
Total synthesis of strictamine: a tutorial for novel and efficient synthesis†
Chao
Wang
 ab,
Shiju
Zhang
c,
Yan
Wang
ab,
Sha-Hua
Huang
*c and
Ran
Hong
*ab
ab,
Shiju
Zhang
c,
Yan
Wang
ab,
Sha-Hua
Huang
*c and
Ran
Hong
*ab
aCAS Key Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry (CAS), 345 Lingling Road, Shanghai 200032, China. E-mail: rhong@sioc.ac.cn
bUniversity of Chinese Academy of Sciences, 19A Yuquan Road, Beijing 100049, China
cCollege of Chemical and Environmental Engineering, Shanghai Institute of Technology, 100 Haiquan Road, Shanghai, 201814, China. E-mail: shahua@sit.edu.cn
First published on 17th October 2017
Abstract
When teaching synthetic design in the classroom, the novelty and efficiency of the overall synthetic route are the key values being imparted to the younger generation of synthetic chemists. The creative synthetic design is of great interest to graduate students who are entering into this exciting field for the first time. Strictamine, an akuammiline alkaloid, bears a congested cage-like structure and has interesting biological activities. Numerous attempts have been made toward the total synthesis of this compound since its discovery five decades ago. The synthesis was not completed until last year when synthetic chemists began to apply novel ring construction strategies. In 2016, two research groups reported the total syntheses of strictamine and four reported the formal syntheses. In this Highlight, the synthesis of strictamine is used as a tutorial case to illustrate novelty and efficiency in total synthesis.
 From right to left: Shiju Zhang, Chao Wang, Yan Wang, Sha-Hua Huang and Ran Hong | Chao Wang received his bachelor's degree in pharmaceutics (2011) and master's degree in medicinal chemistry (2015) from the China Pharmaceutical University. After working in Professor Wei-Sheng Tian's group (SIOC) as a research assistant, he then joined the Hong group in 2016 to pursue his PhD on the total synthesis of natural products. Shiju Zhang received his bachelor's degree in pharmacy from Jining Medical University (2015) and now is the second-year graduate student in the Huang group at the Shanghai Institute of Technology. Yan Wang obtained her bachelor's degree from Sichuan University (2008) and PhD at the Shanghai Institute of Organic Chemistry (CAS). After completing her postdoctoral research in the Scripps Research Institute (Prof. Peter G. Schultz), she returned to SIOC and currently focuses on novel synthetic methods for natural product synthesis. Sha-Hua Huang received her PhD in materials science from the Shanghai Institute of Ceramics (CAS) under the supervision of Prof. Zhao-Yin Wen. After working as a Technical Support in Mettler-Toledo for three years, she joined the Shanghai Institute of Technology in 2010 and focuses on the application of biotransformation in chiral drug synthesis. She was promoted as Associate Professor in 2016. Ran Hong graduated from Sichuan University (1996) and obtained his PhD (2001) under the supervision of Prof. Guo-Qiang Lin at SIOC. After conducting postdoctoral research in the laboratories of late Prof. Jay K. Kochi at the University of Houston and Prof. Li Deng at Brandeis University, he was appointed as a Group Leader at SIOC in 2007 and initiated the research program to apply the biomimetic approach in natural product synthesis as well as synthetic method development. |
The primary intention of this study was to use the preparation of these materials to illustrate synthetic design in the classroom.5 When considering the synthetic route to a specific natural product, efficiency is always the main concern. Prior to initiating any synthetic project, we may ask “are we able to reach this target in a more efficient way than that in the existing synthetic studies?” Moreover, the novelty of the chemical transformations, like the application of newly developed powerful synthetic methods as well as employing rarely utilized reactions on intricate substrates, is also taken into consideration. Ideally, these two facets can be combined into one synthetic work to elevate the current synthesis to a high level of chemical knowledge. The synthesis of strictamine is thus selected as an excellent example for illustration.
Strictamine, first isolated by Ganguli and co-workers in 1966 from Rhazya stricta,6 is a representative congener of the akuammiline alkaloids which have indolenine-fused cage-like structures (Scheme 1). It shows in vitro inhibitory activities against nuclear factor-κB, which plays an important role in regulating gene expression in immune and inflammatory responses.7 The intriguing polycyclic chemical structure includes a highly congested methanoquinolizidine core (highlighted in red). The C–D–E rings of the core are connected via the C7–C16 bond and four stereocenters including a challenging C7 quaternary carbon center. Although, since the early 1970s, numerous attempts have been made to complete the total synthesis,8 the synthesis was not achieved until very recently when several research groups solved the long-standing puzzle of this molecule. Six synthetic groups have accomplished the total or formal synthesis of strictamine, making this molecule a hot synthetic target. Herein, we summarize the innovative strategies and methods developed during the chemical synthesis of strictamine.
 | ||
| Scheme 1 Strictamine, its biosynthetic precursor (geissoschizine), and other representative akuammiline alkaloids. | ||
To synthesize this complex structure, different strategies and methodologies have been adopted for building the congested ring system (Scheme 2). In Garg's work, the C5–N4 bond was chosen to complete the D-ring via an intramolecular substitution.9 Intermediate 6 was built up via a Fischer indolization of ketone 7 bearing the C and E rings. The Zhu group disconnected the C15–C20 bond in the E-ring. This simplified strictamine to vinyl iodide intermediate 8, in which the D-ring could be constructed by an intramolecular nucleophilic substitution of brominated 9 (vide infra) bearing the A and C rings.10 Interestingly, the Ohno, Snyder, and Gaich groups also selected compound 8 as their synthetic target. In the Ohno and Snyder syntheses, they used Au(I)-catalyzed 6-endo-dig cyclizations of dihydro-β-carboline derivatives 10 (X varied between groups) to construct the C ring.11 In Gaich's work, the C ring was established through a [2,3]-Stevens rearrangement of precursor 11 bearing the A and D rings.12 The Blakey group completed the synthesis of the strictamine pentacyclic core with a C21-carboxylic acid group and closed the E-ring via amidation.13 A cascade annulation of compound 13 bearing the A and B rings furnished the C and D rings in one step. In the following section, we will highlight the novelty in each synthetic route.
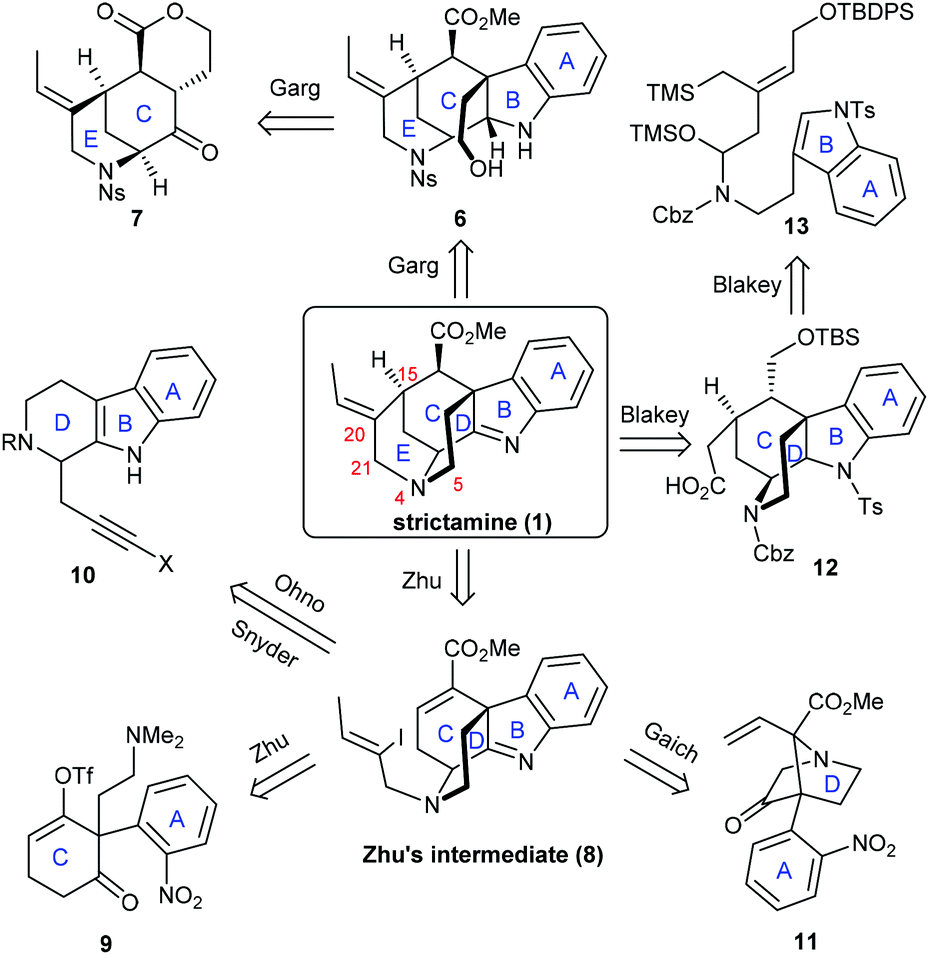 | ||
| Scheme 2 Strategies and tactics in the total synthesis of strictamine. | ||
In 2016, the Garg group at UCLA reported the first enantioselective total synthesis of (+)-strictamine featuring an interrupted Fischer indolization approach (Scheme 3).9 Beginning with dibenzoate 14, desymmetrization, saponification, oxidation and enolization were adopted to prepare silyl enol ether 15. The Toste Au(I)-catalyzed cycloisomerization delivered [3.3.1]-azabicyclic enone 16, which was further elaborated to enal 17 through an epoxidation/Wittig olefination sequence. The full carbon framework in ketolactone 7 was established in seven steps. The subsequent Fischer indolization through intermediate 18 proceeded smoothly, and reduction of the intermediary imine was achieved using a hydride source and afforded 19. The challenging C7 quaternary stereocenter was installed during this step. With 19 in hand, the virtually feasible formation of hydroxyester 6 proved difficult. The C16 stereocenter was readily epimerized under a variety of direct lactone ring-opening conditions. A five-step work-around sequence was eventually developed. It entailed reduction of the lactone, selective silylation of the primary alcohol, and an oxidation/esterification with an in situ deprotection to deliver the requisite hydroxyester 6. Finally, the hydroxyl group was activated by mesylation and chlorination. After PCC oxidation to reveal a ketimine functional group, denosylation and ring closure by the liberated amine completed the total synthesis of strictamine.
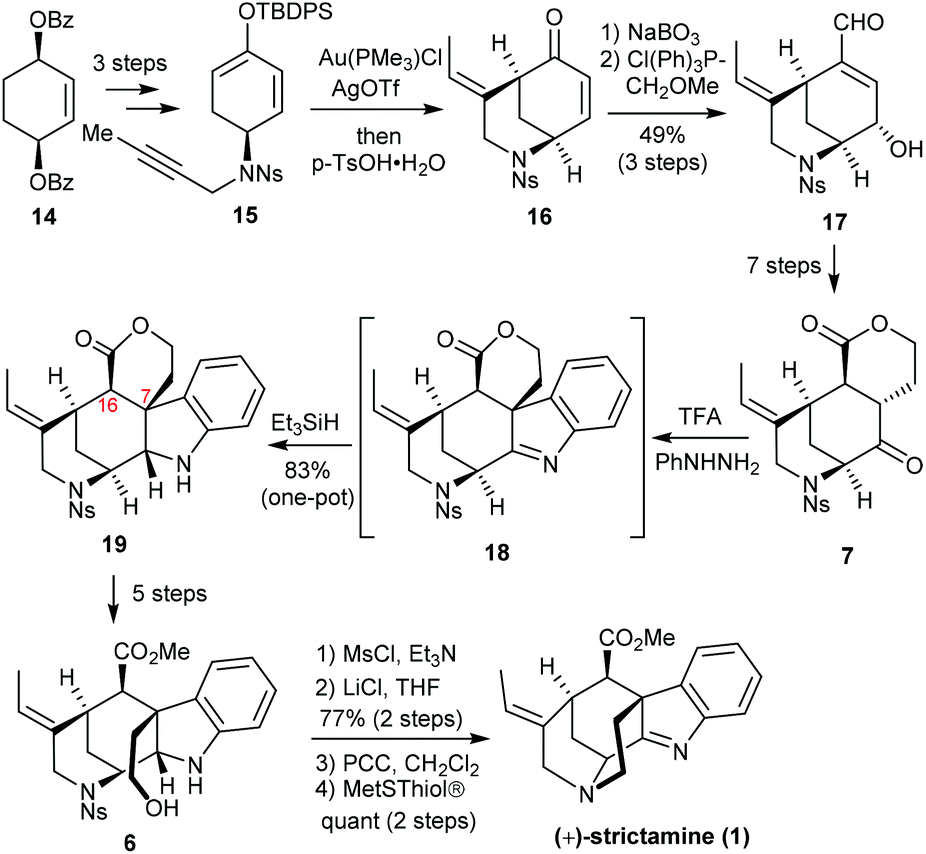 | ||
| Scheme 3 Garg's total synthesis of (+)-strictamine. | ||
The interrupted Fischer indole synthesis with the appropriate precursor smoothly afforded the desired stereochemistry of the quaternary C7 carbon center in the total synthesis of the akuammiline alkaloid. However, the preparation of precursor 7 and elaboration of advanced intermediate 19 to strictamine still required a series of transformations, which attenuates the overall efficiency of this strategy.
The Zhu group at EPFL reported a racemic synthesis of strictamine (Scheme 4).10 In contrast to Garg's approach, they began with the elaboration of 21, which already contained the C7 quaternary stereocenter. 21 was readily prepared from 1,3-cyclohexanedione 20 in five steps based on their previous work.14 The terminal alkene was converted to a tertiary amine via a conventional oxidative cleavage protocol and reductive amination with dimethylamine. The chemoselectivity at this step was essential for preserving the ketone functional group. It was found that if a primary amine was used, condensation with the ketone would compete with the proposed transformation, and the resulting hexahydropyrrole would be a dead-end for the synthesis. After the introduction of bromine at the α-position of ketone 9, a basic aqueous work-up provided the quaternary ammonium salt 23, which was subjected to demethylation for which sodium iodide was found to be the most effective reagent. After four straightforward transformations to set up all the functional groups, the final cyclization of 8 to strictamine turned out to be extremely difficult. An intramolecular Michael addition mediated by lithium–halogen exchange, a Pd-catalyzed reductive Heck reaction, and a radical cyclization were all attempted. The desired product was only obtained when using Ni(cod)2 and Et3N with Et3SiH; under these conditions, vinyl iodide 8 was converted to strictamine in 5–10% yield.
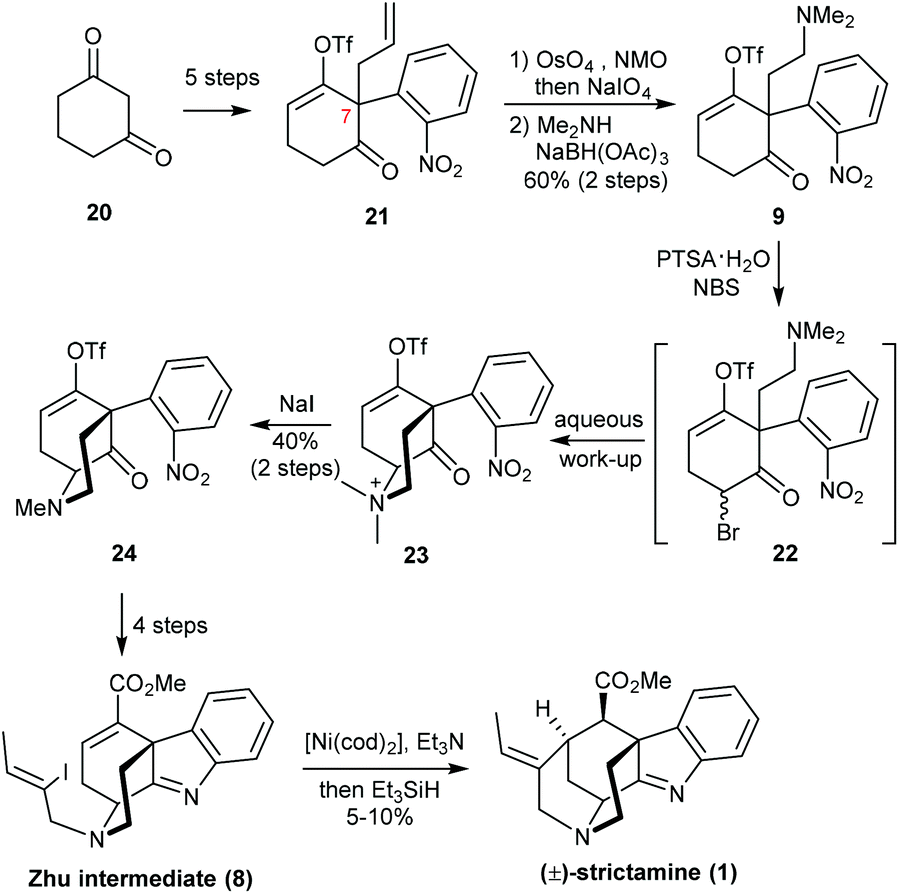 | ||
| Scheme 4 Zhu's total synthesis of (±)-strictamine. | ||
Because Zhu's strategy involved the early-stage creation of the C7 quaternary center and rapid construction of the D-ring, the precursor for the formation of the indolenine unit did not require the tedious redox manipulations seen in other syntheses. Although the yield of the final nickel-catalyzed cyclization step is rather poor, it is still an efficient tactic for constructing the framework and will no doubt be adopted by other groups to enhance the novelty of their own synthetic strategies.
The Blakey group at Emory University published a synthesis of the strictamine pentacyclic core via a creative iminium ion cascade annulation (Scheme 5).13 Starting with hemiaminal 13, an N-acyl iminium ion was first produced in the presence of boron trifluoride diethyl etherate, and then it was attacked by the 2-position of the indole. The corresponding benzylic carbocation was further trapped by the allylsilane, thus establishing the C and D rings with the correct stereochemistry at C7 and C13 in one step. With the key intermediate 26 in hand, the chain elongation of the exocyclic olefin required six steps. To secure the stereocenter at C15, Crabtree's catalyst ([Ir(cod)(py)(PCy3)]BArF4) was found to be the best choice for hydrogenation with good facial selectivity. Subsequent manipulation of 28 provided carboxylic acid 12 in four steps since the primary alcohol at C16 had to be protected. After removal of the Cbz group and amidation with Mukaiyama's reagent, pentacyclic skeleton 29 was obtained in a moderate yield.
 | ||
| Scheme 5 Blakey's synthesis of the strictamine pentacyclic core structure. | ||
Unlike the stepwise annulation strategies used by other groups, this work invented an intriguing cascade reaction to build two rings in a single step. Interestingly, when the Cbz group in 13 was replaced by an electron-donating group (Bn), the corresponding iminium ion would be attacked by the C3-position of indole, leading to the core structure of a different alkaloid.15 However, in this synthesis, the conversion of tetracycle 26 to pentacyclic core 29 required a long synthetic sequence (11 steps). Moreover, the inversion of the C16 stereocenter and installation of the quaternary C7 center as well as the development of an enantioselective variant of the cascade reaction which would be required to complete the synthesis remain a long way off.
The Ohno group at Kyoto University devised a racemic formal synthesis of strictamine based on a novel gold-catalyzed nucleophilic attack of the indole ring on a carbon–carbon triple bond (Scheme 6A).11a The addition of a Grignard reagent to 3,4-dihydro-β-carboline 30 delivered propargyl derivative 31. After some experimentation it was found that a combination of SPhosAuCl and AgNTf2 was the most efficient catalytic system for achieving the 6-endo-dig cyclization of nosylamide 32. To reach Zhu's intermediate 8, they managed to introduce additional 9 steps to install an appropriate ester group at C16, although advanced intermediate 33 was virtually identical to 8.
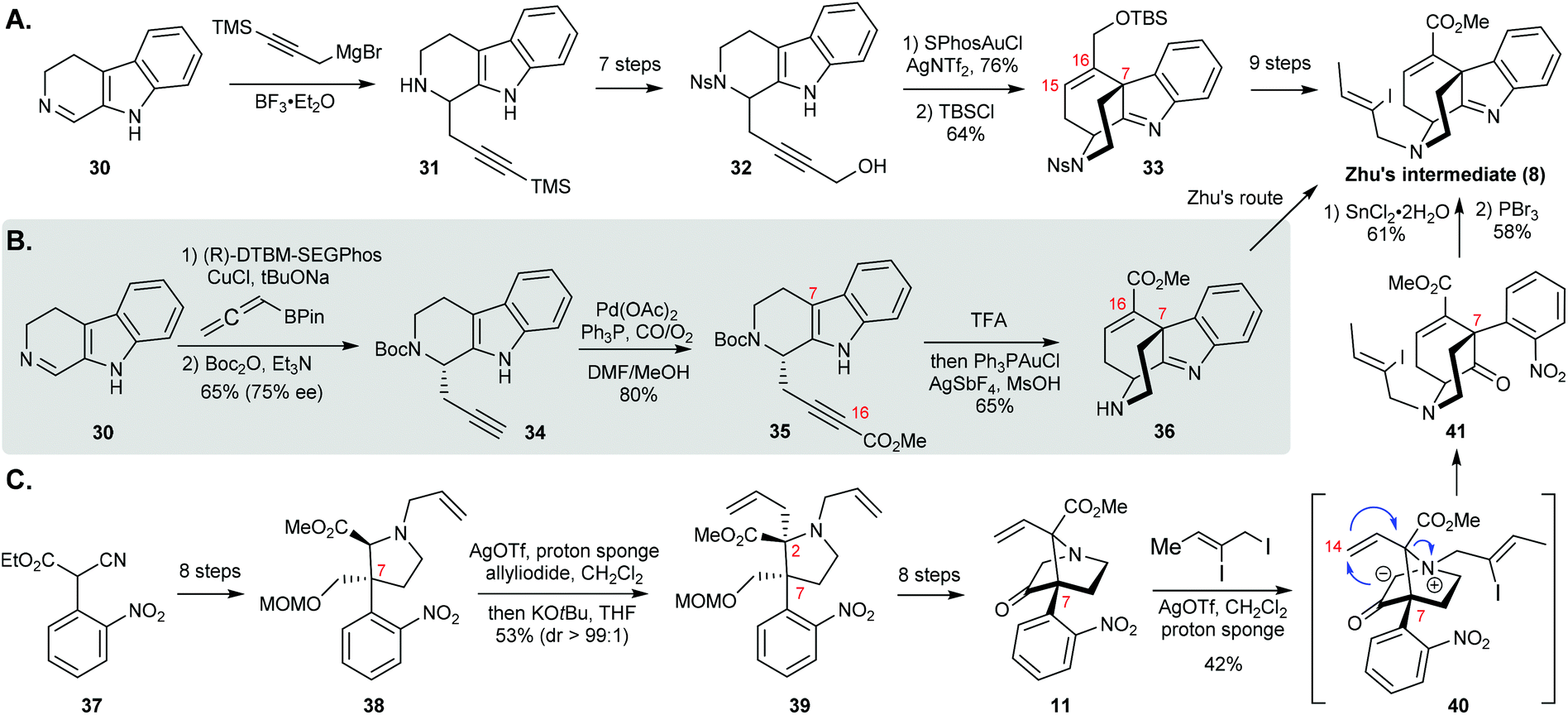 | ||
| Scheme 6 (A) Ohno's formal synthesis of (±)-strictamine. (B) Snyder's formal synthesis of (+)-strictamine. (C) Gaich's formal synthesis of (±)-strictamine. | ||
This strategy was elegant in regard to construction of the framework by the Au-catalyzed cyclization to furnish the C ring and the C7-quaternary carbon center as well as the double bond between C15 and C16 that was required for Zhu's final ring closure step. However, the series of functional group modifications from 31 to 32 and 33 to 8 diminished the efficiency of this work.
In 2017, the Snyder group at the University of Chicago presented an impressive 7-step asymmetric formal synthesis of strictamine via an asymmetric propargylation and metal-mediated cyclization (Scheme 6B).11b They started with the same β-carboline 30 as Ohno. In contrast to Ohno's method, they used allenylboronate to complete the asymmetric propargylation using a Cu(I)-(R)-DTBM-SEGPhos complex as the catalyst to deliver the adduct in good yield and enantioselectivity (as shown for 34). The subsequent CO-insertion into the terminal alkyne proceeded smoothly. After Boc deprotection by trifluoroacetic acid, the resulting ammonium TFA salt bearing an electron-deficient alkyne, 35, was effectively transformed via the key 6-endo-dig cyclization with stoichiometric AgSbF6 and catalytic Ph3PAuCl. From there, only one more step was required to reach Zhu's intermediate 8.
The independent development of the Au(I)-catalyzed 6-endo-dig cyclization in Snyder's synthesis offered several advantages over Ohno's work. This synthetic design allows the introduction of the appropriate functional groups in well-planned steps, which makes this an appealing strategy. Although the final Ni-catalyzed cyclization needs to be further optimized, based on the number of synthetic steps, it clearly merges both novelty and efficiency in one classic synthesis.
The Gaich group at the University of Konstanz reported another formal synthesis of strictamine using [2,3]-Stevens rearrangements as the key transformations (Scheme 6C).12 The synthesis began with α-cyanoacetate 37, which was elaborated to highly substituted pyrrolidine 38 over 8 steps. The allyl group on the nitrogen was transferred to the 2-position of the pyrrolidine ring with excellent stereochemical control through the first Stevens rearrangement. Compound 39 was elaborated to bridged bicyclic system 11 over eight steps via N–H insertion of an α-diazo ketone intermediate. The crucial azabicyclic-[3.3.1]-nonane skeleton, 41, was established through the second Stevens rearrangement via intermediate 40, which was generated in the presence of silver triflate and proton sponge. That was followed by reduction of the nitro group using tin chloride with concomitant imine formation upon treatment with phosphorus tribromide.
Two Stevens rearrangements were the key features in the Gaich synthesis. The outstanding design of the molecular skeleton would be a textbook example for illustrating the application of a Stevens rearrangement in a total synthesis. While several steps in the synthesis of rearrangement precursors 38 and 11 need to be further optimized, this synthesis deserves more attention for its efficiency in an alkaloid synthesis.
Outlook for teaching and learning synthesis
In summary, distinct strategies and synthetic methods have been developed for the total or formal synthesis of strictamine. The Garg group utilized an interrupted Fischer indolization with an appropriate precursor to install the correct stereochemistry. The outstanding contribution of Zhu's synthesis was to introduce the C7-quaternary stereocenter at an early stage, which substantially simplified the synthetic design. The only remaining challenge is further optimization of the nickel-catalyzed Michael-type vinylation to close the E-ring. The impressive cascade reaction to furnish the complex bridged ring system bearing the full carbon framework from the Blakey group relies on a deep appreciation of the molecular architecture; however, the development of an asymmetric variant would be a challenging task. The Ohno group utilized a gold-catalyzed 6-endo-dig cyclization to establish the framework of the target vinyl iodide in Zhu's work. The Snyder group further explored the chemistry stemming from the Ohno study, and their asymmetric propargylation of the imine is noteworthy. The Gaich group again targeted the Zhu's intermediate to complete a formal synthesis. They ingeniously employed [2,3]-Stevens rearrangements to construct the core structure. Even though some transformations and the yield of key reactions require further improvement, these studies exemplify the art of synthesis in terms of both efficiency and novelty in the total syntheses of natural products. The journey of pursuing the ideal synthesis of strictamine provides a good illustration for graduate students to better understand the principles of synthetic design. In the classroom, after a one semester course in synthetic methods, it is time for graduate students to apply the reactions they have learnt to more complex targets and evaluate the merits of various synthetic strategies. It is thus better to group several ingenious synthetic designs together to understand the bond disconnection approach as well as the strategic blueprint. The “art” inherent in synthesis will thus be visible. It is even more exciting to compare chemical synthesis with the elegance of biosynthesis in which strictamine may be constructed through an oxidative coupling between the α-carbon of the ester (C16) and the indole (C7) (Scheme 1). Such oxidative C–C bond forming reaction has been executed,8f,i although without success, and it is believed that the biomimetic approach from naturally abundant geissoschizine would be a truly remarkable synthesis. Graduate students would not be satisfied with the one or two synthetic strategies they initially devised as the only take-home examples; they would be eager to compare different approaches to visualize the novel and efficient pathways to the final product. This creative learning process is valuable to train the next-generation of synthetic chemists.Further reading
The Zu group at Tsinghua University recently reported a unified approach to access several congeners of akuammiline alkaloids and devised an elegant ring-reconstruction approach to complete the synthesis of strictamine.16 On comparing with previous studies, the E-ring formation with concomitant installation of the ester group at C16 with a requisite stereochemistry was outstanding. For the synthetic studies on akuammiline alkaloids in general, we encourage the readers to compare the creativity in individual synthetic designs with previous syntheses in this field.17,18 More generally, the central theme for synthesis could stem from the construction of the morphan core. Several elegant reviews related to this topic are also highly recommended.19Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the financial support from the National Natural Science Foundation of China (21672236 to R. H. and 21402121 to S.-H. H.), the Shanghai Institute of Technology (XTCX2015-16 to S.-H. H.), and the Shanghai Science and Technology Commission for the Shanghai Sailing Program (16YF1414400 to Y. W.). We thank Prof. Hiroaki Ohno (Kyoto University) for sharing with us their recent progress on gold-catalyzed cyclization related to this topic during the ACP Lectureship in Japan (to R. H.). We also thank Prof. Dr Jian Jin (SIOC) and other members in the Hong group for invaluable input during the preparation of the manuscript.Notes and references
- (a) H. Zahreddine and K. L. B. Borden, Front. Pharmacol., 2013, 4, e28 CrossRef PubMed; (b) WHO, Antimicrobial resistance: Global report on surveillance 2014, Geneva, Switzerland, 2014 Search PubMed.
- A. L. Lane and B. S. Moore, Nat. Prod. Rep., 2011, 28, 411–428 RSC.
- (a) J.-Y. Ortholand and A. Ganesan, Curr. Opin. Chem. Biol., 2004, 8, 271–280 CrossRef CAS PubMed; (b) M. Feher and J. M. Schmidt, J. Chem. Inf. Comput. Sci., 2003, 43, 218–227 CrossRef CAS PubMed.
- (a) J. B. Hendrickson, Top. Curr. Chem., 1976, 62, 49–172 CrossRef CAS PubMed; (b) T. Gaich and P. S. Baran, J. Org. Chem., 2010, 75, 4657–4673 CrossRef CAS PubMed.
- (a) E. J. Corey and X. M. Cheng, The logic of Chemical Synthesis, John Wiley & Sons, New York, 1995 Search PubMed; (b) R. W. Hoffmann, Angew. Chem., Int. Ed., 2013, 52, 123–130 CrossRef CAS PubMed.
- H. K. Schnoes, K. Biemann, J. Mokry, I. Kompis, A. Chatterjee and G. Ganguli, J. Org. Chem., 1966, 31, 1641–1642 CrossRef CAS.
- Y. Hou, X. Cao, L. Wang, B. Cheng, L. Dong, X. Luo, G. Bai and W. Gao, J. Chromatogr., B: Biomed. Appl., 2012, 908, 98–104 CrossRef CAS PubMed.
- (a) L. J. Dolby and Z. Esfandiari, J. Org. Chem., 1972, 37, 43–46 CrossRef CAS PubMed; (b) L. J. Dolby and S. J. Nelson, J. Org. Chem., 1973, 38, 2882–2887 CrossRef CAS PubMed; (c) M. L. Bennasar, E. Zulaica, M. López and J. Bosch, Tetrahedron Lett., 1988, 29, 2361–2364 CrossRef CAS; (d) T. Koike, H. Takayama and S.-i. Sakai, Chem. Pharm. Bull., 1991, 39, 1677–1681 CrossRef CAS; (e) M. L. Bennasar, E. Zulaica, A. Ramírez and J. Bosch, J. Org. Chem., 1996, 61, 1239–1251 CrossRef CAS; (f) R. V. Edwankar, C. R. Edwankar, O. A. Namjoshi, J. R. Deschamps and J. M. Cook, J. Nat. Prod., 2012, 75, 181–188 CrossRef CAS PubMed; (g) M. Kawano, T. Kiuchi, S. Negishi, H. Tanaka, T. Hoshikawa, J.-i. Matsuo and H. Ishibashi, Angew. Chem., Int. Ed., 2013, 52, 906–910 CrossRef CAS PubMed; (h) Y. Komatsu, K. Yoshida, H. Ueda and H. Tokuyama, Tetrahedron Lett., 2013, 54, 377–380 CrossRef CAS; (i) W. Ren, N. Tappin, Q. Wang and J. Zhu, Synlett, 2013, 1941–1944 CAS.
- J. Moreno, E. Picazo, L. A. Morrill, J. M. Smith and N. K. Garg, J. Am. Chem. Soc., 2016, 138, 1162–1165 CrossRef CAS PubMed.
- W. Ren, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2016, 55, 3500–3503 CrossRef CAS PubMed.
- (a) D. Nishiyama, A. Ohara, H. Chiba, H. Kumagai, S. Oishi, N. Fujii and H. Ohno, Org. Lett., 2016, 18, 1670–1673 CrossRef CAS PubMed; (b) M. W. Smith, Z. Zhou, A. X. Gao, T. Shimbayashi and S. A. Snyder, Org. Lett., 2017, 19, 1004–1007 CrossRef CAS PubMed.
- (a) R. Eckermann, M. Breunig and T. Gaich, Chem. Commun., 2016, 52, 11363–11365 RSC; (b) R. Eckermann, M. Breunig and T. Gaich, Chem. – Eur. J., 2017, 23, 3938–3949 CrossRef CAS PubMed.
- E. S. Andreansky and S. B. Blakey, Org. Lett., 2016, 18, 6492–6495 CrossRef CAS PubMed.
- W. Ren, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2014, 53, 1818–1821 CrossRef CAS PubMed.
- A. Kong, D. E. Mancheno, N. Boudet, R. Delgado, E. S. Andreansky and S. B. Blakey, Chem. Sci., 2017, 8, 697–700 RSC.
- X. Xie, B. Wei, G. Li and L. Zu, Org. Lett., 2017, 19, 5430–5433 CrossRef CAS PubMed.
- (a) M. Zhang, X. Huang, L. Shen and Y. Qin, J. Am. Chem. Soc., 2009, 131, 6013–6020 CrossRef CAS PubMed; (b) W. Zi, W. Xie and D. Ma, J. Am. Chem. Soc., 2012, 134, 9126–9129 CrossRef CAS PubMed; (c) S.-Z. Jiang, X.-Y. Zeng, X. Liang, T. Lei, K. Wei and Y.-R. Yang, Angew. Chem., Int. Ed., 2016, 55, 4044–4048 CrossRef CAS PubMed; (d) Y. Li, S. Zhu, J. Li and A. Li, J. Am. Chem. Soc., 2016, 138, 3982–3985 CrossRef CAS PubMed; (e) G. Li, X. Xie and L. Zu, Angew. Chem., Int. Ed., 2016, 55, 10483–10486 CrossRef CAS PubMed; (f) T. Wang, X. Duan, H. Zhao, S. Zhai, C. Tao, H. Wang, Y. Li, B. Cheng and H. Zhai, Org. Lett., 2017, 19, 1650–1653 CrossRef CAS PubMed; (g) D. Wang, M. Hou, Y. Ji and S. Gao, Org. Lett., 2017, 19, 1922–1925 CrossRef CAS PubMed.
- (a) R. Eckermann and T. Gaich, Synthesis, 2013, 2813–2823 CAS; (b) J. M. Smith, J. Moreno, B. W. Boal and N. K. Garg, Angew. Chem., Int. Ed., 2015, 54, 400–412 CAS; (c) G. L. Adams and A. B. Smith III, The Chemistry of the Akuammiline Alkaloids, in The Alkaloids, ed. H.-J. Knolker, Elsevier, New York, 2016, vol. 76, pp. 171–257 Search PubMed.
- (a) J. Bonjoch, F. Diaba and B. Bradshaw, Synthesis, 2011, 993–1018 CrossRef CAS; (b) B. Kang, P. Jakubec and D. J. Dixon, Nat. Prod. Rep., 2014, 31, 550–562 RSC; (c) A. K. Chattopadhyay and S. Hanessian, Chem. Rev., 2017, 117, 4104–4146 CrossRef CAS PubMed; (d) Q. Li and H. Zhang, Chin. J. Org. Chem., 2017, 37, 1629–1652 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Complete synthetic routes of all examples discussed in the tutorial review. See DOI: 10.1039/c7qo00837f |
| This journal is © the Partner Organisations 2018 |