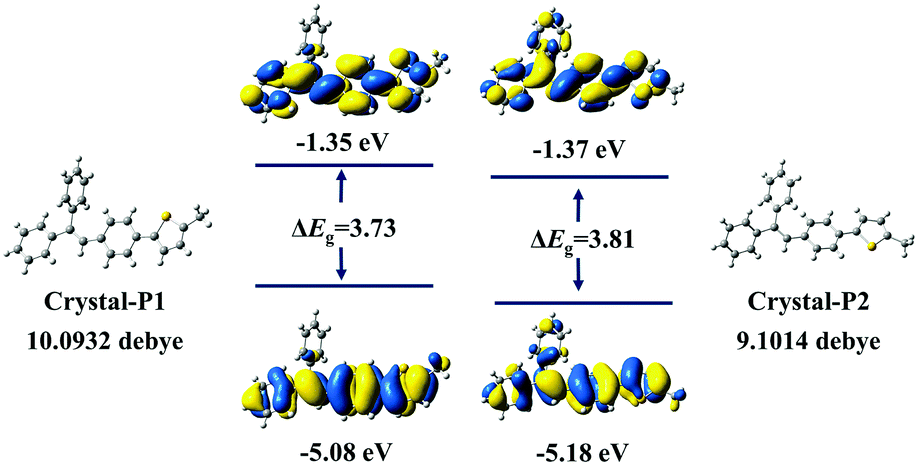DOI:
10.1039/C8QM00411K
(Research Article)
Mater. Chem. Front., 2019,
3, 32-38
Recyclable mechanoluminescent luminogen: different polymorphs, different self-assembly effects of the thiophene moiety and recovered molecular packing via simple thermal-treatment†
Received
18th August 2018
, Accepted 7th September 2018
First published on 7th September 2018
Abstract
We report a purely organic recyclable mechanoluminescent luminogen (tPE-5-MeTh) with the combination of an AIEgen and a thiophene group as the self-assembly unit. As a result of the different self-assembly effects, two crystalline polymorphs were cultured from different solvents, which possess totally different packing modes and various inter/intramolecular interactions with opposite ML properties. Excitingly, the molecular packing status of tPE-5-MeTh could be adjusted through simple thermal-treatment, offering recoverability and controllability of its ML activity. These results provide a new avenue for rational molecular design, convenience of potential applications and deep understanding of the inherent mechanism.
1 Introduction
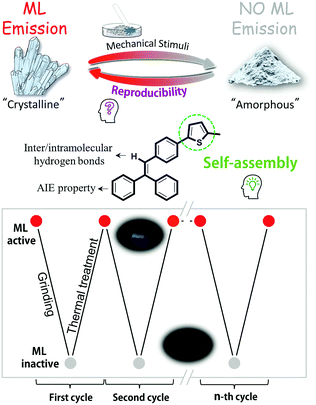
Mechanoluminescence (ML), also named as triboluminescence, is a special light-emission derived from mechanical stimuli in the solid state, frequently in the crystalline state, with huge potential in displays, lighting, bioimaging and stress-sensing applications.1 Although ML was observed from sugar by Francis Bacon earlier in 1605, further development of pure organic ML luminogens remained silent for a very long time, mainly as a result of weak ML signals, intangible inherent mechanisms, and lack of rational molecular design.2 In 2015, the marriage of the ML process and the well-known aggregation induce emission (AIE) offered a new feeling for the molecular design of bright ML luminogens, providing opportunities to explore the mysterious ML excitation process.3a We reported the first example of an AIEgen with fluorescence–phosphorescence dual mechanoluminescence and purely organic ML luminogens with room temperature phosphorescence in 2016 and 2017, respectively, to understand the inherent mechanisms deeply.3 Coupled with excellent studies of other scientists, the ML process is considered to be highly related to the molecular packing in the solid state, and generally occurs accompanying the fracture of crystals upon mechanical stimuli. Accordingly, some more bright ML luminogens have been developed recently.4 However, under continuous stimuli, ML signals become weak and even disappear, in response to the change of luminogens from the crystalline state to an amorphous one, meaning that they can be utilized only once before possible recovery using a tedious dissolution-evaporation process (Chart 1).5 To facilitate convenient practical applications and explore the possible adjustments of the molecular packing of ML luminogens through simple approaches, some new design ideas should be proposed and straightforward methods should be found.
 |
| | Chart 1 The new design of the mechanoluminescence material and chemical structure of tPE-5-MeTh with a diagram of its recyclable utilization. | |
Supramolecular self-assembly is the formation of multimolecular groups held together spontaneously by weak interactions of non-covalent bonds, and has attracted much attention as a hot research topic, in which the key point is that molecules could be rearranged when breaking the thermodynamic equilibrium.6 Thus, under some special stimuli, the molecular status could change from one state to another reversibly. Inspired by this fact, we wondered if the ML property mentioned above could be achieved by utilizing the supramolecular self-assembling effect. Accordingly, the thiophene moiety, frequently used as the building block in semiconductors, organogels and polymer solar cells due to its assembling ability with other groups, was chosen for construction of our dream ML luminogens with recyclable mechanoluminescence (Chart S1, ESI†).7 Thus, also considering the assurance of the AIE characteristic8 and the possible formation of intermolecular/intramolecular hydrogen bonds, tPE-5-MeTh, with the structure shown in Chart 1, was rationally designed, to explore the ML reversibility and possibly the deep inherent mechanism. Interestingly, two crystalline polymorphs of tPE-5-MeTh were cultured from different mixture solvents, which possessed totally different molecular packing partially due to the different assembling behavior of the thiophene moieties. Correspondingly, Crystal-P1 with the P1 space group (CCDC 1846930†) having an extremely ordered spatial packing mode, demonstrates an efficient ML property, while Crystal-P2 with the space group of P212121 (CCDC 1846929†) is ML inactive because of the antiparallel packing mode. Excitingly, the crystalline state of tPE-5-MeTh could be recovered selectively from the amorphous state reversibly by simple thermal-treatment at relatively low temperature. To the best our knowledge, this is the first example of purely organic recyclable mechanoluminescence without melting the solid, which will provide a new avenue for rational molecular design and a deep understanding of the inherent mechanism.
2 Experimental
2.1 Synthesis
The synthetic route to tPE-5-MeTh is illustrated in Scheme S1 (ESI†). tPE-5-MeTh was conveniently synthesized by a typical Wittig and Suzuki reaction, and purified by a silica gel column followed by recrystallization. They were all characterized by mass spectrometry, elemental analysis and 1H/13C NMR spectroscopy.
Synthesis of compound 1.
4-Bromobenzyl bromide (5.00 g, 20 mmol) and a solution of triethyl phosphite (10.34 mL, 60 mmol) were added into a 150 mL flask. The mixture was heated at 150 °C for 24 hours under an atmosphere of nitrogen, and then cooled to room temperature. After removing excess triethyl phosphite by vacuum distillation, the product was obtained in 100% yield and used directly in the next step.
Synthesis of compound tPE-Br.
To a two-necked flask was added the mixture of compound 1 (3.07 g, 10 mmol) and potassium tert-butoxide (1.12 g, 10 mmol) with freshly distilled THF (100 mL). After the addition of the benzophenone (2.00 g, 11 mmol) solution in THF (50 mL), the reaction was stirred for 2 hours at 0 °C under an atmosphere of nitrogen. After warming to room temperature, the reaction was stirred for another 2 hours. After removing the solvent with a rotary evaporator, the reaction mixture was purified by a silica gel column using petroleum ether as an eluent, to give a white solid (3.08 g, 91.8%). 1H NMR (400 MHz, CDCl3, δ): 7.35–7.28 (m, 8H), 7.25 (m, 1H), 7.23 (m, 1H), 7.19–7.16 (m, 2H), 6.88 (m, 2H), 6.86 (m, 1H). 13C NMR (100 MHz, CDCl3, δ): 143.40, 143.03, 139.90, 136.29, 131.07, 131.04, 130.24, 128.75, 128.24, 127.73, 127.63, 127.57, 126.77, 120.53. MS (EI), m/z: 334.62 ([M+], calcd for C20H15Br, 334.04).
Synthesis of compound tPE-5-MeTh.
Into the mixture of tPE-Br (1.01 g, 3 mmol), 5-methylthiophene-2-boronic acid pinacol ester (0.67 g, 3.3 mmol), Pd(PPh3)4 (0.21 g, 0.12 mmol) and K2CO3 (2.49 g, 18 mmol), water (9 mL) and THF (27 mL) were added under an atmosphere of nitrogen. The reaction mixture was stirred at 75 °C for 24 hours. After cooling to room temperature, the resultant reaction mixture was washed with brine and extracted with CH2Cl2 three times. The organic layer was collected and combined. After removing the solvent with a rotary evaporator, the solid was purified by a silica gel column using petroleum ether/CH2Cl2 (v/v = 19/1) as an eluent, to give a pale yellow solid (0.79 g, 74.6%). 1H NMR (400 MHz, CDCl3, δ): 7.38–7.27 (m, 10H), 7.25–7.21 (m, 2H), 7.05 (m, 1H), 6.99 (m, 2H), 6.95 (s, 1H), 6.68 (m, 1H), 2.48 (m, 3H). 13C NMR (100 MHz, CDCl3, δ): 143.35, 142.40, 141.68, 140.39, 139.49, 136.06, 132.93, 130.34, 129.95, 128.74, 128.20, 127.65, 127.55, 127.50, 127.48, 126.22, 124.83, 122.76, 15.50. MS (EI), m/z: 352.00 ([M+], calcd for C25H20S, 352.13). Anal. calcd for C25H20S: C, 85.19; H, 5.72; S, 9.10 found: C, 85.09; H, 5.76; S, 9.04.
2.2 Materials and apparatus
1H NMR and 13C NMR spectra were recorded on a 400 MHz Bruker Advance III spectrometer, using CDCl3 as a solvent and tetramethylsilane (TMS) as the internal standard (δ = 0.00 ppm). Mass spectra were measured on a ZAB 3F-HF mass spectrophotometer. Elemental analyses of carbon, hydrogen and sulfur were performed on a Perkin-Elmer microanalyzer. Photoluminescence spectra were recorded on a Hitachi F-4600 fluorescence spectrophotometer. The UV-vis spectrum was measured on a Shimadzu UV-2550 spectrophotometer. Absolute photoluminescence quantum yield (PLQY) and fluorescence decay were recorded on an Edinburgh FLS980 fluorescencespectrophotometer. The ML spectrum was measured on an Acton SP2750 spectrometer with CCD (SPEC-10, Princeton) as a power detector. The single-crystal X-ray diffraction data were recorded on a Bruker Smart Apex II CCD diffractometer. The powder X-ray diffraction patterns were measured on a Rigaku MiniFlex 600 diffractometer. The Gaussian 09 program was utilized to perform the TD-DFT calculations. Differential scanning calorimetry was performed on a NETZSCHDSC 200 PC instrument. Thermal-treatment was performed using the model of IKA, C-MAG HP 4.
3 Results and discussion
3.1 Optical properties
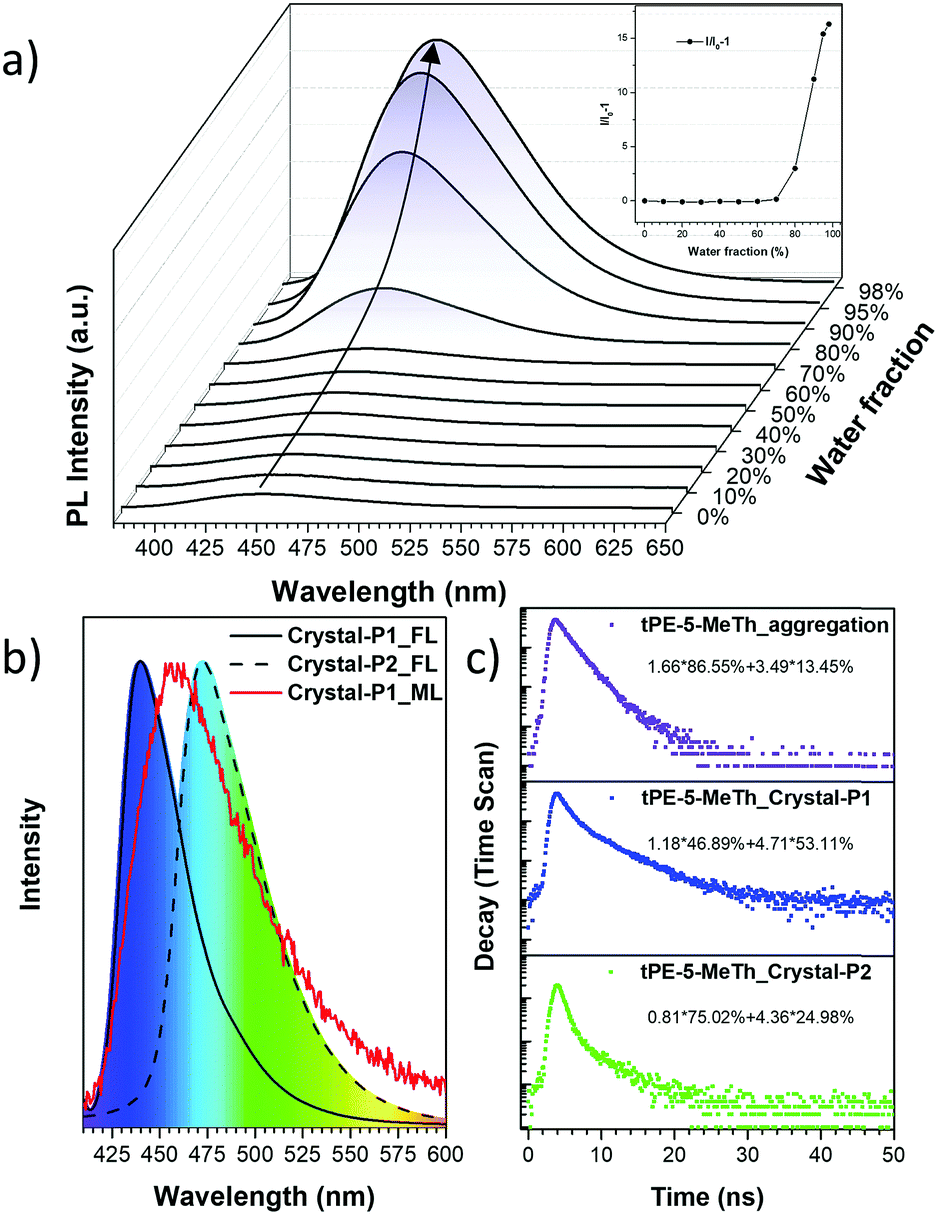
The UV-Visible absorption spectrum was measured in a dilute tetrahydrofuran (THF) solution (10−5 M) with the maximum absorption peak at 346 nm, which should be ascribed to the π–π* electron orbital transition of the conjugated system (Fig. S1a, ESI†). tPE-MeTh exhibited nearly no emission in the THF solution with a PLQY of only 0.38%. However, with an increase in the water fraction (fw) from 0 to 98%, the emission was enhanced 16 times due to gradual molecular aggregation (Fig. 1a). And its PLQY could be as high as 61.46% in the solid state, laying a foundation for its efficient ML emission.9
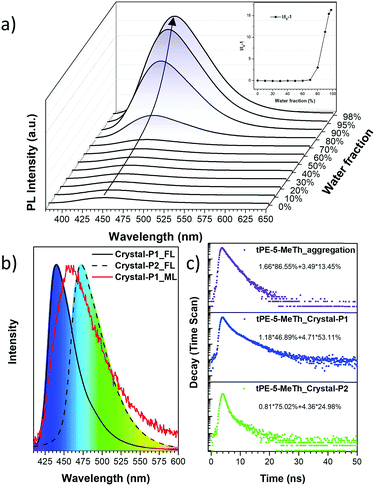 |
| | Fig. 1 (a) PL spectra and relative PL intensities (I/I0 − 1) of tPE-5-MeTh in THF/water mixtures with different water fractions (fw). (b) Normalized PL spectra of Crystal-P1 (line) and Crystal-P2 (dash) and Normalized ML spectrum of Crystal-P1 (red). (c) Emission decay of tPE-5-MeTh in the solid state and different crystals. (I0: the max. emission intensity at 0% water fraction. Excitation wavelength: 343 nm (solution) and 376 nm (aggregation). Concentration: 10−5 M.). | |
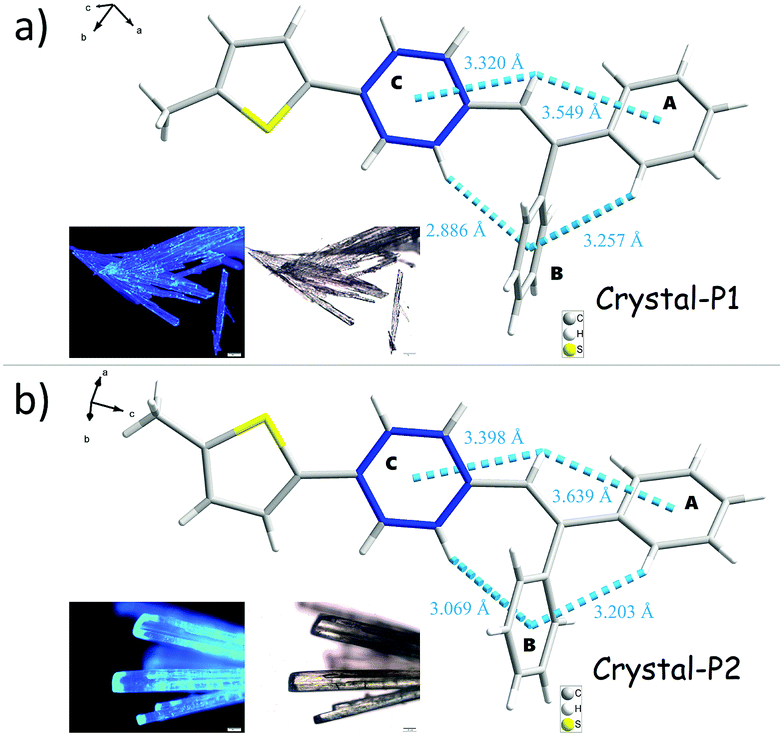
Two crystalline polymorphs of tPE-5-MeTh were cultured by the evaporation of the mixed organic solvents. There were several different conditions chosen to discuss the more effective methods to prepare the polymorphs. As shown in Table S1 (ESI†), Crystal-P2 was always present during the recrystallization in n-hexane, acetonitrile or dimethylsulfoxide and so on. Using the same conditions of temperature or solvent as contrast, several groups with different evaporation rates have been selected to prove that the packing mode of Crystal-P1 needs more time/energy to be cultivated. As shown in Fig. 2, Crystal-P2 has a bigger dimension ratio than Crystal-P1, and could be easily differentiated with the naked eye due to the distinguishable fluorescence under UV illumination. The maximum emission wavelength of Crystal-P2 is centered at 472 nm (ΦF = 25.50%, τ = 1.700 ns), close to the wavelength of the as-prepared powder, while Crystal-P1 exhibits a blue-shift to 440 nm (ΦF = 33.30%, τ = 3.055 ns) (Table 1). Similar to those reported in the literature,9c the ΦF of the amorphous powder is much higher than those of Crystal-P1 and Crystal-P2 (61.46%), disclosing the much different molecular packing status in different solid states. Excitingly, the ML spectrum of tPE-5-MeTh is peaked at 453 nm with a strong signal due to the combination of the AIEgen and crucial self-assembly unit.
 |
| | Fig. 2 The molecular geometries of a single molecule in different crystal structures and their morphology images taken using a Leica M123 optical microscope. | |
Table 1 Optical properties of tPE-5-MeTh
|
|
λ
absmax (nm) |
λ
emmax (nm) |
τ (ns) |
k
r (108 s−1) |
k
nr (108 s−1) |
Φ
F
(%) |
|
Φ
F = photoluminescence quantum yield (PLQY).
Measured in a dilute THF solution, 10−5 M.
Measured in the aggregation state.
|
|
tPE-5-MeTh
|
346b/376c |
452b/468c |
1.906 |
3.22 |
2.02 |
0.38b/61.46c |
|
Crystal-P1
|
— |
440 |
3.055 |
1.09 |
2.18 |
33.30 |
|
Crystal-P2
|
— |
473 |
1.700 |
1.50 |
4.38 |
25.50 |
3.2 Crystallography
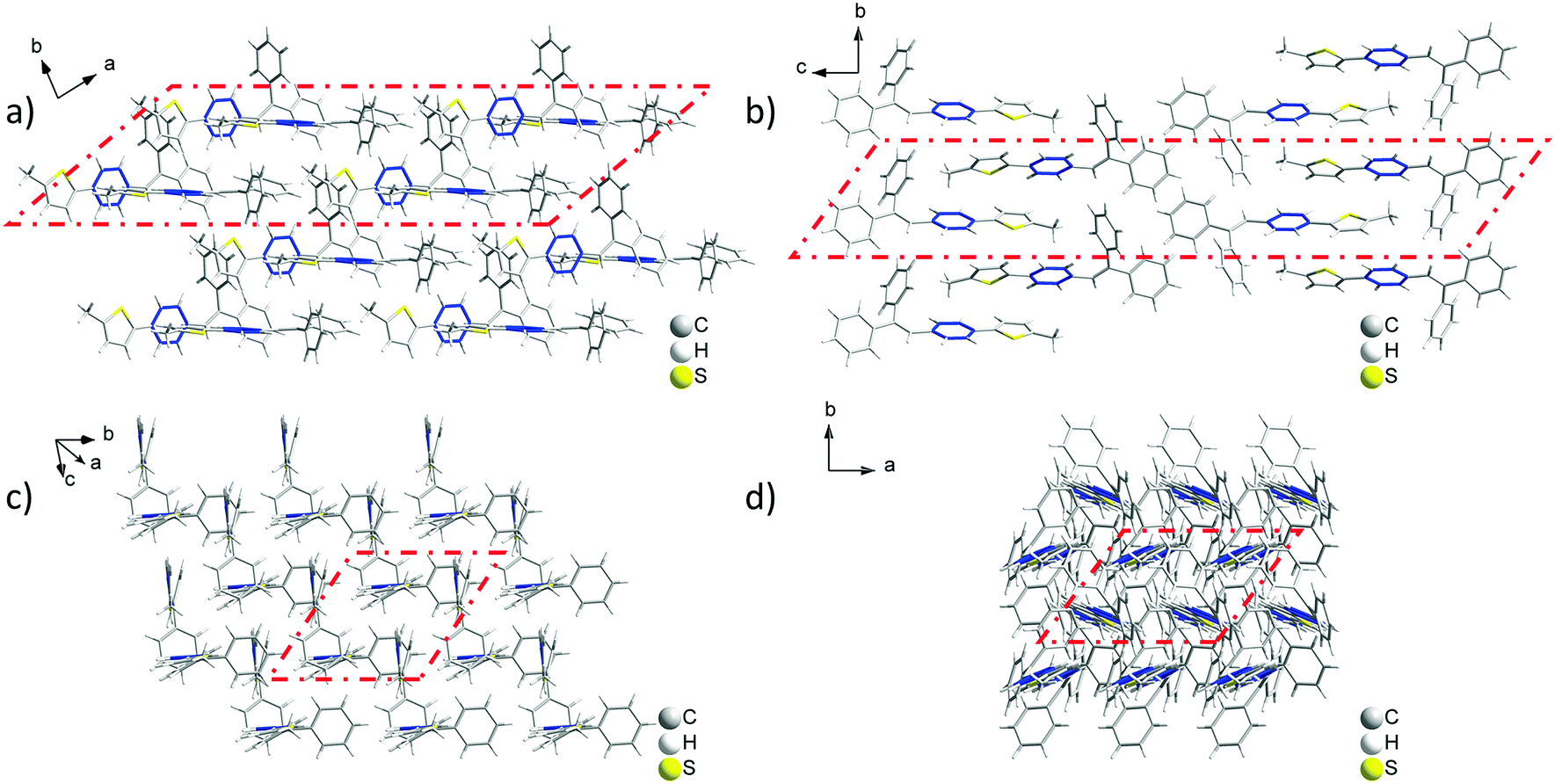
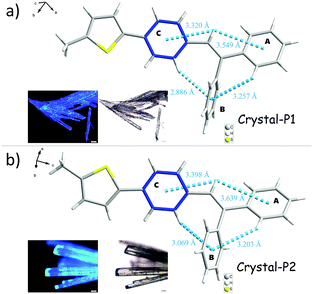
Analyzing the crystal data for a single molecule, there are 4 types of C–H⋯π intramolecular interactions, derived from the introduction of rotatable phenyl rings and an isolated hydrogen atom. For Crystal-P2, the distances of the C–H⋯π intramolecular interactions range from 3.069 to 3.639 Å and the dihedral angle of each phenyl ring is 73.021(198)° (ring a–b), 52.820(183)° (ring a–c) and 63.908(184)° (ring b–c), respectively. However, Crystal-P1 has more compact packing with distances of the C–H⋯π intramolecular interactions ranging from 2.886 to 3.549 Å, and the dihedral angle of each phenyl ring is 89.481(318)° (ring a–b), 26.357(304)° (ring a–c) and 84.305(262)° (ring b–c), respectively. The twisted conformation not only establishes blue emission with a high PLQY in the solid state, but also contributes to an efficient ML property as well. Since the rotation of the thiophene ring is restricted by C–H⋯π interactions from the circumambient tPE unit, some static disorders are observed. The main occupancy ratio of Crystal-P1/P2 is 76.79% and 58.35% in each crystalline state, respectively, which has been selected for subsequent analysis. Further exploring the packing mode of these two polymorphs with opposite ML properties, the space groups of single crystals were triclinic P1 and orthorhombic P212121. As shown in Fig. 3, there is no existence of any type of H/J-aggregation and π–π interaction in either crystal. The molecules in a pair are perpendicular to each other and the whole crystal is tightly packed in the same direction layer by layer in the non-centrosymmetric Crystal-P1, exhibiting an excellent self-assembly property of the thiophene ring. However, the molecules cross each other in pairs and the whole crystal has antiparallel packing in the centrosymmetric Crystal-P2.
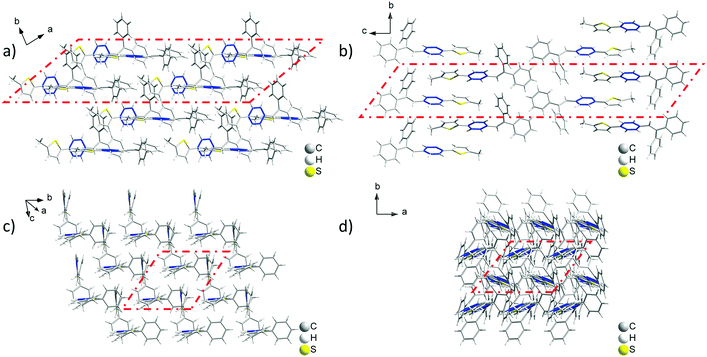 |
| | Fig. 3 Packing mode of the ML-active Crystal-P1 (a and c) and ML-inactive Crystal-P2 (b and d) in different viewing directions. | |
In order to investigate the relationship between mechanoluminescence and packing mode more clearly, the non-repeating unit (the area in red dash in Fig. 3) consisting of eight molecules was chosen to compare the difference between the intermolecular interactions in these polymorphs. As shown in Fig. S3 and Tables S3 and S4 (ESI†), there are 16 + 5 types (19 + 9 numbers) of intermolecular C–H⋯π (C–H⋯Ph + C–H⋯Th) interactions with distances ranging from 3.097 Å to 3.541 Å and 6 types (10 numbers) of intermolecular C–H⋯S interactions with distances ranging from 3.173 Å to 3.590 Å in the ML-active Crystal-P1. In comparison to Crystal-P1, Crystal-P2 has less intermolecular interactions related to the thiophene ring. According to Fig. S4 and Tables S5 and S6 (ESI†), there are 7 + 2 types (10 + 6 numbers) of intermolecular C–H⋯π (C–H⋯Ph + C–H⋯Th) interactions with distances ranging from 2.911 Å to 3.487 Å and 3 types (10 numbers) of intermolecular C–H⋯S interactions with distances ranging from 3.089 Å to 3.531 Å in the ML-inactive Crystal-P2. It is implied that Crystal-P1 has more intense intermolecular/intramolecular interactions than Crystal-P2 between adjacent molecules, and the thiophene ring plays an extremely important role as the self-assembly unit (Table 2). In addition, each molecule interacts to form the closely linked network in Crystal-P1, but each four molecules as a part do not have any interactions between contiguous parts in Crystal-P2. The packing mode with a long-range ordered structure due to the self-assembly effect, has contributed to forming effective meshed connections by intense intermolecular/intramolecular interaction, which is critically important for the ML property.
Table 2 Single crystal information of Crystal-P1 and Crystal-P2
|
|
Intramolecular C–H⋯π |
Intermolecular C–H⋯π |
C–H⋯S |
Space group |
Symmetry |
Crystal system |
|
The quantity of kinds of hydrogen bonds.
The quantity of hydrogen bonds.
|
|
Crystal-P1
|
4a |
4b |
16 + 5a |
19 + 9a |
6a |
10b |
P1 |
Noncentrosymmetric |
Triclinic |
| 2.886–3.549 Å |
3.097–3.541 Å |
3.173–3.590 Å |
|
Crystal-P2
|
4a |
4b |
7 + 2a |
10 + 6b |
3a |
10b |
P212121 |
Centrosymmetric |
Orthorhombic |
| 3.069–3.639 Å |
2.911–3.487 Å |
3.089–3.531 Å |
The powder X-ray diffraction (PXRD) spectra of these two crystalline polymorphs distinctly demonstrate totally different characteristic peaks (Fig. S7, ESI†), with the main characteristic peak of Crystal-P1 and Crystal-P2 at 20° and 25°, respectively. After grinding lightly, the crystals broke into micro-crystals, which still exhibited high and sharp diffraction peaks. Further grinding Crystal-P2 heavily, there was almost no signal in its PXDR spectrum, implying that its crystalline phase was totally destroyed to the amorphous one.10 However, on the contrary, there was still one sharp diffraction peak at 20° in the PXRD spectrum of Crystal-P1 after grinding heavily, showing that its micro-crystals were hard to destroy, which should be ascribed to the strong interactions derived from self-assembly. Based on the crystal structures of these crystalline polymorphs, theoretical calculations were conducted by classical B3LYP density functional theory at the 6-31G level. As shown in Fig. 4, no clear orbital delocalization is marked, mainly due to the nonpolar structure of tPE-5-MeTh. Crystal-P1 has a smaller ΔEg value (3.73 eV) than Crystal-P2 (3.81 eV), indicating that the molecular electrons of Crystal-P1 might be much easily excited. The dipole moments of these two crystals are 10.09 D and 9.10 D, respectively. Generally, a larger dipole moment is beneficial to the piezoelectric effect. Thus, the larger dipole moment of the noncentrosymmetric Crystal-P1 favors its ML effect upon the recombination of the cation and the anion once being stimulated.
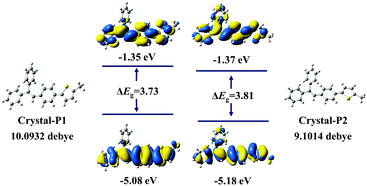 |
| | Fig. 4 The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) density maps of tPE-5-MeTh and their dipole moment in different polymorphs. (Calculated at the B3LYP/6-31G level). | |
In spite of the same molecular structure, these crystalline polymorphs (Crystal-P1 and Crystal-P2) show the opposite phenomenon, implying that the packing mode still acts as the highlight point for the ML property. On the basis of detailed analysis of these two crystals, the introduction of a thiophene ring as the self-assembly unit directly leads to the formation of diverse packing modes, by forming intense intra/intermolecular interactions more handily and efficiently. However, similar to the previous organic ML-active luminogens, the ML signals became weak and then disappeared under continuous stimuli. According to our original design idea, the self-assembled molecules have the inherent ability to rearrange under the influence of the surrounding environment. Combined with the effect of temperature for preparing the crystalline state of tPE-5-MeTh, it enlightens us that there might be an opportunity to explore whether the exciting ML property could be recoverable through thermal-treatment.
3.3 Thermal properties
As shown in Fig. S8 (ESI†), the differential scanning calorimetry (DSC) curve of Crystal-P2 presented its melting point at 112° and no obvious glass-transition temperature. However, two pairs of characteristic peaks were shown in the DSC curve of Crystal-P1. After careful analysis and comparison, it could be that Crystal-P1 transformed during the first phase transition (87°) and melted during the first molten state (104°). Inspiringly, the temperature difference of the phase transition between Crystal-P1 and Crystal-P2 is big enough to be regulated through simple thermal treatment.11
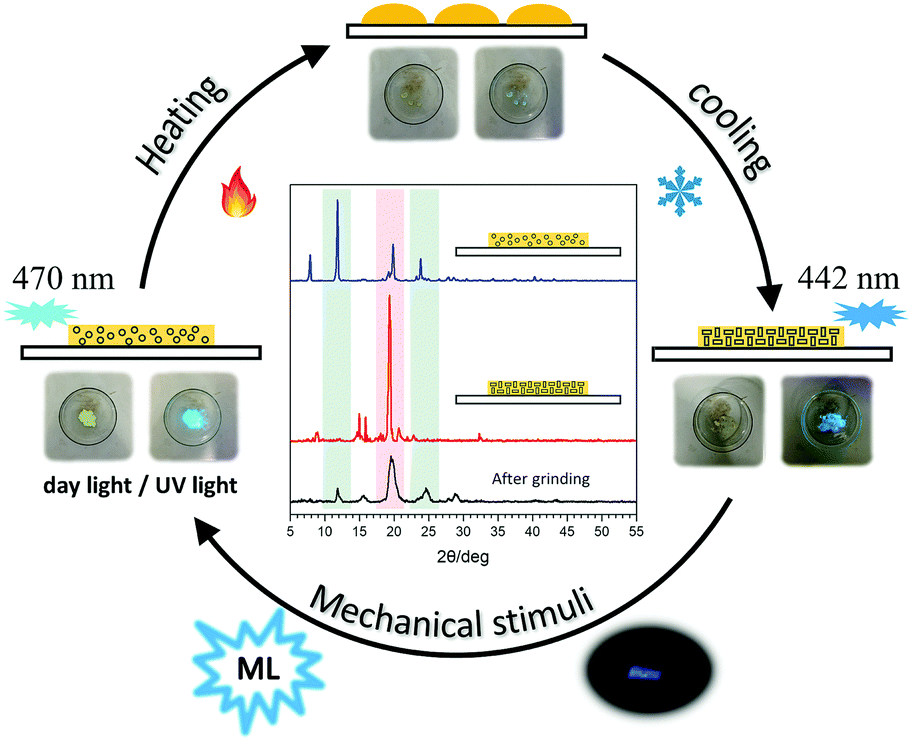
3.4 Recoverable ML property
Accordingly, an experimental setup (Fig. S9, ESI†) has been built to realize the transformation between the different solid states of tPE-5-MeTh with opposite ML properties. Firstly, the powder of tPE-5-MeTh with greenish-blue emission (470 nm) was placed in the center of a petri dish, which was protected by slow-flowing nitrogen. After heating to 90 °C for dozens of seconds, the solid changed during the first phase transition. The emission turned off because the high-temperature increased the motion of the molecules and more energy was expended in non-radiative transitions. Then, the temperature was gradually reduced to 30 °C and was kept stable until the molecules recrystallized. The emission wavelength of the reborn crystals was 442 nm, similar to that of Crystal-P1. Excitingly, there was intense light emission while grinding the crystals in the dish, implying that the reborn crystals could be destroyed to the amorphous form again by mechanical stimuli (Fig. 5). This cycle of phase transitions could be repeated many times, and the ML luminogens could be reused through simple thermal-treatment, thanks to the contribution of self-assembly.
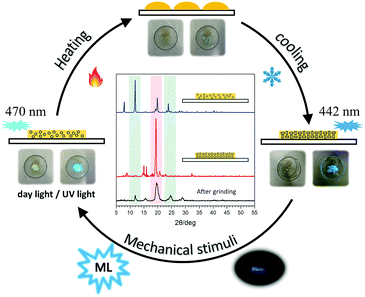 |
| | Fig. 5 Diagram of the cyclic utilization of the ML material (tPE-5-MeTh) and PXRD patterns of tPE-5-MeTh in different states in the circulation. | |
PXRD spectra were measured to further confirm the molecular packing status in each step (Fig. 5). The characteristic peaks of the powder (blue line) were at 12°, 20° and 25°, respectively, the same as the PXRD pattern of ML-inactive Crystal-P2. After simple thermal-treatment, the peak at 20° obviously became stronger and sharper (red line), while the peaks at 12° and 25° nearly did not have any signals, the same as the PXRD pattern of ML-active Crystal-P1. After grinding the reborn crystals heavily, the peak at 20° decreased immediately (black line). This cycle could be repeated endlessly without any sample loss through simple thermal-treatment and the phase transitions from amorphous state to Crystal-P1 could be adjusted conveniently to make the ML property recur, due to the self-assembly ability of Crystal-P1. More interestingly, when the solid underwent the second phase transition (heating temperature higher than 95 °C), the emission of the reborn crystalline state did not change after cooling to recrystallization, but there was no ML emission upon grinding. According to the PXRD patterns in Fig. S10 (ESI†), the opposite result could be obtained. It is implied that the structural rearrangement caused by the self-assembly is sensitive to the environment (e.g. temperature) (Fig. 6). The transition temperatures of the crystalline polymorphs (87 and 92 °C) are important for these very different results. Thus, 90 °C and 95 °C, as crucial temperatures, have been demonstrated to distinguish these two kinds of crystals. For tPE-5-MeTh, the ML property could not only be recovered but also change to the opposite phenomenon through the simple control of the temperature. It is rare that different packing modes can be easily transformed in the solid state at relatively low temperature, emphasizing the key role of the introduced self-assembly effect, which could effectively reduce the energy consumption during the conversion process. Thus, this molecular design could provide valuable information and favorable conditions for related functional materials containing self-assembly units for potential applications.
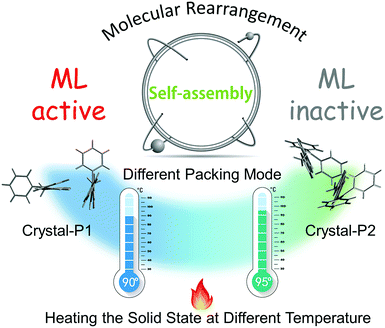 |
| | Fig. 6 The strategy to construct recyclable and controllable ML molecules due to self-assembly. | |
Conclusions
In summary, a new ML luminogen of tPE-5-MeTh has been rationally designed and easily synthesized with a thiophene group as the self-assembly unit. Regardless of the same molecular structure, two crystalline polymorphs were obtained by elaborately preparing different mixture solvents, and their totally different ML properties confirmed the key role of the molecular packing mode once more. Accordingly, the introduction of self-assembly is beneficial to forming intense intra/intermolecular interactions more handily and efficiently, which could induce a tighter packing mode. Excitingly, simple thermal treatment could break the thermodynamic equilibrium of the static system to rearrange the molecules and achieve the recoverability and controllability of ML materials, thanks to the supramolecular self-assembly. Thus, the introduction of self-assembly units opens up a new avenue for further facilitating convenient practical applications and exploring the possible adjustment of the molecular packing of ML materials through simple treatments. Also, the successful case could shed some light on the handling of molecular arrangement of other opto-electronic materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Science Foundation of China (no. 51573140, 21734007), Hubei Province (2017CFA002), and Special funds for basic scientific research services in central colleges and Universities (2042017kf0247).
Notes and references
-
(a) C. N. Xu, T. Watanabe, M. Akiyama and X. Zheng, Appl. Phys. Lett., 1999, 74, 1236–1238 CrossRef CAS;
(b) N. C. Eddingsaas and K. S. Suslick, Nature, 2006, 444, 163 CrossRef CAS;
(c) I. Sage, R. Badcock, L. Humberstone, N. Geddes, M. Kemp and G. Bourhill, Smart Mater. Struct., 1999, 8, 504–510 CrossRef;
(d) D. Tu, C. Xu, A. Yoshida, M. Fujihala, J. Hirots and X. Zheng, Adv. Mater., 2017, 29, 1606914 CrossRef;
(e) N. C. Eddingsaas and K. S. Suslick, J. Am. Chem. Soc., 2007, 129, 6718–6719 CrossRef CAS;
(f) X. Zhang, J. Shi, G. Shen, F. Gou, J. Cheng, X. Zhou and H. Xiang, Mater. Chem. Front., 2017, 1, 1041–1050 RSC.
-
(a) P. Jha and B. P. Chandra, Luminescence, 2014, 29, 977–993 CrossRef CAS;
(b) J. I. Zink, Acc. Chem. Res., 1978, 11, 289–295 CrossRef CAS;
(c) A. J. Walton, Adv. Phys., 1977, 26, 887–948 CrossRef CAS;
(d) R. S. Fontenot, K. N. Bhat, W. A. Hollerman and M. D. Aggarwal, Cryst. Res. Technol., 2012, 47, 573–578 CrossRef CAS;
(e) D. Tu, C. N. Xu, A. Yoshida, M. Fujihala, J. Hirotsu and X. G. Zheng, Adv. Mater., 2017, 29, 1606914 CrossRef.
-
(a) S. Xu, T. Liu, Y. Mu, Y. Wang, Z. Chi, C. Lo, S. Liu, Y. Zhang, A. Lien and J. Xu, Angew. Chem., Int. Ed., 2015, 54, 874–878 CrossRef CAS;
(b) J. Yang, Z. Ren, Z. Xie, Y. Liu, C. Wang, Y. Xie, Q. Peng, B. Xu, W. Tian, F. Zhang, Z. Chi, Q. Li and Z. Li, Angew. Chem., Int. Ed., 2017, 56, 880–884 CrossRef CAS;
(c) J. Yang, X. Gao, Z. Xie, Y. Gong, M. Fang, Q. Peng, Z. Chi and Z. Li, Angew. Chem., Int. Ed., 2017, 56, 15299–15303 CrossRef CAS;
(d) Y. Xie and Z. Li, Chem, 2018, 4, 943–971 CrossRef CAS;
(e) T. Xie, B. Zhang, X. Zhang and G. Zhang, Mater. Chem. Front., 2017, 1, 693–696 RSC;
(f) J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef;
(g) J. Yang, J. Qin, P. Geng, J. Wang, M. Fang and Z. Li, Angew. Chem., Int. Ed., 2018 DOI:10.1002/anie.201809463.
-
(a) B. Xu, J. He, Y. Mu, Q. Zhu, S. Wu, Y. Wang, Y. Zhang, C. Jin, C. Lo, Z. Chi, A. Lien, S. Liu and J. Xu, Chem. Sci., 2015, 6, 3236–3241 RSC;
(b) B. Xu, W. Li, J. He, S. Wu, Q. Zhu, Z. Yang, Y. Wu, Y. Zhang, C. Jin, P. Lu, Z. Chi, S. Liu, J. Xu and M. R. Bryce, Chem. Sci., 2016, 7, 5307–5312 RSC;
(c) S. M. Jeong, S. Song, S. K. Lee and N. Y. Ha, Adv. Mater., 2013, 25, 6194–6200 CrossRef CAS;
(d) P. Chen and Z. Li, Chin. J. Polym. Sci., 2017, 35, 793–798 CrossRef CAS;
(e) C. Zheng, Q. Zang, H. Nie, W. Huang, Z. Zhao, A. Qin, R. Hu and B. Z. Tang, Mater. Chem. Front., 2018, 2, 180–188 RSC;
(f) W. Li, Q. Huang, Z. Mao, Q. Li, L. Jiang, Z. Xie, R. Xu, Z. Yang, J. Zhan, T. Yu, Y. Zhang, M. P. Aldred and Z. Chi, Angew. Chem., Int. Ed., 2018, 57, 12727–12732 CrossRef CAS.
-
(a) F. Liu, J. Tu, X. Wang, J. Wang, Y. Gong, M. Han, X. Dang, Q. Liao, Q. Peng, Q. Li and Z. Li, Chem. Commun., 2018, 54, 5598–5601 RSC;
(b) Y. Gong, P. Zhang, Y. Gu, J. Wang, M. Han, C. Chen, X. Zhan, Z. Xie, B. Zou, Q. Peng, Z. Chi and Z. Li, Adv. Opt. Mater., 2018, 1800198 CrossRef;
(c) X. Gao, Sci. China: Chem., 2018, 61, 641–642 CrossRef CAS;
(d) H. Nie, K. Hu, Y. Cai, Z. Zhao, R. Hu, J. Chen, S. Su, A. Qin and B. Z. Tong, Mater. Chem. Front., 2017, 1, 1125–1129 RSC.
-
(a) C. Richard, F. Balavoine, P. Schultz, T. W. Ebbesen and C. Mioskowski, Science, 2003, 300, 775–778 CrossRef CAS;
(b) M. E. Moussa, S. Evariste, B. Krämer, R. Réau, M. Scheer and C. Lescop, Angew. Chem., Int. Ed., 2018, 57, 795–799 CrossRef;
(c) Y. Liu, C. Zhang, D. Hao, Z. Zhang, L. Wu, M. Li, S. Feng, X. Xu, F. Liu, X. Chen and Z. Bo, Chem. Mater., 2018, 30, 4307–4312 CrossRef CAS;
(d) X. Zhan, J. Zhang, Y. Gong, S. Tang, J. Tu, Y. Xie, Q. Peng, G. Yu and Z. Li, Mater. Chem. Front., 2017, 1, 2341–2348 RSC;
(e) L. Chen, G. Lin, H. Peng, S. Ding, W. Luo, R. Hu, S. Chen, F. Huang, A. Qin, Z. Zhao and B. Z. Tang, Mater. Chem. Front., 2017, 1, 176–180 RSC;
(f) J. Chen, J. Zhao, B. Xu, Z. Yang, S. Liu, J. Xu, Y. Zhang, Y. Wu, P. Lv and Z. Chi, Chin. J. Polym. Sci., 2017, 35, 282–292 CrossRef CAS.
-
(a) J. Han, Y. Chen, W. Chen, C. Yu, X. Song, F. Li and Y. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 32823–32832 CrossRef CAS;
(b) X. Cao, N. Zhao, G. Zou, A. Gao, Q. Ding, G. Zeng and Y. Wu, Soft Matter, 2017, 13, 3802–3811 RSC;
(c) N. Zhou, K. Prabakaran, B. Lee, S. Chang, B. Harutyunyan, P. Guo, M. R. Butler, A. Timalsina, M. J. Bedzyk, M. A. Ratner, S. Vegiraju, S. Yau, C. Wu, R. P. H. Chang, A. Facchetti, M. Chen and T. J. Marks, J. Am. Chem. Soc., 2015, 137, 4414–4423 CrossRef CAS;
(d) Y. Wu, A. Qin and B. Z. Tang, Chin. J. Polym. Sci., 2017, 35, 141–154 CrossRef CAS;
(e) B. Zhang, R. H. Sánchez, Y. Zhong, M. Ball, M. W. Terban, D. Paley, S. J. L. Billinge, F. Ng, M. L. Steigerwald and C. Nuckolls, Nat. Commun., 2018, 9, 1957 CrossRef;
(f) H. Zhao, Y. Luo, L. Liu, Z. Xie and Y. Ma, Mater. Chem. Front., 2017, 1, 1087–1092 RSC;
(g) W. Zheng, G. Yang, S. Jiang, N. Shao, G. Yin, L. Xu, X. Li, G. Chen and H. Yang, Mater. Chem. Front., 2017, 1, 1823–1828 RSC;
(h) Y. Li, Y. Wang, X. Ren and L. Chen, Mater. Chem. Front., 2017, 1, 2599–2605 RSC;
(i) W. Geng, Y. Liu, Z. Zheng, D. Ding and D. Guo, Mater. Chem. Front., 2017, 1, 2651–2655 RSC.
-
(a) Y. N. Hong, J. W. Y. Lama and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC;
(b) H. Wang, E. Zhao, J. W. Y. Lam and B. Z. Tang, Mater. Today, 2015, 18, 365–377 CrossRef CAS;
(c) C. Wang, L. Li, X. Zhan, Z. Ruan, Y. Xie, Q. Hu, S. Ye, Q. Li and Z. Li, Sci. Bull., 2016, 22, 1746–1755 CrossRef;
(d) Y. Yu, J. Yang, Z. Ren, G. Xie, Q. Li and Z. Li, Acta Chim. Sin., 2016, 74, 865–870 CrossRef CAS;
(e) J. Yang, Le. Li, Y. Yu, Z. Ren, Q. Peng, S. Ye, Q. Li and Z. Li, Mater. Chem. Front., 2017, 1, 91–99 RSC;
(f) Q. Li and Z. Li, Adv. Sci., 2017, 4, 1600484 CrossRef PubMed;
(g) Z. Yang, Z. Chi, Z. Mao, Y. Zhang, S. Liu, J. Zhao, M. P. Aldred and Z. Chi, Mater. Chem. Front., 2018, 2, 861–890 RSC;
(h) T. Chen, Z. Chen, W. Gong, C. Li and M. Zhu, Mater. Chem. Front., 2017, 1, 1841–1846 RSC.
-
(a) Y. Xie, J. Tu, T. Zhang, J. Wang, Z. Xie, Z. Chi, Q. Peng and Z. Li, Chem. Commun., 2017, 53, 11330–11333 RSC;
(b) M. Fang, J. Yang, Q. Liao, Y. Gong, Z. Xie, Z. Chi, Q. Peng, Q. Li and Z. Li, J. Mater. Chem. C, 2017, 5, 9879–9885 RSC;
(c) Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC;
(d) J. Huang, P. Chen, X. Yang, R. Tang, L. Wang, J. Qin and Z. Li, Sci. China: Chem., 2013, 56, 1213 CrossRef CAS;
(e) T. Yu, D. Ou, L. Wang, S. Zheng, Z. Yang, Y. Zhang, Z. Chi, S. Liu, J. Xua and M. P. Aldred, Mater. Chem. Front., 2017, 1, 1900–1904 RSC.
-
(a) C. Wang and Z. Li, Mater. Chem. Front., 2017, 1, 2174–2194 RSC;
(b) W. Yuan, X. Shen, H. Zhao, J. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang, Q. Zheng, J. Sun, Y. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 6090–6099 CrossRef CAS.
-
(a) K. Neena, P. Sudhakar, K. Dipak and P. Thilagar, Chem. Commun., 2017, 53, 3641–3644 RSC;
(b) Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS;
(c) W. Wu, R. Yao, X. Huang, R. Chen, K. Li, S. Gao and R. Zou, Mater. Chem. Front., 2017, 1, 1430–1434 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1846929 and 1846930. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qm00411k |
|
| This journal is © the Partner Organisations 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab
*ab