
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluoranthene-based dopant-free hole transporting materials for efficient perovskite solar cells†
Xianglang
Sun‡
a,
Qifan
Xue‡
c,
Zonglong
Zhu
*b,
Qi
Xiao
a,
Kui
Jiang
b,
Hin-Lap
Yip
 c,
He
Yan
*b and
Zhong’an
Li
*a
c,
He
Yan
*b and
Zhong’an
Li
*a
aKey Laboratory for Material Chemistry of Energy Conversion and Storage, Ministry of Education, School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, 430074, Wuhan, P. R. China. E-mail: lizha@hust.edu.cn
bDepartment of Chemistry, Energy Institute and Hong Kong Branch of Chinese National Engineering Research Center for Tissue Restoration and Reconstruction, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China. E-mail: zzhuab@connect.ust.hk; hyan@ust.hk
cInstitute of Polymer Optoelectronic Materials and Devices, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, 510006, Guangzhou, P. R. China
First published on 2nd February 2018
Abstract
Significant efforts have been devoted to developing new dopant-free hole transporting materials (HTMs) for perovskite solar cells (PVSCs). Fluoranthene is one typical cyclopentene-fused polycyclic aromatic hydrocarbon with a rigid planarized structure, and thus could be an ideal building block to construct dopant-free HTMs, which have not been reported yet. Here, we report a new and simple synthetic method to prepare unreported 2,3-dicyano-fluoranthene through a Diels–Alder reaction between dibenzofulvene and tetracyanoethylene, and demonstrate that it can serve as an efficient electron-withdrawing unit for constructing donor–acceptor (D–A) type HTMs. This novel building block not only endows the resulting molecules with suitable energy levels, but also enables highly ordered and strong molecular packing in solid states, both of which could facilitate hole extraction and transport. Thus with dopant-free HTMs, impressive efficiencies of 18.03% and 17.01% which are associated with enhanced stability can be achieved based on conventional n–i–p and inverted p–i–n PVSCs respectively, outperforming most organic dopant-free HTMs reported so far.
Introduction
Organic–inorganic hybrid perovskite solar cells (PVSCs) have triggered worldwide attention due to their impressive research progress within a short time and their very promising market prospects.1–5 Recently, the record-high power conversion efficiency (PCE) of PVSCs has reached the certified value of 22%, almost rivaling that of the crystalline silicon based photovoltaics.6 When fabricating PVSCs, the introduction of suitable interfacial materials, i.e. electron transporting materials (ETMs) and hole transporting materials (HTMs), is critical for achieving high device performance, because they not only improve the charge carrier transport/collection efficiency, but also act as a protective layer for perovskites to enhance device stability.7–11For HTMs applied in PVSCs, organic semiconductors are more popular over their inorganic counterparts which is attributed to their milder processing conditions that are compatible with perovskites. Nevertheless, most organic HTMs exhibit relatively low hole mobility, and thus need to be improved through a chemical doping process by ionic dopants such as Li–bis(trifluoromethanesulfonyl)imide (Li–TFSI).12–16 However, the doping process always induces a negative effect on device stability due to the sophisticated oxidation process associated with undesired ion migration/interactions.17,18 For example, the device PCE derived from the well-known doped HTM, 2,2′,7,7′-tetrakis(N,N-bis(p-methoxyphenyl)amino)-9,9′-spirobifluorene (spiro-OMeTAD), often vanished after 30 days of ambient storage, even though the maximum of its initial PCE was close to 19%.19 Thus, development of efficient dopant-free HTMs is urgently needed. However few dopant-free HTMs can show comparable PCEs to the doped spiro-OMeTAD.18,20–26
Normally, HTMs would not need an additional doping process if they exhibit hole mobility up to 10−4–10−3 cm2 V−1 s−1.27 To this end, two molecular design strategies, donor–acceptor (D–A) type and star-shaped structure, have been mainly used for designing dopant-free HTMs with better intermolecular interactions to ensure sufficient hole mobility.17,28–34 By integrating these two strategies, Nazeeruddin et al. prepared a new class of star-shaped D–A molecules that served as dopant-free HTMs towards ∼19% PCE with enhanced device stability.19,35 Nonetheless, most of the reported D–A type HTMs come from relatively complicated scaffolds with multistep syntheses and purification processes. We recently reported a new dipolar chromophore based dopant-free HTM via a facile synthesis, which afforded a high PCE of 16.9%.36 Following this success, it is crucial to exploit new D–A combinations towards high performance dopant-free HTMs with low synthetic complexity.
One of the key factors for designing high performance HTMs is designing a suitable core structure. Fluoranthene is one typical cyclopentene-fused polycyclic aromatic hydrocarbon, which exhibits a rigid planarized structure that can favor enhanced π–π stacking. More importantly, fluoranthene, as a substructure of fullerene, shows a typical electron-deficient character due to the cyclopenta-fused non-alternant structure.37–40 Thus, fluoranthene may be an ideal electron-withdrawing building block to construct D–A type HTMs for PVSCs, which have not yet been reported, possibly due to a lack of efficient molecular design strategy. In this study, we describe a new and facile synthetic method to prepare an unreported 2,3-dicyano-fluoranthene moiety as an efficient core towards high performance dopant-free HTMs.
Results and discussion
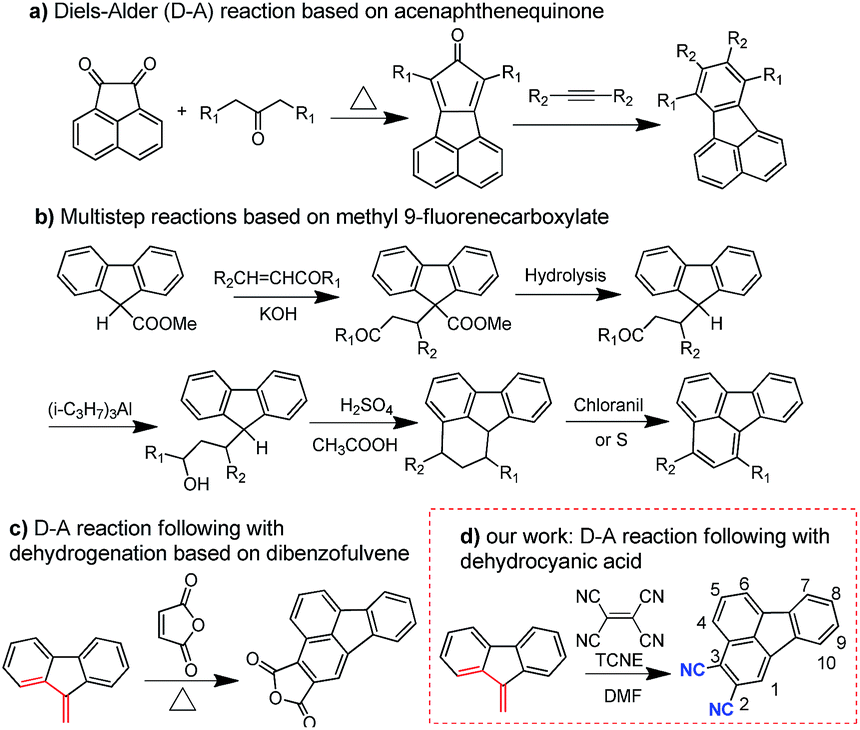
The synthetic route of the HTMs is shown in Scheme 1, while the details are provided in the Experimental section in the ESI.† First, we prepared fluoranthene-cored BTF1–2via a simple Buchwald–Hartwig coupling reaction between the readily accessible 3,8-dibromo-fluoranthene (2)41 and diphenylamine units (3–4). So far, facile synthesis of functionalized fluoranthene derivatives remains a challenge. One of the most widely used methods of affording fluoranthenes is through a typical Diels–Alder (D–A) reaction based on acenaphthenequinone derivatives (Scheme 2a), but the functionalized positions of this method are always limited, mainly to the C7–C10 positions.42,43 Another typical method proposed by Stubbs and Tucker et al. employs fluorene derivatives as the starting materials (Scheme 2b), but the total synthetic efficiency is too poor due to the tedious multistep reactions.44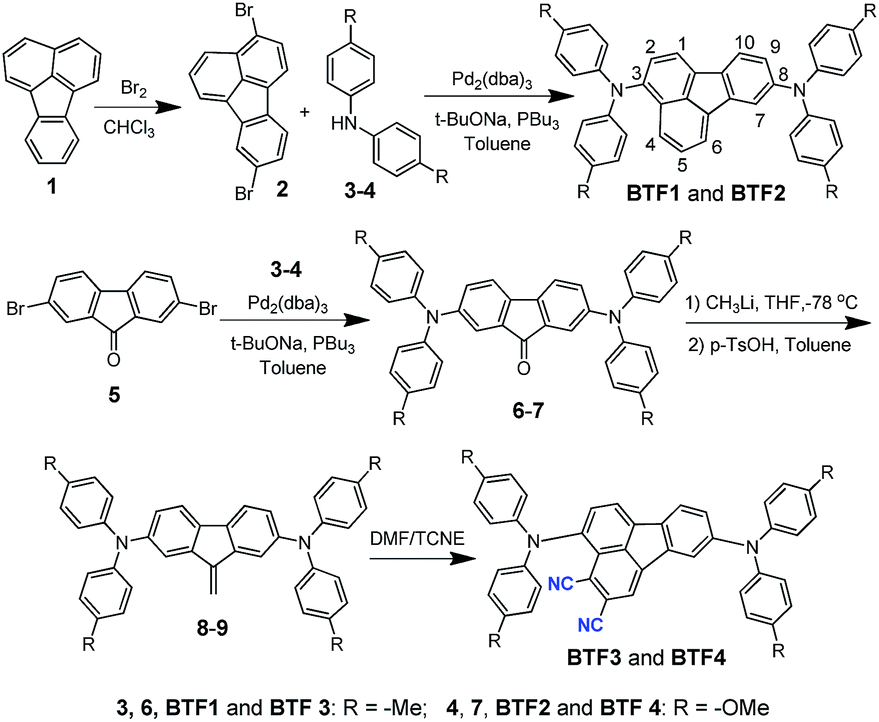 | ||
| Scheme 1 Synthetic route for fluoranthene-cored HTMs (BTF1–4). | ||
 | ||
| Scheme 2 Synthetic methods for functionalized fluoranthenes. | ||
Through careful searching, we encouragingly found that in 1949, dibenzofulvene and maleic anhydride were reported as the starting materials by Wang et al. to yield 2,3-substituted fluoranthene based on the D–A reaction followed by dehydrogenation (Scheme 2c).45 Although the yield of this reaction is low, only ∼10%, the simple and facile procedure is very attractive for us. Thus, with a purpose of synthesizing a more electron-deficient fluoranthene core, tetracyanoethylene (TCNE) was selected as the dienophile to react with dibenzofulvene (Scheme 2d) through the D–A reaction but followed by dehydrocyanic acid. Excitingly, 2,3-dicyano-fluoranthene was successfully obtained as we expected, in spite of a low isolated yield of 3.5%. With an indirect method, we prepared diphenylamine moieties substituted on dibenzofulvene (8–9) first, and then used them as the critical intermediates to react with TCNE as shown in Scheme 1, which successfully obtained our desired products, BTF3–4, with enhanced isolated yields (29.1% for BTF3 and 43.0% for BTF4). The electron-rich substitutions were found to activate the reactivity of fulvene. This can be further evidenced by the higher yield of BTF4, as methoxyl groups in 9 make diene more electron-rich. Moreover, due to the facile synthesis, all synthetic costs, 7.1 $ g−1, 11.4 $ g−1, 53.1 $ g−1 and 62.8 $ g−1 for BTF1–4 (Tables S1–S4†) respectively, are much lower than that of spiro-OMeTAD, making them good material candidates suitable for large scale production.
The ground-state geometric structures of BTF3 (triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group) and BTF4 (monoclinic, P21/c space group) were refined using X-ray crystallography, and the detailed data are shown in Tables S5–S6† in the ESI.†Fig. 1 shows the packing arrangements of BTF3–4. According to the relative arrangement between molecules and contact positions, four types of molecular packing mode are found (Fig. 1a and S3–S4†), including H-type dimeric stacking through dipole–dipole interactions due to dicyano-substitutions (mode 1), H-type dimeric stacking through π–π interactions between two adjacent molecules (mode 2), J-type dimeric stacking through π–π interactions between two adjacent diphenylamine units (mode 3), and herringbone stacking through π–π interactions between two adjacent molecules (mode 4).
space group) and BTF4 (monoclinic, P21/c space group) were refined using X-ray crystallography, and the detailed data are shown in Tables S5–S6† in the ESI.†Fig. 1 shows the packing arrangements of BTF3–4. According to the relative arrangement between molecules and contact positions, four types of molecular packing mode are found (Fig. 1a and S3–S4†), including H-type dimeric stacking through dipole–dipole interactions due to dicyano-substitutions (mode 1), H-type dimeric stacking through π–π interactions between two adjacent molecules (mode 2), J-type dimeric stacking through π–π interactions between two adjacent diphenylamine units (mode 3), and herringbone stacking through π–π interactions between two adjacent molecules (mode 4).
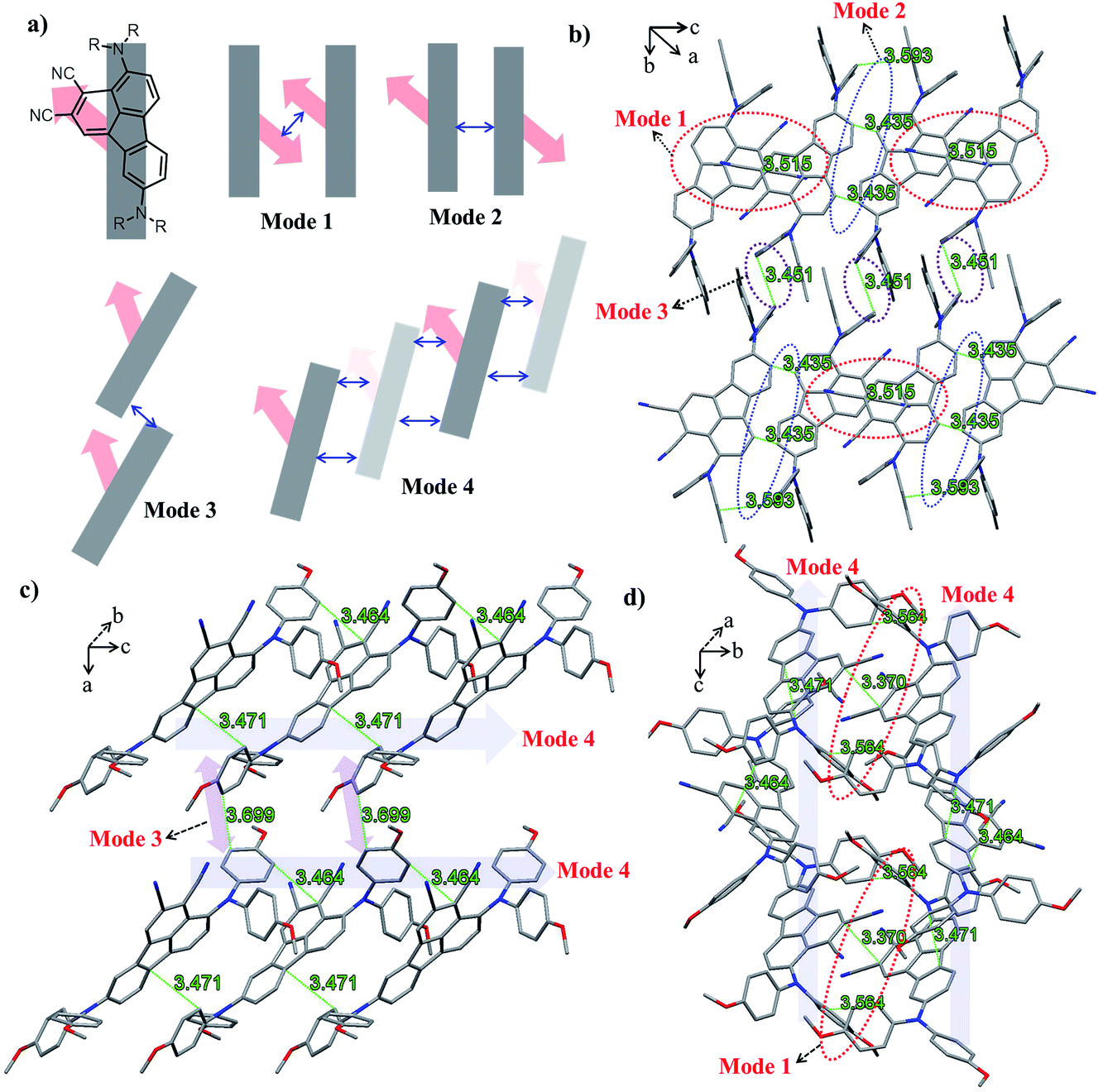 | ||
| Fig. 1 (a) Schematic representations of the π-stacking modes of BTF3–4 based on single-crystal X-ray analyses, where block arrows show the direction of dipole moments, and double arrow lines represent the positions of π-stacking; (b) π-stacks of BTF3 viewed along the a axis; (c) π-stacks of BTF4 viewed along the b axis; and (d) π-stacks of BTF4 viewed along the a axis. For clarity, the hydrogen atoms and solvent molecules in BTF3 are omitted. The π-stacking distances are noted in green. | ||
As shown in Fig. 1b, BTF3 molecules pack into highly ordered and extended H-aggregates along the (0–11) plane, wherein two types of molecular stacking (mode 1 and 2) appear in an alternating fashion. In stacking mode 1, the stacking distance between two dicyano-fluoranthene units is 3.515 Å, while that in stacking mode 2 is 3.435 Å. Except for short stacking distance, the contact area in mode 2 is much larger than that in mode 1, as the diphenylamine units also show a close contact with each other with a distance of 3.593 Å. Moreover, it is encouragingly found that BTF3 molecules in two adjacent H-aggregates can pack into highly ordered J-like aggregates along the (100) plane based on stacking mode 3 with a π–π-distance of 3.451 Å. Thus, the solid state structure of BTF3 can be classified as a two-dimensional “brick-wall”-like assembly.
BTF4 molecules interestingly show a highly ordered herringbone assembly with partial edge-to-face packing along the (101) plane (stacking mode 4, Fig. 2c). Due to the asymmetric nature of cyano-fluoranthene, they make contact in three different positions (Fig. S4†) with a π–π-distance of 3.464 Å, 3.471 Å, and 3.617 Å respectively, making the molecular packing very dense. Differently from those observed in BTF3, the neighboring herringbone assemblies interact with each other along both the (110) and (101) planes, based on stacking mode 3 and 1 respectively. Thus, the packing structure of BTF4 can be classified as a quasi-three-dimensional herringbone assembly. The edge-to-face π–π-distance within the dimer in stacking mode 3 for BTF4 is slightly longer (3.669 Å) than that in BTF3. However, a much shorter dipole–dipole interaction distance of 3.37 Å is obtained for BTF4 in stacking mode 4, attributed to the close edge-to-face π-contact with a distance of 3.564 Å between diphenylamine units that also occurs. In contrast, we only obtained a low-quality single crystal of BTF1 for structure analysis, which indicates an absence of strong π–π interactions (Fig. S5†). Based on these results, we can conclude that cyano-substitution plays a critical role in enabling strong intermolecular interactions in solid states, which is expected to be beneficial for achieving efficient charge transport.
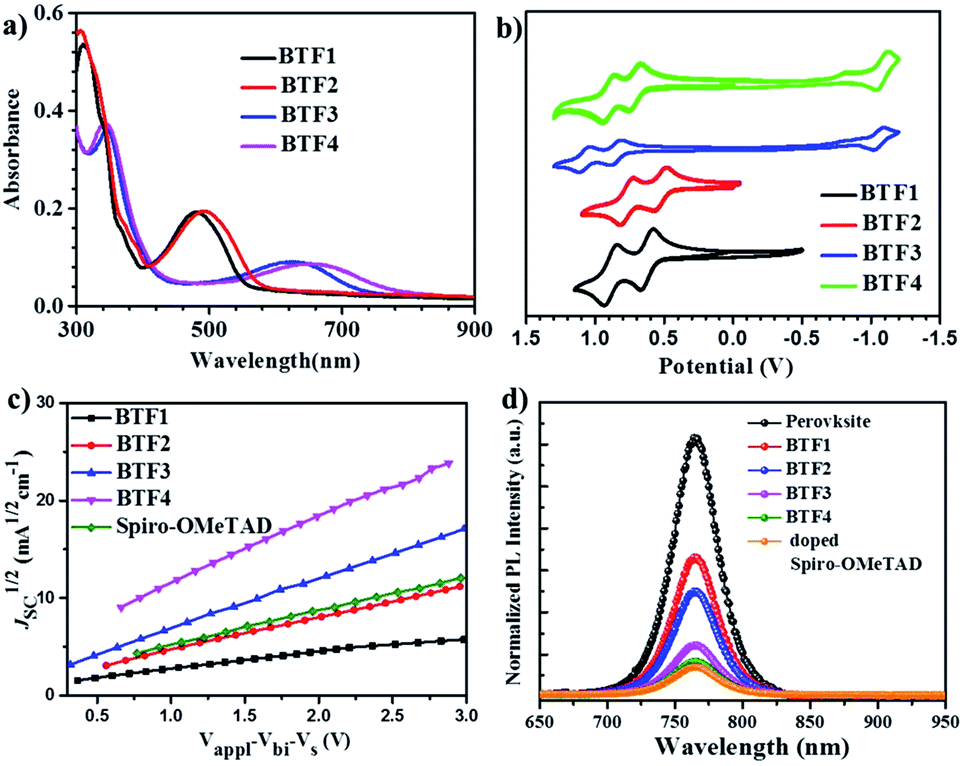 | ||
| Fig. 2 (a) The film absorption spectra. (b) The CV curves versus Fe/Fe+ (0.66 V) measured in DCM solution. (c) The hole injection characteristics measured by the SCLC method based on the device structure of ITO/V2O5/HTMs/V2O5/Al. (d) Steady state PL spectra of the bilayered perovskite films with or without capping with different HTLs. | ||
The absorption spectra of BTF1–4 in diluted solutions and as thin films are shown in Fig. S6† and 2a respectively, with related data listed in Table 1. All spectra show two distinct absorption bands, ascribed to localized π–π* transitions and intramolecular charge transfer (ICT). By comparing the film spectra with their corresponding solution spectra, it is found that the low-energy absorption bands of BTF3–4 are more red-shifted than those of BTF1–2, suggesting that aggregation of the former is more pronounced, which is consistent with the results from the crystal structure analysis. Both methoxy- and cyano-substitutions result in a red-shift in the absorption bands, while the red-shift degree for the latter is much larger, up to 150 nm of the absorption maximum for BTF4, ascribed to more effective ICT. The absorption edges of BTF1–4 in thin films are determined to be 554, 575, 731 and 781 nm respectively, corresponding to the optical bandgaps (Eopt, Table 1) of 2.24, 2.16, 1.70, and 1.59 eV respectively.
| HTMs | λ sol (nm)a | λ fil (nm)b | E opt (eV)c | E HOMO (eV)d | E LUMO (eV) | μ (cm2 V−1 s−1)f |
|---|---|---|---|---|---|---|
| a Absorption maxima in DCM solutions. b Absorption maxima of thin films. c Optical band gap calculated from film absorption edges. d Measured from electrochemistry experiments. e Calculated by an equation of ELUMO = EHOMO + Eopt. f Measured by SCLC method. | ||||||
| BTF1 | 304![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 475 475 |
310481 |
2.24 | −4.94 | −2.68e | 1.46 × 10−5 |
| BTF2 | 305482 |
308493 |
2.16 | −4.80 | −2.59e | 2.13 × 10−5 |
| BTF3 | 344609 |
347629 |
1.70 | −5.19 | −3.46d | 6.36 × 10−5 |
| BTF4 | 344632 |
344654 |
1.59 | −5.02 | −3.38d | 1.17 × 10−4 |
The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels were determined by cyclic voltammetry (CV), and the CV curves in DCM solutions are given in Fig. 2b, with data summarized in Table 1. BTF3–4 display reversible oxidative and reductive processes, while only oxidative processes are observed for BTF1–2. The HOMO levels of BTF1–4 are obtained as −4.94, −4.80, −5.19, and −5.02 eV respectively, versus Fc/Fc+. As shown, the cyano-substitution results in a decrease in the HOMO levels for BTF3–4, making them more compatible with perovskites to facilitate hole extraction (Fig. S10 and S17†). Moreover, we noted that the introduction of electron-rich methoxy groups can induce an increase in the HOMO levels. The LUMO levels of BTF3–4 are calculated from the onset of reduction potential, −3.46 and −3.38 eV respectively, while those of BTF1–2 are determined as −2.68 and −2.59 eV respectively, by subtracting Eopt from the HOMO levels.
The hole mobilities (μ) of BTF1–4 without adding any dopants were measured by the space-charge-limited-current (SCLC) method (Fig. 2c). As shown in Table 1, the μ values of BTF3–4 are 6.36 × 10−5 and 1.17 × 10−4 cm2 V−1 s−1respectively, much higher than those of BTF1–2 and spiro-OMeTAD (2.36 × 10−5 cm2 V−1 s−1). The high mobility of BTF4 would fulfill the needs of dopant-free HTMs. This can be attributed to its quasi-three-dimensional supramolecular assembly associated with much closer molecular stacking. We also characterized the steady-state photoluminescence (PL) spectra (Fig. 2d) of bilayered perovskite/non-doped HTL films to check their ability to extract holes. As seen, the PL of all bilayered films can be quenched by the introduction of hole transporting layers (HTLs), most effectively for BTF4, followed by BTF3, BTF2 and then BTF1. It is worth noting that the quenching effectiveness of dopant-free BTF3–4 is even comparable to that of doped spiro-OMeTAD (Fig. 2d), suggesting that holes are extracted efficiently from perovskites for BTF3–4 even without adding any dopants.
We fabricated conventional n–i–p planar devices with a configuration of FTO/SnO2/PCBM/mixed perovskite/HTL/MoO3/Au to evaluate the efficacy of BTF1–4 as dopant-free HTMs. Mixed perovskite (FAPbI3)0.85(MAPbBr3)0.15 (FA: NH = CHNH3+; MA: CH3NH3+) was used as the active light-harvesting layer. The device fabrication details are described in the ESI.† The morphology of BT1–4 thin films on perovskites has been studied through scanning electron microscopy (SEM), and the images are shown in Fig. S11.† As can be clearly seen, all HTLs possess a smooth surface without pin-holes.
The current density–voltage (J–V) curves of the best-performing conventional PVSCs using dopant-free HTMs are shown in Fig. 3a, with related photovoltaic parameters listed in Table 2. The champion devices based on dopant-free BTF1 and BTF2 showed limited PCEs of 9.97% and 10.45% respectively, which are only slightly enhanced compared to that of spiro-OMeTAD-based dopant-free control devices (9.33%) under the same fabrication conditions. In contrast, the device performance of dopant-free BTF3–4 can be significantly improved, due to their more suitable HOMO levels and enhanced hole mobilities. The champion PVSC based on dopant-free BTF4 delivered a very impressive PCE of 18.03% with an open-circuit voltage (Voc) of 1.06 V, a short-circuit photocurrent (Jsc) of 22.5 mA cm−2, and a FF of 75.6%, which is among the best for dopant-free devices reported so far.25 For BTF3, its dopant-free devices exhibited a slightly decreased device PCE of 16.34%, mainly due to a reduction in the Jsc value caused by its inferior hole mobility compared to that of BTF4. To understand the charge transfer behaviors of these dopant-free HTMs, the electrochemical impedance spectroscopy (EIS) of fabricated cells was further measured at open circuit with a frequency ranging from 1 Hz to 1 MHz, and the Nyquist plots are shown in Fig. S14.† Similar to hole mobility, the obtained recombination resistance (Rrec) also shows an order of Rrec(BTF4) > Rrec(BTF3) > Rrec(BTF2) ∼ Rrec(spiro-OMeTAD) > Rrec(BTF1). This therefore suggests that BTF3 and BTF4 can efficiently reduce the recombination process with higher recombination resistance to produce higher FFs.
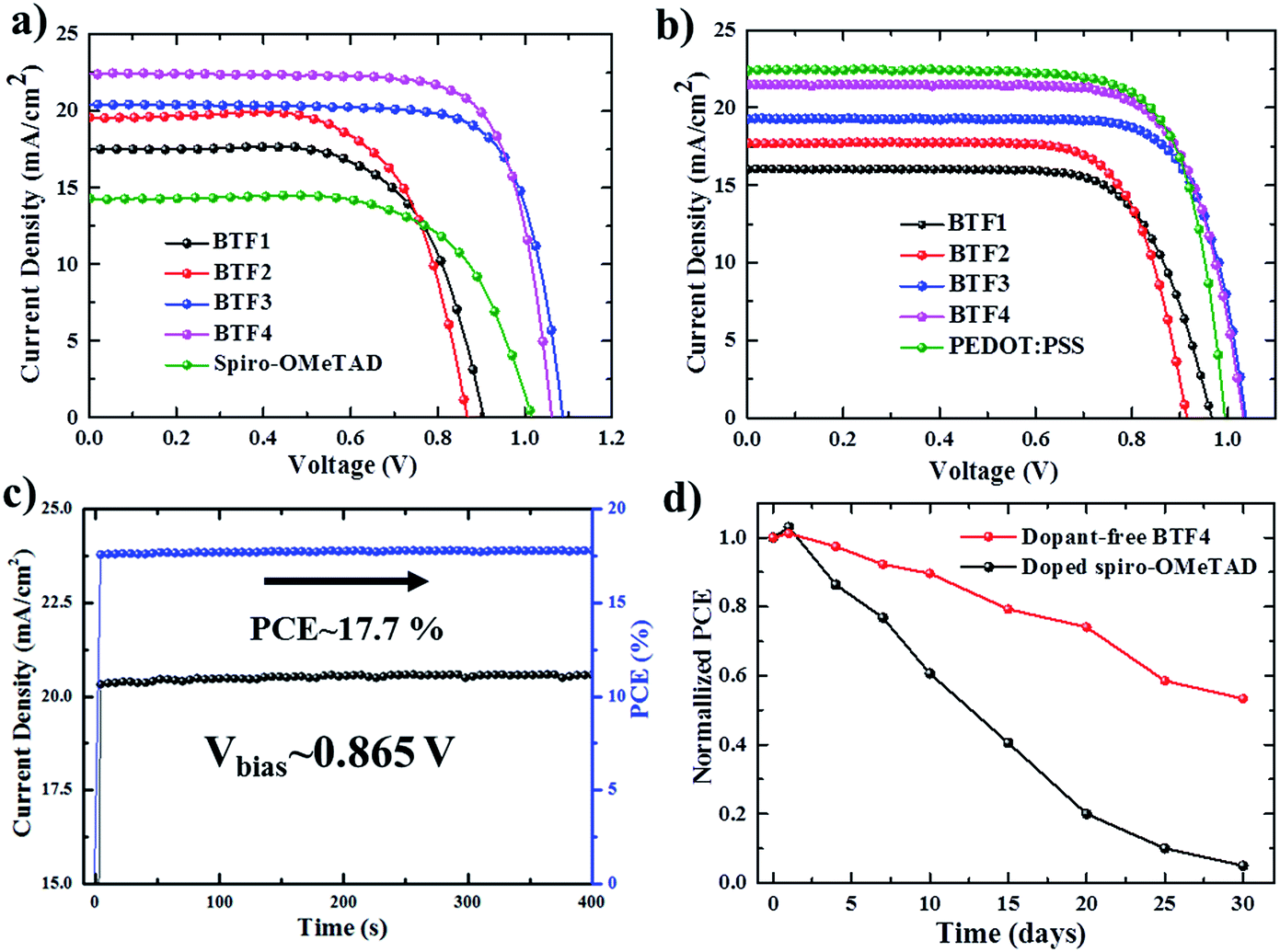 | ||
| Fig. 3 J–V curves of the best-performing conventional (a) and inverted (b) PVSCs with different dopant-free HTMs. (c) Stable output current of a dopant-free BTF4 based conventional PVSC under a constant bias of 0.865 V and (d) the stability test of the conventional PVSCs in ambient air with a humidity of 40–50%. | ||
| HTMs | Device | V oc (V) | J sc (mA cm−2) | FF (%) | PCE (%) |
|---|---|---|---|---|---|
| BTF1 | n–i–p | 0.91(0.90 ± 0.01) | 17.5(16.2 ± 1.3) | 62.6(60.7 ± 1.8) | 9.97(8.84 ± 1.12) |
| BTF2 | n–i–p | 0.85(0.84 ± 0.02) | 19.5(18.0 ± 1.4) | 63.1(60.7 ± 2.1) | 10.45(9.16 ± 1.02) |
| BTF3 | n–i–p | 1.08(1.07 ± 0.02) | 20.4(19.2 ± 1.1) | 74.3(72.4 ± 1.8) | 16.34(15.28 ± 1.13) |
| BTF4 | n–i–p | 1.06(1.05 ± 0.01) | 22.5(21.5 ± 1.1) | 75.6(73.7 ± 1.9) | 18.03(16.97 ± 1.05) |
| Spiro-OMeTAD | n–i–p | 1.02(1.01 ± 0.01) | 14.3(13.0 ± 1.3) | 64.0(62.4 ± 1.5) | 9.33(8.21 ± 1.09) |
| BTF1 | p–i–n | 0.96(0.95 ± 0.01) | 16.1(14.9 ± 1.2) | 72.4(70.7 ± 1.5) | 11.19(10.02 ± 0.98) |
| BTF2 | p–i–n | 0.91(0.90 ± 0.02) | 17.7(16.5 ± 1.1) | 74.3(72.7 ± 1.6) | 11.96(10.51 ± 1.02) |
| BTF3 | p–i–n | 1.03(1.02 ± 0.01) | 19.3(18.3 ± 1.1) | 75.9(74.1 ± 1.7) | 15.09(14.14 ± 0.96) |
| BTF4 | p–i–n | 1.03(1.01 ± 0.02) | 21.5(20.5 ± 1.0) | 76.8(75.3 ± 1.5) | 17.01(16.14 ± 0.85) |
| PEDOT:PSS | p–i–n | 0.96(0.94 ± 0.01) | 22.4(21.5 ± 1.0) | 76.4(74.7 ± 1.6) | 16.42(15.48 ± 0.94) |
Doped conventional devices were also fabricated using Li–TFSI as the dopant with an additive of 4-tert-butyl pyridine (TBP). As shown in Fig. S16,† the device PCEs of doped BTF1–2 can be enhanced by 40% due to an effective improvement on both Voc and FF. Nonetheless, such enhancement is still poor in comparison with that of doped spiro-OMeTAD (18.8%). A slight enhancement on PCEs was observed for the doped BTF3–4 based devices, while the highest PCE of 18.94% was achieved for BTF4. These results, on the other hand, suggest that our dicyano-fluoranthene-cored molecules can serve as highly efficient HTMs without needing dopants.
Today, most HTMs used in inverted p–i–n devices are polymer HTMs such as poly(3,4-ethylenedioxythiophene) polystyrenesulfonate (PEDOT:PSS) and polytriarylamine (PTAA).46–49 A doping process is also required for PTAA to improve device performance.50 PEDOT:PSS belongs to a class of self-doped polymers, but always suffers a large potential loss and inherent acidity-induced stability problem.51,52 Therefore, it is also important to develop dopant-free HTMs suitable for inverted PVSCs.53,54 However, few molecular HTMs have been reported to be applied in both conventional and inverted planar PVSCs. To this end, inverted PVSCs with a configuration of ITO/HTL/mixed perovskite/PCBM/LiF/Ag were also tested using BTF1–4 as the dopant-free HTMs, while a PEDOT:PSS-based control device was fabricated for comparison. The J–V curves of the best-performing devices are shown in Fig. 3b. From BTF1 to BTF4, the resulting PCEs of the inverted devices increase gradually (Table 2), a similar trend to that observed in conventional devices. The champion PCE from BTF4-based inverted PVSCs is 17.01%, higher than that of the PEDOT:PSS-based control device (16.42%).
The stabilized PCE and photocurrent of the champion devices of BTF4 near the maximum power point (Fig. 3c and S18b†) were tested to evaluate the efficacy of the J–V curves. The results clearly show the high reliability of our J–V curve with an absence of current hysteresis, when combined with those J–V curves with forward and reverse scanning directions at different scan rates (Fig. S12b and S18c†). We also measured the incident photon-to-electron conversion efficiency (IPCE) spectra of all champion conventional and inverted devices based on dopant-free BTF1–4 (Fig. S15 and S20†), wherein the integrated Jsc values are consistent with those obtained from the experimental J–V measurements (Table 2).
To check if removing the doping process can improve the device stability of PVSCs over doped spiro-OMeTAD, we compared the environmental stability of PVSCs derived from dopant-free BTF4 and doped spiro-OMeTAD without encapsulation. The PCE decay curves are presented in Fig. 3d, wherein it is easily seen that the performance of both devices decreased quickly at the beginning when being stored in ambient air under a relative humidity of 40–50%. However, the dopant-free BTF4 based device can still retain over 50% of its original PCE after being stored for 30 days, while the PCE of the doped spiro-OMeTAD based device almost vanishes. Moreover, BTF4-based inverted devices also showed an enhanced stability and can retain 55% of their original PCE after being stored in air for 7 days, while 90% of the PCE of the PEDOT:PSS-based control device was lost (Fig. S18e†). Thus, our designed fluoranthene-cored dopant-free HTMs not only deliver a high PCE comparable to doped spiro-OMeTAD, but also show an enhanced device stability, suggesting that they are very promising material candidates towards efficient PVSCs.
Conclusions
In summary, 2,3-dicyano-fluoranthene was first prepared through a new and facile synthetic method based on a Diels–Alder reaction. We found that fluoranthene could be an ideal building block for designing D–A type dopant-free HTMs with low synthetic cost and compatible energy levels with perovskites through rational molecular design. The detailed crystal structure analysis indicates that the resulting molecules with dicyano-substituted fluoranthene as the core present highly ordered and strong molecular packing in solid states, and in particular, BTF4 forms a quasi-three-dimensional herringbone assembly, leading to a much higher hole mobility (up to 10−4 cm2 V−1 s−1) than that of spiro-OMeTAD. Encouragingly, our designed molecules can be applied in planar PVSCs as efficient dopant-free HTMs yielding high device performance, including efficiency and stability. For BTF4, impressive PCEs of 18.03% and 17.01% have been achieved for conventional and inverted cells respectively. Therefore, our work not only develops a general material design strategy to achieve efficient dopant-free HTMs for PVSCs, but also provides a new synthetic method to obtain functionalized fluoranthenes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is funded by the National Science Foundation of China (grant no. 21704030). Z. Li and H.-L. Yip acknowledge the financial support from the National 1000 Young Talents Program hosted by China. We would like to thank the Analytical and Testing Center and Research Core Facilities for Life Science in HUST for using their facilities. Dr Xianggao Meng (Central China Normal University) is also appreciated for his help on X-ray structure refinement.Notes and references
- M. A. Green, A. Ho-Baillie and H. J. Snaith, Nat. Photonics, 2014, 8, 506–514 CrossRef CAS.
- S. T. Williams, A. Rajagopal, C. C. Chueh and A. K. Jen, J. Phys. Chem. Lett., 2016, 7, 811–819 CrossRef CAS PubMed.
- L. Meng, J. You, T.-F. Guo and Y. Yang, Acc. Chem. Res., 2016, 49, 155–165 CrossRef CAS PubMed.
- P. Docampo and T. Bein, Acc. Chem. Res., 2016, 49, 339–346 CrossRef CAS PubMed.
- Z. Yu and L. Sun, Adv. Energy Mater., 2015, 5, 1500213 CrossRef.
- https://www.nrel.gov/pv/assets/images/efficiency-chart.png .
- J. Seo, J. H. Noh and S. I. Seok, Acc. Chem. Res., 2016, 49, 562–572 CrossRef CAS PubMed.
- C.-C. Chueh, C.-Z. Li and A. K. Y. Jen, Energy Environ. Sci., 2015, 8, 1160–1189 CAS.
- S. Ameen, M. A. Rub, S. A. Kosa, K. A. Alamry, M. S. Akhtar, H. S. Shin, H. K. Seo, A. M. Asiri and M. K. Nazeeruddin, ChemSusChem, 2016, 9, 10–27 CrossRef CAS PubMed.
- J. Shi, X. Xu, D. Li and Q. Meng, Small, 2015, 11, 2472–2486 CrossRef CAS PubMed.
- H. Kim, K.-G. Lim and T.-W. Lee, Energy Environ. Sci., 2016, 9, 12–30 CAS.
- K. Rakstys, M. Saliba, P. Gao, P. Gratia, E. Kamarauskas, S. Paek, V. Jankauskas and M. K. Nazeeruddin, Angew. Chem., Int. Ed., 2016, 55, 7464–7468 CrossRef CAS PubMed.
- B. Xu, D. Bi, Y. Hua, P. Liu, M. Cheng, M. Grätzel, L. Kloo, A. Hagfeldt and L. Sun, Energy Environ. Sci., 2016, 9, 873–877 CAS.
- A. Molina-Ontoria, I. Zimmermann, I. Garcia-Benito, P. Gratia, C. Roldan-Carmona, S. Aghazada, M. Graetzel, M. K. Nazeeruddin and N. Martin, Angew. Chem., Int. Ed., 2016, 55, 6270–6274 CrossRef CAS PubMed.
- I. Zimmermann, J. Urieta-Mora, P. Gratia, J. Aragó, G. Grancini, A. Molina-Ontoria, E. Ortí, N. Martín and M. K. Nazeeruddin, Adv. Energy Mater., 2017, 7, 1601674 CrossRef.
- I. García-Benito, I. Zimmermann, J. Urieta-Mora, J. Aragó, A. Molina-Ontoria, E. Ortí, N. Martín and M. K. Nazeeruddin, J. Mater. Chem. A, 2017, 5, 8317–8324 Search PubMed.
- Y. Liu, Z. Hong, Q. Chen, H. Chen, W. H. Chang, Y. M. Yang, T. B. Song and Y. Yang, Adv. Mater., 2016, 28, 440–446 CrossRef CAS PubMed.
- S. Kazim, F. J. Ramos, P. Gao, M. K. Nazeeruddin, M. Grätzel and S. Ahmad, Energy Environ. Sci., 2015, 8, 1816–1823 CAS.
- S. Paek, P. Qin, Y. Lee, K. T. Cho, P. Gao, G. Grancini, E. Oveisi, P. Gratia, K. Rakstys, S. A. Al-Muhtaseb, C. Ludwig, J. Ko and M. K. Nazeeruddin, Adv. Mater., 2017, 29, 1606555 CrossRef PubMed.
- P. Qin, H. Kast, M. K. Nazeeruddin, S. M. Zakeeruddin, A. Mishra, P. Bäuerle and M. Grätzel, Energy Environ. Sci., 2014, 7, 2981–2985 CAS.
- P. Qin, S. Paek, M. I. Dar, N. Pellet, J. Ko, M. Gratzel and M. K. Nazeeruddin, J. Am. Chem. Soc., 2014, 136, 8516–8519 CrossRef CAS PubMed.
- M. Cheng, Y. Li, M. Safdari, C. Chen, P. Liu, L. Kloo and L. Sun, Adv. Energy Mater., 2017, 7, 1602556 CrossRef.
- M. Cheng, B. Xu, C. Chen, X. Yang, F. Zhang, Q. Tan, Y. Hua, L. Kloo and L. Sun, Adv. Energy Mater., 2015, 5, 1401720 CrossRef.
- M. Cheng, C. Chen, X. Yang, J. Huang, F. Zhang, B. Xu and L. Sun, Chem. Mater., 2015, 27, 1808–1814 CrossRef CAS.
- X. Sun, D. Zhao and Z. Li, Chin. Chem. Lett., 2018, 29, 219–231 CrossRef.
- Y. Wang, Z. Zhu, C.-C. Chueh, A. K. Y. Jen and Y. Chi, Adv. Energy Mater., 2017, 7, 1700823 CrossRef.
- L. Cali, S. Kazim, M. Graetzel and S. Ahmad, Angew. Chem., Int. Ed., 2016, 55, 2–26 CrossRef.
- C. Chen, M. Cheng, P. Liu, J. Gao, L. Kloo and L. Sun, Nano Energy, 2016, 23, 40–49 CrossRef CAS.
- M. Cheng, K. Aitola, C. Chen, F. Zhang, P. Liu, K. Sveinbjörnsson, Y. Hua, L. Kloo, G. Boschloo and L. Sun, Nano Energy, 2016, 30, 387–397 CrossRef CAS.
- Y. Liu, Q. Chen, H.-S. Duan, H. Zhou, Y. Yang, H. Chen, S. Luo, T.-B. Song, L. Dou, Z. Hong and Y. Yang, J. Mater. Chem. A, 2015, 3, 11940–11947 CAS.
- J. H. Yun, S. Park, J. H. Heo, H.-S. Lee, S. Yoon, J. Kang, S. H. Im, H. Kim, W. Lee, B. Kim, M. J. Ko, D. S. Chung and H. J. Son, Chem. Sci., 2016, 7, 6649–6661 RSC.
- F. Zhang, C. Yi, P. Wei, X. Bi, J. Luo, G. Jacopin, S. Wang, X. Li, Y. Xiao, S. M. Zakeeruddin and M. Grätzel, Adv. Energy Mater., 2016, 6, 1600401 CrossRef.
- G.-W. Kim, J. Lee, G. Kang, T. Kim and T. Park, Adv. Energy Mater., 2018, 8, 1701935 CrossRef.
- J. Lee, M. Malekshahi Byranvand, G. Kang, S. Y. Son, S. Song, G. W. Kim and T. Park, J. Am. Chem. Soc., 2017, 139, 12175–12181 CrossRef CAS PubMed.
- K. Rakstys, S. Paek, P. Gao, P. Gratia, T. Marszalek, G. Grancini, K. T. Cho, K. Genevicius, V. Jankauskas, W. Pisula and M. K. Nazeeruddin, J. Mater. Chem. A, 2017, 5, 7811–7815 CAS.
- Z. Li, Z. Zhu, C. C. Chueh, S. B. Jo, J. Luo, S. H. Jang and A. K. Jen, J. Am. Chem. Soc., 2016, 138, 11833–11839 CrossRef CAS PubMed.
- Y. Zhou, L. Ding, K. Shi, Y. Z. Dai, N. Ai, J. Wang and J. Pei, Adv. Mater., 2012, 24, 957–961 CrossRef CAS PubMed.
- S. Kumar and S. Patil, J. Phys. Chem. C, 2015, 119, 19297–19304 CAS.
- Y. Q. Zheng, Y. Z. Dai, Y. Zhou, J. Y. Wang and J. Pei, Chem. Commun., 2014, 50, 1591–1594 RSC.
- A. Palmaerts, M. V. Haren, L. Lutsen, T. J. Cleij and D. Vanderzande, Macromolecules, 2006, 39, 2438–2440 CrossRef CAS.
- W. Wu and W. Gao, US.Pat., 2016/0268514 A1, 2016.
- Y. Yuan, G. Q. Zhang, F. Lu, Q. X. Tong, Q. D. Yang, H. W. Mo, T. W. Ng, M. F. Lo, Z. Q. Guo, C. Wu and C. S. Lee, Chem.–Asian J., 2013, 8, 1253–1258 CrossRef CAS PubMed.
- C. M. Thompson, G. Occhialini, G. T. McCandless, S. B. Alahakoon, V. Cameron, S. O. Nielsen and R. A. Smaldone, J. Am. Chem. Soc., 2017, 139, 10506–10513 CrossRef CAS PubMed.
- H. W. D. Stubbs and S. H. Tucker, J. Chem. Soc., 1950, 3288–3292 RSC.
- N. Campbell and H. Wang, J. Chem. Soc., 1949, 1513–1515 RSC.
- J. H. Heo, H. J. Han, D. Kim, T. K. Ahn and S. H. Im, Energy Environ. Sci., 2015, 8, 1602–1608 CAS.
- W. S. Yang, J. H. Noh, N. J. Jeon, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Science, 2015, 348, 1234–1237 CrossRef CAS PubMed.
- W. Yan, S. Ye, Y. Li, W. Sun, H. Rao, Z. Liu, Z. Bian and C. Huang, Adv. Energy Mater., 2016, 6, 1600474 CrossRef.
- E. Edri, S. Kirmayer, D. Cahen and G. Hodes, J. Phys. Chem. Lett., 2013, 4, 897–902 CrossRef CAS PubMed.
- Q. Wang, C. Bi and J. Huang, Nano Energy, 2015, 15, 275–280 CrossRef CAS.
- Q. Xue, G. Chen, M. Liu, J. Xiao, Z. Chen, Z. Hu, X.-F. Jiang, B. Zhang, F. Huang, W. Yang, H.-L. Yip and Y. Cao, Adv. Energy Mater., 2016, 6, 1502021 CrossRef.
- K.-G. Lim, S. Ahn, Y.-H. Kim, Y. Qi and T.-W. Lee, Energy Environ. Sci., 2016, 9, 932–939 CAS.
- C. Huang, W. Fu, C. Z. Li, Z. Zhang, W. Qiu, M. Shi, P. Heremans, A. K. Jen and H. Chen, J. Am. Chem. Soc., 2016, 138, 2528–2531 CrossRef CAS PubMed.
- H. Chen, W. Fu, C. Huang, Z. Zhang, S. Li, F. Ding, M. Shi, C.-Z. Li, A. K. Y. Jen and H. Chen, Adv. Energy Mater., 2017, 7, 1700012 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1589253, 1589258 and 1589261. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc05484j |
| ‡ These authors contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2018 |