
Chirality-activated mechanoluminescence from aggregation-induced emission enantiomers with high contrast mechanochromism and force-induced delayed fluorescence†
Yitong
Chen‡
a,
Chao
Xu‡
a,
Bingjia
Xu
 *ab,
Zhu
Mao
c,
Jian-An
Li
a,
Zhan
Yang
c,
Naga Raju
Peethani
a,
Cong
Liu
a,
Guang
Shi
*a,
Feng Long
Gu
a,
Yi
Zhang
bc and
Zhenguo
Chi
bc
*ab,
Zhu
Mao
c,
Jian-An
Li
a,
Zhan
Yang
c,
Naga Raju
Peethani
a,
Cong
Liu
a,
Guang
Shi
*a,
Feng Long
Gu
a,
Yi
Zhang
bc and
Zhenguo
Chi
bc
aKey Laboratory of Theoretical Chemistry of Environment, Ministry of Education, National and Local Joint Engineering Research Center of MPTES in High Energy and Safety LIBs, Engineering Research Center of MTEES (Ministry of Education), Guangzhou Key Laboratory of Analytical Chemistry for Biomedicine, School of Chemistry and Environment, South China Normal University, Guangzhou 510006, China. E-mail: bingjiaxu@m.scnu.edu.cn; shiguang@scnu.edu.cn
bState Key Laboratory of Optoelectronic Materials and Technologies (Sun Yat-sen Unversity), Guangzhou 510275, China
cSchool of Chemistry, Sun Yat-sen University, Guangzhou 510275, China
First published on 20th June 2019
Abstract
A pair of chiral aggregation-induced emission (AIE) enantiomers and their racemate exhibit not only distinctively different circularly polarized luminescence behaviors, but also completely opposite mechanoluminescence (ML) activities. The data from single crystal analysis identifies that the bright ML of the AIE enantiomers is probably activated by their molecular chirality, which enables unique chiral crystal structures with a polar space group and compact molecular packing. These experimental findings provide a direction for harvesting prominent ML from AIE luminogens. Concurrently, high contrast mechanochromism with larger ΔλPL,max and ΔΦF,s values by up to 140 nm and 0.61 respectively have been achieved from this AIE system, which represents the state-of-the-art mechanochromic performance for ML emitters. Moreover, force-induced white-light emission and thermally activated delayed fluorescence have also been observed. The results of theoretical calculations suggest that the destruction of intermolecular interactions and conversion of molecular conformation account for the significant multiple-variations in emission properties triggered by a single mechanical stimulus.
Introduction
Light emission initiated by fracture or force from crystals, namely, mechanoluminescence (ML), has attracted increasing attention in recent years due to its peculiar photophysical process and potential applications in display, lighting, bioimaging and stress sensing.1 However, materials with bright ML are typically limited to metal-containing inorganic compounds and organometallic complexes, particularly rare-earth phosphors, which are generally toxic, expensive and environmentally unfriendly.2 By contrast, purely organic compounds are ideal alternatives because they hold important advantages of low cost, wide variety, good processability and biocompatibility.3 Unfortunately, aromatic compounds usually experience strong π–π stacking in an aggregated state, resulting in partial or complete emission quenching of the emitters.4 Therefore, exploring purely organic luminophores that can show strong ML at room temperature is quite difficult. Recently, we have found that aggregation-induced emission (AIE) is in favor of increasing the intensity of transient ML and even yielding persistent ML (pML) at room temperature, offering a promising direction for the development of organic compounds with intense mechanofluorescence and/or mechanophosphorescence.2a,5 Although AIE exhibits positive effects on the enhancement of ML, there is no guarantee that AIE luminogens (AIEgens) can produce light emission under the stimulation of mechanical force. In fact, ML-active AIEgens are scarce, and until now, only scattered examples have been reported.6 In this context, how to enable bright ML from AIE luminophores through a simple and useful approach emerges as a critical issue which is urgent to be figured out, so that researchers can fully make use of the inherent potentials of AIEgens to reinvigorate the organic ML materials.Mechanochromic luminophores, which can switch their photoluminescence (PL) in response to mechanical stimuli, are likewise of great importance besides mechanoluminescent emitters from both fundamental and practical viewpoints.4a,7 To achieve high contrast mechanochromism (MC), AIEgens are preferable candidates when compared with conventional organic light-emitting materials, because they usually afford twisted molecular structures as well as high emission efficiencies in the aggregation state and it is easy to alter their molecular conformations to shift the emission maxima and weaken the intermolecular interactions to decrease the luminescence intensities upon force treatments, which may enable more remarkable changes in luminescent signals.8 As a result, lots of AIEgens with MC properties, including derivatives of tetraphenylethene, cyanoethylene and distyrylanthracene, have been sequentially created and intensively studied in the last few years.8c,9 In spite of these advancements, AIE emitters that exhibit significant emission wavelength change and noticeable efficiency variation simultaneously under the stimulation of force are still extremely limited.4a,9c Moreover, most of the MC-active AIEgens switch their luminescence between two different prompt fluorescence bands.7,9b,10 As compared to prompt fluorescence, thermally activated delayed fluorescence (TADF) is more attractive since materials with such fascinating features can harvest triplet excitons to boost the luminescent efficiencies of organic light-emitting diodes (OLEDs) and are promising for oxygen or temperature sensing and time-resolved fluorescence bioimaging.11 Thus, AIEgens that can shift emission maxima and turn on TADF by external force may have more extensive applications. In addition, only a few organic MC luminophores have been documented to be capable of producing prominent ML when treated with mechanical force.5b,6a,12 Indeed, purely organic materials, which display both intense ML and obvious MC, are highly desirable because they have great potential for dual-anticounterfeiting. Consequently, it would be nice for theoretical research and practical usage if purely organic AIEgens could generate striking ML, high contrast MC and TADF emission under a single stimulus of mechanical force. Although great efforts have been devoted to this, the development of such multi-responsive AIE emitters remains a formidable challenge.
In this work, we present a pair of AIE enantiomers that show bright ML together with remarkable variations in photophysical properties under the stimulus of external force. Herein, a phthalimide core was selected as an electron acceptor of the target molecules, because it is capable of building robust intermolecular hydrogen bonds to rigidify the molecular conformation, and subsequently, suppress the nonradiative decay to yield AIE properties.5d,13 Phenothiazine was used as an electron donor, since its derivatives often display variable conformations with different single–triplet and HOMO–LUMO energy gaps, making TADF, distinct MC and even dual-emissive white-light possible upon mechanical stimuli.11c,14 Furthermore, luminescent small molecules with chirality usually give circularly polarized luminescence (CPL) and crystallize into chiral structures that are generally noncentrosymmetric.15 Therefore, chiral emitters with donor–acceptor architectures are likely to form crystals with chiral and polar space groups which are beneficial for inducing the piezoelectric effect to produce ML.2a,12a,16 In view of this, tetrahydronaphthalene, which was substituted by the phthalimide moiety at the 1-carbon position, was introduced to enable the chirality of the compounds.17 Based on this elegant molecular design, notable AIE effects and high contrast mechanochromism, as well as force-induced white-light emission and TADF can be recorded for the resulting enantiomers and their racemate. In contrast to ML annihilation of the racemate, intense luminescence triggered by force and obvious CPL triggered by light were observed for the original samples of the enantiomers, identifying that chirality plays a crucial role in activating ML of the AIEgens.
Results and discussion
Synthetic procedures of the enantiopure target molecules, that are, (R)-ImNT and (S)-ImNT (Fig. 1), are outlined in Scheme S1 (ESI†). The imidizations of phthalic anhydride with (R)-(−)-1,2,3,4-tetrahydro-1-naphthylamine and (S)-(+)-1,2,3,4-tetrahydro-1-naphthylamine generated intermediates (R)-ImBr and (S)-ImBr, respectively, followed by coupling with phenothiazine to furnish the desired enantiomers in high yields above 70%. Their racemate (R,S)-ImNT was prepared by simply adding large amounts of methanol into a concentrated dichloromethane solution that contains (R)-ImNT and (S)-ImNT with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 under the action of ultrasound.
:1 under the action of ultrasound.
 | ||
| Fig. 1 Chemical structures of (R)-ImNT and (S)-ImNT. | ||
The study firstly focused on the possible AIE properties of the racemate and the enantiomers, in which their PL spectra in THF/water mixtures with different water fractions were monitored. Almost no signals were recorded for (R,S)-ImNT, (R)-ImNT and (S)-ImNT when they were well dissolved in pure THF (Fig. S1 (ESI†) and Fig. 2a). However, after massive distilled water (fw > 70%) was added into the solutions, significant emission enhancements were detected and Mie scattering effects in the UV-visible absorption spectra were observed (Fig. S2, ESI†). Obviously, the enhancements in luminescence of the racemate and the enantiomers result from the formation of nanoaggregates, suggesting that (R,S)-ImNT, (R)-ImNT and (S)-ImNT are all AIE-active.18 As compared to the emissive nanoaggregates in THF/water mixtures, the solid powder of (R,S)-ImNT emits even stronger blue-light (λPL = 479 nm, ΦF,s = 46.5%) under the illumination of 365 nm UV light (Table 1), which further manifests its AIE characteristic. The intense luminescence of (R,S)-ImNT was identified to be normal fluorescence and exhibits a prompt decay with a short lifetime of 6.22 ns (Fig. S3, ESI†).
 | ||
| Fig. 2 (a) PL spectra of the dilute solutions of (S)-ImNT in water/THF mixtures with different water fractions (fw) (concentration: 0.1 mM). The insets depict the emission images of (S)-ImNT in pure THF and in water/THF mixtures with 90% water fraction under the illumination of 365 nm UV light. (b) PL spectra of the compounds in the solid state under ambient conditions. | ||
Similarly, excited states of the as-prepared samples of (R)-ImNT and (S)-ImNT also decay rapidly, generating prompt fluorescence with emission maxima at 479 nm (τp = 7.92 ns) and 478 nm (τp = 8.56 ns), respectively. It is noteworthy that the full-width at half-maxima (FWHMs) of the PL spectra of (R)-ImNT and (S)-ImNT were determined to be 57 nm and 58 nm, which are both smaller than the one of (R,S)-ImNT (FWHM = 82 nm). Moreover, higher fluorescence efficiencies were recorded for the solid powders of (R)-ImNT (ΦF,s = 60.0%) and (S)-ImNT (ΦF,s = 63.8%). In consideration of the identical chromophores as well as the similar emission maxima, such a distinct reduction in FWHM and an obvious increase in quantum yield of the enantiomers likely originate from their unique molecular stacking modes and more rigid molecular conformations in the solid states.19
CPL measurements were subsequently conducted to investigate the chiroptical properties of the samples. As expected, (R,S)-ImNT is incapable of emitting CPL under the illumination of UV light on account of its racemism (Fig. 3a). In sharp contrast, (R)-ImNT (λCPL = 479 nm) and (S)-ImNT (λCPL = 478 nm) give clear mirror-image CPL signals with luminescence dissymmetry factors (glum) of 3.42 × 10−3 and −3.65 × 10−3 (Fig. S5, ESI†), confirming their chirality in the excited states.17,20 Meanwhile, shearing the solid powders of (R)-ImNT and (S)-ImNT at room temperature with a spatula produces conspicuous bluish-green light emissions, indicating that these two enantiomers are ML-active (Fig. 3b). The vivid ML phenomena can also be clearly observed by the naked eye, even in daylight (Videos S1 and S2, ESI†), which unambiguously verifies their prominent ML performance. In this regard, (R)-ImNT and (S)-ImNT represent the first pair of AIE enantiomers that can generate strong luminescence under the stimulation of mechanical force under ambient conditions. The ML maxima of the enantiomers locate at around 484 nm and 485 nm, respectively, and are both close to their corresponding PL and CPL peaks, indicating that the excitons may decay from similar excited states. Intriguingly, the bright ML from (R)-ImNT and (S)-ImNT completely annihilated after formation of the racemate, exhibiting coincident behaviors to the CPL. In other words, chirality can not only induce CPL, but also activate ML from the AIEgens of (R)-ImNT and (S)-ImNT. Since photophysical properties of organic materials are related to their aggregation state, measurements of differential scanning calorimetry (DSC) and powder X-ray diffraction (XRD) were performed to examine the phase characteristics of the samples. As depicted in Fig. S6 (ESI†), the intense melting transitions in the DSC curves and the sharp deflection peaks in the XRD patterns of the racemate and enantiomers fully manifest that their as-prepared solid powders are mainly composed of microcrystals. Further elaborative investigation reveals that the melting points and the XRD patterns of (R)-ImNT and (S)-ImNT are almost identical, but distinctly different from those of (R,S)-ImNT. These results imply that the opposite ML behaviors of the enantiomers and racemate probably arise from their different optical activities leading to dissimilar crystal architectures.
 | ||
| Fig. 3 CPL (a) and ML spectra (b) of the as-prepared samples of (R,S)-ImNT, (R)-ImNT and (S)-ImNT under ambient conditions. The insets in (b) depict the ML images of (R)-ImNT and (S)-ImNT in daylight. | ||
| Compound | λ PL a (nm) | λ CPL b (nm) | Φ F,s c | τ p d (ns) | ΔESTe (eV) | ML-Activity | λ PL,g f (nm) | Φ F,g g | ΔλPL,maxh (nm) | τ p,g i (ns) | τ d,g j (μs) | ΔEST,gk (eV) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a, b, c, d and e are the PL maximum, CPL maximum, fluorescence quantum yield, lifetime and single–triplet energy gap of the pristine samples of the enantiomers and racemate under ambient conditions, respectively. f, g, h, i, j and k are the PL maximum, fluorescence quantum yield, lifetime of prompt component, lifetime of delayed component and single–triplet energy gap of the well-ground samples. | ||||||||||||
| (R)-ImNT | 479 | 479 | 0.600 | 7.92 | 0.501 | Active | 618 | 0.018 | 139 | 10.04 | 0.60 | 0.013 |
| (S)-ImNT | 478 | 479 | 0.638 | 8.56 | 0.490 | Active | 618 | 0.019 | 140 | 10.08 | 0.58 | 0.013 |
| (R,S)-ImNT | 479 | — | 0.465 | 6.22 | 0.474 | Inactive | 618 | 0.018 | 139 | 9.92 | 0.61 | 0.009 |
To gain in-depth insight into the effect of chirality on ML performance, single crystals of (R)-ImNT, (S)-ImNT and (R,S)-ImNT were analyzed, especially focusing on the molecular packing mode. The simulated XRD patterns from single crystal data are in accordance with the experimental ones of crystalline powders, suggesting that the microcrystals of the enantiomers and racemate adopt the same molecular arrangements as their corresponding single crystals. As demonstrated in Table S1 (ESI†), the single crystal structure of (R,S)-ImNT belongs to a monoclinic system with a centrosymmetric space group of P21/c (achiral). In the unit cells, there are two (R)-ImNT and two (S)-ImNT chiral molecules, which show identical quasi-axial conformations (Fig. S7, ESI†). Meanwhile, the (R)-ImNT molecules are anti-parallel to the ones of (S)-ImNT (Fig. S8a, ESI†). As a result, the achiral crystal architecture of the racemate becomes non-polar and presents a zero net dipolar moment. By contrast, in the case of (S)-ImNT, each unit cell is composed of four chiral molecules in different quasi-axial conformations (denoted as conformation S1, S2, S3 and S4) (Fig. 4a). Moreover, the crystallographically independent chiral molecules pack in a herringbone manner (Fig. 4b and Fig. S8b, ESI†), thus leading to a chiral crystal structure with a polar space group of P1. The net dipole moment of the four molecules in the unit cell was determined to be 10.01 Debye, which significantly increases as compared to those of single molecules (4.41, 4.19, 4.75 and 4.60 Debye for conformation S1, S2, S3 and S4). Accordingly, the strong piezoelectric effect would probably occur when breaking the crystals, resulting in the production of excitons.5d,16b,21 On the other hand, numerous intermolecular interactions including C–H⋯O, C–H⋯S and C–H⋯π with distance ranging from 2.360 Å to 2.995 Å are observed in the single crystal structure of (S)-ImNT (Fig. S9a, ESI†). Their distribution and strength are simulated by using Multiwfn and visualized in 3D isosurfaces with different gradient colors. As depicted in Fig. 4c, many regions turn into green or even bluish-green, indicating that strong intermolecular interactions probably occur.22 These intense multiple interactions help to solidify the molecular conformations of (S)-ImNT and block the non-radiative pathways via restricting the intramolecular motions of the molecules, which induces a notable AIE effect and gives a high ΦF,s value of (S)-ImNT. The strong intermolecular interactions and the large net dipole moment work synergistically, and thereby makes the bright ML of (S)-ImNT available at room temperature. Similar molecular arrangement and intermolecular interaction strength are involved in the crystal structure of (R)-ImNT (Fig. S8c, S9b and S10, ESI†). Consequently, the crystalline powder of (R)-ImNT can also sparkle conspicuous luminescence upon grinding or shearing. However, for (R,S)-ImNT, the quantity of intermolecular hydrogen bonds obviously reduces (Fig. S9c, ESI†), indicating a relatively loose molecular packing mode. Further evidence is provided by its smaller δginter values from an independent gradient model (IGM) in comparison with those of enantiomers (Fig. 4d).22a,23 The weaker intermolecular interactions combined with a centrosymmetric crystal structure completely quench the ML from (R)-ImNT and (S)-ImNT molecules, even though their racemate remains AIE-active. These results thus suggest that the ML of the polar AIE enantiomers is probably activated by their molecular chirality which leads to a unique chiral crystal structure with a polar space group and compact molecular packing.
 | ||
| Fig. 4 (a) Molecular conformations of (S)-ImNT molecules in a single crystal structure. The digits in the figure represent bent angle, dihedral angle or dipolar moment. Take conformation S1 as an example. Its dipolar moment is 4.41 Debye. While the bent angle between the phenothiazine moiety and its neighboring phenyl ring is 115.89°, the dihedral angle between plane A and plane B is 66.02°. (b) Molecular packing mode of (S)-ImNT molecules in the unit cell. D.M. means net dipolar moment of the molecules in the unit cell. (c) Distribution and strength of intramolecular interactions in the unit cell of (S)-ImNT. (d) Plots of the IGM with isovalue of 0.005 versus the electron density. | ||
Notably, it is found that the photoluminescence of the crystalline powders of the enantiomers and racemate are sensitive to external force. Take (S)-ImNT as an example. When grinding treatment was applied to its pristine sample, a new emission peak at 618 nm emerged (Fig. 5a). Simultaneously, the fluorescent band at around 478 nm gradually weakened, and finally disappeared. That is, the ratios between blue- and red-emission peaks were reversed during grinding. As a result, (S)-ImNT showed remarkable mechanochromism with a large ΔλPL,max value by up to 140 nm, which represents the state-of-the-art mechanochromic performance for ML emitters. In addition, the CIEx,y chromaticity coordinates converted from the proceeding emission changes lie on a straight line with a content correlation coefficient of 0.995. This observation fully demonstrates that the color of (S)-ImNT can be linearly tuned in a wide range from (0.15, 0.27) to (0.54, 0.43) by means of a mild treatment of hand grinding. It is worth noting that the above straight line also crosses the white region of the CIE-1931 chromaticity diagram, and hits the point of (0.31, 0.33) which is close to standard white-light emission. That means (S)-ImNT can also emit white-light with tunable CIEx,y coordinates by simply applying an appropriate mechanical stimulus on its crystalline sample. In contrast to the highly luminescent microcrystals, the well-ground sample of (S)-ImNT exhibits extremely weak red emission under the excitation of 365 nm UV light, as indicated by the fluorescent image in Fig. 5b. The corresponding ΦF,s value is calculated to be 0.019, and reduces over 0.61 in comparison with that of the original sample. Such an alteration in luminescence intensity is sufficiently large as to be readily distinguished by the naked eye. Besides, the red emission peak of (S)-ImNT initially decays promptly with a lifetime of 10.08 ns then followed a delayed decay with a lifetime of 0.58 μs under ambient conditions, exhibiting an interesting phenomenon of force-induced thermally activated delayed fluorescence (Fig. S11a, ESI†). The TADF characteristic of the red emission band is also validated by the oxygen-sensitive PL spectrum (Fig. S11b, ESI†) as well as the temperature dependent emission decay of the delayed component (Fig. 5c) which displays a continuous increase of lifetime from 1.30 μs at 77 K to 2.15 μs at 150 K, and a decrease to 1.17 μs at 300 K. By fuming with dichloromethane vapor, the red TADF emission band of the well-ground sample can be easily erased and the blue prompt fluorescence arises again, exhibiting a reversible mechanochromism (Fig. S11c, ESI†). Similar mechanochromic behaviors are also observed for (R)-ImNT and (R,S)-ImNT, as demonstrated in Fig. S12–S16 (ESI†). Therefore, unlike conventional stimuli-responsive luminescent materials, the enantiomers (R)-ImNT and (S)-ImNT can show strong ML, high contrast MC and TADF emission under a single stimulus of mechanical force. Such a new kind of force-responsive AIE emitter has not yet been achieved before, and may facilitate the applications of ML materials in novel security and data storage systems.
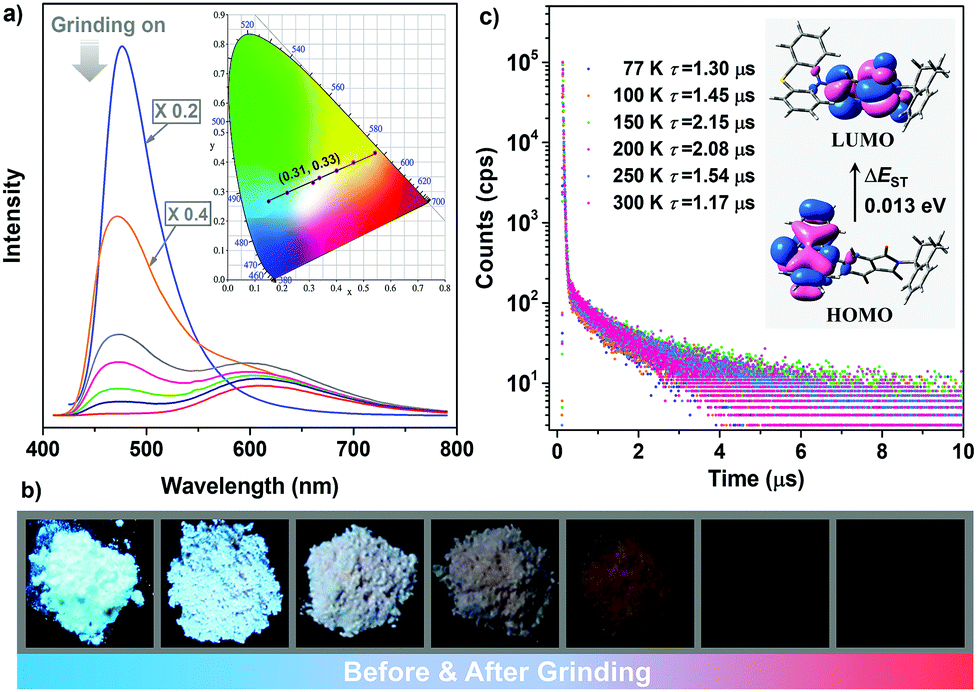 | ||
| Fig. 5 Variations in emission properties of (S)-ImNT triggered by external force. (a) Changes of PL spectra of (S)-ImNT under grinding. “×0.2” and “×0.4” mean that intensities of the spectra presented in the figure are one fifth and two fifths of those of the original ones, respectively. The inset depicts linear changes of CIEx,y coordinates of (S)-ImNT under grinding. (b) Luminescent images of the samples upon grinding treatments (under the illumination of 365 nm UV light). (c) Emission decay curves of the well-ground sample at different temperatures under vacuum. The insets are the Kohn–Sham frontier orbitals of (S)-ImNT in the most possible quasi-equatorial conformation. | ||
Measurements of XRD and DSC were subsequently carried out to investigate the relationship between the changes of emission properties and powder morphologies. The diffraction peaks in the experimental XRD patterns for the powders of the enantiomers and racemate become less defined and more diffuse as the grinding progresses (Fig. S17, ESI†), while exothermic transitions in the DSC curves emerge and gradually enhance (Fig. S18, ESI†). These jointly illustrate that the microcrystals of (R)-ImNT, (S)-ImNT and (R,S)-ImNT are partially destroyed and convert to amorphous states, which would severely weaken the intermolecular interactions and turn on the nonradiative decay to quench the luminescence. To further probe the mechanism of the recorded mechanochromism, theoretical calculations were performed at the density functional theory (DFT) and time-dependent density functional theory (TD-DFT) level. The functional M06-2X and basis set 6-311G** were adopted in all of the calculations. As depicted in Fig. S19 (ESI†), frontier orbitals for the (R)-ImNT and (S)-ImNT molecules with quasi-axial conformations in single crystal structures show obvious overlap at the phthalimide unit. Consequently, ΔEST values obtained from both theoretical and experimental data (0.501 eV for (R)-ImNT, 0.490 eV for (S)-ImNT and 0.474 eV for (R,S)-ImNT) are too large to realize the TADF (Fig. S20, ESI†).11b,24 Conformation searching was then performed for the (R)-ImNT and (S)-ImNT molecules, and stable conformations in quasi-equatorial style are found besides the quasi-axial ones which are in agreement with those in crystals (Fig. S21, ESI†). It is worth noting that the most possible quasi-equatorial conformations of (S)-ImNT and (R)-ImNT show stronger intramolecular charge transfer (ICT) with well-separated HOMO and LUMO in comparison with the quasi-axial molecules in single crystal structures (Fig. 5c and Fig. S21c, ESI†), and thereby they can lower the luminescence intensities and red-shift the emission wavelengths. Furthermore, their ΔEST values are both simulated to be 0.013 eV, which are in accordance with the experimental results (Fig. S22, ESI†) and sufficiently small to enable TADF. Accordingly, the significant variations in emission properties of the enantiomers and racemate under mechanical stimuli are likely caused by the destruction of intermolecular interactions and the formation of quasi-equatorial conformations, leading to the enhancement of nonradiative decay and ICT, as well as the decrease of ΔEST values.
Conclusions
In summary, a pair of purely organic AIE enantiomers, (R)-ImNT and (S)-ImNT, that show bright ML and obvious CPL at room temperature have been developed for the first time. The ML of these two emitters has been identified to be activated by their molecular chirality which enables unique chiral crystal structures with a polar space group and compact molecular packing, providing a direction for harvesting prominent ML from AIEgens. Meanwhile, high contrast mechanochromism, as well as force-induced white-light emission and TADF have been achieved for the enantiomers and racemate. In view of the striking ML together with remarkable multi-changes in intrinsic optical properties triggered by a single stimulus of mechanical force, the AIE-active (R)-ImNT and (S)-ImNT enantiomers may have potential for innovative applications in sensors, displays, security systems and data storage devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51603233), Pearl River S&T Nova Program of Guangzhou (201806010125), Opening Foundation of State Key Laboratory of Optoelectronic Materials and Technologies (Sun Yat-sen University, OEMT-2018-KF-11), Education Department (2016KTSCX027) of Guangdong Province of China and Research Start-up Funds for Outstanding Young Scholars of South China Normal University.Notes and references
- (a) Y. J. Xie and Z. Li, Chem, 2018, 4, 943–971 CrossRef CAS; (b) J. Yang, J. Qin, P. Geng, J. Wang, M. Fang and Z. Li, Angew. Chem., Int. Ed., 2018, 57, 14174–14178 CrossRef CAS PubMed; (c) W. Li, Q. Huang, Z. Mao, Q. Li, L. Jiang, Z. Xie, R. Xu, Z. Yang, J. Zhao, T. Yu, Y. Zhang, M. P. Aldred and Z. Chi, Angew. Chem., Int. Ed., 2018, 57, 12727–12732 CrossRef CAS PubMed; (d) Q. Sun, K. Zhang, Z. Zhang, L. Tang, Z. Xie, Z. Chi, S. Xue, H. Zhang and W. Yang, Chem. Commun., 2018, 54, 8206–8209 RSC; (e) Z. Xie, T. Yu, J. Chen, E. Ubba, L. Wang, Z. Mao, T. Su, Y. Zhang, M. P. Aldred and Z. Chi, Chem. Sci., 2018, 9, 5787–5794 RSC.
- (a) B. Xu, W. Li, J. He, S. Wu, Q. Zhu, Z. Yang, Y.-C. Wu, Y. Zhang, C. Jin, P.-Y. Lu, Z. Chi, S. Liu, J. Xu and M. R. Bryce, Chem. Sci., 2016, 7, 5307–5312 RSC; (b) S. Cai, H. Shi, J. Li, L. Gu, Y. Ni, Z. Cheng, S. Wang, W. W. Xiong, L. Li, Z. An and W. Huang, Adv. Mater., 2017, 29, 1701244 CrossRef PubMed.
- Y. Xiong, Z. Zhao, W. Zhao, H. Ma, Q. Peng, Z. He, X. Zhang, Y. Chen, X. He, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2018, 57, 7997–8001 CrossRef CAS PubMed.
- (a) J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed; (b) Y. Xie, J. Tu, T. Zhang, J. Wang, Z. Xie, Z. Chi, Q. Peng and Z. Li, Chem. Commun., 2017, 53, 11330–11333 RSC.
- (a) J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, B. Z. Tang, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu and D. Zhu, Chem. Commun., 2001, 1740–1741 RSC; (b) B. Xu, J. He, Y. Mu, Q. Zhu, S. Wu, Y. Wang, Y. Zhang, C. Jin, C. Lo, Z. Chi, A. Lien, S. Liu and J. Xu, Chem. Sci., 2015, 6, 3236–3241 RSC; (c) S. Xu, T. Liu, Y. Mu, Y. F. Wang, Z. Chi, C. C. Lo, S. Liu, Y. Zhang, A. Lien and J. Xu, Angew. Chem., Int. Ed., 2015, 54, 874–878 CrossRef CAS PubMed; (d) J. A. Li, J. Zhou, Z. Mao, Z. Xie, Z. Yang, B. Xu, C. Liu, X. Chen, D. Ren, H. Pan, G. Shi, Y. Zhang and Z. Chi, Angew. Chem., Int. Ed., 2018, 57, 6449–6453 CrossRef CAS PubMed.
- (a) E. Ubba, Y. Tao, Z. Yang, J. Zhao, L. Wang and Z. Chi, Chem. – Asian J., 2018, 13, 3106–3121 CrossRef CAS PubMed; (b) P. Thilagar and S. Mukherjee, Angew. Chem., Int. Ed., 2018, 58, 7922–7932 Search PubMed.
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC.
- (a) X. Luo, J. Li, C. Li, L. Heng, Y. Q. Dong, Z. Liu, Z. Bo and B. Z. Tang, Adv. Mater., 2011, 23, 3261–3265 CrossRef CAS PubMed; (b) M. Chen, R. Chen, Y. Shi, J. Wang, Y. Cheng, Y. Li, X. Gao, Y. Yan, J. Z. Sun, A. Qin, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Adv. Funct. Mater., 2018, 28, 1704689 CrossRef; (c) Z. Yang, Z. Chi, Z. Mao, Y. Zhang, S. Liu, J. Zhao, M. P. Aldred and Z. Chi, Mater. Chem. Front., 2018, 2, 861–890 RSC.
- (a) Y. Liu, Q. Zeng, B. Zou, Y. Liu, B. Xu and W. Tian, Angew. Chem., Int. Ed., 2018, 57, 15670–15674 CrossRef CAS PubMed; (b) J. Zhao, Z. Chi, Y. Zhang, Z. Mao, Z. Yang, E. Ubba and Z. Chi, J. Mater. Chem. C, 2018, 6, 6327–6353 RSC; (c) J. Zhao, Z. Chi, Z. Yang, Z. Mao, Y. Zhang, E. Ubbaa and Z. Chi, Mater. Chem. Front., 2018, 2, 1595–1608 RSC.
- J. Mei, Y. Hong, J. W. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- (a) Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS; (b) Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC; (c) Z. Xie, C. Chen, S. Xu, J. Li, Y. Zhang, S. Liu, J. Xu and Z. Chi, Angew. Chem., Int. Ed., 2015, 54, 7181–7184 CrossRef CAS PubMed; (d) Z. He, X. Cai, Z. Wang, D. Chen, Y. Li, H. Zhao, K. Liu, Y. Cao and S.-J. Su, Sci. China: Chem., 2018, 61, 677–686 CrossRef CAS.
- (a) C. Wang, B. Xu, M. Li, Z. Chi, Y. Xie, Q. Li and Z. Li, Mater. Horiz., 2016, 3, 220–225 RSC; (b) C. Arivazhagan, A. Maity, K. Bakthavachalam, A. Jana, S. K. Panigrahi, E. Suresh, A. Das and S. Ghosh, Chem. – Eur. J., 2017, 23, 7046–7051 CrossRef CAS PubMed; (c) Y. B. Gong, P. Zhang, Y. R. Gu, J. Q. Wang, M. M. Han, C. Chen, X. J. Zhan, Z. L. Xie, B. Zou, Q. Peng, Z. G. Chi and Z. Li, Adv. Opt. Mater., 2018, 6, 1800198 CrossRef.
- J. Nishida, H. Ohura, Y. Kita, H. Hasegawa, T. Kawase, N. Takada, H. Sato, Y. Sei and Y. Yamashita, J. Org. Chem., 2016, 81, 433–441 CrossRef CAS PubMed.
- (a) M. Okazaki, Y. Takeda, P. Data, P. Pander, H. Higginbotham, A. P. Monkman and S. Minakata, Chem. Sci., 2017, 8, 2677–2686 RSC; (b) B. J. Xu, Y. X. Mu, Z. Mao, Z. L. Xie, H. Z. Wu, Y. Zhang, C. J. Jin, Z. G. Chi, S. W. Liu, J. R. Xu, Y. C. Wu, P. Y. Lu, A. Lien and M. R. Bryce, Chem. Sci., 2016, 7, 2201–2206 RSC; (c) J. S. Ward, R. S. Nobuyasu, M. A. Fox, J. A. Aguilar, D. Hall, A. S. Batsanov, Z. Ren, F. B. Dias and M. R. Bryce, J. Org. Chem., 2019, 84, 3801–3816 CrossRef CAS PubMed.
- (a) F. Song, Z. Xu, Q. Zhang, Z. Zhao, H. Zhang, W. Zhao, Z. Qiu, C. Qi, H. Zhang, H. H. Y. Sung, I. D. Williams, J. W. Y. Lam, Z. Zhao, A. Qin, D. Ma and B. Z. Tang, Adv. Funct. Mater., 2018, 28, 1800051 CrossRef; (b) S. Feuillastre, M. Pauton, L. Gao, A. Desmarchelier, A. J. Riives, D. Prim, D. Tondelier, B. Geffroy, G. Muller, G. Clavier and G. Pieters, J. Am. Chem. Soc., 2016, 138, 3990–3993 CrossRef CAS PubMed; (c) M. Jin, T. Seki and H. Ito, J. Am. Chem. Soc., 2017, 139, 7452–7455 CrossRef CAS PubMed.
- (a) Q. Sun, L. Tang, Z. Zhang, K. Zhang, Z. Xie, Z. Chi, H. Zhang and W. Yang, Chem. Commun., 2017, 54, 94–97 RSC; (b) H. Nakayama, J.-i. Nishida, N. Takada, H. Sato and Y. Yamashita, Chem. Mater., 2012, 24, 671–676 CrossRef CAS.
- M. Li, S. H. Li, D. Zhang, M. Cai, L. Duan, M. K. Fung and C. F. Chen, Angew. Chem., Int. Ed., 2018, 57, 2889–2893 CrossRef CAS PubMed.
- Y. Hong, J. W. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- (a) S. Zhang, L. Yao, Q. Peng, W. Li, Y. Pan, R. Xiao, Y. Gao, C. Gu, Z. Wang, P. Lu, F. Li, S. Su, B. Yang and Y. Ma, Adv. Funct. Mater., 2015, 25, 1755–1762 CrossRef CAS; (b) J.-Y. Hu, Y.-J. Pu, F. Satoh, S. Kawata, H. Katagiri, H. Sasabe and J. Kido, Adv. Funct. Mater., 2014, 24, 2064–2071 CrossRef CAS.
- J. R. Brandt, X. Wang, Y. Yang, A. J. Campbell and M. J. Fuchter, J. Am. Chem. Soc., 2016, 138, 9743–9746 CrossRef CAS PubMed.
- M. Fang, J. Yang, Q. Liao, Y. Gong, Z. Xie, Z. Chi, Q. Peng, Q. Li and Z. Li, J. Mater. Chem. C, 2017, 5, 9879–9885 RSC.
- (a) C. Lefebvre, G. Rubez, H. Khartabil, J. C. Boisson, J. Contreras-Garcia and E. Henon, Phys. Chem. Chem. Phys., 2017, 19, 17928–17936 RSC; (b) E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed; (c) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- C. Lefebvre, H. Khartabil, J. C. Boisson, J. Contreras-Garcia, J. P. Piquemal and E. Henon, ChemPhysChem, 2018, 19, 724–735 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the synthesis, structural information for the compounds (NMR, elemental analysis and mass spectra), Fig. S1–S36 and Tables S1 and S2. CCDC 1906044–1906046. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qm00312f |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2019 |