DOI:
10.1039/C9QM00178F
(Research Article)
Mater. Chem. Front., 2019,
3, 1159-1167
Rational design of drug delivery systems for potential programmable drug release and improved therapeutic effect†
Received
25th March 2019
, Accepted 24th April 2019
First published on 25th April 2019
Abstract
Nanoparticle-based combination therapy has been reported as one important treatment strategy to improve the therapeutic efficacy in cancer therapy. However, the traditional co-delivery method often suffers from serious coordination issues that fail to account for the differences in the action sites of each of the drugs, greatly counteracting the synergistic effects of combination agents. Herein, we report a pH-reduction dual responsive drug delivery system for the programmable release of combretastatin A-4 (CA4) and cis-platinum (CDDP). According to their spatial and temporal needs, such nanocarriers could firstly release CA4 in the perivascular sites by responding to the acidic microenvironment of the tumor, then further diffuse throughout the tumor and release CDDP upon redox when being taken-up by the cancer cells. With the help of such spatiotemporal release properties, CA4 and CDDP can target the endothelial cells of tumor vessels and the cancer cells in succession, giving rise to temporal and spatial synergisms. On one hand, the first released CA4 provided a complementary effect for improving tumor accumulation of nanocarriers and the delivery effect of CDDP. On the other hand, CA4 and CDDP respectively targeted their action sites, overall maximizing the therapeutic effect. More importantly, such spatiotemporal drug delivery ability is accomplished by the pH-reduction dual responsive design of the nanocarriers, and thus could also serve as a universal approach for programmable release of other combination agents to improve their synergistic effect in cancer therapy.
Introduction
Cancer is the leading cause of death and a major public health problem all around the world.1 Although various treatment modalities including surgery, chemotherapy, radiotherapy, and immunotherapy have been developed and widely used in clinical treatment, the therapeutic effect and the survival rates of patients have not shown significant improvement over the decades.2 At this stage, the development of more efficient therapeutic methods remains the main task in cancer therapy.
Recently, with the rapid development of nanotechnology, nanoparticle-based combination therapy, referring to simultaneous delivery of two or multiple antitumor agents, has been reported as one important strategy to improve the treatment effect for cancer therapy.3–12 Unlike monotherapy, combination therapy, as one of the novel strategies, is characteristic of their numerous advantages. For example, it can modulate different signalling pathways in cancer cells to produce a synergistic response,13 maximize the therapeutic effect while reducing the systematic toxicity,14 and offer promising potential to overcome drug resistance and inhibit tumor relapse.15,16 However, there are still many concerns about its implementation due to the different pharmacokinetics and action sites of different agents.17 For traditional co-delivery systems, the non-controllable and non-targeted release is likely to introduce some new problems. For instance, the systemic toxicity might be enhanced owing to the sum of side effects of separated drugs.8 Thus, the development of a high-efficiency multidrug delivery system with controllable properties and synergistic effects is highly required for combination therapy.18
As a novel concept, programmable combination approaches have emerged as a potential strategy to enhance the antitumor activity in combination therapy.6,7,16,17,19–21 By carefully controlling the release of each of the drugs, programmable treatments based on nanotechnology could potentially minimize drug–drug interactions and enhance the access of drug molecules to their targets,17 thus improving their synergistic effect. Unfortunately, despite the advantages of programmable release methods over co-delivery strategies, the appropriate nanotechnologies and nanocarriers have not been fully developed.17 Due to the different mechanisms and acting sites of anticancer agents, the releases of those drugs need to be accurately controlled in a spatiotemporal manner, making the drugs exert their pharmacological activities only at the target sites.19
In current strategies, one of the typical examples is combined treatment based on vascular disrupting agents (VDAs) and cytotoxic agents. As an elegant concept in cancer therapy, the inhibition of angiogenesis using VDA can produce synergetic responses with many chemotherapeutics such as doxorubicin (DOX), cis-platinum (CDDP), and paclitaxel (PTX) etc.4,19,22–27 When simultaneously treated with VDA and chemotherapy drugs, VDA is able to selectively disrupt the newly formed tumor vasculature and deprive nutrient supply to tumor cells, while chemotherapeutics kill the cancer cells, leading to extensive and comprehensive tumor necrosis.24 However, how to precisely control the release of each agent in their respective action sites to maximize their effectiveness in combination therapy remains a main task and challenge in the design of nanocarriers.
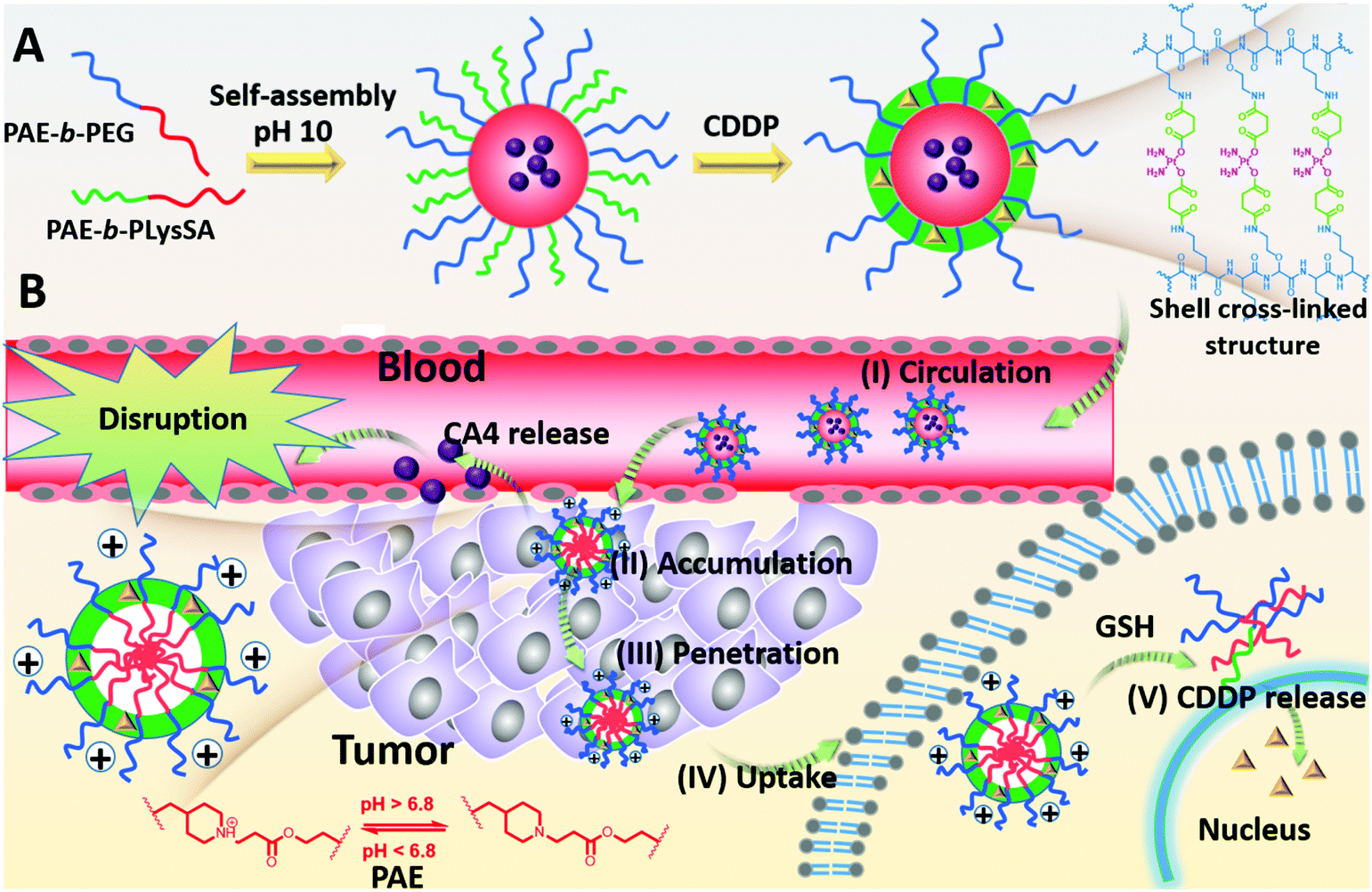
Using combretastatin A4 (CA4) and cis-platinum (CDDP) as an example, the CA4 can cause a rapid vascular shutdown inside a tumor by disrupting the cytoskeletal structures of the endothelial cells,4 implying that CA4 should be released in the regions nearby the tumor vessels. In contrast, CDDP brings about apoptosis by interaction with DNA to form crosslinked DNA adducts only when being transferred into the nucleus of the cancer cells, and requires extensive diffusion in tumor tissues to contact with more cancer cells for sufficient uptake. This opposite requirement may give a great challenge to nanomedicines in combined therapy. To overcome this challenge, we reported a pH-reduction dual-responsive drug delivery system (pH-reduction dual responsive nanocarriers, denoted as DRNs) to achieve programmable releases of CA4 and CDDP. This dual-responsive system comprised a reduction-responsive shell crosslinked by CDDP, a pH-responsive core consisting of poly(β-amine ester) (PAE), and a PEGylated crown. In the bloodstream, the protection of PEG and the crosslinking of CDDP would endow the nanocarriers with outstanding stability. When entering the tumor tissues via the enhanced permeability and retention (EPR) effect, the extracellular low pH microenvironment would trigger the rapid protonation of PAE,28 and give the nanocarriers a positive charge to achieve adequate accumulation in the tumor. At the same time, the CA4 could be quickly released from the pH-responsive PAE core and disrupt tumor vessels, thereby inhibiting the tumor growth by reducing the nutrition supplement from blood, and improving the tumor accumulation of the DRNs resulting from the enhanced retention time in the tumor sites. In addition, the DRNs with positive charge can deeply penetrate into the tumor tissues. When being taken-up by the cancer cells, the intracellular reductive microenvironment, caused by the high level of glutathione (GSH),29–34 would accelerate the release of CDDP, resulting in large apoptosis of the cancer cells and extensive tumor necrosis, which could enhance the therapeutic effect (Scheme 1). In brief, this work reveals the potential of a single programmable nanocarrier to deliver CA4 and CDDP for targeting endothelial cells and cancer cells in succession, giving rise to a temporal and spatial synergism. On one hand, the temporal synergism provides CA4 with a complementary effect to improve tumor accumulation of nanocarriers and enhance the delivery effect of CDDP. On the other hand, spatial synergism would maximize the therapeutic effect because of the accurate targeting of CA4 and CDDP. In a word, this system offers a potential strategy to enhance the treatment effect of combination therapy based on CA4 and CDDP. More importantly, the programmable release property is accomplished by the pH-reduction dual responsive structure of the DRNs, and thus can also provide a universal platform for the spatiotemporal release of other combination agents in combined therapy, enabling their in vivo application and giving a better treatment effect.
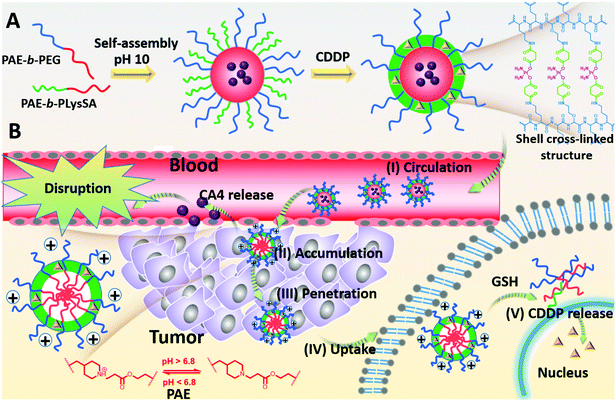 |
| | Scheme 1 (A) Illustration of the preparation and shell crosslinking structure of the DRNs. (B) Illustration of the programmable drug release property of the DRNs in vivo. (I) The PEGlated and CDDP cross-linked stabilization endow the DRNs with outstanding stability in the bloodstream. (II) The acidic microenvironment triggers CA4 release to disrupt the tumor vessel, promoting further tumor accumulation of the DRNs. (III), (IV) The positive surface charge facilitates the tumor penetration of DRNs and uptake. (V) Intracellular reductive conditions, resulting from GSH, trigger the release of CDDP when the DRNs enter the cancer cells. | |
Experimental
Materials
All chemicals were purchased from J&K Chemical Ltd, USA unless otherwise stated. All solvents used in the reaction were distilled after drying with calcium hydride for 12 hours. Poly(ethylene glycol) methyl ether (PEG5k-OH) was purchased from Shanghai YareBio Ltd, China. Cy5.5, Cy3 NHS ester and Cy5.5 NHS ester were purchased from Lumiprobe Co., USA. Succinic anhydride was purchased from Lumiprobe Co., USA. cis-Platinum and combretastatin A4 (CA4) was purchased from Shanghai YareBio Ltd, China.
Preparation and characterization of nanocarriers
Detailed protocols for the synthesis of all block copolymers are described in the Supporting Information (Fig. S1–S3, ESI†). The characterizations of all block copolymers and the products in each step were performed using the 1H NMR spectra. The nanocarriers were prepared through a nanoprecipitation technique referring to previous works.35–37 PAE-b-PEG was dissolved in THF with the concentration of 17.5 mg mL−1. PAE-b-PLysSA solution was prepared in DMSO by the ring-opening reaction of PAE-b-PLys (1 eq.) and SA (25 eq.). After reaction at 35 °C for 48 h, the solution was diluted to 14 mg mL−1. PCL-b-PLysSA solution was prepared using a similar method. According to the predetermined formulation (Table S1, ESI†), a certain amount of the polymer solutions were mixed together and added dropwise into alkaline water (pH 10) under vigorous stirring, followed by stabilization for 2 h. The solution was transferred into a dialysis bag (MWCO 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and dialyzed against PB (pH 9.0, 10 mM) for 1 day to remove excess SA. Then the mixture was allowed to react with CDDP for 24 h and transferred into a dialysis bag (MWCO 3500) and dialyzed against PB (pH 7.4, 10 mM) for 2 days, followed by ultrafiltration to anticipant concentration. Other types of nanocarriers were prepared in a similar method. The formulations are shown in Tables S2 and S3 (ESI†). The characterizations of the nanocarriers were performed using dynamic light scattering (DLS) via laser light scattering spectrometer (BI-200SM), transmission electron microscopy (TEM) and zeta potential via a Brookhaven ZetaPALS (Brookhaven Instrument, USA) at different pH conditions.
000) and dialyzed against PB (pH 9.0, 10 mM) for 1 day to remove excess SA. Then the mixture was allowed to react with CDDP for 24 h and transferred into a dialysis bag (MWCO 3500) and dialyzed against PB (pH 7.4, 10 mM) for 2 days, followed by ultrafiltration to anticipant concentration. Other types of nanocarriers were prepared in a similar method. The formulations are shown in Tables S2 and S3 (ESI†). The characterizations of the nanocarriers were performed using dynamic light scattering (DLS) via laser light scattering spectrometer (BI-200SM), transmission electron microscopy (TEM) and zeta potential via a Brookhaven ZetaPALS (Brookhaven Instrument, USA) at different pH conditions.
Evaluation of the stability and responsiveness of the DRNs via DLS and TEM
To evaluate the stability of the DRNs, phosphate buffer solution (PB, 0.01 M, pH 7.4) was used to simulate the physiological pH value. The stability of the DRNs was determined by measuring the change in particle size over time. Briefly, 0.1 mL of the DRN solution (1 mg mL−1) was diluted with 1.5 mL of PB solution (0.01 M, pH 7.4). The average hydrodynamic diameter distributions of the DRNs over 12 h were acquired from DLS. The responsiveness of the DRNs was evaluated by the measure of DLS and observation of TEM. Briefly, 0.1 mL of the DRN solution (1 mg mL−1) was added into 1.5 mL of three types of phosphate buffer solution (PB) (pH 7.4, pH 6.5 and pH 6.5 + 10 mM GSH). The average hydrodynamic diameter as setting points (0 h, 2 h, 4 h, and 12 h) was acquired from DLS. At the same time, the DRN solutions at different times were collected, and then dropped onto a carbon-coated copper grid and dried slowly at the required temperature, followed by observation via TEM.
In vitro drug release
DRN solution (1 mL, pH 6.5 and 7.4) with an initial concentration of 2 mg mL−1 was added into a dialysis bag (MWCO 5000) and dialyzed against 9 mL of PB solution (10 mM) with the corresponding pH value under vigorous stirring. At set time points, 1 mL of the dialysis fluid was collected and replaced with an equal volume of fresh PB solution. The release of CA4 was determined by measuring the intensity using an ultraviolet spectrometer (at 290 nm). The release of CDDP was evaluated by inductively coupled plasma mass spectrometry (ICP-MS). The release of Cy3 and Cy5.5 was determined by measuring the fluorescence intensity of the emission (570 nm and 690 nm, respectively). The same experiments were repeated three times and the average was taken.
In vitro programmable release
The in vitro programmable release ability of the DRNs was evaluated by cellular uptakes at different pH (pH 7.4 and 6.5) conditions using confocal laser scanning microscopy (CLSM) and flow cytometry (FCM). The culture medium with different pH conditions was prepared in advance. Briefly, 1 mL of PB solution (pH 7.4, 100 mM) was added to 9 mL of DMEM culture medium and its pH was adjusted to 7.4 using HCl solution (1 mM). With a similar method, pH 6.5 medium was prepared. The Cy3-labeled/Cy5.5-loaded DRN (Cy3-DRNs/Cy5.5) solution was respectively diluted five times with the aforementioned culture medium solution. HepG2 cells were cultivated in a confocal dish for 24 h incubation with an initial density of 5000 cells per dish. Whereafter, the culture medium was removed and replaced with 1 mL of Cy3-DRNs/Cy5.5 solution (pH 7.4 and 6.5). After 2 hours, the solution was removed and the cells were washed with 1 mL of PBS solution three times, followed by fixing with 1 mL of 4% paraformaldehyde solution for 15 min and observing with CLSM after staining by 4′,6-diamidino-2-phenylindole (DAPI). Similarly, the 4T1 cells were cultivated in 12-well plates with an initial density of 10000 cells per well for 24 hours incubation, and then treated with the Cy3-DRNs/Cy5.5 solution. After 2 hours, the medium was removed and each well was washed three times. The cells were digested with 0.5 mL of trypsin solution for 1 min, and then dispersed in 0.5 mL of fresh medium. All the cells were transferred into a centrifuge tube, followed by 5 min centrifugation. The supernatant was removed and the cells were washed three times with PBS. Finally, the cells were fixed and dispersed in 1 mL of 4% paraformaldehyde solution for 15 min, and measured by flow cytometry.
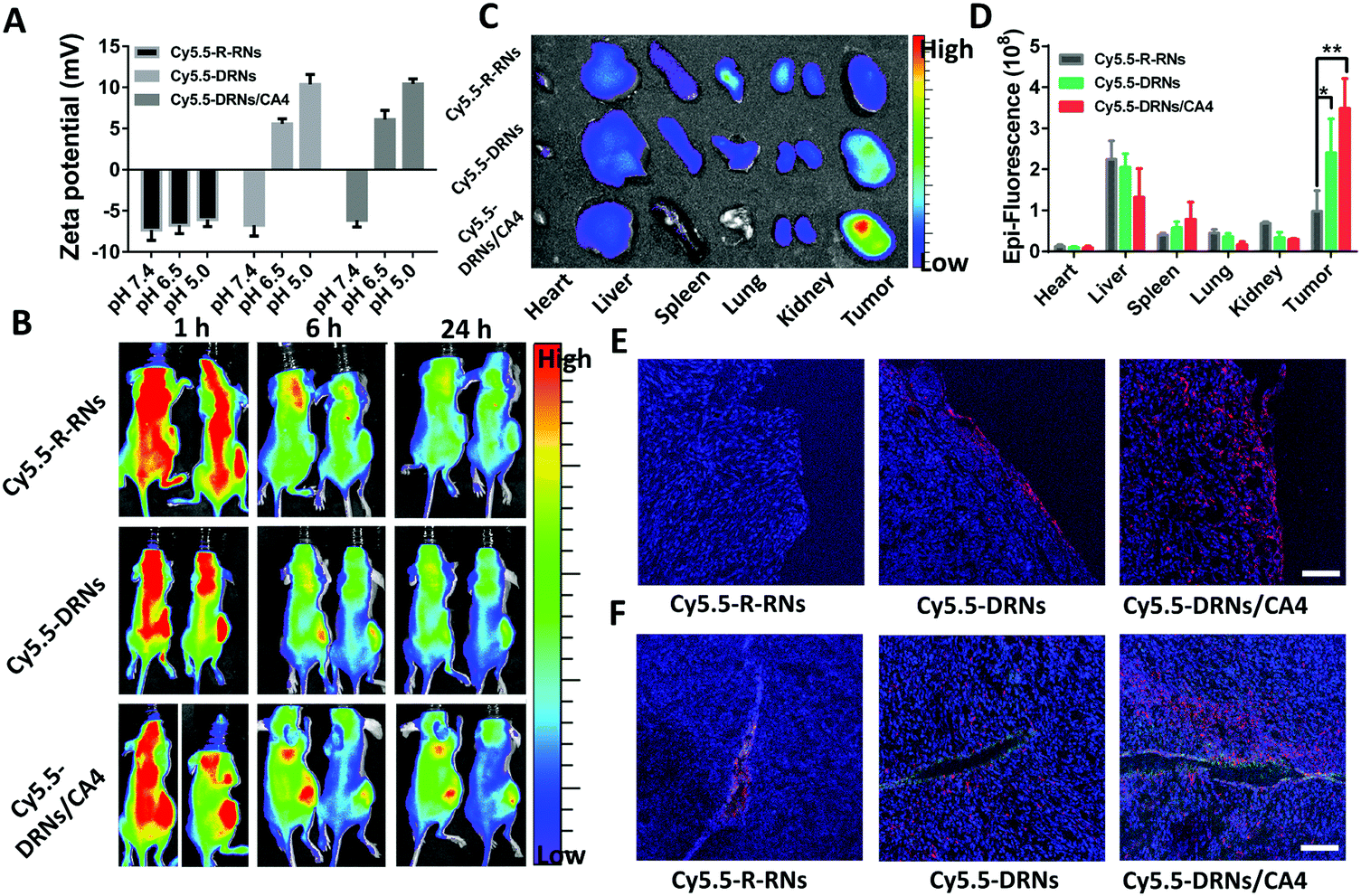
In vivo biodistribution and tumor accumulation
The animal studies were performed in accordance with the Regulations for the Administration of Affairs Concerning Experimental Animals (Tianjin, revised in June 2004) and adhered to the Guiding Principles in the Care and Use of Animals of the American Physiological Society. The BALB/c nude mice were inoculated with 4T1 cells in the left abdomen. 3 weeks later, these mice were randomly divided into three groups and intravenously injected with Cy5.5-labeled reduced-responsive nanocarriers (Cy5.5-R-RNs), Cy5.5-labeled pH-reduction dual responsive nanocarriers (Cy5.5-DRNs) and Cy5.5-labeled CA4-loaded pH-reduction dual responsive nanocarriers (Cy5.5-DRNs/CA4), when the tumors grew to 500 mm3. After three consecutive treatments with the three types of Cy5.5-labeled nanocarriers at days 1, 3 and 5 respectively, the mice were anesthetized and imaged under an in vivo imaging system at 1 h, 6 h, and 24 h post-administration at day 5. At the end (24 h), all mice were sacrificed for harvesting the major organs (heart, liver, spleen, lungs, and kidneys) and tumor. Ex vivo imaging was conducted by a Kodak IS in vivo FX imaging system.
In vivo tumor penetration
The tumors in “In vivo biodistribution and tumor accumulation” were collected, dehydrated by 30% sugar solution for 2 days, fixed with 4% formaldehyde solution for 2 days, and cut into 8 μm-thick sections, followed by staining with DAPI and anti-CD31-FITC, and observation via CLSM.
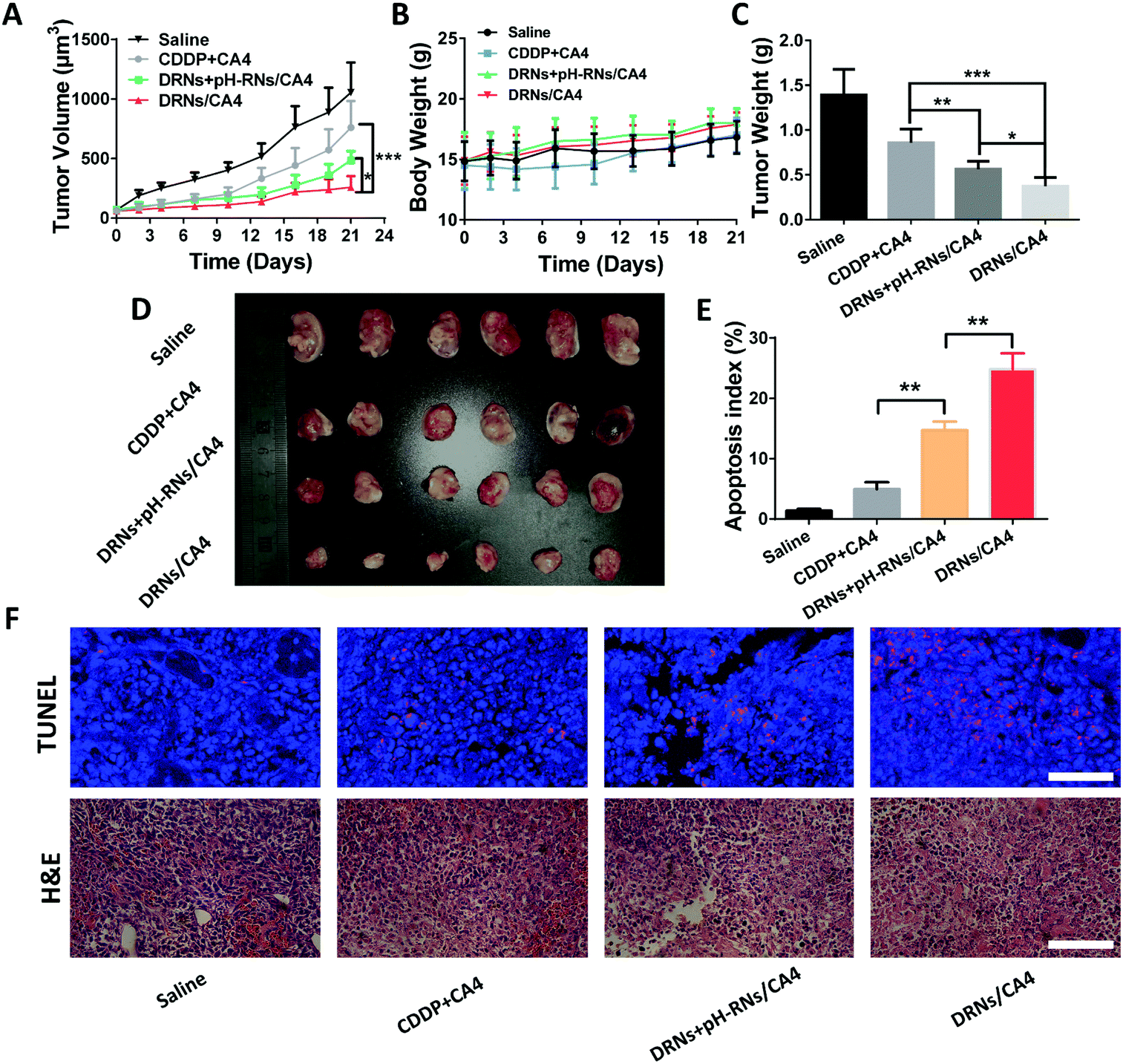
In vivo antitumor efficacy and combination therapy
The BALB/c nude mice were cultivated with 4T1 cancer cells. When the tumor grew to 100 mm3, those tumor-bearing mice were randomly divided into four groups. From day 1, the mice were treated with saline (control), free CA4 + free CDDP, pH-reduction dual responsive nanocarriers (DRNs) + CA4-loaded pH-responsive nanocarriers (pH-RNs/CA4) and CA4-loaded pH-reduction dual responsive nanocarriers (DRNs/CA4) (at days 1, 3, 5, 7 and 9). The doses of CDDP and CA4 were fixed at 5 mg kg−1 and 2 mg kg−1 body weight. Weight of the mice and tumor volume (tumor volume (mm3) = length × width2/2) were measured at a determined time for three weeks.
Hematoxylin/eosin (H&E) and immunohistochemical staining
At the end of the antitumor efficacy experiment, the tumors were collected and fixed in 4% paraformaldehyde for 48 h, embedded with paraffin and 8 μm-thick sections were prepared, followed by hematoxylin/eosin (H&E) and terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end (TUNEL) staining. The resulting sections were observed respectively via an optical microscope (Leica DMI6000 B) and CLSM.
Statistical analysis
Data were shown as means ± standard deviation (SD). Statistical analysis was operated by one-way ANOVA for multiple groups and Student's t-test for two groups. p < 0.05 was considered significant.
Results and discussion
Preparation and characterization of nanocarriers
To prepare nanocarriers, all block copolymers, including PAE-b-PEG, PAE-b-PLys, and PCL-b-PLys, had been synthesized in advance. The 1H NMR spectra of the products in each step are presented in Fig. S4–S7 (ESI†). The representative functional groups could be found in the spectra, suggesting that the products were synthesized successfully. As shown in Fig. S8 (ESI†), the DLS analysis indicated that the diameter of the DRNs was around 75 ± 10 nm, which was very consistent with the TEM observation that the obtained DRNs were spherical and the sizes were around 75 nm (Fig. S9, ESI†).
Evaluation of the stability and responsiveness of the DRNs via DLS and TEM
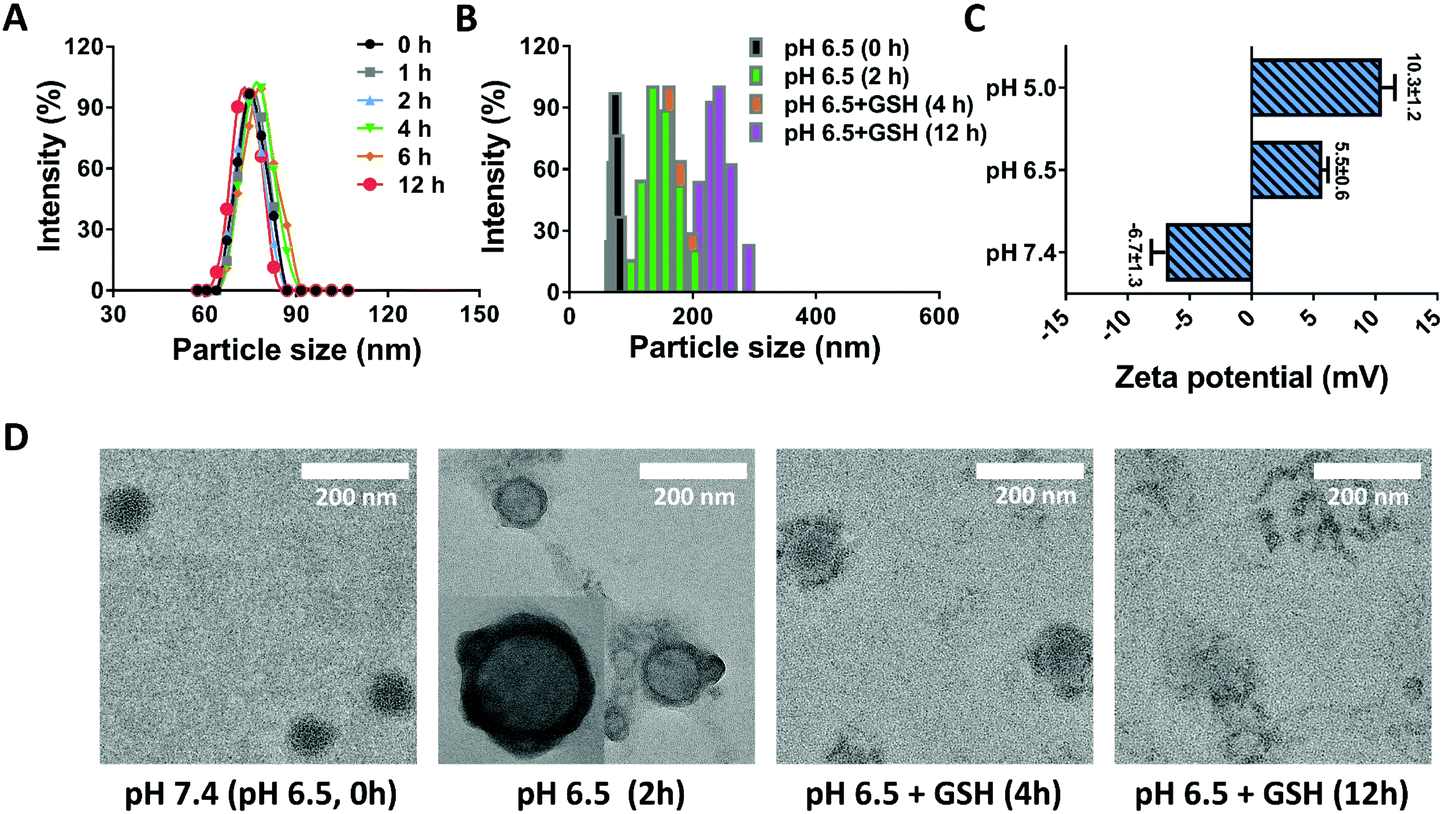
Stability is one of the critical aspects in ensuring the safety of nanomedicines, largely deciding the in vivo fate of nanoparticles.38 For the DRNs, the stabilization of PEG and crosslinking of CDDP would provide them with wonderful stability. To prove this hypothesis, we measured the size change of the DRNs at pH 7.4 over 12 h. As shown in Fig. 1A, the DRNs exhibited a negligible difference in hydrodynamic diameter and distribution in this time period, indicating an outstanding stability of the DRNs under physiological conditions. Once entering tumor tissues, the DRNs were expected to quickly respond to the extracellular acidic microenvironment and the intracellular reductive conditions for achieving programmable drug release. As depicted in Fig. 1B, the particle size increased from 75 ± 10 nm to 150 ± 20 nm with the decrease of pH from 7.4 to 6.5. When glutathione (GSH, 10 mM) was added, the particle size further increased to 170 ± 20 nm at 4 h and 230 ± 30 nm at 12 h, indicating a sensitive acidic and reductive responsiveness of the DRNs. In addition, zeta potential analysis was also employed to further investigate the DRNs’ pH-responsive ability. As shown in Fig. 1C, the DRNs were negatively charged (−6.7 ± 1.3 mV) at pH 7.4, and quickly transformed into positive charge (+5.5 ± 0.6 mV) at pH 6.5 and (+10.3 ± 1.2 mV) at pH 5.0 within 5 min, revealing a robust charge reversal property. Finally, TEM was employed to visually observe the change in DRNs’ structure. In the TEM images, the structure of the DRNs changed from a solid sphere into a hollow vesicle because the core of the DRNs swelled under acidic conditions resulting from the pH-sensitive PAE, which can change from the hydrophobic form into a hydrophilic status at low pH conditions. With the addition of GSH, the cross-linked shell was broken up slowly (4 h) and disassembled in the end (12 h) due to the redox of CDDP (Fig. 1D). These results further confirmed the outstanding pH-reduction dual responsiveness of the DRNs. With such property, the DRNs showed tremendous potential to achieve programmable releases of CDDP and other hydrophobic anticancer agents such as CA4.
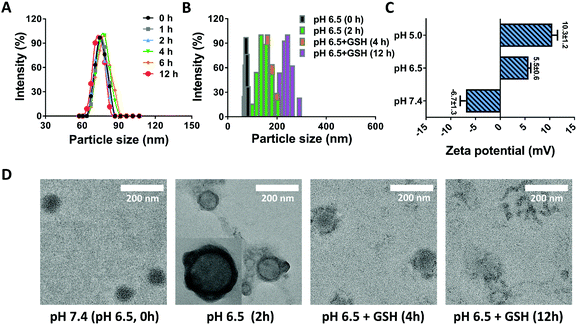 |
| | Fig. 1 The characterization of the DRNs. (A) The stability of the DRNs measured by DLS at pH 7.4. (B) The evaluation of the DRNs’ dual responsiveness via DLS at different conditions. (C) The evaluation of the DRNs’ acidic responsiveness using zeta potentials measured at pH 7.4, 6.5 and 5.0. (D) The TEM images of the DRNs under acidic and reductive conditions. | |
In vitro drug release
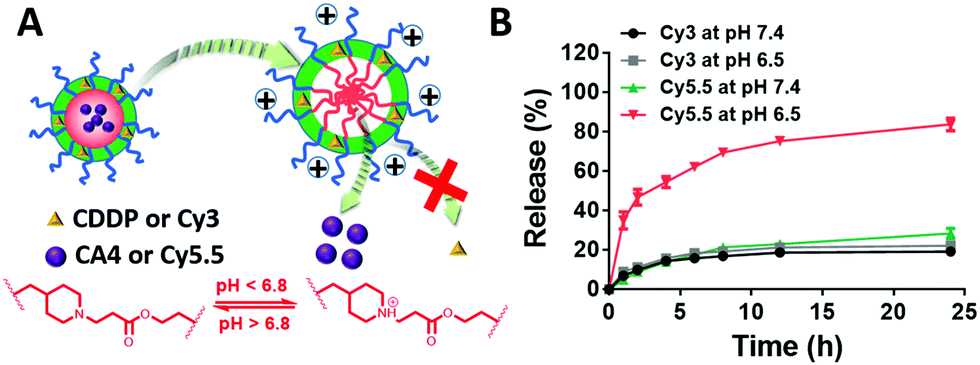
When loaded in the pH-responsive core of the DRNs, CA4 or other agents would be released quickly at acid environments while CDDP shows a lower release rate without reductant (Fig. 2A). To prove this hypothesis, we studied the drug-release behaviour of the DRNs/CA4 at different pH conditions. As shown in Fig. S10 (ESI†), very different release kinetics were observed in CA4, with less than 30% release at pH 7.4 and over 70% release at pH 6.5 after 24 h incubation. In contrast, similar release behaviours at pH 7.4 and 6.5, with less than 30% release, were exhibited in CDDP. Such an acid-promoted drug release is essential to facilitate an effective intracellular release of CA4 to disrupt the tumor vessels. In addition, the unreleased CDDP can be subsequently delivered into the cancer cells by the DRNs to induce cell apoptosis. For a better study and observation of the programmable release ability in cancer cells, we employed Cy3-labeled and Cy5.5-loaded DRNs (Cy3-DRNs/Cy5.5) to stand for DRNs/CA4. In this system, the CDDP and CA4 were respectively represented with Cy3 and Cy5.5. As depicted in Fig. 2B, the similar drug release kinetics with that of DRNs/CA4 further confirmed the wonderful programmable release capability of the DRNs, and also revealed that Cy3-DRNs/Cy5.5 can be used as a model of DRNs/CA4 for the next cell experiment.
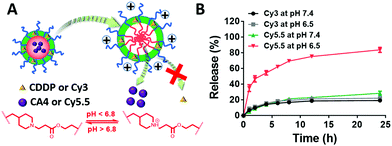 |
| | Fig. 2 The evaluation of the DRNs’ programmable drug release ability via in vitro release kinetics. (A) Illustration of the programmable release combination agents. (B) The release of Cy3 and Cy5.5, which is used to simulate the hydrophobic drug loaded in the core (such as CA4) and crosslinking CDDP, respectively. | |
In vitro programmable release
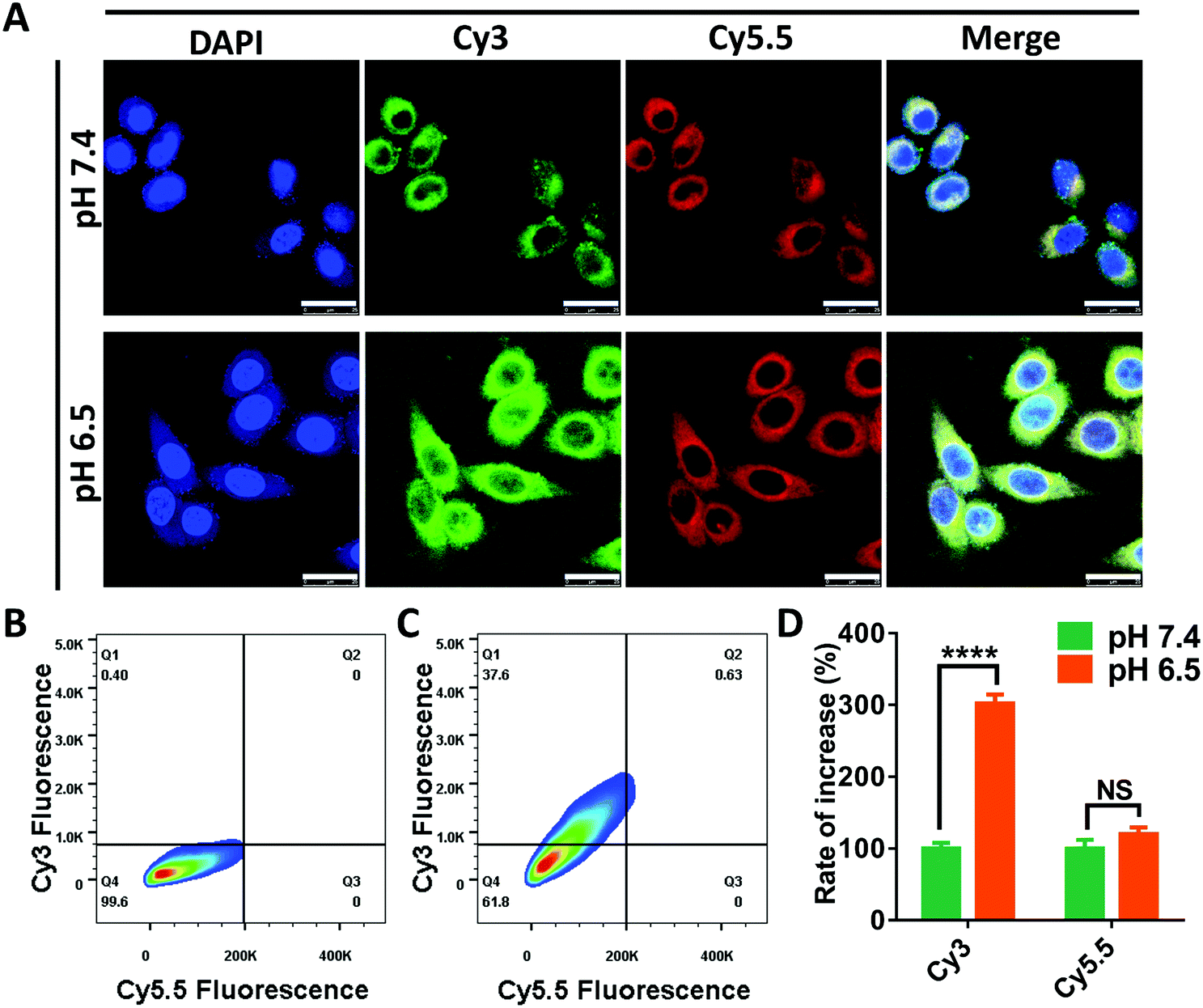
In the following research, we intended to investigate the programmable release ability of the DRNs on a cellular level. In the pilot experiments, we found that the Cy5.5 was more easily taken-up by the cancer cells when loaded in the DRNs compared with free Cy5.5 (Fig. S11 and S12, ESI†). Thus, the cell uptake of Cy5.5 may not significantly enhance when the pH changes from 7.4 to 6.5 because of the rapid release from the DRNs at the extracellular acidic conditions. To prove this hypothesis, we assessed the cellular uptake of Cy3 and Cy5.5 at different pH conditions. As shown in Fig. 3A, the Cy3 fluorescence signal was significantly enhanced with the decrease of pH from 7.4 to 6.5, while the signal of Cy5.5 showed negligible difference. Further quantification was confirmed using flow cytometry analysis (Fig. 2B–D). In agreement with the CLSM observation, the mean fluorescence of Cy3 for the pH 6.5 group enhanced 3 folds compared with that of the pH 7.4 group, while there was only 1.2 times increase for the mean fluorescence of Cy5.5. These results were attributed to the different release of Cy3 and Cy5.5. In acidic environments, the protonation of PAE provided the DRNs with a positive charge, improving the cell uptake of the DRNs as well as the conjugated Cy3. In contrast, because a great mass of Cy5.5 had been released from the DRNs at the extracellular acidic conditions, the positive DRNs showed less impact on the cellular uptake of Cy5.5. Altogether, these results confirmed that the DRNs had huge potential to achieve programmable release of combination agents (such as CA4 and CDDP) according to their temporal and spatial needs, providing them with a strong synergetic effect and a better treatment effect in cancer therapy.
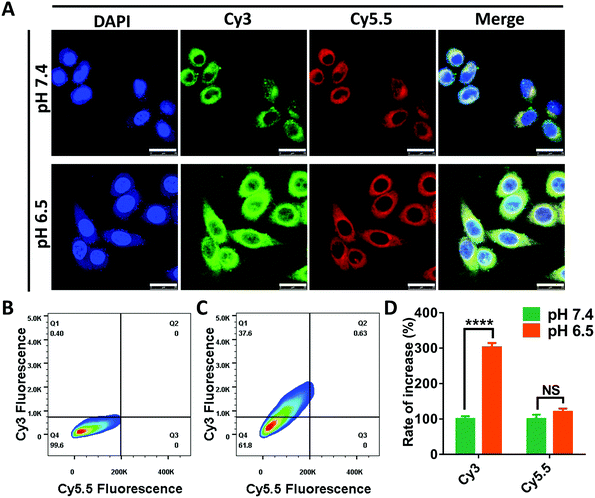 |
| | Fig. 3 The evaluation of DRNs’ programmable drug release ability via in vitro cellular uptake at different pH conditions. (A) The confocal microscopy images of HepG2 cells treated with Cy3-DRNs/Cy5.5 solution (0.1 mg mL−1) under different conditions (scale bar = 20 μm). (B and C) Flow cytometry of 4T1 cells cultured with Cy3-DRNs/Cy5.5 solution (0.1 mg mL−1) for 2 hours at pH 7.4 and pH 6.5, respectively. (D) The quantification analysis of Cy3 and Cy5.5 fluorescence intensity of the cells treated with Cy3-DRNs/Cy5.5 at pH 7.4 and 6.5. Error bars indicate s.d. (n = 3) t-test (NS > 0.05, P* < 0.05, P** < 0.01, P*** < 0.001, P**** < 0.0001). | |
Evaluation of the complementary effect of CA4 for tumor accumulation and penetration in vivo
The CA4, as one of the most common vascular disrupting agents, can shut down the blood vessels and facilitate the accumulation of nanoparticles in the tumor sites.24,39,40 In our system, the DRNs possibly show a higher tumor accumulation capability in comparison with traditional nanoparticles attributed to their charge reversal property.41,42 Moreover, we anticipated that the CA4 might further improve the accumulation effect of nanocarriers to enhance the drug delivery efficiency of CDDP. Thus, we constructed three systems, including the Cy5.5-labeled single reduced-responsive nanocarriers (Cy5.5-R-RNs), Cy5.5-labeled pH-reduction dual responsive nanocarriers (Cy5.5-DRNs) and Cy5.5-labeled CA4-loaded pH-reduction dual responsive nanocarriers (Cy5.5-DRNs/CA4), to certify this hypothesis in 4T1 tumor-bearing nude mice. The zeta potential analysis revealed an obvious change of surface charge in both Cy5.5-DRNs and Cy5.5-DRNs/CA4, but insignificant change in the Cy5.5-R-RNs group while the pH changes from 7.4 to 6.5 and 5.0 due to the different composition of the core (Fig. 4A). After the injection of the formulations three times, the in vivo and ex vivo imaging were monitored via a near-infrared optical imager. As one might expect, it was clearly seen that the pH-reduction dual responsive groups (including Cy5.5-DRNs and Cy5.5-DRNs/CA4) showed stronger red fluorescence signal in tumors compared to that of the single reduced-responsive group (Cy5.5-R-RNs), indicating that the pH-sensitive structure is beneficial to the tumor accumulation for nanocarriers. Furthermore, with the help of CA4, a higher accumulation efficiency was obtained in the Cy5.5-DRNs/CA4 group, implying that CA4 exhibited a wonderful complementary effect for DRNs’ tumor accumulation, because CA4 was capable of enhancing the retention time of the DRNs in the tumor sites (Fig. 4B and C). In addition, the quantification of the biodistributions also revealed more efficient accumulation in the Cy5.5-DRNs/CA4 group, with 3.6 folds versus Cy5.5-R-RNs treatment and 1.45 folds versus Cy5.5-DRNs administration respectively, further confirming the CA4's additive effect for the tumor accumulation of nanoparticles (Fig. 4D). In addition, the protonation of PAE triggered by the acidic tumor microenvironment provided the DRNs with positive charge, and theoretically might endow the DRNs with outstanding tumor penetration ability.42,43 Here, we also evaluated DRNs’ penetration capability via CLSM observations. As shown in Fig. 5E and F, a few red fluorescence emissions could already be observed in the surroundings of the blood vessels and tumor surface in the single reduced-responsive group (Cy5.5-R-RNs). In contrast, deep diffusions were shown in the pH-reduction dual responsive groups (Cy5.5-DRNs and Cy5.5-DRNs/CA4). Moreover, a stronger signal was found in the tumor treated with Cy5.5-DRNs/CA4 compared with Cy5.5-DRNs, further highlighting the importance of CA4 for more valid tumor accumulation. Therefore, with the help of this programmable controlled method, CA4 exhibited a wonderful complementary effect for the tumor accumulation as well as penetration of the DRNs, which could enhance the delivery efficiency of CDDP, achieving the synergistic effect for tumor therapy.
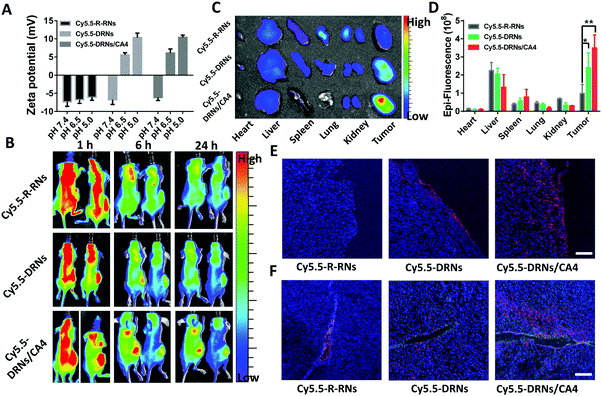 |
| | Fig. 4 The tumor accumulation of DRNs and the complementary effect of CA4 in tumor accumulation. (A) Zeta potentials of Cy5.5-R-RNs, Cy5.5-DRNs, and Cy5.5-DRNs/CA4 at pH 7.4, 6.5, and 5.0. (B) In vivo fluorescence imaging of 4T1 tumor-bearing nude mice at 1, 6, and 24 h post-injection after injection of three types of Cy5.5 labeled nanocarriers three times. (C) Ex vivo fluorescence imaging of the tumor and organs (heart, liver, spleen, lung, and kidney) harvested from the 4T1 tumor-bearing nude mice at 24 h post injection. (D) Quantitative analysis of fluorescent signals from the tissues and the tumor from the ex vivo fluorescence images. Error bar indicates s.d. (n = 3) t-test (P* < 0.05, P** < 0.01). (E and F) The confocal microscopy images of Cy5.5-R-RNs, Cy5.5-DRNs, and Cy5.5-DRNs/CA4 on the surface of the tumor and at the perivascular sites after penetrating into the tumor tissue for 24 h (blue: nucleus stained by DAPI; green: anti-CD31 FITC; red: Cy5.), (scale bar = 200 μm). | |
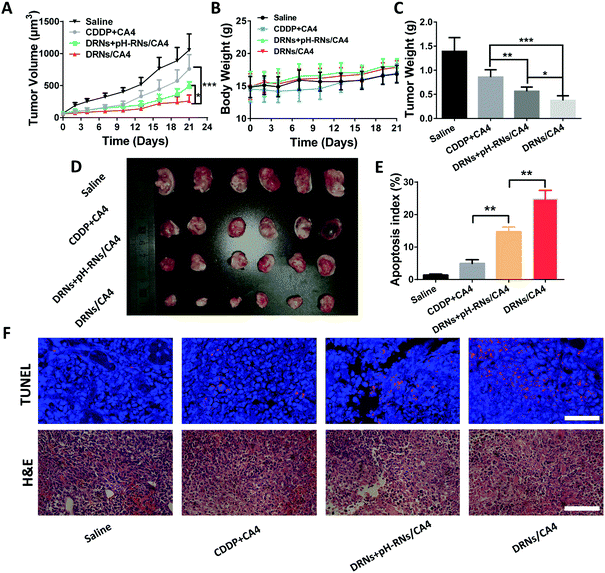 |
| | Fig. 5 The in vivo antitumor efficacy and combination therapy of CA4 and CDDP. (A) The 4T1 tumor growth curves after injection with saline, CA4 + CDDP, DRNs + pH-RNs/CA4, and DRNs/CA4 at a dose of 5 mg (CDDP) kg−1 and 2 mg (CA4) kg−1 body weight, respectively. Error bars indicate s.d. (n = 10) t-test (P* < 0.05, P** <0.01, P*** < 0.001). (B) Relative body weight during treatment for 21 days. Error bars indicate s.d. (n = 10). (C) Tumor weight at day 21. Error bars indicate s.d. (n = 10) t-test (P* < 0.05, P** <0.01, P*** < 0.001). (D) Images of the tumor at day 21. (E) Quantification of the apoptotic cells in tumor tissue after the treatment, obtained through terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end (TUNEL) staining. Error bars indicate s.d. (n = 5) t-test (P* < 0.05, P** <0.01). (F) Histological analysis of tumor sections stained with H&E (scale bar = 100 μm) and TUNEL (scale bar = 50 μm). | |
In vivo antitumor efficacy and combination therapy
The splendid programmable release ability and the additive effect of CA4 in tumor accumulation provided us with a strong rationale to evaluate the antitumor activity of the DRNs/CA4 in vivo. In this study, the mice were administrated with saline, CA4 + CDDP, DRNs + pH-RNs/CA4, and DRNs/CA4, and then the tumor volumes and body weights were recorded for 21 days. As depicted in Fig. 5A and D, the tumor volume increased rapidly for the free drugs group (CDDP + CA4), with only approximately 27.9% inhibition rate versus that of saline treatment. In contrast, the DRNs + pH-RNs/CA4 and DRNs/CA4 groups exhibited significant tumor growth suppression, with 53.8% and 75.4% tumor suppression rates (TSRs), respectively. More importantly, the most effective inhibition of the tumor growth was observed from the mice treated with DRNs/CA4, implying that a synergetic effect could be acquired when CA4 and CDDP were simultaneously loaded in a single delivery system with a programmable release ability. Meanwhile, we measured the weight of tumor mass in each group (Fig. 5C). The DRNs/CA4 group showed the smallest tumor weight, further confirming the better therapeutic effect of this co-delivery system with spatiotemporal release properties. In addition, H&E and TUNEL staining results suggested that the DRNs/CA4 group considerably enhanced necrosis and apoptosis of the cancer cells compared with other groups, further proving the synergetic effect of our designed system for enhanced antitumor therapy.
Safety assessment of the DRNs/CA4
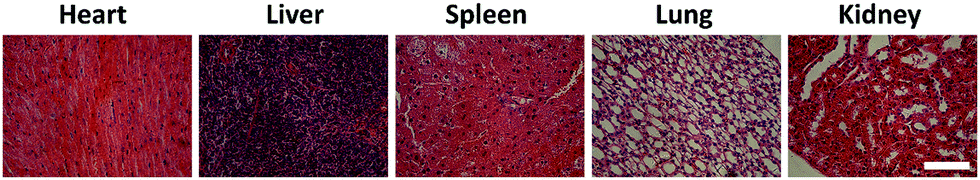
Safety is one of the most critical requirements for nanomedicine.44 Thus, we assessed the in vivo safety of the DRNs/CA4. As shown in Fig. 5B, the treatment of free CA4 + CDDP resulted in obvious body weight loss, while the mice treated with DRNs/CA4 had no body weight loss compared to the saline group, indicating that our designed system possessed good biocompatibility and little adverse reaction in vivo. In addition, we also investigated their in vivo toxicity by histological assessment (Fig. 6). As expected, a clear and compact structure of cells was observed in all organs, further confirming the negligible toxicity of the DRNs/CA4.
 |
| | Fig. 6 H&E staining of the sections of major organs treated with the DRNs/CA4 (heart, lung, liver, spleen, and kidney). Tissue paraffin sections which were 8 μm thick stained with standard hematoxylin and eosin (H&E) and examined with an optical microscope (scale bar = 100 μm). | |
Conclusions
In summary, we have developed a pH-reduction dual responsive drug delivery system for programmable release of CA4 and CDDP according to their spatial and temporal needs. By responding to the acidic tumor microenvironment, such nanocarriers were capable of selectively releasing CA4 in the perivascular sites of tumor tissue to disrupt the blood vessels, then accumulate and further penetrate into the tumor tissues. When taken-up by cancer cells, the intercellular reductive conditions triggered the release of CDDP to promote cancer cell apoptosis. With this programmable release capacity, our designed system was able to carry out precise delivery of the combination agents according to their spatiotemporal needs, further improving the therapeutic effect of combination therapy. In this system, the CA4 can not only achieve a complementary effect for nanocarrier tumor accumulation to improve the delivery efficiency of CDDP but also enhance the synergistic effect of CA4 and CDDP by accounting for their respective spatial and temporal needs, giving rise to a better therapeutic effect in cancer therapy. More importantly, such programmable release strategy is accomplished by the pH-reduction dual response of nanocarriers, so that it is also appropriate for the delivery of other combination agents, providing a universal platform to achieve high-efficiency combination therapy.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 51390483, 91527306, 21620102005 and 51603231), the National Key Research and Development Programs of China (2018YFA0209700), the Thousand Talents Program for Young Professionals, PCSIRT (IRT1257), the Fundamental Research Funds for the Central Universities, the National Natural Science Foundation for Excellent Young Scholars of China (81722026), the CAMS Innovation Fund for Medical Sciences (CIFMS, 2018-I2M-HL-014), and the CAS Interdisciplinary Innovation Team.
Notes and references
- W. Chen, R. Zheng, P. D. Baade, S. Zhang, H. Zeng, F. Bray, A. Jemal, X. Q. Yu and J. He, Ca-Cancer J. Clin., 2016, 66, 115–132 CrossRef PubMed
 .
.
- D. L. Stirland, J. W. Nichols, S. Miura and Y. H. Bae, J. Controlled Release, 2013, 172, 1045–1064 CrossRef CAS PubMed .
- W. Dai, X. Wang, G. Song, T. Liu, B. He, H. Zhang, X. Wang and Q. Zhang, Adv. Drug Delivery Rev., 2017, 115, 23–45 CrossRef CAS PubMed .
- S. Sengupta, D. Eavarone, I. Capila, G. Zhao, N. Watson, T. Kiziltepe and R. Sasisekharan, Nature, 2005, 436, 568–572 CrossRef CAS PubMed .
- X. Xu, W. Ho, X. Zhang, N. Bertrand and O. Farokhzad, Trends Mol. Med., 2015, 21, 223–232 CrossRef CAS PubMed .
- N. Kolishetti, S. Dhar, P. M. Valencia, L. Q. Lin, R. Karnik, S. J. Lippard, R. Langer and O. C. Farokhzad, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17939–17944 CrossRef CAS PubMed .
- D. B. Pacardo, F. S. Ligler and Z. Gu, Nanoscale, 2015, 7, 3381–3391 RSC .
- Q. Hu, W. Sun, C. Wang and Z. Gu, Adv. Drug Delivery Rev., 2016, 98, 19–34 CrossRef CAS PubMed .
- P. Huang, D. Wang, Y. Su, W. Huang, Y. Zhou, D. Cui, X. Zhu and D. Yan, J. Am. Chem. Soc., 2014, 136, 11748–11756 CrossRef CAS PubMed .
- D. Lane, Nat. Biotechnol., 2006, 24, 163–164 CrossRef CAS PubMed .
- D. Peer, J. M. Karp, S. Hong, O. C. FaroKHzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed .
- J. Chen, Z. Guo, L. Lin, Y. Hu, H. Tian, M. Chen and X. Chen, Mater. Chem. Front., 2017, 1, 937–946 RSC .
- T. A. Yap, A. Omlin and J. S. de Bono, J. Clin. Oncol., 2013, 31, 1592–1605 CrossRef CAS PubMed .
- H. P. Gerber and N. Ferrara, Cancer Res., 2005, 65, 671–680 CAS .
- L. Miao, S. Guo, C. M. Lin, Q. Liu and L. Huang, Adv. Drug Delivery Rev., 2017, 115, 3–22 CrossRef CAS PubMed .
- J. Jia, F. Zhu, X. Ma, Z. Cao, Z. W. Cao, Y. Li, Y. X. Li and Y. Z. Chen, Nat. Rev. Drug Discovery, 2009, 8, 111–128 CrossRef CAS PubMed .
- G. Shim, M. G. Kim, D. Kim, J. Y. Park and Y. K. Oh, Adv. Drug Delivery Rev., 2017, 115, 57–81 CrossRef CAS PubMed .
- J. S. Lopez and U. Banerji, Nat. Rev. Clin. Oncol., 2017, 14, 57–66 CrossRef CAS PubMed .
- W. Dai, W. Jin, J. Zhang, X. Wang, J. Wang, X. Zhang, Y. Wan and Q. Zhang, Pharm. Res., 2012, 29, 2902–2911 CrossRef CAS PubMed .
- D. Chen, G. Zhang, R. Li, M. Guan, X. Wang, T. Zou, Y. Zhang, C. Wang, C. Shu, H. Hong and L. J. Wan, J. Am. Chem. Soc., 2018, 140, 7373–7376 CrossRef CAS PubMed .
- H. Wang, H. Xie, J. Wu, X. Wei, L. Zhou, X. Xu and S. Zheng, Angew. Chem., Int. Ed., 2014, 53, 11532–11537 CrossRef CAS PubMed .
- R. S. Kerbel and B. A. Kamen, Nat. Rev. Cancer, 2004, 4, 423–436 CrossRef CAS PubMed .
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed .
- S. Lv, Z. Tang, W. Song, D. Zhang, M. Li, H. Liu, J. Cheng, W. Zhong and X. Chen, Small, 2017, 13, 1600954 CrossRef PubMed .
- J. Chen, J. Ding, Y. Wang, J. Cheng, S. Ji, X. Zhuang and X. Chen, Adv. Mater., 2017, 29, 1701170 CrossRef PubMed .
- F. Fan, S. Gao, S. Ji, Y. Fu, P. Zhang and H. Xu, Mater. Chem. Front., 2018, 2, 2109–2115 RSC .
- L. Niu, F. Zhu, B. Li, L. Zhao, H. Liang, Y. Yan and H. Tan, Mater. Chem. Front., 2018, 2, 1529–1538 RSC .
- M. Kanamala, W. R. Wilson, M. Yang, B. D. Palmer and Z. Wu, Biomaterials, 2016, 85, 152–167 CrossRef CAS PubMed .
- Y. Miura, T. Takenaka, K. Toh, S. Wu, H. Nishihara, M. R. Kano, Y. Ino, T. Nomoto, Y. Matsumoto, H. Koyama, H. Cabral, N. Nishiyama and K. Kataoka, ACS Nano, 2013, 7, 8583–8592 CrossRef CAS PubMed .
- P. a. Ma, H. Xiao, X. Li, C. Li, Y. Dai, Z. Cheng, X. Jing and J. Lin, Adv. Mater., 2013, 25, 4898–4905 CrossRef CAS PubMed .
- H. Cabral, M. Murakami, H. Hojo, Y. Terada, M. R. Kano, U.-i. Chung, N. Nishiyama and K. Kataoka, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11397–11402 CrossRef CAS PubMed .
- M. X. Zhao, B. L. Hu, Z. Gu, K. I. Joo, P. Wang and Y. Tang, Nano Today, 2013, 8, 11–20 CrossRef CAS .
- X. Zhang, L. Han, M. Liu, K. Wang, L. Tao, Q. Wan and Y. Wei, Mater. Chem. Front., 2017, 1, 807–822 RSC .
- S. Feng, Z. X. Wu, Z. Zhao, J. Liu, K. Sun, C. Guo, H. Wang and Z. Wu, ACS Appl. Mater. Interfaces, 2019, 11, 7357–7368 CrossRef CAS PubMed .
- H. Ou, T. Cheng, Y. Zhang, J. Liu, Y. Ding, J. Zhen, W. Shen, Y. Xu, W. Yang, P. Niu, J. Liu, Y. An, Y. Liu and L. Shi, Acta Biomater., 2018, 65, 339–348 CrossRef CAS PubMed .
- Y. Liu, H. J. Busscher, B. R. Zhao, Y. F. Li, Z. K. Zhang, H. C. van der Mei, Y. J. Ren and L. Q. Shi, ACS Nano, 2016, 10, 4779–4789 CrossRef CAS PubMed .
- H. J. Gao, T. J. Cheng, J. F. Liu, J. J. Liu, C. H. Yang, L. P. Chu, Y. M. Zhang, R. J. Ma and L. Q. Shi, Biomacromolecules, 2014, 15, 3634–3642 CrossRef CAS PubMed .
- L. Wu, J. Zhang and W. Watanabe, Adv. Drug Delivery Rev., 2011, 63, 456–469 CrossRef CAS PubMed .
- R. R. Nani, A. P. Gorka, T. Nagaya, H. Kobayashi and M. J. Schnermann, Angew. Chem., Int. Ed., 2015, 54, 13635–13638 CrossRef CAS PubMed .
- W. T. Song, Z. H. Tang, D. W. Zhang, M. Q. Li, J. K. Gu and X. S. Chen, Chem. Sci., 2016, 7, 728–736 RSC .
- T. Cheng, Y. Zhang, J. Liu, Y. Ding, H. Ou, F. Huang, Y. An, Y. Liu, J. Liu and L. Shi, ACS Appl. Mater. Interfaces, 2018, 10, 5296–5304 CrossRef CAS PubMed .
- Y. Ding, J. Liu, Y. Zhang, X. Li, H. Ou, T. Cheng, L. Ma, Y. An, J. Liu, F. Huang, Y. Liu and L. Shi, Nanoscale Horiz., 2019, 4, 658–666 RSC .
- H. J. Li, J. Z. Du, J. Liu, X. J. Du, S. Shen, Y. H. Zhu, X. Wang, X. Ye, S. Nie and J. Wang, ACS Nano, 2016, 10, 6753–6761 CrossRef CAS PubMed .
- O. Veiseh, B. C. Tang, K. A. Whitehead, D. G. Anderson and R. Langer, Nat. Rev. Drug Discovery, 2015, 14, 45–57 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qm00178f |
|
| This journal is © the Partner Organisations 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Xue
Li
a,
Linlin
Xu
a,
Chang
Li
a,
Lin
Ma
b,
Jianfeng
Liu
b,
Rujiang
Ma
b,
Xue
Li
a,
Linlin
Xu
a,
Chang
Li
a,
Lin
Ma
b,
Jianfeng
Liu
b,
Rujiang
Ma