mas-Balleste and F. Zamora, Small, 2011, 7, 1207–1211 CrossRef CAS PubMed
- I. Berlanga, R. Mas-Balleste and F. Zamora, Chem. Commun., 2012, 48, 7976–7978 RSC
- J. W. Colson, A. R. Woll, A. Mukherjee, M. P. Levendorf, E. L. Spitler, V. B. Shields, M. G. Spencer, J. Park and W. R. Dichtel, Science, 2011, 332, 228–231 CrossRef CAS PubMed
- Z. Zha, L. Xu, Z. Wang, X. Li, Q. Pan, P. Hu and S. Lei, ACS Appl. Mater. Interfaces, 2015, 7, 17837–17843 CrossRef CAS PubMed
- J. I. Feldblyum, C. H. McCreery, S. C. Andrews, T. Kurosawa, E. J. G. Santos, V. Duong, L. Fang, A. L. Ayzner and Z. N. Bao, Chem. Commun., 2015, 51, 13894–13897 RSC
- K. Dey, M. Pal, K. C. Rout, H. S. Kunjattu, A. Das, R. Mukherjee, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 13083–13091 CrossRef CAS PubMed
- Y. Chen, H. J. Cui, J. Q. Zhang, K. Zhao, D. F. Ding, J. Guo, L. S. Li, Z. Y. Tian and Z. Y. Tang, RSC Adv., 2015, 5, 92573–92576 RSC
- T. Sick, A. G. Hufnagel, J. Kampmann, I. Kondofersky, M. Calik, J. M. Rotter, A. Evans, M. Doblinger, S. Herbert, K. Peters, D. Bohm, P. Knochel, D. D. Medina, D. Fattakhova-Rohlfing and T. Bein, J. Am. Chem. Soc., 2018, 140, 2085–2092 CrossRef CAS PubMed
- R. K. Yadav, A. Kumar, N. J. Park, K. J. Kong and J. O. Baeg, J. Mater. Chem. A, 2016, 4, 9413–9418 RSC
- Z. Y. Fan, K. Nomura, M. S. Zhu, X. X. Li, J. W. Xue, T. Majima and Y. Osakada, Chem. Commun., 2019, 2, 55 CrossRef
- Y. C. Yuan, B. Sun, A. M. Cao, D. Wang and L. J. Wan, Chem. Commun., 2018, 54, 5976–5979 RSC
- H. Yang, X. P. Cheng, X. X. Cheng, F. S. Pan, H. Wu, G. H. Liu, Y. M. Song, X. Z. Cao and Z. Y. Jiang, J. Membr. Sci., 2018, 565, 331–341 CrossRef CAS
- X. Zhang, Z. Wang, L. Yao, Y. Y. Mai, J. Q. Liu, X. L. Hua and H. Wei, Mater. Lett., 2018, 213, 143–147 CrossRef CAS
- W. Huang, Z. J. Wang, B. C. Ma, S. Ghasimi, D. Gehrig, F. Laquai, K. Landfester and K. A. I. Zhang, J. Mater. Chem. A, 2016, 4, 7555–7559 RSC
- S. Kandambeth, V. Venkatesh, D. B. Shinde, S. Kumari, A. Halder, S. Verma and R. Banerjee, Nat. Commun., 2015, 6, 6786 CrossRef CAS PubMed
- C. Qian, S. Q. Xu, G. F. Jiang, T. G. Zhan and X. Zhao, Chem. – Eur. J., 2016, 22, 17784–17789 CrossRef CAS PubMed
- J. B. Joo, Q. Zhang, I. Lee, M. Dahl, F. Zaera and Y. D. Yin, Adv. Funct. Mater., 2012, 22, 166–174 CrossRef CAS
- C. C. Nguyen, N. N. Vu and T. O. Do, J. Mater. Chem. A, 2015, 3, 18345–18359 RSC
- H. X. Li, Z. F. Bian, J. Zhu, D. Q. Zhang, G. S. Li, Y. N. Huo, H. Li and Y. F. Lu, J. Am. Chem. Soc., 2007, 129, 8406–8407 CrossRef CAS PubMed
- Z. Wang, J. G. Hou, C. Yang, S. Q. Jiao, K. Huang and H. M. Zhu, Energy Environ. Sci., 2013, 6, 2134–2144 RSC
- S. Chandra, T. Kundu, S. Kandambeth, R. BabaRao, Y. Marathe, S. M. Kunjir and R. Banerjee, J. Am. Chem.
Soc., 2014, 136, 6570–6573 CrossRef CAS PubMed
- H. X. Guo, J. H. Wang, Q. R. Fang, Y. Zhao, S. Gu, J. Zheng and Y. S. Yan, CrystEngComm, 2017, 19, 4905–4910 RSC
- Q. Gao, X. Li, G. H. Ning, K. Leng, B. B. Tian, C. B. Liu, W. Tang, H. S. Xu and K. P. Loh, Chem. Commun., 2018, 54, 2349–2352 RSC
- J. Q. Dong, Y. X. Wang, G. L. Liu, Y. D. Cheng and D. Zhao, CrystEngComm, 2017, 19, 4899–4904 RSC
- L. H. Li, X. L. Feng, X. H. Cui, Y. X. Ma, S. Y. Ding and W. Wang, J. Am. Chem. Soc., 2017, 139, 6042–6045 CrossRef CAS PubMed
- G. H. Ning, Z. X. Chen, Q. Gao, W. Tang, Z. X. Chen, C. B. Liu, B. B. Tian, X. Li and K. P. Loh, J. Am. Chem. Soc., 2017, 139, 8897–8904 CrossRef CAS PubMed
- S. Bi, C. Yang, W. B. Zhang, J. S. Xu, L. M. Liu, D. Q. Wu, X. C. Wang, Y. Han, Q. F. Liang and F. Zhang, Nat. Commun., 2019, 10, 2467 CrossRef PubMed
- M. L. Luo, Q. Yang, K. W. Liu, H. M. Cao and H. J. Yan, Chem. Commun., 2019, 55, 5829–5832 RSC
- L. Stegbauer, S. Zech, G. Savasci, T. Banerjee, F. Podjaski, K. Schwinghammer, C. Ochsenfeld and B. V. Lotsch, Adv. Energy Mater., 2018, 8, 1703278 CrossRef
- W. B. Chen, Z. F. Yang, Z. Xie, Y. S. Li, X. Yu, F. L. Lu and L. Chen, J. Mater. Chem. A, 2019, 7, 998–1004 RSC
- W. Huang, Q. He, Y. P. Hu and Y. G. Li, Angew. Chem., Int. Ed., 2019, 58, 8676–8680 CrossRef CAS PubMed
- S. Ghosh, N. A. Kouame, L. Ramos, S. Remita, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P. H. Aubert and H. Remita, Nat. Mater., 2015, 14, 505–511 CrossRef CAS PubMed
- C. C. Wang, Y. Guo, Y. Yang, S. Chu, C. K. Zhou, Y. Wang and Z. G. Zou, ACS Appl. Mater. Interfaces, 2014, 6, 4321–4328 CrossRef CAS PubMed
- J. H. Li, B. A. Shen, Z. H. Hong, B. Z. Lin, B. F. Gao and Y. L. Chen, Chem. Commun., 2012, 48, 12017–12019 RSC
- Z. Cheng, K. Y. Zheng, G. Y. Lin, S. Q. Fang, L. Y. Li, J. H. Bi, J. N. Shen and L. Wu, Nanoscale Adv., 2019, 1, 2674–2680 RSC
- R. Xu, X. S. Wang, H. Zhao, H. Lin, Y. B. Huang and R. Cao, Catal. Sci. Technol., 2018, 8, 2224–2230 RSC
- Y. M. Zhang, Y. M. Hu, J. H. Zhao, E. Park, Y. H. Jin, Q. J. Liu and W. Zhang, J. Mater. Chem. A, 2019, 7, 16364–16371 RSC
- J. Chen, X. P. Tao, L. Tao, H. Li, C. Z. Li, X. L. Wang, C. Li, R. G. Li and Q. H. Yang, Appl. Catal., B, 2019, 241, 461–470 CrossRef CAS
- J. Di, J. X. Xia, H. M. Li, S. J. Guo and S. Dai, Nano Energy, 2017, 41, 172–192 CrossRef CAS
- L. Zhang, W. Z. Wang, S. M. Sun, Y. Y. Sun, E. P. Gao and J. Xu, Appl. Catal., B, 2013, 132, 315–320 CrossRef
- S. Y. Ding, P. L. Wang, G. L. Yin, X. Q. Zhang and G. X. Lu, Int. J. Hydrogen Energy, 2019, 44, 11872–11876 CrossRef CAS
- P. Pachfule, S. Kandambeth, D. D. Diaz and R. Banerjee, Chem. Commun., 2014, 50, 3169–3172 RSC
- J. Thote, H. B. Aiyappa, A. Deshpande, D. D. Diaz, S. Kurungot and R. Banerjee, Chem. – Eur. J., 2014, 20, 15961–15965 CrossRef CAS PubMed
- D. K. Wang, X. Li, L. L. Zheng, L. M. Qin, S. Li, P. Ye, Y. Li and J. P. Zou, Nanoscale, 2018, 10, 19509–19516 RSC
- S. R. Zhu, M. K. Wu, W. N. Zhao, F. Y. Yi, K. Tao and L. Han, J. Solid State Chem., 2017, 255, 17–26 CrossRef CAS
- J. Li, Y. Yu and L. Z. Zhang, Nanoscale, 2014, 6, 8473–8488 RSC
- S. R. Zhu, Q. Qi, Y. Fang, W. N. Zhao, M. K. Wu and L. Han, Cryst. Growth Des., 2018, 18, 883–891 CrossRef CAS
- Y. W. Peng, M. T. Zhao, B. Chen, Z. C. Zhang, Y. Huang, F. N. Dai, Z. C. Lai, X. Y. Cui, C. L. Tan and H. Zhang, Adv. Mater., 2018, 30, 1705454 CrossRef PubMed
- S. S. Yi, J. M. Yan, B. R. Wulan, S. J. Li, K. H. Liu and Q. Jiang, Appl. Catal., B, 2017, 200, 477–483 CrossRef CAS
- M. Zheng, Y. Ding, L. Yu, X. Q. Du and Y. K. Zhao, Adv. Funct. Mater., 2017, 27, 1605846 CrossRef
- C. B. Meier, R. S. Sprick, A. Monti, P. Guiglion, J. S. M. Lee, M. A. Zwijnenburg and A. I. Cooper, Polymer, 2017, 126, 283–290 CrossRef CAS
- D. Wang, H. Zeng, X. Xiong, M.-F. Wu, M. Xia, M. Xie, J.-P. Zou and S.-L. Luo, Sci. Bull., 2019, 65, 113–122 CrossRef
- S. Kuecken, A. Acharjya, L. J. Zhi, M. Schwarze, R. Schomacker and A. Thomas, Chem. Commun., 2017, 53, 5854–5857 RSC
- L. P. Guo, Y. L. Niu, S. Razzaque, B. Tan and S. B. Jin, ACS Catal., 2019, 9, 9438–9445 CrossRef CAS
- T. Ishi-i, K. Yaguma, T. Thiemann, M. Yashima, K. Ueno and S. Mataka, Chem. Lett., 2004, 33, 1244–1245 CrossRef
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1824–1828 CrossRef PubMed
- J. L. Sheng, H. Dong, X. B. Meng, H. L. Tang, Y. H. Yao, D. Q. Liu, L. L. Bai, F. M. Zhang, J. Z. Wei and X. J. Sun, ChemCatChem, 2019, 11, 2313–2319 CrossRef CAS
- F. Li, D. K. Wang, Q. J. Xing, G. Zhou, S. S. Liu, Y. Li, L. L. Zheng, P. Ye and J. P. Zou, Appl. Catal., B, 2019, 243, 621–628 CrossRef CAS
- J. H. Yang, D. G. Wang, H. X. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed
- W. T. Eckenhoff, W. R. McNamara, P. W. Du and R. Eisenberg, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 958–973 CrossRef CAS PubMed
- P. W. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012–6021 RSC
- J. He, L. Chen, F. Wang, Y. Liu, P. Chen, C. T. Au and S. F. Yin, ChemSusChem, 2016, 9, 624–630 CrossRef CAS PubMed
- X. Q. Hao, Z. L. Jin, H. Yang, G. X. Lu and Y. P. Bi, Appl. Catal., B, 2017, 210, 45–56 CrossRef CAS
- Q. Q. Jiang, L. Sun, J. H. Bi, S. J. Liang, L. Y. Li, Y. Yu and L. Wu, ChemSusChem, 2018, 11, 1108–1113 CrossRef CAS PubMed
- J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995–2004 CrossRef CAS PubMed
- T. Banerjee, F. Haase, G. Savasci, K. Gottschling, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2017, 139, 16228–16234 CrossRef CAS PubMed
- J. W. Tang, J. R. Durrant and D. R. Klug, J. Am. Chem. Soc., 2008, 130, 13885–13891 CrossRef CAS PubMed
- Z. A. Lan, Y. X. Fang, Y. F. Zhang and X. C. Wang, Angew. Chem., Int. Ed., 2018, 57, 470–474 CrossRef CAS PubMed
- D. Kong, X. Y. Han, J. J. Xie, Q. S. Ruan, C. D. Windle, S. Gadipelli, K. Shen, Z. M. Bai, Z. X. Guo and J. W. Tang, ACS Catal., 2019, 9, 7697–7707 CrossRef CAS PubMed
- E. Jin, Z. Lan, Q. Jiang, K. Geng, G. Li, X. Wang and D. Jiang, Chem, 2019, 5, 1632–1647 CAS
- J. Chen, X. P. Tao, C. Z. Li, Y. H. Ma, L. Tao, D. Y. Zheng, J. F. Zhu, H. Li, R. G. Li and Q. H. Yang, Appl. Catal., B, 2020, 262, 118271 CrossRef CAS
- A. Dhakshinamoorthy, S. Navalon, A. Corma and H. Garcia, Energy Environ. Sci., 2012, 5, 9217–9233 RSC
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC
- W. G. Tu, Y. Zhou and Z. G. Zou, Adv. Funct. Mater., 2013, 23, 4996–5008 CrossRef CAS
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed
- F. Fresno, R. Portela, S. Suarez and J. M. Coronado, J. Mater. Chem. A, 2014, 2, 2863–2884 RSC
- A. J. Cowan and J. R. Durrant, Chem. Soc. Rev., 2013, 42, 2281–2293 RSC
- W. L. Yu, D. F. Xu and T. Y. Peng, J. Mater. Chem. A, 2015, 3, 19936–19947 RSC
- T. M. Di, B. C. Zhu, B. Cheng, J. G. Yu and J. S. Xu, J. Catal., 2017, 352, 532–541 CrossRef CAS
- M. Lu, Q. Li, J. Liu, F. M. Zhang, L. Zhang, J. L. Wang, Z. H. Kang and Y. Q. Lan, Appl. Catal., B, 2019, 254, 624–633 CrossRef CAS
- J. L. White, M. F. Baruch, J. E. Pander, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed
- J. Agarwal, E. Fujita, H. F. Schaefer and J. T. Muckerman, J. Am. Chem. Soc., 2012, 134, 5180–5186 CrossRef CAS PubMed
- W. F. Zhong, R. J. Sa, L. Y. Li, Y. J. He, L. Y. Li, J. H. Bi, Z. Y. Zhuang, Y. Yu and Z. G. Zou, J. Am. Chem. Soc., 2019, 141, 7615–7621 CrossRef CAS PubMed
- S. Y. Li, S. Meng, X. Q. Zou, M. El-Roz, I. Telegeev, O. Thili, T. X. Liu and G. S. Zhu, Microporous Mesoporous Mater., 2019, 285, 195–201 CrossRef CAS
- P. D. C. Dietzel, R. E. Johnsen, H. Fjellvag, S. Bordiga, E. Groppo, S. Chavan and R. Blom, Chem. Commun., 2008, 5125–5127, 10.1039/b810574j
- J. J. Zhang, H. Wang, X. Z. Yuan, G. M. Zeng, W. G. Tu and S. B. Wang, J. Photochem. Photobiol., C, 2019, 38, 1–26 CrossRef CAS
- H. Yi, M. Yan, D. L. Huang, G. M. Zeng, C. Lai, M. F. Li, X. Q. Huo, L. Qin, S. Y. Liu, X. G. Liu, B. S. Li, H. Wang, M. C. Shen, Y. K. Fu and X. Y. Guo, Appl. Catal., B, 2019, 250, 52–62 CrossRef CAS
- R. Jaiswal, N. Patel, A. Dashora, R. Fernandes, M. Yadav, R. Edla, R. S. Varma, D. C. Kothari, B. L. Ahuja and A. Miotello, Appl. Catal., B, 2016, 183, 242–253 CrossRef CAS
- H. Safajou, H. Khojasteh, M. Salavati-Niasari and S. Mortazavi-Derazkola, J. Colloid Interface Sci., 2017, 498, 423–432 CrossRef CAS PubMed
- J. X. Low, J. G. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Adv. Mater., 2017, 29, 1601694 CrossRef PubMed
- C. Y. Wang, Y. J. Zhang, W. K. Wang, D. N. Pei, G. X. Huang, J. J. Chen, X. Zhang and H. Q. Yu, Appl. Catal., B, 2018, 221, 320–328 CrossRef CAS
- Y. Mi, L. Y. Wen, Z. J. Wang, D. W. Cao, R. Xu, Y. G. Fang, Y. L. Zhou and Y. Lei, Nano Energy, 2016, 30, 109–117 CrossRef CAS
- F. L. Wang, P. Chen, Y. P. Feng, Z. J. Xie, Y. Liu, Y. H. Su, Q. X. Zhang, Y. F. Wang, K. Yao, W. Y. Lv and G. G. Liu, Appl. Catal., B, 2017, 207, 103–113 CrossRef CAS
- N. Xu, R. L. Wang, D. P. Li, X. Meng, J. L. Mu, Z. Y. Zhou and Z. M. Su, Dalton Trans., 2018, 47, 4191–4197 RSC
- F. Niu, L. M. Tao, Y. C. Deng, H. Gao, J. G. Liu and W. G. Song, New J. Chem., 2014, 38, 5695–5699 RSC
- S. J. He, Q. F. Rong, H. Y. Niu and Y. Q. Cai, Chem. Commun., 2017, 53, 9636–9639 RSC
- X. L. Yang, F. F. Qian, G. J. Zou, M. L. Li, J. R. Lu, Y. M. Li and M. T. Bao, Appl. Catal., B, 2016, 193, 22–35 CrossRef CAS
- S. M. Wang, D. L. Li, C. Sun, S. G. Yang, Y. Guan and H. He, Appl. Catal., B, 2014, 144, 885–892 CrossRef CAS
- S. Kumar, A. Baruah, S. Tonda, B. Kumar, V. Shanker and B. Sreedhar, Nanoscale, 2014, 6, 4830–4842 RSC
- S. J. He, B. Yin, H. Y. Niu and Y. Q. Cai, Appl. Catal., B, 2018, 239, 147–153 CrossRef CAS
- Q. H. Liang, Z. Li, Z. H. Huang, F. Y. Kang and Q. H. Yang, Adv. Funct. Mater., 2015, 25, 6885–6892 CrossRef CAS
- J. Q. Pan, L. P. Guo, S. Q. Zhang, N. Wang, S. B. Jin and B. Tan, Chem. – Asian J., 2018, 13, 1674–1677 CrossRef CAS PubMed
- K. Preet, G. Gupta, M. Kota, S. K. Kansal, D. B. Salunke, H. K. Sharma, S. C. Sahoo, P. Van der Voort and S. Roy, Cryst. Growth Des., 2019, 19, 2525–2530 CrossRef CAS
- W. N. Wang, W. J. An, B. Ramalingam, S. Mukherjee, D. M. Niedzwiedzki, S. Gangopadhyay and P. Biswas, J. Am. Chem. Soc., 2012, 134, 11276–11281 CrossRef CAS PubMed
- C. T. Dinh, H. Yen, F. Kleitz and T. O. Do, Angew. Chem., Int. Ed., 2014, 53, 6618–6623 CrossRef CAS PubMed
- S. J. He, Q. F. Rong, H. Y. Niu and Y. Q. Cai, Appl. Catal., B, 2019, 247, 49–56 CrossRef CAS
- S. Gu, S. F. Wu, L. J. Cao, M. C. Li, N. Qin, J. Zhu, Z. Q. Wang, Y. Z. Li, Z. Q. Li, J. J. Chen and Z. G. Lu, J. Am. Chem. Soc., 2019, 141, 9623–9628 CrossRef CAS PubMed
- X. W. Wu, X. Han, Q. S. Xu, Y. H. Liu, C. Yuan, S. Yang, Y. Liu, J. W. Jiang and Y. Cui, J. Am. Chem. Soc., 2019, 141, 7081–7089 CrossRef CAS PubMed
- A. K. Beine, A. J. D. Kruger, J. Artz, C. Weidenthaler, C. Glotzbach, P. J. C. Hausoul and R. Palkovits, Green Chem., 2018, 20, 1316–1322 RSC
- Y. J. Li, S. H. Zheng, X. Liu, P. Li, L. Sun, R. X. Yang, S. Wang, Z. S. Wu, X. H. Bao and W. Q. Deng, Angew. Chem., Int. Ed., 2018, 57, 7992–7996 CrossRef CAS PubMed
- H. Wei, S. Z. Chai, N. T. Hu, Z. Yang, L. M. Wei and L. Wang, Chem. Commun., 2015, 51, 12178–12181 RSC
- S. J. Ren, M. J. Bojdys, R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Adv. Mater., 2012, 24, 2357–2361 CrossRef CAS PubMed
- G. Lin, C. H. Gao, Q. Zheng, Z. X. Lei, H. J. Geng, Z. Lin, H. H. Yang and Z. W. Cai, Chem. Commun., 2017, 53, 3649–3652 RSC
- Y. W. Peng, W. K. Wong, Z. G. Hu, Y. D. Cheng, D. Q. Yuan, S. A. Khan and D. Zhao, Chem. Mater., 2016, 28, 5095–5101 CrossRef CAS
- R. Chen, Z. H. Yan, X. J. Kong, L. S. Long and L. S. Zheng, Angew. Chem., Int. Ed., 2018, 57, 16796–16800 CrossRef CAS PubMed
- W. Y. Lu, T. F. Xu, Y. Wang, H. G. Hu, N. Li, X. M. Jiang and W. X. Chen, Appl. Catal., B, 2016, 180, 20–28 CrossRef CAS
- C. Krishnaraj, A. M. Kaczmarek, H. S. Jena, K. Leus, N. Chaoui, J. Schmidt, R. Van Deun and P. Van der Voor, ACS Appl. Mater. Interfaces, 2019, 11, 27343–27352 CrossRef CAS PubMed
- J. H. Bi, W. Fang, L. Y. Li, J. Y. Wang, S. J. Liang, Y. H. He, M. H. Liu and L. Wu, Macromol. Rapid Commun., 2015, 36, 1799–1805 CrossRef CAS PubMed
Footnote |
| † These authors contribute equally to this article. |
| This journal is © The Royal Society of Chemistry 2020 |