
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent developments in the synthesis and biological activity of acridine/acridone analogues
Monika Gensicka-Kowalewska,
Grzegorz Cholewiński and
Krystyna Dzierzbicka *
*
Department of Organic Chemistry, Chemical Faculty, Gdansk University of Technology, G. Narutowicza 11/12, 80-233 Gdansk, Poland. E-mail: krydzier@pg.gda.pl
First published on 9th March 2017
Abstract
Many people in the world struggle with cancer or bacterial, parasitic, viral, Alzheimer's and other diseases. Therefore, many scientists seek new, more effective, more selective and less toxic drugs. Acridine/acridone derivatives constitute a class of compounds with a broad spectrum of biological activity and are of great interest to scientists. To date, many acridine/acridone analogues have been obtained, which, inter alia, exhibit antitumour (e.g., (1–5)), antimicrobial (e.g., (59)), and antiviral (e.g., (61)) activities and are applicable in the treatment of Alzheimer's disease (e.g., (26)). However, in many cases, their clinical application is limited and excluded because of side effects. In this survey, we describe acridine and acridone derivatives reported since 2013, methods of their synthesis and their potential clinical applications.
Introduction
Acridine derivatives form an important class of heterocycles containing nitrogen due to their broad range of pharmaceutical properties. Acridine derivatives are characterized by unique physical and chemical properties, biological activities, and industrial applications. In the nineteenth century, acridine derivatives were already used industrially as pigments and dyes.1 More critical to the pharmaceutical industry, acridine derivatives have exhibited bioactivities such as anti-inflammatory,2,3 anticancer,4 antimicrobial,5 antitubercular,6,7 antiparasitic,8 antimalarial,9–11 antiviral12,13 and fungicidal activities.14 Acridine derivatives have been shown to be effective as inhibitors of acetylcholinesterase.15 Furthermore, acridines are used as dyes, fluorescent materials for visualization of biomolecules, and in laser technologies.16 These properties of acridines are attributed to their semi-planar heterocyclic structure, which appreciably interacts with different biomolecular targets. Acridine/acridone derivatives are found in natural plants and various marine organisms.17,18 Notably, the anticancer activity of acridine/acridone derivatives has attracted increasing interest. To date, many derivatives of acridine have been synthesized and tested for antitumour activity. The unique planar ring structure allows acridone derivatives to act as DNA intercalators19,20 and to inhibit topoisomerase or telomerase enzymes.21–24 A variety of acridine/acridone derivatives have been synthesized; analogues such as N-(2-(dimethylamino)ethyl)acridine-4-carboxamide (DACA) (1),25–27 triazoloacridone (C-1305) (2)28 and amsacrine (m-AMSA) (3)29 (Fig. 1) have entered clinical studies. Among them, m-AMSA (3) was the first synthetic drug exhibiting clinical efficacy as a topoisomerase inhibitor. Many m-AMSA derivatives (AHMA (4), D3CLP (5)) (Fig. 1) have been developed for stronger anti-cancer properties and removal of many harmful side effects.27,30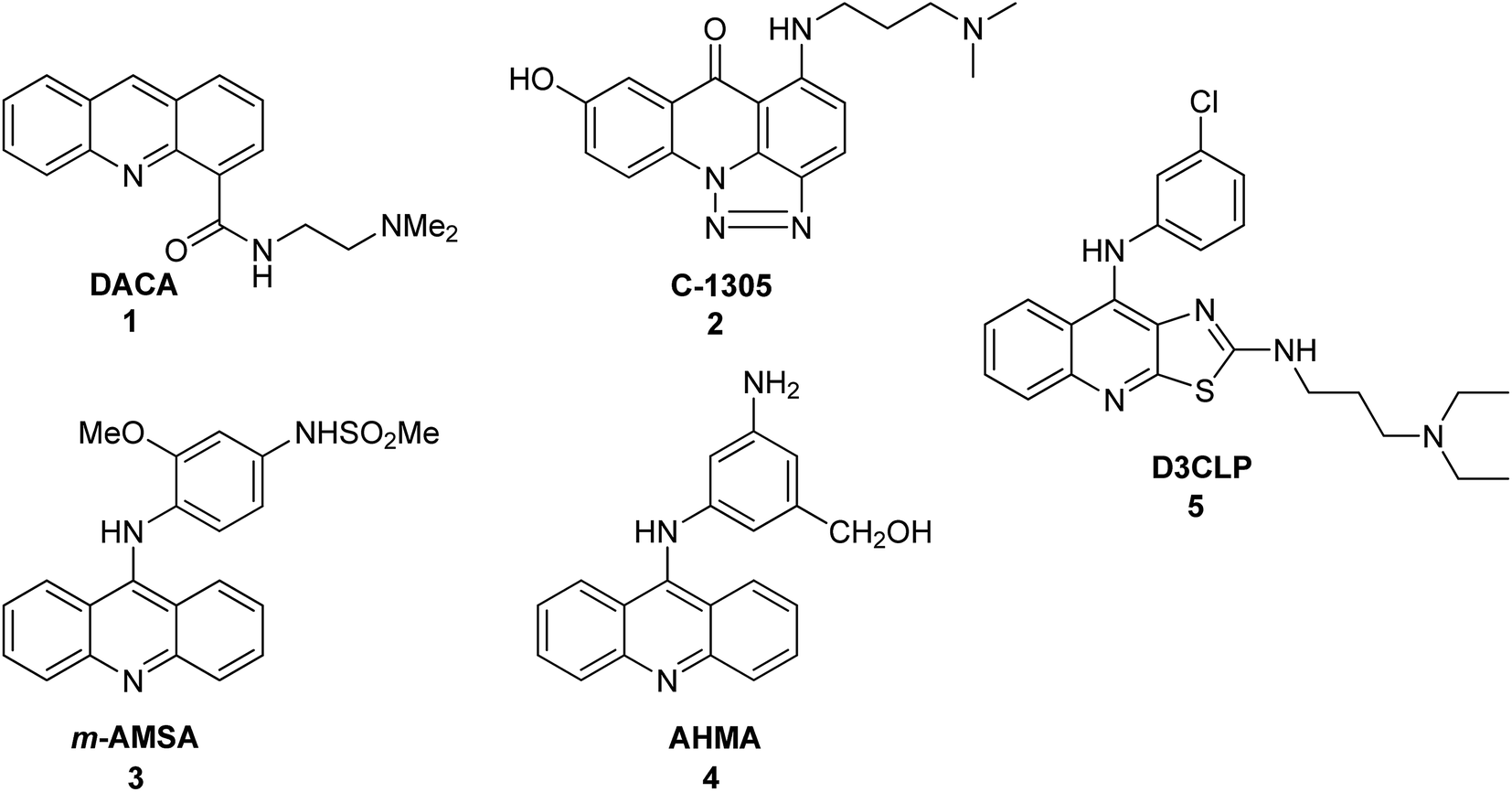 | ||
| Fig. 1 Structures of DACA (1), C-1305 (2), m-AMSA (3), AHMA (4), and D3CLP (5).27–30 | ||
Intermolecular interactions in acridine and acridinium derivatives determine their biological and physical properties including their chemiluminogenic abilities. Therefore, hydrogen bonding and π–π interactions within the Hirshfeld surface have been studied. Recently, Wera and co-workers reported the synthesis and structural investigations of some new acridine and acridinium derivatives.31
Naturally occurring acridine/acridone
Acridine/acridone alkaloids
Alkaloids are naturally occurring chemical compounds, which have a wide range of pharmacological activities (e.g., antimalarial, anticancer, and antibacterial).32 To date, the literature describes a number of acridine/acridone alkaloids, which have been tested as anticancer, antibacterial and antimalarial agents and against Alzheimer's disease.18 Examples of these compounds are cystodytin A (6) (isolated from various marine organisms) and acronycine (7) (isolated from bark of Australian scrub ash tree) (Fig. 2).33–35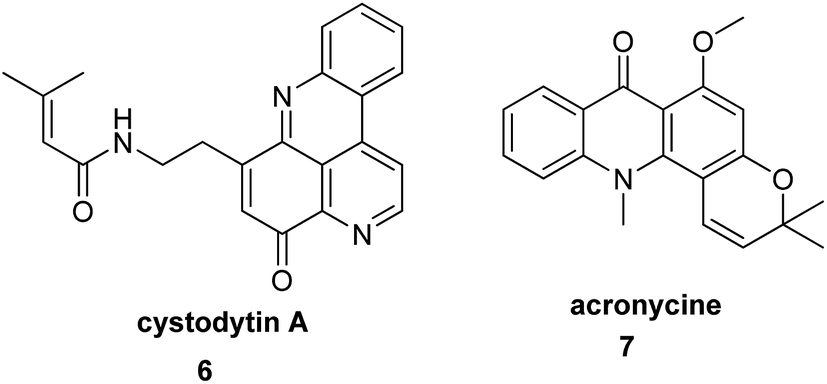 | ||
| Fig. 2 Structure of cystodytin A (6) and acronycine (7).33 | ||
Arai et al. describe the bioassay-guided fractionation and Ngn2 promoter activity of acridine alkaloids (8–10) from an extract of a culture of Streptomyces sp. IFM 11440.17 Inubosin B (9) bearing a hydroxy group at the 4 position showed potent Ngn2 promoter activity, which was dose-dependent. Moreover, compound (9) demonstrated more activity than the positive control baicalin (11), while Insubosin A (8) and Insubosin C (10), which have a hydroxy group at the 5 position, did not show significant activity (Fig. 3).17
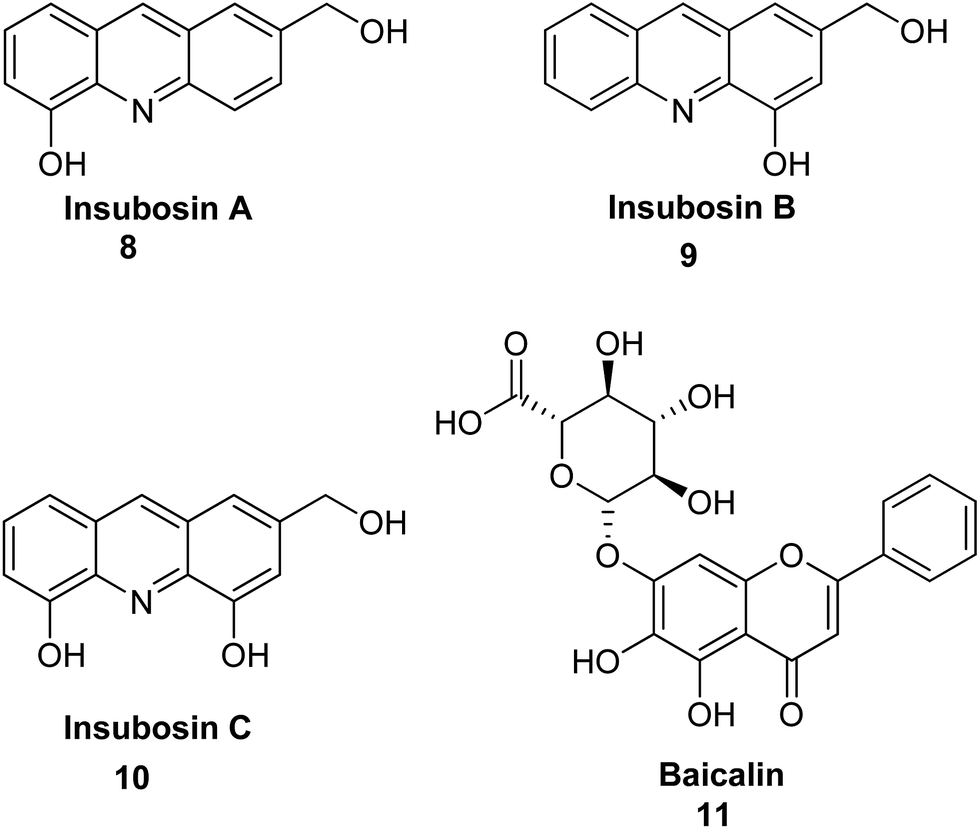 | ||
| Fig. 3 Structure of acridine alkaloids (8–10) and baicalin (11).17 | ||
The Wouatsa group isolated acridone alkaloids (12–21) (Fig. 4) from the MeOH extract of the fruits of Zanthoxylum zanthoxyloides and Zanthoxylum leprieurii, of which six were new (12, 13, 14, 19, 20 and 21). Acridone molecules (19–21) that have a tetracyclic acridone structure as their carbon skeleton have been named as zanthacridone.36
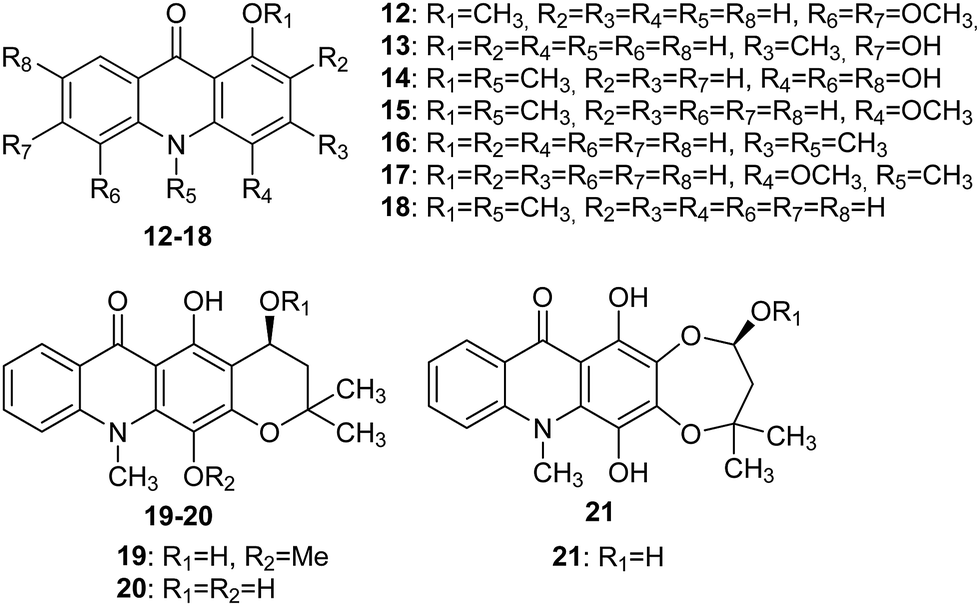 | ||
| Fig. 4 Structures of acridones (12–21).36 | ||
All newly discovered acridones (12, 13, 14, 19, 20 and 21) were tested for antibacterial activity against Bacillus subtilis (MTCC 121), Micrococcus luteus (MTCC 2470), Staphylococcus aureus (MTCC 96), and Pseudomonas aeruginosa (MTCC 741). Compounds (12, 14, 19, and 21) exhibited activity against MTCC 741 with MIC values equal to 500, 125, 250, and 250 μg mL−1, respectively. Furthermore, analogue (14) inhibited MTCC 2470 (MIC 125 μg mL−1), while molecules (15 and 18) failed to show appreciable activity against MTCC 741 but showed good activity in the inhibition of MTCC 96 (MIC 250 and 500 μg mL−1, respectively), MTCC 121 (MIC 250 and 250 μg mL−1, respectively) and MTCC 2470 (MIC 125 and 500 μg mL−1, respectively).36
Acridones (12, 15, 18, and 19) were also tested for in vitro cytotoxicity against liver (WRL-68), colon (Caco2), breast (MCF-7) and prostate (PC-3) cancer cell lines by MTT assay. Studies indicated that compound (12) showed moderate cytotoxic activity against WRL-68 with an IC50 value equal to 86 μM.35 Moreover, quantitative SAR and molecular modelling studies showed that molecules (12, 20, and 21) are inhibitors of aromatase and glycosyltransferase.36
Further acridone alkaloids chlorospermine A (22), chlorospermine B (23) and the known atalaphyllidine (24) and acrifoline (25) (Fig. 5) were isolated from the stem bark of Glycosmis chlorosperma by Beniddir et al.37 They used bioassay-guided isolation for this purpose.
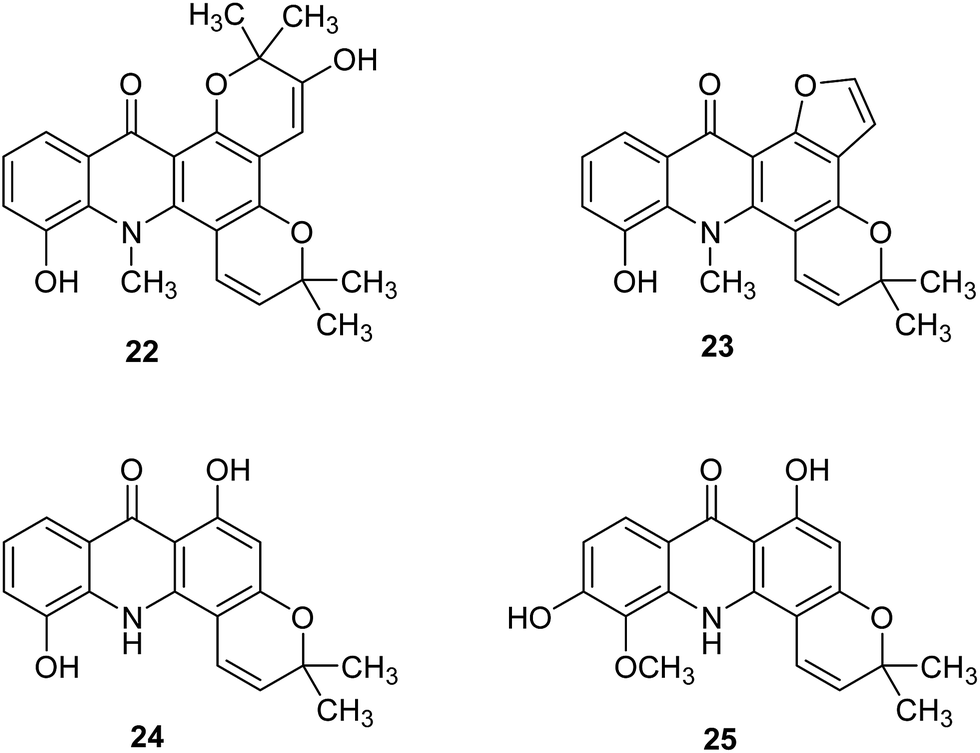 | ||
| Fig. 5 Structure of acridone alkaloids (22–25).37 | ||
Acridone molecules (22–25) were tested for DYRK1A, CLK1, CDK1, CDK5, GSK3, and CK1 activity. Acrifoline (25) showed inhibition activities of DYRK1A, CLK1, GSK3, CDK1, and CDK5 with IC50 values of 0.075, 0.17, 2, 5.3 and 9 μM, respectively. Chlorospermine B (23) and atalaphyllidine (24) exhibited activity against DYRK1A with IC50 values of 5.7 and 2.2 μM, respectively. Furthermore, compound (23) demonstrated inhibition of CLK1 (IC50 = 7 μM), while chlorospermine A (22) failed to show any activity against various kinases. Thus, studies performed suggest that acridine may constitute a molecular skeleton in search of new inhibitors of DYRK1A.37 Moreover, a molecular docking study of (22–35) showed that interactions of the ligand with conserved Lys188 and backbone atoms in the hinge region (Glu239 and/or Leu241) are necessary for good biological activity. The most active compound (25) forms hydrogen bonds between the hydroxy group at C-6 and Glu203 and the conserved Lys188 and interacts with backbone atoms of Glu239 and Leu241 through hydrogen bonds involving the C-1 hydroxy group. Atalaphyllidine (24) is proposed to interact with backbone atoms of Glu239, Leu241, and Ile165 through hydrogen bonds involving the C-9, C-1, and C-5 oxygen atoms, respectively. In turn, derivative (23) showed hydrogen bonds between the hydroxy group in position C-5 and the side chains of Glu203 and Lys188, while inactive chlorospermine A (22) forms hydrogen bonds between its oxygen atoms at C-5, C-9, and C-2′′ and backbone NH of Leu241 and side chains of Asn244 and Asp307 (from the DFG motif), respectively.37
Citrusinine-I (26) (Fig. 6) is an example acridone alkaloid isolated from Swinglea glutinosa. These analogues were described as herbicide models, and their inhibition of photosynthesis was investigated against both photosystem I and II. Measurements in vitro demonstrated significant influence on various stages of reactions in the course of photosynthesis; however, extracts provided only several milligram amounts of tested compounds, and design of multi-stage synthesis would be necessary to accomplish in vivo examinations.38
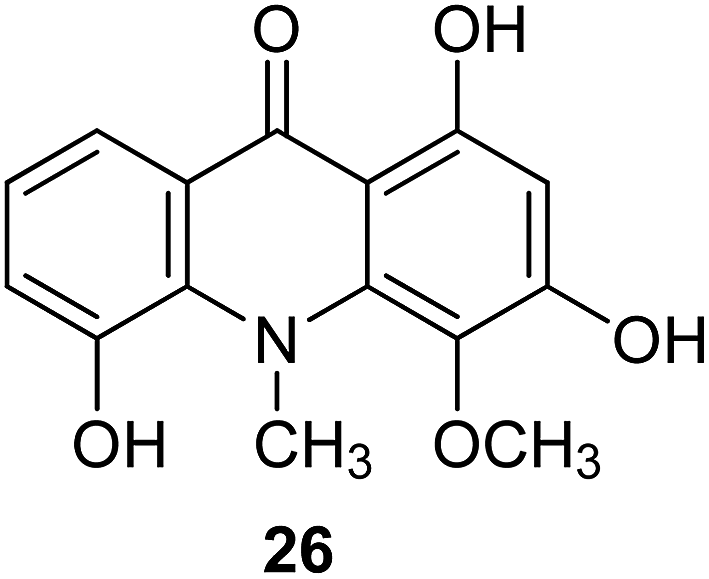 | ||
| Fig. 6 Structure of citrusinine-I (26).38 | ||
Teow presented the methods for treating various liver diseases and certain cancers by used compositions of acridone, xanthone, thioxanthone, tocotrienol and oleanolic triterpenoid compounds.39 He isolated hepatic therapeutic actives from the dried stem, bark and roots of Cratoxylum cochinchinense plant. The acridone molecules are at least one of the anti-viral components in the hepatic pharmaceutical active which probably function by inhibiting the growth and multiplication of the chronic hepatitis HBV and chronic hepatitis HCV in patients afflicted with chronic hepatitis B or chronic hepatitis C.
Synthetic derivatives of acridine/acridone
Acridine/acridone as inhibitors of acetylcholinesterase
Alzheimer's disease (AD) is the most common form of dementia, an incurable and neurodegenerative disease, which manifests via progressive decline in intellectual abilities, caused by the disappearance of nerve cells.38,40 Because AD is currently one of the largest global medical problems, it has attracted the attention of medicinal chemists, who have studied new drugs for this disease.41 The drugs used in AD include cholinesterase inhibitors, which enhance neurotransmission of cholinergic synapses in the brain and thus enhance intellectual activity.42 The acridine derivative used in medicine as an anti-AD drug is tacrine (27) (Fig. 7).43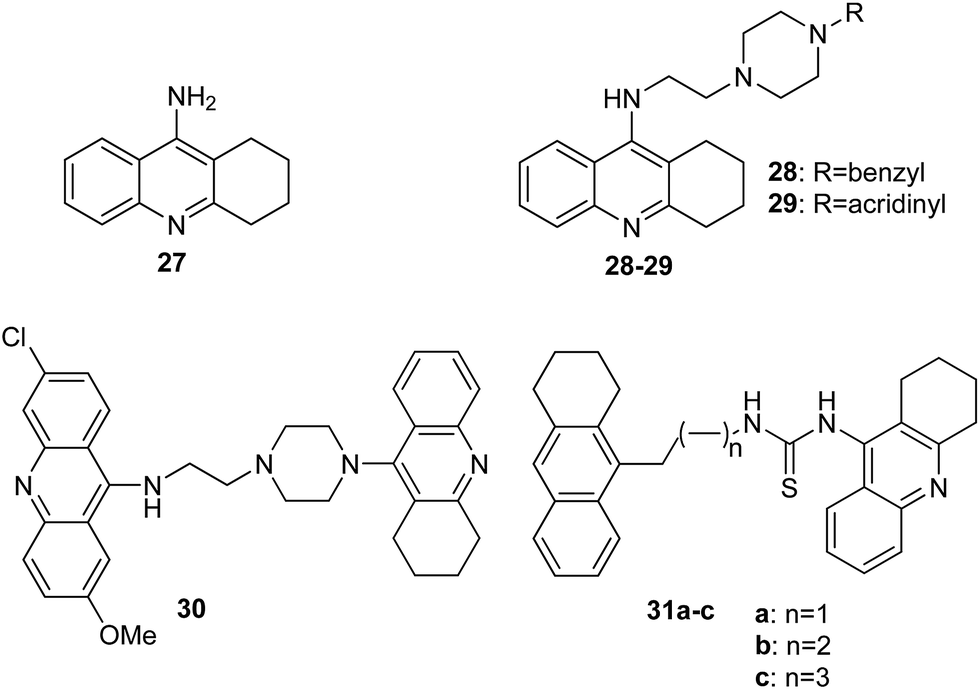 | ||
| Fig. 7 Structure of tacrine (27) and tacrine/acridine derivatives (28–30, 31a–c).44 | ||
Acridines can be also used to diagnosis of neurodegenerative disorders, such as AD. Fuchigami et al. developed 125I-labeled acridines possessing high affinities for Aβ aggregates involved in these pathologies.45 During in vivo experiments on Aβ plaques in the brain slices of Tg2576 mice, the most promising results were obtained in case of 6-iodo-2-methoxy-9-methylaminoacridine and 2,9-dimethoxy-6-iodoacridine as potential imaging agents for amyloid in living brain.
The Hamulakova group reported the potential of novel tacrine/acridines (28–30, 31a–c) (Fig. 7) as human acetylcholinesterase (hAChE) and human butyrylcholinesterase (hBChE) inhibitors.46 In vitro studies have shown that the compounds (29, 30, 31b and 31c) proved to be especially potent against hAChE with IC50 values in the range of 2 to 8 nM, and analogues (28, 29 and 31a–c) were the most effective inhibitors against hBChE with IC50 values 0.4–20 nM; however, tacrine (27) as a reference compound showed less activity against hAChE (IC50 = 500 nM) and hBChE (IC50 = 23 nM).44 Docking models of the binding of compound (29) into hAChE showed cation–π interactions between Tyr341 and Trp286 residues of the peripheral anionic site and the positively charged acridine ring and showed cation–π interactions between Trp86 and the protonated tacrine ring. Additionally, stabilization of compound (29) may be due to a hydrogen bond between the amino group of tacrine and the oxygen atom of Tyr337.44
Furthermore, Janockova et al. studied the interaction of derivative (30) with ctDNA.47 The results of UV-Vis absorption spectroscopy showed that the binding of compound (30) occurred with a binding constant value K = 2.0 × 103 M−1. Thus, the results are indicative of a strong interaction between analogue (30) and ctDNA. Additionally, they demonstrated that (30) is a dual topo inhibitor, acting both as a topo I inhibitor and as a topo II catalytic inhibitor.46
Thiratmatrakul and co-workers synthesized the new tacrine-carbazole hybrids (36a–c) as potential multifunctional agents for the treatment of AD.48 The intermediate imines (34a–c) were prepared by reaction of 7-methoxyheptaphylline (32a) or heptaphylline (32b) with 1,3-diaminopropane (33a) or 1,5-diaminopentane (33b) in MeOH. The reaction of the corresponding imines (34a–c) with 9-chlorotacrine (35) in the presence of pentanol obtained compounds (36a–c) (Scheme 1).47
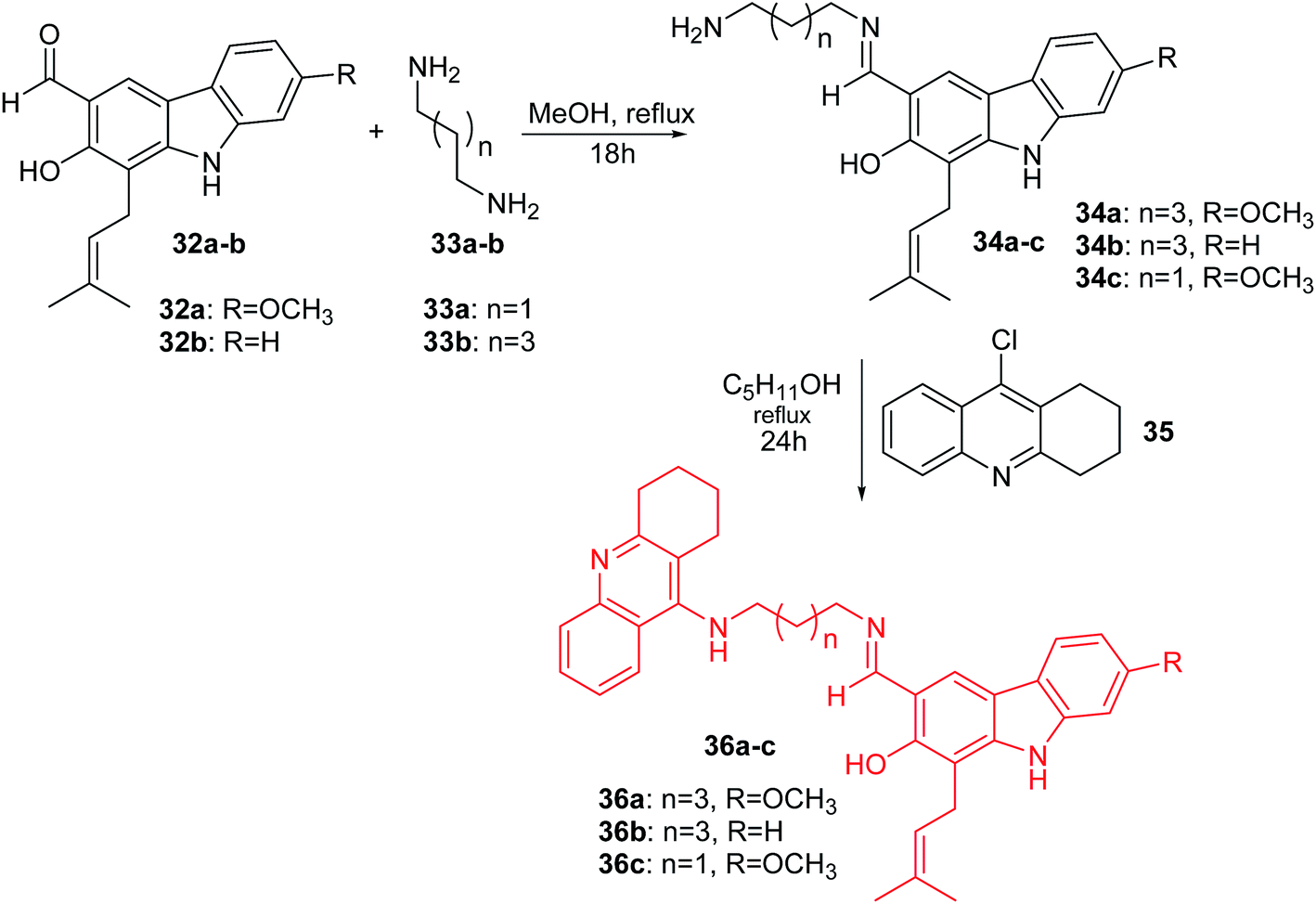 | ||
| Scheme 1 Synthesis of tacrine-carbazole hybrids (36a–c).48 | ||
Derivative (36a) revealed the most potent activity against AChE inhibitory and antioxidant action. Additionally, the molecular modelling study showed that synthesized analogues demonstrated binding with the catalytic active site (CAS) and the peripheral anionic site (PAS) of AChE. Furthermore, tacrine-carbazole hybrids exhibited a neuroprotective effect against oxidative stress induced by H2O2 and Aβ1-42 toxicity. Moreover, analogue (36a) exhibited an ability to improve short-term and long-term memory deficit in mice induced by scopolamine.47
Mohammadi-Khanaposhtani et al. presented the synthesis of triazole derivatives of acridone (44a–n) (Scheme 2).49 In the first step, 2-arylamino benzoic acids (39) were obtained by the Ullmann condensation reaction of 2-bromobenzoic acid (37) and various aniline derivatives (38) in the presence of potassium carbonate and copper in EtOH at reflux. Subsequent cyclization of compound (39) by PPA at 100 °C obtained acridone derivatives (40), which by reaction with propargyl bromide (41) using potassium tert-butoxide in DMSO at room temperature yielded 10-(prop-2-yn-1-yl)acridin-9-one derivatives (42). Then, compounds (42) were converted to the target triazole analogues (44a–n) using CuI and freshly prepared azide derivative (43).48
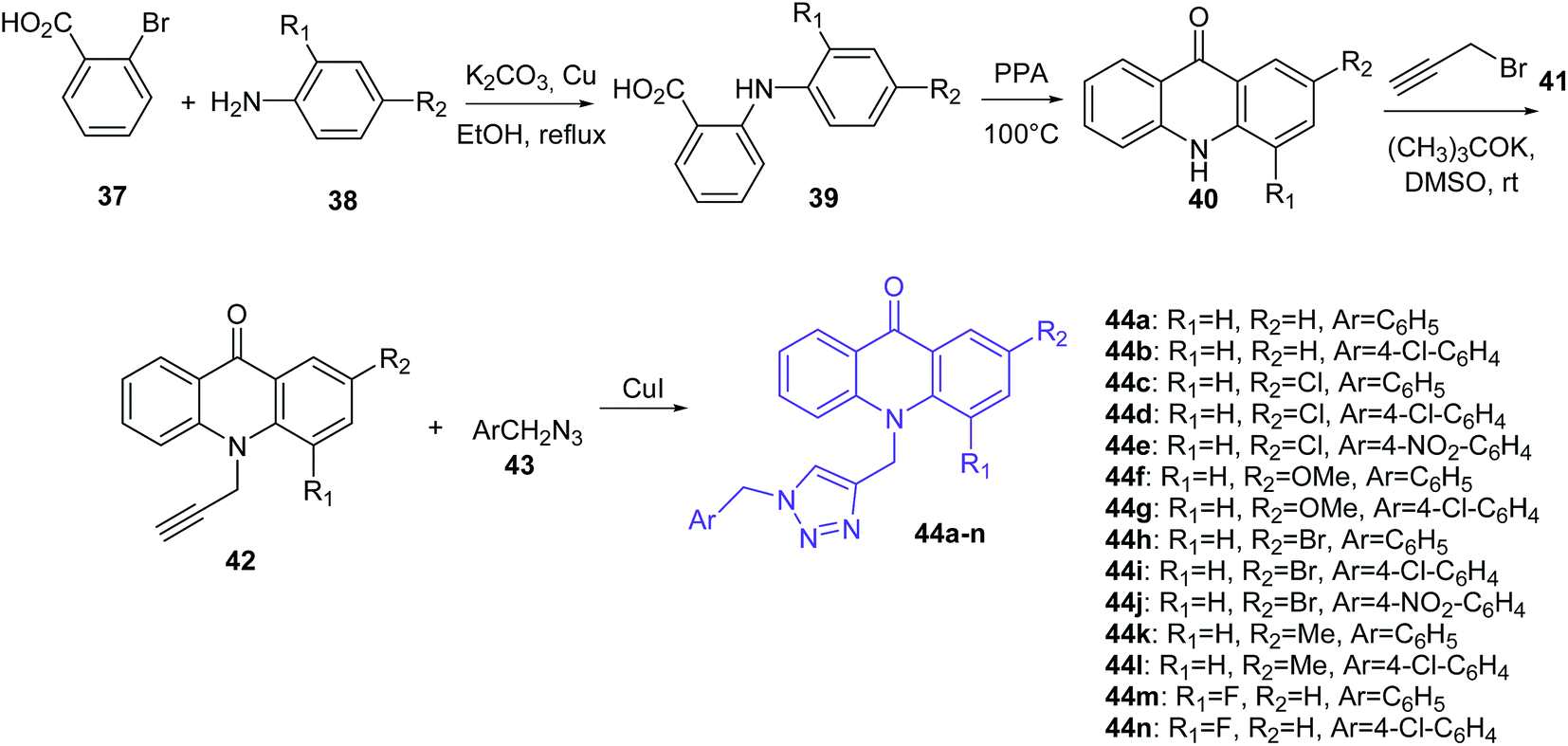 | ||
| Scheme 2 Synthesis of triazole derivatives of acridone (44a–n).49 | ||
Obtained derivatives of acridone (44a–n) were tested in vitro for inhibition of acetylcholinesterase (AChE) and butyrylcholinesterase (BChE). The best ability to inhibit AChE was shown by compound (44g) (IC50 = 7.31 μM), with a 4-substituted chlorine on the pendant benzyl group.48 In turn, derivatives (44e and 44j) with strong electron-withdrawing nitro groups into the pendant benzyl group showed decreased activity. Additionally, the presence of groups in the order of OMe > Me ≥ Cl > Br instead of hydrogen on the 2-substituted acridone moieties provided an increase in inhibitory activity. Thus, the presence of methoxy and chlorine groups contributes to good interactions with the active sites of enzymes and plays an important role in the inhibitory activity. Docking studies of compound (44g) showed π–π interactions between Phe330 and the 1,2,3-triazole moiety and between Trp84 and the acridone moiety. Additionally, derivative (44g) forms hydrogen bonds between the oxygen of OMe and Ser122. Furthermore, a weak interaction between chlorine and the backbone carbonyl group of amino acid is beneficial.48
The Mohammadi-Khanaposhtani group also demonstrated acridone-1,2,4-oxadiazole-1,2,3-triazole hybrids as inhibitors of AChE and BChE.50 In this research, a series of new acridone-1,2,4-oxadiazole-1,2,3-triazoles (47a–p) were obtained according to the synthetic pathway given in Scheme 3. The target compounds (47a–p) were synthesized by the reaction of 10-(prop-2-yn-1-yl)acridin-9-one derivatives (45) in the presence of CuI and the freshly prepared azide derivative (46).50
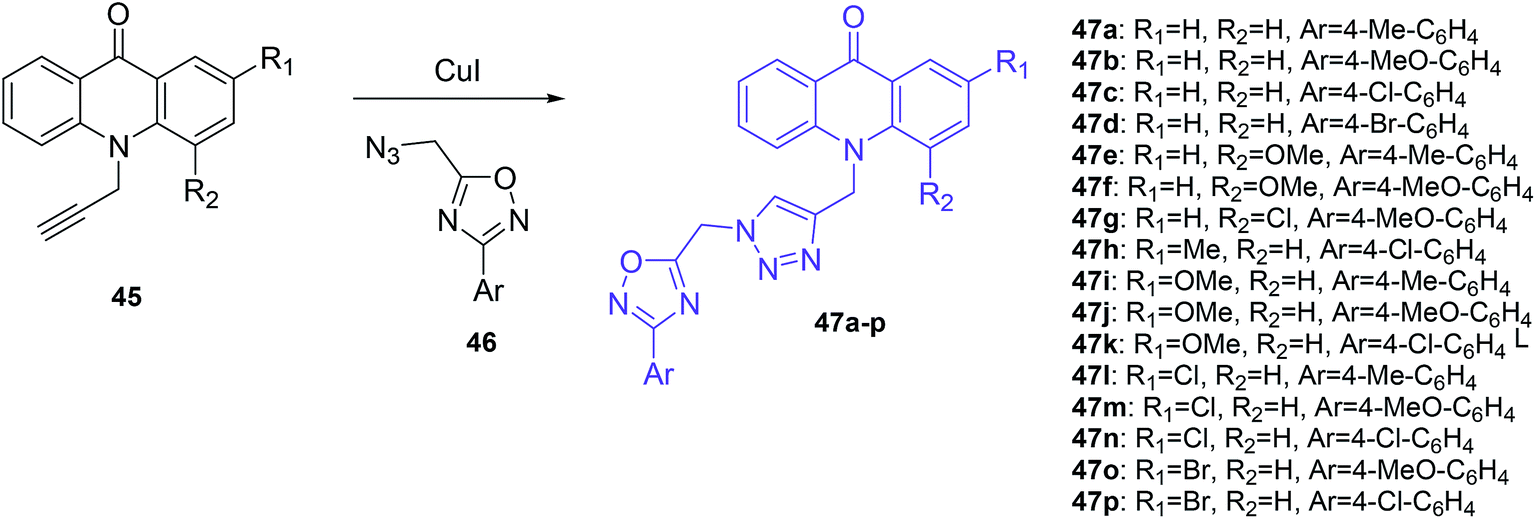 | ||
| Scheme 3 Synthesis of acridone-1,2,4-oxadiazole-1,2,3-triazole derivatives (47a–p).50 | ||
Among these compounds, (47a–d, 47k and 47n) showed an inhibitory effect against AChE with IC50 values ranging between 11.55 and 77.79 μM. The most activity was shown by compound (47b) containing an unsubstituted acridone ring and 4-methoxyphenyl-1,2,4-oxadiazole moieties, while derivatives (47c–j, 47l–m, and 47o–p) containing acridone moieties 2 or 4-substituted by methoxy, chlorine or bromine groups demonstrated no observable activity. Additionally, it may be that a methoxy group into the pendant benzyl group imparted a higher activity than chlorine, methyl, and bromine substituents. Docking studies showed that the orientation of analogue (47b) in the active site of AChE was the same as donepezil. Namely, compound (47b) forms π–π interaction between the acridone moiety and Phe331 and Trp84 in the catalytic anionic site (CAS). Additionally, compound (47b) is proposed to interact with the hydroxyl group of Ser200 in the catalytic triad site through hydrogen bonds involving the carbonyl group of acridone. Further, the 1,2,4-oxadiazole moiety exhibited π–π interaction with a phenyl group of Tyr121 in the peripheral anionic site (PAS).50
Mohammadi-Khanaposhtani et al. presented the synthesis of novel acridone-based 1,2,4-oxadiazoles (57a–n) as potential anticonvulsant agents. In the first step, 2-arylamino benzoic acids (49) were prepared by the Ullmann condensation reaction of 2-bromobenzoic acid (37) and different anilines (48).51 Then, the cyclization of compounds (49) by PPA yielded acridone derivatives (50). The second intermediate 3-aryl-5-(chloromethyl)-1,2,4-oxadiazole derivatives (56) were obtained starting from appropriate benzonitriles (51) and hydroxylamine hydrochloride (52) in the presence of sodium hydroxide in EtOH at reflux. Derivatives (53) and chloroacetyl chloride (54) reacted in dry acetone using potassium carbonate to yield N′-(2-chloroacetoxy)-4-substituted benzimidamides (55). Subsequent cyclization of compounds (55) in refluxing toluene yielded 3-aryl-5-(chloromethyl)-1,2,4-oxadiazole derivatives (56). The desired final products (57a–n) were synthesized by reaction of acridone derivatives (50) with 1,2,4-oxadiazole derivatives (56) in the presence of potassium tert-butoxide in DMSO at room temperature (Scheme 4).51
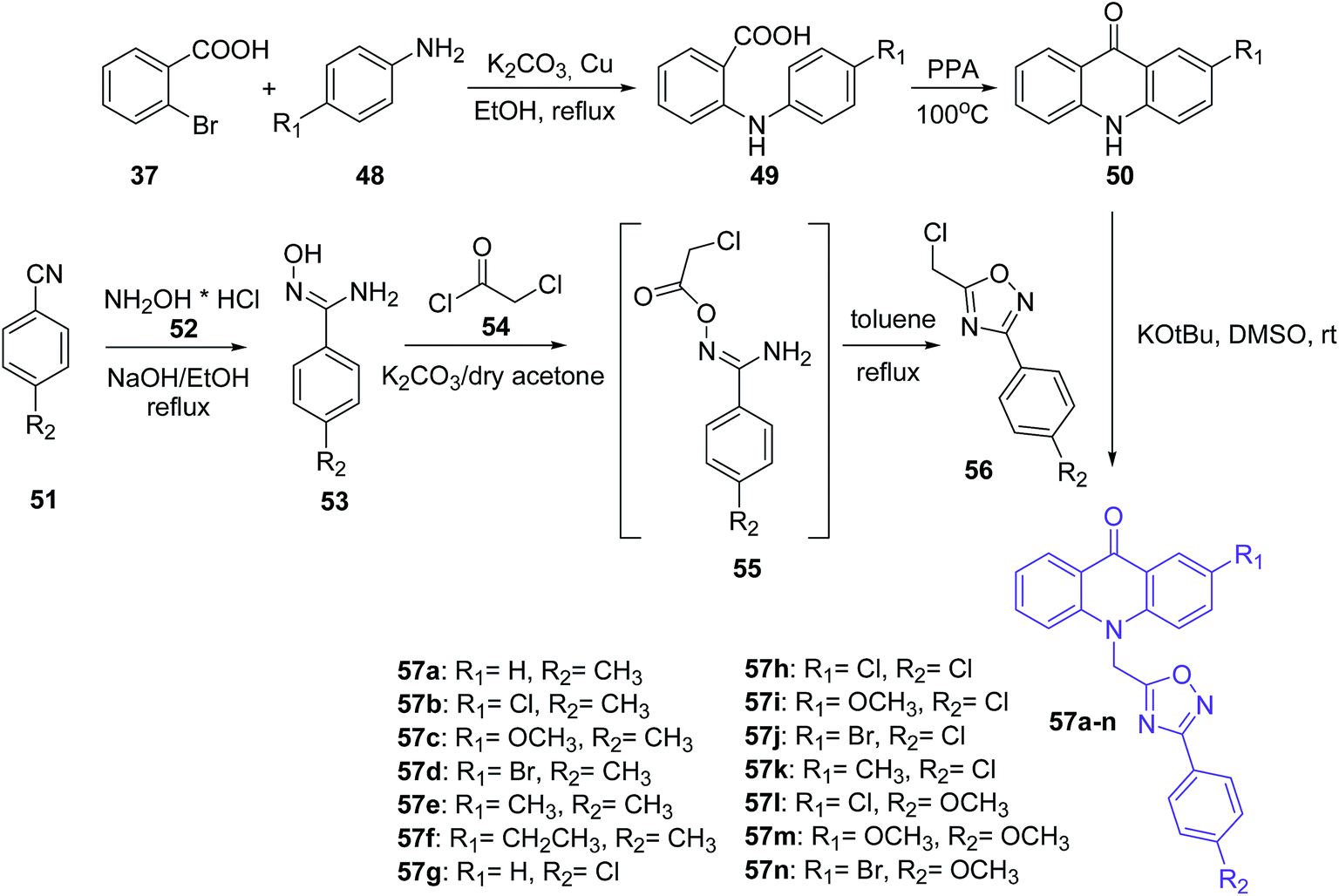 | ||
| Scheme 4 Synthesis of compounds (57a–n).51 | ||
Most prepared compounds showed good anticonvulsant activities. Derivatives (57a–c and 57f–n) had potent anticonvulsant activity in the PTZ-induced seizure test (ED50 = 2.08–18.93 mg kg−1). The most potent anticonvulsant activity against PTZ-induced seizures were demonstrated by 4-methoxyphenyl-oxadiazole derivatives (57l and 57m) bearing a 2-Cl or 2-MeO on the acridone ring, with ED50 values of 2.08 and 2.12 mg kg−1, respectively. Additionally, studies on anticonvulsant activity against MES-induced seizures showed that all prepared compounds (57a–n) had potent anticonvulsant activity (ED50s = 2.24–8.71 mg kg−1). The most potent anticonvulsant activity against MES-induced seizures was shown by compound (57b). Moreover, products (57a–n) were screened for their neurotoxicity in mice using the rotarod test, and these derivatives displayed good anticonvulsant activity with a high safety profile. Furthermore, a docking study of analogue (57l) in the BZD-binding site of the GABAA receptor confirms possible binding of derivative (57l) with BZD receptors.51
Inhibitors of human carbonic anhydrase isozymes based on acridines and acridones analogs
Carbonic anhydrases (CA, EC 4.2.1.1.) are Zn(II) ion-containing metalloenzymes that catalyse the interconversion of carbon dioxide (CO2) and bicarbonate (HCO3−) and the corresponding dehydration of bicarbonate in acidic medium with regeneration of CO2.52,53 Furthermore, they participate in crucial physiological processes associated with, among others, pH homeostasis, electrolyte secretion, respiration, CO2 and ion transport, some biosynthesis reactions (e.g., gluconeogenesis, lipogenesis), and tumourigenicity.54–56 To date, sixteen different α-carbonic anhydrase isozymes have been described in mammals.57 CA isoforms are present in a variety of tissues and are involved in several important biologic processes (e.g., acid–base balance, respiration, carbon dioxide and ion transport, bone resorption, ureagenesis, gluconeogenesis, lipogenesis, and electrolyte secretion). CA II, which is the most active as a catalyst for carbon dioxide hydration, is found primarily in red blood cells and in other secretory tissues of the gastrointestinal tract, kidneys, lungs, eye, and CNS. Therefore, α-carbonic anhydrase isozymes that participate in these processes are important therapeutic targets with the potential to be inhibited/activated for the treatment of a range of disorders such as oedema, glaucoma, obesity, cancer, and epilepsy.58The Yesildag group presented a one-step method for the synthesis of acridine sulfonamide derivatives (61a–n).59 Compounds (61a–n) were prepared by the reaction of dimedone (58), 4-amino-N-[amino(imino)methyl]-benzenesulfonamide (59) and 4-cyano-benzaldehyde (60a–n) in the presence of sulfuric acid as a catalyst in water at room temperature (Scheme 5). This method has high yields and purities of the compounds obtained. In addition, it has advantages of using H2SO4 as an environmentally friendly catalyst and H2O as a green solvent.59
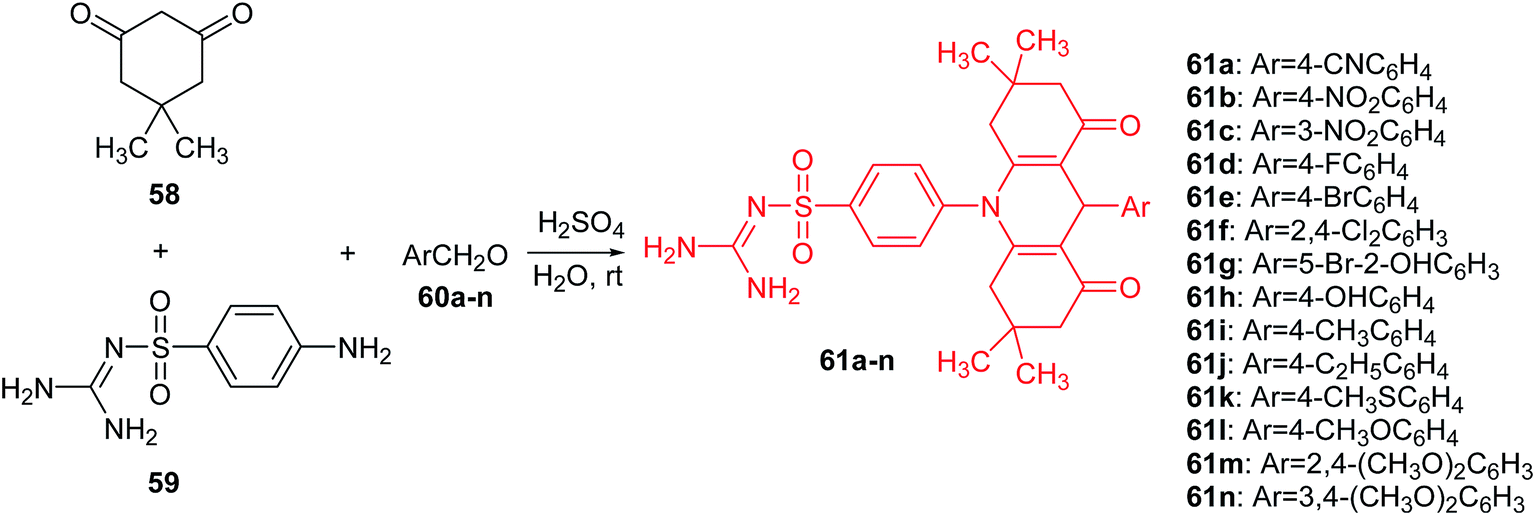 | ||
| Scheme 5 Synthesis of acridine sulfonamide derivatives (61a–n).59 | ||
All synthesized compounds (61a–n) were tested in vitro with respect to inhibition of hydratase and esterase (hCA I and hCA II) isozymes. Analogues (61a–d and 61f–j) showed moderate esterase activities of hCA I and hCA II. Compound (61i), containing a methyl group on the para position of the benzyl group, was the most powerful inhibitor against hCA I (IC50 = 47.2 μM) and hCA II (IC50 = 50.1 μM). In turn, the derivatives containing larger functional groups (–OCH3 and –SCH3) were not active. This may be related as the steric effects of these groups may block the enzyme–inhibitor interaction.59
Other derivatives of acridine bis-sulfonamides (66a–l) were presented by Esirden et al.60 The intermediate nitro-acridine sulfonamides (64a–b) were prepared by a coupling reaction of dimedone (58), 4-aminobenzenesulfonamide (62) and meta- or para-substituted nitrobenzaldehydes (63a or 63b) in the molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 in the presence of DBSA as a phase transfer catalyst in H2O. Then, the reduction of the nitro group was achieved by using excessive reduction-reactive aqueous sodium polysulfide. The final products (66a–l) were synthesized from appropriate sulfonyl chlorides and amino acridines (65a and 65b) in THF in the presence of triethylamine (TEA) (Scheme 6).60 Derivatives (64a–b, 65a–b, and 66a–l) were tested for in vitro inhibition activity against the cytosolic human isoforms (hCA I, II, IX and XII). Compounds (64a–b, 65a–b, and 66a–l) were rather ineffective inhibitors against hCA I (0.69–8.91 μM). These analogues have low activity, probably due to the bulkiness of the tails that they incorporate and that most likely extend out of the active site.59 In the case of hCA II, derivatives (64a–b, 65a–b, and 66a–l) were also rather ineffective inhibitors (0.10–0.96 μM), and no important effects of tail bulkiness were observed. This could be because hCA II has a wide entrance to the active site, and presumably the hydrophobic and hydrophilic interactions of these tails with the active site were sparingly effective. The transmembrane isoforms hCA IX and hCA XII were inhibited better than the cytosolic ones. However, compounds (64a–b, 65a–b, and 66a–l) showed more activity against hCA XII (0.09–1.12 μM) than against hCA IX (0.03–1.02 μM). The low inhibition of these analogues against the transmembrane isoforms (hCA IX and XII) may be because the variations in the structure are distant parts of the tail from the primary sulfonamide, which may result in these parts of the molecules being outside of the active site and causing only small interactions with amino acid residues crucial for the binding of inhibitors.60
:1:1 in the presence of DBSA as a phase transfer catalyst in H2O. Then, the reduction of the nitro group was achieved by using excessive reduction-reactive aqueous sodium polysulfide. The final products (66a–l) were synthesized from appropriate sulfonyl chlorides and amino acridines (65a and 65b) in THF in the presence of triethylamine (TEA) (Scheme 6).60 Derivatives (64a–b, 65a–b, and 66a–l) were tested for in vitro inhibition activity against the cytosolic human isoforms (hCA I, II, IX and XII). Compounds (64a–b, 65a–b, and 66a–l) were rather ineffective inhibitors against hCA I (0.69–8.91 μM). These analogues have low activity, probably due to the bulkiness of the tails that they incorporate and that most likely extend out of the active site.59 In the case of hCA II, derivatives (64a–b, 65a–b, and 66a–l) were also rather ineffective inhibitors (0.10–0.96 μM), and no important effects of tail bulkiness were observed. This could be because hCA II has a wide entrance to the active site, and presumably the hydrophobic and hydrophilic interactions of these tails with the active site were sparingly effective. The transmembrane isoforms hCA IX and hCA XII were inhibited better than the cytosolic ones. However, compounds (64a–b, 65a–b, and 66a–l) showed more activity against hCA XII (0.09–1.12 μM) than against hCA IX (0.03–1.02 μM). The low inhibition of these analogues against the transmembrane isoforms (hCA IX and XII) may be because the variations in the structure are distant parts of the tail from the primary sulfonamide, which may result in these parts of the molecules being outside of the active site and causing only small interactions with amino acid residues crucial for the binding of inhibitors.60
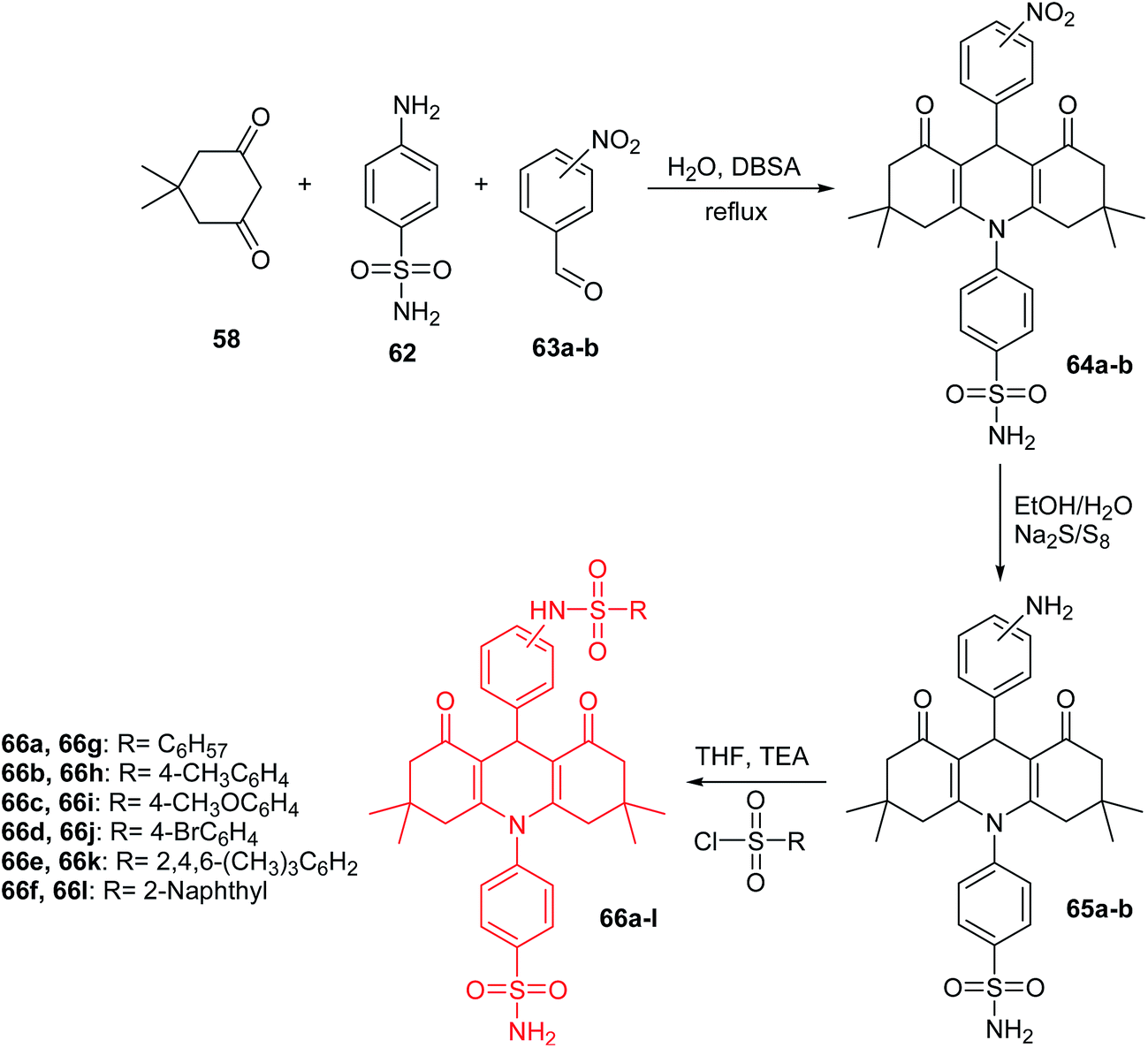 | ||
| Scheme 6 Synthesis of acridine bis-sulfonamide derivatives (66a–l).60 | ||
Ulus et al. presented the synthesis of acridine and bis acridine sulfonamides (71a–r and 73–74) as cytosolic carbonic anhydrase isoforms I, II and VII inhibitors.61 The intermediate (69) were obtained in two steps. Compound (68) was synthesized by reaction of 4-aminobenzenesulfonamide (62) with p-nitrobenzoyl chloride (67) in THF at room temperature in the presence of TEA. The nitro group of (68) was then reduced by aqueous sodium polysulfide in ethanol–water to obtain compound (69) (Scheme 7). Derivatives (71a–r) were prepared by coupling reactions of 4-amino-N-(4-sulfamoylphenyl)benzamide (69) with cyclic-1,3-diketones (58a–b) and aromatic aldehydes (70a–t) in the molar range of 1:2:1 in the presence of p-dodecylbenzenesulfonic acid (DBSA) as a phase transfer catalyst in H2O (Scheme 8). Additionally, bis acridine sulfonamides (73 and 74) were synthesized in the molar range of 1:2:1 ((69):1,3-diketone (58a):1,3-benzenedialdehyde (72a) or 1,4-benzenedialdehyde (72b)) (Scheme 9).61
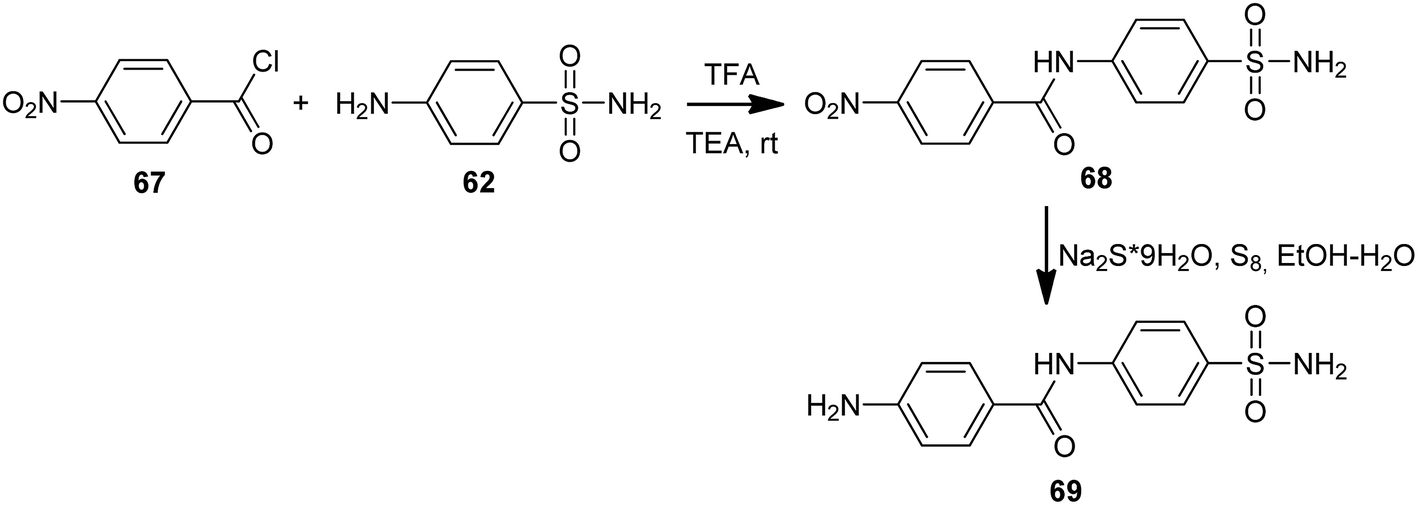 | ||
| Scheme 7 Synthesis of 4-amino-N-(4-sulfamoylphenyl)benzamide (69).61 | ||
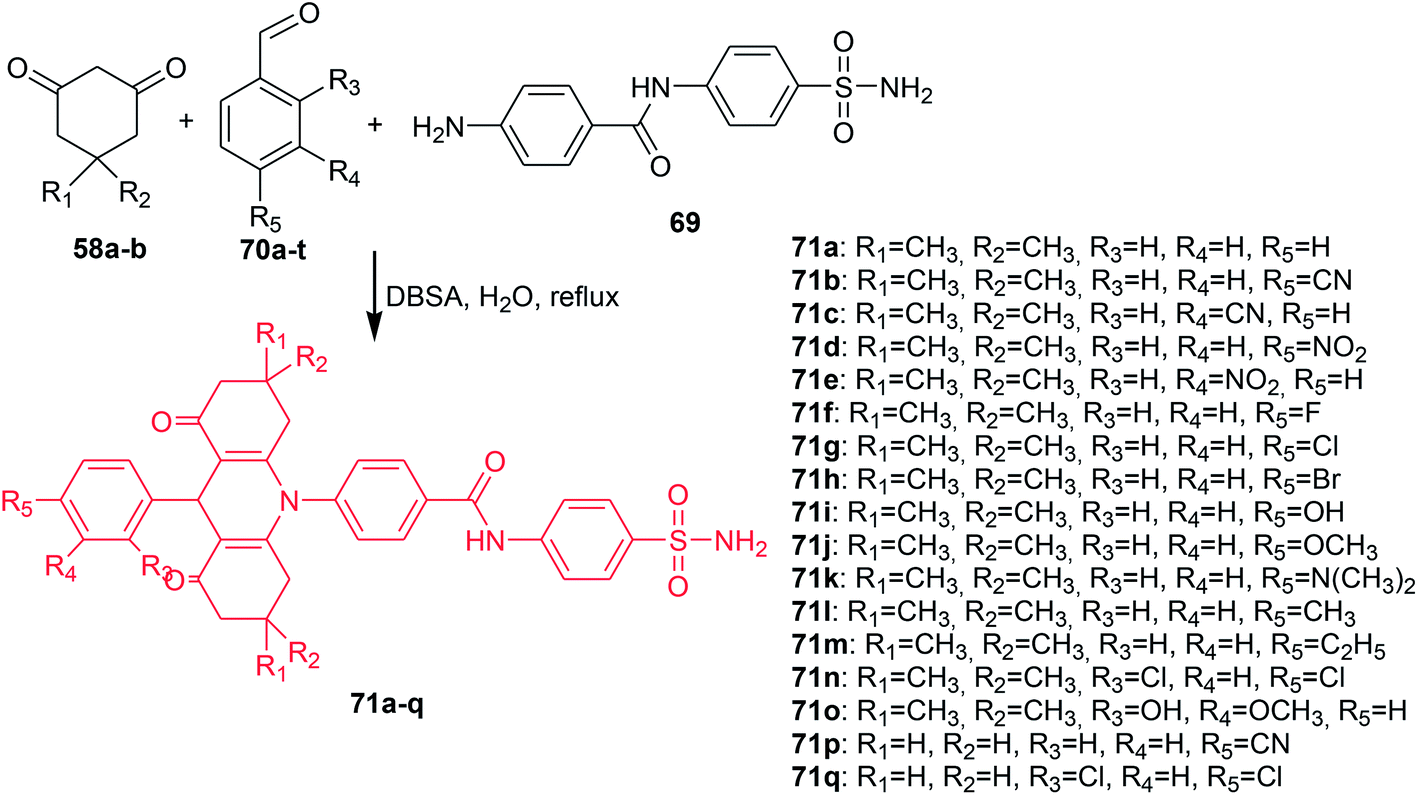 | ||
| Scheme 8 Synthesis of acridine sulfonamides (71a–q).61 | ||
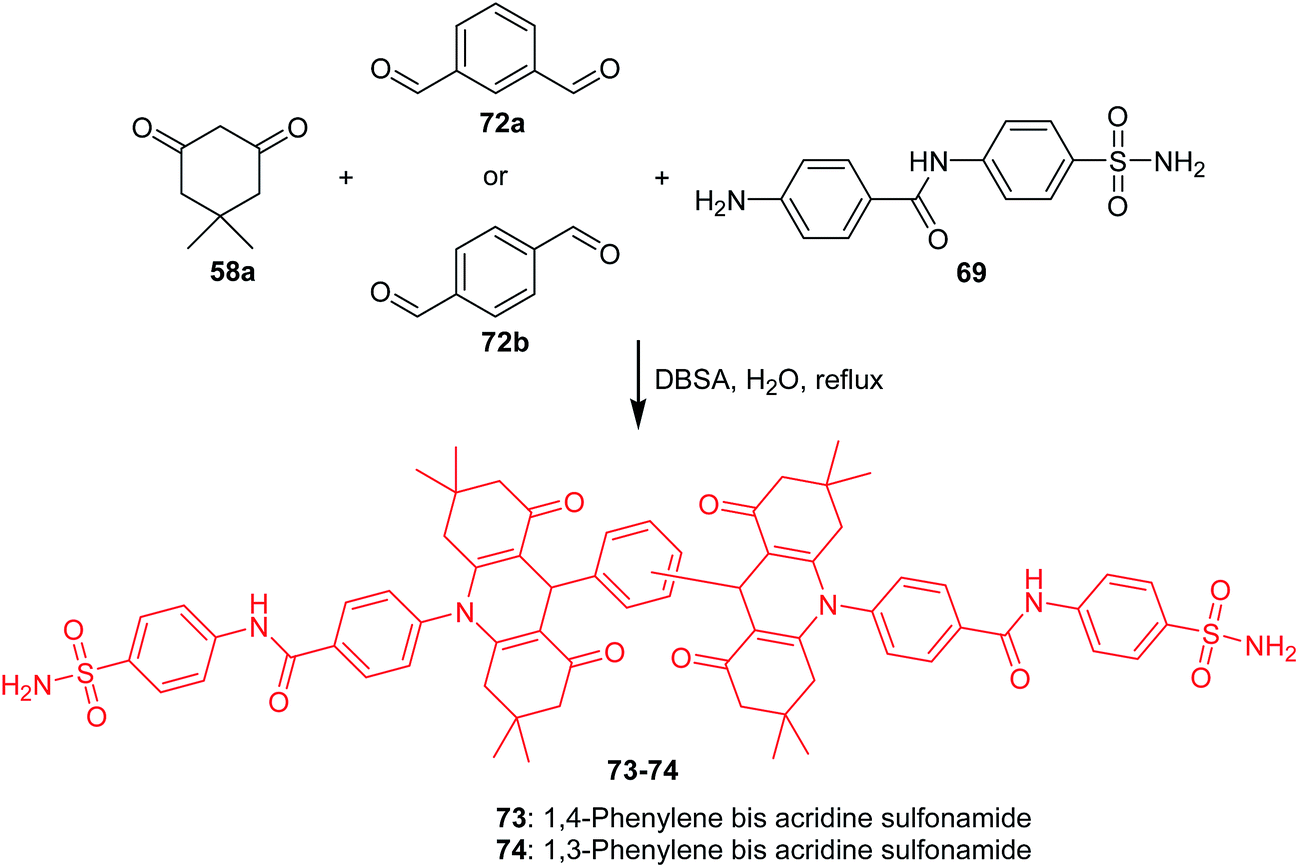 | ||
| Scheme 9 Synthesis of bis acridine sulfonamides (73–74).61 | ||
The prepared compounds (71a–r and 73–74) exhibited good activity results against all three tested isoforms (hCA I, II and IV). Analogue (71a), containing only hydrogen, and derivative (71o), possessing OH and methyl moieties in the aromatic part of the molecule, were found to have the strongest activity against hCA I with KIs values 0.16 and 0.26 μM, respectively. Derivatives (71a, 71c, 71e, 71g, and 71n) showed effective hCA II inhibition with KIs values in the range of 15–23 μM. The best hCA IV inhibitors (KIs of 0.004, 0.005, and 0.022 μM) were compounds (83a, 83o, and 71p), respectively, which were much more effective compared to acetazolamide (a reference compound).61
Acridine/acridone derivatives and their antibacterial and antiparasitic activity
To date, a great number of acridine compounds have been synthesized and clinically used as anti-bacterial drugs (e.g., ethacridine (75)),18 anti-malarial agents (e.g., quinacrine (76)) and anti-viral drugs (e.g. acranil (77)) (Fig. 8).62,63 The ability of acridine to bind to DNA has been used to inactivate genomes of viral and bacterial pathogens present in the cytoplasm and red RBC preparations used for clinical trials and transfusion. An example of such a compound is S-303 (78) (Fig. 8) containing the nitrogen mustard moiety. The ester bond is rapidly hydrolysed, which ensures a short half-life of the compound.64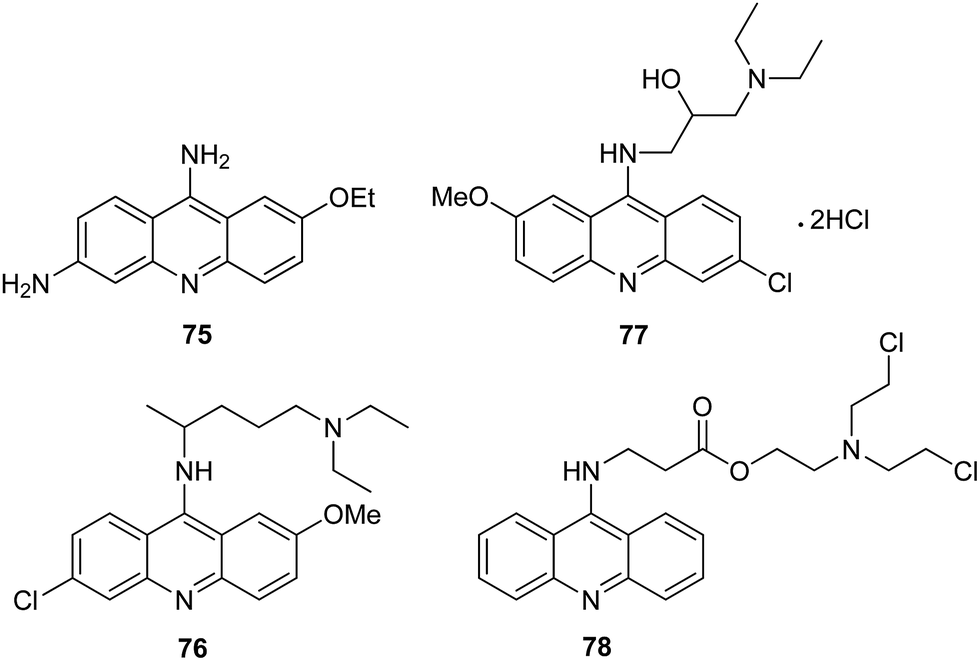 | ||
| Fig. 8 Structure of ethacridine (75), quinacrine (76), acranil (77) and S-303 (78).18,62–64 | ||
Acriflavine (ACF), a mixture of 3,6-diamino-10-methylacridinum chloride (trypaflavine) and 3,6-diaminoacridine (proflavine) showed apart from antibacterial and anticancer, also potent antimalarial in vitro and in vivo properties in the nanomolar range.65
In turn, Chiang proved that ACF can be an effective pharmaceutical for therapy in pathogenic protein diseases.66 Additionally, ACF exhibited potential anticancer activity in mice and was approved by FDA for clinical trials.67
The Joubert group presented the synthesis of artemisinin–acridine hybrids (82a–e).68 The intermediate 9-aminoacridines (80a–e) were prepared by reaction of 6,9-dichloro-2-methoxyacridine (79) with the appropriate amine in the presence of K2CO3 in DMF. The desired final products (82a–e) were obtained using a microwave-assisted radiation method (150 W, 45 °C) by reacting compounds (80a–e) with 2-bromo-(10β-dihydroartemisinoxy)ethane (81) in ACN in the presence of NaHCO3 and KI as catalyst (Scheme 10).68
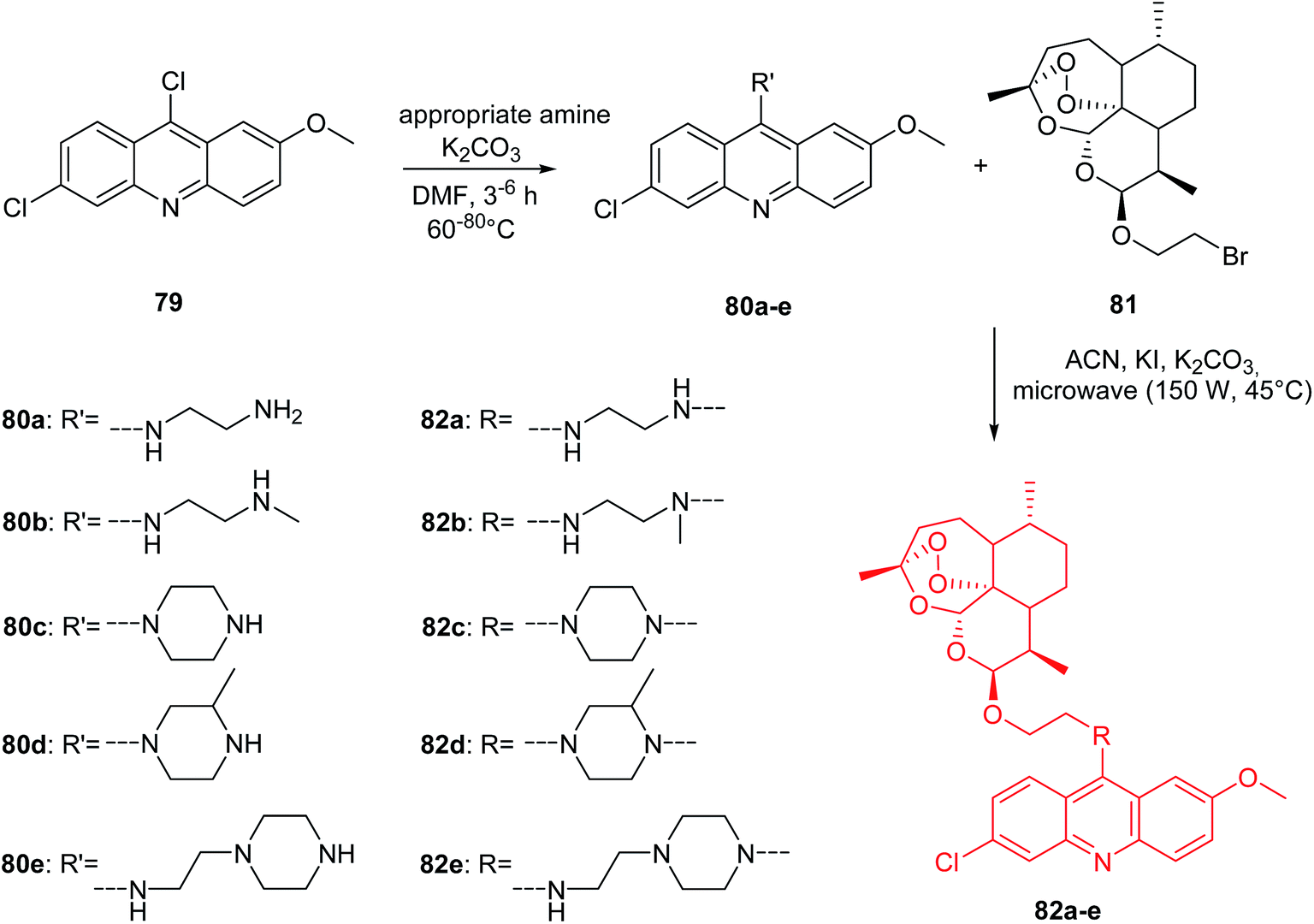 | ||
| Scheme 10 Synthesis of compounds (82a–e).68 | ||
Compounds (80a–e and 82a–e) were assayed in vitro for antimalarial activity against NF54 and Dd2 strains of Plasmodium falciparum. All prepared analogues (80a–e and 82a–e) were active and showed more antimalarial activity against NF54 than Dd2 inhibition. Derivative (82a), containing an ethylenediamine linker, exhibited the most potent activity with IC50 values equal to 2.6 nM against NF54 and 35.3 nM against Dd2. Interestingly, (82a) had 7-fold higher antimalarial potency than chloroquine (CQ) against both the NF54 and Dd2 strains, with high selective action towards the parasitic cells.68
Furthermore, cytotoxic and anticancer effects of analogues (80a–e and 82a–e) on the CHO (Chinese hamster ovarian) and HeLa cell lines, respectively, were assessed by MTT assay. Derivatives (80a–e and 82a–e) have been found to be less cytotoxic compared to reference compound emetine. Hybrids (82a,d) showed stronger anticancer activity against the HeLa cells than the reference compounds (CQ and melphalan).68
Bharathi et al. presented the synthesis, solvatochromic behaviour and larvicidal activity of substituted ethyl 10-chloro-4-(3,4-dimethoxyphenyl)-2-hydroxy-12-phenyl-1,4,5,6-tetrahydro-benzo[a]-acridine-3-carboxylates (85a–e).69 Derivatives (85a–e) were obtained by reaction of (E)-7-chloro-2-(3,4-dimethoxybenzylidene)-9-phenyl-3,4-dihydroacridin-1(2H)-ones (83a–e) with ethyl acetoacetate (84) in the presence of 10% ethanolic NaOH (Scheme 11).69
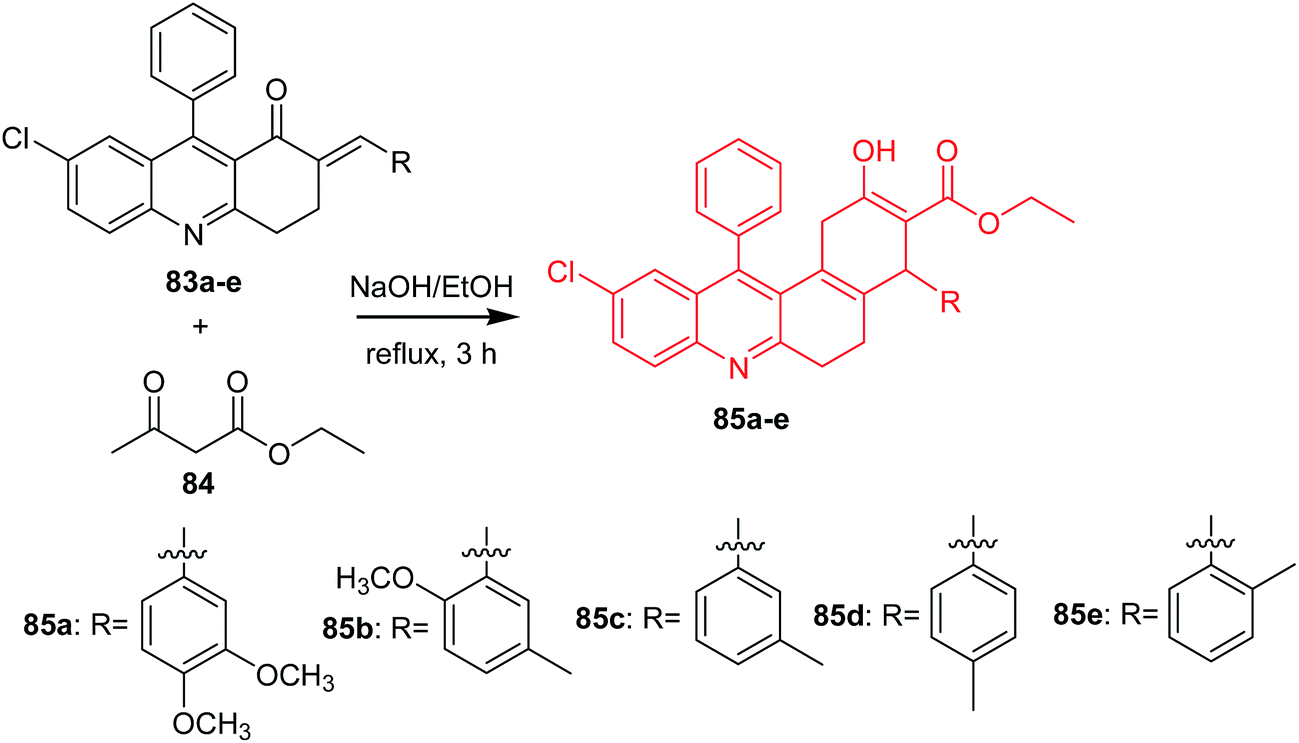 | ||
| Scheme 11 Synthesis of acridine derivatives (85a–e).69 | ||
Derivatives (85b and 85e) showed good larvicidal activities against A. stephensi (LC50 126.3 and 108.17 mg L−1, respectively) and H. maculata (LC50 111.57 and 241.05 mg L−1, respectively). Furthermore, analogues (85b and 85e) were tested against non-target aquatic species (S. annulatum Fabricius, Z. petiolatum Rambur). The results showed that the prepared compounds are non-toxic. Derivatives (85b and 85e) ware active against S. annulatum Fabricius and Z. petiolatum Rambur (with % mortality values equal to 0.4 and 0.8 mg L−1, % mortality values equal to 0.3 and 0.7 mg L−1, respectively).69
Medapi et al. presented the synthesis of p-phenylenediamine-linked acridine derivatives.70 Acridine derivatives (91a–l and 92a–x) were synthesized according to Scheme 12. The synthetic pathway started from the Ullmann condensation reaction of 2-chlorobenzoic acid (86) and different para-substituted anilines (87a–c) to afford 2-(p-tolyl/4-chlorophenyl/phenylamino)benzoic acid derivatives (88a–c).
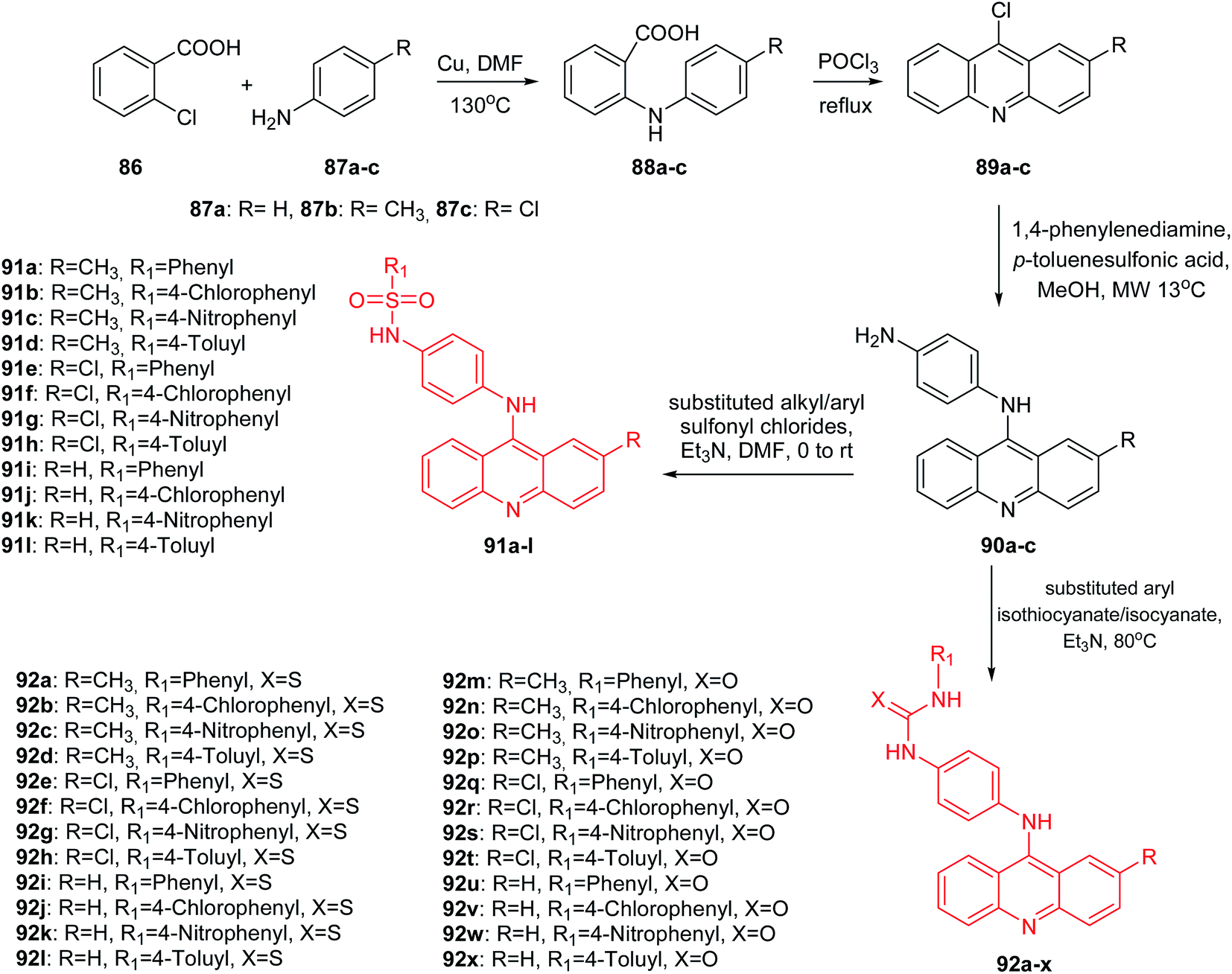 | ||
| Scheme 12 Synthesis of substituted N-[4-(substituted-9-acridinyl)amino]phenyl analogues (91a–l, 92a–x).70 | ||
Compounds (88a–c) easily participated in the cyclization reaction in the presence of POCl3 at reflux and afforded the corresponding 9-chloro-2-substituted acridine derivatives (89a–c). Then, N1-(2-methyl/2-chloro/simple acridin-9-yl)benzene-1,4-diamine derivatives (90a–c) were received in the microwave-assisted reaction of derivatives (89a–c) with p-phenylenediamine in the presence of PTSA (p-toluenesulphonic acid) as the catalyst in methanol. Finally, the acridine derivatives (90a–c) were reacted with different substituted aryl sulfonylchlorides and aryl isothio/isocyanates to afford sulphonamide (91a–l) and thio/urea (92a–l, 92m–x) derivatives.70
These derivatives are structurally similar to m-amsacrine (3), which acts as a eukaryotic topoisomerase II poison and as an MTB topoisomerase I inhibitor with an IC50 of 10 μM. An MTB topoisomerase I relaxation assay indicated that molecules (91a–l and 92a–x) showed inhibition at 50 μM. Thus, a methoxyl group present in the phenyl ring linker of m-amsacrine is of importance. All synthesized compounds (91a–l and 92a–x) were tested for Mycobacterium tuberculosis (MTB) DNA gyrase activity. Derivatives (91a–l and 92a–x) showed good activity with IC50 values in the range of 5.2 to 33.9 μM. The most active were sulphonamide (91a–l) followed by thiourea (92a–l) derivatives, and the least active were urea (92m–x) derivatives. The most potent was derivative (91b) with a CH3 group at the 2-position of the acridine moiety and an unsubstituted phenyl ring. Furthermore, analogue (91b) was shown to be significantly potent with an MTB MIC (IC50 = 6.59 μM) and 11.78% inhibition at 50 μM against mouse macrophage cell line RAW 264.7.70
Muscia et al. described the synthesis of acridine derivatives (95a–b and 96a–b) as potential anti-tuberculosis agents.71 The method is based on the use of the microwave-assisted Friedländer reaction. Compounds (95a–b) were prepared by reaction of cyclohexanone 94 with 2-amino-5-chlorobenzophenone (93a) or 2-amino-5-nitro-benzophenone (93b) in the presence of TFA as a catalyst in an MW oven at constant power (400 W) (Scheme 13). Analogues (96a–b) were obtained by microwave-assisted reaction of 5,5-dimethylcyclohexane-1,3-dione (58) with (93a) or (93b) (Scheme 13).71
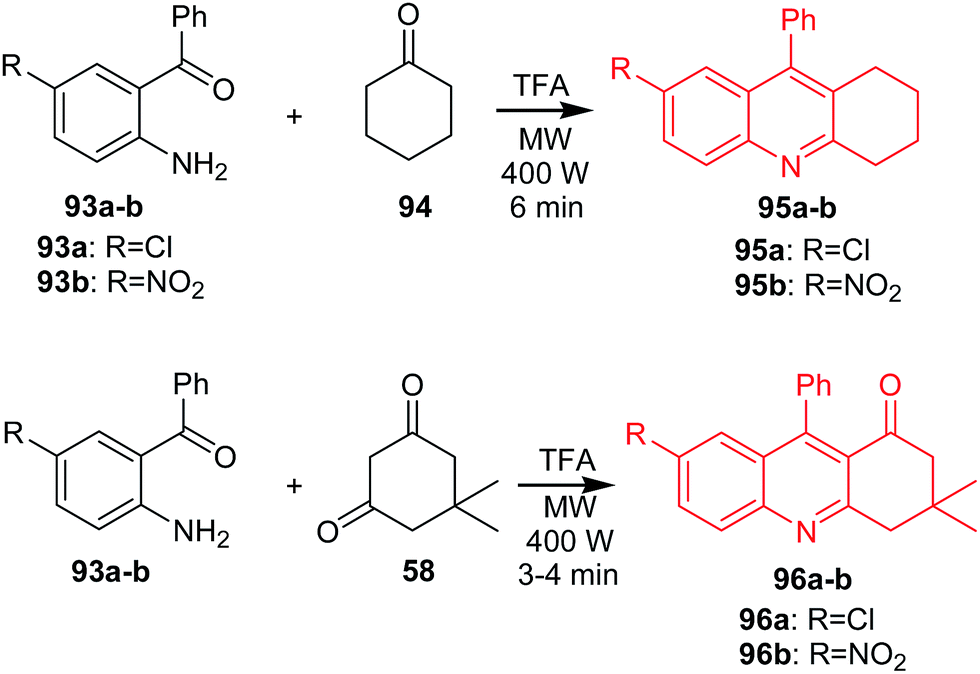 | ||
| Scheme 13 Synthesis of acridine derivatives (95a–b, 96a–b).71 | ||
Synthesized compounds (95a–b) and, synthesized compounds (95a–b and 96a–b) were assayed for inhibition of Mycobacterium tuberculosis H37Rv (Mtb). Derivative (95b) revealed the most potent activity with IC50 values equal to 44.35 μM.71
A different method of obtaining derivatives of 1,8-dioxoacridine (100a–l) was presented by Kaya's group.72 In the first step of the synthesis, the condensation of cyclohexane-1,3-dion (97) or dimedone (58) with p-methoxybenzaldehyde (98) afforded the tetraketone intermediates (99a or 99b). Reaction of tetraketone (99a–b) with primary aromatic amines using the Dean-Stark apparatus in the presence of acetic acid–sodium acetate in toluene gave acridine derivatives (100a–l) (Scheme 14).72
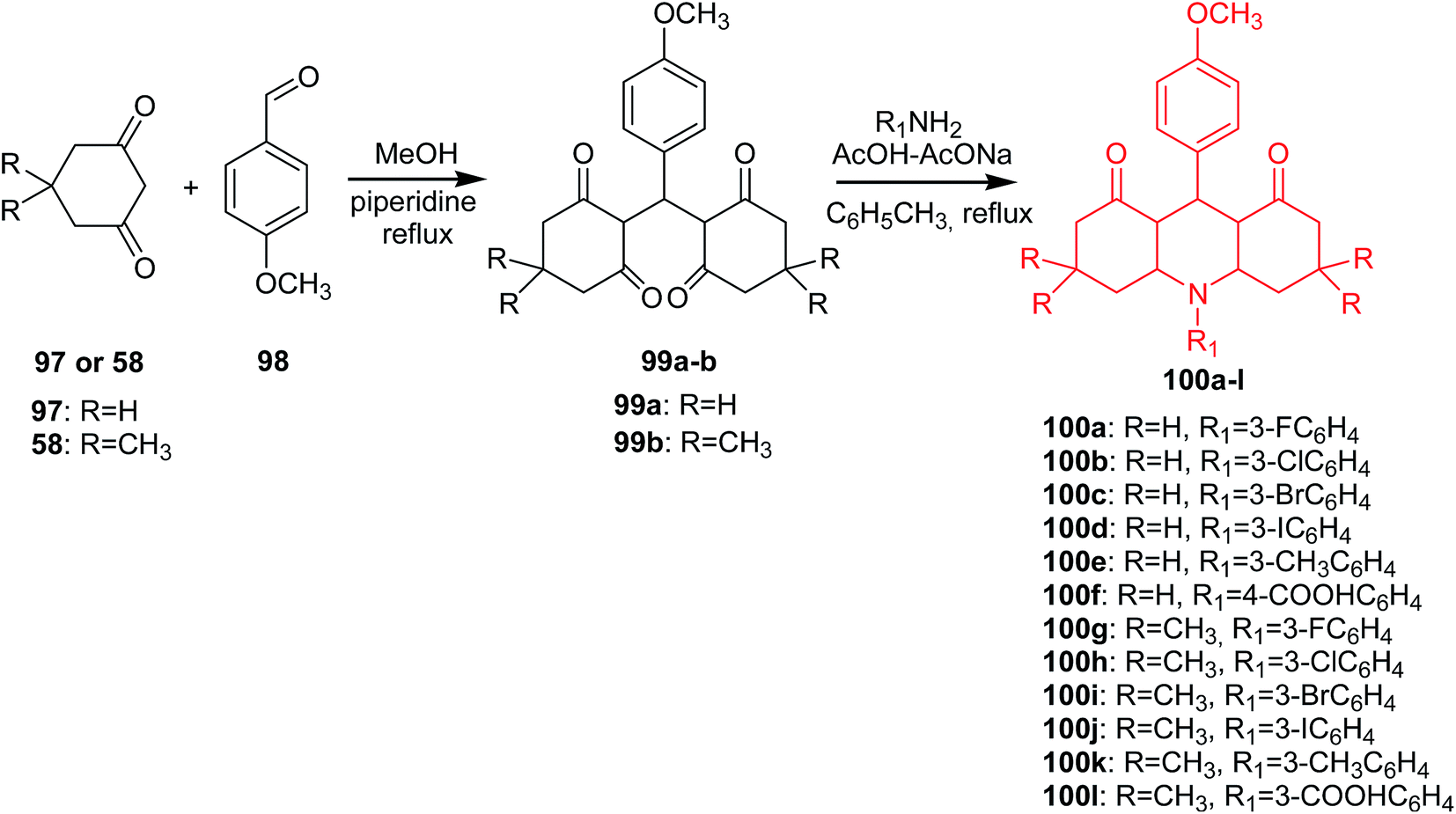 | ||
| Scheme 14 Synthesis of derivatives of 1,8-dioxoacridine (100a–l).72 | ||
All the synthesized compounds (100a–l) were tested for antibacterial and antifungal in vitro activity. Most of the analogues showed significant activity against E. coli, P. aeruginosa, S. enteritidis and S. aureus. Furthermore, compound (100k) showed more effective activity against S. enteritidis than the conventional reference antibiotic erythromycin. Additionally, all derivatives had moderate antifungal activity against C. albicans and C. glabrata.72
Markovich et al. presented the synthesis of 2-(4-methyl-1,3-thiazol-5-yl)ethyl esters of acridone carboxylic acids (103a–g) by the transesterification of the corresponding butyl esters of acridone carboxylic acids (101a–g) with 5-(2-hydroxyethyl)-4-methylthiazole (102) in the presence of sodium methoxide as the catalyst (Scheme 15).73
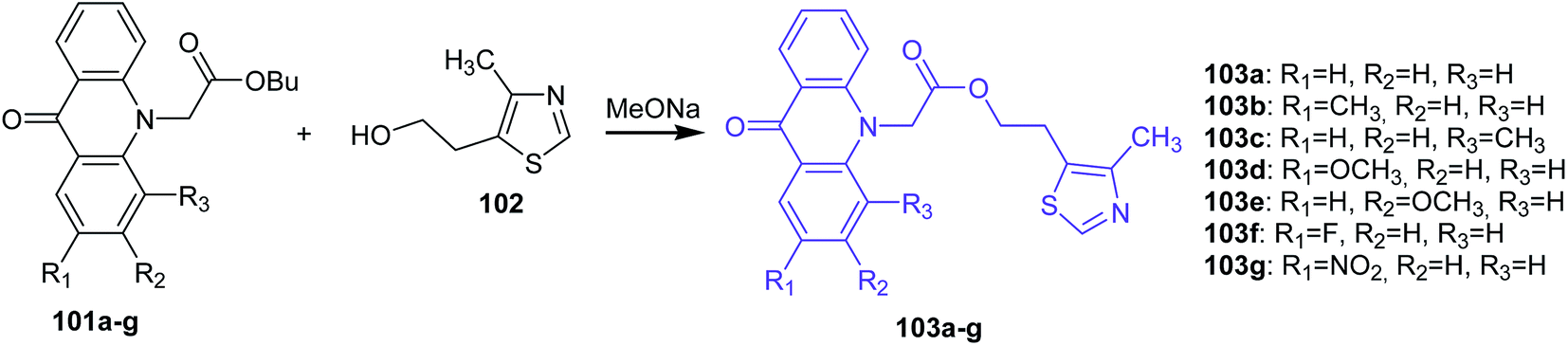 | ||
| Scheme 15 Synthesis of acridone derivatives (103a–g).73 | ||
Analogues of acridone that have a carboxyl group directly bonded to the aromatic ring of acridone also exhibit biological activity.74–76 Therefore, 2-(4-methyl-1,3-thiazol-5-yl)ethyl esters of 2(4)-carboxyacridone (106–107) were prepared.73 The reaction was carried out at 70 °C for 1 h using 2- or 4-carboxyacridone acid chloride (104–105) and 5-(2-hydroxyethyl)-4-methylthiazole (102) (Scheme 16).73
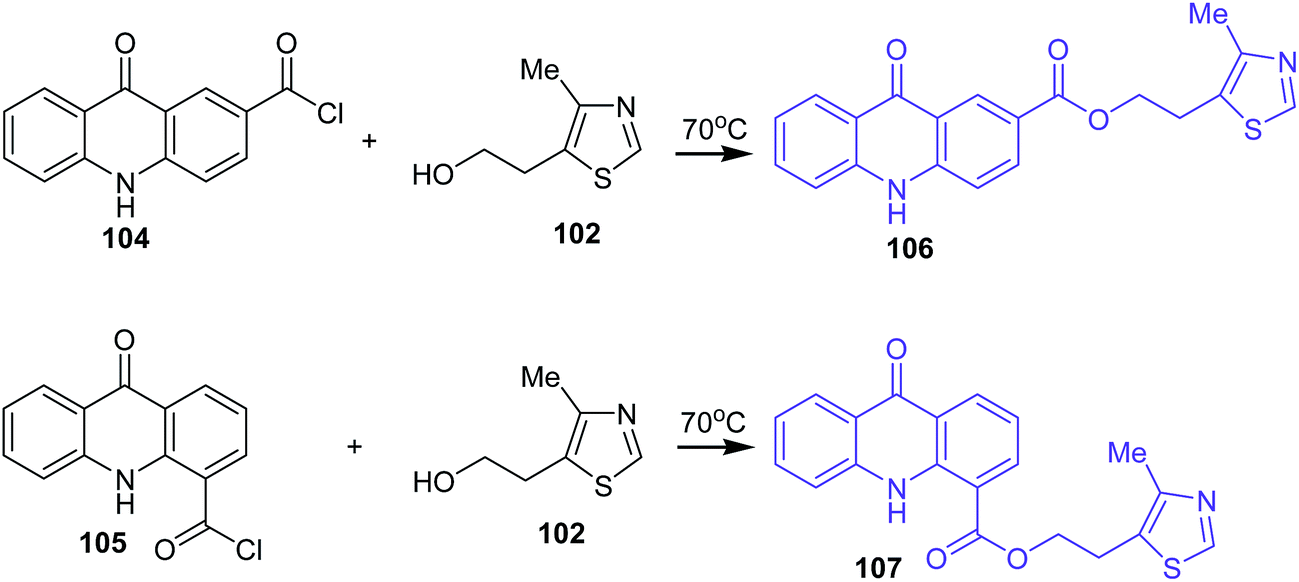 | ||
| Scheme 16 Synthesis of acridone derivatives (106–107).73 | ||
Compounds (103a–b and 107) were tested for antibacterial activity against strains of microorganisms (E. coli, Ps. aeruginosa, Pr. vulgaris, S. aureus, B. subtilis, Candida albicans). The prepared analogues were active and showed inhibition against all microorganisms. Moreover, derivative (103b) with a CH3 group at the 2-position of the acridone moiety showed slightly higher activity against Candida albicans in comparison to Rivanol.73
Bhardwaj et al. synthesized new acridone derivatives. Acridone was coupled via a linker group to an anti-inflammatory (AI) drug such as ibuprofen (109), (S)-naproxen (110), acetyl salicylic acid (108) or chlororofecoxib analogue (114).77 The intermediate N10-(2-hydroxyethyl)acridone (112) was prepared by reaction of acridone (111) with ethylene carbonate in the presence of KOH. Acridone-ibuprofen (113a), acridone-(S)-naproxen (113b), acridone-acetyl salicylic acid (113c) or acridone-chlororofecoxib (115) derivatives were prepared by the condensation of compound (112) with ibuprofen (109), (S)-naproxen (110), acetyl salicylic acid (108) or 4-(trifluoromethyl)benzenesulphonyl chloride (114), respectively, in the presence of N,N′-dicyclohexylcarbodiimide (DCC) with 4-dimethylaminopyridine (DMAP) as catalyst in dry dichloromethane (DCM). Additionally, the Bhardwaj group synthesized analogues (117a–b) by the condensation of acridone (111) with acid chloride derivatives of ibuprofen or (S)-naproxen (116a–b) in the presence of NaH in dry DMSO (Scheme 17).77 Conjugates (113a–c, 115, and 117a–b) were assayed in vitro as COX-1 and COX-2 inhibitors. All prepared analogues (113a–c, 115, and 117a–b) showed more potent inhibition of COX-2 (IC50 = 0.59–21.2 μM range) than COX-1 (IC50 = 18.0 to 88.1 μM range). Furthermore, derivatives (113a–c, 115, and 117a–b) showed a higher COX-2 selectivity index (SI) than AI drugs (108–110 and 114). This may be caused by the fact that the derivatives (113a–c, 115, and 117a–b) have larger molecular volumes (353–412 Å3 range) than the smaller molecular volumes of the AI drugs (155–214 Å3 range). Thus, it is credible that the larger size of the conjugates of acridone may hinder their entry into the smaller COX-1 binding site (316 Å3), thus enabling selective entry into the larger COX-2 binding site (394 Å3). The most potent and selective COX-2 inhibitor was compound (113a) (IC50 = 0.67 μM; SI = 110.6). Additionally, studies have shown that fluorescence emissions of conjugates (113a–c, 115, and 117a–b) (αem = 415–445 nm) were not suitable for fluorescent imaging of cancer cells that over-express the COX-2 isozyme.77
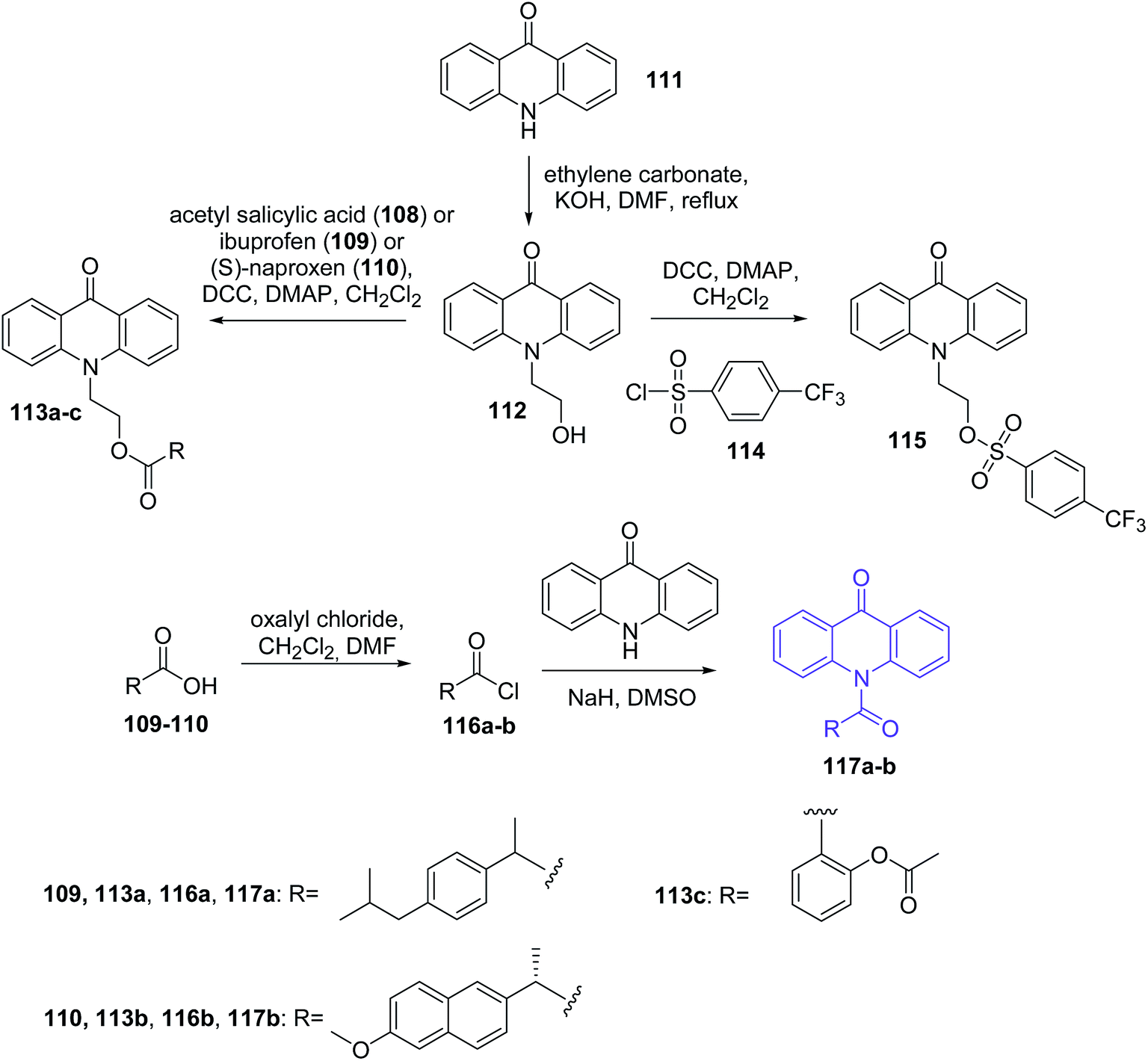 | ||
| Scheme 17 Synthesis of acridone derivatives (113a–c, 115, 117a–b).77 | ||
Anticancer activity of acridine/acridone analogues
The effectiveness of heterocyclic, tricyclic and planar arrays to intercalate between base pairs in the double-stranded DNA structure,78,79 and the ability of acceptor/donor nitrogen are two major considerations for the use of acridines/acridones as potential anti-cancer compounds.80 Additionally, acridine/acridone derivatives prevent proper functioning of tumour cells, including by dysfunction relevant to the enzymes that control the topology of DNA function: topoisomerase, telomerase, and cyclin-dependent kinases.81 There are also reported 125I-radiolabeled acridine derivatives investigated both in vitro and in vivo against melanoma.82In this survey, we describe the latest acridine and acridone derivatives as antitumour agents.
Stark and Dermawan reported a method for treating subjects having cancer that has developed resistance to tyrosine kinase inhibitors by using tyrosine kinase inhibitor (TKI) and NF-κB inhibitor.83 NF-κB activation seems to be involved in the development of resistance to TKIs and other chemotherapeutic agents, and accordingly this resistance can be overcome by inhibiting NF-κB activation. The combination of the TKI and the NF-κB inhibitor provides a synergistic anticancer effect that overcomes the TKI resistance that has developed. For example, researchers administered to a subject a therapeutically effective amount of erlotinib (TKI inhibitor) and quinacrine (76) (NF-κB inhibitor) against erlotinib-resistant NSCLC cell lines (A549, H1975, and H1993). Results showed that the addition of quinacrine (76) to erlotinib treatment inhibits colony formation. Moreover, addition of quinacrine (76) to erlotinib treatment induced significant apoptosis in A549 and H1975 cells.
Arya et al. presented the synthesis of acridine derivatives (120a–h, 121a–d, 122a–d, and 123a–d).15 Compounds (120a–h) were prepared by condensation of appropriate 9-aminoacridine derivatives (118a–h) with 9,10-dihydroanthracene-9,10-α,β-succinic anhydride (119) in tetrahydrofuran at room temperature (Scheme 18). To synthesize analogues (121a–d, 122a–d, and 123a–d), the authors used microwave-assisted condensation of 9-aminoacridine derivatives (118a–d) with phthalic anhydride (124) or cis-1,2,3,6-tetrahydrophthalimide (125) or 2,5-pyrroledione (126) (Scheme 19).15
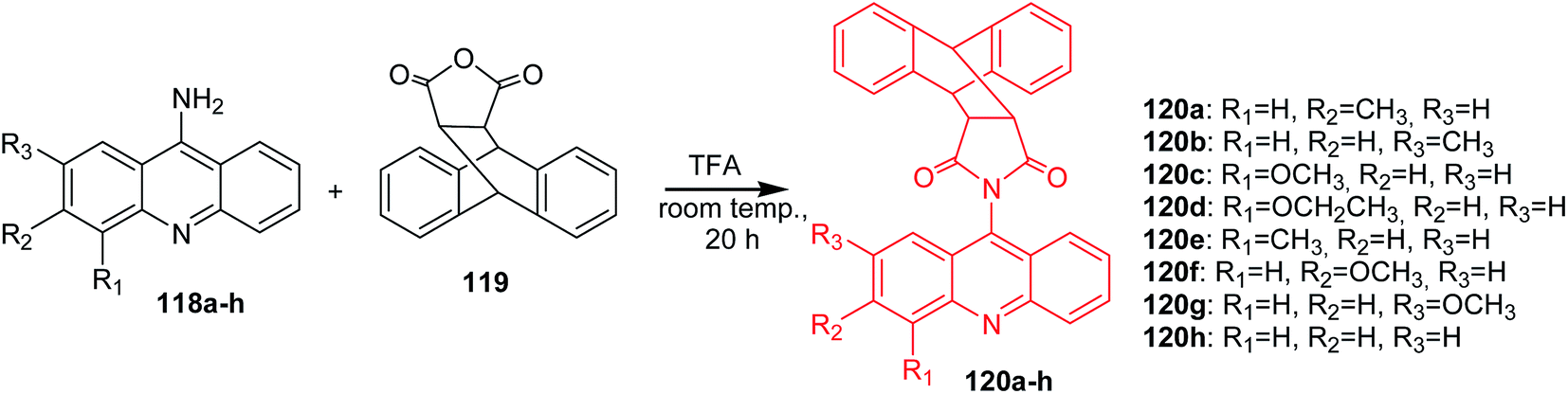 | ||
| Scheme 18 Synthesis of acridine derivatives (120a–h).15 | ||
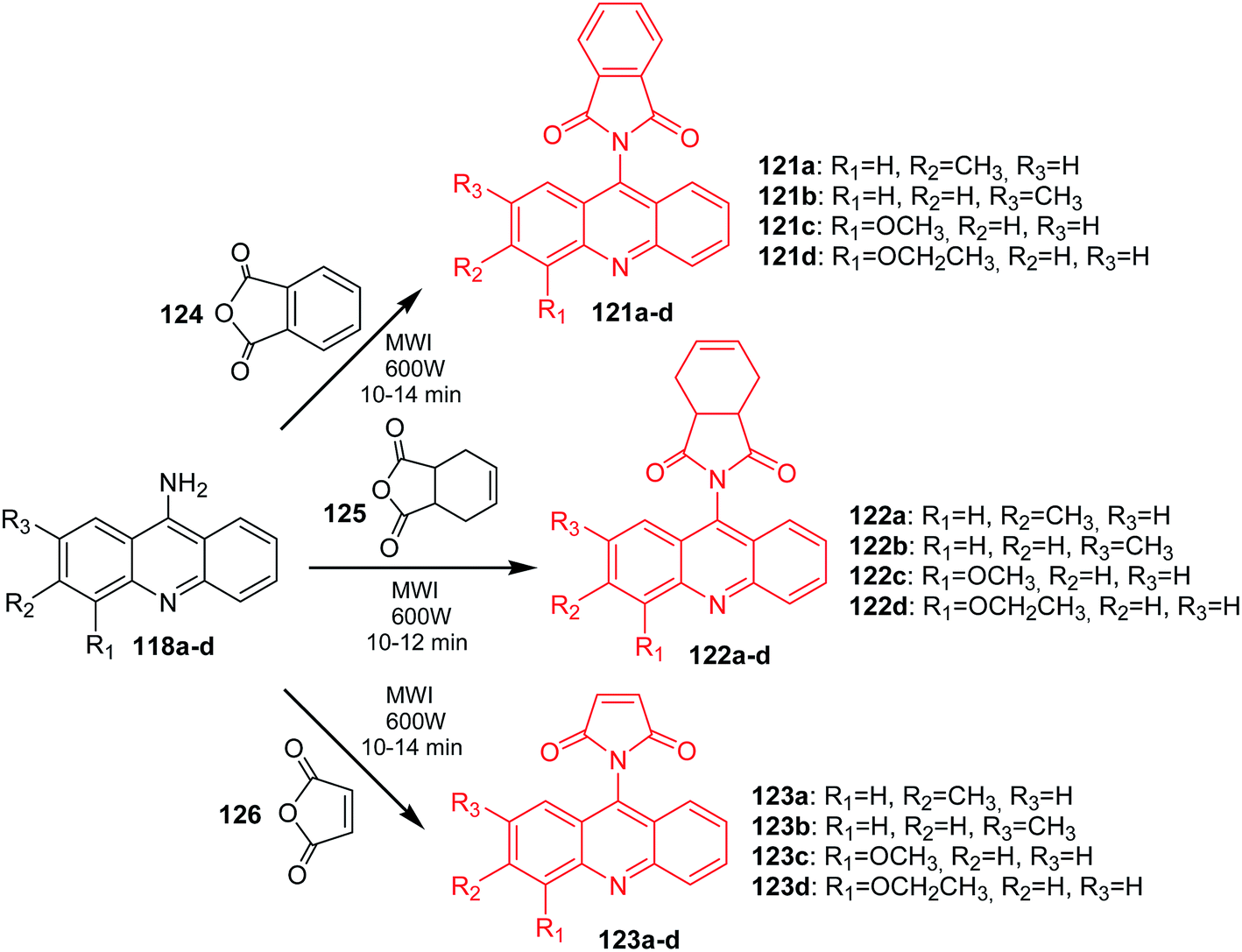 | ||
| Scheme 19 Synthesis of acridine derivatives (121a–d, 122a–d, 123a–d).15 | ||
All the obtained compounds (120a–h, 121a–d, 122a–d, and 123a–d) were tested for in vitro anticancer activity against human cancer cell lines (T47D, NCl H-522, HCT-15, PA-1, HepG-2). The most active was derivative (120a) against T47D (IC50 = 5.4 μM), (120g) against NCl H-522 (IC50 = 4.2 μM), (121a) against HepG-2 (IC50 = 4.5 μM) and (123b) against HCT-15 (IC50 = 2.4 μM). The structure–activity analysis showed that compounds containing a 7- or 8-substituted acridine moiety by an electron-donating group were more active. This may be due to more effective interaction of these molecules with the DNA compared to other derivatives.15
Salem et al. described thiazolidinone-acridines as new potential anticancer agents.84 These compounds exhibited DNA-binding abilities and topoisomerase I and II inhibition. The most active derivative was 2-(acridin-9-yl)imino-3-diphenylamino-1,3-thiazolidin-4-one (130) (Scheme 20), which inhibited topoisomerase II at the concentration of 5 μM. Its synthesis included addition of 1,1-diphenylhydrazine to acridin-9-yl isothiocyanate (127) to form the respective thiosemicarbazide (128). In the next stage, the thiocarbonyl derivative underwent S-alkylation with methyl bromoacetate to intermediate (129) followed by cyclization to the final product (130). Of note, replacement of methyl bromoacetate by bromoacetyl bromide proceeded unselectively, and 3-(acridin-9-yl)-2-(2,2-diphenylhydrazono)-1,3-thiazolidin-4-one (131) was also isolated. The regioisomeric product (131) demonstrated lower activity against HL-60 cancer cells compared to (130).84
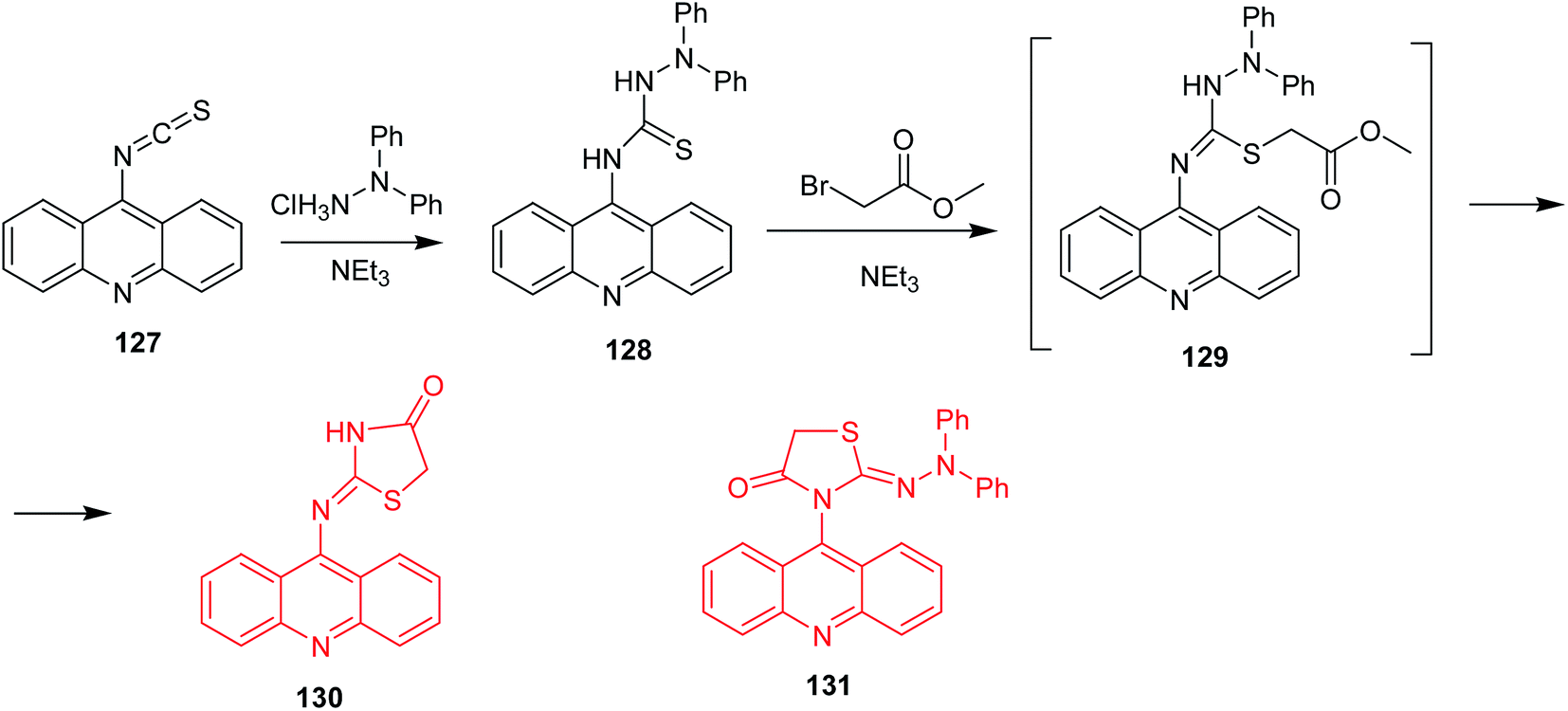 | ||
| Scheme 20 Synthesis of 2-(acridin-9-yl)imino-3-difenylamino-1,3-thiazolidin-4-one (130).84 | ||
Gao et al. reported the synthesis of benzimidazole acridine derivatives (136a–p).85 9-Chloroacridine derivatives (134a–l) were obtained by cyclization of the anthranilic acids (133a–l), which were synthesized by the Ullmann condensation. Then, the reaction of 2-aminomethyl benzimidazole (135) or compounds (137a–b) with the appropriate derivatives of 9-chloroacridine (134a–l) gave the desired final products (136a–p) (Scheme 21).85
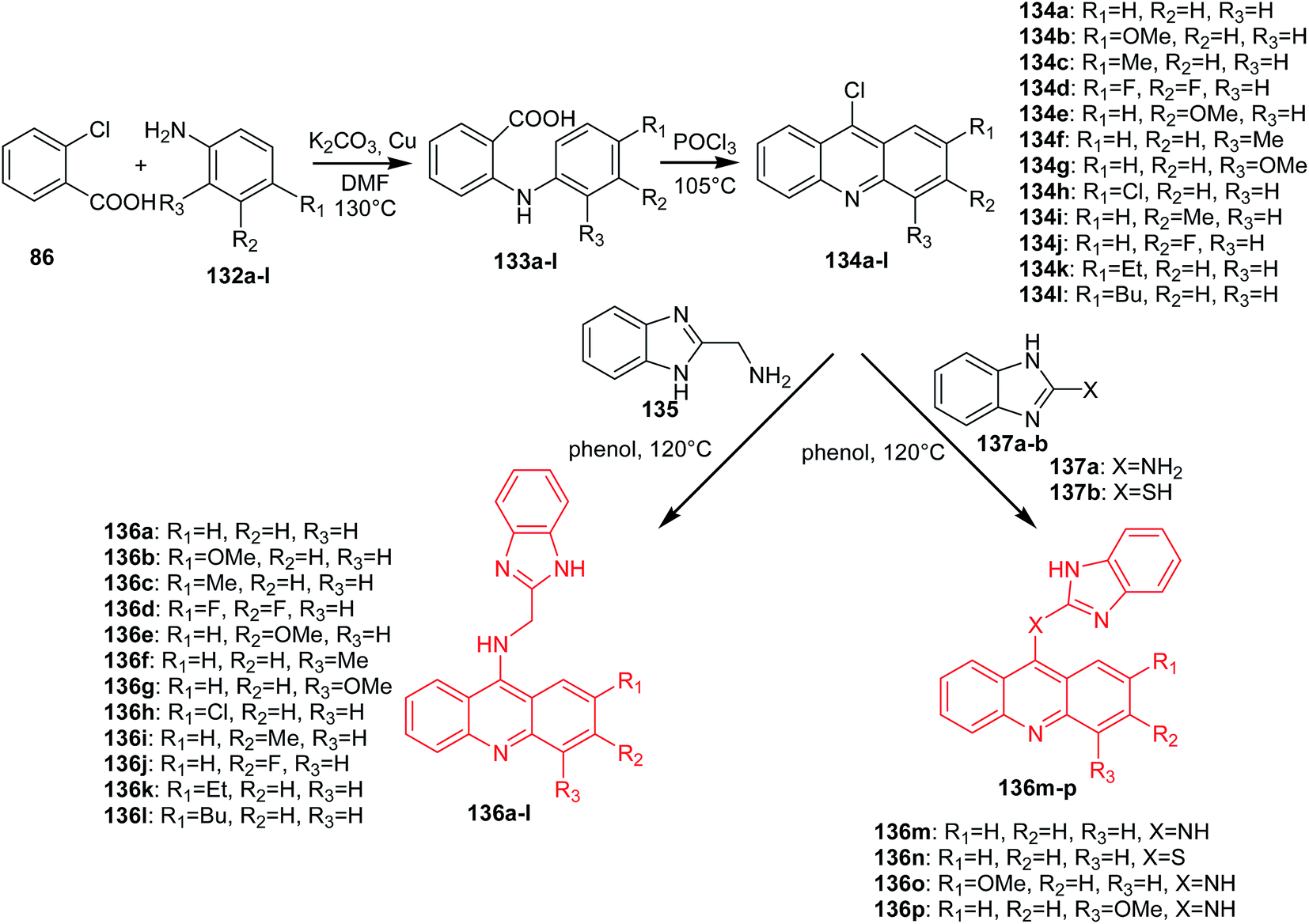 | ||
| Scheme 21 Synthesis of acridine derivatives (136a–p).85 | ||
Analogues (136a–p) were subjected to MTT assay, which showed that most of them had good antiproliferative activity. Compound (136l) had the highest activity against K562 (IC50 = 2.68 μM) and HepG-2 (IC50 = 8.11 μM) cells. Moreover, the derivative (136l) inhibited topo I activity, as well as inducing apoptosis in K562 cell lines through a mitochondrial pathway.81 The structure–activity analysis showed that the substitution of the acridine moiety and the linker between the benzimidazole ring and acridine moiety had a great effect on the antitumour activity. A look at the structures of these molecules shows that substitution at position 2 of the acridine moiety with a butyl group makes this molecule (136l) more active than compounds containing a 2-substituted acridine moiety by ethyl (136k) or methyl (136c) groups. In turn, analogue (136a), with no substituents, displayed similar activity to (136c), with a methyl substituent. Thus, both the size and length of substituents greatly influenced the cytotoxicity. Additionally, the electron-negativity of the substituents also significantly affected the anticancer activity. For example, molecule (136b), with a 2-methoxy substituent, was approximately 3-fold more potent than a compound containing a 2-methyl group (136c) or a 2-chloro group (136h). Moreover, the linker between the benzimidazole ring and acridine moiety is also important in the cytotoxicity, which can be seen from the IC50 value of analogues (136a,m,n, and o), among which derivative (136o), containing an NH-benzene-linker, showed the best anticancer activity.85
Lang et al. synthesized the acridine derivatives (141a–h) as potent DNA-binding antitumour agents.30 First, an Ullmann coupling of benzoic acids (86, 138) with anilines (139a–d) in DMF using Cu as the catalyst produced compounds (140a–f), which were refluxed in POCl3 to afford the 9-chloroacridine derivatives (141a–f). The reaction of (141a–f) with substituted benzylamines (142a–c) in the presence of KI and K2CO3 in absolute ethanol gave compounds (143a–h) (Scheme 22).30
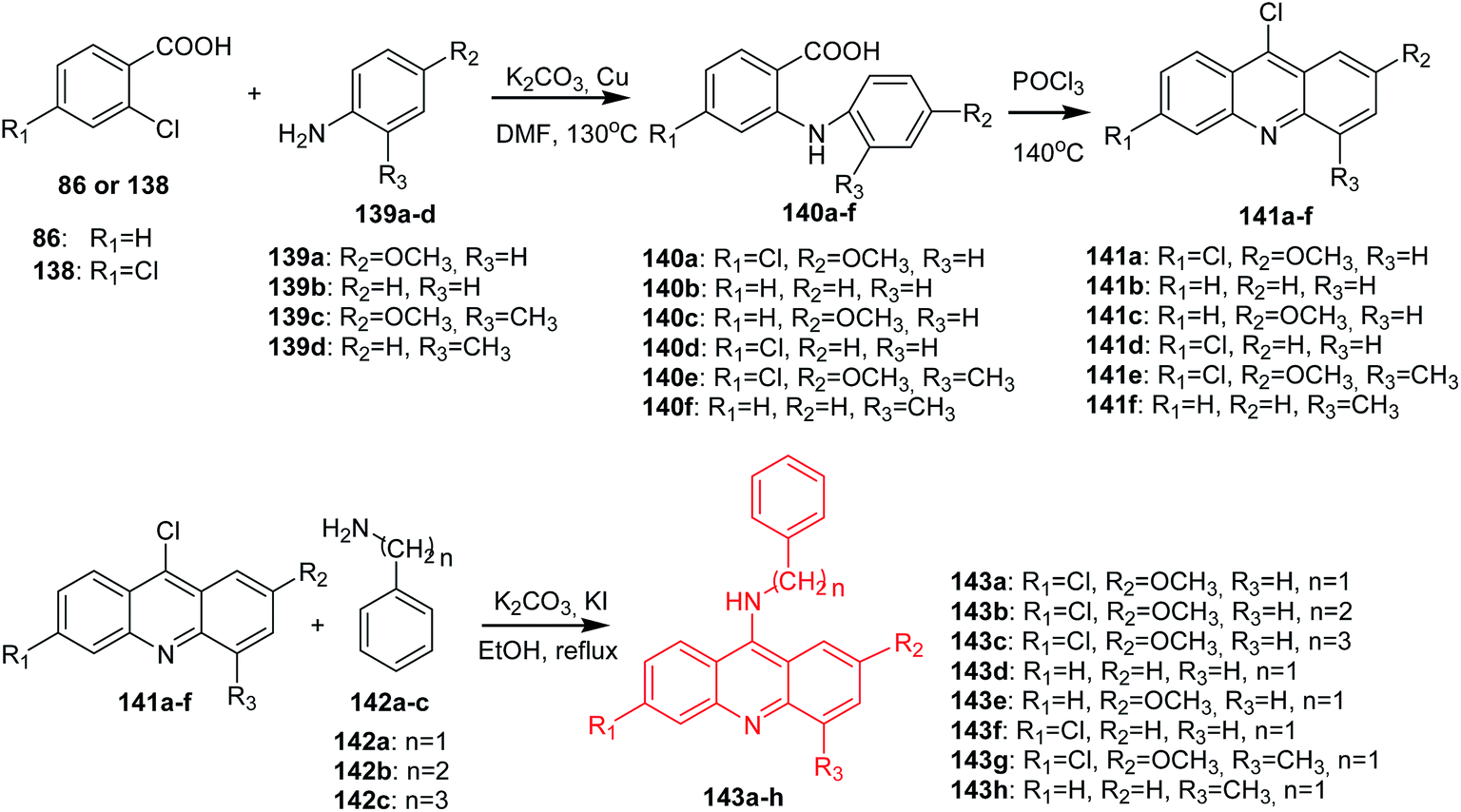 | ||
| Scheme 22 Synthesis of acridine derivatives (143a–h).30 | ||
All synthesized compounds were assayed for inhibition of K562 leukaemia cells and hepatoma HepG-2 cells. These derivatives also exhibited good results in activity against K562 and HepG-2, among which analogue (143e) showed the best antitumour activity against both cell lines. Furthermore, fluorescence spectroscopy studies revealed that most of the derivatives can interact with DNA. Compound (143e) displayed the highest DNA-binding capability and inhibited topoisomerase I activity.30
In turn, Gellerman described the synthesis of N-substituted 9-aminoacridine and bis-acridine derivatives containing electron-withdrawing groups (EWG) or electron-donating groups (EDG), including amino acid residues as potential anticancer agents.86 These derivatives displayed good antitumour activity against the cancer cell lines MDM-MD-A31, MCF-7, HT29, OVCAR8, NCI-ADR, MCF-7mito and HI 299, and comparison with commercial 9-aminoacridine and m-amsacrine (3) drugs. Compound 2-(acridin-9-ylamino)-3-ethoxy-5,8-dihydroxynaphthoquinone revealed the most potent cytotoxic activity against all above mentioned cell lines than the anticancer drug m-amsacrine (3), probably due to enhancement of biologically important chelating properties, possibly leading to formation of more powerful DNA damaging reactive species.
Li et al. developed a procedure for the preparation of novel analogues of 9-amino-acridine (146a–d).87 In a first step, N-phenylanthranilic acid (133a) was prepared by Ullmann reaction. Compound (133a) was stirred in POCl3 to afford the 9-chloro-acridine (134a). Then, acridine (134a) by reaction with phenol was converted to 9-phenoxyacridine (144), which was reacted with the corresponding amines (145) to give derivatives (146a–d) (Scheme 23).87
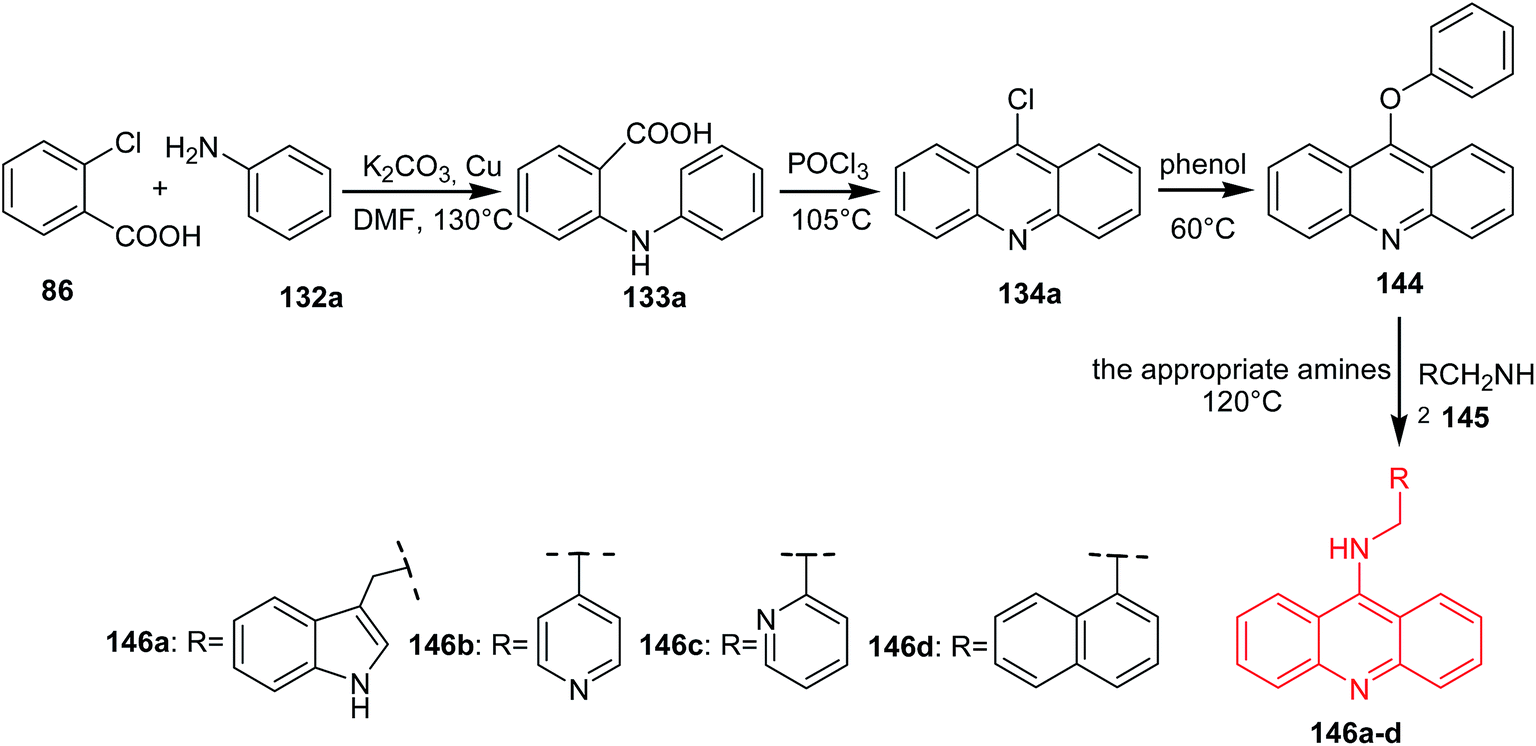 | ||
| Scheme 23 Synthesis of acridine derivatives (146a–d).87 | ||
The synthesized compounds (146a–d) were tested for their antiproliferative activity against K562 and HepG-2 cells. The best activity against both cell lines was shown by derivative (146c) through binding with DNA and inhibiting topoisomerase I activity.87
Roe et al. presented the synthesis of hybrid acridine-HSP90 ligand conjugates as telomerase inhibitors.88 The intermediate G4-azide bearing fragments (149a–d) were prepared by nucleophilic coupling of compound (147) with the amino azides (148a–d) in CHCl3 at reflux. A second intermediate GA-alkyne (151) was synthesized by reacting geldanamycin (150) with propargyl amine in DMF at room temperature. The desired final products (153a–d) were obtained by reaction of the corresponding G4-azide bearing fragments (149a–d) with compound (151) using 1:1 tert-butanol:water, in the presence of CuSO4·5H2O as a catalyst and sodium ascorbate (152) (Scheme 24).88
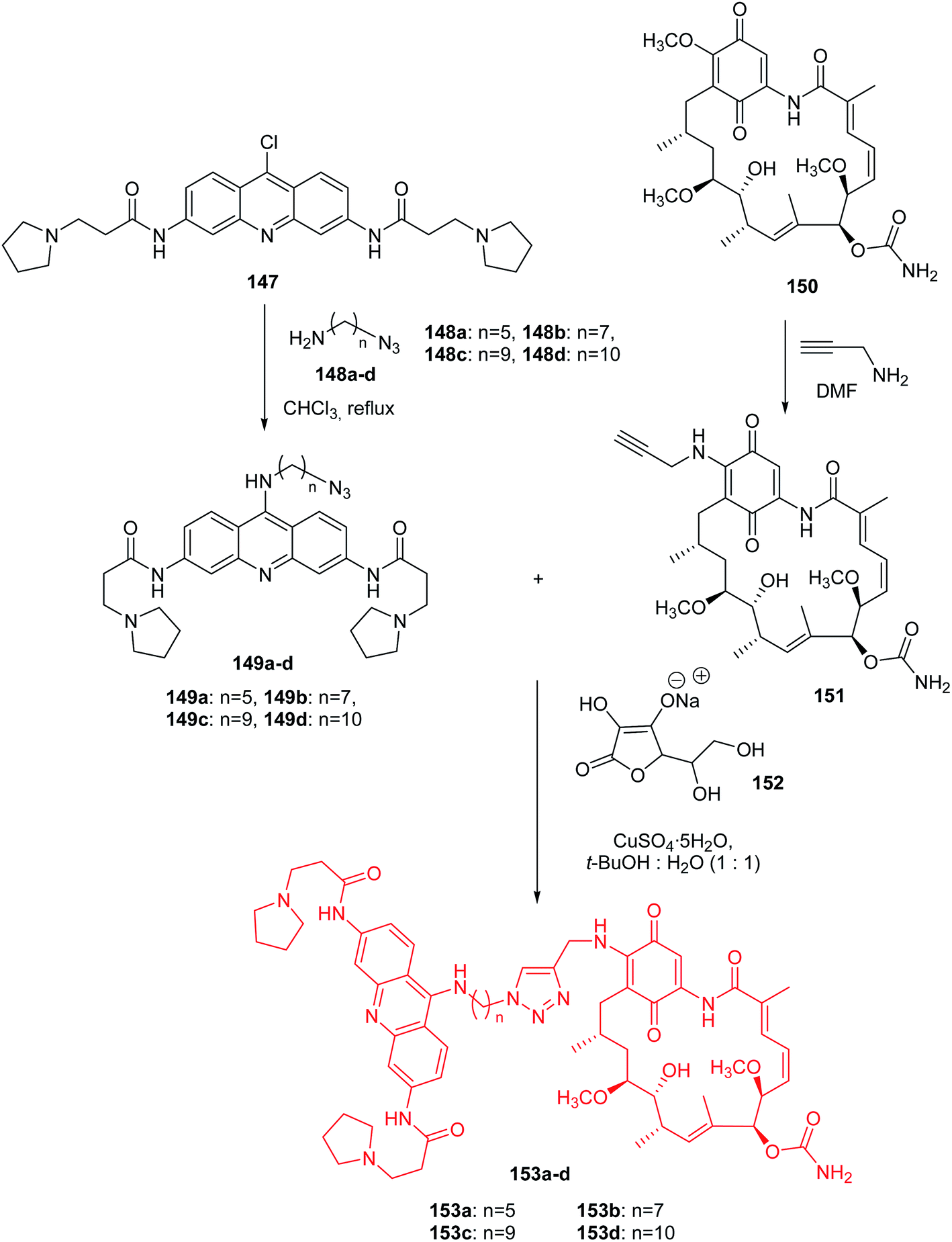 | ||
| Scheme 24 Synthesis of acridine-HSP90 ligand conjugates (153a–d).88 | ||
All synthesized compounds (153a–d) were assayed for inhibition of tumour cell growth including MCF7, A549, GIST48, and WI38. Derivatives (153a–d) showed the best activity against A549 (IC50 = 0.1–0.5 μM range). Compounds (153c–d) containing the longest linkers (n = 9, and 10, respectively) revealed better potent cytotoxic activity than analogue (153a), with the shortest linker (n = 5).88
Acridine derivatives are widely considered as potent photosensitizers for photodynamic therapy in tumour treatment. Recently, acridin-3,6-dialkyldithioureas (155) were developed (Scheme 25), where the propyl analogue (R = n-Pr) revealed the highest photocytotoxicity against the L1221 cell line (IC50 = 0.48 μM).89 The mechanism of action of these compounds was also investigated, and the results strongly suggested that lysosomes of cancer cells were significant targets. Acridin-3,6-dialkyldithioureas (155) were synthesized from 3,6-diaminoacridine (154) through relevant isothiocyanates.89
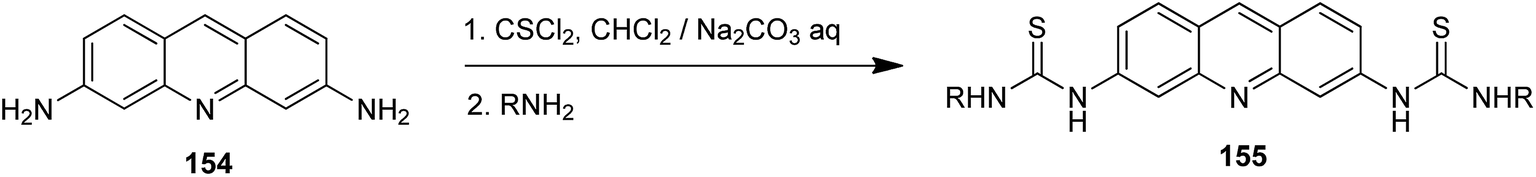 | ||
| Scheme 25 Preparation of acridin-3,6-dialkyldithioureas (155).89 | ||
Similar 3,6-bis(3-alkylguanidino)acridines were synthesized by Plsikova and co-workers.90 These derivatives exhibited DNA-binding abilities (K = 1.25–5.26 × 105 M−1) and are potent DNA-intercalating antitumor agents. It was demonstrated that analogs with longer alkyl chains were able to penetrate cell membranes and suppress cell proliferation effectively. The compound with n-hexyl chains at nitrogen of guanidine moiety proved to be the most cytotoxic.
In turn, Huo et al. presented N-(benzo[b][1,10]phenanthrolin-7-yl)-2-(4-chlorobenzoyl)hydrazinecarbothioamide,91 2-(4-methoxybenzoyl)-N-(2-methylacridin-9-yl)hydrazinecarbothioamide,92 7-p-toluene-aminobenzo[c]acridine hydrochloride93 and N-(4-methoxyphenyl)benzo[c]acridin-7-amine hydrochloride94 as potential antitumor agents against a liver cancer, a lung cancer, a gastric cancer and various tumor cell strains. These compounds revealed strong cytotoxic activity against MGC-803 (IC50 = 8.1, 11.2, 10.4 and 9.7 μg mL−1, respectively), BEL-7404 (IC50 = 24.2, 27.1, 23.1 and 20.3 μg mL−1, respectively) and NCI-H460 (IC50 = 35.3, 25.4, 28.2 and 25.4 μg mL−1, respectively). Moreover, measurements in vivo demonstrated that 7-p-toluene-aminobenzo[c]acridine hydrochloride and N-(4-methoxyphenyl)benzo[c] acridin-7-amine hydrochloride effectively inhibited the division of hepatocarcinoma cells and had a good inhibitory effect in mouse models of liver cancer.
Ramesh and Pasha described an efficient and economical synthesis of 9-aryl-hexahydro-acridine-1,8-diones (158a–k) by cyclocondensation of dimedone (58), aromatic aldehydes (156a–k) and ammonium acetate (157) in the presence of a heterogeneous catalyst SiO2–I in ethanol at 80 °C (Scheme 26).95
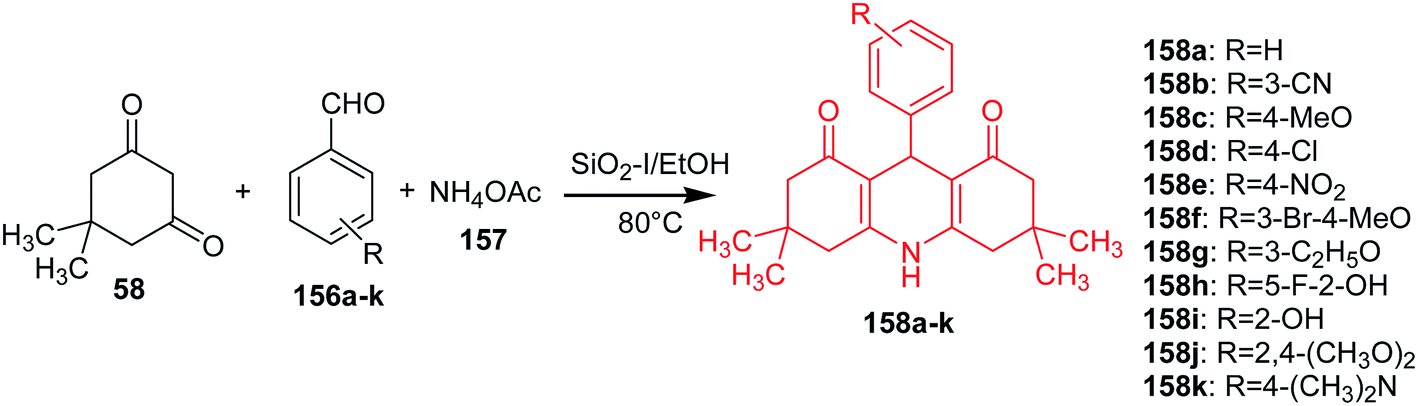 | ||
| Scheme 26 Synthesis of 9-aryl-hexahydro-acridine-1,8-diones (158a–k).95 | ||
The obtained derivatives have shown promising antitumour activity against HepG-2 and MCF-7 cells. Compounds (158b, 158f and 158j) showed good activity against HepG-2 and MCF-7, analogue (158i) towards HepG-2, and the derivative (158g) inhibited MCF-7 cells.95
Synthetic methods leading to desired acridine derivatives are still being developed and optimized. For instance, a ZrO2–TiO2 mixed oxide catalyst enabled a three-component, one-pot condensation of 5,5-dimethylcyclohexane-1,3-dione (58), 3-bromo-5-chloro-2-hydroxybenzaldehyde (159), and ethanolamine (160) to (161) (Scheme 27) in 99% yield.96 Reddy et al. described a synthesis of trans-fused 5H-chromeno[2,3-c]acridine derivatives via intramolecular aza Diels–Alder reaction catalysed by Lewis acid, followed by 1,3-hydrogen shift,97 where 5a,8,8-trimethyl-6,7,7a,8,13,13a-hexahydro-5H-chromeno[2,3-c]acridine (163) was obtained from (162) and aniline in the presence of BF3–OEt2 in 88% yield and 05:95 cis/trans ratio. Distribution of the products was in good agreement with theoretical studies.98 Wang and co-workers reported 3,6-diphenyl-9-aryl-3,4,6,7,9,10-hexahydroacridine-1,8(2H,5H)-dione derivatives received in the course of Knoevenagel, Michael, and cyclization reactions catalysed by L-proline.99 For example, 10-(4-chlorophenyl)-9-(4-methoxyphenyl)-3,6-diphenyl-3,4,6,7,9,10-hexahydroacridine-1,8(2H,5H)-dione (165) was synthesized in the reaction of 5-phenylcyclohexane-1,3-dione (164), p-methoxybenzaldehyde (156c) and p-chloroaniline in an overall yield of 57%. The product (165) gave IC50 = 82.98 μg mL−1 against HepG-2 in the MTT assay.
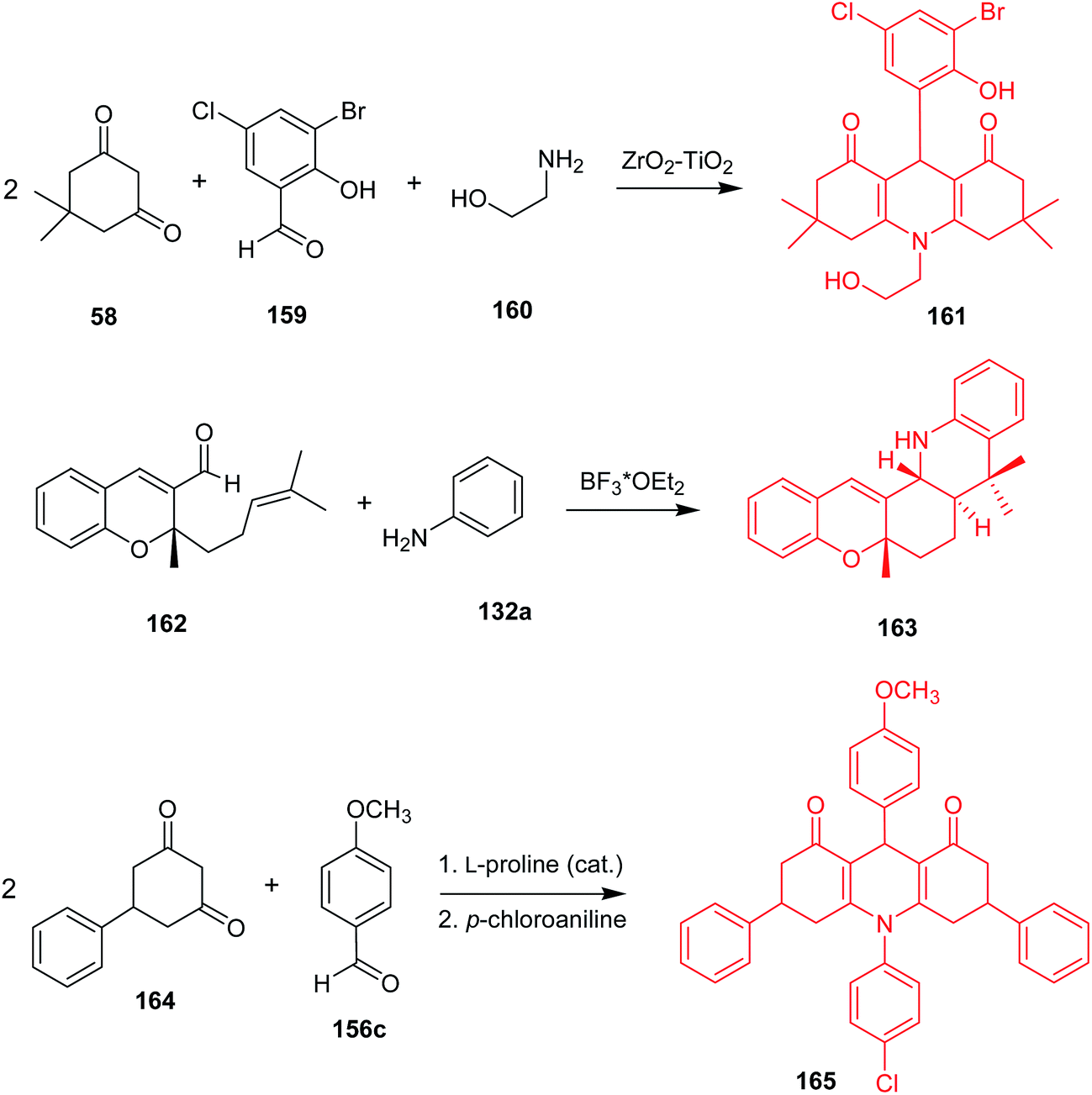
Wang et al. synthesized the 12-(3,4,5-trimethoxyphenyl)benzo-[b][1,3]dioxolo[4,5-i] acridine-6,11(5H,12H)-dione by cyclocondensation of 3,4-methylenedioxy aniline, 3,4,5-methoxybenzaldehyde, 2-hydroxy-1,4-naphthoquinone in the presence of L-proline in ethanol.100 This molecule showed good activity against HepG-2 (IC50 = 11.6 μM).
Pickard et al. developed platinum–acridine anticancer hybrid agents, where replacement of acridine moiety by benz[c]acridine caused less toxicity in vivo in mice.101
Cossio Mora et al. presented the synthesis of 1,2,3,4-tetrahydroacridine derivatives as histone deacetylase inhibitors (HDACs) and therapeutic agents for preventing or treating malignancies associated with aberrant histone acetylation such as cancer, hematological malignancies, proliferative diseases, neurological disorders, and immunological disorders.102 These compounds were tested in vitro with respect to inhibition of histone deacetylase: human isoforms HDAC1, 2, 3, 4, 5, 6, 7, 8, 9, 10 and 11 and HeLa cell line. N-Hydroxy-7-((1,2,3,4-tetrahydroacridin-9-yl)amino)-heptanamide and 7-((2,3-dihydro-1H-cyclopenta[b]quinolin-9-yl)amino)-N-hydroxy-heptanamide showed the best activity against all histone deacetylase isoforms (IC50 = 0.01–1.57 and 0.007–1.28 μM, respectively) and against HeLa (IC50 = 0.023 and 0.019 μM, respectively).
Belmont et al. described tetrahydrocyclopenta[c]acridine derivatives as kinase inhibitors in particular for treating cancer.103 The 5-hydroxy-1-trimethylsilanyl-3,3a,4,5-tetrahydro-2H-cyclopenta[c]acridin-2-one was the most effective inhibitor against CDK1 and CDK5 with IC50 values of 0.56 and 1.6 μM, respectively. In turn, 5-hydroxy-9-methoxy-1-trimethylsilanyl-3,3a,4,5-tetrahydro-2H-cyclopenta[c]acridin-2-one showed the best activity against HT 29 (IC50 = 6.5 μM).
Singh et al. presented the synthesis of amino acid derivatives of acridone as potential leads to anticancer drugs.104 Compound (168) was prepared by the reaction of the 9-oxo-9,10-dihydroacridine-4-carboxylic acid (166) with L-valine methyl ester hydrochloride (167) in the presence of TEA and ethyl chloroformate. A similar reaction of acridone (184) with L-tyrosine methyl ester hydrochloride (169) procured derivative (170). For deprotection of the ester group, analogue (170) was treated with LiOH to give compound (171) (Scheme 28). Then, derivative (171) was reacted with L-proline methyl ester hydrochloride (172) to provide compound (173). Deprotection of the carboxyl function yielded analogue (174), which on treatment with glycine methyl ester hydrochloride (175) gave compound (176). The final product (177) was prepared by hydrolysis of derivative (176) using LiOH (Scheme 29). Similarly, the sequential incorporation of glycine (175) and proline (172) in compound (181) yielded derivative (199).104
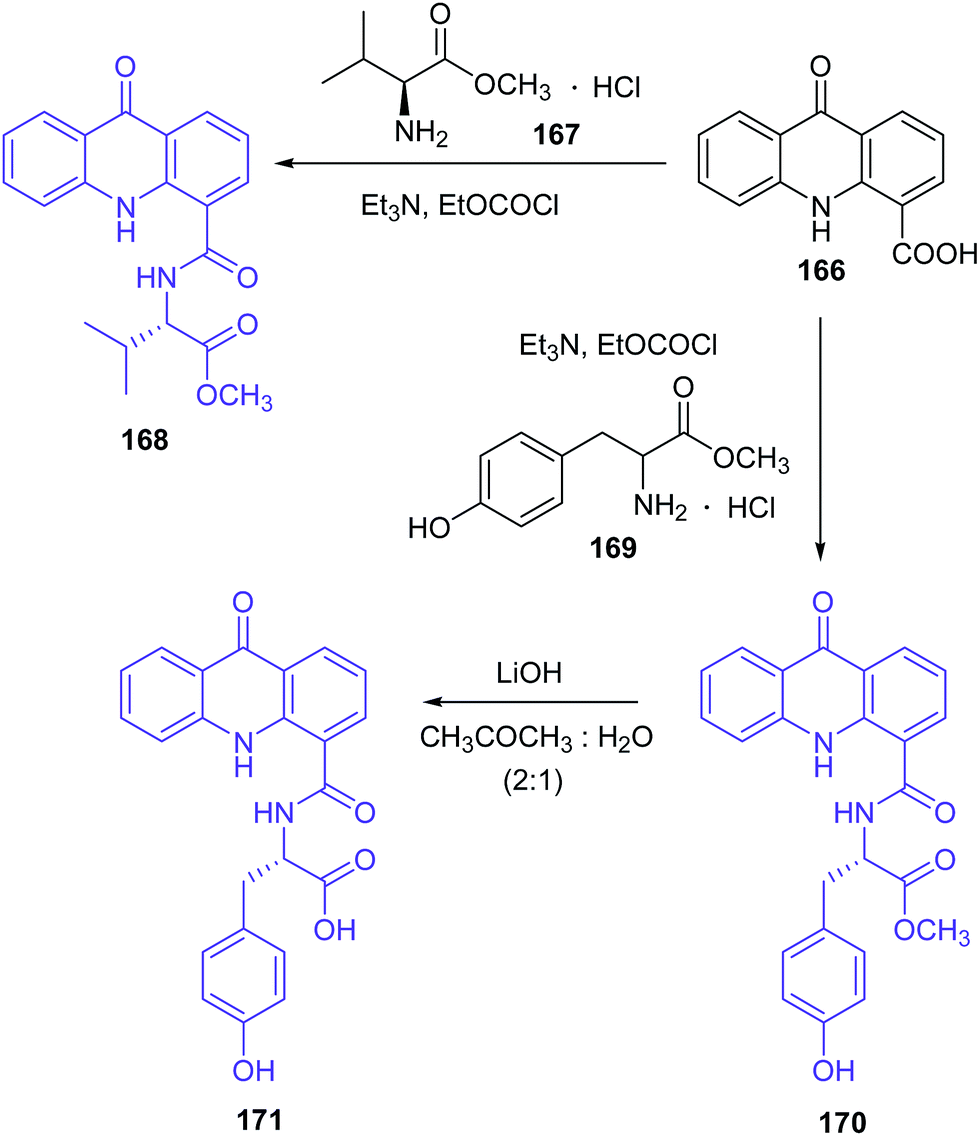 | ||
| Scheme 28 Synthesis of the amino acid derivatives of acridone (168, 170, 171).104 | ||
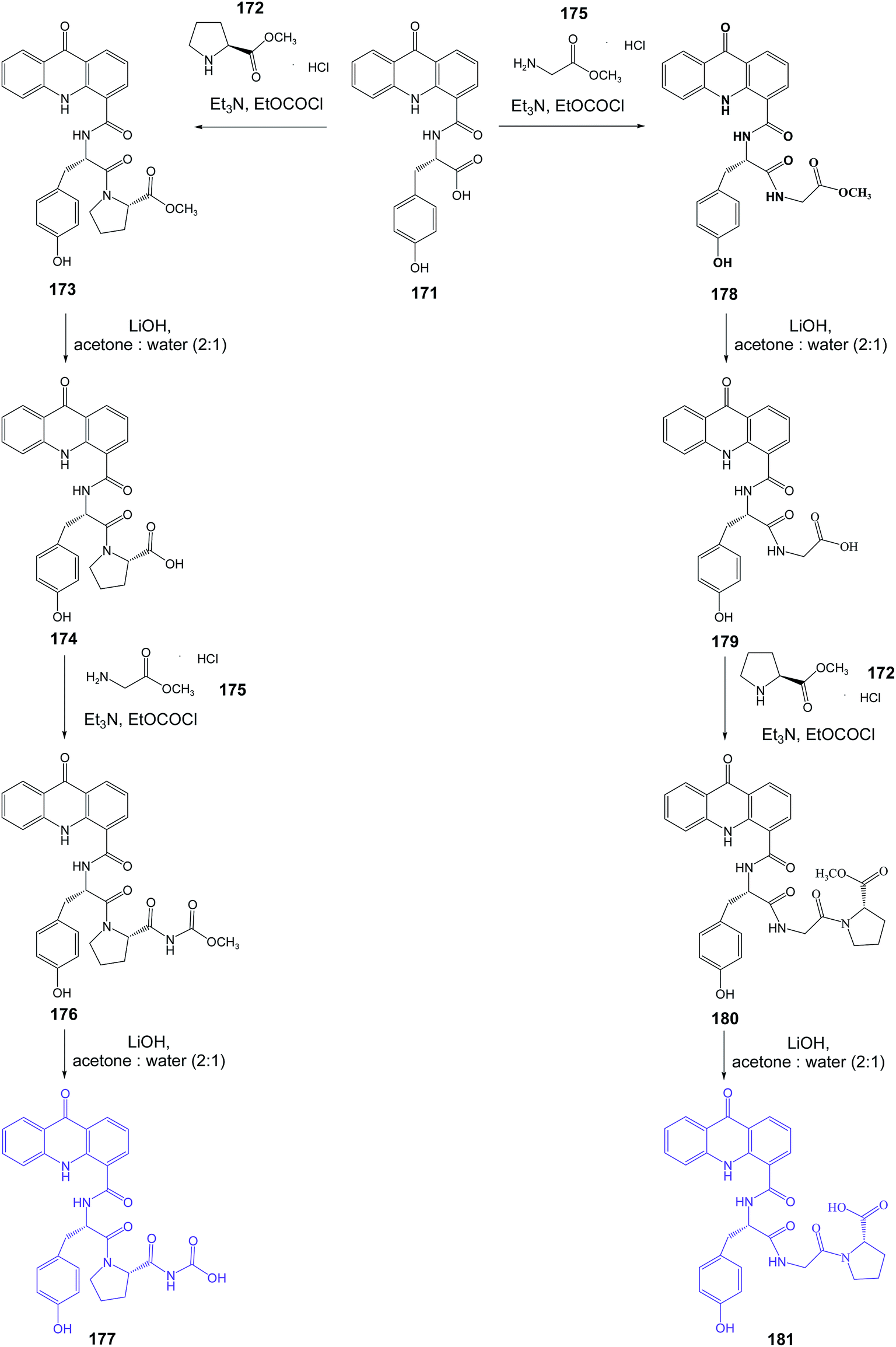 | ||
| Scheme 29 Synthesis of the dipeptide derivatives of acridone (177, 181).104 | ||
Derivatives (186, 188, 189, 195, 196 and 199) were tested for antiproliferative activity against the C6 glioblastoma cell line (IC50 = 14–20 μM range). Further, cell cycle analysis showed that compounds (195, 196 and 199) arrested C6 cells in the G0/G1 phase of the cell cycle. Moreover, analogues (195, 196 and 199) were seen to upregulate mortalin and HSP70 expression thus indicating their senescence-inducing potential.104
Zhang et al. presented the synthesis of novel pyridyl acridones as potent DNA-binding and apoptosis-inducing agents.105 Acridone-4-carboxylic acid derivatives (184a–e) were obtained by cyclization of the anthranilic acids (183a–e), which were prepared from the Ullmann condensation of 2,4-dichlorobenzoic acid (138) with various anthranilic acid derivatives (182a–e) in DMF using Cu as the catalyst (Scheme 30). Then, the reaction of compounds (184a–e) with the corresponding primary aliphatic amines in the presence of N,N′-carbonyldiimidazole (CDI) as the condensation agent gave the intermediate acridone-4-carboxamides (185a–j). Nucleophilic substitution of compounds (185a–j) with appropriate picolylamines or 4-pyridineethanamine obtained the final products (186a–w and 187a–c).105
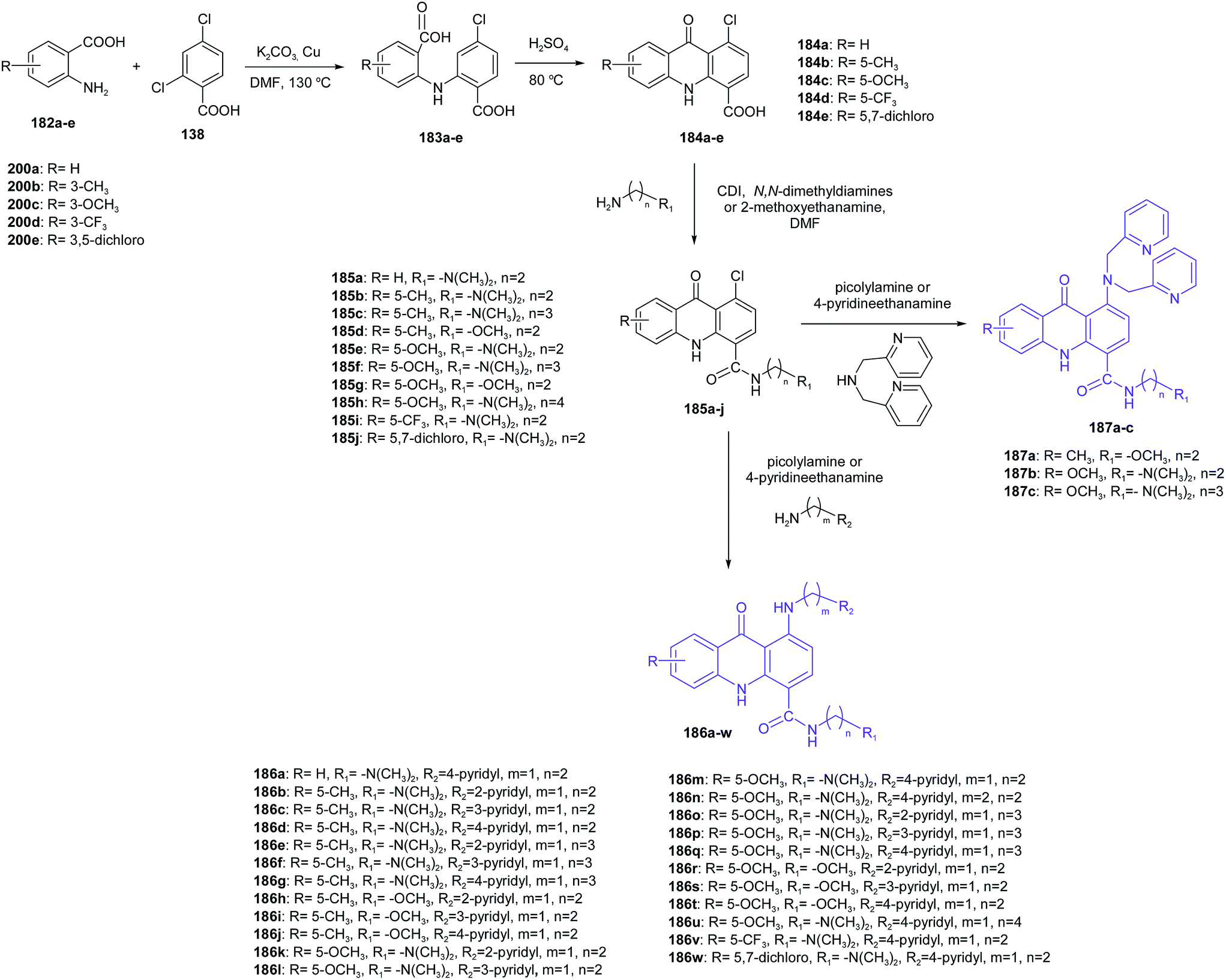 | ||
| Scheme 30 Synthesis of pyridyl acridones (186a–w, 187a–c).105 | ||
Most of the compounds (186a–w and 187a–c) exhibit good cytotoxicity against K562 cells. The best ability to inhibit K562 was shown by the compound containing a methyl group on the C5 position of acridone (186d) (IC50 = 0.46 μM). The structure–activity analysis showed that compounds containing two methylene units between the N,N-dimethylamino group and 4-carboxamide group displayed better antiproliferative activity than derivatives containing three methylene units. Moreover, analogues with an N,N-dimethylamino group on the 4-carboxamide side chain displayed better antitumour activity than compounds with a methoxy group, which showed no cytotoxicity. To evaluate whether the number of pyridyl groups has an effect on antitumour activity (IC50 = 11.2–13.9 μM range), the Zhang group introduced a 2,2′-bis pyridyl group to the acridone scaffold.105 Moreover, compound (186d) was tested for in vitro anticancer activity against cancer cell lines including NCI-H520, U251, A375, A172, HeLa, CNE-2, U118-MG, HepG-2 and MCF-7 cells. Testing results showed that (186d) has broad antitumour activities. Derivative (186d) showed the best antitumour activity against A375 and HepG-2 cells with IC50 values equal to 0.16 and 0.32 μM, respectively. Furthermore, spectrophotometric methods, viscosity measurements and topo I inhibition assays demonstrated that (186d) interacts with DNA, inhibits topo I activity and induces apoptosis through the mitochondrial pathway.105
The Gao group synthesized new derivatives of 10-(3,5-dimethoxy)benzyl-9(10H)-acridone (192a–g).106 Starting from the commercially available acridone (111) by the nitration reaction gave 2,7-dinitro-9(10H)-acridone (188), which in the presence of 3,5-dimethoxybenzyl was converted to 2,7-dinitro-10-(3,5-dimethoxy)benzyl-9(10H)-acridone (189). Then, the reduction of the nitro group by Na2S·9H2O in ethanol gave 2,7-amino-10-(3,5-dimethoxy)benzyl-9(10H)-acridone (190). Subsequently, the derivative of acridone (190) was acylated with 3-chloropropionyl chloride to obtain 2,7-bis(3-chloropropionamido)-10-(3,5-dimethoxybenzyl)-9,10-dihydroacridone (191). As a result of nucleophilic substitution of compound (191) with an appropriate amine, desirable acridone derivatives (192a–g) were synthesized (Scheme 31).106
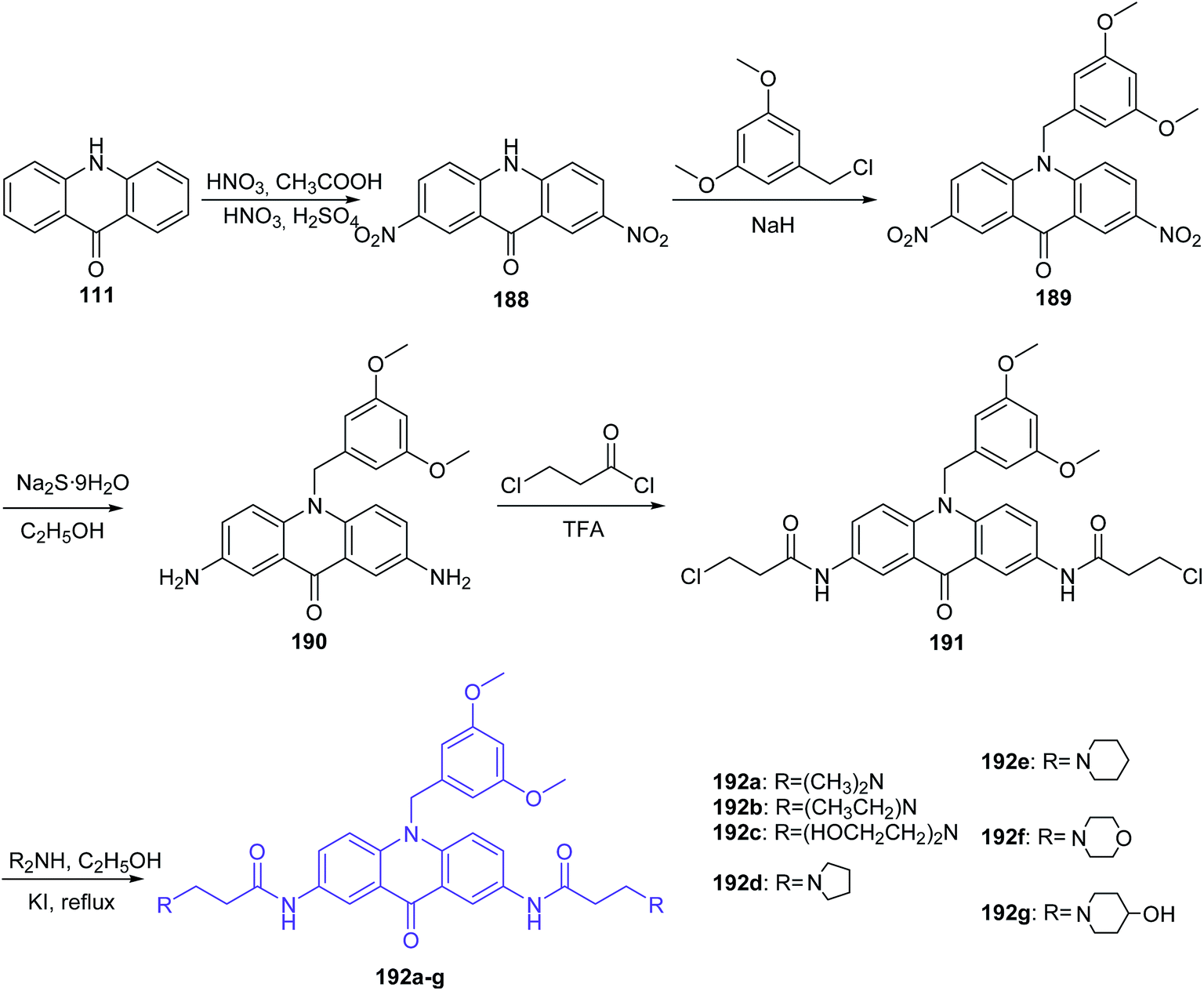 | ||
| Scheme 31 Synthesis of 10-(3,5-dimethoxy)benzyl-9(10H)-acridone derivatives (192a–g).106 | ||
The final products (192a–g) were evaluated for antiproliferative activity. The highest activity (IC50 = 0.3 μM) against leukaemia CCRF-CEM cells was shown by compound (192a), with dimethylamine substituents at the termini of the chains. Compounds (192b and 192c), containing diethylamine and diethanol amine groups, revealed a decreased inhibitory effect. Furthermore, (219a) demonstrated low toxicity against 293T cells (IC50 > 100 μM).106
In search of anticancer compounds, Gao and co-workers developed amino acid acridone analogues possessing a 3,5-dimethoxyphenyl moiety, and the most potent one was N-(10-(3,5-dimethoxybenzyl)-9,10-dihydro-9-oxoacridin-2-yl)-2-aminoacetamide hydrochloride (194) (Scheme 32).107,108 Analogue (194) exhibited significant activity against CCRF-CEM, A549, HepG-2, and MCF-7 cells together with low cytotoxicity to normal cells. The authors have more recently reported more detailed data on the mechanism of action of this compound against CCRF-CEM leukaemia cells, and 55 proteins were identified as involved in the proteomic processes. According to the results, cell death can be caused by multiple mechanisms, such as inhibition of aerobic glycolysis, mitochondrial oxidative phosphorylation, DNA damage, and oxidative stress.108 The synthetic pathway included nitration of acridone (111) followed by N-alkylation with 3,5-dimethoxybenzyl chloride and reduction to the respective amine. Then, 2-amino-10-(3,5-dimethoxy)-benzyl-9(10H)-acridone (193) was coupled with N-(tert-butoxycarbonyl)glycine in the presence of N,N′-diisopropylcarbodiimide (DIC) and 1-hydroxybenzotriazole (HOBt). In the last stage, the target compound (215) was produced in the course of treatment of the respective Boc-protected derivative with HCl.107
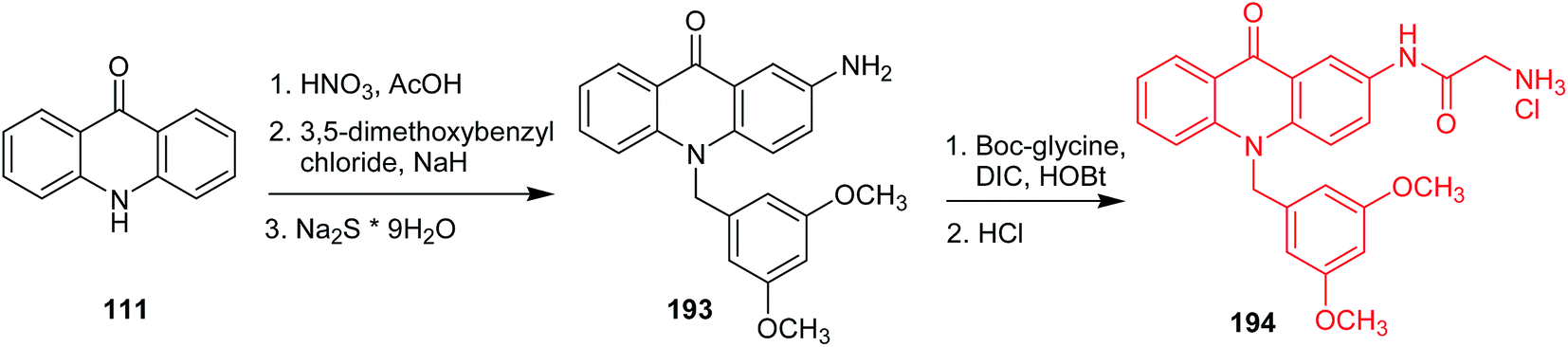 | ||
| Scheme 32 Synthesis of N-(10-(3,5-dimethoxybenzyl)-9,10-dihydro-9-oxoacridin-2-yl)-2-aminoacetamide hydrochloride (194).107 | ||
Prasad et al. presented the synthesis of nitric oxide-releasing acridone carboxamide derivatives as potential anticancer agents.109 Acridone-2-carboxylic acid (197) was prepared by cyclization of the 2,4′-iminodibenzoic acid (196), which was synthesized by the Ullmann condensation reaction of 2-chlorobenzoic acid (86) with p-aminobenzoic acid (195). Then, acridone-2-carboxamides (198a–f) were obtained by the reaction of compound (197) with an appropriate amine. In turn, derivatives (199a–d) were synthesized by stirring acridone-2-carboxamides with the alkylating agent 1-bromo-3-chloropropane or 1-bromo-4-chlorobutane in the presence of the strong base sodium hydride. Subsequently, compounds (199a–d) had introduced the nitric oxide-donating moiety by using silver nitrate in dry acetonitrile to afford the NO-donating acridone carboxamides (200a–d) (Scheme 33).109
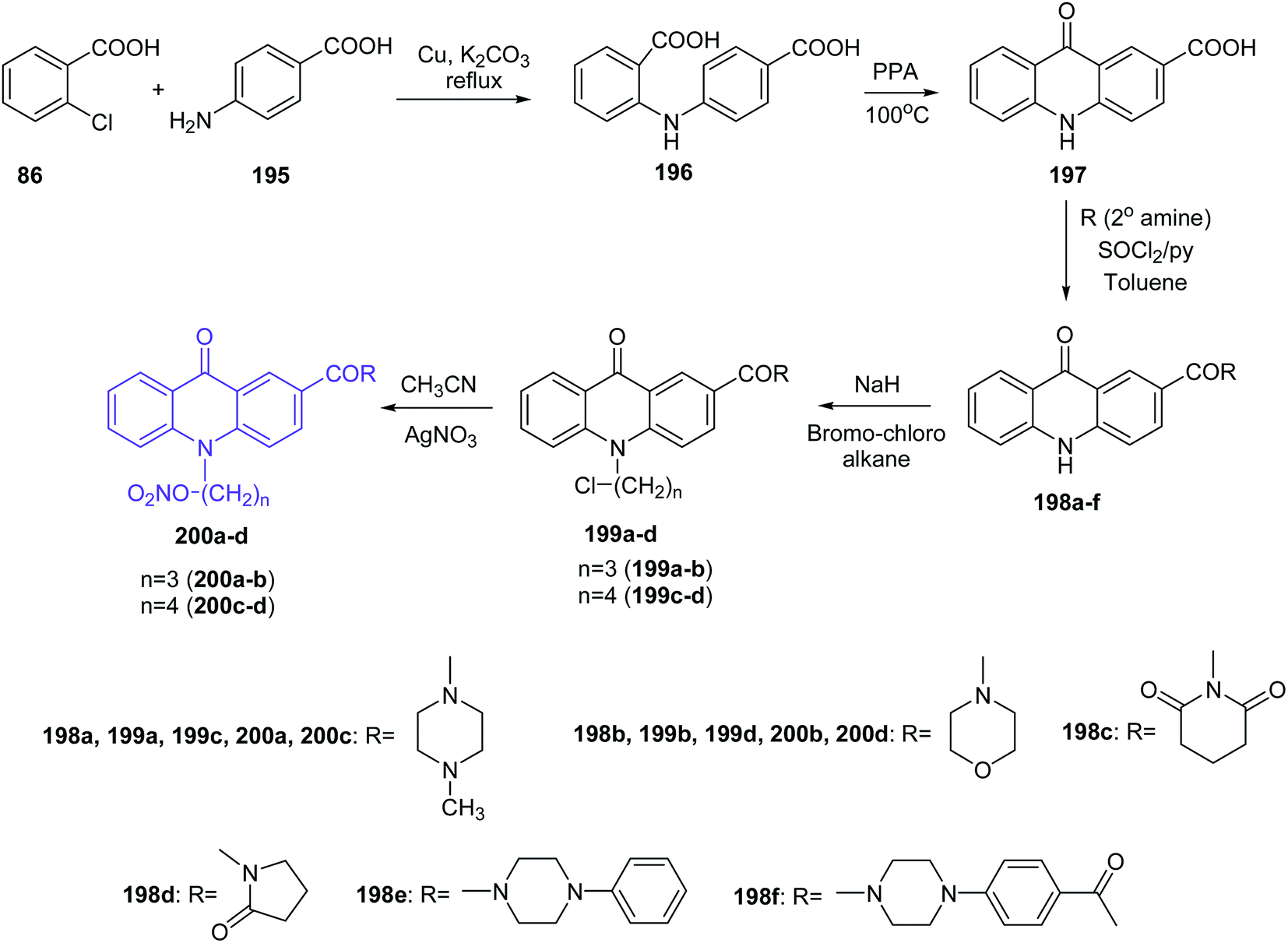 | ||
| Scheme 33 Synthesis of nitric oxide releasing acridone carboxamide derivatives (200a–d).109 | ||
All synthesized compounds were assayed for inhibition of the human breast cancer cell line MCF7 and its two drug cross-resistant phenotype sublines (P-gp and BCRP), doxorubicin-resistant MCF7/Dx (P-gp expression) and mitoxantrone-resistant MCF7/Mr (BCRP expression) cell lines. Compounds fused with NO donors (200a–d) revealed the most potent cytotoxic activity against MCF7, MCF7/Mr and MCF7/Dx cells with IC50 values varying from 0.8 μM to 1.6 μM, 1.7 μM to 2.7 μM and 1.9 μM to 4.2 μM, respectively. Moreover, derivatives (200b–d) were tested for colorectal cancer cell line (SW1398, WiDr and LS174T) activity and showed potent, selective inhibition (IC50 = 1.7–11 μM against SW1398, 2.8–19.1 μM against WiDr and 3.1–18.2 μM against LS174T).109
The Mohammadi-Khanaposhtani group presented the synthesis of new 9(10H)-acridinone-1,2,3-triazole derivatives (207a–n) against human breast cancer cell lines and apoptosis-inducing agents.110 The intermediate acridone derivatives (203) were prepared by cyclization of 2-arylaminobenzoic acids (202), which were obtained from the Ullmann condensation reaction of 2-bromobenzoic acid (37) with the corresponding aniline derivatives (201). Then, compounds (203) were converted to the 10-(prop-2-yn-1-yl)acridin-9-one derivatives (204) using propargyl bromide in the presence of potassium tert-butoxide in DMSO at room temperature. The final products (207a–n) were prepared by the reaction of compounds (204) with in situ prepared 1-(azidomethyl)-4-methoxybenzene derivatives (206) from chloride (205) (Scheme 34).110
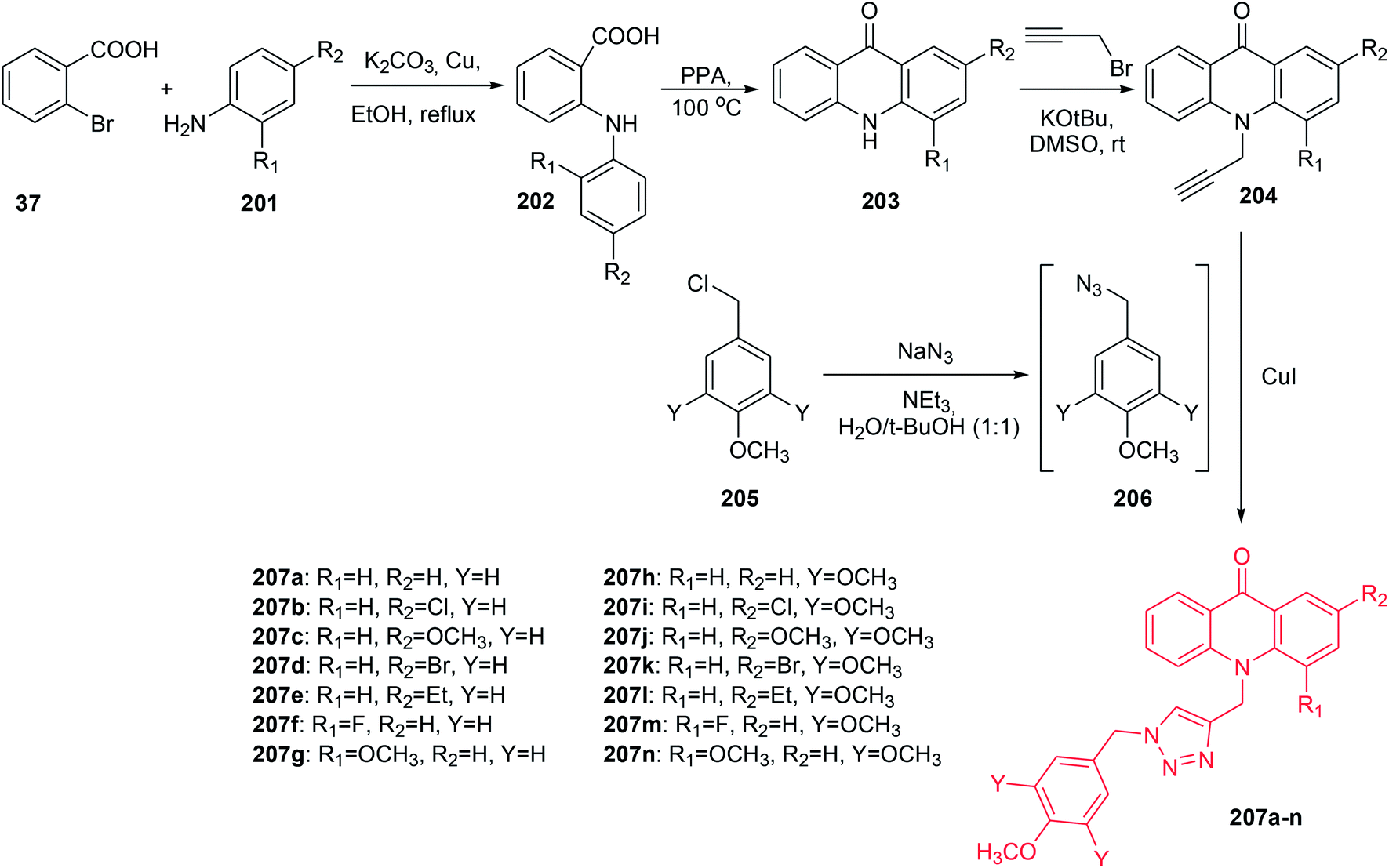 | ||
| Scheme 34 Synthesis of compounds (207a–n).110 | ||
All prepared compounds (207a–n) were assayed for inhibition of three human breast cancer cell lines, MCF-7, T-47D, and MDA-MB-231. Compounds (207a–g) with mono-methoxy substituents on the pendant benzyl group of the 1,2,3-triazole ring displayed better antitumour activity (IC50 = 11.0–60.0 μM) than derivatives (207h–n) with tri-methoxy groups, which showed no cytotoxicity (IC50 > 100 μM). Analogue (207c), with a methoxy group at the 2-position, showed the best activity against MCF-7 (IC50 = 11.0 μM), T-47D (IC50 = 14.5 μM) and MDA-MB-231 (IC50 = 16.6 μM). Moreover, acridine orange/ethidium bromide and Annexin V-FITC/propidium iodide (PI) double staining showed that derivative (207c) can induce apoptosis.110
Kumar and co-workers synthetized new N10-substituted acridone derivatives (209a–n, 210a–b, 211a–b, and 212a–b).111 Compounds (209a–n) were obtained by reaction of the appropriate alkyl halides in aqueous NaOH in the presence of TBAB and 2-butanone with acridin-9(10H)-one (111) or 2-fluoroacridin-9(10H)-one (208) (Scheme 35). Analogues (210a–b) were prepared by reaction of (111) or (208) with hydroxylamine hydrochloride and sodium acetate in water (Scheme 35), while derivatives (211a–b) were synthesized in two steps. Reacting (111) or (208) in DMF and NaH with ethyl bromoacetate produced compounds (211a–b). Further hydrolysis of (211a) or (211b) in dry ethanol, sodium hydroxide in water, and then by acidification HCl yielded derivatives (212a–b) (Scheme 35).111
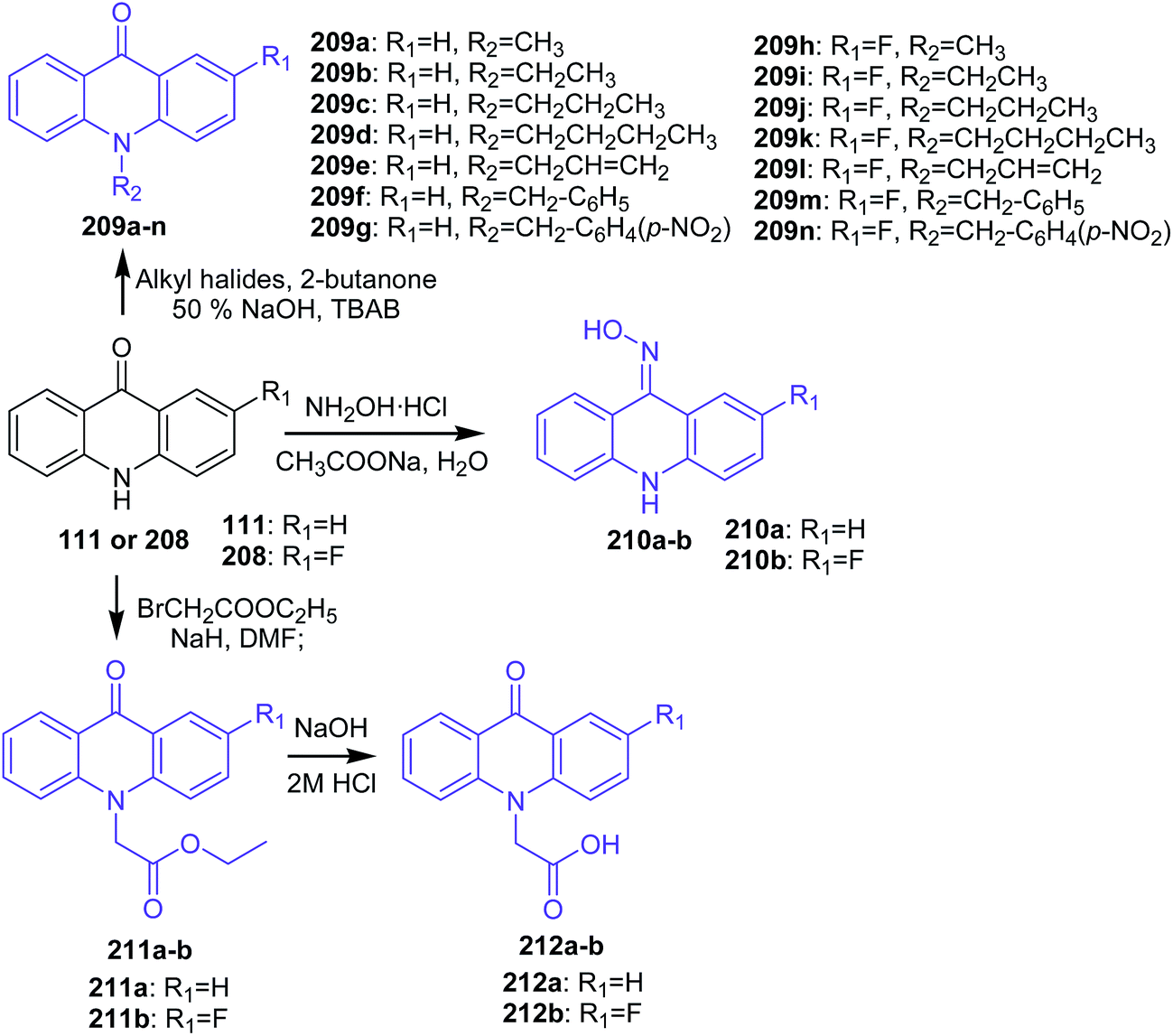 | ||
| Scheme 35 Synthesis of N10-substituted acridone derivatives (209a–n, 210a–b, 211a–b, 212a–b).111 | ||
The cytotoxic effect of analogues (209a–n, 210a–b, 211a–b, and 212a–b) on the four cell lines (MCF-7, A-549, HT-29, and HeLa) were assessed by MTT assay. Compound (209n), containing a p-nitrobenzyl group at the N10 position and a fluoro group at the C2 position, revealed good cytotoxic activity against MCF-7, A-549, and HeLa with IC50 values equal to 57.4, 43.1, and 39.5 μM, respectively. In turn, derivative (209h), with a methyl group at the N10 position of 2-fluoroacridone, showed moderate inhibitory effect, and analogues containing higher alkyl, unsaturated alkyl, ester, and oxime groups were not active. Thus, the N10 position and the C2 position of the acridone ring had a significant influence on the antiproliferative activity of the compounds. Furthermore, the authors conducted a silico study to check the multi-drug resistance modulator (MDR) effect of synthesized derivatives using docking simulations in the ATP binding site of P-gp. Derivatives (209g and 209n) showed similar interactions. These analogues (209g and 209n) showed a hydrogen bond between the carbonyl oxygen at the 9th position of the acridone ring with Tyr1393 and a π–π stacking interaction between the benzyl ring substituted at the 10th position of the acridone moiety with Tyr2352. Furthermore, the nitro group of (209g and 209n) also showed weak non-covalent interactions with positively charged magnesium ion (Mg2+). Analogues (209f, 209g, 209m, 209n, and 212a–b) showed considerable interactions into the ATP binding site of P-gp and can therefore be considered to display MDR modulatory activity as well.111
Kumar et al. presented the synthesis of 2-(9-oxoacridin-10(9H)-yl)-N-phenyl acetamides.112 2-(9-Oxoacridin-10(9H)-yl)-N-phenylacetamide derivatives (215a–o) were prepared by the condensation reaction of acridone (111) with 2-chloro-N-phenylacetamides (214a–o) in the presence of sodium hydride in DMF at reflux (Scheme 36).112
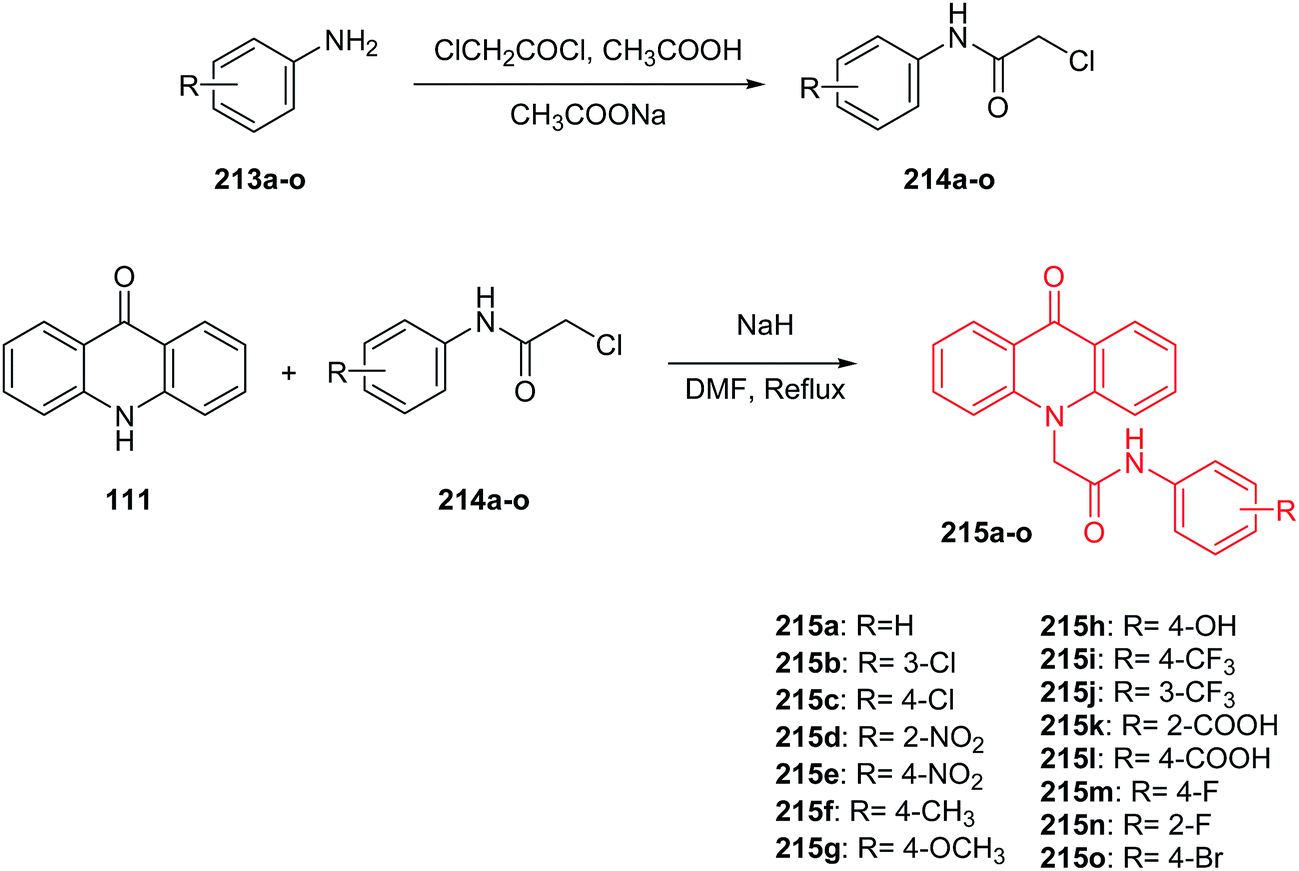 | ||
| Scheme 36 Synthesis of intermediate compounds (214a–o) and final target compounds (215a–o).112 | ||
Compounds (215a–o) were tested for in vitro anticancer activity against three human cancer cell lines (MCF-7, HeLa, and A-549). Derivatives a with carboxylic group at position 2 or 4 of the phenyl ring (215k and 215l) showed the greatest inhibition ability against MCF-7 (IC50 = 6.07 and 7.12 μM, respectively), HeLa (IC50 = 8.80 and 10.66 μM, respectively) and A-549 (IC50 = 7.39 and 8.80 μM, respectively). Moreover, studies performed showed that analogues (215k, 215l, 215d, 215e and 215h), bearing electron-withdrawing groups (–COOH, –NO2, –OH), exhibited better cytotoxic activity compared to unsubstituted (215a) or compound (215f) bearing electron-releasing groups (–CH3). Furthermore, the computational analysis showed that all prepared products display significant interactions with the ATP binding site, and they may therefore serve as MDR modulators.112
Conclusion
Bacterial, parasitic and viral infections and above all neoplastic diseases are still a serious challenge for scientists. Acridine/acridone analogues constitute a group of compounds with antibacterial, antiparasitic, anti-malarial, anti-HIV and antitumour properties. However, the clinical usefulness of acridine/acridone derivatives is limited due to the risk of high toxicity and tumour resistance. Recent research has opened new directions for the design and development of new and more potent inhibitors of acetylcholinesterase, inhibitors of human carbonic anhydrase isozymes and antibacterial, antiparasitic and anticancer drugs.113–117The described compounds showed potent activity against hAChE (IC50 = 3–8 nM) and their derivatives against hBChE (IC50 = 0.4–20 nM). These analogues were more active than the positive control tacrine,46 while compound (44g) (Scheme 2) showed activity against AChE with an IC50 = 7.31 μM.49
Synthesized acridine derivatives have exhibited good results in activity against isoforms (hCA I, II and IV).61 Also described are acridine derivatives as antimalarial agents.68 A novel inhibitor of M. tuberculosis DNA gyrase was prepared that showed good activity with IC50 values in the range of 5.2 to 33.9 μM.70 Derivative (100k, Scheme 14) exhibited more effective activity against S. enteritidis than erythromycin,72 while derivative (103b, Scheme 15) exhibited higher activity against Candida albicans than Rivanol.73
Compounds (121a, 136l, 143e, 146c, 158b, 158f, 158i, 158j, 186d) showed promising antitumour activity against HepG-2 (ref. 15, 30, 85, 87, 95 and 105) Additionally, derivatives (136l, 143e, 146c, 186d) exhibited good activity results against K562 through binding with DNA and inhibiting topo I activity.30,85,87,105 Furthermore, analogue (192a) showed the highest activity (IC50 = 0.3 μM) against leukaemia CCRF-CEM cells.89 Derivatives (153a–d, Scheme 24) showed the best activity against A549 (IC50 = 0.1–0.5 μM)80 and (200a–d) (Scheme 33) revealed the most potent cytotoxic activity against MCF7 (IC50 = 0.8–1.6 μM).92
PEGylated polyacridine peptides are considered as non-viral gene delivery systems.118 Khargharia et al. reported synthesis of DNA binding polyacridine peptides followed by their in vivo gene expression in mice. Length of polyethylene glycol (PEG) moiety influenced DNA polyplexes administration dramatically, whereas its location occurred to be not so crucial. Investigation of the relationship between structure of PEGylated polyacridine peptides and pharmacokinetics, biodistribution and metabolism can lead to novel compounds in gene therapy.
Thus, the acridine/acridone moiety can still serve as a lead structure and can be further modified to increase the specificity of new analogues towards tumour cells and bacterial, parasitic and viral infections.
Acknowledgements
This work was financially supported by the National Science Center (Poland), grant no. 2014/13/B/NZ7/02234.References
- C. D. Geddes, Dyes Pigm., 2000, 45, 243 CrossRef CAS.
- Y. L. Chen, C. M. Lu, I. L. Chen, L. T. Tsao and J. P. Wang, J. Med. Chem., 2002, 45, 4689 CrossRef CAS PubMed.
- L. V. Yartseva, S. G. Isayev and O. M. Svechnikova, Farm Zh., 2003, 3, 60 Search PubMed.
- S. A. Gamega, J. A. Spicer, G. J. Atwell, G. J. Finlay, B. C. Baguley and W. A. Denny, J. Med. Chem., 1999, 42, 2383 CrossRef PubMed.
- M. Kaya, Y. Yildirir and G. Y. Celik, Med. Chem. Res., 2011, 20, 293 CrossRef CAS.
- E. I. Aly and A. H. Abadi, Arch. Pharmacal Res., 2004, 27, 713 CrossRef CAS.
- R. P. Tripathi, S. S. Verma, J. Pandey, K. C. Agarwal, V. Chaturvedi, Y. K. Manju, A. K. Srivastva, A. Gaikwad and S. Sinha, Bioorg. Med. Chem. Lett., 2006, 16, 5144 CrossRef CAS PubMed.
- C. Di Giorgio, M. De Meo, J. Chiron, F. Delmas, A. Nikoyan, S. Jean, G. Dumenil, P. Timon-David and J. P. Galy, Bioorg. Med. Chem., 2005, 13, 5560 CrossRef CAS PubMed.
- A. Kumar, K. Srivastava, S. R. Kumar, S. K. Puri and M. S. Chauhan, Bioorg. Med. Chem. Lett., 2009, 19, 6996 CrossRef CAS PubMed.
- V. Tomar, G. Bhattacharjee, Kamaluddin, S. Rajakumar, K. Srivastava and S. K. Puri, Eur. J. Med. Chem., 2010, 45, 745 CrossRef CAS PubMed.
- X. M. Yu, F. Ramiandrasoa, L. Guetzoyan, B. Pradines, E. Quintino, D. Gadelle, P. Forterre, T. Cresteil, J. P. Mahy and S. Pethe, ChemMedChem, 2012, 7, 587 CrossRef CAS PubMed.
- H. C. Gupta and V. Jaiswal, Indian J. Heterocycl. Chem., 2010, 19, 409 CAS.
- M. Tonelli, G. Vettoretti, B. Tasso, F. Novelli, V. Boido, F. Sparatore, B. Busonera, A. Ouhtit, P. Farci, S. Blois, G. Giliberti and P. La Colla, Antiviral Res., 2011, 91, 133 CrossRef CAS PubMed.
- A. Srivastava and A. Nizamuddin, Indian J. Heterocycl. Chem., 2004, 13, 261 CAS.
- S. Arya, A. Kumar, N. Kumar, P. Roy and S. M. Sondhi, Med. Chem. Res., 2015, 24, 1942 CrossRef CAS.
- K. Niknam and M. Damya, J. Chin. Chem. Soc., 2009, 56, 659 CrossRef CAS.
- M. A. Arai, K. Koryudzu and M. Ishibashi, J. Nat. Prod., 2015, 78, 311 CrossRef CAS PubMed.
- M. Demeunynck, F. Charmantray and A. Martelli, Curr. Pharm. Des., 2001, 7, 1703 CrossRef CAS PubMed.
- G. Minotti, P. Menna, E. Salvatorelli, G. Cairo and L. Gianni, Pharmacol. Rev., 2004, 56, 185 CrossRef CAS PubMed.
- B. C. Baguley, L. P. Wakelin, J. D. Jacintho and P. Kovacic, Curr. Med. Chem., 2003, 10, 2643 CrossRef CAS PubMed.
- A. Raza, B. A. Jacobson, A. Benoit, M. R. Patel, J. Jay-Dixon, H. Hiasa, D. M. Ferguson and R. A. Kratzke, Invest. New Drugs, 2012, 30, 1443 CrossRef CAS PubMed.
- J. Salimon, N. Salih, E. Yousif, A. Hameed and A. Kreem, Arabian J. Chem., 2010, 3, 205 CrossRef CAS.
- C. Gao, S. Li, X. Lang, H. Liu, F. Liu, C. Tan and Y. Jiang, Tetrahedron, 2012, 68, 7920 CrossRef CAS.
- F. Cuenca, M. J. B. Moore, K. Johnson, B. Guyen, A. De Cian and S. Neidle, Bioorg. Med. Chem. Lett., 2009, 19, 5109 CrossRef CAS PubMed.
- R. Zittoun, Eur. J. Cancer Clin. Oncol., 1985, 21, 649 CrossRef CAS PubMed.
- T. L. Su, T. C. Chou, J. Y. Kim, J. T. Huang, G. Ciszewska, W. Y. Ren, G. M. Otter, F. M. Sirotnak and K. A. Watanabe, J. Med. Chem., 1995, 38, 3226 CrossRef CAS PubMed.
- I. Gonzalez-Sanchez, J. D. Solano, M. A. Loza-Mejia, S. Olvera-Vázquez, R. Rodríguez-Sotres, J. Morán, A. Lira-Rocha and M. A. Cerbón, Eur. J. Med. Chem., 2011, 46, 2102 CrossRef CAS PubMed.
- C. H. Chen, Y. W. Lin, X. G. Zhang, T. C. Chou, T. J. Tsai, N. Kapuriya, R. Kakadiya and T. L. Su, Eur. J. Med. Chem., 2009, 44, 3056 CrossRef CAS PubMed.
- X. D. Luan, C. M. Gao, N. N. Zhang, Y. Chen, Q. Sun, C. Tan, H. Liu, Y. Jin and Y. Jiang, Bioorg. Med. Chem., 2011, 19, 3312 CrossRef CAS PubMed.
- X. Lang, L. Li, Y. Chen, Q. Sun, Q. Wu, F. Liu, C. Tan, H. Liu, C. Gao and Y. Jiang, Bioorg. Med. Chem., 2013, 21, 4170 CrossRef CAS PubMed.
- M. Wera, P. Storoniak, I. E. Serdiuk and B. Zadykowicz, J. Mol. Struct., 2016, 1105, 41 CrossRef CAS.
- P. Kittakoop, C. Mahidol and S. Ruchirawat, Curr. Top. Med. Chem., 2014, 14, 239 CrossRef CAS PubMed.
- G. Cholewiński, K. Dzierzbicka and A. M. Kołodziejczyk, Pharmacol. Rep., 2011, 63, 305 CrossRef.
- H. K. H. Fong and B. R. Copp, Mar. Drugs, 2013, 11, 274 CrossRef CAS PubMed.
- P. Belmont, J. Bosson, T. Godet and M. Tiano, Anti-Cancer Agents Med. Chem., 2007, 7, 139 CrossRef CAS PubMed.
- V. Wouatsa, L. Misra, S. Kumar, O. Prakash, F. Khan, F. Tchoumbougnang and R. K. Venkatesh, Chem. Cent. J., 2013, 7, 125 CrossRef PubMed.
- M. A. Beniddir, E. Le Borgne, B. I. Iorga, N. Loaec, O. Lozach, L. Meijer, K. Awang and M. Litaudon, J. Nat. Prod., 2014, 77, 1117 CrossRef CAS PubMed.
- P. H. A. Ferreira, D. A. P. dos Santos, M. F. G. F. da Silva, P. C. Vieira, B. King-Diaz, B. Lotina-Hennsen and T. A. M. Veiga, Chem. Biodiversity, 2016, 13, 100 Search PubMed.
- V. I. Teow, US Pat., 0088137, 2014.
- N. C. Berchtold and C. W. Cotman, Neurobiol. Aging, 1998, 19, 173 CrossRef CAS PubMed.
- C. Ballard, S. Gauthier, A. Corbett, C. Brayne, D. Aarsland and E. Jones, Lancet, 2011, 377, 1019 CrossRef.
- R. E. Becker, N. H. Greig, E. Giacobini, L. S. Schneider and L. Ferrucci, Nat. Rev. Drug Discovery, 2014, 13, 156 CrossRef CAS PubMed.
- M. I. Rodriguez-Franco, M. I. Fernandez-Bachiller, C. Perez, B. Hernandez-Ledesema and B. Bartolome, J. Med. Chem., 2006, 49, 459 CrossRef CAS PubMed.
- L. S. Schneider, Dialogues Clin Neurosci., 2000, 2, 111 Search PubMed.
- T. Fuchigami, N. Kobashi, M. Haratake, M. Kawasaki and M. Nakayama, Eur. J. Med. Chem., 2013, 60, 469 CrossRef CAS PubMed.
- S. Hamulakova, J. Imrich, L. Janovec, P. Kristian, I. Danihel, O. Holas, M. Pohanka, S. Böhm, M. Kozurkova and K. Kuca, Int. J. Biol. Macromol., 2014, 70, 435 CrossRef CAS PubMed.
- J. Janockova, J. Plsikova, J. Kasparkova, V. Brabec, R. Jendzelovsky, J. Mikes, J. Koval, S. Hamulakova, P. Fedorocko, K. Kuca and M. Kozurkova, Eur. J. Pharm. Sci., 2015, 76, 192 CrossRef CAS PubMed.
- S. Thiratmatrakul, C. Yenjai, P. Waiwut, O. Vajragupta, P. Reubroycharoen, M. Tohda and C. Boonyarat, Eur. J. Med. Chem., 2014, 75, 21 CrossRef CAS PubMed.
- M. Mohammadi-Khanaposhtani, M. Saeedi, N. S. Zafarghandi, M. Mahdavi, R. Sabourian, E. K. Razkenari, H. Alinezhad, M. Khanavi, A. Foroumadi, A. Shafiee and T. Akbarzadeh, Eur. J. Med. Chem., 2015, 92, 799 CrossRef CAS PubMed.
- M. Mohammadi-Khanaposhtani, M. Mahdavi, M. Saeedi, R. Sabourian, M. Safavi, M. Khanavi, A. Foroumadi, A. Shafiee and T. Akbarzadeh, Chem. Biol. Drug Des., 2015, 86, 1425 CAS.
- M. Mohammadi-Khanaposhtani, M. Shabani, M. Faizi, I. Aghaei, R. Jahani, Z. Sharafi, N. S. Zafarghandi, M. Mahdavi, T. Akbarzadeh, S. Emami, A. Shafiee and A. Foroumadi, Eur. J. Med. Chem., 2016, 112, 91 CrossRef CAS PubMed.
- C. T. Supuran, Nat. Rev. Drug Discovery, 2008, 7, 168 CrossRef CAS PubMed.
- C. T. Supuran and A. Scozzafava, Bioorg. Med. Chem., 2007, 15, 4336 CrossRef CAS PubMed.
- J. Y. Winum, D. Vullo, A. Casini, J. L. Montero, A. Scozzafava and C. T. Supuran, J. Med. Chem., 2003, 46, 2197 CrossRef CAS PubMed.
- M. Senturk, I. Gulcin, S. Beydemir, O. I. Kufrevioglu and C. Supuran, Chem. Biol. Drug Des., 2011, 77, 494 CAS.
- S. B. O. Sarikaya, F. Topal, M. Senturk, I. Gulcin and C. T. Supuran, Bioorg. Med. Chem. Lett., 2011, 21, 4259 CrossRef PubMed.
- C. T. Supuran and A. Scozzafava, Expert Opin. Ther. Pat., 2002, 12, 217 CrossRef CAS.
- S. B. O. Sarikaya, I. Gulcin and C. T. Supuran, Chem. Biol. Drug Des., 2010, 75, 515 CAS.
- I. Yesildag, R. Ulus, E. Basar, M. Aslan, M. Kaya and M. Bulbul, Monatsh. Chem., 2014, 145, 1027 CrossRef CAS.
- I. Esirden, R. Ulus, B. Aday, M. Tanç, C. T. Supuran and M. Kaya, Bioorg. Med. Chem., 2015, 23, 6573 CrossRef CAS PubMed.
- R. Ulus, I. Yesildag, M. Tanc, M. Bulbul, M. Kaya and C. T. Supuran, Bioorg. Med. Chem., 2013, 21, 5799 CrossRef CAS PubMed.
- M. O. Anderson, J. Sherrill, P. B. Madrid, A. P. Liou, J. L. Weisman, J. L. DeRisi and K. Guy, Bioorg. Med. Chem., 2006, 14, 334 CrossRef CAS PubMed.
- E. T. Glaz, E. Szolgay, I. Stoger and M. Talas, Antimicrob. Agents Chemother., 1973, 3, 537 CrossRef CAS PubMed.
- W. A. Denny, Curr. Med. Chem., 2002, 9, 1655 CrossRef CAS PubMed.
- S. Dana, D. Prusty, D. Dhayal, M. Kumar Gupta, A. Dar, S. Sen, P. Mukhopadhyay, T. Adak and S. Kumar Dhar, Chem. Biol., 2014, 9, 2366 CAS.
- . P. K. Chiang, US Pat., 0005650, 2013.
- K. Lee, H. Zhang, D. Z. Qian, S. Rey, J. O. Liu and G. L. Semenza, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17910 CrossRef CAS PubMed.
- J. P. Joubert, F. J. Smit, L. du Plessis, P. J. Smith and D. D. N'Da, Eur. J. Pharm. Sci., 2014, 56, 16 CrossRef CAS PubMed.
- A. Bharathi, S. M. Roopan, A. A. Rahuman and G. Rajakumar, J. Photochem. Photobiol., B, 2014, 140, 359 CrossRef CAS PubMed.
- B. Medapi, N. Meda, P. Kulkarni, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem., 2016, 24, 877 CrossRef CAS PubMed.
- G. C. Muscia, G. Y. Buldain and E. A. Asís, Eur. J. Med. Chem., 2014, 73, 243 CrossRef CAS PubMed.
- M. Kaya, Y. Yildirir and G. Y. Celik, Pharm. Chem. J., 2015, 48, 722 CrossRef CAS.
- Y. D. Markovich, T. N. Kudryavtseva, K. V. Bogatyrev, P. I. Sysoev, L. G. Klimova and G. V. Nazarov, Russ. Chem. Bull., 2014, 63, 1153 CrossRef CAS.
- A. Stankiewicz-Drogon, L. G. Palchykovska, V. G. Kostina, I. V. Alexeeva, A. D. Shved and A. M. Boguszewska-Chachulska, Bioorg. Med. Chem., 2008, 16, 8846 CrossRef CAS PubMed.
- A. Boumendjel, S. Macalou, A. Ahmed-Belkacem, M. Blanca and A. D. Pietrob, Bioorg. Med. Chem., 2007, 15, 2892 CrossRef CAS PubMed.
- P. Singh, J. Kaur, B. Yadav and S. S. Komath, Bioorg. Med. Chem., 2009, 17, 3973 CrossRef CAS PubMed.
- A. Bhardwaj, J. Kaur, S. K. Sharma, Z. Huang, F. Wuest and E. E. Knaus, Bioorg. Med. Chem. Lett., 2013, 23, 163 CrossRef CAS PubMed.
- J. Joseph, E. Kuruvilla, A. T. Achuthan, D. Ramaiah and G. B. Schuster, Bioconjugate Chem., 2004, 15, 1230 CrossRef CAS PubMed.
- P. Belmont and I. Dorange, Expert Opin. Ther. Pat., 2008, 18, 1211 CrossRef CAS.
- A. Souibgui, A. Gaucher, J. Marrot, F. Bourdreux, F. Aloui, B. B. Hassine and D. Prim, Tetrahedron, 2014, 70, 3042 CrossRef CAS.
- K. Dzierzbicka, A. Kawzowicz, A. Koc and M. Kukowska-Kaszuba, Wiad. Chem., 2009, 63, 107 CAS.
- M. Gardette, C. Viallard, S. Paillas, J. L. Guerquin-Kern, J. Papon, N. Moins, P. Labarre, N. Desbois, P. Wong-Wah-Chung, S. Palle, T. D. Wu, J. P. Pouget, E. Miot-Noirault, J. M. Chezal and F. Degoul, Invest. New Drugs, 2014, 32, 587 CrossRef CAS PubMed.
- G. R. Stark and K. T. Dermawan, US Pat., 0066465 A1, 2014.
- O. M. Salem, M. Vilková, J. Janočková, R. Jendželovský, P. Fedoročko, E. Žilecká, J. Kašpárková, V. Brabec, J. Imrich and M. Kožurková, Int. J. Biol. Macromol., 2016, 86, 690 CrossRef CAS PubMed.
- C. Gao, B. Li, B. Zhang, Q. Sun, L. Li, X. Li, C. Chen, C. Tan, H. Liu and Y. Jiang, Bioorg. Med. Chem., 2015, 23, 1800 CrossRef CAS PubMed.
- G. Gellerman, Eur. Pat., 2493857, 2012.
- B. Li, C. M. Gao, Q. S. Sun, L. L. Li, C. Y. Tan, H. X. Liu and Y. Y. Jiang, Chin. Chem. Lett., 2014, 25, 1021 CrossRef CAS.
- S. Roe, M. Gunaratnam, C. Spiteri, P. Sharma, R. D. Alharthy, S. Neidle and J. E. Moses, Org. Biomol. Chem., 2015, 13, 8500 CAS.
- A. Cisáriková, Z. Barbieriková, L. Janovec, J. Imrich, L. Hunáková, Z. Bačová and H. Paulíková, Bioorg. Med. Chem., 2016, 24, 2011 CrossRef PubMed.
- J. Plsikova, L. Janovec, J. Koval, J. Ungvarsky, J. Mikes, R. Jendzelovsky, P. Fedorocko, J. Imrich, P. Kristian, J. Kasparkova, V. Brabec and M. Kozurkova, Eur. J. Med. Chem., 2012, 57, 283 CrossRef CAS PubMed.
- L. Huo, R. Chen, J. Li and P. Li, CN. Pat., 105399740, 2016.
- L. Huo, R. Chen, P. Li and W. Su, CN. Pat., 104326979, 2015.
- L. Huo, R. Chen, P. Li, W. Su and L. X. Li, CN. Pat., 105399671, 2016.
- L. Huo, R. Chen, P. Li, W. Su and M.-H. Huang, CN. Pat., 105418501, 2016.
- K. B. Ramesh and M. A. Pasha, Bioorg. Med. Chem. Lett., 2014, 24, 3907 CrossRef CAS PubMed.
- M. M. Khalaf and A. A. Abdelhamid, Catal. Lett., 2016, 146, 645 CrossRef CAS.
- B. V. S. Reddy, A. Antony and J. S. Yadav, Tetrahedron Lett., 2010, 51, 3071 CrossRef CAS.
- M. A. Mahi, S. M. Mekelleche, W. Benchouk, M. J. Aurell and L. R. Domingo, RSC Adv., 2016, 6, 15759 RSC.
- F.-M. Wang, D. Bao, B.-X. Hu, Z.-Y. Zhou, D.-D. Huang, L.-Z. Chen and Y.-Y. Dan, J. Heterocycl. Chem., 2017, 54, 784 CrossRef CAS.
- X. Wang, L. Yang, N. Li, Y. Zhang, W. Ma and L. Wu, CN. Pat., 104530062, 2015.
- A. J. Pickard, F. Liu, T. F. Bartenstein, L. G. Haines, K. E. Levine, G. L. Kucera and U. Bierbach, Chem.–Eur. J., 2014, 20, 16174 CrossRef CAS PubMed.
- F. P. Cossio Mora, Y. I. Vara Salazar, M. D. C. Masdeu Margalef, M. R. Alcala Caffarena, S. Villafruela Caneva, D. Otaegui Ansa, E. Aldaba Arevalo, A. Zubia Olascoaga and E. San Sebastian Larzabal, Eur. Pat., 2801569, 2014.
- P. O. Belmont, L. Meijer, P. Cohen, A. Patin, J. Bosson and P. G. Goekjian, US Pat., 8999955, 2015.
- P. Singh, A. Kumar, A. Sharma and G. Kaur, Bioorg. Med. Chem. Lett., 2015, 25, 3854 CrossRef CAS PubMed.
- B. Zhang, K. Chen, N. Wang, C. Gao, Q. Sun, L. Li, Y. Chen, C. Tan, H. Liu and Y. Jiang, Eur. J. Med. Chem., 2015, 93, 214 CrossRef CAS PubMed.
- C. Gao, W. Zhang, S. He, S. Li, F. Liu and Y. Jiang, Bioorg. Chem., 2015, 60, 30 CrossRef CAS PubMed.
- C. Gao, F. Liu, X. Luan, C. Tan, H. Liu, Y. Xie, Y. Jin and Y. Jiang, Bioorg. Med. Chem., 2010, 18, 7507 CrossRef CAS PubMed.
- Y. Wang, D. Gao, B. Chu, C. Gao, D. Cao, H. Liu and Y. Jiang, Proteomics, 2016, 16, 1177 CrossRef CAS PubMed.
- V. V. S. R. Prasad, G. D. Reddy, I. Kathmann, M. Amareswararao and G. J. Peters, Bioorg. Chem., 2016, 64, 51 CrossRef PubMed.
- M. Mohammadi-Khanaposhtani, M. Safavi, R. Sabourian, M. Mahdavi, M. Pordeli, M. Saeedi, S. K. Ardestani, A. Foroumadi, A. Shafiee and T. Akbarzadeh, Mol. Diversity, 2015, 19, 787 CrossRef CAS PubMed.
- R. Kumar, M. S. Bahia and O. Silakari, Med. Chem. Res., 2015, 24, 921 CrossRef CAS.
- R. Kumar, M. Kaur, M. S. Bahia and O. Silakari, Eur. J. Med. Chem., 2014, 80, 83 CrossRef CAS PubMed.
- M. Kukowska-Kaszuba and K. Dzierzbicka, Curr. Med. Chem., 2007, 14, 3079 CrossRef CAS PubMed.
- M. Kukowska-Kaszuba, K. Dzierzbicka, M. Serocki and A. Skladanowski, J. Med. Chem., 2011, 54, 2447 CrossRef CAS PubMed.
- K. Dzierzbicka, A. M. Kołodziejczyk, B. Wysocka-Skrzela, A. Mysliwski and D. Sosnowska, J. Med. Chem., 2001, 44, 3606 CrossRef CAS PubMed.
- K. Dzierzbicka and A. M. Kołodziejczyk, J. Med. Chem., 2003, 46, 183 CrossRef CAS PubMed.
- M. Malachowska-Ugarte, G. Cholewinski, K. Dzierzbicka and P. Trzonkowski, Eur. J. Med. Chem., 2012, 54, 197 CrossRef CAS PubMed.
- S. Khargharia, K. Kizzire, M. D. Ericson, N. J. Baumhover and K. G. Rice, J. Controlled Release, 2013, 170, 325 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |