
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUsing highly emissive and environmentally sensitive o-carborane-functionalized metallophosphors to monitor mitochondrial polarity†
Xiang
Li
a,
Xiao
Tong
b,
Yongheng
Yin
a,
Hong
Yan
 *a,
Changsheng
Lu
*a,
Wei
Huang
b and
Qiang
Zhao
*b
*a,
Changsheng
Lu
*a,
Wei
Huang
b and
Qiang
Zhao
*b
aState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, P. R. China. E-mail: hyan1965@nju.edu.cn; luchsh@nju.edu.cn
bKey Laboratory for Organic Electronics & Information Displays, Institute of Advanced Materials, Nanjing University of Posts and Telecommunications, Nanjing 210023, P. R. China. E-mail: iamqzhao@njupt.edu.cn
First published on 30th May 2017
Abstract
Mitochondria as vital intracellular organelles play critical roles in multiple physiological processes, and their polarity is a crucial characteristic that can reveal the intracellular environment and impact cellular events. In this work, we designed and synthesized a novel series of highly emissive and environmentally sensitive phosphorescent iridium(III) complexes (2a–2e, 3a–3e and 4) functionalized by o-carborane. These complexes showed high emission quantum yields both in solution and in solid state (up to ΦPL = 0.82), long emission lifetime and tunable emission wavelength over 74 nm by introduction of a carboranyl motif in their ligands. Importantly, all the complexes have shown significant solvatochromic effects in contrast to the carborane-free control complex. Among them, complex 2d shows the highest sensitivity to polarity of solvents with a MPPS (maximum peak phosphorescence shift) value of 42 nm and clear dependence of phosphorescence lifetime on solvent polarity. Interestingly, complex 2d can easily penetrate into cells and preferentially distribute in mitochondria. To utilize these properties, the first phosphorescent imaging of mitochondrial polarity has been realized by photoluminescence lifetime imaging microscopy (PLIM), which can monitor mitochondria-relevant cellular processes such as cell apoptosis and distinguish cancer cells from normal cells. Compared to intensity-based sensing, lifetime-based detection is independent of the probe concentration, excitation power and photobleaching of probes, which can show high accuracy and reproducibility.
Introduction
Polarity plays a crucial role in chemistry and chemical biology, and intracellular polarity reflects a lot of complicated physiological and pathological processes in biological systems. Many cellular events, such as adipogenic differentiation, immune response activation, cell migration and death, and molecular transport across cell layers, may phenotypically lead to polarity variation in cells.1 Therefore, abnormal changes in polarity are highly relevant to biological disorders and diseases (e.g., diabetes, liver cirrhosis).2 Mitochondria are vital intracellular organelles and play critical roles in multiple physiological processes, including metabolism, ATP production, cell signaling, and apoptosis. Therefore, mitochondrial polarity assays can reveal information about the cellular internal environment such as transportation of proteins, maintaining of cell function, and homeostasis.3 Thus it is of great significance to monitor mitochondrial polarity at a cellular level.Luminescent bioimaging based on optical probes is a powerful technique for monitoring cellular environmental processing in living systems and has received considerable interest in recent years.4 Currently, some polarity-sensitive fluorescent probes are available.3,5 However, those fluorescent probes are mainly limited to organic dyes, which feature short emission lifetime and suffer from background interference from biosamples and serious photobleaching.6 In contrast, the phosphorescent transition-metal complexes (PTMCs, namely metallophosphors), especially the iridium(III) complexes, exhibit advantageous photophysical properties, such as long phosphorescence lifetime as well as high quantum yield and excellent photostability. These make them promising probes for biological imaging. Especially, their long and sensitive phosphorescence lifetimes are very beneficial for lifetime-based biosensing and bioimaging by photoluminescence lifetime imaging microscopy (PLIM), which can effectively eliminate the unwanted background interference based on the emission lifetime difference between the phosphorescent probe and interference signal. Moreover, the emission lifetime as the sensing signal is independent of the probe concentration, excitation power and photobleaching of probes, which can show high accuracy and reproducibility.7 To date, there have been no reports about polarity-sensitive phosphorescent probes for bioimaging. Therefore, the development of a new type of phosphorescent probes through a combination of high quantum yield, long emission lifetime and excellent polarity sensitivity is highly desired.
Icosahedral o-carborane (1,2-C2B10H12) possesses strong electron-withdrawing ability (C-substitution) and an alterable C–C bond.8o-Carborane cage can serve as a rigid hindrance to prevent probable intermolecular interactions and reduce self-quenching as well as potential concentration quenching.9 Its electron-withdrawing ability can lead to considerable charge redistribution of the phosphorescent core, which can further result in a large emission shift.10 On the other hand, the stretchable C–C bond is quite sensitive to its chemical surroundings, such as substitution at the C–H bonds8–10 and even solvents or media.10 From this viewpoint, o-carborane embedded metallophosphors might be an ideal polarity-sensitive probe in cell imaging for potential biomedical applications.
On the basis of the above hypotheses, a novel series of phosphorescent iridium(III) complexes based on o-carborane functionalized N⁁N ligands have been designed and synthesized for the first time (Scheme 1). The introduction of carborane to the bipyridine ligand of the cationic iridium(III) complexes (2a–2e, 3a–3e and 4) has led to highly improved phosphorescence quantum yields both in solution (from 0.26 to 0.79) and in solid state (from 0.07 to 0.82) in comparison to the carborane-free control complex Model. The emission color has been tuned from green to yellow, or even to red (up to 74 nm). The photophysical properties of these iridium(III) complexes are quite sensitive to the polarity of solvents. Interestingly, the complexes show mitochondria targeting, therefore complex 2d was chosen to develop a phosphorescence probe for monitoring mitochondrial polarity at the cell level through photoluminescence lifetime imaging microscopy. As a result, such a probe can distinguish cancer cells from normal cells, as well as differentiate living cells from dying cells and dead cells.
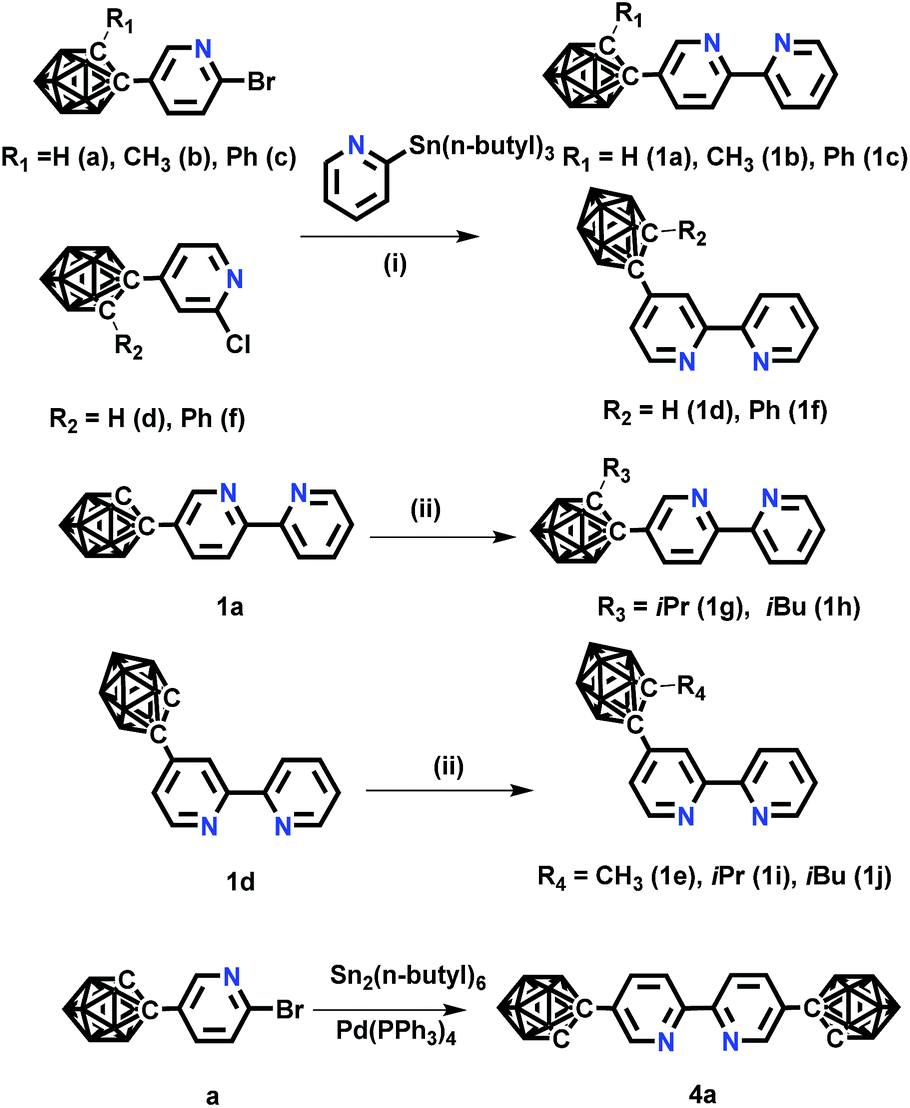 | ||
| Scheme 1 The synthetic routes of o-carborane modified N⁁N ligands. Conditions: (i) Pd(PPh3)4, toluene, 110 °C, 24 h; (ii) NaH, RI (R = CH3, iPr, iBu), DMF, −20 °C. | ||
Results and discussion
Synthesis and characterizations
The novel o-carborane-modified N⁁N ligands (1a–1j) (Schemes 1 and S1†) were synthesized in high yields by using commercially available B10H10(Et4N)2 which is cheap, stable and non-toxic.9f Through the change of the 2-R substituent or the substitution site of the carboranyl unit at bipyridine,11,12 two series of complexes 2a–2e and 3a–3e were prepared by the reactions of the dimeric [(C⁁N)2Ir(μ-Cl)2Ir(C⁁N)2] and the corresponding carborane-functionalized ligands (1a–1j) (Schemes 1 and 2). Complex 4 containing two carboranyl motifs was also synthesized in order to further tune the photophysical properties. Both the new ligands and the newly generated iridium(III) complexes have been fully characterized by NMR, MS spectroscopy, elemental analyses, and X-ray diffraction (Fig. 1, S1 and Table S1†). The crystal structures of complexes Model, 2a, 2b, 2d, 3b, 3c, and 3e exhibit a distorted octahedron geometry around the iridium center (Fig. 1), and the carboranyl units have led to an obvious increase in the volume of the complexes (Tables S1 and S2†). Due to the bulkiness of o-carborane, no intermolecular π–π interactions were observed from the packing structures. The advantage of the bulky carborane cage in inhibiting intermolecular interactions is important to improve the phosphorescent emission of iridium(III) complexes both in solution and in solid state.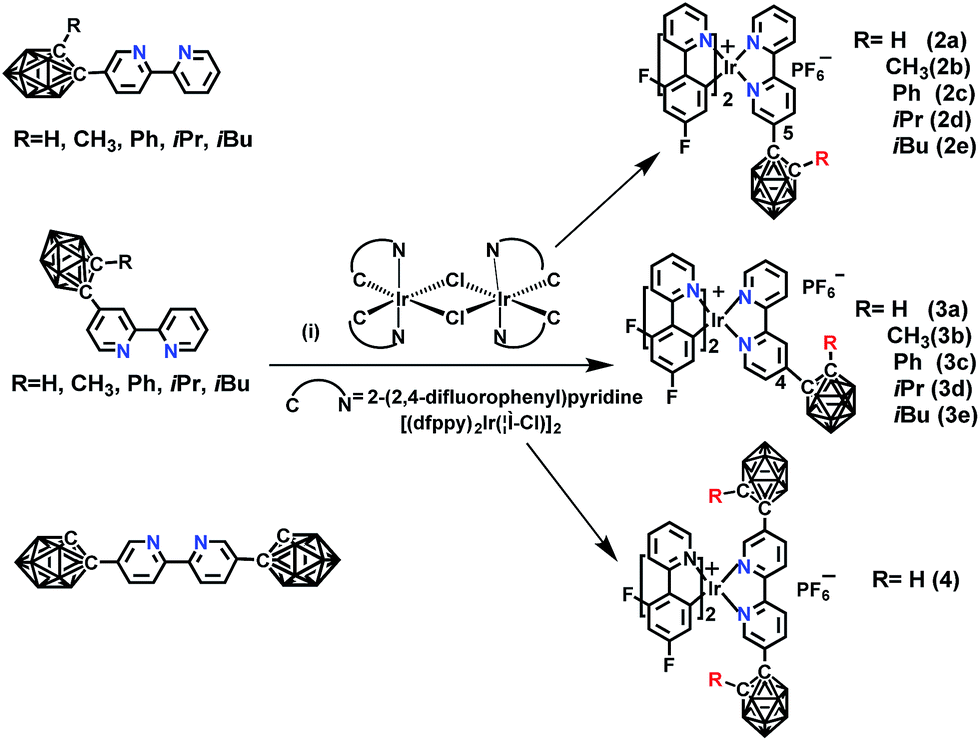 | ||
Scheme 2 The synthetic routes of o-carborane modified iridium(III) complexes: (i) CH2Cl2/CH3OH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), refluxing overnight, KPF6. :1, v/v), refluxing overnight, KPF6. | ||
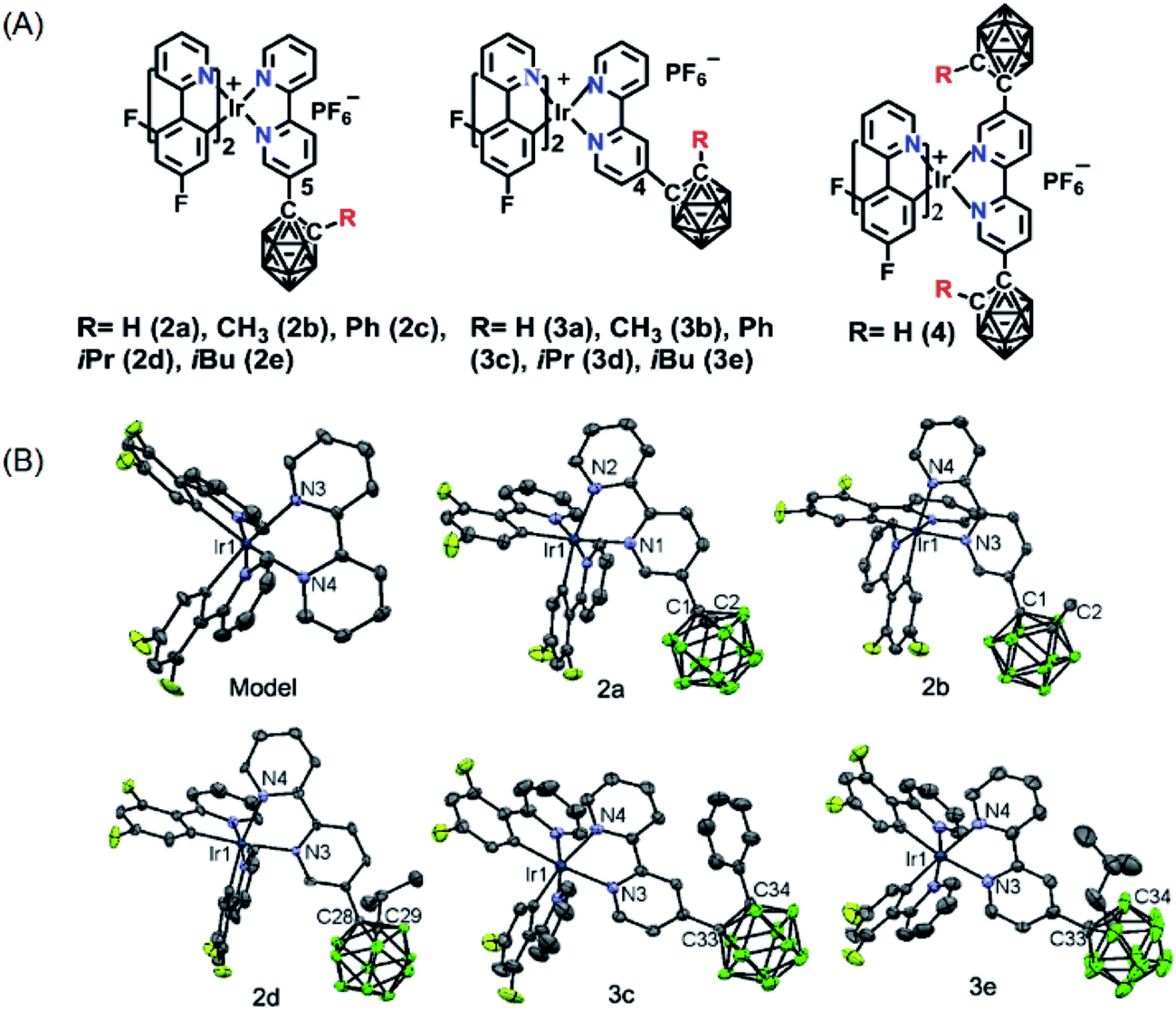 | ||
| Fig. 1 (A) Nomenclatures of carborane-modified iridium(III) complexes; (B) ORTEP diagrams of complexes Model, 2a, 2b, 2d, 3c, and 3e. H atoms, solvent molecules, and PF6− anions are omitted for clarity. | ||
Photophysical properties
The UV/vis absorption and photoluminescence (PL) spectra were measured in CH2Cl2 solution (Table 1). All the complexes show similar absorption bands (Fig. S10 and S11†) compared to the model one. The main strong absorption bands below 350 nm are mainly attributed to the π–π* transitions from the ligands. The weak absorption bands from 350 nm to 450 nm are assigned to metal-to-ligand charge transfer (MLCT) and ligand-to-ligand charge transfer (LLCT) characters, which is referenced by the similar reported ionic iridium(III) complexes. In solution, 2a–2e have similar emission wavelength (λ = 563 nm) (Fig. 2(A)), but a 43 nm bathochromic shift was observed in comparison to the Model, corresponding to the emission color change from green to yellow. The quantum yields have been improved varying from 0.49 to 0.79 (referenced to 0.26 for the Model, see Table 1) dependent on the 2-R substituent at o-carborane, which is higher than the cationic iridium complex (ΦPL = 0.67) containing o-carborane modified cyclometalated ligands.9d The highest quantum yield was observed as 0.79 for 2d owing to the relatively large size of the R (iPr) group. Complexes 3a–3e containing an o-carboranyl unit at a different site of the N⁁N ligand also show highly efficient emission in solution (ΦPL = 0.55–0.76, Table 1). Their emission wavelength is positioned at 556 nm (Fig. 2(B)), having a 36 nm bathochromic shift compared to the Model. A significant red shift of 74 nm has been achieved in complex 4 (594 nm in Fig. 2(C)). Obviously, this is attributed to the cooperative electron-withdrawing property of the two-carboranyl motifs. But the quantum yield of 4 (0.33) in solution has little increase in comparison to the Model (0.26), which may be attributed to the “energy gap law”.13 It is worthy to note that the solid-state quantum yields of these iridium(III) complexes are also significantly increased from 0.07 for Model to 0.16–0.82 for 2a–2e and 0.49–0.71 for 3a–3e, respectively (Table 1 and Fig. S12†), which are among the highest values for metallophosphors. The bulkiness of carborane cage has played an important role in inhibiting intermolecular interactions (Fig. S2–S9†) as indicated by the longer Ir–Ir distances along one or more axes between adjacent metal centers in comparison to the model complex (Fig. S2–S9†). In addition, 2-R substituents decrease the flexibility of molecular structures and increase the volumes of the corresponding unit cells (Tables S1 and S2†), which are important to suppress the interactions between metallophosphors and improve quantum yields. This is clearly attributed to the steric property of the carboranyl motif.| Complex |
λ
abs
[nm] (lgε) |
λ em [nm] | τ [ns] | Φ PL | E oxonset (eV) | E g (eV) | HOMO/LUMOb (eV) |
|---|---|---|---|---|---|---|---|
| a In CH2Cl2. b Data in degassed CH2Cl2 at 298 K (1.0 × 10−5 M) and data measured in solid state are given in parentheses. c HOMO (eV) = −e(Eoxonset + 4.8), Eg = 1240/λ, LUMO (eV) = Eg + HOMO. | |||||||
| 2a | 244(4.0), 296(3.7), 309(3.7), 364(2.9) | 563.0 | 1115.0 | 0.49(0.41) | 1.239 | 2.94 | −6.039/−3.049 |
| 2b | 251(4.0), 300(3.8), 315(3.8), 365(3.1) | 563.0 | 746.9 | 0.59(0.37) | 1.251 | 2.95 | −6.051/−3.101 |
| 2c | 246(4.2), 303(3.9), 315(3.9), 362(3.3) | 560.0 | 794.0 | 0.62(0.16) | 1.270 | 2.95 | −6.070/−3.120 |
| 2d | 246(4.1), 304(3.9), 315(3.9), 363(3.3) | 563.0 | 760.0 | 0.79(0.82) | 1.257 | 2.96 | −6.057/−3.097 |
| 2e | 246(4.1), 304(3.8), 314(3.8), 362(3.3) | 563.0 | 774.6 | 0.72(0.63) | 1.259 | 2.97 | −6.059/−3.087 |
| Model | 247(4.0), 303(3.8), 316(3.8), 362(3.1) | 520.0 | 561.5 | 0.26(0.07) | 1.208 | 3.18 | −6.008/−2.828 |
| 3a | 244(4.1), 298(3.8), 315(3.8), 362(3.4) | 555.0 | 585.3 | 0.55(0.49) | 1.225 | 2.99 | −6.025/−3.039 |
| 3b | 246(4.1), 299(3.8), 315(3.7), 362(3.3) | 555.0 | 733.0 | 0.66(0.67) | 1.240 | 2.99 | −6.040/−3.053 |
| 3c | 266(4.0), 298(3.8), 317(3.7), 363(3.3) | 553.0 | 465.1 | 0.68(0.54) | 1.238 | 2.99 | −6.038/−3.048 |
| 3d | 243(4.2), 304(3.9), 315(3.8), 361(3.5) | 556.0 | 759.3 | 0.76(0.49) | 1.228 | 2.98 | −6.028/−3.046 |
| 3e | 244(4.1), 303(3.8), 314(3.8), 361(3.4) | 556.0 | 793.6 | 0.73(0.71) | 1.232 | 2.98 | −6.032/−3.050 |
| 4 | 249(2.4), 311(3.7), 321(3.8), 364(3.3) | 594.0 | 597.5 | 0.33(0.18) | 1.260 | 2.81 | −6.060/−3.252 |
 | ||
| Fig. 2 PL spectra of iridium(III) complexes: (A) complexes 2a, 2b, 2c, 2d, 2e, and Model; (B) complexes 3a, 3b, 3c, 3d, 3e, and Model in degassed CH2Cl2 (1.0 × 10−5 M) at room temperature. (C) PL spectra of iridium(III) complexes Model, 2a, and 4 in degassed CH2Cl2 (1.0 × 10−5 M) at room temperature (λex = 365 nm and corresponding luminescence photographs are inset). | ||
Cyclic voltammograms and DFT calculations
The electrochemical studies have revealed that all the complexes have reversible oxidation waves with potentials in the range of 1.2–1.3 V (Fig. S13† and Table 1). The oxidation potentials of 2a–2e are similar, independent of the 2-R substituent at the o-carboranyl unit. The same occurs for 3a–3e. This is consistent with the energy gaps and emissive wavelengths. DFT calculations (Fig. S20–S22†) have shown that all the 2-R substituents at carborane are not involved in orbitals, indicating no contribution to the energy levels.14 The HOMOs are mostly distributed over the cyclometalated C⁁N ligands. The LUMOs of complex Model are distributed merely on the bipyridine ligand, whereas those of carborane-embedded complexes are mainly located on bipyridine plus a small fraction on the carboranyl motif. Owing to the inductive electron-withdrawing effect of carborane, the HOMO − LUMO energy gaps of 2a–2e are narrowed by about 0.23 eV because of the greater LUMO stabilization than that of HOMO, in comparison to the narrowed values of about 0.19 eV for 3a–3e (Fig. S20 and S21†). The LUMO level in complex 4 has been further decreased, demonstrating the overlaid inductive electronic effect of the two carboranyl groups. Thus, the DFT calculations are consistent with the observed red-shifted emissions.Solvatochromic effect
Interestingly, all the complexes have shown significant solvatochromic effects in contrast to complex Model. The maximum peak phosphorescence shift (MPPS, denoted as Δν), is defined as the maximum difference between emissions in the low polar solvent toluene (0.36 debye) and in the high polar solvent DMSO (3.96 debye).15 Thus Δν is given as 9 nm for Model, 19 nm for 2a, 32 nm for 2b, 34 nm for 2c, 42 nm for 2d, 18 nm for 2e, 7 nm for 3a, 7 nm for 3b, 24 nm for 3c, 21 nm for 3d, 6 nm for 3e, and 27 nm for 4 (Fig. S14–S19 and Table S3†). Among them, complex 2d exhibits the largest MPPS of 42 nm, corresponding to a distinct color change from green to yellow (Fig. 3(A)). The emission intensity and lifetime of complex 2d show a decreasing trend with the increase of solvent polarity (Fig. S26†).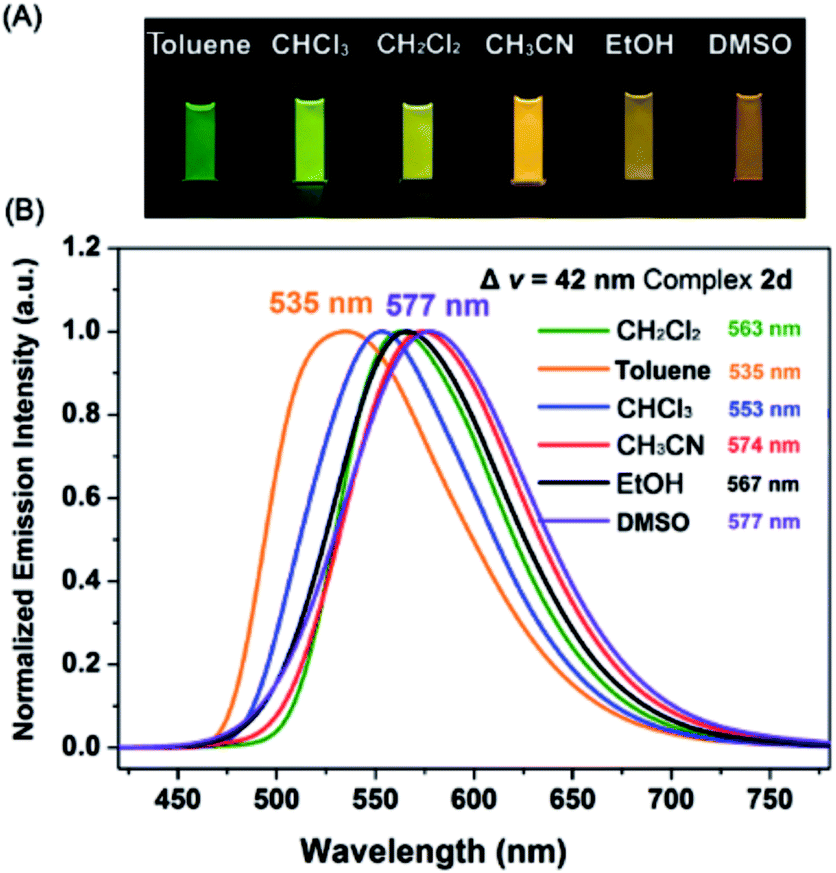 | ||
| Fig. 3 (A) Luminescence photographs of 2d in various solvents. (B) Phosphorescence spectra of complex 2d in toluene (0.36 D), chloroform (1.04 D), dichloromethane (1.60 D), acetonitrile (3.84 D), ethanol (1.69 D), and DMSO (3.96 D). | ||
Studies on MLCT (metal to ligand charge transfer) in iridium(III) complexes have revealed that the solvatochromic effect is related to molecular dipole moments.15 Thus the singlet (S0) and triplet (T1) dipole moments of the complexes in different solvents were calculated (Tables S4 and S5†), and the transition dipole moments (ΔT1–S0) are summarized in Fig. 4 and Tables S6 and S7.† Complexes 2c, 2d, and 3c show big (ΔT1–S0) changes from those in toluene to those in DMSO. This is in accordance with the MPPS shown in emissions (i.e. 34 nm for 2c, 42 nm for 2d, and 24 nm for 3c). Generally, introduction of the electron-withdrawing carboranyl group into the bipyridine ring (N⁁N ligand) has enhanced the transition dipole moments in comparison to complex Model (Tables S6 and S7†). The calculations also demonstrate that the molecular transition dipole moments of the iridium(III) complexes can be finely adjusted by the carboranyl group, as reflected by different emissions. This might shed new light on the utilization of the transition dipole moments of iridium(III) complexes for applications.
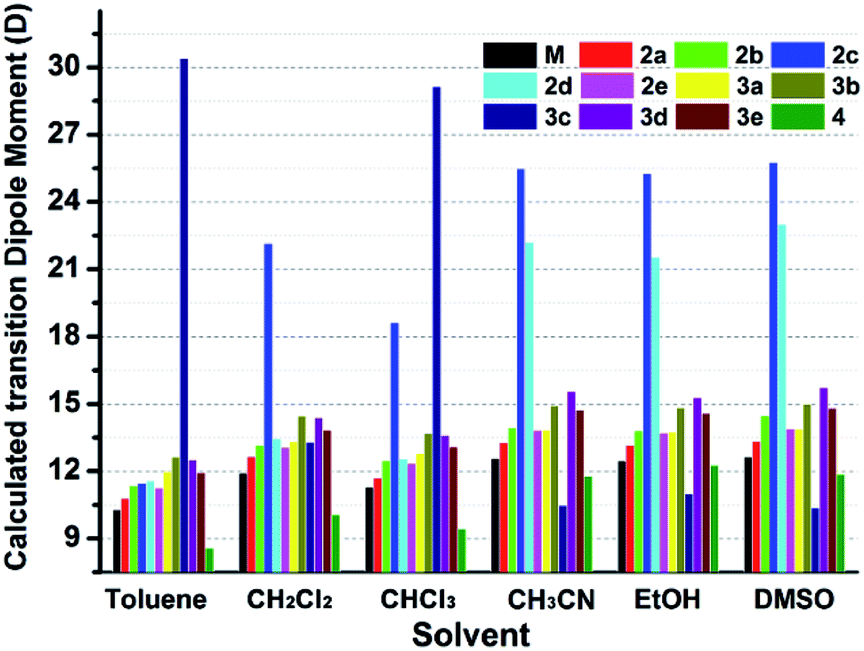 | ||
| Fig. 4 Calculated transition dipole moments (ΔT1–S0) of iridium complexes in different solvents (see data in Tables S7 and S8†). | ||
Cell imaging
Complex 2d shows high quantum yields both in solution (ΦPL = 0.79) and in solid state (ΦPL = 0.82) as well as sensitive emission color and lifetime toward solvent polarity. Therefore, it might be a potential polarity probe by the PLIM technique. As such, the phosphorescent emission in pH 4–10 was measured, which showed no change (Fig. S23†). Next, 2d was found to be more photostable in comparison to the commercially available reference (Mito-Tracker-Red) (Fig. S24†). Moreover, the phosphorescence intensity of 2d was little affected in the presence of common metal ions (Na+, K+: 5 mM; Ca2+, Mg2+: 500 μM; Zn2+, Al3+, Mn2+: 200 μM) and various reactive oxygen species (H2O2: 50 μM; ClO−, O2−, ·OH: 100 μM) (Fig. S25 and S26†). Also, the emission intensity changes little with increasing viscosity from 0.60 cP to 100 cP (Fig. S27†).An MTT assay has shown that complex 2d exhibits no cytotoxicity towards HepG2 cells at concentrations under 10 μM, thus making it suitable for cellular staining experiments (Fig. S28†). Colocalization imaging experiments were performed for liver human hepatoma cells (HepG2) and normal human liver cells (HL-7702) with complex 2d and Mito Tracker Red (mitochondrial dye) to demonstrate that complex 2d can readily penetrate into cells (Fig. S29†). More importantly, complex 2d showed mitochondria targeting with Pearson’s colocalization coefficients of 0.96 and 0.90 in the two cell lines, respectively. It is likely that A549 and HeLa cells also showed mitochondria targeting (Fig. S29†). Other subcellular organelle staining control experiments gave the colocalization coefficients 0.18 for Lyso (lysosomes), 0.68 for Golgi apparatus, 0.72 for ER (endoplasmic reticulum) (Fig. S30†). These experiments further demonstrate that complex 2d can preferentially accumulate in mitochondria. This is probably attributed to its cationic charge and lipophilic carboranyl group3,5 since complex 2d is independent of mitochondrial membrane potential as indicated by the fact that both emission intensity and lifetime of the complex were almost unchanged in the absence or presence of CCCP (carbonyl cyanide 3-chlorophenylhydrazone) (Fig. S31†).
It is well known that cancer cells have different microenvironments from normal cells. In many cases, cancer cells have exhibited mitochondrial disorders. Therefore, we intended to detect the polarity difference in mitochondria in different cell lines by using complex 2d and the PLIM technique. In doing so, the emission spectrum of complex 2d within the cells was measured and was found to be nearly identical to that observed in the extracellular environment (Fig. S32†). The phosphorescence lifetime was also examined and found not to be affected by the dosage of 2d (10, 20, and 30 μM), demonstrating that the probe is stable during cellular imaging (Fig. S33†). Next, HepG2 and HL-7702 cells were incubated with complex 2d (10 μM). The emission lifetime and phosphorescence intensity in HepG2 cells were observed to be much longer and higher than those in HL-7702 cells (Fig. 5(A)).
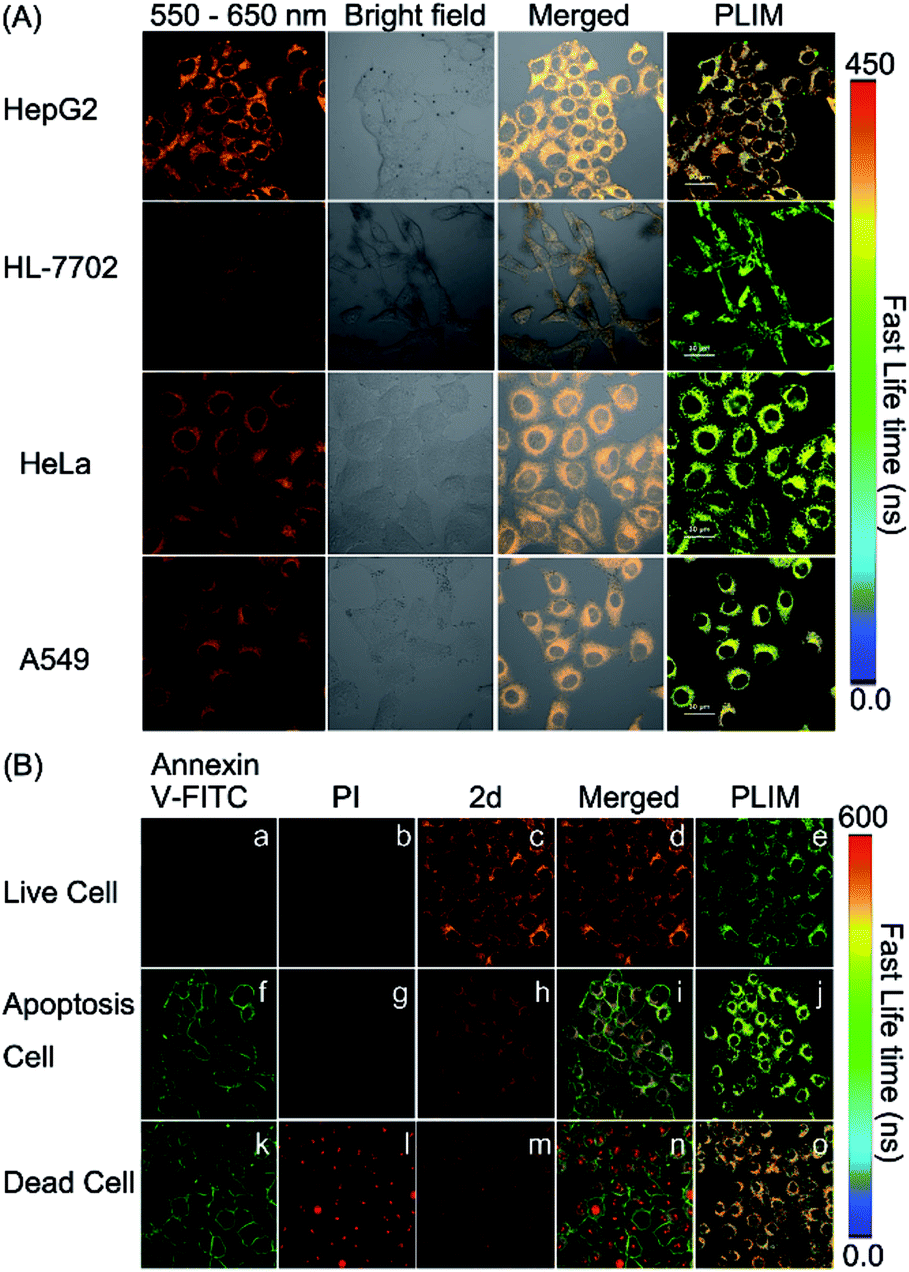 | ||
| Fig. 5 (A) Phosphorescence lifetime imaging of the mitochondrial polarity in HepG2, HL-7702, HeLa, and A549 cells stained with 2d (10 μM) (scale bar: 30 μm). (B) Phosphorescence lifetime imaging of HepG2 cells in different states stained with 2d (10 μM). | ||
According to the polarity–lifetime relationship (Fig. S34†), the cellular environment of HepG2 cells is less polar than that of normal cells (HL-7702), consistent with the reported result.3,4,5a In contrast, HeLa and A549 cell lines exhibited little difference in phosphorescence lifetime in comparison to HL-7702 (Fig. 5(A) and S34†). In particular, both the emission lifetime and phosphorescence intensity of Model remained almost the same in HL-7702 cells, HepG2, A549, and HeLa cell lines (Fig. S35†), indicating insensitivity toward polarity within cells. Hence, the PLIM results demonstrate that complex 2d can be used as a phosphorescent probe to detect the mitochondrial polarity in live cells.
Mitochondrial disorders are well-known to be highly relevant to apoptosis, which is basically defined as a programmed cell-death event in contrast to the unprogrammed cancer cell growing.16,17 During apoptosis process, mitochondria change both in structure and in function.17 Hence, we tried to further use PLIM to monitor the change of mitochondrial polarity during cell apoptosis. Complex 2d and the commercial dye propidium iodide (PI), or Annexin-FITC were incubated together with live HepG2 cells, cells in apoptosis and dead cells, respectively. As illustrated in Fig. 5(B), the emission lifetimes in cells in different states vary. The PLIM signals in living cells show the shortest phosphorescence lifetime, whereas those for dead cells exhibit the longest phosphorescence lifetime. In a sharp contrast, the control Model complex has indicated almost zero-difference in the PLIM signals during cell apoptosis (Fig. S36†). Therefore, the PLIM technique clearly indicates polarity decreasing in mitochondria during cell apoptosis through the use of the phosphorescent probe complex 2d, which can unambiguously differentiate live, apoptotic, and dead cells.
Conclusions
In summary, a novel series of highly emissive phosphorescent iridium(III) complexes containing o-carborane modified N⁁N ligands have been designed and synthesized. Among them, an efficient polarity-sensitive phosphorescent probe (complex 2d) has been developed for the first time. It exhibits a visual color change of phosphorescence emission as a function of polarity and unique mitochondria affinity. This cell-staining complex has shown the ability to discriminate cancer cells and normal cells by PLIM. Moreover, this phosphorescence probe can be used to track cell apoptosis to indicate living cells, cells in apoptosis, and dead cells. Hopefully, this new type of o-carborane-functionalized phosphorescent probe could be further improved to detect mitochondrial disorders in the future.Experimental
General
In this paper, all the synthetic steps were carried out under an inert argon atmosphere using standard Schlenk and glovebox techniques unless otherwise noted. Commercial reagents were used without any further purification. All the solvents are freshly distilled, for example, THF and toluene were distilled on sodium/benzophenone as well as acetonitrile and EtOH on CaH2. Dimeric [(C⁁N)2Ir(μ-Cl)]2 complex was prepared by literature procedures.S1† Intermediate compound B10H12(Et2S)2 was synthesized by a modified method according to literature reports.S2,S3a† Compounds a,S3a†b,S3a†11c,S3b†11d,S3c† and 11fS3b† were synthesized according to the literature. All NMR spectra (1H-, 13C-, and 11B-) were obtained at ambient temperature on Bruker DRX-400 or Bruker DRX-500 spectrometers. Chemical shifts are reported relative to CHCl3/CDCl3 (δ1H = 7.26 ppm, δ13C = 77.0 ppm) and external Et2O·BF3 (δ11B = 0 ppm), respectively. Mass spectra were measured with a Bruker Daltonics Autoflex IITM MALDI-TOF MS spectrometer, Micromass GC-TOF for EI-MS (70 eV) and a ESI-MS (LCQ Fleet, Thermo Fisher Scientific). Melting points were measured with an X4 digital melting point displayer. Phosphorescence measurements were carried out using a Hitachi F-4600 fluorescence spectrophotometer. Electronic absorption spectra were recorded with Shimadzu UV-2550 spectrophotometers. Phosphorescence lifetimes were determined by an Edinburgh Instruments laser impulse fluorometer with picosecond time resolution. Elemental analyses for C, H and N were performed on a Vario MICRO elemental analyzer. IR data were collected on a Bruker Vacuum FT-IR Spectrometer 80 V. X-ray diffraction data were collected on a Bruker Smart CCD Apex DUO diffractometer with graphite monochromated Mo Kα radiation (λ = 0.71073 Å) using the ω–2θ scan mode.Synthesis of 1a
A mixture of a (0.66 g, 2.2 mmol, 1.1 equiv), 2-(tributylstannyl) pyridine (0.74 g, 2 mmol), and Pd(PPh3)4 (0.23 g, 5% mmol) in dry toluene (40 mL) was refluxed for 24 h under argon atmosphere. After cooling down to room temperature, the solvent was evaporated under reduced pressure and the residue was purified by column chromatography on silica gel using ethylacetate/n-hexane (1:8, v/v) as eluent. Drying in vacuum afforded a yellowish white powder of 1a (0.49 g, 83%).
1H NMR (CDCl3): δ 8.80 (d, J = 1.9 Hz, 1H), 8.69 (d, J = 4.2 Hz, 1H), 8.41 (m, 2H), 7.91 (dd, J = 2.2, 8.4 Hz, 1H), 7.85 (t, J = 7.6 Hz, 1H), 7.36 (m, 1H), 4.01 (s, 1H, carborane–CH), 3.40–1.71 (br, 10H, B–H). 13C NMR (CDCl3): δ 157.34, 154.47, 149.36, 147.79, 137.05, 136.13, 129.24, 124.45, 121.38, 120.45, 121.6 (py-C), 73.75 (B–C) and 60.19 (B–C). 11B NMR (CDCl3): δ 1.6 (1B), −0.6 (1B), −5.5 (2B), −7.7 (2B), −8.5 (2B) and −9.4 (2B). IR (KBr): (ν cm−1) 2588. EI-MS (m/z): 298.20.
Synthesis of 1b
This compound was prepared in a manner analogous to the synthesis of 1a using b (0.63 g, 2.0 mmol) and 2-(tributylstannyl) pyridine (1.10 g, 3 mmol), and Pd(PPh3)4 (0.12 g, 5% mmol) to afford a yellowish white powder of 1b (0.51 g, 81%).1H NMR (CDCl3): δ 8.94 (d, J = 2.4 Hz, 1H), 8.72 (d, J = 4.5 Hz, 1H), 8.46 (dd, J = 8.5, 7.8, 2H), 8.07 (dd, J = 2.6, 8.5 Hz, 1H), 7.87 (td, J = 1.7, 7.8 Hz, 1H), 7.38 (ddd, J = 1.1, 4.9, 7.6 Hz, 1H), 1.75 (s, 1H, carborane-CH3), 3.40–1.61 (br, 10H, B–H). 13C NMR (CDCl3): δ 157.81, 154.50, 150.88, 149.44, 139.29, 137.09, 126.88, 124.55, 121.48, 120.54 (py-C), 78.93 (B–C) and 77.21 (B–C), and 23.20 (CH3). 11B NMR (CDCl3): δ 0.6 (2B), −1.2 (2B), −6.3 (3B) and −6.7 (3B). IR (KBr): (ν cm−1) 2588. EI-MS (m/z): 312.00.
Synthesis of 1c
This compound was prepared in a manner analogous to the synthesis of 1a using c (0.38 g, 1.0 mmol) and 2-(tributylstannyl) pyridine (0.55 g, 1.5 mmol), and Pd(PPh3)4 (0.06 g, 5% mmol) to afford a yellowish white powder of 1c (0.29 g, 76%).1H NMR (CDCl3): δ 8.74 (d, J = 2.1 Hz, 1H), 8.65 (d, J = 4.2 Hz, 1H), 8.33 (d, J = 7.9, 1H), 8.22 (d, J = 8.5 Hz, 1H), 7.81 (m, 2H), 7.48 (m, 2H), 7.33 (dd, J = 5.1, 6.6 Hz, 1H), 7.25 (dd, J = 6.9, 7.3 Hz, 1H), 7.17 (dd, J = 4.8, 8.0 Hz, 2H), 3.40–1.80 (br, 10H, B–H). 13C NMR (CDCl3): δ 157.14, 154.48, 150.48, 149.27, 138.71, 137.04, 130.61, 130.09, 128.62, 126.80, 124.42, 121.44, 119.99, 85.22 (B–C) and 82.02 (B–C). 11B NMR (CDCl3): δ 0.9 (2B), −6.1 (3B), 6.9 (3B) and −8.6 (2B). IR (KBr): (ν cm−1) 2596. EI-MS (m/z): 374.30.
Synthesis of 1d
This compound was prepared in a manner analogous to the synthesis of 1a using d (0.52 g, 2.0 mmol) and 2-(tributylstannyl) pyridine (1.10 g, 3 mmol), and Pd(PPh3)4 (0.12 g, 5% mmol) to afford a yellowish white powder of 1d (0.53 g, 90%). 1H NMR (CDCl3): δ 8.69 (d, J = 4.3 Hz, 1H), 8.67 (d, J = 5.2 Hz, 1H), 8.47 (d, J = 1.7 Hz, 1H), 8.43 (d, J = 7.9 Hz, 1H), 7.85 (td, J = 1.7, 7.8 Hz, 1H), 7.43 (dd, J = 2.1, 5.2 Hz, 1H), 7.37 (m, 1H), 4.24 (s, 1H, carborane-CH), 3.30–1.70 (br, 10H, B–H). 13C NMR (CDCl3): δ 157.03, 154.51, 150.88, 149.75, 149.18, 143.00, 137.07, 124.44, 121.83, 121.33, 118.02 (py-C), 74.04 (B–C) and 58.76 (B–C). 11B NMR (CDCl3): δ 1.4 (2B), −0.2 (1B), −5.4 (3B), −8.0 (2B) and −9.6 (2B). IR (KBr): (ν cm−1) 2573. EI-MS (m/z): 298.20.Synthesis of 1f
This compound was prepared in a manner analogous to the synthesis of 1a using f (0.66 g, 2.0 mmol) and 2-(tributylstannyl) pyridine (1.10 g, 3 mmol), and Pd(PPh3)4 (0.12 g, 5% mmol) to afford a yellowish white powder of 1f (0.61 g, 82%).1H NMR (CDCl3): δ 8.70 (d, J = 4.1 Hz, 1H), 8.58 (d, J = 1.4 Hz, 1H), 8.44 (d, J = 5.2 Hz, 1H), 8.30 (d, J = 8.0 Hz, 1H), 7.80 (td, J = 1.9, 7.8 Hz, 1H), 7.51 (d, J = 7.3 Hz, 2H), 7.34 (dd, J = 5.3, 7.7 Hz, 1H), 7.23 (m, 2H), 7.16 (m, 2H), 3.40–1.80 (br, 10H, B–H). 13C NMR (CDCl3): δ 156.79, 154.67, 149.37, 140.03, 136.91, 130.56, 130.11, 128.60, 124.28, 124.04, 122.35, 121.11, 85.20 (B–C) and 82.32 (B–C). 11B NMR (CDCl3): δ 1.6 (2B), 0.7 (3B), −6.9 (3B) and −8.3 (2B). IR (KBr): (ν cm−1) 2597. EI-MS (m/z): 373.20.
Synthesis of 1g
Sodium hydride (60% dispersion in mineral oil, 0.03 g, 0.75 mmol) was suspended in dry DMF (5 mL). After being cooled down to −20 °C, a solution of 1a (0.21 g, 0.68 mmol) in DMF (5 mL) was slowly added to the suspension. The mixture was stirred at room temperature for 1 h, then 2-iodopropane (0.17, 1.0 mmol) was added at −20 °C, and further stirred at room temperature overnight. The reaction was quenched by saturated aqueous NH4Cl solution, and extracted with diethyl ether (30 mL × 3). The organic layer was washed with water (30 mL × 3) and dried over MgSO4. The solvent was evaporated under reduced pressure. The residue was purified by column chromatography on silica gel using ethylacetate/n-hexane (1:4, v/v) as eluent. Drying in vacuum afforded a pale yellow powder of 1g (0.12 g, 51%).
1H NMR (CDCl3): δ 8.91 (d, J = 0.6 Hz, 1H), 8.72 (d, J = 3.5 Hz, 1H), 8.47 (t, J = 9.3 Hz, 2H), 8.05 (d, J = 8.7 Hz, 1H), 7.88 (t, J = 7.5 Hz, 1H), 7.39 (m, 1H), 3.50–1.80 (br, 10H, B–H), 1.73 (sept, 1H, J = 7.0 Hz, –CHCH3), 1.09 (d, J = 7.0 Hz, 3H, –CH3), 1.06 (d, J = 7.0 Hz, 3H, –CH3). 13C NMR (CDCl3): δ 157.83, 154.48, 150.90, 149.41, 139.33, 137.04, 126.73, 124.52, 121.48, 120.52, 121.6 (py-C), 88.60 (B–C), 82.22 (B–C), 31.64, and 23.90 (isopropyl-C). 11B NMR (CDCl3): δ 0.4 (3B), −6.5 (4B) and −8.6 (3B). IR (KBr): (ν cm−1) 2585. EI-MS (m/z): 340.10.
Synthesis of 1h
This compound was prepared in a manner analogous to the synthesis of 1g using 1a (0.24 g, 0.68 mmol) and 1-iodo-2-methylpropane (0.18 g, 1.0 mmol) to afford a yellowish white powder of 1h (0.13 g, 54%).1H NMR (CDCl3): δ 8.92 (s, 1H), 8.73 (s, 1H), 8.50 (d, J = 7.6 Hz, 2H), 8.05 (d, J = 8.7 Hz, 1H), 8.05 (d, J = 8.1 Hz, 1H), 7.91 (t, J = 6.7 Hz, 1H), 7.41 (m, 1H), 3.60–1.80 (br, 10H, B–H), 1.83–1.70 (m, 2H, –CHCH2), 0.85 (d, J = 7.0 Hz, 6H, –CH3). 13C NMR (CDCl3): δ 157.76, 154.52, 151.07, 149.41, 139.45, 137.14, 126.95, 124.57, 121.55, 120.52, 121.6 (py-C), 82.26 (B–C), 80.99 (B–C), 43.88, 28.43, and 23.30 (isobutyl-C). 11B NMR (CDCl3): δ −0.1 (3B), −6.6 (4B) and −7.5 (3B). IR (KBr): (ν cm−1) 2591. EI-MS (m/z): 354.30.
Synthesis of 1e
This compound was prepared in a manner analogous to the synthesis of 1g using 1d (0.21 g, 0.68 mmol) and iodomethane (0.15 g, 1.0 mmol) to afford a yellowish white powder of 1e (0.14 g, 63%).1H NMR (CDCl3): δ 8.74 (d, J = 6.1 Hz, 1H), 8.44 (d, J = 7.9 Hz, 1H), 7.85 (t, J = 7.5 Hz, 2H), 7.55 (d, J = 3.4 Hz, 1H), 7.37 (m, 2H), 1.78 (s, 1H, CH3), 3.50–1.71 (br, 10H, B–H). 13C NMR (CDCl3): δ 157.50, 154.79, 150.04, 149.49, 140.34, 137.14, 124.91, 124.53, 122.48, 121.21, 118.20 (py-C), 79.55 (B–C), 58.95 (B–C), and 23.44 (CH3). 11B NMR (CDCl3): δ 0.9 (2B), −1.4 (2B), and −6.6 (6B). IR (KBr): (ν cm−1) 2590. EI-MS (m/z): 311.20.
Synthesis of 1i
This compound was prepared in a manner analogous to the synthesis of 1g using 1d (0.21 g, 0.68 mmol) and 2-iodopropane (0.17, 1.0 mmol) to afford a yellowish white powder of 1i (0.13 g, 58%).1H NMR (CDCl3): δ 8.73 (s, 1H), 8.44 (d, J = 7.8 Hz, 1H), 7.86 (t, J = 7.2 Hz, 2H), 7.53 (d, J = 3.4 Hz, 1H), 7.38 (m, 2H), 3.50–1.71 (br, 10H, B–H), 1.76 (sept, J = 7.0 Hz, 1H, –CHCH3), 1.09 (d, J = 7.0 Hz, 3H, –CH3), 1.08 (d, J = 7.0 Hz, 3H, –CH3). 13C NMR (CDCl3): δ 157.34, 154.64, 149.84, 149.38, 140.12, 136.96, 124.78, 124.36, 122.46, 121.25 (py-C), 88.58 (B–C), 82.51 (B–C), 31.82, and 24.02 (isopropyl-C). 11B NMR (CDCl3): δ 0.2 (3B), −6.4 (4B) and −8.4 (3B). IR (KBr): (ν cm−1) 2595. EI-MS (m/z): 338.30.
Synthesis of 1j
This compound was prepared in a manner analogous to the synthesis of 1g using 1d (0.24 g, 0.68 mmol) and 1-iodo-2-methylpropane (0.18 g, 1.0 mmol) to afford a yellowish white powder of 1j (0.13 g, 55%).1H NMR (CDCl3): δ 8.73 (d, J = 5.0 Hz, 3H), 8.45 (d, J = 7.9 Hz, 1H), 7.86 (t, J = 8.4 Hz, 1H), 7.52 (dd, J = 1.9, 5.1 Hz, 1H), 7.38 (dd, J = 5.1, 6.8 Hz, 1H), 3.60–1.80 (br, 10H, B–H), 1.79–1.76 (m, 3H, –CHCH2), and 0.84 (d, J = 7.0 Hz, 6H, –CH3). 13C NMR (CDCl3): δ 157.31, 154.60, 149.82, 149.34, 140.19, 136.93, 124.78, 124.34, 122.50, 121.21 (py-C), 82.19 (B–C), 81.35 (B–C), 43.81, 28.39, and 23.23 (isobutyl-C). 11B NMR (CDCl3): δ 0.5 (2B), −0.3 (2B), −6.6 (3B), and −7.3 (3B). IR (KBr): (ν cm−1) 2595. EI-MS (m/z): 353.20.
Synthesis of 4a
5-o-Carboranyl-2-bromopyridine (a) (1.30 g, 4.33 mmol) was charged into a flask which was evacuated and recharged by argon. Anhydrous m-xylene (35 mL) was injected by a syringe, followed by addition of hexa-n-butyldistannane (1.18 mL, 50 mol%). Argon was bubbled through the stirred solution for 1 h before Pd(PPh3)4 (0.12 g, 0.101 mmol) was added from a tip tube. The reaction mixture was heated to 130 °C for 3 days until all starting material was consumed, then poured into aqueous EDTA (1 M, 25 mL). After the mixture was stirred for 15 min, the phases were separated. The aqueous phase was extracted with chloroform, and the combined organic phases were dried over Na2SO4. After evaporation of the solvents, the crude product was flash chromatographed (alumina, 5:1 hexanes/ethylacetate) to afford 4a as a white yellow solid (0.33 g, 35%).
1H NMR (CDCl3) δ (ppm): δ 8.82 (d, J = 2.3 Hz, 2H), 8.41 (d, J = 8.5 Hz, 2H), 7.95 (dd, J = 2.3, 8.5 Hz, 2H), 4.01 (s, 2H, carborane), 3.15–1.70 (br, 20H, B–H). 13C NMR δ (ppm): 155.72, 147.98, 136.33, 130.02, 120.82, 73.46 (B–C) and 60.11 (B–C). 11B NMR δ (ppm): 1.6 (2B), −0.5 (1B), −5.5 (3B), −7.7 (2B), −8.5 (1B) and −8.9 (1B). EI-MS (m/z): 440.20.
Synthesis of 2a
[(dfppy)2Ir(μ-Cl)]2 (0.2432 g, 0.2 mmol), 1a (0.14 g, 0.44 mmol) were dissolved in methanol (10 mL) and CH2Cl2 (10 mL). The resulting mixture was refluxed for 8 h under argon. After cooling down to room temperature, 10-fold excess of KPF6 was added. The mixture was stirred for 2 h, and then filtered to remove insoluble inorganic salts. The resulting solution was evaporated to dryness under reduced pressure. The residue was chromatographed on silica gel with elution by PE/ethyl acetate 1:3 (v/v) to give 2a as a yellow solid. Yield: 166 mg (41%).
1H NMR (500 MHz, 6d-acetone): 8.89 (m, 2H), 8.52 (dd, J = 2.3, 8.7 Hz, 1H), 8.37 (m, 3H), 8.26 (m, 3H), 8.07 (m, 3H), 7.97 (d, J = 5.5 Hz, 1H), 7.91 (d, J = 5.5 Hz, 1H), 7.22 (d, J = 6.2 Hz, 2H), 6.78 (m, 1H), 5.84 (dd, J = 2.2, 8.5 Hz, 2H), 5.15 (s, 1H, carborane-CH), 3.15–1.72 (br, 10H, BH). 13C NMR (6d-acetone): 156.87, 154.07, 150.89, 149.77, 149.67, 149.07, 140.06, 139.67, 139.48, 138.58, 133.17, 129.31, 125.78, 124.58, 124.09, 123.90, 123.42, 123.15, 113.61, 113.41, 98.96, 98.85, 71.63 (B–C) and 60.92 (B–C). 11B NMR (6d-acetone): −0.7 (2B), −0.5 (1B), −5.9 (3B), −8.1 (2B) and −9.5 (2B). C34H30B10N4F4IrPF6 calcd: C, 40.19; N, 5.51; H, 2.97. Found: C, 39.80; N, 4.97; H, 2.85. MALDI-TOF: [M − PF6] (m/z) 871.832. IR (KBr): (ν cm−1) 2589 (B–H). Melting point: 233–235 °C.
Synthesis of 2b
This compound was prepared in a manner analogous to the synthesis of 2a using [(dfppy)2Ir(μ-Cl)]2 (0.2432 g, 0.2 mmol) and 1b (0.13 g, 0.44 mmol) to afford a yellow powder. Yield: 260 mg (63%).1H NMR (500 MHz, 6d-acetone): 8.95 (d, J = 8.6 Hz, 2H), 8.63 (dd, J = 2.2, 8.7 Hz, 2H), 8.38 (m, 3H), 8.28 (m, 2H), 8.07 (m, 2H), 7.93 (d, J = 5.6 Hz, 1H), 7.81 (m, 1H), 7.22 (m, 2H), 6.92 (m, 1H), 6.79 (m, 1H), 5.87 (m, 2H), 3.25–1.70 (br, 10H, BH), 1.68 (s, 3H, –CH3). 13C NMR (6d-acetone): 163.78, 163.68, 163.55, 163.46, 162.51, 162.14, 161.72, 161.63, 161.51, 161.41, 159.44, 159.33, 157.00, 153.32, 152.23, 150.29, 149.93, 149.17, 149.13, 139.09, 138.90, 125.38, 124.48, 123.54, 123.27, 113.01, 112.94, 112.88, 98.24, 98.03, 97.88, 97.67, 71.31 (B–C), 75.81 (B–C), and 21.47 (CH3). 11B NMR (6d-acetone): 1.1 (2B), −1.6 (2B) and −6.8 (6B). C35H32B10F4N4IrPF6 calcd: C, 40.82; N, 5.44; H, 3.13. Found: C, 40.70; N, 5.17; H, 3.05. MALDI-TOF: [M − PF6] (m/z) 885.258. IR (KBr): (ν cm−1) 2587 (B–H). Melting point: 238–240 °C.
Synthesis of 2c
This compound was prepared in a manner analogous to the synthesis of 2a using [(dfppy)2Ir(μ-Cl)]2 (0.2432 g, 0.2 mmol) and 1c (0.16 g, 0.44 mmol) to afford a yellow orange powder. Yield: 253 mg (58%).1H NMR (500 MHz, 6d-acetone): 8.79 (d, J = 8.8 Hz, 2H), 8.51 (d, J = 8.5 Hz, 2H), 8.34 (d, J = 8.3 Hz, 2H), 8.17 (m, 3H), 8.02 (m, 2H), 7.78 (m, 2H), 7.34 (m, 5H), 7.15 (d, J = 5.4 Hz, 2H), 7.00 (d, J = 9.0 Hz, 1H), 6.77 (d, J = 8.9 Hz, 1H), 5.78 (d, J = 5.9 Hz, 2H), 3.20–1.80 (br, 10H, BH). 13C NMR (6d-acetone): 163.03, 162.94, 163.55, 157.55, 154.10, 154.01, 153.90, 153.24, 153.15, 151.05, 149.96, 149.29, 142.34, 140.32, 140.10, 139.79, 131.57, 130.38, 129.76, 129.10, 128.80, 126.31, 124.97, 124.36, 123.90, 123.66, 123.42, 114.30, 114.07, 113.85, 113.61, 99.06, 98.83, 98.70, 98.47, 85.74 (B–C) and 79.40 (B–C). 11B NMR (6d-acetone): 1.7 (2B), −0.4 (2B) and −7.2 (6B). C40H34B10F4N4IrPF6. Calcd: C, 43.99; N, 5.13; H, 3.14. Found: C, 43.60; N, 5.01; H, 3.02. MALDI-TOF: [M − PF6] (m/z) 947.185. IR (KBr): (ν cm−1) 2586 (B–H). Melting point: 237–239 °C.
Synthesis of 2d
This compound was prepared in a manner analogous to the synthesis of 2a using [(dfppy)2Ir(μ-Cl)]2 (0.2432 g, 0.2 mmol) and 1g (0.15 g, 0.44 mmol) to afford a yellow powder. Yield: 267 mg (63%).1H NMR (500 MHz, 6d-acetone): 8.95 (d, J = 8.5 Hz, 2H), 8.62 (dd, J = 2.3, 8.7 Hz, 2H), 8.39 (m, 3H), 8.28 (t, J = 4.2 Hz, 2H), 8.08 (m, 2H), 8.03 (d, J = 5.6 Hz, 1H), 7.93 (d, J = 5.3 Hz, 1H), 7.81 (m, 1H), 7.23 (m, 2H), 6.93 (m, 1H), 6.79 (m, 1H), 5.85 (m, 1H), 3.20–1.80 (br, 10H, BH), 1.64 (sept, J = 7.0 Hz, 1H, –CHCH3), 0.90 (d, J = 7.0 Hz, 3H, –CH3), 0.87 (d, J = 7.0 Hz, 3H, –CH3). 13C NMR (6d-acetone): 163.86, 163.76, 163.61, 163.51, 162.65, 162.59, 162.18, 162.13, 161.81, 161.72, 161.57, 161.41, 159.49, 159.40, 159.34, 157.08, 153.42, 153.31, 152.31, 152.26, 150.34, 149.81, 149.18, 149.06, 139.10, 138.98, 128.87, 125.46, 124.66, 123.27, 112.99, 112.85, 98.30, 98.09, 97.87, 97.71, 88.58 (B–C), 78.84 (B–C), 30.78, 22.32 and 22.06. 11B NMR (6d-acetone): 0.1 (3B), −6.7 (4B) and −8.4 (3B). C37H36B10F4N4IrPF6. Calcd: C, 42.00; N, 5.30; H, 3.43. Found: C, 41.60; N, 5.01; H, 3.22. MALDI-TOF: [M − PF6] (m/z) 921.135. IR (KBr): (ν cm−1) 2588 (B–H). Melting point: 236–237 °C.
Synthesis of 2e
This compound was prepared in a manner analogous to the synthesis of 2a using [(dfppy)2Ir(μ-Cl)]2 (0.2432 g, 0.2 mmol) and 1h (0.16 g, 0.44 mmol) to afford a yellow powder. Yield: 240 mg (56%).1H NMR (500 MHz, 6d-acetone): 8.95 (d, J = 8.5 Hz, 2H), 8.65 (dd, J = 2.3, 8.7 Hz, 2H), 8.40 (m, 3H), 8.28 (dd, J = 3.5, 11.7 Hz, 2H), 8.10 (m, 1H), 8.03 (d, J = 5.6 Hz, 1H), 7.98 (d, J = 5.3 Hz, 1H), 7.82 (m, 1H), 7.27 (m, 1H), 7.20 (ddd, J = 1.2, 6.0, 7.3 Hz, 1H), 6.93 (m, 1H), 6.79 (m, 1H), 5.86 (dd, J = 2.3, 8.4 Hz, 2H), 3.10–1.80 (br, 10H, BH), 0.90–0.82 (m, 3H, –CHCH2), 0.72 (d, J = 7.0 Hz, 3H, –CH3), 0.66 (d, J = 7.0 Hz, 3H, –CH3). 13C NMR (6d-acetone): 163.36, 162.95, 157.79, 154.04, 153.02, 151.13, 150.74, 149.98, 149.71, 139.91, 139.69, 129.63, 126.17, 125.27, 123.95, 113.71, 113.58, 99.05, 98.83, 98.64, 98.43, 82.74 (B–C), 78.17 (B–C), 43.16, 22.22 and 22.07. 11B NMR (6d-acetone): 0.9 (2B), −0.6 (2B) and −6.9 (6B). C38H38B10F4N4IrPF6. Calcd: C, 42.00; N, 5.29; H, 3.43. Found: C, 41.50; N, 5.11; H, 3.21. MALDI-TOF: [M − PF6] (m/z) 926.260. IR (KBr): (ν cm−1) 2589 (B–H). Melting point: 236–238 °C.
Synthesis of 3a
This compound was prepared in a manner analogous to the synthesis of 2a using [(dfppy)2Ir(μ-Cl)]2 (0.2432 g, 0.2 mmol) and 1d (0.14 g, 0.44 mmol) to afford a yellow powder. Yield: 174 mg (43%).1H NMR (500 MHz, 6d-acetone): 9.06 (d, J = 8.2 Hz, 2H), 8.91 (d, J = 1.8 Hz, 1H), 8.36 (m, 3H), 8.25 (dd, J = 5.6, 12.1 Hz, 2H), 8.06 (dd, J = 8.3, 19.3 Hz, 2H), 7.95 (dd, J = 2.1, 5.9 Hz, 1H), 7.90 (dd, J = 5.7, 10.3 Hz, 2H), 7.79 (m, 1H), 7.24 (m, 1H), 7.18 (m, 1H), 6.77 (m, 1H), 5.79 (dd, J = 2.3, 8.5 Hz, 1H), 5.74 (dd, J = 2.3, 8.6 Hz, 1H), 5.48 (s, 1H, carborane CH), 3.10–1.75 (br, 10H, BH). 13C NMR (6d-acetone): 164.23, 163.46, 163.41, 163.35, 162.18, 162.07, 160.10, 160.00, 156.72, 154.58, 153.70, 153.65, 153.60, 151.21, 150.89, 149.64, 149.58, 145.02, 139.85, 139.64, 139.58, 129.28, 127.17, 125.74, 123.87, 122.42, 113.42, 113.39, 113.24, 98.84, 98.79, 98.62, 98.41, 98.36, 73.08 (B–C) and 59.73 (B–C). 11B NMR (6d-acetone): 0.6 (2B), −5.4 (3B), −7.9 (3B) and −9.5 (2B). C34H30B10N4F4IrPF6 calcd: C, 40.19; N, 5.51; H, 2.97. Found: C, 40.07; N, 5.37; H, 2.89. MALDI-TOF: [M − PF6] (m/z) 873.396. IR (KBr): (ν cm−1) 2594 (B–H). Melting point: 230–232 °C.
Synthesis of 3b
This compound was prepared in a manner analogous to the synthesis of 2a using [(dfppy)2Ir(μ-Cl)]2 (0.2432 g, 0.2 mmol) and 1e (0.14 g, 0.44 mmol) to afford a yellow powder. Yield: 263 mg (64%).1H NMR (500 MHz, 6d-acetone): 9.17 (d, J = 8.2 Hz, 1H), 8.98 (d, J = 1.9 Hz, 1H), 8.37 (dd, J = 3.4, 5.9 Hz, 3H), 8.25 (d, J = 5.3 Hz, 1H), 8.07 (m, 3H), 7.91 (dd, J = 6.1, 9.4 Hz, 2H), 7.80 (m, 2H), 7.20 (m, 2H), 6.78 (m, 2H), 5.77 (dd, J = 2.3, 8.5 Hz, 2H), 3.15–1.70 (br, 10H, BH), 1.92 (s, 3H, –CH3). 13C NMR (6d-acetone): 163.25, 163.20, 163.16, 163.11, 162.01, 161.91, 156.97, 154.45, 153.60, 153.55, 153.47, 151.43, 150.58, 149.69, 149.59, 139.76, 139.41, 126.07, 125.97, 123.85, 123.80, 113.35, 113.27, 113.13, 98.70, 98.65, 98.49, 98.44, 98.27, 98.22, 78.45 (B–C), 77.84 (B–C) and 22.24 (CH3). 11B NMR (6d-acetone): 0.9 (2B), −1.8 (2B), and −6.7 (6B). C35H32B10F4N4IrPF6 calcd: C, 40.82; N, 5.44; H, 3.13. Found: C, 40.71; N, 5.27; H, 3.01. MALDI-TOF: [M − PF6] (m/z) 887.635. IR (KBr): (ν cm−1) 2589 (B–H). Melting point: 232–234 °C.
Synthesis of 3c
This compound was prepared in a manner analogous to the synthesis of 2a using [(dfppy)2Ir(μ-Cl)]2 (0.2432 g, 0.2 mmol) and 1f (0.16 g, 0.44 mmol) to afford a yellow powder. Yield: 240 mg (55%).1H NMR (500 MHz, 6d-acetone): 8.98 (d, J = 7.7 Hz, 1H), 8.70 (d, J = 5.8 Hz, 1H), 8.34 (dd, J = 7.5, 14.7 Hz, 3H), 8.10 (m, 4H), 7.77 (m, 3H), 7.58 (d, J = 8.0 Hz, 3H), 7.38 (t, J = 7.4, 1H), 7.22 (m, 4H), 6.75 (t, J = 7.7 Hz, 2H), 5.70 (ddd, J = 1.8, 8.5, 10.8 Hz, 2H), 3.10–1.80 (br, 10H, BH). 13C NMR (6d-acetone): 163.71, 162.75, 162.58, 161.57, 159.53, 155.80, 153.53, 153.08, 152.77, 150.34, 150.23, 148.68, 148.54, 139.49, 139.16, 129.99, 128.08, 123.29, 122.82, 112.95, 112.82, 98.24, 98.05, 85.33 (B–C) and 79.94 (B–C). 11B NMR (6d-acetone): 1.9 (2B), −0.4 (2B) and −6.9 (6B). C40H34B10F4N4IrPF6. Calcd: C, 43.99; N, 5.13; H, 3.14. Found: C, 43.58; N, 5.00; H, 3.03. MALDI-TOF: [M − PF6] (m/z) 949.685. IR (KBr): (ν cm−1) 2590 (B–H). Melting point: 231–233 °C.
Synthesis of 3d
This compound was prepared in a manner analogous to the synthesis of 2a using [(dfppy)2Ir(μ-Cl)]2 (0.2432 g, 0.2 mmol) and 1i (0.15 g, 0.44 mmol) to afford a yellow powder. Yield: 262 mg (62%).1H NMR (500 MHz, 6d-acetone): 9.15 (d, J = 8.2 Hz, 1H), 8.94 (d, J = 1.7 Hz, 1H), 8.36 (m, 3H), 8.22 (d, J = 5.2 Hz, 1H), 8.06 (m, 4H), 7.88 (t, J = 6.5 Hz, 2H), 7.79 (m, 1H), 7.20 (m, 2H), 6.77 (m, 2H), 5.76 (ddd, J = 2.3, 8.5, 16.6 Hz, 2H), 3.10–1.70 (br, 10H, BH), 1.95 (sept, J = 7.0 Hz, 1H, –CHCH3), 1.05 (d, J = 7.0 Hz, 3H, –CH3), 1.01 (d, J = 7.0 Hz, 3H, –CH3). 13C NMR (6d-acetone): 163.69, 163.61, 162.70, 161.66, 161.55, 161.44, 159.47, 159.37, 156.61, 153.93, 153.01, 151.05, 150.13, 149.18, 149.05, 139.38, 139.01, 125.56, 125.51, 123.32, 122.69, 113.02, 112.86, 112.71, 98.24, 98.02, 97.81, 89.27 (B–C), 80.28 (B–C), 31.09, 27.68 and 22.58. 11B NMR (6d-acetone): 0.1 (3B), −6.7 (4B) and −8.4 (3B). C37H36B10F4N4IrPF6. Calcd: C, 42.00; N, 5.30; H, 3.43. Found: C, 41.60; N, 5.11; H, 3.26. MALDI-TOF: [M − PF6] (m/z) 915.525. IR (KBr): (ν cm−1) 2581 (B–H). Melting point: 235–237 °C.
Synthesis of 3e
This compound was prepared in a manner analogous to the synthesis of 2a using [(dfppy)2Ir(μ-Cl)]2 (0.2432 g, 0.2 mmol) and 1j (0.16 g, 0.44 mmol) to afford a yellow powder. Yield: 231 mg (54%).1H NMR (500 MHz, 6d-acetone): 9.17 (d, J = 8.2 Hz, 1H), 8.98 (s, 1H), 8.38 (m, 3H), 8.24 (d, J = 5.1 Hz, 1H), 8.07 (m, 3H), 7.92 (dd, J = 5.5, 5.4 Hz, 3H), 7.80 (m, 2H), 7.23 (t, J = 6.2 Hz, 1H), 7.17 (t, J = 6.6 Hz, 1H), 6.78 (m, 1H), 5.78 (ddd, J = 2.2, 5.8, 8.2 Hz, 2H), 3.15–1.80 (br, 10H, BH), 1.90–1.62 (m, 3H, –CHCH2), 0.73 (d, J = 7.0 Hz, 3H, –CH3), 0.70 (d, J = 7.0 Hz, 3H, –CH3). 13C NMR (6d-acetone): 164.27, 164.18, 163.32, 163.27, 162.24, 162.14, 160.02, 159.95, 157.09, 154.43, 153.62, 153.57, 153.51, 153.47, 151.54, 150.76, 149.66, 149.53, 141.63, 139.89, 139.58, 130.07, 129.26, 126.27, 126.05, 123.96, 123.76, 123.26, 113.59, 113.46, 113.32, 98.78, 98.56, 98.35, 83.28 (B–C), 79.48 (B–C), 43.14, 27.99 and 22.04. 11B NMR (6d-acetone): 0.7 (2B), −0.7 (2B) and −6.8 (6B). C38H38B10F4N4IrPF6. Calcd: C, 42.00; N, 5.29; H, 3.43. Found: C, 41.60; N, 5.01; H, 3.11. MALDI-TOF: [M − PF6] (m/z) 929.514. IR (KBr): (ν cm−1) 2583 (B–H). Melting point: 236–237 °C.
Synthesis of 4
This compound was prepared in a manner analogous to the synthesis of 2a using [(dfppy)2Ir(μ-Cl)]2 (0.2432 g, 0.2 mmol) and 4a (0.19 g, 0.44 mmol) to afford a yellow powder. Yield: 240 mg (52%).1H NMR (500 MHz, 6d-acetone): 8.94 (d, J = 8.5 Hz, 2H), 8.54 (d, J = 8.3 Hz, 2H), 8.42 (d, J = 8.4 Hz, 2H), 8.28 (d, J = 1.6 Hz, 2H), 8.09 (t, J = 7.8 Hz, 2H), 7.98 (d, J = 5.4 Hz, 2H), 6.91 (m, 4H), 5.91 (dd, J = 2.2, 8.4 Hz, 2H), 5.18 (s, 2H, carborane CH), 3.02–1.60 (br, 20H, BH). 13C NMR (6d-acetone): 163.63, 163.54, 162.26, 162.22, 161.92, 161.84, 161.50, 161.42, 159.78, 159.69, 155.00, 152.17, 152.13, 149.49, 148.67, 139.30, 138.30, 133.31, 127.15, 125.07, 123.66, 122.88, 113.40, 98.45, 98.27, 98.09, 70.96 (B–C) and 60.40 (B–C). 11B NMR (6d-acetone): 0.9 (4B), 0.1 (2B), −5.7 (6B), −8.2 (4B), and −9.3 (4B). C36H40B20N4F4IrPF6 calcd: C, 37.33; N, 4.84; H, 3.48. Found: C, 37.01; N, 4.67; H, 3.31. MALDI-TOF: [M − PF6] (m/z) 1013.225. IR (KBr): (ν cm−1) 2589 (B–H). Melting point: 245–247 °C.
Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (21531004, 21472086, 21271102), National Program for Support of Top-Notch Young Professionals, and high-performance computational center of Nanjing University.Notes and references
- D. G. Drubin and W. J. Nelson, Cell, 1996, 84, 335 CrossRef CAS PubMed.
- (a) Y. D. Zhuang, P. Y. Chiang, C. W. Wang and K. T. Tan, Angew. Chem., Int. Ed., 2013, 52, 8124 CrossRef CAS PubMed; (b) L. Huang and S. W. Tam-Chang, J. Fluoresc., 2011, 21, 213 CrossRef CAS PubMed.
- H. B. Xiao, P. Li, W. Zhang and B. Tang, Chem. Sci., 2016, 7, 1588 RSC.
- (a) Z. G. Yang, J. F. Cao, Y. X. He, J. H. Yang, T. Kim, X. J. Peng and J. S. Kim, Chem. Soc. Rev., 2014, 43, 4563 RSC; (b) H. Zhu, J. L. Fan, J. J. Du and X. J. Peng, Acc. Chem. Res., 2016, 49, 2115 CrossRef CAS PubMed.
- (a) N. Jiang, J. L. Fan, F. Xu, X. J. Peng, H. Y. Mu, J. Y. Wang and X. Q. Xiong, Angew. Chem., Int. Ed., 2015, 54, 2510 CrossRef CAS PubMed; (b) E. Yamaguchi, C. G. Wang, A. Fukazawa, M. Taki, Y. Sato, T. Sasaki, M. Ueda, N. Sasaki, T. Higashiyama and S. Yamaguchi, Angew. Chem., Int. Ed., 2015, 54, 4539 CrossRef CAS PubMed; (c) C. G. Wang, A. Fukazawa, M. Taki, Y. Sato, T. Higashiyama and S. Yamaguchi, Angew. Chem., Int. Ed., 2015, 54, 15213 CrossRef CAS PubMed.
- M. Chen, Y. Q. Wu, Y. Liu, H. R. Yang, Q. Zhao and F. Y. Li, Biomaterials, 2014, 35, 8748 CrossRef CAS PubMed.
- (a) Q. Zhao, F. Y. Li and C. H. Huang, Chem. Soc. Rev., 2010, 39, 3007 RSC; (b) Q. Zhao, C. H. Huang and F. Y. Li, Chem. Soc. Rev., 2011, 40, 2508 RSC; (c) Y. M. Yang, Q. Zhao, W. Feng and F. Y. Li, Chem. Rev., 2013, 113, 192 CrossRef CAS PubMed; (d) X. L. Yang, G. J. Zhou and W. Y. Wong, Chem. Soc. Rev., 2015, 44, 8484 RSC; (e) W. Lv, T. Yang, Q. Yu, Q. Zhao, K. Zhang, H. Liang, S. Liu, F. Li and W. Huang, Adv. Sci., 2015, 2, 1500107 CrossRef PubMed; (f) H. Sun, S. Liu, W. Lin, K. Y. Zhang, W. Lv, X. Huang, F. Huo, H. Yang, G. Jenkins, Q. Zhao and W. Huang, Nat. Commun., 2014, 5, 3601 Search PubMed; (g) W. Y. Wong and C. L. Ho, Coord. Chem. Rev., 2009, 253, 1709 CrossRef CAS; (h) X. B. Xu, X. L. Yang, J. Zhao, G. J. Zhou and W. Y. Wong, Asian J. Org. Chem., 2015, 4, 394 CrossRef CAS; (i) X. J. Liu, S. M. Wang, B. Yao, B. H. Zhang, C. L. Ho, W. Y. Wong, Y. X. Cheng and Z. Y. Xie, Org. Electron., 2015, 21, 1 CrossRef CAS.
- (a) R. N. Grimes, in Carboranes, Academic Press Inc, 2nd edn, 2011, vol. 9, p. 301 Search PubMed; (b) B. P. Dash, R. Satapathy, J. A. Maguire and N. S. Hosmane, New J. Chem., 2011, 35, 1955 RSC; (c) B. P. Dash, R. Satapathy, E. R. Gaillard, J. A. Maguire and N. S. Hosmane, J. Am. Chem. Soc., 2010, 132, 6578 CrossRef CAS PubMed; (d) A. Ferrer-Ugalde, A. Gonzalez-Campo, C. Vinas, J. Rodriguez-Romero, R. Santillan, N. Farfan, R. Sillanpaa, A. Sousa-Pedrares, R. Nunez and F. Teixidor, Chem.–Eur. J., 2014, 20, 9940 CrossRef CAS PubMed; (e) F. Teixidor, R. Nunez, C. Vinas, R. Sillanpaa and R. Kivekas, Angew. Chem., Int. Ed., 2000, 39, 4290 CrossRef CAS; (f) R. Nunez, P. Farras, F. Teixidor, C. Vinas, R. Sillanpaa and R. Kivekas, Angew. Chem., Int. Ed., 2006, 45, 1270 CrossRef CAS PubMed; (g) N. S. Hosmane, Boron Science: New Technologies and Applications, CRC Press, Boca Raton, FL, 2011 Search PubMed; (h) N. S. Hosmane, Boron and Gadolinium Neutron, Capture Therapy for Cancer Treatment, World Scientific Publishers, Hackensack, NJ, USA, 2012, vol. 36, ch. 28, pp. 877–900 Search PubMed; (i) V. I. Bregadze, Chem. Rev., 1992, 92, 209 CrossRef CAS; (j) D. Zhao and Z. W. Xie, Coord. Chem. Rev., 2016, 314, 14 CrossRef CAS; (k) A. Ferrer-Ugalde, E. J. Juarez-Perez, F. Teixidor, C. Vinas, R. Sillanpaa, E. Perez-Inestrosa and R. Nunez, Chem.–Eur. J., 2012, 18, 544 CrossRef CAS PubMed; (l) J. X. Guo, D. Q. Liu, J. H. Zhang, J. J. Zhang, Q. Miao and Z. W. Xie, Chem. Commun., 2015, 51, 12004 RSC.
- (a) Y. H. Lee, J. Park, J. Lee, S. U. Lee and M. H. Lee, J. Am. Chem. Soc., 2015, 137, 8018 CrossRef CAS PubMed; (b) T. Kim, H. Kim, K. M. Lee, Y. S. Lee and M. H. Lee, Inorg. Chem., 2013, 52, 160 CrossRef CAS PubMed; (c) H. J. Bae, J. Chung, H. Kim, J. Park, K. M. Lee, T. W. Koh, Y. S. Lee, S. Yoo, Y. Do and M. H. Lee, Inorg. Chem., 2014, 53, 128 CrossRef CAS PubMed; (d) C. Shi, H. B. Sun, Q. B. Jiang, Q. Zhao, J. X. Wang, W. Huang and H. Yan, Chem. Commun., 2013, 49, 4746 RSC; (e) C. Shi, D. S. Tu, Q. Yu, H. Liang, Y. H. Liu, Z. H. Li, H. Yan, Q. Zhao and W. Huang, Chem.–Eur. J., 2014, 20, 16550 CrossRef CAS PubMed; (f) X. Li, H. Yan and Q. Zhao, Chem.–Eur. J., 2016, 22, 1888 CrossRef CAS PubMed.
- (a) K. R. Wee, W. S. Han, D. W. Cho, S. Kwon, C. Pac and S. O. Kang, Angew. Chem., Int. Ed., 2012, 51, 2677 CrossRef CAS PubMed; (b) C. Shi, H. B. Sun, X. Tang, W. Lv, H. Yan, Q. Zhao, J. X. Wang and W. Huang, Angew. Chem., Int. Ed., 2013, 52, 13434 CrossRef CAS PubMed; (c) K. Kokado and Y. Chujo, J. Org. Chem., 2011, 76, 316 CrossRef CAS PubMed; (d) K. Kokado, M. Tominaga and Y. Chujo, Macromol. Rapid Commun., 2010, 31, 1389 CrossRef CAS PubMed; (e) S. Y. Kim, Y. J. Cho, G. F. Jin, W. S. Han, H. J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2015, 17, 15679 RSC; (f) K. Nishino, H. Yamamoto, K. Tanaka and Y. Chujo, Org. Lett., 2016, 18, 4064 CrossRef CAS PubMed.
- (a) Y. H. Lee, J. Park, S. J. Jo, M. Kim, J. Lee, S. U. Lee and M. H. Lee, Chem.–Eur. J., 2015, 21, 2052 CrossRef CAS PubMed; (b) J. Park, Y. H. Lee, J. Y. Ryu, J. Lee and M. H. Lee, Dalton Trans., 2016, 45, 5667 RSC.
- X. Li, X. Tong, H. Yan, C. S. Lu, Q. Zhao and W. Huang, Chem.–Eur. J., 2016, 22, 17282 CrossRef CAS PubMed.
- (a) J. V. Caspar and T. J. Meyer, Inorg. Chem., 1983, 22, 2444 CrossRef CAS; (b) J. S. Wilson, N. Chawdhury, M. R. A. Al-Mandhary, M. Younus, M. S. Khan, P. R. Raithby, A. Kohler and R. H. Friend, J. Am. Chem. Soc., 2001, 123, 9412 CrossRef CAS PubMed.
- (a) M. Sarma, T. Chatterjee, R. Bodapati, K. N. Krishnakanth, S. Hamad, S. V. Rao and S. K. Das, Inorg. Chem., 2016, 55, 3530 CrossRef CAS PubMed; (b) L. Zhu, X. Tang, Q. Yu, W. Lv, H. Yan, Q. Zhao and W. Huang, Chem.–Eur. J., 2015, 21, 4721 CrossRef CAS PubMed.
- (a) H. A. Al-Attar, G. C. Griffiths, T. N. Moore, M. Tavasli, M. A. Fox, M. R. Bryce and A. P. Monkman, Adv. Funct. Mater., 2011, 21, 2376 CrossRef CAS; (b) E. M. Kober, B. P. Sullivan and T. J. Meyer, Inorg. Chem., 1984, 23, 2098 CrossRef CAS; (c) A. P. Wilde and R. J. Watts, J. Phys. Chem., 1991, 95, 622 CrossRef CAS.
- (a) D. R. Green and J. C. Reed, Science, 1998, 281, 1309 CrossRef CAS PubMed; (b) H. Yoshida, Y. Y. Kong, R. Yoshida, A. J. Elia, A. Hakem, R. Hakem, J. M. Penninger and T. W. Mak, Cell, 1998, 94, 739 CrossRef CAS PubMed.
- (a) G. Kroemer and J. C. Reed, Nat. Med., 2000, 6, 513 CrossRef CAS PubMed; (b) T. Xiao, J. K. Fan, H. L. Huang, J. F. Gu, L. Y. Li and X. Y. Liu, Cell Res., 2010, 20, 367 CrossRef CAS PubMed; (c) S. L. Luo, X. Tan, S. T. Fang, Y. Wang, T. Liu, X. Wang, Y. Yuan, H. Q. Sun, Q. R. Qi and C. M. Shi, Adv. Funct. Mater., 2016, 26, 2826 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthesis and spectra data, biological studies and tables. CCDC 1496512–1496520. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc00160f |
| This journal is © The Royal Society of Chemistry 2017 |