
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWhite-light emission from a single organic compound with unique self-folded conformation and multistimuli responsiveness†
Dengfeng
Li‡
,
Wende
Hu‡
,
Jie
Wang
,
Qiwei
Zhang
,
Xiao-Ming
Cao
 ,
Xiang
Ma
* and
He
Tian
,
Xiang
Ma
* and
He
Tian
Key Laboratory for Advanced Materials, Institute of Fine Chemicals, Center for Computational Chemistry, Research Institute of Industrial Catalysis, School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai 200237, P. R. China. E-mail: maxiang@ecust.edu.cn
First published on 21st May 2018
Abstract
White-light emitting organic materials attract broad attention which are ascribed to their potential for applications in lighting devices and display media. Most reported organic white-light emitters rely on the combination of several components that emit different colors of light (red/green/blue or orange/blue), which may cause problems to stability, reproducibility and device fabrication. By contrast, white-light emission from single-molecule systems offers opportunities to overcome these disadvantages, meanwhile engendering white-light with high quality. Nevertheless, limited cases of white-light emission at the molecular scale reported principally concentrate on organic solvents. Herein, we designed and synthesized new bi-functional organic molecules with a symmetric donor–acceptor–donor (D–A–D) type structure with the aim to construct a single-molecule white-light emitting system in aqueous solution. Further experiments and calculations demonstrate the possibility of stacking between the pyridinium-naphthalene (PN) core and coumarin groups in the designed molecules, ascribed to hydrophobic effects, π–π stacking and donor–acceptor interactions, which could dramatically enhance the intramolecular charge transfer (ICT) efficiency along with remarkable charge transfer (CT) emission. Based on this, multicolor photoluminescence including white-light can be finely tuned in various modes including excitation wavelength, solvent polarity, temperature, and host–guest interactions. A white-light emitting (WLE) hydrogel was also facilely prepared through the dispersion of one of the compounds in a commercial agarose gelator. This innovative study helps enrich the strategies to construct single-molecule organic white-light emitting materials in aqueous medium using the self-folding behavior.
Introduction
White-light emitting organic materials have attracted increasing attention owing to their potential for application in lighting devices and display media.1–7 The generation of white-light emission commonly requires the simultaneous emission of three primary RGB (red, green and blue) colors or at least two complementary colors. A variety of approaches have been adopted to develop efficient white-light emitting systems, including polymers,8–10 metal–organic frameworks,11–14 quantum dots,15,16 nanoparticles,17–19 self-assemblies,20–24 and small molecules.25–28 So far, most organic white-light emitters reported in the literature rely on (1) a combination of several components that emit different colors of light to cover the visible spectrum (400 to 700 nm) or (2) a single organic molecule emitting white-light. In comparison with multicomponent emitters, white-light emission from a single molecule offers advantages over the former including improved stability, excellent reproducibility, and a simplified fabrication process. Nevertheless, few examples of white-light emission from single molecule systems have been reported, since fluorophores tended to attain the lowest vibrational states resulting in monochromatic emission according to Kasha's rule.29,30 Simple grafting of fluorophores in a single molecular backbone often causes an intramolecular Förster resonance energy transfer (FRET) process along with the enhancement of long-wavelength emission from fluorescent acceptors. Therefore, the exploration of molecules that are able to emit pure white-light is of great significance, though it remains a big challenge to scientists and researchers in the field.Several single molecule systems have been reported to display dual emissions based on various principles such as excited-state intramolecular proton transfer (ESIPT),31–33 twisted intramolecular charge transfer (TICT),34,35 tautomerization,36–39 self-assembly40,41 and other mechanisms.42,43 Strongin reported anovel benzo[a]xanthene, seminaphtho[a]fluorine, which possessed the ability to display multicolor emission including the white-light one by adjusting the solution pH to generate different emissive tautomers.36 Additionally, ESPIT molecules possess a stable enol form in the ground state but are easily converted to the keto form in the excited state through intramolecular proton transfer resulting in a larger Stokes shift in the emission spectrum. Based on the ESIPT concept, Park presented the example of a white-light emitting molecular dyad composed of blue and orange light emitters between which the energy transfer was frustrated.33 The self-assembly behaviors of π-conjugated chromophores usually wield the high directional nature of noncovalent interactions to build precisely hierarchical structures with different luminescence characteristics. Ghosh demonstrated white-light emission from the single component naphthalene diimide chromophore with J-aggregation mediated by hydrogen bonding.40
Since most reported cases of white-light emission at the molecular scale principally concentrated on organic solvents, we herein designed and synthesized a series of new bi-functional organic molecules (NP2C, NP4C and NP6C; Fig. 1) with symmetric D–A–D structures to achieve white-light emission in aqueous solution. These compounds comprised a central PN unit connected with two coumarin moieties in common but linked by flexible alkyl chains of different lengths. Among them, NP4C and NP6C could induce dual emission in water, which could emit white-light with a CIE coordinate of (0.30, 0.33). The reference compounds NPM, PC and NPMC were prepared as detailed in the ESI.† To the best of our knowledge, the design of single organic molecules emitting white-light in aqueous solution based on the self-folded pattern has been rarely reported to date.
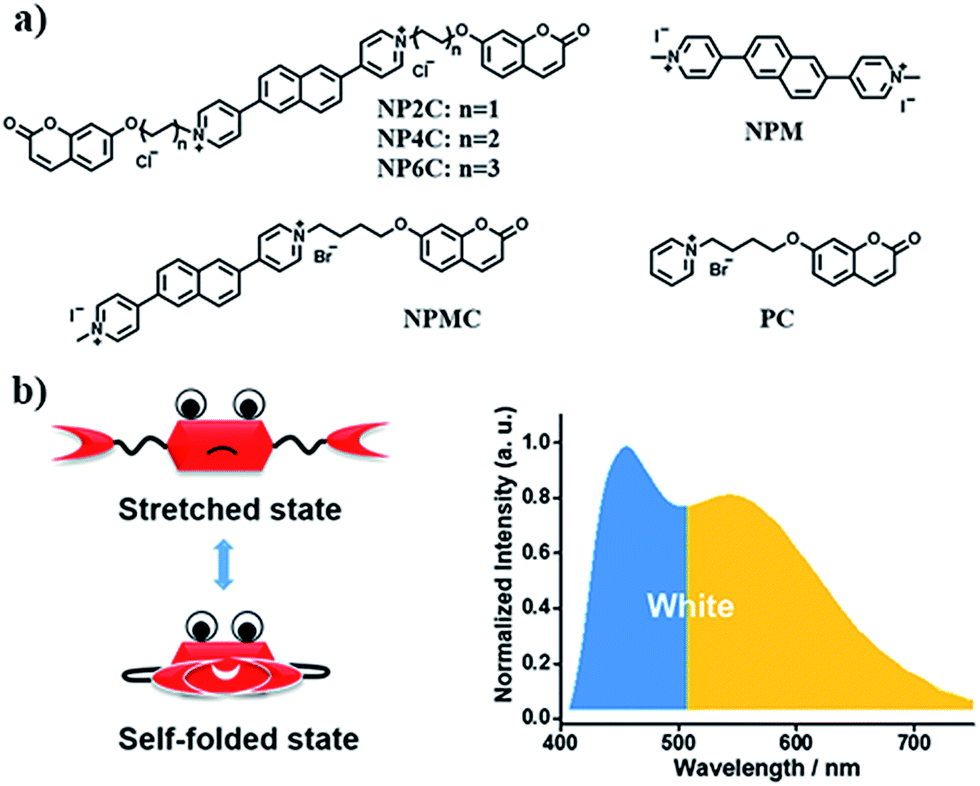 | ||
| Fig. 1 (a) Molecular structures of white-light emitting organic molecules (NP4C and NP6C) and the reference compounds (NP2C, NPM, NPMC and PC); (b) the schematic cartoon illustration of single-molecule white-light emitters. | ||
Results and discussion
To better explore this phenomenon, the absorption properties of all compounds were investigated. The UV-Vis spectrum of NP4C (Fig. 2a) in aqueous solution displayed two significant absorption bands centered approximately at 280 and 330 nm with one shoulder at 388 nm, attributed to the chromophores contained in NP4C compared with NPM and PC. Moreover, a remarkable bathochromic shift was observed for NP4C, especially for the peak at around 388 nm, which implied the possibility of a CT process. To further confirm this finding, DFT calculations were performed to calculate the frontier molecular orbitals of NPM and NP4C. The highest occupied molecular orbital (HOMO) of NP4C was primarily located in the coumarin groups, while the lowest-unoccupied molecular orbital (LUMO) was distributed on the PN unit in favor of the charge-transfer process. Nevertheless, the occupied and virtual orbitals of NPM completely differed from those of NP4C and were concentrated mostly on the PN core (Fig. 2e), lacking in the condition inducing the CT process. These results further indicated that white-light emission was developed in the D–A–D type. | ||
| Fig. 2 (a) Absorption spectra and (b) PL emission spectra of aqueous NP2C, NP4C, NP6C, PC, NPM and NPMC solutions; (c) PL excitation spectra for dual emission of NP4C, respectively, monitored at 455 and 550 nm. (d) PL emission spectra of NP4C measured at various excitation wavelengths. (e) Calculated frontier molecular orbitals of NP4C and NPM. [NP2C] = [NP4C] = [NP6C] = [NPM] = [PC] = [NPMC] = 25 μM. | ||
With the aim of distinguishing the occurrence of the CT character in the molecule itself or in adjacent molecules, a series of tests were implemented. Electrical conductivity measurements at various concentrations (from 1 μM to 1 mM; Fig. S3†) were conducted to confirm that NP4C dispersed in aqueous solution without any kind of aggregates. The concentration-dependent fluorescence (Fig. S4†) and UV-Vis absorption spectra (Fig. S5†) of NP4C itself were also measured. The shape and position of emission bands in the fluorescence spectra remained almost unchanged in spite of its concentrations ranging from 1 μM to 1 mM. Fig. S4† shows that the character of dual emission was observed in an extremely dilute solution, which indicated that the luminescence properties originated from the molecule itself rather than from intermolecular interactions. The absorption spectra showed no obvious red shift when the concentration of NP4C was increased. The absorption intensity at 328 nm rose linearly with the increased concentration, which neatly fitted the Beer–Lambert law and manifested no obvious interactions between the solute molecules (Fig. S5†). Hence, we may conclude that this interesting optical property originated from the NP4C itself rather than from any aggregates between molecules, which verifies that the white-light emitters belonged to a single molecule system.
TD-DFT calculations were also performed to investigate the excited state properties of NP4C (Table 1). The results showed that the S1 state of NP4C was calculated at 2.08 eV. However, radiative transition was forbidden (f = 0), which was against the fluorescence emission signal as shown in Fig. 2. It was assumed that the flexible alkyl chain was beneficial to the rotation and motion of the coumarin groups at ends, which might lead to stacking with the PN core due to hydrophobic effects, π–π stacking and donor–acceptor interactions. The ROESY spectrum of NP4C showed weak but clearly visible cross peaks between the protons (Ha, Hb, Hc and He) of the coumarin unit and protons (Hj and Hk) of the pyridium group, which indicated the adjacency of these two fractions in spatial distance (Fig. 3). To further verify this finding, another two compounds (NP2C and NP6C) containing different lengths of alkyl chains were synthesized, and their fluorescence spectra were measured at various excitation wavelengths (Fig. S6†). Fig. 2b and S7† show that they all possessed a blue light band in common but only NP4C and NP6C could simultaneously generate CT emission, which was attributed to the steric hindrance from the short chain that restricted the self-folding behavior. Meanwhile, similar but weaker ROE signals implying folding conformation were only observed in NP6C by analysing the ROESY spectra of NP2C and NP6C (Fig. S7 and S8†). Moreover, the fact that NP4C tended to lie at the self-folded state has been further confirmed by the theoretical calculations by comparing with the Gibbs free energy of the stretched state in water (Fig. 4b).
| Compound | State | Excitation energy (eV) | Oscillator strength | MO composition |
|---|---|---|---|---|
| NP4C | S1 | 2.08 | 0.0000 | H → L (94.7%) |
| S2 | 2.09 | 0.0000 | H-1 → L (93.9%) | |
| S3 | 3.56 | 0.5205 | H-2 → L (94.0%) | |
| Folded NP4C | S1 | 3.22 | 0.0451 | H → L (85.4%) |
| S2 | 3.29 | 0.0494 | H-1 → L (75.7%) | |
| S3 | 3.42 | 0.3477 | H-2 → L (72.3%) | |
| NPM | S1 | 3.53 | 0.3860 | H → L (94.1%) |
| NPMC | S1 | 3.34 | 0.0080 | H → L (76.0%) |
| S2 | 3.42 | 0.2781 | H-1 → L (85.5%) |
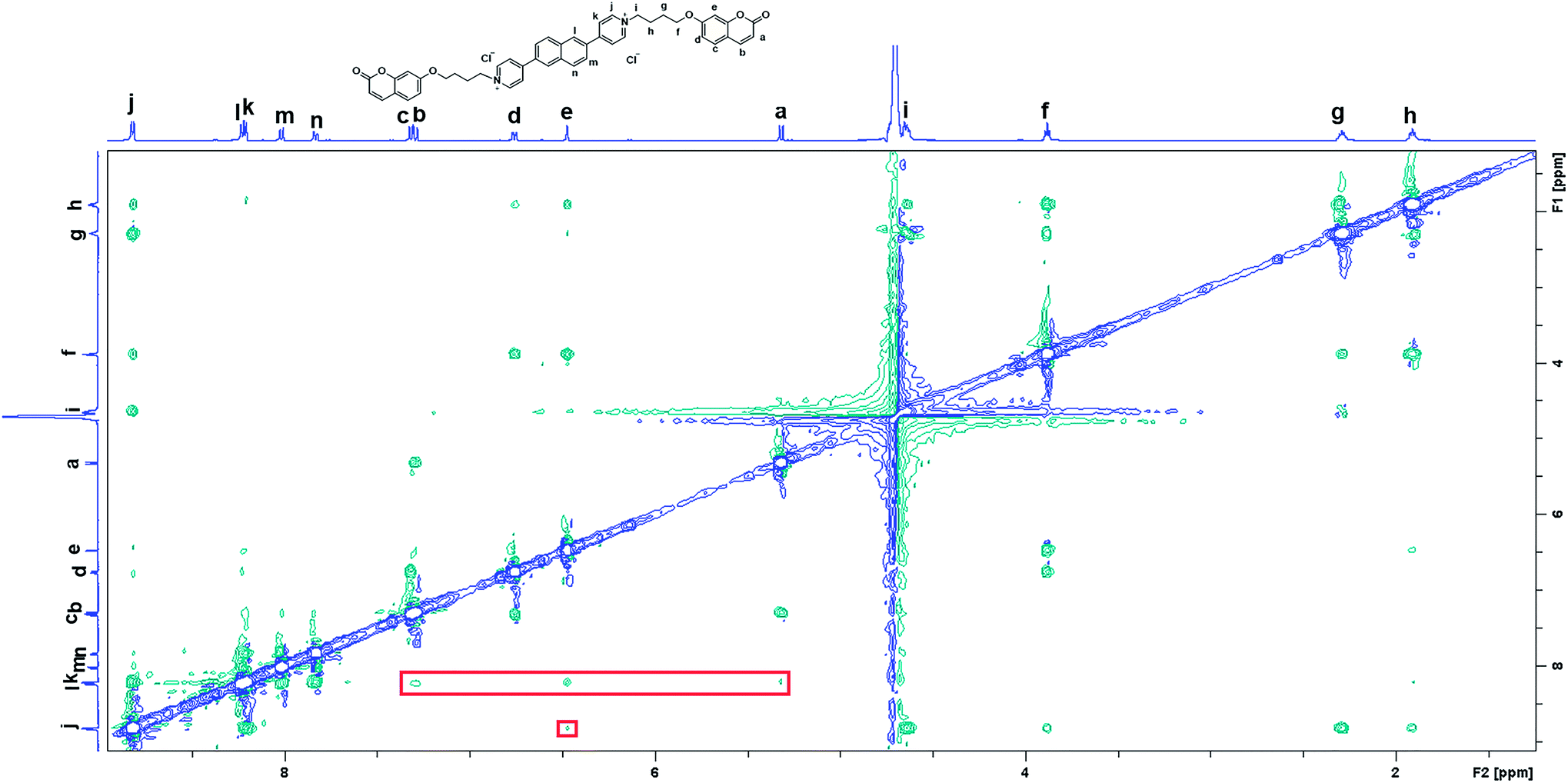 | ||
| Fig. 3 2D ROESY spectrum of NP4C in D2O with water suppression. [NP4C] = 1.0 mM, 25 °C. | ||
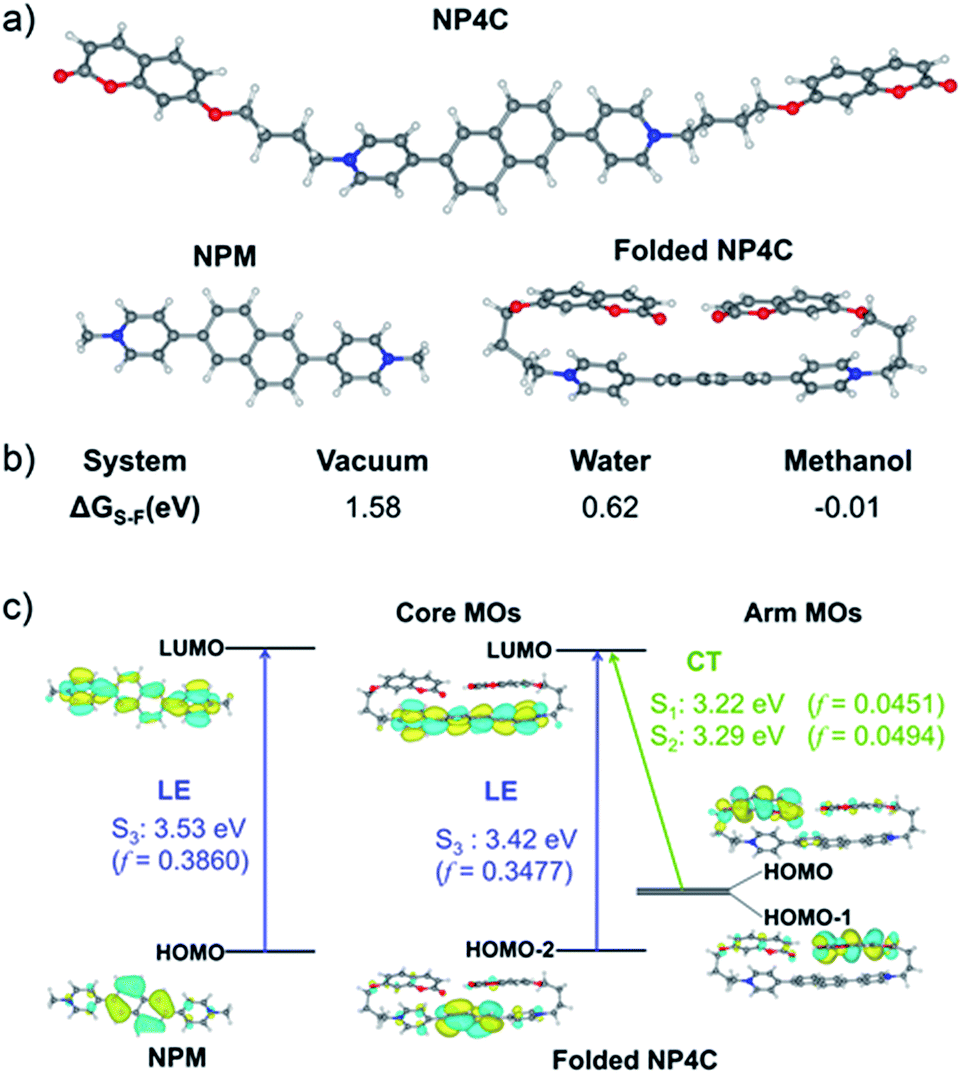 | ||
| Fig. 4 (a) Optimized structures of NP4C and NPM calculated at the B3LYP/6-311+G(d,p) level; (b) Gibbs free energy difference (ΔGS–F) between NP4C and folded NP4C in the different mediums (ΔGS–F = G(NP4C) − G(folded NP4C)); (c) theoretical calculations for PL mechanistic investigation. | ||
Next, the photoluminescence (PL) spectra of NP4C were measured at different excitation wavelengths. As shown in Fig. 2d, NP4C displayed apparent dual emissions in water at 455 nm and 550 nm, respectively, close to white emission with a CIE coordinate of (0.30, 0.33). In contrast with PC, the emission from the coumarin group in NP4C disappeared due to the FRET or self-absorption quenching effect, which is reflected in the overlap between the emission spectrum of PC and the excitation and UV-Vis spectra of NP4C (Fig. 2a and b). It was obvious that the blue band came from the PN unit referred to the observed optical properties of NPM and NPMC (Fig. 2b and S9†). Given the different maximum excitation wavelengths, it could be safely inferred that yellow-green light emission couldn't be induced from a coumarin monomer of its excimer.44 High temperature intensified the intramolecular motions and further accelerated the transformation between the self-folded and stretched states of NP4C. Additionally, the temperature-dependent fluorescence spectra revealed that yellow-green emission decreased remarkably with the increment of external temperature (Fig. 5a). Besides, the UV-Vis spectra of the NP4C aqueous solution at different temperatures also suggested that the existence of the reversible blue shift was attributed to the weakened ICT process at high temperature (Fig. S10†). On the basis of the experimental results above, we assumed that yellow-green luminescence belonged to CT emission. TD-DFT calculations also afforded helpful conclusions to illustrate the emission mechanism (Fig. 4c). NPM and NP4C possess the analogical locally excited (LE) state through the electron transitions HOMO−2 → LUMO and HOMO → LUMO, respectively, attributed to the excitation and emission transition of the blue-light band. For NP4C, its two approximate transitions with lower excitation energy reflected yellow-green emission in the corresponding PL spectra. The electrons traveled a long distance from the arms of NP4C to its PN core in these two transitions, in accordance with a typical ICT process.
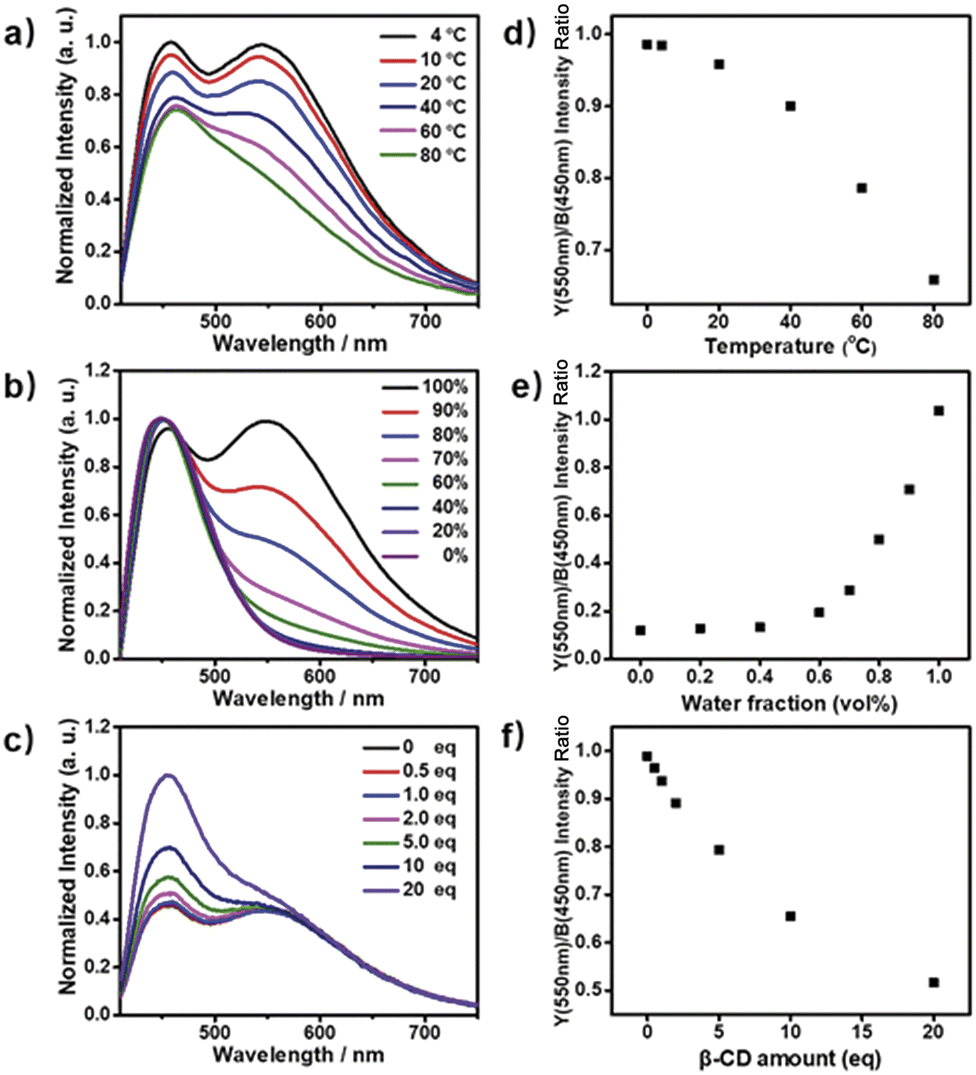 | ||
| Fig. 5 (a) PL emission spectra of NP4C aqueous solution measured at different temperatures ranging from 4 to 80 °C; (b) PL emission spectra of NP4C aqueous solution at different volume fractions of H2O (fw, from 0 to 100%) and MeOH, the intensity of the blue light band was unified to the same level; (c) PL emission spectra of NP4C aqueous solution when added with different amounts of the β-CD host molecules (from 0 to 20 eq.). The yellow/blue (Y/B) emission intensity ratio as a function of (d) temperature, (e) water fraction fw and (f) the amount of β-CD. All the measurements were taken using aqueous NP4C solutions with a unified concentration ([NP4C] = 25 μM) and excited at 400 nm. | ||
Color-tunable emission could be conveniently achieved through regulating the ICT efficiency of NP4C. One of the methods available was using the methanol (MeOH)/water (H2O) mixed solvent since MeOH was a desirable solvent for solvating extended π systems. Fig. 5b shows the normalized fluorescence spectra of NP4C in the MeOH–H2O solution of different water ratios (fw). However, the CT emission of NP4C declined with the reduction of water, and NP4C only emitted mono emission when fw = 40%, due to the elimination of self-folded conformation. Since the distance between the donors and acceptors enlarged in the high MeOH fraction, the ICT process could be overtly impaired and restricted as the molecules preferably resided on the stretched state. This hypothesis could also be testified by the theoretical calculations. As shown in Fig. 4b, NP4C in MeOH is more likely to stretch itself out to maintain at the lower free energy (ΔGS–F = −0.01 eV) compared to water medium (ΔGS–F = −0.62 eV).
β-Cyclodextrin (β-CD) consisting of seven glucopyranose units has been widely used in supramolecular chemistry because its hydrophobic cavity could selectively accommodate the guest molecules in water. The cavity of β-CD is big enough to encapsulate the coumarin unit in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding stoichiometry.45–48 When β-CD captured the coumarin groups of NP4C, the CT process could be efficiently disturbed due to the isolation between the donor and acceptor. As presented in Fig. S11,† the protons of aromatic rings underwent downfield shifts after the addition of β-CD. The π–π stacking between the coumarin group and the PN unit could induce the upfield movement of their chemical shifts. The host–guest inclusion from β-CD impeded the π–π stacking from the self-folding behavior, and then the protons shifted downfield, although the shielding effect of β-CD on the included coumarin group could generate upfield shifts of its corresponding protons. Besides, the 2D ROESY NMR spectrum of the complex NP4C@β-CD (Fig. S12†) in D2O clearly showed the visible ROE signals between the coumarin protons and β-CD, which indicated the formation of inclusions. Fig. 5c shows the proportion of the blue light band which rose dramatically as the relative amount of β-CD increased from 0 to 20 equivalent of NP4C because of the host–guest interaction.
:1 binding stoichiometry.45–48 When β-CD captured the coumarin groups of NP4C, the CT process could be efficiently disturbed due to the isolation between the donor and acceptor. As presented in Fig. S11,† the protons of aromatic rings underwent downfield shifts after the addition of β-CD. The π–π stacking between the coumarin group and the PN unit could induce the upfield movement of their chemical shifts. The host–guest inclusion from β-CD impeded the π–π stacking from the self-folding behavior, and then the protons shifted downfield, although the shielding effect of β-CD on the included coumarin group could generate upfield shifts of its corresponding protons. Besides, the 2D ROESY NMR spectrum of the complex NP4C@β-CD (Fig. S12†) in D2O clearly showed the visible ROE signals between the coumarin protons and β-CD, which indicated the formation of inclusions. Fig. 5c shows the proportion of the blue light band which rose dramatically as the relative amount of β-CD increased from 0 to 20 equivalent of NP4C because of the host–guest interaction.
As a control experiment, the reference compound NPM was also titrated with β-CD but no similar experimental phenomena emerged compared with its PL emission (Fig. S13†) and 1H NMR (Fig. S14†) spectra. Furthermore, the almost unaltered absorption spectra of the NP4C aqueous solution added with β-CD demonstrated no correlations between the PN unit and β-CD (Fig. S15†). The analogous compounds NP2C and NP6C also exhibited the abilities to occupy the cavity of β-CD with their hydrophobic coumarin units by analyzing their corresponding ROESY spectra (Fig. S16 and S17†). Therefore, it could be reasonable to deduce that the hydrophobic cavity of β-CD was inclined to incorporate the coumarin group rather than the charged PN core of NP4C.
Based on the aforementioned attributes, colorful luminescence ranging from blue-purple to yellow-green could be readily switched (Fig. 6b). A number of PL spectra were obtained, and certain corresponding luminescent color coordinates were calculated and plotted in the CIE 1931 chromaticity diagram (Fig. 6a). Interestingly, white-light emission (0.30, 0.33) could be achieved by multiple modes including the excitation wavelength, temperature, solvent polarity and host molecules.
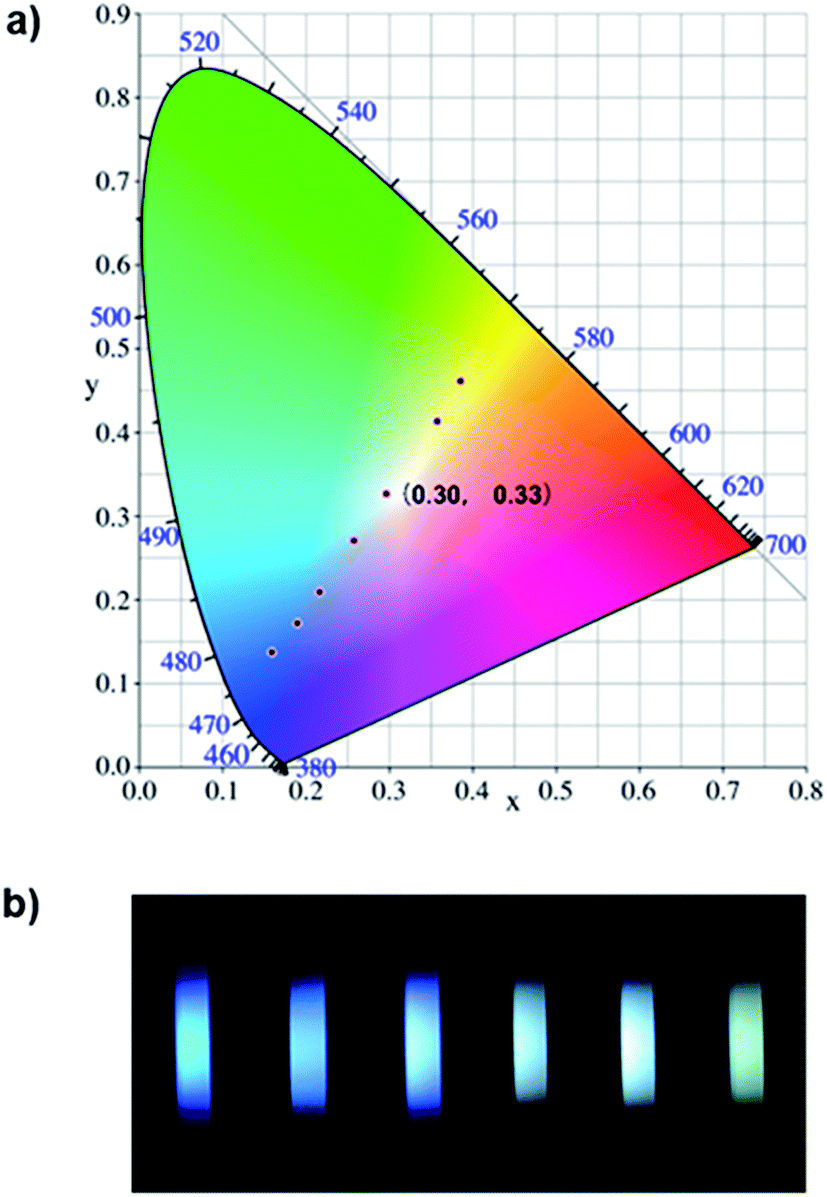 | ||
| Fig. 6 (a) Several luminescent color coordinates for NP4C plotted in the CIE 1931 chromaticity diagram corresponding to the PL emission for multifarious control strategies. (b) Several fluorescence photographs of NP4C solutions in combination with multiple conditions including temperature, solvent polarity and β-CD, [NP4C] = 25 μM. | ||
A white-light emitting (WLE) hydrogel was prepared through the dispersion of NP4C in an agarose gelator. When NP4C was embedded into the hydrogel, a blue-shifted phenomenon occurred to the CT emission of NP4C. Meanwhile, the PL intensity corresponding to the CT emission decreased (Fig. 7). It could be explained that the polyhydroxy structures of agarose might contribute to the hydrogen bonding between its own hydroxyl groups and the coumarin groups of NP4C, which hindered the folding behavior to some extent. Even so, the resultant hydrogel still showed broad dual emissions through the white region upon the variation of excitation wavelength. As expected, it engendered nearly white-light with color coordinates of (0.29, 0.35) when excited at 410 nm (Fig. S18†) and was considered as a perfect candidate for organic white-light emitting materials.
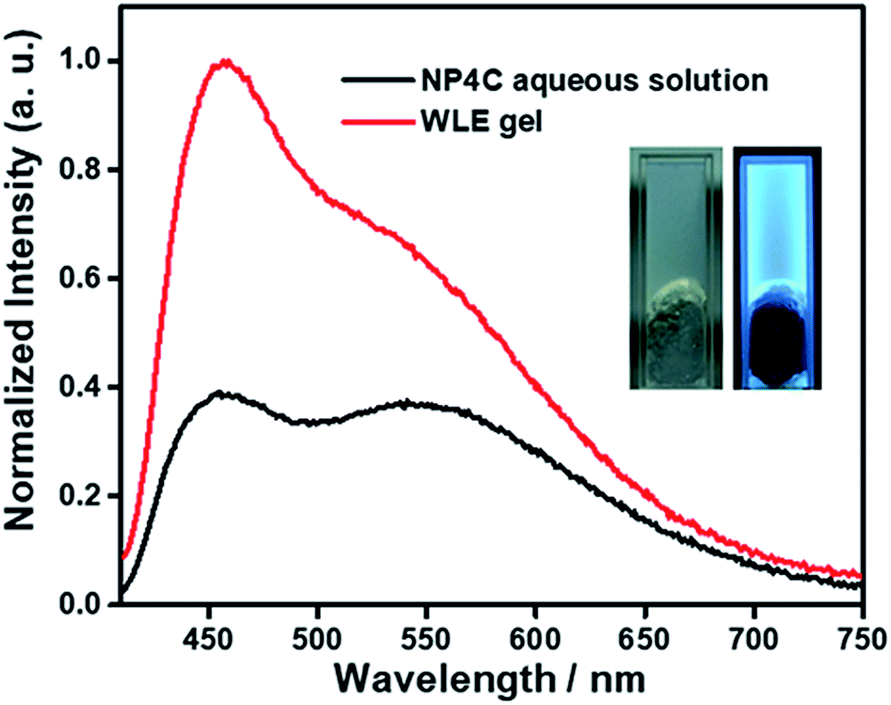 | ||
| Fig. 7 PL emission spectra of the WLE hydrogel (∼2 wt%) and its adopted NP4C aqueous solution excited at 400 nm, [NP4C] = 0.1 mM, and 25 °C. Inset: photographs of the WLE hydrogel in daylight and 365 nm UV light, respectively. | ||
Conclusions
In summary, we have successfully developed white-light-emitting systems in water at the single-molecule scale utilizing a new class of organic molecules with natural self-folded conformation. Our results demonstrated that this particular folding behavior could enhance the ICT efficiency and lead to pure white-light emission. Multicolor photoluminescence including the white-light one could be finely tuned in various ways: (1) by regulating the distance between the donor and acceptor groups by increasing the methanol fraction in the solvent; (2) by using high temperature to expedite the speed of structural transition from the self-folded state to the stretched state; and (3) by hindering the donor units from the acceptor core by the host–guest interaction. A WLE hydrogel was also facilely prepared through the dispersion of NP4C in a commercial agarose gelator. This innovative design of single-molecule compounds with white-light emission in aqueous solution is pioneering in the field, paving the pathway for future chemical engineering of white-light-emitting systems in the aqueous phase.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from NSFC (21788102, 21722603, 214210004 and 21476075), Programme of Introducing Talents of Discipline to Universities (B1607), the Innovation Program of Shanghai Municipal Education Commission and the Fundamental Research Funds for the Central Universities. Dr X. Rao is also thanked for refining the manuscript.Notes and references
- J. Kido, M. Kimura and K. Nagai, Science, 1995, 267, 1332–1334 CrossRef PubMed.
- S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Lüssem and K. Leo, Nature, 2009, 459, 234 CrossRef PubMed.
- M. C. Gather, A. Koehnen, A. Falcou, H. Becker and K. Meerholz, Adv. Funct. Mater., 2007, 17, 191–200 CrossRef.
- G. M. Farinola and R. Ragni, Chem. Soc. Rev., 2011, 40, 3467–3482 RSC.
- C. Vijayakumar, V. K. Praveen and A. Ajayaghosh, Adv. Mater., 2009, 21, 2059–2063 CrossRef.
- M. Chen, Y. Zhao, L. Yan, S. Yang, Y. Zhu, I. Murtaza, G. He, H. Meng and W. Huang, Angew. Chem., Int. Ed., 2017, 56, 722–727 CrossRef PubMed.
- S. Santhosh Babu, J. Aimi, H. Ozawa, N. Shirahata, A. Saeki, S. Seki, A. Ajayaghosh, H. Moehwald and T. Nakanishi, Angew. Chem., Int. Ed., 2012, 51, 3391–3395 CrossRef PubMed.
- S. Shinde and S. K. Asha, Macromolecules, 2016, 49, 8134–8145 CrossRef.
- H. Wu, L. Ying, W. Yang and Y. Cao, Chem. Soc. Rev., 2009, 38, 3391–3400 RSC.
- P. T. Furuta, L. Deng, S. Garon, M. E. Thompson and J. M. J. Frechet, J. Am. Chem. Soc., 2004, 126, 15388–15389 CrossRef PubMed.
- M. Zhang, S. Yin, Z. Zhou, M. L. Saha, P. J. Stang, S. Yin, J. Zhang and C. Lu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3044–3049 CrossRef PubMed.
- X. Yan, P. Wang, F. Huang, T. R. Cook and P. J. Stang, Nat. Chem., 2015, 7, 342–348 CrossRef PubMed.
- M.-S. Wang, S.-P. Guo, Y. Li, L.-Z. Cai, J.-P. Zou, G. Xu, W.-W. Zhou, F.-K. Zheng and G.-C. Guo, J. Am. Chem. Soc., 2009, 131, 13572–13573 CrossRef PubMed.
- D. F. Sava, L. E. S. Rohwer, M. A. Rodriguez and T. M. Nenoff, J. Am. Chem. Soc., 2012, 134, 3983–3986 CrossRef PubMed.
- J. H. Park, J. Y. Kim, B. D. Chin, Y. C. Kim, J. K. Kim and O. O. Park, Nanotechnology, 2004, 15, 1217–1220 CrossRef.
- S. Huo, T. Jiao, Q. Peng, S. Huo, P. Duan, M. Liu and M. Liu, Angew. Chem., Int. Ed., 2017, 56, 12174–12178 CrossRef PubMed.
- J. Malinge, C. Allain, A. Brosseau and P. Audebert, Angew. Chem., Int. Ed., 2012, 51, 8534–8537 CrossRef PubMed.
- A. Layek, P. C. Stanish, V. Chirmanov and P. V. Radovanovic, Chem. Mater., 2015, 27, 1021–1030 CrossRef.
- X. Zhang, S. Rehm, M. M. Safont-Sempere and F. Würthner, Nat. Chem., 2009, 1, 623 CrossRef PubMed.
- Y. Sun, Y. Lei, L. Liao and W. Hu, Angew. Chem., Int. Ed., 2017, 56, 10352–10356 CrossRef PubMed.
- Q.-W. Zhang, D. Li, X. Li, P. B. White, J. Mecinovic, X. Ma, H. Agren, R. J. M. Nolte and H. Tian, J. Am. Chem. Soc., 2016, 138, 13541–13550 CrossRef PubMed.
- X.-L. Ni, S. Chen, Y. Yang and Z. Tao, J. Am. Chem. Soc., 2016, 138, 6177–6183 CrossRef PubMed.
- R. Abbel, C. Grenier, M. J. Pouderoijen, J. W. Stouwdam, P. E. L. G. Leclere, R. P. Sijbesma, E. W. Meijer and A. P. H. J. Schenning, J. Am. Chem. Soc., 2009, 131, 833–843 CrossRef PubMed.
- D. Li, F. Lu, J. Wang, W. Hu, X.-M. Cao, X. Ma and H. Tian, J. Am. Chem. Soc., 2018, 140, 1916–1923 CrossRef PubMed.
- Z. Xie, C. Chen, S. Xu, J. Li, Y. Zhang, S. Liu, J. Xu and Z. Chi, Angew. Chem., Int. Ed., 2015, 54, 7181–7184 CrossRef PubMed.
- D. Tu, P. Leong, S. Guo, H. Yan, C. Lu and Q. Zhao, Angew. Chem., Int. Ed., 2017, 56, 11370–11374 CrossRef PubMed.
- Q.-Y. Yang and J.-M. Lehn, Angew. Chem., Int. Ed., 2014, 53, 4572–4577 CrossRef PubMed.
- C. Hang, H.-W. Wu and L.-L. Zhu, Chin. Chem. Lett., 2016, 27, 1155–1165 CrossRef.
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- S. Mukherjee and P. Thilagar, Dyes Pigm., 2014, 110, 2–27 CrossRef.
- K.-C. Tang, M.-J. Chang, T.-Y. Lin, H.-A. Pan, T.-C. Fang, K.-Y. Chen, W.-Y. Hung, Y.-H. Hsu and P.-T. Chou, J. Am. Chem. Soc., 2011, 133, 17738–17745 CrossRef PubMed.
- J. Zhao, S. Ji, Y. Chen, H. Guo and P. Yang, Phys. Chem. Chem. Phys., 2012, 14, 8803–8817 RSC.
- S. Park, J. E. Kwon, S. H. Kim, J. Seo, K. Chung, S.-Y. Park, D.-J. Jang, B. M. Medina, J. Gierschner and S. Y. Park, J. Am. Chem. Soc., 2009, 131, 14043–14049 CrossRef PubMed.
- W. Rettig, Angew. Chem., Int. Ed., 1986, 25, 971–988 CrossRef.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
- Y. Yang, M. Lowry, C. M. Schowalter, S. O. Fakayode, J. O. Escobedo, X. Xu, H. Zhang, T. J. Jensen, F. R. Fronczek, I. M. Warner and R. M. Strongin, J. Am. Chem. Soc., 2006, 128, 14081–14092 CrossRef PubMed.
- H. V. Huynh, X. He and T. Baumgartner, Chem. Commun., 2013, 49, 4899–4901 RSC.
- J. Liu, Z. Diwu and W.-Y. Leung, Bioorg. Med. Chem. Lett., 2001, 11, 2903–2905 CrossRef PubMed.
- D. Liu, Z. Zhang, H. Zhang and Y. Wang, Chem. Commun., 2013, 49, 10001–10003 RSC.
- M. R. Molla, D. Gehrig, L. Roy, V. Kamm, A. Paul, F. Laquai and S. Ghosh, Chem.–Eur. J., 2014, 20, 760–771 CrossRef PubMed.
- M. R. Molla and S. Ghosh, Chem.–Eur. J., 2012, 18, 1290–1294 CrossRef PubMed.
- Z. Zhang, Y.-S. Wu, K.-C. Tang, C.-L. Chen, J.-W. Ho, J. Su, H. Tian and P.-T. Chou, J. Am. Chem. Soc., 2015, 137, 8509–8520 CrossRef PubMed.
- Z. He, W. Zhao, J. W. Y. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, Nat. Commun., 2017, 8, 416 CrossRef PubMed.
- T. Liu and S. Wu, Wuli Huaxue Xuebao, 1996, 12, 677–683 Search PubMed.
- T. M. Guardado-Alvarez, L. Sudha Devi, M. M. Russell, B. J. Schwartz and J. I. Zink, J. Am. Chem. Soc., 2013, 135, 14000–14003 CrossRef PubMed.
- J. N. Moorthy, K. Venkatesan and R. G. Weiss, J. Org. Chem., 1992, 57, 3292–3297 CrossRef.
- H. Takakusa, K. Kikuchi, Y. Urano, T. Higuchi and T. Nagano, Anal. Chem., 2001, 73, 939–942 CrossRef PubMed.
- M. A. Hossain, H. Mihara and A. Ueno, J. Am. Chem. Soc., 2003, 125, 11178–11179 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experiments, additional spectroscopic data and computational simulation details. See DOI: 10.1039/c8sc01915k |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |