
Single nanoparticle analysis by ICPMS: a potential tool for bioassay†
Jianyu
Hu
a,
Dongyan
Deng
a,
Rui
Liu
 a and
Yi
Lv
*ab
a and
Yi
Lv
*ab
aCollege of Chemistry, Sichuan University, Chengdu, Sichuan 610064, China. E-mail: lvy@scu.edu.cn; Fax: +86-28-85412798; Tel: +86-28-85412798
bAnalytical & Testing Center, Sichuan University, Chengdu, Sichuan 610064, China
First published on 13th November 2017
Abstract
Inductively coupled plasma mass spectrometry (ICPMS) has already been demonstrated as a promising technique for metallic nanoparticle tagged bioassays due to its high sensitivity, wide dynamic linear range, and more importantly multiplex and absolute quantification ability. Besides, single nanoparticle analysis by ICPMS has also recently been applied for many metal nanoparticles. Moreover, its short data acquisition dwell times (serval hundred microseconds) lead to an extremely high signal to noise ratio for metal nanoparticles (i.e., low detection limits). This perspective focuses on single nanoparticle analysis-based ICPMS bioassays, which provide high sensitivity without any sophisticated signal amplification procedures. Herein, the recent development of single nanoparticle analysis, ICPMS instrument design, and single molecule analysis is discussed. Considering the vast types of metallic nanoparticles currently available and simultaneous multiplex detection capability of TOF-ICPMS, single nanoparticle analysis-based bioassays may open a new avenue for multiplex single molecule analysis.
 Jianyu Hu | Jianyu Hu is a PhD candidate supervised by Professor Yi Lv in the College of Chemistry, Sichuan University. He received his BEng degree from Sichuan University (China) in 2011. His current research is on mass spectrometry based bioassays, including nanoparticle label based multiplex bioassays. |
 Dongyan Deng | Dongyan Deng has been working at the College of Chemistry, Sichuan University (China) since 2014. She received her B.S. degree in Chemistry (2009) and her PhD degree in Analytical Chemistry (2014) from Sichuan University (China). Her research focuses on the development of novel nanomaterial-based spectrometry. |
 Rui Liu | Rui Liu is an associate professor of analytical chemistry in Sichuan University. He received his B. S. and Ph.D. degrees from Sichuan University in 2004 and 2012, respectively. He was a postdoctoral fellow in the Department of Chemistry in Tsinghua University in 2013–2016. He joined the faculty of the College of Chemistry in Sichuan University in 2016. His current research interest is metal stable isotope tagging-based bioassays. |
 Yi Lv | Yi Lv is a full professor in the College of Chemistry and director of the Analytical & Testing Center at Sichuan University, China. He obtained his PhD degree in 2005 from Southwest China Normal University (currently Southwest University), China. After a two-year stay at Tsinghua University as a postdoctoral researcher in Professor Xinrong Zhang's research group, he joined the faculty of the College of Chemistry at Sichuan University in 2005. His research interests are mainly in the areas of analytical spectrometry and nano-materials for analytical chemistry. He is the author and co-author of ∼120 journal articles and one book chapter. He is a member of the International Advisory Board of the Journal of Analytical Atomic Spectrometry (JAAS). |
Introduction
Inductively coupled plasma mass spectrometry (ICPMS) is the most powerful technique available to determine trace or ultra-trace metal elements. The hard ionization source of ICP provides an average temperature of 7000–8000 K. At this high temperature, metal-containing samples are readily dried and vaporized, and metal isotopes in the sample are also effectively atomized and ionized, which means the mass-based response of the isotopes is almost independent of the chemical species present in the sample. This great detection power has resulted in the renaissance of radioimmunoassays for the multiplex and absolute quantification of biomolecules by metal stable isotope tagging.3 It is worth mentioning the following intriguing features of current ICPMS-based bioassay by metal stable isotope tagging:4–10 most metal isotope tags can be simultaneously detected at the lower part per trillion level with 9 orders of dynamic linear range and low matrix effects; the metal isotopes inside tags are directly detected without the need of any special properties; and metal nanoparticle tags can provide high sensitivity due to the internal large amount of detectable stable isotopes.Highly sensitive techniques for bioassay are crucial for the early diagnosis of diseases and molecular mechanism elucidation. To further improve the detection sensitivity, many signal amplification techniques have been developed for ICPMS bioassay, including immunogold-silver enhancement,11 rolling circle amplification,12,13 ligation-mediated amplification,14 and duplex-specific nuclease amplification.15 The signal amplification techniques are effective, which brought significantly improved detection sensitivity and detection limits, while higher reagent and labor consumption, longer reaction time, and lower system stability.
Recently, single nanoparticle analysis by ICPMS has gained great research interest.16–18 Single nanoparticle analysis by ICPMS can provide data on size, size distribution, nanoparticle number concentration, nanoparticle mass concentration, agglomeration, and composition for low concentration metallic nanoparticles. Additionally, the short data acquisition dwell times of ICPMS lead to extremely high signal to noise ratios (e.g., 0.03 atto-gram gold is detectable),19 which is distinct from conventional integral mode detection for nanoparticles. Combined with the excellent features of ICPMS bioassay (including multiplex and absolute quantification), single nanoparticle analysis provides great potential for extremely sensitive bioassay without the need for any signal amplification.
Despite the obvious potential of single nanoparticle analysis-based ICPMS bioassay, there are few reports on it. In this perspective, to propagate and advocate single nanoparticle analysis bioassay, we summarize the recent publications on this topic. The single nanoparticle counting-based heterogenous bioassay, single nanoparticle counting-based homogenous bioassay, and digital-based bioassay are discussed in detail with emphasis on the future development of highly multiplex single molecule bioassay. Single cell bioassays,20–24 which are quite similar to single nanoparticle bioassay, are introduced in many excellent reviews,25–27 and thus are not included in this perspective.
Single nanoparticle analysis by ICPMS
Nanoparticles are objects with three dimensions in the nanoscale level (1–100 nm). Due to their vast and important applications, many techniques have been applied to determine their characteristics such as size, composition and concentration, including dynamic light scattering, field flow fractionation, ultraviolet-visible spectroscopy, and electron microscopy (transmission electron microscopy and scanning electron microscopy).28 However, these characterization techniques have some disadvantages. For instance, nanoparticle number concentration is hard to measure in suspension. Besides, all these detection techniques are unable to discriminate mixtures of monometallic and/or bimetallic particles.Single nanoparticle analysis by ICPMS was first reported by Degueldre et al.,29 and has gained great attention in recent years. More and more research laboratories and ICPMS instrument manufacturers are dedicated to providing hardware and software updates for facile single nanoparticle analysis.30–32Fig. 1 illustrates the nanoparticles detectable by ICPMS. In single nanoparticle analysis, only one nanoparticle is measured during each reading period. Very diluted nanoparticle suspensions and adequate data acquisition frequency are the prerequisites to guarantee single nanoparticle analysis. Once the nanoparticle is introduced into the ICP, it is vaporized and ionized as a gaseous ion cloud in plasma, which can be measured as a signal pulse. Parameters including mean size, size distribution, nanoparticle number concentration, nanoparticle mass concentration, agglomeration, and composition, can be measured with single nanoparticle analysis by ICPMS. Nanoparticles in environmental samples can be detected and characterized, which are existing at extremely low levels and are challenging for other techniques including electron microscopy (e.g., number concentrations of 103 cm−3 to 105 cm−3).33
 | ||
| Fig. 1 Elements that form nanoparticles detectable by ICPMS (in yellow) whereas that in pink does not exhibit the nanoparticle form. | ||
A large number of nanoparticles have been successfully investigated and determined by single nanoparticle analysis ICPMS (Table 1 presents the typical nanoparticles analyzed by single nanoparticle ICPMS in recent years), which provides potential tags for single nanoparticle analysis-based sensitive bioassay (Fig. 1). Despite the great success of analytical methods as well as instrument designs, single nanoparticle analysis still poses great challenges to mass spectrometry instruments.
| ICP-MS approach | Analyte nanoparticles | LOD | Particle diameters | Ref. |
|---|---|---|---|---|
| Q-ICPMS (single particle mode) | Monometallic and bimetallic (core–shell) nanoparticles | — | Au 60 nm, Ag 60 nm and AuAg 60 nm | 40 |
| Q-ICPMS (single particle mode) | Rare earth oxides | — | Average 35 ± 3 nm | 37 |
| Mass cytometer | Au NPs at the single-cell level | 10 AuNPs in a single cell | 3 nm core size | 41 |
| Q-ICPMS (single particle mode) | Silver and gold nanoparticles in environmental water | 8 × 104 particles per L | 20 and 19 nm respectively | 42 |
| Q-ICPMS (coupled with ETV) | Gold nanoparticles in environmental water | 2.2 ng L−1 | — | 43 |
| Q-ICPMS (single particle mode) | Silver nanoparticle in lake water | — | 33.8 ± 0.4 nm–46.6 ± 0.4 nm | 44 |
| ICP-MS (coupled with HPLC) | Silver nanoparticles | 0.09 μg L−1 | 18–55 nm | 45 |
| Q-ICPMS (coupled with magnetic solid phase extraction) | Silver sulfide nanoparticles in environmental waters | 0.068 μg L−1 | 4.3 ± 0.9 nm–17.5 ± 3.0 nm | 46 |
| Q-ICPMS (single particle mode) | Platinum nanoparticles uptake and bioaccumulation by Lepidium sativum and Sinapis alba plants | 0.002 mg g−1 | Average 67.1 nm | 47 |
| ICP-MS (with laser ablation) | Gold nanoparticle | 10 ng g−1 | 13 and 50 nm | 48 |
| Q-ICPMS (single particle mode) | Silver nanoparticles internalized by Arabidopsis plants | 10 nm | Initial 12.84 nm/final 20.70 nm | 49 |
| ICP-SFMS | Ag NPs suspended in natural lake water | ∼10 nm & 2.7 ng L−1 | 15–18 nm | 50 |
| Q-ICPMS (single particle mode) | Silver nanoparticles | 30 nm | 30, 40, 50 and 80 nm | 51 |
| Q-ICPMS (coupled with SdFFF) | TiO2 nanoparticles | 10 ng L−1 | 21 and 50 nm | 52 |
| ICP-SFMS | Iron oxide nanoparticles | 2.4 μg L−1 | 24.7 ± 1.5 nm | 53 |
| Q-ICPMS (coupled with CER-CVG) | Silver nanoparticles | 0.3 ng L−1 | — | 54 |
| Q-ICPMS (single particle mode) | Silver nanoparticles in plasma and blood of burn patients | 13 ng L−1 | 9.2 ± 3.2 nm–99.0 ± 5.1 nm | 55 |
| Q-ICPMS (single particle mode) | Zinc oxide and cerium dioxide nanoparticles during drinking water treatment | 0.20 μg L−1, 0.10 μg L−1 | 35 to 40 nm, 18 to 20 nm | 56 |
| Q-ICPMS | Lanthanide nanoprobe | 0.5 ng mL−1 | 13.6 nm to 16.2 nm | 57 |
| ICP-MS (with laser ablation) | Nanoparticles (Al2O3, Ag, and Au) in Daphnia magna and zebrafish embryos | 500–1000 fg Al, 35.7 fg Ag, 12.5 fg Au/50 μm spot | 35 ± 11 nm–503 ± 367 nm | 58 |
| Q-ICPMS (single particle mode) | NIST gold nanoparticle | 20 nm | 30 and 60 nm | 59 |
| Q-ICPMS coupled with CE | Gold nanoparticles | 0.8–1.0 μg L−1 | 5 and 10 nm/20 and 50 nm | 60 |
| Q-ICPMS (single particle mode) | Au NPs, AgNPs, TiO2 NPS, SiO2 NPs | 1 ng L−1 for 60 nm Au NPs to 0.1 mg L−1 for 500 nm SiO2 particles | 20 nm (Au and Ag), 50 (TiO2) and 200 nm (SiO2) | 61 |
| Q-ICPMS | Superparamagnetic iron oxide (SPIO) nanoparticles with a unique lanthanide dopant | — | 33.54 nm, 33.47 nm, 35.57 nm and 34.84 nm | 62 |
| ICP-MS (with laser ablation) | SiO2 nanoparticle | — | 31 ± 5 nm and 57 ± 20 nm | 63 |
| Q-ICPMS | GO/Au and GO/Ag | — | 13.3 nm and 48.8 nm | 64 |
| Q-ICPMS (coupled with micellar electrokinetic chromatography) | Engineered nanoparticles (ENPs) | 0.27–0.55 μg L−1 | 5 nm, 20 nm and 50 nm | 65 |
I. The typical ion cloud produced from individual nanoparticles exists in the plasma and detection channel as a transient signal of 200–500 μs, which generally requires fast data acquisition (sampling frequency of around 105 Hz, i.e., detector dwell time of around 10 μs).
II. Another challenge is the multi-isotope analysis of transient signals from individual nanoparticles,34 which requires simultaneous or fast scanning mass spectrometers that are able to provide full spectra of the mass range of interest at frequencies of around 105 Hz.
III. The sample introduction efficiency is low by common solution nebulization (typically lower than 5%). To determine transportation efficiencies, the particle frequency and particle size methods produce similar efficiencies, while the waste collection method is consistently around 50% higher.35 Referring to particle number concentrations and particle size distributions, the efficiency value from the waste collection method consistently underestimates particle number concentration, suggesting an overestimation of transport efficiency. Liu et al. used characterized RM 8017 (polyvinylpyrrolidone coated nominal 75 nm silver nanoparticles) and NIST RM 8013 Gold Nanoparticles to build the calibration and demonstrate that the particle size method is more robust to measure transportation efficiency.36
IV. The detection power for small size nanoparticles is still somewhat lacking, which needs further optimization of instrument design for higher sensitivity. The typical limits of detection, expressed as equivalent spherical diameter, range from 10 nm (Ta, U, Ir, Rh, Th, Ce, and Hf) to 200 nm (Se, Ca, and Si) for monoisotopic nanoparticles37,38 (for instance, single gold nanoparticle analysis allows a size detection limit of around 15 nm,39i.e., 0.03 fg gold). Additionally, the size-based detection limits are poorer when the nanoparticles are composed of more than one element or isotope.
Current ICPMS instrument design
ICPMS has been successfully commercialized in many forms, offering a direct means for trace element and isotope determination in solution. Among them, quadrupole ICPMS (Q-ICPMS), sector field ICPMS (SF-ICPMS), multi-collector ICPMS (MC-ICPMS), and time of flight ICPMS (TOF-ICPMS) are most often applied in metal nanoparticle analysis. Different mass analyzers give them varied features for single nanoparticle analysis (Table 2).| ICPMS instrument | Data acquisition frequency | Full mass ranged acquisition | Multi-isotope measurement in short transient | Sensitivity |
|---|---|---|---|---|
| Q-ICPMS | 104–105 Hz | Yes | No | Medium |
| SF-ICPMS | 104 Hz | Yes | No | High |
| MC-ICPMS | 10 Hz | No | Yes | Low |
| TOF-ICPMS | 104–105 Hz | Yes | Yes | Medium |
The Q-ICPMS system is the most commonly used mass analyzer in ICPMS. The first concept of coupling an ICP to MS was demonstrated by Houk et al. in 1980 as Q-ICPMS.66 Fast data acquisition67 is now realized by commercial Q-ICPMS instruments (for instance, 105 Hz by Perkin Elmer NexION 350 Q-ICPMS and NexION 2000 Q-ICPMS, and 104 Hz by Agilent 7900 Q-ICPMS). However, the settling time on the ms level between isotopes is the limitation for the determination of multi-isotopes in individual nanoparticles. As a triple quadrupole ICPMS (ICP-QQQ), the Agilent 8900 has transformed interference removal in ICPMS, using MS/MS to control reaction chemistry and deliver consistent, reliable results in the reaction mode. The tandem ICPMS uses two quadrupole mass filters (Q1 and Q2), with an octopole reaction cell located between them to provide a double mass selection capability. The ICP-QQQ technique can resolve matrix interferences and provides excellent single particle detection capability.
SF-ICPMS is a commercialized double focusing sector field ICPMS.68 There is series of magnetic and electric fields to continuously yield different m/z ions on the way to a single detector, which provides a high resolution of mass discrimination and high data acquisition frequency of 104 Hz.69 The application of high resolution SF-ICPMS totally solved the interference problems of atomic ions and disturbing polyatomic ions at the same nominal mass. The SF-ICPMS instrument has been recognized as the most sensitive and powerful inorganic mass spectrometer. The current SF-ICPMS instrument (Thermo Scientific Element XR) extends its linear dynamic range using a Faraday detector combined with a dual mode SEM detector. However, isotopic ratio measurement can only be performed sequentially.
To overcome the limitation of SF-ICPMS in slow multiple isotope detection, an ICP source was coupled to a double focusing magnetic analyzer with multiple Faraday detectors, thereby allowing the isotopic components of an element to be measured simultaneously.68 This system turns SF-ICPMS into multiple collector ICPMS (MC-ICPMS), which provides accurate and precise multiple isotope simultaneous determination.70 The signal acquisition frequency (typically 10 Hz) of MC-ICPMS is restricted due to the necessary parallel use of fast ion counters and relatively slow Faraday cups.71 MC-ICPMS does not allow the acquisition of full mass spectra, even though this technique provides the most precise isotope ratios.72
Multiple isotope simultaneous measurement is one of the superiorities of TOF-ICPMS. The principle of TOF-ICPMS is to utilize ions in same momentum but at different masses get different traveling times through the field. TOF-ICPMS is capable of offering full mass spectra at a dwell time of 33 μs, which allows multi-isotope monitoring of short transient signals generated from individual droplets and nanoparticles.73
Generally, the current scanning ICPMS instruments (Q-ICPMS and SF-ICPMS) provide fast data acquisition (from the duration of the transient signal generated by an individual NP, i.e., hundreds of microseconds) and sufficient sensitivity for single nanoparticle analysis (with size detection limit typically in the range of 10 to 20 nm). MC-ICPMS is mainly aimed at high precision and accurate isotope ratio simultaneous measurements of one to three nearby elements. However, for the multi-isotope detection of transient signals generated by individual NPs, MC-ICPMS still exhibits some drawbacks in multiplex single nanoparticle analysis due to its limited detection mass range and simultaneous detection capability. Additionally, for the multi-element detection of transient signals generated by individual NPs, the simultaneous use of TOF-ICPMS instruments is involved.
However, the current MC-ICPMS instrument can only analyse multi-isotopes of adjacent mass to charge ratio, and do not permit the continuous monitoring at acquisition times below several milliseconds. Accordingly, the implement of TOF-ICPMS becomes mandatory for multi-element and multi-isotope analysis in individual nanoparticles.
Single nanoparticle analysis for bioassay: towards multiplex single molecule analysis
ICPMS-based bioassays have gained great success in the past few years, and have been successfully applied in the sensitive, multiplex, and absolute quantification of small biomolecules, proteins, nucleic acids, and cells. Metal isotope labels can be detected with a low limit of detection (LOD, pg mL−1 level for most metal isotopes), wide dynamic linear range (up to 9 orders of magnitude), low matrix effect, and high mass spectral resolution. High sensitivity is feasible by employing nanoparticle labels instead of metal ion labels since a large number of metal atoms are contained in all nanoparticle labels. Nanoparticles tags are often dissolved in solution by nitric acid and detected using traditional integral mode detection. The limits of detection of analyte biomolecules by these bioassays are often in the ng mL−1 level, which limits their applications in sensitive bioassay, especially in early disease diagnosis.Single nanoparticle analysis by ICPMS has been already demonstrated for many nanoparticles (i.e., Au nanoparticles, Ag nanoparticles, TiO2, SiO2, and rare earth element nanoparticles). During the ionization of nanoparticles in the ICP, the transient signals produced from the flush of ions are measured by the mass spectrometer, which gives one transient signal in each given dwell time. Due to the low noise in short integral times (200–500 μs) for individual nanoparticles, an extremely high signal to noise ratio is obtained by single nanoparticle analysis. Therein, ICPMS in single-particle mode (spICPMS) has unique bioanalytical application due to its special sample introduction system. With the capability of single particle counting, spICPMS is able to provide size, size distribution and major elemental composition information for analyte nanoparticles. By utilizing spICPMS, the study of nanoparticle uptake and transformation in biological systems has gotten to a new level. Gray et al. combined spICPMS with tissue extraction to quantify and characterize metallic engineering nanoparticles (ENPs) in environmentally relevant biological tissues for the first time.74 ENPs were extracted from tissues using TMAH, before detection by spICPMS. By using ground beef, the ENPs investigated included 100 and 60 nm Au and Ag. The size distributions and particle number concentrations were obtained for ENPs extracted by spICPMS. Besides Au and Ag nanoparticles, Jiménez-Lamana et al. utilized spICPMS to detect platinum nanoparticle uptake and bioaccumulation in two model organisms.47 Information about nanoparticle size, nanoparticle number concentration and the presence of dissolved platinum in the samples was provided by spICPMS analysis, which was the first study to investigate the uptake and fate of PtNPs in plants.
When nanoparticles were employed as tags in bioassay, this single nanoparticle analysis strategy affords the possibility of developing highly sensitive bioassays by individual nanoparticle tag counting.
Single nanoparticles analysis heterogenous assay
A wide variety of bioassay designs and platforms have been developed with low detection limits in recent years. Generally, regarding the bio-recognition taking place on a solid surface or in solution phase, bioassays are often divided into heterogeneous and homogeneous formats. Heterogeneous bioassays need the conjugated bio-complex to be physically separated from the free tagged biomolecules. To realize the physical separation, solid phases such as microtiter plates, membranes, and tissue sections are usually employed. Heterogeneous bioassays are the most frequently applied schemes, which usually offer low detection limits.According to whether bio-specific binding is recoded directly or through the replacement of a tagged analyte, bioassays can also be divided into non-competitive and competitive formats. For instance, in non-competitive immunoassays (usually sandwich type immunoassays), excessive antibody over antigen is used. Non-competitive immunoassays feature – low detection limits, large dynamic linear ranges and high accuracy compared with competitive immunoassays. In contrast, for competitive immunoassays, the tagged analyte competes with the analyte to bind with the antigen reactive sites of the antibody. The sensitivity of the competitive immunoassay is determined by the affinity between the antigen and antibody. Furthermore, due to their one-step immunoreaction, competitive immunoassays are simple and fast.
Based on mass spectrometric detection in time-resolved analysis (TRA), the first single nanoparticle analysis ICPMS bioassay was demonstrated in a competitive heterogeneous immunoassay (Fig. 2).75
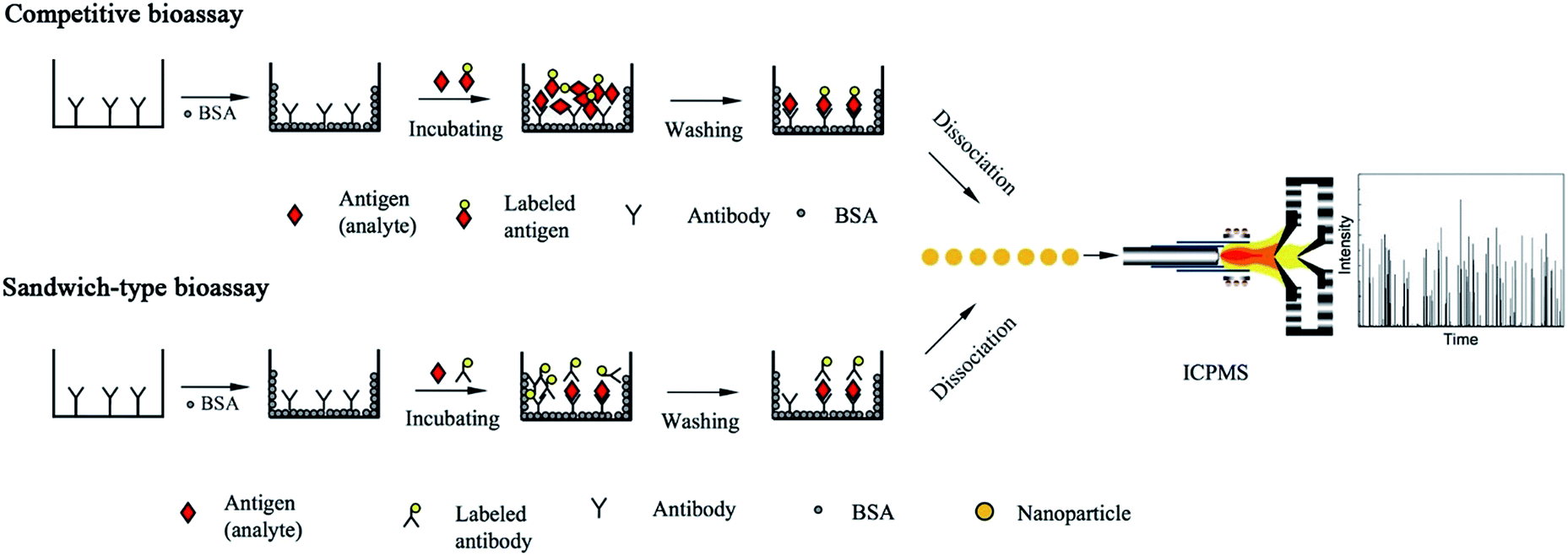 | ||
| Fig. 2 Schematic of single nanoparticle analysis-based bioassay (typical competitive bioassay and sandwich-type bioassay). | ||
The purified antibody solution was added to microtiter plates and incubated overnight. After incubation, the plate was washed three times with phosphate-buffered saline (PBS) to remove any unbound antibody. The unbound sites were blocked with BSA by incubation at 37 °C for 1 h.
After the immunoreaction, 10 min sonication in pure water caused the Au NPs to dissociate from the bottom of the microtiter plates, which exhibited a better performance than acid-based dissociation.19 Au NPs tend to aggregate under the conditions of acidic media due to hydrogen-bonding. Additionally, Au NPs served as nanoparticle tags for signal pulse counting. Au NPs of 20, 45, and 80 nm in diameter were introduced to ICPMS and investigated in the TRA mode. A good correlation between the transient signal frequency and the analyte concentration was observed. α-Fetoprotein (AFP), which is an important tumor biomarker, was selected as the model analyte. The limit of detection (the concentration of 20 percent signal inhibition, IC20), was calculated to be 0.016 μg L−1 of AFP. Non-competitive bioassay (sandwich immunoassay) was also investigated for obtaining high sensitivity (Fig. 2). The analytical performances of Au-labeled antibody were compared in detail using ICPMS in conventional integral mode and single particle mode. The limit of detection was calculated to be 0.02 pg mL−1 (100 μL of 0.13 fM IgG, i.e., 1.3 × 10−20 mol) for detecting Au NPs tagged antibodies using a 1 min data acquisition time. A comparison of conventional integral mode and single particle mode ICPMS was made for Au-labeled IgG detection.19 With single particle mode ICPMS detection and 1 min data acquisition time, the limit of detection showed about one order magnitude improvement compared with the conventional integral mode, which demonstrates the possibility of developing highly sensitive immunoassays. However, the precision and linear range of TRA mode detection was slightly worse than the conventional integral mode. The possible aggregation of the Au-NPs may explain the results of the precision study, which caused an uncertainty in frequency counting. Finally, the proposed method was successfully applied in rabbit-anti-human serum samples. The limit of detection can be further lowered by increasing the data acquisition time. Ideally, with a 100% sample introduction efficiency (for instance, using a monodisperse droplet generator or high-performance concentric nebulizer),76,77 there will be a signal flush pulse once there is a nanoparticle tagged molecule.
Single nanoparticles analysis homogenous assay
Homogeneous bioassays are based on the alternation of the activity of a tagged reagent which occurs when its binding with the analyte to form a conjugated bio-complex, and thus does not need any additional separation step. Homogeneous bioassays are very intriguing bioassay formats due to their easy automation, low contamination, and minimal washing/handling steps. The basic principle of homogeneous bioassays is sensitively distinguishing the properties of antigen–antibody complexes or nucleic acid hybridization products from their unreacted counterpart.A one step homogeneous DNA bioassay was developed using ICPMS single nanoparticle analysis (Fig. 3).2 During the bioassay, citrate protected Au NPs were first functionalized with DNA probe 1 and DNA probe 2. The target DNA was then hybridized with Au NPs-DNA probe 1 and Au NPs-DNA probe 2 in buffer solution. Dimers, trimers, or large aggregates of Au NPs were formed due to the hybridization of the target DNA and DNA probes conjugated on Au NPs. The Au NP number decreased during the DNA hybridization, while the nanoparticle size increased. By counting the single nanoparticle number, the amount of target DNA was detected quantitatively. The increased signal for individual Au nanoparticles also indicated that the DNA hybridization caused aggregation of the Au NPs. This simple and sensitive strategy can be easily extended to other bispecific assays, such as immunoassays and aptamer-based bioassays.
 | ||
| Fig. 3 (a) Schematic of homogenous DNA hybridization assay by single particle analysis ICPMS. (b)–(e) TEM images of Au NPs after the addition of 10 μL target DNA of different concentrations to the Au NPs probe solution: (b) no target DNA; (c) 10 pM target DNA; (d) 100 pM target DNA; and (e) 1 nM target DNA.2 Reproduced with permission from John Wiley and Sons. | ||
Potential for multiplex single molecule analysis
Biological research and clinical practice often rely on the development of powerful analytical methods for the accurate and precise quantification of biomolecules. For instance, the concentrations of a great number of disease-related biomarkers lie in the range of 10−16 to 10−12 M in serum. This low concentration is a great challenge for conventional immunoassays (for instance, enzyme-linked immunosorbent assays often have detection limits higher than 10−12 M).78 Consequently, the design of new bioassays to improve detection sensitivity has gained great research interest. Single molecule analysis strategies have great potential in the development of ultrasensitive bioassays. Single molecule analysis is to identify 1/NA (1.66 × 10−24) mol or 1.66 yoctomole of a substance. By detecting single molecules individually, the ultimate detection limit is obtained, while the signal to noise ratio remains constant with decreasing analyte concentration.79One of the mainstream techniques for single molecule analysis is nanoparticle direct counting. The first single molecule strategy used for quantitative analysis was the nanopore developed by Bayley et al.80 for counting metal ions and organic molecules. The nanopore design is quite similar to the pores and channels in cellular membranes. To measure conductivity across the membrane, electrodes were placed on each side of the nanopore. When the analyte passes through and somewhat blocks the nanopore, the conductivity across the membrane decreases. Modification of the nanopore with affinity substances results in analyte selectivity. Consequently, resistive pulses indicate the presence of the analyte, and the frequency of pulses reflects the concentration of the analyte. Similarly, single-molecule fluorescence counting is also applied in the quantification of target molecules.78 In this case, after fluorescence tagging, the fluorescence pulse indicates the presence of target molecules, and the frequency of the fluorescence pulse represents the target molecule concentration.
Another mainstream technique for single molecule analysis is digital bioassay with binary property.81 Distinct from conventional bioassays performed in a single react vessel, the solution is divided into many microreactors in digital bioassays. The microreactors are filled with 0 or 1 target molecule. For digital nucleic acid analysis (digital PCR), target sequences are amplified by polymerase. Digital protein bioassays are usually dependent on the combined use of enzymes and fluorogenic substrates. Despite their great success, these strategies often suffer from drawbacks such as low fluorescent tag stability and limited spectral window capacity for multiplex detection. Also, a few studies have been reported on non-fluorescent digital bioassays.
Due to excellent isotopic resolution of ICPMS, highly-multiplexed detection can be realized, thus opening the door for multiplexed single molecule sensing which may yield valuable insight into complex systems.3 More than 100 metal stable isotopes and their nanoparticles are potentially applicable as tags for single nanoparticle ICPMS bioassay without the need for any special optical or electrochemical properties. To demonstrate a sensitive and multiplex DNA assay, single nanoparticle ICPMS analysis was employed as the readout for three DNA targets associated with clinical diseases (HIV, HAV, and HBV) in a heterogeneous sandwich bioassay (Fig. 4).1 The LODs were 1 pM (10 seconds sampling time) using DNA probes tagged with Au NPs, Ag NPs, and Pt NPs via DNA sandwich hybridization assay. Moreover, the high sensitivity and high throughput DNA assay was achieved without chain reaction amplification or any other signal amplification. Besides, single nucleotide polymorphisms in genes were also successfully discriminated. Furthermore, single nanoparticle homogenous assays can be also extended to multiplex bioassays (Fig. 5). One of the most important reasons for this is that the signal of single nanoparticle ICPMS is not sensitive to the sample matrix, thus this method has the potential to provide accurate and multiplex determination of low abundant DNA in real samples. To realize the fast screening of multiple isotopes in individual nanoparticles, TOF-ICPMS is required.
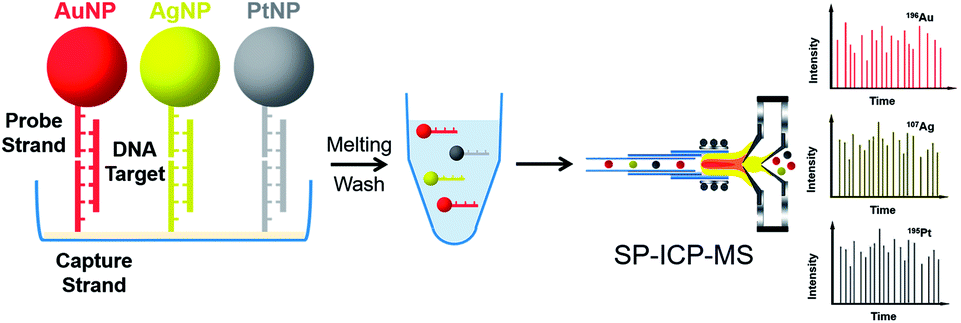 | ||
| Fig. 4 Schematic of multiplex DNA assay based on nanoparticle probes by single particle ICPMS.1 Reproduced with permission from American Chemical Society. | ||
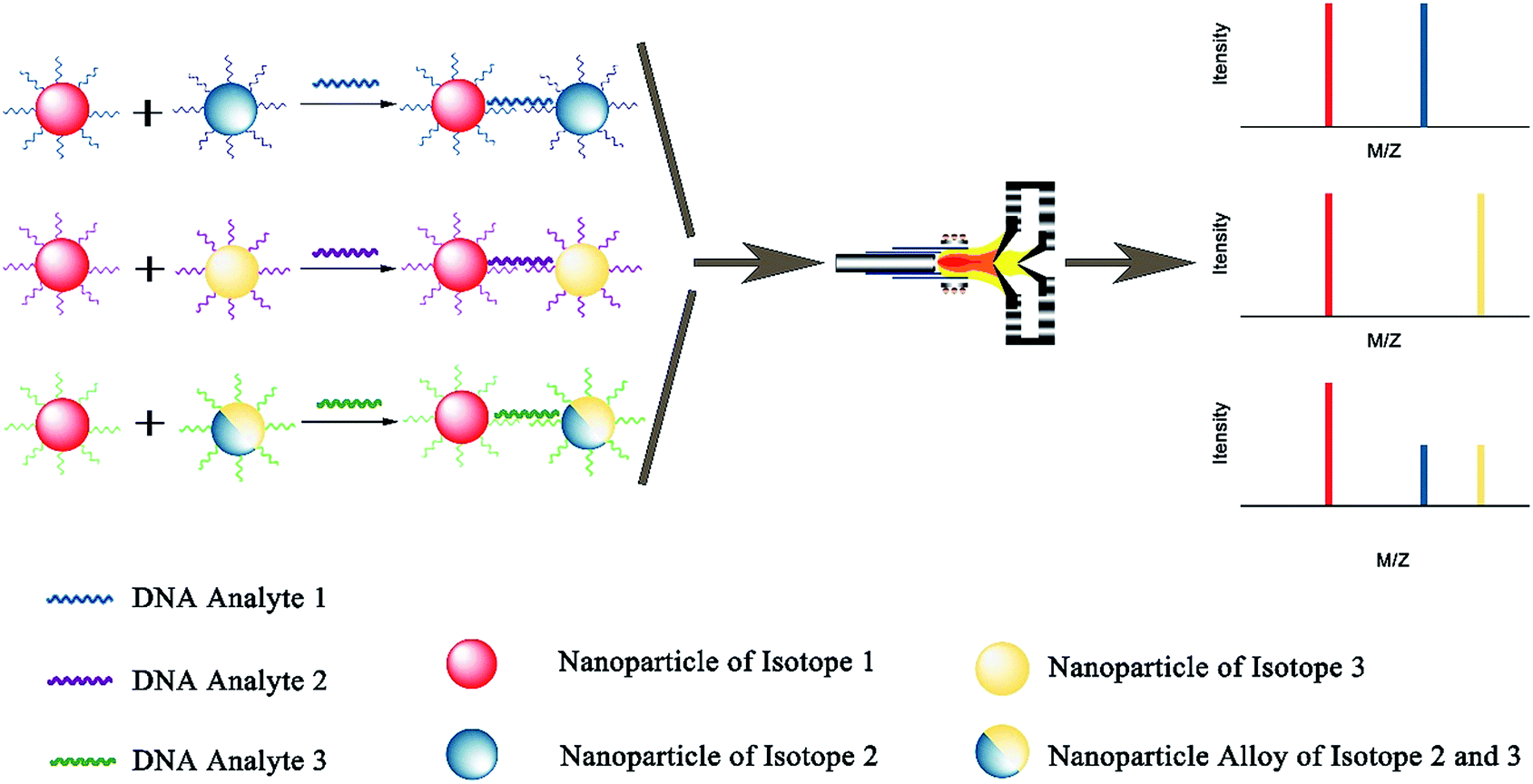 | ||
| Fig. 5 Schematic of multiplex and homogenous single particle analysis bioassay. | ||
TOF-ICPMS is capable of measuring multiple elements simultaneously in a short transient, while MC-ICPMS is capable of monitoring multi-isotopes of adjacent mass to charge ratios. MC-ICPMS in single particle mode has been utilized to analyse isotope ratios in individual particles.82 A 130 nm diameter as the low limit of detectable particle size was acquired by this method. Besides, by using the linear regression slope method, the precision and accuracy of erbium isotope ratios have been improved significantly, where its size detection limit proves that this technique is suitable for the sensitive detection and precise isotopic measurement of individual nanoparticles. However, MC-ICPMS in single particle mode has drawbacks. Tanner et al. observed isotope ratios drifting in transient signals, which limit the precision.71 To combine TOF-ICPMS and single nanoparticle bioassay, Tanner et al. designed a commercial mass cytometer.20 This high-throughput mass cytometer allows 44 or even more markers to be analyzed in thousands of single cells in every minute.83 This invention was selected as one of the top cutting edge technologies of 2011 by The Scientist Magazine – Life Sciences.
Similar to the current fluorescence digital ELISA design,84 this ICPMS single nanoparticle analysis technique can be also employed in the digital format with additional multiplex potential. Already, laser ablation (LA-ICPMS) has been successfully used for the detection of multiple stable isotope labeled biomolecules in immuno-microarrays.85 AFP, CEA, and human IgG, as model analytes, were detected by sandwich type immunoassays on the microarray with Sm3+-labeled AFP antibody, Eu3+-labeled CEA antibody, and Au NPs-labeled goat-antihuman IgG as labeled antibodies. The spatial resolution was at the micrometer level, which is potentially suitable for the fabrication of high-density microarrays. Further, the simultaneous imaging of 32 proteins realized discrimination of cell subpopulations and cell to cell interactions by LA-ICPMS at subcellular resolution, which highlighted tumor heterogeneity.86 As schematically illustrated in Fig. 6, high resolution imaging mass spectrometry methods may provide a next generation digital ELISA technique for multiplex single molecule analysis.
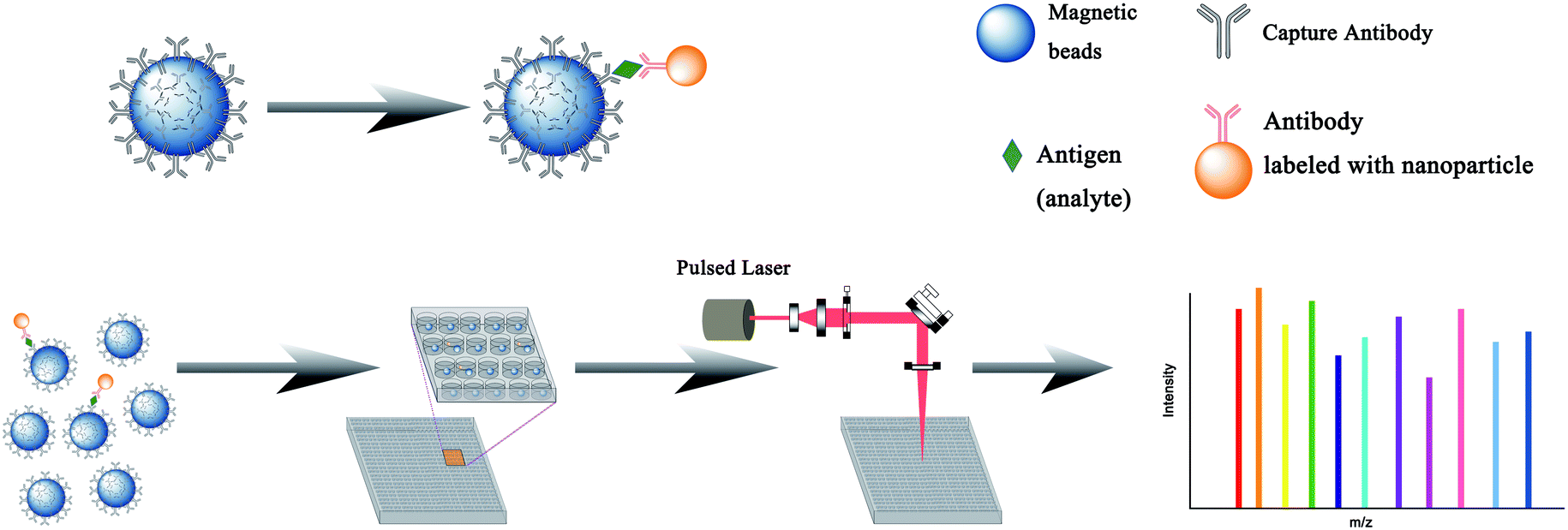 | ||
| Fig. 6 Schematic of laser ablation ICPMS detection of single nanoparticles for digital assay. | ||
Conclusion
The development of new strategies for sensitive bioassay is one of the hottest topics in recent years. The combination of single nanoparticle analysis and ICPMS represents a platform of high potential, highly sensitive bioassays without any signal amplification procedure. The single nanoparticle analysis homogenous assay, single nanoparticle analysis homogenous assay, and multiplex single molecule analysis have been demonstrated in several reports. However, complex sample matrices (e.g. serum) containing many different biomolecules and salts may cause matrix effects. Thus, the further development of single nanoparticle analysis bioassays in real sample matrices for the detection of disease biomarkers with low abundance is necessary. Complex sample preparation is still a major challenge to single nanoparticle analysis and bioassays. ICPMS can perform sensitive and quantitative single nanoparticle analysis and TOF-ICPMS can further provide multiplex single nanoparticle analysis. Therefore, single nanoparticle ICPMS analysis approaches may well satisfy the requirement and hold promising potential for the development of multiplex immunoassays, DNA hybridization assays, and other biomolecule assays at the single molecule level.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the National Natural Science Foundation of China (No. 21375089, 21505095 and 21505008) and the Fundamental Research Funds for the central Universities for financial support. Dr Shixi Zhang of Institute of Geographic Sciences and Natural Resources Research, CAS is gratefully thanked for the help in the design of figures.References
- S. Zhang, G. Han, Z. Xing, S. Zhang and X. Zhang, Anal. Chem., 2014, 86, 3541–3547 CrossRef CAS PubMed.
- G. J. Han, Z. Xing, Y. H. Dong, S. C. Zhang and X. R. Zhang, Angew. Chem., Int. Ed., 2011, 50, 3462–3465 CrossRef CAS PubMed.
- R. Liu, S. Zhang, C. Wei, Z. Xing, S. Zhang and X. Zhang, Acc. Chem. Res., 2016, 49, 775–783 CrossRef CAS PubMed.
- Y. Liang, L. M. Yang and Q. Q. Wang, Appl. Spectrosc. Rev., 2016, 51, 117–128 CrossRef CAS.
- H. Wang, M. He, B. Chen and B. Hu, J. Anal. At. Spectrom., 2017, 32, 1650–1659 RSC.
- Y. He, Y. Zhang, C. Wei, C. Li, Y. Gao and R. Liu, Appl. Spectrosc. Rev., 2014, 49, 492–512 CrossRef CAS.
- R. Liu, P. Wu, L. Yang, X. Hou and Y. Lv, Mass Spectrom. Rev., 2014, 33, 373–393 CrossRef CAS PubMed.
- J. S. Becker and N. Jakubowski, Chem. Soc. Rev., 2009, 38, 1969–1983 RSC.
- J. Bettmer, M. M. Bayon, J. R. Encinar, M. L. F. Sanchez, M. D. F. de la Campa and A. S. Medel, J. Proteomics, 2009, 72, 989–1005 CrossRef CAS PubMed.
- R. Lobinski, D. Schaumloffel and J. Szpunar, Mass Spectrom. Rev., 2006, 25, 255–289 CrossRef CAS PubMed.
- R. Liu, X. Liu, Y. R. Tang, L. Wu, X. D. Hou and Y. Lv, Anal. Chem., 2011, 83, 2330–2336 CrossRef CAS PubMed.
- Y. Luo, X. Yan, Y. Huang, R. Wen, Z. Li, L. Yang, C. J. Yang and Q. Wang, Anal. Chem., 2013, 85, 9428–9432 CrossRef CAS PubMed.
- Y. He, D. Chen, M. Li, L. Fang, W. Yang, L. Xu and F. Fu, Biosens. Bioelectron., 2014, 58, 209–213 CrossRef CAS PubMed.
- K. Bruckner, K. Schwarz, S. Beck and M. W. Linscheid, Anal. Chem., 2014, 86, 585–591 CrossRef PubMed.
- S. Zhang, R. Liu, Z. Xing, S. Zhang and X. Zhang, Chem. Commun., 2016, 52, 14310–14313 RSC.
- M. D. Montano, J. W. Olesik, A. G. Barber, K. Challis and J. F. Ranville, Anal. Bioanal. Chem., 2016, 408, 5053–5074 CrossRef CAS PubMed.
- F. Laborda, E. Bolea and J. Jimenez-Lamana, Anal. Chem., 2014, 86, 2270–2278 CrossRef CAS PubMed.
- P. Krystek, A. Ulrich, C. C. Garcia, S. Manohar and R. Ritsema, J. Anal. At. Spectrom., 2011, 26, 1701–1721 RSC.
- R. Liu, Z. Xing, Y. Lv, S. Zhang and X. Zhang, Talanta, 2010, 83, 48–54 CrossRef CAS PubMed.
- D. R. Bandura, V. I. Baranov, O. I. Ornatsky, A. Antonov, R. Kinach, X. D. Lou, S. Pavlov, S. Vorobiev, J. E. Dick and S. D. Tanner, Anal. Chem., 2009, 81, 6813–6822 CrossRef CAS PubMed.
- S. C. Bendall, E. F. Simonds, P. Qiu, E. A. D. Amir, P. O. Krutzik, R. Finck, R. V. Bruggner, R. Melamed, A. Trejo, O. I. Ornatsky, R. S. Balderas, S. K. Plevritis, K. Sachs, D. Pe'er, S. D. Tanner and G. P. Nolan, Science, 2011, 332, 687–696 CrossRef CAS PubMed.
- A. P. Frei, F.-A. Bava, E. R. Zunder, E. W. Y. Hsieh, S.-Y. Chen, G. P. Nolan and P. F. Gherardini, Nat. Methods, 2016, 13, 269–275 CrossRef CAS PubMed.
- X. Wei, L. L. Hu, M. L. Chen, T. Yang and J. H. Wang, Anal. Chem., 2016, 88, 12437–12444 CrossRef CAS PubMed.
- M. Wang, L. N. Zheng, B. Wang, H. Q. Chen, Y. L. Zhao, Z. F. Chai, H. J. Reid, B. L. Sharp and W. Y. Feng, Anal. Chem., 2014, 86, 10252–10256 CrossRef CAS PubMed.
- S. Tanner, V. Baranov, O. Ornatsky, D. Bandura and T. George, Cancer Immunol. Immunother., 2013, 62, 955–965 CrossRef PubMed.
- D. R. Winter, G. Ledergor and I. Amit, Nat. Biotechnol., 2015, 33, 931–932 CrossRef CAS PubMed.
- N. Zivanovic, A. Jacobs and B. Bodenmiller, Curr. Top. Microbiol. Immunol., 2014, 377, 95–109 CAS.
- J. Shang and X. H. Gao, Chem. Soc. Rev., 2014, 43, 7267–7278 RSC.
- C. Degueldre and P. Y. Favarger, Colloids Surf., A, 2003, 217, 137–142 CrossRef CAS.
- A. R. M. Bustos and M. R. Winchester, Anal. Bioanal. Chem., 2016, 408, 5051–5052 CrossRef CAS PubMed.
- J. J. Tan, J. Y. Liu, M. D. Li, H. El Hadri, V. A. Hackley and M. R. Zechariah, Anal. Chem., 2016, 88, 8548–8555 CrossRef CAS PubMed.
- R. P. Lamsal, G. Jerkiewicz and D. Beauchemin, Anal. Chem., 2016, 88, 10552–10558 CrossRef CAS PubMed.
- H. E. Pace, N. J. Rogers, C. Jarolimek, V. A. Coleman, C. P. Higgins and J. F. Ranville, Anal. Chem., 2011, 83, 9361–9369 CrossRef CAS PubMed.
- M. Tanner and D. Günther, Anal. Chim. Acta, 2009, 633, 19–28 CrossRef CAS PubMed.
- H. E. Pace, N. J. Rogers, C. Jarolimek, V. A. Coleman, C. P. Higgins and J. F. Ranville, Anal. Chem., 2011, 83, 9361–9369 CrossRef CAS PubMed.
- J. Liu, K. E. Murphy, M. R. Winchester and V. A. Hackley, Anal. Bioanal. Chem., 2017, 409, 6027–6039 CrossRef CAS PubMed.
- L. Frechette-Viens, M. Hadioui and K. J. Wilkinson, Talanta, 2017, 163, 121–126 CrossRef CAS PubMed.
- S. Lee, X. Bi, R. B. Reed, J. F. Ranville, P. Herckes and P. Westerhoff, Environ. Sci. Technol., 2014, 48, 10291–10300 CrossRef CAS PubMed.
- R. Liu, Z. Xing, Y. Lv, S. C. Zhang and X. R. Zhang, Talanta, 2010, 83, 48–54 CrossRef CAS PubMed.
- R. C. Merrifield, C. Stephan and J. R. Lead, Talanta, 2017, 162, 130–134 CrossRef CAS PubMed.
- Y. S. Yang, P. U. Atukorale, K. D. Moynihan, A. Bekdemir, K. Rakhra, L. Tang, F. Stellacci and D. J. Irvine, Nat. Commun., 2017, 8, 14069 CrossRef CAS PubMed.
- Y. Yang, C. L. Long, H. P. Li, Q. Wang and Z. G. Yang, Sci. Total Environ., 2016, 563–564, 996–1007 CrossRef CAS PubMed.
- Y. Liu, M. He, B. Chen and B. Hu, Spectrochim. Acta, Part B, 2016, 122, 94–102 CrossRef CAS.
- J. Jimenez-Lamana and V. I. Slaveykova, Sci. Total Environ., 2016, 573, 946–953 CrossRef CAS PubMed.
- C. A. Sötebier, S. M. Weidner, N. Jakubowski, U. Panne and J. Bettmer, J. Chromatogr. A, 2016, 1468, 102–108 CrossRef PubMed.
- X. Zhou, J. Liu, C. Yuan and Y. Chen, J. Anal. At. Spectrom., 2016, 31, 2285–2292 RSC.
- J. Jiménez-Lamana, J. Wojcieszek, M. Jakubiak, M. Asztemborska and J. Szpunar, J. Anal. At. Spectrom., 2016, 31, 2321–2329 RSC.
- S. G. Elci, B. Yan, S. T. Kim, K. Saha, Y. Jiang, G. A. Klemmer, D. F. Moyano, G. Y. Tonga, V. M. Rotello and R. W. Vachet, Analyst, 2016, 141, 2418–2425 RSC.
- D. Bao, Z. G. Oh and Z. Chen, Front. Plant Sci., 2016, 7, 32 Search PubMed.
- K. Newman, C. Metcalfe, J. Martin, H. Hintelmann, P. Shaw and A. Donard, J. Anal. At. Spectrom., 2016, 31, 2069–2077 RSC.
- C. A. Sötebier, D. J. Kutscher, L. Rottmann, N. Jakubowski, U. Panne and J. Bettmer, J. Anal. At. Spectrom., 2016, 31, 2045–2052 RSC.
- J. Soto-Alvaredo, F. Dutschke, J. Bettmer, M. Montes-Bayón, D. Pröfrock and A. Prange, J. Anal. At. Spectrom., 2016, 31, 1549–1555 RSC.
- B. Meermann, K. Wichmann, F. Lauer, F. Vanhaecke and T. A. Ternes, J. Anal. At. Spectrom., 2016, 31, 890–901 RSC.
- K. Huang, K. Xu, J. Tang, L. Yang, J. Zhou, X. Hou and C. Zheng, Anal. Chem., 2015, 87, 6584–6591 CrossRef CAS PubMed.
- M. Roman, C. Rigo, H. Castillo-Michel, I. Munivrana, V. Vindigni, I. Micetic, F. Benetti, L. Manodori and W. R. Cairns, Anal. Bioanal. Chem., 2016, 408, 5109–5124 CrossRef CAS PubMed.
- A. R. Donovan, C. D. Adams, Y. Ma, C. Stephan, T. Eichholz and H. Shi, Anal. Bioanal. Chem., 2016, 408, 5137–5145 CrossRef CAS PubMed.
- D. Feng, F. Tian, W. Qin and X. Qian, Chem. Sci., 2016, 7, 2246–2250 RSC.
- S. Böhme, H. J. Stärk, D. Kühnel and T. Reemtsma, Anal. Bioanal. Chem., 2015, 407, 5477–5485 CrossRef PubMed.
- A. R. Montoro Bustos, E. J. Petersen, A. Possolo and M. R. Winchester, Anal. Chem., 2015, 87, 8809–8817 CrossRef CAS PubMed.
- M. Matczuk, K. Anecka, F. Scaletti, L. Messori, B. K. Keppler, A. R. Timerbaev and M. Jarosz, Metallomics, 2015, 7, 1364–1370 RSC.
- R. Peters, Z. Herrera-Rivera, A. Undas, M. van der Lee, H. Marvin, H. Bouwmeester and S. Weigel, J. Anal. At. Spectrom., 2015, 30, 1274–1285 RSC.
- A. Elias, S. H. Crayton, R. Warden-Rothman and A. Tsourkas, Sci. Rep., 2014, 4, 5840 CrossRef CAS PubMed.
- D. Drescher, I. Zeise, H. Traub, P. Guttmann, S. Seifert, T. Büchner, N. Jakubowski, G. Schneider and J. Kneipp, Adv. Funct. Mater., 2014, 24, 3765–3775 CrossRef CAS.
- X. Zhou, M. Dorn, J. Vogt, D. Spemann, W. Yu, Z. Mao, I. Estrela-Lopis, E. Donath and C. Gao, Nanoscale, 2014, 6, 8535–8542 RSC.
- B. Franze and C. Engelhard, Anal. Chem., 2014, 86, 5713–5720 CrossRef CAS PubMed.
- R. S. Houk, V. A. Fassel, G. D. Flesch, H. J. Svec, A. L. Gray and C. E. Taylor, Anal. Chem., 1980, 52, 2283–2289 CrossRef CAS.
- A. J. Managh, D. N. Douglas, K. Makella Cowen, H. J. Reid and B. L. Sharp, J. Anal. At. Spectrom., 2016, 31, 1688–1692 RSC.
- A. J. Walder and P. A. Freedman, J. Anal. At. Spectrom., 1992, 7, 571–575 RSC.
- K. Shigeta, G. Koellensperger, E. Rampler, H. Traub, L. Rottmann, U. Panne, A. Okino and N. Jakubowski, J. Anal. At. Spectrom., 2013, 28, 637–645 RSC.
- L. Yang, Mass Spectrom. Rev., 2009, 28, 990–1011 CrossRef CAS PubMed.
- M. Tanner and D. Gunther, Anal. Chim. Acta, 2009, 633, 19–28 CrossRef CAS PubMed.
- K. G. Heumann and F. Vanhaecke, Anal. Bioanal. Chem., 2007, 390, 433–435 CrossRef.
- O. Borovinskaya, B. Hattendorf, M. Tanner, S. Gschwind and D. Günther, J. Anal. At. Spectrom., 2013, 28, 226–233 RSC.
- E. P. Gray, J. G. Coleman, A. J. Bednar, A. J. Kennedy, J. F. Ranville and C. P. Higgins, Environ. Sci. Technol., 2013, 47, 14315–14323 CrossRef CAS PubMed.
- S. H. Hu, R. Liu, S. C. Zhang, Z. Huang, Z. Xing and X. R. Zhang, J. Am. Soc. Mass Spectrom., 2009, 20, 1096–1103 CrossRef CAS PubMed.
- S. Gschwind, M. Aja Montes and D. Günther, Anal. Bioanal. Chem., 2015, 407, 4035–4044 CrossRef CAS PubMed.
- K. Inagaki, S.-i. Fujii, A. Takatsu and K. Chiba, J. Anal. At. Spectrom., 2011, 26, 623–630 RSC.
- F. Ma, Y. Li, B. Tang and C. Y. Zhang, Acc. Chem. Res., 2016, 49, 1722–1730 CrossRef CAS PubMed.
- J. J. Gooding and K. Gaus, Angew. Chem., Int. Ed., 2016, 55, 11354–11366 CrossRef CAS PubMed.
- O. Braha, B. Walker, S. Cheley, J. J. Kasianowicz, L. Song, J. E. Gouaux and H. Bayley, Chem. Biol., 1997, 4, 497–505 CrossRef CAS PubMed.
- Y. Zhang and H. Noji, Anal. Chem., 2017, 89, 92–101 CrossRef CAS PubMed.
- Y. Su, W. Wang, Z. Li, H. Deng, G. Zhou, J. Xu and X. Ren, J. Anal. At. Spectrom., 2015, 30, 1184–1190 RSC.
- S. C. Bendall, K. L. Davis, E. D. Amir, M. D. Tadmor, E. F. Simonds, T. J. Chen, D. K. Shenfeld, G. P. Nolan and D. Pe'er, Cell, 2014, 157, 714–725 CrossRef CAS PubMed.
- D. M. Rissin, C. W. Kan, T. G. Campbell, S. C. Howes, D. R. Fournier, L. Song, T. Piech, P. P. Patel, L. Chang, A. J. Rivnak, E. P. Ferrell, J. D. Randall, G. K. Provuncher, D. R. Walt and D. C. Duffy, Nat. Biotechnol., 2010, 28, 595–599 CrossRef CAS PubMed.
- S. H. Hu, S. C. Zhang, Z. C. Hu, Z. Xing and X. R. Zhang, Anal. Chem., 2007, 79, 923–929 CrossRef CAS PubMed.
- C. Giesen, H. A. O. Wang, D. Schapiro, N. Zivanovic, A. Jacobs, B. Hattendorf, P. J. Schuffler, D. Grolimund, J. M. Buhmann, S. Brandt, Z. Varga, P. J. Wild, D. Gunther and B. Bodenmiller, Nat. Methods, 2014, 11, 417–422 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ja00235a |
| This journal is © The Royal Society of Chemistry 2018 |